Submitted:
02 June 2025
Posted:
05 June 2025
You are already at the latest version
Abstract
The prevailing fungal infections, especially with Candida albicans are serious health concern
particularly in immunocompromised patients. Considering the menace of drug resistance and
tremendous mortality ratio of Candida-related infections, consequential efforts are ongoing to
characterize new drug targets or agents for therapeutic intervention. As a consequence, in the
present study, we targeted Rrp9 protein from Candida albicans which plays an essential role in
ribosome biogenesis and it has role in the cellular response to certain drugs. Sequence analysis
using BLAST protein search and 3D modeling using Discovery Studio (DS) program for Rrp9
protein prediction were carried out. The antifungal activity of few selected ligands,
antimuscarinc acetylcholine receptor drugs (Dicyclomine, Solifenacin, Hydroxyzine,
Promethazine and Flavoxate) on the predicted 3D model of Rrp9 protein through molecular
docking studies using GOLD software was done. Our in silico analysis afforded insight
information of structure and binding interactions of 3D modeling C. albicans Rrp9 protein with
these selected ligands. All compounds showed good docking interaction with Rrp9 protein. The
antimuscarinic, Solifenacin succinate presented better interactions with good binding energy. We
suggested that these antimuscarinic drugs can be used as anti-Candidal albicans as well as
antifungal agents.
Keywords:
Dicyclomine
; Solifenacin
; Hydroxyzine
; Promethazine
; Flavoxate
; Candida albicans Rrp9 protein
; Docking
Introduction
Microbial infections are considered as a major public health problem over the past decades as they have a serious and continuous threat to humans and animals [1]. The incidence of -threating infections of life caused by pathogenic microbes like bacteria, fungi, and viruses is very high due to the increased number of multi-drug resistant of microbial strains [2]. Of all the pathological microbes and the prevalent fungal infections are became alarming in the medical field [3]. There are 600 fungal species are estimated as human pathogens that may be endogeneous opportunistic pathogens or environmentally acquired pathogens [1,4,5]. Fungal infections caused by pathogenic fungus are range from cutaneous skin infections to lethal acute or chronic infections to deep tissues [4, 6]. Most of the fungal infections occurring worldwide are due to the Candida species, which are commensal microbes of human. Candida species are present as commensal microbe on skin, oropharynx, upper respiratory tract, gastrointestinal tract, genitouninary system, tract without causing harm to healthy individuals. Candida albicans is the most opportunistic pathogen of Candida species when occurs breakdown of host immune and defense system [6,7,8]. Candida albicans, is one of the frequent causative agent of candidiasis in human, causing superficial infections of vulvovaginal, esophageal or oropharyngeal and complicated invasive diseases which include candidemia and deep tissue candidiasis [6,8,9]. Among of these, the widespread invasive infections may lead to increased morbidity and mortality in immunocompromised patients suffering from acquired immune deficiency syndrome (AIDS), cancer, tuberculosis, , and in organ transplant cases [6,10,11]. The high mortality of Candida-related infections in immunocompromised patients is a perpetual problem due to difficulty of diagnosis and invasive treatment or systemic candidiasis; thus there is a dire need for the development of appropriate strategies for candidiasis treatment. This opportunistic pathogen is often linked to using of wide-ranging antibiotics, immunosuppressive agents, anticancer, and anti-AIDS drugs [12]. One of the complications in the treatment of Candida albicans infections is the expansion of antifungal drug resistance such as antimycotic therapy in HIV-affected individuals [13]. As the intended threat of drug resistance, the research is ongoing on to recognize new drug targets for therapeutic intervention of these infections, while simultaneously with low toxicity profile. Many of these proceeds with the recognition appliances of proteomic era. Proteomic approaches in yeast have identified many factors such as U3 snoRNA-associated proteins and proteins are derived from open reading frames [14]. The U3-specific Rrp9 protein plays an essential role in the posttranscriptional maturation of rRNAs with the association of the U3 small nucleolar ribonucleoprotein complex (U3 snoRNP) and it has role in in the cellular response to certain drugs [15, 16]. A U3-specific Rrp9 protein is consist of a WD repeat domain that is preceded by an N-terminal region. The WD repeats in the protein are thought to mediate dynamic protein–protein interactions and provide a platform for the assembly of multicomponent complexes [17]. Any alterations in these repeats leads to the disassociation of the protein complexes suggesting the perturbations in ribosome biogenesis providing a cellular mechanism for the inhibition of the pathogenicity caused by the yeast. Targeting this protein may provide a promising approach for the management of microbial infections. In this study, the sequence analysis, 3D structure modeling and validation of the target protein Rrp9 from Candida albicans are carried out. Further with the validated modeled protein, the molecular docking is applied to study the binding conformations and structural specificity of the selected antimuscarinic acetylcholine receptor drugs using GOLD. All selected antimuscarinic drugs presented good binding interactions with 3D modeling C. albicans Rrp9 protein while Solifenacin succinate had better binding. This study discovered that these selected antimuscarinic receptor drugs can be redirected as anti-Candida albicans and antifungal agents. Also these observations may be of great help in the designing of novel antifungal medications based on the structural and functional basis of ligand binding to C. albicans Rrp9 protein.
Materials and Methods
Retrieve and analysis of protein sequence
The amino acid sequence of hyphothetical protein Rrp9 from Candida albicans was retrieved from the SwissProt database website using the accession No. Q5A213. By using ProtParam tool the primary structure of Rrp9 protein was predicted. The physiochemical properties were computed which are included the molecular weight, theoretical isoelectric point (pI), amino acid composition, atomic composition, total number of positive and negative residues, extinction coefficient, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). The Secondary structure of this protein was predicted using fasta sequence using SOPMA (Self Optimized Prediction Method with Alignment) program which uses multiple alignments. The secondary structural feature was calculated and analyzed which are alpha helixes, extended strand, random coil and beta turns of the selected protein sequence.
Modeling of 3D Structure
The 3D structure of protein Rrp9 from C. albicans was determined using homology modeling methods of Discovery Studio (DS) program. Homology modeling method was relied on the basis of alignment of target sequence and the sequences of known structures of one or more proteins that can be used as templates which is resemble the structure of the query sequence. The sequence alignment and template structure are then used to generate the structural model of target protein. The retrieved fasta sequence as query, by using BLAST against PDB, the homologous sequences with known related protein 3D structures that can be used as templates are searched. From the obtained BLAST results, the sequence with maximum identity score and lowest e-value was retrieved. The query and the template sequences are aligned using align sequences program of DS. Based on this sequence alignment, using crystal structural coordinates of templates, three 3D models of our target protein was generated with the homology model building program of DS Modeler. The best model of target was selected on the basis of the analysis results of the internal scoring functions, PDF Total Energy, PDF Physical Energy, DOPE Scores of DS. The best homology model with least energy function was taken for further refinement using DS energy minimization methods. In the first, all the hydrogen atoms are allowed while performing the calculations. Energy minimization was carried out under CHARMmforce fields with 1000 steps of the steepest descent algorithm, then followed by 1000 steps of the conjugate gradient algorithm, with minimization criteria set at 0.001 root mean square gradients, respectively to obtain a stable and low energy conformation. After the optimization procedure, the final 3D model was chosen for further validation.
Model Evaluation:
After the optimization procedure, the final 3D model of Rrp9 was chosen for further validation such as PROCHECK, Verify 3D, ProSA and RMSD. PROCHECK are used in validation of modeled structure by verifying the Ramachandran plot quality through analyzing the dihedral angles Ψ and Φ of amino acid residues in protein structure. It showed the possible conformations of Φ and Ψ angles for the assessment of the stereo chemical quality of the protein structure. Verify 3D was analyzed the compatibility of an atomic model (3D) with its own amino acid sequence (1D). The scores of a sliding 21-residue window (from -10 to +10) are added and plotted for individual residues. ProSA was used to analyzes the energy criteria and gave the Z-score values for the comparison of reliability. The model Z score should be comparable to the Z scores was determined experimentally of similar structures. Additionally, the Root Mean Square Deviation value indicated the 3D structures degree of similarity. In this, both the query and template structures are superimposed for RMSD calculation. Lower RMSD value represented more similarity in the structures.
Identification of Active Site
The possible binding site of the modeled Candida albicans Rrp9 was predicted according to the receptor cavity method (Eraser algorithm) using the DS Analyze Binding Site tool. The receptor molecule is first defined by using define receptor molecule module of DS. The proteins active site-Search was done using ‘find sites from the receptor cavities’ which identified the protein active sites by locating cavity in the modeled structure. When the search was completed, the largest site, Site 1 was automatically displayed on the structure. The predicted site 1 was chosen as the most favorable binding site for docking with the ligands.
