Submitted:
17 May 2025
Posted:
19 May 2025
You are already at the latest version
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) has emerged as the leading cause of chronic liver disease worldwide, posing a substantial public health burden and underscoring the urgent need for effective therapeutic and preventive strategies. MASLD is defined by excessive hepatic lipid accumulation and, in a subset of patients, progresses to metabolic dysfunction-associated steatohepatitis (MASH), a pro-inflammatory and pro-fibrotic condition associated with increased risk of fibrosis, cirrhosis, and hepatocellular carcinoma. Although the mechanisms driving MASLD progression remain incompletely understood, dysregulation of key metabolic pathways, including triglyceride handling, cholesterol catabolism, bile acid metabolism, mitochondrial function, and autophagy, has been consistently observed in MASLD livers. Human antigen R (HuR), a ubiquitously expressed mRNA-binding protein, is a post-transcriptional regulator of diverse cellular processes, including nutrient metabolism, cell survival, and stress responses. Recent evidence highlights HuR’s critical role in maintaining hepatic homeostasis, particularly under conditions of metabolic stress, such as those found in MASLD. Moreover, comorbid metabolic diseases, including obesity, type 2 diabetes mellitus, and cardiovascular disease, not only exacerbate MASLD severity but also involve HuR dysregulation in extrahepatic tissues, further contributing to hepatic dysfunction. This review explores the dynamic and cell-type–specific roles of HuR in MASLD pathogenesis and its associated metabolic comorbidities. A deeper understanding of HuR’s post-transcriptional regulatory networks across metabolic tissues may inform the development of targeted therapies aimed at preventing or reversing MASLD progression.
Keywords:
RNA binding protein
; Human antigen R
; metabolic dysfunction-associated steatotic liver disease (MASLD)
; metabolic syndrome
1. Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) is now recognized as the most common cause of chronic liver disease globally [1]. Its growing prevalence poses a substantial socio-economic burden, prompting medical societies and public health agencies to prioritize the development of effective interventions and preventive strategies [2,3]. MASLD encompasses a spectrum of disease severity, ranging from simple, non-inflammatory metabolic dysfunction-associated steatotic liver (MASL) to metabolic dysfunction-associated steatohepatitis (MASH), which is characterized by liver inflammation and fibrosis [4]. Progression to MASH significantly increases the risk of end-stage liver disease, cardiovascular events, and extrahepatic malignancies [5,6]. Despite the high prevalence of MASLD, only 10–30% of affected individuals develop MASH [1]. While lifestyle modifications such as dietary intervention and increased physical activity are effective in reversing MASLD phenotypes, poor adherence limits their long-term success [7]. Understanding the mechanisms by which hepatic steatosis leads to inflammation, hepatocyte death, and fibrogenesis in a subset of patients is therefore critical to reducing MASLD-related morbidity and mortality. Moreover, MASLD frequently coexists with other metabolic disorders, including obesity and type 2 diabetes mellitus (T2DM), both of which are associated with increased MASLD severity [8,9]. These associations underscore the importance of investigating how metabolic dysfunction in one tissue can exacerbate disease in another.
Human antigen R (HuR), encoded by the embryonic lethal abnormal vision-like 1 (Elavl1) gene, is a member of the Hu family of RNA-binding proteins (RBPs) [10]. The Elavl gene family was first identified in Drosophila as essential for embryonic and ocular development [11]. In humans, Hu proteins selectively bind adenine- and uridine-rich elements (AREs) within the 3’ untranslated regions (3’ UTRs) of mature mRNAs and in intronic sequences of pre-mRNAs [10,12,13]. AREs are present in approximately 22% of human 3’ UTRs and 25% of introns [14], and are typically found in transcripts encoding proteins involved in development, inflammation, metabolism, and signal transduction [15]. While Hu family members HuB, HuC, and HuD are predominantly expressed in the nervous system [16], HuR is ubiquitously expressed and essential for normal development and homeostasis [10,17]. HuR was initially identified as a regulator of early-response gene mRNA stability [18], but has since emerged as a dynamic, context-dependent modulator of numerous biological processes, including cell cycle progression [19,20,21], differentiation [22,23], apoptosis [24,25], and senescence [26,27]. HuR deficiency in mice is embryonically lethal [28], underscoring its essential role in development. HuR activity is regulated by various factors, including cell type, subcellular localization, and environmental cues. For example, in liver-resident hepatic stellate cells (HSCs), HuR induction and cytoplasmic translocation promote HSC activation and liver fibrosis [29,30], while reduced HuR expression in adipose tissue has been linked to obesity and insulin resistance [31,32]. Given its role in regulating thousands of transcripts [13,33], much remains to be learned about HuR’s functions in tissue-specific homeostasis and disease progression.
The rationale for this review stems from HuR’s emerging role as a key post-transcriptional regulator of metabolic homeostasis, inflammation, and fibrosis, hallmark features of MASLD and its progression to MASH. Given HuR’s cell-type–specific and stimulus-responsive activity, its function in different tissues may either promote or protect against MASLD depending on the biological context. This review provides a comprehensive synthesis of current evidence on HuR’s role in liver pathophysiology, focusing on its regulation of lipid metabolism, bile acid synthesis, mitochondrial function, oxidative stress, autophagy, inflammation, and fibrogenesis. We also highlight HuR’s involvement in extrahepatic metabolic tissues, including adipose, cardiac, and immune cells, where its dysregulation contributes to comorbid conditions such as obesity, cardiovascular disease, and T2DM, all of which exacerbate MASLD. Ultimately, we discuss the therapeutic potential and challenges of targeting HuR in MASLD and related metabolic disorders, emphasizing the need for cell-specific strategies that preserve HuR’s protective roles while mitigating its pathogenic effects.
2. HuR Structure and Subcellular Localization
As a ubiquitously expressed RNA binding protein with diverse roles in physiological and pathological processes [17], HuR is comprised of multiple domains that facilitate high affinity and specificity binding to AREs found within target transcripts [10,34]. HuR was first cloned and characterized by who used degenerate oligonucleotide-directed PCR to amplify a 140 nucleotide product from HeLa cells [10]. This gene product was called HuR and was highly similar, but distinct, in sequence to mRNAs encoding HuB, HuC, and HuD. The protein encoded by HuR/Elavl1 is comprised of a short 20 amino acid long N-terminus that precedes three RNA recognition motifs (RRMs) (Figure 1). RRM1 and RRM2 are connected via a 12 amino acid linker domain, while a much longer 60 amino acid hinge domain connects RRM2 to RRM3 [10,34,35]. All three RRMs are highly conserved among Hu protein family members and have high affinity and specificity for AREs that are prevalent within the 3’ UTR and intronic regions of many labile and stress-inducible mRNAs [36]. Interdomain cooperation is essential to RNA binding protein function and localization within the cell [37]. Based on the crystal structure of RNA-bound HuR, binding of RRM2 or RRM3 to an ARE is dependent upon the confirmational changes to HuR that accompany initial binding of RRM1 to the ARE [34,35]. Although non-covalent interactions between RRM1/2 and AREs act as adhesive to temporarily anchor HuR to target transcripts, maximum HuR function cannot be achieved without involvement of RRM3. For instance, both binding of the RRM3 domain of HuR to the poly(A) tail of mRNAs [38] and HuR dimerization via the RRM3 domain have been shown to increase the binding affinity of HuR for its target transcripts, in part by facilitating binding of multiple HuR proteins to tandem AREs [34]. HuR binding to other proteins, including SETα, SETβ, and APRIL, is also RRM3 dependent [39]. Specific interactions between the structural domains of HuR and other cellular factors, including RNA and protein, dictate HuR function, localization, and stability.
Early on, subcellular localization of HuR in both human and mouse cell lines was established as predominantly nuclear [40]. For instance, immunofluorescence staining of HuR revealed that only 2% of the HuR protein detected was localized to the cytoplasm of the HeLa cell line under normal culture conditions [41]. However, ARE-mediated mRNA decay is believed to be primarily cytosolic, leaving question as to how HuR could perform its mRNA stabilizing function if retained within the nucleus [36]. To address this question, Fan et. al. used a heterokaryon fusion experiment to show that flag-tagged HuR can shuttle back and forth between the nucleus and cytoplasm [40]. Later, this same group generated deletion mutants of HuR to identify both the nuclear localization signal (NLS) and the nuclear export signal (NES) of HuR [42]. Deletion of RRM3 and the hinge domain, but not RRM3 alone, inhibited nucleocytoplasmic shuttling of HuR, indicating that the hinge domain, connecting RRM2 and RRM3 of HuR, was necessary for nucleocytoplasmic shuttling. Further study of HuR deletion mutants revealed the HuR nucleocytoplasmic shuttling sequence (HNS) to be a 32 amino acid stretch within the hinge domain that is necessary and sufficient for HuR nucleocytoplasmic shuttling [42]. Initially, shuttling of HuR from the nucleus to the cytoplasm was thought to play an important role in protecting HuR-bound mRNAs from ARE-associated degradation machinery [42]. Association of HuR with poly(A) tail-enriched mRNA in both the nucleus and cytoplasm, as well as association of HuR with polysome fractions provided further support for HuR’s mRNA stabilizing role [41]. However, nucleocytoplasmic shuttling of both RNAs and proteins, such as HuR, through the nuclear pore complex (NPC) is largely nuclear transport factor, or karyopherin, dependent [43,44]. Therefore, identifying karyopherin-HuR interactions is essential to understanding mechanisms underlying HuR nucleocytoplasmic shuttling. Both A proliferation-inducing ligand (APRIL) and pp32 were found to directly bind HuR and mediate an interaction between HuR and the nuclear exportin, chromosomal region maintenance 1 (CRM1) [39,45]. Following heat shock, nuclear export of HuR into the cytoplasm of HeLa cells was shown to be CRM1-dependent [45]. Likewise, multiple pathways mediate import of HuR into the nucleus, including transport by nuclear importins, including transportin 1 and 2 (TRN1/2) and the karyopherin adaptor protein, importin-α (IMPα) [46,47]. Interestingly, AMPK activation and negative energy balance were associated with importin-mediated transport of HuR into the nucleus, while CRM1-mediated export of HuR into the cytoplasm occurred under conditions of heightened cellular stress [45,47], in both cases intracellular HuR localization impacting HuR-mediated regulation of gene expression.
3. Regulation of HuR Expression
Various regulatory mechanisms govern HuR expression, localization, and function. Transcriptional regulation of HuR expression has important implications for the cellular stress response and tumorigenesis. There are four annotated HuR transcripts in mice (NCBI Gene ID: 15568). However, all four transcripts encode the same 326 amino acid protein, with transcript variation due to differences in sequence and secondary structure of both the 3’ and 5’ untranslated regions (UTR) [48,49,50]. The HuR gene contains three putative transcriptional start sites located upstream of the methionine start codon. These sites are flanked by transcription factor binding motifs, including c-ets, CREB, and AP1 elements [51]. Several transcription factors have been shown to bind directly to the HuR promoter (Figure 1). For example, nuclear factor κβ (NFκβ) enhances transcription of the long HuR variant by direct binding of the p65 subunit to the promoter, promoting cell survival and proliferation in MKN74 gastric cancer cells [52]. Similarly, ATP depletion activates SMAD-mediated transcription of the short HuR variant in LLC-PK1 porcine kidney proximal tubule cells, also contributing to cell survival [53]. The presence and differential regulation of both short and long HuR transcript variants enable context-specific fine-tuning of HuR expression [50].
Beyond transcriptional control, HuR expression is further regulated at the post-transcriptional level by RNA-binding proteins and microRNAs (miRNAs) that modulate mRNA stability, translation, and subcellular localization through direct interaction with AU-rich elements in the HuR transcript [54,55] (Figure 1). In fact, HuR can positively autoregulate its own expression by binding to its cognate mRNA, enhancing its stability, cytoplasmic localization, and translation [27,54]. In contrast, decay-promoting RNA-binding proteins such as AUF1 (ARE/poly(U)-binding degradation factor 1) bind to the same ARE regions to destabilize HuR mRNA and simultaneously modulate broader gene expression patterns [54,56]. Several miRNAs, including miR-22, miR-29, and miR-519, have also been identified as key post-transcriptional regulators of HuR expression [57,58,59]. More specifically, miR-519 binds directly to both the 3’ untranslated region (UTR) and coding sequence of HuR mRNA, repressing translation without affecting mRNA stability across multiple cancer cell lines, including HeLa cells [59].
While transcriptional and post-transcriptional mechanisms regulate HuR expression, its functional activity is more strongly influenced by post-translational modifications, protein–protein interactions, and interactions with noncoding RNAs [34,55,60] (Figure 1). Phosphorylation of HuR by kinases such as protein kinase C (PKC), cyclin-dependent kinase 1 (Cdk1), p38 mitogen-activated protein kinase (MAPK), and checkpoint kinase 2 (Chk2) modulates both its nucleocytoplasmic shuttling and its binding affinity for target transcripts, ultimately impacting cell survival [61,62,63,64,65,66]. Methylation of HuR enhances the stability of its target mRNAs and promotes cytoplasmic accumulation in cancer cells [26]. In contrast, ubiquitination marks HuR for proteasomal degradation under cellular stress, while caspase-mediated cleavage during apoptosis generates HuR fragments with distinct RNA-binding profiles compared to the full-length protein [24,67]. Biophysical and biochemical analyses using techniques such as fluorescence resonance energy transfer (FRET), X-ray crystallography, and immunoprecipitation have shown that HuR engages in non-covalent interactions with itself and other proteins to enhance binding to longer AREs and promote expression of target transcripts through oligomerization [34,68,69,70]. Additionally, in intestinal epithelial cells, transient interactions between HuR and various noncoding RNAs, including long noncoding RNAs, miRNAs, and circular RNAs, can modulate HuR’s affinity for its mRNA targets [55]. Together, these multilayered regulatory mechanisms underscore the complexity of HuR function, enabling dynamic and context-specific control of its activity in response to diverse cellular signals.
4. Overview of HuR Function During Physiological and Pathological Conditions
Under physiological conditions, HuR is predominantly localized to the nucleus, where it plays key roles in splicing, 3’ end processing, pre-mRNA stabilization, and nuclear export of target transcripts [33] (Figure 1). Although HuR was originally characterized for its cytoplasmic role in stabilizing mature mRNAs [71], its nuclear functions in gene regulation are increasingly recognized. For instance, approximately 35% of HuR binding sites identified in unstressed HeLa cells by photoactivatable ribonucleotide crosslinking and immunoprecipitation (PAR-CLIP) were located within intronic regions, suggesting a major role in pre-mRNA processing [13]. Consistent with this, HuR knockdown in human embryonic kidney (HEK293) cells induces widespread changes in exon usage, further supporting its involvement in alternative splicing [33]. More specifically, nuclear HuR has been implicated in regulating transcripts involved in metabolism and mitochondrial function. In HEK293 cells, HuR binds to the long noncoding RNA (lncRNA) RNA component of mitochondrial RNA processing endoribonuclease (RMRP), facilitating its export to the cytoplasm where it regulates mitochondrial DNA replication and RNA processing [72]. Similarly, in activated B cells, HuR regulates expression of Dihydrolipoamide S-Succinyltransferase (Dlst), a critical component of the alpha-ketoglutarate dehydrogenase (α-KGDH) complex in the tricarboxylic acid (TCA) cycle, by binding to introns within Dlst pre-mRNA, thereby preventing the production of non-functional transcript variants [73].
