Submitted:
08 May 2025
Posted:
09 May 2025
You are already at the latest version
Abstract
Mitochondrial dysfunction is a hallmark in a variety of disease conditions, including ischemia/reperfusion (I/R) injury, stroke, and myocardial infarction (MI), metabolic syndrome, and aging. As with all biological organelles, the function of mitochondria is tightly linked with their structure. The inner mitochondrial membrane is a highly regulated, large surface area membrane that hosts the electron transport chain (ETC) machinery, generates membrane potential necessary for ATP generation, and forms the signature cristae folds of mitochondria. Mitochondrial inner membrane protein (Mitofilin/Mic60) is part of a large complex that constitutes the mitochondrial inner membrane organizing system (MICOS or MINOS), which plays a critical role in maintaining mitochondrial architecture and function. Recent evidence has highlighted the importance of cristae morphology in mitochondrial function and cell survival. Mic60/Mitofilin elimination during reperfusion was reported to determine the extent of myocardial infarct size after I/R. Here, we investigated the effects and mechanism of a novel Mic60/Mitofilin inhibitor, Miclxin, in cell viability and death using H9c2 cardiomyoblasts. Cultured rat H9c2 cardiomyoblasts were incubated in the presence of different concentrations (0, 5, 10, and 20 mM) of Miclxin. Cell viability was determined using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay, and cell death determined by flow cytometry using Propidium iodide dye. Mitochondrial membrane potential measurement was assessed using MitoTracker Red CMXROS assay kit, and Mitophagy in mitochondria was detected by using a Mitophagy Detection Kit. Mitochondrial morphology was assessed using electron microscopy, and proteins quantification was measured by Western blot analysis and immunofluorescence staining. After 24 hours of treatment, Miclxin concentration-dependently decreased cell viability and reduced the number of viable cells measured using MTT assay. This effect was associated with pronounced reduction of Mic60 protein levels measured by Western blot, and immunocytochemistry. We found that the mechanism of Miclxin-reduction of cell viability was related to inhibition of the elimination of mitochondria by mitophagy. Our finding suggests that treatment of cells with Miclxin decreases the levels of Mic60, which reduces cell viability that is associated with cell death by increasing mitochondria structural damage and dysfunction via impairment of mitophagy.
Keywords:
Mic60/Mitofilin protein
; Miclxin
; Mitophagy
; H9C2 rat cardiomyomyoblasts
; mitochondria
; cell viability
Introduction
Mitochondria are essential organelles whose function is crucial to homeostasis, energy generation, and cellular fate. Mitochondria are recognized as the “powerhouse” of eukaryotic cells, especially in those that require high-energy demand such as cardiomyocytes [1]. Therefore, mitochondrial dysfunction is a hallmark in a variety of disease conditions, including in ischemia/reperfusion (I/R) injury, stroke, and myocardial infarction, metabolic syndrome, neurodegeneration, and aging [2,3,4,5]. In the human heart, mitochondria occupy around 30% of the total volume of the cardiomyocytes [6], and these organelles supply, through oxidative phosphorylation (OXPHOS), approximately 6 kg of adenosine triphosphate (ATP) per day [7], which is required to sustain cardiomyocyte function in the heart [4,8]. In addition to their pivotal role in energy production, mitochondria are also play an important role in cell signaling, reactive oxygen species (ROS) generation, and Ca2+ buffering [9,10]. In addition, mitochondria are the central hub of cellular metabolism providing metabolites for biosynthesis and producing ROS [11]. As with all biological organelles, the function of mitochondria is tightly linked with their structure. The mitochondrial inner membrane (IMM) has a unique composition of protein[8]s and phospholipids, whose interdependence is crucial for mitochondrial function. It is highly enriched in proteins specific to this membrane, the majority of which are encoded by the nuclear genome and imported from the cytosol [12]. The IMM is a highly regulated, large surface area membrane that hosts the electron transport chain (ETC) machinery, generates membrane potential necessary for ATP generation, and forms the signature folds of mitochondria, known as cristae.
Recent evidence has highlighted the importance of cristae morphology in mitochondrial function and cell survival, with a particular focus on the mitochondrial inner membrane organizing system (MINOS or MICOS) [13] in which Mic60 is the “core” of MICOS [14]. MINOS or MICOS system is composed of many subunits, such as Mic60/Mitofilin, Mic10, coiled-coil-helix-coiled-coil-helix domain containing 3 (Mic19/CHCHD3), coiled-coil-helix-coiled-coil-helix domain containing 6 (Mic25/CHCHD6), Mic13/Qil1, coiled-coil-helix-coiled-coil-helix domain containing 10 (Mic14/CHCHD10), Mic23/ApoO, and apolipoprotein O like Mic27/ApoOL. Mic60 and Mic10 are considered the most critical components of MICOS. Mic60 is recognized as the “core” of MINOS and a critical transmembrane IMM protein whose downregulation, modification, or destruction results in mitochondrial dysfunction and cell death [15,16]. This suggests a critical role of Mic60 in the mechanism of cell death, a major hallmark of many mammalian diseases such as neurodegenerative disease, stroke, and myocardial infarction [17]. We recently reported that after heart I/R injury, depletion of Mic60 impairs cardiac functional recovery, increases myocardial infarct size, and facilitates mitochondrial structural damage and dysfunction [18]. After acute renal I/R injury, we have shown that the receptor-interacting protein kinase 3 (RIP3) translocates into mitochondria to promote Mic60 degradation, which initiates an increase in kidney inflammation and injury [19]. Our group further revealed that Parkin interacts with Mic60 in response to Parkinson's disease stressor neurotoxicity, which leads to the degradation of Mic60, resulting in mitochondrial structural damage and dysfunction that is responsible for neuronal death by apoptosis [20].
