Submitted:
01 May 2025
Posted:
07 May 2025
You are already at the latest version
Abstract
In bioinformatics, pathway analyses are used to interpret biological data by mapping measured molecules with known pathways to discover their functional processes and relationships. Pathway analysis has become an essential tool for interpreting large-scale omics data, translating complex gene sets into actionable experimental insights. However, issues inherent to pathway databases and misinterpretations of pathway relevance often result in ‘pathway fails,’ where findings, though statistically significant, lack biological applicability. For example, the Tumor Necrosis Factor (TNF) pathway was originally annotated based on its association with observed tumor necrosis, while it is actually a multifunctional biological pathway across diverse physiological processes in the body. This review broadly evaluates pathway analysis interpretation, including embedding-based, semantic similarity-based, and network-based approaches. Each method for interpretation is assessed for its strengths, such as high-quality visualizations and ease of use, as well as its limitations, including data redundancy and database compatibility challenges. Despite advancements in the field, the principle of "garbage in, garbage out" (GIGO) shows that the reliability of any analysis ultimately hinges on high- quality input data. Methodological standardization, scalability improvements, and integration with diverse data sources remain areas for further development. By providing critical guidance with contextual examples such as the TNF pathway, we aim to help researchers align their objectives with the appropriate method. Advancing pathway analysis interpretation will further enhance the utility of pathway analysis, ultimately propelling progress in systems biology and personalized medicine.
Keywords:
omics interpretation
; pathway analysis
; gene ontology
; embeddings
; semantic similarity
Introduction
The development of high throughput multiomics technologies—such as genomics, transcriptomics and proteomics—has revolutionized biological research, enabling the exploration of cellular processes with unprecedented depth and scale [1]. These advances have catalyzed the rise of systems biology, an integrative approach that focuses on the interactions within biological systems rather than isolated molecular components [2]. By combining diverse omics layers, researchers are now driving personalized medicine forward, tailoring treatments to individual biological signatures derived from patient-specific data [3]. In this context, pathway analysis has become an indispensable tool for translating omic datasets into actionable clinical insights by targeting key pathways and mechanisms for further investigation and manipulation [4]. As multiomics data continue to expand, pathway analyses offer a crucial method for gleaning meaningful biological conclusions from raw data, linking molecular changes to functional outcomes across various experimental conditions [5]. This approach grows our understanding of disease pathogenesis and also accelerates the development of targeted therapies [6].
Despite pathway analyses providing instrumental insights into biological functioning, inherent challenges often prevent effective application. Translating these analyses into actionable experimental targets and candidate treatments remains a persistent bottleneck in omics-driven research [7,8]. Further deciphering clinically relevant mechanisms proposed by pathway analysis is difficult given the unpredictable interactions within biological systems [9,10,11,12,13]. The extensive lists of results generated by pathway analysis, while promising, are difficult to curate manually, which also increases the risk of bias during pathway selection [14,15,16]. Moreover, even when pathways may appear promising, validation is frequently hindered due to the variability among experimental models [17,18,19] and the potential mismatch between statistical confidence and biological significance [20,21].
Another key challenge lies in the complexity of new biological data types, such as kinome array data, continually outpacing their capability to be analyzed, leaving researchers without techniques to translate their findings in meaningful ways [22,23]. The diversity of available tools further complicates this process. Varying algorithms and databases may yield inconsistent results which often makes the harmonization of outputs impractical [24], thus researchers carry the responsibility of evaluating the accuracy and practical utility of selected tools to ensure proper integration of multiomics data and sensible outputs [25,26]. An example of this is the correct prioritization and validation of selected pathways for targeted therapies [27]. Misinterpretation or incomplete analysis of pathway data may lead to erroneous biological conclusions, undermining the reliability of personalized treatments [28].
While effective management of research data is a cornerstone of rigorous and reproducible science, it remains fraught with challenges. The prevalence of data-management errors, such as coding mistakes and ambiguous documentation, undermine scientific reliability and have led to numerous article retractions [29]. These errors, which may occur at any stage of the research workflow, waste resources and highlight the vulnerability of current systems to human fallibility [30]. Retractions due to honest mistakes have also caused immense personal stress and professional consequences for researchers, prompting calls to improve data workflows and prevent similar errors in the future [30]. Emerging frameworks like FAIR (Findable, Accessible, Interoperable, Reusable) data principles aim to address these issues by promoting robust, scalable and automatable systems that ensure interoperability, standardization and accessibility of data generated from pathway analyses [31]. By integrating multiomics datasets into unified frameworks, these principles can enhance the efficiency and reliability of pathway analysis at scale [7,32,33]. As pathway analysis tools evolve, their capacity to offer high-confidence outputs that align with real-world applicability is now becoming increasingly important.
Here, we address a critical gap by focusing on the interpretation of pathway analysis results—a novel approach distinct from previous work [34,35,36,37,38]—which primarily examines different methods for performing pathway analyses. In this review, we evaluate the strengths, limitations and practical applicability of various pathway analysis interpretation methods, offering guidelines to optimize their use across diverse research settings. Ultimately, we argue that the future of pathway analysis lies in enhancing automation, scalability and the development of tools that produce interpretable outputs, bridging the gap between computational predictions and experimental validation.
Key Challenges to Pathway Analysis
Pathway Annotation
One of the primary issues in pathway annotation arises from the provenance in how pathways were first identified and named [39]. Pathway names typically reflect the initial experimental conditions in which they were discovered, rather than encompassing their broader roles. A notable example of this is the Tumor Necrosis Factor (TNF) pathway. Despite its name, TNF is not solely a mechanism for tumor necrosis. Early researchers linked TNF with tumor suppression, as necrosis of tumors was observed in vivo under specific pathological experimental conditions [40]. Subsequent studies, however, revealed TNF as a multipotent cytokine involved in numerous physiological processes, including the immune response, inflammation and apoptosis across many different tissues [41]. This represents an example of a domain-specific anchor bias [42], wherein the initial characterization of a pathway becomes a fixed reference point influencing subsequent perspectives. With TNF, the original association with tumor necrosis has anchored its perception, overshadowing its other roles in normal physiology. Surprisingly, the so-called “TNF pathway” also mediates NMDA receptor activity in neurons and glial cells [43]. Such semantic mismatches obscure a pathway’s true biological functions, perpetuating narrow interpretations and hindering a comprehensive understanding of pathways beyond the conditions in which they were originally discovered [44,45].
Interpreting the function of a pathway is also highly context-dependent, requiring careful consideration of experimental design and biological domain. For instance, in cancer research, activation of apoptosis pathways is typically associated with programmed cell death, the mechanism for eliminating cancerous cells [46]. However, in the brain, similar pathway activation likely indicates synaptic pruning or neurite retraction, which are critical for neurodevelopment and synaptic plasticity [47,48,49]. Similarly, inflammation pathways activated during injury or infection might signal tissue damage and repair processes [50,51], whereas in the brain, these pathways may reflect immune activation in response to neuroinflammation or other neural stimuli [52,53]. A notable example of this context dependence is the NF-κB pathway, which has distinct canonical and non-canonical activation mechanisms. The canonical NF-κB pathway is typically associated with acute inflammatory responses and innate immunity [54], while the non-canonical pathway governs processes like lymphoid organ development and adaptive immune signaling [55]. Misinterpreting pathway activation could lead to flawed conclusions, such as conflating an immune response with developmental signaling or vice-versa. Without accounting for the biological context, researchers may draw incorrect conclusions about pathways’ roles, potentially leading to flawed experimental designs or misdirected therapeutic strategies.
Bias, redundancy and overlap in pathway annotation databases also present significant challenges for interpreting pathway enrichment results [53]. Databases like Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, WikiPathways and others commonly describe similar biological functions each using slightly different gene sets or interaction details [56]. Within databases, these pathways may be annotated as multiple distinct terms with close variations in the genes and regulatory mechanisms involved, complicating interpretation. For example, in GO, "cell adhesion" (GO:0007155) is annotated as the attachment of cells to other cells or the extracellular matrix, while "cell-cell adhesion" (GO:0098609) specifies adhesion between cells only. Similarly, "cell migration" (GO:0016477) refers specifically to directed cell movement, while "cell motility" (GO:0048870) includes directed movement and also broader movements. These subtle distinctions result in overlapping pathway enrichment results, hindering prioritization of the most relevant pathways [57,58].
