Submitted:
12 May 2025
Posted:
13 May 2025
You are already at the latest version
Abstract
Sporadic Parkinson’s Disease (PD) affects 3% of people over 65 years of age. People are living longer, thanks in large part to improvements in global health technology and health access for non-neurological diseases. Consequently, neurological diseases of senescence such as PD are represent-ing an ever- increasing share of global disease burden. There is an intensifying research focus on the processes which underlie these conditions in the hope that neurological decay may be arrested at the earliest timepoint.
The concept of neuronal death linked to ageing- neural senescence- first emerged in the 1800s. By the late 20th century, it was recognised that neurodegeneration was common to all ageing human brains, but in most cases this process did not lead to clinical disease during life. Conditions such as PD are the result of accelerated neurodegeneration in particular brain foci. In the case of PD, de-generation of the substantia nigra pars compacta (SNpc) is especially implicated. Exactly why neu-ral degeneration accelerates in such specific regions remains a point of contention though current evidence implicates a complex interplay between a vast array of neuronal cell functions, neuron bio-energetic failure, and a dysfunctional brain immunological response. Their complexity is a consider-able barrier to disease modification trials which seek to intercept these maladaptive cell processes.
This paper reviews current evidence in the domain of neurodegeneration in Parkinson’s disease, with a focus on alpha-synuclein accumulation and deposition and the role of oxidative stress and inflammation in progressive brain changes. Recent approaches to disease modification are discussed, including the prevention or reversal of alpha-synuclein accumulation and deposition, modification of oxidative stress, alteration of maladaptive innate immune processes and reactive cascades, and regeneration of lost neurons using stem cells and growth factors. The limitations of past research methodologies are interrogated, including the difficulty of recruiting patients in the clinically quies-cent prodromal phase of sporadic Parkinson’s disease. Recommendations are provided for future studies seeking to identify novel therapeutics with disease modifying properties.
Keywords:
parkinson's disease
; oxidative stress
; mitochondria
; disease modification
Parkinson’s Disease – Epidemiology, Aetiology, Current Treatments and the Search for Disease Modification
Parkinson’s disease is the second most common neurodegenerative disease worldwide and is only exceeded by Alzheimer’s dementia. As global populations age, many clinicians are predicting an impending ‘Parkinson pandemic,’ with the incidence of Parkinson’s Disease predicted to double in 20 years and triple by 2050 [3]. Over 90% of PD cases are thought to arise because of a complex interaction between a patient’s genetic profile across numerous loci and unidentified environmental factors (sporadic PD). Recent genome-wide association studies have identified over 90 independent risk variants for PD [4]. The remaining 5–10% of cases have been linked to single gene mutations in LRRK2, SNCA, PARKIN, PINK-1, MAPT, VPS35, and GBA genes. Clinical manifestations and histopathological findings vary considerably between these mono-allelic forms, and likely represent distinct pathophysiological mechanisms to the more prevalent sporadic form [5]. Nevertheless, each of these mutations leads to selective destruction of dopaminergic neurons and provides invaluable insights into the mechanisms driving neural degeneration in the more common sporadic form.
The classic motor features of Parkinson’s Disease- bradykinesia, rigidity, and tremor- are a consequence of degeneration within dopamine-producing neural systems within the midbrain and basal ganglia. By the time motor features are manifested, anywhere between 40 and 60% of dopaminergic neurons within the substantia nigra pars compacta have already disappeared [6]. Historically, there has been little focus on the ‘non-motor’ features of Parkinson’s disease which may predate the emergence of motor symptoms by decades. These non-motor features include anosmia, constipation, autonomic dysfunction and sleep disorders such as REM-sleep behaviour disorder. Subclinical changes in visual processing, cognitive dexterity, and mood are now identified in patients with prodromal PD.[7,8] As the disease progresses, cognitive features including cognitive impairment or dementia as well as hallucinations, personality change and executive dysfunction become increasingly disabling and greatly contribute to morbidity [9].
Currently available treatments for PD act principally on motor dysfunction by upregulating dopamine within the midbrain and basal ganglia [10]. Eventually patients lose responsiveness to dopamine therapy necessitating increasing doses which in turn results in dopamine-related side effects such as dyskinesia, autonomic dysfunction, impulsivity and psychosis.
Disease modifying therapy represents an elusive “Holy Grail” in the treatment of neurodegenerative diseases such as PD. Disease modification pertains to therapies that could slow disease progression or promote neural recovery or compensation, thereby preventing or reversing the disease mechanism itself rather than relying on crude dopamine-based therapies [11]. Over the last 30 years hundreds of novel therapeutics have been evaluated as potential disease-modifying agents for PD. Unfortunately while many have shown promise in animal models and ex vivo studies, none have demonstrated the ability to arrest or slow the progression of PD in humans [3,12]. This article reviews these agents and discusses contemporary approaches in the search for disease-modifying agents in PD.
Methodology
PubMed and MEDLINE were searched using the following criteria: Parkinson’s Disease + Disease modification + neurodegeneration. Publications published prior to 2015 were excluded unless they were especially contributory to modern understanding. Reviews and summary articles were included where relevant. Highly relevant articles referenced within these papers that were not detected using the above search criteria were also included in the discussion.
Neuropathophysiology of Parkinson’s Disease - What We Know
Alpha-Synuclein Aggregates, Known as Lewy Bodies, Are the Histopathological Hallmark of PD
Parkinson’s Disease is caused by a stereotyped pattern of neurodegeneration across multiple brain regions. Intracytoplasmic aggregates of fibrillar alpha-synuclein (aS), called lewy bodies, are the histopathological hallmark of Parkinson’s disease and the closely related lewy body dementias (DLB) [13]. Early studies point towards a close relationship between CNS protein status and clinical outcome, with the ratio of fibrillar aS to Abeta1-42 in CSF not only predicting the development of sporadic PD and DLB but also indicating which patients are prone to motoric symptoms and which will develop cognitive impairment.
Braak and colleagues first described fibrillar aS deposition spreading in a caudal-rostral pattern [14]. They hypothesized that the pattern of neurodegeneration in PD followed this course of lewy body spread. In their model, the pathological process of sporadic PD begins with aggregation of lewy bodies within two peripheral nerve sites, namely the enteric plexus and olfactory nerve. This process advances to lower brain regions over time including the dorsal motor nucleus, reticular formation and olfactory nucleus (Braak stages 1 and 2). These early stages of the disease may correspond to the emergence of early premotor features including autonomic dysfunction, anosmia, and REM sleep behavior disorder (RBD). Over time, aS deposition spreads to the rostral midbrain and basal ganglia, infiltrating the dopaminergic pathways within the substantia nigra and ventral tegmental area (Braak stages 3 and 4). The neurodegeneration that accompanies aS deposition in this region is insidious and initially silent, with PET studies demonstrating neurodegeneration occurring many years before the onset of motor symptoms [15,16]. In the more advanced stages of PD(Braak stages 5 and 6), aS deposits can be detected in higher cortical regions, notably in the mesial temporal lobe, primary motor and sensory cortices, and prefrontal regions. It is hypothesized that this stage corresponds to the emergence of higher-order cognitive changes in memory, visuospatial processing, and executive function. This apparent chain-like spread of Lewy bodies could indicate that abnormal aS aggregation is a self-propagating process akin to a prion disease [17]. Indeed, preliminary cell studies have demonstrated this prion-like behaviour of aS and its ability to self-propagate between neighbouring neurons [18].
The Braak Hypothesis for aS propagation within the peripheral and central nervous system
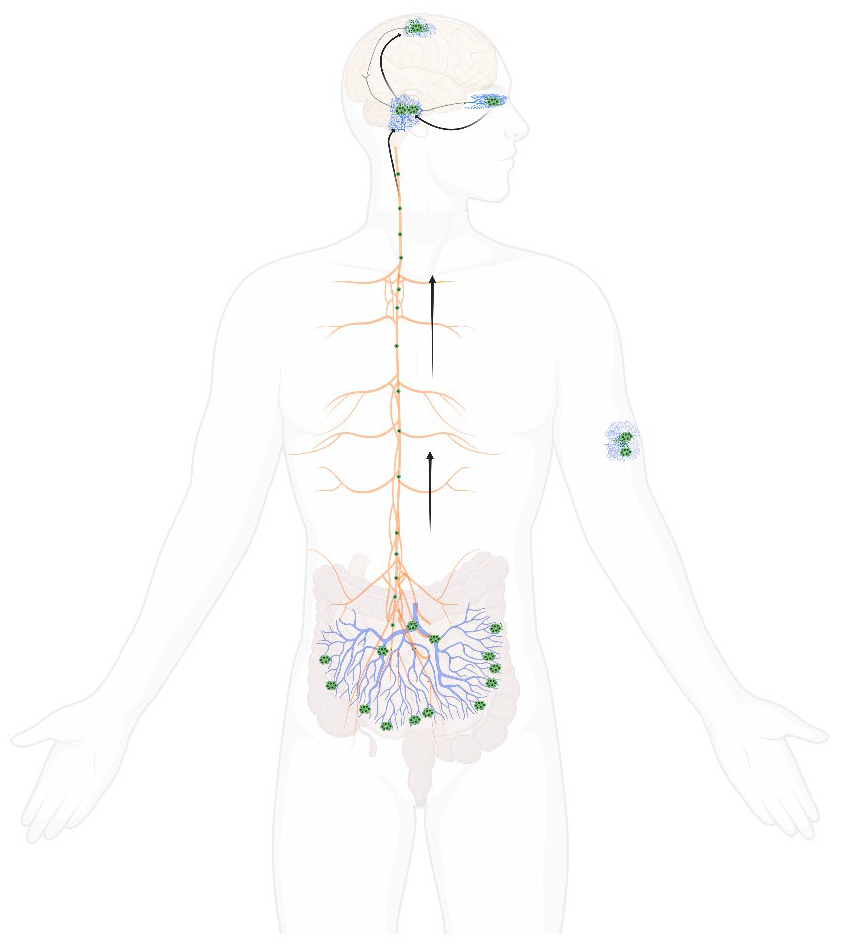
Arrows indicating the proposed pathway of lewy body changes, from peripheral centres including the enteric plexus and olfactory nucleus to central regions including the brainstem, midbrain and higher cortical regions. This diagram also demonstrates how lewy bodies have been demonstrated in skin tissue.