During cellular stress, HuR plays an increasingly prominent role in regulating mature mRNA stability within the cytoplasm [17]. Translocation of HuR from the nucleus to the cytoplasm is observed in various pathological and stress-related contexts, including cancer spprogression [74], oxidative injury [75], inflammation [76,77], fibrosis [29], and cell death [24]. Once in the cytoplasm, HuR stabilizes and enhances the translation of thousands of ARE-containing transcripts, many of which encode cell survival proteins and pro-inflammatory cytokines [13,71]. Although a basal level of cytoplasmic HuR is present under homeostatic conditions [41], both its upregulation and increased cytoplasmic localization have been implicated in the pathogenesis of inflammatory diseases such as rheumatoid arthritis [78], atherosclerosis [79], and inflammatory bowel disease [80], as well as in cancer progression [74]. Stabilized mRNAs include those encoding key regulators of proliferation (e.g., c-Fos, p21, Cyclins B1, D1, E1, mdm2) and inflammation (e.g., iNOS, GM-CSF, TNFα, COX-2) [17]. In some cases, HuR enhances the translation of target mRNAs without altering their stability, as seen with X-linked inhibitor of apoptosis (XIAP) [81], while in rare instances such as with internal ribosome entry site (IRES)-containing transcripts like p27, HuR binding suppresses translation [82].
Beyond its pro-inflammatory and pro-tumorigenic targets, HuR also regulates mRNAs encoding other RNA-binding proteins, mRNA processing factors, transcription factors, and cytokines, reflecting its broad influence on gene expression across disease states [33]. For instance, HuR stabilizes forkhead box Q1 (FOXQ1) mRNA in MDA-MB-231 triple negative breast cancer cells, promoting tumorignesis and metastases [83], and stabilizes transforming growth factor beta (TGF-β) mRNA in activated hepatic stellate cells (HSCs), contributing to fibrosis in chronic liver disease [30]. Importantly, cytoplasmic HuR is not always pathogenic. It also supports adaptive cellular responses by stabilizing transcripts that encode metabolic enzymes [31], structural proteins [84], and signaling mediators [85]. In fact, reduced HuR expression is linked to the progression of certain disorders, including obesity and MASLD [31,85,86,87], underscoring its potential protective role in maintaining cellular homeostasis. Despite these advances, the full scope of HuR’s function in cellular homeostasis and disease remains to be fully elucidated.
5. Role of HuR in Liver Homeostasis and MASLD
5.1. MASLD Epidemiology and Prevalence
MASLD is the most common cause of chronic liver disease today, with MASLD incidence continuing to increase across all populations [1,2]. In 2019, the global prevalence of MASLD was estimated at 23.4-30% of adults, with incidence also increasing in children and adolescents [2,88,89,90]. Although originally referred to as Non-Alcoholic Fatty Liver Disease (NAFLD), the term MASLD was adopted in 2023 by major international liver societies and patient advocacy groups to better reflect the heterogeneity of steatotic liver disease and its close association with other metabolic disorders [91]. Importantly, >96% of individuals who originally met NAFLD-diagnostic criteria also meet MASLD-diagnostic criteria, allowing for continuity between NAFLD-related and MASLD-related studies [92]. MASLD is defined as excessive hepatic triglyceride accumulation (>5% liver weight) in the presence of at least one additional cardiometabolic risk factor and absence of excess alcohol consumption [91]. The adult diagnostic criteria for MASLD-related cardiometabolic risk factors include 1) Body mass index (BMI) ≥ 25 kg/m2 or waist circumference > 94 cm in men and > 80 cm in women, with ethnicity taken into account, 2) Fasting blood glucose ≥ 100 mg/dl or 2-hrs post-load glucose level ≥ 140 mg/dl or HbA1c ≥ 5.7%, 3) Blood pressure ≥ 130/85 mmHg, 4) Plasma triglycerides ≥ 150 mg/dl, and 5) Plasma high-density lipoprotein (HDL) cholesterol < 40 mg/dl for men or <50 mg/dl for women [91]. The MASLD spectrum ranges in severity from simple, non-inflammatory hepatic steatosis (>5% liver lipid), termed MASL, to the pro-inflammatory and pro-fibrotic phenotype of MASH [4]. Liver fibrosis is associated with increasing MASH severity and progression to cirrhosis and/or hepatocellular carcinoma (HCC) characterizes the progressive end of the MASLD spectrum [1].
MASLD presents an increasingly significant socioeconomic burden [88]. For instance, in 2016 alone, direct medical costs associated with MASLD were estimated to be about $103 billion in the United States [3]. Likewise, despite the effectiveness of lifestyle interventions, such as weight loss, increased exercise, and dietary restrictions, on reversing deleterious MASLD phenotypes, poor adherence to lifestyle interventions results in continued MASLD progression in many [1,7]. Although MASLD progresses to MASH in only 10-30% of human MASLD patients [1], MASH -and its subsequent fibrotic manifestations- greatly increases the risk for development of end-stage liver diseases, such as liver cirrhosis and hepatocellular carcinoma, and both fatal and non-fatal cardiovascular events [1,5,6]. Indicative of the increasing impact of MASLD on human health outcomes, MASH was the second most common cause of liver transplantation within the United States in 2019, and was also the most rapidly increasing liver transplantation etiology at that time [93]. Also indicative of the urgency to address the growing MASLD problem, as of January 2025, over 500 MASLD/MASH related clinical trials were currently ongoing or actively recruiting participants (clinicaltrials.gov). However, despite significant efforts, in 2024, approval of the β thyroid hormone receptor agonist, resmetirom marked the first -and only- Food and Drug Administration (FDA) approved pharmacotherapeutic for use in MASLD patients [94]. Importantly, resmetirom use is restricted to a small subset of MASLD patients and much uncertainty remains regarding its long term efficacy and side effect profile, pointing to the need for further MASLD-related pharmacotherapeutic development [94].
The pathogenesis and progression of MASLD are complex and multifactorial. Although significant progress has been made in understanding the molecular mechanisms underlying MASLD, the factors that drive the progression from non-inflammatory, simple steatosis to MASH in some individuals—but not others—remain poorly understood [4]. This knowledge gap continues to hinder effective therapeutic development. In this context, exploring the role of HuR, a dynamic and multifaceted RNA-binding protein, in a heterogeneous and multisystem disease like MASLD may offer valuable insights. Importantly, the loss of hepatic HuR expression observed in both MASLD patients and preclinical mouse models suggests a potential role for HuR in maintaining liver homeostasis and protecting against MASLD development [86]. Consistently, liver-specific HuR knockout mice show increased susceptibility to diet-induced MASLD, highlighting HuR and its downstream pathways as promising therapeutic targets [85,86,87,95]. The remainder of this review will explore HuR’s regulatory functions in key pathways commonly disrupted in MASLD (Figure 2).
5.2. Role of HuR in Hepatic Steatosis
In MASLD, hepatic steatosis arises from an imbalance between lipid acquisition and elimination in the liver [96]. In human MASLD patients, approximately 60% of hepatic lipids are derived from adipose tissue lipolysis, 25% from hepatic de novo lipogenesis (DNL), and 15% from dietary intake [97]. In mouse models, liver-specific HuR deficiency exacerbates high-fat diet-induced steatosis, suggesting a protective role for HuR in lipid homeostasis [85,86,87,95]. In the following section, we will examine the potential mechanisms contributing to steatosis development in HuR-deficient livers.
5.2.1. HuR Regulates Insulin Signaling and Hepatic Steatosis
MASLD is strongly associated with insulin resistance and hyperinsulinemia [98]. More than 60% of individuals with type 2 diabetes mellitus (T2DM) also develop MASLD, placing them at significantly elevated risk for progression to pro-fibrotic MASH [99]. Insulin resistance in adipose tissue, liver, and skeletal muscle contributes to the metabolic disturbances that drive hepatic steatosis in MASLD [100]. In adipose tissue, insulin resistance impairs the anti-lipolytic effects of insulin, resulting in sustained lipolysis and increased circulating free fatty acids (FFAs), which promote ectopic lipid deposition in the liver [101]. In parallel, hepatic insulin resistance enhances DNL [100], while insulin resistance in skeletal muscle reduces glucose uptake and glycogen synthesis, leading to systemic hyperglycemia and further stimulating hepatic DNL [102].
Paradoxically, liver-specific HuR-deficient mice display enhanced insulin sensitivity and lower blood glucose levels compared to wild-type controls following 24 weeks of high-fat diet (HFD) feeding, despite exhibiting more pronounced hepatic steatosis [85]. To elucidate the underlying mechanisms, hepatic expression and activation of insulin signaling mediators including components of the phosphoinositide 3-kinase (PI3K)/Akt pathway were assessed in HuR-deficient mice [85]. Under normal conditions, hepatic insulin signaling via the PI3K/Akt axis inhibits glucose production and promotes glucose utilization through glycogenesis, DNL, and triglyceride export [100]. In HuR-deficient livers, Akt activation was elevated, and the expression of genes involved in glycolysis and DNL was increased, while genes related to hepatic glucose production were suppressed—indicating heightened sensitivity to insulin signaling [85]. Phosphatases such as phosphatase and tensin homolog (PTEN) act as negative regulators of the PI3K/Akt pathway and thereby reduce hepatic insulin sensitivity [103]. Like HuR-deficient mice, liver-specific Pten knockout mice show increased insulin sensitivity but develop more severe hepatic steatosis [104]. PTEN expression was decreased in HuR-deficient livers, and Pten mRNA was shown to associate with HuR in ribonucleoprotein immunoprecipitation (RNP-IP) assays from hepatocytes [85]. These findings suggest that HuR stabilizes Pten transcripts and that its loss leads to reduced PTEN expression, enhanced PI3K/Akt signaling, and increased hepatic insulin sensitivity, thereby promoting DNL. However, overexpression of PTEN in HuR-deficient livers did not reduce serum alanine aminotransferase (ALT) levels following HFD feeding, indicating that loss of HuR-mediated PTEN stabilization does not fully explain the susceptibility to liver injury in this model [85]. Supporting this, two additional studies using liver-specific HuR knockout mice reported significant upregulation of DNL-related genes, such as acetyl-CoA carboxylase 1 (Acc1) and fatty acid synthase (Fasn), compared to wild-type controls, under both normal chow and Western diet conditions (42 kcal% fat, 0.1% cholesterol) [86,87]. These findings collectively suggest that hepatic HuR plays a critical role in modulating insulin signaling and lipid metabolism, where its loss enhances insulin sensitivity but simultaneously promotes hepatic lipogenesis and steatosis.
5.2.2. HuR Regulation of Very Low-Density Lipoprotein Secretion
The assembly and secretion of very low-density lipoprotein (VLDL) particles represent the primary pathway for hepatic lipid export and are essential for maintaining liver lipid homeostasis [105]. Impaired VLDL secretion leads to lipid accumulation, lipotoxicity, and exacerbation of MASLD [106,107,108]. In hepatocytes, VLDL particles are synthesized through microsomal triglyceride transfer protein (MTTP)–mediated lipidation of apolipoprotein B-100 (APOB-100), followed by the incorporation of storage lipids such as triglycerides and cholesterol esters [105]. Additional apolipoproteins, including apolipoprotein E (APOE), contribute to the stabilization and distribution of VLDL particles [109]. Enhanced VLDL secretion is generally protective against hepatic steatosis, as evidenced by the increased prevalence of MASLD in individuals with loss-of-function mutations in MTTP or APOB [110]. However, chronic overproduction of VLDL in MASLD patients is also linked to dyslipidemia and heightened cardiovascular risk [6,106].
In a recent study using liver-specific HuR-deficient mice, increased hepatic steatosis was accompanied by reduced expression of both APOB-100 and APOE, as well as decreased serum lipid levels [95]. Supporting a direct regulatory role for HuR, the authors demonstrated HuR binding to Apob pre-mRNA, which enhanced APOB expression in a human hepatoma cell line [95]. Similarly, another study using the same Alb-Cre-driven HuR knockout model found elevated hepatic steatosis under normal chow conditions, though this effect was not observed after 6 weeks of choline-deficient high-fat diet (HFD-CD) feeding [86]. Because choline deficiency impairs hepatic VLDL secretion [111], this dietary context may have masked the effect of HuR loss on lipid export. Further supporting a role for HuR in VLDL regulation, shotgun lipidomic analysis revealed increased levels of major VLDL components such as various triglyceride and cholesterol ester species in HuR-deficient livers, with lipid profiles resembling those observed in human MASLD [86]. In addition, ER stress and dysregulated FXR signaling, both known to impair VLDL secretion and promote steatosis in MASLD [105], were also observed in HuR-deficient livers. Bulk RNA-seq analysis confirmed disruption of FXR target gene expression, lipid export pathways, and ER stress response genes, further supporting the hypothesis that impaired VLDL secretion contributes to lipid accumulation in the absence of hepatic HuR [86].
5.3. Role of HuR in Cholesterol Metabolism
Hepatic steatosis is considered the “first hit” in MASLD pathogenesis [112]. However, only 10-30% of individuals with MASLD progress to the inflammatory and fibrotic stage of MASH [1]. As shown in human clinical trials [1], improvements in hepatic steatosis do not necessarily correlate with reductions in liver injury or disease severity. This highlights the importance of identifying additional molecular “hits” that drive disease progression and may serve as therapeutic targets [113]. While overall hepatic fat accumulation defines MASLD, dysregulation of cholesterol metabolism is increasingly recognized as a critical driver of disease progression [114,115]. Cholesterol homeostasis in the liver is governed by a balance between assimilation (uptake, synthesis, esterification, storage) and elimination (lipoprotein secretion, bile acid synthesis, and excretion) [114]. In human MASLD, hepatic cholesterol accumulation is frequently linked to increased endogenous cholesterol synthesis [114,116]. This process is regulated by sterol regulatory element-binding protein 2 (SREBP-2/Srebf2), an ER-resident transcription factor activated under low sterol conditions [117]. Hepatic cholesterol biosynthesis involves a multi-step pathway, beginning with the conversion of acetyl-CoA to mevalonate by HMG-CoA reductase (HMGCR/Hmgcr), the rate-limiting step of the pathway and a major control point [118]. Cholesterol metabolites, glucagon, and statins inhibit HMGCR, whereas insulin and SREBP-2 activate [118,119]. While hepatic cholesterol accumulation normally suppresses SREBP-2 activation, inflammation and insulin resistance can override this feedback, resulting in sustained cholesterol synthesis during MASLD [120,121].
Paradoxically, although HuR-deficient mouse livers show increased hepatic cholesterol content under MASLD-inducing diets [86,87], transcriptomic analysis revealed that cholesterol synthesis pathways were significantly downregulated in HuR-deficient livers under normal dietary conditions [86]. In the same study, RNP-IP sequencing (GEO access ID: GSE143703) in healthy murine liver tissue revealed direct binding of HuR to the Hmgcr mRNA, indicative of HuR-mediated post-transcriptional regulation of Hmgcr. These findings suggest that HuR loss alters the regulatory landscape of cholesterol metabolism even before overt MASLD develops, though further investigation is needed to clarify how HuR influences Hmgcr expression and cholesterol synthesis under different dietary conditions.
Following synthesis, cholesterol can be stored or utilized in various cellular processes, including bile acid (BA) synthesis and steroidogenesis. Excess free cholesterol is esterified by acyl-CoA:cholesterol acyltransferase (ACAT) for storage in lipid droplets [122]. Alternatively, unesterified cholesterol is distributed to cellular membranes to maintain membrane fluidity and serve as a precursor for cholesterol derivatives [122]. In hepatocytes, free cholesterol predominantly accumulates in the plasma membrane, while its accumulation in organelle membranes, particularly the mitochondrial and ER membranes, is tightly restricted to preserve membrane integrity [118]. During MASLD, excess accumulation of free cholesterol in the mitochondrial and ER membranes leads to mitochondrial dysfunction and ER stress, promoting inflammation, cell death, and MASLD progression [123]. Although membrane-specific free cholesterol distribution has not yet been assessed in HuR-deficient mouse livers, elevated total hepatic cholesterol levels in these mice are accompanied by increased mitochondrial dysfunction [95] and ER stress [86], raising the possibility that disrupted cholesterol compartmentalization, including excess free cholesterol, contributes to liver injury in this model. This warrants further investigation into the role of HuR in regulating intracellular cholesterol distribution and its implications for MASLD progression.