Structural differences between pathway annotation databases compound these challenges, obscuring shared biological function and contributing to inconsistencies in pathway coverage. For example, overlapping gene sets among pathways labeled as “Wnt signaling” in KEGG, Reactome and WikiPathways have significant divergence, with only 73 overlapping genes out of 148, 312, and 135 total genes respectively across these databases despite their focus on the same function [59]. Furthermore, total gene coverage is heavily disparate, with databases like Reactome annotating over 10,000 human genes, while others like WikiPathways cover only about 6,000 [59]. Study biases further exacerbate this problem, as genes frequently studied in predominant fields like cancer research are counterintuitively underrepresented in gene-set annotations [60]. Similarly, GO, originally developed for model organisms, may overemphasize highly conserved cellular processes at the expense of species-specific functions [61]. Despite this stringency, even curated GO annotations have error rates ranging from 18% to 49% [61].
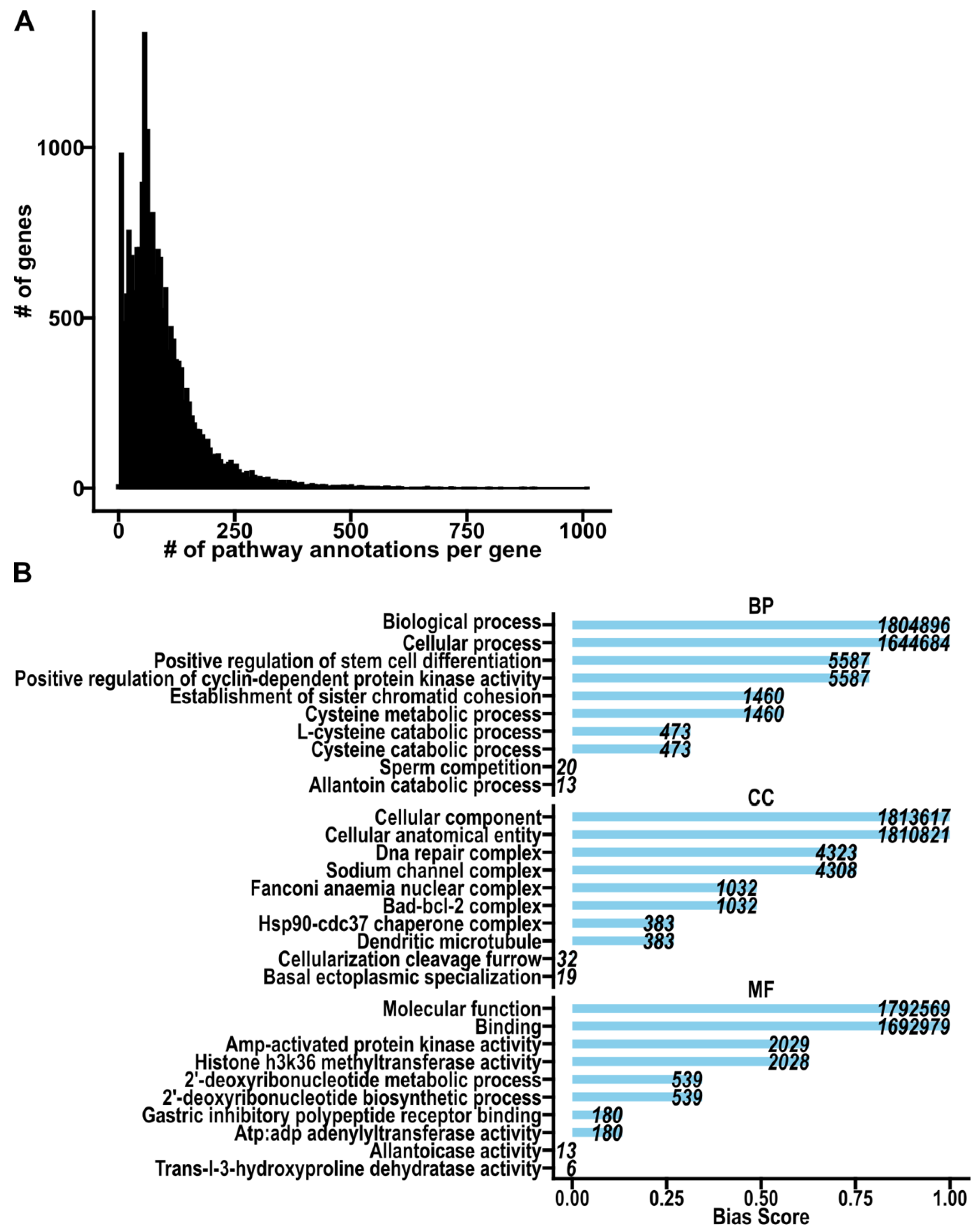
To explore potential biases in pathway annotations, we surveyed gene-sets presently annotated to GO terms, i.e. pathways, which illustrated how these challenges may manifest as patterns of redundancy and bias. We found that certain genes, like transforming growth factor beta 1, are annotated to over 1,000 pathways, while others like chromosome 6 open reading frame 62 are annotated to just 2 pathways (Table 1, Supplemental Table 1). Moreover, a substantial number of genes, including 611 genes coding for protein products lack any known pathway annotations entirely (Table 2). Therefore, if a researcher were to investigate the function of any of these unannotated genes (Supplemental Table 2) using GO, they would be excluded from any pathway analyses. These disparities result in highly skewed gene coverage, where a small subset of genes dominates pathway annotations (Figure 1A). Similarly, a handful of highly annotated pathways disproportionately account for the majority of all known gene-pathway associations (Figure 1B). These patterns highlight the broader challenges of redundancy and bias in practice, which obscures biological significance of underrepresented genes and hinders the prioritization of relevant pathways by researchers. Efforts to mitigate these issues, such as set-theory-based approaches to reduce pathway overlap, have shown promise but require further refinement to ensure comprehensive and equitable representation within pathway databases [62]. Awareness of these domain-specific challenges is essential to improve the clarity and utility of pathway annotation in pathway analysis.

Table 1.
Representative Top 2 Genes Nearest Tukey’s Five-Number Summary of Gene Ontology Annotations (GOALL w/ IEA, Bader Lab, Oct 2024). The complete list is available in the supplement (Table S1).
Table 1.
Representative Top 2 Genes Nearest Tukey’s Five-Number Summary of Gene Ontology Annotations (GOALL w/ IEA, Bader Lab, Oct 2024). The complete list is available in the supplement (Table S1).
| Gene | # of Pathways |
|---|---|
| TGFB1 transforming growth factor beta 1 | 1010 |
| CTNNB1 catenin beta 1 | 894 |
| ACADL acyl-CoA dehydrogenase long chain | 120 |
| ACTBL2 actin beta like 2 | 120 |
| ABCA6 ATP binding cassette subfamily A member 6 | 72 |
| ACKR1 atypical chemokine receptor 1 (Duffy blood group) | 72 |
| ABCF3 ATP binding cassette subfamily F member 3 | 44 |
| ADISSP adipose secreted signaling protein | 44 |
| C6orf62 chromosome 6 open reading frame 62 | 2 |
| CTAGE3P CTAGE family member 3, pseudogene | 2 |
Table 2.
Locus Types of All HGNC Genes with Zero Gene Ontology Annotations (GOALL w/ IEA, Bader Lab, Oct 2024).
Table 2.
Locus Types of All HGNC Genes with Zero Gene Ontology Annotations (GOALL w/ IEA, Bader Lab, Oct 2024).
| Locus Type | Count |
|---|---|
| pseudogene | 13940 |
| RNA, long non-coding | 5640 |
| RNA, micro | 1912 |
| gene with protein product | 611 |
| RNA, transfer | 591 |
| RNA, small nucleolar | 568 |
| immunoglobulin pseudogene | 202 |
| readthrough | 143 |
| RNA, cluster | 119 |
| fragile site | 116 |
| endogenous retrovirus | 92 |
| T cell receptor gene | 67 |
| RNA, ribosomal | 58 |
| immunoglobulin gene | 55 |
| RNA, small nuclear | 51 |
| region | 46 |
| unknown | 46 |
| T cell receptor pseudogene | 38 |
| RNA, misc | 29 |
| virus integration site | 8 |
| complex locus constituent | 6 |
| RNA, vault | 4 |
| RNA, Y | 4 |
HGNC: HUGO Gene Nomenclature Committee.
Table 3.