Generation of Ligand data set and their optimization
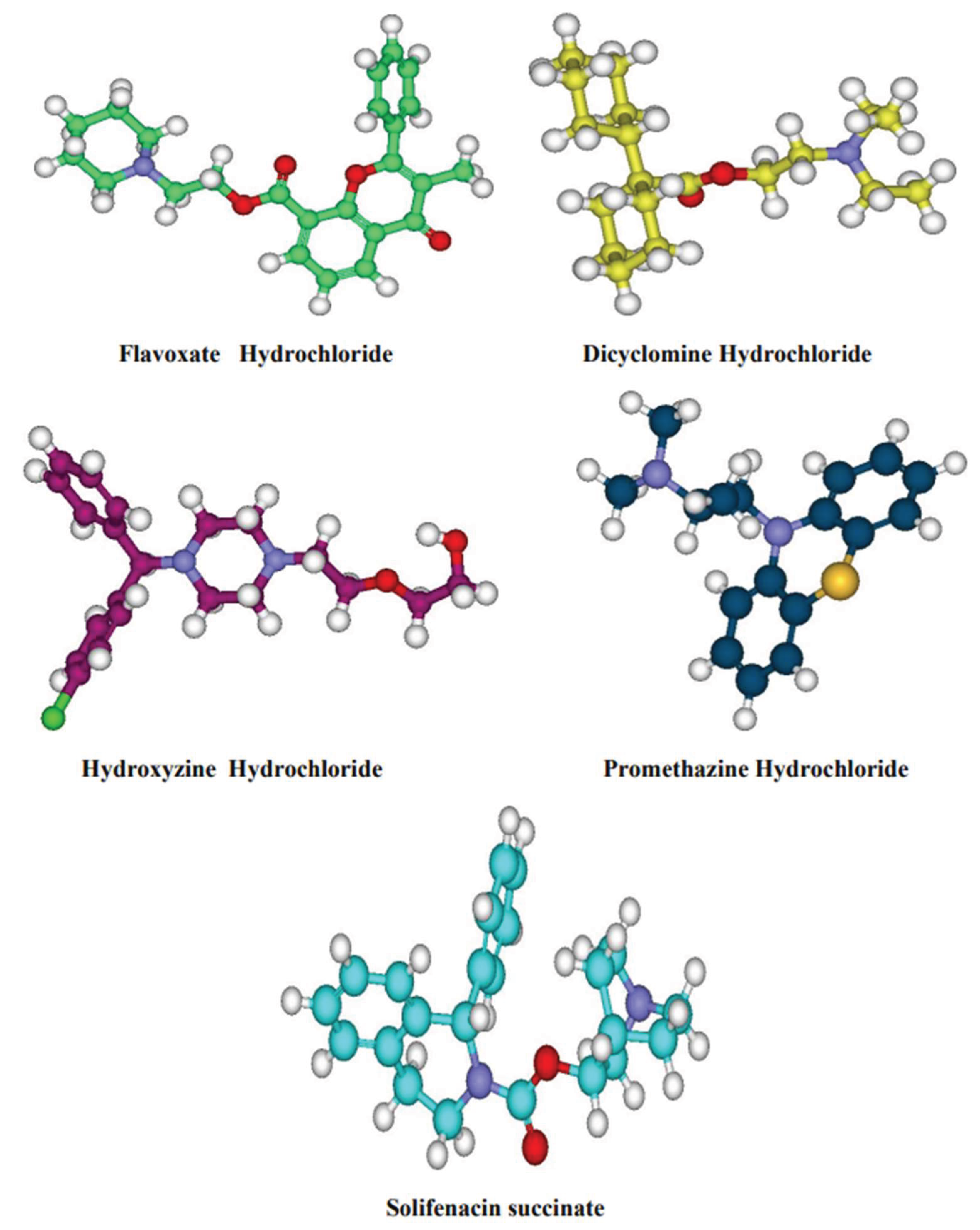
Five compounds structures are retrieved from Pubchem database in SDF format, i.e Dicyclomine hydrochloride, Flavoxate hydrochloride, Hydroxyine hydrochloride, Promethazine hydrochloride, Solifenacin succinate. Their 2D structures were sketched using ACD Chemsketch software and converted to 3D representation using catalyst algorithm of DS. Prepare ligands module of DS was used for Ligand preparation which corrected for hydrogen bonds addition, bond lengths, bond angles, isomer and tautomer generation and filters the ligands by removing the duplicate structures. Further followed by minimization and optimization in CHARMm force field with the smart minimizer algorithm which followed by the conjugant gradient algorithm, till it satisfied with the convergence gradient of 0.001 kcal/mol, for attaining the low energy conformational structures. The three dimensional structures of the compounds were depicted in figure 1.
Figure 1.
3D structures of the compounds.
Molecular docking
Docking simulations of the ligands and their interactions with the homology model of Candida albicans Rrp9 was generated by molecular docking using the Genetic Optimization for Ligand Docking (GOLD) software from Cambridge Crystallographic Data Center, UK. This program uses Genetic Algorithm which allows partial flexibility of protein and full flexibility of ligand. In GOLD docking was carried out using the wizard with default parameters population size (100); selection- pressure (1.1); number of operations (10,000); number of islands (1); niche size (2); and operator weights for migrate (0), mutate (100), and crossover (100) were applied. The active site with a 10 Å radius sphere was defined by selecting an active site residue of protein. Default Genetic Algorithm settings were used for all calculations and a set of 10 solutions were saved for each ligand. The interactions of the ligand and the receptor were evaluated and ranked via GOLD Fitness score implemented in GOLD, and the orientation with the highest docking score was returned for further analysis.
Results and discussion
Retrieve and analysis of sequence
The sequence of hypothetical protein Rrp9 from C. albicans was retrieved in fasta format from SWISS PROT database with accession No: Q5A213 and entry name: Q5A213_CANAL. The primary structure was predicted using ProtParam tool and the physicochemical parameters computed are presented in Table 1 . Results showed that the protein has 549 amino acid residues with an estimated molecular weight of 62414.05 daltons. The maximum number of amino acids present in the sequence was found to be Asn (12.5%) and the least was Trp (0.3%). The total numbers of negatively charged residues (Asp+Glu) are 78 and the total numbers of positively charged residues (Arg+Lys) are 75. The isoelectric point pI was 6.59 revealed the acidic nature of this protein. The instability index is 44.74 which classify the protein as unstable and the, aliphatic index is 76.03 which indicated that this protein is thermotable. The estimated half-life is 30 hours (mammalian reticulocytes, in vitro), greater than 20 hours (yeast, in vivo) and greater than 10 hours (Escherichia coli, in vivo). Negative GRAVY value indicated the hydrophilicity of the protein. The calculated GRAVY is -0.753 indicated that this protein is hydrophilic and soluble in nature.
Secondary structure analysis was predicted by SOPMA program as shown in Figure 2. The result showed random coil was predominant (39.89%), followed by alpha helix (26.96%) and extended strand (25.68%). Also, beta turn was found as 7.47%. The percentage of alpha helix and extended strand were almost equal. The high percentage of random coils indicated the flexibility of the protein which is responsible for more interactions.
3. D Structure modeling
The predicted homology model of the target protein’s accuracy was closely related to the percentage of identity and the optimal alignment of the sequences between the template and target. The results from BLASTP against PDB in search of template structure have revealed the similar structures with high sequence identity. RNA binding Protein of Saccharomyces cerevisiae (PDB ID: 4J0X) had 45% identity and 66% similarity with the sequence of our target protein Rrp9 from C. albicans (Q5A213_CANAL). On the basis of sequence alignment of the selected template RNA binding Protein of Saccharomyces cerevisiae (PDB ID: 4J0X) and target sequence Rrp9 from C. albicans, the modeling of the protein was carried out to generate high quality models. In the inbuilt programs of DS perform sequence alignment and homology modeling, the sequence alignment is done using sequence analysis protocols called “Align multiple sequences”. The protocol inputs are target sequence and 4J0X template, which aligns them and gave the alignment output as shown in Figure 3. The initial models of the Rrp9 was built using protein modeling protocol called “build homology model” which uses modeler to build homology models. The modeler options are kept default while running ended up with a loop refinement by default modeling process. Of all the developed three homology models of Rrp9 the model with least internal scoring functions, of DS was selected. The build models were B99990001, B99990002 and B99990003. Out of the three models, Model of B99990001 as shown in Figure.4 had the lowest value in PDF (Probability Density Functions) Total Energy (6141.58), PDF Physical Energy (-556.69) and DOPE (Discrete Optimized Protein Energy) score (-40198.65) , which indicated it as the best model. The energy refinement method gave the best conformation to the model. For optimization, the CHARMm forcefield and steepest descent algorithm were applied with 0.001 minimizing RMS gradient and 1000 minimizing steps. Following the above steps of minimization, the protein was minimized using conjugate gradient algorithm preceded by smart minimizer algorithm until the convergence gradient of 0.001 kcal mol-1 is satisfied. The finally refined protein generated was further taken for model validation by PROCHECK, Verify 3D, ProSA and RMSD.