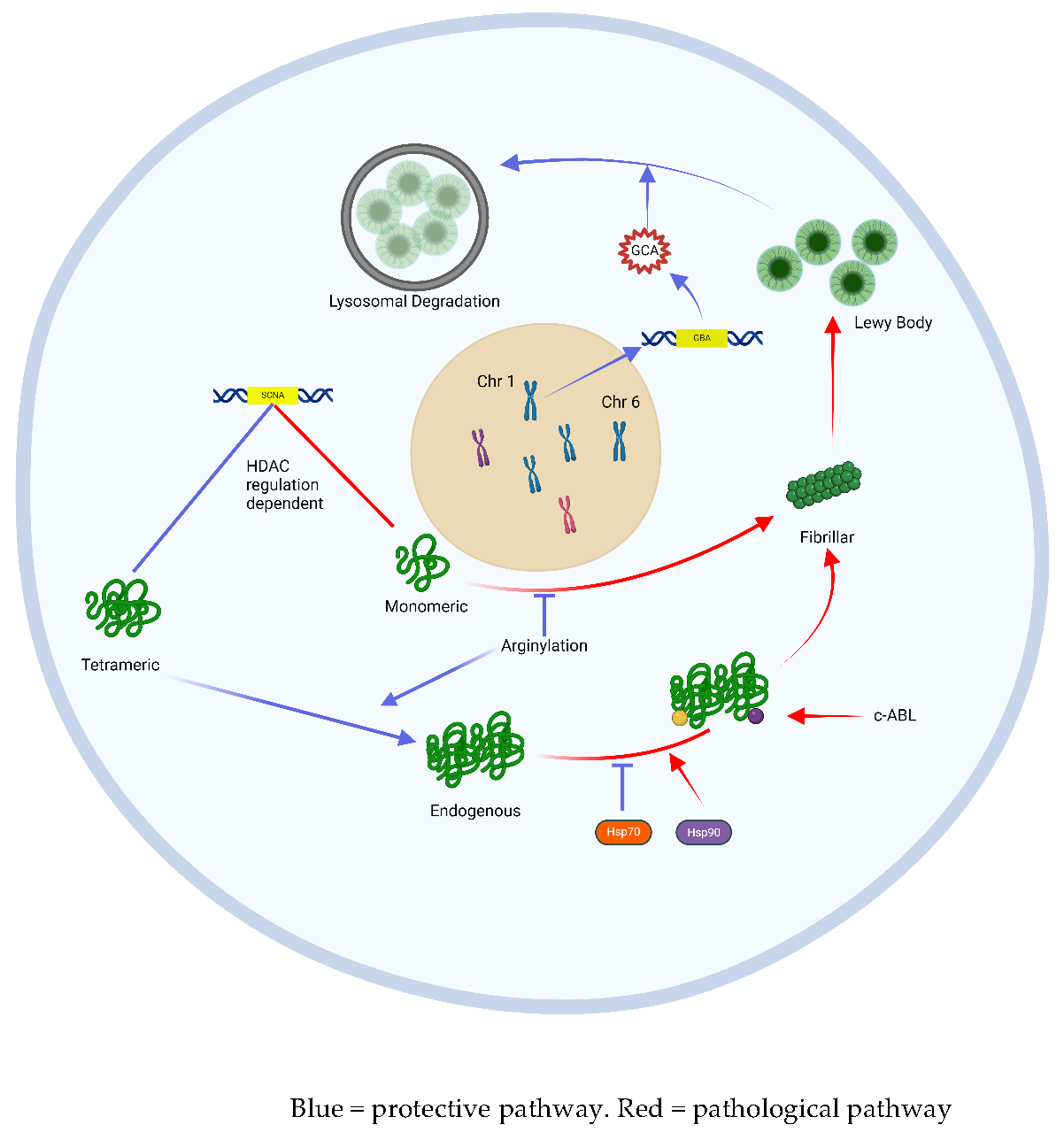
Neurons incorporate several mechanisms to prevent the accumulation of pathological aS, a process referred to as ‘proteostasis’ [19]. Failure of these mechanisms may trigger and/or exacerbate this accumulation of aS as Lewy bodies. One such mechanism is epigenetic regulation through histone acetylation of DNA chromatin [20,22]. A study by Park et al. demonstrated changes in histone acetylation (HATs) and deacetylation enzymes (HDACs) involved in the regulation of the alpha-synuclein gene (SNCA) that were unique to the SNpc of Parkinson’s patients, suggesting a potential dysregulation of gene transcription underlying the accumulation of aS in this cohort [21]. Other studies have highlighted the importance of post-translational stabilization and transport of aS via the chaperone proteins Hsp70 and Hsp90 [22]. More specifically, Hsp70 appears to protect against lewy body aggregation by binding to endogenous aS thereby preventing its accumulation and promoting lewy body disassembly [23]. Other proteins, called HSF-1 modulators, regulate the activity of these Hsp chaperone proteins and may influence aS proteostasis in this way [26]. The most common gene associated with early onset PD, GBA, encodes a lysosomal enzyme glucocerebrosidase (GCA). GCA is critical for lysosome-mediated sequestration and degradation of aS, and loss-of-function mutations in the GBA gene lead to the accumulation of aS in affected carriers.[24,25] A knock-in GBA1 mouse model has demonstrated that increased GCA expression may also regulate SCNA expression of aS from its aggregation-prone monomeric form to a benign tetrameric form that is less prone to Lewy body aggregation and this cellular function appears protective against motoric parkinsonism [26].
Post-translational modification of aS can alter its propensity to aggregate into pathological Lewy bodies. Arginylation (the addition of the amino acid arginine to the N-terminus of amino acids) within the peptide chain can reduce aS aggregation [27]. On the other hand, c-abl enzyme-driven phosphorylation of tyrosine 39 of the aS peptide leads to abnormal protein folding and aggregation. This enzyme is indeed overexpressed in diseased neurons in PD [3,28]. Phosphorylation of serine 129 (S129) is the dominant post-translational modification found in Lewy-body-aggregated aS, present in up to 90% of Lewy bodies, compared with 4% in healthy samples [29,30,31].
In addition to Lewy bodies, up to 50% of patients with PD eventually develop a sufficient burden of beta-amyloid plaques as well as tau-containing neurofibrillary tangles to justify a second diagnosis of Alzheimer’s dementia [32]. Individuals with AD-PD overlap are pathologically distinct from the pure Parkinson’s Disease Dementia (PDD) cohort, with a recent antibody study indicating different tau species expressed between the two groups [33]. This suggests that while aS is central to the pathological process of PD, synergistic accumulation of different pathological protein bodies may precipitate more rapid neurological and cognitive decline in many sufferers and may have real-world implications for disease-modifying therapies.
Alpha-Synuclein in the Neuron – Protective and Pathological Pathways
Oxidative Stress and Mitochondrial Dysfunction Induce Innate Immune Processes that Drive Cell Death in PD
While abnormal alpha-synuclein aggregates (Lewy bodies) are the histopathological hallmark of idiopathic Parkinson’s disease, their precise role in neural degeneration remains unclear. While it is possible that Lewy bodies, or the fibrillar aS peptides that form them, cause neural degeneration, they may also represent a biochemical byproduct of another fundamental cell death pathway.
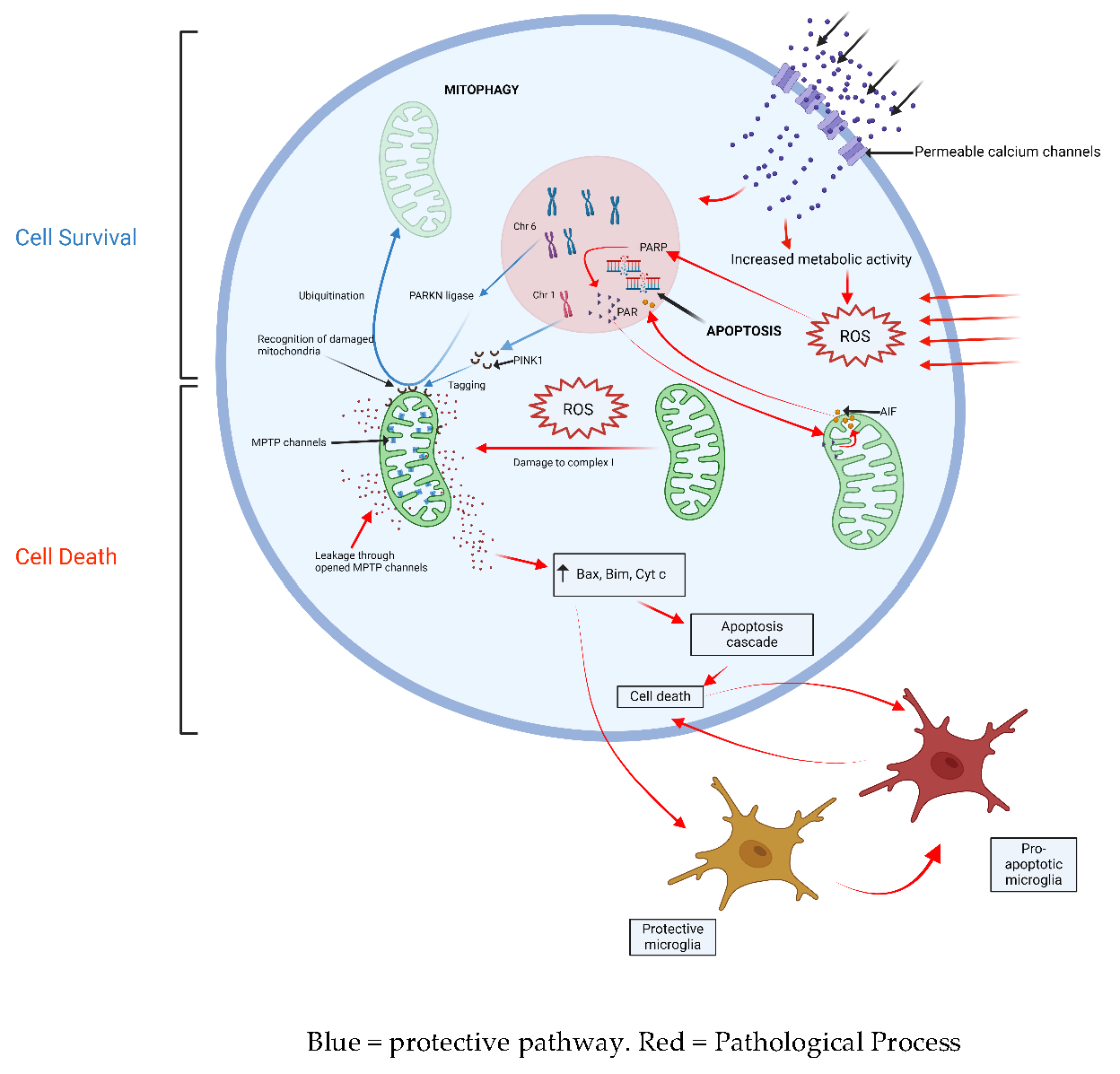
The innate immune response, triggered by oxidative stress, is fundamental to disease progression [12,34]. Degenerating neurons in the substantia nigra are highly complex, unmyelinated fibers with an unusually high metabolic demand. SNpc neurons also have pacemaker Ca2+ channels that regulate their background activity. It is likely that these properties make these cell types particularly vulnerable to the production of reactive oxygen species and their harmful downstream effects. Oxidative stress is harmful to mitochondria, causing dysfunction in complex 1 of the electron transport chain [35]. Diseased mitochondria can trigger apoptosis through processes such as abnormal activation of cytochrome C, upregulation of pro-apoptotic proteins Bax and Bim, and opening of mtPTP channels within the mitochondrial membrane which trigger pro-apoptotic cascades within the cell cytoplasm [36]. Parthanatos, another mitochondria-mediated mechanism of cell death, may also be triggered when reactive oxygen species up-regulate poly (ADP-ribose) polymerase (PARP-1) activity, which in turn triggers the release of apoptosis-inducing factor (AIF1) from the mitochondrial membrane [37]. The removal of damaged mitochondria is fundamental to protecting neurons against downstream cellular processes that lead to apoptosis. Mutations in the PARKN gene can cause early-onset PD. PARKN encodes a ubiquitin ligase that drives the adaptive breakdown of dysfunctional mitochondria, a protective process called ‘mitophagy’. The loss of this important cell function leads to neuronal death and results in Parkinsonism in teenage years or early adulthood [38]. A related gene mutation in PINK1 is another genetic cause of early-onset PD. The PINK1 protein product has been shown to tag pathological mitochondria for recognition by PARKN, lending further support to the notion that pathological mitochondria damaged by reactive oxygen species are central to neuron death [41]. LRRK2- a mutation in which causes the most common form of mono-allelic PD- encodes a protein involved in diverse cellular functions including oxidative stress, inflammatory cascades, protein stabilization, mitochondrial stabilization, and mitophagy, which provides even further evidence of the centrality of these cell functions in the genesis of PD [39].
