
Submitted:
22 April 2025
Posted:
24 April 2025
You are already at the latest version
Abstract
Clinical data, animal and cell studies show that some antidiabetic drugs, including glucagon-like peptide 1 receptor agonists (GLP-1RAs), exert therapeutic effects in Alzheimer's disease (AD) via modulation of amyloid-β peptide (Aβ) metabolism. Meanwhile, the direct interactions of GLP-1RAs with Aβ and their functional consequences remain unexplored. The interaction with monomeric Aβ40/Aβ42 of GLP-1(7-37) and its several analogues (semaglutide (Sema), liraglutide (Lira), exenatide (Exen)) was studied by biolayer interferometry and surface plasmon resonance spectroscopy. Quaternary structure of the GLP-1RAs was investigated by dynamic light scattering. The effect of GLP-1RAs on Aβ fibrillation was assessed by Thioflavin T assay and electron microscopy. The impact of GLP-1RAs on Aβ cytotoxicity was studied using the MTT assay. Monomeric Aβ40 and Aβ42 directly bind to GLP-1(7-37), Sema, Lira, Exen, with highest affinity for Lira (the lowest estimates of equilibrium dissociation constants are 42-60 nM). The GLP-1RAs are prone to oligomerization, which may affect their binding to Aβ. GLP-1(7-37) and Exen inhibit Aβ40 fibrillation, whereas Sema shows the opposite effect. The GLP-1 analogues decreased Aβ cytotoxicity towards SH-SY5Y cells, while GLP-1(7-37) enhanced the Aβ40 cytotoxicity, not affecting the cytotoxic effect of Aβ42. Overall, the GLP-1RAs interact with Aβ, differentially modulating its fibrillation and cytotoxicity, suggesting the need for further clinical studies of GLP-1RAs to exclude the possibility of Aβ-mediated neuronal injury.
Keywords:
diabetes mellitus
; glucagon-like peptide-1
; liraglutide
; exenatide
; semaglutide
; Alzheimer's disease
; amyloid-β peptide
; Aβ fibrillation
; cytotoxicity
; bio-layer interferometry
; surface plasmon resonance
; dynamic light scattering
; electron microscopy
; molecular docking
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by a gradual decline in cognitive abilities and memory impairment, which significantly complicates social and professional activities. Worldwide, approximately 416 million individuals are affected by AD dementia, prodromal AD, or preclinical AD, accounting for 22% of the population aged 50 years and older [1]. To date, U.S. Food and Drug Administration (FDA) has approved nine drugs for treatment of AD, of which only three (aducanumab, lecanemab, donanemab) are used for pathogenetic therapy and target the reduction of amyloid-β peptide (Aβ) deposits [2,3]. At the same time, the use of these drugs is associated with side effects, such as brain edema and microhemorrhages [4]. Aβ plays a central role in the AD pathology [5]. It is derived from a transmembrane amyloid precursor protein (APP)), through sequential proteolytic cleavage by β-secretase and γ-secretase [6]. The predominant forms of Aβ are the peptides comprising of 38, 40 or 42 residues, Aβ38, Aβ40 and Aβ42, respectively [7,8]. Monomeric Aβ is intrinsically disordered and therefore prone to aggregation with formation of short fibrillar oligomers, most cytotoxic globular nonfibrillar oligomers, and mature amyloid fibrils [9].
Epidemiological data indicate a strong link between AD and diabetes mellitus (DM): patients with diabetes have a 65% increased risk of developing AD [10,11]. DM is a severe chronic disease that has a serious impact on the life and well-being of individuals, families and society. Globally, at least 529 million people suffer from diabetes [12]. Type 2 diabetes mellitus (DM2) is the most prevalent form of DM (90% of all cases), characterized by high blood glucose levels (hyperglycemia) and insulin resistance [13]. The last one, along with neuroinflammation, oxidative stress, increased levels of advanced glycosylation end products, mitochondrial dysfunction, metabolic syndrome, and the accumulation of Aβ and tau protein in the brain, are common features of AD and DM2 (reviewed in [14]). Therefore, type 3 diabetes, which manifests as insulin resistance in brain tissue, affects cognitive function and contributes to AD progression, has recently been proposed as a brain-specific type of DM [15].