Figure 3.
Alignment between the RNA binding Protein of Saccharomyces cerevisiae (PDB ID: 4J0X) and query sequence Rrp9 from C. albicans. Thick blue color represents the conserved regions and light blue color represents variable regions of template and query.
Figure 3.
Alignment between the RNA binding Protein of Saccharomyces cerevisiae (PDB ID: 4J0X) and query sequence Rrp9 from C. albicans. Thick blue color represents the conserved regions and light blue color represents variable regions of template and query.

Figure 4.
Representation of Modeled Rrp9 structure in N-C terminal representation contains number of helices 4, Strands 47, turns 54.
Figure 4.
Representation of Modeled Rrp9 structure in N-C terminal representation contains number of helices 4, Strands 47, turns 54.
Model Validation
The stereo chemical quality and accuracy of the final refined modeled Rrp9 protein was evaluated by Ramachandran plot calculations computed with the PROCHECK, described a good quality model which had over 90% residues in most favored region. The results showed that 93.1% residues were in favored region (Figure 5). Of the total sequence length, 549 amino acid residues in favorable and additionally allowed and only five residues were in generously allowed regions which indicated a good quality model.
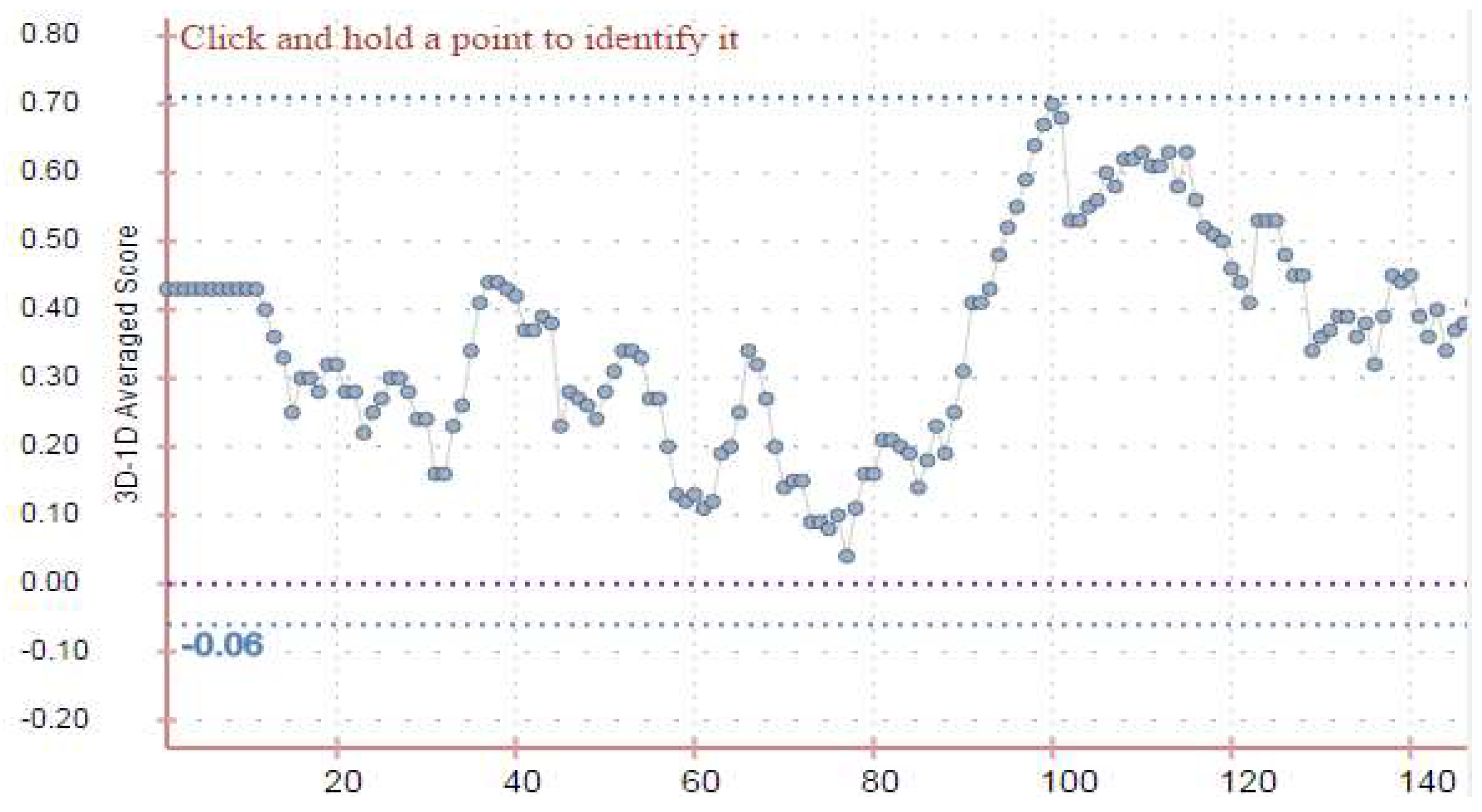
In Verify 3D, the certainty of the 3D model was determined in contrast with its own sequence adopting a 3D profile estimated using atomic coordinates of its structure. 3D profiles of equitable protein structure possessing high S scores in contrast with poor structures with low scores. The S score/3D-1D profile score is the sum over all residue positions of 3D-1D score for amino acid sequence of the protein. The validation results of Verify 3D for this model demonstrated that at least 86.51% of the amino acids have scored > = 0.2 in the 3D/1D profile, which is also satisfactory.
Figure 6.
The Verify 3D score profile of the homology model (target) protein.
The overall protein quality was calculated using ProSA based on the scores of all experimentally determined protein chains found in PDB. The program was gave z-score and a plot of the residue energies. The Z-Score plot showed spot of Z score values of proteins determined by NMR (represented in dark blue color) and by X ray (represented in light blue color). The Z-scores of the obtained modeled Structure of Candida Rrp9 was –6.82 (shown in Figure 7). The black dot represent Z-scores of our model that was within the range of scores of similar proteins determined by X ray crystallography derived structures. .
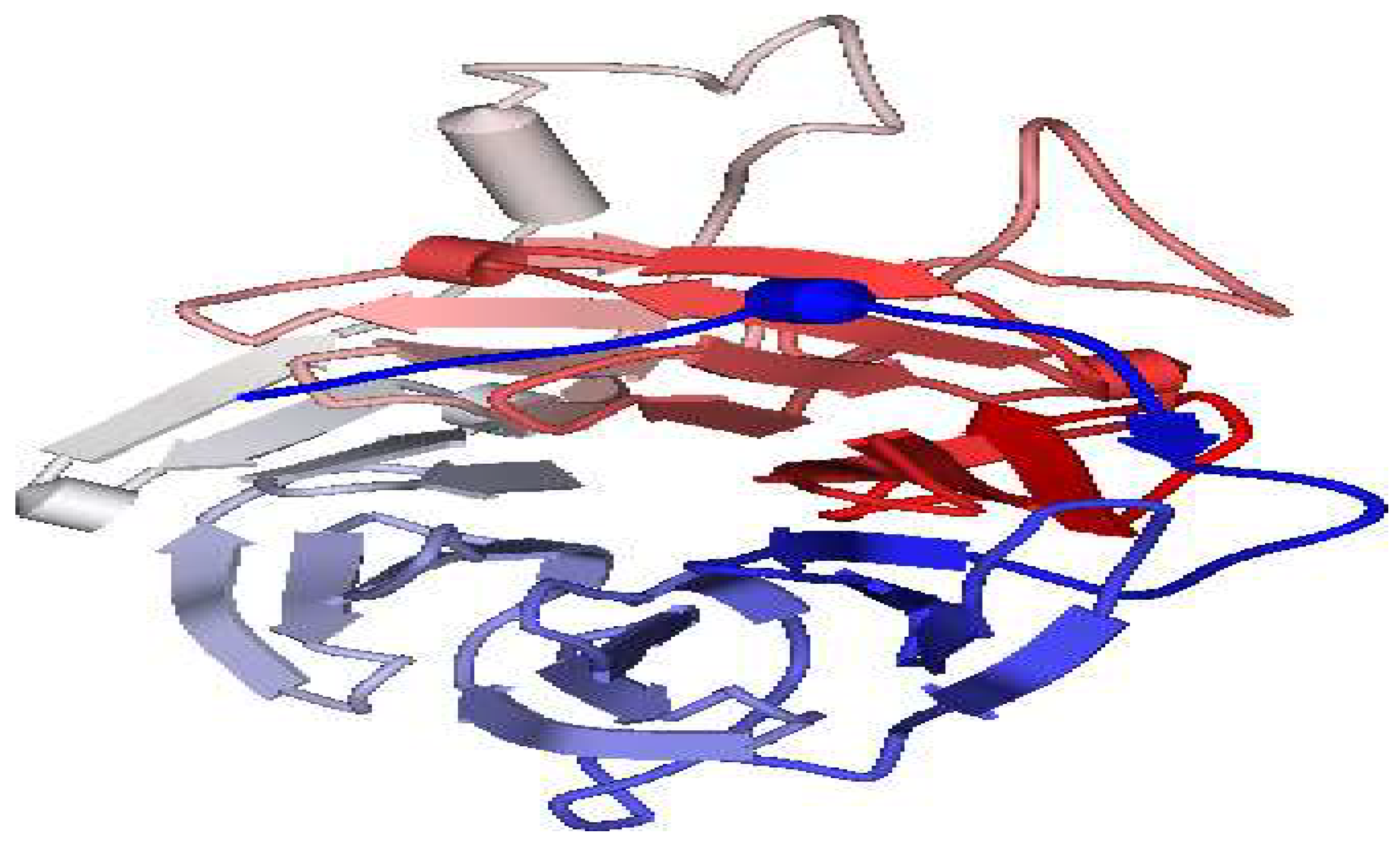
Finally, this was also confirmed by superposition of optimized homology model against the template and the RMSD is estimated using SPDBV. The structural super imposition of the Rrp9 from C.albicans and the template RNA binding Protein of Saccharomyces cerevisiae (PDB ID: 4J0X) is shown in Figure 8. The weighted root mean square deviation was 0.75 Å. This final refined model was used for the identification of active site and for docking of the ligands with the homology model of Rrp9.