Microglia, the resident immune cells in the central nervous system, can modify the downstream cellular effects of oxidative stress and mitochondrial dysfunction, and determine the innate immune response against affected neurons. In non-PD brain tissue, protective microglia suppress the immune attack of dopaminergic neurons through neuron-glia crosstalk, histone modification and mRNA regulation [40]. The transition to pro-apoptotic microglia through an unidentified signaling mechanism may promote cell death in response to oxidative stress and mitochondrial dysfunction. Supporting the role of the immune response in the genesis of PD, studies have demonstrated elevated serum levels of inflammatory cytokines such as IL-2, IL-6, IL-8, TNF-α, and IFN-γ in PD sufferers.[41,42] PD sufferers also demonstrate a lower systemic lymphocyte count and higher neutrophil-to-lymphocyte ratio (NLR) than healthy controls [43]. More recently, there has been an increasing focus on the role of the signaling molecule sphingosine-1-phosphate (SIP) in attenuating microglial-driven neuronal apoptosis [44]. An association study of 392 participants demonstrated under-expression of S1P in the serum of PD sufferers compared to matched controls. In 64 PD participants who were followed up prospectively, advancing PD severity was correlated with a progressive reduction in serum S1P levels [45].
Interactions Between Oxidative Stress, Mitochondria and Innate Immunity
The Vicious Cycle of Oxidative Stress and Alpha-Synuclein Deposition
As well as being a driver of mitochondrial injury and inflammation, it has been demonstrated that oxidative stress can trigger the pathological phosphorylation and nitration of tyrosine residues within aS oligomers which leads to the formation of lewy bodies [46]. In addition, model studies suggest that excessive Ca2+ influx- a known cause of oxidative stress- is a key driver of overproduction of s129 aS, the aS form most associated with lewy body deposition as mentioned above [33]. In other words, oxidative stress may directly drive the aggregation of aS as lewy bodies in diseased neurons.
While pathological alpha-synuclein deposits may indeed be caused by oxidative stress, some evidence also suggests the opposite. That is, misfold aS accumulation may in turn drive oxidative stress. A central function of endogenous aS is the stabilization of SNARE proteins, which aggregate dopamine in presynaptic vesicles and allow vesicular fusion with the presynaptic endplate for its release into the synapse. Therefore, loss of this function may lead to the accumulation of cytoplasmic dopamine, a recognized generator of oxidative stress, within the cell cytoplasm [49]. The mechanism by which cytoplasmic dopamine induces oxidative stress is likely multifactorial, although a key mechanism involves the formation of an unstable redox couplet with ferrous/ferric cations (Fe2+/Fe3+) [47]. This relationship between Fe2+/Fe3+ and cytoplasmic dopamine is made all the more compelling by studies demonstrating that the PD brain undergoes disproportionate iron deposition, as quantified by brain imaging, in areas of known degeneration where dopamine stores are highest, namely the SNpc [48]. The propagation of pathological aS may also lead to oxidative stress through the loss of other protective properties of functioning aS. For example, endogenous aS may play a role in sequestering early peroxidation products of fatty acids. On the other hand, fibrillar aS- the form associated with lewy bodies- demonstrates an 80% reduction in this scavenging property which increases cell exposure to these reactive fatty acid metabolites [49]. To summarise, the cell processes underlying neuronal death in sporadic PD may involve a complex reciprocal interaction between oxidative stress cell pathways and the accumulation of pathological proteins such as aS.
Targets for Disease Modification
These mechanisms of neural degeneration present a large array of targets for disease modification. Prior studies have focused on four primary strategies (1) attenuation, prevention, or reversal of alpha-synuclein deposition (2) modification of oxidative stress and immune responses leading to cell death (3) regeneration or replacement lost neurons and (4) repurposing old drugs hypothesized to have disease modifying properties.
Prevention and Attenuation of Alpha-Synuclein Aggregation
Treatments that prevent or reverse the accumulation of aS and formation of lewy bodies could provide a pathway to disease modification.
Any intervention that suppresses the expression of SCNA, the gene encoding aS, could theoretically prevent aS aggregation. Stimulation of the beta-2 receptor may confer this very advantage [50]. A large cohort study in Norway demonstrated that patients who regularly received salbutamol, a sympathomimetic agent that acts via selective beta-2 adrenergic receptor stimulation, had a reduced risk of developing IPD compared to the general population [51]. Similar findings were observed for other beta-2 agonists such as clenbuterol and metaproterenol. In contrast, patients who used propranolol, a beta receptor antagonist, demonstrated a modest increase in relative risk of IPD. This link is only an association, however, and critics point out the possibility of reverse causation because prodromal PD patients are more likely to present with non-specific action tremors which may in fact herald the onset of PD, and that are commonly treated with propranolol [52].
One fundamental way SCNA transcription can be attenuated is by targeting the specific epigenetic processes that control gene transcription. Mazzocchi et al. suggested that class 2 HDACs, molecules involved in histone acetylation, represent compelling targets for epigenetic suppression of SCNA expression [22]. Preliminary studies indicate that modification of upstream SCNA gene regulators, namely FTO, can achieve a similar effect on aS expression. Introduction of m6A demethylase FTO (siFTO) to neurons via mesenchymal stem cell-derived exosomes has been shown to slow dopaminergic neuron death in PD mouse models [53].
As previously discussed, GCA is an enzyme involved in the lysosomal sequestration of misformed aS. It also modifies SCNA gene expression to transcribe a benign form of aS with a low propensity for aggregation. A phase 1 trial of LTI-291, an allosteric activator of GCA, has demonstrated good tolerability among 40 GBA-PD sufferers, with a long-term RCT to follow [54].
As highlighted in Braak’s hypothesis discussed above, aS deposition is known to first accumulate in the peripheral nervous system, particularly the enteric plexus innervating the gastrointestinal tract. The gut microbiota is a complex ecosystem of bacteria that is in constant interface with enteric neurons and has been the focus of PD research for many years. PD is associated with higher gut levels of verrucomicrobiaceae (gram negative, obligate anaerobes) and lower levels of prevotellaceae (gram negative anaerobes) compared with healthy controls [55]. The relative concentrations of these organisms affect toll-like receptor (TLR) expression in gut dendritic cells which impacts mucosal permeability to aS, influencing its aggregation and propagation throughout the body. In MPTP mice models, fecal micro-transplantation can reconstitute a protective gut microbiota profile and has been shown to protect against striatal neuron loss [56].
Other studies have focused on altering the post-translational modifications, aggregation, and degradation of aS. UCB0599 is a novel small molecule inhibitor of aS misfolding and aggregation which demonstrates good brain penetrance. Phase 1 trials have suggested good tolerability in human subjects [57]. ABT-888, a small molecule inhibitor of PARP-1 designed to dampen parthanatos-mediated cell death, has been shown to in fact prevent misfolding of aS into its aggregation-prone fibrillar form, demonstrating the intimate relationship between these pathological cell processes [40]. Several studies have explored the manipulation of endogenous chaperones, or heat shock proteins, which can prevent the aggregation of aS by facilitating its ubiquitination and upregulating proteosomal activity within cells (see above) [58]. Geldanamycin is one such agent that inhibits Hsp90. Pretreatment with geldanamycin protects against dopaminergic neurodegeneration in MPTP mouse models of Parkinson's Disease [59]. Other novel therapeutics targeting the upregulation of the protective Hsp70 demonstrate a similar effect in these mouse models [25]. FTY720 (fingolimod) is another enzyme involved in aS degradation, and one study has demonstrated the practicality of using a chitosan nanocarrier to introduce FTY720 into neurons [60]. A nanoscavenger molecule (NanoCA) delivered nasally has been shown to upregulate exosomal clearance of aS in mouse models by upregulating transcription factor EB, a major autophagy regulator, thereby protecting against MPP+-induced dopaminergic toxicity [61]. Iron sequestration has been theorized as a mechanism of neuroprotection in Parkinson’s Disease, by interrupting the interaction between aS deposition and potentially toxic iron accumulation. A phase one study of PBT434, a small molecule with a partial affinity iron-binding motif, reduced iron load and iron-mediated aS aggregation in 6-OHDA, MPTP and transgenic PD mouse models, and resulted in the rescue of motor function [62]. Disappointingly, a recent human study of another iron chelator, deferiprone, demonstrated no clinical benefit over a 36-week treatment period, with a signal towards harm with more participants in the treatment arm progressing to increased levodopa requirements [63]. GYY4137, a novel therapeutic agent, has been shown to prevent aS nitration in mouse models by releasing hydrogen sulphide (H2S) which sequesters reactive oxygen species; however, no study demonstrates its efficacy in humans [64]. As previously mentioned, the tyrosine kinase c-abl phosphorylates aS and potentiates its misfolding and aggregation. Additionally, it appears to inhibit the neuroprotective function of PARKN. Several c-abl inhibitors, including nilotinib and radotinib, have shown neuroprotective properties in animal models of PD (Karuppagounder et al., 2018, Karuppagounder et al., 2014). Unfortunately, multiple human trials on nilotinib have failed to demonstrate any meaningful clinical benefit over 12 month treatment periods.[65,66] Nilotinib poorly traverses the blood brain barrier, and many researchers still believe an alternative c-abl inhibitor with good CSF bioavailability represents a promising therapeutic pathway [67]. One such candidate c-abl inhibitor, vodobatinib, has so far demonstrated reasonable tolerability in phase 1 human trials [68].
