
Submitted:
30 September 2025
Posted:
01 October 2025
You are already at the latest version
Abstract
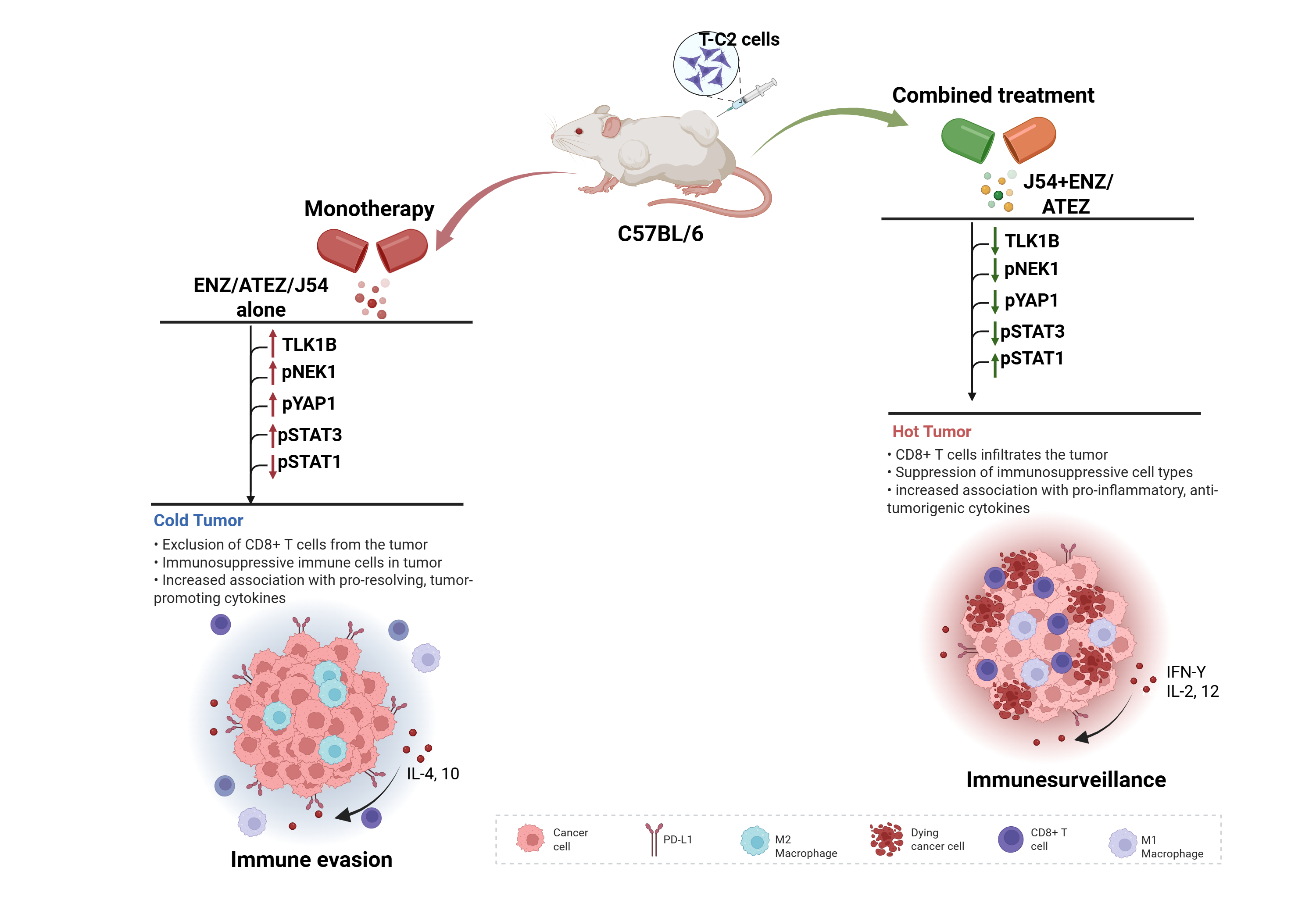
Our recent studies on the activation of the TLK1B>NEK1>YAP1 axis following ADT/ARSI have revealed the outlines of an innate immunity integration response that includes PD-L1 expression and alterations in cGAS-pSTING and pSTAT1/3, which drive the immunoevasion of PCa cells after prolonged treatment with ARSI and can thus contribute to CRPC progression. These observations are consistent with the general assessment that human PCa is immunologically ‘cold’ and unresponsive in the long term to ICB treatments aimed at combating mCRPC once it is established. Here, we utilized a TLK1 inhibitor (J54) and a PD-L1 monoclonal antibody to suppress tumor growth in a syngeneic PCa model (TC2 in C57/Bl6 mice), in addition to Enzalutamide (ENZ). Remarkably, the combinations of ENZ+J54 or Atez+J54 resulted in durable suppression of tumor growth, partly explained by a strong antitumor immune response. These observations were further generalized using a genetic model (TRAMPxNEK1+/- mice), in which we occasionally observed progression to genuine PRAD with age. In cases when it did progress, there was clear evidence of a strong immunoreactive response, spatially discernible with VISIUM (10XG) of PEFF prostate sections. We also defined the mechanism of YAP-dependent expression of PD-L1 using a number of common PCa cell lines, OE plasmids for YAP-wt and inactive Y407F mutant, Luc reporters containing proximal and distal regulatory elements of the PD-L1 gene, and occupancy of those same elements by YAP/TEAD by ChIP.
Keywords:
advanced CRPC therapy
; CRPC
; ICB
; PD-L1
; TLK1
; NEK1
; YAP
Introduction
The standard of care for advanced Prostate Cancer (PCa) consists of Androgen Deprivation Therapy (ADT) and the use of ever more effective therapies targeting the Androgen Receptor (AR) under different mechanisms (rev in [1]). Unfortunately, these treatments ultimately fail due to various PCa adaptation pathways [2] resulting in the incurable phase of the disease: metastatic castration resistant prostate cancer (mCRPC). These pathways often participate in normal cellular processes but become oncogenic after ADT [3]. One of the most common mechanisms is the ability to integrate AR signals with ever-diminishing residual testosterone, with the frequent participation of Yes-associated protein (YAP) as AR co-activator (rev in [4]).
We previously uncovered the upregulation of Tousled-like kinase 1B (TLK1B) through mTOR-dependent translational derepression after ADT, which is critical for promoting the flux through TLK1>Nek1-T141>ATR>Chk1 axis in mediating the DNA damage response (DDR) and cell cycle checkpoint while transiting from Androgen Sensitive (AS) to Insensitive (AI) growth for LNCaP, VCaP, TRAMP-C2 cells, and in preliminary experiments in a AR+/PDX model [5,6]. In addition, we found that overexpression of wt-Nek1 (but not the T141A kinase-hypoactive mutant that TLK1 cannot phosphorylate) hastens the progression of LNCaP cells to AI growth. The protective TLK1>Nek1 DDR-induced cell cycle arrest does not explain the rapid growth recovery observed in ARSI-treated cells when Nek1 is overexpressed[7,8] and suggests that Nek1 may have additional functions. We have ample evidence that it regulates the Hippo pathway because we found that Nek1 interacts with and activates YAP through Y407 phosphorylation [6,9]. It was reported that ectopic expression of YAP is sufficient to convert LNCaP cells from AS to AI growth[10]. It was also determined that the ETS-related gene (ERG) (and the common TMPRSS2-ERG rearrangement) activates the transcriptional program regulated by YAP and that prostate-specific activation of either ERG or YAP in mice induces similar transcriptional changes and results in age-related prostate tumors[11]. This work provided direct genetic evidence of a causal role for ERG in prostate cancer and revealed a connection between ERG and the Hippo pathway. However, in human PCa, the key components/regulators of the Hippo pathway are still unknown. Recently, we found that SCID xenografts of the VCaP model that carry such common translocation initially respond well (with shrinkage of the tumors) to the combination of Enzalutamide (ENZ) and J54 (TLKi). Still, ultimately, in about 50% of the mice, the tumors will re-grow. This is despite the fact that J54[12] is still capable of suppressing pNek1-T141 and its target, pYAP-Y407[13,14], the active/nuclear form of the co-activator. We favored the hypothesis that the TMPRSS2-ERG oncogenic translocation can implement a parallel AI (CRPC) conversion than the one mediated by the AR/YAP integration, with elevated expression of their target genes, including epithelial to mesenchymal transition (EMT) and androgen independence determinants[13].
Evidence is accumulating that YAP contributes to cancer immunosuppression[15,16]. An important distinction that characterizes YAP’s co-opted regulation of cancer progression genes is its capacity to mediate YAP-induced programmed death ligand 1 (PD-L1) expression, which drives immune evasion [17,18,19]. PD-L1, a type I transmembrane surface glycoprotein encoded by the CD274 gene, promotes T-cell tolerance and inhibits its effector functions[20]. Its expression correlates with dysregulation of the immune response that characterizes tumor progression and therapy resistance [21]. Immune checkpoint inhibitors (ICIs) are primarily used for tumors. However, their efficacy is significantly reduced due to therapy resistance[22]. The link between treatment-induced YAP-Y407 phosphorylation-mediated influence on immune cell recruitment to the tumor site remains unexplored. Our findings show that pIRF3 in recidive VCaP xenografts was suppressed with J54, thus curtailing a key branch of innate immunity through the expression of INF-g, which is also critical for PDL-1/2 expression and avoiding immune surveillance[23]. At the same time, pSTAT1 was dramatically increased, likely through activation of cGAS/STING from the release of nuclear and mitochondrial DNA upon implementation of apoptosis [6,24,25,26]. Conversely, pSTAT3 was elevated in tumors of mice treated with ENZ but not concomitantly with J54 – all this suggesting prolonged treatment with ARSI suppresses anti-tumor-induced, YAP- and NFKB-modulated inflammatory responses[27]. In fact, our single-cell VISIUM analysis of advanced age TRAMP PCas revealed that in TRAMP mice crossed to be haploinsufficient for Nek1, there was a considerable infiltration of CTLs after castration of the animals, which was found to elevate expression of TLK1B. Hence, we hypothesize that the addition of ICB modulators could still work well for PCa patients who may not be responding optimally to our ENZ+J54 treatment, as we anticipate it may happen for cases harboring TMPRSS2-ERG translocation (reported to be as many as 45% of patients). We thus initiated xenograft studies in syngeneic/immunocompetent mice to investigate this proposition using the TRAMP-C2 cells model in C57Bl/6 mice[28], which is already relatively well characterized for its immune-inducing responses. Our findings suggest a change of an overall immunologically ‘cold’ tumor, the typical condition of most PCa [29,30], into a ‘hot’ one with the combination of ICIs and TLK1 axis inhibition.
Materials and Methods
J54 was purchased from Probechem (TLK1 inhibitor J54|TLK1 inhibitor|Probechem Biochemicals, China) while Enzalutamide (AR inhibitor) and Atezolizumab (PD-L1 inhibitor) were purchased from BOC Sciences, NY, USA, and Targetmol Chemicals, MA, respectively.
Animal Studies
All animals involved in this study received humane care following the guidelines of the American Veterinary Medical Association. The Institutional Animal Care and Use Committee of the LSU Health Sciences Center at Shreveport approved all testing protocols. Although the number of mice was kept to a minimum, we adhered to ARRIVE guidelines thought the work and its analysis. Male C57BL/6 mice (Charles River, Skokie, IL, USA) were utilized to host TRAMP-C2 prostate cancer tumors, with 0.5 × 10^6 TRAMP-C2 cells grafted subcutaneously into the lower back flanks of the mice. Once the tumors reached approximately 100 mm³ in size, the tumor-bearing mice were randomized into eight treatment groups. The treatments included a vehicle control (PBS), an AR inhibitor ENZ (10 mg/ml), a PD-L1 inhibitor ATEZ (400 µg/100 µl), a TLK1-NEK1 inhibitor J54 (10 mg/kg), combinations of J54 with ENZ or ATEZ, and a triple drug combination. All treatments were administered orally and given bi-weekly. The dosing for J54 was determined based on our previous research. Tumor sizes were measured every other day using calipers. The inhibition treatment continued for 28 days, equivalent to roughly nine bi-weekly cycles. Mice were sacrificed at the experiment’s conclusion using CO2 asphyxiation, and the tumors were excised for tissue Western blots or immunohistochemistry (IHC). Blood samples were also collected for further analysis. Whole blood was centrifuged at 3000 rpm for 15 minutes at 4 °C, and the supernatant was stored at -80 degrees for future use. The packed cell pellet was utilized for RNA extraction using the Trizol-chloroform method.
