Submitted:
03 June 2025
Posted:
03 June 2025
You are already at the latest version
Abstract
RNA editing sites and functional analysis were firstly predicted and verified in the chloroplast genes of Chelidonium majus. The results found that nine genes have the same number of editing sites, while 40 genes have different numbers of editing sites. There are 28 types of codons and 13 types of amino acid mutations in the chloroplast genes of C. majus, among which serine mutations to leucine (S→L) have the highest number. All the molecular weight, extinction coefficients, and hydropathicity are increasing, while the value of theoretical isoelectric points (PI) are decreasing or constant after RNA editing. The five types of proteins, including ndhF, ndhD, ndhB, psaA, and psaB have more than 11 transmembrane regions, as well as they have the most conserved domains and open reading frames (ORFs). The number of ORFs by each gene is decreasing, whereas the length of ORFs is becoming more longer. The signal peptide was found within one protein of the petA gene. The rpoB protein had the approximate three-dimensional domain of Ricin-type beta-trefoil lectin with one more increase of conserved domain through mutation. After experiment validation, the two new mutated proteins were found within the rpoB protein. These results provide a solid basis for exploring functional expression regulation and the origin of evolution.
Keywords:
Chelidonium majus
; chloroplast RNA editing
; transmembrane zone
; conserved domains
; signal peptide
; ORFs
; homologous proteins
; tertiary structure
; biological functions and pathway
; alkaloids
1. Introduction
Plant gene editing technologies mainly include the first-generation Zinc-finger nucleases (ZFN) technology [1], the second-generation TALENs gene editing technology [2], and the third-generation CRISPR/Cas9 technology [3]. RNA editing is a phenomenon of base differences in the process of DNA transcription in organisms, which can cause changes in codons and the function of encoding proteins before and after editing, thereby changing the genetic information, regulatory functions of growth, and development of species [4]. RNA editing in organelles is common in chloroplasts and mitochondrial transcripts, with the presence of common cytosine to uracil conversion and the reverse uracil to cytosine conversion form in horns and ferns [5], mainly at the first and second bases of codons, which alters the physicochemical properties of amino acids [6], so variation in the intrinsic function of biological organisms affects the extrinsic traits and reproductive characteristics of biological organisms. RNA editing in the plant chloroplast genome has been observed in a variety of plants, including maize [7], Arabidopsis thaliana[8], tobacco [9], Hordeum vulgare var. Coeleste [10], barley [11], and Aegilops tauschii [12], etc. A standard method for studying RNA editing is rRNA-depleted total RNA-seq sequencing analysis, which is compared using commercial Ribo-off-seq and standard mRNA-seq [13]. The main tools for plant RNA editing research are JACUSA, SPRINT, and REDO. The central databases are REDIdb, PED, and dbRES, which elucidate the mechanisms and roles in RNA editing by studying RNA editing sites, pentapeptide repeat proteins (PPRs) involved in RNA editing, and RNA editing factors [14]. It was reported that the expression level of CsMORF 9.2 in the “Huabiao albino 1” tea cultivar was significantly down-regulated, which provided a basis for the role of chloroplast RNA editing in the process of tea albinism [15].
The species of Chelidonium majus, in appearance and historic names, it is similar to the plants from the species of Stylophorum lasiocarpum, Hylomecon japonica, and Coptis genus, which is a perennial dry whole grass of the Papaveraceae family. The distribution of C. majus L. is located in the most areas of China [16]. Fresh plants have a strong orange-yellow emulsion that contains a variety of alkaloids, including Chelidonine, fumarine, stylopine, allocryptopine, berberine, chelerythrine, sanguinarine, and sparteine [17]. In addition, it also contains chlorogenic acid, malic acid, citric acid, succinic acid, choline, methylamine, histamine, tyramine, saponin, flavonol, chlorogenic alcohol, and cardiac glycoside, especially in the whole flowering period, and the content of various components in roots, stems and leaves is higher. The fruit mainly contains flavonoids and volatile oils [18]. The whole herb of C. majus is used in medicine, with a bitter taste, a cool nature, and poisonous. It exhibits anti-tumor, antiviral, anti-inflammatory, antibacterial, and analgesic pharmacological effects and is used for gastric pain, cough, asthma, and whooping cough [19]. At present, the basic information, main contents, simple repeats, various repeats, and evolutionary and developmental relationships within the chloroplast genome of C. majus have been reported in many research papers, which showed that the species of C. majus had a close relationship to the plants of Coreanomecon genus, Meconopsis genus, and Papaver genus in the Papaveraceae family [20,21]. In a study on the chloroplast genome and developmental relationship of Eomecon chionantha, it was found that it also has a close evolutionary relationship with C. majus, Cordyceps cicadae, Macleaya microcarpa, and H. japonica [22]. Moreover, the root and leaf transcriptomes in C. majus were comparatively analyzed, revealing that the main transcript factor originated from the bHLH family, five unigenes encoding enzymes within 10 SSRs in the pathway of benzylisoquinoline alkaloids, and the new 10 identified conserved miRNAs [23]. In the transcriptome database of leaf in C. majus, more than 23,004 CDS should be further analyzed the proteomic types, together with the 334 different putative proteins identified in milky sap and 1155 in whole plant extract [24]. In addition, previous studies have also identified the cytostreptovirus CheYMaV identified in the chloroplast genome of C. majus using high-throughput sequencing and reverse transcription-polymerase chain reaction (RT-PCR) method. Phylogenetic analysis showed that CheYMaV was most closely related to TpVA andGllV1 [25].
In this study, the RNA editing sites and function application of chloroplast genomes in C. majus were predicted and compared by using the bioinformatics methods to those of different plants, hoping to lay a foundation for studying the biological function of chloroplast RNA editing influenced the structures and functions of proteins and RNAs in C. majus, which provides scientific data for further revealing the molecular mechanism of gene and protein on plant appearance, chemicals, functional regulation, and development.
2. Results
2.1. Plant Appearance Characteristics of the C. majus
The plant appearance of C. majus was identified from different tissues on the left of Figure 1 in Xiangshan Park in Beijing (photo credit: Ou Chonglie), Hebei North University in Zhangjiakou, Hebei Province (photo credit: Fan Yingxin), and Shenyang Wetland in Liaoning Province (photo credit: Zhao Yan) (photo credit: Zhao Yan) (photo credit). As can be seen from Figure 1, a herbaceous plant of C. majus develops into phytobiocoenose with a stout, conical, dense fibrous roots showing earthy yellow or dark brown (Figure 1a and 1g). The stems are erect, cylindrical, hollow, easily broken, clustered umbel-like and multi-branched. The branches are often pubescent with white elongated pubescence (Figure 1b). The leaves are fully lobed, all lobes 2-4 pairs, obovate-oblong having irregular deep or shallow lobes with rounded toothed margins, green on the surface, glabrous, white powder on the back, and sparsely pubescent (Figure 1c). The umbel-shaped inflorescence is ovate, yellow, 4-lobed petals with numerous stamens of filamentous filigree, oblong anthers, pistil composed of two carpels, and green, glabrous, linear ovary of 2-lobed stigma (Figure 1d). The capsule is narrowly cylindrical with a stalk shorter than the fruit (Figure 1e). There are numerous small, dark brown or black, ovate seeds with shiny, honeycomb-like lattices (Figure 1f).
2.2. Prediction and Comparative Analysis of RNA Editing Sites
The similarities and differences among the chloroplast genomic RNA editing sites of C. majus, A. tauschii, and G. x gandavensis were found by reference comparison with the that of chloroplast genome in database of A. thaliana (Table S1), as shown in Figure 2. The number of 590, 560 and 505 RNA editing sites in the whole chloroplast genomes of C. majus, G. x gandavensis, and A. tauschii, respectively within 62 genes (Table S2), in which the most number of 420, 272, and 408 RNA editing sites were found in the conversion of base C to base U, respectively (Table S3). From Table 1 and Figure 2, in the chloroplasts of three species, nine genes had the same number of RNA editing sites, namely atpF (2), ndhJ (3), atpB (3), petB (1), petG (1), psaB (1), psbN (1), psbZ (1), and rpl16 (1) (Figure 2a); forty genes had different RNA editing sites (Figure 2b), and in particular, four genes ycf2, matK, rpoC2, and ndhB had more than 19 editing sites (Table S4). No RNA editing sites were found in the genes at 21 established positions in the chloroplast genome of C. majus (Table S5), namely psbA (base range 1558-497), psbI (base range 8183-8293), atpH (base range 14025-13780). ), petN (base range 29381-29470), psbM (base range 30748-30644), psbD (base range 34723-35784), ycf3 (base range 45422-45195), atpE (base range 55885-55487), rbcL (base range 58111-59535), psbL (base range 67435-67319), rpl33 (base range 70890-71090), rps12 (base range 73044-72931), clpP (base range 75283-75215), psbB (base range 77872-78093), psbH (base range 77872-78093), rpl36 (base range 83042-82929), rps19 (base range 159661-159729), rps7 (base range 101540-101073 and 145891-146358), rpl32 (base range 117874-118023), and psaC (122205-121960). By comparison, in the chloroplast genome of A. tauschii, no RNA editing sites were found within the other 11 genes at 18 position areas, including ycf3, psbJ, psbF, psbE, and psaJ, rps12 (two positions), psbT, ndhH, and ycf1 (two positions, Table S5).
2.3. Codon Changes Caused By RNA Editing
Among the sites of conversion of RNA-editing base C to base U in the chloroplast genome of C. majus, 266 editing sites (63.33%) occurred at the second base of the codon, 154 editing sites (36.67%) occurred at the first base of the codon, and no editing site was found at the third base, i.e., no synonymous editing was found (Table S3). In the two-way conversion of base C and U in the chloroplast genomes of C. majus and A. tauschii, 54 codon pair types were found, while 55 codon pair types were detected in the cp genomes of G. x gandavensis. Moreover, there are specific types of codon pair transitions in the chloroplast genome of each plant, such as the conversion of four codon pairs in the chloroplast genome RNA editing of C. majus including UGU to CGU, CAA to UAA, CUA to UUA, and UUG to CUG. Conversion of four pairs of codons containing UUA to CUA, CUG to UUG, UGC to CGC, and UGA to CGA were special in the chloroplast genome of A. tauschii. Only the one-pair conversion of codons UAA to CAA was especially predicated in the chloroplast genome of G. x gandavensis (Table S6). After the one-way conversion of RNA editing bases C and U, the specific two types of codon conversion were screened in the cp genome of C. majuss consisting of CAA to UAA and CUA→UUA (Table 1). Comparatively, the conversion of one pair of codons CAA to UAA was found in the chloroplast genome of G. x gandavensis. In contrast, that of codon conversion of CUG→UUG was found in the chloroplast genome of A. tauschii (Table S7).
