Submitted:
07 May 2025
Posted:
08 May 2025
You are already at the latest version
Abstract
Chronic Kidney Disease (CKD) is a group of clinical symptoms characterized by progressive loss of kidney function, defined as Glomerular Filtration Rate (GFR) below 60 mL/min/1.73 m². For more than three months. The harm of CKD is extensive and has become a global public health problem. The harm of CKD is not only limited to the kidney itself, but also causes a variety of complications, among which Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) is one of the important complications of CKD. It is characterized by abnormal mineral metabolism, bone disease, and vascular calcification, which seriously affects the long-term prognosis of patients. The pathophysiological mechanism of CKD-MBD is complex and involves a variety of factors, including dysregulation of phosphate and calcium metabolism, elevated Parathyroid Hormone (PTH) levels, and abnormal vitamin D metabolism. These disorders interact to cause abnormal bone metabolism and calcification of blood vessels, increasing the risk of cardiovascular disease. Although significant advances have been made in the understanding and treatment strategies for CKD-MBD, deficiencies remain. Current treatments focus on regulating mineral metabolism and inhibiting hyperparathyroidism, but these treatments have limitations. In addition, early diagnosis and prevention strategies for CKD-MBD are not yet perfect, and further research is needed to improve the accuracy of diagnosis and the effectiveness of treatment. This review aims to systematically review the latest advances in pathophysiology, diagnosis, clinical management and treatment of CKD-MBD. This paper focuses in particular on recent scientific discoveries and assesses their potential impact on improving patient management and clinical outcomes. Future research should focus on further exploring the molecular mechanisms of CKD-MBD, validating new biomarkers, developing personalized medicine strategies. These studies will provide new insights into the clinical treatment of CKD-MBD, help improve long-term patient outcomes, and lay the foundation for personalized medicine in the future.
Keywords:
chronic kidney disease
; chronic kidney disease-mineral and bone disorder
; mineral metabolism disorders
; skeletal abnormalities
; vascular calcification
Introduction
Chronic Kidney Disease (CKD) is a global public health problem with increasing morbidity and mortality. As Chronic Kidney Disease progresses to advanced stages, patients are often accompanied by Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD), which not only affects patients' quality of life, but also significantly increases the risk of cardiovascular events and mortality[1]. Chronic Kidney Disease-Mineral and Bone metabolism Disorder is one of the common complications in patients with chronic kidney disease[2]. Chronic Kidney Disease - Mineral and Bone Disorders can be caused by one or more of the following abnormalities: (1) abnormal metabolism of calcium, phosphorus, Parathyroid Hormone (PTH), or vitamin D; (2) abnormal bone metabolism, mineralization, linear volume growth, or strength; (3) calcification of blood vessels or other soft tissues (Figure 1)[3,4]. Chronic Kidney Disease - Mineral and Bone Disorders have complex pathophysiological mechanisms involving disturbances in calcium, phosphorus, and vitamin D metabolism, as well as abnormal regulation of parathyroid hormones. Studies in recent years have shown that these metabolic disorders are associated with abnormal bone turnover, vascular calcification, or soft tissue calcification, further exacerbating the condition in CKD patients[5]. Nr Hill et al. found that the global prevalence of chronic kidney disease was between 11% and 13% through random effects model evaluation in eight databases, and most patients were stage 3, of which a significant proportion of patients may develop CKD-MBD[6]. A study conducted in 2024 collected information on the prevalence of chronic kidney disease in 161 countries. The analysis revealed that the global median prevalence of chronic kidney disease was 9.5% (interquartile range 5.9-11.7). The highest prevalence rates were found in Eastern and Central Europe (12.8%, 11.9-14.1)[7]. In addition, the prevalence rate among adults in mainland China was 8.2%[8].
Clinical trials have validated the efficacy of phosphorus-binding agents and vitamin D analogs in regulating mineral metabolism, while calcium-sensitive receptor agonists and innovative PTH analogs have brought new options for CKD-MBD treatment. Nonetheless, challenges such as the need for individualized treatment, drug side effects, and patient compliance remain. In addition, early diagnosis and prevention strategies for CKD-MBD have yet to be perfected, and in-depth studies are urgently needed to unravel the mechanisms and develop intervention strategies. The aim of this review is to synthesize and analyze the new findings on the pathophysiological mechanisms of CKD-MBD, the improvement of diagnostic methods, the update of therapeutic strategies, and the development of clinical practice guidelines, so as to better understand the complexity of CKD-MBD and to guide the future direction of research and clinical practice.
Materials and Methods
In order to comprehensively collect relevant academic information, we systematically searched a number of well-known online academic databases, including but not limited to PubMed, Web of Science, CNKI, Wanfang Database and VIP database. We used a range of keywords including "chronic kidney disease", "osteoporosis", "chronic kidney disease - mineral and bone diseases", "vascular calcification", "pathophysiology", "management" and "treatment". Through their combined use, the aim is to comprehensively capture the research literature related to the pathological mechanisms of chronic kidney disease - mineral and bone diseases and their complications. In particular, we looked at literature published between 2020 and 2024 to ensure that the most recent research findings were covered. To supplement important historical background and basic research data, we have also included some landmark research before 2020. This careful search strategy ensures the depth and breadth of our review, providing readers with a comprehensive perspective on the pathological mechanisms and complications of chronic kidney diseases-mineral and bone diseases.
Pathophysiology
1. Abnormalities in the Metabolism of Calcium, Phosphorus, Vitamin D, Parathyroid Hormone, and FGF23
The effects of chronic kidney disease on bone and mineral metabolism are multidimensional, mainly due to the impaired central role of the kidneys in maintaining the balance of minerals such as calcium and phosphorus (Figure 2)[9,10].
- 1.1.
- Disorders of Calcium and Phosphorus Balance
As chronic kidney disease progresses, a decrease in Glomerular Filtration Rate (GFR) leads to a decrease in the ability of the kidneys to excrete phosphate, resulting in the development of overt hyperphosphatemia. Simultaneously active vitamin D and Klotho protein levels decline. The body compensatorily increases fibroblast growth factor 23 (FGF23) and parathyroid hormone levels in order to maintain stable blood calcium and phosphorus levels. However, this compensatory mechanism often leads to pathophysiologic changes such as abnormal bone metabolism and ectopic calcification[11]. Hyperphosphatemia exacerbates hyperparathyroidism by inducing hypocalcemia, decreasing 1, 25-dihydroxyvitamin D3 production, and increasing PTH gene expression. In addition, elevated plasma phosphate concentrations significantly inhibit parathyroid calcium sensitive receptor (CaSR) activity, which directly affects PTH synthesis and further exacerbates the development of hyperparathyroidism[9]. High blood phosphorus levels bind to calcium in the blood, causing blood calcium levels to drop. Hypocalcemia stimulates parathyroid hormone secretion to increase blood calcium levels. Chronic high PTH levels can lead to secondary hyperparathyroidism (SHTP), which in turn causes bone calcium mobilization and increased bone resorption[12]. The colloidal mineral-protein complex of crystalline calcium phosphate and serum protein fetuin A is called secondary calciprotein particles (CPP2), and CPP2-induced tubular injury and interstitial inflammation or fibrosis reduce the number of functional renal units. Further increases in phosphate excretion per renal unit are required to maintain phosphate homeostasis unless phosphate intake is reduced[13]. In addition, deficiency of active vitamin D further affects calcium absorption and utilization, exacerbating disturbances in calcium and phosphorus metabolism[14].
- 1.2.
- Disorders of Vitamin D Metabolism
Vitamin D is essential in bone formation and remodeling, promoting intestinal calcium absorption, renal tubular calcium reabsorption, osteoblast proliferation and differentiation, and significantly affecting bone matrix mineralization. Its active form, 1, 25-dihydroxyvitamin D3, regulates calcium and phosphate homeostasis in the small intestine, kidney, and bone[15,16]. Vitamin D synthesis and metabolism are finely regulated processes in which the kidney plays a central role. Vitamin D is first converted to 25-hydroxyvitamin D by the action of 25-hydroxylase in the liver, and then this inactive form is further converted to the biologically active 1, 25-dihydroxyvitamin D3 in the mitochondria of renal proximal tubular epithelial cells by the action of 1α-hydroxylase (CYP27B1)[16,17]. 1, 25-dihydroxyvitamin D3 indirectly promotes bone mineralization by facilitating intestinal absorption of calcium and phosphate as well as renal tubular reabsorption of calcium, while inducing the expression of osteocalcin, activating the Receptor Activator of Nuclear Factor-κ B Ligand(RANKL), facilitating bone resorption, and releasing calcium salts from old bone into the circulation[18]. Meyer et al. showed that CYP27B1 and CYP24A1 (24-hydroxylase) in the kidney work together to regulate blood levels of 1, 25-dihydroxyvitamin D3, which is essential for calcium and phosphate homeostasis[19]. However, in CKD, 1α-hydroxylase activity is inhibited and 1, 25-dihydroxyvitamin D3 production is blocked, which affects calcium and phosphate homeostasis, and vitamin D deficiency has been associated with loss of vitamin D-binding proteins and 1, 25-dihydroxyvitamin D3 associated with proteinuria and uremia[20]. 1, 25-dihydroxyvitamin D3 and its analogs effectively inhibit PTH synthesis and secretion through transcriptional and post-transcriptional mechanisms. This biological response is mediated by the binding of 1, 25-dihydroxyvitamin D3 to intracellular receptors belonging to the steroid receptor superfamily[21]. 1, 25-dihydroxyvitamin D3, PTH and FGF23 together constitute an endocrine regulatory system that maintains calcium and phosphorus homeostasis. However, in CKD, the function of this system is impaired, leading to disturbances in mineral metabolism[22]. In addition, vitamin D levels are negatively associated with cardiovascular risk in men and women. 1, 25-dihydroxyvitamin D3 favors cardiovascular disease through a mechanism of down-regulation of renin expression, renin-angiotensin-aldosterone system activity, and its interaction with vitamin D receptors[23].
- 1.3.
- Disorders of Parathyroid Hormone Metabolism
As kidney function decreases, blood phosphorus rises and blood calcium decreases, stimulating the parathyroid glands to secrete more PTH. Excess parathyroid hormone causes serum levels of calcium and phosphorus to rise, a change that triggers the conversion of preosteoclasts and mesenchymal cells to osteoclasts, which accelerates the process of dissolution of bone matrix and bone salts. In addition, the excessive action of PTH leads to a decrease in the density of the outer layer of bone (cortical bone) and the formation of a large number of pores, which increases the porosity of cortical bone. This structural change weakens the strength and stability of the bone, putting it at increased risk for osteoporosis, which in turn may lead to fractures[24,25]. Under normal conditions, osteoblasts can secrete osteoid to fill the traps formed by osteoclast bone resorption, followed by mineralization of osteoid (deposition of inorganic salts such as calcium and phosphorus) into new bone. In chronic kidney disease, osteoblasts are inhibited due to disturbances in calcium and phosphorus metabolism and elevated levels of PTH, resulting in decreased bone formation[24,26,27]. PTH regulates FGF23 secretion, reduces blood phosphorus, promotes bone calcium mobilization, elevates blood calcium, and has a bidirectional regulatory effect on bone metabolism[12]. PTH secretion is negatively regulated by serum calcium levels through the parathyroid calcium sensitive receptor (CaSR)[28]. In the kidney, PTH increases distal tubular calcium reabsorption and down-regulates the proximal tubular sodium-phosphate cotransporter proteins, NPT2a and NPT2c, thereby increasing urinary phosphate excretion. Under the influence of parathyroid hormone, by increasing renal tubular reabsorption of calcium and tubular excretion of phosphorus, which would otherwise enter the circulation from the skeleton and intestine[29]. In addition, PTH enhances the synthesis of 1, 25-dihydroxyvitamin D3 through activation of the 1-alpha-hydroxylase gene, which further promotes intestinal absorption of calcium and phosphorus, which is essential for the correction of hypocalcemia and maintenance of mineral metabolic homeostasis[9]. PTH also inhibits the expression of 24-hydroxylase and slows down the inactivation of 1, 25-dihydroxyvitamin D3, ensuring its effective concentration in the body, which together promote calcium and phosphorus metabolic homeostasis[29]. These regulatory mechanisms are essential for the treatment and mineral homeostasis of patients with CKD-MBD.
- 1.4.