Western Blot
Western blots were performed in three biological replicates utilizing tumors excised from various treatment groups, and total protein assay was performed as described in [31]. The sample supernatant was denatured with 1X Laemmli Buffer for 10 minutes at 95 °C and subsequently separated using a 10% PROTEAN TGX protein gel (Bio-Rad, Hercules, CA, USA). Proteins were transferred to the Immun-Blot PVDF membrane (BioRad) with a Criterion Blotter at 100 volts on ice. The membrane was blocked using 5% non-fat dry milk in 1X Tris-buffered saline with Tween-20 (TBST) for 1 hour at room temperature. Following this, the membrane was washed three times with 1X TBST and incubated overnight at 4 °C with gentle rocking in a solution containing mouse anti-PD-L1 antibody (Thermo Scientific; Cat. No. 14-5983-82), anti-pSTING (Cell Signaling Technology; Cat. No. 19781S), anti-pSTAT3 (Cell Signaling Technology; Cat. No. 9145S), anti-Pstat1 (Cell Signaling Technology; Cat. No. 9167S), along with anti-pYAP1-Y407, anti-pNek1-T141, and anti-TLK1 (Thermo Scientific) antibodies diluted in 5% BSA in 1X TBST. After washing, the membranes were incubated for 1 hour at room temperature with either horse anti-mouse or anti-rabbit antibodies (Cell Signaling Technology) conjugated with horseradish peroxidase in 5% BSA in 1X TBST. Once incubation was complete, the membrane was washed, and bands were visualized using a Pierce™ ECL Western Blotting Substrate (Thermo Scientific) Cat. No. 32106) on the ChemiDoc MP Imaging System.
ELISA
This was done using the mouse immune response ELISA Strip (Signosis Inc., CA, USA, Catalog No: EA-1171). Briefly, 100 μL of serum was incubated per well for 2 hours at room temperature with gentle shaking. Each well was aspirated and washed thrice with 200 μL of wash buffer. 100 μL of biotin-labeled antibody was added to each well and incubated for 1 hour with gentle shaking. The washing with wash buffer was repeated three times. 100 μL of streptavidin-HRP was added to each well and incubated for 45 minutes at room temperature. The wash step was repeated afterward. 100 μL of substrate was added to each well and incubated for 30 minutes, after which 50 μL of stop solution was added. The optical density was determined at 450 nm using a microplate reader. Finally, the concentrations were calculated using the appropriate conversions.
Immunohistochemistry
Tissues were sectioned and mounted on slides, dried, deparaffinized, and rehydrated using xylene and a graded ethanol series (100% - 70%, followed by water). Antigen retrieval was performed, followed by a peroxidase block. Slides were incubated with blocking buffer for 30 minutes. Overnight incubation with primary antibodies (F4-80 OR CD3) was conducted at 4 °C. The slides were washed three times with PBS for 5 minutes each and incubated with an HRP-conjugated secondary antibody for 30 minutes. 3,3’-diaminobenzidine (DAB) was added and incubated for about 8 minutes to develop color. Counterstaining with hematoxylin was performed, followed by dehydration using graded ethanol to xylene. The slides were mounted with a resin-based medium. Images were captured under the microscope. Immune cell quantification was conducted using QuPath Open Software.
RT-PCR
RT-PCR was carried out using SYBR Green Master Mix in a 20 μL reaction. Gene-specific primers were designed and synthesized by IDT. Below is a list of the primer sequences for the respective genes used for RT-PCR studies in 5′–3′: F—forward (sense primer), R—reverse (antisense primer). Relative gene expression levels were calculated using the ddCt method, with the housekeeping gene (GAPDH) used as an internal control. Each sample was analyzed in triplicate.
| GENES | PRIMER SEQUENCES |
| CD3 | F: GCGTCTGGTGCCTTCTTCAG R: CAATGTTCTCGGCATCGTCCT |
| CD4 | F: TTCTGGCAACCTGACTCTGAC R: ACCCCTCTGGATAAAACCTGGA |
| CD8 | F: ACTTCAGTTCTGTCGTGCCA R: GCAAACACGCTTTCGGCTC |
| F4/80 | F: CCTCTGTGCCTTTGGCTATGG R: TGAAGGTCAGCAACCTCGTG |
Spatial Transcriptomics
Spatial transcriptomics data were generated from four mouse prostate tissue sections using the 10x Genomics Visium platform. For each sample, raw data were processed using Space Ranger. The resulting outs directory, which includes the high-resolution tissue image, was used for downstream analysis in the R environment with Seurat (https://github.com/satijalab/seurat). Spots with low spatial UMI counts (nCount_Spatial <= 1000) were filtered out. The data were then normalized and variance stabilized using SCTransform with the “Spatial” assay. Principal component analysis (PCA) was performed on the SCTransformed data (RunPCA), and the top 50 principal components were used for downstream analyses. Neighborhood graph construction (FindNeighbors) and Louvain clustering (FindClusters with resolution = 2) were completed, followed by Uniform Manifold Approximation and Projection (UMAP) for visualization (RunUMAP).
A previously annotated single-cell RNA sequencing (scRNA-seq) dataset (MoPSA PMID: 39095562) was utilized as a reference for cell type deconvolution. The reference dataset was then downsampled to 1500 cells (subset(x = data, downsample = 1500)) and normalized using SCTransform. To infer cell type composition within each Visium spot, label transfer was performed using the prepared scRNA-seq reference. Anchors between the reference and each Visium query dataset were identified using FindTransferAnchors with the “SCT” normalization method. Cell type probabilities were subsequently transferred to the Visium spots using TransferData. From the prediction scores for each spot, the cell type with the highest probability was assigned as Dominant_type1, while the cell type with the second highest probability was designated as Dominant_type2. These dominant cell type annotations were included in the metadata of each Visium Seurat object. The spatial distribution of clusters and cell types was visualized using SpatialDimPlot. Quality control metrics (nCount_Spatial, nFeature_Spatial) and gene expression were visualized using SpatialFeaturePlot and VlnPlot. Gene set module scores were calculated to assess pathway activity. Epithelial-Mesenchymal Transition (EMT) scores were computed using the AddModuleScore_UCell function with a gene set derived from “HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION.v2023.1.Mm.grp.csv”. Similarly, YAP activity scores were determined using AddModuleScore_UCell with a gene set from “CORDENONSI_YAP_CONSERVED_SIGNATURE.v2023.2.Hs.grp.csv”; human gene symbols in this set were converted to their mouse orthologs using nichenetr::convert_human_to_mouse_symbols prior to scoring. The resulting module scores (e.g., “EMT_UCell”, “YAP_UCell”) were visually represented spatially using SpatialFeaturePlot and across clusters/dominant types using VlnPlot. ImageJ quantification of area was used to quantify specific cell area.
Luciferase Reporter Assay