2.4. Types of Mutations in Proteins Induced by RNA Editing
The RNA editing in the chloroplast genomes of the three comparison plants, there were 24 and 13 types of amino acid transitions in the bidirectional conversion of bases C and U and unidirectional conversion of C and U , respectively (Figure 2c and Table 1), of which the 13 types of protein mutations in the unidirectional conversion of bases were found, including Alanine (A) to Valine (V), Histidine (H) to Tyrosine (Y), Leucine(L) to Phenylalanine (F), Leucine (L) to Leucine (L, C. majus and A. tauschii), Proline (P) to Leucine (L), Proline (P) to Serine (S), Glutamine(Q) to Terminator (*, C. majus and G. gandavensis), Arginine (R) to C, Arginine (R) to Tryptophan (W), Serine (S) to Phenylalanine (F), Serine (S) to Leucine (L), Threonine (T) to Isoleucine (I), and Threonine (T) to Methionine (M) (Table S7).
2.5. Effects on Physicochemical Properties and Conserved Domain of Sixteen Mutant Proteins by Chloroplast RNA Editing
Although the chloroplast ycf gene has the most RNA editing sites in the chloroplast of C. majus, its functions are not well understood. From the predicted results, sixteen genes with more than five well-defined editing sites of chloroplast RNA were identified in C. majus and further studied. The molecular weight and average hydrophilicity (Grand average of hydropathicity, GRAVY) of the 16 proteins are increasing, while the isoelectric point is decreasing, indicating that they were more stable in an acidic environment and unstable in an alkaline environment, and the extinction coefficient is increasing within nine proteins except the constant for the other eight proteins (Table S8). RNA editing can result in the replacement of specific amino acids with those that carry different charges. In this study, replacing a positively charged amino acid (such as histidine) with an uncharged amino acid (such as tyrosine) or replacing a polar amino acid with a nonpolar amino acid can result in changes in amino acids may lead to changes in the three-dimensional conformation of proteins. In turn, it affects the distribution of surface charge and the overall charge state of the protein, which lowers the isoelectric point and influences other performance.
Regarding the conserved domains of 16 types protein structures before and after mutation influenced by RNA editing (Table S8), among the five proteins of ccsA, ndhB, rpoC2, accD, and rpoB, the former four kinds of proteins have notable transformation within the two positions, especially the two former proteins containing the transmembrane region, and a new domain (RNA_pol_Rpb2_1) has been added within the latter (rpoB) one protein after RNA editing (Figure 3a and 3b). The 14 protein mutations within rpoB protein sequences were predicated during RNA editing, the conserved domain is belong to CHL00001 members of the protein superfamily CL47023, which is approximately 137 amino acids. Its structure is similar to that of the ricin-type beta-trefoil lectin domain, which present in the database of PFAM00652 and IPR000772, which structural proteins of A0A5J4LQ17 (Streptomyces angustmyceticus, Figure 3c) and Q8X123 (Amanita phalloides/Marasmius oreades, Figure 3d) were predicted by AlphaFold. The coincident domain indicates that plants and fungi share a common protein active domain, providing a scientific basis for their good symbiosis to effectively promote the regulation of plant growth and development. In addition, the two proteins, ndhD and ndhF, have the most conserved domains, including the transmembrane region, which remains unchanged. Furthermore, the conserved domains of remaining nine proteins still keep inalterability during RNA editing.
2.6. Transmembrane Proteins and Signal Peptides by the RNA Editing
Through comparative analysis of the transmembrane performance of the 85 CDS proteins in the chloroplast genome of C. majus by RNA-editing, the results showed forty-four (51.8%) proteins have 1 to 16 transmembrane proteins (Figure S1), the seven types of proteins including ndhF, ndhD, ndhB (two positions), psaA, psaB, ndhA, and ccsA have above eight transmembrane proteins, among which 34 proteins were found the homologous proteins (Table S9). The transmembrane proteins of five proteins, consisting of ndhD, ndhB, petA, ndhG, and rpoC2, were influenced by RNA editing, in particular, each of the two proteins (ndhD and rpoC2) has a new transmembrane protein after RNA editing (Figure 4 and Table S10). There were similarities and differences in the number and location of transmembrane segments among protein structures identified using different methods. Being predicted by TOPCONS and PHILIUS methods, the transmembrane segments of ndhD protein increased by one after mutation (TM12:359-379), and by way of OCTOPUS, PolyPhobius, SCAMPI, and SPOCTOPUS, the number of transmembrane segments before and after the mutation was 11, 14, 14, and 11, respectively. The position and number of the transmembrane segment regarding the homologous protein was 3rkoM and 14 without variation, similar to that by the SCAMPI method (Figure S2). For the ndhB protein with the homologous protein of 3rkoA, by the methods of TOPCONS, OCTOPUS, PHILIUS, SCAMPI, and SPOCTOPUS, the number of transmembrane segments were increased by 3 (11/14, TM5: 149-169, TM8: 264-284, and TM10: 322-342), 3 (8/11, TM5: 180-200, TM6: 226-246, and TM11: 483-503), 1 (13/14, TM5: 149-168), 3 (11/14, TM5: 146-166, TM8: 264-284, and TM10: 323-343), 4 (8/11, TM4: 149-169, TM5: 180-200, TM6: 226-246, and TM11: 483-503), while by the PolyPhobius method that of keeps constant 14 (Figure 4). The number and position of transmembrane regions within ndhD remained unchanged at 16, 14, 16, 16, 16, 16 and 14, respectively, indicating that the protein was conserved, and the homologous protein was 3rkoL. Three transmembrane regions of rpoC2 proteins were only identified by SCAMPI method (TM1:238-258/TM1:234-254,TM2:1177-1197/TM2:1173-1193,TM3:1261-1281/TM3:1257-1277) before and after mutation, and no homologous proteins were predicted (Table S10). As can be seen from the results, the stability and functional diversity of the protein increased due to the augmentation of hydrophilicity and transmembrane regions within the proteins, thereby prolonging the action time of its functionality.
Specifically, only one or two transmembrane regions and one signal peptide were found to exhibit noticeable differences within the proteins of petA due to mutations in the CDS proteins of C. majus chloroplast genome resulting from RNA editing (Table 2, Table S10, and Figure 5a). The number of two transmembrane regions within petA protein were diversely predicated by the way of PolyPhobius and SCAMPI. with the position of from 14 to 34, including signal peptide and one mutation protein at 32 sites marked by blue frame, and from 285 to 305 from the starting sequence of petA protein. The type of signal peptides (SPs) is standard secretion signal peptide of Sec/SPI (transported by Sec translocation, cleaved by signal peptidase I (Leptin, Lep)) with the value of 0.9434 and 0.9179 before and after RNA editing. The signal peptide sliding cleavage sites are between 35 and 36 amino acids, including the N-terminal region, center hydrophobic region, and c-terminal region from the n-terminal region of beginning protein with the possibility of 0.887037 (Figure 5b and 5c). No sequence of signal peptide was found within the other 84 CDS proteins, although the proteins of cemA and atpH are other possibilities, as shown in the forecast figure (Figure S3).
2.7. Comparative Analysis of RNA Editing Results Between Prediction and Experimental Validation
In order to verify the accuracy of the prediction results, the RNA editing sites of five selected protein-coding genes of matK, ndhB, ndhD, rpoB, and accD, with most variation in the chloroplast genome of C. majus were analyzed by sequencing. The DNA and RNA from the leaves of C. majus were extracted and detected by the electrophoretic gel and Nanospectrophotometer (Table S11). After that, nucleotide amplification of the five genes was executed using DNA and cDNA templates, and the resulting products were identified with the primer pairs by the PCR and RT-PCR method (Table S12 and Figure S4). From the detection and verification results (Table S13), the RNA editing sites within the four genes tested, including matK, ndhB, ndhD, and accD were entirely consistent with the predicted variant sites through the comparison of nucleotide sequences from the method of PCR and RT-PCR amplification (Table S14), whereas within the gene of rpoB, editing sites of the new five variation sites within the nucleotide sequence and the new two variations within the protein sequence were identified (Table S14 and Figure 6a and 6b). The RNA editing sites resulted in the mutation of amino acids, and the two pairs of mutation proteins, from Phenylalanine (F) to Cysteine (C) and Isoleucine (I) to Lysine (K), involved the transformation from non-polar hydrophobic to polar hydrophilic amino acids with positive charge and alkalinity. The mutation of the rpoB protein induced secondary structure variation in the protein within five areas, designed as A, B, C, D, and E (Figure 6c). The increasing of hydrophilic residues tends to form hydrogen bonds with water molecules, making the protein more soluble in water. This facilitates its diffusion and distribution in the cell, cytoplasm, or nucleus and enables it to participate in gene expression regulation, playing a crucial role in maintaining the overall conformational stability of proteins.
2.8. Structure Variety of RNA and Proteins Before and After RNA Editing
Comparing the structural mutations of the sixteen proteins before and after RNA editing, it was found that all the secondary structure of 16 proteins and RNA are influenced by the variation (Table 2). Among them, the detailed secondary structures of six proteins, matK, rpoC2, ndhB, ndhD, ndhF, and rpoB, were comparatively analyzed before and after RNA editing. Compared to before RNA editing, undergoing after RNA editing, the secondary structure of the matK protein increases by three β folds (Figure 7b, A1, C1 and F1), decreases by two β folds (Figure 7b, D1), increases one α helix (Figure 7b, E1), decreases one α helix (Figure 7b, B1), and shortens the α helix region of G1 (Figure 7b). The secondary structure of the rpoC2 protein adds two α helix regions (Figure 7d, A2 and B2), reduces one α helix region (Figure 7d, D2) and extends the C2 region α helix region (Figure 7d), adding two β fold regions (Figure 7d, D2). The secondary structure of the ndhB protein adds three α helix regions (Figure 7f, B3, and C3), reduces one α helix region (Figure 7f, D3) and the A3 region extends α helix region (Figure 7f), reducing two β pleated regions (Figure 7f, B3, and C3) while the β pleated region of E7 (Figure 3f, E3). The secondary structure of the ndhF protein adds two α-helix regions (Figure 7h, A4, and C4) and decreases two β-folded regions (Figure 7h, A4, and B4). The secondary structure of the ndhD protein adds four α-helix regions (Figure 7j, C5, D5, E5, and F5), reduces one α-helix region (Figure 7j, A5), and extends the F5 region α helix region, adds three β-folded regions (Figure 7j, A5, B5, and E5), and reduces one β-folded region (Figure 7j, F5). The secondary structure of rpoB protein was increased by two α helix regions (Figure 7l, B6) and the β fold extension of the A6 region (Figure 7l, A6).
The tertiary structures of the 16 proteins have been predicted the best model, diversified from the variation of secondary structures influenced by RNA editing (Table 2). Regarding the three proteins, ndhB, ndhD, and rpoB, we investigated their diverse tertiary structures due to the numerous mutation sites identified during RNA editing through experimental verification. The best models of tertiary structure for ndhB and ndhD proteins, before and after mutation, are closely related to the proteins of P0CC94.1.A (99.22%, Nandina domestica) and P0CC99.1.A (98.43%, Nicotiana tabacum); A0A6G7MZ33.1.A (100%, Chelidonium majus) and A0A6B9PH19.1.A (91%, Actaea biternata), while the three types of rpoB proteins are all similar to the temple protein of 8xzv.1.B (Table S15). The Root mean square deviation (RMSD) of the tertiary structures within the three proteins of ndhB (Figure 8a), ndhD (Figure 8b), and rpoB (Figure 8c) between original and mutation structures is 0.176, 0.166, and 0.098 at the super alignment method, which indicated that the mutation sites (Figure 8 marked blue and rose colors)of each pair protein may adapt to different environments and exerted active functions by influencing the formation of its functional domain, subcellular localization, and interaction.