Immunotherapies are the most advanced and targeted therapies to accelerate the removal of pathological aS. An Austrian phase 1 trial demonstrated the safety of PDO1A, a novel 8-amino acid-peptide that mimics an epitope of the C-terminus of aS and induces active immunity against endogenous aS [69]. Doses between 15 and 75 mg showed good tolerability and triggered an active humoral response in PD patients. Those who received the 75 mg dose showed reduced CSF concentrations of oligomeric aS [72]. Passive immunotherapies, which involves exogenously produced antibodies against aS, have also shown early promise. Hung et al., in their review ‘Approaches to Disease Modification for Parkinson’s Disease: Clinical Trials and Lessons Learned’, provide an exhaustive list of passive immunotherapies currently in early phase trials, some of which have demonstrated good tolerability [3]. These agents include prasinezumab (PRX002/RG7935), a humanized monoclonal antibody against the C-terminus of aS. This agent demonstrated good tolerability in a phase 1 trial. A recent phase 2 trial did not reached its primary endpoint of improvement in clinical status among participants, although an extended 2b trial has demonstrated modest attenuation of motor decline as measured by the MDS-UPDRS part 3 scale.[70,71,72] More recent work by the Pagano group did not demonstrate a benefit to prasinezumab, with a phase 2 clinical trial over 1 year failing to meet its primary or secondary clinical endpoints, and SPECT imaging not demonstrating protection of dopamine transporter density in the putamen (PASADENA trial) [73]. Similarly, cinpanemab, another monoclonal antibody to aS, showed no clinical benefit over a 52-week treatment period [74].
Modification of Oxidative Stress, Mitochondrial Dysfunction and Neuroinflammation
Other studies have focused on modifying oxidative stress, mitochondrial dysfunction and the resultant immune responses which can lead to neuronal death. Fat-soluble vitamins are a valuable source of antioxidants and have been considered for disease modification for decades. Vitamin E, vitamin C, and polyphenols interact with reactive oxygen species and terminate oxidative chain reactions [75]. Multiple studies have demonstrated that higher dietary vitamin E intake correlates inversely with the risk of PD [76,78]. The first clinical trial using vitamin E for treating early symptomatic PD was carried out over 25 years ago (DATATOP study) and it failed to demonstrate a sustained clinical benefit [3,77]. A randomized controlled trial in 2017 demonstrated a modest but statistically significant improvement in UPDRS scores after 12 weeks of treatment with high-dose vitamin E and omega-3 fatty acids [78]. Further studies that incorporate a clinical washout period are needed to investigate the long-term effects of high-dose vitamin E supplementation on PD progression. However, many researchers doubt the clinical utility of vitamin E because it poorly traverses the blood-brain barrier. N-acetyl cysteine (NAC) is a precursor of glutathione, a powerful antioxidant. Many studies have explored the potential of NAC to dampen reactive oxygen species and inflammation in neurodegenerative diseases [79]. NAC administration in 6-OHDA PD mouse models proved to be neuroprotective [80]. A trial of 42 patients with IPD demonstrated that combined intravenous infusion and oral supplementation of NAC led to increased dopamine transporter (DAT) binding in the caudate and putamen [81]. Unfortunately, intranasal glutathione treatment did not alter the clinical outcomes of PD participants [82]. Urate, a byproduct of purine metabolism, is another powerful endogenous antioxidant that activates the Nrf2 antioxidant pathway. In 2017, Crotty et al., demonstrated that treatment with purine inosine could elevate CSF urate concentrations and was well-tolerated in PD participants [83]. Unfortunately, a 2 year trial of inosine in 298 participants with PD failed to meet its primary endpoint of slowing UPDRS clinical disease progression [84]. Celastrol is another potent antioxidant agent that also activates the Nrf2 pathway and has shown disease-modifying effects in the MPTP PD mouse model [85]. Other novel scavenger antioxidants are currently under investigation for their neuroprotective effects, including two scavenger antioxidants (lipophilic metalloporphyrins), which have been shown to preserve ventral midbrain dopaminergic neurons in mouse models [86].
Removal of diseased mitochondria can circumvent downstream cell death pathways. Rapamycin reduces mitochondrial stress in LRRK2 mutant mice but has not yet been shown to confer advantage in human patients [87]. An interesting trial from 2020 indicates the activation of mu-opioid receptors can protect cells against mitochondrial dysfunction by upregulating PARKN-mediated mitophagy [88]. This may prove significant as hydrocortisone, the ubiquitous anti-inflammatory steroid, has been shown to increase PARKN levels in mice brains leading to increased dopaminergic neuron survival [89].
Targeted anti-immune treatments can circumvent the common pathway by which oxidative stress and mitochondrial dysfunction lead to neuronal death. Long-term caffeine intake is negatively correlated with risk of PD. It has been postulated that caffeine may confer this advantage by preventing microglial activation via the inhibition of adenosine A2A receptors [90]. Intriguingly, activation of the endocannabinoid system through the use of targeted THC and CBD phytocannabinoids may confer a similar advantage by redirecting M1 microglia to the neuroprotective M2 phenotype [91]. Novel ligands that modify microglial reactivity, such as the nuclear receptor-4A2 ligand C-DIM12, have demonstrated a neuroprotective effect in MPTP knockout mice, whereas a liposome vector that delivers dexamethasone directly to CD163+ resident macrophages in the basal ganglia can protect dopaminergic neurons in the 6-OHDA Parkinson’s mouse model.[92,93] Finally, other novel drug candidates have demonstrated increased cell survival by targeting the Src/phosphatase and PPARγ pathways.[94,95] Hence, although a multitude of mechanisms have been shown to circumvent destructive processes in diseased neurons, further studies are required to investigate their potential to confer disease modification in human patients.
Regeneration of Lost Neurons
Given that symptoms of PD manifest only after years of neuronal degeneration, a challenge for the above treatment modalities is that by the time patients are symptomatic, vast neuronal populations have already been lost [96]. Leaving aside the challenges of identifying neural degeneration before symptom onset, for those who already suffer from clinical disease, replacing lost neuronal populations may be the only hope for meaningful disease recovery. For decades, stem cell therapy has been regarded as the great hope for a cure. Dopamine progenitor cells can be derived from pluripotent fetal cells; however, the ethical quandary of harvesting fetal tissue remains a barrier to ongoing research. Recent advances in adult-derived pluripotent stem cells provide hope that stem cell research in PD may yet advance [97]. Mesenchymal stem cells are the largest and most easily accessible reservoir of adult stem cells, but research into their efficacy in PD remains limited [93,98]. Song et al. demonstrated that adult neural progenitor cells may be introduced to the basal ganglia through astrocyte co-grafting [99]. As of 2023, there are 13 human trials investigating the use of stem cells for the treatment of PD [100]. Stoddard-Bennet and Pera point out the inherent risk of this process, with pluripotent stem cells presenting the theoretical risk of tumors and also severe immune reactions within the host [101].
The human adult brain retains a reservoir of pluripotent neural progenitor cells that could be stimulated to regenerate the patients’ own dopaminergic neurons, thereby circumventing the need for stem cell grafting. These cells are responsive to neurotrophic molecular signals such as glial cell line-derived neurotrophic factor, fibroblast growth factor 2 and cerebral dopamine neurotrophic factor (CDNF), all of which have been shown to change concentration in PD brains [102]. Cell and animal-based trials have demonstrated the practicality of viral vectors to introduce genes expressing glial cell line-derived neurotrophic factor and neurturin into dopaminergic cells of the SNpc [103]. A phase 1 trial of direct intraputamenal injections of CDNF showed good tolerability, though a 12-month treatment of moderate to severe PD sufferers did not meet the secondary endpoint of improved PD motor scores [104]. It is still hoped that increased expression of neuronal growth factors could protect against neuronal death and promote neuronal arborisation.
Other neural regeneration signaling pathways have also shown promise. Pirodipine is a small molecule under development for the treatment of Huntington’s disease that has also been shown to stimulate dopamine neuron regeneration through activation of the sigma-1 receptor [105]. As an interesting side note, it has been postulated that deep brain stimulation, a symptomatic therapy for advanced PD, may in fact stimulate STN neuron regeneration through increasing brain-derived neurotrophic factor (BDNF) concentrations [106]. Further trials are needed to explore the potential of these and other neurotrophic factors to alter PD trajectory.
Repurposing Old Drugs
Repurposing available drug therapies represents the fastest and most cost-effective approach to disease modification. This paper has already discussed the potential advantages of some current immune modulators, beta-agonists, caffeine, dietary anti-oxidants and endocannabinoids.
Until recently, it was posited that monoamine oxidase (MAO) inhibitors, drugs commonly used to inhibit dopamine metabolism to treat symptomatic PD, may also protect dopaminergic neurons [107]. Recent longitudinal studies however have not demonstrated this advantage of MAO inhbitors. On the other hand, an anti-cancer (anti-VEGF) agent SU4312 protects against MPTP-associated neurotoxicity, at least in part, through its inhibition of MAO-B [108]. A meta-analysis from 2016 looked at the apparent advantage of anti-hypertensives to protect against PD [109]. Intriguingly, in this study it was calcium channel blockers, rather than beta-blockers, which were most associated with reduced PD risk. Dopaminergic fibers within the basal ganglia have self-generating Ca2+ channels that increase their metabolic demand. Unfortunately a recent study of isradipine did not demonstrate any disease-modifying effect in adult volunteers [110]. In a recent cohort study, Vitamin B12, a critical precursor in myelin production and other nerve cell functions, did not demonstrate an advantage for PD sufferers [111]. Exanatide, a GLP-1 agonist used for diabetes management, has been shown to modify neuron survival through various signaling cascades including microglial activity modification.[112,113] A small, well-designed, randomized, double-blind trial incorporating 62 participants and a 12-week washout period failed to demonstrate a meaningful change in disease trajectory [116]. A more recent study of exendin-4, another GLP-1 analogue, demonstrated disease-modifying effects in a PD mouse model [114]. NLY01, a brain-penetrating, long-acting version of exanatide failed to demonstrate a disease-modifying effect in a 36-week randomized controlled trial of 255 participants with early IPD [115]. Finally, it has been proposed that statin therapy may confer neuronal protection by upregulating synapse complexity and VMAT receptor density in dopaminergic neurons, though evidence for this effect is limited [116].