A 900 bp fragment of human genomic DNA encompassing the YAP-TEAD binding sites in the 6 kb (distal) and 1 kb (proximal) upstream regions of the PDL1 transcription start site was amplified with primers designed to include restriction sites compatible with the multiple cloning site of the pGL4.26 luciferase vector (Promega). The amplified products and the vector were digested with KpnI and BamH1, ligated using T4 DNA ligase (NEB), and the resulting constructs were used to transform E. coli DH5 competent cells. Positive clones were identified by colony PCR. LNCaP, 22RV1, and PC3 cells, expressing YAP-wt or Y407F mutant, were co-transfected with the pGL4.26 containing the cloned regions and a RFP-expressing plasmid as a transfection efficiency control. Control cells were transfected with an empty vector for 24 hours using Lipofectamine LTX. Afterward, cells were lysed and washed with 1x Passive Lysis Buffer (Promega) for 15 minutes at room temperature. Luciferase activity was measured using the Luciferase Assay System (Promega), following the manufacturer’s instructions. RFP fluorescence was also measured using a microplate reader to normalize for transfection efficiency. Luciferase activity was normalized to GFP fluorescence and compared to cells transfected with the empty vector.
Statistical Analysis
All statistical analyses were conducted using GraphPad Prism 10.0. Data quantifications are expressed as mean ± standard deviation (SD). Statistical significance was calculated by a one-way ANOVA followed by Tukey’s post hoc analysis when comparing more than two groups. All p-values < 0.05 and < 0.001 were deemed statistically significant.
Results
TLK1i and ICI Combination Reversed Aggressive PCa Tumor Growth Phenotypes with Improved Immune Signaling in Mice
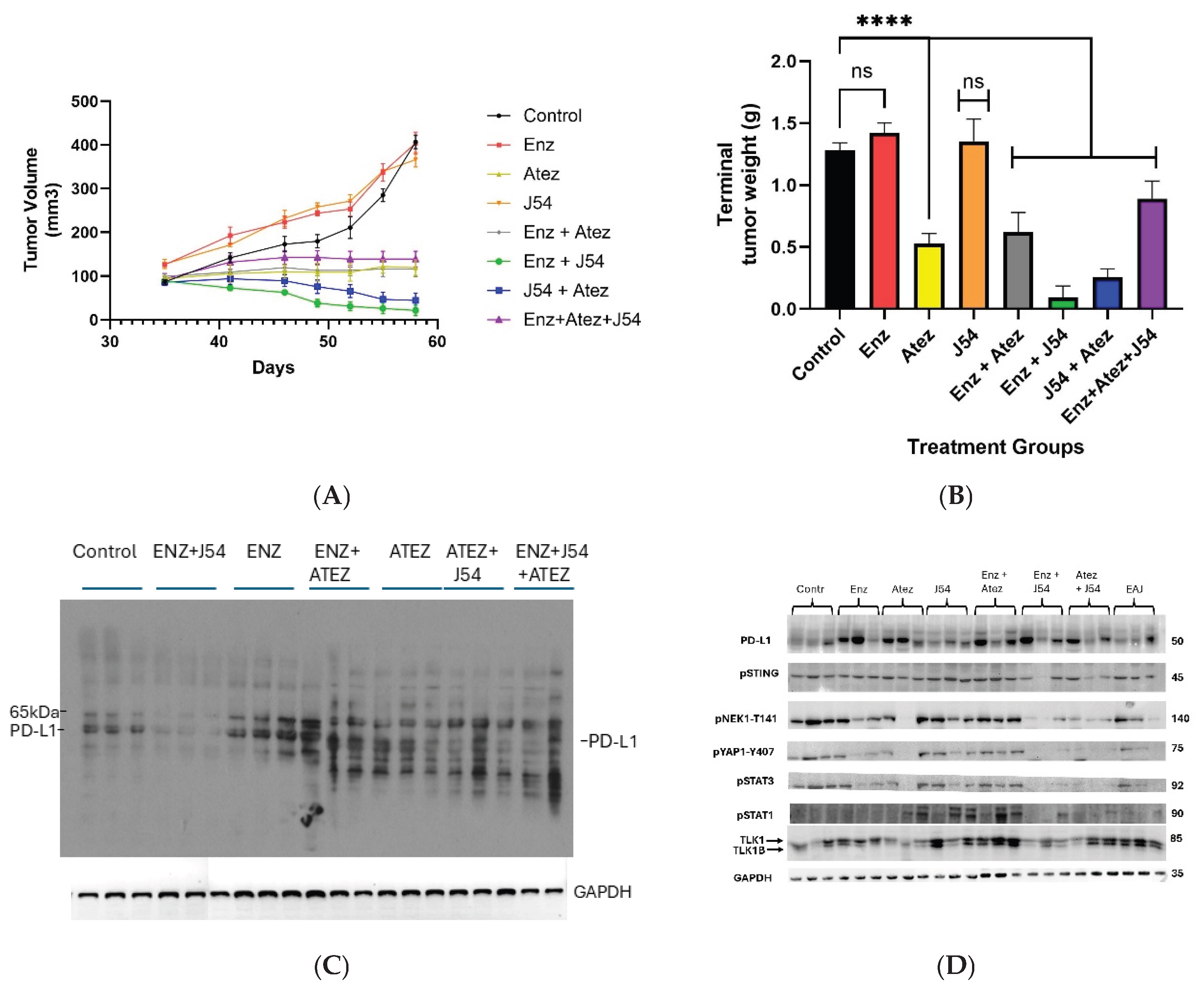
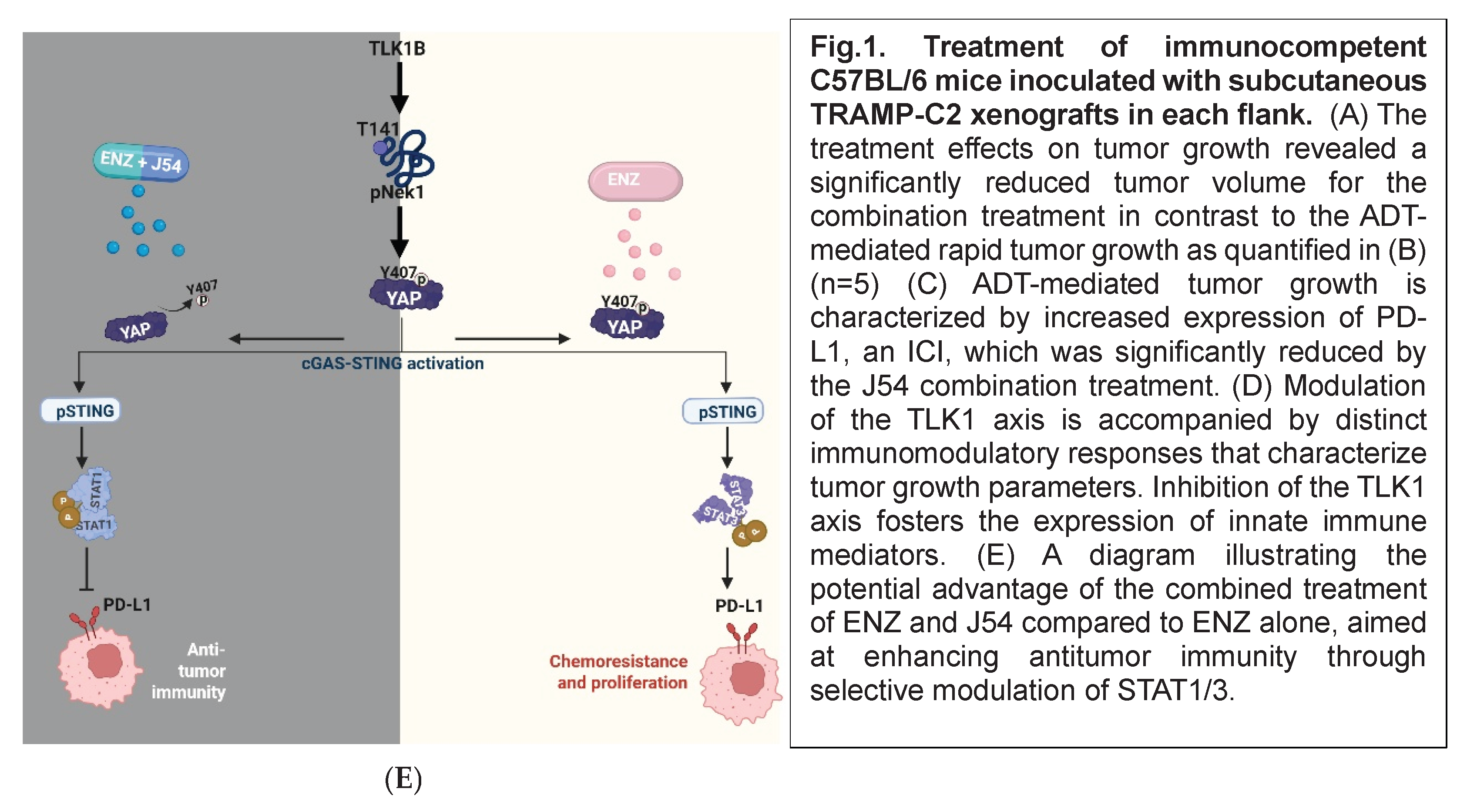
To evaluate the treatment efficacy of ADT, ICIs, and TLK1i alone or in combination, we introduced TRAMP-C2 cell lines subcutaneously into C57BL/6 immunocompetent mice and administered treatment biweekly. Tumor growth measurements taken during the four weeks of treatment revealed a significant reduction in tumor volume for the ENZ+J54 and J54+Atez groups, compared to the progressive increase in tumor volume of the control, ENZ, and J54 groups (Figure 1A). Atez alone, ENZ+Atez, and the ENZ+Atez+J54 treatments exhibited no significant changes. Consistent with tumor volume, immunoblot of the excised tumor from the respective groups showed a significantly reduced expression of PD-L1 (Figure 1B) for the ENZ+J54 and J54+Atez groups in contrast to the elevated PD-L1 expression found in the control and other treatment groups (a slower mobility band may be a PD-L1 ubiquitinated product possibly by SPOP [32], which is one of most mutated genes in human PCa; similarly smaller immunoreactive bands could be proteasome products). ENZ alone (reported to lag C2 tumors growth by ~10 days if injected at 20mg/kg [33]) did not appear to have an impact at the effective dose we tested. Rather we observed the rapid development of resistance to ENZ (CRPC phase due to the previously noted overexpression of PD-L1 after ENZ treatment). The combination of ENZ+J54 (suppressing PD-L1) resulted in the best tumor regression, whereas ATEZ alone or with ENZ had primarily cytostatic effects. The triple combination was no more effective than ENZ + J54, as expected, targeting the same PD-L1 suppression effect. To determine the immunomodulatory effects characterizing the tumor volume and PD-L1 expression dynamics in mice, we performed immunoblot analyses for innate immune mediators in tumors derived from the respective treatment groups. Increased PD-L1 in ENZ-treated groups was associated with elevated levels of TLK1B, pNek1-T141, pYAP1-Y407 with Atez alone, J54 alone and Enz+Atez showing similar expression patterns for these mediators (Figure 1C). In contrast, J54 In combination with Enz/Atez significantly reduced TLK1B, pNek1-T141, pYAP1-Y407 protein expression while J54 + Atez + Enz treatment exhibited mixed effects, potentially due to complex drug interaction that diminish their net positive effect. Notably, similar expression of pSTING was biased towards pSTAT3 expression in control, Enz alone, Atez alone, J54 alone and Enz+Atez treatment groups but not in the J54 + Enz/Atez groups where pSTAT1 was preferentially expressed (Figure 1C), This indicates a suppressive inflammatory response induced by prolonged treatment with ENZ through reciprocal inhibition of YAP/NFKB and ENZ>YAP-mediated suppression of apoptotic activation that typically results in cGAS/STING>pSTAT1, which is unmasked by J54 – Note that the success of ICB in combinatorial testing with different chemotherapeutics has been generally not that great [34], making J54 even more remarkable.
Figure 1.
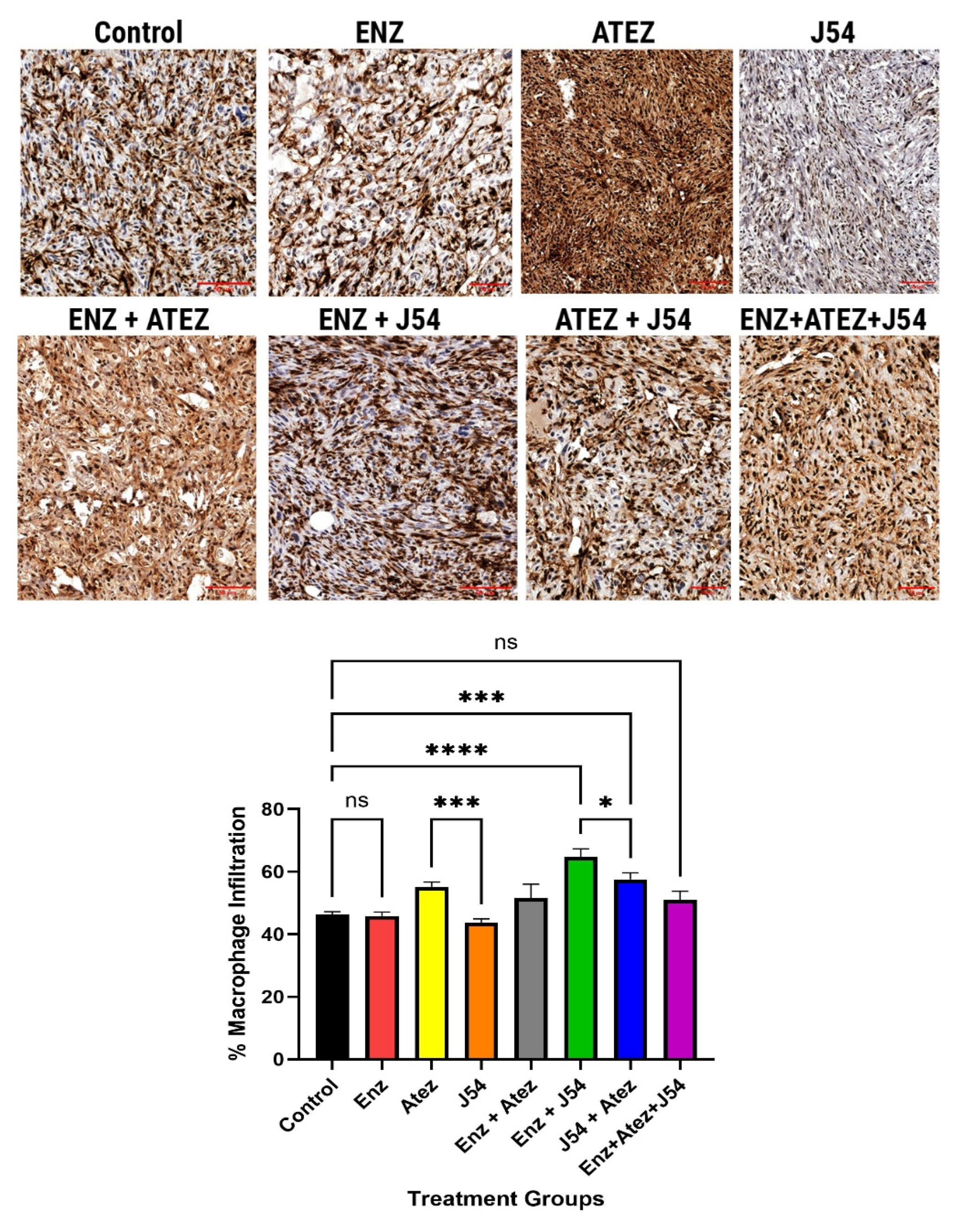
Prostate tumors are generally regarded as immunologically cold due to their low immune cell infiltration, often exemplified by the TRAMP-C2 models, which are well-characterized in terms of their immune properties. To evaluate the implications of differential expression of innate immune mediators on immune cell infiltration in these tumors, we inoculated 10^6 TRAMP-C2 cells into each flank of C57BL/6 male mice. We treated them biweekly with ENZ, ATEZ, and/or J54 over four weeks. We performed immunohistochemical analysis to detect T-cells (CD3) and macrophages (F4-80) in tumor sections. Tumor sections from control and ENZ-alone groups showed sparse T-cell populations (Figure 2A). While the combination treatment of Atez and ENZ yielded a moderate increase in T-cell infiltration, treatment with J54 and ATEZ/ENZ resulted in a significant (p< 0.001) increase in T-cell populations, with relative infiltrations of 35% and 45%, respectively, compared to ENZ treatment alone (Figure 2A,B). Notably, the T-cell infiltration observed with the combination of ENZ and J54 was markedly more significant (p< 0.001) than other treatments, suggesting that J54 in combination with ENZ mediates a more robust mechanism for increased T-cell infiltration. The combination of all three drugs was only mildly effective in enhancing T-cell infiltration. Macrophage infiltration, assessed through F4-80 antibody detection, was significantly (p< 0.001) lower in the control, ENZ, and J54-treated groups (Figure 2C,D). In contrast, drug combinations increased the relative proportion of intratumoral macrophage densities (Figure 2C). Staining quantification of the infiltrated macrophages indicated a significant (p< 0. 001) increase of approximately 65% infiltration in the J54 + ENZ treatment and 59% for the J54 and Atez treatment compared to 35% macrophage infiltration in the control group and those treated with ENZ (Figure 2D). Representative images (Figure 2A,C) displayed dense T- cell and macrophage aggregates in the J54 and ENZ/Atez combinations compared to the sparse populations in the ENZ- treated groups, suggesting an active conversion of a cold tumor to a hot tumor that is densely populated with immune cells. This presents great potential for an improved antitumor immune response; however, the functional activity of the T-cell and macrophage subtypes within the tumor needs further analysis.