The role of Mic60 in cell death has been abundantly studied using cell lines transfected with siRNA and Mic60-overexpressed plasmids [16,20]. More recently, a drug, referred to as Miclxin, has been reported to inhibit Mic60 function [21]. In fact, using Miclxin-immobilized beads, Imoto’s group has identified Mic60 as a target protein of this compound. In that study, author concluded that targeting Mic60 with Miclxin represents a potential strategy with which tumor cells can be killed through induction of severe mitochondrial damage in a mutant β-catenin-dependent manner. However, further studies determining the role and mechanisms of Miclxin in cell viability and death in normal cells (non-tumor cells) were still needed. In addition, whether Miclxin induces only inhibition of Mic60, but not its elimination still need to be clarified, as Mic60 elimination is well known to induce cell death in several cell lines. In this study, we unveiled the effects of Miclxin in cell viability and death using cultured rat H9c2 cardiomyoblasts. We report that Miclxin induces H9c2 cardiomyoblasts death by increasing elimination of Mic60 protein level, which contributes to mitochondria structural damage and dysfunction through impaired mitochondria removal by mitophagy.
Materials and Methods
Institutional Approval
Protocols followed the Guide for the Care, and Use of Laboratory Animals (US Department of Health, NIH), and received UT Health Science Center at San Antonio Institutional Animal Care and Use Committee (IACUC) institutional approval. Animals were housed in the animal specific pathogen free facility at UTHSCSA main campus in cages with standard wood bedding and space for five mice or two rats. The animals had free access to food and drinking water and a 12-hour shift between light and darkness. The animals were selected randomly, and the data analysis was performed by a blinded investigator.
Cell Culture
Rat H9c2 cardiomyoblast line was purchased from the American Type Culture Collection (ATCC no. CRL-1446), and HEK 293 cell line was obtained from ATCC (no. CRL-3216). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen Life Technologies) supplemented with 10% fetal bovine serum (FBS; GIBCO-BRL, Grand Island, NY), 100 U/ml penicillin-streptomycin and grown in an atmosphere of 5% CO2-95% humidified air at 37°C. The culture medium was changed every second day. Cells were used between passage 4 and 7, at 70–80% confluence. The cells were selected randomly, and the data analysis was performed by a blinded investigator.
Treatment of Cells with Miclxin
Rat H9c2 cardiomyoblasts were incubated for 24 hours in the presence of Miclxin (5, 10 20 μM), or Vehicle (0.01% DMSO in media). 24 hours post treatment; cells were trypsinized and collected for further applications.
Cell Viability
Cell viability was determined using the tetrazolium dye 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay by following standard protocols. Briefly, cells were cultured in a 96 well plate and treated with either Vehicle or Miclxin as described above. At the end of the treatment, cells were placed in 50 µL of serum-free media supplemented with 50 µL of MTT solution into each well for 3 h incubation. After that, 150 µL of DMSO was added into each well before absorbance was read at 590 nm.
Western Blot Analysis
Equal amounts of the whole lysate protein were loaded in each well of 4–20% Tris-glycine gels (Bio-Rad) as recently described in [19]. After electrophoresis for 90 min at 125 V of constant voltage, the gel was blotted onto a nitrocellulose membrane by electrophoretic transfer at 70 V of constant voltage for 1–2 h. The membrane was washed, blocked with 5% blocking solution, and probed with various primary antibodies: anti-Mic60 (Abcam, catalog no. ab110329), and anti-GAPDH (CST, catalog no. 2118S) at 4°C overnight. The immunoreactive bands were visualized using secondary Li-Cor antibodies (LI-COR Biotechnologies, Lincoln, NE): Ire 800CW goat anti-rabbit antibody, (catalog no. 926-32211) and IRDye 680RD goat anti-mouse antibody, (catalog no. 926-68070).
Antibodies and Reagents
All materials were purchased from Sigma-Aldrich, unless otherwise stated. Studies utilized antibodies against the following targets (Table 1):
Mitochondrial Membrane Potential Measurement
Mitochondrial membrane potential measurement (ΔΨm) was assessed using MitoTracker Red CMXROS assay kit (ThermoFisher Scientific, catalog no. M7512) by fluorescent microscopy according to manufacturer’s protocol. MitoTracker red dye is a red-fluorescent dye that stains mitochondria in live cells and its accumulation is dependent upon membrane potential. H9c2 myoblasts were plated in six-well plates for MitoTracker Red assay or on coverslips for labeling and allowed to reach 70–80% confluence, after which cells were treated with vehicle, or Miclxin for 24 h. Live cells were harvested and incubated with MitoTracker Red (150 nM) for 1 h at 37°C. Carbonyl cyanide m-chlorophenylhydrazone (CCCP) was used as a control. Mitochondrial membrane potential was qualitatively assessed in normal and Miclxin-treated rat myoblasts using JC-1 dye (Cayman, 15003) [18,22]. After, 24h transfection, media were carefully removed from twenty-four well plates and cells washed twice with PBS, 0.5 ml of DMEM containing FBS. Cells were resuspended in DMEM containing FBS, stained by adding JC-1 (10 μg/ml) and cultured for 30 min at 37 °C with continuous gently shaking. At the end of the incubation, the media was removed, and cells were washed twice with normal PBS. To image the cells, 0.5 ml of DMEM containing FBS was added to each well and image taken with fluorescence microscopy.