5.4. Role of HuR in Bile Acid Metabolism
Metabolism of cholesterol into bile acids represents the primary route for cholesterol elimination in the body [124]. Bile acid synthesis occurs mainly in the liver and involves two distinct pathways: the classical and the alternative pathways [124]. The classical pathway predominates in the adult human liver, while the alternative pathway is more active in infants and becomes increasingly relevant in MASLD [125]. In the classical synthesis pathway, both microsomal and mitochondrial cytochrome P-450s (CYPs), including 7α-hydroxylase (CYP7A1), 3β-hydroxy-Δ5-C27-steroid dehydrolase (3β-HSD), sterol 12α-hydroxylase (CYP8B1), aldo-keto reductase 1D1 (AKR1D1) and sterol 27-hydroxylase (CYP27A1), oxidize cholesterol into the bile acids, cholic acid (CA) and chenodeoxycholic acid (CDCA). In contrast, the alternative pathway is initiated by sterol hydroxylases that metabolize cholesterol into various oxysterols, including 24-, 25-, and 27-hydroxycholesterol [126]. Mitochondrial CYP7B1 then further hydroxylates these oxysterols into 3β, 7α-dihydroxy-5-cholestenoic acid, which subsequently enters the classical synthesis pathway for further metabolism by AKR1D1 and CYP27A1 to generate CDCA [124]. In humans, most CA and CDCA are conjugated with glycine or taurine at a 3:1 ratio by the peroxisomal enzymes bile acid-CoA synthase (BACS) and bile acid N-acyltransferase (BAAT) [127]. In mice, however, CDCA is further converted to α- and β muricholic acids (MCAs) by Cyp2c70 before conjugation [128]. Additionally, murine BAAT preferentially conjugates bile acids with taurine, resulting in nearly all primary bile acids being taurine-conjugated [129]. These species-specific differences in bile acid synthesis and conjugation significantly influence bile acid pool composition and signaling, which limits the direct applicability of findings from murine models to human physiology [124].
MASLD development is associated with a progressive increase in serum bile acid levels and alterations in bile acid pool composition [130,131]. However, the underlying mechanisms driving bile acid induction in MASLD remain controversial and incompletely understood [132]. For instance, upregulation of bile acid synthesis genes, including CYP7A1, CYP8B1, and CYP27A1 [133], along with elevated levels of bile acid synthesis marker 7-alpha-hydroxy-4-cholesten-3-one (C4) [134], has been linked to increased bile acid pool size in two human MASLD cohorts. In contrast, another study reported that although serum primary bile acids increased with higher MASH activity scores and fibrosis progression, C4 levels remained unchanged [130]. Similar discrepancies have been observed in diet-induced rodent MASLD models. In rats, MASLD induction led to increased expression of Cyp7a1 and Cyp8b1 [133], whereas a MASLD mouse model showed elevated hepatic bile acid levels despite decreased expression of classical bile acid synthesis genes, including Cyp27a1 and Cyp8b1 [135].
HuR appears to play a regulatory role in hepatic BA synthesis [86,87]. In hepatocyte- and cholangiocyte-specific HuR-deficient mouse livers, key genes involved in both classical and alternative BA synthesis pathways such as Cyp7a1 and Cyp7b1 are downregulated. RNP-IP from healthy murine liver tissue confirms that HuR directly binds to both Cyp7a1 and Cyp7b1 transcripts, suggesting post-transcriptional regulation by HuR [86]. Cyp7b1 repression is commonly observed during MASLD and is associated with induction of the alternative BA synthesis pathway, leading to oxysterol accumulation, which contributes to hepatic inflammation and fibrosis [126,136,137]. Whether Cyp7b1 suppression in HuR-deficient livers promotes oxysterol accumulation during MASLD remains to be determined. In addition to gene repression, hepatocyte- and cholangiocyte-specific HuR-deficient livers show reduced levels of conjugated primary bile acids, including tauro-CDCA (TCDCA) and tauroursodeoxycholic acid (TUDCA) [86]. Contrastingly, hepatocyte-specific HuR-deficient mice exhibit increased levels of conjugated primary and secondary bile acids in both liver and serum following MASLD-inducing diet feeding [87]. The authors of that study hypothesized that this accumulation may promote MASH fibrosis by activating sphingosine-1-phosphate receptor 2 (S1PR2) and upregulating H19 expression [87]. The discrepancies between these two studies may be attributed to differences in the scope of HuR deletion (hepatocyte-specific vs. hepatocyte-and-cholangiocyte-specific) as well as variations in diet feeding conditions.
Enterohepatic bile acid circulation that recycles bile acids between the liver and the intestine is essential to not only disposition of lipid-soluble metabolites, but also to bile acid-mediated regulation of metabolic homeostasis [125]. Around 95% of the bile acids that enter the small intestine are reabsorbed by ileal enterocytes. Prior to recycling, some bile acids are metabolized by gut bacteria in the distal ileum and large intestine into secondary BAs, including DCA, hyocholic acid (HCA), lithocholic acid (LCA), ursodeoxycholic acid (UDCA) and ω-MCA. Indicative of impaired enterohepatic bile acid circulation, conjugated bile acids including TCA are elevated in the liver and serum of hepatocyte-specific HuR-deficient mice compared to wild-type controls, while primary and secondary bile acids are reduced in the cecum following MASLD-inducing diet feeding [87]. These findings suggest that HuR plays a multifaceted role in regulating bile acid metabolism by modulating the expression of key synthetic enzymes and maintaining proper enterohepatic circulation. Its deficiency disrupts BA synthesis, alters BA pool composition, and impairs metabolic signaling, collectively contributing to MASLD progression.
5.5. HuR Regulates Mitochondrial Function and Oxidative Stress
Mitochondrial dysfunction is a key contributor to MASLD progression toward MASH [138]. As central regulators of lipid metabolism and cellular energy homeostasis, mitochondria act both as protectors of liver function and as drivers of MASLD under metabolic stress [139,140]. For instance, mitochondrial fatty acid oxidation (FAO) is initially upregulated in response to increased intrahepatic lipid accumulation during early MASLD development [139]. However, as the disease progresses to MASH, mitochondrial oxidative metabolism becomes impaired, resulting in reduced ATP production and increased reactive oxygen species (ROS) generation [138,139,140,141,142].
HuR appears to play a hepatocyte-specific role in regulating mitochondrial function. In liver-specific HuR-deficient mice, increased hepatic steatosis is associated with significantly reduced ATP levels and downregulation of electron transport chain (ETC) proteins, including cytochrome c (Cycs), NADH: ubiquinone oxidoreductase subunit B6 (Ndufb6), and ubiquinol-cytochrome c reductase binding protein (Uqcrb), following four weeks of high-fat diet feeding [95]. Direct binding of HuR to the mRNAs of Cycs, Ndufb6, and Uqcrb was also demonstrated in Hepa1-6 cells, supporting a post-transcriptional regulatory role for HuR [95]. However, overexpression of Cycs alone did not restore mitochondrial ATP production in HuR-deficient livers, suggesting that HuR may influence mitochondrial function through multiple pathways [95].
Accumulation of ROS in dysfunctional mitochondria contributes to oxidative injury in MASLD [143]. To mitigate oxidative stress, the nuclear factor erythroid 2–related factor 2 (Nrf2, Nfe2l2) pathway is activated, inducing the expression of key cytoprotective genes such as superoxide dismutase 2 (Sod2) and heme oxygenase-1 (HO-1) [144]. HuR has been shown to enhance this antioxidant response; HuR-mediated upregulation of HO-1 is associated with improved survival in liver transplant recipients [75]. Furthermore, in rats fed a methionine- and choline-deficient (MCD) diet for six weeks, a positive correlation between hepatic HuR expression and both HO-1 and Sod2 expression was observed, further supporting HuR’s role in regulating oxidative stress responses in vivo [145].
In summary, HuR helps preserve mitochondrial function and protects against oxidative injury by regulating components of the electron transport chain and key antioxidant defense pathways, underscoring its potential role in preventing MASLD progression.
5.6. Role of HuR in Autophagy
Autophagic degradation of lipids (lipophagy) and damaged mitochondria (mitophagy) plays a protective role in preventing MASLD progression to MASH [146]. Although autophagy is constitutively active in all cell types, it is strongly induced under conditions of cellular stress and energy depletion [147]. During autophagy, autophagy-related proteins including Atg8/ microtubule-associated protein 1A/1B light chain (LC3) and selective autophagy receptors (SARs) such as sequestosome 1 (SQSTM1/p62) target cellular components for sequestration into double-membraned vesicles called autophagosomes [146]. Fusion of autophagosomes with lysosomes enables degradation of the sequestered materials [147]. In autophagy-deficient livers, clearance of lipid droplets and dysfunctional mitochondria is impaired, contributing to increased hepatic steatosis and mitochondrial dysfunction during MASLD progression [147,148].
While HuR’s specific role in autophagy during MASLD remains to be defined, growing evidence suggests that HuR functions as a positive regulator of autophagy in various cell types and disease contexts, particularly under oxidative and metabolic stress [149]. In liver cancer cell lines such as L-02 and Hep3B, HuR deficiency reduces autophagosome formation and autophagic flux. RNP-IP revealed direct HuR binding to the 3’ UTRs of Atg5, Atg12, and Atg16 mRNAs in Hep3B cells [150]. Similarly, in HuR-deficient mouse livers, reduced expression of Atg3, Atg5, and Atg7 was observed alongside more severe liver injury following acetaminophen (APAP) overdose and the direct binding of HuR to the 3’ UTRs of these genes was also confirmed in Hepa1-6 cells [151]. Given HuR’s established role in regulating autophagy across multiple tissues and models of liver injury, further investigation into its function in autophagy during MASLD development is highly warranted.
5.7. HuR Regulates Apoptosis and Cell Survival
As MASLD progresses to MASH, worsening liver dysfunction triggers activation of cell death pathways, particularly apoptosis. Apoptosis is considered the predominant mode of cell death in MASLD livers, and caspase substrates, such as cytokeratin-18, have been proposed as potential serum biomarkers for human MASH [4,152]. HuR, widely recognized as a survival-promoting factor in various cancers, regulates the stability of both pro-apoptotic transcripts (e.g., p53, p27, caspase-8, caspase-9, Fas, c-Myc) and anti-apoptotic transcripts (e.g., Bcl-2, SIRT1, Prothymosin α) [153,154,155]. However, under conditions of lethal cellular stress, HuR itself is cleaved and adopts a pro-apoptotic role by directly binding to and stabilizing Caspase-9 mRNA [24,25].
Apoptosis induction in MASLD is strongly associated with the accumulation of non-storage lipids, including FFAs and free cholesterol, in the liver [156,157]. Although HuR’s specific role in hepatocyte apoptosis during MASLD remains to be fully elucidated, elevated hepatic FFA and cholesterol levels have been observed in HuR-deficient mouse livers, correlating with increased liver injury after 24 weeks of high-fat diet feeding [85]. These non-storage lipids contribute to hepatocyte apoptosis by activating both intrinsic (organelle-mediated) and extrinsic (death receptor-mediated) apoptotic pathways [123,158,159]. Intrinsic apoptosis is initiated when FFAs and free cholesterol accumulate in organelle membranes, particularly the ER, mitochondria, and lysosomes, triggering ER stress, mitochondrial dysfunction, and release of lysosomal proteases [158]. RNP-IP analysis from healthy murine livers revealed direct binding of HuR to transcripts involved in the ER stress response, suggesting a potential role for HuR in regulating organelle stress pathways during MASLD [86]. In parallel, non-storage lipids also activate extrinsic apoptosis signaling. FFAs sensitize hepatocytes to apoptosis via JNK activation, upregulation of death receptor 5 (DR5) and Fas receptor, and activation of Bax [160,161]. Similarly, excessive free cholesterol accumulation within mitochondrial membranes increases mitochondrial sensitivity to oxidative stress and enhances hepatocyte susceptibility to TNFα- and Fas ligand-mediated apoptosis [123]. Both DR5 and Fas are highly expressed in human MASH livers, which also exhibit greater degrees of mitochondrial dysfunction [141,156,160]. In liver cancer models, HuR has been shown to inhibit apoptosis by directly binding to the 3′ UTR of Fas mRNA in HepG2 cells and repressing its translation [162]. Whether HuR plays a similar survival-promoting and anti-apoptotic role in hepatocytes during MASLD progression remains an important area for future investigation.
5.8. HuR in Hepatic Inflammation
Chronic liver injury and cell death during MASLD progression to MASH are accompanied by sustained immune activation, contributing to aberrant wound healing, inflammation, and fibrosis [4]. In MASLD, damage-associated molecular patterns (DAMPs) released from stressed or dying hepatocytes and pathogen-associated molecular patterns (PAMPs) from gut-derived microbial products activate Toll-like receptors (TLRs) on various hepatic cell types to initiate immune responses [163]. For instance, TLR4 activation by FFAs and unesterified cholesterol triggers pro-inflammatory signaling cascades, particularly in macrophages [164]. This leads to increased expression of pro-inflammatory cytokines and chemokines such as tumor necrosis factor alpha (Tnfα), interleukin 6 (IL-6), interleukin 1β (IL-1β), and C-C motif chemokine ligand 2 (Ccl2), the molecules tightly linked to MASLD progression and fibrosis [163]. TNFα acts on both parenchymal and non-parenchymal cells to promote immune cell recruitment, activation, and fibrogenic remodeling in the liver [163,165]. HuR has been shown to bind directly to the Tnf mRNA 3′ UTR in RAW 264.7 macrophages, and its expression is essential for LPS-induced Tnfα production in bone marrow-derived macrophages (BMDMs) [166,167,168]. Additional pro-inflammatory targets of HuR include Ccl2, inducible nitric oxide synthase (iNOS, Nos2), vascular endothelial growth factor (VEGF, vegfa), and cyclo-oxygenase 2 (COX-2, Ptgs2) [169]. Although HuR is generally regarded as a positive regulator of inflammation, studies in myeloid-specific HuR knockout models suggest a more nuanced role. In these models, HuR depletion exacerbates inflammation and mortality in response to LPS-induced endotoxemia and chemically induced colitis [170,171,172]. While the role of myeloid HuR in MASLD has not yet been studied, hepatocyte-specific HuR-deficient mice fed a WD diet for four weeks exhibit increased liver inflammation, characterized by F4/80-positive macrophage infiltration and upregulation of Ccl2, Tnf, Il6, and Il1b [87].
Beyond macrophages, the roles of other immune cells, including natural killer T (NKT) cells and adaptive immune populations, are gaining recognition in MASLD pathogenesis [173]. NKT cells exert both pro- and anti-inflammatory effects but are enriched in the liver during MASLD progression to MASH and fibrosis [173,174]. Adaptive immune subsets, including Th1, Th17, and B cells, also increase in abundance and activity with worsening disease severity [173]. HuR has been shown to influence the adaptive immune response; HuR-deficient T and B cells exhibit impaired activation and proliferation upon inflammatory stimulation [73,175,176]. Moreover, postnatal HuR depletion results in apoptosis of immune progenitor cells in the bone marrow and thymus, highlighting HuR’s essential role in immune cell survival [23]. In the context of MASLD, inflammation and tumorigenesis are exacerbated in hepatocyte- and cholangiocyte-specific HuR-deficient mice after 14 months of HFD-CD feeding [86]. These livers show increased infiltration of NKT cells and CD8+ T cells, immune subsets commonly associated with severe inflammation and fibrosis in MASLD [86,173,174].
In summary, HuR exerts a context-dependent and multifaceted influence on inflammation, regulating both innate and adaptive immune responses. However, its specific roles within individual immune cell populations in the context of MASLD remain unexplored. Further investigation is warranted to elucidate how immune cell–specific HuR activity contributes to immune-mediated mechanisms underlying MASLD progression.