Figure 2.
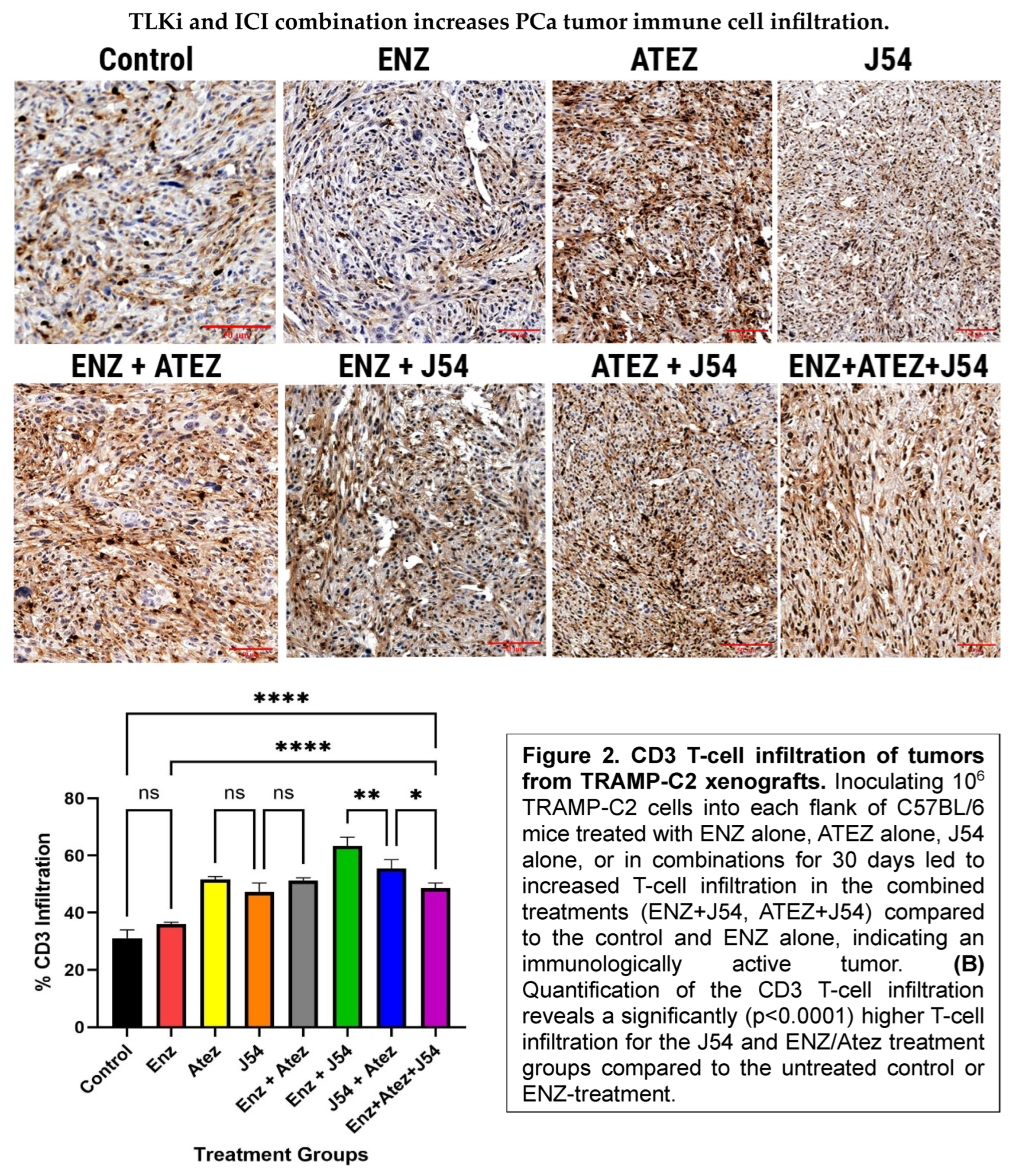
Figure 3.
Macrophage infiltration of tumors from TRAMP-C2 xenografts. Inoculating 106 TRAMP-C2 cells into each flank of C57BL/6 mice treated with ENZ alone, ATEZ alone, J54 alone, or in combinations for 30 days resulted in increased infiltration of macrophages to varying degrees in the respective treatments as detected using F4/80 antibody. (B) Quantification of the F4-80-cell infiltration reveals a significantly (p<0.0001) increased macrophage population for the J54 and ENZ/Atez treatment groups compared to the untreated control or ENZ-treatment.
Figure 3.
Macrophage infiltration of tumors from TRAMP-C2 xenografts. Inoculating 106 TRAMP-C2 cells into each flank of C57BL/6 mice treated with ENZ alone, ATEZ alone, J54 alone, or in combinations for 30 days resulted in increased infiltration of macrophages to varying degrees in the respective treatments as detected using F4/80 antibody. (B) Quantification of the F4-80-cell infiltration reveals a significantly (p<0.0001) increased macrophage population for the J54 and ENZ/Atez treatment groups compared to the untreated control or ENZ-treatment.
TLKi and ICI Combination Modulates Circulating Cytokine Expression for Tumor Regression
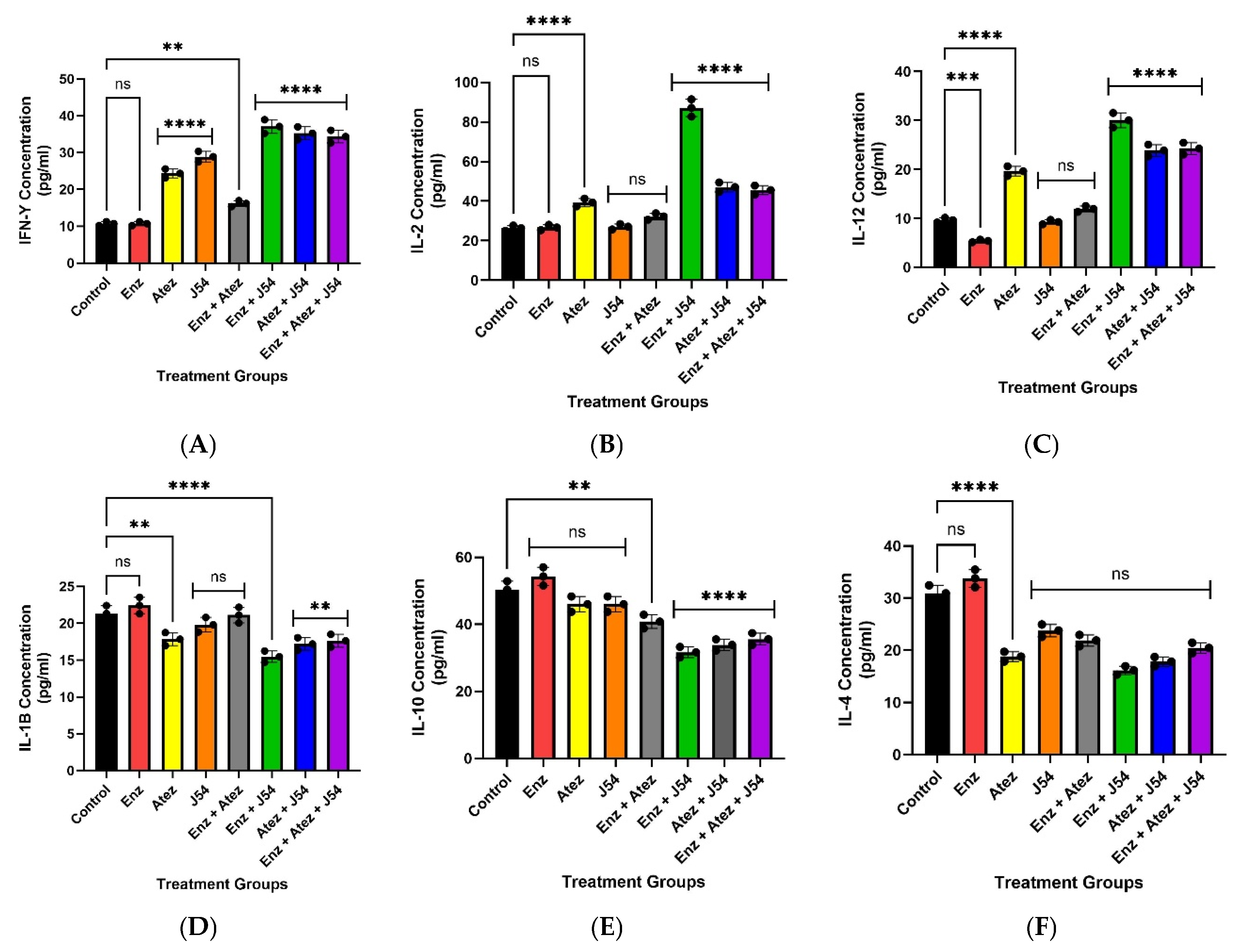
To assess the levels of inflammatory response associated with tumor growth and immune response, we quantified the levels of pro-inflammatory (IL-6, IL-10, IL-4, and IL-1β ) and anti-inflammatory (IFN-γ, IL-2, and IL-12) cytokines in circulating blood samples collected at the end of the study. The circulating levels of anti-inflammatory cytokines (IFN-γ, IL-2, and IL-12), which were reduced in control, ENZ, and ENZ + Atez treatments, significantly increased (p<0.001) with the J54 and ENZ/Atez treatments (Figure 4). Conversely, the J54 and ENZ/Atez treatments significantly reduced the expression of the anti-inflammatory cytokines IL-4, IL10[35] compared to the control and ENZ groups (Figure 4). No other particular trend was observed for the other cytokines. This suggests a possible cytokine-mediated regulation of immune homeostasis that supports tumor suppression or regression. Specifically, these data demonstrate J54 and ENZ/Atez-mediated shift towards pro-inflammatory cytokine profiles and reduced anti-inflammatory pathways that inhibit tumor regrowth or recurrence.
Figure 4.
Treatment-induced expression of cytokines. The J54 combination treatment increases the expression of pro-inflammatory cytokines (IFN-γ, IL-12, and IL-2), which are reduced by ADT. In contrast, the increased expression of pro-resolving cytokines, IL-10 and IL-4, which are immunosuppressive, induced by ADT, was significantly reduced by J54 combination treatment. Data represent mean±SE.
Figure 4.
Treatment-induced expression of cytokines. The J54 combination treatment increases the expression of pro-inflammatory cytokines (IFN-γ, IL-12, and IL-2), which are reduced by ADT. In contrast, the increased expression of pro-resolving cytokines, IL-10 and IL-4, which are immunosuppressive, induced by ADT, was significantly reduced by J54 combination treatment. Data represent mean±SE.
Identification of qPCR-Based Immune Cell Phenotyping in the Blood of Tumor-Bearing Mice
Due to the scarce white cells component of the mice in the different treatment group for characterization by the standard FACS methods, we set out to address this issue via qRT-PCR of total RNA isolated from the blood cell pellets in order to assess particularly the mean levels of circulating T-cells and establish if there is an overt increase in tumor-attacking leukocytes. Although such a method was established before [36], we wanted to validate it for our study by comparing whether the results based on IHC of tumor slices could be matched by an analysis of CD3-expressing cells at the transcript level from total RNA isolated from a small amount (~5mg) of tumor tissue. As shown in Figure 5, the same treatment groups that displayed the highest infiltration of T-cells by IHC also displayed the highest presence of CD3-encoding mRNA, similar to a collective sc identification.
Figure 5.
Determination of infiltrating CD3 cells in tumors via qRT-PCR.