Immunofluorescence Staining
Cells were cultured on coverslips and treated as described above. After treatment, cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.25% Triton X-100. After blocking in 3% BSA for 30 min, slides were incubated with first antibody diluted in 1% BSA for overnight. Coverslips were incubated with primary antibodies for Mic60 antibody (Proteintech Cat# 10179-1-AP, RRID:AB_2127193), followed by secondary antibodies Alexa Fluor 488 Goat anti-rabbit (Abcam Cat# ab150077, RRID:AB_2630356) and Goat anti-mouse (Abcam Cat# ab150113, RRID:AB_2576208). Images were taken on a Zeiss Axiovert 200M inverted motorized fluorescence microscope (Carl Zeiss Microscope, Jena, Germany).
Detection of Mitophagy
Mitophagy in mitochondria was detected by use of a Mitophagy Detection Kit according to the manufacturer (Dojindo Molecular Technology, Rockville, MD, USA) as described in [23]. This kit is composed of Mtphagy Dye, reagent for detection of mitophagy, and Lyso Dye, reagent for staining of lysosomes allowing accurate quantification of the damaged mitochondria fusing to the lysosomes. The signal was detected in cultured H9c2 cardiomyomyoblasts using confocal microscopy analysis at the following wavelengths: Mtphagy Dye: 561 nm (Ex) and 650 nm (Em); Lyso Dye: 488 nm (Ex) and 550 nm (Em).
Transmission Electron Microscopy
To analyze mitochondrial morphology in normal and I/R mice hearts, samples were fixed in a phosphate buffered solution of 4% formaldehyde with 1% glutaraldehyde, and stored at 4°C overnight as described in [23]. Tissue sections were washed with PBS, post-fixed in 2% (wt/vol) osmium tetroxide for 2 h at room temperature, and dehydrated in a graded alcohol series before being embedded in Eponate 12 medium. The blocks were cured at 60°C for 48 h, and 70 nm sections were cut using an ultramicrotome, mounted on Formvar-coated grids and double-stained with uranyl acetate and lead citrate. The resulting samples were analyzed and imaged using a JEOL 1230 transmission electron microscope. Mitochondria were classified as ‘damaged’ if they had more than 50% disorganized/destroyed cristae structure. Mitochondrial area was calculated using ImageJ software.
Flow Cytometry Analysis of Cell Death
Cell death was determined using Propidium iodide (BD PharMingen, catalog no. 556547) according to the manufacturer's instructions. Briefly, the harvested cells were washed twice in PBS and resuspended in 500 μl binding buffer. The cells were treated with 5 μl propidium iodide and immediately analyzed using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA). Propidium iodide (PI) is a membrane-impermeable fluorescent dye that stains DNA and cells used to label dead cells.
Statistical Analysis
Data presented in bar graphs are expressed as means, and error bars are the standard errors of the mean (± SD) for a minimum of three independent trials (n ≥ 3). Comparisons were conducted using the Student’s t-test and one-way ANOVA with post-hoc Dunnett’s or Tukey’s corrections for multiple comparisons, where appropriate, using Prism 8 (Graphpad Software). A difference of P < 0.05 was considered to be statistically significant.
Results
Miclxin Induces Mic60 Elimination
Miclxin has been reported to inhibit Mic60 [21]. To determine the effect of Miclxin on Mic60 regulation, we treated cultured rat H9c2 cardiomyoblasts with different concentrations of Miclxin: 0, 5, and 10 μM. Figure 1A shows that Western blot analysis performed on the whole cell lysate with anti-Mic60 antibody exhibit a concentration-dependent reduction of the level of Mic60 was compared to untreated cells. In fact, the level of Mic60 normalized to normal cells (100%) was reduced to 80.36±7% in cells treated with Miclxin (5 μM), and to 45.02%±7% in cells treated with Miclxin (10 μM), n=4/group. We further determined Miclxin effects on Mic60 expression in function of the duration of treatment. Cells were treated with Miclxin at 10 μM for 0, 24, 48, and 72 h. Western blot analysis show a time-dependent reduction of the level of Mic60 was compared to untreated cells. In fact, the level of Mic60 normalized to normal cells (100%) (Miclxin 0 μM) was reduced to 45.01±7% in cells treated with Miclxin (for 24h), to 42.54±13% in cells treated with Miclxin (48 h) and to 27.78±10% in cells treated with Miclxin (72 h), n=4/group. (Figure 1B). These results indicate that Miclxin inhibits Mic60 function by increasing its reduction (elimination) in cultured H9c2 cardiomyoblasts. We also measured the levels of Mic60 and VDAC1 by fluorescence microscopy using their respective antibodies after treatment with Miclxin (10 μM) in H9c2 cardiomyoblasts. We found that Miclxin (10 μM) decreased the levels of both Mic60 and VDAC1 in H9c2 cardiomyoblasts compared to untreated cells (Figure 1C and D). In fact, the levels of both Mic60 and VDAC1 in H9c2 cardiomyoblast death normalized to untreated cells 100% were decreased to 39%±8%, and 50%±3%, respectively in Miclxin 10 μM, n = 3/group, (Figure 1C and D).