- Disorders of FGF23-Klotho Metabolism
FGF23 possesses the ability to stimulate the proliferation and differentiation of a wide range of cells that play a crucial regulatory role in the process of osteogenesis, which includes bone progenitor cells and osteoblasts. By promoting the proliferation and differentiation of these cells, FGF23 regulates the process of osteogenesis and plays a decisive role in bone development and maintenance[30]. Elevated serum FGF23 levels in patients with chronic kidney disease-mineral and bone disorders may be a compensatory response to increased serum phosphorus load[31]. Increased levels of FGF23 can be seen in the circulation of patients with early chronic kidney disease, and early rises in FGF23 are protective and essential for maintaining phosphate homeostasis. FGF23 increases renal phosphorus excretion by down-regulating sodium-dependent phosphate co-transporter proteins in the proximal renal tubule; inhibition of proximal tubular 1-alpha-hydroxylase expression leads to decreased 1, 25-dihydroxyvitamin D3 synthesis, resulting in a relatively stable calcium-phosphorus metabolism in patients with early chronic kidney disease, and influences the process of bone remodeling by signaling through the dependence or non-independence of the transmembrane protein Klotho. With a decrease in estimated glomerular filtration rate (eGFR), FGF23-induced phosphorus excretion is diminished, leading to hyperphosphatemia[32,33,34]. Shimada et al. demonstrated this in transgenic mice expressing human FGF23[35]. FGF23 and soluble α-Klotho may affect the skeleton by inhibiting osteoblast mineralization and osteoblast marker gene expression through FGFR1 signaling and increased FGFR1 and FGFR3 expression in MC3T3-E cells[31]. In another study, increased Lipocalin 2 (LCN2) in the kidney of patients with chronic kidney disease was found to target stimulation of cyclic AMP-mediated signaling in osteocytes to stimulate FGF23 transcription[36]. Klotho is a membrane protein, classified as αKlotho and βKlotho, and is a major part of the endocrine fibroblast growth factor receptor complex. Target organ signaling by FGF23 is mediated by its binding to a heterodimeric complex of FGF receptors and α-Klotho co-receptors, which are significantly expressed in kidney, parathyroid, bone and other tissues [29]. The possibility that the Klotho-FGFR complex regulates its own production through a negative feedback loop and an autocrine feedback loop controlling FGF23 synthesis and secretion is high. It has been shown that bone mineralization defects in Klotho-deficient mice are caused by 1, 25-dihydroxyvitamin D3-driven up-regulation of bone pyrophosphate and bone bridging proteins[37,38,39]. FGF23 reduces 1, 25-dihydroxyvitamin D3 concentrations by inhibiting CYP27B1 and stimulating CYP24A1. This is in contrast to the action of parathyroid hormone[29]. FGF23 inhibits parathyroid hormone secretion by activating the intracellular mitogen-activated protein kinase (MAPK) signaling pathway through binding to the Klotho-FGFRl complex on parathyroid cell membranes[40]. Therefore, FGF23 is considered a promising therapeutic target for improving the prognosis of CKD patients with associated complications[41].
In conclusion, the reduced renal function in patients with chronic kidney disease leads to the inability of patients to properly excrete waste products and excess electrolytes such as phosphorus and calcium, which disrupts the original mineral balance[42]. The changes in these minerals and their effects are summarized in Table 1. As a result, patients with chronic kidney disease are often accompanied by osteoporosis, which is manifested by decreased bone density, weakened bone strength, and increased fracture risk. It has been shown that high fracture risk is caused by a combination of CKD-induced changes in bone and mineral metabolism[43].
2. Skeletal Changes
In patients with chronic kidney disease (CKD), bone renewal capacity suffers significantly, leading to more fragile bones and increased fracture risk. Patients with CKD G3a-G5D have a higher fracture rate compared to the general population[44]. Patients with CKD often have abnormal metabolic conditions such as elevated blood phosphorus, decreased blood calcium, and vitamin D deficiency, which interfere with the process of bone mineralization (Figure 3)[45]. Phosphate is the main component of skeletal hydroxyapatite, which provides strength and stiffness to the bone. It plays a role as a signaling molecule in bone metabolism, influencing osteoblast activation, differentiation and bone resorption activities of osteoclasts. Hyperphosphatemia can lead to soft tissue calcification but at the same time inhibits normal mineralization of bone[46]. Phosphorus and calcium easily react in biological fluids to produce calcium phosphate and calciprotein particles (CPPs), which destroy tubular cells by binding to TLR4 expressed on the tubules, and sustained tubular injury induces interstitial fibrosis and reduces the number of renal units, with progression of chronic kidney disease ensuing[47]. Vitamin D is also essential for maintaining bone health, and its inadequate supply can lead to renal osteodystrophy, osteochondrosis, and hyperparathyroidism, whereas an oversupply may cause ectopic calcification and increase cardiovascular risk. In patients with chronic kidney disease, monitoring vitamin D status and adjusting treatment regimens are key to managing mineral and bone abnormalities[20]. Bone conversion rate affects the cancellous and dense portions of the skeleton and is regulated by several factors, among which parathyroid hormone plays a key role in bone remodeling. In patients with CKD, the loss of cortical bone becomes particularly prominent in the long term due to a higher proportion of cortical bone and a sustained major effect of PTH on cortical bone, leading to thinning, cancellous and porous bones, which considerably increases the fragility of the skeleton, especially in long bones, where the integrity of cortical bone is crucial for skeletal health[45]. The inability to effectively regulate calcium and phosphorus levels due to decreased renal function leads to increased PTH secretion. Elevated parathyroid hormone promotes osteoblasts and osteocytes mediating indirect effects on osteoclast precursor cells and osteoclasts, thereby promoting their proliferation and differentiation and inducing bone resorption, while inhibiting osteoblasts' function, leading to a further decrease in bone mineral density and bone mass[48]. Parathyroid hormone regulates the signaling pathways of Bone Morphogenetic Protein (BMP), Transforming growth factor beta (TGFβ), and Wnts, thereby directing the differentiation of mesenchymal stem cells (MSCs). During bone resorption, the release and activation of stromal TGFβ recruits MSCs. MSCs differentiate into osteoblasts through signaling pathways including BMP, Wnt, Hedgehog and Notch[49].
In the management of CKD-MBD, an in-depth understanding of bone conversion abnormalities is essential for the development of effective treatment strategies. By regulating calcium-phosphorus balance, supplementing with vitamin D and its analogs, and controlling sHPT, it is possible to ameliorate abnormal bone conversion, slow the progression of osteoporosis, and improve patients' bone health and overall prognosis.
3. Vascular Calcification
Disturbed mineral metabolism in chronic kidney disease promotes the conversion of vascular smooth muscle cells to osteoblasts and the formation of calcium and phosphorus deposits in the vessel wall, known as vascular calcification. Vascular calcification is one of the major causes of cardiovascular complications in CKD patients[50]. It leads to vascular stiffness, vessel wall thickening and lumen narrowing, which in turn affects cardiovascular function and increases the risk of cardiovascular disease[4]. Osteoporosis, mineral quality abnormalities and vascular calcification often coexist in CKD patients and contribute to each other (Figure 4)[51]. Osteoporosis and abnormal mineral quality lead to disturbances in calcium and phosphorus metabolism, which in turn promote vascular calcification. In turn, vascular calcification exacerbates the progression of disturbed mineral metabolism and osteoporosis[52]. These pathologic changes interact with each other and severely affect the quality of life and prognosis of patients[51]. In patients with CKD, vascular calcification is not only a manifestation of vascular pathology, but also a key factor contributing to increased cardiovascular events. The main event in vascular calcification is the transformation of vascular smooth muscle cells (VSMCs) into osteoblasts and chondrocyte-like cells. As CKD progresses, changes in the bone-vascular axis increase the risk of renal and myocardial fibrosis, left ventricular hypertrophy, fractures, vascular and soft tissue calcification, bone loss, and fractures, all of which increase morbidity and mortality in patients with CKD[50]. Disorders of mineral metabolism, inflammation, oxidative stress, and vascular endothelial dysfunction all play a crucial role in the development of vascular calcification. In particular, hyperphosphatemia and vitamin D abnormalities due to imbalances in calcium and phosphorus metabolism exacerbate vascular calcification by promoting osteoblastic phenotypic shifts in VSMCs[53,54,55]. In addition, patients with chronic kidney disease are often associated with secondary hyperparathyroidism, which further by increasing parathyroid hormone levels. PTH is a major regulator of the Receptor Activator of Nuclear Factor-κB (RANK)/RANK Ligand (RANKL)/Osteoprotegerin (OPG) system, which is essential for bone maintenance and plays an important role in vascular smooth muscle cell calcification. A new RANKL receptor, the leucine-rich repeat-containing G-protein-coupled receptor 4 (LGR4), is important for osteoblast differentiation. It has been found that high PTH increases LGR4 and RANKL and decreases the expression of OPG in the aorta, thereby favoring vascular calcification[56]. In addition to RANKL, LGR4 also interacts with R-spondins (RSPO). New studies have found that RSPOs have little effect on VSMC calcification, but rather RANKL interacts with LGR4 to drive VSMC osteogenic differentiation[57]. Vascular calcification increases arterial stiffness and diminishes vascular adaptation to hemodynamic changes, leading to increased left ventricular load and myocardial ischemia, ultimately increasing the risk of cardiovascular events. Therefore, vascular calcification is not only an important predictor of cardiovascular complications in patients with CKD, but also a key factor affecting patient prognosis.
Diagnostic Methods
The diagnosis of CKD-MBD begins with a thorough clinical evaluation of the patient, including detailed history taking and physical examination. Laboratory tests play a central role in the diagnosis of CKD-MBD. Serum levels of calcium, phosphorus, and parathyroid hormone are important indicators for assessing a patient's bone metabolic status. Abnormal serum calcium levels are usually associated with hypocalcemia or hypercalcemia, whereas elevated phosphorus levels may indicate the risk of bone disease and vascular calcification. Abnormally elevated levels of PTH, a key hormone that regulates calcium and phosphorus homeostasis, are usually associated with secondary hyperparathyroidism[58,59].
In addition to traditional biochemical markers, novel biomarkers such as fibroblast growth factor 23 (FGF23), osteoprotegerin, and osteocalcin (OC) have provided new perspectives for the diagnosis and treatment of CKD-MBD. A case-cohort study demonstrated a linear correlation between FGF23 and fracture incidence, while α-Klotho, PTH, and phosphate levels were u-correlated with it[60]. Elevated levels of FGF23 are associated with an increased risk of cardiovascular events in patients with CKD. In progressive CKD, elevated circulating FGF23 levels combine with decreased renal klotho expression to result in klotho-independent effects of FGF23 on the heart, promoting left ventricular hypertrophy, heart failure, atrial fibrillation and death[61]. Osteoprotegerin (OPG) is a soluble RANKL decoy receptor produced primarily by osteoblasts that reduces bone resorption by preventing osteoclast formation and osteoclast bone resorption through inhibition of RANKL-RANKL receptor interactions[62]. A growing body of data suggests that OPG is involved in the regulation of vascular endothelial cell function. Under basal conditions, OPG is present in vascular endothelial cells and vascular smooth muscle cells (VSMCs) and is significantly increased in response to various stimuli ( for example, cytokines or hormones). TNF-alpha and interleukin-1beta (IL-1B) are strong stimulators of OPG production in endothelial cells and VSMCs. These cytokines can increase OPG expression 5-40-fold[63]. Osteocalcin (OC), on the other hand, is a non-collagenous protein secreted by osteoblasts, which is involved in the process of bone matrix mineralization and is a sensitive indicator of the rate of bone formation. Wei-Chen Lin et al. took blood biochemical studies of fasting blood samples from renal transplant recipients and found that osteocalcin concentration was negatively correlated with lumbar spine bone density in renal transplant recipients[64]. In addition, measurements of Alkaline Phosphatase (ALP) and Bone-specific alkaline phosphatase (BALP) are useful in assessing bone metabolic status. ALP is an independent risk marker for cardiovascular disease and death in the general population and in CKD. The hydrolytic activity of ALP regulates the mineralization inhibitor PPi, which plays a central role in mechanisms of dysregulated calcium phosphate metabolism, inflammation, osteogenic gene expression, and transdifferentiation of vascular smooth muscle cells. The ability of these circulating bone markers to replace bone biopsy in the clinical setting has been repeatedly examined[65].
Traditional techniques such as X-ray, CT scan and MRI have been widely used to evaluate bone structure. With advances in technology, imaging techniques such as dual-energy X-ray absorptiometry (DXA) and high-resolution peripheral quantitative computed tomography (HR-pQCT) have provided a more accurate bone assessment for the diagnosis of CKD-MBD. DXA is commonly used to assess bone density, while HR-pQCT provides detailed information about bone microarchitecture[66,67]. MRI is uniquely suited to assess soft tissue and vascular calcification[68]. Bone quality can be assessed by non-invasive methods such as Trabecular Bone Score (TBS), high-resolution bone imaging methods and invasive bone biopsy[69]. In some cases, a bone biopsy may be necessary to directly assess bone tissue, especially if the bone disease is considered to be poorly responsive to treatment or if other bone diseases need to be excluded. Bone biopsy can provide detailed information about bone mineralization, bone formation, and bone resorption[70]. The summary summarizes the diagnostic methods of CKD-MBD in Table 2. In conclusion, early diagnosis and treatment of CKD-MBD is essential to slow the progression of CKD and improve patient prognosis.
Clinical Management and Treatment
1. Development of Therapeutic Strategies to Maintain Calcium and Phosphorus Balance
Patients with CKD-MBD should follow a low-phosphorus diet to minimize phosphorus intake, which usually includes limiting high-phosphorus foods. It is recommended that people with CKD avoid excessive amounts of dietary protein, mixing animal and vegetable proteins, and avoid processed foods rich in phosphate-containing additives. In contrast, only 50% of the organophosphates in vegetables and about 70-80% of the organophosphates in animal protein-rich products are absorbed[71]. In addition to dietary modifications, additional treatments may be needed. This may include vitamin D supplements to promote calcium absorption and calcium supplements to maintain blood calcium levels. However, calcium supplements need to be used with caution to avoid the risk of hypercalcemia. Calcium supplements are commonly used in the treatment of CKD-MBD, especially in cases of hypocalcemia or hyperphosphatemia. The use of calcium agents needs to be individualized according to the patient's blood calcium and blood phosphorus levels[72]. In a multicenter double-blind randomized clinical trial, Obi Y et al. studied the effect of oral cholecalciferol supplementation on serum ferritin and parameters related to anemia and CKD-MBD in hemodialysis patients and found that supplementation with cholecalciferol increased serum ferritin-25 levels in hemodialysis patients in the short term and may increase erythropoietin resistance in the long term[73]. Phosphate-binding agents are a class of drugs that bind to phosphate in the gut and reduce its absorption, and these drugs are critical for controlling hyperphosphatemia. A post hoc analysis of a randomized phase 3 study evaluating the effect of 1 year of treatment with the phosphate-binding agents iron hydroxide or sevelamer carbonate (sevelamer) on the CKD-MBD index in dialysis patients with hyperphosphatemia found that 1 year of treatment with iron hydroxide or sevelamer not only substantially reduced serum phosphorus levels, but also significantly reduced serum FGF23[74]. Tenapanor, a novel selective inhibitor of the intestinal sodium/hydrogen exchanger 3 transporter protein, was found to result in an adequate reduction in serum phosphorus levels when administered in a phase 3 clinical trial and was considered safe and tolerable[75]. Although phosphate binders are used to reduce phosphate, the effects of specific phosphate-binding agent types on vitamin D metabolism were found to be inconsistent by Ginsberg C et al. 24,25-dihydroxyvitamin D and vitamin D metabolite ratio (VMR) were increased in the calcium acetate group, and 24,25-dihydroxyvitamin D3 and vitamin D metabolite ratio (VMR) were decreased in the sevelamer carbonate group, respectively[76]. However, in a study of cats, a magnesium-rich phosphate-restricted diet (PRD) was found to further stabilize plasma FGF23 and prevent hypercalcemia, suggesting that a magnesium-rich PRD is a novel therapeutic strategy[77]. Further studies are therefore necessary to evaluate the effects of other phosphate-lowering drugs, including iron-based phosphate binders, on vitamin D metabolism as well as on clinical outcomes including fractures and vascular calcification.