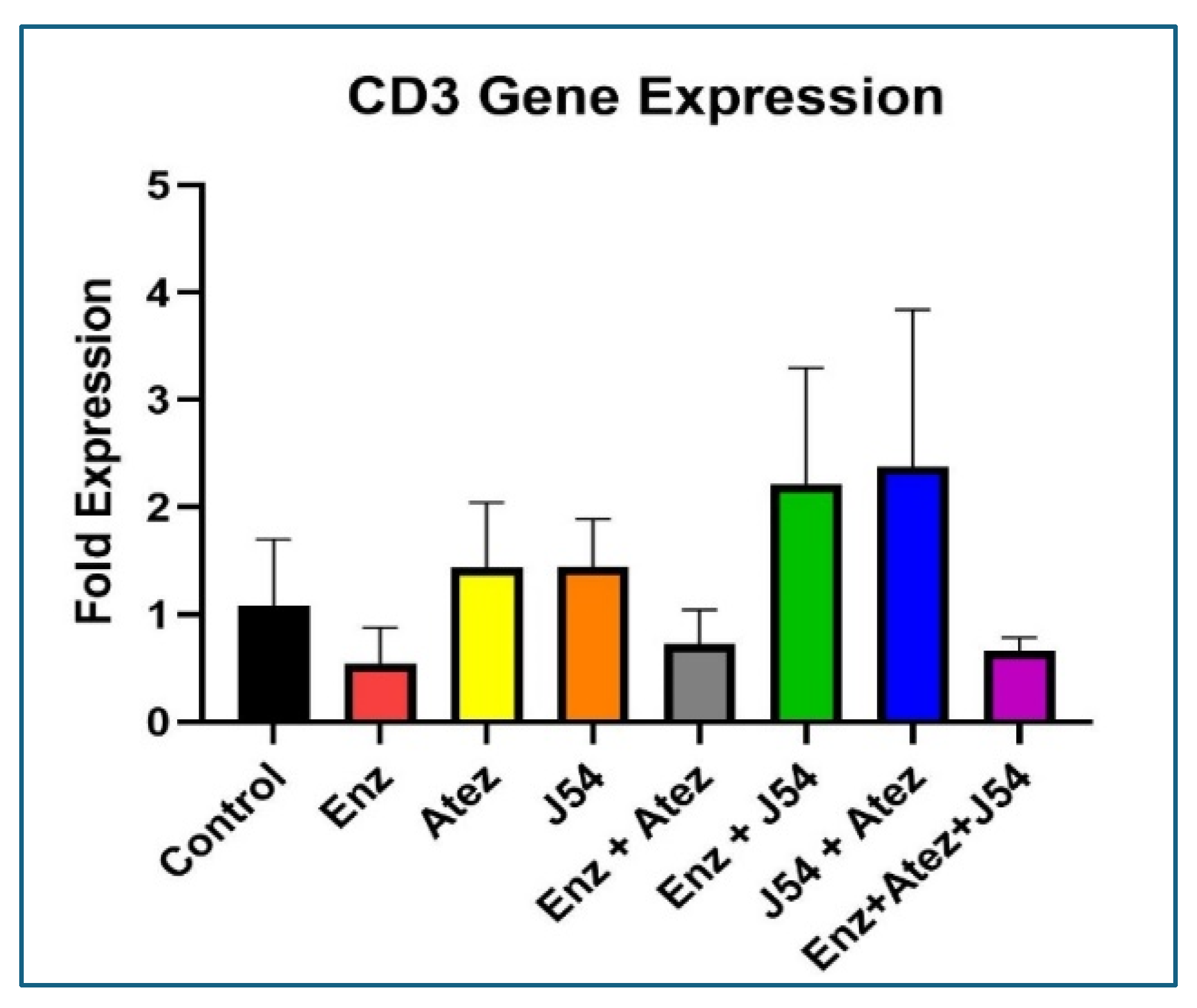
Figure 6.
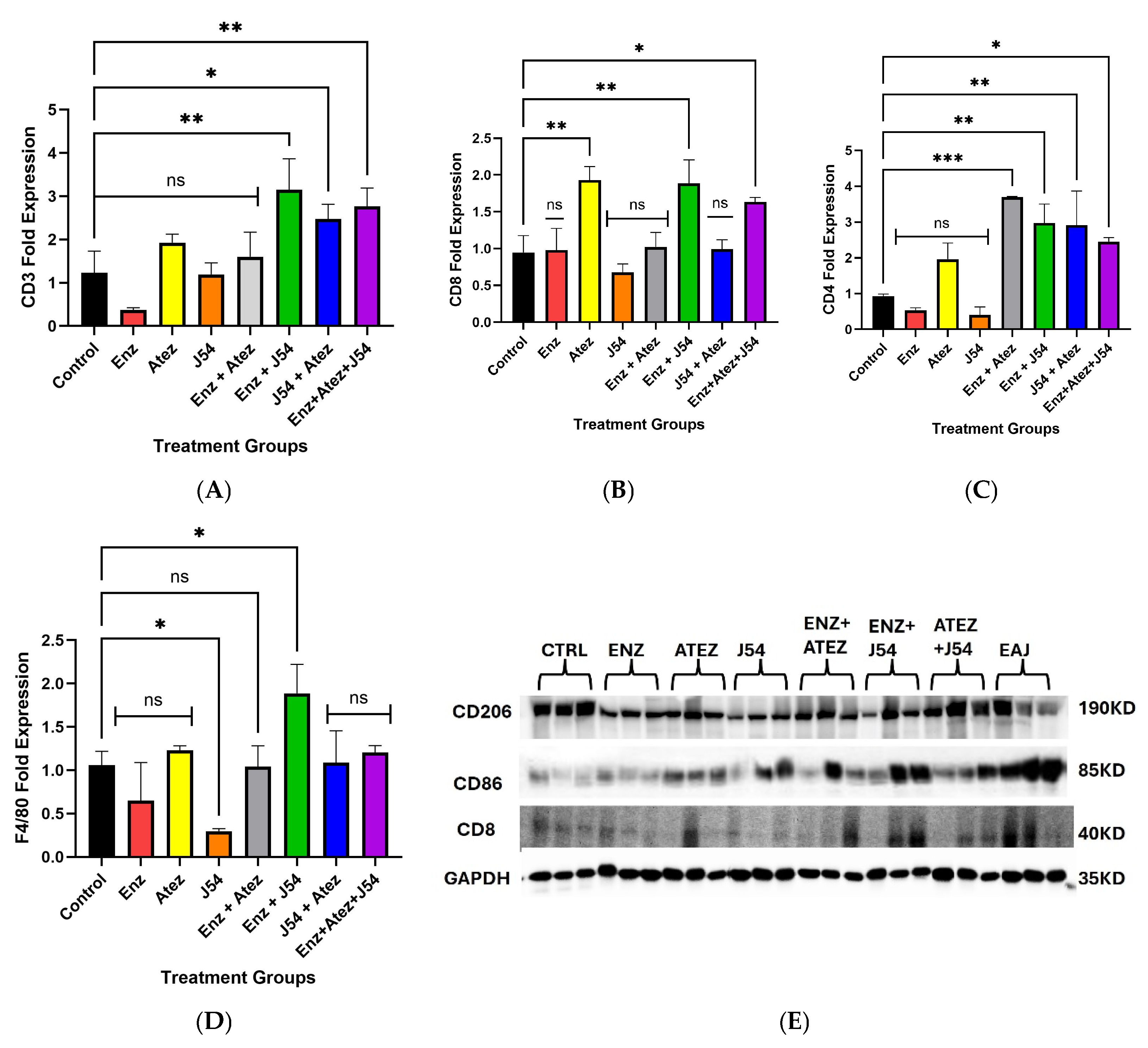
Assessment of circulating leukocytes in tumor-bearing mice from different treatment groups revealed changes in CD3, CD4, CD8, and F4/80 mRNA, as determined by qRT-PCR. Western blot analysis demonstrated treatment-induced variations in markers for intratumoral immune cell subsets. (A) Combination treatments showed increased levels of CD3 mRNA compared to ADT and single treatments, indicating a potential rise in circulating T-cell markers. (B) Atezolizumab, combined with ENZ and J54 treatments, as well as the triple treatment combinations, induced CD8 gene expression, suggesting an enhanced presence or activation of CD8 T-cells. (C) The upregulation of CD4 gene expression implies an increased presence of circulating CD4+ T-cells following combination treatments. (D) qRT-PCR revealed elevated expression of F4/80 after treatment with the ENZ and J54 combination, reflecting enhanced signatures associated with circulating macrophages. (E) Combined treatments exhibited increased expression of the antitumoral macrophages (M1) marker (CD86) alongside a corresponding rise in CD8 markers, in contrast to the low expression seen in the control and ADT mice groups, where the tumor-associated macrophage (M2) marker (CD206) is more prominent. Data represent mean ± SD from n=3 replicates.
Figure 6.
Assessment of circulating leukocytes in tumor-bearing mice from different treatment groups revealed changes in CD3, CD4, CD8, and F4/80 mRNA, as determined by qRT-PCR. Western blot analysis demonstrated treatment-induced variations in markers for intratumoral immune cell subsets. (A) Combination treatments showed increased levels of CD3 mRNA compared to ADT and single treatments, indicating a potential rise in circulating T-cell markers. (B) Atezolizumab, combined with ENZ and J54 treatments, as well as the triple treatment combinations, induced CD8 gene expression, suggesting an enhanced presence or activation of CD8 T-cells. (C) The upregulation of CD4 gene expression implies an increased presence of circulating CD4+ T-cells following combination treatments. (D) qRT-PCR revealed elevated expression of F4/80 after treatment with the ENZ and J54 combination, reflecting enhanced signatures associated with circulating macrophages. (E) Combined treatments exhibited increased expression of the antitumoral macrophages (M1) marker (CD86) alongside a corresponding rise in CD8 markers, in contrast to the low expression seen in the control and ADT mice groups, where the tumor-associated macrophage (M2) marker (CD206) is more prominent. Data represent mean ± SD from n=3 replicates.
Mapping the Immune Infiltrate Component in the TRAMPxNek1+/- PCa Model
We have previously reported preliminary observations that TRAMP mice haploinsufficient for Nek1, even at sexual maturity and beyond (24 w of age) usually do not progress beyond PIN and into invasive PRAD, but rarely, if castrated, develop NEPC more readily than wt-TRAMP (castrated or not) at 24 w [13]. We suggested with the inclusion of IHCs that these processes (or lack thereof) in the Nek+/- mice seem to depend on pYAP1-Y407 and its now demonstrated stabilization[13,14]. Indeed, IHC for chromogranin in aged+castrated mice, only in this group, occasionally displays more than minimal, sporadic staining (SI-file1 and summarized in Table 1). We have also previously reported that YAP1 loss in the TRAMP lesions correlates with the development of NEPC features at this age [37].