Miclxin-Induced Mic60 Loss Decreases Cell Viability and Increases Cell Death
We previously reported that Mic60 knockdown enhances cell death by apoptosis. Here, we determined whether Mic60 knockdown reduces cell viability, rat H9c2 cardiomyoblasts were treated with different concentrations of Miclxin: 0, 5, and 10 μM. To measure the cell viability, we performed MTT assay, which determines the level of metabolic activity in eukaryotic cells and can be used as an estimate of live cells, on cells treated with Miclxin. We found that Miclxin concentration-dependently reduced H9c2 myoblast viability compared to untreated cells (Figure 2A). In fact, the H9c2 myoblast viability normalized to untreated cells 100% was reduced to 96.52%±7% in Miclxin 5 μM, 65.24±3% in Miclxin 10 μM, and 27.53±2% in Miclxin 20 μM, n = 4/group, (Figure 2B). We also determined Miclxin effects on cell viability in function of the duration of treatment. Cells were treated with Miclxin at 10 μM for 0, 24, 48, and 72 h. We found a time-dependent decrease in cell viability in Miclxin-treated cells compared to normal cells (Miclxin 0 μM). The H9c2 myoblast viability normalized to untreated cells 100% was reduced to 64.41%±3% in Miclxin (24 h), 43.25±6% in Miclxin (48 h), and 13.33%±7% in Miclxin (72 h), n = 6/group, (Figure 2C). We also measured the effect of Miclxin in H9c2 cardiomyoblast death by performing flow cytometry analysis using propidium iodide, which determines the level of cell death. We found that Miclxin (10 μM) increased H9c2 cardiomyoblast death compared to untreated cells (Figure 2D). In fact, the H9c2 myoblast death normalized to untreated cells 100% was increased to 149.42%±2% in Miclxin 10 μM, n = 3/group. Taken together, these results indicate that Miclxin treatment reduces rat H9c2 cardiomyoblast viability and increases cell death.
Miclxin Reduces Cell Viability by Promoting Mitochondrial Dysfunction
To assess the mechanism responsible for Miclxin-induced reduction of cell viability, we determined whether the mitochondrial membrane potential (MMP) is impacted by Miclxin treatment. Cells treated with different concentrations of Miclxin were harvested and the MMP was measured by fluorescence microscopy using MitoTracker Red (CMXRos) dye, a cell-permeant dye that accumulates in mitochondria with intact membrane potentials. We found a concentration-response reduction in the MMP in cells transfected with Miclxin. In fact, the number of MitoTracker red positive cells was decreased with the increase in Miclxin concentration (Figure 3).
We also assessed mitochondrial membrane potential (MMP) using the JC-1 dye. As shown in Figure 4, the ratios of green (Aggregate)/red (Monomer) fluorescence intensity was concentration-dependently increased in Miclxin treated cells compared with normal cells. The ratio of green/red fluorescence intensity normalized to 100 was increased to 784.15±84 (arbitrary unit) in cells treated with Miclxin (5 μM), and to 2184.48±156 (arbitrary unit) in Miclxin (10 μM)-treated cells, n=3/group. Since an increase in the green (Monomer)/red (Aggregate) ratio of JC-1 indicates reduction of MMP (Figure. 4), this result indicates that Miclxin-treated mitochondria are more uncoupled than untreated mitochondria. Together, these results indicate that treatment with Miclxin induces dissipation of MMP in H9c2 myoblasts, which induces mitochondrial dysfunction.
Miclxin Induces Mitochondrial Dysfunction by Altering Mitophagy
As we found that Miclxin treatment in H9c2 cells increases mitochondrial damage and dysfunction, we focused on investigating the mechanism underlying Miclxin effects. Thus, we studied whether the Miclxin actions on mitochondria via Mic60 elimination are due to the alteration of the removal of damaged mitochondria by mitophagy. To this end, H9c2 cells were treated with Miclxin at 0, 5, and 10 μM for 24 hours and mitophagy was evaluated by the number of engulfed mitochondria. Using electron microscopy, we observed that mitochondrial structural integrity was preserved with untreated cell as compared to Miclxin-treated cells. We also noticed that although the number of mitochondria was not very different between the groups, mito-phagosomes were more observed after Miclxin treatment versus untreated cells (Figure 5). In fact, as shown in Figure 5, the number of engulfed mitochondria was dramatically increased in cells treated with Miclxin as compared to non-treated cells, suggesting alteration of removal of the damaged mitochondria. To determine the mechanism by which Miclxin alters mitophagy in H9c2 cultured cells, we studied the impact of the PINK1 pathway, which is known to be involved in mitophagy via autophagy adaptors [24], in Miclxin action. Using Western blot analysis, we found that in the Miclxin-treated cells, PINK1 protein levels were decreased when compared to untreated cells (Figure 6). We also found that Miclxin treatment increased the Microtubule-associated protein 1A/1B-light chain 3 (LC3) LC3II/LC3I ratio and decreased p- protein p62/Sequestosome 1 (p62) and total ubiquitin levels compared with untreated cells (Figure 6). These results indicate that the mechanism by Miclxin alters mitophagy involves deactivation of the PINK1 pathway. To confirm this observation, the flux of mitophagy was evaluated by using mitodye and lysodye. Mitochondrial autophagosomes fuse with lysosomes during mitophagy, and the fluorescence intensity of Mtphagy Dye increases. Alteration of mitophagy is associated with increase in lysodye that measure mitochondria engulfed in autophagosomes. As shown in Figure 7, accumulation of autophagosomes was dramatically increased in cells treated with Miclxin (10 μM) similar to cells treated with CCCP as compared to untreated cells (control). Together, these data support the postulate that Miclxin action via Mic60 elimination involves the alteration of mitophagy resulting in accumulation of damaged mitochondria that is responsible for mitochondria dysfunction.