5.9. HuR in Liver Fibrosis
Fibrosis is the major predictor of liver-related morbidity and mortality in MASLD patients, significantly increasing the risk of liver failure and hepatocellular carcinoma (HCC) [177,178]. Hepatic fibrosis develops when chronic inflammation, sustained cell death, and aberrant wound healing activate normally quiescent fibroblasts, primarily hepatic stellate cells (HSCs), into highly proliferative, contractile, and collagen-secreting myofibroblasts [179,180]. HSC activation is driven by various profibrotic signals, including transforming growth factor β1 (TGFβ1) and platelet-derived growth factor (PDGF) [180,181]. In MASLD, free cholesterol accumulation within HSCs enhances their activation, while other pro-fibrotic cues, such as Notch signaling activation in hepatocytes and lncRNA H19 induction in cholangiocytes, further amplify fibrogenic responses [87,182,183,184].
HuR plays a complex and cell type-specific role in liver fibrosis. In hepatocytes, HuR appears protective, as its deficiency exacerbates fibrosis progression in MASLD mouse models [86,87]. Hepatocyte- and cholangiocyte-specific HuR deficiency leads to hepatic cholesterol accumulation and marked dysregulation of cholesterol metabolism, conditions known to promote HSC activation [86]. These HuR-deficient mice also show elevated expression of the Notch signaling mediator osteopontin (OPN) [86] and, under WDSW diet feeding, increased H19 expression [87], both associated with more severe fibrosis.
In contrast, HuR expression in hepatic myofibroblasts (activated HSCs) promotes both bile duct ligation and CCl4-induced fibrosis development [29,30]. In murine HSCs, TGFβ1 and PDGF both induce cytoplasmic localization of HuR, while PDGF specifically increases HuR expression [29,30]. In HuR-deficient rat HSCs, PDGF-induced proliferation and migration are significantly impaired [29]. RNP-IP experiments in the activated HSC line CFSC-8B revealed that HuR binds directly to transcripts encoding pro-proliferative (e.g., cyclin D1, cyclin B1) and motility-related proteins (e.g., MMP9, actin) following PDGF stimulation [29]. Similarly, HuR directly binds to sphingosine kinase 1 (Sphk1) mRNA, which is required for TGFβ1-mediated induction of Col1a1 and α-SMA in both human HSCs LX-2 cells and murine primary HSCs [30]. In addition to promoting HSC activation, HuR may also contribute to fibrosis resolution. In a recent study, sorafenib-induced ferroptosis in HSCs was dependent on HuR-mediated stabilization of the autophagy gene beclin 1 (Becn1), suggesting a role for HuR in promoting autophagy and cell death in activated HSCs under therapeutic stress [185].
The transition of HSCs from a quiescent to a myofibroblast-like phenotype is a critical determinant of both fibrosis progression and resolution in the liver [177]. HuR has been shown to regulate differentiation in various cell types, including enterocytes and immune cells, and is implicated in Wnt/β-catenin signaling during cancer progression [179,186,187]. Although HuR’s role in HSC differentiation remains incompletely defined, there is growing interest in its interaction with key developmental signaling pathways such as Notch, Wnt, and Hedgehog, which are known to influence fibrogenesis. In summary, HuR functions as a pivotal post-transcriptional regulator of liver fibrosis, with cell-type–specific effects that may drive both the progression and resolution of fibrogenic responses in MASLD. Future research should aim to dissect the signaling networks through which HuR modulates HSC plasticity, with the goal of identifying new therapeutic strategies to prevent or reverse hepatic fibrosis.
6. Role of HuR in Extrahepatic Metabolic Comorbidities
MASLD is closely linked to a range of extrahepatic metabolic disorders, including obesity, T2DM, and cardiovascular disease [1]. Reflecting the central role of systemic metabolic dysfunction in MASLD pathogenesis, current diagnostic criteria require the presence of at least one additional cardiometabolic risk factor, such as obesity or insulin resistance, in conjunction with hepatic steatosis to establish a MASLD diagnosis [91]. In this section, we provide a brief overview of HuR’s emerging roles in extrahepatic metabolic diseases commonly associated with MASLD, highlighting its potential contribution to the broader metabolic syndrome context.
6.1. Obesity
6.1.1. Overview of Obesity and Its Association with MASLD
Obesity is a well-established independent risk factor for MASLD progression to MASH, even in the absence of other metabolic comorbidities [9]. The global prevalence of obesity has increased dramatically over the past 50 years, driven by a complex interplay of genetic, behavioral, environmental, and socioeconomic factors [188]. Understanding the mechanisms linking obesity to metabolic disease progression is essential for improving both survival and quality of life in affected individuals. While chronic low-grade inflammation, insulin resistance, and dyslipidemia are recognized contributors to MASLD development in obese individuals, the molecular drivers of these pathophysiological changes remain incompletely defined [188]. For instance, although adipokine signaling dysregulation is known to exacerbate hepatic steatosis and insulin resistance [9], the specific regulatory networks involved are still not fully understood [189]. Given these knowledge gaps, investigating the role of HuR in adipose tissue function and obesity development may offer novel insights into the mechanisms driving MASLD and identify potential therapeutic targets.
6.1.2. Role of HuR in Adipocyte Differentiation and Obesity
HuR is a key post-transcriptional regulator of adipocyte function, and its expression is dynamically regulated during adipocyte differentiation and obesity development. In vitro differentiation of 3T3-L1 pre-adipocytes is associated with increased HuR expression and cytoplasmic localization [22]. In contrast, in vivo studies show that mature adipocytes express lower levels of HuR compared to pre-adipocytes residing in the stromal vascular fraction [31]. Importantly, obesity is associated with further suppression of HuR expression in adipose tissue in both humans and mice [31]. In this section, we highlight HuR’s role in adipocyte differentiation and dysfunction, with an emphasis on mechanisms relevant to MASLD [22,31,32,190,191,192].
Adipocyte differentiation from pre-adipocytes into mature and lipid-storing adipocytes is essential for maintaining lipid homeostasis. HuR supports this process by stabilizing mRNAs encoding key adipogenic regulators, including CCAAT/enhancer-binding protein beta (C/EBPβ) and solute carrier family 2 member 1 (Slc2a1, also known as GLUT1) [22]. In addition, HuR directly stabilizes transcripts encoding peroxisome proliferator-activated receptor gamma (Pparγ) and adiponectin (Adipoq), both of which are critical for adipocyte function and insulin sensitivity [190]. The activity and localization of HuR during adipocyte differentiation are modulated by regulatory signals, including Pparγ and the long non-coding RNA CAAlnc1. Binding of CAAlnc1 to HuR interferes with HuR-mediated stabilization of adipogenic transcription factor mRNAs in C3H10 cells [192].
In vivo, HuR is essential for maintaining adipose tissue health and systemic metabolic balance. Adipocyte-specific HuR deletion in mice leads to exacerbation of obesity, MASLD, and cardiovascular disease [31,32,191]. Following 16 weeks of high-fat diet feeding, adipocyte-specific HuR-deficient mice exhibited significantly greater hepatic steatosis than wild-type controls. This was partly attributed to reduced expression of patatin-like phospholipase domain-containing 2 (Pnpla2), which encodes adipose triglyceride lipase (ATGL), a key enzyme in adipocyte lipolysis [31]. Impaired ATGL activity resulted in adipocyte hypertrophy, inflammation, and systemic insulin resistance, all of which promote MASLD progression. Further studies demonstrated that HuR deficiency selectively impacted abdominal (epididymal) adipose depots, but not subcutaneous (inguinal) depots. This regional HuR loss was associated with increased adipogenesis and pro-inflammatory cytokine expression, a pattern closely linked to obesity-associated MASLD in humans [32]. In the context of cardiovascular disease, HuR deficiency in adipocytes led to cardiac hypertrophy and fibrosis, despite unaltered HuR expression in heart tissue. Transcriptomic analyses revealed significant alterations in both adipose and cardiac gene expression, and the pro-inflammatory environment in HuR-deficient adipose tissue likely contributed to a systemic inflammatory state, promoting pathological cardiac remodeling [191].
In summary, adipocyte-specific HuR is a critical regulator of adipocyte differentiation, lipid metabolism, and systemic inflammation. Its loss not only promotes adipose tissue dysfunction and obesity but also contributes to the development of MASLD and cardiovascular disease, underscoring HuR’s central role in metabolic health [31,32,191].
6.2. Cardiovascular Disease and Type II Diabetes Mellitus
6.2.1. Overview of Cardiovascular Disease, Diabetic Cardiomyopathy and Their Link to MASLD
Similar to obesity, T2DM and MASLD are closely interconnected metabolic disorders [193]. While approximately 30% of the global adult population is estimated to have MASLD, the prevalence rises to nearly 60% among individuals with T2DM, underscoring their frequent coexistence [1]. Key pathophysiological contributors to this relationship include systemic insulin resistance and hyperglycemia, both hallmark features of T2DM [100]. Conversely, growing evidence suggests that MASLD itself may act as a precursor to T2DM, rather than merely a consequence [194]. Mechanistically, hepatic steatosis contributes to systemic insulin resistance, chronic low-grade inflammation, and dyslipidemia, all of which drive T2DM pathogenesis. Moreover, progression of MASLD to MASH and advanced fibrosis further increases the risk for developing T2DM [194].
Cardiovascular disease (CVD), including myocardial infarction, ischemic stroke, and heart failure, is the leading cause of death in patients with both MASLD [195] and T2DM [196]. In T2DM, chronic hyperglycemia-induced vascular injury underlies multiple cardiovascular complications, including atherosclerotic cardiovascular disease and diabetic cardiomyopathy [197]. In diabetic cardiomyopathy, insulin resistance and hyperglycemia contribute to persistent inflammation, oxidative stress, cardiomyocyte death, and cardiac remodeling, leading to heart failure independent of atherosclerosis [198]. However, atherosclerosis frequently co-occurs with T2DM and contributes to the 2-fold or greater increase in CVD risk observed in this population [196]. Similarly, MASLD contributes to CVD development through shared mechanisms, including hepatic steatosis–induced insulin resistance, chronic systemic inflammation, and atherogenic dyslipidemia [5]. These overlapping metabolic disturbances suggest that MASLD is not only a liver disease but also a significant contributor to cardiometabolic risk.
6.2.2. Role of HuR in Metabolic Dysfunction-Associated Cardiovascular Disease
HuR expression and cytoplasmic localization in cardiac tissue are associated with CVD development in both human patients and experimental animal models [199]. While some discrepancies in the literature exist regarding the direction of HuR’s effects [200], the majority of studies suggest that HuR induction contributes to cardiac dysfunction in conditions such as ischemia/reperfusion (I/R) injury [201,202,203], pressure overload–induced cardiac remodeling [204,205], and myocardial infarction (MI) [206]. In diabetic (db/db) mice, HuR is upregulated not only in cardiomyocytes but also in infiltrating immune cells within the heart [203]. Correspondingly, elevated HuR expression has been observed in myocardial tissue from heart failure patients [203,204], including those with diabetes [67].
HuR contributes to CVD pathogenesis through several mechanisms, including cardiomyocyte death, inflammatory cytokine production, and immune cell infiltration, all of which drive adverse cardiac remodeling and progression to heart failure [198]. Following ischemic injury, HuR induction in the infarct zone is associated with increased expression of pro-inflammatory cytokines such as TNFα, IL-1β, CCL2, and the pro-apoptotic factor p53. In mice with cardiomyocyte-specific HuR depletion, infarct-associated cell death and inflammation were significantly reduced compared to controls [206]. In a coronary artery–ablation model of murine I/R injury, pharmacological inhibition of HuR led to reduced Il6 and Tnfa expression in the infarcted region [201]. Interestingly, while cardiomyocyte apoptosis (as measured by TUNEL staining) remained unchanged between treatment groups, CD68+ immune cell infiltration into the infarct site was reduced in HuR-inhibited mice, correlating with improved cardiac function and attenuated fibrosis [201]. Prolonged inflammation contributes to fibrotic remodeling and heart failure. In diabetic patients with heart failure, an increased number of HuR+ and F4/80+ macrophages has been observed in failing hearts [203]. In mouse models, macrophage-specific HuR deletion led to reduced pro-inflammatory cytokine expression and fibrosis in an angiotensin II–induced model of cardiac injury [203]. Similarly, fibroblast-specific HuR deletion resulted in decreased myofibroblast activation (e.g., reduced α-SMA expression), less fibrosis, and preserved cardiac function following pressure overload–induced injury [205]. In a separate study, cardiomyocyte-specific HuR deletion protected against cardiac hypertrophy and fibrosis in a pressure overload model, in part by suppressing TGF-β expression in cardiomyocytes [204].
In summary, HuR contributes to the development and progression of cardiovascular disease under conditions of metabolic stress by promoting inflammation, cell death, immune infiltration, and fibrotic remodeling in the heart. Its cell-specific functions in cardiomyocytes, macrophages, and fibroblasts highlight HuR as a multifaceted regulator of cardiac pathology in metabolic disease contexts such as MASLD and T2DM.
7. Conclusions and Future Perspectives
As a ubiquitously expressed and highly dynamic regulator of post-transcriptional gene expression, HuR plays a critical role in numerous cellular processes. Expanding our understanding of HuR function in the context of MASLD holds significant potential for informing the development of novel therapeutic strategies. However, HuR’s essential role in maintaining homeostasis across multiple tissues and cell types presents a substantial challenge for its therapeutic targeting. This complexity is exemplified by the opposing effects of HuR in different liver cell populations. While hepatocyte-specific HuR protects against diet-induced MASLD progression [86], HuR activation in HSCs promotes fibrogenesis and chronic liver injury [29]. As such, indiscriminate modulation of HuR in both hepatocytes and HSCs could inadvertently exacerbate disease progression or produce limited therapeutic benefit. Similarly, although reduced hepatic HuR expression is associated with worsening MASLD [85], HuR induction and cytoplasmic localization are poor prognostic markers in both hepatocellular carcinoma [207] and heart failure [203], underscoring the need for context- and cell-specific therapeutic approaches.
To address this challenge, several strategies have been proposed to enhance therapeutic specificity. One approach involves cell-targeted delivery, such as conjugating small-molecule inhibitors to retinol for selective uptake by retinol-storing HSCs, thereby limiting off-target effects while inhibiting fibrosis progression [208]. Additionally, rather than targeting HuR expression broadly, interventions could focus on specific functional aspects of HuR, including nuclear-to-cytoplasmic translocation, dimerization, post-translational modifications, or mRNA-binding activity—each of which may contribute differentially to HuR’s role in disease [209]. Importantly, genetic HuR depletion in metabolic tissues such as the liver, heart, and adipose tissue typically does not disrupt basal tissue function unless triggered by metabolic stressors like lipid overload [31,95,204]. This observation suggests that therapeutic inhibition of HuR may be selectively toxic under pathological conditions, allowing for dose titration strategies to minimize effects on healthy tissue. For example, KH-3, a small-molecule HuR inhibitor that blocks mRNA binding, has shown promise in selectively inhibiting HuR in tumor cells with high HuR expression, while sparing normal tissues [83].
Although further research is necessary to develop safe and effective HuR-targeted therapies, a deeper understanding of HuR’s multifaceted role in metabolic disease offers new opportunities for precision medicine. At the very least, this insight highlights the need for caution in the use of HuR inhibitors for treating cancer or age- and inflammation-related diseases. Ultimately, unraveling the mechanisms by which HuR contributes to metabolic homeostasis and dysfunction may yield novel therapeutic targets for MASLD and related cardiometabolic disorders.
Author Contributions
Conceptualization, N.E. and Y.Z.; writing—original draft preparation, N.E. and Y.Z.; writing—review and editing, N.E., E. J., F.A., Y.Z.; funding acquisition, Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIH grants R01DK119131 and K22CA184146.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
BMDMs: Bone Marrow-Derived Macrophages; CCL2: Chemokine (C-C Motif) Ligand 2; DAMP: Damage-Associated Molecular Patterns; HCC: hepatocellular carcinoma; HSC: hepatic stellate cell; IL: interleukin; LPS: lipopolysaccharide; MASH: metabolic dysfunction-associated steatohepatitis; MASL: metabolic dysfunction-associated steatotic liver; MASLD: metabolic dysfunction-associated steatotic liver disease; MCD: Methionine Choline Deficient; NAFLD: nonalcoholic fatty liver disease; NF-κB: Nuclear Factor Kappa B; NLRP3: NLR Family Pyrin Domain Containing 3; NOS2: Nitric Oxide Synthase 2; PPAR: Peroxisome Proliferator-Activated Receptor; ROS: reactive oxygen species; T2DM: Type II Diabetes Mellitus; TGF-b: Transforming Growth Factor Beta; TLR: Toll-like receptor; TNFa: Tumor Necrosis Factor Alpha.