2.9. Prediction and Identification of Open Reading Frame (ORF) and Transcription Factors (TFs)
After the functional annotation of ORFs within seven genes in the chloroplast genome of C. majus, which includes matK, rpoC2, ndhB, ndhD, ndhF, accD, and rpoB with much more than eight variation sites, the number of ORFs within all of the analyzed genes are decreased through RNA editing. On the contrary, the ORF length increases and becomes longer because of the short ORF merging into the long bigger ORFs (Table S16) within these genes. For instance, the most number of ORFs is 54 within rpoC2 protein before RNA editing, while that of reduces to 10 after RNA editing. Similarly, in comparison within the rest six of the proteins during the course of RNA editing, the number of ORFs varied from 18 to 5 (matK and ndhD), from 31 to 13 (ndhB, Figure 9), from 20 to 6 (ndhF), from 44 to 12 (ropB), and from 17 to 4 (accD). Although new nucleotide sequence differences were identified after the verification of rpoB gene, the results of ORF prediction and experimental verification only affected the extension of the position of the 8th ORF endpoint. Three more nucleotides were added to the 8th ORF, and one more protein was expressed (Table S16). Therefore, the length and position of ORFs may cause for the sequences that are not in the reading frame to be included or excluded, or remove stop codons and extend protein translation.
The transcription factors within 85 CDS genes in the chloroplast genome of C. majus were performed by homology searches against PlantTFDB, which controls the synthesis of active components, particularly secondary metabolism and regulation of gene expression. The results showed that the five TF encoding transcripts existed in the five genes (rpoB, accD, petA, rps19, and ndhD) in the CDS genes of C. majus chloroplast genome were identified and further classified into five different families (B3, C3H, GRAS, AP2, and bZIP) for the species of Brassica rapa, Vitis vinifera, Medicago truncatula, Malus domestica, and Medicago truncatula as the reference genomes, respectively (Table S17). Among the transcription factor families, the primary function of the B3 and bZIP domains in transcription factors is to activate or inhibit the transcription and regulation of target genes by binding to DNA sequences; C3H transcription factors, a kind of zinc finger (ZnF) transcription factors, regulate gene expression by binding to the mRNA of genes; GRAS transcription factors are plant-specific, while AP2/ERF transcription factors are also involved in hormone regulation in plants, all of which play crucial roles in plant growth, development, and stress resistance.
2.10. GO, KOG, and KEGG Pathway
The genes in the chloroplast genome of each plant play a crucial role in regulating biological processes and pathways due to their unique sequence and position differences. After annotation of CDS genes in the chloroplast genome of C. majus from the functional database of Gene Oncology (GO), eukaryotic Orthologous Groups (KOG), and Kyoto Encyclopedia of Genes and Genomes (KEGG), all these genes can make actions for the cellular component of cell, membrane, protein-containing complex, and organelle part; molecular function of catalytic activity, structural molecule activity, transporter activity, and binding; biological process of metabolism, response to stimulus, localization, biological regulation, and cellular component organization or biogenesis (Table S18 and Figure S6). Especially the functions of these genes are mainly concentrated on translation, ribosomal structure and biogenesis, energy production, and conversion (Table S19 and Figure S7). They can mainly influence the pathway of plants during the biological metabolism of carbon, carbohydrate, glyoxylate and dicarboxylate, propanoate, pyruvate, and microorganism in diverse environments; fatty acid biosynthesis of secondary metabolites; the course of oxidative phosphorylation and photosynthesis, transcription of RNA polymerase and translation of ribosome; cell growth and death of cell cycle, and longevity regulating pathway (Table S20 and Figure S8).
3. Discussion
As a herbaceous plant of C. majus in the Papaveraceae family, it has bright yellow four petals and yellow milk as its characteristics during the flowering and ripening season, as well as S. lasiocarpum and Oreomecon radicata, but the diversified features of S. lasiocarpum have a blood-red sap, 4-7 flowers, and the leaves are broadly ovate; while the species of O. radicata grows in a limited area and is a wild plant with yellowish-white or yellowish cup-shaped flowers to adequately absorb and use sunlight through adapting to the cold Arctic environment. Although plants can be identified to a certain extent through their appearance, it is still necessary to explore the reasons for the changes that occur from the inside out in the growth and development of plants, driven by internal factors.
3.1. Significance of RNA Editing Occurring Within the CDS Genes in the Chloroplast Genome
Among the induced factors for plant mutations, RNA editing is a crucial post-transcriptional gene processing and modification that produces different functional proteins in plant organelles, such as mitochondria and chloroplasts, thereby affecting the function of plant organs and tissues and influencing plant growth and development. In this study, six proteins encoded by chloroplast RNA editing in C. majus were found in more than eight mutation sites. The performance and biochemical function of the encoded proteins changed due to different structure-activity effects after mutation. As a highly conserved protein kinase, matK plays a crucial role in intracellular signaling. More than 20 matK genes have been identified, which are distributed across plants with diverse characteristics. This is because matK genes encode mature enzymes related to plant chloroplast class II intron splicing, and their evolution rate is relatively fast. It is often used to study the phylogenetic profile of plants and species identification. For example, the sequence data of the transcriptional spacer region (nrITS), trnH-psbA, and matK in the nucleosome were comprehensively used to analyze the phylogenetic relationships of associated plants, such as Bupleurum, within the Apiaceae family. It was found that the species of Bupleurum were divided into two main lineages, which should be classified into the Bupleurum subgenera of Neves and Watson. The grades of the three species, including Bupleurum parviflora, were also changed [26]. The antimalarial alkaloid quinine can usually be acquired from the plants of Cinchona pubescens and Cinchona calisaya in Brazil. The two genes, matK, and rbcL, were detected in the regions of these samples to distinguish and identify the 23 samples among the 28 collected bark samples [27]. In addition, all nine matK proteins had a conserved domain X functional region, and there were three differential amino acids in this region, but the amino acid variation did not cause changes in the molecular force of the corresponding amino acid residues [28]. Although there are multiple amino acid variations in the secondary structure of proteins, these variations have not led to the discovery or change of protein transmembrane segments and signal peptides, indicating that the protein sequence is highly conserved in terms of both the evolutionary process and specific functions. The plant chloroplast rpoC2 gene encodes a subunit of RNA polymerase, which is responsible for transcribing approximately 80% of chloroplast genes and is a key enzyme in the process of photosynthesis, essential for plant growth and development. Previous studies have shown that the deletion-mutated rpoC2 gene interferes with chloroplast biogenesis and the development of thylakoid membranes by downregulating the transcription of 28 chloroplast genes, resulting in the yellow streaks of Clivia miniata variety, which suggests its role in plant hybrid breeding [29]. The protein encoded by the ndh gene in plant chloroplasts is part of the NDH complex and plays a crucial role in photosynthesis, primarily involved in the electron transport process of photosystem I (PSI). For instance, in the case of the deletion of the quantitative trait locus qKW9 in maize, the C-U editing of ndhB in maize chloroplasts was also significantly reduced, which reduced photosynthesis and led to the reduction of maternal photosynthetic products in maize at the grain-filling stage[30]. Compared with the comparative verification results of chloroplast RNA editing of A. tauschii, it was found that the chloroplast of C. majus also had two more transmembrane protein structures between amino acids 200~300 [12]. It was also predicted to have two more transmembrane protein structures than the chloroplast RNA editing of A. tauschii before and after amino acids 100 and 500. The ndhD and ndhF genes are mainly involved in the complex NDH and electron transport processes in the photosynthetic electron transport chain, and the complex NDH-mediated CEF1 plays a positive role in improving the salt tolerance of soybeans [31]. The protein encoded by the chloroplast rpoB gene is the β subunit of RNA polymerase (RpoB), which recognizes promoters, binds nucleotides, and catalyzes the synthesis of RNA strands. It was reported that OsPPR16 was responsible for a single editing event at position 545 of chloroplast rpoB messenger RNA (mRNA), resulting in the change of amino acids in the PEPβ subunit from serine to leucine. The knockout of OsPPR16 resulted in impaired RpoB accumulation, decreased PEP-dependent gene expression, and an early phenotype characterized by pallor, which affected normal plant development [32]. The rpoB protein identified in this study contains a ricin-type Trifolium lectin domain [33] that may play a role in cytotoxicity, cell recognition, and signaling. This domain can be utilized in cell biology research and drug development to explore cell recognition and signaling mechanisms.
Different software for predicting the transmembrane region of proteins yielded different results, and the accuracy of the methods for predicting protein topology in the study varied [34]. Therefore, it is necessary to combine various methods to verify the effects of RNA editing before and after editing. From the results of ORF predication in the cp genome of C. majus, the significance of RNA editing cannot be ignored. After RNA editing, the longer open reading frames (ORFs) are more likely to encode complete proteins, which can help better understand the expression regulation mechanism of genes, and this is of great significance in the study of gene expression and protein function. The identified TFs differ from those in the transcriptome of leaves in C. majus, which will facilitate the further development and verification of their functions and distinctive significance.
3.2. RNA Editing of Chloroplast Genes Influenced the Alkaloid Chemicals
Alkaloids are a large class of secondary metabolites produced by plants to adapt to their long-term ecological environment, and they play an active role in regulating the functions of organisms and facilitating ecological adaptation. Alkaloids are synthesized and stored in specific tissues, organs, and cell types. During different developmental stages of plants, they may be transported, converted, accumulated, and metabolized in various tissues [35]. Many alkaloids are found in the leaves of plants. From a subcellular structure perspective, alkaloids may be synthesized in organelles such as chloroplasts. For example, the alkaloids of vindoline and catharanthine found in Vinca rosea are primarily distributed in the tender leaves [36]. Research has shown that these compounds are mainly synthesized and metabolized in this area, and the synthesis process is light-induced [37]. The quinolizidine synthase and its precursor, lysine (lysine decarboxylase and 17-oxosparteine synthase), are present in chloroplasts [38,39]. The activity and formation of alkaloid synthase are regulated by light and accumulate in the leaves to synthesize alkaloids. Alkaloids are found in all parts of tobacco, with the highest content in leaves [40]. Therefore, genes in the chloroplast genome play a crucial role in the synthesis and utilization of alkaloids. Previous research has shown that the genes encoding ndhA and ndhB catalyze the oxidation of nicotine to 6-hydroxy-nicotine in a novel degradation pathway for the alkaloid nicotine [41]. Moreover, chloroplasts can also be utilized in genetic transformation and other gene engineering techniques, offering advantages such as efficient expression of foreign genes, precise integration, no position effect, and genetic stability [42]. The technology has been successfully applied to a variety of plants, including A. thaliana, potato, tomato, rice, and carrot. Thus, through chloroplast-based gene editing technologies, including RNA editing, various issues can be addressed, such as the transformation and expression of chemical components, as well as the sterility of regenerated plants [43]. The interaction between chloroplast genes and chemical components is also bidirectional. The study reported that benzylisoquinoline alkaloids could inhibit photosynthetic metabolism by downregulating ndhF expression during the dark-light transition (DLT) [44].