Discussion
In this paper, we report that Miclxin treatment decreases cultured H9c2 cardiomyoblast viability by enhancing Mic60 elimination. The mechanism of this deleterious effect is associated with an increase in alteration of the removal of damaged mitochondria by mitophagy.
Mitochondria are well known to play a crucial role in the cardiac cells. The roles of mitochondria in the cell include but are not limited to energy production, calcium homeostasis, and regulation of cell death [25,26]. There is a consensus that mitochondrial dysfunction is responsible for cell death, and is a hallmark of several disease including ischemia/reperfusion injury, stroke, neurodegenerative disease and metabolic syndrome. The structure of mitochondria and the protein composition of its inner membrane have now received increasing importance in determining the function of mitochondria in health and pathophysiology conditions [27,28]. Mic60 is now recognized as the core unit of the MINOS complex that is the critical organizer of mitochondrial cristae morphology and, thus, essential for normal mitochondrial function [29]. Our group has demonstrated that Mic60 knockdown in H9c2 cardiomyoblasts by siRNA induces mitochondrial structural damage leading to increased cell apoptosis via an Apoptosis-Inducing Factor (AIF)-Poly (ADP-ribose) polymerase (PARP) pathway [16]. More recently, we have reported that knockdown of Mic60 in mice increases mitochondrial structural damage and dysfunction, which results in critical failure of mitochondria to regulate Ca2+ homeostasis leading to increased mitochondrial sensitivity to Ca2+ overload that favors mPTP opening and, subsequently, causes cardiomyocyte death. Furthermore, after I/R, we found that loss of Mic60 during the early moment of reperfusion [18] induces dysregulation of Members of the mitochondrial carrier family (SLC25) As solute carriers that promote an increase in reactive oxygen species (RO)S generation that facilitates the release of Mitochondrial DNA (mtDNA) release into the cytosol, where it activates the signaling pathway that leads to augmented transcription of a nuclear transcription of pro-inflammatory cytokines that subsequently exacerbates I/R injury. However, studies investigating the role of Mic60 using pharmacological tools were needed.
Recently, a drug, referred to as Miclxin, was identified as an inhibitor of Mic60 that induces apoptosis through mitochondrial stress in β-Catenin mutant tumor cells [21]. In fact, using Miclxin-immobilized beads, Mic60 was identified as a target protein of Miclxin. Interestingly, Mic60 dysfunction caused by Miclxin induced a mitochondrial stress response in a mutant β-catenin-dependent manner. However, more studies supporting Miclxin as an inhibitor of Mic60 function were necessary in other cell lines. To this end, we treated different concentrations of Miclxin in cultured H9c2 cardiomyoblasts. We found that Miclxin induces a concentration-dependent reduction of Mic60 protein level (Figure 1). This observation, similar to those previously reported with Mic60 knockdown in H9c2 and HEK 293 cells transfected with Mic60siRNA [30], indicate that the mechanism of Miclxin includes dysfunction of Mic60 via its elimination. Concomitantly, Miclxin reduced cell viability time and concentration-dependently (Figure 2B-C) and increases cell death (Figure 2D) suggesting that Miclxin induces cytotoxic effects by reducing Mic60. We found that the mechanism of Miclxin-induced