References
- Wong, V.W.S.; Ratziu, V.; Bugianesi, E.; Francque, S.; Zelber-Sagi, S.; Valenti, L.; Roden, M.; Schick, F.; Yki-Järvinen, H.; Gastaldelli, A.; et al. EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). Journal of Hepatology 2024, 81, 492–542. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. In Annual Review of Pathology: Mechanisms of Disease, Vol 13, Abbas, A.K., Aster, J.C., Eds.; Annual Review of Pathology-Mechanisms of Disease; 2018; Volume 13, pp. 321-350.
- Targher, G.; Byrne, C.D.; Tilg, H. MASLD: a systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef]
- Mantovani, A.; Csermely, A.; Petracca, G.; Beatrice, G.; Corey, K.E.; Simon, T.G.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: an updated systematic review and meta-analysis. Lancet Gastroenterology & Hepatology 2021, 6, 903–913. [Google Scholar] [CrossRef]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized Controlled Trial Testing the Effects of Weight Loss on Nonalcoholic Steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef]
- Golabi, P.; Paik, J.M.; Kumar, A.; Al Shabeeb, R.; Eberly, K.E.; Cusi, K.; Gundurao, N.; Younossi, Z.M. Nonalcoholic fatty liver disease (NAFLD) and associated mortality in individuals with type 2 diabetes, pre-diabetes, metabolically unhealthy, and metabolically healthy individuals in the United States. Metabolism-Clinical and Experimental 2023, 146. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism-Clinical and Experimental 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Ma, W.J.; Cheng, S.; Campbell, C.; Wright, A.; Furneaux, H. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. Journal of Biological Chemistry 1996, 271, 8144–8151. [Google Scholar] [CrossRef]
- Campos, A.R.; Grossman, D.; White, K. Mutant alleles at the locus elav in Drosophila melanogaster lead to nervous system defects. A developmental-genetic analysis. J Neurogenet 1985, 2, 197–218. [Google Scholar] [CrossRef]
- Zhu, H.; Hasman, R.A.; Barron, V.A.; Luo, G.B.; Lou, H. A nuclear function of Hu proteins as neuron-specific alternative RNA processing regulators. Molecular Biology of the Cell 2006, 17, 5105–5114. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, S.; Jens, M.; Theil, K.; Schwanhäusser, B.; Selbach, M.; Landthaler, M.; Rajewsky, N. Transcriptome-wide Analysis of Regulatory Interactions of the RNA-Binding Protein HuR. Molecular Cell 2011, 43, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, T.; Hitti, E.; Khabar, K.S.A. ARED-Plus: an updated and expanded database of AU-rich element-containing mRNAs and pre-mRNAs. Nucleic Acids Research 2018, 46, D218–D220. [Google Scholar] [CrossRef]
- Khabar, K.S.A. The AU-rich transcriptome: More than interferons and cytokines, and its role in disease. Journal of Interferon and Cytokine Research 2005, 25, 1–10. [Google Scholar] [CrossRef]
- Hinman, M.N.; Lou, H. Diverse molecular functions of Hu proteins. Cellular and Molecular Life Sciences 2008, 65, 3168–3181. [Google Scholar] [CrossRef]
- Srikantan, S.; Gorospe, M. HuR function in disease. Frontiers in Bioscience-Landmark 2012, 17, 189–205. [Google Scholar] [CrossRef]
- Myer, V.E.; Fan, X.H.C.; Steitz, J.A. Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. Embo Journal 1997, 16, 2130–2139. [Google Scholar] [CrossRef]
- Guo, X.; Connick, M.C.; Vanderhoof, J.; Ishak, M.A.; Hartley, R.S. MicroRNA-16 Modulates HuR Regulation of Cyclin E1 in Breast Cancer Cells. International Journal of Molecular Sciences 2015, 16, 7112–7132. [Google Scholar] [CrossRef]
- Haga, Y., et al. Increased expression of long non-coding RNA FIRRE promotes hepatocellular carcinoma by HuR-CyclinD1 axis signaling. Journal of Biological Chemistry 2024, 300. [CrossRef]
- Wang, W.G.; Caldwell, M.C.; Lin, S.K.; Furneaux, H.; Gorospe, M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. Embo Journal 2000, 19, 2340–2350. [Google Scholar] [CrossRef]
- Gantt, K.; Cherry, J.; Tenney, R.; Karschner, V.; Pekala, P.H. An early event in adipogenesis, the nuclear selection of the CCAAT enhancer-binding protein β (C/EBPβ) mRNA by HuR and its translocation to the cytosol. Journal of Biological Chemistry 2005, 280, 24768–24774. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Aguila, H.L.; Michaud, J.; Ai, Y.X.; Wu, M.T.; Hemmes, A.; Ristimaki, A.; Guo, C.Y.; Furneaux, H.; Hla, T. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. Journal of Clinical Investigation 2009, 119, 3530–3543. [Google Scholar] [CrossRef] [PubMed]
- Mazroui, R.; Di Marco, S.; Clair, E.; von Roretz, C.; Tenenbaum, S.A.; Keene, J.D.; Saleh, M.; Gallouzi, I.E. Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. Journal of Cell Biology 2008, 180, 113–127. [Google Scholar] [CrossRef]
- von Roretz, C.; Lian, X.J.; Macri, A.M.; Punjani, N.; Clair, E.; Drouin, O.; Dormoy-Raclet, V.; Ma, J.F.; Gallouzi, I.E. Apoptotic-induced cleavage shifts HuR from being a promoter of survival to an activator of caspase-mediated apoptosis. Cell Death and Differentiation 2013, 20, 154–168. [Google Scholar] [CrossRef]
- Pang, L.J.; Tian, H.Y.; Chang, N.; Yi, J.; Xue, L.X.; Jiang, B.; Gorospe, M.; Zhang, X.W.; Wang, W.G. Loss of CARM1 is linked to reduced HuR function in replicative senescence. Bmc Molecular Biology 2013, 14. [Google Scholar] [CrossRef]
- Yi, J.; Chang, N.; Liu, X.W.; Guo, G.; Xue, L.X.; Tong, T.J.; Gorospe, M.; Wang, W.G. Reduced nuclear export of HuR mRNA by HuR is linked to the loss of HuR in replicative senescence. Nucleic Acids Research 2010, 38, 1547–1558. [Google Scholar] [CrossRef]
- Katsanou, V.; Milatos, S.; Yiakouvaki, A.; Sgantzis, N.; Kotsoni, A.; Alexiou, M.; Harokopos, V.; Aidinis, V.; Hemberger, M.; Kontoyiannis, D.L. The RNA-Binding Protein Elavl1/HuR Is Essential for Placental Branching Morphogenesis and Embryonic Development. Molecular and Cellular Biology 2009, 29, 2762–2776. [Google Scholar] [CrossRef]
- Woodhoo, A.; Iruarrizaga-Lejarreta, M.; Beraza, N.; García-Rodríguez, J.L.; Embade, N.; Fernández-Ramos, D.; Martínez-López, N.; Gutiérrez-De Juan, V.; Arteta, B.; Caballeria, J.; et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology 2012, 56, 1870–1882. [Google Scholar] [CrossRef]
- Ge, J.J.; Chang, N.; Zhao, Z.X.; Tian, L.; Duan, X.H.; Yang, L.; Li, L.Y. Essential Roles of RNA-binding Protein HuR in Activation of Hepatic Stellate Cells Induced by Transforming Growth Factor-β1. Scientific Reports 2016, 6. [Google Scholar] [CrossRef]
- Li, J.Y.; Gong, L.; Liu, S.Z.; Zhang, Y.J.; Zhang, C.M.; Tian, M.; Lu, H.X.; Bu, P.L.; Yang, J.M.; Ouyang, C.H.; et al. Adipose HuR protects against diet-induced obesity and insulin resistance. Nature Communications 2019, 10. [Google Scholar] [CrossRef]
- Siang, D.T.C.; Lim, Y.C.; Kyaw, A.M.M.; Win, K.N.; Chia, S.Y.; Degirmenci, U.; Hu, X.; Tan, B.C.; Walet, A.C.E.; Sun, L.; et al. The RNA-binding protein HuR is a negative regulator in adipogenesis. Nature Communications 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative Regulatory Mapping Indicates that the RNA-Binding Protein HuR Couples Pre-mRNA Processing and mRNA Stability. Molecular Cell 2011, 43, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Pabis, M.; Popowicz, G.M.; Stehle, R.; Fernández-Ramos, D.; Asami, S.; Warner, L.; García-Mauriño, S.M.; Schlundt, A.; Martínez-Chantar, M.L.; Díaz-Moreno, I.; et al. HuR biological function involves RRM3-mediated dimerization and RNA binding by all three RRMs. Nucleic Acids Research 2019, 47, 1011–1029. [Google Scholar] [CrossRef]
- Wang, H.; Zeng, F.X.; Liu, Q.; Liu, H.H.; Liu, Z.X.; Niu, L.W.; Teng, M.K.; Li, X. The structure of the ARE-binding domains of Hu antigen R (HuR) undergoes conformational changes during RNA binding. Acta Crystallographica Section D-Structural Biology 2013, 69, 373–380. [Google Scholar] [CrossRef]
- Barreau, C.; Paillard, L.; Osborne, H.B. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Research 2005, 33, 7138–7150. [Google Scholar] [CrossRef]
- Kenan, D.J.; Query, C.C.; Keene, J.D. RNA recognition: towards identifying determinants of specificity. Trends Biochem Sci 1991, 16, 214–220. [Google Scholar] [CrossRef]
- Ma, W.J.; Chung, S.; Furneaux, H. The Elav-like proteins bind to AU-rich elements and to the poly(A) tail of mRNA. Nucleic Acids Research 1997, 25, 3564–3569. [Google Scholar] [CrossRef]
- Brennan, C.M.; Gallouzi, I.E.; Steitz, J.A. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. Journal of Cell Biology 2000, 151, 1–13. [Google Scholar] [CrossRef]
- Fan, X.C.; Steitz, J.A. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J 1998, 17, 3448–3460. [Google Scholar] [CrossRef]
- Gallouzi, I.E.; Brennan, C.M.; Stenberg, M.G.; Swanson, M.S.; Eversole, A.; Maizels, N.; Steitz, J.A. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proceedings of the National Academy of Sciences of the United States of America 2000, 97, 3073–3078. [Google Scholar] [CrossRef]
- Fan, X.H.C.; Steitz, J.A. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proceedings of the National Academy of Sciences of the United States of America 1998, 95, 15293–15298. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.H.; Hoelz, A. The Structure of the Nuclear Pore Complex (An Update). In Annual Review of Biochemistry, Vol 88, Kornberg, R.D., Ed.; Annual Review of Biochemistry; 2019; Volume 88, pp. 725-783.