2. Therapeutic Advances in Controlling PTH Levels
Secondary hyperparathyroidism is a common complication in patients with chronic kidney disease and is characterized by persistent abnormally elevated parathyroid hormone levels. Treatment to control PTH levels is critical because SHPT is associated with a variety of adverse clinical outcomes, including osteodystrophy, vascular calcification, and cardiovascular events. Pharmacologic therapy is preferred in the treatment of SHPT and includes the use of vitamin D analogs, calcimimetics, and phosphorus binding agents[78]. Parathyroidectomy (PTX) is an effective treatment for patients for whom pharmacologic therapy is ineffective. Surgery can effectively reduce PTH levels, improve the calcium and phosphorus metabolic status of patients, and reduce the risk of bone disease and vascular calcification[79].
Cinacalcet, a calcium-sensitive receptor agonist that treats SHPT by decreasing PTH secretion, has been shown to significantly reduce blood calcium and blood phosphorus levels without increasing alkaline phosphatase (ALP) levels[80]. Wang AY et al. found in a randomized controlled trial that both oral cinacalcet and parathyroidectomy were effective in improving a variety of biochemical abnormalities in CKD-MBD, but did not reduce left ventricular mass, coronary artery and cardiac valve calcification, or arterial stiffness in patients with advanced SHPT, nor did they improve patient-centered HRQOL measures. However, the conclusions ultimately suggest that cinacalcet may be used as an alternative to surgery for advanced SHPT[81]. Etelcalcetide reduces parathyroid hormone levels in hemodialysis (HD) patients. A recent study, analyzed by the cumulative incidence of discontinuation method and linear generalized estimating equations for trajectory analysis, found a substantial and sustained reduction in patients' PTH levels over the 12 months following initiation of etelcalcetide. This supports the utility of etelcalcetide as an effective therapy for CKD-MBD[82]. Tsujita M's study found that cholecalciferol significantly reduced whole PTH concentrations compared to placebo, suggesting a significant role in the treatment of SHPT[83]. Pharmacologic therapy may have limitations, such as price issues and intolerable side effects for patients, which may limit its widespread use. Surgery also has certain risks and contraindications, such as being contraindicated in advanced patients with combined renal failure or those with parathyroid cancer that has developed distant metastases. When formulating a treatment strategy, physicians need to consider the patient's overall condition, PTH level, calcium and phosphorus metabolism status, and complications. In addition, postoperative management is also important, and the patient's calcium and phosphorus levels need to be closely monitored and the treatment regimen needs to be adjusted in order to maintain the balance of mineral metabolism.
3. Progress in the Maintenance of Bone Health
Bone health is an important public health issue, especially in patients with CKD, where the prevalence of osteoporosis is significantly increased. Therapeutic strategies for osteoporosis need to be directed not only at improving BMD but also include overall fracture risk management. Dudar I et al. demonstrated the efficient role of etelcalcetide in the treatment of SHPT in hemodialysis patients in a prospective study and showed that treatment of SHPT with etelcalcetide improved clinical outcomes such as high incidence of fractures[84]. Tsujita M's study found that 4000 IU/d cholecalciferol not only significantly reduced PTH levels, but also attenuated lumbar spine bone mineral density loss after kidney transplantation. Additionally the authors noted that this regimen has the potential to eliminate vitamin D deficiency and can also have beneficial effects on bone health under glucocorticoid therapy[83]. In a prespecified secondary endpoint analysis of a 48-week randomized, placebo-controlled, double-blind trial, Stathi D et al. found that in patients with type 2 diabetes mellitus (T2DM) and stage 3 CKD, osteotriol reduced levels of CTX, OCN, PINP, and iPTH[85]. A study by Ketteler M et al. found that 1 year of treatment with ferric hydroxide or sevelamer not only drastically reduced serum phosphorus and FGF23 levels, but also found an increase in the levels of bone formation markers in the later stages of treatment, which was associated with a clinical benefit in patients with CKD-MBD[74]. A post hoc analysis of data from a 12-month randomized, double-blind, phase III study by Sugimoto T et al. found that once-monthly administration of risedronate 75 mg tablets showed consistent safety and efficacy in inhibiting bone turnover and increasing BMD in Japanese patients with mild-to-moderate chronic kidney disease-primary osteoporosis[86]. Metabolic acidosis associated with CKD may lead to muscle dysfunction and bone disease. Melamed ML et al. tested whether sodium bicarbonate therapy improved muscle and bone outcomes. Sodium bicarbonate treatment in patients with CKD stages 3 and 4 was found to significantly increase serum bicarbonate and decrease potassium levels, but no differences were found in muscle function or bone mineral density between randomly assigned groups. Larger trials are therefore needed to assess the effects on renal function[87]. In addition, management of osteoporosis involves lifestyle modifications such as appropriate physical activity, smoking cessation, alcohol restriction, and calcium and vitamin D supplementation to support bone health.
4. Advances in the Prevention and Treatment of Vascular Calcification
Vascular calcification is a common and serious complication in patients with chronic kidney disease, and it is strongly associated with an increased risk of cardiovascular events in patients. Strategies adopted to prevent and treat vascular calcification include strict management of traditional cardiovascular risk factors, which involves control of conditions such as hypertension, diabetes, and dyslipidemia. In terms of dietary management, a recent study by Goraya N and colleagues found that in patients with chronic kidney disease, treatment with either base-rich fruits and vegetables or oral sodium bicarbonate improved metabolic acidosis to some extent and kept estimated glomerular filtration rate (eGFR) stable. However, increased intake of fruits and vegetables performed better in improving cardiovascular disease risk markers compared with sodium bicarbonate therapy, suggesting that dietary modification, particularly increased intake of alkaline foods, may be a more effective treatment option for reducing cardiovascular disease risk[88]. In addition, targeting CKD-specific risk factors, such as disorders of mineral metabolism and inflammation, are keys to reducing cardiovascular risk[89]. New vasoprotective therapeutic approaches are being explored, including the use of vitamin K antagonists, phosphorus-binding agents, and FGF23 inhibitors, aimed at preserving vascular health by modulating calcium and phosphorus metabolism and inhibiting the vascular calcification process[90]. Serum Klotho is thought to have heart and kidney protective effects, and altering its levels may be important in chronic kidney disease. Milovanova LY et al. found that successful control of parathyroid hormone levels with vitamin D receptor activators was associated with higher serum Klotho, with selective drugs showing a greater effect, suggesting that long-term treatment with selective vitamin D receptor activators may help prevent cardiovascular calcification by altering Klotho levels[91]. Supplementation with vitamin K2, particularly its most potent form, MK7, has been shown to reduce circulating levels of dephosphorylated uncarboxylated matrix gamma-carboxyglutamate proteins in patients with end-stage renal disease, which may be beneficial in cardiovascular disease[92]. It has also been shown that MK7 has a beneficial role in reducing the rate of progression of arterial stiffness in diabetic patients on chronic hemodialysis[93]. Magnesium and phosphate are both considered modulators of vascular calcification and chronic inflammation, both features of CKD that contribute to arterial stiffness. In a multicenter, placebo-controlled, randomized controlled trial, magnesium citrate supplementation and phosphate-lowering therapy were found to be a combined intervention with iron hydroxide in patients with stage 3-4 CKD who did not have significant hyperphosphatemia, and the results of this combined intervention may be useful for future early interventions in patients with CKD in order to reduce the risk of cardiovascular disease and death[94]. Magnesium inhibits vascular calcification, which is exacerbated by the uremic toxin indole sulfate. Sakaguchi Y et al. evaluated the efficacy of magnesium oxide and the oral carbon adsorbent AST-120 in slowing the progression of coronary artery calcification in CKD. It was found that the median change in coronary artery calcification scores was significantly smaller in MgO compared to controls, while the percentage change in coronary artery calcification scores was not significantly different between the AST-120 and control groups[95]. Aldosterone is thought to enhance the progression of vascular calcification, and a clinical study has further investigated the role of aldosterone inhibition by spironolactone, a hydrocorticoid receptor antagonist, on vascular calcification in CKD, and found that spironolactone treatment improved the propensity for vascular calcification in patients undergoing hemodialysis in the MiREnDa trial[96]. In a randomized controlled study, Gao Y et al. found that oral activated charcoal was effective in delaying the onset of hyperphosphatemia in patients with chronic kidney disease and appeared to delay the development of vascular calcification in patients with stage 3-4 CKD[97].
Emerging Research and Future Directions
Precision medicine is an important direction for the future development of CKD-MBD treatment. Through precision medicine, patients can be provided with more personalized and effective treatment plans. Precision medicine provides personalized treatment by taking into account the patient's genetic background, lifestyle and disease characteristics (Figure 6). Adam E Gaweda et al. combined quantitative systems pharmacology (QSP) methodology with machine learning, especially reinforcement learning (RL), to simulate in Matlab the simultaneous administration of a phosphate-binding agent, a vitamin D analog, and a calcimimetic to achieve serum calcium, phosphorus, and parathyroid hormone prescribed targets. Computer simulation results show that the application of QSP modeling in combination with RL can be used to achieve therapeutic goals more efficiently and rapidly, even in simulated subjects with poor adherence to medication, and to identify key decision variables for treatment recommendations[98]. Despite the great potential of precision medicine in the treatment of CKD-MBD, it also faces several challenges. First, precision medicine requires a large amount of individualized data and complex analysis methods, which puts higher demands on clinical practice. Second, the implementation of precision medicine requires multidisciplinary cooperation and coordination, which puts higher demands on medical resources and professionals. In addition, the ethical and legal issues of precision medicine need to be further explored and resolved. In order to realize precision therapy, the exploration of biomarkers and the application of multi-omics technologies are indispensable.
1. Biomarker Research: Clinical Value and Research Progress of Novel Biomarkers
Biomarkers are of great significance in the research of various diseases, and they can be used for early diagnosis of diseases, monitoring of treatment response, assessment of disease progression and prognosis. Through the study of biomarkers, the key processes of disease occurrence and development can be identified more precisely, and personalized solutions can be provided for the treatment of diseases.
In the study of CKD-MBD, the discovery and validation of novel biomarkers have provided new tools for early diagnosis and therapeutic monitoring of CKD-MBD. Biomarkers such as FGF23 and fibroblast growth factor receptor (FGFR) have been shown to be closely associated with mineral metabolism and bone health in CKD-MBD[34,99]. In a Meta-analysis, Liu C et al. found that five plasma biomarkers including FGF23 showed high correlation in assessing the prognosis of CKD patients, namely TNFR1, FGF23, TNFR2, KIM-1 and suPAR[100]. In recent years, Graziella D'Arrigo et al. investigated the association of CKD-MBD biomarkers with composite renal outcomes in patients with stage 2-5 CKD, and found that PTH was the only variable that showed a linear increase over time and was directly correlated with the same outcomes, suggesting that PTH is an important biomarker for CKD-MBD[101]. A study by Laster M et al. found elevated serum sclerostin levels in both early and advanced CKD and identified sclerostin as a potential biomarker or therapeutic target in pediatric renal osteodystrophy[102]. A study by Ouyang L et al. found that ALKBH1-mediated DNA N6-methyladenine demethylation modification promotes vascular calcification in chronic kidney disease through osteogenic reprogramming, suggesting that ALKBH1 may also be a biomarker for CKD-MBD[103]. Li W et al. found that SIRT6 inhibits osteogenic transdifferentiation of vascular smooth muscle in chronic kidney disease through the regulation of Runx2, which provides a potential target for VC therapy in CKD patients[104].
2. Application of Multi-Omics Technologies: Genomics, Proteomics and Metabolomics in CKD-MBD Research
The use of histologic techniques in disease research is becoming widespread, and multiple histologies can complement each other to provide a comprehensive understanding from genes to the final phenotype. Genomics can identify disease-associated genes, transcriptomics can reveal the regulation of the expression of these genes, proteomics can study how the proteins encoded by these genes are involved in the disease process, and metabolomics can reflect the impact of these changes on cellular metabolism. By integrating these histologic data, a more comprehensive understanding of the molecular mechanisms of disease can be achieved, providing a scientific basis for disease prevention, diagnosis and treatment.
