Submitted:
22 May 2025
Posted:
22 May 2025
You are already at the latest version
Abstract
The Inhibitor of DNA-binding (ID) proteins are dominant-negative regulators of basic helix-loop-helix (bHLH) transcription factors, playing a pivotal role in cellular differentiation, proliferation, and lineage plasticity. The Polycomb Repressive Complex 2 (PRC2), through trimethylation of histone H3 at lysine 27 (H3K27me3), mediates long-term transcriptional repression essential for development and cancer. Here, we hypothesize that ID proteins indirectly modulate PRC2 recruitment and function by altering chromatin accessibility and transcription factor occupancy, particularly during transitions between stem-like and differentiated states. We propose a mechanistic model whereby ID proteins prime the chromatin landscape for PRC2 activity, promoting a permissive environment for epigenetic silencing in stem/progenitor populations and tumor cells. This functional support could cause phenotypic plasticity in cancer and offer novel epigenetic targets for therapeutic intervention.
Keywords:
cancer stem cells
; chromatin
; epigenetics
; EZH2
; ID proteins
; PRC2
Introduction
Cellular identity is established and maintained through a complex interplay between transcriptional regulators and epigenetic machinery. This coordinated regulation ensures that developmental genes are activated or repressed in the appropriate spatiotemporal context, enabling stem cell self-renewal, lineage commitment, and terminal differentiation. Disruption of these processes underlies various developmental disorders and contributes to the pathogenesis of cancer. Among the key players in these regulatory networks are the Inhibitor of DNA-binding (ID) protein family and the Polycomb Repressive Complex 2 (PRC2). Despite being mechanistically distinct, both ID proteins and PRC2 are crucial in maintaining the balance between pluripotency and differentiation. Here, we propose a hypothetical model in which ID proteins indirectly modulate PRC2 activity and chromatin occupancy, contributing to lineage plasticity and tumorigenesis.
The ID protein family (ID1–ID4) consists of helix-loop-helix (HLH) transcriptional regulators that lack a DNA-binding domain. These proteins act as dominant-negative inhibitors of basic HLH (bHLH) transcription factors, such as E-proteins (e.g., E2A/TCF3), by forming non-DNA-binding heterodimers.[1,2] This interaction prevents bHLH-mediated transcriptional activation of target genes that are typically involved in cell cycle exit, differentiation, and lineage specification.[3,4] ID proteins are highly expressed in embryonic stem cells (ESCs) and adult progenitor populations, where they help preserve an undifferentiated, proliferative state.[5,6] In the hematopoietic and nervous systems, for example, ID expression marks stem/progenitor compartments and is rapidly downregulated upon differentiation.[7,8] In cancer, ID proteins are often aberrantly expressed and have been implicated in promoting tumor progression, metastasis, and therapeutic resistance. High levels of ID1 and ID3 have been associated with poor prognosis in breast cancer, glioblastoma, and pancreatic cancer, where they promote stem-like properties and plasticity.[9,10,11] ID proteins have also been connected with other disease properties such as vascular and neurovascular.[12,13,14,15] Moreover, ID proteins facilitate epithelial-to-mesenchymal transition (EMT), angiogenesis, and immune evasion—key features of aggressive, treatment-refractory malignancies.[16,17,18]
Parallel to these findings, PRC2 has emerged as a master regulator of epigenetic silencing. As a core Polycomb group (PcG) complex, PRC2 catalyzes the trimethylation of histone H3 at lysine 27 (H3K27me3), a mark that contributes to the repression of developmental and lineage-specific genes.[19,20] The catalytic activity of PRC2 is mediated by EZH2 or its homolog EZH1, in association with other essential subunits such as SUZ12, EED, and RBBP4/7.[21] PRC2 plays a pivotal role in early embryonic development and is required to maintain stem cell identity by repressing genes associated with differentiation.[22,23] In cancer, PRC2 is frequently dysregulated, with EZH2 overexpression or gain-of-function mutations driving epigenetic reprogramming in lymphomas, prostate cancer, and other malignancies.[24,25,26] Interestingly, both ID proteins and PRC2 are co-expressed in various stem-like and tumorigenic contexts. For example, in glioma and breast cancer models, high levels of ID1 and EZH2 are associated with enhanced tumor-initiating capacity and resistance to differentiation cues.[27,28] Despite this co-occurrence, the functional relationship between these two regulatory axes has not been clearly defined. PRC2 does not bind DNA directly but is recruited to chromatin through a combination of accessory proteins, RNA molecules, and chromatin features, such as unmethylated CpG islands and prior histone modifications.[29,30,31] Therefore, the transcriptional and chromatin state of a cell—shaped in part by ID proteins—may critically influence PRC2 recruitment and function.