Identification of Active Site
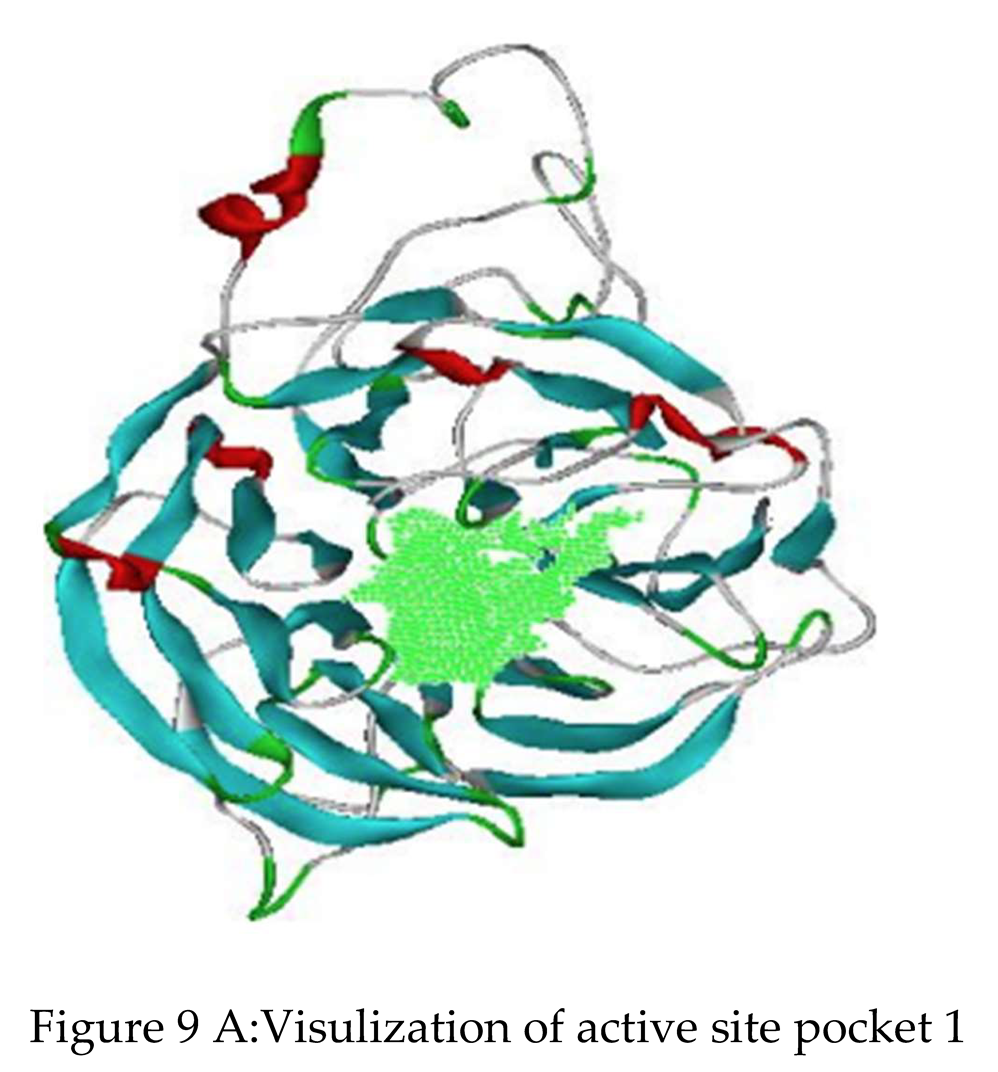
The active site residues of the modeled Rrp9 were identified by using DS which employs cavity detection method using eraser algorithm. The predicted active site pocket 1 shown in figure 9 A had a average volume of 500 and had coordinates as X-1.648, Y-14.419, Z—28.890.
Molecular Docking
The binding mode for the taken ligands into the predicted binding site of the homology model Rrp9 was generated by molecular docking using GOLD software. GOLD Fitness score was used to estimate the ligand binding energies. The orientation of protein–ligand complex with the highest GOLD score was chosen for further analysis. Apart from these, hydrogen bond formations are also considered to evaluate the binding efficacy of the ligands with the protein. The dock scores computed by the scoring algorithm are given in Table 3. All the docked compounds showed fitness scores with a range of 18.75 to 62.17 (Table 3). Almost all the compounds showed hydrogen bonding with the binding site residues of the target protein Rrp9, but Promethazine Hydrochloride doesn't form any hydrogen bonds. Even though the compound Promethazine Hydrochloride had good dock scores it does not show any hydrogen bond interactions. The compounds, Dicycloamine Hydrochloride and Solifenacin Succinate showed single hydrogen bond and the compound Hydroxyine hydrochloride had two hydrogen bond formations with the target protein Rrp9. Considering the dock scores and hydrogen bond interactions, the Solifenacin succinate have chosen as the best compound and Hydroxyzine hydrochloride next to that among all the compounds. Though Hydroxyzine hydrochloride formed two hydrogen bonds, it showed least binding affinity with a fitness score of 54.55 compared to that of the Solifenacin succinate having a fitness score of 62.17. The hydrogen bonding interactions and the interacting amino acids along with the hydrogen bond distances were depicted in the Table 4. Figure 9B-9H showed the binding poses predicted for the compounds against Rrp9 protein model.