Overview of pathway analysis interpretation tools and their features. This table lists various tools categorized by their method of analysis (e.g., semantic, network, embedding), detailing their year of release, description, access platform, supported databases and visualization capabilities. This comparison serves as a comprehensive resource for researchers to identify tools best suited for their specific pathway analysis needs.
Table 3.
Overview of pathway analysis interpretation tools and their features. This table lists various tools categorized by their method of analysis (e.g., semantic, network, embedding), detailing their year of release, description, access platform, supported databases and visualization capabilities. This comparison serves as a comprehensive resource for researchers to identify tools best suited for their specific pathway analysis needs.
| Tool | Year | Method | Access | Database | Visualization | Description |
|---|---|---|---|---|---|---|
| REVIGO [91] | 2011 | Semantic | Web | GO | Scatterplots, interactive graph, tree maps | Summarizes GO term lists using semantic similarity and clustering |
| clusterProfiler [94] | 2013 | Semantic | R package | GO, KEGG, DO | Dot plot | Enrichment analysis for GO/KEGG terms and visualization |
| ReCiPa [58] | 2018 | Semantic | R package | KEGG, Reactome | Data tables | Controls redundancy in pathway databases |
| GOGO [93] | 2018 | Semantic | Web, Perl | GO | Data tables | Calculates semantic similarity of GO terms using improved algorithms |
| FunSet [129] | 2019 | Semantic | Web, Standalone | GO | 2D plots | Performs GO enrichment analysis with interactive visualizations |
| GeneSetCluster [130] | 2020 | Semantic | R package | Any | Network graph, dendogram, heatmap | Groups gene-sets post-analysis based on shared genes |
| GOMCL [131] | 2020 | Semantic | Python | GO | Heatmap, Network graph | Clusters GO terms using Markov clustering algorithm |
| GoSemSim [132] | 2020 | Semantic | R package | GO | Data tables | Computes semantic similarity among GO terms for comparison |
| GO-FIGURE! [15] | 2021 | Semantic | Python | GO | Scatterplot | Visualizes GO term similarity with custom scatterplots |
| SimplifyEnrichment [133] | 2022 | Semantic | R package | GO | Heatmap | Clusters with a unique binary cut algorithm. |
| RICHNET [98] | 2019 | Network | R protocol | MSigDB | Network graph | Automated gene-set network creation |
| EnrichmentMap [66] | 2019 | Network | Cytoscape | Any | Interactive network | Detailed enrichment mapping |
| Gscluster [99] | 2019 | Network | Web, R Package | MSigDB | Interactive network | Network-weighted gene-set clustering integrating PPI data |
| aPEAR [101] | 2019 | Network | R package | Any | Network graph | Clustering with automated naming |
| GeneFEAST [100] | 2023 | Network | Web, Python | Any | Heatmap, Dot plot, Upset plot | Highlights multi-enrichment genes |
| vissE [102,103] | 2023 | Network | R package | MSigDB, Any | Network graph | Visualizes higher-order interactions |
| pathlinkR [97] | 2024 | Network | R package | Reactome, MSigDB, InnateDB | Network graph, Volcano plot, Dot plot | Integrated PPI network construction |
| PAVER [110] | 2024 | Embedding | Web, R package | Any | UMAP, Heatmap, Dot plot | Embedding-based clustering with UMAP for clear pathway visualization |
| Mondrian-Map [113] | 2024 | Embedding | Python | WikiPathways | Mondrian Map | Embedding visualizations highlighting pathway interactions and crosstalk |
| GOsummaries [134] | 2015 | Word Cloud | R package | GO | PCA, Boxplot | Visualizes GO analyses as word clouds and overlays results |
| genesetSV [135] | 2023 | Game Theory | Python | KEGG, MSigDB | Scatterplot | Uses Shapley values for ranking and reducing pathway sets |
| Archetype-Discovery [136] | 2024 |
Non-negative matrix factorization (NMF) |
MATLAB | MSigDB, Any | Radar, scatter & boxplot, Heatmap | Uses NMF to derive compact archetypal gene-set patterns and their pathway associations |
Visualizing Pathway Findings
Another critical challenge in pathway analysis is effectively visualizing high-dimensional data, which is essential for interpreting results and communicating findings [10,63]. The complexity of multiomics datasets often makes illustrating informative relationships between data difficult [25]. Without specialized computational expertise, this complexity may hinder an experimental biologist’s ability to derive meaningful conclusions [64]. Visualization tools must therefore balance simplifying data for clarity while retaining necessary detail for accurate interpretation [65]. Moreover, pathway visualization is further complicated by the aforementioned pathway redundancy, leading to visual clutter that obscures key findings. Heatmaps, Uniform Manifold Approximation and Projection (UMAP) plots, and network-based plots attempt to address these challenges by offering more intuitive representations. Notably, these approaches all have limitations, such as artifacts introduced by dimensionality reduction or network overcrowding and sparsity. [66,67]. A lack of standardized and widely accepted visualization practices exacerbates this challenge, leaving researchers with fragmented outputs. Thus, developing more effective and user-friendly visualization strategies remains a key priority for advancing pathway analysis and improving accessibility to a broader research audience.
Limitations to Pathway Analysis Utility
Despite the potential of pathway analysis to generate new insights, it often falls short in providing actionable leads, resulting in cases where results are uninformative. In studies using gene expression data, analyses may overrepresent genes in canonical pathways, such as “immune function,” even when biologically irrelevant to the experimental conditions [68]. For example, in an RNAseq study of postmortem DLPFC tissue exploring gene co-expression networks related to schizophrenia [69], pathways such as eye development was identified within astrocyte modules. Such results lack readily apparent insights into schizophrenia-specific biological mechanisms. Similarly, pathways associated with learning and memory, highlighted in neuron modules, presented challenges as their relevance to schizophrenia risk and clinical state is extremely vague. Such “pathway fails” are not uncommon. For example, the kynurenine pathway’s role in psychiatric conditions such as schizophrenia and major depressive disorder is well characterized [70], however the precise mechanisms by which it contributes to changes in cognitive function remain poorly understood. This lack of clarity complicates the development of targeted therapeutic interventions, exemplifying how even well-studied pathways can yield results that are uninformative for real-world translational applications. These instances highlight a common pitfall in pathway analysis when statistical significance does not necessarily equate to biological relevance [20,21].
However, not all analyses fall short; there are notable examples of “pathway successes” that have led to significant clinical advancements: nearly 2/3rds of recent FDA-approved drugs were shown a priori that their gene or protein targets had a significant phenotypic association with the targeted disease [71]. Anifrolumab, an IFNAR1 antagonist approved for systemic lupus erythematosus (SLE) lacks itself direct association with SLE, however; variants in TYK2—a kinase that physically interacts with IFNAR1—have a pathogenic role acting via the TYK2/JAK pathways in a pathway association study [72,73]. Similarly, PCSK9, a target for hyperlipidemia therapies, was identified through pathway analysis despite lacking direct genetic association with the disease; instead, it was implicated via protein interaction networks whose interacting nodes were enriched for hyperlipidemia-related pathways using pathway analysis [74]. These successes underscore the potential of pathway analysis to yield actionable insights, even as challenges persist in ensuring biological relevance and clinical utility. However, selecting appropriate pathway analysis methods and interpreting results within the appropriate biological context remains crucial to avoid uninformative or misleading conclusions.
Discrepancies in Molecular Biology Mislead Validation
Traditionally, mRNA (i.e., gene) expression levels and protein abundance have been relied on as proxies for biological activity [75,76,77]. While mRNA levels are often used as convenient inferences of protein expression, substantial evidence shows that mRNA and protein abundances are not well-correlated [78,79]. Gene expression levels often do not correlate with protein abundance, and protein abundance does not necessarily reflect functional activity due to factors such as post-translational modifications, protein-protein interactions and subcellular localization [80,81]. These discrepancies present significant challenges in pathway analyses based solely on transcriptomics or proteomics data, as they may not fully reflect functional states of biological pathways. Researchers unaware of these limitations risk drawing incomplete conclusions about cell processing and signaling [82]. To address this challenge, there has been a shift toward functionally informed methods [83,84,85,86], such as phosphoproteomics and kinome reporter phosphopeptide arrays. These methods enable active profiling by directly assessing protein activity to provide a more functional understanding of signaling networks [87]. By capturing the dynamic nature of biological systems, these approaches mitigate the inherent pitfalls of traditional omics-driven pathway predictions and enhance the overall reliability of pathway analyses [88].