The application of multi-omics research techniques in CKD-MBD has taken off. Proteomics and metabolomics studies have contributed to the discovery of biomarkers and therapeutic targets in CKD-MBD, and Taherkhani A et al. summarized the potential biomarkers for each chronic kidney disease subtype of disease analyzed by proteomics and metabolomics[105]. Si et al. revealed 32 proteins associated with CKD, renal function, or certain CKD clinical types by integrating the plasma proteome and transcriptome[106]. Through proteomic analysis, changes in protein expression and modification associated with CKD-MBD can be found, thus providing new clues for disease diagnosis and treatment. Using SomaScan V.4.0 (SomaLogic, Boulder, CO), a large-scale aptamer proteomic platform, the largest proteomic analysis of CKD progression to date has revealed multiple individual protein risk factors for CKD progression that have not been previously described. Drug targets in protein risk models and important MR Findings may provide momentum for the development of therapies, and biological pathways and individual proteins have been identified, including BMP antagonists, liver ligand signaling, and prothrombin activation[107]. Genomics and metabolomics in metabolite genome-wide association analysis is used to elucidate the underlying mechanisms of human metabolism, and Köttgen A et al. argued that genome-wide association analysis of metabolites using quantified metabolite concentrations from blood and/or urine can better elucidate the physiological and pathophysiological functions of the kidney and provide more resolved pathways for the study of CKD pathogenesis[108]. Recent studies have shown that by integrating data from different histologic levels, scientists are able to understand the pathophysiological process of CKD-MBD more comprehensively. Shen J et al. explored the effects and mechanisms of salt dulcimer cortex on CKD-MBD by integrating network pharmacology, transcriptomics, and metabolomics, and found that salt dulcimer cortex is highly likely to be effective in alleviating the negative effects of CKD-MBD on renal injury and bone damage induced in 5/6 nephrectomy-induced mice on a low-calcium/high-phosphorus diet through the PPARG/AMPK signaling pathway[109].
In conclusion, future studies need to further explore integrated analysis methods of multi-omics data, discover more biomarkers, and apply them to clinical practice to achieve accurate diagnosis and treatment of CKD-MBD.
Conclusion
Chronic Kidney Disease-Mineral and Bone Disorders (CKD-MBD) is a complex cluster of clinical conditions characterized by abnormalities in mineral metabolism and bone lesions. In recent years, significant progress has been made in the study of CKD-MBD, particularly in understanding its pathophysiologic mechanisms. The complex interactions of FGF23 with phosphate regulation, abnormalities in vitamin D metabolism, and the role of parathyroid hormone in bone metabolism[9,45]. The discovery of novel biomarkers provides new tools for early diagnosis and risk stratification. These biomarkers help to more accurately assess disease activity and fracture risk in patients with CKD-MBD. The detection of new biomarkers has gradually become part of routine clinical practice, helping physicians to recognize and intervene in the disease earlier[58,110]. In addition, a deeper understanding of calcium and phosphorus metabolism and parathyroid hormone has facilitated the development of therapeutic regimens, including the use of vitamin D analogs, phosphate-binding agents, calcimimetics, parathyroidectomy, and novel agents such as cinacalcet. Optimization of these therapeutic strategies has helped to improve the mineral metabolic status of patients, reduce the risk of cardiovascular events, and improve quality of life[111,112]. However, challenges remain in clinical practice: individualization of treatment regimens, adherence to long-term treatment, and monitoring of treatment effects. Despite the advances in CKD-MBD research, there are still limitations in existing studies. First, many studies have limited sample sizes, which may affect the generalizability and extrapolation of results. Second, there may be differences in results between studies, which may be related to differences in study design, patient populations, and interventions. In addition, the relative lack of long-term follow-up studies limits understanding of the natural history of the disease and long-term assessment of treatment effects. Therefore, future studies need larger sample sizes, more rigorous study designs, and longer-term follow-up to provide higher levels of evidence.
The management of CKD-MBD is a major challenge in current medicine. Despite the availability of global and regional guidelines, the majority of CKD patients are still affected by CKD-MBD abnormalities. The management of CKD-MBD requires a comprehensive consideration of the patient's mineral metabolic status, bone lesions, and cardiovascular risk[113]. Current therapeutic strategies focus on regulating mineral metabolism and suppressing hyperparathyroidism[20]. However, these treatments often have limitations, such as narrow therapeutic windows, side effects, and patient compliance issues. Therefore, future management strategies need to be more personalized and precise to improve patients' quality of life and improve prognosis. International collaboration and knowledge sharing are important in advancing CKD-MBD research and treatment. Through collaborative research and data sharing on a global scale, new biomarkers and therapeutic targets can be identified more quickly, accelerating the clinical application of new therapies. In addition, international collaboration can help to develop more uniform diagnostic and treatment guidelines and standards, and improve the treatment outcomes of CKD-MBD patients worldwide[114]. Future CKD-MBD research should delve into the molecular mechanisms of the disease, including disorders of mineral metabolism, bone lesions, and interactions with cardiovascular disease. These studies will help us to more fully understand the complexity of CKD-MBD and provide a theoretical basis for the development of new treatments. Clinical application and evaluation of the efficacy of new treatments will also be a focus of future research. This includes improvements to existing treatments, clinical trials of new drugs, and the development of personalized medicine strategies. Rigorous clinical studies will allow us to evaluate the safety, efficacy and cost-effectiveness of these new treatments to ensure that they can be widely used in the clinic and provide real benefits to people with CKD-MBD.
In conclusion, the management of CKD-MBD requires continuous exploration and innovation. By improving our understanding of disease mechanisms, developing new therapeutic strategies, and collaborating and sharing knowledge internationally, we can expect to improve the quality of life and prognosis of CKD-MBD patients in the future.
Funding
The present study received fnancial support from the Luzhou Science and Technology Bureau Key Research and Development Project: Study on the Therapeutic Effects and Mechanism of Shenwei Compound in Treating Chronic Kidney Disease-Induced Osteoporosis by Inhibiting Osteoclasts (2022-SYF-76) Project leader: Lin Luo
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Figures were created with biorender.com.
Acknowledgements
Not applicable.
Author contributions
Lin Luo: Conceptualization, Methodology, Validation, Investigation, Formal analysis, Writing-original draft. Jiaru Lin: Writing-review & editing, Supervision.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors made contributions to the article and authorized the submitted version.
Competing interests
The authors announce that there are no potential competing interests in this research.
Abbreviations
| CKD | Chronic Kidney Disease |
| GFR | Glomerular Filtration Rate |
| CKD-MBD | Chronic Kidney Disease-Mineral and Bone Disorder |
| PTH | Parathyroid Hormone |
| TCM | Traditional Chinese Medicine |
| FGF23 | Fibroblast growth factor 23 |
| SHTP | Secondary hyperparathyroidism |
| CaSR | Calcium sensitive receptor |
| CPP2 | Secondary calciprotein particles |
| RANKL | Receptor Activator of Nuclear Factor-κ B Ligand |
| NPT2a | Sodium Phosphate Transporter 2a |
| NPT2c | Sodium Phosphate Transporter 2c |
| EGFR | Estimated glomerular filtration rate |
| LCN2 | Lipocalin 2 |
| MAPK | Mitogen-activated protein kinase |
| TLR4 | Toll-like receptor 4 |
| BMP | Bone Morphogenetic Protein |
| TGFβ | Transforming growth factor beta |
| Wnts | Wingless and Int-1 related proteins |
| MSCs | Mesenchymal stem cells |
| Hedgehog | Hedgehog signaling pathway |
| Notch | Notch signaling pathway |
| VSMCs | Vascular smooth muscle cells |
| RANK | Receptor Activator of Nuclear Factor-κB |
| OPG | Osteoprotegerin |
| LGR4 | Leucine-rich repeat-containing G-protein-coupled receptor 4 |
| RSPO | R-spondins |
| OC | Osteocalcin |
| TNF-alpha | Tumor Necrosis Factor-alpha |
| IL-1B | Interleukin-1beta |
| ALP | Alkaline Phosphatase |
| BALP | Bone-specific alkaline phosphatase |
| DXA | Dual-energy X-ray absorptiometry |
| HR-pQCT | High-resolution peripheral quantitative computed tomography |
| TBS | Trabecular Bone Score |
| VMR | Vitamin D metabolite ratio |
| PRD | Phosphate-restricted diet |
| PTX | Parathyroidectomy |
| HD | Hemodialysis |
| T2DM | Type 2 diabetes mellitus |
| CTX | C-terminal telopeptide of type I collagen |
| OCN | Osteocalcin |
| PINP | Procollagen type I N-terminal propeptide |
| IPTH | Intact parathyroid hormone |
| AST-120 | Adsorptive Sorbent for Toxic Substance |
| QSP | Quantitative systems pharmacology |
| RL | Reinforcement learning |
| TNFR1 | Tumor Necrosis Factor Receptor 1 |
| TNFR2 | Tumor Necrosis Factor Receptor 2 |
| KIM-1 | Kidney Injury Molecule-1 |
| SuPAR | Soluble Urokinase Plasminogen Activator Receptor |
| ALKBH1 | AlkB Homolog 1 |
| SIRT6 | Sirtuin 6 |
| Runx2 | Runt-related transcription factor 2 |
| PPARG | Peroxisome Proliferator-Activated Receptor Gamma |
| AMPK | AMP-activated protein kinase |
References
- Aguilar A, Gifre L, Ureña-Torres P, Carrillo-López N, Rodriguez-García M, Massó E, et al. Pathophysiology of bone disease in chronic kidney disease: from basics to renal osteodystrophy and osteoporosis. Front. Physiol. 2023, 14, 1177829. [Google Scholar]
- Liu B H, Chong F L, Yuan C C, Liu Y L, Yang H M, Wang W W, et al. Fucoidan ameliorates renal injury-related calcium-phosphorus metabolic disorder and bone abnormality in the ckd-mbd model rats by targeting fgf23-klotho signaling axis. Front. Pharmacol. 2020, 11, 586725. [Google Scholar]
- Liu Z H, Li G, Zhang L, Chen J, Chen X, Zhao J, et al. Executive summary: clinical practice guideline of chronic kidney disease – mineral and bone disorder (ckd-mbd) in china. Kidney Dis. 2019, 5, 197–203. [Google Scholar] [CrossRef]
- Yamada S, Nakano T. Role of chronic kidney disease (ckd)–mineral and bone disorder (mbd) in the pathogenesis of cardiovascular disease in ckd. J. Atheroscler. Thromb. 2023, 30, 835–850. [Google Scholar] [CrossRef]
- Sun L, Huang Z, Fei S, Ni B, Wang Z, Chen H, et al. Vascular calcification progression and its association with mineral and bone disorder in kidney transplant recipients. Ren. Fail. 2023, 45, 2276382. [Google Scholar] [CrossRef]
- Nr H, St F, Jl O, Ja H, Ca O, Ds L, et al. Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PloS One.
- Bello A K, Okpechi I G, Levin A, Ye F, Damster S, Arruebo S, et al. An update on the global disparities in kidney disease burden and care across world countries and regions. Lancet Glob. Health. 2024, 12, e382–e395. [Google Scholar]
- Wang L, Xu X, Zhang M, Hu C, Zhang X, Li C, et al. Prevalence of chronic kidney disease in china: results from the sixth china chronic disease and risk factor surveillance. JAMA Intern. Med. 2023, 183, 298–310. [Google Scholar]
- Hu L, Napoletano A, Provenzano M, Garofalo C, Bini C, Comai G, et al. Mineral bone disorders in kidney disease patients: the ever-current topic. Int. J. Mol. Sci. 2022, 23, 12223. [Google Scholar] [CrossRef]
- Pazianas M, Miller P D. Osteoporosis and chronic kidney disease-mineral and bone disorder (ckd-mbd): back to basics. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2021, 78, 582–589. [Google Scholar] [CrossRef]
- Lan Y, Pei W. Advances of chronic kidney disease-mineral and bone disorder. Chin. J. Nephrol.
- Ni Lihua, Liu Bicheng, Tang Rining. Research progress of bone mineral density decrease in chronic kidney disease. Chin. Med. J. (Engl.). 2018, 98, 317–320. [Google Scholar]
- Kuro-o, M. Calcium phosphate microcrystallopathy as a paradigm of chronic kidney disease progression. Curr. Opin. Nephrol. Hypertens. 2023, 32, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Fleet J, C. Vitamin d-mediated regulation of intestinal calcium absorption. Nutrients.
- Amarnath S S, Kumar V, Barik S. Vitamin d and calcium and bioavailability of calcium in various calcium salts. Indian J. Orthop. 6: 1).