Clinical studies in patients with mild cognitive impairment and AD have demonstrated that administration of certain antidiabetic medications, including intranasal insulin, metformin, incretins, and thiazolidinediones, can improve cognition and memory (reviewed in [16]). Incretins (glucagon-like peptide 1 (GLP-1) and gastric inhibitory peptide) are gut hormones that are secreted after nutrient intake and act on the pancreatic β-cells to enhance glucose-stimulated insulin secretion [17]. Incretin-based therapy (including truncated GLP-1 and its derivatives) is playing an increasingly important role in treatment of DM2 due to its efficacy and safety [18,19].
The N-terminally truncated forms of GLP-1, GLP-1(7-36)/(7-37), secreted from intestinal L cells [20], control meal-related glycemic excursions by augmentation of insulin expression and secretion and inhibition of glucagon release (reviewed in [21]). Some population of neurons in the nucleus tractus solitarii of the brainstem can also express GLP-1 [22,23]. GLP-1 can cross the blood-brain barrier (BBB) [24] and acts through GLP-1 receptor, GLP-1R, which is expressed in several brain regions, including the hypothalamus, cerebral cortex, amygdala, hippocampus, caudate putamen, and globus pallidum [25]. GLP-1 signaling is important for cognition, and preclinical studies evidence neuroprotective action of GLP-1 [26,27]. Murine GLP-1R contributes to control of synaptic plasticity and memory formation [28]. GLP-1R-deficient mice have a learning-deficient phenotype which can be rescued through hippocampal GLP1R gene transfer, while the rats overexpressing GLP-1R in the hippocampus show improved memory and learning abilities [29]. GLP-1(7-36) has been shown to reduce Aβ levels in the mouse brain in vivo and to decrease levels of APP in cultured neuronal cells [30]. Similarly, GLP-1(7-36) protects cultured hippocampal neurons against Aβ/iron-induced death [30], while mutated GLP-1 rescues SH-SY5Y cells from Aβ42-induced apoptosis [31].
GLP-1 is efficiently inactivated by dipeptidyl peptidase-4 (DPP-4) and neutral endopeptidase 24.11, resulting in a plasma half-life of GLP-1 of approximately 1.5-5 minutes [32,33,34]. To overcome this limitation, DPP-4 inhibitors and long-acting GLP-1R agonists (GLP-1RAs) resistant to proteolysis by DPP-4 have been developed for clinical use (reviewed in [32,35]): Exenatide (Exen), Liraglutide (Lira), Semaglutide (Sema), etc.
Exen (trade name Byetta) consists of 39 amino acid residues with 53% homology to human GLP-1(7-37) (Figure 1A, Figure 1C), and is resistant to DPP-4-mediated inactivation [36]. Exen decreases Aβ toxicity and oxidative stress in primary neuronal cultures and SH-SY5Y cells, interferes with the development of cognitive impairments and significantly reduces brain levels of APP and Aβ in animal AD models [37,38,39,40]. Reduced Aβ accumulation in response to Exen has been shown in both mouse and worm AD models [40,41]. Evaluation of the Exen’s effect in patients with moderate Parkinson’s disease showed sustained improvements in cognitive and motor measures [42]. Meanwhile, a pilot study of Exen in AD did not reveal significant differences in clinical, cognitive, or biomarker outcomes compared with placebo, except for a reduction in Aβ42 levels in extracellular vesicles [43].