Methods for Pathway Analysis Interpretation
Semantic Similarity Based Methods
Semantic similarity-based methods are a foundational approach in pathway analysis interpretation, designed to quantify the functional relationships among complex pathway annotation terms. For example, semantic similarity-based methods are often applied to interpret the extensive and often redundant lists of GO terms [89]. These methods leverage the hierarchical structure of GO, where terms are organized into parent-child relationships that span from general biological processes to gradually more specific functions [90]. By calculating a “semantic similarity” score between terms, these methods identify and group related GO terms [91], reducing redundancy and enabling researchers to focus on overarching biological themes.
The calculation of semantic similarity typically involves information content metrics based on shared ancestors within the GO hierarchy, reflecting the functional overlap between terms [92]. For instance, terms that share a highly specific ancestor within the GO tree yield higher similarity scores due to their closer functional relationship. Popular metrics include Resnik's and Lin’s similarity measures, which quantify similarity by evaluating the specificity of shared ancestors and their positions within the GO structure [93]. Tools like clusterProfiler implement these methods by grouping semantically similar GO terms into clusters. Representative terms are selected based on similarity scores and user-defined thresholds, with results visualized through scatter plots or dot plots for clarity [94]. This grouping provides a summarized and non-redundant list of terms, making interpretation of large pathway results more feasible.
However, these methods are inherently tied to the GO framework and can fail to generalize to other pathway databases like KEGG, Reactome or WikiPathways [90,95]. Additionally, some studies suggest that these similarity scores may not fully capture the nuanced meanings of GO terms, highlighting an area for further refinement [96]. The combination of functional clustering and visualization described offers a powerful means of simplifying GO term analysis, but ongoing developments are needed to broaden applicability and enhance interpretive precision.
Network Based Methods
Network-based methods provide a powerful approach for pathway analysis by visualizing pathways as interconnected networks, where pathways are represented as nodes and edges denote shared genes or similar functional annotations [97]. These methods rely on graph theory principles to quantify relationships between pathways based on shared content, using metrics like gene overlap, the Jaccard index or semantic similarity [98]. This network structure captures relationships between pathways, identifies clusters of related pathways, and reveals broad biological themes that may be obscured in list-based approaches.
Clustering algorithms, such as modularity-based or hierarchical clustering, group these pathways into distinct modules based on their connectivity, reducing redundancy and highlighting cohesive functional groups [99]. In pathway networks, nodes with above-average connections, or “hubs,” represent key biological functions that interact in communities of multiple pathways, potentially indicating regulatory roles [100]. Visualization techniques position these pathways close together, with edge weights reflecting the strength of their relationships, thereby emphasizing direct and indirect associations across broader biological processes [101].
Tools like EnrichmentMap enhance these analyses by constructing pathway networks based on gene overlap, using significance thresholds to reduce visual complexity and emphasize biologically relevant connections [66]. These clustering and visualization approaches allow researchers to interpret enrichment results into visually accessible maps, highlighting major biological processes and their relationships. Similarly, Visualization of Set Enrichment (vissE) extends the utility of network-based methods by integrating data across modalities, including single-cell and spatial transcriptomics [102,103]. vissE clusters pathways based on their content similarity and links them to specific cellular phenotypes, providing insights into pathway interactions within complex biological contexts.
Together, these approaches offer researchers a dynamic, systems-level framework for interpreting pathway data by revealing functional relationships at the network level, clustering pathways into interpretable communities, and accordingly simplifying large datasets for actionable insights.
Embedding Based Methods
Embedding-based methods offer a cutting-edge approach to pathway analysis by transforming biological entities, like pathways, into high-dimensional vectors, known as embeddings [104,105]. These embeddings numerically encode the semantic meaning of pathways, capturing complex relationships among genes and pathways within the context of large biomedical datasets. Originating in natural language processing, embeddings are widely used to represent words in hundreds of numerical dimensions, allowing models to mathematically quantify relationships between concepts (e.g., "Proteome - Protein + Kinases = Kinome") [106]. In biomedical research, this technique has become instrumental for clustering, visualization and predictive modeling [107].
In pathway analysis interpretation, embedding-based methods reduce redundancy and enhance interpretability by grouping pathways based on their semantic similarity [96]. Tools like Pathway Analysis Visualization with Embedding Representations (PAVER) leverage cosine similarity between pathway embeddings to cluster similar pathways and identify a “most representative term” (MRT) for each group based on the average embedding [108,109,110]. This results in streamlined and summarized output, which is especially useful for managing large datasets. PAVER also employs dimensionality reduction techniques, such as UMAP, to convert high-dimensional embeddings into two-dimensional visualizations. These layouts visually group related pathways while preserving semantic relationships, enabling researchers to more easily identify biological themes and generate publication-ready visualizations [111,112].
Another innovative tool, MondrianMap, draws inspiration from abstract art to spatially arrange pathways on a grid, where proximity reflects functional similarity, and color indicates regulatory states, such as upregulation or downregulation [113]. This intuitive visualization method facilitates the exploration of pathway relationships, revealing functional clusters and crosstalk patterns across complex datasets. By going beyond traditional node-edge diagrams or heatmaps, MondrianMap provides an interactive and visually accessible representation of pathway data.
While embedding-based methods are promising, they are limited by the information encoded in pathway descriptions or hierarchical structures [111]. These pre-trained language models can capture broad semantic relationships, but they may lack context-specific details [96]. As these models evolve, embedding-based approaches are likely to become even more sophisticated and scalable for interpreting pathway analysis results across diverse datasets.
Applications of Tools for Pathway Interpretation
The practical utility of exemplar pathway analysis tools can be best appreciated through real-world case studies that illustrate the impact of their specific feature set on advancing biological research.
clusterProfiler: clusterProfiler is widely used as a standard for pathway enrichment analysis and visualization in many fields. Chen et al. employed clusterProfiler to identify differentially expressed genes in acute myocardial infarction, identifying key enrichment of cytokine-cytokine receptor interaction and TNF signaling pathways via GO and KEGG pathway analyses [114]. Jia et al. applied clusterProfiler to distinguish unique gene expression profiles between luminal A and basal-like subtypes of breast cancer, identifying pathways involved in subtype-specific progression and novel therapeutic targets, such as neuromedin U receptor 1, neural cell adhesion molecule 1 and STIL centriolar assembly protein [115]. In neurodegenerative research, Niu et al. utilized clusterProfiler to study differentially expressed genes in varying stages of Alzheimer's disease, uncovering the role of mitochondrial components and proteasome subunits in disease progression [116]. Additionally, Gamazon et al. employed clusterProfiler in a multi-tissue transcriptome study to link gene expression with neuropsychiatric traits, demonstrating its utility in mapping complex genetic influences across brain and non-brain tissues [117]. clusterProfiler's semantic similarity-based method was particularly valued in these studies for its simplicity, ease of use, and capability to effectively identify key pathways in diverse datasets with minimal computational overhead. However, despite its strengths, the semantic similarity-based method of clusterProfiler may seemingly fail to be effective with the inherent bias and redundancy in pathway annotation databases used like GO or KEGG, potentially leading to the overrepresentation of certain pathways while underrepresenting others with biological relevance. Additionally, the tool's reliance on predefined pathway databases limits its applicability in contexts where novel, poorly annotated, or species-specific pathways are of interest, potentially overlooking critical insights in less-studied biological systems.
vissE: vissE excels in handling complex multiomics data, enabling researchers to identify pathway relationships within diverse contexts at the network level. Kulasinghe et al. utilized vissE to analyze transcriptomic profiles of cardiac tissues from patients who succumbed to SARS-CoV-2. Visualization of enriched pathways related to DNA damage and immune responses was able to pinpoint the molecular impact of COVID-19 on cardiac health [118]. In immune research, Dalit et al. applied vissE to map divergent cytokine and transcriptional signatures across T follicular helper cell populations, revealing how different signaling environments guide immune cell heterogeneity and B cell output during various pathogen exposures [119]. In colorectal cancer, Lee et al. leveraged vissE to explore serotonin-mediated signaling pathways, identifying key pathway interactions linked to tumor growth suppression through ERK signaling [120]. In these studies, vissE's network-based approach facilitated the identification of pathway-pathway relationships and communities, allowing for a deeper understanding of the complex interactions they observed. However, despite its ability to explain these higher-order phenotypic patterns, vissE’s reliance on network-based visualization can become unwieldy when dealing with highly interconnected or very large datasets, potentially leading to information overload and obscured insights for researchers lacking advanced computational expertise. Moreover, the tool's dependence on comprehensive input data from multiomics experiments may amplify the impact of incomplete datasets or biases within them, thereby influencing the reliability and interpretability of the visualized pathways in these specific research contexts.