- Saponaro F, Saba A, Zucchi R. An update on vitamin d metabolism. Int. J. Mol. Sci. 2020, 21, 6573. [Google Scholar] [CrossRef]
- Shen, Y. Role of nutritional vitamin d in chronic kidney disease-mineral and bone disorder: a narrative review. Medicine (Baltimore). 2023, 102, e33477. [Google Scholar] [CrossRef]
- Charoenngam N, Holick M F. Immunologic effects of vitamin d on human health and disease. Nutrients. 2020, 12, 2097. [Google Scholar] [CrossRef] [PubMed]
- Meyer M B, Pike J W. Mechanistic homeostasis of vitamin d metabolism in the kidney through reciprocal modulation of cyp27b1 and cyp24a1 expression. J. Steroid Biochem. Mol. Biol. 2020, 196, 105500. [Google Scholar]
- Brandenburg V, Ketteler M. Vitamin d and secondary hyperparathyroidism in chronic kidney disease: a critical appraisal of the past, present, and the future. Nutrients. 2022, 14, 3009. [Google Scholar] [CrossRef]
- Xiang Z, Wang M, Miao C, Jin D, Wang H. Mechanism of calcitriol regulating parathyroid cells in secondary hyperparathyroidism. Front. Pharmacol. 2022, 13, 1020858. [Google Scholar]
- Portales-Castillo I, Simic P. PTH, fgf-23, klotho and vitamin d as regulators of calcium and phosphorus: genetics, epigenetics and beyond. Front. Endocrinol. 2022, 13, 992666. [Google Scholar]
- Renke G, Starling-Soares B, Baesso T, Petronio R, Aguiar D, Paes R. Effects of vitamin d on cardiovascular risk and oxidative stress. Nutrients. 2023, 15, 769. [Google Scholar] [CrossRef]
- Wein M N, Kronenberg H M. Regulation of bone remodeling by parathyroid hormone. Cold Spring Harb. Perspect. Med. 2018, 8, a031237. [Google Scholar] [CrossRef]
- Ulmer C Z, Kritmetapak K, Singh R J, Vesper H W, Kumar R. High-resolution mass spectrometry for the measurement of pth and pth fragments: insights into pth physiology and bioactivity. J. Am. Soc. Nephrol. JASN. 2022, 33, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- DeMambro V E, Tian L, Karthik V, Rosen C J, Guntur A R. Effects of pth on osteoblast bioenergetics in response to glucose. Bone Rep. 2023, 19, 101705. [Google Scholar] [CrossRef]
- Rendina-Ruedy E, Rosen C J. Parathyroid hormone (pth) regulation of metabolic homeostasis: an old dog teaches us new tricks. Mol. Metab. 2022, 60, 101480. [Google Scholar] [CrossRef] [PubMed]
- Mantovani G, Elli F M. PTH resistance. Mol. Cell. Endocrinol. 2021, 531, 111311. [Google Scholar] [CrossRef]
- Musgrove J, Wolf M. Regulation and effects of fgf23 in chronic kidney disease. Annu. Rev. Physiol. 2020, 82, 365–390. [Google Scholar] [CrossRef]
- Aikawa T, Segre G V, Lee K. Fibroblast growth factor inhibits chondrocytic growth through induction of p21 and subsequent inactivation of cyclin e-cdk2*. J. Biol. Chem. 2001, 276, 29347–29352. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub V, Ward S C, Sun B, Stevens J, Renshaw L, Hawkins N, et al. Fibroblast growth factor 23 (fgf23) and alpha-klotho stimulate osteoblastic mc3t3.e1 cell proliferation and inhibit mineralization. Calcif. Tissue Int. 2011, 89, 140–150. [Google Scholar] [CrossRef]
- Bellorin-Font E, Rojas E, Martin K J. Bone disease in chronic kidney disease and kidney transplant. Nutrients. 2022, 15, 167. [Google Scholar] [CrossRef]
- Graciolli F G, Neves K R, Barreto F, Barreto D V, Dos Reis L M, Canziani M E, et al. The complexity of chronic kidney disease–mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017, 91, 1436–1446. [Google Scholar] [CrossRef]
- Sun T, Yu X. FGF23 actions in ckd-mbd and other organs during ckd. Curr. Med. Chem. 2023, 30, 841–856. [Google Scholar] [CrossRef]
- Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type iia. Biochem. Biophys. Res. Commun. 2004, 314, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Courbon G, Francis C, Gerber C, Neuburg S, Wang X, Lynch E, et al. Lipocalin 2 stimulates bone fibroblast growth factor 23 production in chronic kidney disease. Bone Res. 2021, 9, 35. [Google Scholar] [CrossRef]
- Murali S K, Roschger P, Zeitz U, Klaushofer K, Andrukhova O, Erben R G. FGF23 regulates bone mineralization in a 1,25(oh)2 d3 and klotho-independent manner. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2016, 31, 129–142. [Google Scholar] [CrossRef]
- Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt K P, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [PubMed]
- Kawai M, Kinoshita S, Kimoto A, Hasegawa Y, Miyagawa K, Yamazaki M, et al. FGF23 suppresses chondrocyte proliferation in the presence of soluble α-klotho both in vitro and in vivo*. J. Biol. Chem. 2013, 288, 2414–2427. [Google Scholar] [CrossRef]
- Wu Q, Fan W, Zhong X, Zhang L, Niu J, Gu Y. Klotho/fgf23 and wnt in shpt associated with ckd via regulating mir-29a. Am. J. Transl. Res. 2022, 14, 876–887. [Google Scholar]
- Vogt I, Haffner D, Leifheit-Nestler M. FGF23 and phosphate-cardiovascular toxins in ckd. Toxins. 2019, 11, 647. [Google Scholar] [CrossRef] [PubMed]
- Cernaro V, Longhitano E, Calabrese V, Casuscelli C, Di Carlo S, Spinella C, et al. Progress in pharmacotherapy for the treatment of hyperphosphatemia in renal failure. Expert Opin. Pharmacother. 2023, 24, 1737–1746. [Google Scholar]
- Pimentel A, Ureña-Torres P, Bover J, Luis Fernandez-Martín J, Cohen-Solal M. Bone fragility fractures in ckd patients. Calcif. Tissue Int. 2021, 108, 539–550. [Google Scholar] [CrossRef]
- KDIGO 2017 clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease–mineral and bone disorder (ckd-mbd). Kidney Int. Suppl. 2017, 7, 1–59. [Google Scholar]
- Cannata-Andía J B, Martín-Carro B, Martín-Vírgala J, Rodríguez-Carrio J, Bande-Fernández J J, Alonso-Montes C, et al. Chronic kidney disease-mineral and bone disorders: pathogenesis and management. Calcif. Tissue Int. 2021, 108, 410–422. [Google Scholar] [CrossRef]
- Wagner C, A. The basics of phosphate metabolism. Nephrol. Dial. Transplant. 2023, 39, 190–201. [Google Scholar] [CrossRef]
- Shiizaki K, Tsubouchi A, Miura Y, Seo K, Kuchimaru T, Hayashi H, et al. Calcium phosphate microcrystals in the renal tubular fluid accelerate chronic kidney disease progression. J. Clin. Invest.131,e145693.
- Chen T, Wang Y, Hao Z, Hu Y, Li J. Parathyroid hormone and its related peptides in bone metabolism. Biochem. Pharmacol. 2021, 192, 114669. [Google Scholar]
- Wang L T, Chen L R, Chen K H. Hormone-related and drug-induced osteoporosis: a cellular and molecular overview. Int. J. Mol. Sci. 2023, 24, 5814. [Google Scholar] [CrossRef] [PubMed]
- Kaur R, Singh R. Mechanistic insights into ckd-mbd-related vascular calcification and its clinical implications. Life Sci. 1: B), 1211.
- Hsu C Y, Chen L R, Chen K H. Osteoporosis in patients with chronic kidney diseases: a systemic review. Int. J. Mol. Sci. 2020, 21, 6846. [Google Scholar] [CrossRef]
- Aaltonen L, Koivuviita N, Seppänen M, Kröger H, Tong X, Löyttyniemi E, et al. Association between bone mineral metabolism and vascular calcification in end-stage renal disease. BMC Nephrol. 2022, 23, 12. [Google Scholar]
- Bao S, Guo Y, Diao Z, Guo W, Liu W. Genome-wide identification of lncrnas and mrnas differentially expressed in human vascular smooth muscle cells stimulated by high phosphorus. Ren. Fail. 2020, 42, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Jin D, Lin L, Xie Y, Jia M, Qiu H, Xun K. NRF2-suppressed vascular calcification by regulating the antioxidant pathway in chronic kidney disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2022, 36, e22098. [Google Scholar]
- Cutini P H, Campelo A E, Massheimer V L. Vascular response to stress: protective action of the bisphosphonate alendronate. Vasc. Med. Lond. Engl. 2022, 27, 425–432. [Google Scholar] [CrossRef]
- Carrillo-López N, Martínez-Arias L, Alonso-Montes C, Martín-Carro B, Martín-Vírgala J, Ruiz-Ortega M, et al. The receptor activator of nuclear factor κβ ligand receptor leucine-rich repeat-containing g-protein-coupled receptor 4 contributes to parathyroid hormone-induced vascular calcification. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. - Eur. Ren. Assoc. 2021, 36, 618–631. [Google Scholar]
- Fernández-Villabrille S, Martín-Vírgala J, Martín-Carro B, Baena-Huerta F, González-García N, Gil-Peña H, et al. RANKL, but not r-spondins, is involved in vascular smooth muscle cell calcification through lgr4 interaction. Int. J. Mol. Sci. 2024, 25, 5735. [Google Scholar] [CrossRef]
- Fusaro M, Barbuto S, Gallieni M, Cossettini A, Re Sartò G V, Cosmai L, et al. Real-world usage of chronic kidney disease - mineral bone disorder (ckd-mbd) biomarkers in nephrology practices. Clin. Kidney J. 2024, 17, sfad290. [Google Scholar] [CrossRef] [PubMed]
- Galassi A, Fasulo E M, Ciceri P, Casazza R, Bonelli F, Zierold C, et al. 1,25-dihydroxyvitamin d as predictor of renal worsening function in chronic kidney disease. results from the pascal-1,25d study. Front. Med. 2022, 9, 840801. [Google Scholar]
- Desbiens L C, Sidibé A, Ung R V, Mac-Way F. FGF23-klotho axis and fractures in patients without and with early ckd: a case-cohort analysis of cartagene. J. Clin. Endocrinol. Metab. 2022, 107, e2502–e2512. [Google Scholar] [CrossRef] [PubMed]
- Edmonston D, Grabner A, Wolf M. FGF23 and klotho at the intersection of kidney and cardiovascular disease. Nat. Rev. Cardiol. 2024, 21, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Udagawa N, Koide M, Nakamura M, Nakamichi Y, Yamashita T, Uehara S, et al. Osteoclast differentiation by rankl and opg signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Dutka M, Bobiński R, Wojakowski W, Francuz T, Pająk C, Zimmer K. Osteoprotegerin and rankl-rank-opg-trail signalling axis in heart failure and other cardiovascular diseases. Heart Fail. Rev. 2022, 27, 1395–1411. [Google Scholar] [CrossRef]
- Lin W C, Lee M C, Chen Y C, Hsu B G. Inverse association of serum osteocalcin and bone mineral density in renal transplant recipients. Tzu-Chi Med. J. 2022, 35, 165–170. [Google Scholar]
- Haarhaus M, Cianciolo G, Barbuto S, Manna G L, Gasperoni L, Tripepi G, et al. Alkaline phosphatase: an old friend as treatment target for cardiovascular and mineral bone disorders in chronic kidney disease. Nutrients.
- Rasmussen N H H, Dal J, Kvist A V, Van den Bergh J P, Jensen M H, Vestergaard P. Bone parameters in t1d and t2d assessed by dxa and hr-pqct - a cross-sectional study: the diafall study. Bone. 2023, 172, 116753. [Google Scholar] [CrossRef]
- Shevroja E, Cafarelli F P, Guglielmi G, Hans D. DXA parameters, trabecular bone score (tbs) and bone mineral density (bmd), in fracture risk prediction in endocrine-mediated secondary osteoporosis. Endocrine. 2021, 74, 20–28. [Google Scholar] [CrossRef]
- Nagata M, Minami M, Yoshida K, Yang T, Yamamoto Y, Takayama N, et al. Calcium-binding protein s100a4 is upregulated in carotid atherosclerotic plaques and contributes to expansive remodeling. J. Am. Heart Assoc. 2020, 9, e016128. [Google Scholar] [CrossRef]
- Abdalbary M, Sobh M, Elnagar S, Elhadedy M A, Elshabrawy N, Abdelsalam M, et al. Management of osteoporosis in patients with chronic kidney disease. Osteoporos. Int. J. Establ. Result Coop. Eur. Found. Osteoporos. Natl. Osteoporos. Found. USA. 2022, 33, 2259–2274. [Google Scholar] [CrossRef] [PubMed]
- The role of bone biopsy in the management of ckd-mbd - pubmed[EB].