Materials and Methods
4.1. Plant Photo and Materials
Three photographs of the original plant of C. majus were bought from the China Plant Image Library (https://ppbc.iplant.cn/) of Botanical Intelligence. After that, the fresh plant and photo were collected and taken in the Zuojia Town, Changyi District, Jilin City, Jilin Province (44.054°N, 126.108°E, 2166889875@qq.com). The characteristics of C. majus were described from different tissues of the roots, stems, leaves, flowers, fruits and seeds of the original C. majus plant to determine the specificity and authenticity of species. The fresh leaves of the three C. majus plantlets were collected, rinsed with RO water in a pot, dried the surface with absorbent paper, and ground in liquid nitrogen for the experiments of PCR and RT-PCR technology. The samples and specimens were stored at the Qinghai-Tibetan Plateau Museum of Biology in the Xining Botanical Garden of Qinghai Province, with voucher numbers DQbqc2025-1, DQbqc2025-2, and DQbqc2025-3.
4.2. Sources of Chloroplast Genomes
The chloroplast genome reference sequences of Chelidonium majus, A. thaliana, G. gandavensis, and A. tauschii are downloaded from NCBI with the serial numbers MN518846.1, NC_000932.1, OQ351928.1, and KJ614412.1.
4.3. Prediction of RNA Editing Sites and Protein Variation
Through the PREPACT [45] computational tools online platform for RNA editing, prediction, and analysis calculation of plants (http://www.prepact.de/prepact-main.php), using chloroplast genome of A. thaliana as the reference genome, RNA editing sites and protein variation were compared and predicted by BLASTX alignment within the plant chloroplast genomes of C. majus, G. gandavensis, and A. tauschii. The RNA editing sites, specifically from base C to U and reverse U to C, were predicted and statistically analyzed through the calculated parameters: E-value ≥ 0.001 and a result extension and filtration threshold of 2 amino acids (30%).
4.4. Protein Feature and Structure, RNA Structure, and Functional Changes
The CPGAVAS2 [46] online platform was used to annotate and extract the original sequence of the encoded protein in the chloroplast genome of the research species of proteins were predicted through . The physicochemical properties, secondary structures, conserved domains, signal peptides, transmembrane topology, and tertiary structures of proteins were predicated by the server platforms of Expasy (https://web.expasy.org/protparam/) [47], CFSSP (https://cfpred.sourceforge.net/)[48], NCBI conserved domain database (CDD, https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) [49] , SignalP 6.0 (https://services.healthtech.dtu.dk/services/SignalP-6.0/) [50], DeepTMHMM (v.1.0.13) (https://services.healthtech.dtu.dk/services/DeepTMHMM-1.0/) [51], and SWISS-MODEL (https://swissmodel.expasy.org/) [52]. The bound membrane proteins of chloroplast CDS genes before and after chloroplast RNA editing were analyzed by using diverse platforms of TOPCONS、OCTOPUS、PHILIUS、PolyPhobius、SCAMPI, and SPOCTOPUS (https://topcons.net/pred/,https://phobius.sbc.su.se/index.html) [53,54]. The protein mutations resulting from three pairs of RNA editing were compared and visualized using PyMOL software.
4.5. Validation of RNA Editing by Using PCR and RT-PCR Experiment
Total genomic DNA and RNA of C. majus were solely extracted from fresh young leaves of three plantlets by using SteadyPure Plant Genomic DNA Extraction Kit (CTAB) and RNA Extraction Kit (Vazyme, Nanjing, China). The cDNA Synthesis from RNA solution was performed using the Reverse transcription kit (PrimeScript IV 1st strand cDNA Synthesis Mix) with the Following Reverse transcription PCR reaction conditions: 30.0℃, 10 s; 42.0℃, 20 s; 70.0℃, 15 s; and stored at 4.0℃ indefinitely. The purity of DNA, RNA, cDNA, and amplification products were detected by 1.0% agarose gel electrophoresis stained with ethidium bromide alongside a 100 bp ladder (Vazyme, Nanjing, China) by using the DNA marker as the reference (Vazyme, Nanjing, China). The concentrations of DNA, RNA, and cDNA were determined using a Nanodrop spectrophotometer 2000 (Thermo Fisher Scientific, Waltham, MA, USA). The primer pairs for PCR amplification were designed using BioXM software (v 2.7.1), and the cloning fragments of the target sequences were performed using forward and reverse primers (Table S13). The PCR mixture consisted of 25.0 µL PowerPol 2× PCR Mix with Dye V2, 0.5 µL template DNA or cDNA, 1.0 µL forward primer and reverse primer, and ddH2O to a total volume of 50 µL. The PCR program consisted of a predenaturation step (98℃, 3 min), followed by 30 cycles of denaturation (98℃, 10 sec), primer annealing (60℃, 30 sec) and extension (72℃, 1 min), followed by a final extension step (72℃, 5 min). The amplification products were saved 4℃. A negative control (Milli-Q water in place of the DNA template) was included in each PCR to ensure there was no contamination. The presence of amplification products was confirmed by analyzing 5.0 µL of the PCR product with agarose gel electrophoresis and staining with ethidium bromide. The DNA amplification for sequence analysis positions was sequenced through the platform (www.generalbiol.com) on the ABI Prism 3730 Genetic Analyzer (Applied Biosystems, USA) by at least one forward and one reverse sequence read using the primers given (Table S12). The peak graph and sequence comparison were analyzed separately using Chromas and BioXM software.
4.6. Analysis of ORF and Transcription Factor
The nucleotide sequences of six mutation proteins with more RNA editing sites in the chloropalst genomes of C. majus were translated by the EMBOSS Backtranseq server (https://www.ebi.ac.uk/jdispatcher/st/emboss_backtranseq) [55]. Moreover, the open reading frames (ORFs) of the seven genes before and after the mutation were predicted through the ORF finder within the NCBI database (https://www.ncbi.nlm.nih.gov/orffinder/) [56] by using the Standard genetic code and Blast Database (UniProtKB/Swiss-Prot(SwissProt) [57]. The heatmap comparison of ORF mutation by each gene was visualized by using the Weishengxin online platform (https://www.bioinformatics.com.cn) and Adobe Photoshop (Adobe Systems, San Jose, Calif)). The identification of transcription factors in the chloroplast genome of C. majus was predicted using the CDS sequences through the PlantTFDB database (https://planttfdb.gao-lab.org/) [58], which links to the A. thaliana database.
4.7. GO, KOG, and KEGG Analysis
The annotations and classification of Gene Ontology (GO) [59], Eukaryotic Orthologous Groups of protein (KOG) [60], and Kyoto Encyclopedia of Genes and Genomes (KEGG) [61] were treated, visualized, and analyzed by using Genepioneer information platform (http://cloud.genepioneer.com:9929/), KEGG database (https://www.kegg.jp/kegg/), and Weishengxin online platform (https://www.bioinformatics.com.cn).
5. Conclusions
RNA editing has a significant impact on the functional and performance differences of proteins. The mechanism of protein functional variations needs to be continuously discovered through RNA editing, as it will play a non-negligible role in the synthesis and metabolism of chemicals, plant breeding, and systematic evolution in the species of C. majus from the Papaveraceae family.
Supplemental materials
Supplementary materials(Figures S1-S8 and Table S1-S20) can be downloaded at: figshare. Dataset. https://doi.org/10.6084/m9.figshare.29161274.
Author Contributions
Conceptualization, project administration, methodology, software, and visualization, Q.D.; formal analysis, validation, and data curation, D.Q., R.L., L. C., and K.L.; writing original draft preparation, review and editing manuscript, Q.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was not supported by any fund.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The verified sequencing data of RNA editing openly available in the GenBank database with accession numbers PV707905-PV707934.
Acknowledgments
We give the sincere thanks to Professor Zhao Yucheng for his unselfish assistance regarding the guidance of PCR and RT-PCR verification. Thanks to the sequencing technology support from geeneral biology (Anhui) Co., LTD.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| PI | Isoelectric points |
| ORFs | Open reading frames |
| ZFN | Zinc-finger nucleases |
| PPRs | Pentapeptide repeat proteins |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| GRAVY | Grand average of hydropathicity |
| SPs | Signal peptides |
| RMSD | Root Mean Square Deviation |
| TFs | Transcription factors |
| DLT | Dark-light transition |
References
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Hensel, G.; Kumlehn, J. Genome Engineering Using TALENs: Methods and Protocols. Mol. Biol. 2019, 195–215. [Google Scholar]
- Gupta, D.; Bhattacharjee, O.; Mandal, D.; Sen, M.K.; Dey, D.; Dasgupta, A.; Kazi, T.A.; Gupta, R.; Sinharoy, S.; Acharya, K.; Chattopadhyay, D.; Ravichandiran, V.; Roy, S.; Ghosh, D. CRISPR-Cas9 system: A new-fangled dawn in gene editing. Life Sci, 2019, 232, 116636. [Google Scholar] [CrossRef]
- Hao, L.; Chen, M.; Li, D.; Lijavetzky, D. Editorial:Plant RNA processing: discovery, mechanism and function. Front. Plant Sci. 2024, 15, 1359415. [Google Scholar] [CrossRef] [PubMed]
- Knoop, V. C-to-U and U-to-C: RNA editing in plant organelles and beyond. J. Exp. Bot. 2023, 74, 2273–2294. [Google Scholar] [CrossRef]
- Hao, W.; Liu, G.; Wang, W.; Shen, W.; Zhao, Y.; Sun, J.; Yang, Q.; Zhang, Y.; Fan, W.; Pei, S.; Chen, Z.; Xu, D.; Qin, T. RNA Editing and Its Roles in Plant Organelles. Front. Genet. 2021, 12, 757109. [Google Scholar] [CrossRef]
- Hoch, B.; Maier, R.M.; Appel, K.; Igloi, G.L.; Kössel, H. Editing of a chloroplast mRNA by creation of an initiation codon. Nature. 1991, 353, 178–180. [Google Scholar] [CrossRef]
- Maier, U.G.; Bozarth, A.; Funk, H.T.; Zauner, S.; Rensing, S.A.; Schmitz-Linneweber, C.; Börner, T.; Tillich, M. Complex chloroplast RNA metabolism: just debugging the genetic programme? BMC Biol. 2008, 6, 36. [Google Scholar] [CrossRef]
- Wang, H.; Yang, JK. Identification and analysis of RNA editing sites in the tobacco chloroplast genome. Mol. Plant Breed, 2020, 18, 6649–6656. [Google Scholar]
- Chang, X; Pei, Y; Hu, F.; Ren, M.M.; Wang, T.; Nie X.J. Predication, identification and characterization of the chloroplast RNA editing sites of Hulless barley. J. of triticeae crops. 2019, 39, 1316–1325.