PAVER: PAVER’s embedding-based method, coupled with UMAP visualizations, has proven effective in simplifying complex datasets and revealing critical biological insights. In the brain, Nguyen et al. used PAVER to analyze transcriptomic and kinomic data from mice exposed to pyrethroid pesticides during development, uncovering disruptions in pathways related to MAP kinase signaling and circadian rhythms that may underlie neurodevelopmental disorders [121]. Similarly, Curtis et al. used PAVER to integrate metabolomic and transcriptomic data from the brains of male mice developmentally exposed to deltamethrin, effectively visualizing pathway clusters related to folate biosynthesis, dopamine synapses, and MAPK signaling to highlight the multi-modal impact of environmental exposure on adult brain metabolism [122]. O'Donovan et al. characterized transcriptional changes in the orbitofrontal cortex across psychiatric disorders such as schizophrenia and bipolar disorder, identifying immune-related pathways and sex-specific gene expression patterns that distinguished diagnoses and contributed to understanding disease mechanisms [123]. In toxicology research, Hu et al. applied PAVER to interpret kidney transcriptomics in mice exposed to microcystin-LR, demonstrating its use in identifying pathways modulated by probiotic treatment, which offered protective effects against toxin-induced damage [124]. In these studies, PAVER simplified interpretation by visually identifying clusters of similar pathways and highlighting functional groups in their multiomic datasets. However, PAVER necessitates the use of pre-computed embeddings, which may limit flexibility in real-time analyses or exploration of novel datasets. Additionally, the tool’s effectiveness is heavily dependent on the quality and diversity of the input datasets, which may restrict utility in studies where such data are incomplete or unbalanced, potentially impacting the robustness of its pathway clustering and functional interpretations.
Collectively, these tools highlight how different pathway analysis interpretation methods cater to distinct research needs. By choosing the right tool for generating interpretations and deliverables of pathway analysis results, researchers may maximize the impact of their pathway analyses on the understanding of complex biological systems they study.
Choosing the Right Tool for Your Research
Choosing the most appropriate pathway analysis method depends on aligning one’s research goals with the specific strengths and limitations of each approach available. Semantic similarity-based, network-based and embedding-based methods (Table 2) qualitatively compared below may guide researchers in selecting the method that best fits their study’s objectives.
Visualization quality and usability differ across methods. Semantic similarity-based methods, as seen with clusterProfiler, provide straightforward and accessible output of data, such as via bar charts, for researchers seeking simple visualization. Network-based methods, such as vissE, offer interactive and detailed visualizations that map complex relationships between pathways, ideal for the exploration of pathway interrelations at a deeper level. Embedding-based approaches, such as those employed by PAVER, excel in generating high-quality visualizations to simplify pathway clusters for clearer interpretation.
Ease of use varies as well. Semantic similarity-based methods offer simple workflows, appealing to experimental biologists who are new to bioinformatics. In contrast, network-based methods can demand more technical expertise, appealing to researchers who are comfortable navigating intricate visualizations and data relationships. Embedding-based methods generally automate visualization, requiring minimal input, which is beneficial for quick insights and user-friendly experiences.
Effectiveness in handling redundancy and integrating multiomics data also sets these methods apart. Semantic similarity-based methods, while excellent at reducing GO term redundancy, are more limited when integrating non-GO pathway data. Network-based approaches stand out in integrating multiomics data by mapping interconnected pathways and revealing functional interactions across different biological layers. Embedding-based methods reduce redundancy effectively by clustering similar pathways, facilitating interpretation of high-dimensional datasets.
Computational efficiency and scalability are further practical considerations. Semantic similarity-based methods are lightweight and run efficiently for smaller-scale analyses. Network-based methods, however, can be more computationally demanding, especially when visualizing extensive networks or handling dense data. Embedding-based methods are often efficient and scalable, making them suitable for large-scale studies involving dimensionality reduction.
Accessibility also varies across methods. Semantic similarity-based methods are widely accessible as R packages or command-line tools with strong community support. Network-based methods might require specific software installations but are supported by active, albeit more niche, user communities. Embedding-based tools are often web-based with minimal setup requirements.
In summary, semantic similarity-based methods are most appropriate for GO-focused studies requiring straightforward analysis. Network-based methods are best suited for complex analyses that need detailed mapping of pathway interactions. Embedding-based methods are ideal for those seeking quick, visually intuitive summaries and data-driven redundancy reduction. By understanding the unique features of each method, researchers can better align their pathway analysis strategies with their study goals. This thoughtful approach provides a guide for future advancements and adaptations in pathway analysis interpretation as the field continues to evolve.
Conclusions & Future Directions
This review highlights the strengths and limitations of the three main pathway analysis interpretation methods: semantic similarity-based, network-based and embedding-based approaches. Each method offers unique advantages, from the straightforward, GO-focused analyses provided by semantic similarity-based techniques to the detailed interaction maps facilitated by network-based methods, and the high-quality, visually intuitive outputs of embedding-based methods. However, no single approach is without limitations. Challenges remain in integrating diverse data types, minimizing redundancy, and ensuring compatibility across pathway databases. Recognizing these strengths and limitations helps researchers select the most appropriate method for their specific objectives and experimental contexts.
We have highlighted areas of improvement for pathway analysis interpretation tools. Ultimately, the principle of "garbage in, garbage out" (GIGO) remains paramount. High-quality, standardized datasets are essential for obtaining reliable results and maximizing the value of any analysis tool [125]. Poor input data or inconsistent standards can significantly limit the effectiveness of these methods and lead to erroneous conclusions, highlighting the need for rigorous data curation and adherence to open data standards. Future developments should focus on enhancing scalability to accommodate increasingly large datasets and ensuring compatibility with so-called non-model model organisms. The influence of pathway size and database choice on enrichment outcomes must be carefully considered, as this may impact the interpretation of results [126]. Furthermore, consistent standards for functional enrichment analysis, such as proper p-value corrections and background gene list selection, are necessary to ensure reliable findings [127,128].
Looking ahead, researchers are encouraged to adopt tools that streamline and automate pathway analysis, thus enhancing reproducibility and scalability. These advancements are particularly vital as multiomics data grows in complexity and requires more accurate interpretations. Continued innovation is essential to bridge the gap between computational predictions and experimental validation, driving deeper insights and supporting the advancement of systems biology and precision medicine. As the field evolves, the development of more adaptable, comprehensive, and user-friendly pathway analysis tools will empower researchers to fully leverage omics data, uncover complex biological relationships, and inform therapeutic strategies. By addressing current challenges and promoting methodological rigor, the pathway analysis field will continue to be a robust and indispensable component of modern biological research.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
This work was supported by NIH grant 1T32GM144873-01, R01MH107487, R01MH121102, R01AG057598, and R01AG083628.
Glossary
Pathway Analysis: A method for identifying biological pathways enriched with differentially expressed genes or proteins in datasets.
Dimensionality Reduction: Techniques used to visualize high-dimensional data in simpler forms for clearer pathway analysis.
Pathway Redundancy: The occurrence of overlapping or repeated pathways in analysis, which can complicate interpretation and reduce clarity.
Embedding-Based Methods: Computational approaches that represent biological pathways as high-dimensional numerical vectors for analysis.
Semantic Similarity: A metric that quantifies the functional similarity between different biological terms or pathways.
Network-Based Analysis: A method that visualizes relationships between pathways as interconnected networks, highlighting shared functions or genes.