- Favero C, Carriazo S, Cuarental L, Fernandez-Prado R, Gomá-Garcés E, Perez-Gomez M V, et al. Phosphate, microbiota and ckd. Nutrients. 2021, 13, 1273. [Google Scholar] [CrossRef] [PubMed]
- Rastogi A, Bhatt N, Rossetti S, Beto J. Management of hyperphosphatemia in end-stage renal disease: a new paradigm. J. Ren. Nutr. 2021, 31, 21–34. [Google Scholar] [CrossRef]
- Obi Y, Yamaguchi S, Hamano T, Sakaguchi Y, Shimomura A, Namba-Hamano T, et al. Effect of cholecalciferol on serum hepcidin and parameters of anaemia and ckd-mbd among haemodialysis patients: a randomized clinical trial. Sci. Rep. 2020, 10, 15500. [Google Scholar]
- Ketteler M, Sprague S M, Covic A C, Rastogi A, Spinowitz B, Rakov V, et al. Effects of sucroferric oxyhydroxide and sevelamer carbonate on chronic kidney disease-mineral bone disorder parameters in dialysis patients. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. - Eur. Ren. Assoc. 2019, 34, 1163–1170. [Google Scholar]
- Nakayama M, Kobayashi S, Kusakabe M, Ohara M, Nakanishi K, Akizawa T, et al. Tenapanor for peritoneal dialysis patients with hyperphosphatemia: a phase 3 trial. Clin. Exp. Nephrol. 2024, 28, 153–164. [Google Scholar] [CrossRef]
- Ginsberg C, Zelnick L R, Block G A, Chertow G M, Chonchol M, Hoofnagle A, et al. Differential effects of phosphate binders on vitamin d metabolism in chronic kidney disease. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. - Eur. Ren. Assoc. 2020, 35, 616–623. [Google Scholar]
- Tang P K, Van den Broek D H N, Jepson R E, Geddes R F, Chang Y M, Lötter N, et al. Dietary magnesium supplementation in cats with chronic kidney disease: a prospective double-blind randomized controlled trial. J. Vet. Intern. Med. 2024, 38, 2180–2195. [Google Scholar] [CrossRef]
- Rodzoń-Norwicz M, Norwicz S, Sowa-Kućma M, Gala-Błądzińska A. Secondary hyperparathyroidism in chronic kidney disease: pathomechanism and current treatment possibilities. Endokrynol. Pol. 2023, 74, 490–498. [Google Scholar] [CrossRef]
- Hiramitsu T, Hasegawa Y, Futamura K, Okada M, Goto N, Narumi S, et al. Treatment for secondary hyperparathyroidism focusing on parathyroidectomy. Front. Endocrinol. 2023, 14, 1169793. [Google Scholar]
- Bernardor J, De Mul A, Bacchetta J, Schmitt C P. Impact of cinacalcet and etelcalcetide on bone mineral and cardiovascular disease in dialysis patients. Curr. Osteoporos. Rep. 2023, 21, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Wang A Y M, Lo W K, Cheung S C W, Tang T K, Yau Y Y, Lang B H H. Parathyroidectomy versus oral cinacalcet on cardiovascular parameters in peritoneal dialysis patients with advanced secondary hyperparathyroidism (proceed): a randomized trial. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. - Eur. Ren. Assoc. 2023, 38, 1823–1835. [Google Scholar]
- Karaboyas A, Muenz D, Fuller D S, Desai P, Lin T C, Robinson B M, et al. Etelcalcetide utilization, dosing titration, and chronic kidney disease-mineral and bone disease (ckd-mbd) marker responses in us hemodialysis patients. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2022, 79, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Tsujita M, Doi Y, Obi Y, Hamano T, Tomosugi T, Futamura K, et al. Cholecalciferol supplementation attenuates bone loss in incident kidney transplant recipients: a prespecified secondary endpoint analysis of a randomized controlled trial. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2022, 37, 303–311. [Google Scholar]
- Dudar I, Shifris I, Dudar S, Kulish V. Current therapeutic options for the treatment of secondary hyperparathyroidism in end-stage renal disease patients treated with hemodialysis: a 12-month comparative study. Pol. Merkur. Lek. Organ Pol. Tow. Lek. 2022, 50, 294–298. [Google Scholar]
- Stathi D, Fountoulakis N, Panagiotou A, Maltese G, Corcillo A, Mangelis A, et al. Impact of treatment with active vitamin d calcitriol on bone turnover markers in people with type 2 diabetes and stage 3 chronic kidney disease. Bone. 2023, 166, 116581. [Google Scholar] [CrossRef]
- Sugimoto T, Inoue D, Maehara M, Oikawa I, Shigematsu T, Nishizawa Y. Efficacy and safety of once-monthly risedronate in osteoporosis subjects with mild-to-moderate chronic kidney disease: a post hoc subgroup analysis of a phase iii trial in japan. J. Bone Miner. Metab. 2019, 37, 730–740. [Google Scholar] [CrossRef]
- Melamed M L, Horwitz E J, Dobre M A, Abramowitz M K, Zhang L, Lo Y, et al. Effects of sodium bicarbonate in ckd stages 3 and 4: a randomized, placebo-controlled, multicenter clinical trial. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2020, 75, 225–234. [Google Scholar] [CrossRef]
- Goraya N, Munoz-Maldonado Y, Simoni J, Wesson D E. Fruit and vegetable treatment of chronic kidney disease-related metabolic acidosis reduces cardiovascular risk better than sodium bicarbonate. Am. J. Nephrol. 2019, 49, 438–448. [Google Scholar] [CrossRef]
- Kratky V, Valerianova A, Hruskova Z, Tesar V, Malik J. Increased cardiovascular risk in young patients with ckd and the role of lipid-lowering therapy. Curr. Atheroscler. Rep. 2024, 26, 103–109. [Google Scholar] [CrossRef]
- Singh A, Tandon S, Tandon C. An update on vascular calcification and potential therapeutics. Mol. Biol. Rep. 2021, 48, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Milovanova L Y, Beketov V D, Milovanova S Y, Taranova M V, Kozlov V V, Pasechnik A I, et al. Effect of vitamin d receptor activators on serum klotho levels in 3b-4 stages chronic кidney disease patients: a prospective randomized study. Ter. Arkh. 2021, 93, 679–684. [Google Scholar]
- Kaesler N, Schurgers L J, Floege J. Vitamin k and cardiovascular complications in chronic kidney disease patients. Kidney Int. 2021, 100, 1023–1036. [Google Scholar] [CrossRef]
- Naiyarakseree N, Phannajit J, Naiyarakseree W, Mahatanan N, Asavapujanamanee P, Lekhyananda S, et al. Effect of menaquinone-7 supplementation on arterial stiffness in chronic hemodialysis patients: a multicenter randomized controlled trial. Nutrients. 2023, 15, 2422. [Google Scholar] [CrossRef]
- Vermeulen E A, Eelderink C, Hoekstra T, Van Ballegooijen A J, Raijmakers P, Beulens J W, et al. Reversal of arterial disease by modulating magnesium and phosphate (roadmap-study): rationale and design of a randomized controlled trial assessing the effects of magnesium citrate supplementation and phosphate-binding therapy on arterial stiffness in moderate chronic kidney disease. Trials. 2022, 23, 769. [Google Scholar]
- Sakaguchi Y, Hamano T, Obi Y, Monden C, Oka T, Yamaguchi S, et al. A randomized trial of magnesium oxide and oral carbon adsorbent for coronary artery calcification in predialysis ckd. J. Am. Soc. Nephrol. JASN. 2019, 30, 1073–1085. [Google Scholar] [CrossRef]
- Hammer F, Buehling S S, Masyout J, Malzahn U, Hauser T, Auer T, et al. Protective effects of spironolactone on vascular calcification in chronic kidney disease. Biochem. Biophys. Res. Commun. 2021, 582, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Gao Y, Wang G, Li Y, Lv C, Wang Z. Effects of oral activated charcoal on hyperphosphatemia and vascular calcification in chinese patients with stage 3-4 chronic kidney disease. J. Nephrol. 2019, 32, 265–272. [Google Scholar] [CrossRef]
- Gaweda A E, Lederer E D, Brier M E. Artificial intelligence-guided precision treatment of chronic kidney disease-mineral bone disorder. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 1305–1315. [Google Scholar] [CrossRef]
- Vervloet, M. Renal and extrarenal effects of fibroblast growth factor 23. Nat. Rev. Nephrol. 2019, 15, 109–120. [Google Scholar]
- Liu C, Debnath N, Mosoyan G, Chauhan K, Vasquez-Rios G, Soudant C, et al. Systematic review and meta-analysis of plasma and urine biomarkers for ckd outcomes. J. Am. Soc. Nephrol. JASN. 2022, 33, 1657–1672. [Google Scholar] [CrossRef] [PubMed]
- D’Arrigo G, Mallamaci F, Pizzini P, Versace M C, Tripepi G L, Zoccali C, et al. MO499CKD-mbd biomarkers and ckd progression: an analysis by joint models. Nephrol. Dial. Transplant. 0019.
- Laster M, Pereira R C, Noche K, Gales B, Salusky I B, Albrecht L V. Sclerostin, osteocytes, and wnt signaling in pediatric renal osteodystrophy. Nutrients. 2023, 15, 4127. [Google Scholar] [CrossRef]
- Ouyang L, Su X, Li W, Tang L, Zhang M, Zhu Y, et al. ALKBH1-demethylated dna n6-methyladenine modification triggers vascular calcification via osteogenic reprogramming in chronic kidney disease. J. Clin. Invest. 2021, 131, e146985–146985. [Google Scholar] [CrossRef]
- Li W, Feng W, Su X, Luo D, Li Z, Zhou Y, et al. SIRT6 protects vascular smooth muscle cells from osteogenic transdifferentiation via runx2 in chronic kidney disease. J. Clin. Invest. 2022, 132, e150051. [Google Scholar] [CrossRef] [PubMed]
- Taherkhani A, Farrokhi Yekta R, Mohseni M, Saidijam M, Arefi Oskouie A. Chronic kidney disease: a review of proteomic and metabolomic approaches to membranous glomerulonephritis, focal segmental glomerulosclerosis, and iga nephropathy biomarkers. Proteome Sci. 2019, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Si S, Liu H, Xu L, Zhan S. Identification of novel therapeutic targets for chronic kidney disease and kidney function by integrating multi-omics proteome with transcriptome. Genome Med. 2024, 16, 84. [Google Scholar] [CrossRef]
- Dubin R F, Deo R, Ren Y, Wang J, Zheng Z, Shou H, et al. Proteomics of ckd progression in the chronic renal insufficiency cohort. Nat. Commun. 2023, 14, 6340. [Google Scholar] [CrossRef]
- Köttgen A, Raffler J, Sekula P, Kastenmüller G. Genome-wide association studies of metabolite concentrations (mgwas): relevance for nephrology. Semin. Nephrol. 2018, 38, 151–174. [Google Scholar] [CrossRef]
- Shen J, Liu Y, Wang Q, Chen H, Hu Y, Guo X, et al. Integrated network pharmacology, transcriptomics, and metabolomics analysis to reveal the mechanism of salt eucommiae cortex in the treatment of chronic kidney disease mineral bone disorders via the pparg/ampk signaling pathway. J. Ethnopharmacol. 2023, 314, 116590. [Google Scholar] [CrossRef]
- Novel biomarkers of bone metabolism - pmc[EB].
- Thadhani R I, Rosen S, Ofsthun N J, Usvyat L A, Dalrymple L S, Maddux F W, et al. Conversion from intravenous vitamin d analogs to oral calcitriol in patients receiving maintenance hemodialysis. Clin. J. Am. Soc. Nephrol. CJASN. 2020, 15, 384–391. [Google Scholar] [CrossRef]
- Mathur A, Ahn J B, Sutton W, Chu N M, Gross A L, Segev D L, et al. Secondary hyperparathyroidism (ckd-mbd) treatment and the risk of dementia. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. - Eur. Ren. Assoc. 2022, 37, 2111–2118. [Google Scholar]
- Evenepoel P, Cunningham J, Ferrari S, Haarhaus M, Javaid M K, Lafage-Proust M H, et al. European consensus statement on the diagnosis and management of osteoporosis in chronic kidney disease stages g4–g5d. Nephrol. Dial. Transplant. 2021, 36, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Karam S, Wong M M Y, Jha V. Sustainable development goals: challenges and the role of the international society of nephrology in improving global kidney health. Kidney360. 2023, 4, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pathologic features of CKD-MBD. CKD-MBD has three major pathologic features, which are biochemical metabolic abnormalities, osteoporotic bone loss, and vascular and soft tissue calcification.
Figure 1.
Pathologic features of CKD-MBD. CKD-MBD has three major pathologic features, which are biochemical metabolic abnormalities, osteoporotic bone loss, and vascular and soft tissue calcification.
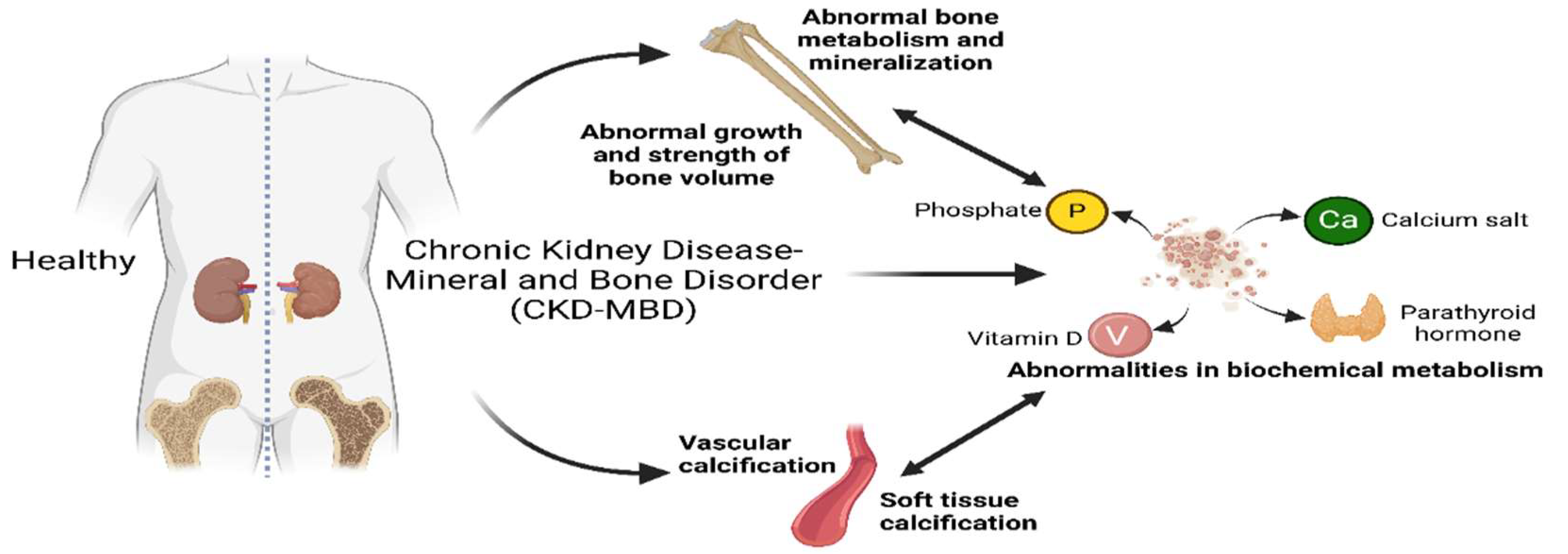
Figure 2.
Disorders of mineral metabolism in CKD-MBD. In patients with chronic kidney disease, decreased GFR leads to hyperphosphatemia, hypocalcemia, and decreased active vitamin D. This triggers elevated levels of parathyroid hormone and FGF23, leading to abnormal bone metabolism and vascular calcification. Hyperphosphatemia exacerbates CKD progression by promoting soft tissue calcification and inhibiting bone mineralization. Hypocalcemia stimulates parathyroid hormone secretion to increase blood calcium levels. Chronic high PTH levels lead to secondary hyperparathyroidism, which in turn causes bone calcium mobilization, promotes bone resorption, and decreases bone density. Vitamin D deficiency affects calcium absorption and bone mineralization. FGF23 interacts with Klotho and affects bone and kidney health.