We hypothesize that ID proteins reshape the chromatin and transcription factor landscape in a way that facilitates PRC2 binding to key regulatory loci. By sequestering bHLH transcription factors, ID proteins not only suppress differentiation-promoting transcriptional programs but may also indirectly prevent recruitment of chromatin remodelers that oppose Polycomb repression (e.g., SWI/SNF complexes).[32,33] This creates a permissive environment for PRC2-mediated gene silencing, stabilizing undifferentiated states and contributing to lineage infidelity in cancer. Such cooperation may represent a convergent mechanism that underlies both normal developmental plasticity and pathological reprogramming in cancer. In this manuscript, we first review the individual functions of ID proteins and PRC2 in developmental and oncogenic contexts. We then describe a hypothetical model in which ID proteins facilitate PRC2 recruitment and stabilization at chromatin targets during lineage specification. Finally, we outline experimental approaches to test this model and explore its potential implications in cancer biology, regenerative medicine, and epigenetic therapy.
Hypothesis with Rationale:
We hypothesize that ID proteins modulate PRC2 activity indirectly by reshaping chromatin accessibility and transcription factor binding at lineage-specific genes, thereby facilitating PRC2 recruitment and stabilization at target loci. This support is especially critical in maintaining stem-like states and enabling phenotypic plasticity in cancer. The regulatory mechanisms that maintain cellular plasticity and prevent premature differentiation are essential in development and frequently co-opted in tumorigenesis. ID proteins and the Polycomb Repressive Complex 2 (PRC2) play central, notwithstanding mechanistically distinct roles in this process. Here, we outline evidence supporting a hypothetical model in which ID proteins prime the chromatin landscape to facilitate PRC2-mediated silencing of differentiation-associated genes. This interaction could reinforce both normal stem cell behavior and pathological states such as cancer.
Figure 1.
Hypothetical model with ID proteins, bHLH transcription factors, PRC2, and H3K27me3.
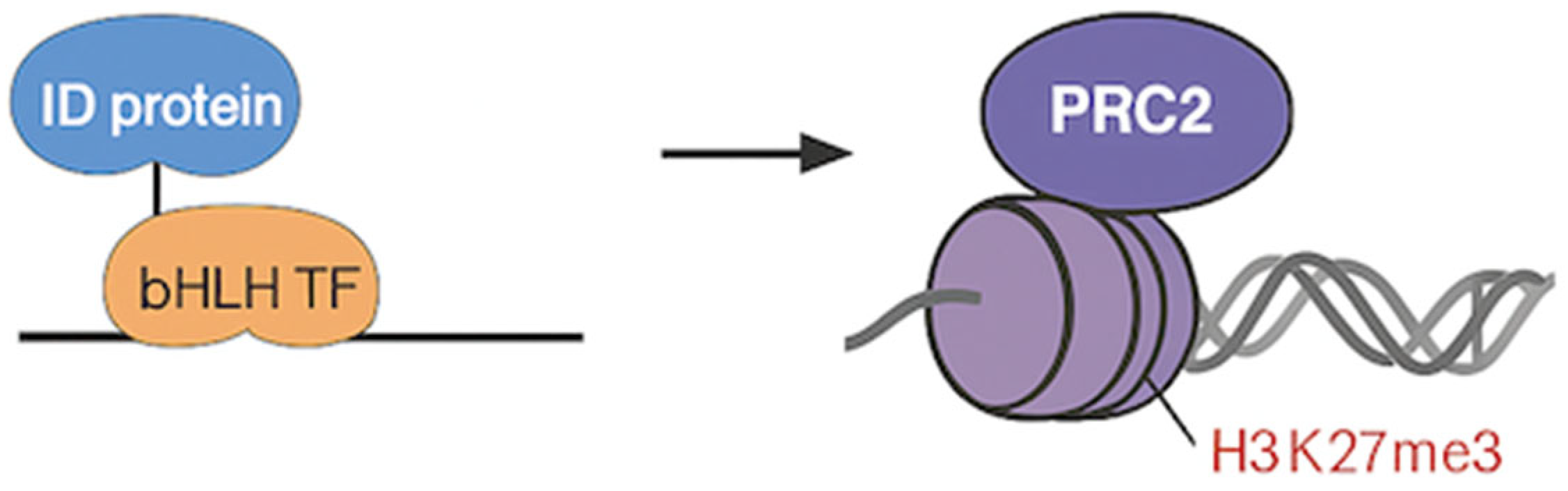
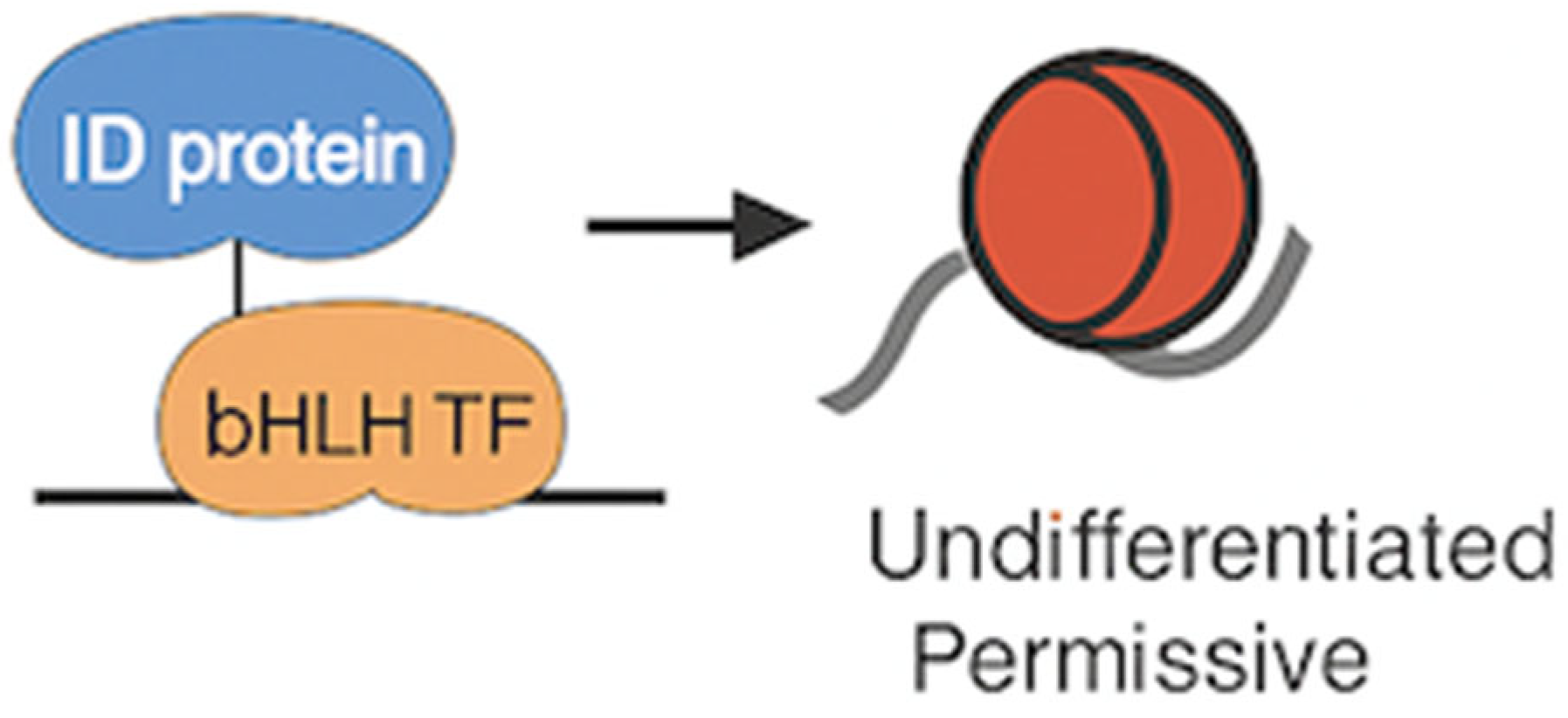
1. ID Proteins Promote an Undifferentiated, Permissive Chromatin State