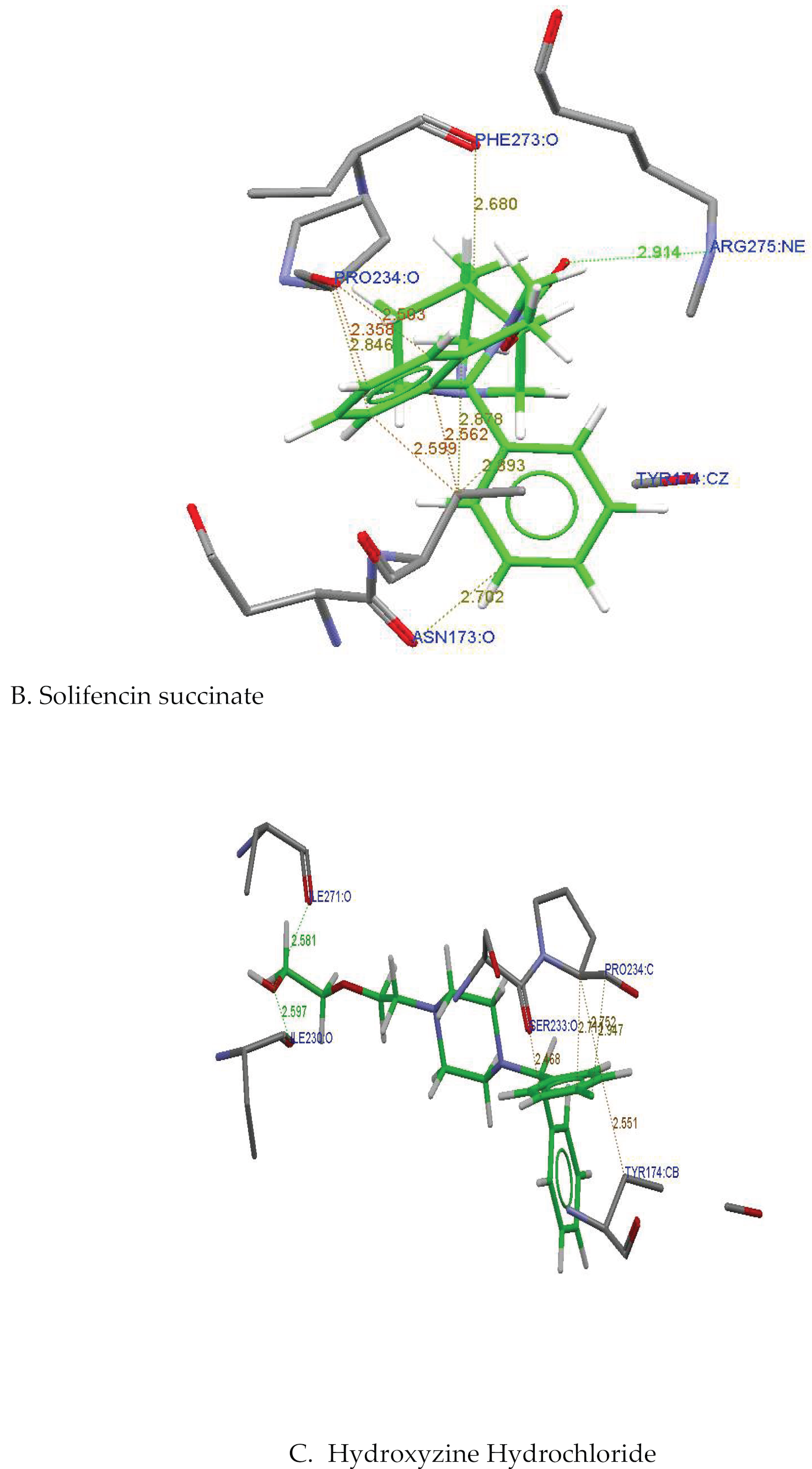
Figure 9B presented the binding pose of compound Solifenacin succinate against Rrp9, indicating that the compound interacts with Rrp9 by forming hydrogen bond with Arginine275. The N-atom of Arginine275 interacted with the oxygen atom of the compound Solifenacin Succinate(Arg275:NE----O2: Solifenacin Succinate) with a hydrogen bond distance of 2.914 Å . the Solifenacin Succinate compound also showed additional interactions with residues Phe273, Asn173 and Pro234 in binding pocket. They are formed with the oxygen atom of phenyl alanine 273 and eight carbon atom of the compound (Phe273: O---C8: Solifenacin Succinate), between the alpha carbon atom of amino acid proline234 and 24th carbon atom of the compound (Pro234:CA---C24: Solifenacin Succinate), between the oxygen atom of amino acid aspargine173 and 17th carbon atom of compound (Asn173:O---C17: Solifenacin Succinate) with a distances of 2.503 Å, 2.356 Å and 2.702 Å respectively.
From the figure 9C, it was revealed that two hydrogen bonds are formed between the amino acid Isoleucine271 and isoleucine 230 and the Hydroxyzine Hydrochloride compound with a hydrogen bond distances of 2.581 Å and 2.597 Å respectively. One hydrogen bond was formed between the oxygen atom of isoleucine 271 and hydroxy group of the compound (Ile271:O---OH: Hydroxyzine Hydrochloride ) and the other between the oxygen atom of isoleucine 230 and hydroxyl group of the compound (Ile230:O---OH: Hydroxyzine Hydrochloride ). The other interactions were formed with the oxygen atom of serine233 and the carbon atom C21 of the compound (Ser233:O—C21: Hydroxyzine Hydrochloride) and proline 234 interacted with C17 of the compounds (Pro234:C---C17: Hydroxyzine Hydrochloride) with a distances of 2.468 Å and 2.752 Å respectively.
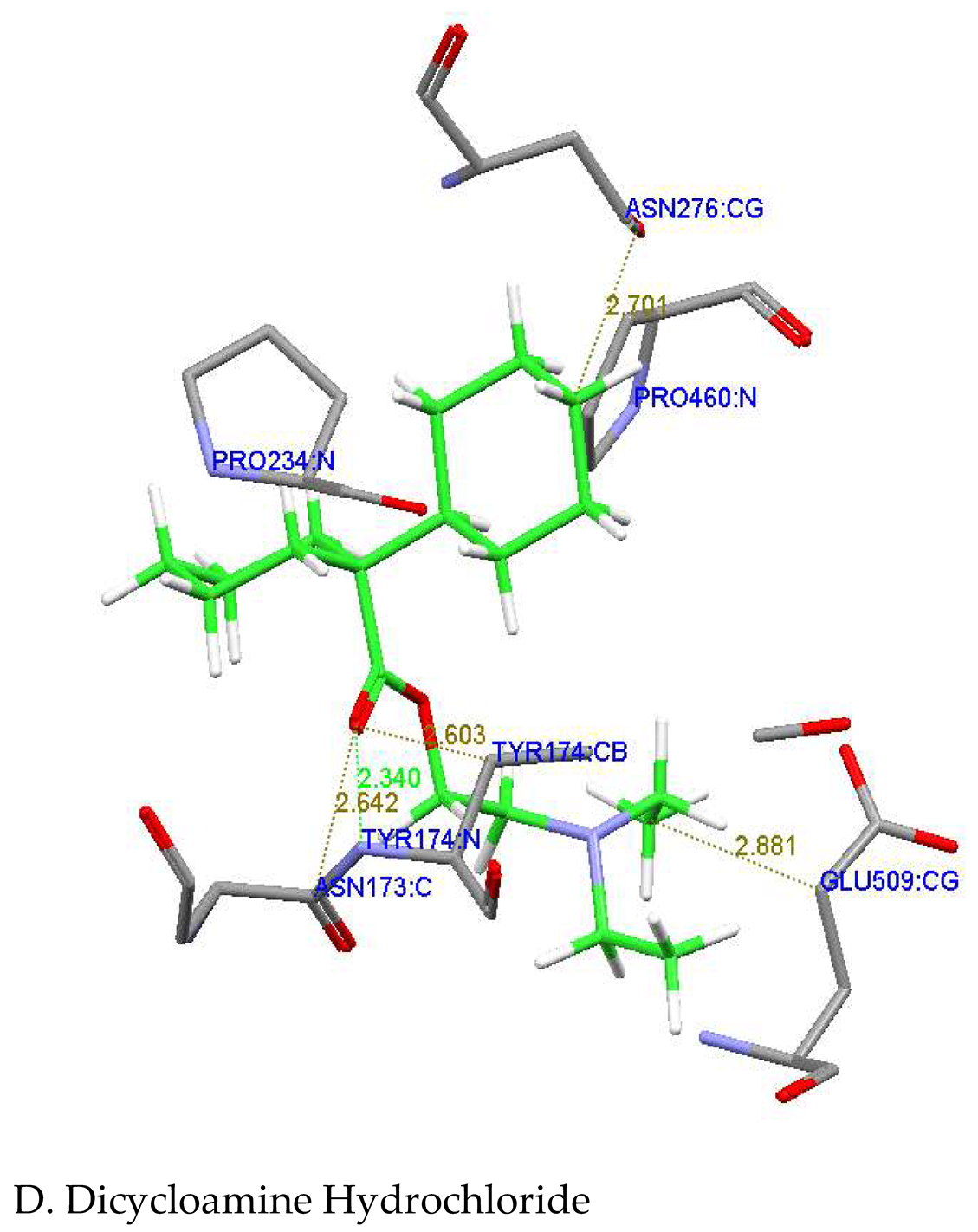
From the Figure 9D it was revealed that the hydrogen bond was formed between nitrogen atom of Tyrosine 174 and the oxygen atom of the compound Dicycloamine (Tyr174:N---O2:Dicycloamine) with a hydrogen bond distance of 2.340 Å. Some close contacts were also formed between the protein and the compound. They were formed with the carbon atom of Tyrosine174 and carbon atom number 8 of the compound (TYR174: CB---C8: Dicycloamine), between the carbon atom of amino acid aspargine173 and oxygen atom of the compound (Asn173: C...O2: Dicycloamine) and between the carbon atom of amino acid glutamine509 and 21st carbon atom of compound (Glu509: CG...C21: Dicycloamine) with a bond distances of 2.603 Å, 2.642 Å and 2.881 Å respectively.