- Wing, C.E.; Fung, H.Y.J.; Chook, Y.M. Karyopherin-mediated nucleocytoplasmic transport. Nature Reviews Molecular Cell Biology 2022, 23, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Gallouzi, I.E.; Brennan, C.M.; Steitz, J.A. Protein ligands mediate the CRM1-dependent export of HuR in response to heat shock. Rna 2001, 7, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Güttinger, S.; Mühlhäusser, P.; Koller-Eichhorn, R.; Brennecke, J.; Kutay, U. Transportin2 functions as importin and mediates nuclear import of HuR. Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 2918–2923. [Google Scholar] [CrossRef]
- Wang, W.G.; Yang, X.L.; Kawai, T.; de Silanes, I.L.; Mazan-Mamczarz, K.; Chen, P.L.; Chook, Y.M.; Quensel, C.; Köhler, M.; Gorospe, M. AMP-activated protein kinase-regulated phosphorylation and acetylation of importin α1 -: Involvement in the nuclear import of RNA-binding protein HuR. Journal of Biological Chemistry 2004, 279, 48376–48388. [Google Scholar] [CrossRef]
- Al-Ahmadi, W.; Al-Ghamdi, M.; Al-Haj, L.; Al-Saif, M.; Khabar, K.S.A. Alternative polyadenylation variants of the RNA binding protein, HuR: abundance, role of AU-rich elements and auto-Regulation. Nucleic Acids Research 2009, 37, 3612–3624. [Google Scholar] [CrossRef]
- Govindaraju, S.; Lee, B.S. Kruppel -Like Factor 8 is a Stress-Responsive Transcription Factor that Regulates Expression of HuR. Cellular Physiology and Biochemistry 2014, 34, 519–532. [Google Scholar] [CrossRef]
- Jeyaraj, S.C.; Dakhlallah, D.; Hill, S.R.; Lee, B.S. Expression and distribution of HuR during ATP depletion and recovery in proximal tubule cells. American Journal of Physiology-Renal Physiology 2006, 291, F1255–F1263. [Google Scholar] [CrossRef]
- King, P.H.; Fuller, J.J.; Nabors, L.B.; Detloff, P.J. Analysis of the 5’ end of the mouse Elavl1 (mHuA) gene reveals a transcriptional regulatory element and evidence for conserved genomic organization. Gene 2000, 242, 125–131. [Google Scholar] [CrossRef]
- Kang, M.J.; Ryu, B.K.; Lee, M.G.; Han, J.; Lee, J.H.; Ha, T.K.; Byun, D.S.; Chae, K.S.; Lee, B.H.; Chun, H.S.; et al. NF-κB Activates Transcription of the RNA-Binding Factor HuR, via PI3K-AKT Signaling, to Promote Gastric Tumorigenesis. Gastroenterology 2008, 135, 2030–2042. [Google Scholar] [CrossRef]
- Ayupova, D.A.; Singh, M.; Leonard, E.C.; Basile, D.P.; Lee, B.S. Expression of the RNA-stabilizing protein HuR in ischemia-reperfusion injury of rat kidney. American Journal of Physiology-Renal Physiology 2009, 297, F95–F105. [Google Scholar] [CrossRef] [PubMed]
- Pullmann, R.; Kim, H.H.; Abdelmohsen, K.; Lal, A.; Martindale, J.L.; Yang, X.L.; Gorospe, M. Analysis of turnover and translation regulatory RNA-Binding protein expression through binding to cognate mRNAs. Molecular and Cellular Biology 2007, 27, 6265–6278. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Xiao, L.; Wang, J.Y. HuR and Its Interactions with Noncoding RNAs in Gut Epithelium Homeostasis and Diseases. Frontiers in Bioscience-Landmark 2023, 28. [Google Scholar] [CrossRef]
- Lal, A.; Mazan-Mamczarz, K.; Kawai, T.; Yang, X.L.; Martindale, J.L.; Gorospe, M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. Embo Journal 2004, 23, 3092–3102. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Chen, X.R.; Cheng, R.J.; Yang, F.; Yu, M.C.; Wang, C.; Cui, S.F.; Hong, Y.T.; Liang, H.W.; Liu, M.H.; et al. The Jun/miR-22/HuR regulatory axis contributes to tumourigenesis in colorectal cancer. Molecular Cancer 2018, 17. [Google Scholar] [CrossRef]
- Tang, Y.J.; Wu, W.; Chen, Q.Q.; Liu, S.H.; Zheng, Z.Y.; Cui, Z.L.; Xu, J.P.; Xue, Y.; Lin, D.H. miR-29b-3p suppresses the malignant biological behaviors of AML cells via inhibiting NF-κB and JAK/STAT signaling pathways by targeting HuR. Bmc Cancer 2022, 22. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Srikantan, S.; Kuwano, Y.; Gorospe, M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 20297–20302. [Google Scholar] [CrossRef]
- Grammatikakis, I.; Abdelmohsen, K.; Gorospe, M. Posttranslational control of HuR function. Wiley Interdisciplinary Reviews-Rna 2017, 8. [Google Scholar] [CrossRef]
- Liu, L.; Rao, J.N.; Zou, T.T.; Xiao, L.; Wang, P.Y.; Turner, D.J.; Gorospe, M.; Wang, J.Y. Polyamines Regulate c-Myc Translation through Chk2-dependent HuR Phosphorylation. Molecular Biology of the Cell 2009, 20, 4885–4898. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Pullmann, R.; Lai, A.; Kim, H.H.; Galban, S.; Yang, X.L.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.A.; et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Molecular Cell 2007, 25, 543–557. [Google Scholar] [CrossRef]
- Doller, A.; Huwiler, A.; Müller, R.; Radeke, H.H.; Pfeilschifter, J.; Eberhardt, W. Protein kinase Cα-dependent phosphorylation of the mRNA-stabilizing factor HuR:: Implications for posttranscriptional regulation of cyclooxygenase-2. Molecular Biology of the Cell 2007, 18, 2137–2148. [Google Scholar] [CrossRef] [PubMed]
- Fernau, N.S.; Fugmann, D.; Leyendecker, M.; Reimann, K.; Grether-Beck, S.; Galban, S.; Ale-Agha, N.; Krutmann, J.; Klotz, L.O. Role of HuR and p38MAPK in ultraviolet B-induced post-transcriptional regulation of COX-2 expression in the human keratinocyte cell line HaCaT. J Biol Chem 2010, 285, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Lafarga, V.; Cuadrado, A.; Lopez de Silanes, I.; Bengoechea, R.; Fernandez-Capetillo, O.; Nebreda, A.R. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol 2009, 29, 4341–4351. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalaf, H.H.; Aboussekhra, A. ATR controls the UV-related upregulation of the CDKN1A mRNA in a Cdk1/HuR-dependent manner. Mol Carcinog 2014, 53, 979–987. [Google Scholar] [CrossRef]
- Jeyabal, P.; Thandavarayan, R.A.; Joladarashi, D.; Babu, S.S.; Krishnamurthy, S.; Bhimaraj, A.; Youker, K.A.; Kishore, R.; Krishnamurthy, P. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochemical and Biophysical Research Communications 2016, 471, 423–429. [Google Scholar] [CrossRef]
- David, P.S.; Tanveer, R.; Port, J.D. FRET-detectable interactions between the ARE binding proteins, HuR and p37AUF1. Rna 2007, 13, 1453–1468. [Google Scholar] [CrossRef]
- Fialcowitz-White, E.J.; Brewer, B.Y.; Ballin, J.D.; Willis, C.D.; Toth, E.A.; Wilson, G.M. Specific protein domains mediate cooperative assembly of HuR oligomers on AU-rich mRNA-destabilizing sequences. Journal of Biological Chemistry 2007, 282, 20948–20959. [Google Scholar] [CrossRef]
- Cho, S.J.; Zhang, J.; Chen, X.B. RNPC1 modulates the RNA-binding activity of, and cooperates with, HuR to regulate p21 mRNA stability. Nucleic Acids Research 2010, 38, 2256–2267. [Google Scholar] [CrossRef]
- de Silanes, I.L.; Zhan, M.; Lal, A.; Yang, X.L.; Gorospe, M. Identification of a target RNA motif for RNA-binding protein HuR. Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 2987–2992. [Google Scholar] [CrossRef]
- Noh, J.H.; Kim, K.M.; Abdelmohsen, K.; Yoon, J.H.; Panda, A.C.; Munk, R.; Kim, J.; Curtis, J.; Moad, C.A.; Wohler, C.M.; et al. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev 2016, 30, 1224–1239. [Google Scholar] [CrossRef]
- Diaz-Munoz, M.D.; Bell, S.E.; Fairfax, K.; Monzon-Casanova, E.; Cunningham, A.F.; Gonzalez-Porta, M.; Andrews, S.R.; Bunik, V.I.; Zarnack, K.; Curk, T.; et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nature Immunology 2015, 16, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Q.; Xu, L. The RNA-binding protein HuR in human cancer: A friend or foe. Advanced Drug Delivery Reviews 2022, 184. [Google Scholar] [CrossRef] [PubMed]
- Dery, K.J.; Nakamura, K.; Kadono, K.; Hirao, H.; Kageyama, S.; Ito, T.; Kojima, H.; Kaldas, F.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Human Antigen R (HuR): A New Regulator of Heme Oxygenase-1 Cytoprotection in Mouse and Human Liver Transplantation. American Journal of Transplantation 2020, 20, 721–721. [Google Scholar] [CrossRef]
- Matsye, P.; Zheng, L.; Si, Y.; Kim, S.; Luo, W.Y.; Crossman, D.K.; Bratcher, P.E.; King, P.H. HuR promotes the molecular signature and phenotype of activated microglia: Implications for amyotrophic lateral sclerosis and other neurodegenerative diseases. Glia 2017, 65, 945–963. [Google Scholar] [CrossRef]
- Peng, W.D.; Furuuchi, N.; Aslanukova, L.; Huang, Y.H.; Brown, S.Z.; Jiang, W.; Addya, S.; Vishwakarma, V.; Peters, E.; Brody, J.R.; et al. Elevated HuR in Pancreas Promotes a Pancreatitis-Like Inflammatory Microenvironment That Facilitates Tumor Development. Molecular and Cellular Biology 2018, 38. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, Y.; Hu, W.R.; Liu, X.Y.; Hao, W.J.; Xing, J.; Ding, J.; Xu, Y.C.; Yao, F.; Zhao, Y.J.; et al. TRPM7 facilitates fibroblast-like synoviocyte proliferation, metastasis and inflammation through increasing IL-6 stability via the PKCα-HuR axis in rheumatoid arthritis. International Immunopharmacology 2024, 132. [Google Scholar] [CrossRef]
- Wang, Y.X.; Zheng, H.Y.; Zhou, K.; Xie, H.L.; Ren, Z.; Liu, H.T.; Liu, H.; Zhou, Z.X.; Jiang, Z.S. Multifaceted Nature of HuR in Atherosclerosis Development. Current Medicinal Chemistry 2024. [Google Scholar] [CrossRef]
- Christodoulou-Vafeiadou, E.; Ioakeimidis, F.; Andreadou, M.; Giagkas, G.; Stamatakis, G.; Reczko, M.; Samiotaki, M.; Papanastasiou, A.D.; Karakasiliotis, I.; Kontoyiannis, D.L. Divergent Innate and Epithelial Functions of the RNA-Binding Protein HuR in Intestinal Inflammation. Frontiers in Immunology 2018, 9. [Google Scholar] [CrossRef]
- Durie, D.; Lewis, S.M.; Liwak, U.; Kisilewicz, M.; Gorospe, M.; Holcik, M. RNA-binding protein HuR mediates cytoprotection through stimulation of XIAP translation. Oncogene 2011, 30, 1460–1469. [Google Scholar] [CrossRef]
- Kullmann, M.; Göpfert, U.; Siewe, B.; Hengst, L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes & Development 2002, 16, 3087–3099. [Google Scholar] [CrossRef]
- Wu, X.Q.; Gardashova, G.; Lan, L.; Han, S.; Zhong, C.C.; Marquez, R.T.; Wei, L.J.; Wood, S.; Roy, S.; Gowthaman, R.; et al. Targeting the interaction between RNA-binding protein HuR and FOXQ1 suppresses breast cancer invasion and metastasis. Communications Biology 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Srivastava, O.P.; Pfister, R.R. Downregulation of β-actin and its regulatory gene HuR affect cell migration of human corneal fibroblasts. Molecular Vision 2014, 20, 593–605. [Google Scholar] [PubMed]
- Tian, M.; Wang, J.J.; Liu, S.M.; Li, X.Y.; Li, J.Y.; Yang, J.M.; Zhang, C.; Zhang, W.C. Hepatic HuR protects against the pathogenesis of non-alcoholic fatty liver disease by targeting PTEN. Cell Death & Disease 2021, 12. [Google Scholar] [CrossRef]
- Subramanian, P.; Gargani, S.; Palladini, A.; Chatzimike, M.; Grzybek, M.; Peitzsch, M.; Papanastasiou, A.D.; Pyrina, I.; Ntafis, V.; Gercken, B.; et al. The RNA binding protein human antigen R is a gatekeeper of liver homeostasis. Hepatology 2022, 75, 881–897. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Tai, Y.L.; Way, G.; Zeng, J.; Zhao, D.; Su, L.Y.; Jiang, X.X.; Jackson, K.G.; Wang, X.; Gurley, E.C.; et al. RNA binding protein HuR protects against NAFLD by suppressing long noncoding RNA H19 expression. Cell and Bioscience 2022, 12. [Google Scholar] [CrossRef]
- Paik, J.M.; Henry, L.; Younossi, Y.; Ong, J.; Alqahtani, S.; Younossi, Z.M. The burden of nonalcoholic fatty liver disease (NAFLD) is rapidly growing in every region of the world from 1990 to 2019. Hepatology Communications 2023, 7. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, M.S.; Liu, Z.Q.; Yuan, H.B.; Wu, X.F.; Shi, T.T.; Chen, X.D.; Zhang, T.J. Increasing prevalence of NAFLD/NASH among children, adolescents and young adults from 1990 to 2017: a population-based observational study. Bmj Open 2021, 11. [Google Scholar] [CrossRef]
- Rupasinghe, K.; Hind, J.; Hegarty, R. Updates in Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD) in Children. Journal of Pediatric Gastroenterology and Nutrition 2023, 77, 583–591. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Journal of Hepatology 2023, 79, 1542–1556. [Google Scholar] [CrossRef]
- Hagstroem, H.; Vessby, J.; Ekstedt, M.; Shang, Y. 99% of patients with NAFLD meet MASLD criteria and natural history is therefore identical. Biocatalysis and Agricultural Biotechnology 2024, 56, E76–E77. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Ong, J.; Trimble, G.; AlQahtani, S.; Younossi, I.; Ahmed, A.; Racila, A.; Henrys, L. Nonalcoholic Steatohepatitis Is the Most Rapidly Increasing Indication for Liver Transplantation in the United States. Clinical Gastroenterology and Hepatology 2021, 19, 580. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Resmetirom: First Approval. Drugs 2024, 84, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Zong, C.; Jiang, M.Y.; Hu, H.; Cheng, X.L.; Ni, J.H.; Yi, X.; Jiang, B.; Tian, F.; Chang, M.W.; et al. Hepatic HuR modulates lipid homeostasis in response to high-fat diet. Nature Communications 2020, 11. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Molecular Metabolism 2021, 50. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. Journal of Clinical Investigation 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. American Journal of Medicine 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Cho, E.E.L.; Ang, C.Z.; Quek, J.; Fu, C.E.; Lim, L.K.E.; Heng, Z.E.Q.; Tan, D.J.H.; Lim, W.H.; Yong, J.N.; Zeng, R.B.C.; et al. Global prevalence of non-alcoholic fatty liver disease in type 2 diabetes mellitus: an updated systematic review and meta-analysis. Gut 2023, 72, 2138–2148. [Google Scholar] [CrossRef]
- Titchenell, P.M.; Lazar, M.A.; Birnbaum, M.J. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends in Endocrinology and Metabolism 2017, 28, 497–505. [Google Scholar] [CrossRef]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proceedings of the National Academy of Sciences of the United States of America 2007, 104, 12587–12594. [Google Scholar] [CrossRef]
- Wong, J.T.; Kim, P.T.W.; Peacock, J.W.; Yau, T.Y.; Mui, A.L.F.; Chung, S.W.; Sossi, V.; Doudet, D.; Green, D.; Ruth, T.J.; et al. Pten (phosphatase and tensin homologue gene) haploinsufficiency promotes insulin hypersensitivity. Diabetologia 2007, 50, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.; Wang, Y.; Stahl, A.; Bassilian, S.; Lee, W.P.; Kim, Y.J.; Sherwin, R.; Devaskar, S.; Lesche, R.; Magnuson, M.A.; et al. Live-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity. Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Ginsberg, H.N. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends in Endocrinology and Metabolism 2011, 22, 353–363. [Google Scholar] [CrossRef]
- Adiels, M.; Taskinen, M.R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Hakkinen, A.; Olofsson, S.O.; Yki-Jarvinen, H.; et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef]
- Eaton, S.; Bartlett, K.; Pourfarzam, M. Mammalian mitochondrial beta-oxidation. Biochemical Journal 1996, 320, 345–357. [Google Scholar] [CrossRef]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. Journal of Gastroenterology 2013, 48, 434–441. [Google Scholar] [CrossRef]
- Marais, A.D. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 2019, 51, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Welty, F.K. Hypobetalipoproteinemia and abetalipoproteinemia: liver disease and cardiovascular disease. Current Opinion in Lipidology 2020, 31, 49–55. [Google Scholar] [CrossRef]
- Vacca, M.; Kamzolas, I.; Harder, L.M.; Oakley, F.; Trautwein, C.; Hatting, M.; Ross, T.; Bernardo, B.; Oldenburger, A.; Hjuler, S.T.; et al. An unbiased ranking of murine dietary models based on their proximity to human metabolic dysfunction-associated steatotic liver disease (MASLD). Nature Metabolism 2024, 6, 1178. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of Inflammation in Nonalcoholic Fatty Liver Disease: The Multiple Parallel Hits Hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Horn, C.L.; Morales, A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatology Communications 2022, 6, 12–35. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends in Endocrinology and Metabolism 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Simonen, P.; Kotronen, A.; Hallikainen, M.; Sevastianova, K.; Makkonen, J.; Hakkarainen, A.; Lundbom, N.; Miettinen, T.A.; Gylling, H.; Yki-Järvinen, H. Cholesterol synthesis is increased and absorption decreased in non-alcoholic fatty liver disease independent of obesity. Journal of Hepatology 2011, 54, 153–159. [Google Scholar] [CrossRef]
- Sato, R. Sterol metabolism and SREBP activation. Archives of Biochemistry and Biophysics 2010, 501, 177–181. [Google Scholar] [CrossRef]
- Botham KM, M.P. Harper’s Illustrated Biochemistry, 31 ed.; Rodwell, V., Bender, DA, Botham, KM, Kennelly PJ, Weil P., Ed.; McGraw-Hill Education: 2018.