ID proteins function as dominant-negative inhibitors of basic helix-loop-helix (bHLH) transcription factors by forming non-DNA-binding heterodimers, thereby blocking their ability to activate lineage-specific gene expression programs.[1,2] By preventing transcriptional activation of differentiation genes, ID proteins help preserve an undifferentiated, proliferative state, particularly in embryonic and adult stem cells.[5,6] Recent studies suggest that ID-mediated transcriptional repression is accompanied by changes in chromatin structure. In embryonic stem cells and neural progenitors, overexpression of ID1 or ID3 is associated with a more open, dynamic chromatin state characterized by increased chromatin accessibility and reduced heterochromatin compaction.[8,34] This permissive chromatin environment may paradoxically enhance the accessibility of PRC2 to previously active or poised regulatory regions that are rendered transcriptionally silent due to the absence of bHLH factor binding. As a result, PRC2 may gain access to and methylate newly vacated sites, locking them in a silenced state via H3K27me3 deposition.35
Figure 2.
ID proteins promote an undifferentiated chromatin state.
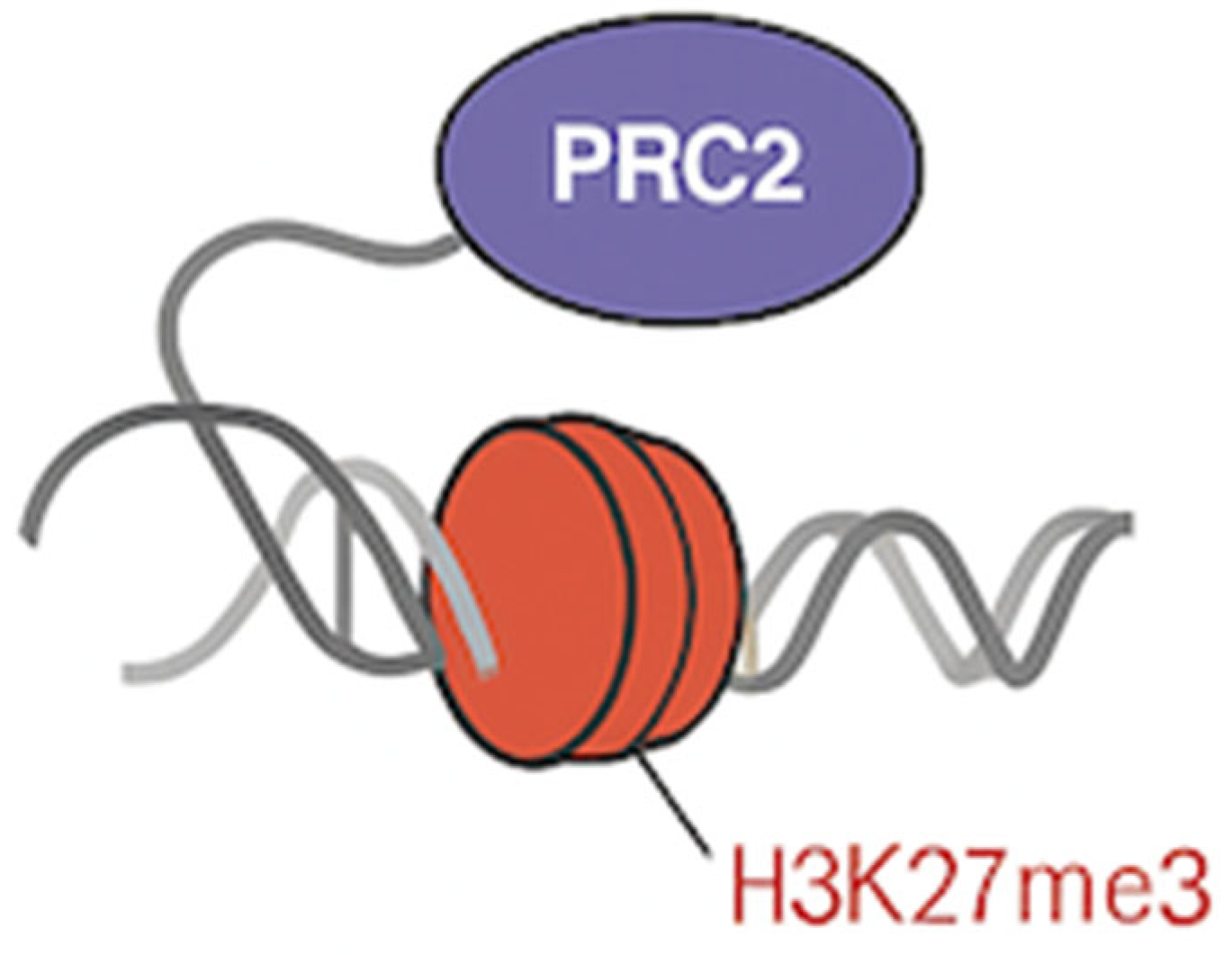
2. PRC2 Requires Pre-patterned Chromatin for Efficient Targeting
Unlike many transcription factors, PRC2 lacks sequence-specific DNA-binding ability and is instead recruited to target loci through pre-existing chromatin features, including unmethylated CpG islands, histone modifications (e.g., H2A ubiquitination), and interactions with accessory proteins or noncoding RNAs [29,30,31]. PRC2 preferentially targets regions that are transcriptionally inactive or weakly transcribed, as active transcription can evict PRC2 and inhibit its enzymatic function.[36] Regions regulated by bHLH transcription factors are typically transcriptionally active or poised. When ID proteins sequester these TFs and inhibit their activity, such regions may transition into a low-transcription or nucleosome-rich state, favoring PRC2 occupancy. Supporting this, several studies have demonstrated that PRC2 is recruited to newly silenced developmental genes following withdrawal of transcription factor activity or epigenetic remodeling.[37,38] ID-induced TF sequestration could therefore function as an upstream event that derepresses PRC2-susceptible loci.