Figure 2.
Disorders of mineral metabolism in CKD-MBD. In patients with chronic kidney disease, decreased GFR leads to hyperphosphatemia, hypocalcemia, and decreased active vitamin D. This triggers elevated levels of parathyroid hormone and FGF23, leading to abnormal bone metabolism and vascular calcification. Hyperphosphatemia exacerbates CKD progression by promoting soft tissue calcification and inhibiting bone mineralization. Hypocalcemia stimulates parathyroid hormone secretion to increase blood calcium levels. Chronic high PTH levels lead to secondary hyperparathyroidism, which in turn causes bone calcium mobilization, promotes bone resorption, and decreases bone density. Vitamin D deficiency affects calcium absorption and bone mineralization. FGF23 interacts with Klotho and affects bone and kidney health.
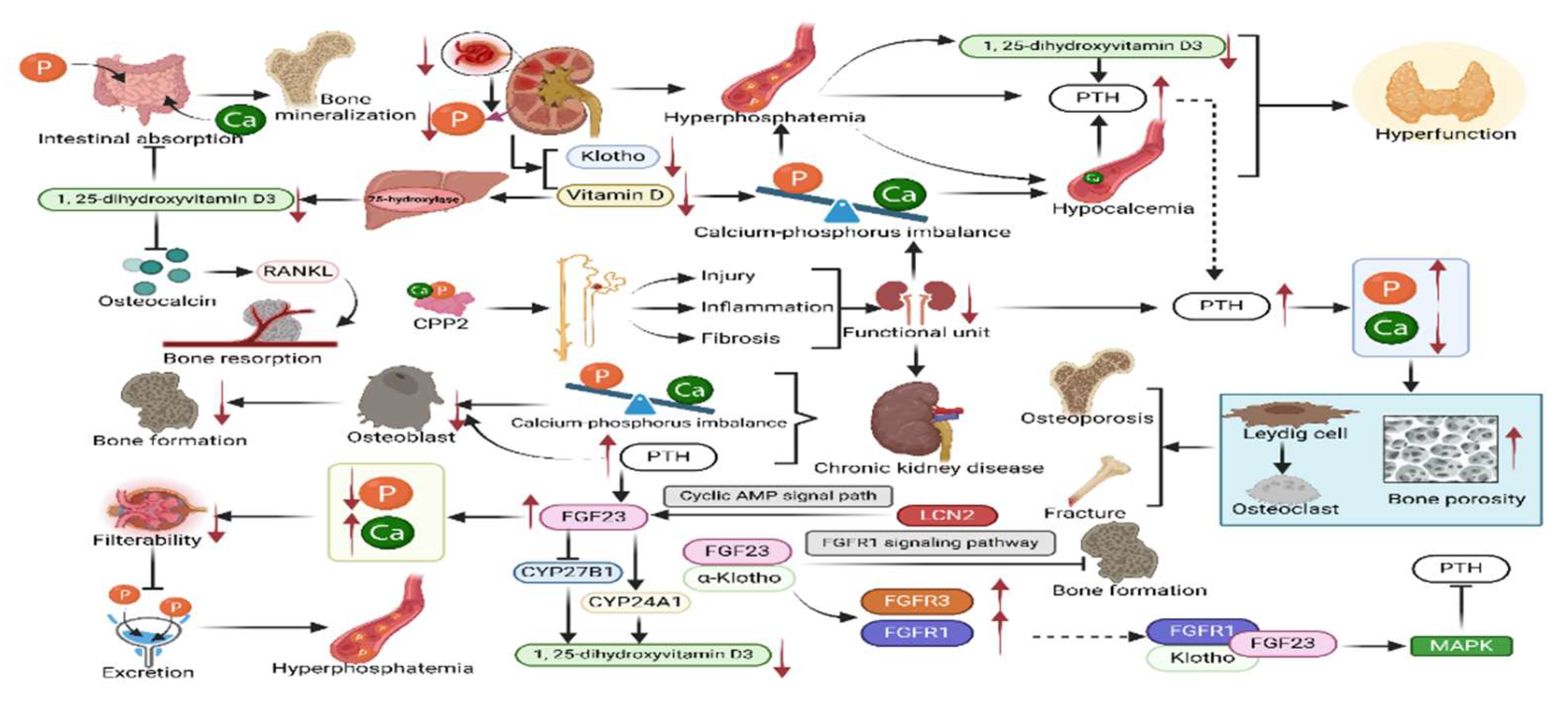
Figure 3.
Skeletal changes in CKD-MBD. Patients with chronic kidney disease face a higher risk of fracture due to impaired bone renewal. Metabolic abnormalities such as elevated blood phosphorus, decreased blood calcium and vitamin D deficiency interfere with bone mineralization and affect bone strength. Hyperphosphatemia inhibits bone mineralization while promoting soft tissue calcification, exacerbating kidney injury and CKD progression. Vitamin D deficiency can lead to renal osteodystrophy. Parathyroid hormone is critical in bone remodeling. Elevated PTH in CKD promotes bone resorption and inhibits osteoblast function, leading to decreased bone density. Bone morphogenetic protein (BMP), transforming growth factor β(TGFβ) and Wnts signaling pathways play a guiding role in osteoblast differentiation. Management of CKD-MBD requires an in-depth understanding of abnormal bone turnover to improve bone health and prognosis through regulation of calcium-phosphorus balance, vitamin D supplementation, and control of secondary hyperparathyroidism.
Figure 3.
Skeletal changes in CKD-MBD. Patients with chronic kidney disease face a higher risk of fracture due to impaired bone renewal. Metabolic abnormalities such as elevated blood phosphorus, decreased blood calcium and vitamin D deficiency interfere with bone mineralization and affect bone strength. Hyperphosphatemia inhibits bone mineralization while promoting soft tissue calcification, exacerbating kidney injury and CKD progression. Vitamin D deficiency can lead to renal osteodystrophy. Parathyroid hormone is critical in bone remodeling. Elevated PTH in CKD promotes bone resorption and inhibits osteoblast function, leading to decreased bone density. Bone morphogenetic protein (BMP), transforming growth factor β(TGFβ) and Wnts signaling pathways play a guiding role in osteoblast differentiation. Management of CKD-MBD requires an in-depth understanding of abnormal bone turnover to improve bone health and prognosis through regulation of calcium-phosphorus balance, vitamin D supplementation, and control of secondary hyperparathyroidism.
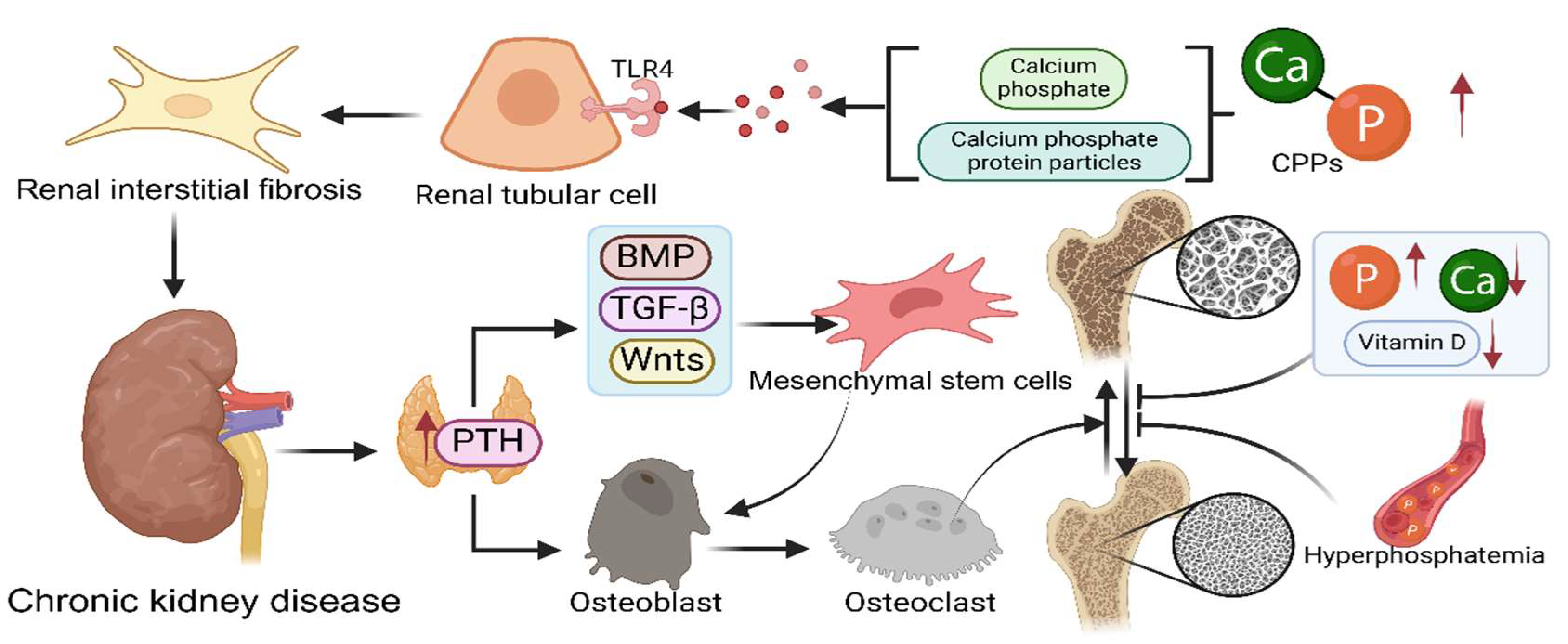
Figure 4.
Effect of vascular calcification in CKD-MBD. Chronic kidney disease patients are often associated with disturbances in mineral metabolism, leading to the conversion of vascular smooth muscle cells to osteoblasts and the formation of vascular calcification, a process that increases the risk of cardiovascular events. Vascular calcification is associated with osteoporosis and mineral quality abnormalities, exacerbating the condition of CKD patients. Hyperphosphatemia promotes the phenotypic shift of vascular smooth muscle cells to osteoblasts, exacerbating vascular calcification. Secondary hyperparathyroidism further promotes vascular calcification by affecting the RANK/RANKL/OPG system through PTH. Vascular calcification is an important predictor of cardiovascular complications in CKD patients and has a significant impact on patient prognosis.
Figure 4.
Effect of vascular calcification in CKD-MBD. Chronic kidney disease patients are often associated with disturbances in mineral metabolism, leading to the conversion of vascular smooth muscle cells to osteoblasts and the formation of vascular calcification, a process that increases the risk of cardiovascular events. Vascular calcification is associated with osteoporosis and mineral quality abnormalities, exacerbating the condition of CKD patients. Hyperphosphatemia promotes the phenotypic shift of vascular smooth muscle cells to osteoblasts, exacerbating vascular calcification. Secondary hyperparathyroidism further promotes vascular calcification by affecting the RANK/RANKL/OPG system through PTH. Vascular calcification is an important predictor of cardiovascular complications in CKD patients and has a significant impact on patient prognosis.
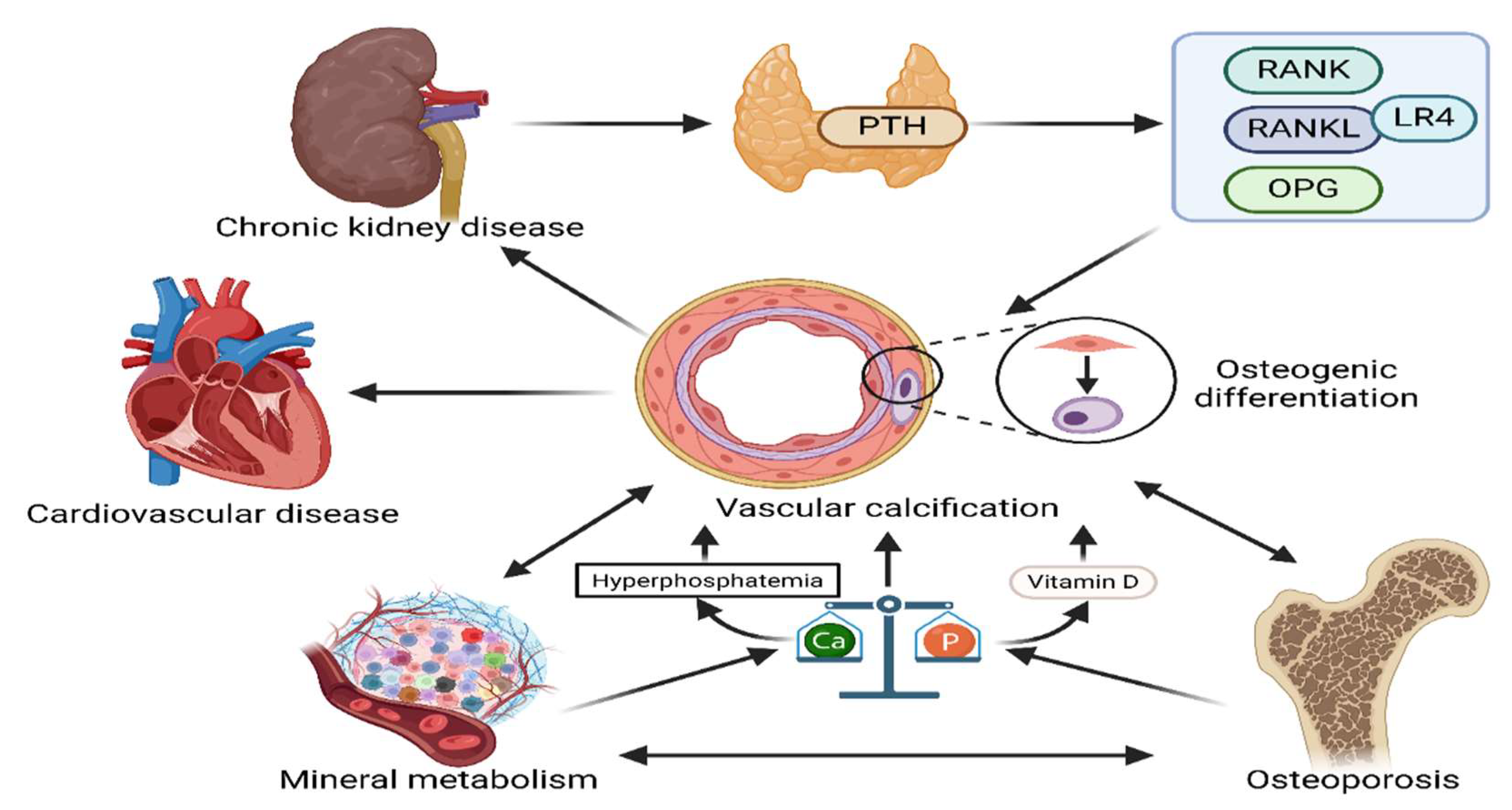
Figure 5.