reduction of cell viability and increase in the rate of cell death via Mic60 dysfunction to be associated with a reduction in mitochondrial dysfunction related alteration of the removal of damaged mitochondria by mitophagy. In fact, Miclxin treatment concentration-dependently reduced mitochondrial membrane potential measure with MitoTracker red (Figure 3) and JC-1 dye (Figure 4). This finding supports the assumption that dysregulation of Mic60 by Miclxin contributes mitochondrial depolarization, as Miclxin effect is similar to CCCP, a mitochondrial uncoupler [21]. Supporting this observation Miclxin treatment reduces VDAC1 protein levels much similar to Mic60 (Figure 1)
The decrease in mitochondrial membrane potential triggers removal of dysfunctional mitochondria by mitophagy via autophagy adaptors [31]. We thus defined whether Miclxin induces mitochondrial depolarization by causing alteration of mitophagy that ultimately exacerbates mitochondrial stress and accumulation of damaged and dysfunctioned mitochondria. In fact, autophagy is a process by which cells degrade proteins and organelles via a lysosome-dependent pathway. The overall mechanism that eliminate mitochondria via autophagy is called mitochondrial autophagy (mitophagy). Mitophagy is known as a specific process of autophagic elimination of its own mitochondria, therefore it is a crucial regulatory mechanism for cells to maintain homeostasis and energy production. As stated by Wang et al [32], removal of mitochondria by mitophagy exerts a dual function; it favors cell survival or death depending on the cellular context and microenvironment. The degradation of mitochondria via the mitophagy mechanism is critical for cell survival under physiological conditions [33]. However, alteration or hyperactivation of mitophagy impairs mitochondrial function. E3 ubiquitin ligase, Parkin, plays a key role in mitophagy via autophagy adaptors. [34] Within mitochondria, Parkin induces protein ubiquitination, which triggers mitophagy [35]. In mitophagy, p62 protein binds to ubiquitinated proteins via its ubiquitin-associated domain [36] and to LC3 via its LC3-interacting region [37]. LC3I and LC3II are known to interact with p62 to tether mitochondria to the autophagosome [38]. We found that Miclxin decreased Pink1, phosphorylation of P62, and mitochondrial protein ubiquitination. This effect is associated with an increase in the ration LC3II/LC3I (Figure 6). Interestingly, accumulation of autophagosomes measured with lysodye was drastically increased in Miclxin-treated cells compared to untreated cells (Figure 7). These results suggest the involvement of the alteration of mitophagy in the mechanism of Miclxin actions. As Miclxin initiates mitochondrial depolarization, which is known promoting sequestration of mitochondria into autophagosomes [39], our results indicate that Miclxin alters removal of damaged mitochondria by inhibiting the PINK1-pathway dependent mitophagy.
Conclusions
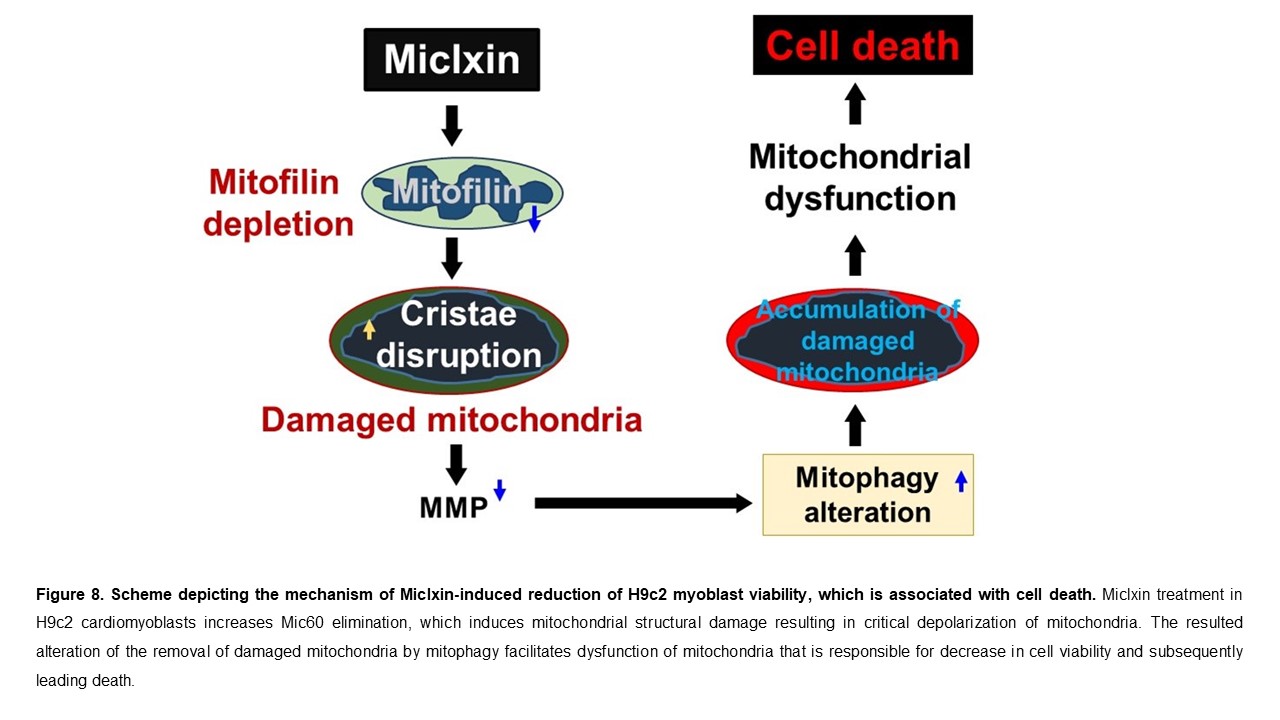
We report that the Miclxin treatment in H9c2 cardiomyoblasts increases Mic60 elimination that increases mitochondrial structural damage, which results in critical depolarization of mitochondria. We propose for the first time that Miclxin-induced alteration of removal of damaged mitochondria by mitophagy contributes to dysfunction of mitochondria that is responsible for reduction of cell viability that is associated with increased cell death as represented in Figure 8.