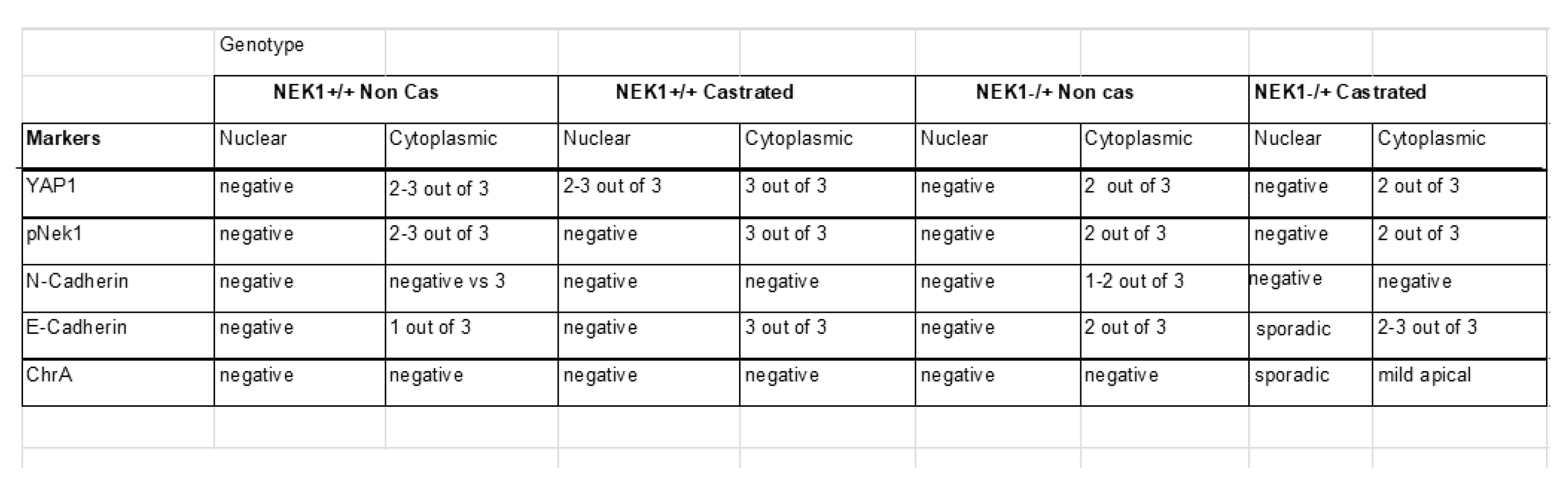
VISIUM Spatial Analysis of FFPE Sections in the TRAMP/Nek1 Haploinsufficient Model
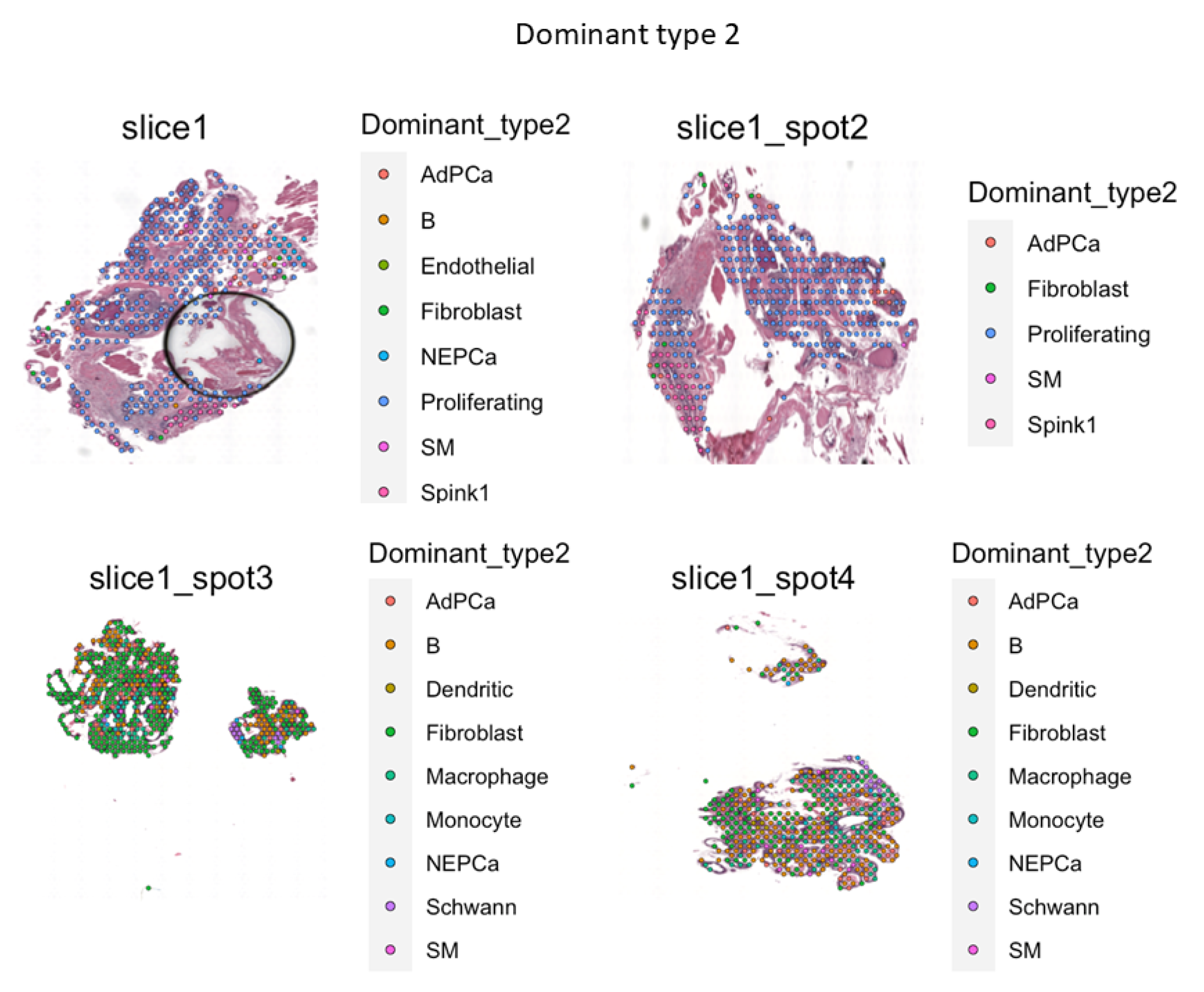
In order to study these features more accurately at the single-cell transcriptional level, we employed the use of VISIUM on a pair of randomly selected samples from the ‘rare’ PRAD that were identified via IHC in the castrated TRAMPxNek1+/- sections, compared to the matched Nek1+/+ animals. Two key sets of data were of particular interest to us from these specimens: 1) the proportion of infiltrating immune cells; 2) the proportion of putative NEPC cells in those sections (Figure 7 and SI-File S2). We also aimed to solidify the hypothesis that Nek1 is critical for PCa progression in TRAMP via upregulation of YAP, and more so after castration because of increased TLK1B expression [38], all this by generating TRAMP/Nek1kat-2j mice. We earlier reported that while intact TRAMP/Nek1+/- mice develop PCa rather normally with much enlargement of the prostate at 18-20 weeks, if mice are castrated, the tumors usually fail to grow [13]. IHC of prostates from castrated TRAMP/Nek1+/- was unable to reveal bona fide tumor areas (only hyperplasia/PIN), and pNek1 and YAP1 staining was strongly reduced while EMT markers were curtailed [13]. Nek1 is essential for YAP1 accumulation [9,14], CRPC progress [39,40,41], and immune evasion[18] but its expression is decreased during (often therapy-driven) NEPC transdifferentiation [37,42]. TRAMP mice haploinsufficient for Nek1 revealed a greatly reduced YAP1 expression and YAP and EMT genes signatures (SI-file 2-3). We suggested with the inclusion of IHCs that these processes (or lack thereof) in the Nek+/- mice seem to depend on pYAP1-Y407 and its now demonstrated stabilization[13,14]. Very notable is significant proportion of B cells, dendritic, and macrophages in the TME of castrated TRAMPxNek1+/- sections. The high importance of a considerable infiltration of B cells in the TME and in attacking the tumor cells has been often downplayed [43]. The difference in the volumes of these prostate adenocarcinomas from matching random sets of castrated animals is also quite noticeable as is number of proliferating PRAD cells based primarily on PCNA and Ki67 expression, with the TRAMP-wt exhibiting large tumors and the Nek1+/- presenting much smaller ones. Additionally, the increased presence of NEPC cells and ‘fibroblasts tissue replacement’ in the Nek1 haploinsufficient mice is very noticeable, as they display reduced YAP1 expression.
Figure 7.
Spatial rendering of main cells in castrated TRAMP-wt (top slices) and Nek1+/-(bottom slices).
Figure 7.
Spatial rendering of main cells in castrated TRAMP-wt (top slices) and Nek1+/-(bottom slices).
The PD-L1 OE After Activation by the TLK1>NEK1>YAP Axis May Be a General Feature of EMT Transition and CRPC Progression
We previously described in details how ultimately the phosphorylation of YAP-Y407 by NEK1 leads to its nuclear retention and activation of several of its target genes[13], progression to EMT and AI growth, and provisionally (but critically) also PD-L1[14]; a finding recently confirmed by another group[44]. However, a mechanistic/transcriptional confirmation is only beginning to emerge. In Figure 8 and Figure 9, we now present a summary of a set of studies, using overexpressed GFP-YAP (wt and Y407F) in the case of LNCaP 22RV1 (which are considered AR+ PRAD) demonstrating its occupancy by ChIP, as is the case for the reported conversion of LNCaP-GFP-YAP to AI-growth [13]cell, but much less for PC3 that are AR- and present features that are NEPC-like; and by enhancing and Luc reporters that this is likely a general phenomenon.
Figure 8.
YAP1 expression correlates with PD-L1 expression and enhancer occupancy. A) Western blot analysis shows that YAP1-wt expression is linked to increased PD-L1 protein levels in treatment-resistant prostate cancer (PCa) cell lines (22RV1), which typically express AR variants. B) Hi-C chromatin conformation data reveal a spatial interaction between the PD-L1 promoter and the enhancer region containing two high-affinity YAP1-TEAD binding motifs, with the interaction becoming stronger as PCa is featured as AR+ adenocarcinoma (LNCaP[PRAD] vs. PC3[~NEPC]. C & D) Distal YAP1-TEAD is involved in Enhancer-promoter looping to facilitate PDL1 transcription. Chip-PCR confirms increased enrichment of YAP1-TEAD at the distal enhancer region in cells (LNCaP and 22RV1) overexpressing YAP1-wt, but not in cells expressing the mutant YAP1, supporting a model in which YAP1-Y407 phosphorylation drives PD-L1 expression via direct enhancer binding, particularly at the distal enhancer.
Figure 8.
YAP1 expression correlates with PD-L1 expression and enhancer occupancy. A) Western blot analysis shows that YAP1-wt expression is linked to increased PD-L1 protein levels in treatment-resistant prostate cancer (PCa) cell lines (22RV1), which typically express AR variants. B) Hi-C chromatin conformation data reveal a spatial interaction between the PD-L1 promoter and the enhancer region containing two high-affinity YAP1-TEAD binding motifs, with the interaction becoming stronger as PCa is featured as AR+ adenocarcinoma (LNCaP[PRAD] vs. PC3[~NEPC]. C & D) Distal YAP1-TEAD is involved in Enhancer-promoter looping to facilitate PDL1 transcription. Chip-PCR confirms increased enrichment of YAP1-TEAD at the distal enhancer region in cells (LNCaP and 22RV1) overexpressing YAP1-wt, but not in cells expressing the mutant YAP1, supporting a model in which YAP1-Y407 phosphorylation drives PD-L1 expression via direct enhancer binding, particularly at the distal enhancer.