Figure 3.
PRC2 requires pre-patterned chromatin for efficient targeting.
3. Co-occurrence in Stem and Cancer Contexts
The co-expression of ID proteins and PRC2 components in pluripotent and tumorigenic contexts suggests a possible convergence in their function. In embryonic stem cells, high levels of both ID proteins and PRC2 subunits (e.g., EZH2, SUZ12) maintain the repression of lineage-specific genes while preserving a plastic, self-renewing phenotype.[22,23] Notably, PRC2 targets many of the same differentiation-associated genes that are transcriptionally repressed by ID-sequestered bHLH factors, such as NeuroD, MyoD, and E2A.[20] In cancers, both ID proteins and PRC2 are frequently overexpressed and have been independently implicated in the acquisition of stem-like properties, resistance to differentiation, and poor clinical outcomes (Figure 4). For instance, high ID1 and EZH2 expression correlates with increased tumor-initiating capacity and therapy resistance in glioblastoma, breast, and prostate cancers.[10, 24, 28] This phenotypic overlap supports the notion that ID proteins and PRC2 may cooperate to stabilize undifferentiated, proliferative cell states in malignancy.
4. Functional Cross-Talk Between Chromatin Regulators
An emerging body of evidence highlights the interplay between transcription factors and chromatin remodelers in controlling gene expression (Figure 5). Several bHLH transcription factors, including Ascl1, MyoD, and TCF3, have been shown to recruit chromatin-modifying complexes such as SWI/SNF (BAF) to activate lineage-specific enhancers.[39,40,41] These complexes are known to antagonize Polycomb-mediated silencing by promoting open chromatin and displacing repressive marks.[33] By sequestering bHLH transcription factors, ID proteins may block this recruitment of SWI/SNF, thereby preventing activation of differentiation genes and indirectly favoring the establishment or maintenance of Polycomb-repressed chromatin states. In this context, ID proteins not only act as transcriptional inhibitors but may also serve as upstream regulators of chromatin state by modulating the balance between opposing chromatin regulators.[32] This suggests a model in which ID proteins create a chromatin landscape that passively or actively promotes PRC2 occupancy and activity, particularly during transitions in cell identity (Figure 5).
