
Submitted:
04 June 2025
Posted:
04 June 2025
You are already at the latest version
Abstract
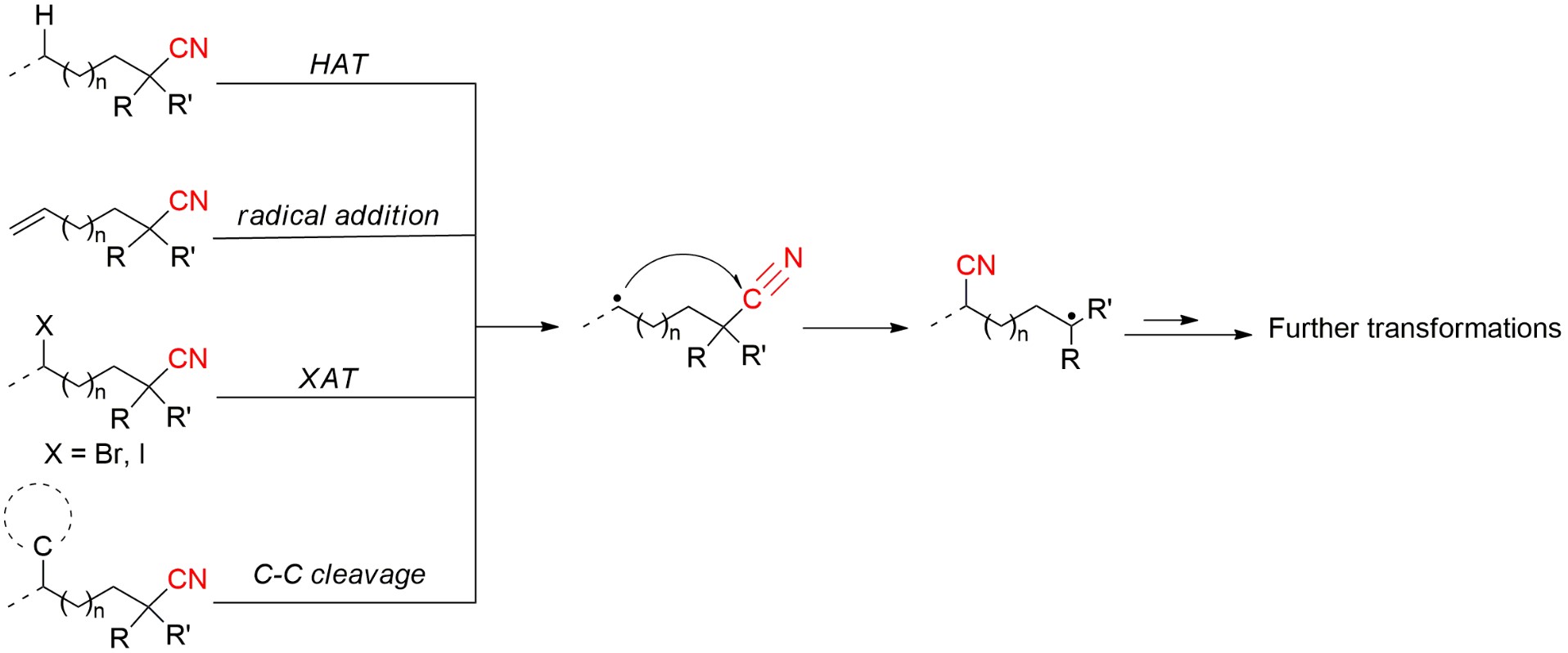
Radical-mediated intramolecular translocation of cyano groups has been recognized as a useful tool for the site-selective functionalization of organic molecules. The process is believed to proceed through the addition of an in-situ generated carbon-centered radical to the nitrile triple bond, followed by β-scission of the resulting cyclic iminyl radical intermediate to relocate the cyano group and produce a more stable carbon radical for further elaboration. Beginning in the early 1960’s and continuing for the next forty years, the research in this particular area has seen a surge of growth during the past two decades with advances in radical chemistry and application of photocatalysis. The present article attempts to conduct a comprehensive review of existing studies on this topic by covering the literature from 1961 to 2025. The procedures developed for the purpose are grouped and discussed in four sections according to the strategies used to generate the initial carbon radicals, which include i) hydrogen atom transfer (HAT); ii) radical addition to π system, iii) halogen atom transfer (XAT); and iv) homolytic fission of C-C single bond. In each section, a specific emphasis will be placed on reaction conditions, substrate scopes and mechanisms.
Keywords:
intramolecular migration
; translocation
; radicals
; cyano group
; nitrile
; site-selective functionalization
; photocatalysis
1. Introduction
The cyano group is one of the most versatile functional groups in organic chemistry [1]. The unique reactivity imparted by the nucleophilic nitrogen atom, the electropositive carbon center and π-coordinating ability of the triple bond allows it to be easily converted into carbonyl [2,3,4,5], amino [5,6,7] or heterocyclic [5,8,9,10,11] functionalities under suitable conditions. It can also serve as a directing, stabilizing, activating or leaving group in many transformations, such as C-H functionalization [12], α-functionalization [13], Diels-Alder cycloaddition [14,15], cyclopropanation [16], and transition-metal-catalyzed hydrodecyanation [17]. Additionally, the cyano group has been recognized as an important pharmacophore in drug discovery field, and often required to be incorporated into the structures of lead compounds for improving pharmacological properties or combating drug resistance [18,19]. The broad utility has promoted the development of numerous methods for the preparation of cyano compounds, which can be classified into several categories according to the way of introducing the cyano group. One type of commonly used methods is based on the conversion of a pre-installed functionality, e.g. an amide [20] or aldoxime [21], into the cyano group [22]. Another category of methods involves the cyanation of various substrates with external cyano-containing reagents, such as trimethylsilyl cyanide (Me3SiCN) [23], N-cyano-N-phenyl-p-toluenesulfonamide (NCTs) [24], butyronitrile [25], CuCN [26] or K4[Fe(CN)6] [27], that typically act as nucleophiles [23], electrophiles [24] or donors of CN ligand [25,26,27]. In some cases, NaN3 or TMSN3 was instead used as the nitrogen source for preparing aryl, alkenyl or oxo-nitriles through the C-H or C-C bond cleavage of the corresponding hydrocarbons [28]. However, even with the proven effectiveness, these traditional methods are generally not applicable for the selective installation of the cyano group at an unactivated or a sterically congested carbon site with concomitant introduction other functional group(s) of interest into a molecule. To this end, one may resort to another approach known as intramolecular cyano group migration (or translocation), which is usually implemented with readily available cyano-containing substrates and has proven to be useful for accessing nitrile derivatives that are non-trivial to obtain by conventional methods.
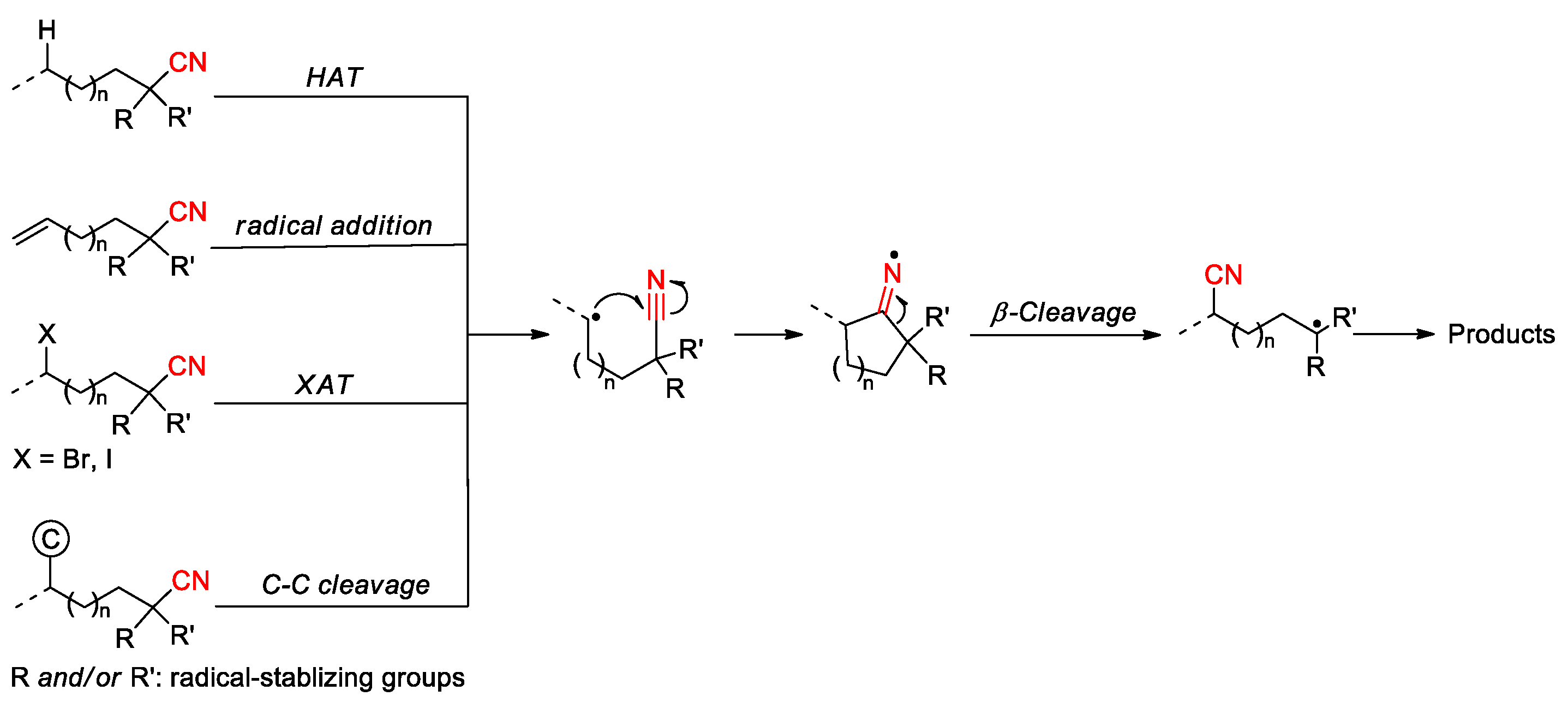
The radical-mediated translocation of cyano groups has emerged as a powerful tool for the site-selective functionalization of organic molecules. The process is believed to proceed through the addition of an in-situ generated carbon radical to the nitrile triple bond, followed by β-cleavage of the resulting cyclic iminyl radical to relocate the site of the cyano group (Scheme 1). The β-cleavage step is usually driven by the formation of a more stable carbon-centered radical, which then undergoes various transformations to yield different types of products. There are four general strategies that can be used to generate the initial carbon radical, including i) hydrogen atom transfer (HAT) from C(sp3)-H scaffolds; ii) radical addition to unsaturated π system, iii) halogen atom transfer (XAT); and iv) homolytic fission of C-C single bond. The research in this particular area has begun since the early 1960s [29,30], and benefited tremendously from advances in the fields of radical chemistry and photocatalysis over the past twenty years. This article intends to provide a comprehensive review of existing studies on this topic by covering the literature from 1961 to 2025. The procedures developed for the purpose will be discussed in four sections according to the strategies for producing the initiating carbon radical, with specific emphasis on mechanism and substrate scope. Some research works discussed herein were also mentioned in several published accounts, but only as a small part of reviewing the migrations of a large array of functional groups [31,32,33,34,35,36]. Moreover, the non-radical methods reported for CN migration, mostly involving the release and recombination of cyanide anion or the formation of cyanide complex, are not included in this article.
Scheme 1.
General Schemes for radical-mediated cyano group migration.
2. Radical-Mediated Translocation of Cyano Groups
2.1. Site Selectivity in CN Migration
Before we discuss the detailed synthetic strategies, it is instructive to clarify the origin of the site selectivity regarding the migratory distance of a cyano group from one carbon center to another. After the generation of the initial carbon radical, the site selectivity will be directed by the type of subsequent cyclization onto the nitrile group. It has been realized that the 1,4- or 1,5-cyano migration caused by a 5- or 6-exo-dig cyclization is generally preferred over other migration processes, e.g., 1,3-, or 1,2-migration [34,35], which is consistent with the results from a recently reported density-functional theory (DFT) study on azido radical-addition-induced cyano migration of several homologous alkenyl cyanohydrins [37]. After the addition of the azido radical to C-C double bonds, the relative free energies calculated for the cyclization step are outlined in Figure 1. The calculations suggest that 3- or 4-exo-dig cyclization (n = 0 or 1) accounting for the 1,2- or 1,3-migration is either impossible or formidable due to the high energy associated with the three- or four-membered cyclic radical intermediate (or transition state), whereas 5- or 6-exo-dig cyclization (n = 2 or 3) is energetically favored to produce the corresponding cyclic iminyl radical intermediate. The energy profile of the 7-exo-dig cyclization (n = 4) appears to be more appealing when compared with that of the 3- or 4-exo-dig cyclization. However, the 1,6-cyano migration resulting from the very process is rare in the literature possibly due to an unfavorable distance between the reaction centers for intramolecular cyclization. It is also noteworthy that although 4-exo-dig cyclization is generally disfavored, the 1,3-cyano migration has been achieved in several studies by using specific substrates and/or reaction conditions as encountered in the following sections.
2.2. CN Group Translocation via HAT
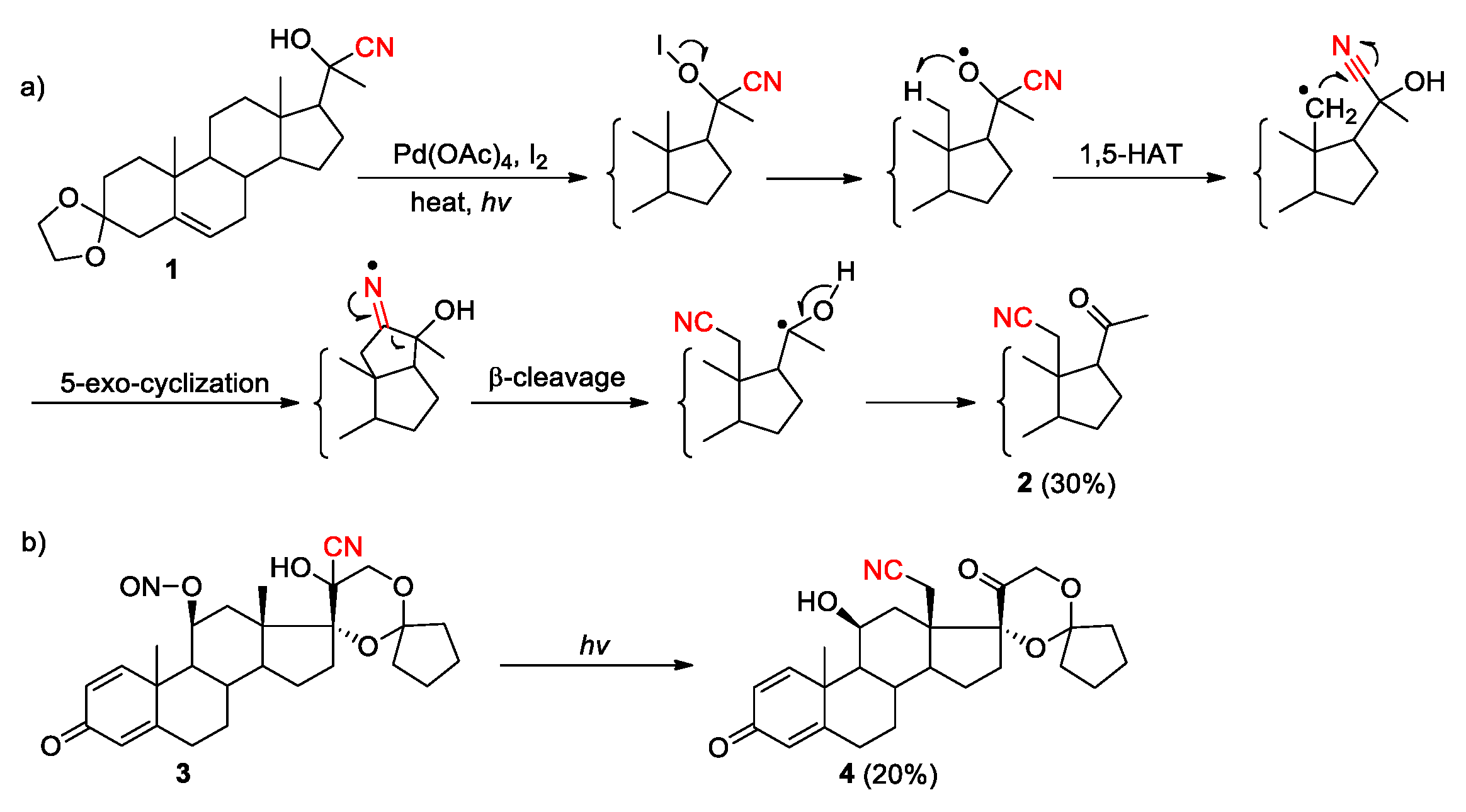
In 1961, Kalvoda reported the cyanohydrin-ketonitrile rearrangement of steroid 1 as the first known example to demonstrate the migratory aptitude of cyano group (Scheme 2, a) [29]. Mechanistically, the generation of an alkyoxy radical under the Barton hypoiodite reaction conditions triggered a 1,5-HAT process, allowing the creation of a carbon radical from unactivated C(sp3)-H bond in the C-18 angular methyl group. Subsequent 5-exo cyclization onto the nitrile followed by β-scission of the resulting iminyl radical provided a carbon-centered radical stabilized by the adjacent OH group. Further loss of a hydrogen atom from this intermediate gave the rearrangement product 2. In the following report, Kalvoda also described that the 1,4-cyano migration could also be initiated by the N-O cleavage of the 11β-nitrite group in 3, giving the formation of the product 4 upon irradiation (Scheme 2, b) [38].
Scheme 2.
1,4-Cyano migrations of steroidal cyanohydrins via HAT (Kalvoda, 1961 and 1970).
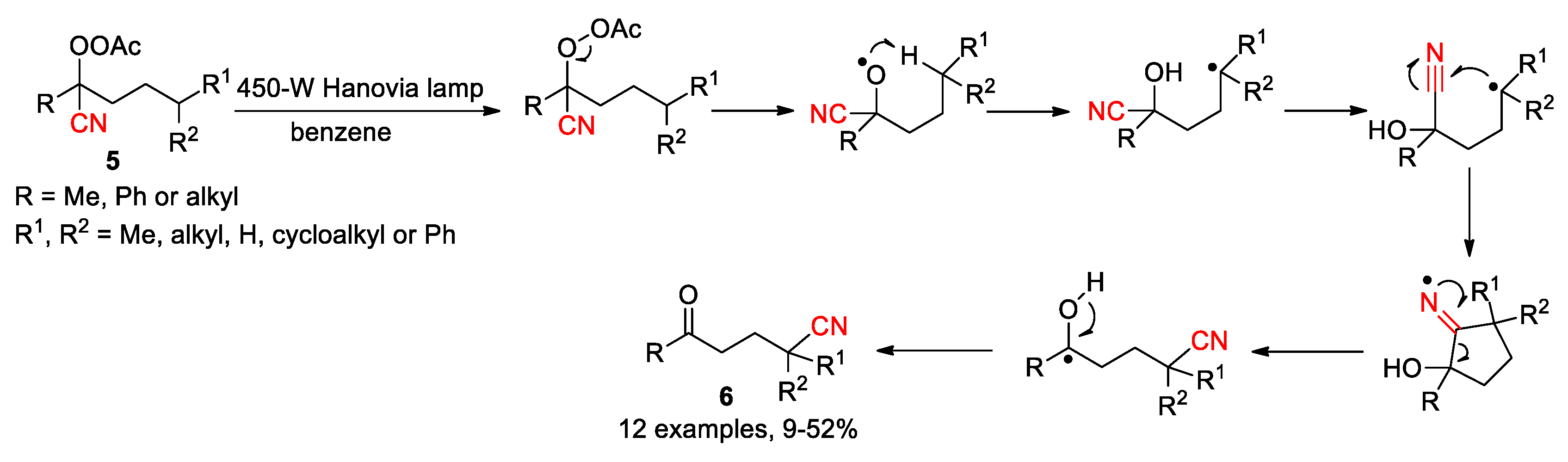
A similar cyano-migration was reported by Watt in 1976, after performing photolysis experiments on α-peracetoxynitriles 5 (Scheme 3) [39]. The reaction was initiated by the generation of alkoxy radical through the homolytic cleavage of the O-O bond and afforded the δ-ketonitrile products 6 in low to moderate yields. Under the employed conditions, the competitive decarboxylation of 5 and/or Norrish type II fragmentation of 6 mainly accounted for the low yields encountered in most cases. In a separate report, Watt described that this photolytic protocol was comparable to Kalvoda’s method, like delivering 2 in 21% yield from the corresponding 20-peracetoxy-20-cyanosteroid [40]. The Kalvoda and Watt procedures have not found much application since they were reported [41], possibly due to the inconvenience of preparing substrates and lower yields associated with harsh reaction conditions. Despite this, these pioneering studies had provided valuable mechanistic insight into the migration process, thus creating a foundation for the future development.
Scheme 3.
Conversion of α-peracetoxynitriles into δ-ketonitriles via HAT-mediated 1,4-CN migration (Watt, 1976).
Scheme 3.
Conversion of α-peracetoxynitriles into δ-ketonitriles via HAT-mediated 1,4-CN migration (Watt, 1976).
Organic photocatalysis, including photoredox catalysis, has aroused great attention over the past two decades as a platform for the development of new synthetic strategies [42]. The photocatalysts, such as the commonly employed polypyridyl ruthenium or iridium complexes, can pass to excited states after absorbing light in the visible region of the electromagnetic spectrum. The excited species then interacts with organic molecules through energy transfer or single-electron-transfer (SET), generating reactive intermediates, such as radical, radical anion or diradical, to perform organic transformations. The excited catalysts will fall back to the ground state via unimolecular or bimolecular chemical processes, to complete a photocatalytic cycle.
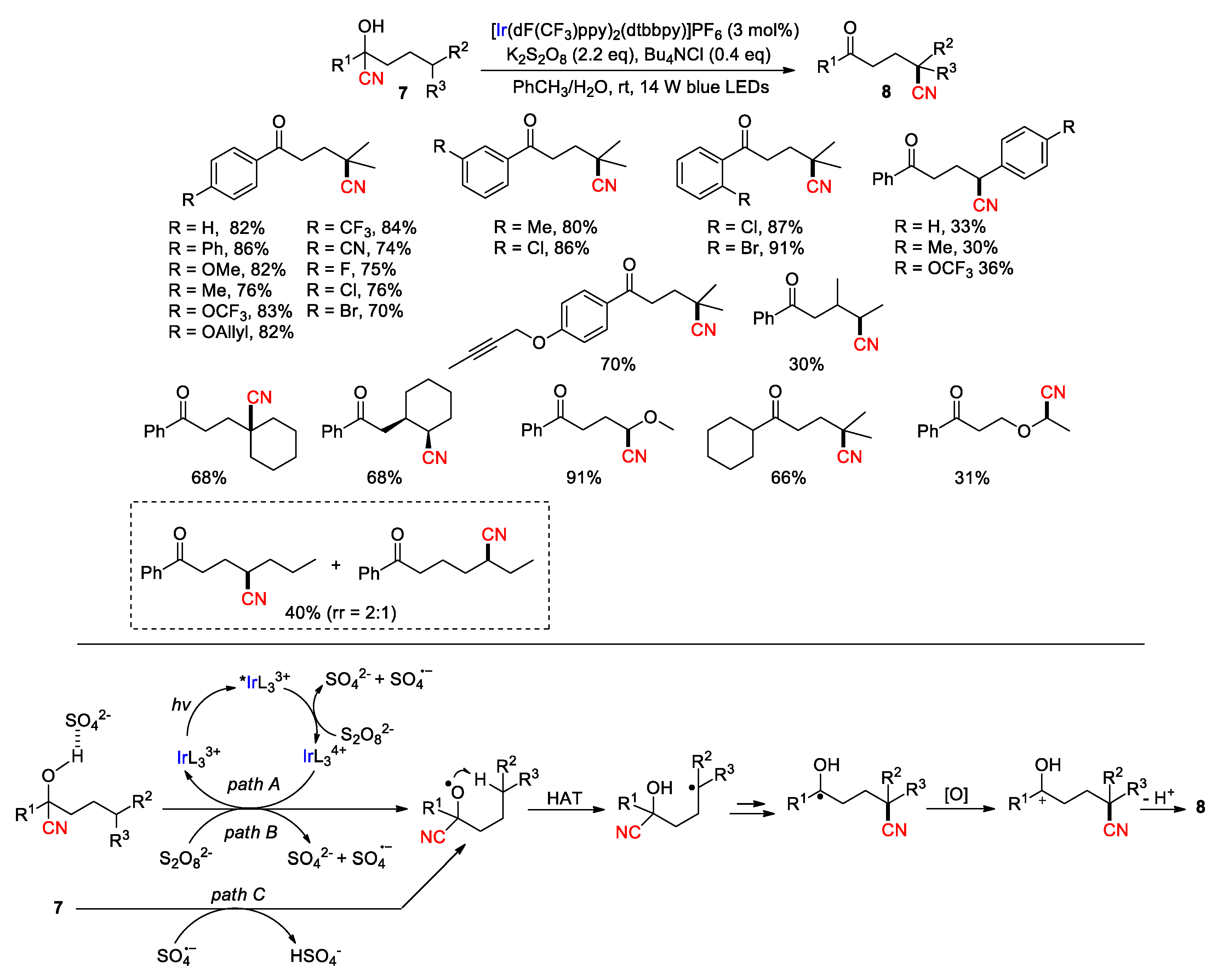
Photocatalysis has also been widely used for functional group translocations [32,36], with some studies being directed to HAT-mediated CN migration. In 2019, Zhu and co-workers reported a photoredox catalytic protocol for the direct conversion of cyanohydrins 7 into the corresponding δ-ketonitriles 8 without any pretreatment (Scheme 4) [43]. During the model study, the iridium photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine)] combined with K2S2O8 oxidant and Bu4NCl were found to be optimal among the tested variants, giving the best yield of the desired product (82%) upon visible-light irradiation (82%). The reaction was applicable to a range of cyanohydrins containing various functional groups, leading to the desired products in moderate to good yields. Besides, the selective generation of the δ-ketonitrile over its ε-regioisomer was observed in the presence of multiple reactive methylene sites (rr: 2:1), ascribing to kinetically more favorable six-membered cyclic transition state of 1,5-HAT.
As in all photocatalytic reactions, the process is initiated by photoexcitation of the IrL33+ catalyst to the *IrL33+ that can be further oxidized by persulfate, giving IrL34+ along with sulfate ion (SO42-) and sulfate radical anion (SO4•– ) [44]. The IrL34+ species then converts the hydroxyl group into an oxy radical via a proton-coupled electron transfer (PCET) process, which is probably facilitated by the interaction with a conjugate base, e.g., sulfate ion, to reduce the oxidation barrier of the O-H bond. This event also reduces the photocatalyst back to IrL33+ to complete the catalytic cycle (pathway A). Moreover, the formation of the desired product, albeit in lower yield, was still observed when the reaction was irradiated in the absence of photocatalyst, suggesting that a persulfate-mediated chain reaction may concurrently engage in the PCET process to produce the alkoxy radical (path B). In addition, the possibility of abstracting the alcoholic hydrogen atom by SO4•– released from path A and/or B cannot be ruled out (path C). Subsequent 1,5-HAT generates a carbon radical to trigger the cyano group migration, leading to a ketyl radical intermediate. Finally, oxidation of this intermediate followed by deprotonation of the resulting carbocation may account for the formation of the product.
Scheme 4.
Conversion of cyanohydrins into δ-ketonitriles under photoredox catalytic conditions (Zhu, 2019).
Scheme 4.
Conversion of cyanohydrins into δ-ketonitriles under photoredox catalytic conditions (Zhu, 2019).
The alcoholic function in the aforementioned reaction did not take part directly in the CN transfer step, but instead serving as an auxiliary group to form the first carbon radical via HAT and to stabilize the second one. The overall process results in the migration of a cyano group accompanied by the conversion of the hydroxyl group into a carbonyl and the loss of two hydrogen atoms. In a conceptually distinct contribution, Xu et al. recently reported an elegant photocatalytic, reversible C-H sampling strategy that enables the direct positional exchange between a CN group and an unactivated C-H bond without introducing any accompanying variations to a molecule (100% atom economy) [45,46]. The transformation is conducted by merging hydrogen-atom donation (HAD) catalysis with hydrogen-atom abstraction (HAA) catalysis in one reaction system, and begins with non-specific cleavage of C-H bonds by HAA to generate a series of carbon radicals (Figure 2). The resulting radicals are then sorted based on the delicately established kinetic reversibility and the rate differences for the consequence step, permitting the C-4 radical to undergo the cyclization with the nitrile to form the five-membered iminium radical whereas restoring other unfunctionalized radicals to the C-H bonds through HAD.
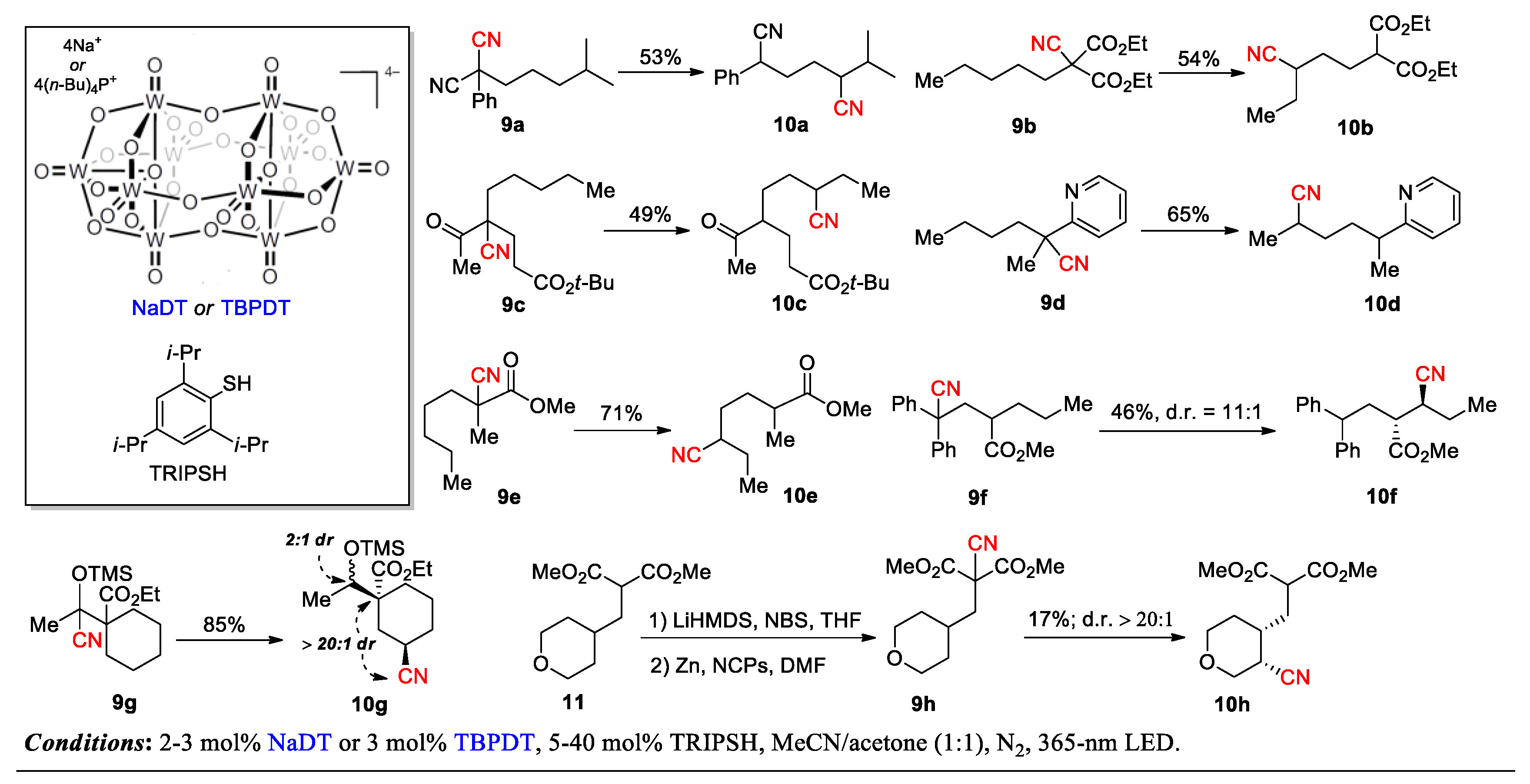
This strategy presents an unconventional paradigm to achieve site-selective C-H transformations without necessitating a site-selective C-H cleavage step. As described, a large array of malononitriles and mono-nitriles was smoothly converted to the desired 1,4-CN translocation products by using sodium or tetra-n-butylphosphonium decatungstate (NaDT or TBPDT) as the photo-HAA catalyst [47] and 2,4,6-triisopropylbenzenethiol (TRIPSH) as the HAD catalyst under 365-nm irradiation. Notably, the presence of a radical-stabilizing substituent at the α position of the migrating cyano group (e.g., CN, Ar, CO2R, COR or OTMS) is essential for the transformation, which is consistent with the mechanism shown in Scheme 1. The selected examples in Scheme 5 demonstrate remarkably high functional group tolerance and site selectivity between the 1,4- and 1,5-translocation (> 20:1 r.r.) (9a-g→10a-g). Also outlined is a strategic sequence for the diastereoselective synthesis of compound 10h, involving the initial introduction a cyano group to the readily accessible C-H site in 11 followed by relocating the CN group to a hard-to-target site in 9h to give 10 h. The readers are encouraged to refer to the original paper for more interesting results and detailed discussions. More advances are expected with applying this ‘radical sampling’ strategy to the functionalization of other types of C-H bonds
Scheme 5.
Selected examples for 1,4-CN migration of nitriles via radical sampling strategy (Xu, 2023).
Scheme 5.
Selected examples for 1,4-CN migration of nitriles via radical sampling strategy (Xu, 2023).
2.3. Cyano Group Migration via Radical Addition to Unsaturated CC Bonds
2.3.1. Early Reported Works
The addition of a radical species to a CC double or triple bond can serve as another approach to trigger the cyano group migration. The reactions in this category are abundant in the literature, and those reported from the late 1970s to the early 2010s are discussed in this section.