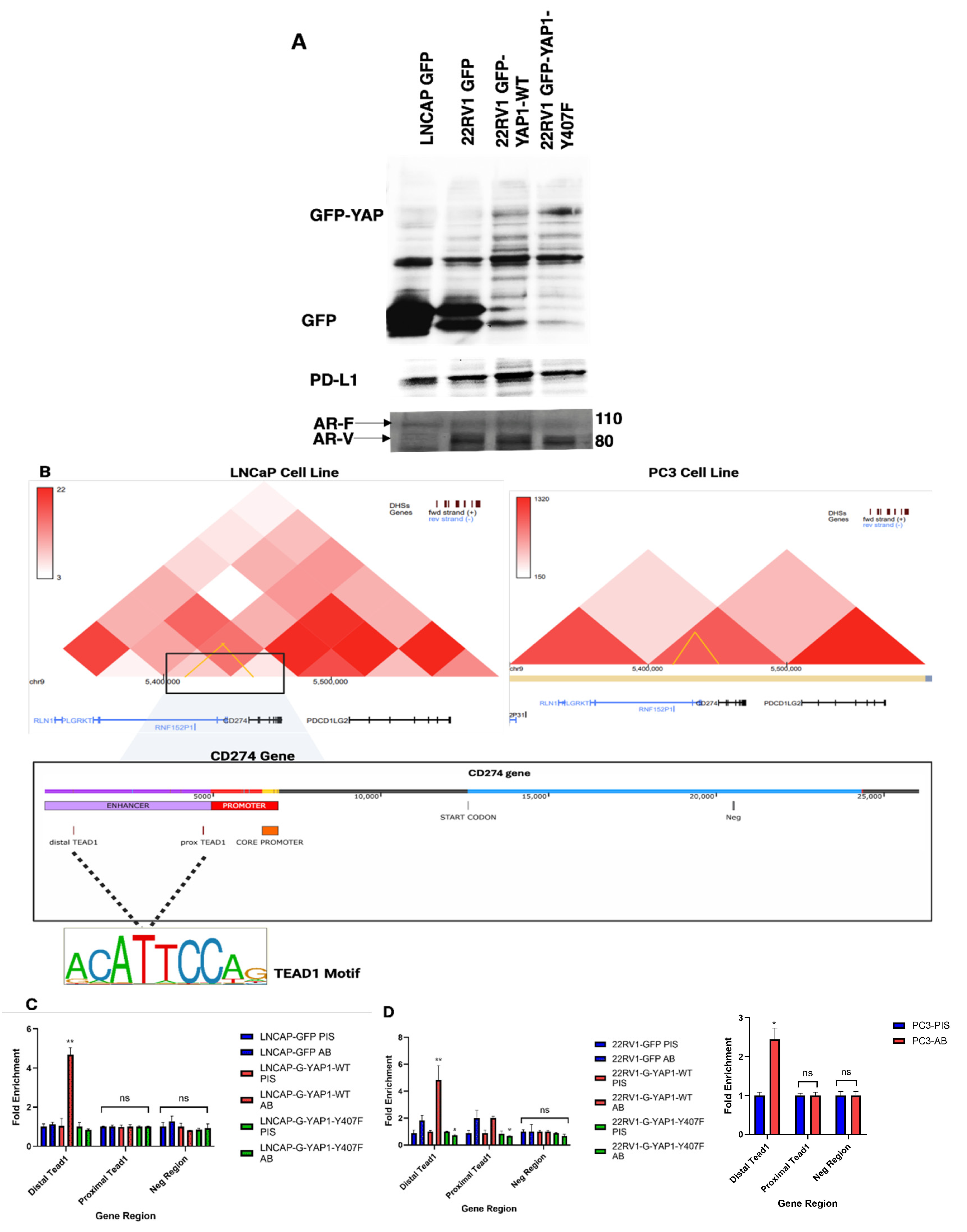
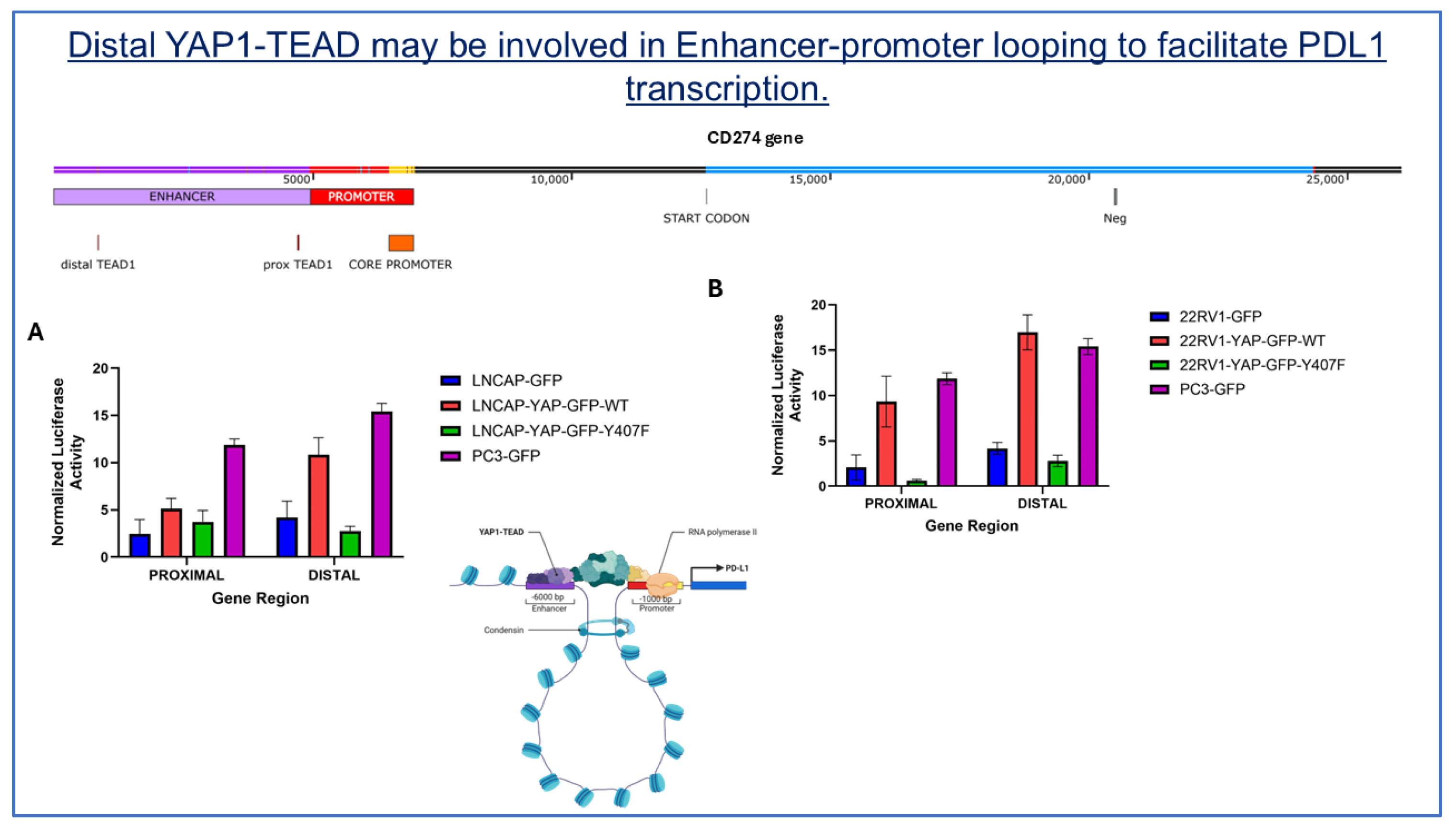
Figure 9.
The distal YAP1-TEAD enhancer region is a key regulator of PDL1 transcription. A luciferase reporter assay compares the proximal and distal YAP1-TEAD regions in the wild-type, mutant, and parental constructs of (A) LNCaP and (B) 22RV1 cells. Cells were co-transfected with a luciferase reporter plasmid (pGL4.26) containing either the distal or proximal regions of the YAP1-TEAD regions of the PDL1 gene, along with a RFP normalization plasmid. Control cells were transfected with an empty vector. Luciferase activity was measured after 24 hours and normalized to GFP fluorescence. Bars represent the mean ± SEM of experiments performed in triplicate.
Figure 9.
The distal YAP1-TEAD enhancer region is a key regulator of PDL1 transcription. A luciferase reporter assay compares the proximal and distal YAP1-TEAD regions in the wild-type, mutant, and parental constructs of (A) LNCaP and (B) 22RV1 cells. Cells were co-transfected with a luciferase reporter plasmid (pGL4.26) containing either the distal or proximal regions of the YAP1-TEAD regions of the PDL1 gene, along with a RFP normalization plasmid. Control cells were transfected with an empty vector. Luciferase activity was measured after 24 hours and normalized to GFP fluorescence. Bars represent the mean ± SEM of experiments performed in triplicate.
Discussion
Our study demonstrates that the combination of J54 and ENZ exerts the most potent anti-tumor effects, characterized by a significant reduction in tumor volume, enhanced immune cell infiltration, favorable shifts in the tumor immune microenvironment, and increased levels of anti-inflammatory cytokines. These findings provide compelling insights into the therapeutic benefits of this combination regimen and predict a complementary mechanism that appears to have improved immunomodulatory functions.
The observed suppression of tumor growth in the J54 and ENZ combination treatment groups, which outweighs that of either the drug alone or the control group, suggests a synergistic interaction between J54 and ENZ. This is particularly significant given that either drug alone worsens tumor growth, while Atez was primarily cytostatic when used alone or combined with ENZ, perhaps due to the activation of compensatory resistance mechanisms that drive tumor growth in each case. J54 + Atez also proved to be effective against tumor volume, while the combination of the three drugs was cytostatic, an effect that could have resulted from a complicated drug-drug interaction. The significant tumor regression observed with J54 and ENZ/Atez may reflect complementary modes of action whereby J54 exerts a cytotoxic effect via increased apoptosis and aids the other drugs in priming the tumor immune microenvironment to facilitate sustained immune-mediated control of tumor growth. The variability in the effects of J54 + ENZ and J54 + Atez suggests that there is an additional mechanism beyond the modulation of PD-L1 expression that is mediated by the J54 + ENZ, which accounts for the improved effect.
The expression of TLK1B is regulated translationally via the mTOR>4EBP pathway, most commonly in response to survival ‘emergency’ situations. It is conceivable that ICB treatment, which unmasks the existence of the tumor and makes it accessible to immune attack, could be one such situation for the cancer cells. Conversely, the concomitant inhibition of TLK1/1B (a mediator of survival response) would be expected to elicit a high-level apoptotic response, resulting in a reduction of tumor mass and cells, and leading to the preferential loss of high turnover proteins like TLK1B. Direct evidence that tumors become immunologically hot is provided through IHC determination of infiltrating immune cells and the detection of specific cytokines present in the plasma.
Interestingly, a central finding of this study is the increased infiltration of immune effector cells – particularly T-cells and macrophages – into the tumor mass following combination treatment. The components of the tumor microenvironment, including a dense stroma, immunosuppressive myeloid-derived suppressor cells, and T regulatory cells (Tregs), contribute to the scarcity of intratumoral immune cells. Effective therapies often aim to enhance immune cell infiltration, thereby facilitating immune-mediated control of tumor growth. IHC revealed both elevated immune cell density and percentage infiltration of T-cells and macrophages with J54 and ENZ/Atez treatment, a hallmark of improved tumor immunogenicity. This can be interpreted as converting the originally cold tumor into a hot one, making it susceptible to immune cell-mediated control of tumor growth. This J54 combination therapy overcame immune exclusion or stromal barriers often associated with immunologically “cold” tumors. Macrophages present within the tumor microenvironment can be either proinflammatory, tumoricidal M1 macrophages or the immunosuppressive, tumor-promoting M2 subtypes. Beyond enhancing the quantity of T-cell infiltrates, our combination therapies also appear to target the M1/M2 reprogramming without significantly affecting the number of intratumoral macrophages. This likely transforms the tumor-associated macrophages into allies of the immune system, facilitating an immune-controlled attack on the tumor. The increased immune cell infiltration correlates with significant tumor regression, suggesting that these cells may exert cytotoxic mechanisms that prevent tumor growth.
Mechanistically, PD- L 1 reduction in the J54 and ENZ/Atez groups, compared to other groups, correlates with reduced flux through the TLK 1> Nek 1> YAP 1 axis and is characterized by increased expression of STAT 1 but not STAT 3. The balance of STAT 1 and STAT 3 involves shaping tumor immune responses toward immunosuppression or an antitumor response. The increased STAT 1 in the J54 and ENZ/Atez treatment groups, which correlates with tumor regression, suggests the initiation of signaling that enhances cytotoxic function and effector T-cell activity, likely resulting from both direct tumor cell priming and the reversal of immunosuppressive signals within the tumor environment. Hence, reducing the ICI molecule – PD- L 1 facilitates immune surveillance. In contrast, STAT 3, which is highly expressed in control and monotherapy groups, correlates with increased PD-L1, likely activating immunosuppressive signals or creating a pro-tumorigenic environment that drives tumor growth. Interestingly, the combination treatment also led to elevated expression of pro-inflammatory cytokines such as IFN- γ, IL- 2, and IL- 12, which were predicted to rise by analogy to the VCaP model after observing the profound increase in pSTAT 1, without the concomitant increase in antinflammatory cytokines, especially IL- 10 and IL- 4, which are crucial for immunosuppression. Furthermore, PD- L 1 expression is known to be regulated by INFᵞ and IL- 2 through STAT 1 and STAT 3 signaling [27]. Over the past two decades, interleukin- 12 (IL- 12) has emerged as one of the most potent cytokines in mediating antitumor activity in various preclinical models [45]. In contrast, the fact that IL-10 doesn’t appear to change with the various treatments of interest is actually more significant than it is often credited for, as it typically limits inflammation [46]. This observation suggests that J54 and ENZ/Atez promote the resolution of inflammation while enhancing antigen presentation. This also correlates limiting tumor-promoting inflammation with improved cytotoxic responses. Perhaps the reduction in IL- 10, which normally favors T- regulatory cells, accompanied by the increased IL- 12 and IFN- γ, may serve as a regulatory role that promotes immune-mediated control and sustains effective anti-tumor immunity.