Despite the suspected immune basis of neurodegeneration in PD, there has been surprisingly little focus on well-established immune modulatory drugs used for treating neuroinflammatory conditions, particularly those used to treat multiple sclerosis (MS). As previously mentioned, S1P is a regulator of microglial activity and aS deposition and fingolimod (FTY720) is a moderate to high efficacy treatment for MS which is a modulator of S1P activity. FTY720 treatment of 6-OHDA Parkinson’s mouse models protects against dopaminergic neuron loss [117]. Given its proven safety in humans, this is a compelling approach for future trials in PD.
Summary of trials
| Agent/Intervention | Mechanism of action | Efficacy in preclinical models | Participant profile/human trial form | Status |
| alpha-synuclein | ||||
| Salbutamol (beta2AR agonist) | ?suppression of SCNA gene expression | Large Norwegian cohort of salbutamol users | Cohort data demonstrating 34% lifetime risk reduction | |
| Clenbuterol (beta2AR agonist) | ?suppression of SCNA gene expression | Protection of SNPc neurons in MPTP mouse models | ||
| siFTO (upstream SCNA gene regulator) | ?suppression of SCNA gene expression through upstream moodlation of FTO gene function | Neuroprotective in mouse models of PD | ||
| UCB0599 | Inhibition of aS misfolding and aggregation | Phase 1/1b trial | Good tolerability amongst 94 volunteers | |
| ABT888 | Inhibition of as misfiling and aggregation (?through inhibition of PARP-1) | Reduced deposition of fibrillar aS in mouse brains | ||
| Geldanamycin | Upregulation of HsP mediated proteosomal activity | Prevention of dopaminergic neuron degeneration in MPTP mouse models | ||
| NanoCA | Upregulation of tfEB mediated aS clearance | Protection against MPP+ induced neuron toxicity | ||
| GY4137 | Prevention of aS nitration | Reduced aS aggression in mouse models | ||
| Nilotinib | c-abl tyrosine kinase inhibitor | Reduced dopaminergic neuronal degeneration in mouse models | Phase 1: 6 month triall of 76 participants | No clinical benefit on UPDRS motor score Poor CSF bioavailability |
| Vodobatinib | Novel c-abl tyrosine kinase inhibitor | Phase 1 tolerability study | Good CSF bioavailability, good tolerability | |
| PDO1A | Active immunization against aS | Phase 1: 32 participants, mild IPD (within 4 years of diagnosis) | Reduced CSF aS concentration in human PD participants | |
| Prasinezumab | Passive immunotherapy | Phase 2: 316 participants, early stage IPD | Negative result. | |
| Cinpanemab | Passive immunotherapy | Phase 2: 357 participants with early stage IPD | Negative result | |
| Mitochondrial dysfunciton/oxidative stress | ||||
| Vitamin E | Termination of oxidative chain reactions | Cohort meta-analysis —> DATATOP study —> 800 participants, early IPD |
Reduced IPD incidence Modest benefit in UPDRS score over 12 week treatment period |
|
| IV + oral NAC (N-acetyl choline) | Antioxidant | Enhanced dopaminergic neuronal viability in 6-OHDA PD mouse model | Phase 1: 42 participants, mild IPD | Increased DAT binding in caudate and putamen |
| Intranasal glutathione | Antioxidant | Phase 2 | Negative trial | |
| Inosine | Increases urate concentration | Phase 1 —> Phase 2 —> 298 participants, 2 year treatment |
Increases CSF urate concentration, well tolerated Negative trial |
|
| Lipophilic metalloporphyrins | Novel scavenger antioxidant | Preserves ventral midbrain dopaminergic neurons in mice models | ||
| Rapamycin | Reduced mitochondrial stress | Dopaminergic neuron protection in LRRK2 mutant mice | ||
| Immune modulation | ||||
| C-DIM12 | Microglial nuclear receptor-4A2 ligand | Neuroprotective in MPTP knockout mice | ||
| Dexamethasone liposome vector | Dexamethasone delivery to CD163+ macrophages | Neuroprotective in 6-OHDA mouse model | ||
| NLY01 | Modification of microglia activity | 36 week trial of 255 participants with early PD | Negative result | |
| Regeneration of lost dopaminergic neurons | ||||
| Stem cell therapies | Regeneration of lost neurons | Pending | Pending | |
| Intraputamenal injection of CDNF | Neurotrophic stimulation of puatmenal stem cells | Phase 1 trial | Good tolerability | |
| Repurposing of old drugs | ||||
| Isradipine | Calcium channel blocker ?reduced metabolic toxicity to dopaminergic neurons | Protective in mouse models | Phase 3 —> 336 participants, early stage IPD (<3yrs) | Negative result |
| SU4312 | Anti-cancer agent, ?MAO-B inhibition | Neuroprotective in MPTP mouse models | ||
| Exanatide | GLP-1 agonist | Cohort study —> Phase 2 clinical trial —> 62 participants, early PD |
Neuroprotective in 6-OHDA mouse model Negative trial |
|
| Fingolimod | S1P modulator AND aS modulator | Neuroprotective in 6-OHDA mouse model |
COVID-19: A Unique Window into the Pathogenesis of IPD?
COVID-19 is a once-in-a-century pandemic that, at the time of writing this paper, has infected well over a billion people worldwide. Acute and chronic complications of COVID-19 infection have been reported in almost every organ system, and the nervous system is no exception [118]. For these reasons, the COVID-19 pandemic presents a unique opportunity to investigate the mechanisms by which neurodegenerative diseases such as Parkinson’s Disease may arise [119].
If there is indeed a link between COVID-19 and Parkinson’s Disease, it will emerge over time as previously infected patients age [120]. If we examine the already recognized relationship between Parkinson’s Disease and viral infections, then there is indeed cause for concern. In 2009, H5N1, the ‘Avian flu’, was shown to induce inflammatory cascades and protein aggregation in mouse neural tissue mimicking the changes shown in PD, and these mice subsequently demonstrated neural degeneration specifically in the SNpc [121]. Encephalitis lethargica is an inflammatory CNS condition characterized by hypersomnolence, akinesis and abulia that followed the emergence of the Spanish flu [122]. Patients who recovered often suffered from long-term Parkinsonism, a syndrome referred to as ‘Post-Encephalitic Parkinsonism’ (PEP), suggesting these patients’ extrapyramidal symptoms may have been a direct result of the CNS changes triggered by the H1N1 virus [122,123]. Indeed, studies have demonstrated that the H1N1 virus can induce CNS auto-immunity through the induction of pro-apoptotic microglia; a mechanism already thought to be involved in the genesis of PD [124]. Intriguingly, posthumous biopsies of PEP patients demonstrated tau-rich neurofibrillary tangles and an absence of Lewy bodies, indicating the underlying disease process of PEP is in some ways pathologically distinct from sporadic PD, and histologically is more reminiscent of Progressive Supranuclear Palsy [125].
Tissue biopsies in most cases of COVID-19-related neurological injury have not demonstrated direct virus infiltration of neuronal tissue. Despite this, it is now widely accepted that in rare cases coronaviruses such as SARS-Cov2 can also access the CNS and generate harmful downstream cell processes, though the mechanism by which they access the CNS is still not completely understood. Proposed mechanisms include direct neuron-to-neuron communication via the olfactory bulb, lymphatic spread, or direct haematogenous spread following the breakdown of the blood-brain barrier during the acute inflammatory phase [122]. The avidity of certain neuronal populations for COVID-19 invasion can be explained by the expression of cell surface ACE2 receptors, the receptors to which the COVID spike protein binds and thereby gains entry into cells [126]. Mouse models suggest COVID-19 can access the vertebrate CNS via the nasal route and take up residence in neuronal regions known to have a high density of these receptors, including the basal ganglia, brainstem and hippocampus (Chana-Cuevas et al. 2020). Of some concern, coronaviruses similar to COVID-19 have been shown to remain latent within resident CNS leucocytes and neurons for years after infection, potentially influencing cellular processes decades after patient recovery [127,129]. Alpha-synuclein has been shown to be upregulated following exposure to the West Nile virus, which some interpret as evidence this protein is operating as a resident anti-viral defence mechanism [129]. By extension, it could be possible that exposure to other viruses, including COVID-19, could lead to alpha-synuclein accumulation and the subsequent emergence of Parkinson’s Disease. Such a finding would lend further support to the hypothesis that alpha-synuclein is indeed pathogenic in the development of PD. On the other hand, cell models have also shown that SARS-COV2 (COVID-19) can induce the death of dopamine brainstem neurons through the direct activation of destructive cell processes, including the activation of caspases 2, 3 and 8, as well as the NFkappaB cascade within dopaminergic cells [128].
Conclusions and ‘Where to from Here’
Much has been learnt about the pathogenesis of Parkinson’s Disease. While the fundamental mechanism of neuron death remains debated, it is becoming clear that it is driven by a complex interplay between alpha-synuclein aggregation, reactive oxygen species, mitochondrial dysfunction and innate immune processes. These discoveries have opened the door to a range of potential avenues to disease modification by arresting these neuron death processes. To date, no therapy has been shown to produce a profound and sustained improvement in human participants, and much more must be done to explore the potential therapeutic avenues discussed above.
One major barrier to disease-modifying treatments is that by the time patients are symptomatic, neural degeneration has been occurring for decades. More must be done to identify patients in the earliest stages of neural degeneration to introduce treatments before patients become symptomatic. Such an achievement will require more foundational knowledge of the factors predisposing patients to PD so that appropriate candidates can be identified for surveillance. Surveillance programs could monitor for the emergence of early premotor features, including anosmia, autonomic dysregulation or REM sleep behaviour disorder. Of these, RBD has emerged as the frontrunner, as it demonstrates a high degree of specificity for conversion to a synucleinopathy (PD, MSA, DLB or PDD), with a 7% annual conversion and 90% lifetime incidence for those diagnosed with RBD by polysomnography (Hogl et al., 2017). The link between RBD and PD is likely an underlying failure of REM sleep atonia, which is challenging to identify in the absence of frequent, typical RBD-related sleep movements and in patients without a bed partner. For this reason RBD lacks the required sensitivity of a prodromal disease marker.