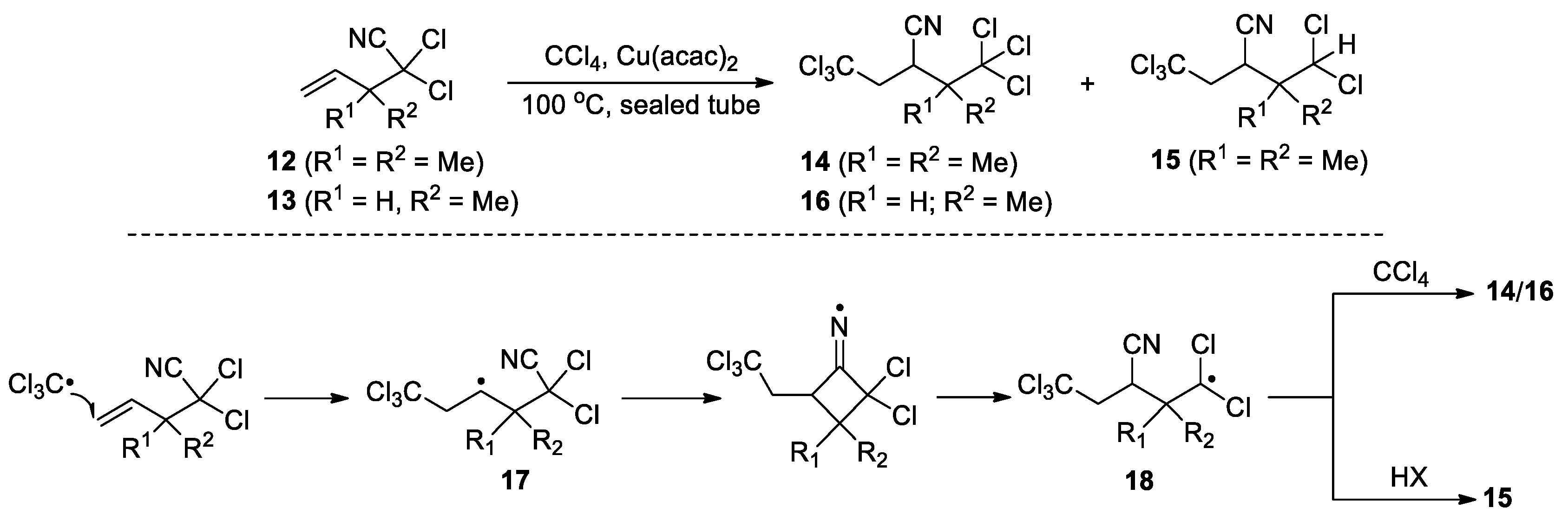
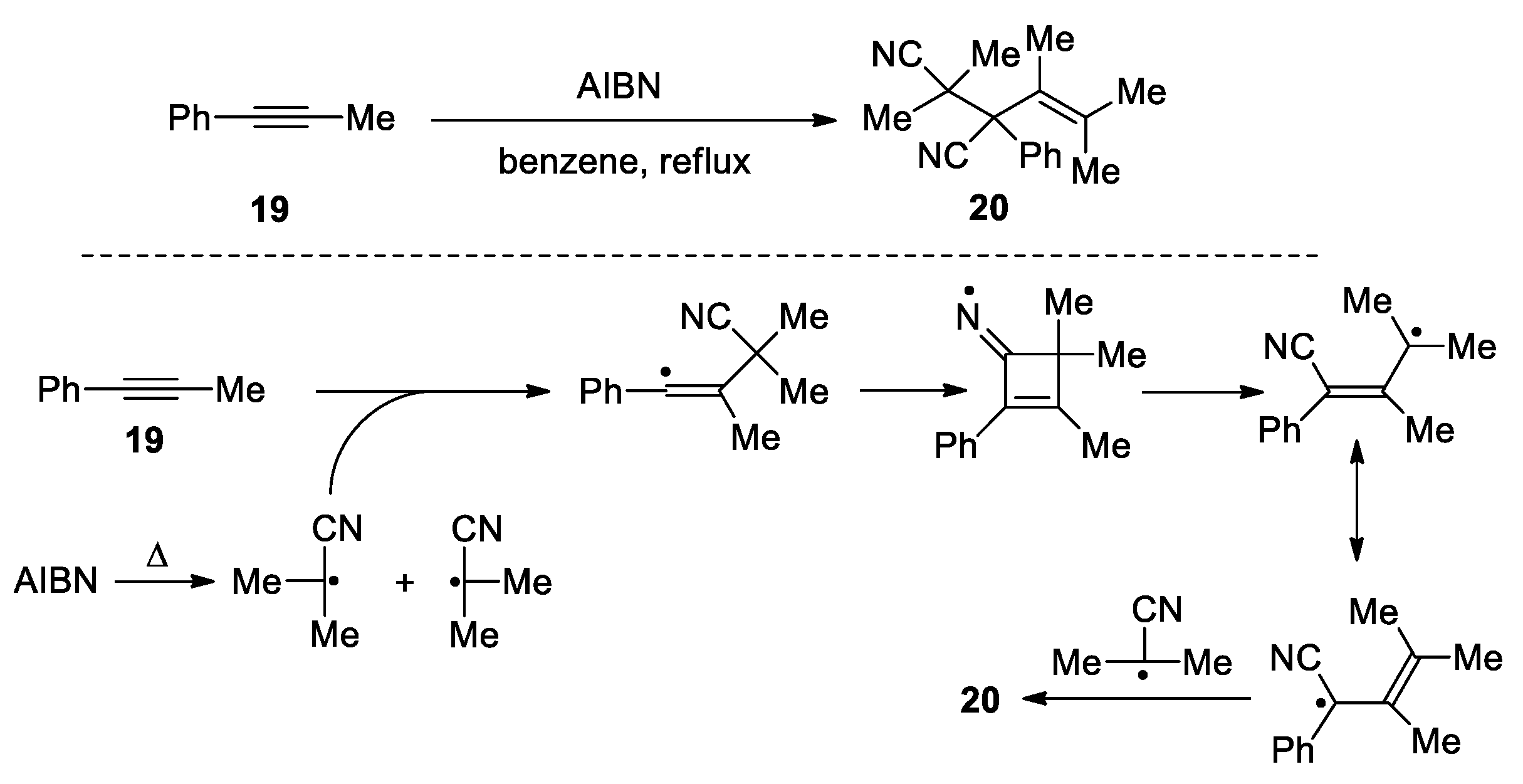
The radical used for the addition step can be directly generated from a reagent. For instance, Johnson et al. described that a trichloromethyl radical generated from CCl4 could add to the C=C double bonds of pent-4-enenitriles 12 and 13 to trigger a 1,3-cyano migration and lead to the formation of the polychlorinated products 14-16 (Scheme 6) [48]. The driving force for the migration is the higher stability of the dichloroalkyl radical 18 than that of the secondary alkyl radical 17. Moreover, the methyl substituents in the 3-position were found to be critical for the reaction, suggesting the favorable Thorpe-Ingold effect in the cyclization step. In 1997, Montevecehi et al. reported the formation of the product 20 via the reaction of phenylpropyne 19 with AIBN in reflux benzene. The proposed mechanism involves the addition of 2-cyano-isopropyl radical (CIR) generated by the thermal decomposition of AIBN to the C≡C triple bond. The resulting vinyl radical then cyclizes onto the C≡N bond to form a cyclobutenyl iminium species. Subsequent ring opening with β-scission of the C-CMe2 bond affords a resonance-stabilized radical and thence 20 after trapping by another CIR (Scheme 7) [49]. The authors also postulated that the cyclization of the vinyl radical was facilitated by the β-methyl group, as similar treatment on phenylacetylene did not give any CN migration product. More recently, a Cu(I)-catalyzed carbocyanation reaction of trisubstituted olefins 21 with chlorinated cyanides 22 was reported by Inoue’s group, giving the formation a range of adducts 23 in 24 to 78% yield (11 examples) (Scheme 8) [50]. The process begins with Cu(I)-promoted reductive C-Cl bond cleavage to give an electron-deficient carbon radical along with Cu(II)Cl. Addition of the radical to the C=C double bond in 21 from the less hindered side yields a tertiary carbon radical. Subsequent cyclization followed by β-cleavage lead to a Cl-stablized radical through the 1,3-cyano migration. Finally, abstraction of the chloride from Cu(II)Cl furnishes the products with regeneration of the Cu(I) catalyst. Notably, these reactions all involved 1,3-cyano migration and consequently required elevated temperatures to overcome the high energy barrier associated with four-membered iminyl radicals and transition states.
Scheme 6.
Trichloromethyl radical-triggered 1,3-cyano migration (Johnson, 1982).
Scheme 7.
2-Cyano-isopropyl radical-triggered 1,3-CN migration (Montevecehi, 1997).
Scheme 8.
Radical-mediated carbocyanation of olefins (Inoue, 2012).
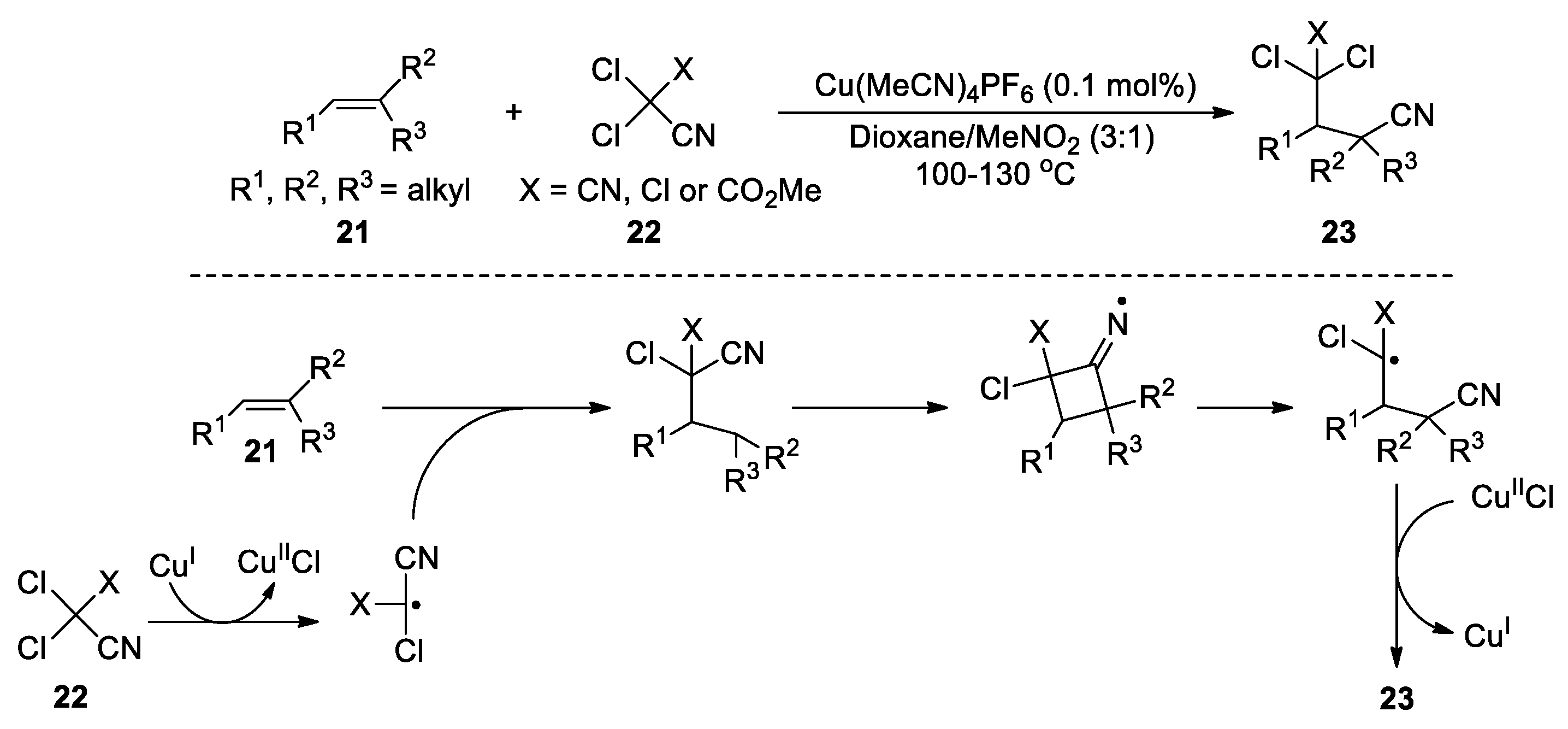
The CN migration strategy is amenable to the development of cascade cyclization reactions, in which a reactive intermediate or a substrate itself acts as the radical precursor. Curran et al. reported an annulation reaction by heating iodomalononitriles with alkenes, e.g., 24 with 25, in the presence of tributyltin hydride (Scheme 9) [51]. The thermal coupling between 24 and 25 first produces an iodane intermediate, which serves as a radical precursor and undergoes the dehalogenation upon the action of Bu3Sn• to yield a secondary alkyl radical. The addition of the radical to the terminal alkene triggered the 1,4-nitrile migration, leading to a tertiary carbon radical after 5-exo-dig-cyclization and β-cleavage. Hydrogen atom abstraction, likely from Bu3SnH, then furnishes the cyclized product 26. In this case, the β-cleavage is aided by relieving the ring-strain of the bicyclic iminyl radical in addition to the radical stabilising nitrile group. Zard et al. developed another cyclization process terminating on a nitrile, in which the employed cyanide substrate 27 serves as a radical precursor via a tributyltin radical-promoted N-O bond cleavage. The addition of the resulting amidyl radical to the olefin is followed by the cyclization and β-scission, yielding a tertiary carbon radical stabilized by the adjacent nitrogen atom and thence the product 28 (Scheme 10) [52].
Scheme 9.
Annulation involving radical-addition-triggered 1,4-CN migration (Curran, 1992).
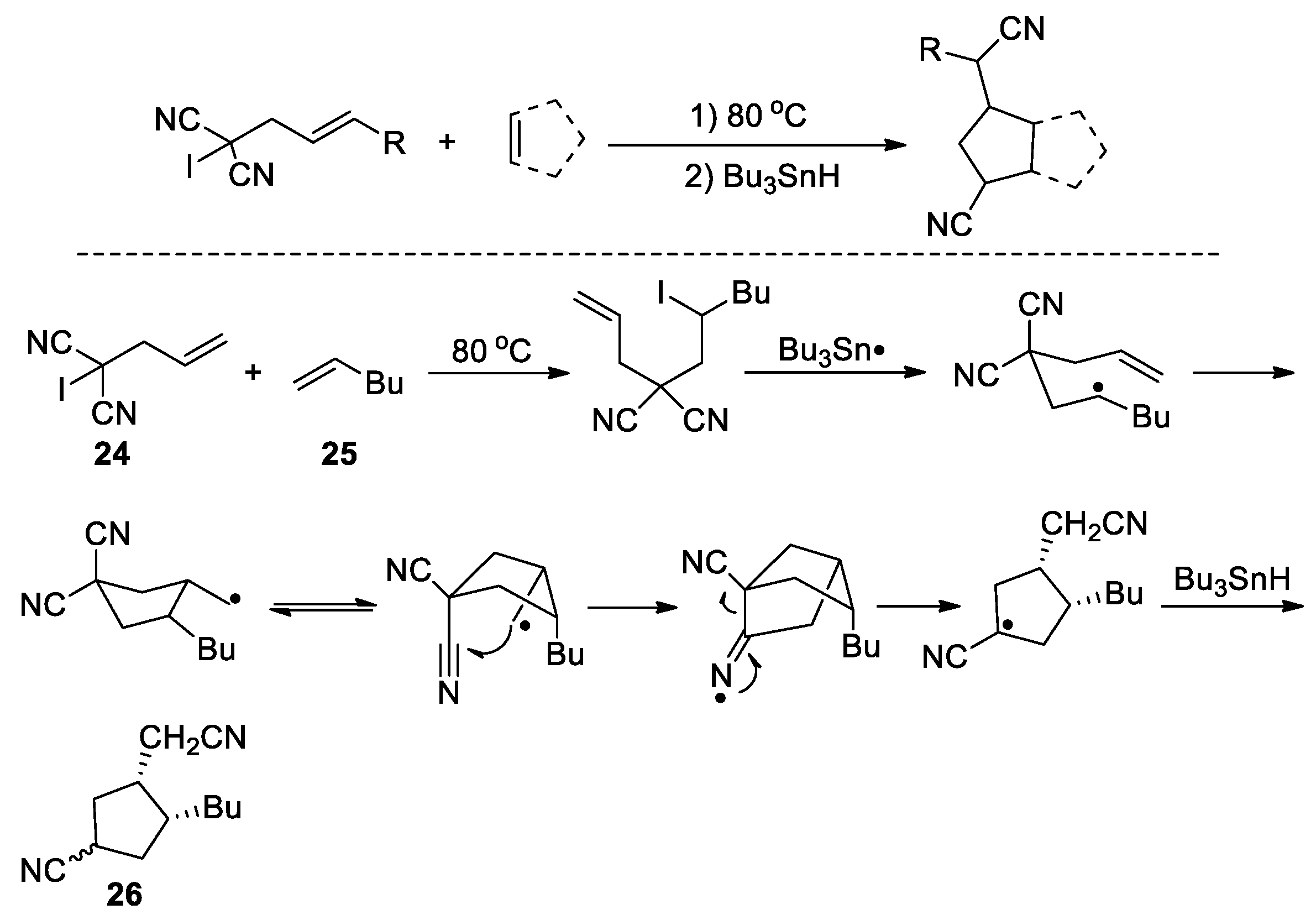
Scheme 10.
Amidyl radical-induced cyclization involving 1,4-CN migration (Zard, 1994).
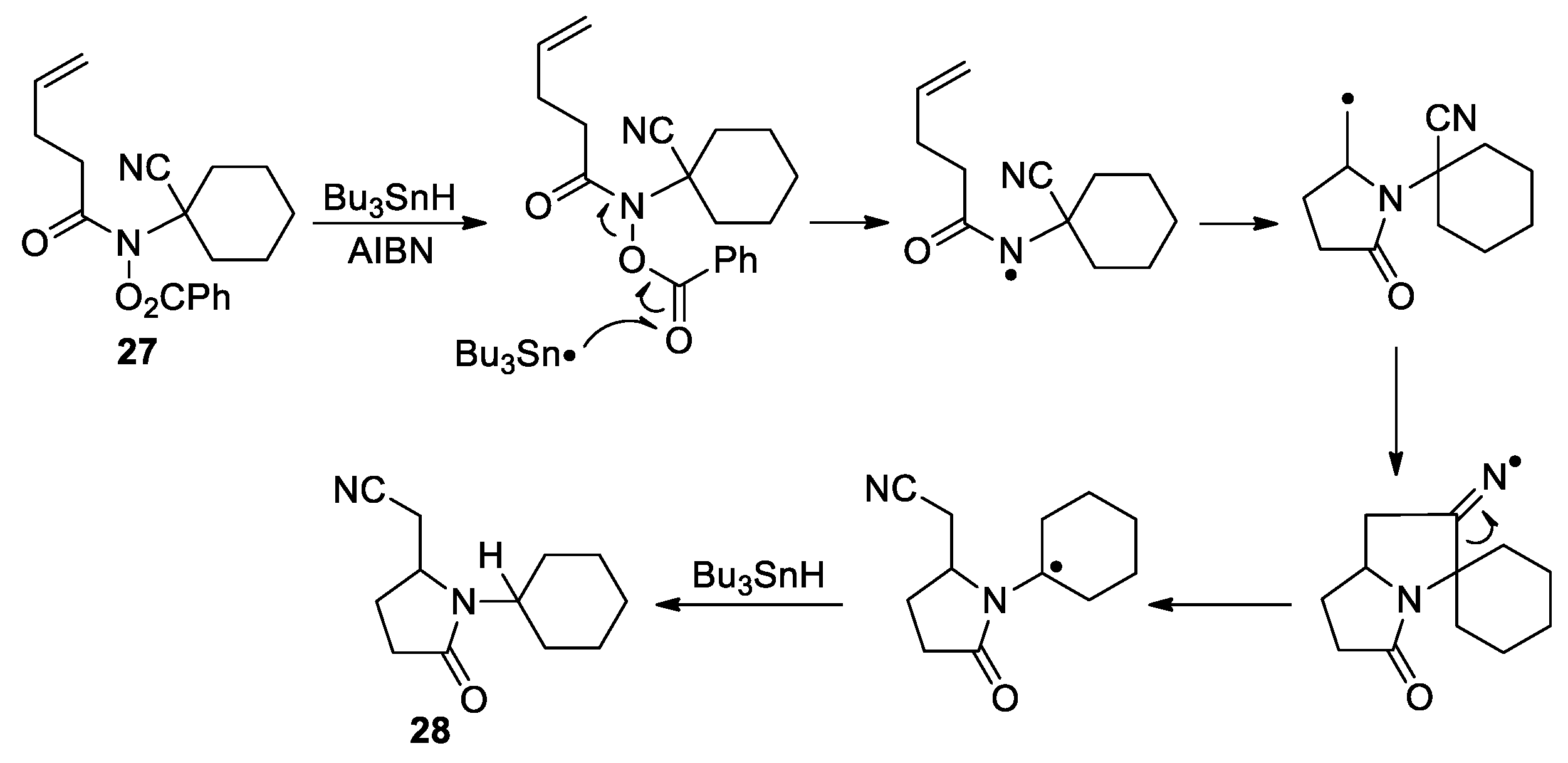
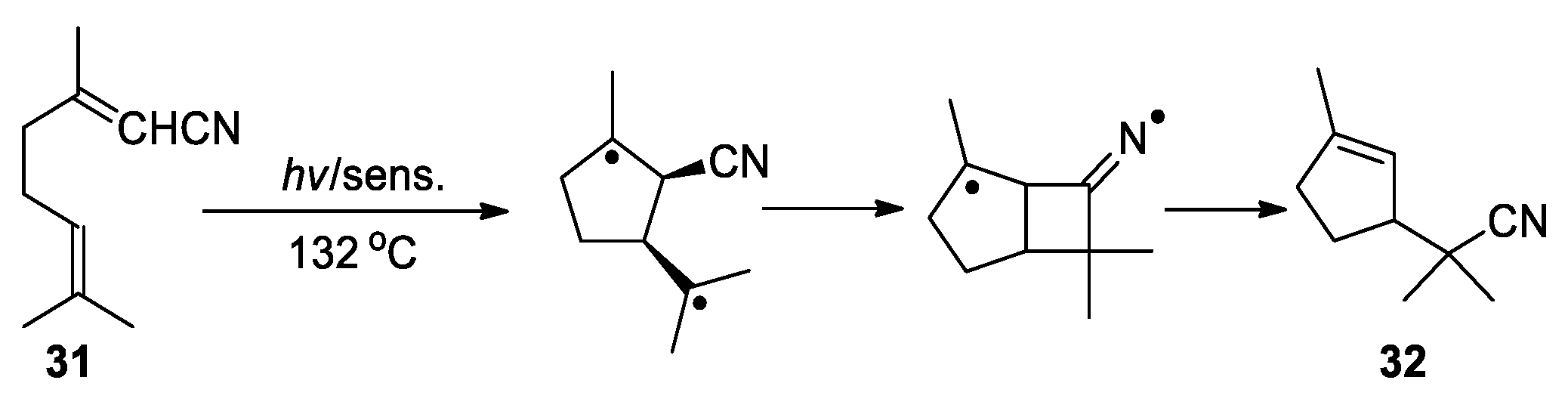
In two particular cases, the CN migration was proposed to proceed through biradical intermediate. Saito and Matsuura reported the formation of 5-substituted uracil 30 upon irradiating 6-cyano-1,3-dimethyluracil 29 with 2-methyl-2-butene in acetonitrile (Scheme 11) [53]. The result was rationalized by assuming that a biradical intermediate resulting from the photoaddition of 29 with the alkene could engage in a 5-exo cyclization to afford a bicyclic iminium species. Subsequent 1,2-hydrogen shift followed by β-cleavage could furnish the rearranged adduct 30. Furthermore, Wolff and Agosta described the photolytic conversion of geranonitrile 31 to the cyclopentene product 32 through 1,3-shift of the cyano group from a biradical intermediate (Scheme 12) [54]. This is an overall 1,6-nitrile migration reaction, and was not observed at lower temperatures.
Scheme 11.
Formation of 5-substituted 1,3-dimethyluracil via photoaddition of alkene to 6-cyanouracil (Saito, 1980).
Scheme 11.
Formation of 5-substituted 1,3-dimethyluracil via photoaddition of alkene to 6-cyanouracil (Saito, 1980).
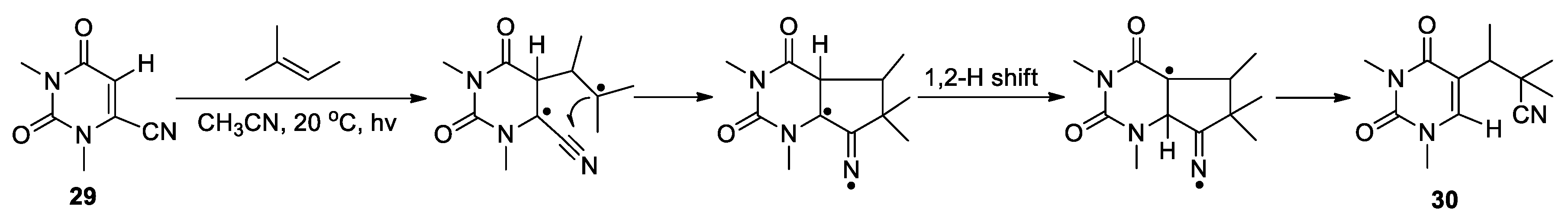
Scheme 12.
1,3-Migration of cyano group via photolysis of geranonitrile (Wolff, 1978).
The above examples have demonstrated the diversity of substrates that are eligible for radical-addition-induced cyano migration. In recent years, such an approach has been mainly used to achieve the multi-site functionalization of the unsaturated cyanohydrin or nitrile compounds. These studies, together with two recently reported cyano migration processes involving diradical or radical cation intermediates, will be discussed in the following sections (2.3.2-2.24).
2.3.2. CN Migration with Unsaturated Cyanohydrins
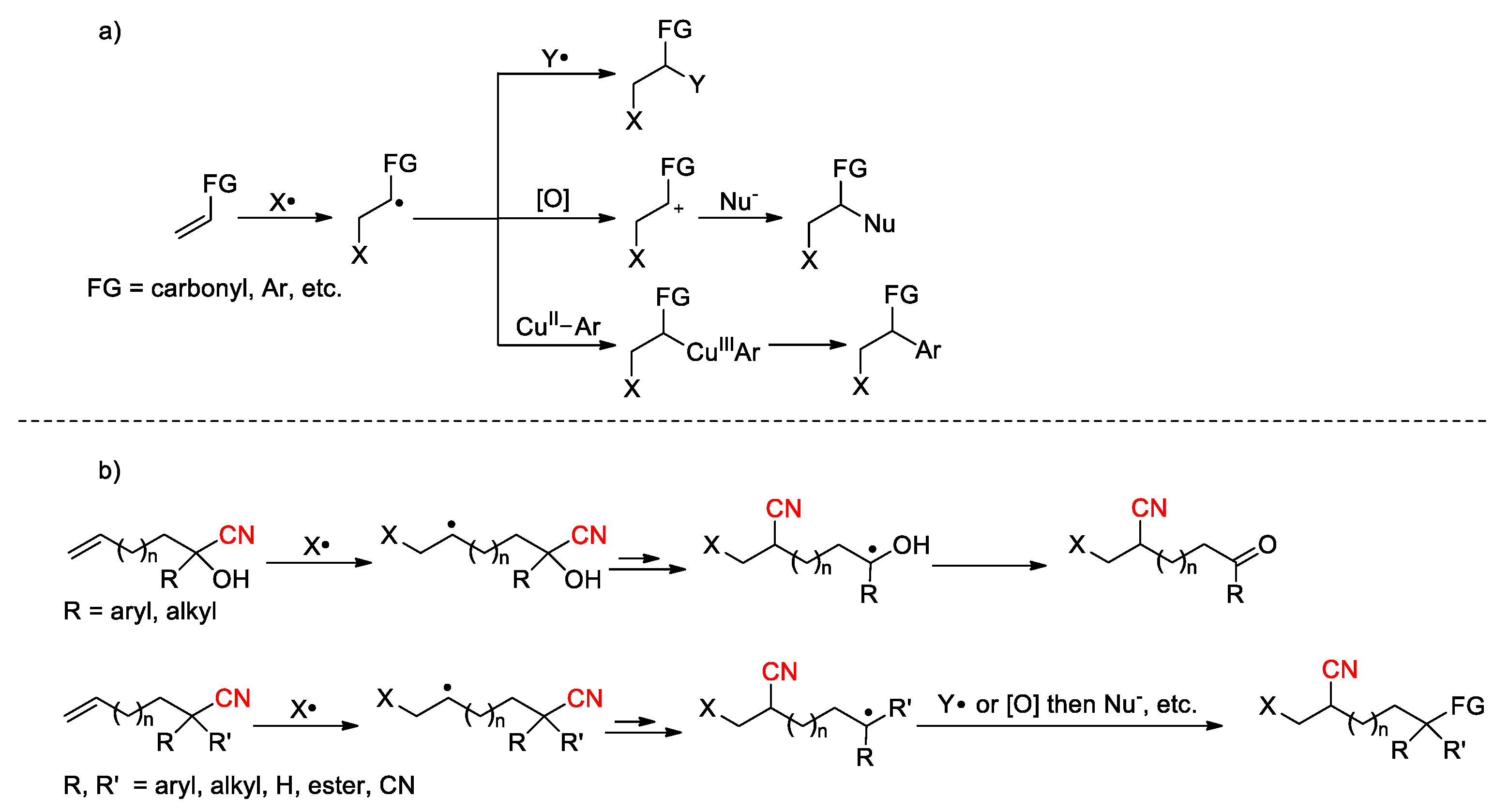
Radical-mediated olefin difunctionalization has provided a powerful tool for enhancing the molecular complexity of simple olefins. In a common paradigm, the addition of a radical (X•) to the C-C double bond generates an alkyl radical, which can be trapped by another radical specie (Y•) [55] or oxidized into a carbocation to facilitate the incorporation of second functional group via nucleophilic attack (Nu-) (Scheme 13, a) [55,56]. In some cases, the carbon-centered radical can be sequestered by organometallic species [57,58] (e.g., CuII-Ar), and functionalized through reductive elimination of the resulting complex. However, the scope of alkenes in these transformations has been largely restricted to activated alkenes, including those containing aryl [55], carbonyl [59] or heteroatom [60] substituents to confer suitable electronic properties to the double bond for efficient reaction with the incoming radical and stabilize the nascent carbon-centered radical through p-π conjugation. In comparison, the difunctionalization of unactivated alkenes (e.g., FG = alkyl) in a one-step fashion is more challenging and less explored [61,62]. Among several approaches to be considered, the radical-mediated distal functional group migration (FGM) has emerged as an attractive option, allowing the incorporation of diverse functionalities, such as aryl, heteroaryl, carbonyl, alkynyl, alkenyl, imino or cyano group, onto an alkene in addition to the initiating radical species [35,63,64]. Due to the pivotal roles of cyano group in organic chemistry and related fields [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19], direct cyanofunctionlization of olefin via FGM strategy is unarguably an important subset of the alkene difunctionalization platform, which has been mostly performed with olefinic cyanohydrins or nitriles. During the processes, the ketyl radicals generated from cyanohydrins are readily oxidized to the corresponding ketones, while the radical intermediates derived from alkenyl nitriles undergo varies transformations, such as radical coupling or oxidation combined with nucleophilic addition, affording different kinds of products (Scheme 13, b). Despite known for decades (Scheme 6), extensive studies on such transformations have only been carried out in recent fifteen years with flourishing developments in radical chemistry and photocatalysis that enable efficient reagent systems to become increasingly available [63].
Scheme 13.
Strategies for radical-mediated alkene difunctionalization.
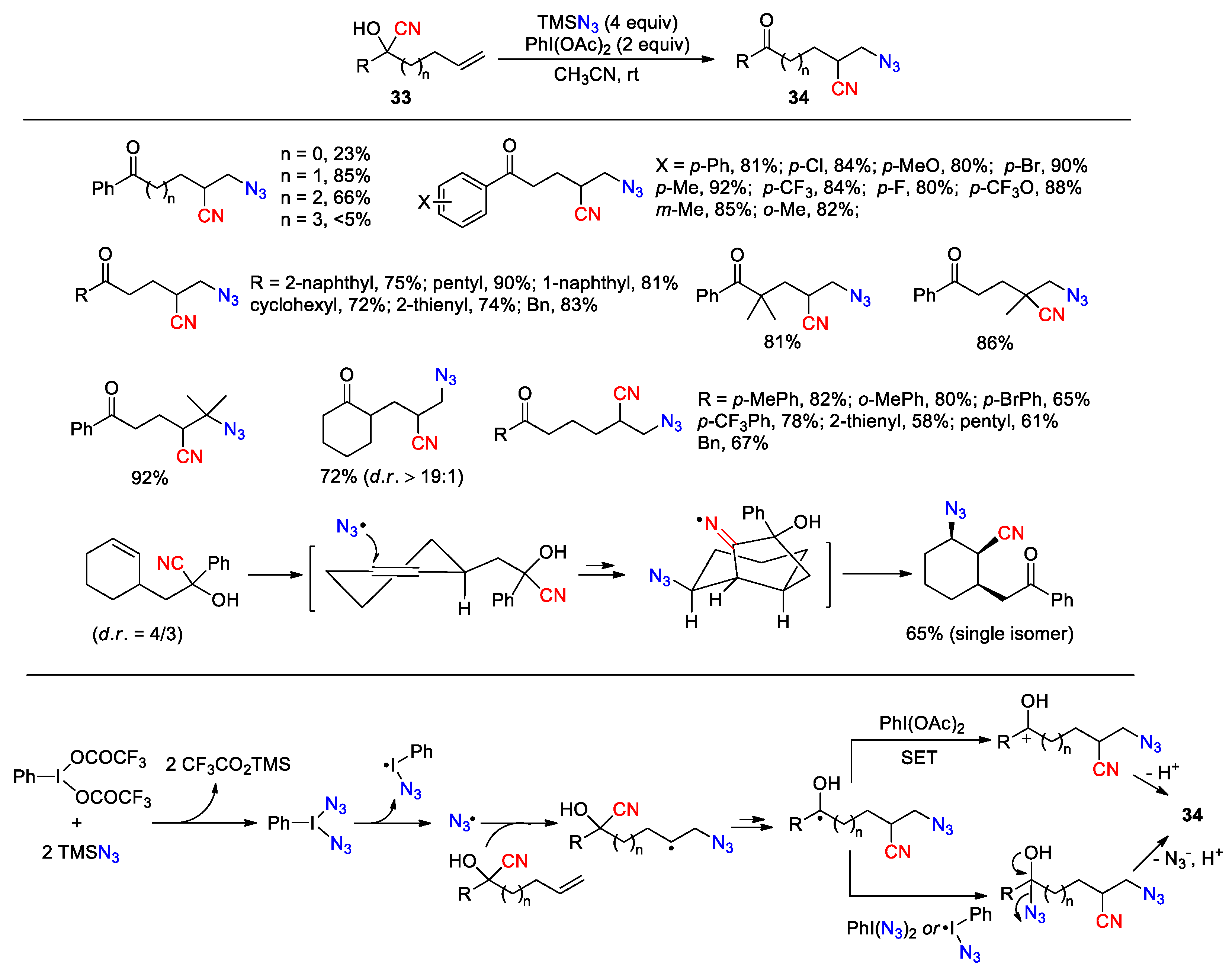
The studies performed with unsaturated cyanohydrins are first discussed. In an early contribution, Zhu et al. reported a method for the azidocyanation of unactiavted alkenes by using TMSN3 and PhI(OAc)2 (PIDA) as the reagents (Scheme 14) [65]. During the initial assessment with homologues cyanohydrins, the 1,4- and 1,5-nitrile transfer reactions (n = 1, 2) were found to occur more efficiently than the corresponding 1,3- and 1,6-nitrile transfer reactions (n = 0, 3) as reflected by the yields of the obtained products. These results are also consistent with the DFT calculation results shown in Figure 1. In this context, a variety of bis- and trihomoallylic cyanohydrins 33 were further evaluated, leading to the corresponding 1,2-azidonitriles 34 in good to high yields with complete regioselectivity. Particularly interesting is the generation of a single cyclohexane isomer with all-cis 1,2,3-substituents from a 4:3 diastereomeric mixture. It would seem that this could be attributed to the facial selectivity in the radical addition step directed by the conformation of the cyclohexene ring and an equatorial orientation of the side chain. Mechanistically, the interaction of TMSN3 with PhI(OAc)2 produces PhI(N3)2, which then decomposes giving azido radical. The radical addition to the olefin followed by 1,4(5)-cyano migration produce an α-hydroxy alkyl radical intermediate. As proposed, the conversion of this intermediate to the product may occur in two separate events: i) PIDA-mediated single-electron oxidation to a carbonium ion followed by deprotonation, or ii) capture by the azido radical releasing from PhI(N3)2 or Ph(N3)I• [66] followed by the collapse of the resulting azidohydrin.