References
- Manzoni, C., et al., Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Briefings in Bioinformatics, 2016. 19(2): p. 286-302. [CrossRef]
- Veenstra, T.D., Omics in Systems Biology: Current Progress and Future Outlook. Proteomics, 2021. 21(3-4): p. e2000235. [CrossRef]
- Herr, T.M., et al., A conceptual model for translating omic data into clinical action. Journal of Pathology Informatics, 2015. 6(1): p. 46. [CrossRef]
- García-Campos, M.A., J. Espinal-Enríquez, and E. Hernández-Lemus, Pathway Analysis: State of the Art. Front Physiol, 2015. 6: p. 383. [CrossRef]
- Wegman-Points, L., et al., Subcellular partitioning of protein kinase activity revealed by functional kinome profiling. Scientific Reports, 2022. 12(1): p. 17300. [CrossRef]
- Ramanan, V.K., et al., Pathway analysis of genomic data: concepts, methods, and prospects for future development. Trends Genet, 2012. 28(7): p. 323-32. [CrossRef]
- Krassowski, M., et al., State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front Genet, 2020. 11(610798): p. 610798. [CrossRef]
- Sboner, A., et al., The real cost of sequencing: higher than you think! Genome Biol, 2011. 12(8): p. 125. [CrossRef]
- D'Adamo, G.L., J.T. Widdop, and E.M. Giles, The future is now? Clinical and translational aspects of "Omics" technologies. Immunol Cell Biol, 2021. 99(2): p. 168-176. [CrossRef]
- Denecker, T. and G. Lelandais, Omics Analyses: How to Navigate Through a Constant DataData Deluge, in Yeast Functional Genomics: Methods and Protocols, F. Devaux, Editor. 2022, Springer US: New York, NY. p. 457-471.
- Bell, G., T. Hey, and A. Szalay, Computer science. Beyond the data deluge. Science, 2009. 323(5919): p. 1297-8. [CrossRef]
- Stead, W.W., et al., Biomedical informatics: changing what physicians need to know and how they learn. Acad Med, 2011. 86(4): p. 429-34. [CrossRef]
- Pita-Juárez, Y., et al., The Pathway Coexpression Network: Revealing pathway relationships. PLOS Computational Biology, 2018. 14(3): p. e1006042. [CrossRef]
- Chicco, D. and G. Agapito, Nine quick tips for pathway enrichment analysis. PLoS Comput Biol, 2022. 18(8): p. e1010348. [CrossRef]
- Reijnders, M.J. and R.M. Waterhouse, Summary visualizations of gene ontology terms with GO-Figure! Frontiers in Bioinformatics, 2021. 1: p. 6. [CrossRef]
- Yu, C., et al., A strategy for evaluating pathway analysis methods. BMC Bioinformatics, 2017. 18(1): p. 453. [CrossRef]
- Searson, P.C., The Cancer Moonshot, the role of in vitro models, model accuracy, and the need for validation. Nature Nanotechnology, 2023. 18(10): p. 1121-1123. [CrossRef]
- Durinikova, E., K. Buzo, and S. Arena, Preclinical models as patients’ avatars for precision medicine in colorectal cancer: past and future challenges. Journal of Experimental & Clinical Cancer Research, 2021. 40(1): p. 185. [CrossRef]
- Diaz-Uriarte, R., et al., Ten quick tips for biomarker discovery and validation analyses using machine learning. PLOS Computational Biology, 2022. 18(8): p. e1010357. [CrossRef]
- Grabowski, T., et al. Between Biological Relevancy and Statistical Significance - Step for Assessment Harmonization. 2021.
- Committee, E.S., et al., Guidance on the assessment of the biological relevance of data in scientific assessments. EFSA Journal, 2017. 15(8): p. e04970. [CrossRef]
- Perez-Riverol, Y., et al., Quantifying the impact of public omics data. Nat Commun, 2019. 10(1): p. 3512. [CrossRef]
- Misra, B.B., et al., Integrated omics: tools, advances and future approaches. Journal of Molecular Endocrinology, 2019. 62(1): p. R21-R45. [CrossRef]
- Domingo-Fernández, D., et al., PathMe: merging and exploring mechanistic pathway knowledge. BMC Bioinformatics, 2019. 20(1): p. 243. [CrossRef]
- Wieder, C., et al., PathIntegrate: Multivariate modelling approaches for pathway-based multi-omics data integration. PLOS Computational Biology, 2024. 20(3): p. e1011814. [CrossRef]
- Canzler, S. and J. Hackermüller, multiGSEA: a GSEA-based pathway enrichment analysis for multi-omics data. BMC Bioinformatics, 2020. 21(1): p. 561. [CrossRef]
- Ivanisevic, T. and R.N. Sewduth, Multi-Omics Integration for the Design of Novel Therapies and the Identification of Novel Biomarkers. Proteomes, 2023. 11(4): p. 34. [CrossRef]
- Mohr, A.E., et al., Navigating Challenges and Opportunities in Multi-Omics Integration for Personalized Healthcare. Biomedicines, 2024. 12(7): p. 1496. [CrossRef]
- Conroy, G., Retractions caused by honest mistakes are extremely stressful, say researchers. Nature, 2025. [CrossRef]
- Kovacs, M., et al., Opening the black box of article retractions: exploring the causes and consequences of data management errors. R Soc Open Sci, 2024. 11(12): p. 240844. [CrossRef]
- Wilkinson, M.D., et al., The FAIR Guiding Principles for scientific data management and stewardship. Sci Data, 2016. 3: p. 160018. [CrossRef]
- Doniparthi, G., T. Mühlhaus, and S. Deßloch, Integrating FAIR Experimental Metadata for Multi-omics Data Analysis. Datenbank-Spektrum, 2024. 24(2): p. 107-115. [CrossRef]
- Jan, M., et al., A multi-omics digital research object for the genetics of sleep regulation. Scientific Data, 2019. 6(1): p. 258. [CrossRef]
- Khatri, P., M. Sirota, and A.J. Butte, Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol, 2012. 8(2): p. e1002375. [CrossRef]
- Nguyen, T.-M., et al., Identifying significantly impacted pathways: a comprehensive review and assessment. Genome Biology, 2019. 20(1): p. 203. [CrossRef]
- Nam, D. and S.Y. Kim, Gene-set approach for expression pattern analysis. Brief Bioinform, 2008. 9(3): p. 189-97. [CrossRef]
- Maghsoudi, Z., et al., A comprehensive survey of the approaches for pathway analysis using multi-omics data integration. Briefings in Bioinformatics, 2022. 23(6). [CrossRef]
- García-Campos, M.A., J. Espinal-Enríquez, and E. Hernández-Lemus, Pathway Analysis: State of the Art. Frontiers in Physiology, 2015. 6. [CrossRef]
- Winston, J.E., Twenty-First Century Biological Nomenclature—The Enduring Power of Names. Integrative and Comparative Biology, 2018. 58(6): p. 1122-1131. [CrossRef]
- Vassalli, P., The pathophysiology of tumor necrosis factors. Annu Rev Immunol, 1992. 10: p. 411-52. [CrossRef]
- Webster, J.D. and D. Vucic, The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front Cell Dev Biol, 2020. 8: p. 365. [CrossRef]
- Wang, D., et al., Designing Theory-Driven User-Centric Explainable AI, in Proceedings of the 2019 CHI Conference on Human Factors in Computing Systems. 2019, Association for Computing Machinery: Glasgow, Scotland Uk. p. Paper 601.
- Jara, J.H., et al., Tumor necrosis factor alpha stimulates NMDA receptor activity in mouse cortical neurons resulting in ERK-dependent death. J Neurochem, 2007. 100(5): p. 1407-20. [CrossRef]
- Sebastian-Leon, P., et al., Understanding disease mechanisms with models of signaling pathway activities. BMC Systems Biology, 2014. 8(1): p. 121. [CrossRef]
- Lee, J., et al., Prioritizing biological pathways by recognizing context in time-series gene expression data. BMC Bioinformatics, 2016. 17(17): p. 477. [CrossRef]
- Sjöström, J. and J. Bergh, How apoptosis is regulated, and what goes wrong in cancer. BMJ, 2001. 322(7301): p. 1538-1539. [CrossRef]
- Nguyen, T.T.M., G. Gillet, and N. Popgeorgiev, Caspases in the Developing Central Nervous System: Apoptosis and Beyond. Frontiers in Cell and Developmental Biology, 2021. 9. [CrossRef]
- Ryu, J.R., et al., Control of adult neurogenesis by programmed cell death in the mammalian brain. Molecular Brain, 2016. 9(1): p. 43. [CrossRef]
- Anosike, N.L., et al., Necroptosis in the developing brain: role in neurodevelopmental disorders. Metabolic Brain Disease, 2023. 38(3): p. 831-837. [CrossRef]
- Saini, S., P. Kakati, and K. Singh, Role of Inflammation in Tissue Regeneration and Repair, in Inflammation Resolution and Chronic Diseases, A. Tripathi, et al., Editors. 2024, Springer Nature Singapore: Singapore. p. 103-127.