- Ma, H. Z, Zhang, B.J.; Fan, H.L. Predication and identification of RNA editing sites in the chloroplast genomes of Barley (Hordeumvulgare L.). J. of triticeae crops. 2013, 33, 1071–1077. [Google Scholar]
- Wang, M.X.; Zhan, H.S.; Lu, M.L; Liu, S.Y.; Song, W. N. Identification and analysis of RNA editing sites in chloroplast transcripts of Aegilops tauschii. J. of triticeae crops. 2014, 34, 1341–1349. [Google Scholar] [CrossRef]
- Liu, K.; Xie, B.; Peng, L.; Wu, Q.; Hu, J. Profiling of RNA editing events in plant organellar transcriptomes with high-throughput sequencing. Plant J. 2024, 118, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, Y.; Zhang, G.; Miao, Y.; Liu, C.; & Huang, L. A Bibliometric Study for Plant RNA Editing Research:Trends and Future Challenges. Mol Biotechnol, 2023, 65, 1207-1227.
- Zhao, Y.; Gao, R.; Zhao, Z.; Hu, S.; Han, R.; Jeyaraj, A.; Arkorful, E.; Li, X.; Chen, X. Genome-wide identification of RNA editing sites in chloroplast transcripts and multiple organellar RNA editing factors in tea plant (Camellia sinensis L.): Insights into the albinism mechanism of tea leaves. Gene, 2023, 848, 146898. [Google Scholar] [CrossRef] [PubMed]
- Atlas of China's higher plants D, 1972, 2, 3, Figure 1735.
- Li, X. L.; Sun, Y. P.; Wang, M.; Wang, Z. B.; Kuang, H. X. Alkaloids in Chelidonium majus L: a review of its phytochemistry, pharmacology and toxicology. Front. Pharmacol. 2024, 15, 1440979. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.Q.; Zou, X.; Qu, Z.Y.; Ji, Y.B. The research progress of chemical composition and pharmacological effects of Chelidonium majus. Chin. Trad. and Herb. Drugs, 2009, 40, 38–40. [Google Scholar]
- Dou, S.L.; Li, L.R.; Gu, S.; Cai, M.C.; Li, X. Comprehensive research on the pharmacological effects of Chelidonium majus. Jilin traditional Chinese medicine, 2022, 42, 84–87. [Google Scholar]
- Wu, J.Y.; He, J.; Peng, L. L.; Wang, A.; Zhao, L.C. The complete chloroplast genome sequence of Chelidonium majus (Papaveraceae). Mitochondrial DNA Part B, 2019, 4, 1206–1207. [Google Scholar] [CrossRef]
- Guan, Y.J. Whole genome feature sequence and phylogenetic analysis of the chloroplast Chelidonium majus. Xiangnan College, 2023.
- Zhang, Z.; Zhang, G.; Zhang, X.; Zhang, H.; Xie, J.; Zeng, R.; Guo, B.; Huang, L. The complete chloroplast genome sequence and phylogenetic relationship analysis of Eomecon chionantha, one species unique to China. J. Plant Res. 2024, 137, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Pourmazaheri, H.; Soorni, A.; Kohnerouz, B.B.; Dehaghi, N.K.; Kalantar, E.; Omidi, M.; Naghavi, M.R. Comparative analysis of the root and leaf transcriptomes in Chelidonium majus L. PloS one, 2019, 14, e0215165.
- Nawrot, R.; Barylski, J.; Lippmann, R.; Altschmied, L.; Mock, H.P. Combination of transcriptomic and proteomic approaches helps to unravel the protein composition of Chelidonium majus L. milky sap. Planta, 2016, 244, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cui, X.; An, W.; Li, C.; Zhang, S.; Cao, M.; Yang, C. The complete genome sequence of a putative novel cytorhabdovirus identified in Chelidonium majus in China. Arch Virol. 2024, 169, 56. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.B.; Xiang, G.M.; Xing, J.H. A taxonomic re-assessment in the Chinese Bupleurum (Apiaceae): Insights from morphology, nuclear ribosomal internal transcribed spacer, and chloroplast (trnH-psbA, matK) sequences. J. of Plant Taxonomy. 2011, 49, 32. [Google Scholar] [CrossRef]
- Palhares, R. M.; Drummond, M. G.; Brasil, B. S.; Krettli, A. U.; Oliveira, G. C.; Brandão, M. G. The use of an integrated molecular-, chemical- and biological-based approach for promoting the better use and conservation of medicinal species: a case study of Brazilian quinas. Journal of ethnopharmacology, 2014, 155, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Huang, X.Y.; Huang, M.Y.; Zhang, X.; Fan, J. Sequence variations of matK gene in Spiranthes plants and the effect on protein molecular interaction and function. Mol. Plant Breed, 2019, 17, 7017–7030. [Google Scholar]
- Wu, Y.; Zheng, Y.; Xu, W.; Zhang, Z.; Li, L.; Wang, Y.; Cui, J.; Wang, Q.M. Chimeric deletion mutation of rpoC2 underlies the leaf-patterning of Clivia miniata var. variegata. Plant Cell Rep. 2023, 42, 1419–1431. [Google Scholar] [CrossRef]
- Yan, Y.; Duan, F.; Li, X.; Zhao, R.; Hou, P.; Zhao, M.; Li, S.; Wang, Y.; Dai, T.; Zhou, W. Photosynthetic capacity and assimilate transport of the lower canopy influence maize yield under high planting density. Plant Physiology. 2024, 195, 2652–2667. [Google Scholar] [CrossRef]
- He, Q. The soybean NDH complex and its role in salt stress. Zhe Jiang University. 2012.
- Huang, W.; Zhang, Y.; Shen, L.; Fang, Q.; Liu, Q.; Gong, C.; Zhang, C.; Zhou, Y.; Mao, C.; Zhu, Y.; Zhang, J.; Chen, H.; Zhang, Y.; Lin, Y.; Bock, R.; Zhou, F. Accumulation of the RNA polymerase subunit RpoB depends on RNA editing by OsPPR16 and affects chloroplast development during early leaf development in rice. New Phytol. 2020, 228, 1401–1416. [Google Scholar] [CrossRef]
- Gupta, G.S. Mannose Receptor Family: R-Type Lectins. Springer Vienna. 2012, 331–347. [Google Scholar]
- Duart, G.; Graña-Montes, R.; Pastor-Cantizano, N.; & Mingarro, I. Experimental and computational approaches for membrane protein insertion and topology determination. Methods. 2024, 226, 102–119.
- Facchini P., J. ALKALOID BIOSYNTHESIS IN PLANTS: Biochemistry, Cell Biology, Molecular Regulation, and Metabolic Engineering Applications. Annual review of plant physiology and plant molecular biology. 2001, 52, 29–66. [Google Scholar] [CrossRef] [PubMed]
- Yahyazadeh, M.; Selmar, D.; Jerz, G. Electrospray-Mass Spectrometry-Guided Targeted Isolation of Indole Alkaloids from Leaves of Catharanthus roseus by Using High-Performance Countercurrent Chromatography. Molecules (Basel, Switzerland). 2025, 30, 2115. [Google Scholar] [CrossRef]
- Mall, M.; Shanker, K.; Samad, A.; Kalra, A.; Sundaresan, V.; Shukla, A. K. Stress responsiveness of vindoline accumulation in Catharanthus roseus leaves is mediated through co-expression of allene oxide cyclase with pathway genes. Protoplasma. 2022, 259, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.; Schoofs, G.; Wink, M. A chloroplast-localized lysine decarboxylase of Lupinus polyphyllus: the first enzyme in the biosynthetic pathway of quinolizidine alkaloids. FEBS letters, 1980, 115, 35–38. [Google Scholar] [CrossRef]
- Wink, M.; Hartmann, T. Activation of chloroplast-localized enzymes of quinolizidine alkaloid biosynthesis by reduced thioredoxin. Plant cell reports. 1981, 1, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Tian, Y. X.; Zhang, F.; Chen, Q.; Zhang, Y.; Luo, Y.; Wang, X. R.; Lin, F. C.; Yang, J.; Tang, H. R. Variations of Alkaloid Accumulation and Gene Transcription in Nicotiana tabacum. Biomolecules. 2018, 8, 114. [Google Scholar] [CrossRef]
- Li, H.; Xie, K.; Yu, W.; Hu, L.; Huang, H.; Xie, H.; Wang, S. Nicotine dehydrogenase complexed with 6-Hydroxypseudooxynicotine oxidase involved in the hybrid nicotine-degrading pathway in Agrobacterium tumefaciens S33. Applied and environmental microbiology. 2016, 82, 1745–1755. [Google Scholar] [CrossRef]
- Fuentes, P.; Armarego-Marriott, T.; Bock, R. Plastid transformation and its application in metabolic engineering. Curr. Opin. Biotechnol. 2018, 49, 10–15. [Google Scholar] [CrossRef]
- Ruf, S.; Forner, J.; Hasse, C.; Kroop, X.; Seeger, S.; Schollbach, L.; Schadach, A.; Bock, R. High-efficiency generation of fertile transplastomic Arabidopsis plants. Nature plants. 2019, 5, 282–289. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, T.; Jiang, J.; Deng, L.; Liu, Y.; Zhong, Z.; Fu, H.; Yang, B.; Zhang, L. The chloroplast GATA-motif of Mahonia bealei participates in alkaloid-mediated photosystem inhibition during dark to light transition. J. of plant physiology. 2023, 280, 153894. [Google Scholar] [CrossRef]
- Lenz, H.; Hein, A.; Knoop, V. Plant organelle RNA editing and its specificity factors: enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinformatics. 2018, 19, 255. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic acids research. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Wilkins, M. R.; Gasteiger, E.; Bairoch, A.; Sanchez, J. C.; Williams, K. L.; Appel, R. D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods in molecular biology (Clifton, N.J.). 1999, 112, 531–552. [Google Scholar] [PubMed]
- Chen, H.; Gu, F.; Huang, Z. Improved Chou-Fasman method for protein secondary structure prediction. BMC Bioinformatics. 2006, 7, S14. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M. K.; Gonzales, N. R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; Yang, M.; Zhang, D.; Zheng, C.; Lanczycki, C. J.; Marchler-Bauer, A. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Nielsen, H.; Teufel, F.; Brunak, S.; von Heijne G. SignalP: The Evolution of a Web Server. In: Lisacek, F. (eds) Protein Bioinformatics. Methods in Molecular Biology. 2024, 2836.
- Hallgren, J.; Tsirigos, K. D.; Pedersen, M. D.; Armenteros, J. J. A.; Marcatili, P.; Nielsen, H.; Krogh, A.; and Winther, O. Deeptmhmm predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv. 2022. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F. T.; de Beer, T. A. P.; Rempfer, C.; Bordoli, L.; Lepore, R.; Schwede, T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Tsirigos, K. D.; Peters, C.; Shu, N.; Käll, L.; & Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Research. 2015, 43, W401–W407.