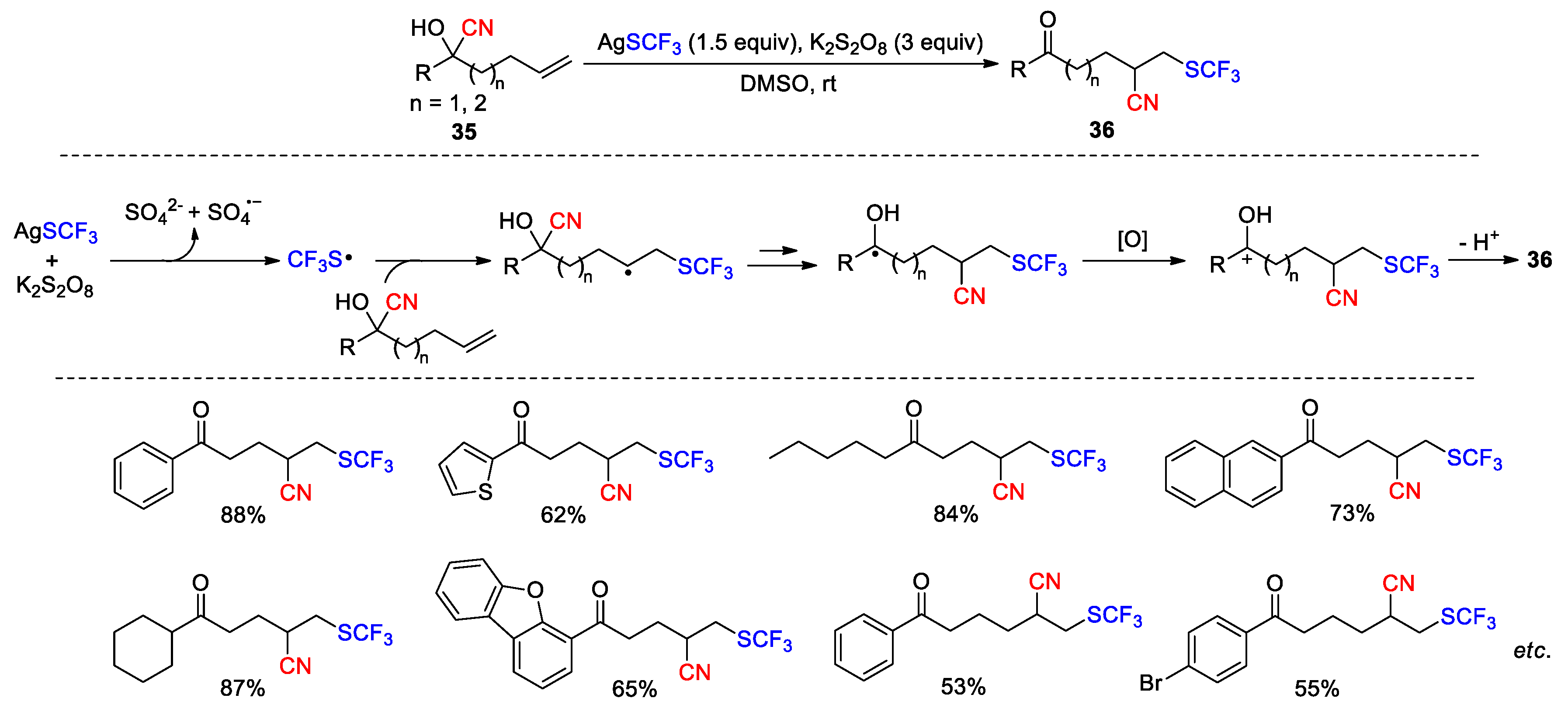
Subsequent to their initial report, Zhu and co-workers further disclosed several methods for the functionalization of unsaturated cyanohydrins using similar strategies (Scheme 15, Scheme 16, Scheme 17 and Scheme 18). In 2017, they reported the conversion of cyanohydrins 35 to CF3S-substituted ketonitriles 36 through the treatment with AgSCF3 and K2S2O8 (Scheme 15) [67]. In the process, trifluoromethyl sulfide is oxidized by persulfate to generate a trifluoromethylthio radical, which then adds to the alkene to trigger the cyano migration and produce a ketyl radical intermediate. The single-electron oxidation of this intermediate by persulfate, followed by the deprotonation of the resulting carbocation affords the final product. The reaction is applicable to a variety of substrates containing alkyl, naphthyl, thiophenyl, dibenzofuryl or phenyl groups with diverse substitution patterns, and provides the desired products in moderate to good (26 examples, 35-88%).
Scheme 14.
Radical-mediated azidocyanation of alkenes via 1,4(5)-CN migration (Zhu, 2016).
Scheme 15.
Radical-mediated cyanotrifluoromethylthiolation of alkenes via 1,4(5)-CN migration (Zhu, 2017).
Scheme 15.
Radical-mediated cyanotrifluoromethylthiolation of alkenes via 1,4(5)-CN migration (Zhu, 2017).
Photoredox catalysis has been utilized by Zhu’s group to achieve the polyfunctionalization of alkenyl cyanohydrins. In 2017, they reported a reaction of compounds 37 with fluoro-substituted bromide reagents 38a-e, including bromodifluoroacetate, 2-bromo-2,2-difluoro-1-morpholinoethanone, bromodifluoromethanesulfonylbenzene, dibromodifluoromethane and bromofluoroacetate, for the synthesis di- or mono-fluorinated ketonitriles 39 (Scheme 16) [68]. Mechanistically, the employed iridium catalyst fac-Ir(ppy)3 (ppy =2-phenylpyridine) (simplified as IrL33+) engages in single-electron transfer with 38 from its photoexcited state *IrL33+, thereby giving a IrL34+ complex. Upon the reduction, 38 may undergo mesolysis, the fragmentation of a radical anion to afford a bromide anion and a carbon radical 40 [69]. Addition of 40 to 37 followed by 5-exo-dig cyclization and β-cleavage lead to ketyl radical intermediate 41. Oxidation or 41 by the IrL34+ species provides intermediate 42, returning the catalyst to its Ir(III) oxidation state. Finally, deprotonation of 42 furnished the products 39. Various substrates bearing naphthyl, thiophenyl, benzyl, alkyl, or phenyl group with diverse substitution patterns were allowed to react with 38a-e, yielding over 40 products in good to high yields (50-91%). Besides, increased catalyst loading and more intense radiation were required for the reactions of 38e to improve the yields of mono-fluorinated products.
In another work, Zhu and co-workers described the preparation of 1,8-diketone derivatives 45 via the photoredox coupling between cyanohydrins 43 and cyclopropanols 44 (30 examples, Scheme 17) [70]. A reagent system comprising Ir(dtbbpy)(ppy)2PF6 catalyst, K2S2O8 and BF3Et2O was found to be optimal for the reaction, while the function of BF3.Et2O had remained unknown to the authors. In the process, the photoexcited catalyst *IrL33+ is oxidized by K2S2O8 to yield the IrL34+ complex, which then converts 44 to the cyclopropyloxy radical 46 with the regeneration of the IrL33+ catalyst. The formation of the desired product was still observed (in a lower yield) when the reaction was conducted in the absence of photocatalyst, suggesting that the oxidation of 44 by persulfate may concurrently contribute to the formation of 46. The ring-opening of 46 provides a β-carbonyl radical 47, which then adds to the olefin to produce another alkyl radical 48. The addition step may be reversible due to similar thermodynamic stability of intermediates 47 and 48. Nevertheless, the rapid interception of the carbon radical in 48 by the cyano group can drive the process forward to yield the iminium radical 49, and thence the more stable ketyl radical 50 after the β-cleavage. Single-electron oxidation of 50 followed by deprotonation furnished the product.
Scheme 16.
Synthesis fluorinated ketonitriles via photoredox-promoted 1,4-cyano migration (Zhu, 2017).
Scheme 16.
Synthesis fluorinated ketonitriles via photoredox-promoted 1,4-cyano migration (Zhu, 2017).
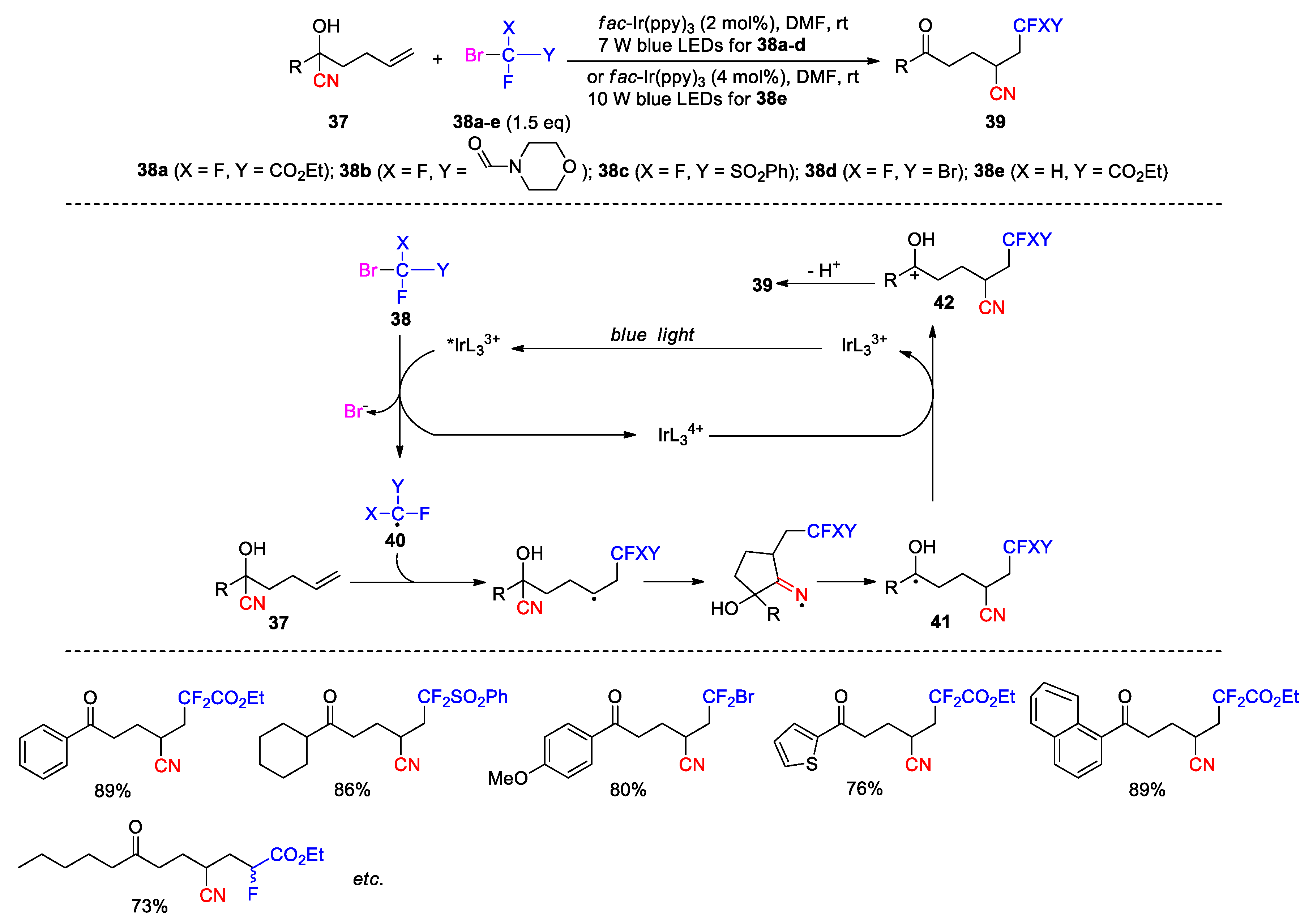
Scheme 17.
Synthesis of 1,8-diketones via photoredox-promoted coupling and 1,4-cyano migration (Zhu, 2019).
Scheme 17.
Synthesis of 1,8-diketones via photoredox-promoted coupling and 1,4-cyano migration (Zhu, 2019).
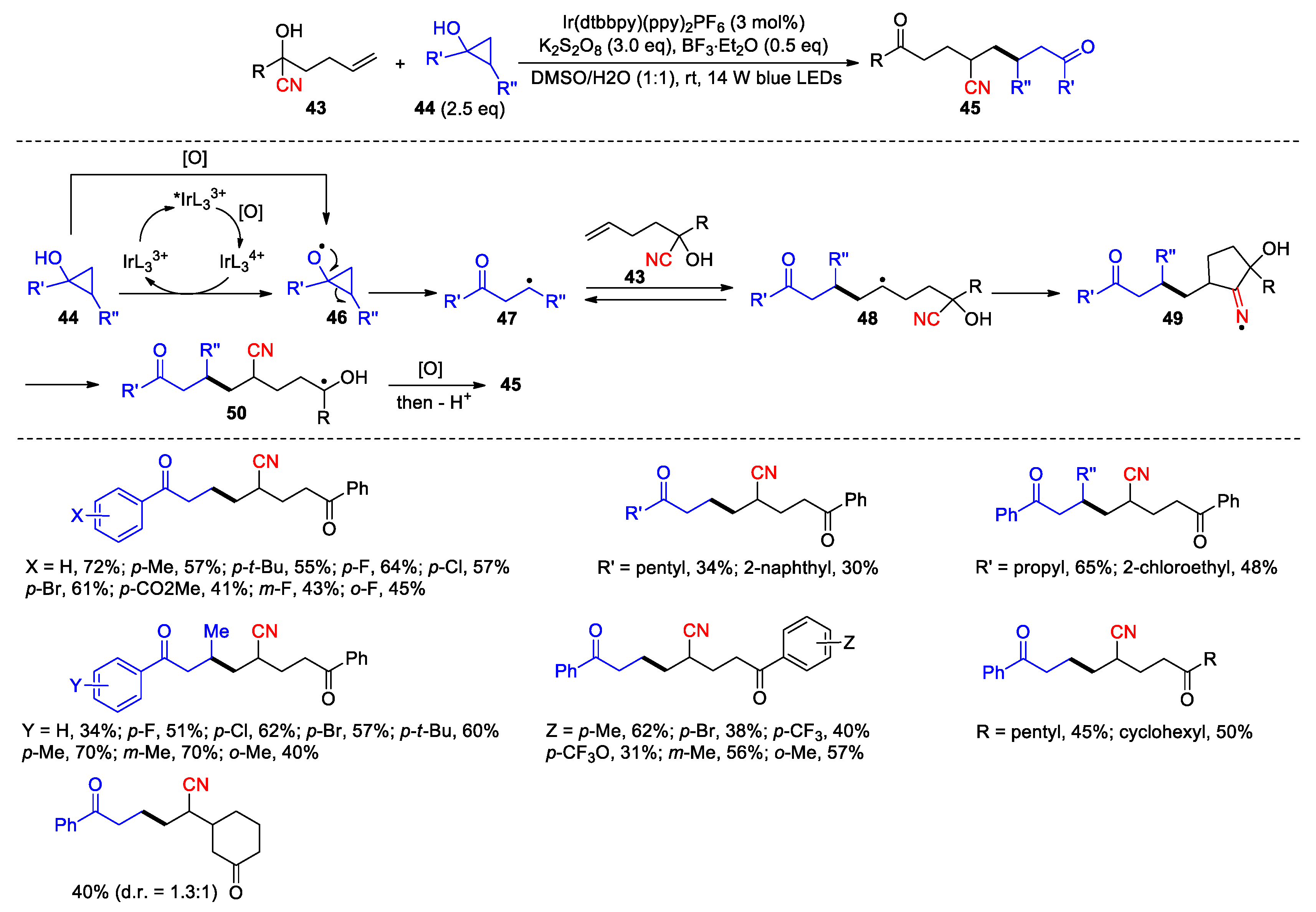
Moreover, the 1,2-difunctionalization of internal alkynes was realized in a study by Zhu et al. It was found that cyanohydrins 51, upon the treatment with AgSCF3 (1.5 equiv) and K2S2O8 (3 equiv), could be converted to the acrylonitrile products 52a and 52b as the mixtures of E-regioisomers (25 examples, 52a/52b = 2:1 to 8:1, 40-83%) (Scheme 18) [71]. In the event, addition of the in-situ generated CF3S radical occurs at both end of the alkyne. The resulting alkenyl radicals then engage in the 1,4- or 1,5-cyano migration to yield the ketyl radicals via cyclic iminium species. Oxidation of these intermediates followed by deprotonation lead to the isomeric products. The ratios between 52a and 52b indicated that the 1,4-cyano migration resulting from the outer-carbon addition (a) should occur more readily than the 1,5-migration triggered by the addition on the inner carbon atom (b).
Scheme 18.
Cyanotrifluoromethylthiolation of alkynes via radical-mediated CN migration (Zhu, 2018).
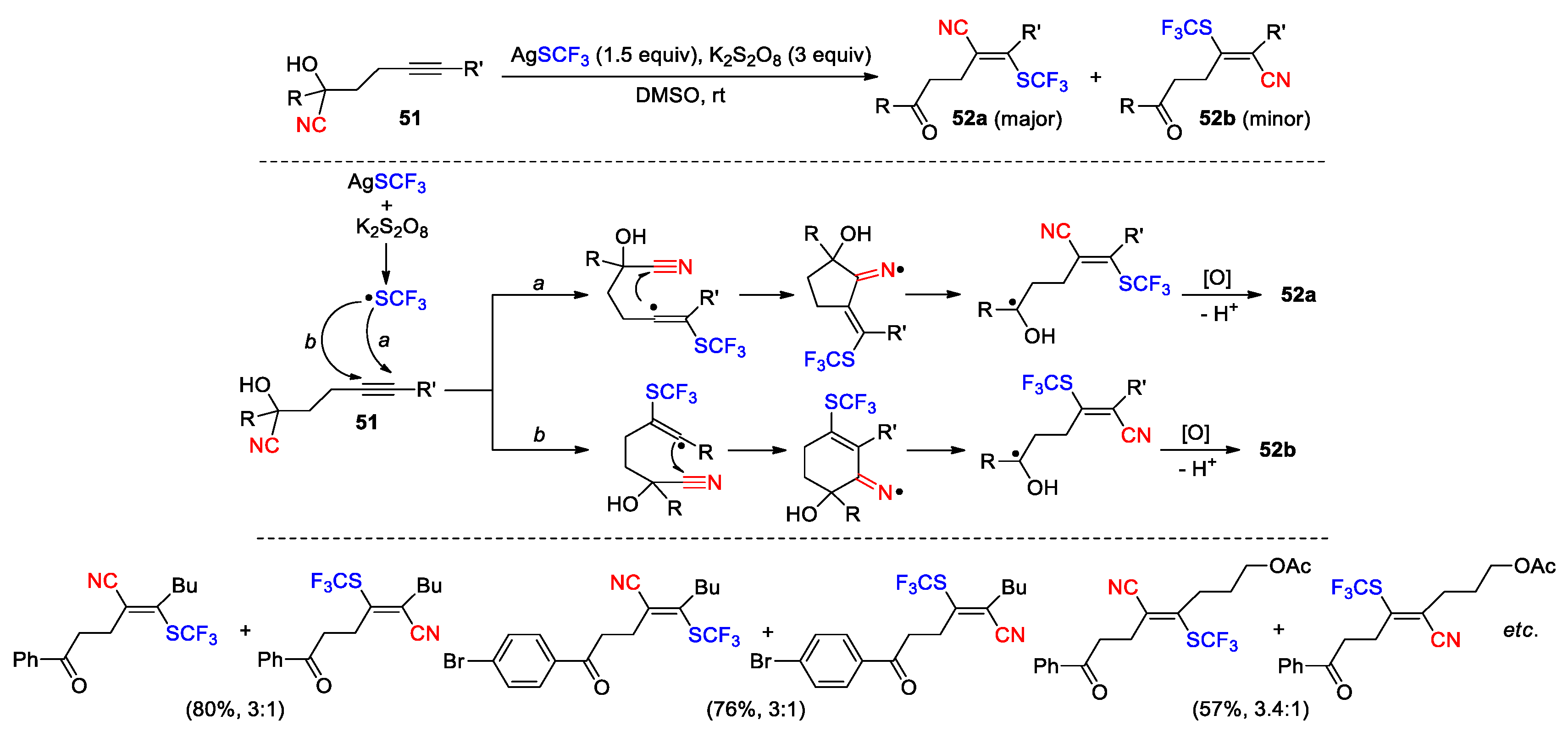
Given the efficient generation of carbon- or heteroatom-centered radicals, the aforementioned studies by Zhu et al. has demonstrated the feasibility of achieving the cyanofunctionalization of unsaturated CC bonds in cyanohydrins. In addition, the contributions from several other groups have also been noticed in the literature. In 2016, Liu et al. collectively reported a series of reactions of acyclic or aryl-tethered trimethylsilyl-protected cyanohydrines 53, enabling the generation of the corresponding β-functionalized nitriles 54-58 via the trifluoromethyl, azido, phosphonyl, sulfonyl or perfluoroalkyl radical-triggered 1,4(5)-cyano migration followed by oxidation and TMS-deprotection (Scheme 19) [72]. The trifluoromethylation and azidation reactions were conducted with Togni’s and Zhdankin reagents in the presence of CuI catalyst, while diphenyl phosphine oxide and AgNO3 were employed to effect the phosphonylation. The sulfonylation and perfluoroalkylation were both performed under photoredox catalysis by using p-toluenesulfonyl chloride or fluorinated sulfonyl chlorides as radical sources. Although not providing in the report, the detailed mechanism for the generation of initiating radical species as well as the rational for catalysis can be found in or deduced from other literature [73,74,75,76,77,78]. In a more recent report (2024), Li et. al. similarly employed CF3SO2Cl as radical precursor to achieve the trifluoromethylation of cyanohydrins as in the production of 58 (Rf = CF3) but without using metal catalyst (455 nm Blue LED/diethyl ether/rt). The photo-induced C-Cl cleavage followed by desulfonylation was proposed to account for the generation of CF3 radical [79].
Scheme 19.
Diverse difunctionalization of alkenes via radical-mediated 1,4(5)-CN migration (Liu, 2016).
Scheme 19.
Diverse difunctionalization of alkenes via radical-mediated 1,4(5)-CN migration (Liu, 2016).
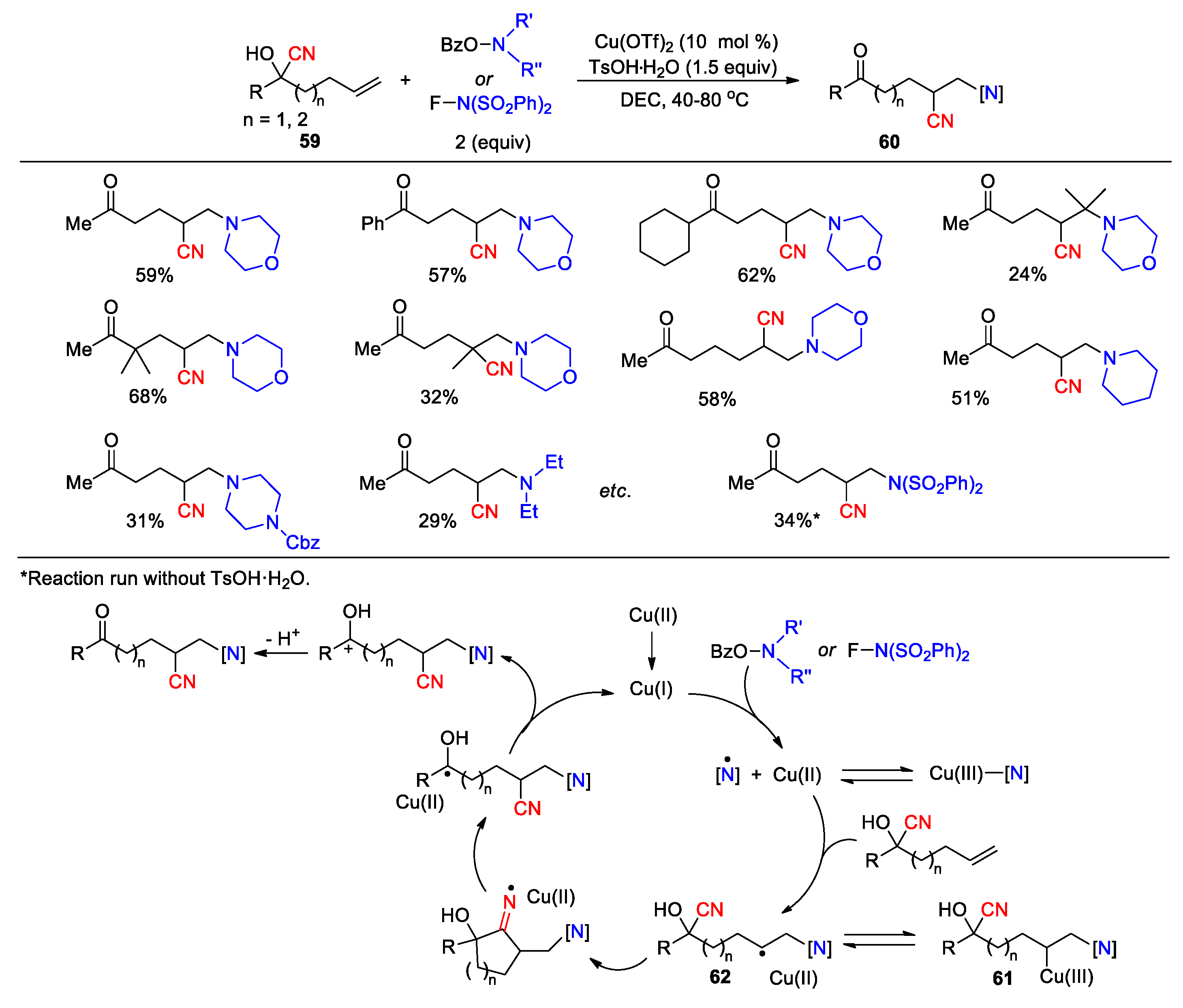
Wang and co-worker have developed another copper-catalyzed method for the 1,2-aminocyanation by using O-benzoylhydroxylamines and N-fluorobenzenesulfonimide (NFSI) as nitrogen precursors. Treatment of cyanohydrins 59 with these reagents in the presence of 10 mol% of Cu(OTf)2 and 1.5 equiv of TsOH afforded β-amino and β-sulfonimido nitriles 60 in varying yields (29 examples, 24-68%) (Scheme 20) [80]. The positive effect of p-TsOH on the formation of the desired product was observed but had not been fully understood by the authors. In the proposed mechanisms, the employed Cu(OTf)2 is first activated into the Cu(I) catalyst presumably through the action of a nitrogen-containing ligand (O-benzyolhydroxylamine or NFSI) [81]. The resulting Cu(I) catalyst further engages in the oxidative addition with O-benzyolhydroxylamine or NFSI to generate an amino-Cu(III) complex. Addition of this complex to the alkene affords the complex 61, which then undergoes reversible single-electron transfer to yield copper(II) and the secondary carbon radical 62. Alternatively, intermediate 62 can be produced through the addition of a N-centered radical released from the amino-Cu(III) complex to the olefin. Subsequent cyano migration through the cyclic iminium radical intermediate provides a more stable ketyl radical. Finally, the SET oxidation by Cu(II) can transfer this radical to a carbocation, affording the product after deprotonation and regenerating the copper(I) catalyst.
Scheme 20.
Copper-catalyzed 1,2-aminocyanation of alkenes via 1,4-CN migration (Wang, 2020).
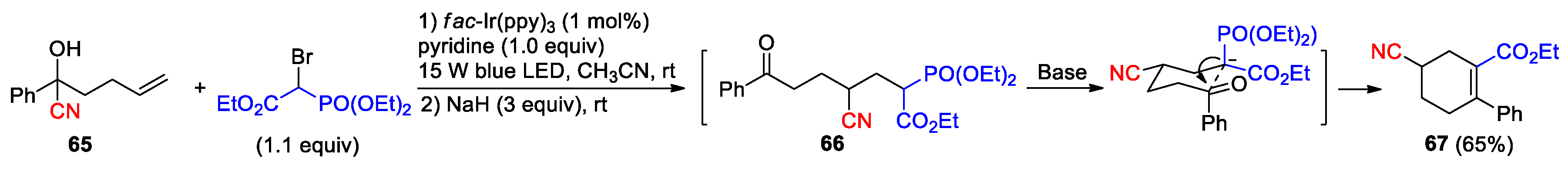
Two isolated photoredox reactions have also been reported by using fac-Ir(ppy)3 as catalyst. As a part of investigating the distal migration of a variety of functional groups, Ngai et al. demonstrated that an aroyl radical, generated upon the reduction of 2,4,6-trichlorobenzoyl by the excited photoredox catalyst, could add to the alkene in the TMS-protected cyanohydrin 63 to trigger the 1,4-cyano migration and give the formation of the β-cyanated-1,6-diketone 64 after a further oxidation-deprotection sequence (Scheme 21) [82]. Feng et al. described the construction of cyclohexene 67 by merging the catalytic reaction between the cyanohydrin 65 and 2-bromo-2-phosphorylacetate with the one-pot HWE olefination of the resulting product 66 (Scheme 22) [83]. The proposed pathway leading to 66 is similar to that shown in Scheme 16 for the generation of 39.
Scheme 21.
Benzoylcyanation of alkene via Photoredox-promoted 1,4-CN migration (Ngai, 2019).

Scheme 22.
Construction of cyclohexene via radical-mediated cyanofunctionlization of alkene and HWE olefination (Feng, 2021).
Scheme 22.
Construction of cyclohexene via radical-mediated cyanofunctionlization of alkene and HWE olefination (Feng, 2021).
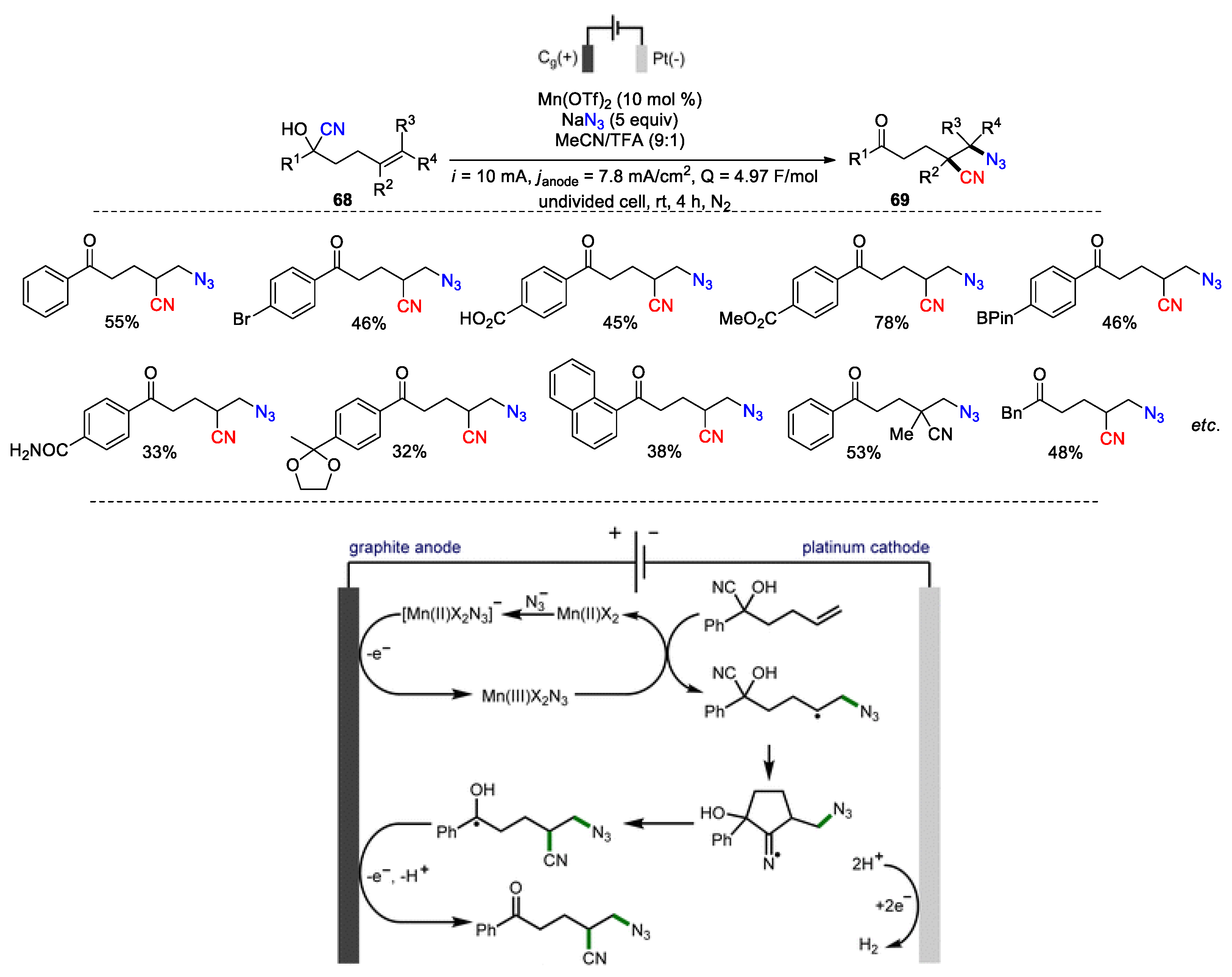
The azidocyanation reaction shown in Scheme 14 utilizes PIDA as a high molecular weight oxidant, generating stoichiometric organic waste that requires separation from the desired products. In a conceptually distinct contribution, Morrill and co-workers have developed an alternative oxidant-free protocol to achieve the azidocyanation of alkenes under electrochemical conditions. It was found that a system composed of Mn(OTf)2 and NaN3 as both azide source and electrolyte in MeCN/TFA was capable of converting aromatic (R1 = aryl) and aliphatic (R1 = alkyl) cyanohydrins 68 into the 1,2-azidonitriles 69 upon galvanostatic cycling (28 examples, 33-55%) (Scheme 23). [84]. Besides, an array of useful functional groups, such as halogen, carboxylic acid, ester, borane, amide, and ketal in the 4-position of phenyl ring were tolerated, giving the potential for further derivatization. Based on the results obtained from the radical clock and cyclic voltammetry studies, the authors proposed that the process is initiated by the formation of [Mn(II)X2N3] from Mn(II)X2 and NaN3, which is oxidized at the anode to form Mn(III)X2N3. This intermediate may deliver an azide radical to the cyanohydrin, giving a secondary alkyl radical. After the 1,4-nitrile migration, a second oxidation of the resulting ketyl radical at the anode, followed by proton loss provides the final product. Meanwhile, hydrogen gas is evolved via proton reduction at the cathode.