Research into prodromal disease biomarkers has intensified recently with the discovery of real-time quaking-induced conversion (RT-QuiC) as a validated method of detecting aS in many different tissues including CSF, serum and skin.[129,130] Subsequent studies in RBD participants validate RT-QUiC as a method of prodromal disease detection, providing hope that a simple blood, skin or other non-invasive tissue sample could be used for widespread screening of prodromal PD and related synucleinopathies [131]. Another more costly approach could utilize either VMAT PET or SPECT imaging to look for early reduction in dopamine activity, though again, by the time such changes are observed much neuronal degeneration has likely already occurred. Furthermore, up to 20% of patients with clinical IPD do not demonstrate evidence of dopamine deficiency on imaging (SWEDD, ‘scans without evidence of dopamine deficiency’) [3].
Many of the candidate interventions discussed in this review were first tried on mouse models of PD, such as the MPTP model. MPTP is especially toxic to dopaminergic neurons, thereby mimicking many of the motor features of PD. However, direct toxicity of dopaminergic neurons does not reflect the physiological processes which cause the loss of dopaminergic neurons in human PD, nor does it replicate the extra-dopaminergic changes demonstrated in humans [132]. It is, therefore, of little surprise that disease-modifying effects in animal models have not been replicated in human subjects.
Another major challenge to disease modification research in human trials relates to study design. There is currently no agreed-upon objective measure of neuronal loss, so treatment efficacy relies on subjective measures of clinical status such as the UPDRS Parkinson’s scale [12]. Further to this, most phase 2 trials are typically no more than six months in duration. PD moves slowly, with often-times an almost imperceptible progression in mot impairment over any six-month period. For this reason, demonstrating a separation in outcomes between control and treatment arms over this short time is inherently problematic. Apart from the problem of inter and intra-rater reliability such scales introduce, these scales are unable to differentiate between a treatment’s symptomatic benefit and true disease modification. Studies have attempted to get around this issue by introducing a clinical ‘washout period’ prior to clinical assessment, but this adds significantly to the study duration, and there is currently no way of knowing how long such washout periods should be. The consensus opinion remains that the UPDRS Parkinson’s scale is the gold standard for assessing motor outcomes in long-term disease-modifying trials, though experts now advocate the inclusion of non-motor parameters to provide a broader measure of PD severity and progression.[133,134] The identification of a clinical biomarker of active disease would represent an elegant solution to these limitations [135]. Trials on CSF aS levels have provided confounding results with lower CSF aS modestly correlating with the risk of PD [136]. McGhee et al. provide a thorough review of recent clinical trials of disease modification and argue that the current gold standard trial design is a long-term follow-up study aiming to demonstrate a sustained and robust divergence in clinical outcome measures between cases and controls [137].
When we consider the above challenges, it could be argued that the greatest hope of a cure for sporadic PD lies in neural regeneration utilizing stem cell therapy. Such therapies could theoretically reverse nerve cell degeneration, leading to a rapid and sustained clinical improvement that does not rely on early detection of symptoms or protracted studies looking for improved clinical outcomes over decades. Grafted stem cells carry the theoretical risk of tumors and host rejection, so a focus on neurotrophic factors to stimulate autologous neural stem cells presents a compelling approach.
As global populations age, neurodegenerative diseases are emerging as one of the major drivers of human suffering and healthcare burden. For sporadic PD, dopamine-based therapies eventually lose efficacy and patients suffer from mounting non-motor features that significantly contribute to patient morbidity and mortality. For these reasons, there is a critical urgency for further research into disease modification in PD. The limited success of past efforts is sobering. On the other hand, the diversity and international reach of current efforts to discover new therapeutics provides much hope for the future.
References
- Fearnley, J. M. and A. J. Lees (1991). "Ageing and Parkinson's disease: substantia nigra regional selectivity." Brain 114 ( Pt 5): 2283-2301. [CrossRef]
- Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565-81. [CrossRef]
- Hung AY, Schwarzschild MA. Approaches to Disease Modification for Parkinson's Disease: Clinical Trials and Lessons Learned. Neurotherapeutics. 2020;17(4):1393-405. [CrossRef]
- Grenn FP, Kim JJ, Makarious MB, Iwaki H, Illarionova A, Brolin K, et al. The Parkinson's Disease Genome-Wide Association Study Locus Browser. Movement Disorders. 2020;35(11):2056-67.
- Kim CY, Alcalay RN. Genetic Forms of Parkinson's Disease. Semin Neurol. 2017;37(2):135-46.
- Poewe W, Seppi K, Marini K, Mahlknecht P. New hopes for disease modification in Parkinson's Disease. Neuropharmacology. 2020;171:108085. [CrossRef]
- Jackson H, Anzures-Cabrera J, Simuni T, Postuma RB, Marek K, Pagano G. Identifying prodromal symptoms at high specificity for Parkinson's disease. Front Aging Neurosci. 2023;15:1232387. [CrossRef]
- Fengler S, Liepelt-Scarfone I, Brockmann K, Schaffer E, Berg D, Kalbe E. Cognitive changes in prodromal Parkinson's disease: A review. Mov Disord. 2017;32(12):1655-66. [CrossRef]
- Fang C, Lv L, Mao S, Dong H, Liu B. Cognition Deficits in Parkinson's Disease: Mechanisms and Treatment. Parkinsons Dis. 2020;2020:2076942. [CrossRef]
- Poewe W, Antonini A, Zijlmans JC, Burkhard PR, Vingerhoets F. Levodopa in the treatment of Parkinson's disease: an old drug still going strong. Clin Interv Aging. 2010;5:229-38. [CrossRef]
- Kordower JH, Burke RE. Disease Modification for Parkinson's Disease: Axonal Regeneration and Trophic Factors. Mov Disord. 2018;33(5):678-83. [CrossRef]
- Kalia LV, Kalia SK, Lang AE. Disease-modifying strategies for Parkinson's disease. Mov Disord. 2015;30(11):1442-50.
- Engelhardt E, Gomes MDM. Lewy and his inclusion bodies: Discovery and rejection. Dement Neuropsychol. 2017;11(2):198-201. [CrossRef]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24(2):197-211. [CrossRef]
- Hilker R, Schweitzer K, Coburger S, Ghaemi M, Weisenbach S, Jacobs AH, et al. Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch Neurol. 2005;62(3):378-82. [CrossRef]
- Athauda D, Foltynie T. Challenges in detecting disease modification in Parkinson's disease clinical trials. Parkinsonism Relat Disord. 2016;32:1-11. [CrossRef]
- Burre J, Sharma M, Sudhof TC. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb Perspect Med. 2018;8(3).
- Melki, R. Alpha-synuclein and the prion hypothesis in Parkinson's disease. Rev Neurol (Paris). 2018;174(9):644-52. [CrossRef]
- Mazzocchi M, Collins LM, Sullivan AM, O'Keeffe GW. The class II histone deacetylases as therapeutic targets for Parkinson's disease. Neuronal Signal. 2020;4(2):Ns20200001. [CrossRef]
- Thomas EA, D'Mello SR. Complex neuroprotective and neurotoxic effects of histone deacetylases. J Neurochem. 2018;145(2):96-110. [CrossRef]
- Park G, Tan J, Garcia G, Kang Y, Salvesen G, Zhang Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J Biol Chem. 2016;291(7):3531-40. [CrossRef]
- Friesen EL, De Snoo ML, Rajendran L, Kalia LV, Kalia SK. Chaperone-Based Therapies for Disease Modification in Parkinson's Disease. Parkinsons Dis. 2017;2017:5015307. [CrossRef]
- Tao J, Berthet A, Citron YR, Tsiolaki PL, Stanley R, Gestwicki JE, et al. Hsp70 chaperone blocks alpha-synuclein oligomer formation via a novel engagement mechanism. J Biol Chem. 2021;296:100613. [CrossRef]
- Riboldi GM, Di Fonzo AB. GBA, Gaucher Disease, and Parkinson's Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells. 2019;8(4).
- Gan-Or Z, Bar-Shira A, Gurevich T, Giladi N, Orr-Urtreger A. Homozygosity for the MTX1 c.184T>A (p.S63T) alteration modifies the age of onset in GBA-associated Parkinson's disease. Neurogenetics. 2011;12(4):325-32. [CrossRef]
- Glajch KE, Moors TE, Chen Y, et al. Wild-type GBA1 increases the alpha-synuclein tetramer-monomer ratio, reduces lipid-rich aggregates, and attenuates motor and cognitive deficits in mice. Proc Natl Acad Sci U S A. 2021;118(31).
- Pan B, Kamo N, Shimogawa M, Huang Y, Kashina A, Rhoades E, et al. Effects of Glutamate Arginylation on alpha-Synuclein: Studying an Unusual Post-Translational Modification through Semisynthesis. J Am Chem Soc. 2020;142(52):21786-98. [CrossRef]
- Mahul-Mellier AL, Fauvet B, Gysbers A, Dikiy I, Oueslati A, Georgeon S, et al. c-Abl phosphorylates alpha-synuclein and regulates its degradation: implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson's disease. Hum Mol Genet. 2014;23(11):2858-79.
- Anderson JP, Walker DE, Goldstein JM, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281(40):29739-52. [CrossRef]
- Arawaka S, Sato H, Sasaki A, et al. Mechanisms underlying extensive Ser129-phosphorylation in alpha-synuclein aggregates. Acta Neuropathol Commun. 2017;5(1):48. [CrossRef]
- Ghanem SS, Majbour NK, Vaikath NN, et al. α-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc Natl Acad Sci U S A. 2022;119(15):e2109617119. [CrossRef]
- Irwin DJ, Lee VM, Trojanowski JQ. Parkinson's disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci. 2013;14(9):626-36.
- van der Gaag BL, Deshayes NAC, Breve JJP, Bol J, Jonker AJ, Hoozemans JJM, et al. Distinct tau and alpha-synuclein molecular signatures in Alzheimer's disease with and without Lewy bodies and Parkinson's disease with dementia. Acta Neuropathol. 2024;147(1):14.