Limitations
In this paper, we only used rat H9c2 cardiomyoblasts, it would be necessary to confirm the effects of this drug in another cell line. Also, studies investigating the effects of Miclxin in vivo would support the possible use of this drug in clinic. The premise is built upon previously published studies and compelling preliminary data. Outcomes were assessed by a blinded investigator. In this study, we report that Miclxin induces Mic60 reduction, however, it is possible that effect is the consequence of its initial inhibition. Further studies are needed to examine the effect of Miclxin in acute conditions.
Author Contributions
M.S.K., and J.C.B. conceived and designed the experiments. M.S.K., S.N., and M.O. performed the experiments. M.S.K. and J.C.B. analyzed the data. M.S.K. and J.C.B. drafted the manuscript. S.N., M.O., K.S., S.B., M.B., and J.C.B. scrutinized and discussed the results. KS, S.B., M.B., and J.C.B. and J.C.B. revised the paper. J.C.B. supervised the project. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health [grant HL138093 (JCB), NIH/NHLBI T32 HL007446-41], and NINDS R25 grant [NS115552].
Institutional Review Board Statement
All protocols followed the Guide for the Care and Use of Laboratory Animals (US Department of Health, NIH) and received UT Health Science Center at San Antonio Institutional Animal Care and Use Committee (IACUC) institutional approval. Protocol number 20140022AR, approval date: 10-23-2023.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author/s.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
Mic60, Inner mitochondrial membrane protein; ROS, reactive oxygen species; MICOS, mitochondrial contact site and cristae organizing system; MMP, mitochondrial membrane potential; CCCP, Carbonyl cyanide m-chlorophenylhydrazone; LC3, Microtubule-associated protein 1A/1B-light chain 3; p62, The protein p62/Sequestosome 1; AIF, Apoptosis-Inducing Factor; PARP, Poly (ADP-ribose) polymerase; mtDNA, Mitochondrial DNA; SLC25A, Members of the mitochondrial carrier family (SLC25); ETC, electron transport chain; APOOL, cardiolipin-binding component of the MINOS protein complex; CHCHD coiled-coil-helix-coiled-coil-helix domain, RIP3, receptor-interacting protein kinase 3; MTT, 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase
References
- Nan J, Zhu W, Rahman MS, Liu M, Li D, Su S, Zhang N, Hu X, Yu H, Gupta MP and Wang J. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta Mol Cell Res 2017; 1864: 1260-1273.
- Saleh MC, Wheeler MB and Chan CB. Uncoupling protein-2: evidence for its function as a metabolic regulator. Diabetologia 2002; 45: 174-187.
- Christophe M and Nicolas S. Mitochondria: a target for neuroprotective interventions in cerebral ischemia-reperfusion. Curr. Pharm. Des 2006; 12: 739-757.
- Li A, Gao M, Jiang W, Qin Y and Gong G. Mitochondrial Dynamics in Adult Cardiomyocytes and Heart Diseases. Front Cell Dev Biol 2020; 8: 584800.
- Bernardi P, Colonna R, Costantini P, Eriksson O, Fontaine E, Ichas F, Massari S, Nicolli A, Petronilli V and Scorrano L. The mitochondrial permeability transition. Biofactors 1998; 8: 273-281.
- Ramaccini D, Montoya-Uribe V, Aan FJ, Modesti L, Potes Y, Wieckowski MR, Krga I, Glibetic M, Pinton P, Giorgi C and Matter ML. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front Cell Dev Biol 2020; 8: 624216.
- Bhullar SK and Dhalla, NS. Status of Mitochondrial Oxidative Phosphorylation during the Development of Heart Failure. Antioxidants (Basel) 2023; 12:.
- Cao YP and Zheng, M. Mitochondrial dynamics and inter-mitochondrial communication in the heart. Arch Biochem Biophys 2019; 663: 214-219.
- Bopassa JC, Ferrera R, Gateau-Roesch O, Couture-Lepetit E and Ovize M. PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc. Res 2006; 69: 178-185.
- Gomez L, Thibault H, Gharib A, Dumont JM, Vuagniaux G, Scalfaro P, Derumeaux G and Ovize M. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am. J. Physiol Heart Circ. Physiol 2007; 293: H1654-H1661.
- Spinelli JB and Haigis, MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 2018; 20: 745-754.
- Akbar M, Essa MM, Daradkeh G, Abdelmegeed MA, Choi Y, Mahmood L and Song BJ. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res 2016; 1637: 34-55.
- Kozjak-Pavlovic, V. The MICOS complex of human mitochondria. Cell Tissue Res 2017; 367: 83-93.
- John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ and Zha J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell 2005; 16: 1543-1554.
- Li H, Ruan Y, Zhang K, Jian F, Hu C, Miao L, Gong L, Sun L, Zhang X, Chen S, Chen H, Liu D and Song Z. Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ 2016; 23: 380-392.
- Madungwe NB, Feng Y, Lie M, Tombo N, Liu L, Kaya F and Bopassa JC. Mitochondrial inner membrane protein (mitofilin) knockdown induces cell death by apoptosis via an AIF-PARP-dependent mechanism and cell cycle arrest. Am J Physiol Cell Physiol 2018; 315: C28-C43.
- van der Laan M, Horvath SE and Pfanner N. Mitochondrial contact site and cristae organizing system. Curr Opin Cell Biol 2016; 41: 33-42.