Scheme 23.
Electrochemical alkene azidocyanation via 1,4-nitrile migration (Morrill, 2022).
2.3.3. CN Migration with Alkenyl Nitriles
Radical-addition-triggered CN migration has also been widely used for the multi-site functionalization of alkenyl nitriles, mostly as 5-enenitriles. The resulting carbon radicals generated in this fashion were shown undergo a variety of subsequent transformations (Scheme 13, b), which are discussed below in four aspects: i) radical coupling, ii) oxidation to carbocations followed by nucleophilic attack, iii) formation of CC double bonds via oxidation/deprotonation, coupling/elimination or HAT sequence, and iv) reduction to a carboanions followed by protonation or nucleophilic substitution.
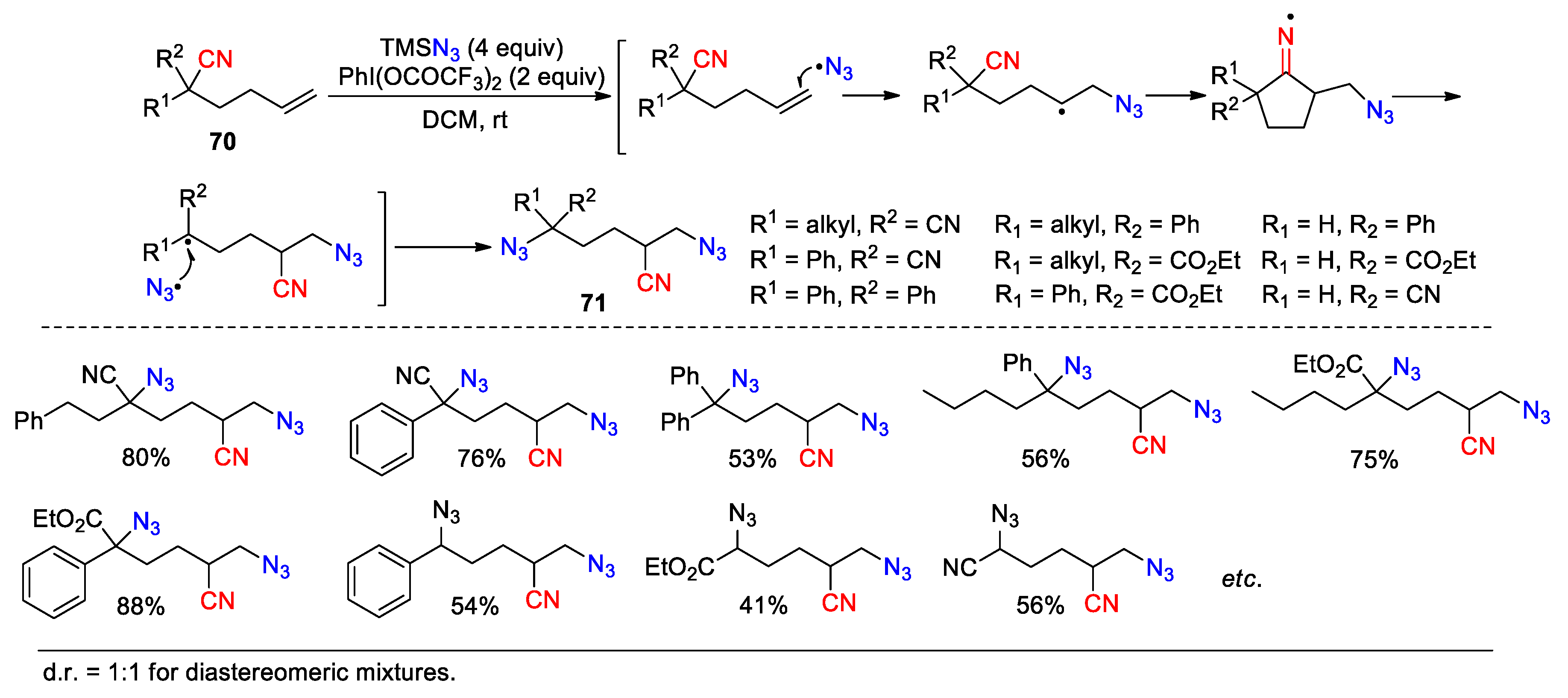
First, they can be captured by external radical species, either identical to or different from those used for the addition step (Scheme 24, Scheme 25, Scheme 26 and Scheme 27). Zhu et al. in 2022 reported the conversion of a variety of hexenenitriles 70 into the diazidation products 71 by using TMSN3 as the radical source and PIFA as a stoichiometric agent (30 examples, 41-90%) (Scheme 24) [85]. In the process, azido radicals should be generated in similar way as shown in Scheme 14. After this, one azido radical triggers the 1,4-cyano migration through the addition to 70, while the other one intercepts the resulting carbon radical to give the product. Once again, the presence of at least one radical stabilizing substituent (R1 and/or R2 = CN, Ph, CO2Et) is required to facilitate the β-C-C bond cleavage in the cyclic iminyl radical intermediate.
Scheme 24.
Trifunctionalization of hexenenitriles via azido radical-mediated cyano migration (Zhu, 2022).
Scheme 24.
Trifunctionalization of hexenenitriles via azido radical-mediated cyano migration (Zhu, 2022).
Yang et al. have developed a photolytic protocol for the trifluoromethylimination of alkenes by using (diphenylmethylene)-1,1,1-trifluoromethanesulfonamide 72 as a bifunctional reagent. In the event, a benzophenone (BP)-mediated photosensitized energy transfer (EnT) promotes the successive N-S-C bond cleavage in 72 to generate a diphenyl iminyl and trifluoromethyl radical pair after the extrusion of SO2. Consecutive delivery of these radicals to an alkene provides the β-CF3 imine product. Moreover, KOH was used as an external base to remove the released SO2. When the reactions were performed with the alkenes 73 tethered to a cyano group, the 1,4-CN migration occurred prior to the radical coupling to give the trifunctionalization products 74 as shown in Scheme 25 [86].
Scheme 25.
Photochemical alkene trifluoromethylimination involving 1,4-CN migration (Yang, 2023).
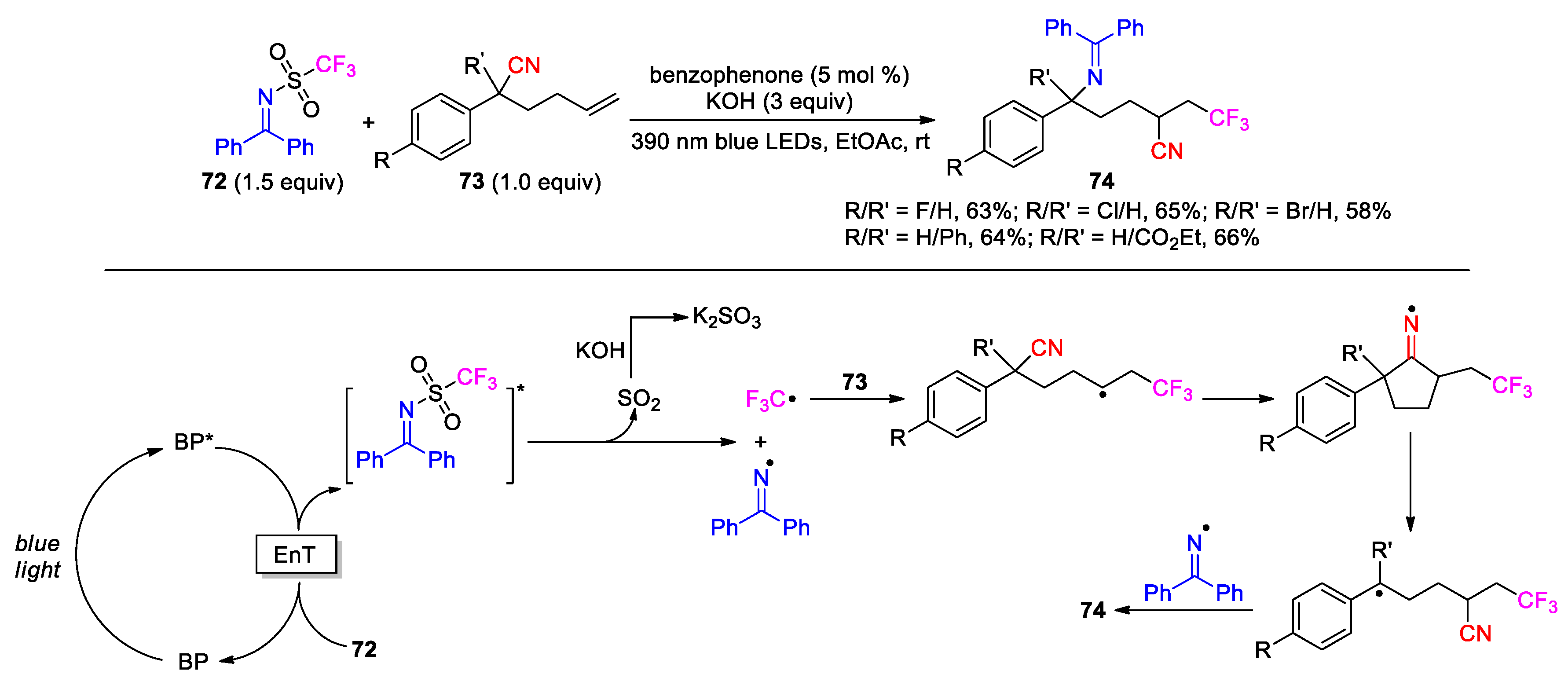
The radical-mediated trifunctionalization of alkenyl nitriles has also been realized via N-heterocyclic carbene (NHC) organocatalysis. In 2023, Du et al. reported an NHC-catalyzed three-component reaction of hexenenitriles 75 with aromatic aldehydes 76 and trifluoromethyl iodide, which provided the products 77 (33 examples, 55-87%) by using a thiazolium salt as precatalyst and K2CO3 as base (Scheme 26) [87]. In the proposed pathway, the carbene 78 derived from the precatalyst reacts with 76 to form enaminol 79 (so called Breslow intermediate) and further enolate 80 after proton abstraction. A single electron transfer (SET) from 80 [88] to CF3-I would produce NHC-bound ketyl anion radicals 81 along with a trifluoromethyl radical. Subsequent addition of the CF3 radical to 75 followed by the 1,4-CN migration provides carbon radical 82, which then undergo the coupling with 81 to give intermediates 83. Finally, collapse of 83 leads to the product 77 and regeneration of carbene 78 for the next cycle. In addition to CF3I, this radical-relay protocol was also applicable to several other fluorinated reagents including BrCF2CO2Et, CF2Br2 and C6F13I as well as ICH2Ts. However, it was not compatible with the tested aliphatic aldehyde as a replacement for 76.
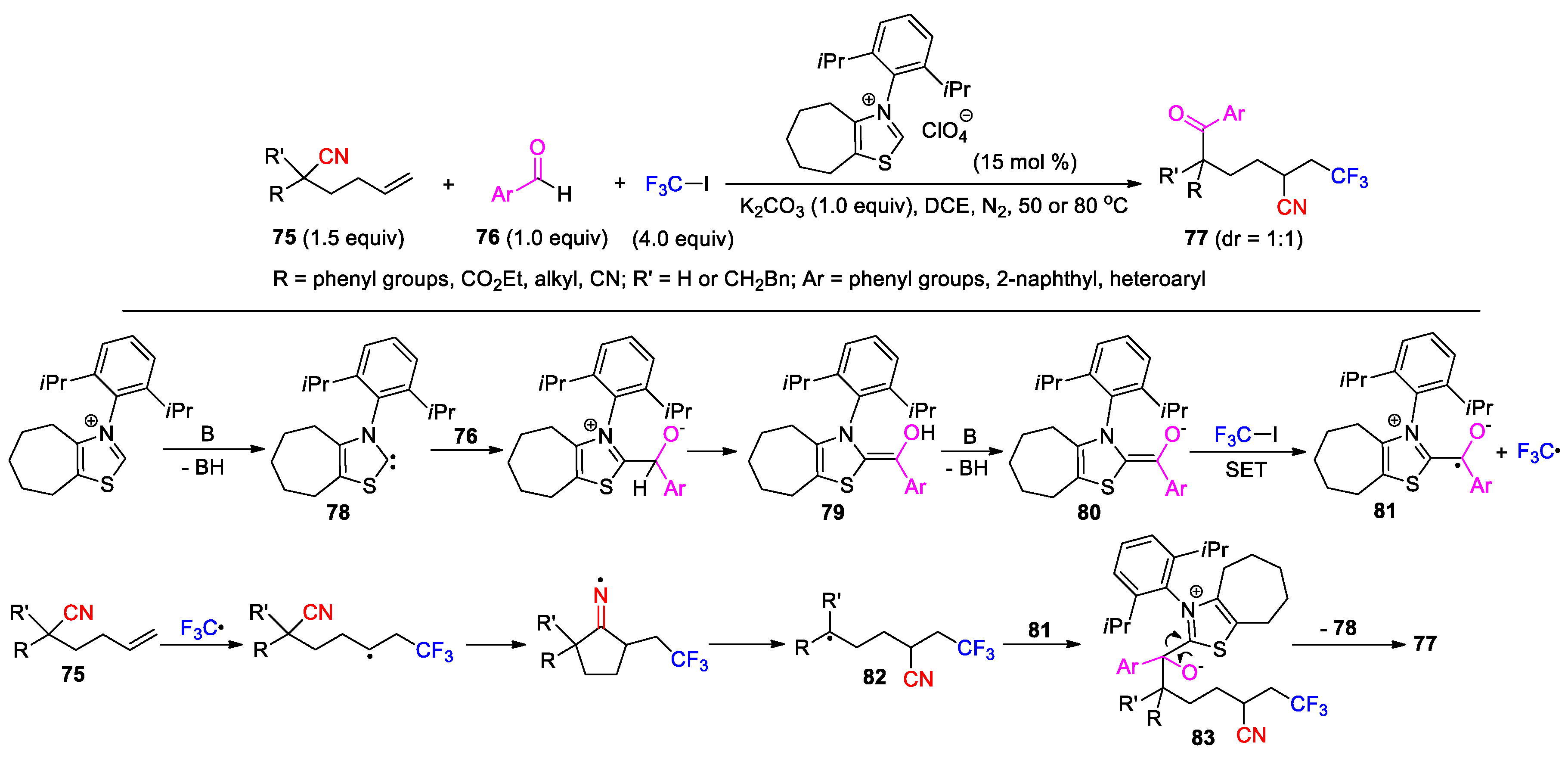
More recently, the same research group has described an alternative method for the trifluoromethylation-acylation of hexenenitriles by instead using aromatic acids and CF3SO2Na as the reactants. A dual NHC/photoredox catalysis system comprising a triazolium salt, Ir(dtbbpy)(ppy)2PF6 catalyst and Cs2CO3 was capable of effecting the transformation upon visible-light irradiation [89]. Besides, this method also requires the pre-activation of the acids into the corresponding benzoylimidazoles through the in-situ treatment with 1,1’-carbonyldiimidazole. As a typical example, the reaction of hexenenitrile 75a with CF3SO2Na and acid 84 under the developed conditions affords the product 77a in good yield (Scheme 27). A likely mechanism involves the addition of NHC 85 to the carbonyl group of benzoylimidazole 86, giving acyl azolium 87. Meanwhile, the single-electron oxidation of CF3SO2Na by the excited catalyst produces a CF3 radical along with the Ir(II) species that is sufficiently reducing to donate an electron to 87, thereby generating NHC-bounded radical anion 88. Addition of the CF3 radical to 75a followed by cyano migration provides benzylic radical 89, which can couple with 88 to yield a highly congested intermediate 90. Finally, decomposition of 90 furnishes the product 77a with regeneration of 85 to complete the cycle.
Scheme 26.
NHC-catalyzed radical relay trifluoromethylation-acylation of hexenenitriles via cyano migration (Du, 2023).
Scheme 26.
NHC-catalyzed radical relay trifluoromethylation-acylation of hexenenitriles via cyano migration (Du, 2023).
Scheme 27.
Dual NHC/photoredox catalytic trifluoromethylation-acylation of hexenenitriles via cyano migration (Du, 2024).
Scheme 27.
Dual NHC/photoredox catalytic trifluoromethylation-acylation of hexenenitriles via cyano migration (Du, 2024).
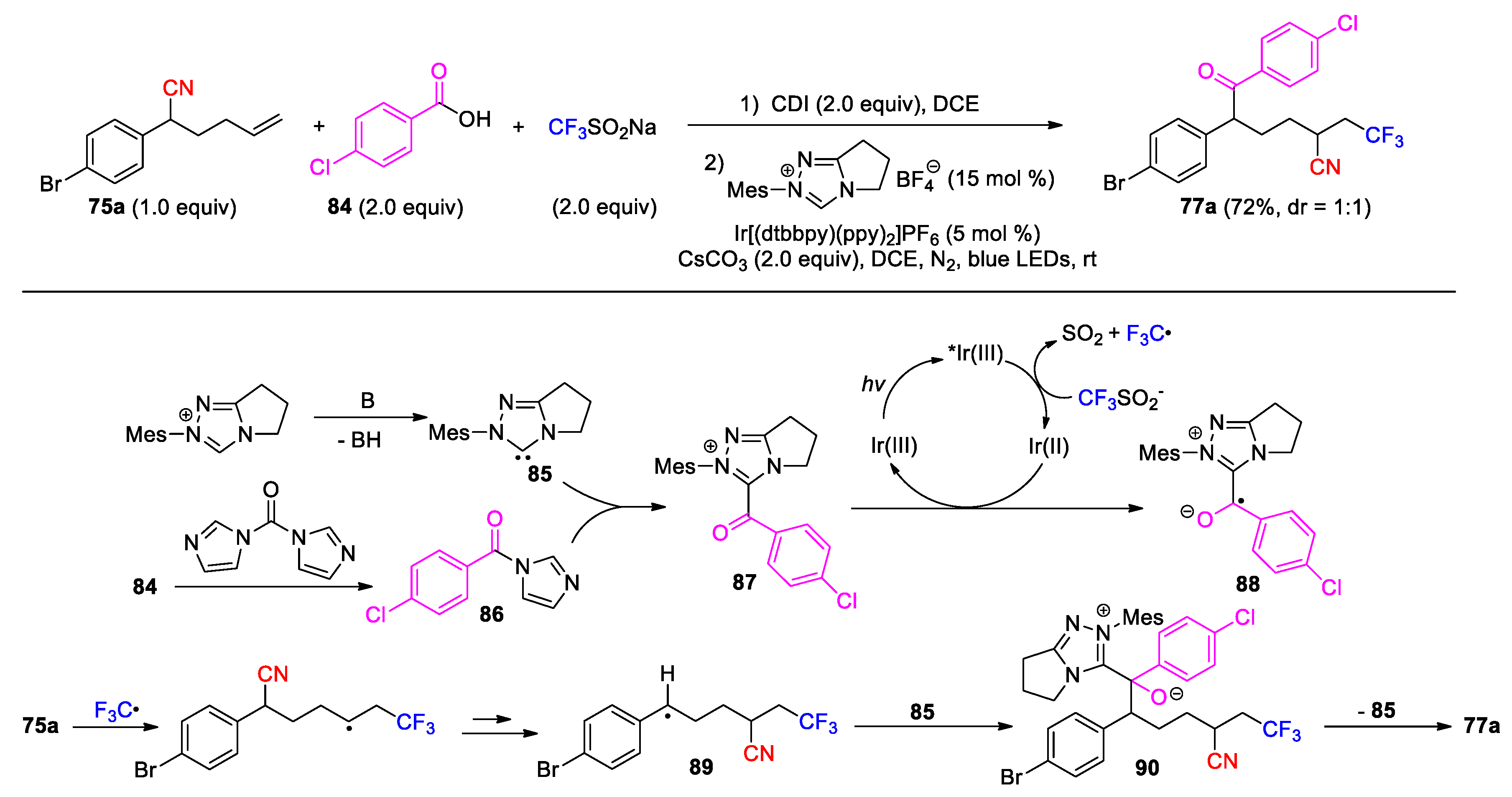
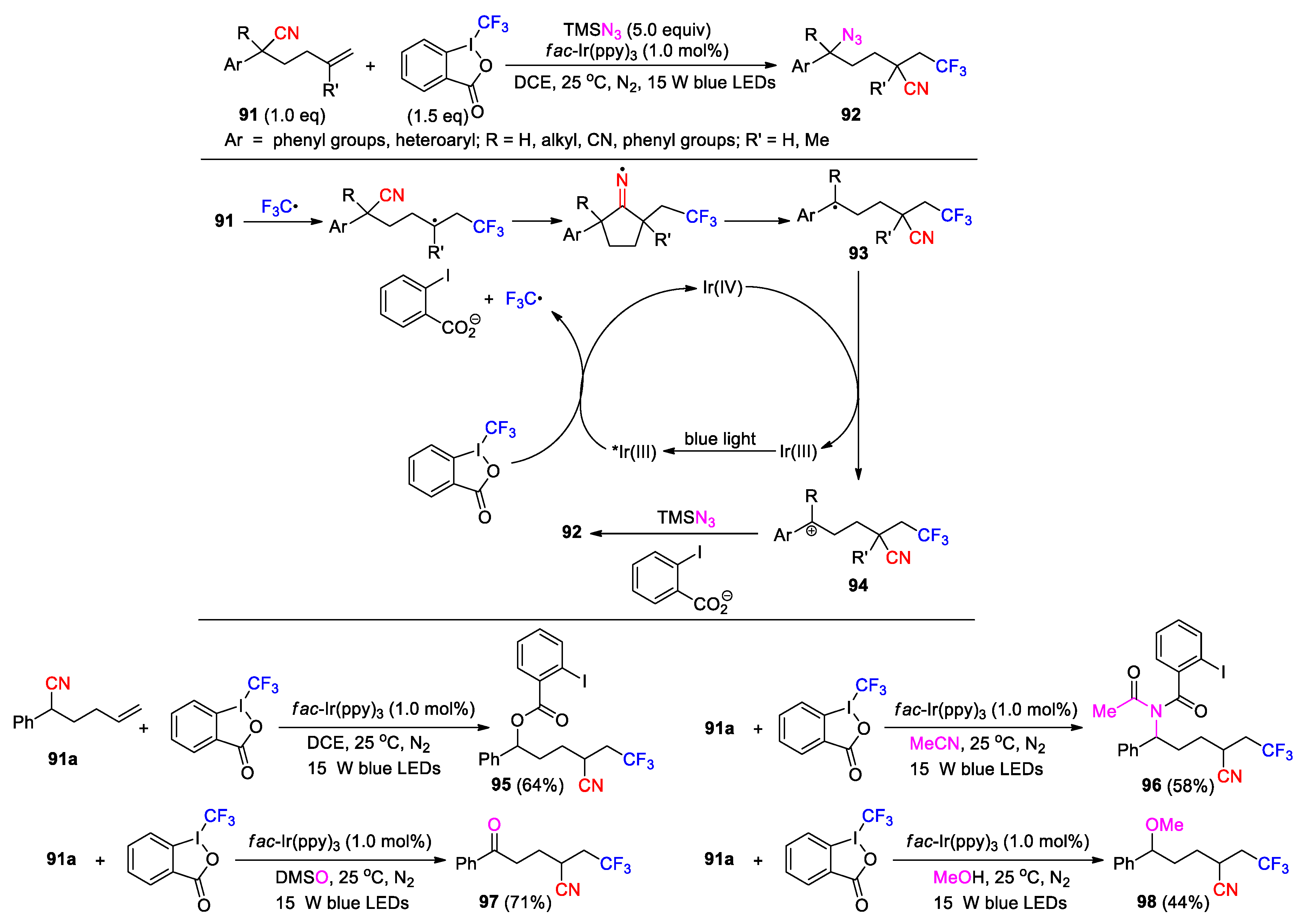
The carbon radical resulting from the CN migration can be oxidized into a carbocation, thus offering an opportunity for the incorporation of second functional group via nucleophilic attack (Scheme 28, Scheme 29 and Scheme 30). Chen and Zhu’s group in 2022 reported the photoredox trifunctionalization of 5-hexenenitriles 91 using Togni’s reagent II as radical source and TMSN3 as nucleophile precursor. The reaction was conducted with fac-Ir(ppy)3 catalyst to give the polyfunctionalized products 92 upon visible light irradiation in DCE (31 examples, 32-86%) (Scheme 28) [90]. In the process, reduction of Togni’s reagent by the excited Ir(III) catalyst gives a trifluoromethyl radical and 2-iodobenzoate. The addition of the CF3 radical to 91 triggers the 1,4-CN migration to provide a benzyl radical 93 after the cyclization and β-cleavage of the iminyl radical intermediate. Then, single-electron oxidation of 93 by Ir(IV) species proceeds to afford the corresponding carbocation 94 with the regeneration of ground state Ir(III) catalyst. Finally, nucleophilic attack of the azide anion on 94 furnishes the product. It was additionally found that the reaction could be carried out in concert with other external nucleophiles. In the absence of TMSN3, Togni’s reagent served as both the radical source and the nucleophile precursor to give the ester product 95. When MeCN was employed as the solvent, a Ritter-type addition dominated to yield the amidation product 96 via subsequent nucleophilic attack by 2-iodobenzoate and Mumm rearrangement. DMSO and MeOH were also attempted as the nucleophiles, leading to the formation of the products 97 and 98, respectively.
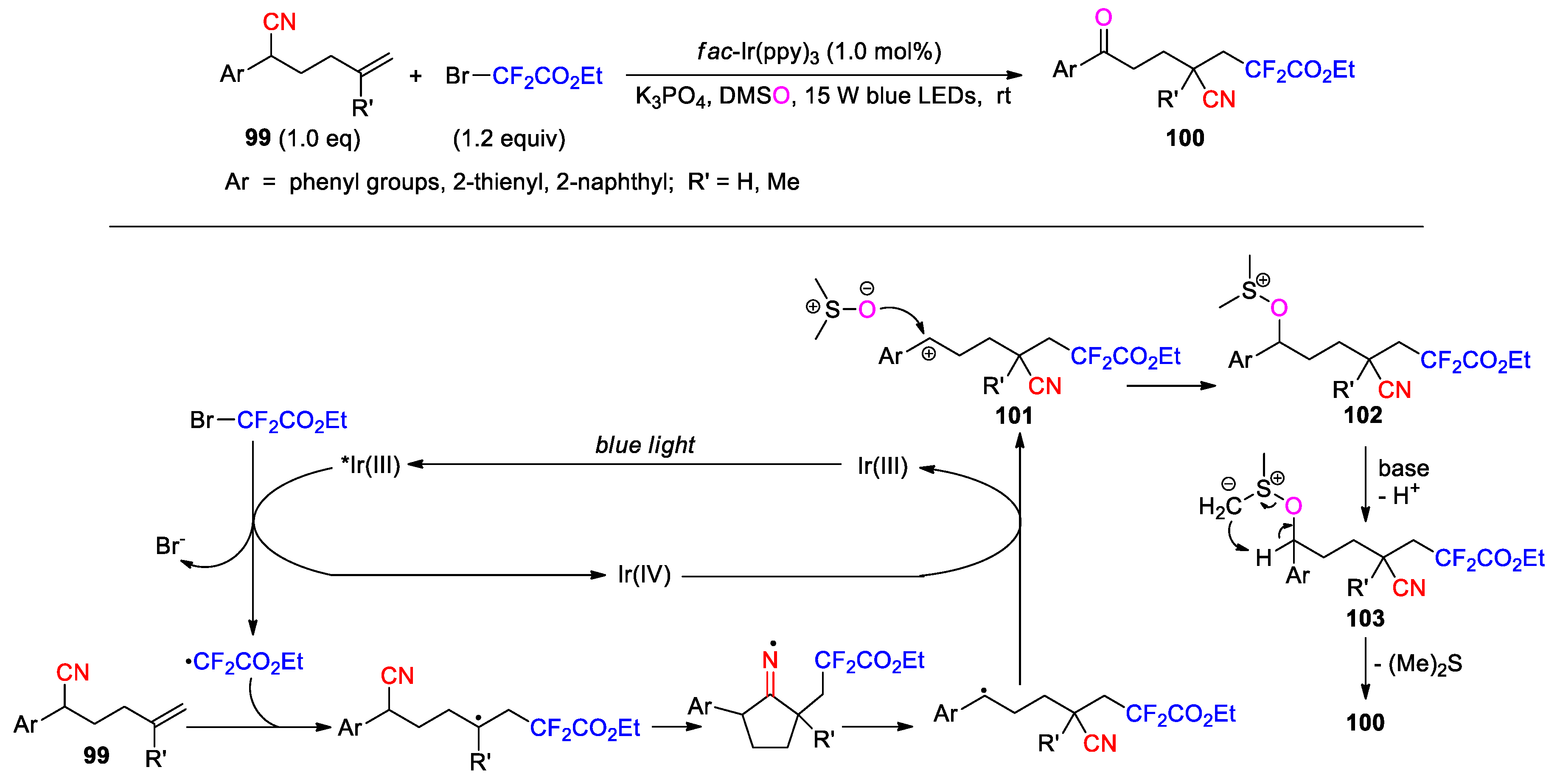
Guo later described the photoredox conversion of 5-hexenenitriles 99 to difluoroalkylated ketonitriles 100 through the reaction with BrCF2CO2Et in DMSO using fac-Ir(ppy)3 as catalyst and K3PO4 as a base (Scheme 29) [91], in which DMSO acts as both solvent and nucleophile as in the formation of 97. Once the carbocation intermediate 101 is formed via the oxidation of the benzylic radical, it will be captured by DMSO to give an alkoxysulfonium ion 102. Further deprotonation affords an alkoxysulfonium ylide 103, which then converts to the product through a [2,3]-sigmatropic shift.
More recently, O-acyl hydroxylamines, such as 105, have been employed as bifunctional reagents to achieve the diamidation of 5-hexenonitriles 104 under photoredox catalysis. The reaction was conducted with 1 mol % of fac-Ir(ppy)3 in acetonitrile (as in the reaction to give 96) to afford the distal-imido β-amino nitriles 106 in varying yields (22 example, 21-81%) (Scheme 30) [92]. Mechanistically, the O-acyl hydroxylamine undergoes the cleavage upon the single-electron reduction by the excited Ir(III) catalyst, affording a N-centered radical and a carboxylate anion. The 1,4-CN migration induced by the radical addition provides a benzylic radical that can be further oxidized into a carbocation by Ir(IV). The carbocation is then intercepted by acetonitrile to give a nitrilium ion. Attack of the nitrilium ion by the carboxylate anion combined with Mumm rearrangement furnish the desired product.
Scheme 28.
Photoredox functionalization of hexenenitriles with Togni II reagent and external nucleophiles (Chen, 2022).
Scheme 28.
Photoredox functionalization of hexenenitriles with Togni II reagent and external nucleophiles (Chen, 2022).
Scheme 29.
Photoredox functionalization of hexenenitriles with BrCF2CO2Et in MeCN (Guo, 2023).
Scheme 30.
Photoredox diamidation of hexenenitriles using O-acyl hydroxylamine in MeCN (Akondi, 2025).
Scheme 30.
Photoredox diamidation of hexenenitriles using O-acyl hydroxylamine in MeCN (Akondi, 2025).