- Skaper SD, Facci L, Zusso M, Giusti P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front Cell Neurosci. 2018;12:72. [CrossRef]
- Bjorklund G, Dadar M, Anderson G, Chirumbolo S, Maes M. Preventive treatments to slow substantia nigra damage and Parkinson's disease progression: A critical perspective review. Pharmacol Res. 2020;161:105065. [CrossRef]
- Park HA, Ellis AC. Dietary Antioxidants and Parkinson's Disease. Antioxidants (Basel). 2020;9(7). [CrossRef]
- Hastings L, Sokratian A, Apicco DJ, Stanhope CM, Smith L, Hirst WD, et al. Evaluation of ABT-888 in the amelioration of alpha-synuclein fibril-induced neurodegeneration. Brain Commun. 2022;4(2):fcac042. [CrossRef]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85(2):257-73.
- Wallings R, Manzoni C, Bandopadhyay R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 2015;282(15):2806-26. [CrossRef]
- Le W, Wu J, Tang Y. Protective Microglia and Their Regulation in Parkinson's Disease. Front Mol Neurosci. 2016;9:89. [CrossRef]
- Reynolds A, Laurie C, Mosley RL, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol. 2007;82:297-325.
- Schwab AD, Thurston MJ, Machhi J, Olson KE, Namminga KL, Gendelman HE, et al. Immunotherapy for Parkinson's disease. Neurobiol Dis. 2020;137:104760.
- Grillo P, Sancesario GM, Bovenzi R, Zenuni H, Bissacco J, Mascioli D, et al. Neutrophil-to-lymphocyte ratio and lymphocyte count reflect alterations in central neurodegeneration-associated proteins and clinical severity in Parkinson Disease patients. Parkinsonism Relat Disord. 2023;112:105480. [CrossRef]
- Wang W, Zhao Y, Zhu G. The role of sphingosine-1-phosphate in the development and progression of Parkinson's disease. Front Cell Neurosci. 2023;17:1288437. [CrossRef]
- Schwedhelm E, Englisch C, Niemann L, Lezius S, von Lucadou M, Marmann K, et al. Sphingosine-1-Phosphate, Motor Severity, and Progression in Parkinson's Disease (MARK-PD). Mov Disord. 2021;36(9):2178-82.
- Barrett PJ, Timothy Greenamyre J. Post-translational modification of alpha-synuclein in Parkinson's disease. Brain Res. 2015;1628(Pt B):247-53.
- Pezzella A, d'Ischia M, Napolitano A, Misuraca G, Prota G. Iron-mediated generation of the neurotoxin 6-hydroxydopamine quinone by reaction of fatty acid hydroperoxides with dopamine: a possible contributory mechanism for neuronal degeneration in Parkinson's disease. J Med Chem. 1997;40(14):2211-6. [CrossRef]
- Barbosa JH, Santos AC, Tumas V, Liu M, Zheng W, Haacke EM, et al. Quantifying brain iron deposition in patients with Parkinson's disease using quantitative susceptibility mapping, R2 and R2. Magn Reson Imaging. 2015;33(5):559-65. [CrossRef]
- De Franceschi G, Fecchio C, Sharon R, Schapira AHV, Proukakis C, Bellotti V, et al. alpha-Synuclein structural features inhibit harmful polyunsaturated fatty acid oxidation, suggesting roles in neuroprotection. J Biol Chem. 2017;292(17):6927-37.
- Abdelmotilib H, West AB. Breathing new life into an old target: pulmonary disease drugs for Parkinson's disease therapy. Genome Med. 2017;9(1):88. [CrossRef]
- Mittal S, Bjørnevik K et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson's disease. Science. 2017;357(6354):891-8.
- Hopfner F, Höglinger GU et al. β-adrenoreceptors and the risk of Parkinson's disease. Lancet Neurol. 2020;19(3):247-54. [CrossRef]
- Geng Y, Long X et al. FTO-targeted siRNA delivery by MSC-derived exosomes synergistically alleviates dopaminergic neuronal death in Parkinson's disease via m6A-dependent regulation of ATM mRNA. J Transl Med. 2023;21(1):652.
- den Heijer JM, Kruithof AC et al. A Phase 1B Trial in GBA1-Associated Parkinson's Disease of BIA-28-6156, a Glucocerebrosidase Activator. Mov Disord. 2023;38(7):1197-208.
- Wang Q, Luo Y, Ray Chaudhuri K, Reynolds R, Tan EK, Pettersson S. The role of gut dysbiosis in Parkinson's disease: mechanistic insights and therapeutic options. Brain. 2021;144(9):2571-93. [CrossRef]
- Sun MF, Zhu YL, Zhou ZL et al. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson's disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav Immun. 2018;70:48-60.
- Smit JW, Basile P, Prato MK et al. Phase 1/1b Studies of UCB0599, an Oral Inhibitor of α-Synuclein Misfolding, Including a Randomized Study in Parkinson's Disease. Mov Disord. 2022;37(10):2045-56.
- Török N, Majláth Z, Szalárdy L, Vécsei L. Investigational α-synuclein aggregation inhibitors: hope for Parkinson's disease. Expert Opin Investig Drugs. 2016;25(11):1281-94. [CrossRef]
- Shen HY, He JC, Wang Y et al. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem. 2005;280(48):39962-9. [CrossRef]
- Sardoiwala MN, Karmakar S, Choudhury SR. Chitosan nanocarrier for FTY720 enhanced delivery retards Parkinson's disease via PP2A-EzH2 signaling in vitro and ex vivo. Carbohydr Polym. 2021;254:117435. [CrossRef]
- Liu J, Liu C, Zhang J, Zhang Y, Liu K, Song JX, et al. A Self-Assembled alpha-Synuclein Nanoscavenger for Parkinson's Disease. ACS Nano. 2020;14(2):1533-49.
- Finkelstein DI, Billings JL, Adlard PA et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson's disease. Acta Neuropathol Commun. 2017;5(1):53. [CrossRef]
- Devos D, Labreuche J, Rascol O et al. Trial of Deferiprone in Parkinson's Disease. N Engl J Med. 2022;387(22):2045-55. [CrossRef]
- Hou X, Yuan Y, Sheng Y et al. GYY4137, an H(2)S Slow-Releasing Donor, Prevents Nitrative Stress and alpha-Synuclein Nitration in an MPTP Mouse Model of Parkinson's Disease. Front Pharmacol. 2017;8:741.
- Pagan F, Hebron M, Valadez EH et al. Nilotinib Effects in Parkinson's disease and Dementia with Lewy bodies. J Parkinsons Dis. 2016;6(3):503-17. [CrossRef]
- Pagan FL, Hebron ML, Wilmarth B et al. Nilotinib Effects on Safety, Tolerability, and Potential Biomarkers in Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020;77(3):309-17.
- Werner, M.H. , Olanow, C.W., 2022. Parkinson's Disease Modification Through Abl Kinase Inhibition: An Opportunity. Movement Disorders 37, 6–15. [CrossRef]
- Walsh RR, Damle NK, Mandhane S et al. Plasma and cerebrospinal fluid pharmacokinetics of vodobatinib, a neuroprotective c-Abl tyrosine kinase inhibitor for the treatment of Parkinson's disease. Parkinsonism Relat Disord. 2023;108:105281. [CrossRef]
- Volc D, Poewe W, Kutzelnigg A et al. Safety and immunogenicity of the alpha-synuclein active immunotherapeutic PD01A in patients with Parkinson's disease: a randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020;19(7):591-600.
- Masliah E, Rockenstein E, Mante M et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011;6(4):e19338. [CrossRef]
- Jankovic J, Goodman I, Safirstein B et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-alpha-Synuclein Monoclonal Antibody, in Patients With Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018;75(10):1206-14.
- Pagano G, Boess FG, Taylor KI et al. A Phase II Study to Evaluate the Safety and Efficacy of Prasinezumab in Early Parkinson's Disease (PASADENA): Rationale, Design, and Baseline Data. Front Neurol. 2021;12:705407. [CrossRef]
- Pagano G, Taylor KI, Anzures-Cabrera J, Marchesi M, Simuni T, Marek K, et al. Trial of Prasinezumab in Early-Stage Parkinson's Disease. N Engl J Med. 2022;387(5):421-32. [CrossRef]
- Lang AE, Siderowf AD, Macklin EA et al. Trial of Cinpanemab in Early Parkinson's Disease. N Engl J Med. 2022;387(5):408-20. [CrossRef]
- Park HA, Ellis AC. Dietary Antioxidants and Parkinson's Disease. Antioxidants (Basel). 2020;9(7). [CrossRef]
- Etminan M, Gill SS, Samii A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson's disease: a meta-analysis. Lancet Neurol. 2005;4(6):362-5.
- Parkinson Study, G. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med. 1993;328(3):176-83.
- Taghizadeh M, Tamtaji OR, Dadgostar E et al. The effects of omega-3 fatty acids and vitamin E co-supplementation on clinical and metabolic status in patients with Parkinson's disease: A randomized, double-blind, placebo-controlled trial. Neurochem Int. 2017;108:183-9. [CrossRef]
- Tardiolo G, Bramanti P, Mazzon E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules. 2018;23(12). [CrossRef]
- Virel A, Johansson J, Axelsson J et al. N-acetylcysteine decreases dopamine transporter availability in the non-lesioned striatum of the 6-OHDA hemiparkinsonian rat. Neurosci Lett. 2022;770:136420. [CrossRef]
- Monti DA, Zabrecky G, Kremens D et al. N-Acetyl Cysteine Is Associated With Dopaminergic Improvement in Parkinson's Disease. Clin Pharmacol Ther. 2019;106(4):884-90. [CrossRef]
- Mischley LK, Lau RC, Shankland EG et al. Phase IIb Study of Intranasal Glutathione in Parkinson's Disease. J Parkinsons Dis. 2017;7(2):289-99. [CrossRef]
- Crotty GF, Schwarzschild MA. Chasing Protection in Parkinson's Disease: Does Exercise Reduce Risk and Progression? Front Aging Neurosci. 2020;12:186.
- Investigators TPSGS-P. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA. 2021;326(10):926-39.
- Zhang C, Zhao M, Wang B et al. The Nrf2-NLRP3-caspase-1 axis mediates the neuroprotective effects of Celastrol in Parkinson's disease. Redox Biol. 2021;47:102134. [CrossRef]
- Liang LP, Fulton R, Bradshaw-Pierce EL, Pearson-Smith J, Day BJ, Patel M. Optimization of Lipophilic Metalloporphyrins Modifies Disease Outcomes in a Rat Model of Parkinsonism. J Pharmacol Exp Ther. 2021;377(1):1-10. [CrossRef]
- Cooper O, Seo H, Andrabi S et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson's disease. Sci Transl Med. 2012;4(141):141ra90. [CrossRef]
- Xu Y, Zhi F, Mao J et al. delta-opioid receptor activation protects against Parkinson's disease-related mitochondrial dysfunction by enhancing PINK1/Parkin-dependent mitophagy. Aging (Albany NY). 2020;12(24):25035-59.