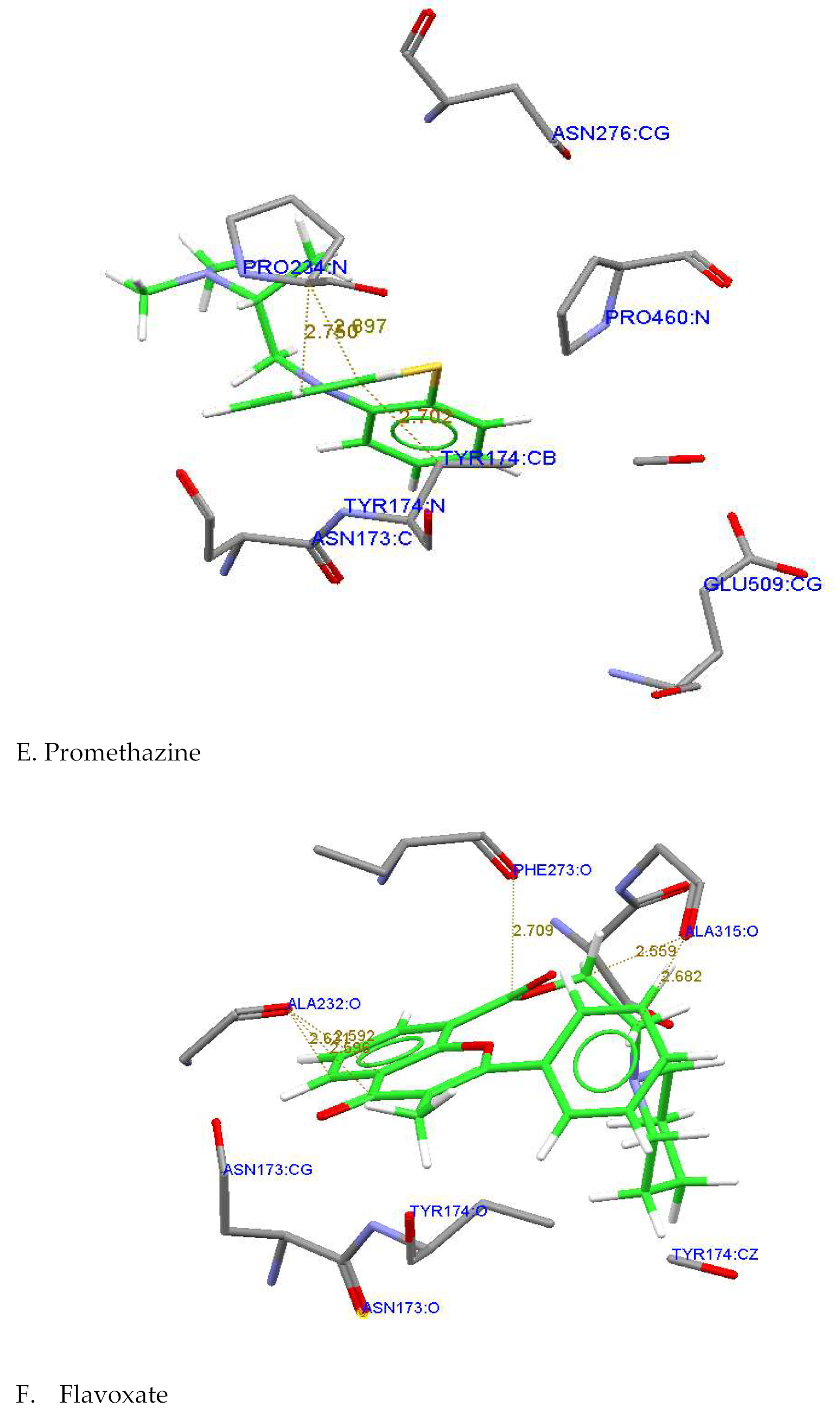
Figure 9E and 9F showed the interactions of the protein Rrp9 with the compounds, Promethazine and Flavoxate indicating some close interactions were formed between the protein and the compounds. From the figure 9E, it was revealed that the 13th carbon atom of the compound, Promethazine interacted with the carbon atom of tyrosine174 (Tyr174: CB---C13: Promethazine) and nitrogen atom of proline234 (Pro:234: N....C13 : Promethazine) with a distances of 2.750Å and 2.897Å respectively. Figure 9F showed that the oxygen atom of the residue Alanine 232 interacted with 21st carbon atom and oxygen atom of the compound, Flavoxate (Ala232: O---C21: Flavoxate and Ala232;O---O4: Flavoxate) with a bond distances of 2.621Å and 2.596 Å respectively. And also the oxygen atom and 11th carbon atom of Flavoxate interacted with the oxygen atoms of phenylalanine273 and Alanine315 (Phe273: O---O3: Flavoxate Hydrochloride and Ala315: O---C11: Flavoxate) with a bond distances of 2.709Å and 2.559 Å respectively.
Figure 9: A. Visualization of active site pocket 1 and Binding poses predicted by GOLD against the homology model Rrp9 with B. Solifencin C. Dicyclomine D. Hydroxygene E. Promethazine F. Flavoxate. Hydrogen bonds are depicted by a green dotted line.
Conclusion
In the current study, we attempted to explore the structural determination and interactions of few selected ligands, antimuscarinic drugs against 3D modeling Candida abicans Rrp9 protein via homology modeling and molecular docking study using GOLD software. All selected ligands during molecular docking showed good binding interactions with good binding energy. Whereas Solifenacin succinate appeared a high binding energy along with hydrogen bonding. These observations may facilitate a promising approach to design novel antifungal agents depending on the structural and functional basis of ligand binding to the C. albicans Rrp9 protein for further research. According to this study, we revealed that the antimuscarinic acetylcholine receptor drugs could be used as anti-Candida albicans as well as antifungal agents.
Acknowledgement
My special and great thanks go to Prof. Mohammed Ahmed Ameen AL-Ahdal (Present Vice Chancellor of Hodeidah University) for his support and facilities. My deepest appreciation goes to Dr. Mohammed Suhail, Director of Medical College and Medical Sciences and Dr. Izz alddin, Head of Academic affairs for their help and support.
Author contributions
Awad Ali designed this study and wrote the manuscript
Competing interest
The author is declare no competing interest
References
- F.M Garibotto, A.D.Garro, M.F.Masman, A.M.Rodriguez, P.G.Luiten, M.M.Raimondi, S.A.Zacchino, C.Somlai, B.Penke, R.D.Enriz, New small-size peptides possessing antifungal activity. Bioorg. Med. Chem., 2010;18: 158–167.
- Narang R, Narasimhan B, Sharma S, Sriram D, Yogeeswari P, Clercq ED, Balzarini J, Nicotinic acid benzylidene hydrazides: Synthesis, antitubercular, antiviral, antimicrobial evaluation and QSAR studies. Med Chem Res, 2012; 21:1557-1576.
- Kankate, R.S. et al., Design, synthesis and antifungal evaluation of novel benzimidazole tertiary amine type of fluconazole analogues. Arabian Journal of Chemistry (2015). [CrossRef]
- Brown, G.D.; Denning, D.W.; Levitz, S.M. Tackling human fungal infections. Science 2012, 336. [Google Scholar] [CrossRef] [PubMed]
- D.Barrett, From natural products to clinically useful antifungals. Biochim. Biophys. Acta. 2002;1587: 224–233.
- Yapar, N. Epidemiology and risk factors for invasive candidiasis. Ther Clin Risk Manag 10,95–105 (2014).
- Salgado, Paula S., et al. "Structural basis for the broad specificity to host-cell ligands by the pathogenic fungus Candida albicans." Proceedings of the National Academy of Sciences 108.38 (2011): 15775-15779.
- Ge SH, Wan Z, Li J, Xu J, Li RY, Bai FY (2010) Correlation between azole susceptibilities, genotypes, and ERG11 mutations in Candida albicans isolates associated with vulvovaginal candidiasis in China. Antimicrob Agents Chemother 54(8):3126–3131.
- Lum, Kah Yean, et al. "Activity of novel synthetic peptides against Candida albicans." Scientific reports 5 (2015).
- Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the national healthcare safety network at the centers for disease control and prevention, 2009–2010. Infect Control Hosp Epidemiol. 2013;34:1–14.
- Pfaller, MA. Antifungal drug resistance: mechanisms, epidemiology and consequences for treatment. Am J Med. 2012;125:3–13.
- Chin VK, Lee TY, Rusliza B, Chong PP. Dissecting Candida albicans Infection from the Perspective of C. albicans Virulence and Omics Approaches on Host–Pathogen Interaction: A Review. Woo PCY, ed. International Journal of Molecular Sciences. 2016;17(10):1643. [CrossRef]
- Xu, Kehan, et al. "Design, synthesis, and antifungal activities of novel triazole derivatives containing the benzyl group." Drug design, development and therapy 9 (2015): 1459.