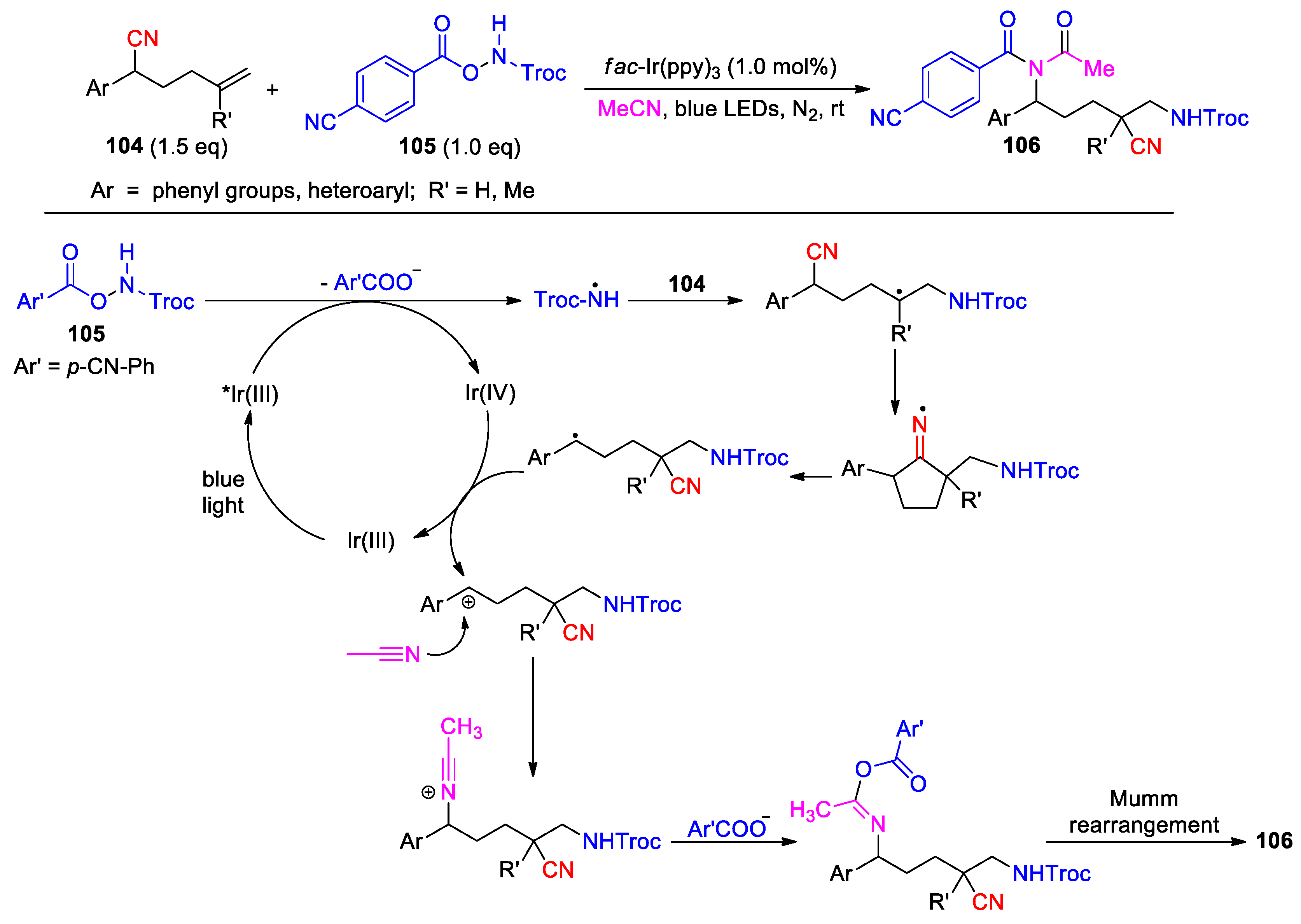
In some reported cases, the employed reaction conditions enabled the in-situ creation a carbon-carbon double bond after distal cyano group migration (Scheme 31, Scheme 32 and Scheme 33). Zhu and co-workers have developed a method allowing for the cyanosulfonylation of terminal alkenes with concurrent incorporation of a new C=C bond into the structure [93]. By using fac-Ir(ppy)3 as catalyst and Na2CO3 as base, the photo-reaction of diphenyl hexenenitriles 107 with sulfonyl chlorides 108 afforded olefinic β-cyanosulfones 109 via consecutive sulfonylation, cyano migration, single-electron oxidation of the benzyl radical followed by deprotonation (26 examples, 35-88%) (Scheme 31). The reaction accommodates both aryl and aliphatic sulfonyl chlorides containing a broad range of substituents, but is not compatible to the alkyl-phenyl- or dialkyl-substituted hexenenitrile due to the weakened stability of radical or cation intermediate. Moreover, the desired products were obtained as the mixtures of Z/E isomers from unsymmetrically-substituted substrates (R1 ≠ R2).
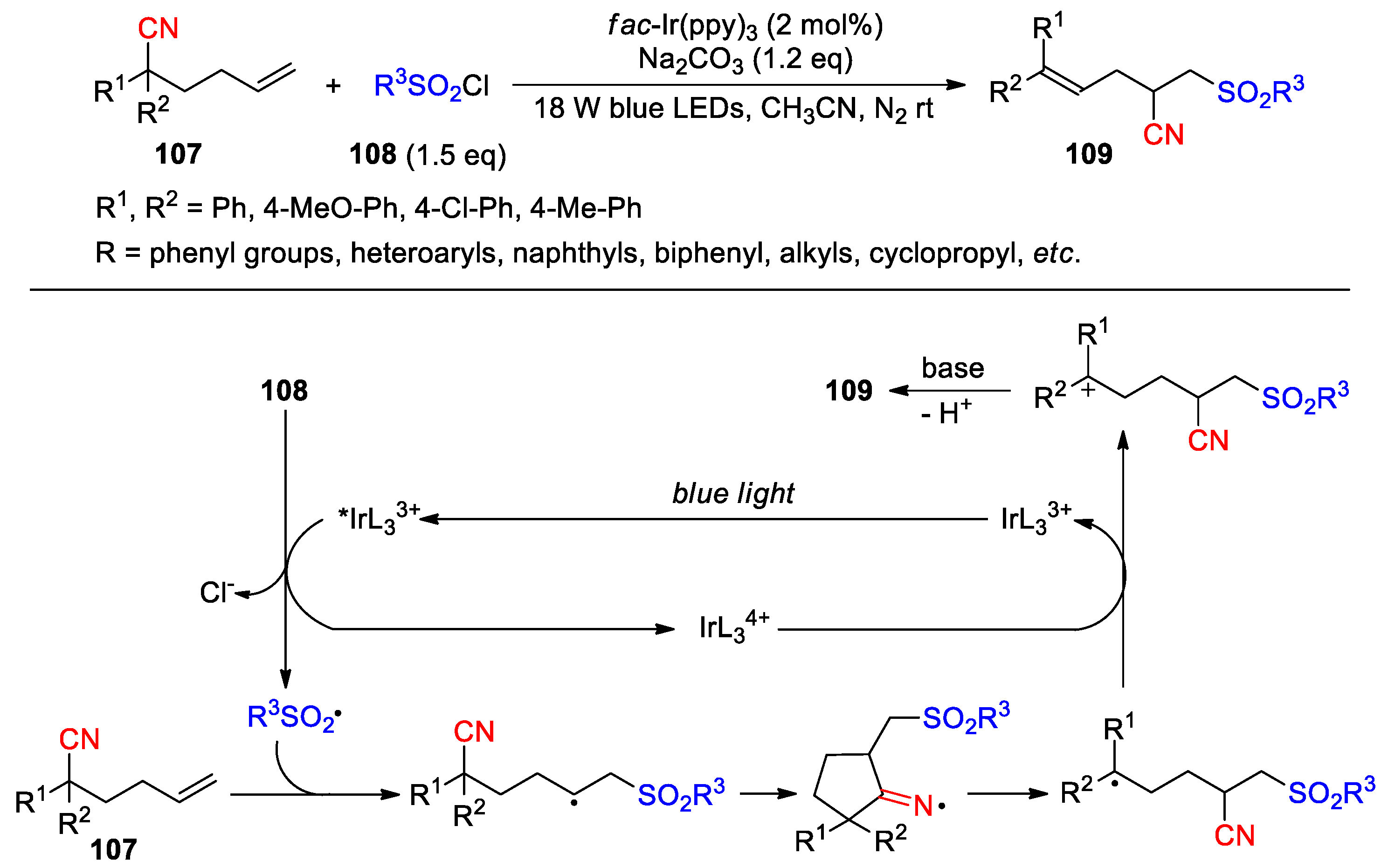
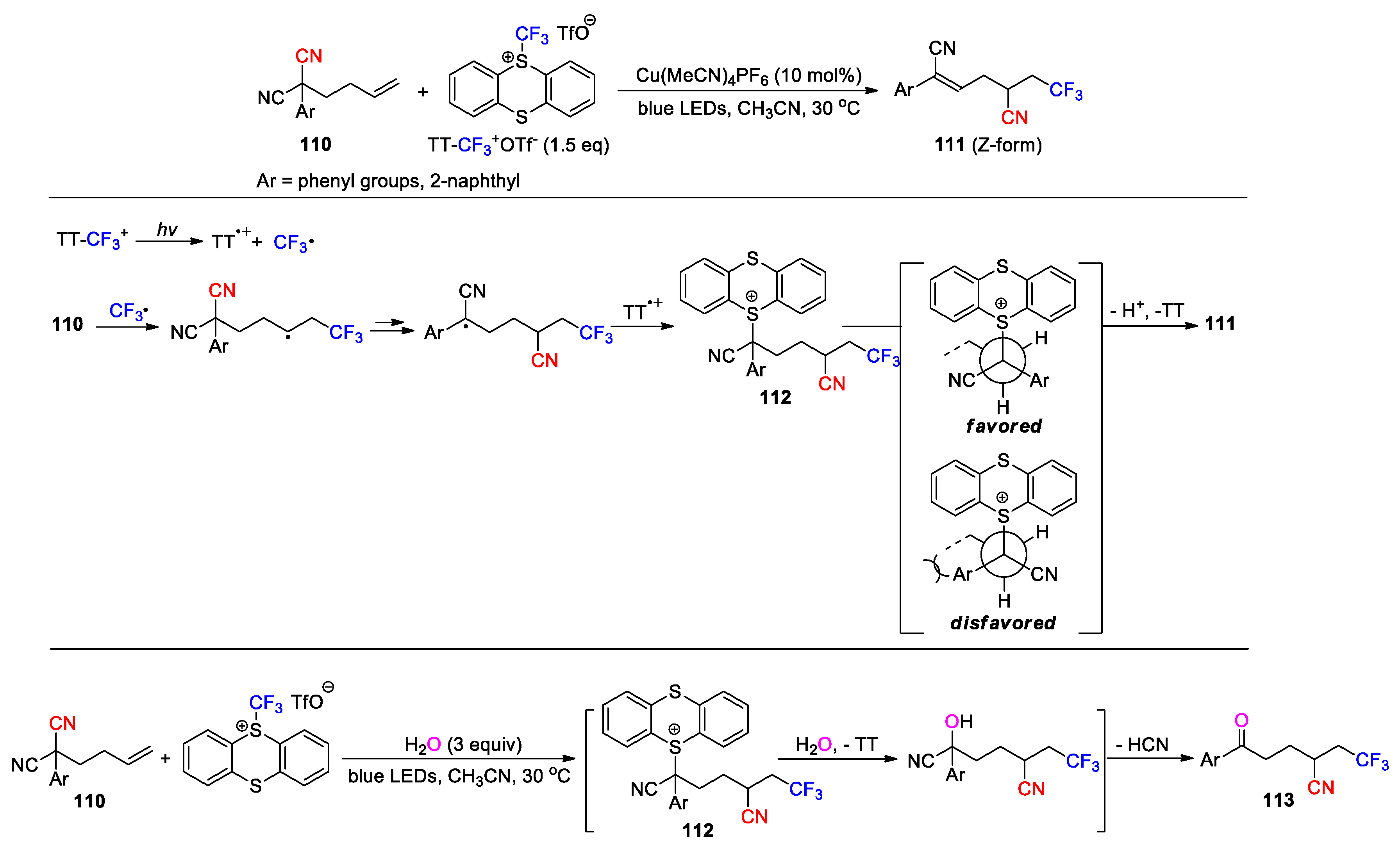
Huang et al. in 2023 demonstrated the conversion of 2-aryl- 2-(but-3-en-1-yl)malononitriles 110 into trifluoromethylated (Z)-alkenyl products 111 using trifluoromethyl thianthrenium triflate (TT-CF3+ OTf−) as reagent and Cu(MeCN)4PF6 as catalyst under the illumination of blue LEDs (19 examples, 49-82%) (Scheme 32) [94]. The reaction was carried out with phenyl- or 2-naphthyl-substituted substrates, which worked particularly well for the those possessing para-electron-donating groups at the phenyl ring. It is postulated that TT-CF3+ can fragment into TT·+ and trifluoromethyl radical upon absorption of blue light. Addition of the CF3 radical to the terminal olefin followed by the 1,4-cyano migration leads to a tertiary radical intermediate, which is captured by TT·+ to afford an α-thianthrenium cyano species 112 as ascertained through high-resolution mass spectrometry. Intermediate 112 then undergoes a trans-elimination, possibly with the assistance of the copper catalyst, to provide the product and release TT. The remarkable Z-selectivity observed in this reaction is rationalized by the conformation adopted for the trans-elimination, in which the steric interaction between the bulky aryl group and alkyl chain can be significantly minimized. Furthermore, addition of 1 mol % of Ir[dF(CF3)ppy]2(dtbbpy)PF6 to the reaction mixture was shown to improve the yield of the desired product (Ar = Ph, 93% vs. 70%), but result in poorer diastereoselectivity (Z/E ratio = 3:1) due to an alkene isomerization enabled by the energy transfer photocatalyst. The report also revealed that the reaction pathway of intermediate 112 was altered in the presence of water, yielding the ketone products 113 via a nucleophilic substitution process (12 examples, 48-77%).
Scheme 31.
Formation of olefinic β-cyanosulfones via radical-mediated functionalization of diphenyl hexenenitrile (Zhu, 2023).
Scheme 31.
Formation of olefinic β-cyanosulfones via radical-mediated functionalization of diphenyl hexenenitrile (Zhu, 2023).
Scheme 32.
Photolytic reactions of alkenyl malononitriles with trifluoromethyl thianthrenium salt (Huang, 2023).
Scheme 32.
Photolytic reactions of alkenyl malononitriles with trifluoromethyl thianthrenium salt (Huang, 2023).
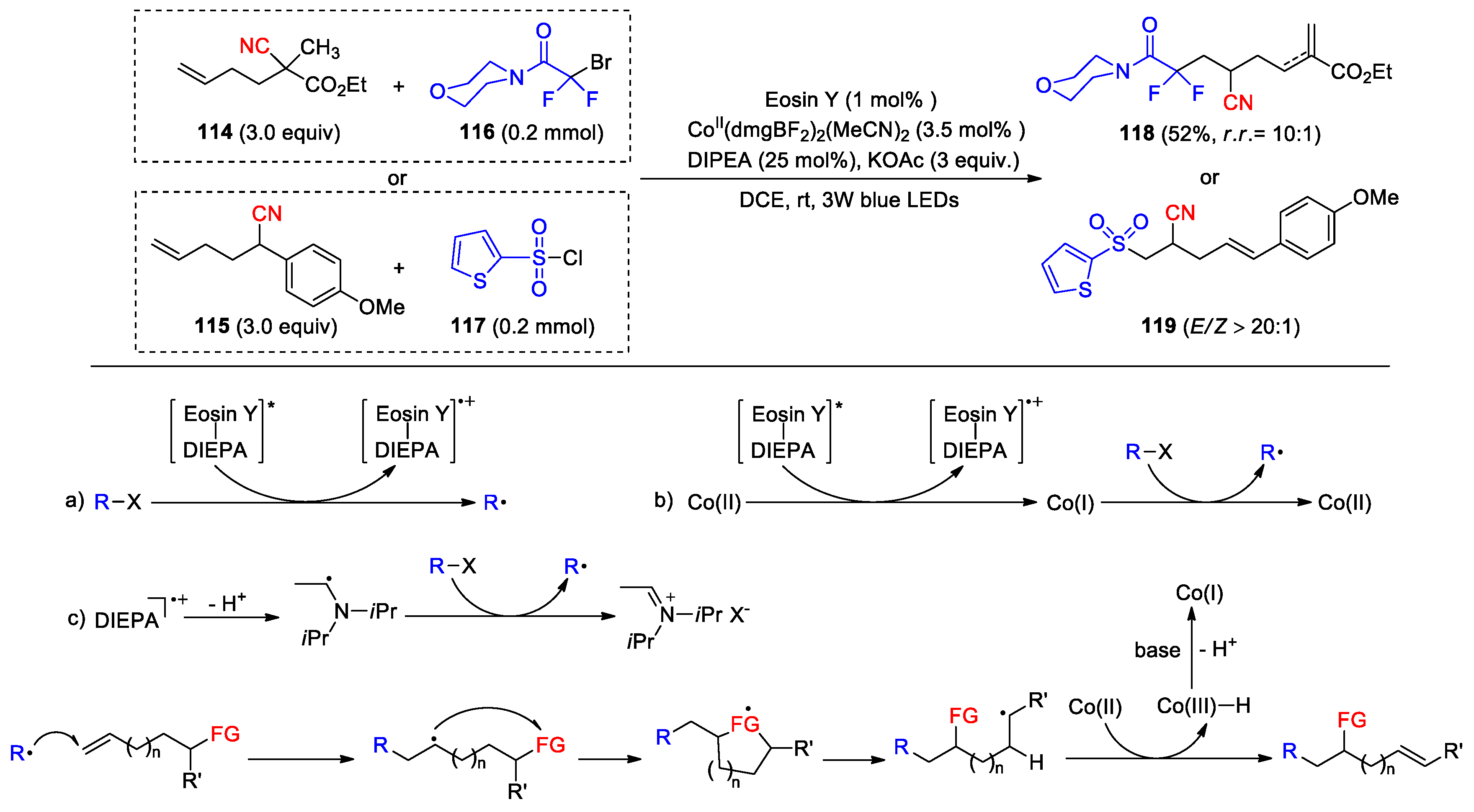
Employing photo/cobalt dual catalysis, Lei and co-workers has recently reported an alternative method for alkene difunctionalization with simultaneous creation of a C-C double bond via radical-triggered translocation of functional groups and Co-promoted remote C(sp3)-H desaturation [95]. It shows good compatibility with diverse fluoroalkyl and sulfonyl radical precursors, enabling the migration of benzoyloxy, acetoxy, formyl, heteroaryl and cyano groups in different types of alkenes to achieve the modular synthesis of polyfunctionalized products. In the study, ethyl 2-cyano-2-methylhex-5-enoate 114 and several phenyl-substituted 5-enenitriles (e.g., 115) were investigated for the 1,4- cyano migration using 2-bromo-2,2-difluoroacetamides such as 116 and sulfonyl chlorides such as 117 as radical precursors. The optimum reaction conditions involve the use of 1 mol% Eosin Y as the photocatalyst, 3.5 mol% CoII(dmgBF2)2(MeCN)2 as the HAT catalyst (dmg = dimethylglyoxime), 25 mol% DIPEA and 3 equiv. of KOAc as the base in a DCE solution under blue-LED illumination. The reactions proceeded smoothly to afford a total of 20 products with a remarkable site- and stereo-selectivity (E:Z > 20:1) as exemplified by 118 and 119 (Scheme 33).
On the basis of detailed mechanistic studies and DFT calculations, a general pathway involving the migration of functional groups (FG) was proposed. There may be several ways to generate radicals to trigger this reaction. Ultraviolet–visible and other experiments suggest the formation of a complex between Eosin Y and DIPEA. This complex, upon photoexcitation, could be reductively quenched by radical precursors R-X to form radicals (a) [96] or reduce cobaloxime(II) to Co(I). The Co(I) species then leads to radical generation via a halogen atom transfer (XAT) (b). In these processes, DIPEA may as a reducing agent to yield an aminoalkyl radical after an oxidative deprotonation. Thus, participation of the aminoalkyl radical in XAT activation to generate radicals along with the iminium salt is also feasible (c) [97]. The addition of the resulting radicals to olefin triggers the FG migration, yielding thermodynamically more stable radicals. With the formation of these carbon-centered radicals, the adjacent C(sp3)-H bonds can be effectively weakened, allowing for a cobaloxime(II)-assisted HAT process to give the products.
Scheme 33.
Photo/cobalt-catalyzed functionalization of alkenes via FGM and Co-promoted HAT (Lei, 2024).
Scheme 33.
Photo/cobalt-catalyzed functionalization of alkenes via FGM and Co-promoted HAT (Lei, 2024).
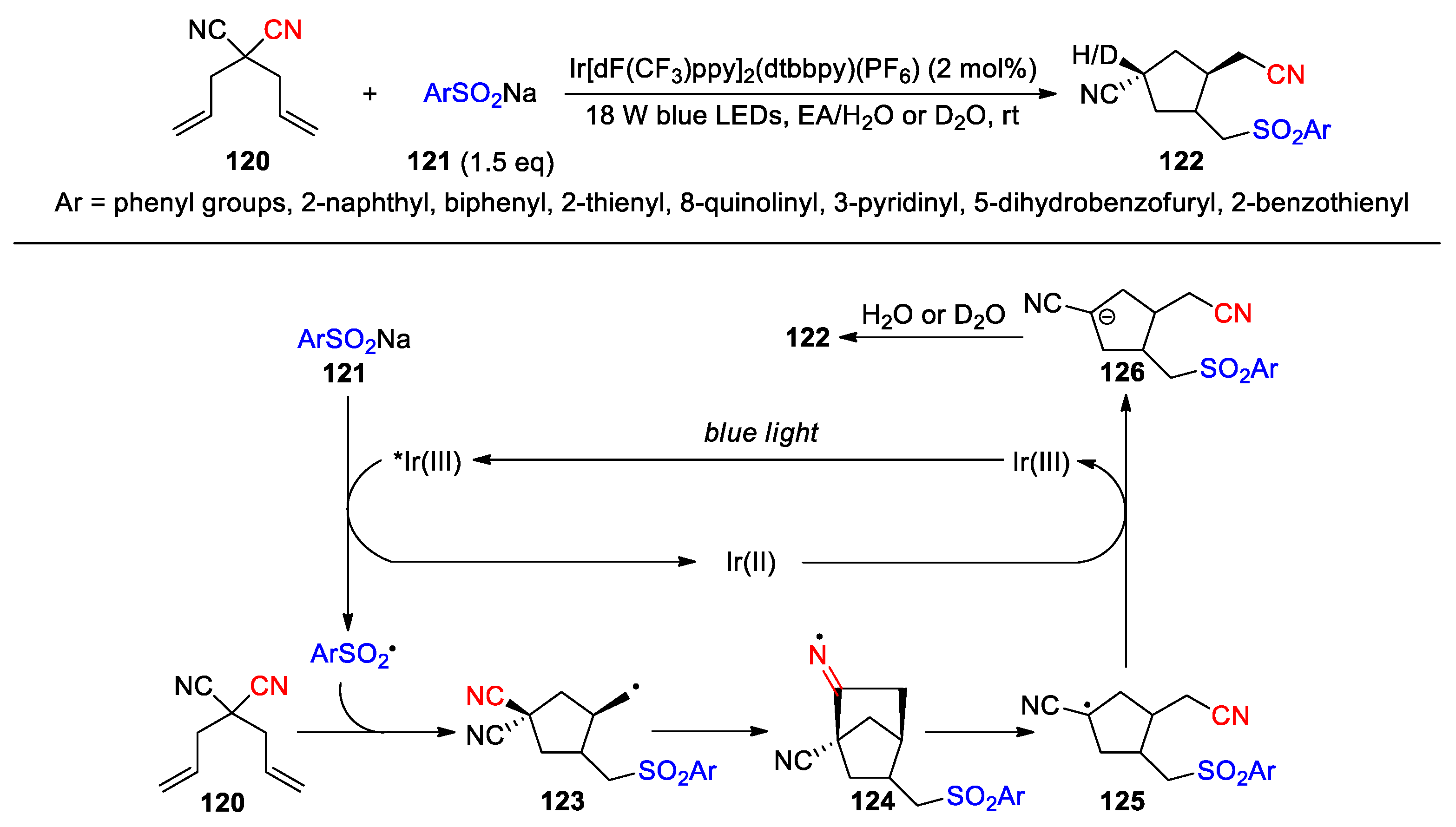
Finally, the carbon radicals resulting from 1,4-cyano migration can be in-situ reduced to carbanions that are capable of undergoing protonation or nucleophilic substitution (Scheme 34, Scheme 35 and Scheme 36). Zhu and co-workers have demonstrated the construction of trisubstituted cyclopentanes 122 via the photoredox catalytic reaction of 4,4-dicyano-1,6-diene 120 with sodium (hetero)aryl sulfinates 121 in the presence of H2O or D2O (Scheme 34) [98]. Mechanistically, the addition of an extra sulfonyl radical triggers the intramolecular cyclization of 120 and produces cyclopentane intermediate 123 bearing a primary alkyl radical. Intramolecular addition of the alkyl radical to the suprafacial cyano group affords the bicyclic iminyl radical 124, which subsequently undergoes a β-scission yielding intermediate 125. The β-scission is considered to be driven by the relief of ring-strain in 124 in addition to the radical stabilising nitrile group [99]. Reduction of the alkyl radical in 125 by Ir(II) gives carbanion 126 and regenerates the ground-state Ir(III). Finally, intermediate 126 can be trapped by a proton or a deuterium ion to furnish the product. An array of sodium sulfinates containing various aryl and heteroaryl groups was employed for the reaction, and the desired products were all obtained as diastereomeric mixtures in about 1:1 ratio (25 examples, 51-96%).
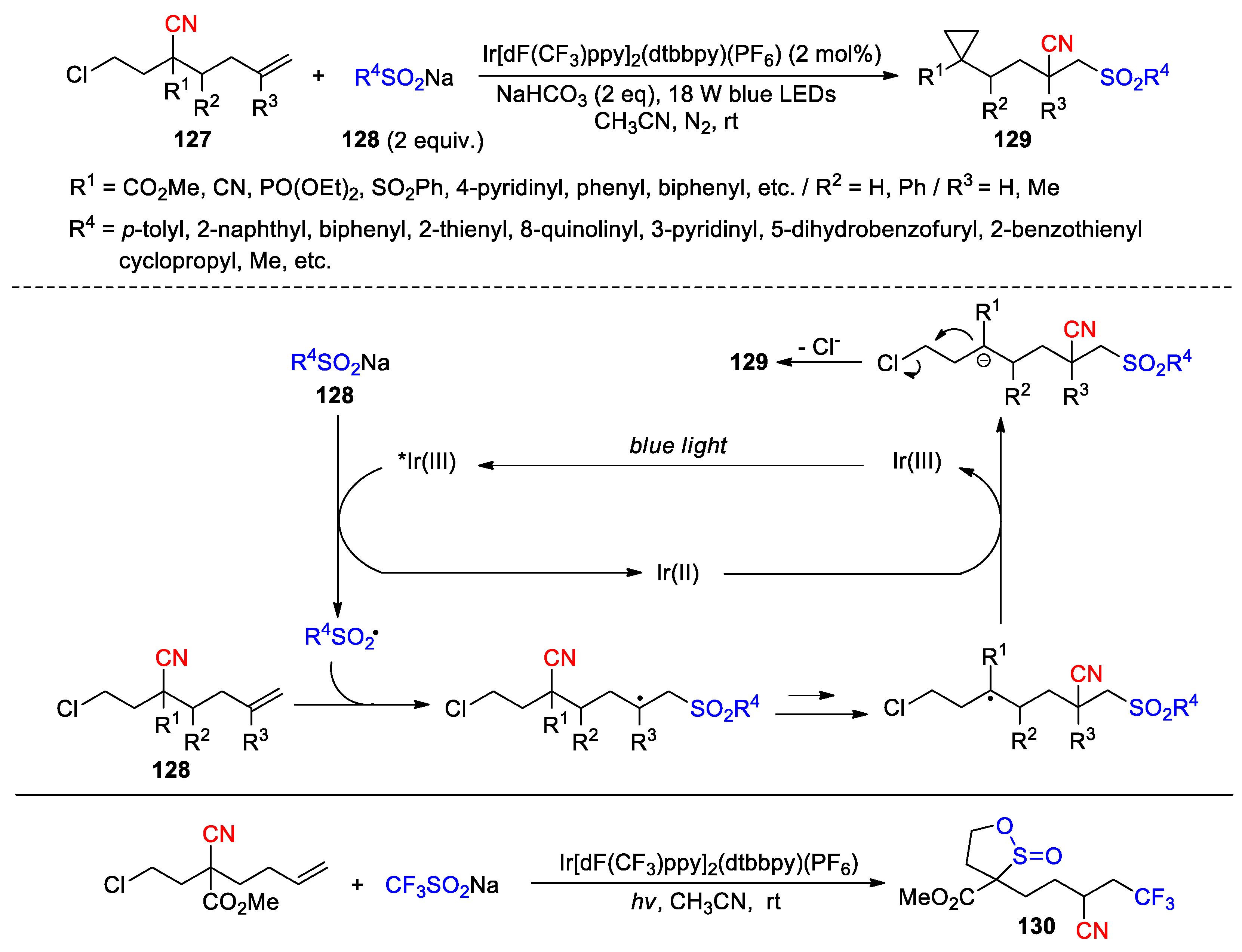
In another report, Zhu’s group disclosed a radical-polar crossover (RPC) reaction of 2-(2-chloroethyl)hex-5-enenitriles 127 with sodium sulfonates 128 under photocatalytic conditions, in which, the carboanions generated through the reduction of the radical intermediates were shown to displace the terminal chloride to form multisubstituted cyclopropanes 129 (40 examples, 22-98%) (Scheme 35) [100]. Both aryl and aliphatic sodium sulfonates could be used for the reaction, while the compatibility with aliphatic sulfinates is particularly noteworthy as they are susceptible to free radical desulfonylation under harsh conditions. The protocol also exhibited good tolerance to a wide range of functional groups present in 127 (R1), thus further enriching the diversity of the cyclopropane structures. Interestingly, when the reaction was conducted with Langlois’ reagent, the formation of a distinct sultine product 130 was observed through the insertion of SO2 during the cyclopropanation process.
Scheme 34.
Construction of substituted cyclopentanes via radical-triggered transannular cyano migration (Zhu, 2025).
Scheme 34.
Construction of substituted cyclopentanes via radical-triggered transannular cyano migration (Zhu, 2025).
Scheme 35.
Cyano migration-mediated radical-polar crossover cyclopropanation and formation of sultine product (Zhu, 2024).
Scheme 35.
Cyano migration-mediated radical-polar crossover cyclopropanation and formation of sultine product (Zhu, 2024).
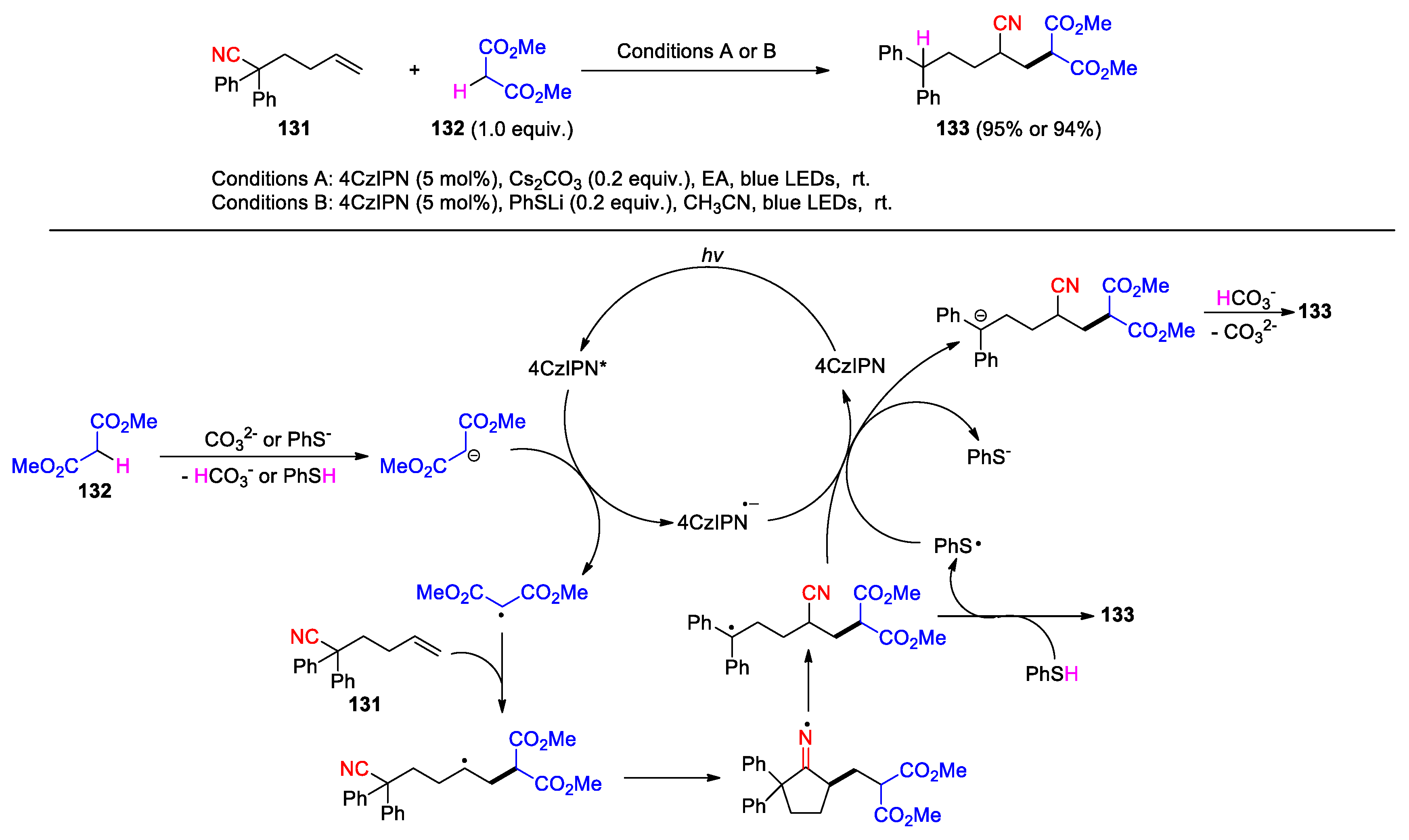
Using photoredox/ Brønsted–Lowry base dual catalysis, Deng et al. have developed a protocol for the alkylcyanation of unactivated alkenes with protic C(sp3)-H feedstocks via 1,4-cyano group migration [101]. As described in the report, several mono- or diaryl-substituted 5-enenitriles were allowed to react with dimethyl malonate, triethyl methanetricarboxylate, methyl-Meldrum’s acid, methyl acetoacetate or 1,3-cycloheptanedione in the presence of 2 mol% of 4CzIPN as the photocatalyst and 20 mol% of Cs2CO3 (Conditions A) or PhSLi (Conditions B) as base, leading to the generation of the alkylated nitrile products in varying yields depending on the reactivity of the substrates and radical precursors (13 examples, 24-95%). The reaction of 2,2-diphenylhex-5-enenitrile 131 with dimethyl malonate 132 to yield product 133 is illustrated as an example for all cases (Scheme 36). Mechanistically the carbanion generated via the deprotonation of 132 is oxidized by the photoexited 4CzIPN to give the malonate radical. Addition of the radical to 131 triggers the cyano migration leading to the benzylic radical intermediate that could be converted into 133 in two different ways. Under the conditions A, the benzylic radical may react with the reduced photocatalyst to deliver the benzylic anion. Further proton exchange with the conjugate acid furnishes 133 along with the neutral base. Under the conditions B, the product is achieved directly from the benzylic radical through the hydrogen atom transfer with PhSH. Reduction of the resulting phenyl sulfide radical by 4CzIPN•– species completes the photocatalytic cycle with regeneration of phenyl sulfide anion.
Scheme 36.
Alkylcyanation of unactivated Alkenes with protic C(sp3)-H feedstocks via cyano group migration (Deng, 2025).
Scheme 36.