- Tombo N, Imam Aliagan AD, Feng Y, Singh H and Bopassa JC. Cardiac ischemia/reperfusion stress reduces inner mitochondrial membrane protein (mitofilin) levels during early reperfusion. Free Radic Biol Med 2020; 158: 181-194.
- Feng Y, Imam Aliagan A, Tombo N, Draeger D and Bopassa JC. RIP3 Translocation into Mitochondria Promotes Mitofilin Degradation to Increase Inflammation and Kidney Injury after Renal Ischemia-Reperfusion. Cells 2022; 11:.
- Imam Aliagan AD, Ahwazi MD, Tombo N, Feng Y and Bopassa JC. Parkin interacts with Mitofilin to increase dopaminergic neuron death in response to Parkinson's disease-related stressors. Am J Transl Res 2020; 12: 7542-7564.
- Ikeda H, Muroi M, Kondoh Y, Ishikawa S, Kakeya H, Osada H and Imoto M. Miclxin, a Novel MIC60 Inhibitor, Induces Apoptosis via Mitochondrial Stress in beta-Catenin Mutant Tumor Cells. ACS Chem Biol 2020; 15: 2195-2204.
- Elefantova K, Lakatos B, Kubickova J, Sulova Z and Breier A. Detection of the Mitochondrial Membrane Potential by the Cationic Dye JC-1 in L1210 Cells with Massive Overexpression of the Plasma Membrane ABCB1 Drug Transporter. Int J Mol Sci 2018; 19:.
- Feng Y, Madungwe NB, da Cruz Junho CV and Bopassa JC. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br J Pharmacol 2017; 174: 4329-4344.
- Kubli DA and Gustafsson, AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 2012; 111: 1208-1221.
- El-Hattab AW, Suleiman J, Almannai M and Scaglia F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases. Mol Genet Metab 2018; 125: 315-321.
- El-Hattab AW and Scaglia, F. Mitochondrial cytopathies. Cell Calcium 2016; 60: 199-206.
- Van Laar VS, Otero PA, Hastings TG and Berman SB. Potential Role of Mic60/Mitofilin in Parkinson's Disease. Front Neurosci 2018; 12: 898.
- Feng Y, Madungwe NB and Bopassa JC. Mitochondrial inner membrane protein, Mic60/mitofilin in mammalian organ protection. J Cell Physiol 2019; 234: 3383-3393.
- von der MK, Muller JM, Bohnert M, Oeljeklaus S, Kwiatkowska P, Becker T, Loniewska-Lwowska A, Wiese S, Rao S, Milenkovic D, Hutu DP, Zerbes RM, Schulze-Specking A, Meyer HE, Martinou JC, Rospert S, Rehling P, Meisinger C, Veenhuis M, Warscheid B, van dK, I, Pfanner N, Chacinska A and van der LM. Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev. Cell 2011; 21: 694-707.
- Madungwe NB, Feng Y, Lie M, Tombo N, Liu L, Kaya F and Bopassa JC. Mitochondrial Inner Membrane Protein (Mitofilin) Knockdown Induces Cell Death by Apoptosis Via an AIF-PARP-Dependent Mechanism and Cell Cycle Arrest. Am J Physiol Cell Physiol 2018;
- Takagi H, Matsui Y and Sadoshima J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid Redox Signal 2007; 9: 1373-1381.
- Wang DD, Jin MF, Zhao DJ and Ni H. Reduction of Mitophagy-Related Oxidative Stress and Preservation of Mitochondria Function Using Melatonin Therapy in an HT22 Hippocampal Neuronal Cell Model of Glutamate-Induced Excitotoxicity. Front Endocrinol (Lausanne) 2019; 10: 550.
- Cesarini E, Cerioni L, Canonico B, Di Sario G, Guidarelli A, Lattanzi D, Savelli D, Guescini M, Nasoni MG, Bigini N, Cuppini R, Stocchi V, Ambrogini P, Papa S and Luchetti F. Melatonin protects hippocampal HT22 cells from the effects of serum deprivation specifically targeting mitochondria. PLoS One 2018; 13: e0203001.
- Suen DF, Narendra DP, Tanaka A, Manfredi G and Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A 2010; 107: 11835-11840.
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ and Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12: 119-131.
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR and Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 2004; 24: 8055-8068.
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G and Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282: 24131-24145.
- Ashrafi G and Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013; 20: 31-42.
- Elmore SP, Qian T, Grissom SF and Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 2001; 15: 2286-2287.

Table 1.
List of antibodies used in this study.
| Antibody Target | Supplier | Catalog # | Concentration |
|---|---|---|---|
| Mitofilin/Mic60 | Proteintech Group | 10179-1-AP | 1 µg/mL |
| LC3I/LC3II | 1 µg/mL | ||
| p-p62 | 1 µg/mL | ||
| Pink1 | 1 µg/mL | ||
| Ubiquitin | 1 µg/mL | ||
| VDAC1 and VDAC3 | 1 µg/mL | ||
| GAPDH | 1  g/ml g/ml |
||
| IRDye 800CW Goat anti-Rabbit | LI-COR | 926-32211 | 0.1 µg/mL |
| IRDye 680RD Goat anti-Mouse | LI-COR | 926-68070 | 0.1 µg/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