- Viklund, H.; Bernsel, A.; Skwark, M.; Elofsson, A. SPOCTOPUS: a combined predictor of signal peptides and membrane protein topology. Bioinformatics (Oxford, England) 2008, 24, 2928–2929. [Google Scholar] [CrossRef]
- Olson S., A. EMBOSS opens up sequence analysis. European Molecular Biology Open Software Suite. Briefings in bioinformatics. 2002, 3, 87–91. [Google Scholar] [CrossRef]
- Wheeler, D. L.; Church, D. M.; Federhen, S.; Lash, A. E.; Madden, T. L.; Pontius, J. U.; Schuler, G. D.; Schriml, L. M.; Sequeira, E.; Tatusova, T. A.; Wagner, L. Database resources of the National Center for Biotechnology. Nucleic acids research. 2003, 31, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Boutet, E. , Lieberherr, D., Tognolli, M., Schneider, M., Bansal, P., Bridge, A. J., Poux, S., Bougueleret, L., & Xenarios, I. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Methods in molecular biology (Clifton, N.J.). 2016, 1374, 23–54. [Google Scholar] [PubMed]
- Blanc-Mathieu, R.; Dumas, R.; Turchi, L.; Lucas, J.; & Parcy, F. Plant-TFClass: a structural classification for plant transcription factors. Trends in plant science. 2024, 29, 40–51.
- Harris, M. A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; Richter, J.; Rubin, G. M.; Blake, J. A.; Bult, C.; Dolan, M.; Drabkin, H.; Eppig, J. T.; Hill, D. P.; Ni, L.; Ringwald, M. Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic acids research. 2004, 32, D258–D261. [Google Scholar]
- Fu, C.; Yang, Y. iCAZyGFADB: an insect CAZyme and gene function annotation database. Database : the journal of biological databases and curation 2023, baad086.
- Nguyen, H.; Pham, V. D.; Nguyen, H.; Tran, B.; Petereit, J.; Nguyen, T. CCPA: cloud-based, self-learning modules for consensus pathway analysis using GO, KEGG and Reactome. Briefings in bioinformatics. 2024, 25, bbae222. [Google Scholar] [CrossRef]
Figure 1.
Plant characteristics of C. majus. (a) Root (b) Stem (c)Leaf (d) Flower (e) Fruit (f) Seed (g) Phytobiocoenose.
Figure 1.
Plant characteristics of C. majus. (a) Root (b) Stem (c)Leaf (d) Flower (e) Fruit (f) Seed (g) Phytobiocoenose.

Figure 2.
RNA editing sites and amino acid mutation types in the chloroplast genome of C. majus, G. X gandavensis, and A. tauschii.(a)Nine genes with the same number of RNA editing sites.(b) The 40 genes with a different number of RNA editing sites. (c) Mutation types of the 13 amino acids.
Figure 2.
RNA editing sites and amino acid mutation types in the chloroplast genome of C. majus, G. X gandavensis, and A. tauschii.(a)Nine genes with the same number of RNA editing sites.(b) The 40 genes with a different number of RNA editing sites. (c) Mutation types of the 13 amino acids.
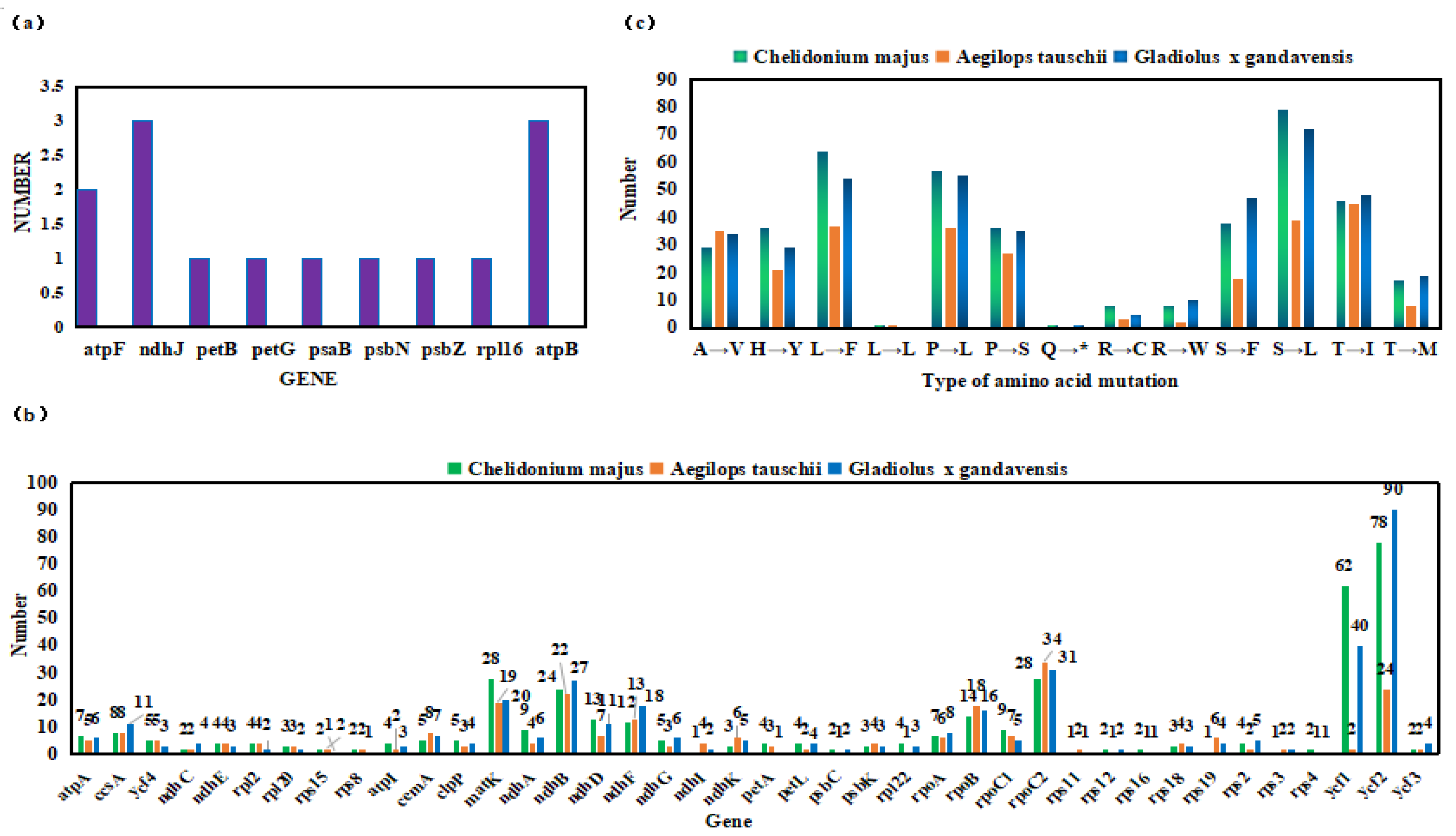
Figure 3.
The conserved domain of rpoB protein during the RNA editing. (a) original protein (b) protein after RNA editing (c) pfam00652 protein (No. A0A5J4LQ17, Streptomyces hyphurium) (d) IPR000772 protein (No. Q8X123, Amanita phalloides/Marasmius oreades).
Figure 3.
The conserved domain of rpoB protein during the RNA editing. (a) original protein (b) protein after RNA editing (c) pfam00652 protein (No. A0A5J4LQ17, Streptomyces hyphurium) (d) IPR000772 protein (No. Q8X123, Amanita phalloides/Marasmius oreades).
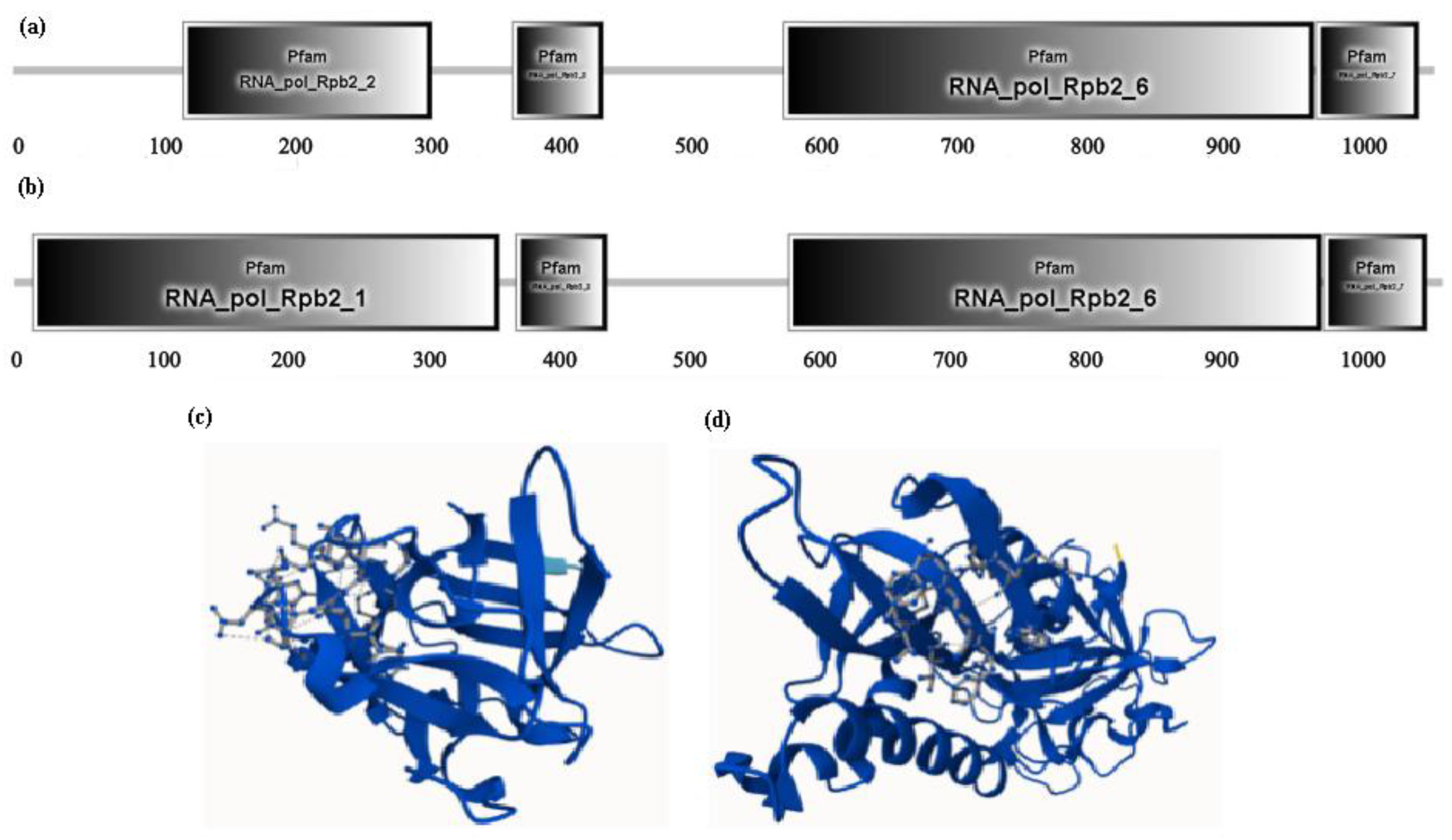
Figure 4.
Effect of RNA editing from the ndhB gene on the transmembrane region of the protein-encoding in the chloroplast genome of C. majus. (a) Before mutation in the ndhB encoding protein. (b) After mutation in the ndhB encoding protein. The red pentagon star ( ) represents differential sites in the transmembrane region before and after mutation in the protein encoding ndhB gene.