Alkylcyanation of unactivated Alkenes with protic C(sp3)-H feedstocks via cyano group migration (Deng, 2025).
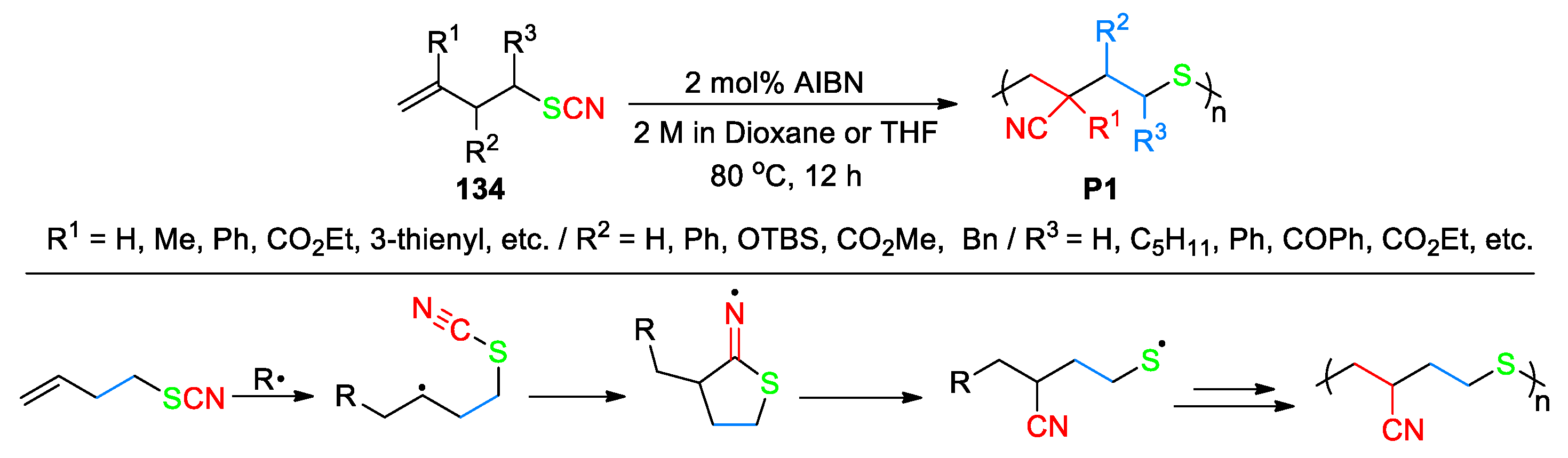
In addition to the aforementioned modifications, the radical addition-triggered cyano group migration has also been applied to polymer synthesis [102,103,104,105]. It is well-accepted that α-olefins and their functionalized derivatives are nearly impossible undergo homopolymerization under radical conditions due to the interference from chain-transfer side reactions. For instance, the highly reactive propagating secondary carbon-centered radicals tend to abstract an allylic hydrogen atom from another monomer to generate more stable allylic radicals, instead of adding to the double bond. To cope with this problem, Li et al. have recently reported a study on the homopolymerization of thiocyanate functionalized linear α-olefins enabled by radical-mediated 1,4-cyano transfer [104]. A vast array of monomers 134 with different functionalities could undergo such polymerization, allowing access to a library of ABC sequence-regulated polymers P1 with high molecular weights and unprecedented structures (Scheme 37). By taking 4-thiocyanato-1-butene as an example, the pathway proposed for its polymerization involves a 5-exo-dig addition of the initial carbon-centered radical onto the cyano group followed by β-fragmentation of the created iminyl radical intermediate to generate a relatively stable thiyl radical. The thiyl radical then performs rapid addition with another monomer, and so on to eventually lead to the polymerized product [Conv. > 95%; Mn = 28.7 k (Dioxane), 20.3 k (THF)]. In another noteworthy report, Sun et al. revealed the preparation of several ABC sequence-defined polymers from the corresponding functionalized hept-6-enenitriles via 1,5-cyano migration under radical conditions [105].
Scheme 37.
Homopolymerization of linear α-olefins enabled by radical-mediated 1,4-cyano group migration (2024, Li).
Scheme 37.
Homopolymerization of linear α-olefins enabled by radical-mediated 1,4-cyano group migration (2024, Li).
2.3.4. Recently Reported Cyano Migrations Involving Diradical or Radical Cation Intermediates
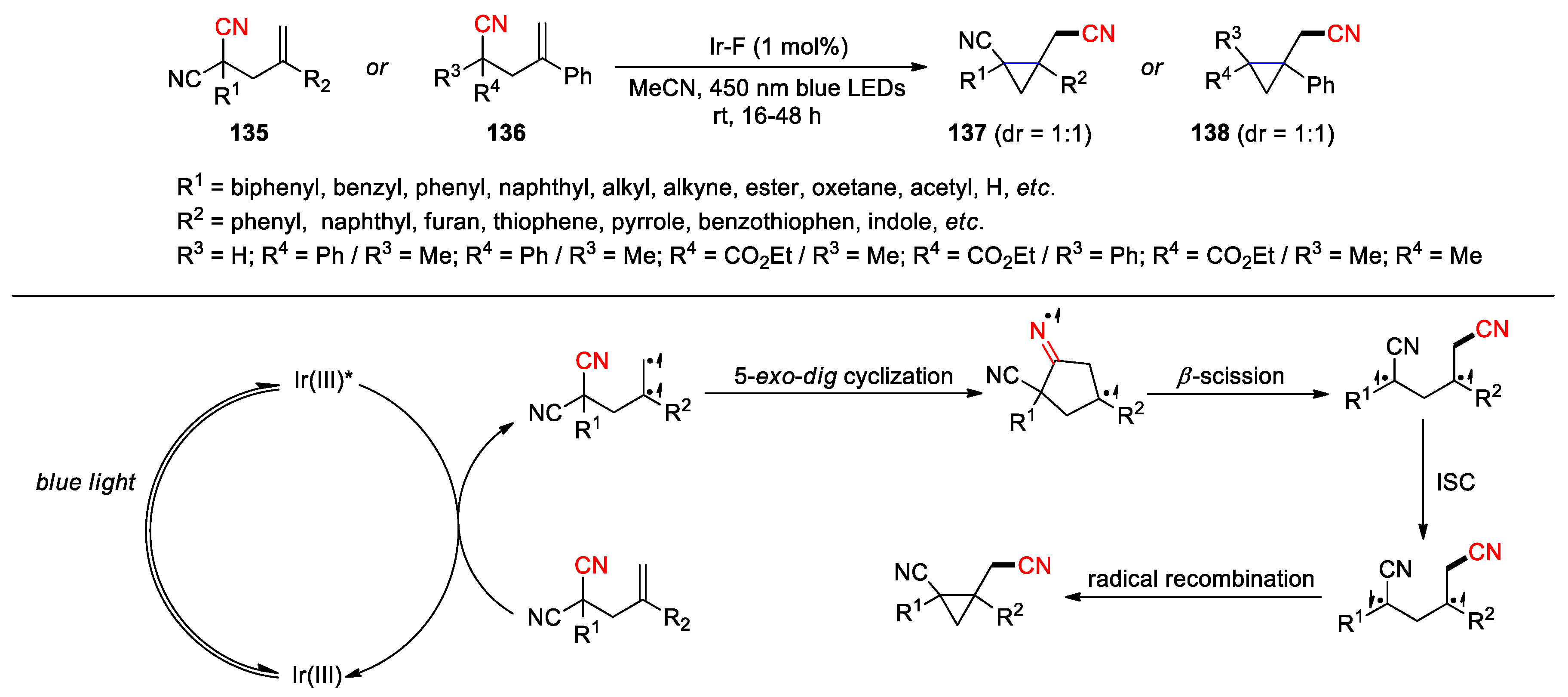
Recent advance in the study of photocatalysis has led to the development of several strategies to achieve the cyano migration of alkenyl nitrile compounds without altering their overall composition. Huang and co-workers in 2024 reported the rearrangement of 2-allylmalononitriles 135 and 4-pentenenitriles 136 into the cyclopropane products 137 and 138, respectively, using 1 mol% Ir-F as photocatalyst in CH3CN under blue-light irradiation (Scheme 38) [106]. In the proposed mechanism, a diradical intermediate is first generated from the C-C double bond through energy-transfer catalysis. Subsequent 5-exo-dig cyclization followed by β-scission of the resulting imido radical could produce another diradical intermediate. After intersystem crossing (ISC), the newly formed diradical intermediate may undergo a radical-radical recombination to give the three-membered-ring products. This unprecedented approach was defined as a di-π-ethane rearrangement in order to differentiate from the known di-π-methane rearrangement (Zimmerman rearrangement) wherein two π systems engaging in photochemical rearrangement are separated by one sp3 carbon atom (methane-like). Over fifty substrates 135 containing various R1 and R2 substituents, together with the indicated 136, underwent the reaction smoothly, achieving good to excellent yields under the optimal conditions (48-98%, mostly > 90%). In a few exceptional cases, the desired products were not obtained from the 4-pentenenitriles with hydrogen atom(s) or a piperidinyl ring (R3 = R4 = H; R3 =Me/R4 = H; R3 = CO2Et/R4 = H or R3 = R4 = -CH2CH2N(Boc)CH2CH2-) as well as the malononitriles bearing tri- or tetrasubstituted alkenyl groups probably due to altered reactivity of in-situ formed radical species or steric hindrance. The author also demonstrated that this protocol was not applicable to the translocation of a cyano group through a three- or six-membered-ring transition state. Shortly after the report from Huang’s group, Zhong et al. published a similar method involving the migration of cyano and aryl functional groups by using 0.01 to 1 mol% 2-chlorothioxanthone as photosensitizer [107].
Scheme 38.
Di-π-ethane rearrangement of cyano groups via energy-transfer catalysis (Huang, 2024).
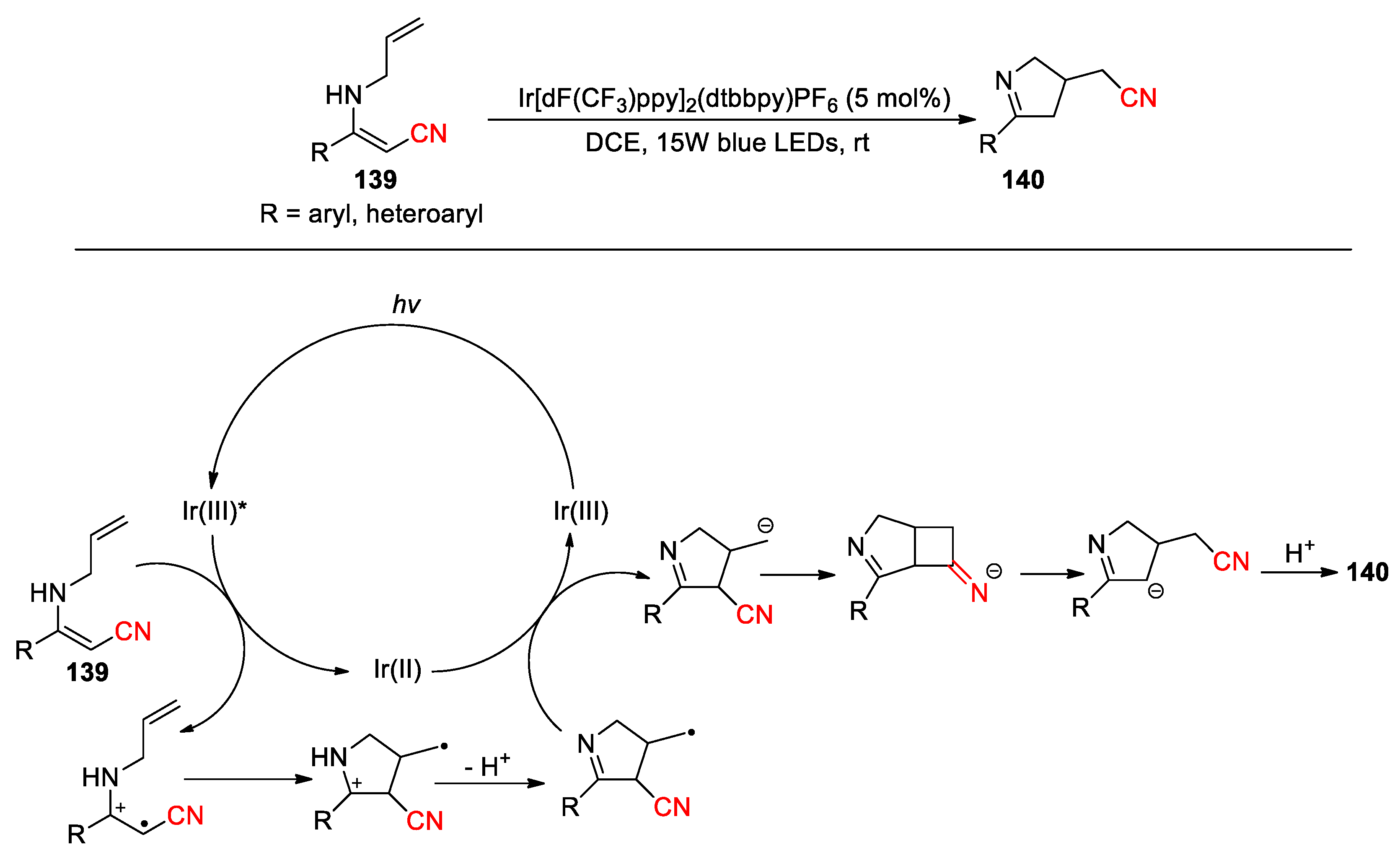
The earlier reported 1,3-cyano migration reactions generally required elevated temperatures to overcome the energy barrier associated with highly strained four-membered ring intermediates (Scheme 6, Scheme 7 and Scheme 8 and Scheme 12). Pan et al. recently reported a visible-light-induced tandem cyclization/1,3-cyano migration process, which allowed the conversion of nitrile-substituted N-allyl enamines 139 into the corresponding cyclic imines 140 under relatively mild conditions (26 example, 44-93%) (Scheme 39) [108]. Moreover, a 1,3-acyl migration could also be realized with replacing the cyano moiety in 139 by an ester group. Mechanistic studies and DFT calculations suggest that the reaction may proceed through SET oxidation of N-allyl enamine by the photoexcited Ir(III) catalyst to generate a radical cation. Subsequent 5-exo-trig cyclization followed by deprotonation allows the formation of a cyclic imine intermediate bearing a methylene radical. This intermediate could be further reduced into a carbanion by Ir(II) species. Then, the nucleophilic addition to the cyano group is followed by the ring-opening and protonation to afford the products.
Scheme 39.
Synthesis of cyclic imines via radical-triggered 1,3-cyano migration of N-allyl enamines (Pan, 2024).
Scheme 39.
Synthesis of cyclic imines via radical-triggered 1,3-cyano migration of N-allyl enamines (Pan, 2024).
2.4. Cyano Group Migrations via Halogen Atom Transfer
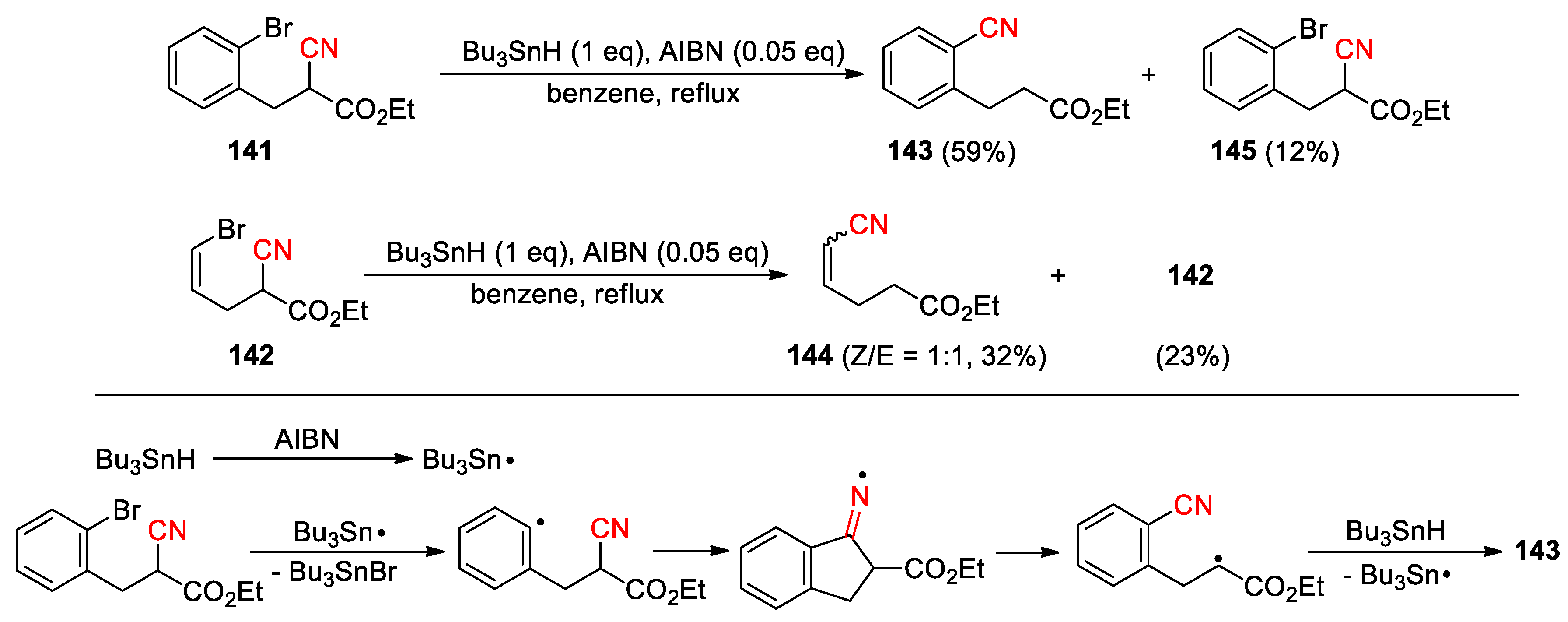
The generation of carbon-centered radical via halogen atom transfer (XAT) between carbon-halogen bonds and tributyltin radical has been used for many years to promote cyano migrations. In the earlier reports (1987 and 1988), Beckwith demonstrated that heating of cyanoacetate 141 or 142 with n-Bu3SnH and AIBN initiator in refluxing benzene could produce the l,4-transfer products 143 and 144, along with the simple dehalogenation product 145 or recovered starting material (Scheme 40) [109,110]. The most probable route for the translocation involves the cyclisation of the initially formed aryl (or vinyl) radical onto the nitrile to give a cyclic iminyl radical. This intermediate undergoes rapid β-scission to yield 143 (or 144) after hydrogen atom abstraction. In these cases, the β-scission is aided by the presence of a radical stabilizing ester substituent.
Scheme 40.
XAT-mediated CN migration of o-bromophenyl and bromovinyl cyanoacetates (Beckwith, 1987, 1988).
Scheme 40.
XAT-mediated CN migration of o-bromophenyl and bromovinyl cyanoacetates (Beckwith, 1987, 1988).
About a decade later, Cossy et al. shown that the similar nitrile transfer reaction could proceed in moderate yields with α-(bromophenylamino)nitriles such as 146 upon the treatment with Bu3SnH/AIBN in refluxing toluene, affording 2-(alkylamino)benzonitriles such as 147 as the only isolated products (Scheme 41, a) [111]. However, a subsequent report by Sulsky and co-workers described a very different outcome from those reported by Cossy [112]. For example, compound 146, treated under apparently identical conditions, led to the spiroindoxylimine 148 as the major product (58%), together with a 24:1 mixture of 147 and the direct reduction product 149 in 35% yield (Scheme 41, b). They speculated that the highly polar 148 could not have been isolated under the chromatography conditions described by Cossy, and may well have been present in the unpurified reaction mixture. Through single-crystal X-ray analyses, it was also discovered that the relative configuration of the anilino nitrogen in precursor 150 (equatorial/trans to the tert-butyl group) had been inverted during the cyclization into its spiroimine derivative 151 (axial/cis to the tert-butyl group). This suggests that ring opening and configuration inversion, followed by ring closure must be rapid and reversible relative to the quenching of intermediates with Bu3SnH, and that the cis isomer is more stable than the trans isomer (Figure 3).
Scheme 41.
Reactions reported for α-(bromophenylamino)nitrile 146 under same conditions (Cossy, 1995; Sulsky, 1999).
Scheme 41.
Reactions reported for α-(bromophenylamino)nitrile 146 under same conditions (Cossy, 1995; Sulsky, 1999).
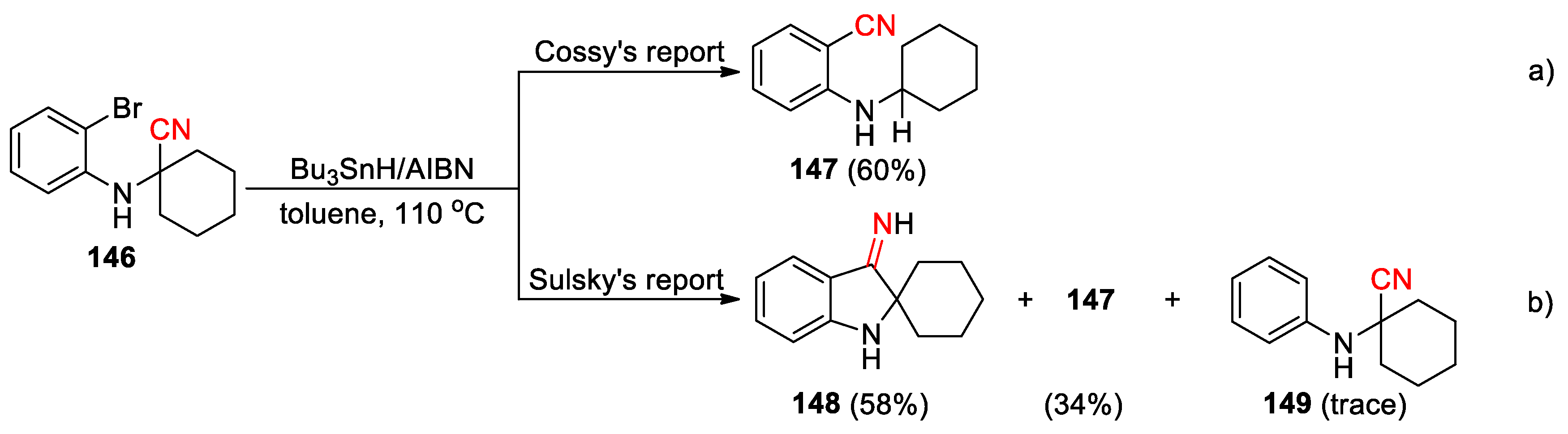
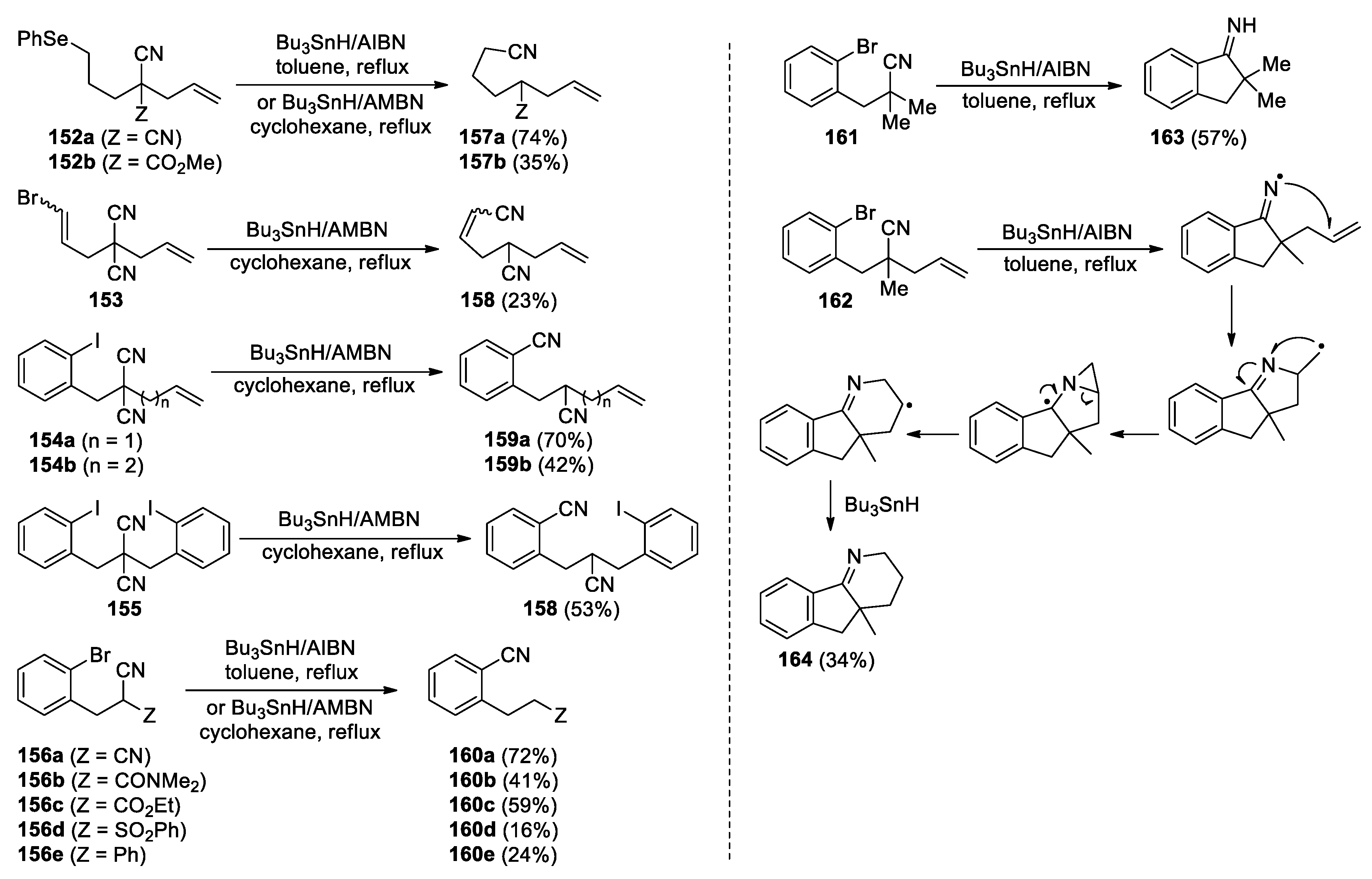
The inconsistent results illustrated above suggest that the reactivity of intermediate iminyl radicals, generated by 5-exo cyclisation of C-centred radicals onto nitriles, could be influenced by α-substituent, i.e., ester, amino or alkyl group. A study carried out by Bowman et al. has further validated this concept by using a set of phenylselenyl, bromo and iodo radical precursors [113]. With the presence of α-CN, CO2R, CONMe2, SO2Ph or Ph substituent, the iminyl radicals derived from 152-156 tended to undergo β-scission to yield stable/irreversible ring-opened radicals and thus the nitrile translocation products 157-160 (Scheme 42). Note that an unusual C-Se bond cleavage was utilized to generate the alkyl carbon radicals to perform the addition with the nitriles in the production of 157. When the α-substituent was an alkyl group, the formation of nitrile transfer products was not observed but rather cyclised imines or triheterocycle derivative, as is shown with the conversion of 161 and 162 into 163 and 164.
Scheme 42.
Nitrile transfer or cyclization reactions influenced by α-substituents (Bowman, 2000).
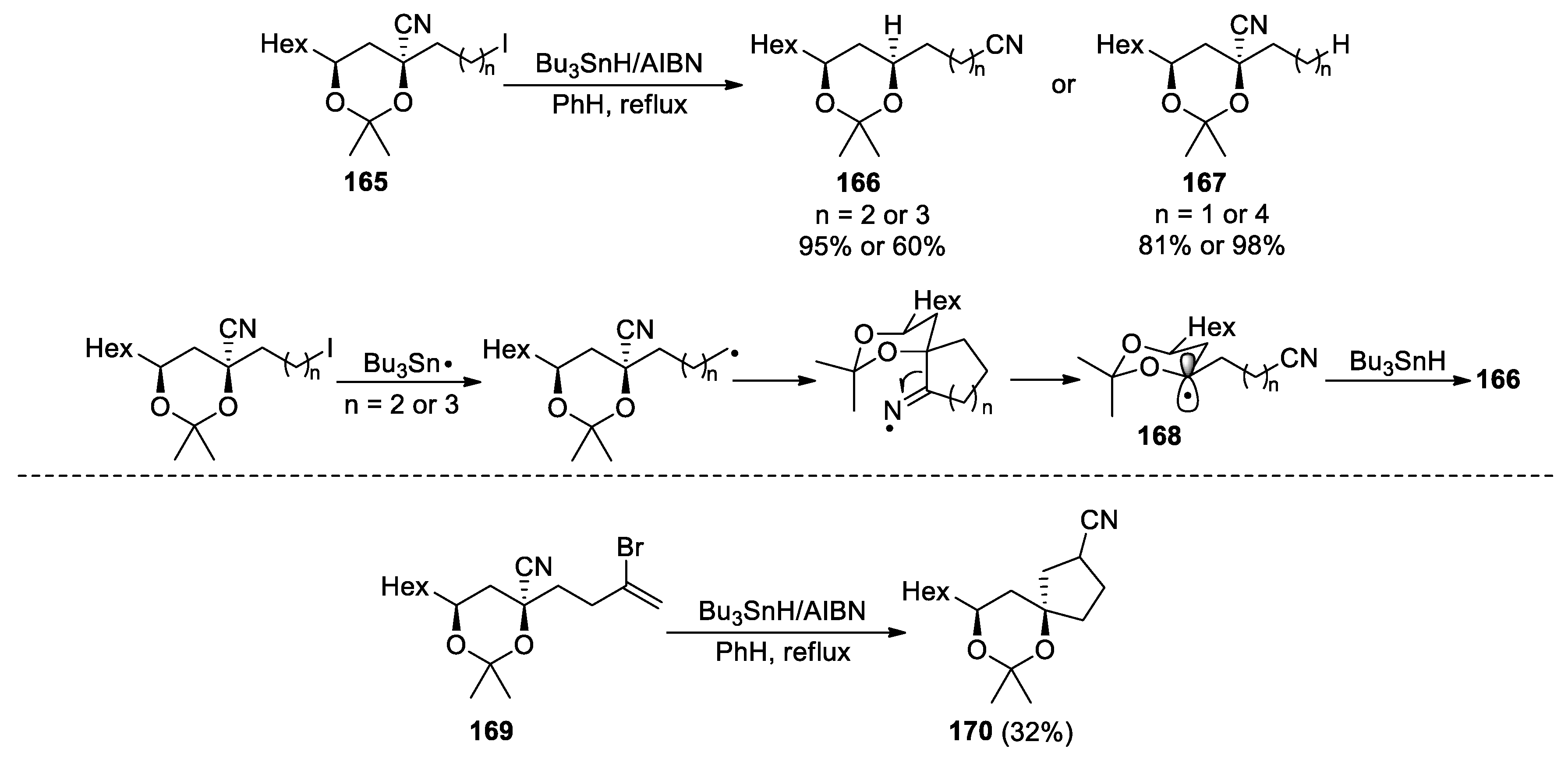
For 1,3-dioxane-4-nitriles 165, Ryclmovsky demonstrated that the 1,4- and 1,5-nitrile transfer reactions (n = 2, 3) could take place efficiently to give the products 166, whereas the corresponding 1,3- and 1,6-nitrile transfer reactions (n = 1, 4) were not observed and instead led to the dehalogenation products 167 (Scheme 43) [114]. During the nitrile transfer, the ring-opening of the iminyl radical should be encouraged by the formation of the stabilised α-alkoxy radical. Besides, products 166 were exclusively obtained as the syn-l,3-diol isomers, which is presumably a result of the stereoselective reduction of intermediate 168 with Bu3SnH. The authors also revealed that the α-alkoxy radicals produced in nitrile transfer could participate in a cyclization with appropriate unsaturated substituents. For instance, treatment of vinyl bromide 169 with Bu3SnH and catalytic AIBN in refluxing benzene provided the spirocyclic compound 170 via a tandem transfer-cyclization process. Although a 5-endo-trig cyclization like this one is generally disfavored according to Baldwin's rules, the complementary electronic distributions of the α-alkoxy radical and β-cyano alkene are supposed to favor the cyclization.
Scheme 43.
XAT-mediated cyano migration of 1,3-dioxane-4-nitriles (Ryclmovsky, 1997).