- Ham S, Lee YI, Jo M et al. Hydrocortisone-induced parkin prevents dopaminergic cell death via CREB pathway in Parkinson's disease model. Sci Rep. 2017;7(1):525.
- Chen Y, Shen J, Ke K, Gu X. Clinical potential and current progress of mesenchymal stem cells for Parkinson's disease: a systematic review. Neurol Sci. 2020;41(5):1051-61. [CrossRef]
- Cooray R, Gupta V, Suphioglu C. Current Aspects of the Endocannabinoid System and Targeted THC and CBD Phytocannabinoids as Potential Therapeutics for Parkinson's and Alzheimer's Diseases: a Review. Mol Neurobiol. 2020;57(11):4878-90. [CrossRef]
- Hammond SL, Popichak KA, Li X et al. The Nurr1 Ligand,1,1-bis(3'-Indolyl)-1-(p-Chlorophenyl)Methane, Modulates Glial Reactivity and Is Neuroprotective in MPTP-Induced Parkinsonism. J Pharmacol Exp Ther. 2018;365(3):636-51. [CrossRef]
- Tentillier N, Etzerodt A, Olesen MN et al. Anti-Inflammatory Modulation of Microglia via CD163-Targeted Glucocorticoids Protects Dopaminergic Neurons in the 6-OHDA Parkinson's Disease Model. J Neurosci. 2016;36(36):9375-90. [CrossRef]
- Lecca D, Nevin DK, Mulas G et al. Neuroprotective and anti-inflammatory properties of a novel non-thiazolidinedione PPARγ agonist in vitro and in MPTP-treated mice. Neuroscience. 2015;302:23-35. [CrossRef]
- Wang YD, Bao XQ, Xu S et al. A Novel Parkinson's Disease Drug Candidate with Potent Anti-neuroinflammatory Effects through the Src Signaling Pathway. J Med Chem. 2016;59(19):9062-79. [CrossRef]
- Ross GW, Petrovitch H, Abbott RD et al. Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann Neurol. 2004;56(4):532-9. [CrossRef]
- Kirkeby A, Parmar M, Barker RA. Strategies for bringing stem cell-derived dopamine neurons to the clinic: A European approach (STEM-PD). Prog Brain Res. 2017;230:165-90.
- Unnisa A, Dua K, Kamal MA. Mechanism of Mesenchymal Stem Cells as a Multitarget Disease- Modifying Therapy for Parkinson's Disease. Curr Neuropharmacol. 2023;21(4):988-1000. [CrossRef]
- Song JJ, Oh SM, Kwon OC et al. Cografting astrocytes improves cell therapeutic outcomes in a Parkinson's disease model. J Clin Invest. 2018;128(1):463-82. [CrossRef]
- Marsili L, Sharma J, Outeiro TF et al. Stem Cell Therapies in Movement Disorders: Lessons from Clinical Trials. Biomedicines. 2023;11(2). [CrossRef]
- Stoddard-Bennett T, Pera RR. Stem cell therapy for Parkinson's disease: safety and modeling. Neural Regen Res. 2020;15(1):36-40. [CrossRef]
- Virachit S, Mathews KJ, Cottam V et al. Levels of glial cell line-derived neurotrophic factor are decreased, but fibroblast growth factor 2 and cerebral dopamine neurotrophic factor are increased in the hippocampus in Parkinson's disease. Brain Pathol. 2019;29(6):813-25.
- Kirik D, Cederfjäll E, Halliday G et al. Gene therapy for Parkinson's disease: Disease modification by GDNF family of ligands. Neurobiol Dis. 2017;97(Pt B):179-88. [CrossRef]
- Huttunen HJ, Booms S, Sjogren M et al. Intraputamenal Cerebral Dopamine Neurotrophic Factor in Parkinson's Disease: A Randomized, Double-Blind, Multicenter Phase 1 Trial. Mov Disord. 2023;38(7):1209-22. [CrossRef]
- Francardo V, Geva M, Bez F et al. Pridopidine Induces Functional Neurorestoration Via the Sigma-1 Receptor in a Mouse Model of Parkinson's Disease. Neurotherapeutics. 2019;16(2):465-79. [CrossRef]
- Fischer DL, Sortwell CE. BDNF provides many routes toward STN DBS-mediated disease modification. Mov Disord. 2019;34(1):22-34. [CrossRef]
- Hauser RA, Li R, Pérez A et al. Longer Duration of MAO-B Inhibitor Exposure is Associated with Less Clinical Decline in Parkinson's Disease: An Analysis of NET-PD LS1. J Parkinsons Dis. 2017;7(1):117-27. [CrossRef]
- 1Guo B, Hu S, Zheng C, Wang H, Luo F, Li H, et al. Substantial protection against MPTP-associated Parkinson's neurotoxicity in vitro and in vivo by anti-cancer agent SU4312 via activation of MEF2D and inhibition of MAO-B. Neuropharmacology. 2017;126:12-24. [CrossRef]
- Mullapudi A, Gudala K, Boya CS et al. Risk of Parkinson's Disease in the Users of Antihypertensive Agents: An Evidence from the Meta-Analysis of Observational Studies. J Neurodegener Dis. 2016;2016:5780809. [CrossRef]
- Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann Intern Med. 2020;172(9):591-8.
- Dietiker C, Kim S, Zhang Y et al. Characterization of Vitamin B12 Supplementation and Correlation with Clinical Outcomes in a Large Longitudinal Study of Early Parkinson's Disease. J Mov Disord. 2019;12(2):91-6. [CrossRef]
- Athauda D, Foltynie T. Insulin resistance and Parkinson's disease: A new target for disease modification? Prog Neurobiol. 2016;145-146:98-120.
- Athauda D, Wyse R, Brundin P et al. Is Exenatide a Treatment for Parkinson's Disease? J Parkinsons Dis. 2017;7(3):451-8.
- Bu LL, Liu YQ, Shen Y et al. Neuroprotection of Exendin-4 by Enhanced Autophagy in a Parkinsonian Rat Model of α-Synucleinopathy. Neurotherapeutics. 2021;18(2):962-78. [CrossRef]
- McGarry A, Rosanbalm S, Leinonen M et al. Safety, tolerability, and efficacy of NLY01 in early untreated Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2024;23(1):37-45. [CrossRef]
- Schmitt M, Dehay B, Bezard E et al. Harnessing the trophic and modulatory potential of statins in a dopaminergic cell line. Synapse. 2016;70(3):71-86. [CrossRef]
- Zhao P, Yang X, Yang L et al. Neuroprotective effects of fingolimod in mouse models of Parkinson's disease. FASEB J. 2017;31(1):172-9. [CrossRef]
- Roy D, Ghosh R, Dubey S et al. Neurological and Neuropsychiatric Impacts of COVID-19 Pandemic. Can J Neurol Sci. 2021;48(1):9-24. [CrossRef]
- Sulzer D, Antonini A, Leta V et al. COVID-19 and possible links with Parkinson's disease and parkinsonism: from bench to bedside. NPJ Parkinsons Dis. 2020;6:18. [CrossRef]
- Brundin P, Nath A, Beckham JD. Is COVID-19 a Perfect Storm for Parkinson's Disease? Trends Neurosci. 2020;43(12):931-3.
- Jang H, Boltz D, Sturm-Ramirez K et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A. 2009;106(33):14063-8. [CrossRef]
- Hoffman LA, Vilensky JA. Encephalitis lethargica: 100 years after the epidemic. Brain. 2017;140(8):2246-51. [CrossRef]
- Victorino DB, Guimarães-Marques M, Nejm M, Scorza FA et al. COVID-19 and Parkinson's Disease: Are We Dealing with Short-term Impacts or Something Worse? J Parkinsons Dis. 2020;10(3):899-902.
- Sadasivan S, Zanin M, O'Brien K et al. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS One. 2015;10(4):e0124047.
- Jellinger, KA. Absence of alpha-synuclein pathology in postencephalitic parkinsonism. Acta Neuropathol. 2009;118(3):371-9. [CrossRef]
- Pavel A, Murray DK, Stoessl AJ. COVID-19 and selective vulnerability to Parkinson's disease. Lancet Neurol. 2020;19(9):719. [CrossRef]
- Desforges M, Le Coupanec A, Dubeau P et al. Human Coronaviruses and Other Respiratory Viruses: Underestimated Opportunistic Pathogens of the Central Nervous System? Viruses. 2019;12(1). [CrossRef]
- Chaudhry ZL, Klenja D, Janjua N et al. COVID-19 and Parkinson's Disease: Shared Inflammatory Pathways Under Oxidative Stress. Brain Sci. 2020;10(11). [CrossRef]
- Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3(10):812-8.
- Okuzumi A, Hatano T, Matsumoto G et al. Propagative α-synuclein seeds as serum biomarkers for synucleinopathies. Nat Med. 2023;29(6):1448-55. [CrossRef]
- Iranzo A, Mammana A, Munoz-Lopetegi A et al. Misfolded alpha-Synuclein Assessment in the Skin and CSF by RT-QuIC in Isolated REM Sleep Behavior Disorder. Neurology. 2023;100(18):e1944-e54. [CrossRef]
- Vijiaratnam N, Simuni T, Bandmann O et al. Progress towards therapies for disease modification in Parkinson's disease. Lancet Neurol. 2021;20(7):559-72. [CrossRef]
- Gonzalez-Robles C, Bartlett M, Burnell, C.S. et al. Embedding Patient Input in Outcome Measures for Long-Term Disease-Modifying Parkinson Disease Trials. Mov Disord. 2024;39(2):433-8. [CrossRef]
- Lenka A, Jankovic J. How should future clinical trials be designed in the search for disease-modifying therapies for Parkinson's disease? Expert Rev Neurother. 2023;23(2):107-22.
- Ko WKD, Bezard E. Experimental animal models of Parkinson's disease: A transition from assessing symptomatology to alpha-synuclein targeted disease modification. Exp Neurol. 2017;298(Pt B):172-9. [CrossRef]
- Gao L, Tang H, Nie K et al. Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson's disease diagnosis: a systematic review and meta-analysis. Int J Neurosci. 2015;125(9):645-54. [CrossRef]
- McGhee DJ, Ritchie CW, Zajicek JP et al. A review of clinical trial designs used to detect a disease-modifying effect of drug therapy in Alzheimer's disease and Parkinson's disease. BMC Neurol. 2016;16:92. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