- Choi, B., C. Lee, and J.-W. Yu, Distinctive role of inflammation in tissue repair and regeneration. Archives of Pharmacal Research, 2023. 46(2): p. 78-89. [CrossRef]
- Wyss-Coray, T. and L. Mucke, Inflammation in neurodegenerative disease--a double-edged sword. Neuron, 2002. 35(3): p. 419-32. [CrossRef]
- Gasque, P., et al., Roles of the complement system in human neurodegenerative disorders: pro-inflammatory and tissue remodeling activities. Mol Neurobiol, 2002. 25(1): p. 1-17. [CrossRef]
- Shih, R.-H., C.-Y. Wang, and C.-M. Yang, NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Frontiers in Molecular Neuroscience, 2015. 8. [CrossRef]
- Sun, S.-C., The non-canonical NF-κB pathway in immunity and inflammation. Nature Reviews Immunology, 2017. 17(9): p. 545-558. [CrossRef]
- Adriaens, M.E., et al., The public road to high-quality curated biological pathways. Drug Discovery Today, 2008. 13(19): p. 856-862. [CrossRef]
- Shin, M.-G. and A.R. Pico, Using published pathway figures in enrichment analysis and machine learning. BMC Genomics, 2023. 24(1): p. 713. [CrossRef]
- Vivar, J.C., et al., Redundancy control in pathway databases (ReCiPa): an application for improving gene-set enrichment analysis in Omics studies and "Big data" biology. Omics, 2013. 17(8): p. 414-22. [CrossRef]
- Pastrello, C., Y. Niu, and I. Jurisica, Pathway Enrichment Analysis of Microarray Data. Methods Mol Biol, 2022. 2401: p. 147-159.
- Gable, A.L., et al., Systematic assessment of pathway databases, based on a diverse collection of user-submitted experiments. Briefings in Bioinformatics, 2022. 23(5). [CrossRef]
- Maertens, A., et al., Functionally Enigmatic Genes in Cancer: Using TCGA Data to Map the Limitations of Annotations. Sci Rep, 2020. 10(1): p. 4106. [CrossRef]
- Stoney, R.A., et al., Using set theory to reduce redundancy in pathway sets. BMC Bioinformatics, 2018. 19(1): p. 386. [CrossRef]
- Krassowski, M., et al., State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front Genet, 2020. 11: p. 610798. [CrossRef]
- Hanspers, K., et al., Ten simple rules for creating reusable pathway models for computational analysis and visualization. PLOS Computational Biology, 2021. 17(8): p. e1009226. [CrossRef]
- He, C., et al., Interactive visual facets to support fluid exploratory search. Journal of Visualization, 2023. 26(1): p. 211-230. [CrossRef]
- Reimand, J., et al., Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nature Protocols, 2019. 14(2): p. 482-517. [CrossRef]
- Ovchinnikova, S. and S. Anders, Exploring dimension-reduced embeddings with Sleepwalk. Genome Res, 2020. 30(5): p. 749-756. [CrossRef]
- Li, Y., et al., Exaggerated false positives by popular differential expression methods when analyzing human population samples. Genome Biology, 2022. 23(1): p. 79. [CrossRef]
- Radulescu, E., et al., Identification and prioritization of gene sets associated with schizophrenia risk by co-expression network analysis in human brain. Mol Psychiatry, 2020. 25(4): p. 791-804. [CrossRef]
- Sapienza, J., et al., Importance of the dysregulation of the kynurenine pathway on cognition in schizophrenia: a systematic review of clinical studies. Eur Arch Psychiatry Clin Neurosci, 2023. 273(6): p. 1317-1328. [CrossRef]
- Rusina, P.V., et al., Genetic support for FDA-approved drugs over the past decade. Nat Rev Drug Discov, 2023. 22(11): p. 864. [CrossRef]
- Ochoa, D., et al., Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs. Nat Rev Drug Discov, 2022. 21(8): p. 551. [CrossRef]
- Diogo, D., et al., TYK2 protein-coding variants protect against rheumatoid arthritis and autoimmunity, with no evidence of major pleiotropic effects on non-autoimmune complex traits. PLoS One, 2015. 10(4): p. e0122271. [CrossRef]
- MacNamara, A., et al., Network and pathway expansion of genetic disease associations identifies successful drug targets. Sci Rep, 2020. 10(1): p. 20970. [CrossRef]
- de la Fuente van Bentem, S., et al., Towards functional phosphoproteomics by mapping differential phosphorylation events in signaling networks. PROTEOMICS, 2008. 8(21): p. 4453-4465. [CrossRef]
- Ponomarenko, E.A., et al., Workability of mRNA Sequencing for Predicting Protein Abundance. Genes, 2023. 14(11): p. 2065. [CrossRef]
- Prabahar, A., et al., Unraveling the complex relationship between mRNA and protein abundances: a machine learning-based approach for imputing protein levels from RNA-seq data. NAR Genomics and Bioinformatics, 2024. 6(1). [CrossRef]
- de Sousa Abreu, R., et al., Global signatures of protein and mRNA expression levels. Molecular BioSystems, 2009. 5(12): p. 1512-1526. [CrossRef]
- Upadhya, S.R. and C.J. Ryan, Experimental reproducibility limits the correlation between mRNA and protein abundances in tumor proteomic profiles. Cell Rep Methods, 2022. 2(9): p. 100288. [CrossRef]
- Arshad, O.A., et al., An Integrative Analysis of Tumor Proteomic and Phosphoproteomic Profiles to Examine the Relationships Between Kinase Activity and Phosphorylation*. Molecular & Cellular Proteomics, 2019. 18(8, Supplement 1): p. S26-S36. [CrossRef]
- Liu, Y., A. Beyer, and R. Aebersold, On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell, 2016. 165(3): p. 535-550. [CrossRef]
- Handly, L.N., J. Yao, and R. Wollman, Signal Transduction at the Single-Cell Level: Approaches to Study the Dynamic Nature of Signaling Networks. J Mol Biol, 2016. 428(19): p. 3669-82. [CrossRef]
- Creeden, J.F., et al., Kinome Array Profiling of Patient-Derived Pancreatic Ductal Adenocarcinoma Identifies Differentially Active Protein Tyrosine Kinases. Int J Mol Sci, 2020. 21(22). [CrossRef]
- Litichevskiy, L., et al., A Library of Phosphoproteomic and Chromatin Signatures for Characterizing Cellular Responses to Drug Perturbations. Cell Syst, 2018. 6(4): p. 424-443.e7. [CrossRef]
- Reinecke, M., et al., Kinobeads: A Chemical Proteomic Approach for Kinase Inhibitor Selectivity Profiling and Target Discovery, in Target Discovery and Validation. 2019. p. 97-130.
- Patricelli, M.P., et al., In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol, 2011. 18(6): p. 699-710. [CrossRef]
- Alganem, K., et al., The active kinome: The modern view of how active protein kinase networks fit in biological research. Curr Opin Pharmacol, 2022. 62: p. 117-129. [CrossRef]
- Cowen, L., et al., Network propagation: a universal amplifier of genetic associations. Nat Rev Genet, 2017. 18(9): p. 551-562. [CrossRef]
- Supek, F. and N. Skunca, Visualizing GO Annotations. Methods Mol Biol, 2017. 1446: p. 207-220.