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Progress in Lipid Research 2013, 52, 175–191. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, Y.X.; Tang, R.K.; Chen, Y.; Li, Q.; Gong, J.P.; Huang, A.L.; Varghese, Z.; Moorhead, J.F.; Ruan, X.Z. Inflammatory stress exacerbates hepatic cholesterol accumulation via increasing cholesterol uptake and de novo synthesis. Journal of Gastroenterology and Hepatology 2011, 26, 875–883. [Google Scholar] [CrossRef]
- van Rooyen, D.M.; Larter, C.Z.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Hepatic Free Cholesterol Accumulates in Obese, Diabetic Mice and Causes Nonalcoholic Steatohepatitis. Gastroenterology 2011, 141, 1393–U1850. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nature Reviews Molecular Cell Biology 2008, 9, 125–138. [Google Scholar] [CrossRef]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metabolism 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Choudhuri, S.; Klaassen, C.D. Molecular Regulation of Bile Acid Homeostasis. Drug Metabolism and Disposition 2022, 50, 425–455. [Google Scholar] [CrossRef] [PubMed]
- Li, T.G.; Chiang, J.Y.L. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacological Reviews 2014, 66, 948–983. [Google Scholar] [CrossRef]
- Pandak, W.M., Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Research 2019, 3, 88–98. [CrossRef]
- Linnet, K. Postprandial plasma concentrations of glycine and taurine conjugated bile acids in healthy subjects. Gut 1983, 24, 249–252. [Google Scholar] [CrossRef]
- Takahashi, S.; Fukami, T.; Masuo, Y.; Brocker, C.N.; Xie, C.; Krausz, K.W.; Wolf, C.R.; Henderson, C.J.; Gonzalez, F.J. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. Journal of Lipid Research 2016, 57, 2130–2137. [Google Scholar] [CrossRef]
- Falany, C.N.; Fortinberry, H.; Leiter, E.H.; Barnes, S. Cloning, expression, and chromosomal localization of mouse liver bile acid CoA:amino acid N-acyltransferase. Journal of Lipid Research 1997, 38, 1139–1148. [Google Scholar] [CrossRef]
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.; Siddiqui, M.S.; Boyett, S.; Min, H.K.; et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef]
- Grzych, G.; Chávez-Talavera, O.; Descat, A.; Thuillier, D.; Verrijken, A.; Kouach, M.; Legry, V.; Verkindt, H.; Raverdy, V.; Legendre, B.; et al. NASH-related increases in plasma bile acid levels depend on insulin resistance. Jhep Reports 2021, 3. [Google Scholar] [CrossRef]
- Chávez-Talavera, O.; Haas, J.; Grzych, G.; Tailleux, A.; Staels, B. Bile acid alterations in nonalcoholic fatty liver disease, obesity, insulin resistance and type 2 diabetes: what do the human studies tell? Current Opinion in Lipidology 2019, 30, 244–254. [Google Scholar] [CrossRef]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.S.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Schreuder, T.; Marsman, H.A.; Lenicek, M.; van Werven, J.R.; Nederveen, A.J.; Jansen, P.L.M.; Schaap, F.G. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. American Journal of Physiology-Gastrointestinal and Liver Physiology 2010, 298, G440–G445. [Google Scholar] [CrossRef] [PubMed]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.; Reily, M.D.; Lu, Z.Q.; Lehman-McKeeman, L.D.; Cherrington, N.J. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicology and Applied Pharmacology 2013, 268, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Hyogo, H.; Honda, A.; Miyazaki, T.; Tokushige, K.; Hashimoto, E.; Inui, K.; Matsuzaki, Y.; Tazuma, S. Increased serum liver X receptor ligand oxysterols in patients with non-alcoholic fatty liver disease. Journal of Gastroenterology 2012, 47, 1257–1266. [Google Scholar] [CrossRef]
- Raselli, T.; Hearn, T.; Wyss, A.; Atrott, K.; Peter, A.; Frey-Wagner, I.; Spalinger, M.R.; Maggio, E.M.; Sailer, A.W.; Schmitt, J.; et al. Elevated oxysterol levels in human and mouse livers reflect nonalcoholic steatohepatitis. Journal of Lipid Research 2019, 60, 1270–1283. [Google Scholar] [CrossRef]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. Journal of Hepatology 2023, 78, 415–429. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metabolism 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022, 76, 1452–1465. [Google Scholar] [CrossRef]
- Gancheva, S.; Kahl, S.; Pesta, D.; Mastrototaro, L.; Dewidar, B.; Strassburger, K.; Sabah, E.; Esposito, I.; Weiss, J.; Sarabhai, T.; et al. Impaired Hepatic Mitochondrial Capacity in Nonalcoholic Steatohepatitis Associated With Type 2 Diabetes. Diabetes Care 2022, 45, 928–937. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends in Pharmacological Sciences 2006, 27, 639–645. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cellular and Molecular Life Sciences 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, A.; Campagnoli, L.I.M.; Barbieri, A.; Marchesi, N.; Pascale, A.; Croce, A.C.; Vairetti, M.; Di Pasqua, L.G. MCD Diet Modulates HuR and Oxidative Stress-Related HuR Targets in Rats. International Journal of Molecular Sciences 2023, 24. [Google Scholar] [CrossRef]
- Ma, X.W.; McKeen, T.; Zhang, J.H.; Ding, W.X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Filali-Mouncef, Y.; Hunter, C.; Roccio, F.; Zagkou, S.; Dupont, N.; Primard, C.; Proikas-Cezanne, T.; Reggiori, F. The menage a trois of autophagy, lipid droplets and liver disease. Autophagy 2022, 18, 50–72. [Google Scholar] [CrossRef]
- Yu, S.H.; Palanisamy, K.; Sun, K.T.; Li, X.; Wang, Y.M.; Lin, F.Y.; Chen, K.B.; Wang, I.K.; Yu, T.M.; Li, C.Y. Human antigen R regulates hypoxia-induced mitophagy in renal tubular cells through PARKIN/BNIP3L expressions. Journal of Cellular and Molecular Medicine 2021, 25, 2691–2702. [Google Scholar] [CrossRef]
- Ji, E.; Kim, C.; Kang, H.; Ahn, S.; Jung, M.; Hong, Y.; Tak, H.; Lee, S.; Kim, W.; Lee, E.K. RNA Binding Protein HuR Promotes Autophagosome Formation by Regulating Expression of Autophagy-Related Proteins 5, 12, and 16 in Human Hepatocellular Carcinoma Cells (vol 39, e00508-18, 2020). Molecular and Cellular Biology 2021, 41. [Google Scholar] [CrossRef]
- Lu, L.L.; Chen, J.C.; Jiang, H.; Li, N.; Li, X.J.; Liu, Y.H.; Zong, C. Hepatocyte-Specific HuR Protects Against Acetaminophen-Induced Liver Injury in Mice. Journal of Cellular and Molecular Medicine 2024, 28. [Google Scholar] [CrossRef]
- Arab, J.P.; Hernández-Rocha, C.; Morales, C.; Vargas, J.I.; Solís, N.; Pizarro, M.; Robles, C.; Sandoval, D.; Ponthus, S.; Benítez, C.; et al. Serum cytokeratin-18 fragment levels as noninvasive marker of nonalcoholic steatohepatitis in the chilean population. Gastroenterologia Y Hepatologia 2017, 40, 388–394. [Google Scholar] [CrossRef]
- Simion, V.; Zhou, H.Y.; Haemmig, S.; Pierce, J.B.; Mendes, S.; Tesmenitsky, Y.; Pérez-Cremades, D.; Lee, J.F.; Chen, A.F.; Ronda, N.; et al. A macrophage-specific lncRNA regulates apoptosis and atherosclerosis by tethering HuR in the nucleus. Nature Communications 2020, 11. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Gorospe, M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdisciplinary Reviews-Rna 2010, 1, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Lal, A.; Kim, H.H.; Gorospe, M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle 2007, 6, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Caballero, F.; Fernández, A.; De Lacy, A.M.; Fernández-Checa, J.C.; Caballería, J.; García-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. Journal of Hepatology 2009, 50, 789–796. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Kohli, R.; Gores, G.J. Mechanisms of Lipotoxicity in NAFLD and Clinical Implications. Journal of Pediatric Gastroenterology and Nutrition 2011, 53, 131–140. [Google Scholar] [CrossRef]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Review of Gastroenterology & Hepatology 2011, 5, 201–212. [Google Scholar] [CrossRef]
- Malhi, H.; Barreyro, F.J.; Isomoto, H.; Bronk, S.F.; Gores, G.J. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut 2007, 56, 1124–1131. [Google Scholar] [CrossRef]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. Journal of Biological Chemistry 2006, 281, 12093–12101. [Google Scholar] [CrossRef]
- Zhu, H.F.; Berkova, Z.; Mathur, R.; Sehgal, L.; Khashab, T.; Tao, R.H.; Ao, X.; Feng, L.; Sabichi, A.L.; Blechacz, B.; et al. HuR Suppresses Fas Expression and Correlates with Patient Outcome in Liver Cancer. Molecular Cancer Research 2015, 13, 809–818. [Google Scholar] [CrossRef]
- Vachliotis, I.D.; Polyzos, S.A. The Role of Tumor Necrosis Factor-Alpha in the Pathogenesis and Treatment of Nonalcoholic Fatty Liver Disease. Current Obesity Reports 2023, 12, 191–206. [Google Scholar] [CrossRef]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. Journal of Lipid Research 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Cerami, A. The biology of cachectin/TNF--a primary mediator of the host response. Annu Rev Immunol 1989, 7, 625–655. [Google Scholar] [CrossRef] [PubMed]
- Sedlyarov, V.; Fallmann, J.; Ebner, F.; Huemer, J.; Sneezum, L.; Ivin, M.; Kreiner, K.; Tanzer, A.; Vogl, C.; Hofacker, I.; et al. Tristetraprolin binding site atlas in the macrophage transcriptome reveals a switch for inflammation resolution. Molecular Systems Biology 2016, 12. [Google Scholar] [CrossRef]
- Tiedje, C.; Ronkina, N.; Tehrani, M.; Dhamija, S.; Laass, K.; Holtmann, H.; Kotlyarov, A.; Gaestel, M. The p38/MK2-Driven Exchange between Tristetraprolin and HuR Regulates AU-Rich Element-Dependent Translation. Plos Genetics 2012, 8. [Google Scholar] [CrossRef]
- Dean, J.L.E.; Wait, R.; Mahtani, K.R.; Sully, G.; Clark, A.R.; Saklatvala, J. The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Molecular and Cellular Biology 2001, 21, 721–730. [Google Scholar] [CrossRef]
- Abdelsam, S.S.; Ghanem, S.K.; Zahid, M.A.; Abunada, H.H.; Bader, L.; Raiq, H.; Khan, A.; Parray, A.; Djouhri, L.; Agouni, A. Human antigen R: Exploring its inflammatory response impact and significance in cardiometabolic disorders. Journal of Cellular Physiology 2024, 239. [Google Scholar] [CrossRef]
- Yiakouvaki, A.; Dimitriou, M.; Karakasiliotis, I.; Eftychi, C.; Theocharis, S.; Kontoyiannis, D.L. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. Journal of Clinical Investigation 2012, 122, 48–61. [Google Scholar] [CrossRef]
- Rhee, W.J.; Ni, C.W.; Zheng, Z.L.; Chang, K.; Jo, H.; Bao, G. HuR regulates the expression of stress-sensitive genes and mediates inflammatory response in human umbilical vein endothelial cells. Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 6858–6863. [Google Scholar] [CrossRef]
- Fan, J.S.; Ishmael, F.T.; Fang, X.; Myers, A.; Cheadle, C.; Huang, S.K.; Atasoy, U.; Gorospe, M.; Stellato, C. Chemokine Transcripts as Targets of the RNA-Binding Protein HuR in Human Airway Epithelium. Journal of Immunology 2011, 186, 2482–2494. [Google Scholar] [CrossRef]
- Sawada, K.; Chung, H.K.; Softic, S.; Moreno-Fernandez, M.E.; Divanovic, S. The bidirectional immune crosstalk in metabolic dysfunction-associated steatotic liver disease. Cell Metabolism 2023, 35, 1852–1871. [Google Scholar] [CrossRef]
- Syn, W.K.; Oo, Y.H.; Pereira, T.A.; Karaca, G.F.; Jung, Y.M.; Omenetti, A.; Witek, R.P.; Choi, S.S.; Guy, C.D.; Fearing, C.M.; et al. Accumulation of Natural Killer T Cells in Progressive Nonalcoholic Fatty Liver Disease. Hepatology 2010, 51, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, O.; Milatos, S.; Grammenoudi, S.; Mukherjee, N.; Keene, J.D.; Kontoyiannis, D.L. Control of Thymic T Cell Maturation, Deletion and Egress by the RNA-Binding Protein HuR. Journal of Immunology 2009, 182, 6779–6788. [Google Scholar] [CrossRef]
- Chen, J.; Martindale, J.L.; Abdelmohsen, K.; Kumar, G.; Fortina, P.M.; Gorospe, M.; Rostami, A.; Yu, S.G. RNA-Binding Protein HuR Promotes Th17 Cell Differentiation and Can Be Targeted to Reduce Autoimmune Neuroinflammation. Journal of Immunology 2020, 204, 2076–2087. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wong, V.W.S.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Sanz, M.A.Q.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443. [Google Scholar] [CrossRef]
- Angulo, P.; Machado, M.V.; Diehl, A.M. Fibrosis in Nonalcoholic Fatty Liver Disease: Mechanisms and Clinical Implications. Seminars in Liver Disease 2015, 35, 132–145. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Gandhi, C.R. Hepatology Snapshot: Hepatic stellate cell activation and pro-fibrogenic signals. Journal of Hepatology 2017, 67, 1104–1105. [Google Scholar] [CrossRef]
- Zhu, C.Y.; Kim, K.; Wang, X.B.; Bartolome, A.; Salomao, M.; Dongiovanni, P.; Meroni, M.; Graham, M.J.; Yates, K.P.; Diehl, A.M.; et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Science Translational Medicine 2018, 10. [Google Scholar] [CrossRef]
- Li, X.J.Y.; Liu, R.P.; Yang, J.; Sun, L.X.; Zhang, L.Y.; Jiang, Z.Z.; Puri, P.; Gurley, E.C.; Lai, G.H.; Tang, Y.P.; et al. The Role of Long Noncoding RNA H19 in Gender Disparity of Cholestatic Liver Injury in Multidrug Resistance 2 Gene Knockout Mice. Hepatology 2017, 66, 869–884. [Google Scholar] [CrossRef]
- Liu, R.P.; Li, X.J.Y.; Zhu, W.W.; Wang, Y.Y.; Zhao, D.; Wang, X.; Gurley, E.C.; Liang, G.; Chen, W.D.; Lai, G.H.; et al. Cholangiocyte-Derived Exosomal Long Noncoding RNA H19 Promotes Hepatic Stellate Cell Activation and Cholestatic Liver Fibrosis. Hepatology 2019, 70, 1317–1335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.L.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.J.; Chen, A.P.; Zhang, F.; Zheng, S.Z. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.; Karschner, V.; Jones, H.; Pekala, P.H. HuR, an RNA-binding protein, involved in the control of cellular differentiation. In Vivo 2006, 20, 17–23. [Google Scholar]
- Kim, I.; Hur, J.; Jeong, S. HuR represses Wnt/β-catenin-mediated transcriptional activity by promoting cytoplasmic localization of β-catenin. Biochemical and Biophysical Research Communications 2015, 457, 65–70. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: global epidemiology and pathogenesis. Nature Reviews Endocrinology 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Zhang, P.P.; Wu, W.Y.; Ma, C.F.; Du, C.Y.; Huang, Y.R.; Xu, H.X.; Li, C.C.; Cheng, X.F.; Hao, R.J.; Xu, Y.J. RNA-Binding Proteins in the Regulation of Adipogenesis and Adipose Function. Cells 2022, 11. [Google Scholar] [CrossRef]
- Hwang, J.S.; Lee, W.J.; Hur, J.; Lee, H.G.; Kim, E.; Lee, G.H.; Choi, M.J.; Lim, D.S.; Paek, K.S.; Seo, H.G. Rosiglitazone-dependent dissociation of HuR from PPAR-γ regulates adiponectin expression at the posttranscriptional level. Faseb Journal 2019, 33, 7707–7720. [Google Scholar] [CrossRef]
- Guarnieri, A.R.; Anthony, S.R.; Gozdiff, A.; Green, L.C.; Fleifil, S.M.; Slone, S.; Nieman, M.L.; Alam, P.; Benoit, J.B.; Owens, A.P.; et al. Adipocyte-specific deletion of HuR induces spontaneous cardiac hypertrophy and fibrosis. American Journal of Physiology-Heart and Circulatory Physiology 2021, 321, H228–H241. [Google Scholar] [CrossRef]
- Shen, L.; Han, J.; Wang, H.Y.; Meng, Q.Y.; Chen, L.L.; Liu, Y.G.; Feng, Y.; Wu, G.H. Cachexia-related long noncoding RNA, CAAlnc1, suppresses adipogenesis by blocking the binding of HuR to adipogenic transcription factor mRNAs. International Journal of Cancer 2019, 145, 1809–1821. [Google Scholar] [CrossRef]
- Vanni, E.; Marengo, A.; Mezzabotta, L.; Bugianesi, E. Systemic Complications of Nonalcoholic Fatty Liver Disease: When the Liver Is Not an Innocent Bystander. Seminars in Liver Disease 2015, 35, 236–249. [Google Scholar] [CrossRef]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The complex link between NAFLD and type 2 diabetes mellitus - mechanisms and treatments. Nature Reviews Gastroenterology & Hepatology 2021, 18, 599–612. [Google Scholar] [CrossRef]
- Targher, G.; Day, C.P.; Bonora, E. Risk of Cardiovascular Disease in Patients with Nonalcoholic Fatty Liver Disease. New England Journal of Medicine 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.D.; Lin, J.N.C.; Mahoney, T.; Ume, N.; Yang, G.C.; Gabbay, R.A.; ElSayed, N.A.; Bannuru, R.R. Economic Costs of Diabetes in the US in 2022. Diabetes Care 2024, 47, 26–43. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. MECHANISMS OF DIABETIC COMPLICATIONS. Physiological Reviews 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.G.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nature Reviews Cardiology 2020, 17, 585–607. [Google Scholar] [CrossRef]
- de Bruin, R.G.; Rabelink, T.J.; van Zonneveld, A.J.; van der Veer, E.P. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. European Heart Journal 2017, 38, 1380–1388D. [Google Scholar] [CrossRef]
- Hu, H.; Jiang, M.Y.; Cao, Y.P.; Zhang, Z.J.; Jiang, B.; Tian, F.; Feng, J.; Dou, Y.L.; Gorospe, M.; Zheng, M.; et al. HuR regulates phospholamban expression in isoproterenol-induced cardiac remodelling. Cardiovascular Research 2020, 116, 944–955. [Google Scholar] [CrossRef]
- Slone, S.; Anthony, S.R.; Green, L.C.; Parkins, S.; Acharya, P.; Kasprovic, D.A.; Reynolds, K.; Jaggers, R.M.; Nieman, M.L.; Alam, P.; et al. HuR inhibition reduces post-ischemic cardiac remodeling by dampening myocyte-dependent inflammatory gene expression and the innate immune response. Faseb Journal 2025, 39. [Google Scholar] [CrossRef]
- Green, L.C.; Slone, S.; Anthony, S.R.; Guarnieri, A.R.; Parkins, S.; Shearer, S.M.; Nieman, M.L.; Roy, S.; Aube, J.; Wu, X.Q.; et al. HuR-dependent expression of Wisp1 is necessary for TGFβ-induced cardiac myofibroblast activity. Journal of Molecular and Cellular Cardiology 2023, 174, 38–46. [Google Scholar] [CrossRef]
- Govindappa, P.K.; Patil, M.; Garikipati, V.N.S.; Verma, S.K.; Saheera, S.; Narasimhan, G.; Zhu, W.Q.; Kishore, R.; Zhang, J.Y.; Krishnamurthy, P. Targeting exosome-associated human antigen R attenuates fibrosis and inflammation in diabetic heart. Faseb Journal 2020, 34, 2238–2251. [Google Scholar] [CrossRef]
- Green, L.C.; Anthony, S.R.; Slone, S.; Lanzillotta, L.; Nieman, M.L.; Wu, X.Q.; Robbins, N.; Jones, S.M.; Roy, S.; Owens, A.P.; et al. Human antigen R as a therapeutic target in pathological cardiac hypertrophy. Jci Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Singh, S.; Dubey, P.K.; Tousif, S.; Umbarkar, P.; Zhang, Q.K.; Lal, H.; Sewell-Loftin, M.K.; Umeshappa, C.S.; Ghebre, Y.T.; et al. Fibroblast-Specific Depletion of Human Antigen R Alleviates Myocardial Fibrosis Induced by Cardiac Stress. Jacc-Basic to Translational Science 2024, 9, 754–770. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.; Lambers, E.; Verma, S.; Thorne, T.; Qin, G.J.; Losordo, D.W.; Kishore, R. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. Faseb Journal 2010, 24, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Lachiondo-Ortega, S.; Delgado, T.C.; Baños-Jaime, B.; Velázquez-Cruz, A.; Díaz-Moreno, I.; Martinez-Chantar, M.L. Hu Antigen R (HuR) Protein Structure, Function and Regulation in Hepatobiliary Tumors. Cancers 2022, 14. [Google Scholar] [CrossRef]
- Vyas, K.; Patel, M.M. Insights on drug and gene delivery systems in liver fibrosis. Asian Journal of Pharmaceutical Sciences 2023, 18. [Google Scholar] [CrossRef]
- Majumder, M.; Chakraborty, P.; Mohan, S.; Mehrotra, S.; Palanisamy, V. HuR as a molecular target for cancer therapeutics and immune-related disorders. Advanced Drug Delivery Reviews 2022, 188. [Google Scholar] [CrossRef]
Figure 1.