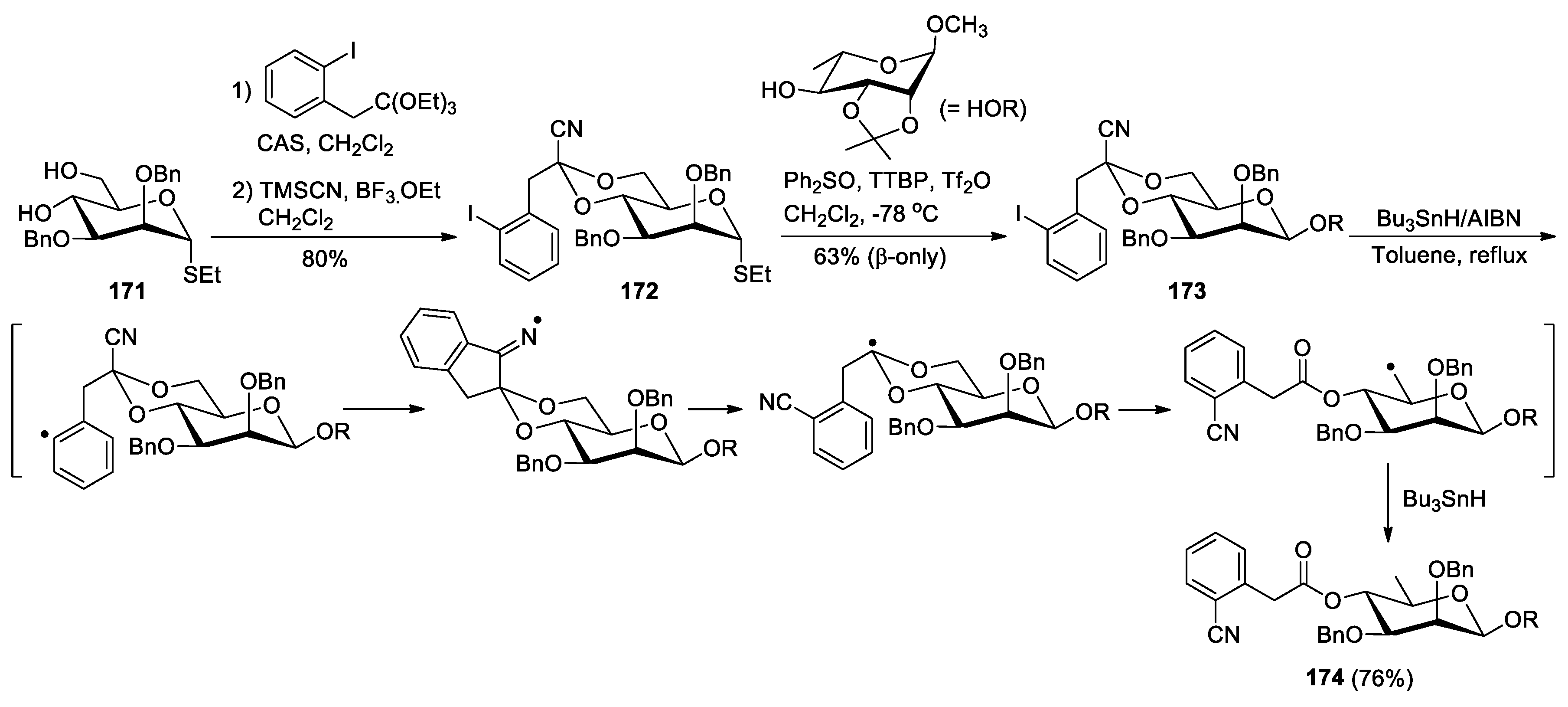
The strategy described in this section has been utilized in the syntheses of complex and biologically important natural molecules. Crich and Bowers reported a method for the preparation of β-rhamnopyranosides from thioglycosides, as exemplified by the conversion of 171 into 174 (Scheme 44) [115]. In the sequence, the 4,6-O-[1-cyano-2-(2-iodophenyl)-ethylidene] group was first introduced to 171 as a single diastereomer via the acid-catalyzed reaction of the 4,6-diol with triethyl (2-iodophenyl)orthoacetate, followed by BF3.OEt2-promoted cyanation. The acetal-protecting group in the resulting product 172 conveys strong β-selectivity to allow the stereocontrolled formation of β-mannoside 173 through the coupling with methyl 2,3-O-isopropylidene-R-L-rhamnopyranoside. As a key operation, treatment of 173 with tributyltin hydride and AIBN in toluene at reflux triggered a cyano transfer/fragmentation process to produce the product 174. The utility of this method was further demonstrated by the concise synthesis of a naturally occurring β-(1→3)-D-rhamnotetraose via a one-pot quadruple radical fragmentation [116]. To this end, a suitably protected monomer 176 was first synthesized from 4,6-O-benzylidene-protected thiomannoside 175 in 6 steps (Scheme 45) [116]. After this, the sequential coupling reactions of 176 were carried out, first with cyclohexanol and then with 3-OH-deprotected oligomer formed in each step, to give the formation of a β-mannotetraose 177. Subjection of 177 to conditions of radical fragmentation in refluxing xylenes, followed by NaBH4 reduction to facilitate removal of the tin residues, and then saponification provided the precursor tetraol 178. Finally, deprotection of 178 via hydrogenation eventually furnished the target molecule 179 in 90% yield.
Scheme 44.
Preparation of β-rhamnopyranoside from thioglycoside via nitrile transfer/fragmentation (Crich, 2006).
Scheme 44.
Preparation of β-rhamnopyranoside from thioglycoside via nitrile transfer/fragmentation (Crich, 2006).
Scheme 45.
Total synthesis of a natural tetrasaccharide via one-pot quadruple radical fragmentation (Crich, 2006).
Scheme 45.
Total synthesis of a natural tetrasaccharide via one-pot quadruple radical fragmentation (Crich, 2006).
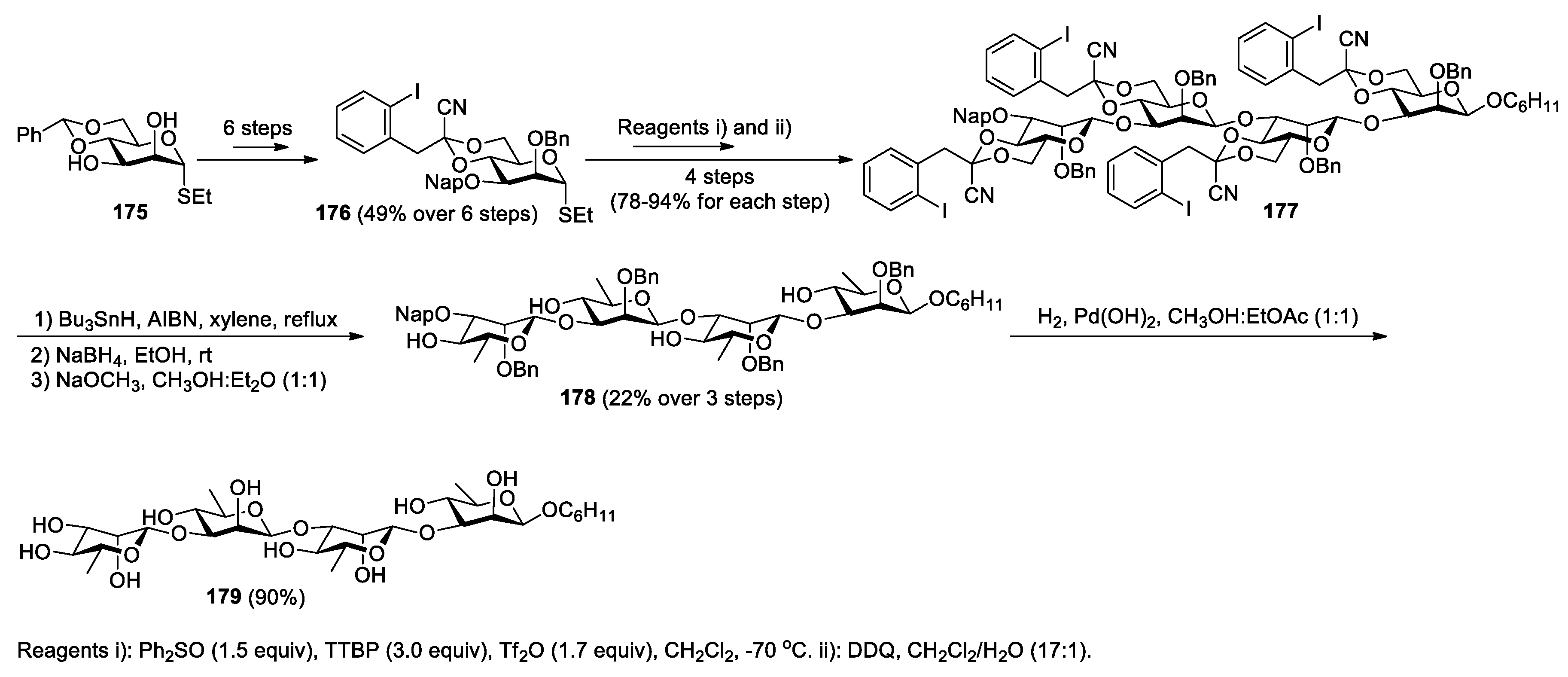
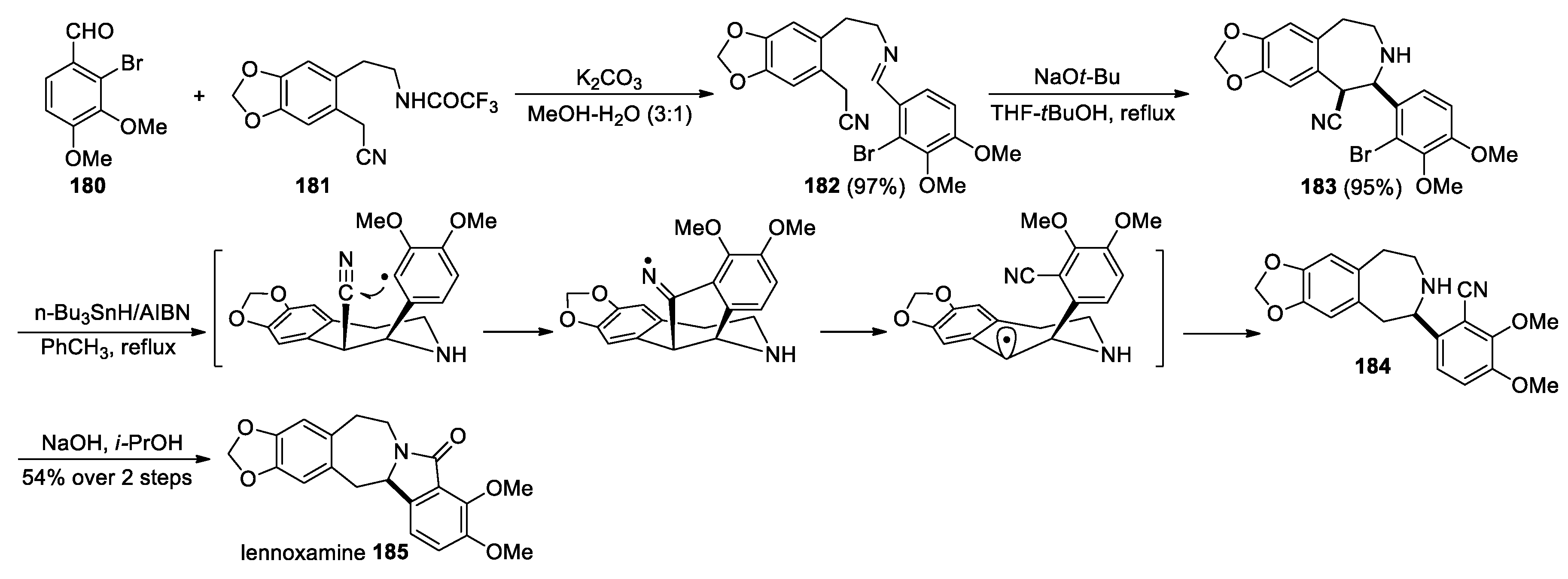
Orito and co-workers reported the total synthesis of a natural isoindolobenzazepine alkaloid, lennoxamine, based on a radical migration of a cyano group (Scheme 46) [117]. Their route involved the condensation of bromobenzaldehyde 180 with trifluoroacetamide 181 to give imine 182, which then underwent cyclization under basic conditions yielding benzazepine 183. Subsequent migration of the cyano group under radical conditions, followed by basic cyclization of the resulting precursor 184 competed the synthesis of lennoxamine 185.
Scheme 46.
Synthesis of lennoxamine through cyano migration (Orito, 2009).
2.5. Cyano Group Migrations via C-C Bond Fragmentation
Radical-mediated C-C bond fragmentation can be used as another approach for producing the carbon-centered radical to trigger nitrile migration, yet it has only been limited to a few reports including one of ours.
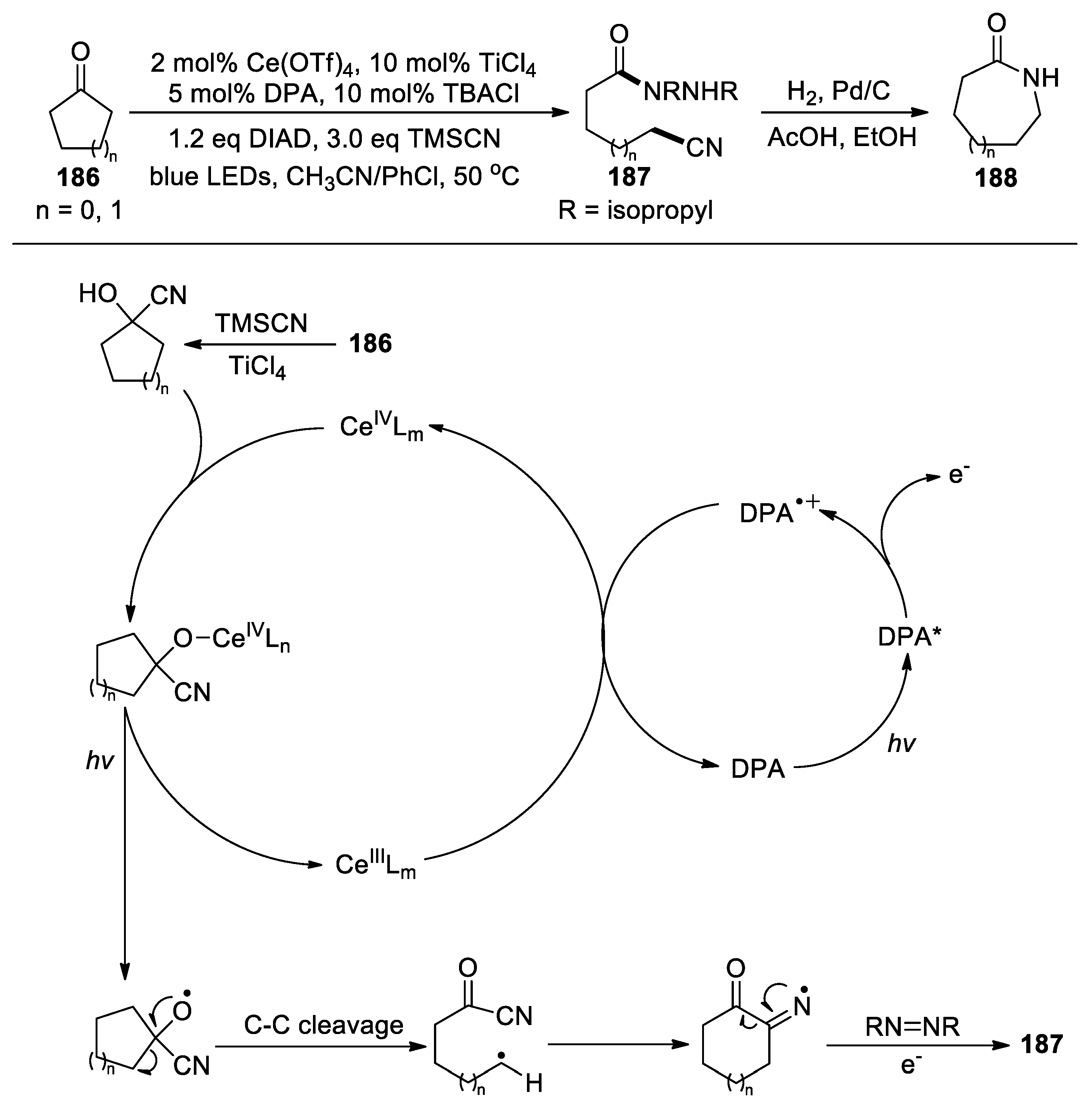
Zuo and colleagues exploited the selective C-C bond cleavage of ketones through a cooperative utilization of Lewis acid catalysis and ligand-to-metal charge transfer (LMCT) excitation [118]. During the study, a migration of a cyano group was observed for the reactions of clobutanones and cyclopentanones 186, yielding the cyanated hydrazides 187 under the conditions shown in Scheme 47 (18 examples, 51-97%). The resulting products could be further elaborated into the corresponding lactam 188 by a simple hydrogenation.
The proposed pathway to 187 involves the generation of a cyanohydrin by the nucleophilic addition with TMSCN. In addition to cerium triflate, titanium tetrachloride catalyst is required in this step to convert inactive cyanohydrin silyl ether into the cyanohydrin. Under blue light irradiation, the cerium(IV)-cyanohydrin coordination complex can be excited to promote the homolysis of the Ce-O bond, generating an alkoxy radical. Subsequent scission of the α-C-C bond produces a carbon-centered radical, which will trigger the migration of the cyano group to give a carbonyl radical intermediate. Trapping of this intermediate by diisopropyl azodicarboxylate (DIAD), followed by a photo-induced single-electron reduction with 9,10-diphenylanthracene (DPA) provides the product 187 along with a radical cation of DPA. The radical cation then engages in single-electron transfer with cerium(III) to complete the catalytic cycle. In the cases of α-substituted cyclic ketones, the C-C bond between the most substituted α-carbon and the carbonyl group was shown to be selectively cleaved to form more stable carbon radical. Moreover, the radical intermediates derived from 6-membered and lager cyclic ketones, as well as acyclic ketones, did not undergo cyano migration after the C-C bond cleavage, but instead reacted with DIAD and introduced methanol to afford aminated carboxylic esters as the products.
Scheme 47.
Functionalization of cyclic ketones via LMCT-mediated C-C bond cleavage and cyano migration (Zou, 2020).
Scheme 47.
Functionalization of cyclic ketones via LMCT-mediated C-C bond cleavage and cyano migration (Zou, 2020).
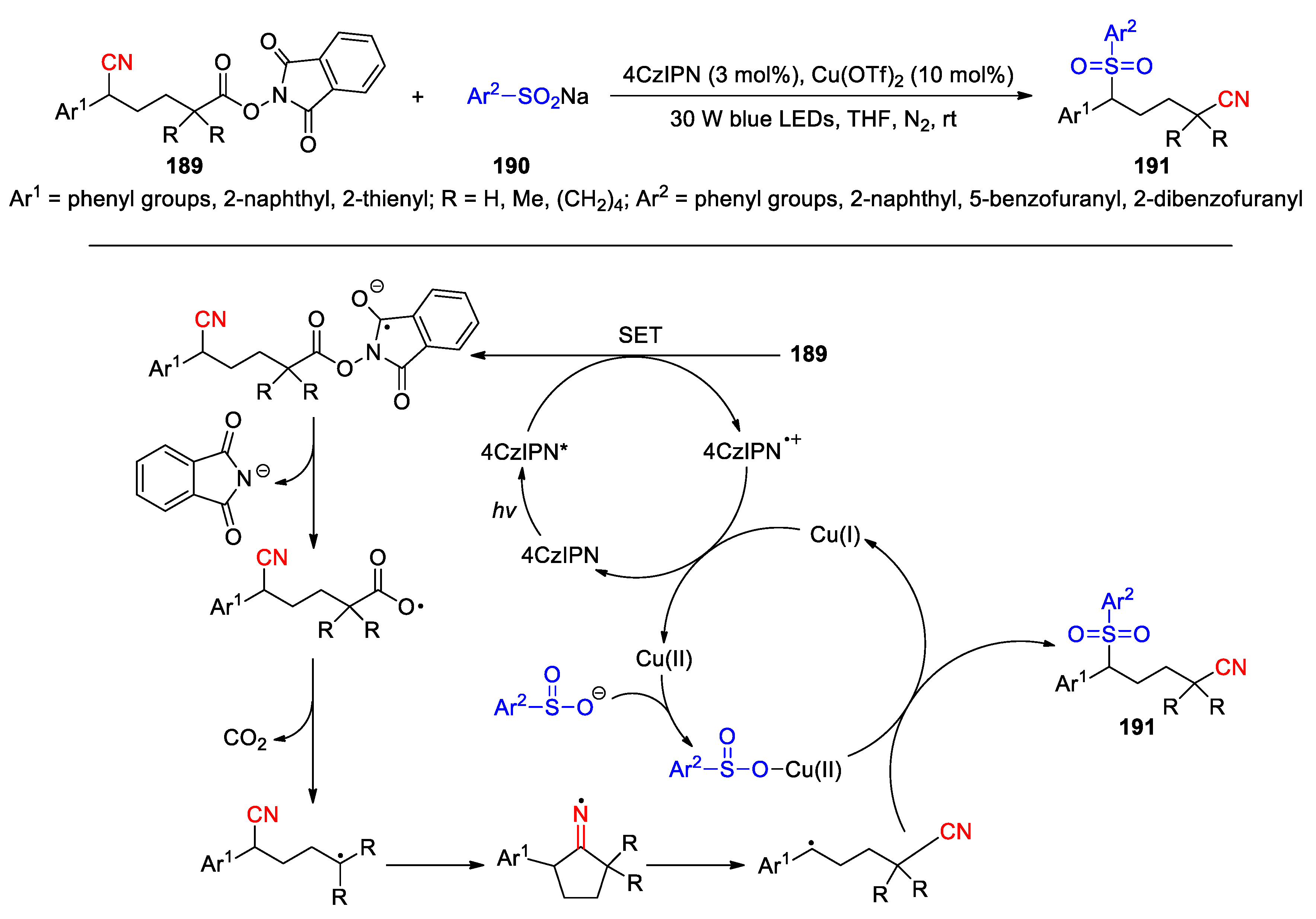
A photoredox/Cu dual catalyzed 1,4-cyanosulfonylation of NHPI esters 189 with sodium arylsulfinates 190 has been established by Chen et al., which provides a variety of δ-sulfonyl nitriles 191 via a decarboxylation-triggered remote cyano migration under the conditions shown in Scheme 48 (30 examples, 30-72%) [119]. In their proposed mechanism, the SET reduction of 189 by the excited photocatalyst 4CzIPN* takes place to produce intermediate 192 and the 4CzIPN radical cation. Elimination of the imide anion from 192 via a N-O bond cleavage gives the carboxyl radical 193, which subsequently undergoes a decarboxylation yielding an alkyl radical to trigger the 1,4-cyano migration. Afterwards, the resulting benzylic radical 194 could be trapped by the sulfonate radical released from the in-situ formed Cu(II)-arylsulfinate complex to furnish the product 191. The Cu(I) generated in this step can be re-oxidized by the 4CzIPN radical cation, leading to the regeneration of both Cu(II) and 4CzIPN catalysts.
Scheme 48.
Preparation of δ-sulfonyl nitriles via decarboxylation-triggered 1,4-cyano migration (Chen, 2024).
Scheme 48.
Preparation of δ-sulfonyl nitriles via decarboxylation-triggered 1,4-cyano migration (Chen, 2024).
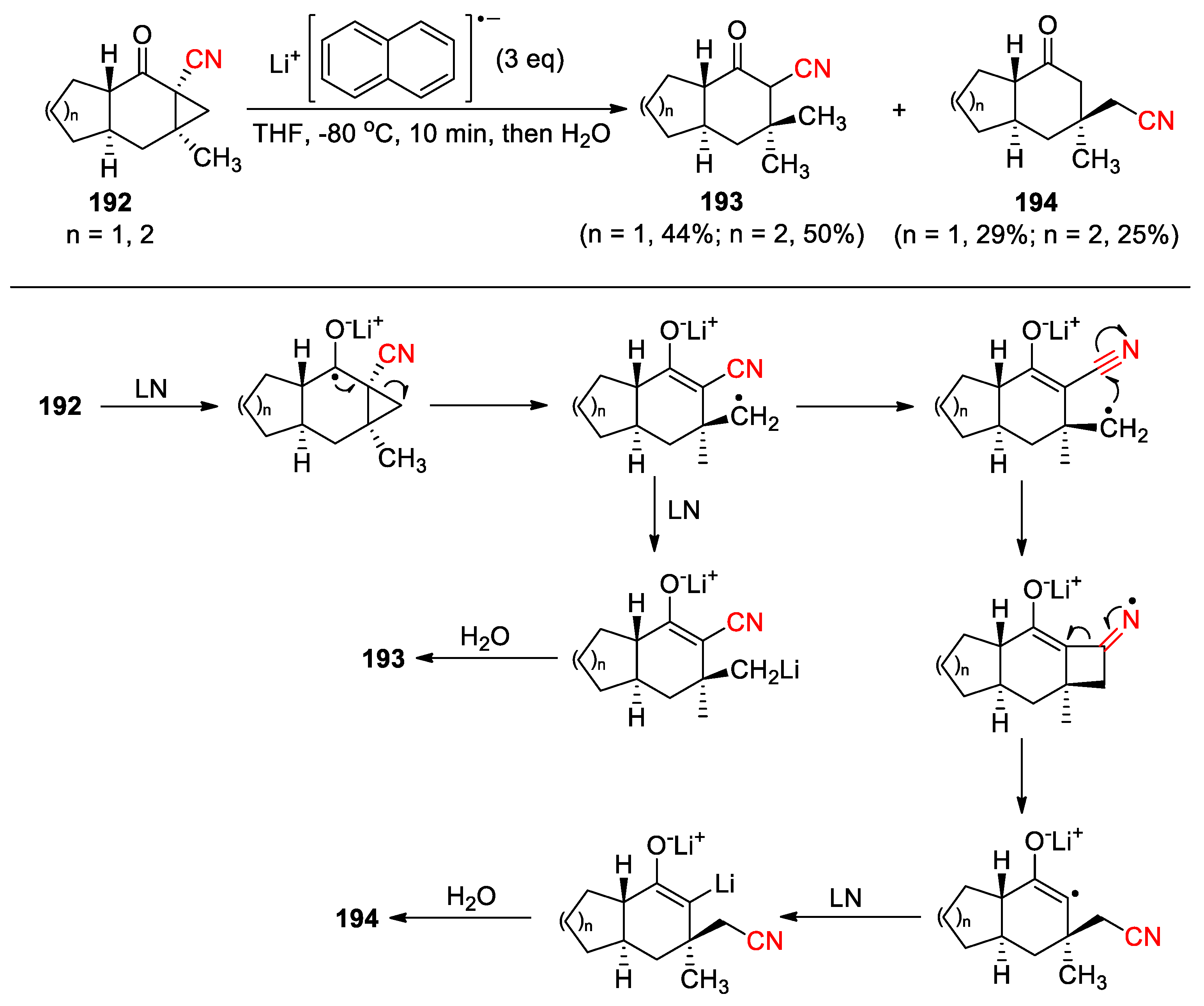
We previously synthesized the fused tricyclic α-keto cyclopropyl nitriles 192 via the rhodium-catalyzed intramolecular cyclopropanation. Treatment of 192 with lithium naphthalenide (LN) as an electron-donating agent caused the ring-opening of the cyclopropane to give α-cyano ketones 193 as the major products, along with γ-cyano ketones 194 resulting from an additional 1,3-cyano migration (Scheme 49) [120]. We proposed that the carbonyl group in 192 was first reduced to a ketyl radical anion by LN, which then triggered consecutive ring opening and cyano migration to yield a vinyl radical anion intermediate. Further reduction followed by hydrolysis gave the formation of 194. The carbon radical derived from the ring-opening could also be reduced into a carbanion in the presence of excess of LN to provide 193, and the reduction seemed to occur faster than the addition to the nitrile group as suggested by the yields obtained for the products. Moreover, exclusive formation of α-cyano ketone products was observed when the angular methyl groups in 192 was replaced by a hydrogen atom, implying that the methyl groups might play a role in directing reaction pathway possibly through steric effects.
Scheme 49.
1,3-Cyano migration triggered by reductive ring opening (Zhu, 2017).
3. Conclusions
The radical-mediated distal cyano group migration has served as an important tool for the site-selective functionalization of organic molecules. The process requires the initial generation of a carbon-centered radical to trigger the CN migration and provide a more stable carbon radical for further elaboration. Decades of studies on this topic have led to the discovery of a plethora of procedures for the migration, which are systematically reviewed in four sections according to the strategies used to generate the initial carbon radicals. As we have shown, the early reported reactions were often conducted under harsh thermal conditions, resulting in relatively low yields for desired products. In recent twenty years, advances in radical chemistry together with application of photocatalysis have stimulated the development of a number of mild and efficient methods to achieve the migration with various nitrile substrates. Particularly remarkable is the diversity of photocatalytic techniques that have been used for such transformations, such as photoredox catalysis, organo-photocatalysis, dual NHC/photoredox catalytsis, dual metal/photoredox catalysis, and photo-induced ligand-to-metal charge transfer (LMCT). The utility of these catalytic systems arises from their ability to effectively generate reactive intermediates, e.g., radical, radical cation, or radical anion, via single-electron transfer, thus facilitating the migration along with the incorporation of other functional group(s) into a molecule. Overall, this article provides a comprehensive assessment of all achievements in this area, which may inspire the researchers to develop more useful synthetic tools for radical-mediated functional group migration.
References
- Seth, A.; Mehta, P.K. The Chemistry of Nitriles, 1st ed.; Seth, A., Ed.; Nova Science Publishers, Inc.: New York, 2024. [Google Scholar]
- Xia, Y.; Jiang, H.; Wu, W. Recent Advances in Chemical Modifications of Nitriles. Eur. J. Org. Chem. 2021, 6658–6669. [Google Scholar] [CrossRef]
- Harnett, G.J.; Hauck, U.; Hayes, J.J.; Hoffmann, U.; Lohri, B.; Meade, M.; Morawitz, F.; Plesniak, M.P.; Stahr, H.; Veits, J.; Walsh, A. Zogg, A.; Reents, R.; Smith, D.A; Martin, R.E. Development and Demonstration of a High-Volume Manufacturing Process for a Key Intermediate of Dalcetrapib: Investigations on the Alkylation of Carboxylic Acids, Esters, and Nitriles. Org. Process Res. Dev. 2023, 27, 2329–2346. [Google Scholar] [CrossRef]
- Fan, T.; Qin, J.; Du, X.; Yuan, F.; Zhao, H.; Long, J.; Dong, L.; Chen, J.; Long, Y.; Ma, J. Amorphous Fe0.7Cu0.3Ox as a Bifunctional Catalyst for Selective Hydration of Nitriles to Amides in Water. ACS Sustainable Chem. Eng. 2024, 12, 7791–7801. [Google Scholar] [CrossRef]
- Rakshit, A.; Dhara, H.N.; Sahoo, A.H.; Patel, B.K. The Renaissance of Organo Nitriles in Organic Synthesis. Chem. Asian J. 2022, 17, e202200792. [Google Scholar] [CrossRef]
- Yamada, T.; Park, K.; Furugen, C.; Jiang, J.; Shimizu, E.; Ito, N.; Sajiki, H. Highly Selective Hydrogenative Conversion of Nitriles into Tertiary, Secondary, and Primary Amines under Flow Reaction Conditions. ChemSusChem 2022, 15, e202102138. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.V.; Alawaed, A.A. Room Temperature Reduction of Titanium Tetrachloride-Activated Nitriles to Primary Amines with Ammonia-Borane. Molecules 2023, 28, 60. [Google Scholar] [CrossRef]
- Kovela, S.; Karad, S.; Tatipudi, V.V.G.; Arumugam, K.; Somwanshi, A.V.; Muthukumar, M.; Mathur, A.; Tester, R. Synthesis of diversely substituted quinazoline-2,4(1H,3H)-diones by cyclization of tert-butyl (2-cyanoaryl)carbamates. Org. Biomol. Chem. 2024, 22, 6495–6499. [Google Scholar] [CrossRef]
- Mazloumi, M.; Shirini, F. Acidic Ionic Liquid Bridge Supported on Nano Rice Husk Ash: An Efficient Promoter for the Conversion of Nitriles to Their Corresponding 5-Substituted 1H-Tetrazoles and Amides. ChemistrySelect 2023, 8, e202203554. [Google Scholar] [CrossRef]
- Ghodse, S.K.; Barde, P.D. Catalyst free, one-pot green synthesis of 2-aryl-2-oxazoline derivatives from aryl nitrile using ionic liquid. Syn. Commun. 2023, 53, 1616–1622. [Google Scholar] [CrossRef]
- Facchinetti, V.; Gomes, C.R.B.; de Souza, M.V.N. Application of nitriles on the synthesis of 1,3-oxazoles, 2-oxazolines, and oxadiazoles: An update from 2014 to 2021. Tetrahedron 2021, 102, 132544. [Google Scholar] [CrossRef]
- Ramesh, P.; Sreenivasulu, C.; Gorantla, K.R.; Mallik, B.S.; Satyanarayana, G. A simple removable aliphatic nitrile template 2-cyano-2,2-di-isobutyl acetic acid for remote meta-selective C–H functionalization. Org. Chem. Front. 2021, 8, 1959–1969. [Google Scholar] [CrossRef]
- Lýpez, R.; Palomo, C. Cyanoalkylation: Alkylnitriles in Catalytic C-C Bond-Forming Reactions. Angew. Chem. Int. Ed. 2015, 54, 13170–13184. [Google Scholar] [CrossRef] [PubMed]
- Amancha, P.K.; Lai, Y.J.; Chen, I.C.; Liu, H.J.; Zhu, J.L. Terahedron 2010, 66, 871-877.
- Zhu, J.L.; Huang, P.W.; You, R.Y.; Lee, F.Y.; Tsao, S.W.; Chen, I.C. Total Syntheses of (±)-(Z)- and (±)-(E)-9-(Bromomethylene)-1,5,5-trimethylspiro[5.5]undeca-1,7-dien-3-one and (±)-Majusculone. Synthesis 2011, 715–722. [Google Scholar] [CrossRef]
- Chen, M.L.; Chou, C.w.; Zhu, J.L.; Tsai, M.H. Access to cyclohexadiene and benzofuran derivatives via catalytic arene cyclopropanation of α-cyanodiazocarbonyl compounds. Org. Biomol. Chem. 2024, 22, 5552–5560. [Google Scholar] [CrossRef]
- Nakao, Y. Metal-mediated C−CN Bond Activation in Organic Synthesis. Chem. Rev. 2021, 121, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Bonatto, V.; Lameiro, R.F.; Rocho, F.R.; Lameira, J.; Leitão, A.; Montanari, C.A. Nitriles: an attractive approach to the development of covalent inhibitors. RSC Med. Chem. 2023, 14, 201–217. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Li, X.; Yu, Z.; Song, C.; Du, Y. Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies. RSC Med. Chem. 2021, 11, 1650–1671. [Google Scholar] [CrossRef]
- Ganesan, M.; Nagaraaj, P. Recent developments in dehydration of primary amides to nitriles. Org. Chem. Front. 2020, 7, 3792–3814. [Google Scholar] [CrossRef]
- Singh, M.K.; Lakshman, M.K. A Simple Synthesis of Nitriles from Aldoximes. J. Org. Chem. 2009, 74, 3079–3084. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; Thadathil, D.A.; Ponmudi, K.; George, A.; Varghese, A. Recent Advances in Electrochemical Synthesis of Nitriles: A Sustainable Approach. ChemistrySelect 2022, 7, e202200081. [Google Scholar] [CrossRef]
- Makino, K.; Hasebe, M.; Sueki, S.; Anada, M. Brønsted Acid Catalyzed Dehydroxylative Cyanation of Benzylic Alcohols with Trimethylsilyl Cyanide Using1,1,1,3,3,3-Hexafluoro-2-propanol as a Solvent. Eur. J. Org. Chem. 2024, 27, e202400474. [Google Scholar] [CrossRef]
- Ren, X.; Shen, C.; Wang, G.; Shi, Z.; Tian, X.; Dong, K. Access to α-Cyano Carbonyls Bearing a Quaternary Carbon Center by Reductive Cyanation. Org. Lett. 2021, 23, 2527–2532. [Google Scholar] [CrossRef]
- Yu, P.; Morandi, B. Nickel-Catalyzed Cyanation of Aryl Chlorides and Triflates Using Butyronitrile: Merging Retro-hydrocyanation with Cross-Coupling. Angew. Chem. 2017, 129, 15899–15903. [Google Scholar] [CrossRef]
- Ellis, G.P.; Romney-Alexander, T.M. Cyanation of Aromatic Halides. Chem. Rev. 1987, 87, 779–794. [Google Scholar] [CrossRef]
- Yan, G.; Kuang, C.; Zhang, Y.; Wang, J. Palladium-Catalyzed Direct Cyanation ofIndoles with K4[Fe(CN)6]. Org. Lett. 2010, 12, 1052–1055. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jiao, N. Direct Approaches to Nitriles via Highly Efficient Nitrogenation Strategy through C−H or C−C Bond Cleavage. Acc. Chem. Res. 2014, 47, 1137–1145. [Google Scholar] [CrossRef]
- Meystre, C.; Heusler, K.; Kalvoda, J.; Wieland, P.; Anner, G.; Wettstein, A. New substitution reactions in steroids. Experientia 1961, 17, 475–480. [Google Scholar] [CrossRef]
- Kalvoda, J.; Meystre, C.; Anner, G. Reaktionen von Steroid-Hypojoditen VIII 1,4-Verschiebung der Nitrilgruppe (18-Cyano-pregnane). Helv. Chim. Acta 1966, 49, 424–436. [Google Scholar] [CrossRef]
- Robertson, J.; Pillai, J.; Lush, R.K. Chem. Soc. Rev. 2001, 30, 94-103.