Figure 4.
Effect of RNA editing from the ndhB gene on the transmembrane region of the protein-encoding in the chloroplast genome of C. majus. (a) Before mutation in the ndhB encoding protein. (b) After mutation in the ndhB encoding protein. The red pentagon star ( ) represents differential sites in the transmembrane region before and after mutation in the protein encoding ndhB gene.
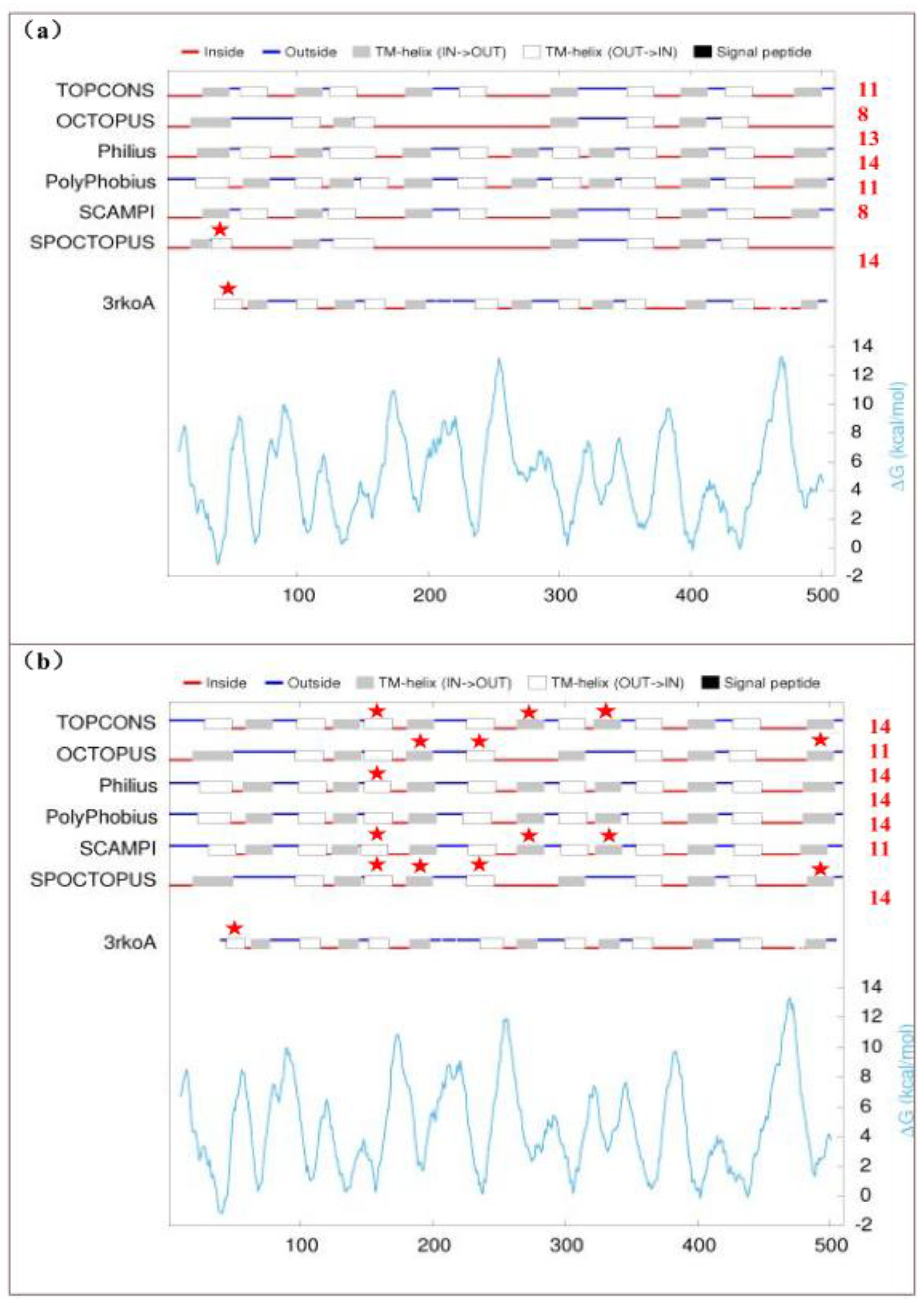
Figure 5.
Transmembrane proteins and signal peptides within petA protein by RNA editing. (a)Before and after RNA editing by the software of DeepTMHMM (b) Signal peptides before RNA editing by the online platform SignalP 6.0 (c) Signal peptides after RNA editing by the online platform SignalP 6.0. The blue frames show the mutation of one paris protein.SP (Sec/SPI): Signal peptide sequence; CS: Cleavage site; OTHER: No signal peptide sequence.
Figure 5.
Transmembrane proteins and signal peptides within petA protein by RNA editing. (a)Before and after RNA editing by the software of DeepTMHMM (b) Signal peptides before RNA editing by the online platform SignalP 6.0 (c) Signal peptides after RNA editing by the online platform SignalP 6.0. The blue frames show the mutation of one paris protein.SP (Sec/SPI): Signal peptide sequence; CS: Cleavage site; OTHER: No signal peptide sequence.
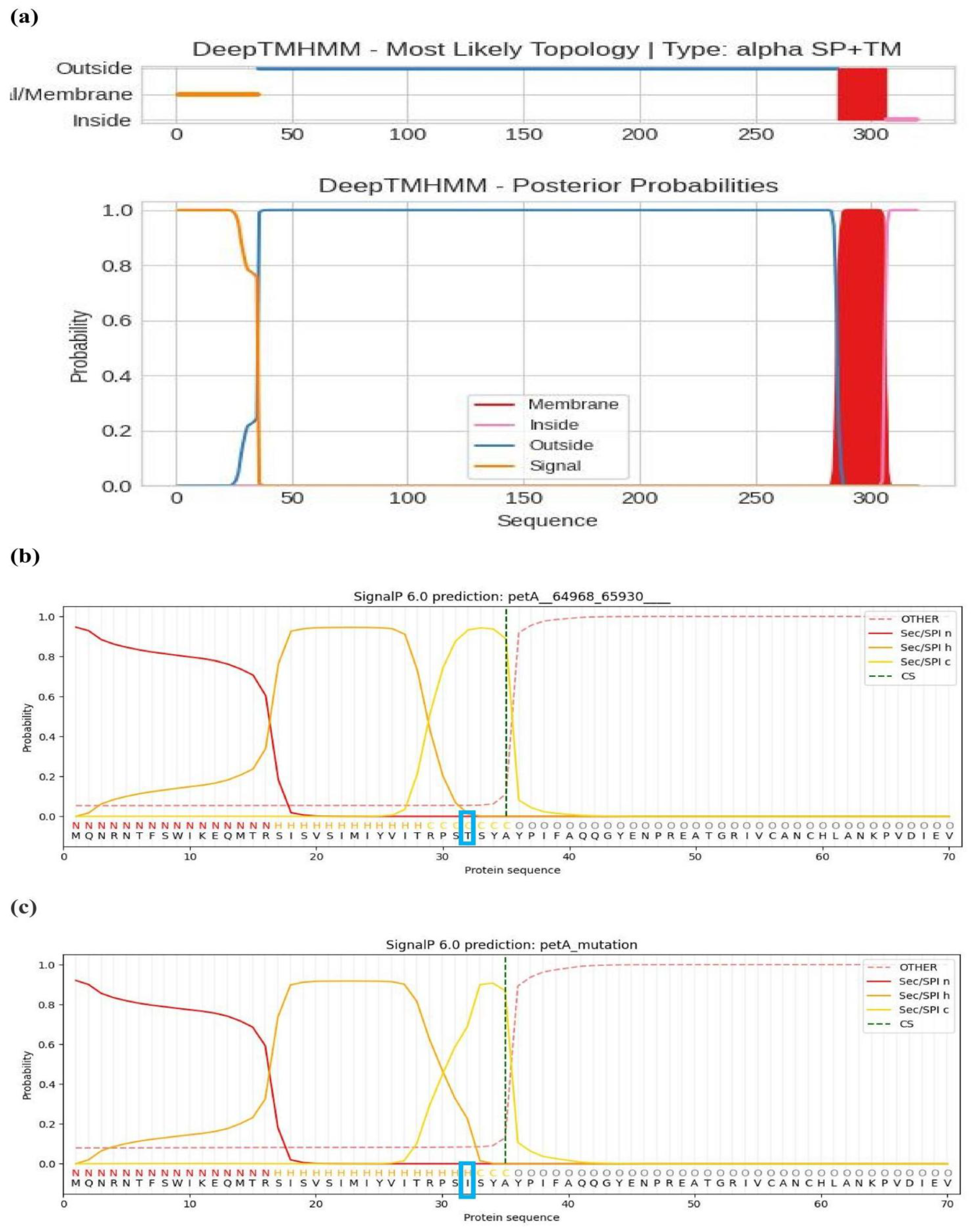
Figure 6.
The sequences and structures of the rpoB gene and protein between prediction and verification by experiment for the RNA editing in the chloroplast genome of C. majus. (a) The comparative mutation nucleotide sequences of predicted DNA and cDNA amplification for the rpoB gene. (b) The comparative mutation protein sequences of predicted, DNA, and cDNA amplification for the rpoB gene. (c) The secondary structure variation of rpoB protein. The marked letters A, B, C, D, and E with blue frames indicate the variation sites.
Figure 6.
The sequences and structures of the rpoB gene and protein between prediction and verification by experiment for the RNA editing in the chloroplast genome of C. majus. (a) The comparative mutation nucleotide sequences of predicted DNA and cDNA amplification for the rpoB gene. (b) The comparative mutation protein sequences of predicted, DNA, and cDNA amplification for the rpoB gene. (c) The secondary structure variation of rpoB protein. The marked letters A, B, C, D, and E with blue frames indicate the variation sites.

Figure 7.
Effect of chloroplast RNA editing on the secondary structure of six gene-encoded proteins in the chloroplast genome of C. majus. Marked the letters a, c, e, g, i, and k as the secondary structures of the proteins encoded by the matK, rpoC2, ndhB, ndhD, ndhF, and rpoB genes before RNA editing, respectively; Marked the letters b, d, f, h, j, and l as the secondary structures of the proteins encoded by the matK, rpoC2, ndhB, ndhD, ndhF, and rpoB genes after RNA editing, respectively.
Figure 7.
Effect of chloroplast RNA editing on the secondary structure of six gene-encoded proteins in the chloroplast genome of C. majus. Marked the letters a, c, e, g, i, and k as the secondary structures of the proteins encoded by the matK, rpoC2, ndhB, ndhD, ndhF, and rpoB genes before RNA editing, respectively; Marked the letters b, d, f, h, j, and l as the secondary structures of the proteins encoded by the matK, rpoC2, ndhB, ndhD, ndhF, and rpoB genes after RNA editing, respectively.

Figure 8.
Comparisons of tertiary structures in three proteins of the best model by RNA editing. (a)ndhB (green) and ndhB mutation (pink) (b) ndhB (green) and ndhB mutation (pink). (b) rpoB (green) , rpoB mutation (pink), and rpoB experiment (orange). The blue stick represents the comparative mutation sites of the proteins resulting from RNA editing by each pair. The two rose spheres within rpoB (c) represent the new mutation sites identified after the experiment.