The TC2/C57Bl6 was the simplest syngeneic mouse PCa syngeneic model available to us for rapid analysis, but we wanted to expand on its generality and utility with a more genetically tractable with the inclusion of our previously developed TRAMPxNek1+/- GEMM. In this model, the normally increased TLK1B expression after castration or ARSI and typically leading to the activation of NEK1>YAP stabilization axis, is curtailed due to Nek1 haploinsufficiency, and indeed the expression of YAP was demonstrated to be strongly reduced (Table 1 and [6]). The consequence of this was not fully appreciated in that report, although we had observed the significant decrease in frequency of overt adenocarcinomas of their prostates (PRAD) and the better life expectancy of those mice (beyond 30 weeks of age – endpoint when we sacrificed all animals to investigate their prostates). As shown in Figure 7 for the few animals (~15%) in which PRAD was confirmed, the tumors volumes were noticeable smaller, and the subsequent FFPE VISUM sections of two randomly selected mice, vs control (also castrated) wt-TRAMP mice, displayed a vastly different map of the main cell types present in the lesions, with a much stronger presence of various immune cells infiltrates in the TME and much reduced representation of PRAD cells. It is difficult to escape the general conclusion that the TLK1B.NEK1.YAP axis is of critical importance for development and progression of PRAD after castration, although not to NEPC.
Conclusions
The combined effects of J54 and ENZ/Atez on tumor shrinkage, immune infiltration, and anti-inflammatory cytokine expression could be therapeutically beneficial for single-agent immunotherapies for PCa. Its dual action on tumor intrinsic signals and the tumor microenvironment is critical for overcoming immune resistance to achieve a robust response. In the context of immune cold tumors (most PCa), the ability of this combination therapy to enhance immune cell infiltration without inducing systemic cytokine toxicity may be highly beneficial for improving PCa patient survival.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Ethics approval and consent to participate
The study does not include human participants. Institutional Review Board Statement: The animal study protocol was approved by the Institutional ACUC – protocols P-20-24 TARGETING THE TLK1/NEK1 AXIS IN PROSTATE CANCE, and S-24-004 COMBINING ANTIANDROGEN+J54 WITH AVELUMAB FOR PCA TREATMENT. Approved 5-29-2024, for studies involving animals.
Consent of publication
All authors have approved the manuscript.
Availability of Data and material
Description of all data and materials can be found in the referenced article. No additional data has been withheld from the public. Plasmids and cell lines will be made available upon request to all qualified investigators.
Competing Interest
Authors declare no conflicts of interest. A patent: Targeting the TLK1/NEK1 axis in prostate cancer”: United States Patent, Patent Application No.: US16/834,061 that includes J54 was awarded 07-24-2023 to LSUHS on behalf of ADB, but there are currently no licensing agreements.
Funding
This work was supported by a DoD-PCRP grant W81XWH-17-1-0417 and grants from Feist-Weiller Cancer Center (FWCC) of LSU Health Shreveport to ADB.
Author Contributions
Conceptualization by A.D.B. and D.M.O, manuscript writing: A.D.B., D.M.O., O.C.F, and X.Y.; methodology: A.D.B, O.F. and D.M.O.: experiment analysis and interpretation: A.D.B., D.M.O., O.C.F, and X.Y.: support for the VISUM and IHC analysis; A.D.B., O.C.F, and X.Y.: acquisition of resources for the project.
Acknowledgments
We would like to thank the INLET facility of LSU Health Shreveport, especially Camille Canon, for expert assistance preparation of the IVIS libraries. We also would like to thank the Research Core Facility Genomics Core and animal facility of LSU Health Shreveport for help with the qPCR analysis and animal work. A particular thanks to Siyuan Cheng for developing algorithms in R, expert Bioinformatics analysis, interpretation and visualization of theVISIUM datasets.
References
- Chen, Y.; Clegg, N.J.; Scher, H.I. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol 2009, 10, 981–991. [Google Scholar] [CrossRef]
- Chism, D.D.; De Silva, D.; Whang, Y.E. Mechanisms of acquired resistance to androgen receptor targeting drugs in castration-resistant prostate cancer. Expert Rev Anticancer Ther 2014, 14, 1369–1378. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Salem, O.; Hansen, C.G. The Hippo Pathway in Prostate Cancer. Cells 2019, 8. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. International journal of cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer letters 2019, 453, 131–141. [Google Scholar] [CrossRef]
- Liu, S.; Ho, C.K.; Ouyang, J.; Zou, L. Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc Natl Acad Sci U S A 2013, 110, 2175–2180. [Google Scholar] [CrossRef]
- Wengner, A.M.; Scholz, A.; Haendler, B. Targeting DNA Damage Response in Prostate and Breast Cancer. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Khalil, M.I.; Ghosh, I.; Singh, V.; Chen, J.; Zhu, H.; De Benedetti, A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12, 3666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol Cell Biol 2015, 35, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell 2015, 27, 797–808. [Google Scholar] [CrossRef]
- Singh, V.; Bhoir, S.; Chikhale, R.V.; Hussain, J.; Dwyer, D.; Bryce, R.A.; Kirubakaran, S.; De Benedetti, A. Generation of Phenothiazine with Potent Anti-TLK1 Activity for Prostate Cancer Therapy. iScience 2020, 23, 101474. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Khalil, M.I.; Mirza, R.; King, J.; Olatunde, D.; De Benedetti, A. NEK1-Mediated Phosphorylation of YAP1 Is Key to Prostate Cancer Progression. Biomedicines 2023, 11, 734. [Google Scholar] [CrossRef]
- Olatunde, D.; De Benedetti, A. TLK1>Nek1 Axis Promotes Nuclear Retention and Activation of YAP with Implications for Castration-Resistant Prostate Cancer. Cancers 2024, 16, 2918. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res 2018, 78, 1457–1470. [Google Scholar] [CrossRef]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef]
- Collak, F.K.; Demir, U.; Sagir, F. YAP1 Is Involved in Tumorigenic Properties of Prostate Cancer Cells. Pathol Oncol Res 2020, 26, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, Y.; Yu, H.; Zhao, Y.; Sun, X.; Li, Q.; Wang, Y. The role of YAP1 in survival prediction, immune modulation, and drug response. A pan-cancer perspective. Frontiers in Immunology 2022, 13. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.A.; Park, S.H.; Shin, E.C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol Res 2018, 6, 255–266. [Google Scholar] [CrossRef]
- Miao, J.B.; Hsu, P.C.; Yang, Y.L.; Xu, Z.D.; Dai, Y.Y.; Wang, Y.C.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.T.; Chen, H.J.; Luo, S.M.; Li, L.N.; Zhou, S.J.; Shen, R.F.; Lin, H.; Xie, X.H. The correlation between programmed death-ligand 1 expression and driver gene mutations in NSCLC. Oncotarget 2017, 8, 23517–23528. [Google Scholar] [CrossRef]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front Immunol 2022, 13, 964442. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep 2017, 19, 1189–1201. [Google Scholar] [CrossRef]
- Wang, M.; Ran, X.; Leung, W.; Kawale, A.; Saxena, S.; Ouyang, J.; Patel, P.S.; Dong, Y.; Yin, T.; Shu, J.; et al. ATR inhibition induces synthetic lethality in mismatch repair-deficient cells and augments immunotherapy. Genes Dev 2023, 37, 929–943. [Google Scholar] [CrossRef]
- Zheng, W.; Liu, A.; Xia, N.; Chen, N.; Meurens, F.; Zhu, J. How the Innate Immune DNA Sensing cGAS-STING Pathway Is Involved in Apoptosis. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Singh, V.; Khalil, M.I.; De Benedetti, A. The TLK1/Nek1 axis contributes to mitochondrial integrity and apoptosis prevention via phosphorylation of VDAC1. Cell Cycle 2020, 19, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lu, J.; Li, W.; Wu, A.; Zhang, X.; Tong, W.; Ho, K.K.; Qin, L.; Song, H.; Mak, K.K. Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nature Communications 2018, 9, 4564. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Wang, I.; Song, L.; Wang, B.Y.; Rezazadeh Kalebasty, A.; Uchio, E.; Zi, X. Prostate cancer immunotherapy: a review of recent advancements with novel treatment methods and efficacy. Am J Clin Exp Urol 2022, 10, 210–233. [Google Scholar]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer and Prostatic Diseases 2021, 24, 697–717. [Google Scholar] [CrossRef] [PubMed]
- Olatunde, D.; De Benedetti, A. TLK1>Nek1 Axis Promotes Nuclear Retention and Activation of YAP with Implications for Castration-Resistant Prostate Cancer. Cancers 2024, 16. [Google Scholar] [CrossRef]
- Ding, P.; Ma, Z.; Fan, Y.; Feng, Y.; Shao, C.; Pan, M.; Zhang, Y.; Huang, D.; Han, J.; Hu, Y.; et al. Emerging role of ubiquitination/deubiquitination modification of PD-1/PD-L1 in cancer immunotherapy. Genes Dis 2023, 10, 848–863. [Google Scholar] [CrossRef]
- Cerasuolo, M.; Maccarinelli, F.; Coltrini, D.; Mahmoud, A.M.; Marolda, V.; Ghedini, G.C.; Rezzola, S.; Giacomini, A.; Triggiani, L.; Kostrzewa, M.; et al. Modeling Acquired Resistance to the Second-Generation Androgen Receptor Antagonist Enzalutamide in the TRAMP Model of Prostate Cancer. Cancer Res 2020, 80, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Minn, A.J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef]
- Nikovics, K.; Favier, A.L.; Rocher, M.; Mayinga, C.; Gomez, J.; Dufour-Gaume, F.; Riccobono, D. In Situ Identification of Both IL-4 and IL-10 Cytokine-Receptor Interactions during Tissue Regeneration. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Demmler, R.; Mohamed Abdou, M.; Thoma, O.-M.; Weigmann, B.; Waldner, M.J.; Stürzl, M.; Naschberger, E. Rapid qPCR-based quantitative immune cell phenotyping in mouse tissues. Journal of Investigative Medicine 2024, 72, 47–56. [Google Scholar] [CrossRef]
- Cheng, S.; Prieto-Dominguez, N.; Yang, S.; Connelly, Z.M.; StPierre, S.; Rushing, B.; Watkins, A.; Shi, L.; Lakey, M.; Baiamonte, L.B.; et al. The expression of YAP1 is increased in high-grade prostatic adenocarcinoma but is reduced in neuroendocrine prostate cancer. Prostate Cancer Prostatic Dis 2020, 23, 661–669. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett 2019, 453, 131–141. [Google Scholar] [CrossRef]
- Lee, H.-C.; Ou, C.-H.; Huang, Y.-C.; Hou, P.-C.; Creighton, C.J.; Lin, Y.-S.; Hu, C.-Y.; Lin, S.-C. YAP1 overexpression contributes to the development of enzalutamide resistance by induction of cancer stemness and lipid metabolism in prostate cancer. Oncogene 2021, 40, 2407–2421. [Google Scholar] [CrossRef] [PubMed]
- Cinar, B.; Al-Mathkour, M.M.; Khan, S.A.; Moreno, C.S. Androgen attenuates the inactivating phospho-Ser-127 modification of yes-associated protein 1 (YAP1) and promotes YAP1 nuclear abundance and activity. J Biol Chem 2020, 295, 8550–8559. [Google Scholar] [CrossRef]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nature Communications 2015, 6, 8126. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Matsubara, D.; Tanaka, I.; Makiya, K.; Tanei, Z.I.; Kumagai, Y.; Shiu, S.J.; Nakaoka, H.J.; Ishikawa, S.; Isagawa, T.; et al. Loss of YAP1 defines neuroendocrine differentiation of lung tumors. Cancer Sci 2016, 107, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Largeot, A.; Pagano, G.; Gonder, S.; Moussay, E.; Paggetti, J. The B-side of Cancer Immunity: The Underrated Tune. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Pavinato, L.; Villamor-Payà, M.; Sanchiz-Calvo, M.; Andreoli, C.; Gay, M.; Vilaseca, M.; Arauz-Garofalo, G.; Ciolfi, A.; Bruselles, A.; Pippucci, T.; et al. Functional analysis of TLK2 variants and their proximal interactomes implicates impaired kinase activity and chromatin maintenance defects in their pathogenesis. J Med Genet 2022, 59, 170–179. [Google Scholar] [CrossRef]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; vom Berg, J.; Kulig, P.; Becher, B. New insights into IL-12-mediated tumor suppression. Cell Death & Differentiation 2015, 22, 237–246. [Google Scholar]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol 2012, 32, 23–63. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