Proposed Experimental Approach:
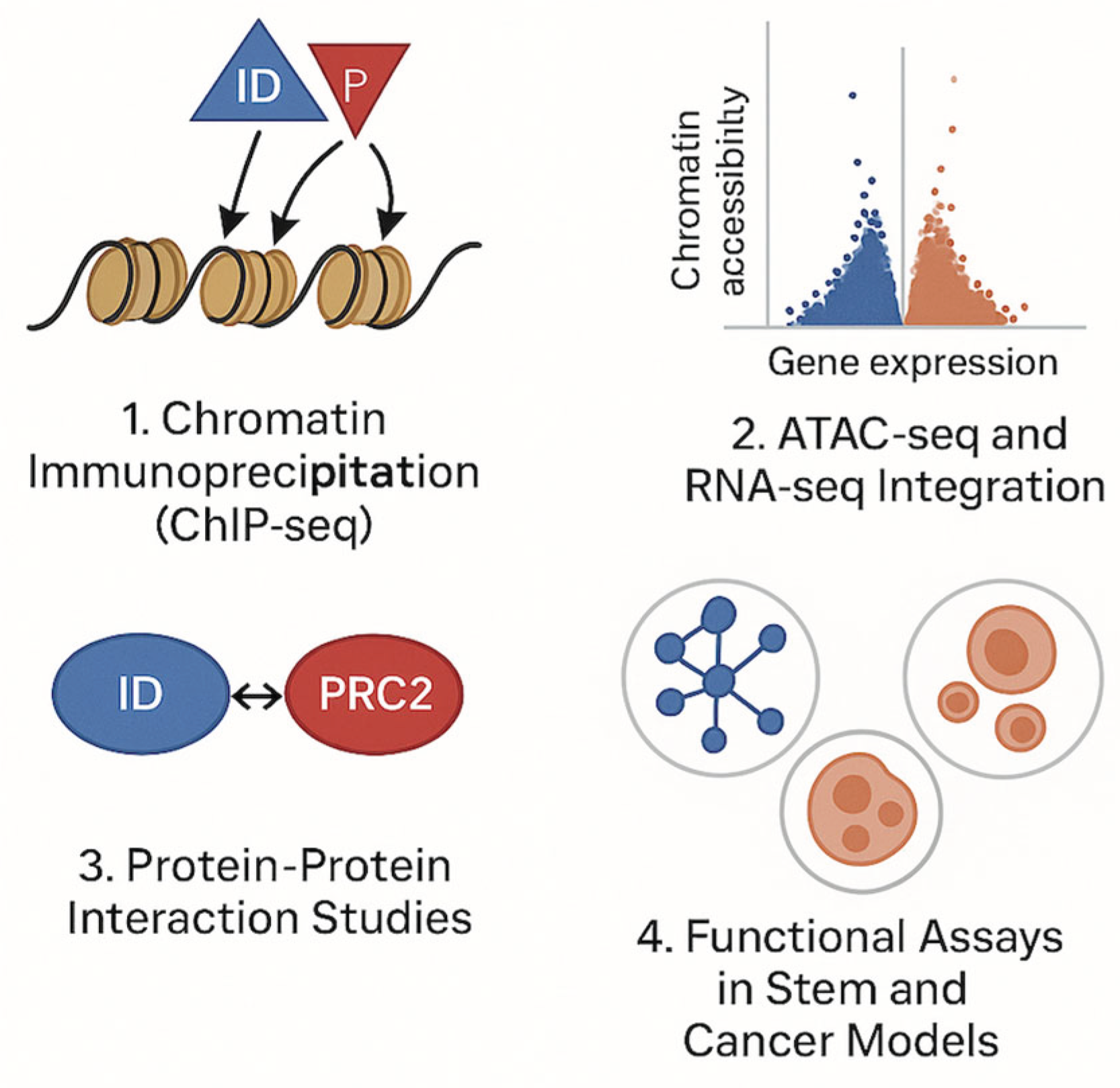
To investigate the mechanistic interplay between ID proteins and PRC2 in modulating chromatin landscapes during lineage specification and tumorigenesis, we propose a multifaceted experimental strategy. These approaches aim to dissect the functional, epigenomic, and molecular associations that underpin our hypothetical model (Figure 6).
1. Chromatin Immunoprecipitation Sequencing (ChIP-seq)
The objective is to determine whether ID protein levels affect PRC2 recruitment and histone methylation patterns genome-wide. By performing ChIP-seq for H3K27me3 and EZH2, the catalytic subunit of PRC2, in cells with gain- or loss-of-function of ID proteins (e.g., ID1, ID3), one can assess whether PRC2 is redirected to novel genomic loci as a consequence of altered ID expression. This experiment would test the prediction that ID-induced inhibition of bHLH transcription factors permits PRC2 targeting to regions previously occupied by active transcriptional complexes. For instance, in neural progenitor cells or embryonic stem cells, overexpression of ID proteins may lead to increased H3K27me3 deposition at genes associated with neuronal differentiation (e.g., Neurod1, Ascl1), supporting the model of ID-PRC2 crosstalk.[42,43] These ChIP-seq datasets should be compared against known bHLH target gene lists and Polycomb target regions to identify overlaps and shifts in occupancy.