Schematic overview of HuR structure, function, and regulatory mechanisms. HuR (Human antigen R) contains three highly conserved RNA recognition motifs (RRMs) connected by flexible hinge and linker domains. The HuR nucleocytoplasmic shuttling sequence (HNS) enables HuR to dynamically move between the nucleus and cytoplasm. HuR binds directly to target mRNAs by recognizing adenine- and uridine-rich elements (AREs) through its RRMs, classically exemplified by its interaction with transcripts such as c-fos. HuR expression and activity are regulated at multiple levels, including transcriptional, post-transcriptional, post-translational, and protein–protein interactions, which collectively govern its localization and functional output. Under homeostatic conditions, HuR is predominantly localized in the nucleus, where it binds to introns and splice sites of pre-mRNAs to regulate pre-mRNA processing. In response to cellular stress, HuR translocates to the cytoplasm, where it enhances stability and translation of mature mRNA targets involved in stress response, survival, inflammation, and metabolism.
Figure 1.
Schematic overview of HuR structure, function, and regulatory mechanisms. HuR (Human antigen R) contains three highly conserved RNA recognition motifs (RRMs) connected by flexible hinge and linker domains. The HuR nucleocytoplasmic shuttling sequence (HNS) enables HuR to dynamically move between the nucleus and cytoplasm. HuR binds directly to target mRNAs by recognizing adenine- and uridine-rich elements (AREs) through its RRMs, classically exemplified by its interaction with transcripts such as c-fos. HuR expression and activity are regulated at multiple levels, including transcriptional, post-transcriptional, post-translational, and protein–protein interactions, which collectively govern its localization and functional output. Under homeostatic conditions, HuR is predominantly localized in the nucleus, where it binds to introns and splice sites of pre-mRNAs to regulate pre-mRNA processing. In response to cellular stress, HuR translocates to the cytoplasm, where it enhances stability and translation of mature mRNA targets involved in stress response, survival, inflammation, and metabolism.
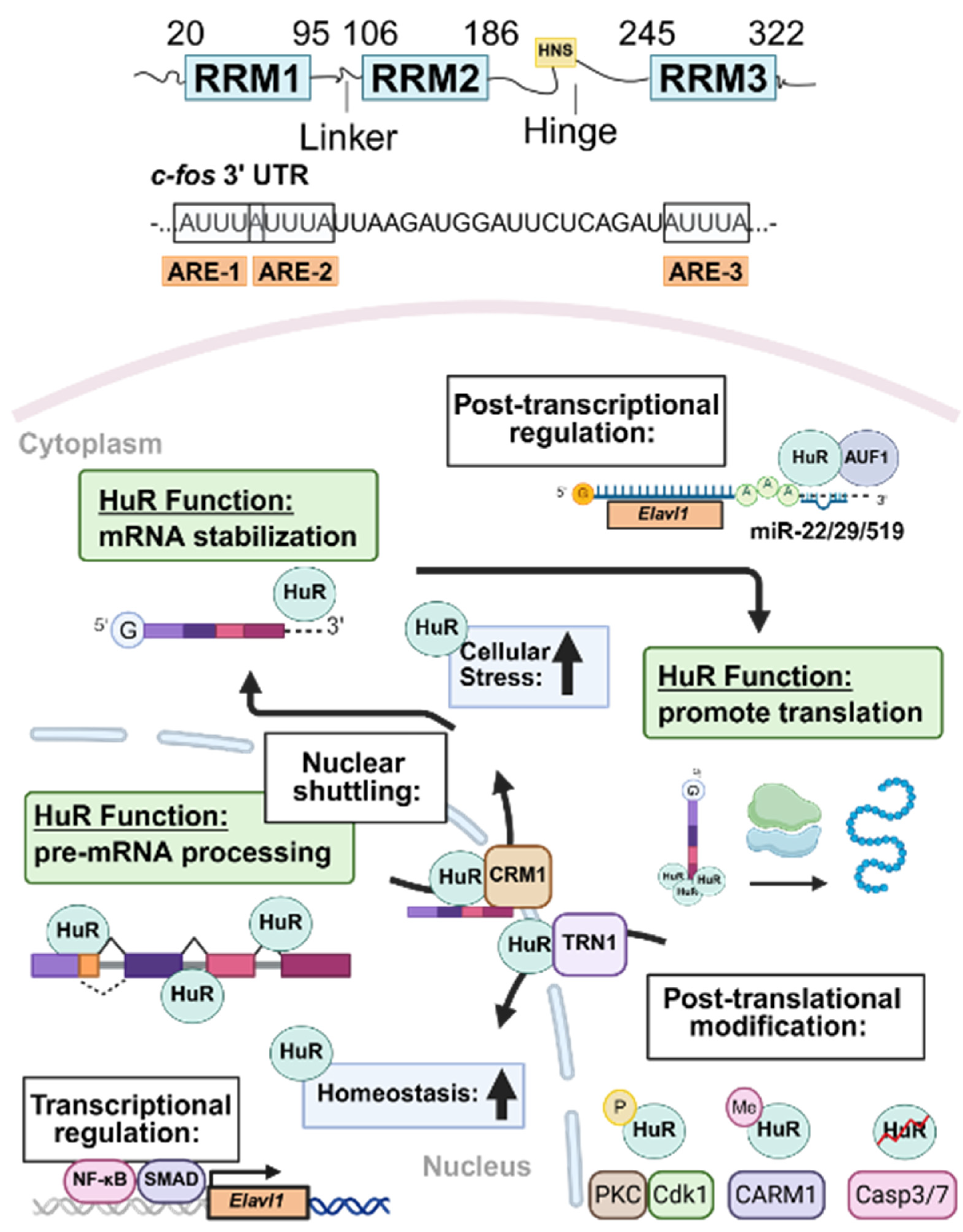
Figure 2.
Graphical summary of HuR’s roles in liver homeostasis and MASLD pathogenesis. HuR plays a multifaceted role in maintaining hepatic homeostasis and regulating key processes implicated in MASLD development. These include electron transport chain activity and mitochondrial function, hepatic stellate cell (HSC) activation and fibrogenesis, insulin signaling via the PI3K/Akt pathway, lipoprotein assembly and VLDL secretion, and bile acid synthesis and metabolism. HuR directly or indirectly regulates the expression of a wide array of target transcripts involved in these pathways, including Ndufb6, Uqcrb, Cycs (mitochondrial function), Opn, H19 (fibrosis signaling), PTEN (insulin signaling), Apob, Apoe (lipid export), and Cyp7a1, Cyp7b1 (bile acid synthesis). Collectively, these regulatory activities position HuR as a key post-transcriptional mediator in both physiological liver function and MASLD progression.
Figure 2.
Graphical summary of HuR’s roles in liver homeostasis and MASLD pathogenesis. HuR plays a multifaceted role in maintaining hepatic homeostasis and regulating key processes implicated in MASLD development. These include electron transport chain activity and mitochondrial function, hepatic stellate cell (HSC) activation and fibrogenesis, insulin signaling via the PI3K/Akt pathway, lipoprotein assembly and VLDL secretion, and bile acid synthesis and metabolism. HuR directly or indirectly regulates the expression of a wide array of target transcripts involved in these pathways, including Ndufb6, Uqcrb, Cycs (mitochondrial function), Opn, H19 (fibrosis signaling), PTEN (insulin signaling), Apob, Apoe (lipid export), and Cyp7a1, Cyp7b1 (bile acid synthesis). Collectively, these regulatory activities position HuR as a key post-transcriptional mediator in both physiological liver function and MASLD progression.
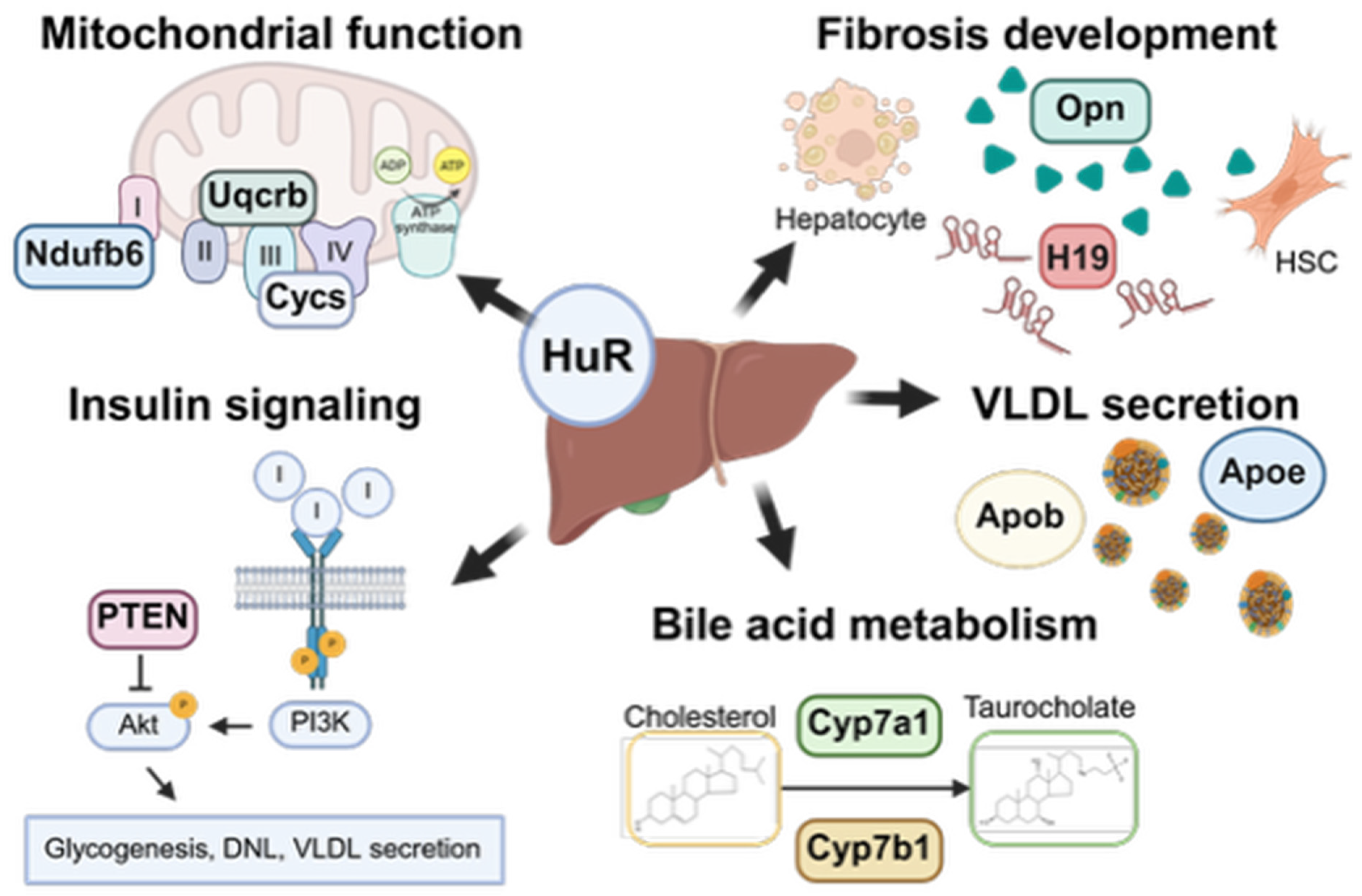
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