Figure 8.
Comparisons of tertiary structures in three proteins of the best model by RNA editing. (a)ndhB (green) and ndhB mutation (pink) (b) ndhB (green) and ndhB mutation (pink). (b) rpoB (green) , rpoB mutation (pink), and rpoB experiment (orange). The blue stick represents the comparative mutation sites of the proteins resulting from RNA editing by each pair. The two rose spheres within rpoB (c) represent the new mutation sites identified after the experiment.
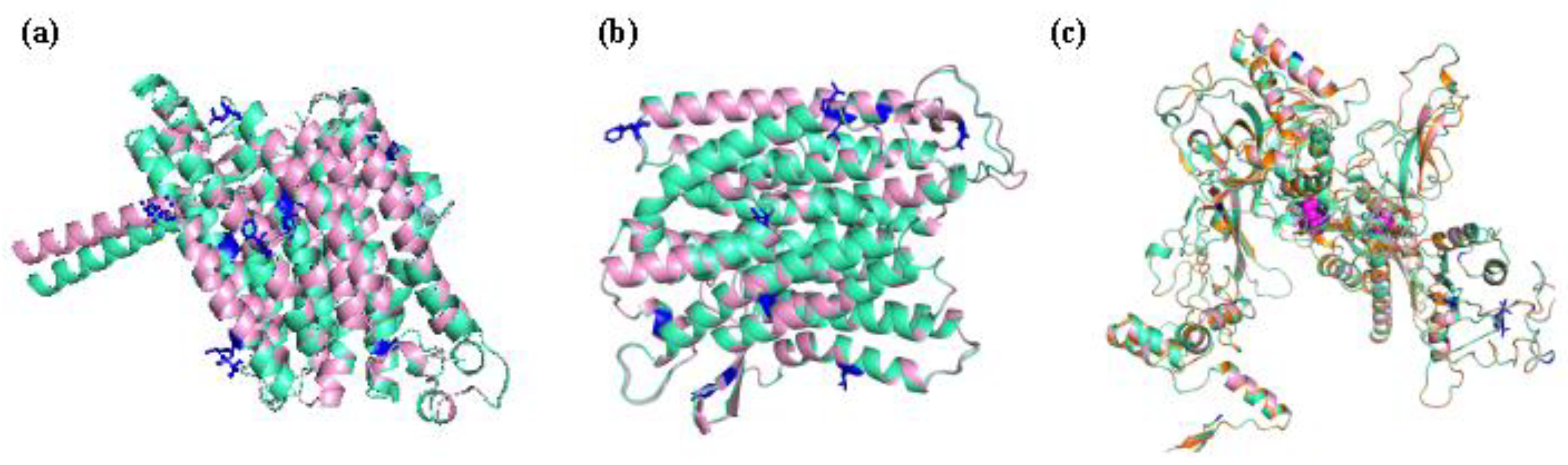
Figure 9.
The number and length of ORFs within ndhB gene in the chloroplast genome of Chelidonium majus.(a) Predicted ORFs before RNA editing (b) Predicted ORFs after RNA editing.
Figure 9.
The number and length of ORFs within ndhB gene in the chloroplast genome of Chelidonium majus.(a) Predicted ORFs before RNA editing (b) Predicted ORFs after RNA editing.
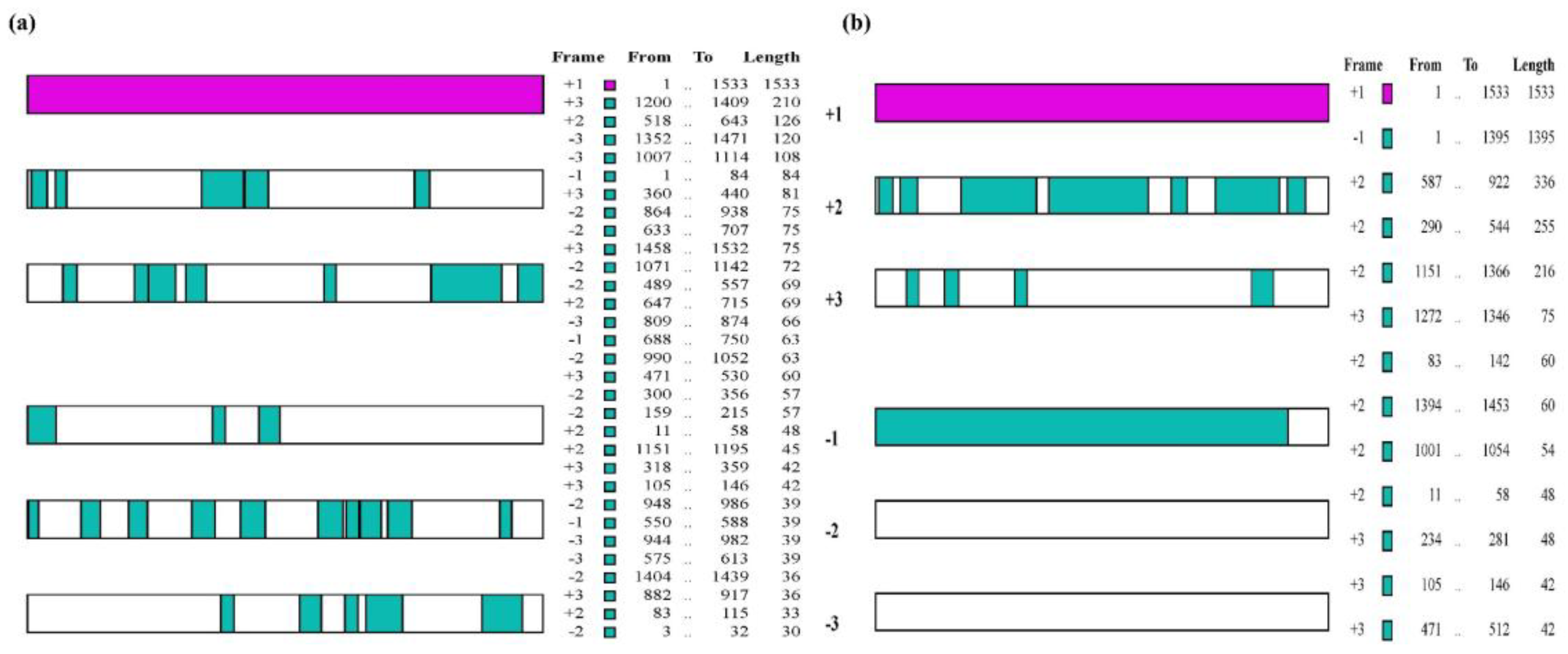

Table 1.
Genes, codon types and amino acid mutation types (base conversion from cytosine C to uracil U) predicted for RNA editing in chloroplast genome of C. majus.
Table 1.
Genes, codon types and amino acid mutation types (base conversion from cytosine C to uracil U) predicted for RNA editing in chloroplast genome of C. majus.
| The genes and numbers of RNA editing | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(18) | 2(11) | 3(7) | 4(8) | 5(4) | 7(2) | 8(2) | 9(2) | 12(1) | 13(1) | 14(1) | 24(1) | 27(1) | 28(1) | 62(1) | 78(1) |
| ndhI, petB, petD, petG, psaB, psaI, psaJ, psbE, psbF, psbN, psbT, psbZ, rpl14, rpl16, rps11, rps14, rps19, rps3 | atpF, ndhC, psaA, psbC, psbJ, rps12, rps15, rps16, rps4, rps8, ycf3 | atpB, ndhH, ndhJ, ndhK, psbK, rpl20, rps18 | atpI, ndhE, petA, petL, rpl2, rpl22, rpl23, rps2 | cemA, clpP, ndhG, ycf4 | atpA, rpoA | accD, ccsA | ndhA, rpoC1 | ndhF | ndhD | rpoB | ndhB | matK | rpoC2 | ycf1 | ycf2 |
| Codon types and number | |||||||||||||||
| 28 | ACA→AUA, ACC→AUC, ACG→AUG, ACU→AUU, CAA→UAA, CAC→UAC, CAU→UAU, CCA→CUA, CCA→UCA, CCC→CUC, CCC→UCC, CCG→CUG, CCG→UCG, CCU→CUU, CCU→UCU, CGG→UGG, CGU→UGU, CUA→UUA, CUC→UUC, CUU→UUU, GCA→GUA, GCC→GUC, GCG→GUG, GCU→GUU, UCA→UUA, UCC→UUC, UCG→UUG, UCU→UUU | ||||||||||||||
| Types and number of amino acid mutations | |||||||||||||||
| 29 | 36 | 64 | 1 | 57 | 36 | 1 | 8 | 8 | 38 | 79 | 46 | 17 | |||
| A→V | H→Y | L→F | L→L | P→L | P→S | Q→* | R→C | R→W | S→F | S→L | T→I | T→M | |||
Table 2.
RNA editing impact on protein transmembrane, RNA secondary structures, protein structures, signal peptide in the cp genome of C. majus.
Table 2.
RNA editing impact on protein transmembrane, RNA secondary structures, protein structures, signal peptide in the cp genome of C. majus.
| Genes containing RNA editing sites | RNA secondary structure | Protein structure | Signal Peptide (Sec/SPI) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| transmembrane domain number(before/after) | transmembrane domain affected | Homologous protein | affected | ɑ-helix | β-strand | Tertiary structure (best model) | Before mutation | After mutation | ||
| atpA | + | - | - | - | + | + | - | c6fkfA | - | - |
| ccsA | + | 8 | - | - | + | +1 | -1 | c7s9yA | - | - |
| ycf4 | + | 2 | - | - | + | + | +1 | c8keiD | - | - |
| cemA | + | 4 | - | - | + | -1 | +1 | c6ynyB | - | - |
| matK | + | - | - | - | + | -2 | + | c8h2hC | - | - |
| ndhA | + | 8 | - | 4heaH | + | +1 | -1 | c7eu3A/c7wffA | - | - |
| ndhB | + | 8~14/11~14 | + | 3rkoA | + | +1 | -2 | c7wffB | - | - |
| ndhD | + | 11~14/11~15 | + | 3rkoM | + | +4 | +2 | c7wffD | - | - |
| ndhF | + | 14~16 | - | 3rkoL | + | +2 | -2 | c7wffF | - | - |
| ndhG | + | 5 | + | 3rkoJ | + | + | -1 | c7wffG | - | - |
| rpoA | + | - | - | - | + | + | + | c8w9zB | - | - |
| rpoB | + | - | - | - | + | +2 | -2 | c8w9zC | - | - |
| rpoC1 | + | - | - | - | + | - | -2 | c8w9zD | - | - |
| rpoC2 | + | 3 | + | - | + | + | +1 | c1gprA | - | - |
| petA | + | 1 | - | 4h44C | + | - | -2 | c7zyvK | 0.9434 | 0.9179 |
| accD | + | - | - | - | + | -2 | +2 | c2f9iB | - | - |
| “+”representative the structures were affected by RNA editing. | ||||||||||
| “-”representative the structures were not affected by RNA editing or "No existing". | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