2. Integrated ATAC-seq and RNA-seq Analysis
The goal is to map chromatin accessibility changes and transcriptional responses following ID protein perturbation and correlate them with PRC2 targeting. ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) provides a powerful means to evaluate chromatin accessibility across the genome. When coupled with RNA-seq, this strategy enables the identification of differentially accessible regions and gene expression changes upon ID overexpression or knockdown. The expectation is that ID overexpression would decrease chromatin accessibility at lineage-specifying genes by displacing bHLH transcription factors and facilitating PRC2 recruitment and silencing. Integration of ATAC-seq and RNA-seq data with ChIP-seq for H3K27me3 would identify candidate loci where loss of accessibility, gain of repression marks, and transcriptional downregulation co-occur in an ID-dependent manner.[5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44]
3. Protein-Protein Interaction Studies
To determine whether ID proteins directly or indirectly associate with PRC2 components or regulatory cofactors. While ID proteins lack known enzymatic activity or canonical chromatin-binding domains, their ability to modulate transcription factors may influence the assembly of chromatin regulatory complexes. Co-immunoprecipitation (co-IP) and proximity-dependent labeling techniques such as BioID or APEX2 can be employed to investigate whether ID proteins form complexes with PRC2 members (e.g., EZH2, SUZ12) or interact with accessory proteins that scaffold PRC2 recruitment (e.g., JARID2, MTF2). BioID-tagged ID1 constructs expressed in stem-like or cancer cells would allow for biotin labeling of proximal proteins, followed by mass spectrometry-based identification.[46,47] These results could reveal novel molecular links or scaffolding factors bridging ID function and Polycomb activity.
4. Functional Assays in Stem and Cancer Models
To test the biological relevance of ID-PRC2 interplay in self-renewal, lineage restriction, and tumorigenicity. Using in vitro and in vivo models, one can evaluate how modulation of ID proteins and PRC2 affects cell fate and transformation potential. Neural stem cells, hematopoietic progenitors, or established cancer cell lines (e.g., glioblastoma, breast cancer) can be engineered to express ID proteins constitutively or inducibly, alone or in combination with PRC2 inhibitors (e.g., GSK126 or EPZ-6438 targeting EZH2).[48] Key assays would include: sphere-forming capacity (self-renewal), differentiation assays (lineage marker expression), tumor xenografts in immunodeficient mice, and epigenetic profiling of key differentiation genes. Rescue experiments, where differentiation is restored by co-inhibiting PRC2 in ID-overexpressing cells, would provide functional evidence for ID-PRC2 cooperation in maintaining the undifferentiated state.[48,49]
This integrated experimental framework allows for the evaluation of chromatin dynamics, transcriptional regulation, molecular interactions, and phenotypic outcomes of ID protein function in the context of PRC2 modulation. If validated, this model would offer a new paradigm for understanding how transcriptional and epigenetic programs are coordinated during cell fate transitions. This would position ID proteins as upstream chromatin modulators, integrating environmental or developmental signals with PRC2-mediated gene repression. In cancer, this interaction could underlie stemness, resistance, and metastasis—making the ID-PRC2 axis a compelling target for epigenetic therapy.
Conclusions
The interplay between ID proteins and the Polycomb Repressive Complex 2 represents a potentially undervalued axis in the regulation of chromatin dynamics and cell fate decisions. While ID proteins are traditionally studied as inhibitors of lineage-specific transcription factors, our proposed model suggests a broader epigenetic function: that ID proteins help configure the chromatin landscape in a way that favors PRC2 recruitment and the establishment of repressive histone modifications such as H3K27me3. This could occur through several, non-mutually exclusive mechanisms, including the sequestration of transcription factors that generally antagonize PRC2 activity, the modulation of chromatin accessibility, and possibly the recruitment of chromatin-associated cofactors that bridge ID and Polycomb pathways. This hypothesis has significant implications in both developmental biology and oncology. By defining how ID proteins may prime or potentiate PRC2-mediated repression, we can better understand how transcriptional and epigenetic mechanisms converge to control plasticity. This could illuminate key nodes in the regulatory network that, when perturbed, contribute to developmental disorders or malignancy. From a translational perspective, targeting the ID–PRC2 interaction axis offers a dual advantage: it may allow for more precise modulation of epigenetic states while simultaneously affecting transcription factor networks that drive disease. Future studies that integrate chromatin profiling, transcription factor mapping, and functional assays in both normal and disease contexts are essential to validate this model. Ultimately, explaining this relationship will not only fill a gap in our understanding of chromatin biology but also pave the way for novel therapeutic strategies.
Data Availability
Not applicable.
Statement of Ethics
An ethics statement was not required.