- Granneman, Sander, et al. "Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs." Proceedings of the National Academy of Sciences 106.24 (2009): 9613-9618.
- Enjalbert B, Moran GP, Vaughan C, Yeomans T, MacCallum DM, et al. (2009) Genome-wide gene expression ƉƌŽĮůŝnŐ and a forward ŐĞnĞtic screen show that ĚŝīĞƌĞntiĂů expression of the sodium ion transporter Ena21 contributes to the ĚŝīĞƌĞntiĂů tolerance of Candida albicans and Candida dubliniensis to ŽƐmŽtic stress. Mol Microbiol 72: 216-228.
- Aoyama T, Nakayama H, Ueno K, Inukai T, Tanabe K, et al. (2014) Genome-wide survey of ƚƌĂnƐcƌŝƉtiŽnĂů ŝnŝtiĂtiŽn in the pathogenic fungus, Candida glabrata. Genes Cells 9: 478-503.
- Zhang, L., Lin, J., & Ye, K. (2013). Structural and functional analysis of the U3 snoRNA binding protein Rrp9 . RNA, 19(5), 701–711. [CrossRef]
Figure 2.
Secondary structure of hypothetical protein Rrp9 from C.albicans (Q5A213_CANAL) by SOPMA.

Figure 5.
Ramachandran Plot.
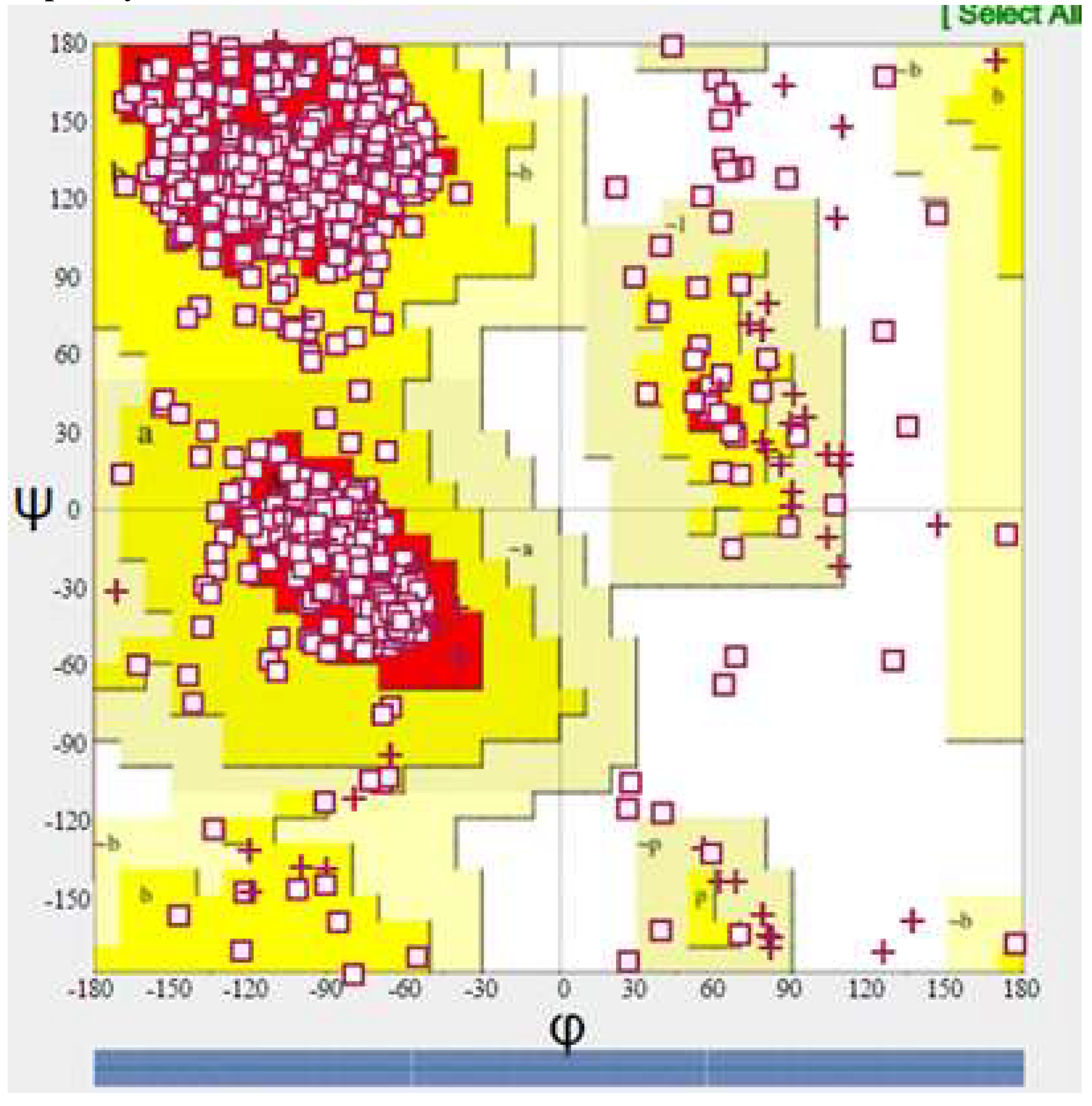
Figure 7.
Plot of Z –Score of from ProSA-web server showing the quality of predicted Structure of Rrp9. .
Figure 7.
Plot of Z –Score of from ProSA-web server showing the quality of predicted Structure of Rrp9. .
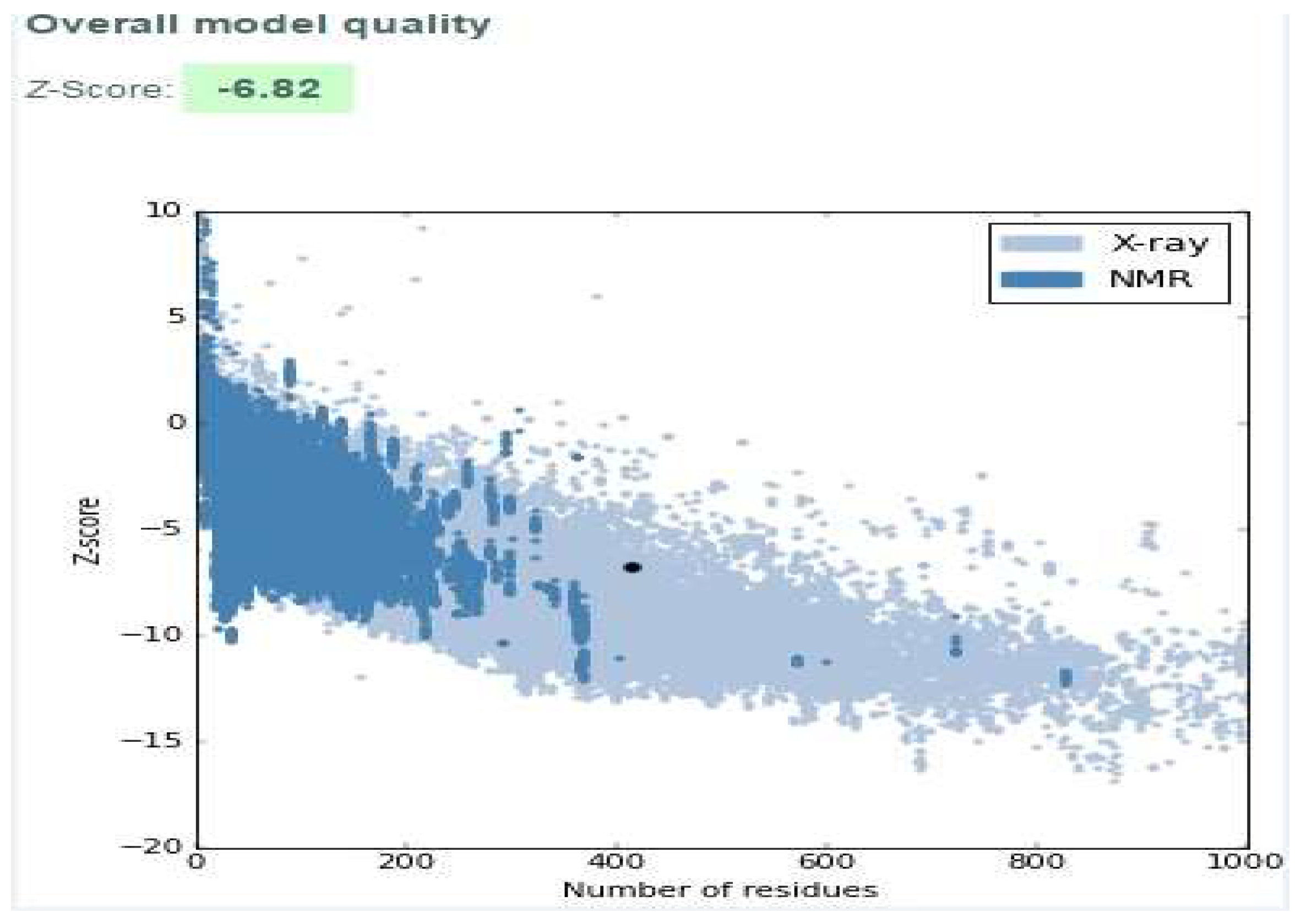
Figure 8.