- Gan, M., X. Dou, and R. Jiang, From ontology to semantic similarity: calculation of ontology-based semantic similarity. ScientificWorldJournal, 2013. 2013: p. 793091. [CrossRef]
- Supek, F., et al., REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One, 2011. 6(7): p. e21800. [CrossRef]
- Pesquita, C., et al., Semantic similarity in biomedical ontologies. PLoS Comput Biol, 2009. 5(7): p. e1000443. [CrossRef]
- Zhao, C. and Z. Wang, GOGO: An improved algorithm to measure the semantic similarity between gene ontology terms. Sci Rep, 2018. 8(1): p. 15107. [CrossRef]
- Yu, G., et al., clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS, 2012. 16(5): p. 284-7. [CrossRef]
- Galeota, E., K. Kishore, and M. Pelizzola, Ontology-driven integrative analysis of omics data through Onassis. Sci Rep, 2020. 10(1): p. 703. [CrossRef]
- Duong, D., et al., Word and Sentence Embedding Tools to Measure Semantic Similarity of Gene Ontology Terms by Their Definitions. J Comput Biol, 2019. 26(1): p. 38-52. [CrossRef]
- Blimkie, T.M., A. An, and R.E.W. Hancock, Facilitating pathway and network based analysis of RNA-Seq data with pathlinkR. PLOS Computational Biology, 2024. 20(9): p. e1012422. [CrossRef]
- Prummer, M., Enhancing gene set enrichment using networks. F1000Res, 2019. 8: p. 129. [CrossRef]
- Yoon, S., et al., GScluster: network-weighted gene-set clustering analysis. BMC Genomics, 2019. 20(1): p. 352. [CrossRef]
- Taylor, A., et al., GeneFEAST: the pivotal, gene-centric step in functional enrichment analysis interpretation. arXiv preprint arXiv:2309.00061, 2023. [CrossRef]
- Kerseviciute, I. and J. Gordevicius, aPEAR: an R package for autonomous visualization of pathway enrichment networks. Bioinformatics, 2023. 39(11). [CrossRef]
- Bhuva, D.D., et al., vissE: a versatile tool to identify and visualise higher-order molecular phenotypes from functional enrichment analysis. BMC Bioinformatics, 2024. 25(1): p. 64. [CrossRef]
- Mohamed, A., et al., vissE.cloud: a webserver to visualise higher order molecular phenotypes from enrichment analysis. Nucleic Acids Res, 2023. 51(W1): p. W593-W600. [CrossRef]
- Major, V., A. Surkis, and Y. Aphinyanaphongs, Utility of General and Specific Word Embeddings for Classifying Translational Stages of Research. AMIA Annu Symp Proc, 2018. 2018: p. 1405-1414.
- Chiu, B., et al. How to train good word embeddings for biomedical NLP. in Proceedings of the 15th workshop on biomedical natural language processing. 2016.
- Mikolov, T., et al., Efficient estimation of word representations in vector space. arXiv preprint arXiv:1301.3781, 2013. [CrossRef]
- Ofer, D., N. Brandes, and M. Linial, The language of proteins: NLP, machine learning & protein sequences. Comput Struct Biotechnol J, 2021. 19: p. 1750-1758. [CrossRef]
- Xenos, A., et al., Linear functional organization of the omic embedding space. Bioinformatics, 2021. 37(21): p. 3839-3847. [CrossRef]
- Asgari, E. and M.R. Mofrad, Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics. PLoS One, 2015. 10(11): p. e0141287. [CrossRef]
- Ryan, V.W., et al., Interpreting and visualizing pathway analyses using embedding representations with PAVER. Bioinformation, 2024. 20(7): p. 700-704. [CrossRef]
- Kulmanov, M., et al., Semantic similarity and machine learning with ontologies. Brief Bioinform, 2021. 22(4). [CrossRef]
- Lerman, G. and B.E. Shakhnovich, Defining functional distance using manifold embeddings of gene ontology annotations. Proc Natl Acad Sci U S A, 2007. 104(27): p. 11334-9. [CrossRef]
- Al Abir, F. and J.Y. Chen, Mondrian Abstraction and Language Model Embeddings for Differential Pathway Analysis. bioRxiv, 2024.
- Chen, D.-Q., et al., Identification of Differentially Expressed Genes and Signaling Pathways in Acute Myocardial Infarction Based on Integrated Bioinformatics Analysis. Cardiovascular Therapeutics, 2019. 2019(1): p. 8490707. [CrossRef]
- Jia, R., et al., Identification of key genes unique to the luminal a and basal-like breast cancer subtypes via bioinformatic analysis. World Journal of Surgical Oncology, 2020. 18(1): p. 268. [CrossRef]
- Niu, Y., et al., Bioinformatics to analyze the differentially expressed genes in different degrees of Alzheimer’s disease and their roles in progress of the disease. Journal of Applied Genetics, 2024. [CrossRef]
- Gamazon, E.R., et al., Multi-tissue transcriptome analyses identify genetic mechanisms underlying neuropsychiatric traits. Nature Genetics, 2019. 51(6): p. 933-940. [CrossRef]
- Kulasinghe, A., et al., Transcriptomic profiling of cardiac tissues from SARS-CoV-2 patients identifies DNA damage. Immunology, 2023. 168(3): p. 403-419. [CrossRef]
- Dalit, L., et al., Divergent cytokine and transcriptional signatures control functional T follicular helper cell heterogeneity. bioRxiv, 2024: p. 2024.06.12.598622. [CrossRef]
- Lee, J.-Y., et al., Inhibition of HTR2B-mediated serotonin signaling in colorectal cancer suppresses tumor growth through ERK signaling. Biomedicine & Pharmacotherapy, 2024. 179: p. 117428. [CrossRef]
- Nguyen, J.H., et al., Developmental pyrethroid exposure disrupts molecular pathways for MAP kinase and circadian rhythms in mouse brain. bioRxiv, 2024. [CrossRef]
- Curtis, M.A., et al., Developmental pyrethroid exposure in mouse leads to disrupted brain metabolism in adulthood. NeuroToxicology, 2024. 103: p. 87-95. [CrossRef]
- O'Donovan, S., et al., Shared and unique transcriptional changes in the orbitofrontal cortex in psychiatric disorders and suicide. Translation: The University of Toledo Journal of Medical Sciences, 2024. 12. [CrossRef]
- Hu, Y., et al., Probiotic Protects Kidneys Exposed to Microcystin-LR. Translation: The University of Toledo Journal of Medical Sciences, 2024. 12(1). [CrossRef]
- Hodgman, C., A. French, and D. Westhead, BIOS Instant Notes in Bioinformatics. 2009: Taylor & Francis.
- Karp, P.D., et al., Pathway size matters: the influence of pathway granularity on over-representation (enrichment analysis) statistics. BMC Genomics, 2021. 22(1): p. 191. [CrossRef]
- Wijesooriya, K., et al., Urgent need for consistent standards in functional enrichment analysis. PLoS Comput Biol, 2022. 18(3): p. e1009935. [CrossRef]
- Ziemann, M., B. Schroeter, and A. Bora, Two subtle problems with over-representation analysis. Bioinformatics Advances, 2024. [CrossRef]
- Hale, M.L., I. Thapa, and D. Ghersi, FunSet: an open-source software and web server for performing and displaying Gene Ontology enrichment analysis. BMC Bioinformatics, 2019. 20(1): p. 359. [CrossRef]
- Ewing, E., et al., GeneSetCluster: a tool for summarizing and integrating gene-set analysis results. BMC Bioinformatics, 2020. 21(1): p. 443. [CrossRef]
- Wang, G., D.H. Oh, and M. Dassanayake, GOMCL: a toolkit to cluster, evaluate, and extract non-redundant associations of Gene Ontology-based functions. BMC Bioinformatics, 2020. 21(1): p. 139. [CrossRef]
- Yu, G., Gene Ontology Semantic Similarity Analysis Using GOSemSim. Methods Mol Biol, 2020. 2117: p. 207-215.
- Gu, Z. and D. Hübschmann, SimplifyEnrichment: A Bioconductor Package for Clustering and Visualizing Functional Enrichment Results. Genomics, Proteomics & Bioinformatics, 2022. 21(1): p. 190-202. [CrossRef]
- Kolde, R. and J. Vilo, GOsummaries: an R Package for Visual Functional Annotation of Experimental Data. F1000Res, 2015. 4: p. 574. [CrossRef]
- Balestra, C., et al., Redundancy-aware unsupervised ranking based on game theory: Ranking pathways in collections of gene sets. PLoS One, 2023. 18(3): p. e0282699. [CrossRef]
- Weistuch, C., et al., Normal tissue transcriptional signatures for tumor-type-agnostic phenotype prediction. Scientific Reports, 2024. 14(1): p. 27230. [CrossRef]
Figure 1.
Gene Ontology Gene-Term Annotations (GOALL w/ IEA, Bader Lab, Oct 2024). A. Histogram showing the frequency of all annotated genes and the number of pathways, i.e. GO terms, annotated to them. B. Bar plot showing representative terms in each GO subontology after percentile-ranking all terms by their cumulative gene annotations (bias score) and using Tukey’s five-number summary. BP: biological process; CC: cellular component; MF: molecular function.
Figure 1.
Gene Ontology Gene-Term Annotations (GOALL w/ IEA, Bader Lab, Oct 2024). A. Histogram showing the frequency of all annotated genes and the number of pathways, i.e. GO terms, annotated to them. B. Bar plot showing representative terms in each GO subontology after percentile-ranking all terms by their cumulative gene annotations (bias score) and using Tukey’s five-number summary. BP: biological process; CC: cellular component; MF: molecular function.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