Author Contributions
V. A. conceptualized, designed, conducted, and wrote the manuscript
Funding
This study was not supported by any sponsor or funding source.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Norton, J.D. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000, 113, 3897–905. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; et al. Id proteins at the cross-road of development and cancer. Oncogene. 2014, 33, 4659–67. [Google Scholar] [CrossRef] [PubMed]
- Avecilla, V. Effect of Transcriptional Regulator ID3 on Pulmonary Arterial Hypertension and Hereditary Hemorrhagic Telangiectasia. Int J Vasc Med. 2019, 2019, 2123906. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ling, F.; et al. Id proteins: small molecules, mighty regulators. Curr Top Dev Biol. 2014, 110, 189–229. [Google Scholar]
- Yokota, Y. Id and development. Oncogene. 2001, 20, 8290–8. [Google Scholar] [CrossRef]
- Ruzinova, M.B.; Benezra, R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003, 13, 410–8. [Google Scholar] [CrossRef]
- Jankovic, V.; et al. Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc Natl Acad Sci USA. 2007, 104, 12682–7. [Google Scholar] [CrossRef]
- Niola, F.; et al. Id proteins synchronize stemness and anchorage to the niche of neural stem cells. Nat Cell Biol. 2012, 14, 477–87. [Google Scholar] [CrossRef]
- Perk, J.; et al. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005, 5, 603–14. [Google Scholar] [CrossRef]
- Anido, J.; et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell. 2010, 18, 655–68. [Google Scholar] [CrossRef]
- Wen, Y.H.; et al. The role of ID1 in cancer progression and therapy resistance. Front Cell Dev Biol. 2020, 8:186.
- Avecilla, V.; Doke, M.; Das, M.; Alcazar, O.; Appunni, S.; Rech Tondin, A.; Watts, B.; Ramamoorthy, V.; Rubens, M.; Das, J.K. Integrative Bioinformatics-Gene Network Approach Reveals Linkage between Estrogenic Endocrine Disruptors and Vascular Remodeling in Peripheral Arterial Disease. Int J Mol Sci. 2024, 25, 4502. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Avecilla, V.; Doke, M.; Appunni, S.; Rubens, M.; Ramamoorthy, V.; Das, J.K. Pathophysiological Features of Remodeling in Vascular Diseases: Impact of Inhibitor of DNA-Binding/Differentiation-3 and Estrogenic Endocrine Disruptors. Med Sci (Basel). 13, 2. [CrossRef] [PubMed] [PubMed Central]
- Avecilla, V. Microarray Analysis Shows Important ID3 – BMP9 Driven Gene Signatures. Preprints 2025, 2025041283. [Google Scholar] [CrossRef]
- Avecilla, V. Gene Interactions in Cerebral Cavernous Malformations: A Brief Report. Preprints 2024, 2024050746. [Google Scholar] [CrossRef]
- Stankic, M.; et al. TGF-β–Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Reports. 2013, 5, 1228–42. [Google Scholar] [CrossRef]
- Niola, F.; et al. Id2 mediates tumor initiation, proliferation, and angiogenesis in glioblastoma. Cancer Res. 2013, 73, 3611–21. [Google Scholar]
- Benezra, R. The elusive tumor suppressor function of ID proteins. Oncogene. 2014, 33, 5564–5. [Google Scholar]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature. 2011, 469, 343–9. [Google Scholar] [CrossRef]
- Schuettengruber, B.; et al. Genome regulation by Polycomb and Trithorax proteins. Cell. 2007, 128, 735–45. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004, 15, 57–67. [Google Scholar] [CrossRef]
- Boyer, L.A.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006, 441, 349–53. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006, 125, 301–13. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat Med. 2016, 22, 128–34. [Google Scholar] [CrossRef]
- Morin, R.D.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010, 42, 181–5. [Google Scholar] [CrossRef]
- Gan, L.; et al. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res. 2018, 6, 10. [Google Scholar] [CrossRef]
- Liu, Y.; et al. ID1 cooperates with oncogenic Ras to inhibit senescence in primary murine fibroblasts by antagonizing p16INK4a expression. Mol Cell Biol. 2004, 24, 2570–82. [Google Scholar]
- Lawson, D.A.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015, 526, 131–5. [Google Scholar] [CrossRef]
- Oksuz, O.; et al. Capturing the onset of PRC2-mediated repressive domain formation. Mol Cell. 2018, 70, 1149–1162e5. [Google Scholar] [CrossRef]
- Wang, X.; et al. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat Struct Mol Biol. 2017, 24, 1028–1038. [Google Scholar] [CrossRef]
- Mendenhall, E.M.; et al. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010, 6(12)\:e1001244.