Superpose of the designed optimised homology model (white) with template 4JOX (light blue).
Figure 8.
Superpose of the designed optimised homology model (white) with template 4JOX (light blue).
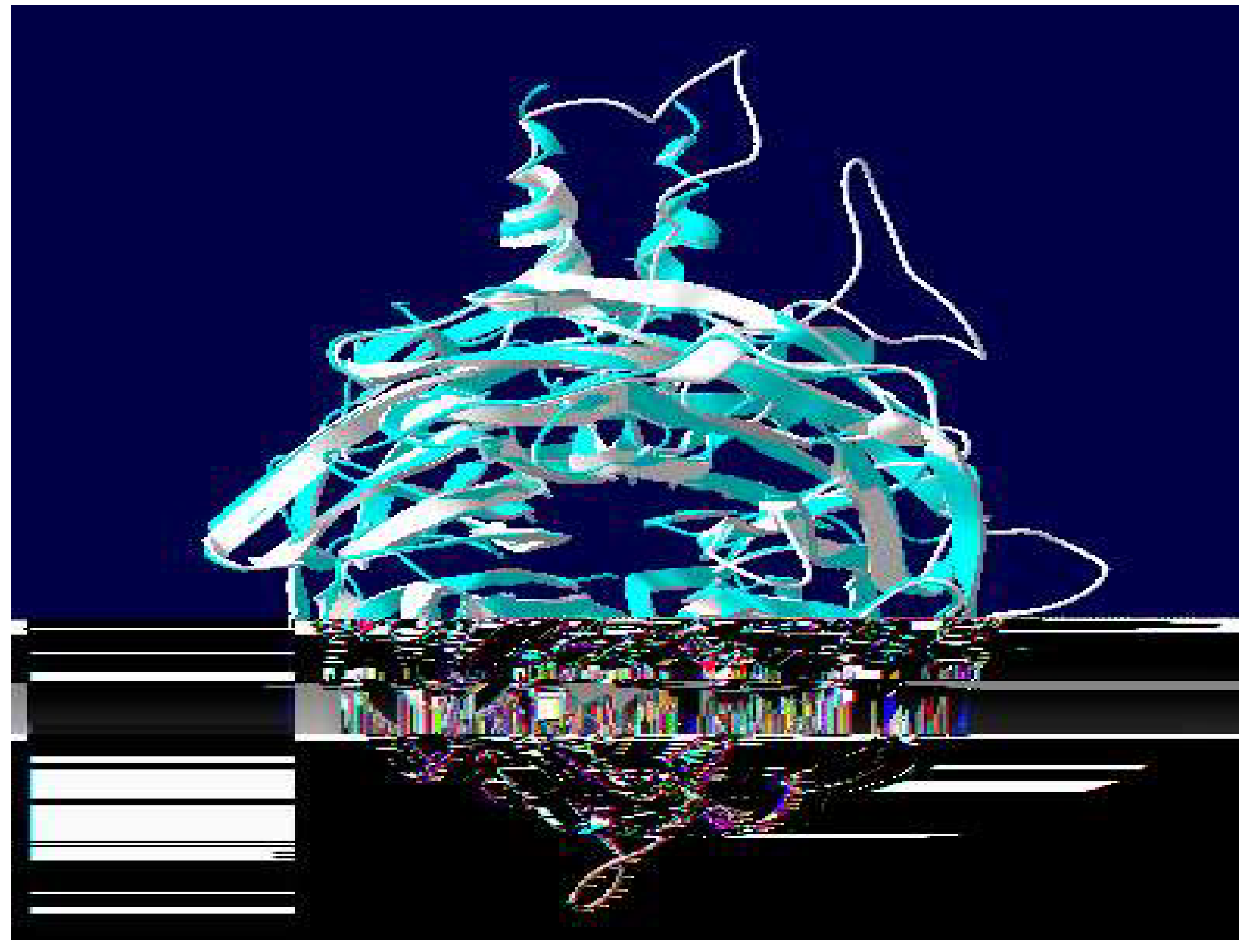

Table 1.
The predicted physicochemical properties of the hypothetical protein Rrp9 from C. albicans (Q5A213_CANAL).
Table 1.
The predicted physicochemical properties of the hypothetical protein Rrp9 from C. albicans (Q5A213_CANAL).
| Parameter | Value | |
| Amino Acid Length | 549 | |
| Molecular Weight (M.wt.) | 62414.05 | |
| PI | 6.59 | |
| Total number of negatively charged residues (Asp + Glu) | 78 | |
| Total number of positively charged residues (Arg + Lys) | 75 | |
| Instability index (II) | 44.74 | |
| Aliphatic index (AI) | 76.03 | |
| Half-life | Mammalian reticulocytes | 30 hours |
| Yeast | >20 hours | |
| E. coli | >10 hours | |
| GRAVY | -0.753 | |
Table 2.
Secondary structure analysis of hypothetical protein Rrp9 from C.albicans (Q5A213_CANAL) by SOPMA.
Table 2.
Secondary structure analysis of hypothetical protein Rrp9 from C.albicans (Q5A213_CANAL) by SOPMA.
| Parameters | Number of amino acids | Amino acids (%) |
| Alpha helix (Hh) | 148 | 26.96 |
| 310 helix (Gg) | 0 | 0.00 |
| Pi helix (Ii) | 0 | 0.00 |
| Beta bridge (Bb) | 0 | 0.00 |
| Extended strand (Ee) | 141 | 25.68 |
| Beta turn (Tt) | 46 | 7.47 |
| Bend region (Ss) | 0 | 0.00 |
| Random coil (Cc) | 219 | 39.89 |
| Ambigous states | 0 | 0.00 |
| Other states | 0 | 0.00 |
Table 3.
Docking summary of the compounds using GOLD software.
| Name | Fitness score | S(hb_ext) | S(Vdw_ext) | S(hb_int) | S(Int) |
| Dicyclomine hydrochloride | 42.14 | 0.00 | 37.83 | 0.00 | -9.88 |
| Hydroxyine hydrochloride | 54.55 | 0.00 | 44.39 | 0.00 | -6.49 |
| Promethazine Hydrochloride |
45.91 | 0.00 | 36.84 | 0.00 | -4.75 |
| Solifenacin succinate | 62.17 | 0.00 | 44.26 | 0.00 | 1.32 |
| Flavoxate Hydrochloride | 57.67 | 0.00 | 48.75 | 0.00 | -9.36 |
Table 4.
summary of the compounds dock score, interaction data and distance between the protein residues.
Table 4.
summary of the compounds dock score, interaction data and distance between the protein residues.
| Ligand name | Interacting aminoacids | Interacting atoms | H-distance |
| Solifenacin Succinate | Pro234,Arg275, tyr174,Asn173, Phe273 |
Arg275:NE----O2: Solifenacin Succinate Phe273:O---C8: Solifenacin Succinate Pro234:CA---C24: Solifenacin Succinate Asn173:O---C17: Solifenacin Succinate |
2.914 2.503 2.356 2.702 |
| Dicycloamine |
Glu509,Asn173, Tyr174,Asn276, Pro460,Pro234 |
Tyr174:N---O2:Dicycloamine TYR174:CB---C8: Dicycloamine Asn173:C..O2: Dicycloamine Glu509:CG...C21: Dicycloamin |
2.340 2.603 2.642 2.881 |
| Hydroxyzine hydrochloride |
ILE271,ILE230, Pro234,Ser232, Tyr174,Asp506 Ser233 |
Ile271:O---OH: Hdroxygene Hydrochloride Ile230:O—OH:Hydroxygene Hydrochloride Ser233:O—C21: Hydroxygene Hydrochloride Pro234:C---C17: Hydroxygene Hydrochloride |
2.581 2.597 2.468 2.752 |
| Promethazine Hydrochloride |
Pro234,Phe273 Ser233,Ala232 Ty174,Arg275 |
Tyr174:CB---C13:Promethazine Hydrochloride Pro:234:N....C13 : Promethazine Hydrochloride |
2.730 2.897 |
| Flavoxate |
Pro234,Phe273 Ser233,Ala232 Ty174,Arg275 |
Phe273:O---O3:Flavoxate Ala232:O---C21: Flavoxate Ala315:O---C11: Flavoxate Ala232;O---O4: Flavoxate |
2.709 2.621 2.559 2.596 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