CKD-MBD clinical management program. CKD-MBD patients are categorized into four major groups clinically based on the pathological features of CKD-MBD, including regulation of calcium-phosphorus metabolic balance, regulation of PTH levels, maintenance of bone health, and prevention and treatment of vascular and soft tissue calcification.
Figure 5.
CKD-MBD clinical management program. CKD-MBD patients are categorized into four major groups clinically based on the pathological features of CKD-MBD, including regulation of calcium-phosphorus metabolic balance, regulation of PTH levels, maintenance of bone health, and prevention and treatment of vascular and soft tissue calcification.
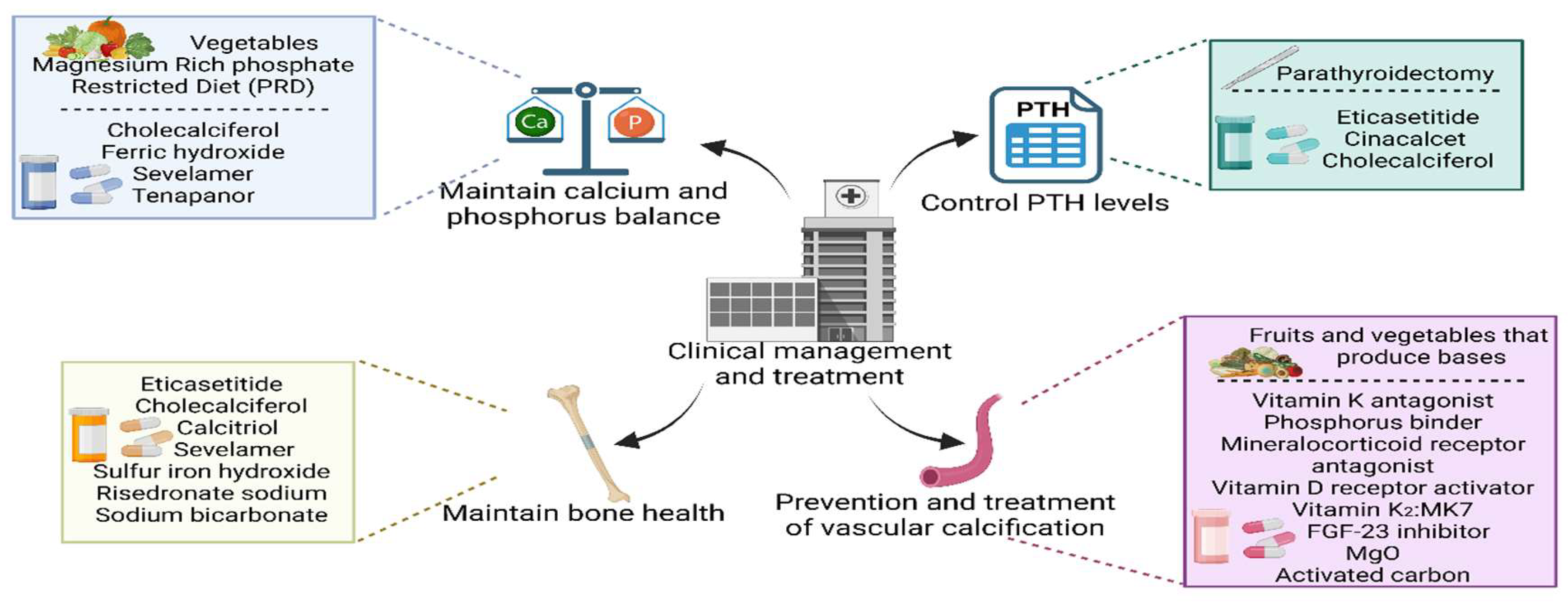
Figure 6.
Precision medicine is an emerging future research direction for CKD-MBD. By integrating transcriptomics, proteomics and metabolomics, more biomarkers can be explored and more valuable biomarkers can be used for drug therapy research to realize precise targeted therapy and bring better prognosis.
Figure 6.
Precision medicine is an emerging future research direction for CKD-MBD. By integrating transcriptomics, proteomics and metabolomics, more biomarkers can be explored and more valuable biomarkers can be used for drug therapy research to realize precise targeted therapy and bring better prognosis.
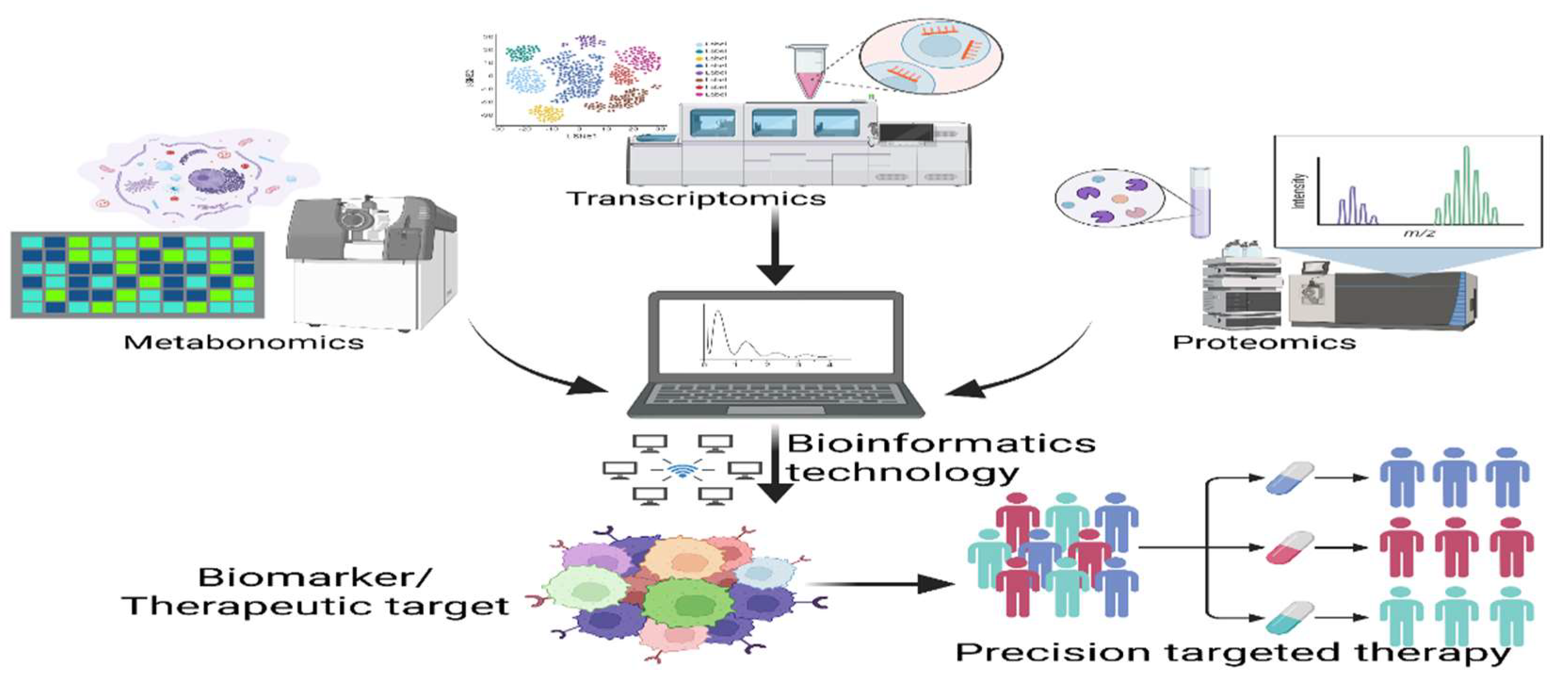

Table 1.
Factors affecting bone strength in chronic kidney disease-mineral and bone disorder (CKD-MBD).
Table 1.
Factors affecting bone strength in chronic kidney disease-mineral and bone disorder (CKD-MBD).
| Factor | Source | Main effect | Refs. |
|---|---|---|---|
| ↓Calcium | Kidney Intestine |
↑SPTH ↑Phosphate excretion |
[12,13] |
| ↑Phosphate | Kidney | ↑SPTH ↓1, 25-dihydroxyvitamin D3 synthesis ↓Calcium |
[9] |
| ↓Vitamin D | Kidney | ↑Parathyroid hormone secretion ↑Phosphate ↓Calcium |
[18,20,21] |
| ↑Parathyroid hormone | Parathyroid gland | ↓Calcium ↑Phosphate ↑FGF23 levels |
[12,28,29] |
| ↑FGF23 | Bone | ↑Phosphate excretion ↓1, 25-dihydroxyvitamin D3 synthesis |
[32,33,34] |
| ↓Klotho | Kidney | ↑FGF23 levels | [29] |
↓: decrease, ↑: increase.
Table 2.
Summary of diagnostic methods for CKD-MBD.
| Styles | Mechanisms | Effects | Refs. |
|---|---|---|---|
| FGF23 | Increases in circulating FGF23 levels combined with decreases in renal klotho expression lead to klotho-independent effects of FGF23 on the heart. | Left ventricular hypertrophy, heart failure, atrial fibrillation and death. | [61] |
| Osteoprotegerin | Inhibits RANKL-RANKL receptor interaction to prevent osteoclast formation and osteoclast bone resorption. | Reduces bone resorption. | [62] |
| Osteocalcin | Participates in the mineralization of bone matrix and is a sensitive indicator of the rate of bone formation. | Negative correlation with lumbar spine bone density in kidney transplant recipients. | [64] |
| Alkaline Phosphatase | Hydrolytic activity of ALP regulates the mineralization inhibitor PPi. | Regulates dysregulation of calcium phosphate metabolism, inflammation, osteogenic gene expression, and transdifferentiation of vascular smooth muscle cells. | [65] |
| Dual-energy X-ray absorptiometry | Areal BMD measurements. | Assess Bone Density. | [66] |
| High-resolution peripheral quantitative computed tomography | Trabecular architecture Volumetric BMD. | Detailed information on bone microstructure. | [66,67] |
| MRI | Cortical porosity Marrow perfusion, and molecular diffusion. | Provides unique advantages in assessing soft tissue and vascular calcifications. | [68] |
| Trabecular Bone Score | Lumbar Spine DXA Images. | Assesses the condition of bone microarchitecture. | [69] |
| Bone biopsy | Observes and analyzes bone tissue samples directly. | Provides detailed information on bone mineralization, bone formation and bone resorption. | [69,70] |
Table 3.
Management and treatment of CKD-MBD.
| Treatments | Ways | Influences | Refs. |
|---|---|---|---|
| Cholecalciferol supplementation | Oral | Increasing serum ferritin-25 levels in hemodialysis patients in the short term and erythropoietin resistance in the long term, reducing whole PTH concentrations, suggesting a significant role in the treatment of SHPT, attenuating mineral density loss in lumbar vertebrae after renal transplantation and eliminating vitamin D deficiency. | [73,83] |
| Phosphate-binding agents iron hydroxide or sevelamer carbonate | Oral | Reducing serum phosphorus levels, but also significantly reducing serum FGF23, rising levels of bone formation markers. | [74] |
| Tenapanor | Oral | Reducing phosphorus levels in serum. | [75] |
| A magnesium-rich Phosphate-restricted diet | Oral | Stabilizing plasma FGF23 and prevent hypercalcemia. | [77] |
| Cinacalcet | Oral | Decreasing PTH secretion to treat SHPT and lowering blood calcium and phosphorus levels without increasing alkaline phosphatase. | [80] |
| Etelcalcetide | Intravenous injection | Reducing parathyroid hormone levels in hemodialysis patients, declining fracture incidence. | [82,84] |
| Risedronate Sodium | Oral | Suppressing Bone Conversion and Increasing BMD in Japanese Patients with Mild to Moderate Chronic Kidney Disease Primary Osteoporosis. | [86] |
| Sodium bicarbonate | Oral or intravenous injection | Increasing serum bicarbonate and decreasing potassium levels, Improving metabolic acidosis and stabilizing estimated glomerular filtration rate. | [87,88] |
| Vitamin D Receptor Activator | Oral or inject | Controlling parathyroid hormone levels and higher serum Klotho. | [91] |
| Vitamin K2 | Oral | Decreasing circulating levels of dephosphorylated uncarboxylated matrix gamma-carboxyglutamate protein and decreasing progression of arterial stiffness in diabetic patients on chronic hemodialysis were shown in patients with end-stage renal disease. | [92,93] |
| Spironolactone | Oral | Improving vascular calcification. | [96] |
| Activated Carbon | Oral | Delaying the onset of hyperphosphatemia in patients with chronic kidney disease and delaying the development of vascular calcification in patients with stage 3-4 CKD. | [97] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