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011, 11, 481–92. [Google Scholar] [CrossRef]
- Kadoch, C.; et al. Dynamics of BAF–Polycomb complex opposition on heterochromatin in normal and malignant pediatric brain tumors. Cell. 2017, 171, 216–229e19. [Google Scholar]
- O'Brien, C.A.; et al. The E2A gene is required for normal B-cell development and suppresses alternative lineage development. Immunity. 2004, 20, 349–60. [Google Scholar]
- Li, G.; et al. Chromatin accessibility dynamics reveal novel functional enhancers in response to IL-4 stimulation in macrophages. Nucleic Acids Res. 2016, 44, e27. [Google Scholar]
- Kaneko, S.; et al. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell. 2014, 53, 290–300. [Google Scholar] [CrossRef]
- Riising, E.M.; et al. Gene silencing triggers Polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell. 2014, 55, 347–60. [Google Scholar] [CrossRef]
- Schwartz, Y.B.; Pirrotta, V. Polycomb complexes and epigenetic states. Curr Opin Cell Biol. 2008, 20, 266–73. [Google Scholar] [CrossRef]
- Chai, R.; et al. NeuroD1 coordinates chromatin remodeling and transcription factor recruitment to regulate differentiation of granule cells. Sci Rep. 2017, 7, 6136. [Google Scholar]
- Maves, L.; et al. The BAF complex interacts with Pax7 in myogenic progenitors. Development. 2007, 134, 859–69. [Google Scholar]
- Hnisz, D.; et al. Transcriptional super-enhancers are linked to cell identity and disease. Cell. 2013, 155, 934–47. [Google Scholar] [CrossRef]
- Choi, J.; et al. Histone demethylase KDM6B regulates the timing of neural differentiation in human ESCs. Cell Stem Cell. 2015, 16, 249–60. [Google Scholar]
- Pasini, D.; et al. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb Repressive Complex 2. Genes Dev. 2008, 22, 1345–55. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin. Nat Methods. 2013, 10, 1213–8. [Google Scholar] [CrossRef] [PubMed]
- Lareau, C.A.; et al. Chromatin accessibility dynamics reveal novel functional enhancers in response to hypoxia. Mol Cell. 2019, 75, 865–878e8. [Google Scholar]
- Kim, D.I.; et al. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci USA. 2014, 111, E2453–61. [Google Scholar] [CrossRef]
- Blackledge, N.P.; et al. PRC1 catalyzes monoubiquitylation of histone H2A to promote H3K27 methylation by PRC2. Mol Cell. 2014, 53, 644–57. [Google Scholar]
- McCabe, M.T.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012, 492, 108–12. [Google Scholar] [CrossRef]
- Suvà, M.L.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014, 157, 580–94. [Google Scholar] [CrossRef]
Figure 4.
The left and right panels show how ID proteins and PRC2 interact/behave within embryonic stem cells and cancer, respectively.
Figure 4.
The left and right panels show how ID proteins and PRC2 interact/behave within embryonic stem cells and cancer, respectively.

Figure 5.
Model of functional cross – talks between chromatin regulators.
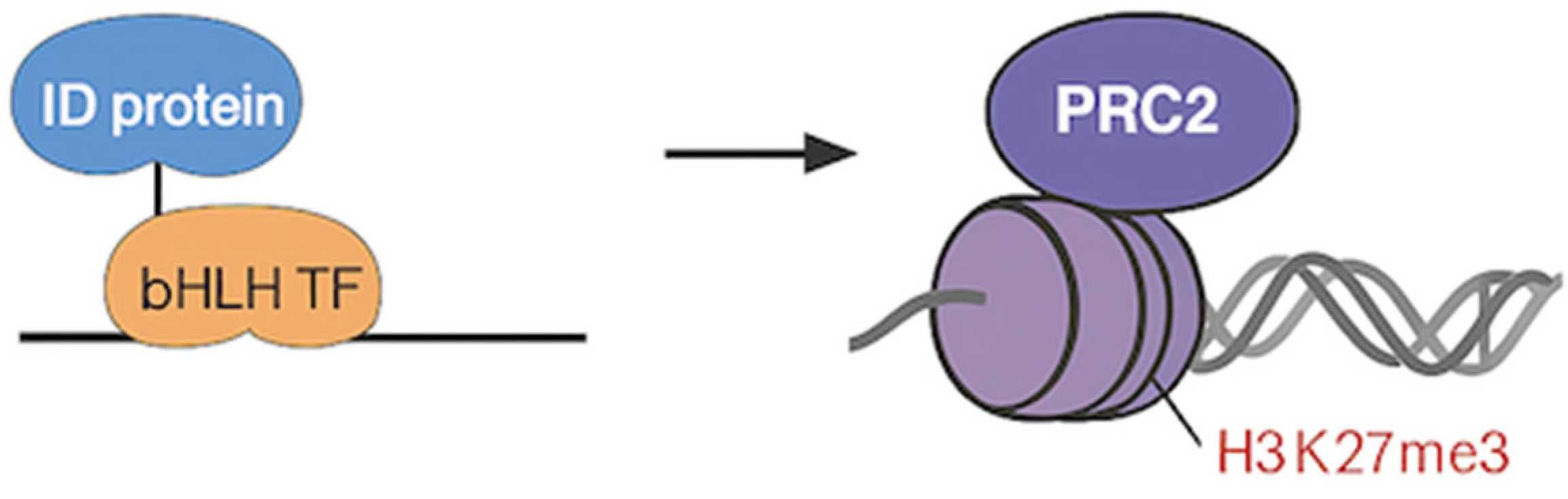
Figure 6.
Different approaches that aim to examine the functional, epigenomic, and molecular connections that support our hypothetical model.
Figure 6.
Different approaches that aim to examine the functional, epigenomic, and molecular connections that support our hypothetical model.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