- Li, W.; Xu, W.; Xie, J.; Yu, S.; Zhu, C. Distal radical migration strategy: an emerging synthetic means. Chem. Soc. Rev. 2018, 47, 654–667. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, C. Recent Advances in Radical-Mediated C-C Bond Fragmentation of Non-Strained Molecules. Chin. J. Chem. 2019, 37, 171–182. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, C. Radical-Mediated Remote Functional Group Migration. Acc. Chem. Res. 2020, 53, 1620–1636. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ma, Z.; Feng, T.; Zhu, C. Radical-mediated rearrangements: past, present, and future. Chem. Soc. Rev. 2021, 50, 11577–11613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ma, X.; Zhi, S.; Zhang, W. Radical-Mediated Trfunctionalization Reactions. Molecules 2024, 29, 3620. [Google Scholar] [CrossRef] [PubMed]
- Lafzi, F.; Eşsiz, S. Computational Study on Chemoselective Difunctionalization of Unactivated Alkenes with Radical-mediated Remote Functional Group Migration. ChemPhysChem 2023, 24, e202200886. [Google Scholar] [CrossRef]
- Kalvoda, J. A New Type of Intramolecular Group-transfer in Steroid Photochemistry. A Contribution to the Mechanism of the Oxidative Cyanohydrin-Cyano-ketone Rearrangement. Chem. Commun 1970, 1002–1003. [Google Scholar] [CrossRef]
- Watt, D.S. A Reiterative Functionalization of Unactivated Carbon-Hydrogen Bonds. Photolysis of α-Peracetoxynitriles. J. Am. Chem. Soc. 1976, 98, 271–273. [Google Scholar] [CrossRef]
- Freerksen, R.W.; Pabst, W.E.; Raggio, M.L.; Sherman, S.A.; Wroble, R.R.; Watt, D.S. Photolysis of α-Peracetoxynitriles. 2. A Comparison of Two Synthetic Approaches to 18-Cyano-20-ketosteroids. J. Am. Chem. Soc. 1977, 99, 1536–1542. [Google Scholar] [CrossRef]
- Jiang, X.; Manion, B.D.; Benz, A.; Rath, N.P.; Evers, A.S.; Zorumski, C.F.; Mennerick, S.; Covey, D.F. Neurosteroid Analogues. 9. Conformationally Constrained Pregnanes: Structure-Activity Studies of 13,24-Cyclo-18,21-dinorcholane Analogues of the GABA Modulatory and Anesthetic Steroids (3α,5α)- and (3α,5α)-3-Hydroxypregnan-20-one. J. Med. Chem. 2003, 46, 5334–5348. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Wang, M.; Huan, L.; Zhu, C. Cyanohydrin-Mediated Cyanation of Remote Unactivated C(sp3)-H Bonds. Org. Lett. 2019, 21, 821–825. [Google Scholar] [CrossRef]
- For the production of sulfate radical anion (SO4•– ) along with SO42- from S2O82- , see: Jin, J.; MacMillan, D.W.C. Direct α-Arylation of Ethers through the Combination of Photoredox-Mediated C-H Functionalization and the Minisci Reaction. Angew. Chem. Int. Ed. 2015, 54, 1565-1569.
- Lei, Z.; Wu, J. Terminal-selective C(sp3)-H borylation of unbranched alkanes enabled by intermolecular radical sampling and LMCT photocatalysis. Natl. Sci. Rev. 2024, 11, nwae105. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zeng, Q.; Xie, L.; Xue, Z.; Wang, J.; Xu, Y. Functional-group translocation of cyano groups by reversible C-H sampling. Nature 2023, 620, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Sinha, S.; Gahtori, P.; Tivaric, S.; Srivastava, V. Recent advances of decatungstate photocatalyst in HAT process. Org. Biomol. Chem. 2024, 22, 2523–2538. [Google Scholar] [CrossRef]
- Bury, A.; Bougeard, P.; Corker, S.J.; Johnson, M.D.; Perlmann, M. 1,3-Migration of a Cyano-group in Substituted 3-Cyanopropyl Radicals. J. Chem. Soc. Perkin Trans. II 1982, 1367–1373. [Google Scholar] [CrossRef]
- Montevecehi, P.C.; Navacehia, M.L.; Spagnolo, P. 2-Cyano-isopropyl radical addition to alkynes. Tetrahedron 1997, 53, 7929–7936. [Google Scholar] [CrossRef]
- Kamijo, S.; Yokosaka, S.; Inoue, M. Carbocyanation of trisubstituted olefins via Cu-catalyzed atom transfer radical addition. Tetrahedron Lett. 2012, 53, 4324–4327. [Google Scholar] [CrossRef]
- Curran, D.P.; Seong, C.M. Radical annulation reactions of allyl iodomalononitriles. Tetrahedron 1992, 48, 2175–2190. [Google Scholar] [CrossRef]
- Callier, A.C.; Quiclet-Sire, B.; Zard, S.Z. Amidyl and carbamyl radicals by stannane mediated cleavage of O-benzoyl hydroxamic acid derivatives. Tetrahedron Lett. 1994, 35, 6109–6112. [Google Scholar] [CrossRef]
- Saito, I.; Shimozono, K.; Matsuura, T. A Novel Photoaddition of 6-Cyanouracils to Alkenes and Alkynes Involving Migration of a Cyano Group. J. Am. Chem. Soc. 1980, 102, 3948–3950. [Google Scholar] [CrossRef]
- Wolff, S.; Agosta, W.C. Triplet-Sensitized Photochemical Rearrangement of Geranonitrile at Elevated Temperature. J. Org. Chem. 1978, 43, 3627–3628. [Google Scholar] [CrossRef]
- Bao, X.; Li, J.; Jiang, W.; Huo, C. Radical-Mediated Difunctionalization of Styrenes. Synthesis 2019, 51, 4507–4530. [Google Scholar] [CrossRef]
- Yao, H.; Hu, W.; Zhang, W. Difunctionalization of Alkenes and Alkynes via Intermolecular Radical and Nucleophilic Additions. Molecules 2021, 26, 105. [Google Scholar] [CrossRef]
- Liang, Q.; Walsh, P.J.; Jia, T. Copper-Catalyzed Intermolecular Difunctionalization of Styrenes with Thiosulfonates and Arylboronic Acids via a Radical Relay Pathway. ACS Catal. 2020, 10, 2633–2639. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Wan, X.; Shen, Q. Cobalt-Catalyzed Enantioselective Dicarbofunctionalization of Acrylates. ACS Catal. 2024, 14, 15221–15236. [Google Scholar] [CrossRef]
- Wang, Q.-L. Zhou, Q.; Liao, J.; Chen, Z.; Xiong, B.-Q.; Deng, G.-J.; Tang, K.-W.; Liu, Y. Cu-Catalyzed Oxidative Dual Arylation of Active Alkenes: Preparation of Cyanoarylated Oxindoles through Denitrogenation of 3-Aminoindazoles. J. Org. Chem. 2021, 86, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.H.R.; Csiba, M.A.; Jaszberenyi, J.C. Ru(bpy)32+-mediated addition of Se-phenyl p-tolueneselenosulfonate to electron rich olefins. Tetrahedron Lett. 1994, 35, 2869–2872. [Google Scholar] [CrossRef]
- Courant, T.; Masson, G. Recent Progress in Visible-Light Photoredox-Catalyzed Intermolecular 1,2-Difunctionalization of Double Bonds via an ATRA Type Mechanism. J. Org. Chem. 2016, 81, 6945–6952. [Google Scholar] [CrossRef]
- Bian, K.J.; Nemoto Jr., D.; Kao, S.C.; He, Y.; Li, Y.; Wang, X.-S.; West, J.G. Modular Difunctionalization of Unactivated Alkenes through Bio-Inspired Radical Ligand Transfer Catalysis. J. Am. Chem. Soc. 2022, 144, 11810–11821, and references cited therein. [Google Scholar] [CrossRef]
- Wu, X.; Wu, S.; Zhu, C. Radical-mediated difunctionalization of unactivated alkenes through distal migration of functional groups. Tetrahedron. Lett. 2018, 59, 1328–1336. [Google Scholar] [CrossRef]
- Meyer, S.; Claraz, A. Vicinal Difunctionalization of Unactivated Alkenes Through Radical Addition/Remote (Hetero)Aryl Migration Cascade. Eur. J. Org. Chem. 2024, 27, e202400504. [Google Scholar] [CrossRef]
- Wu, Z.; Ren, R.; Zhu, C. Combination of a Cyano Migration Strategy and Alkene Difunctionalization: The Elusive Selective Azidocyanation of Unactivated Olefins. Angew. Chem. Int. Ed. 2016, 55, 10821–10824. [Google Scholar] [CrossRef] [PubMed]
- Matcha, K.; Narayan, R.; Antonchick, A.P. Metal-Free Radical Azidoarylation of Alkenes: Rapid Access to Oxindoles by Cascade C-N and C-C Bond-Forming Reactions. Angew. Chem. Int. Ed. 2013, 52, 7985–7989. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Wu, Z.; Yu, J.; Wan, X.; Zhu, C. Cyanotrifluoromethylthiolation of Unactivated Olefins through Intramolecular Cyano Migration. Adv. Synth. Catal. 2017, 359, 1959–1962. [Google Scholar] [CrossRef]
- Ren, R.; Wu, Z.; Huan, L.; Zhu, C. Synergistic Strategies of Cyano Migration and Photocatalysis for Difunctionalization of Unactivated Alkenes: Synthesis of Di- and Mono-Fluorinated Alkyl Nitriles. Adv. Synth. Catal. 2017, 359, 3052–3056. [Google Scholar] [CrossRef]
- Nguyen, J.D.; D’Amato, E.M.; Narayanam, J.M.R.; Stephenson, C.R.J. Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions. Nature Chem. 2012, 4, 854. [Google Scholar] [CrossRef]
- Ji, M.; Wu, Z.; Zhu, C. Visible-light-induced consecutive C-C bond fragmentation and formation for the synthesis of elusive unsymmetric 1,8-dicarbonyl compounds. Chem. Commun. 2019, 55, 2368–2371. [Google Scholar] [CrossRef]
- Ji, M.; Yu, J.; Zhu, C. Cyanotrifluoromethylthiolation of unactivated dialkyl-substituted alkynes via cyano migration: synthesis of trifluoromethylthiolated acrylonitriles. Chem. Commun. 2018, 54, 6812–6815. [Google Scholar] [CrossRef]
- Wang, N.; Li, L.; Li, Z.-L.; Yang, N.-Y.; Guo, Z.; Zhang, H.-X.; Liu, X.-Y. Catalytic Diverse Radical-Mediated 1,2-Cyanofunctionalization of Unactivated Alkenes via Synergistic Remote Cyano Migration and Protected Strategies. Org. Lett. 2016, 18, 6026–6029. [Google Scholar] [CrossRef]
- For producing trifluoromethyl radical form Togni’s reagent and Cu(I) catalyst, see: Barata-Vallejo, S; Lantaño, B.; Postigo, A. Recent Advances in Trifluoromethylation Reactions with Electrophilic Trifluoromethylating Reagents. Chem. Eur. J. 2014, 20, 1-25.
- For producing azido radical form Zhdankin reagent and Cu(I) catalyst, see: Rabet, P.T.G.; Fumagalli, G.; Boyd, S.; Greaney, M.F. Benzylic C-H Azidation Using the Zhdankin Reagent and a Copper Photoredox Catalyst. Org. Lett. 2016, 18, 1646-1649.
- For the SET reduction of Cu(II) species by ketyl radical intermediate to regenerate Cu(I) catalyst, see: Gao, P.; Shen, Y.-W.; Fang, R.; Hao, X.-H.; Qiu, Z.-H.; Yang, F.; Yan, X.-B.; Wang, Q.; Gong, X.-J.; Liu, X.-Y.; Liang, Y.-M. Copper-catalyzed one-pot trifluoromethylation/aryl migration/carbonyl formation with homopropargylic alcohols. Angew. Chem. Int. Ed. 2014, 53, 7629–7633.
- For producing phosphonyl radical from diphenyl phosphine oxide and AgNO3, see: Liu, Y.; Chen, X.-L.; Zeng, F.-L. Sun, K.; Qu, C.; Fan, L.-L.; An, Z.-L.; Li, R.; Jing, C.-F.; Wei, S.-K.; Qu, L.-B.; Yu, B.; Sun, Y.-Q.; Zhao, Y.-F. Phosphorus Radical-Initiated Cascade Reaction to Access 2-Phosphoryl-Substituted Quinoxalines. J. Org. Chem. 2018, 83, 11727-11735.
- For producing p-toluenesulfonyl radical from p-toluenesulfonyl chloride under photoredox catalytic conditions, see: Ding, R.; Li, L.; Yu, Y.-T.; Zhang, B.; Wang, P.-L. Photoredox-Catalyzed Synthesis of 3-Sulfonylated Pyrrolin-2-ones via a Regioselective Tandem Sulfonylation Cyclization of 1,5-Dienes. Molecules 2023, 28, 5473.
- For producing perfluoroalkyl radicals from fluorinated sulfonyl chlorides under photoredox catalytic conditions, see: Kliś, T. Visible-Light Photoredox Catalysis for the Synthesis of Fluorinated Aromatic Compounds. Catalysts 2023, 13, 94.
- Hu, J.; Yang, C.; Qin, X.; Liu, H.; Ma, T.; Shi, A.-t.; Lv, Q.-L.; Liu, X.; Yang, J.; Li, D. Catalyst- and base-free visible light-enabled radical relay trihalomethylation/functional group-migration/carbonylation with CX3SO2Cl. Org. Biomol. Chem. 2024, 22, 4488–4493. [Google Scholar] [CrossRef]
- Kwon, Y.; Wang, Q. Copper-Catalyzed 1,2-Aminocyanation of Unactivated Alkenes via Cyano Migration. Org. Lett. 2020, 22, 4141–4145. [Google Scholar] [CrossRef]
- Cheng, C.; Chen, D.; Li, Y.; Xiang, J.-N.; Li, J.-H. Fluoroamide-driven intermolecular hydrogen-atom-transfer-enabled intermolecular 1,2-alkylarylation of alkenes. Org. Chem. Front. 2023, 10, 943–950. [Google Scholar] [CrossRef]
- Sarkar, S.; Banerjee, A.; Yao, W.; Patterson, E.V.; Ngai, M.-Y. Photocatalytic Radical Aroylation of Unactivated Alkenes: Pathway to β-Functionalized 1,4-, 1,6-, and 1,7-Diketones. ACS Catal. 2019, 9, 10358–10364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-G.; Li, X.; Zhang, C.; Feng, C. Multisubstituted Cyclohexene Construction through Telescoped Radical-Addition Induced Remote Functional Group Migration and Horner-Wadsworth-Emmons (HWE) Olefination. Org. Lett. 2021, 23, 9611–9615. [Google Scholar] [CrossRef] [PubMed]
- Seastram, A.C.; Hareram, M.D.; Knight, T.M.B.; Morrill, L.C. Electrochemical alkene azidocyanation via 1,4-nitrile migration. Chem. Commun. 2022, 58, 8658–8661. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Zhang, H.; Wu, X.; Zhu, C. Radical trifunctionalization of hexenenitrile via remote cyano migration. Chem. Commun. 2022, 58, 1005–1008. [Google Scholar] [CrossRef]
- Zheng, Y.; Liao, Z.; Xie, Z.; Chen, H.; Chen, K.; Xiang, H.; Yang, H. Photochemical Alkene Trifluoromethylimination Enabled by Trifluoromethylsulfonylamide as a Bifunctional Reagent. Org. Lett. 2023, 25, 2129–2133. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Li, J.; Wei, Z.; Feng, J.; Du, D. Organocatalytic radical relay trifunctionalization of unactivated alkenes by a combination of cyano migration and alkylacylation. Chem. Commun. 2023, 59, 5395–5398. [Google Scholar] [CrossRef]
- Delfau, L.; Nichilo, S.; Molton, F.; Broggi, J.; Tomás-Mendivil, E.; Martin, D. Critical Assessment of the Reducing Ability of Breslow-type Derivatives and Implications for Carbene-Catalyzed Radical Reactions. Angew. Chem. Int. Ed. 2021, 60, 26783–26789. [Google Scholar] [CrossRef]
- Feng, J.-Q.; Li, L.; Wang, J.; Ni, A.; Wei, Z.; Du, D.; Feng, J. Dual visible-light and NHC-catalyzed radical relay trifunctionalization of unactivated alkenes. Chem. Synth 2024, 4, 5. [Google Scholar] [CrossRef]
- Guo, K.; Gu, C.; Li, Y.; Xie, X.; Zhang, H.; Chen, K.; Zhu, Y. Photoredox Catalyzed Trifluoromethyl Radical-Triggered Trifunctionalization of 5-Hexenenitriles via Cyano Migration. Adv. Synth. Catal. 2022, 364, 1388–1393. [Google Scholar] [CrossRef]
- Guo, K. Photoredox-Catalyzed Trifunctionalization of 5-Heteroaryl-Substituted 1-Hexenes or 5-hexenenitriles via Remote Heteroaryl or Cyano Group Migration. Adv. Synth. Catal. 2023, 365, 3616–3621. [Google Scholar] [CrossRef]
- Ghouse, A.M.; Maheswari, B.; Akondi, S.M. Visible-Light Photoredox-Catalyzed Trifunctionalization of Unactivated Alkenes via Diamidation and Cyano Migration. Adv. Synth. Catal. 2025, 367, e202400987. [Google Scholar] [CrossRef]
- Wang, Z.; Chang, C.; Chen, Y.; Wu, X.; Li, J.; Zhu, C. Remote desaturation of hexenenitriles by radical-mediated cyano migration. Tetrahedron 2023, 131, 133228. [Google Scholar] [CrossRef]
- Li, B.; Xing, D.; Li, X.; Chang, S.; Jiang, H.; Huang, L. Chemo-divergent Cyano Group Migration: Involving Elimination and Substitution of the Key α-Thianthrenium Cyano Species. Org. Lett. 2023, 25, 6633–6637. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Luo, X.; Wang, Y.; Liu, Z.; Yu, Y.; Wang, X.; Ren, D.; Wang, P.; Chen, Y.-H.; Qi, X.; Yi, H.; Lei, A. Radical-triggered translocation of C-C double bond and functional group. Nat. Chem. 2024, 16, 1621–1629. [Google Scholar] [CrossRef]
- Shigenaga, S.; Shibata, H.; Tagami, K.; Kanbara, T.; Yajima, T. Eosin Y-Catalyzed Visible-Light-Induced Hydroperfluoroalkylation of Electron-Deficient Alkenes. J. Org. Chem. 2022, 87, 14923–14929. [Google Scholar] [CrossRef]
- Zhao, H.; McMillan, A.J.; Constantin, T.; Mykura, R.C.; Juliá, F.; Leonori, D. Merging Halogen-Atom Transfer (XAT) and Cobalt Catalysis to Override E2-Selectivity in the Elimination of Alkyl Halides: A Mild Route toward contra-Thermodynamic Olefins. J. Am. Chem. Soc. 2021, 143, 14806–14813. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Y.; Zhu, C. Radical Functionalization of 1,6-Diene via Transannular Cyano Migration: Synthesis of Polysubstituted Cyclopentanes. Chin. J. Chem. 2025, 43, 437–442. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, H. Fluoroalkylative Ketonization of Malononitrile-Tethered Alkenes via Nickel Electron-Shuttle and Lewis Acid Catalysis. Org. Lett. 2024, 26, 4532–4536. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Y.; Li, J.; Zhu, C. Cyano migration-mediated radical-polar crossover cyclopropanation. Sci. China Chem. 2024, 67, 241–248. [Google Scholar] [CrossRef]
- Hong, Y.; Deng, H.-P. Alkylcyanation of Unactivated Alkenes with Protic C(sp3)-H Feedstocks via Radical-Initiated Intramolecular Cyano Group Migration Enabled by Photoredox/Brønsted Base Dual Catalysis. ChemPhotoChem 2025, 9, e202400282. [Google Scholar] [CrossRef]
- Sato, T.; Miki, K.; Seno, M. Radical Group-Transfer Polymerization of 2-Thiocyanatoethyl Vinyl Ether. Macromolecules 1999, 32, 4166–4172. [Google Scholar] [CrossRef]
- Uchiyama, M.; Imai, M.; Kamigaito, M. Synthesis of degradable polymers via 1,5-shift radical isomerization polymerization of vinyl ether derivatives with a cleavable bond. Polym. J. 2024, 56, 359–368. [Google Scholar] [CrossRef]
- An, B.; Zhou, L.; Liu, S.; Zheng, Y.; Li, C.; Cui, F.; Yue, C.; Liu, H.; Sui, Y.; Ji, C.; Yan, J.; Li, Y. Radical Homopolymerization of Linear α-Olefins Enabled by 1,4-Cyano Group Migration. Angew. Chem. Int. Ed. 2024, 63, e202402511. [Google Scholar] [CrossRef]
- Song, S.; Wang, S.; Wang, Z.; Sun, H.; Wang, X.; Zhu, C. Switchable Radical Polymerization of α-Olefins via Remote Hydrogen Atom or Group Transfer for Enhanced Battery Performance. Angew. Chem. Int. Ed. 2024, 63, e202418350. [Google Scholar]
- Zheng, Y.; Dong, Q.-X.; Wen, S.-Y.; Ran, H.; Huang, H.-M. Di-π-ethane Rearrangement of Cyano Groups via Energy-Transfer Catalysis. J. Am. Chem. Soc. 2024, 146, 18210–18217. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, J.; Pang, T.; Wu, G.; Zhong, F. Norrish-Yang-type cyclopropanation via functional group migration with photosensitizer at ppb loading. Chem Catal. 2024, 4, 101099. [Google Scholar] [CrossRef]
- Zheng, B.; Zhi, J.; Wang, N.; Zhang, D.; Shimakoshi, H.; Li, Y.; Liu, Q.; Pan, L. Radical-triggered base-free 1,3-C → C migrations: chemodivergent synthesis of cyclic imines from N-allyl enamines. Org. Chem. Front. 2024, 11, 500–507. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; O'Shea, D.M.; Gerba, S.; Westwood, S.W. Cyano or Acyl Group Migration by Consecutive Homolytic Addition and β-Fission. J. Chem. Soc. Chem. Commun. 1987, 666–667. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; O'Shea, D.M.; Westwood, S.W. Rearrangement of Suitably Constituted Aryl, Alkyl, or Vinyl Radicals by Acyl or Cyano Group Migration. J. Am. Chem. Soc. 1988, 110, 2565–2575. [Google Scholar] [CrossRef]
- Cossy, J.; Poitevin, C.; Pardo, D.G.; Peglion, J.L. Synthesis of 2-(Alkylamino)benzonitriles from α-(Bromoarylamino)nitriles. Synthesis 1995, 1368–1370. [Google Scholar] [CrossRef]
- Sulsky, R.; Gougoutas, J.Z.; DiMarco, J.; Biller, S.A. Conformational Switching and the Synthesis of Spiro[2H-indol]-3(1H)-ones by Radical Cyclization. J. Org. Chem. 1999, 64, 5504–5510. [Google Scholar] [CrossRef] [PubMed]
- Bowman, W.R.; Bridge, C.F.; Brookes, P. Radical cyclisation onto nitriles. Tetrahedron Lett. 2000, 41, 8989–8994. [Google Scholar] [CrossRef]
- Ryclmovsky, S.D.; Swenson, S.S. Tandem Radical Nitrile Transfer-Cyclization Reactions of 1,3-Dloxane-4-Nltriles: Synthesis of Spirocyclic Systems. Tetrahedron 1997, 53, 16489–16502. [Google Scholar] [CrossRef]
- Crich, D.; Bowers, A.A. 4,6-O-[1-Cyano-2-(2-iodophenyl)ethylidene] Acetals. Improved Second-Generation Acetals for the Stereoselective Formation ofβ-D-Mannopyranosides and Regioselective Reductive Radical Fragmentation to β-D-Rhamnopyranosides. Scope and Limitations. J. Org. Chem. 2006, 71, 3452–3463. [Google Scholar] [CrossRef]
- Crich, D.; Bowers, A.A. Synthesis of a β-(1→3)-D-Rhamnotetraose by a One-Pot, Multiple Radical Fragmentation. Org. Lett. 2006, 8, 4327–4330. [Google Scholar] [CrossRef]
- Onozaki, Y.; Kurono, N.; Senboku, H.; Tokuda, M.; Orito, K. Synthesis of Isoindolobenzazepine Alkaloids Based on Radical Reactions or Pd(0)-Catalyzed Reactions. J. Org. Chem. 2009, 74, 5486–5495. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Du, J.; Zou, Z. Selective C-C Bond Scission of Ketones via Visible-Light-Mediated Cerium Catalysis. Chem. 2020, 6, 266–279. [Google Scholar] [CrossRef]
- Xie, X.; Li, Y.; Bo, Z.; Zhu, Y.; Chen, K. Photoredox/Cu dual catalyzed 1,4-cyanosulfonylation enabled by remote cyano migration. Org. Chem. Front., 2024, 11, 4857–4861. [Google Scholar] [CrossRef]
- Zhu, J.-L.; Wu, Y.-P. Rhodium-Catalyzed Intramolecular Cyclopropanation of α-Diazo β-Keto Nitriles Containing an Unsaturated Substituted Cycloalkyl Group. Synlett 2017, 28, 1467–1472. [Google Scholar] [CrossRef]
Figure 1.
DFT energy calculation for radical cyclization onto nitrile group (Eşsiz, 2023).
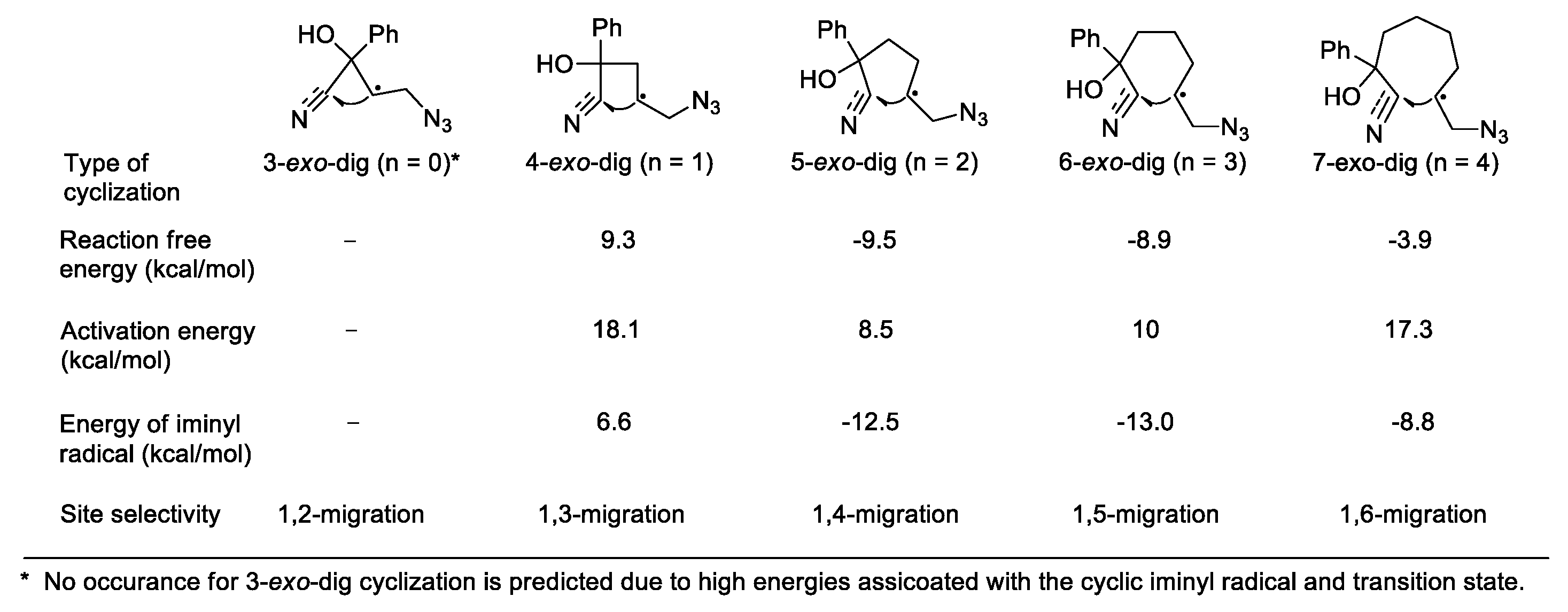
Figure 2.
Concept of using radical sampling strategy to achieve 1,4-cyano translocation.
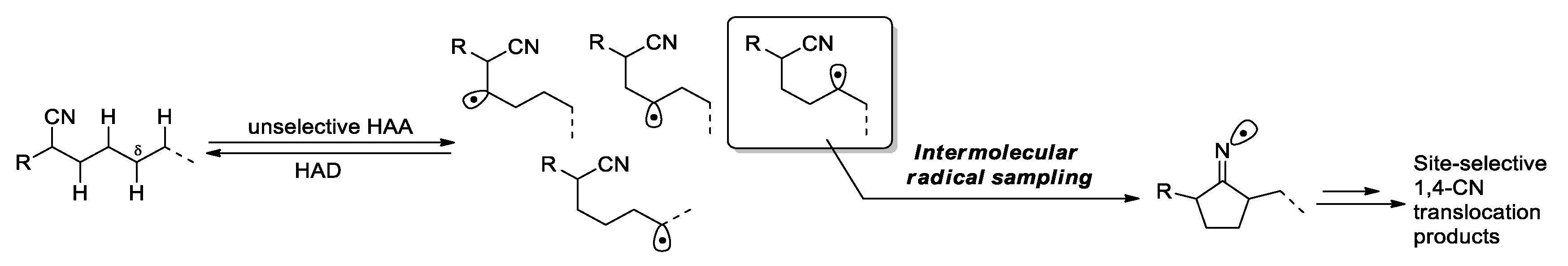
Figure 3.
Configuration inversion during the cyclization of 150 into 151 (Sulsky, 1999).
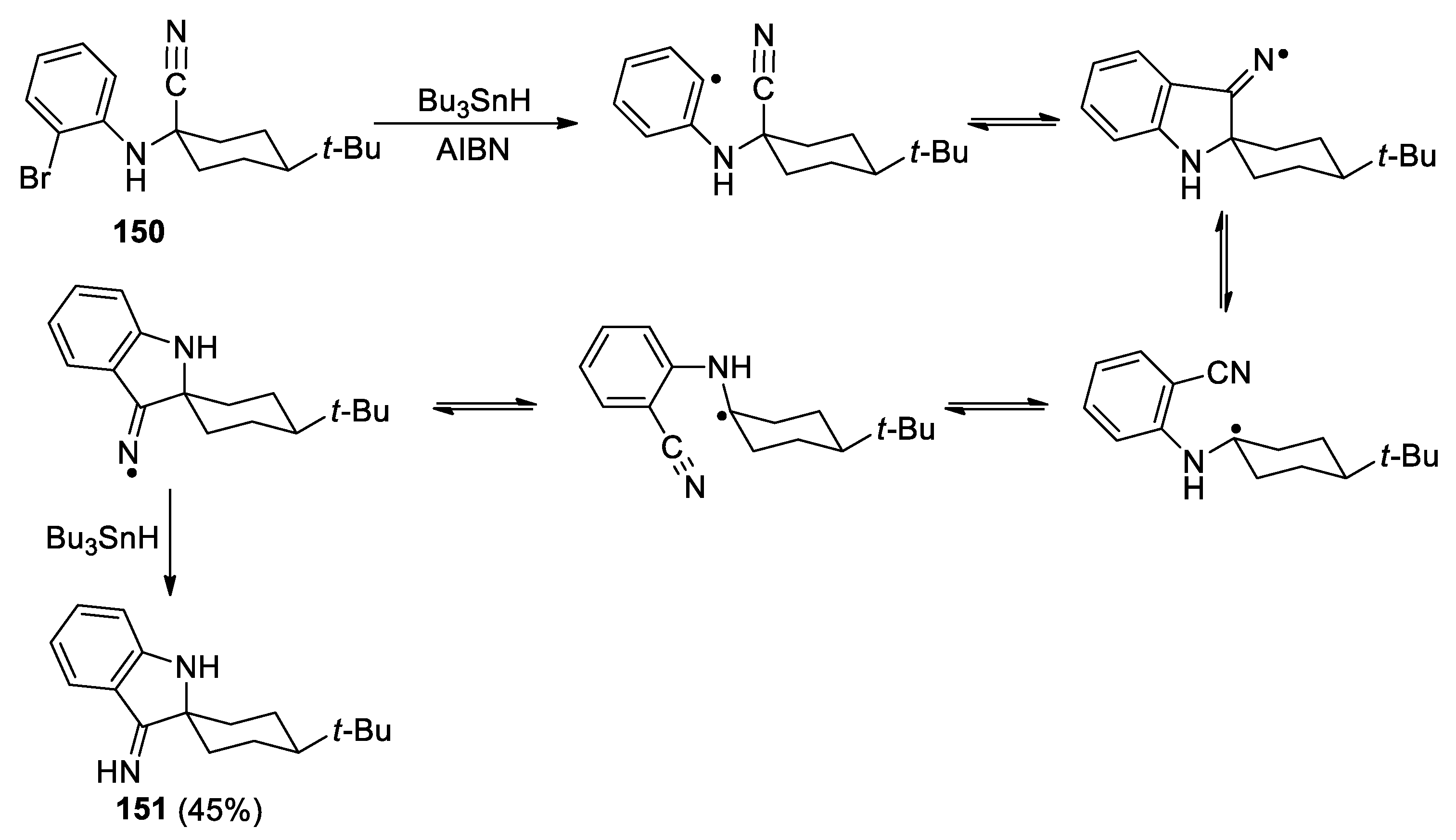
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