Another long-acting GLP-1 derivative, Lira (brand names Victoza and Saxenda), differs from GLP-1(7-37) by K34R substitution and palmitic acid attached to K26 residue through a glutamic acid spacer (Figure 1A, Figure 1E). The attached fatty acid chain favors binding to serum albumin thereby slowing the clearance of Lira [44,45]. Lira has been shown to alleviate neuronal insulin resistance and to reduce Aβ formation and tau hyperphosphorylation in SH-SY5Y cells [46]. Tests of Lira in APP/PS1 AD mice showed that it crosses the BBB, prevents memory loss and hippocampal deterioration, increases the number of young neurons in the dentate gyrus, and reduces neuronal inflammation, Aβ oligomer,APP levels, and Aβ plaque formation [47,48,49]. In clinical trials involving AD patients, Lira was found to prevent the decline of brain glucose metabolism, however, it did not significantly affect Aβ accumulation or cognition [50]. Functional magnetic resonance imaging revealed significant improvement in intrinsic connectivity in the default mode network in the group of persons at risk for AD taking Lira, but without detectable cognitive differences between the study groups [51].
Sema (trade names Ozempic, Wegovy, etc.) is a prolonged-release form of Lira with increased affinity for HSA, suitable for once-weekly administration [52]. Compared to Lira, Sema contains 2-aminoisobutyric acid at position 2 (prevents breakdown by DPP-4) and differs in structure of the fatty acid chain (C18 di-acid chain) and its linker (Figure 1A, Figure 1B). Sema protects SH-SY5Y cells from Aβ25-35 by enhancement of autophagy and inhibition of apoptosis [53]. The neuroprotective and anti-inflammatory properties of Sema were shown in a rat model of stroke [54]. Recent studies using human AD brain organoids have shown that Sema decreases levels of Aβ and phosphorylated tau levels. Additionally, in APP/PS1 transgenic mice, Sema improves cognitive performance, particularly learning and memory, and reduces amyloid plaque [55]. Oral form of Sema is currently being tested in patients with early AD in phase 3 clinical trials (NCT04777396 and NCT04777409) [56,57].
Despite encouraging clinical data, animal and cellular studies on the role of GLP-1 and its analogues in AD progression, information on their direct interaction with Aβ is lacking. To fill this gap, in the present study we probe the interaction of several GLP-1RAs with monomeric Aβ40/42 using biolayer interferometry (BLI) and surface plasmon resonance (SPR) spectroscopy. Furthermore, we assess the effects of, of the GLP-1RAs on Aβ fibrillation and Aβ cytotoxicity towards SH-SY5Y cells.
2. Results
2.1. Interaction of GLP-1RAs with Monomeric Aβ
Aβ40/Aβ42 was immobilized of the surface of BLI sensor by amine coupling using EDAC/sulfo-NHS, followed by removal of the non-covalently bound Aβ molecules with 0.5% SDS solution, which ensured monomeric state of Aβ. Passage over the sensor of 4-50 µM solutions of GLP-1(7-37), Exen, Lira and Sema in the buffer simulating conditions of the extracellular space results in the concentration-dependent sensograms characteristic of association/dissociation phases (Figure 2). Some of the resulting kinetic curves were successfully fitted using either a single binding site model (1) or a heterogeneous ligand scheme (2) (Figure 2). The resulting parameters of the GLP-1RA ‒ Aβ interactions are summarized in Table 1. Meanwhile, some of the kinetic data are not consistent with these interaction models. Nevertheless, the clear signs of these interactions evidence that their equilibrium dissociation constants, KD, reach the level of the analyte concentrations used in the BLI experiments, i.e., 25–50 µM for GLP-1(7–37) and 4–15 µM for Exen. The highest affinity for Aβ40/Aβ42 is observed for Lira with KD values of 42-60 nM at protein concentrations of 5-10 µM (Table 1). Sema is 2.4-2.6 orders of magnitude less specific to Aβ40/Aβ42 (KD values of 11-22 µM) at protein concentrations of 17-38 µM.
The analogous examination of Aβ40/Aβ42 affinity for the GLP-1RAs using SPR spectroscopy and Aβ as a ligand (Figure 3) yielded 1-1.5 orders of magnitude higher lowest KD estimates for Sema and Lira (Table 2), which may be due to differences in the buffer conditions or analyte concentrations used in the BLI and SPR experiments. For Exen, , the quality of the SPR data (Figure 3C, Figure 3F) was insufficient for a reliable kinetic analysis, but it can be concluded that the corresponding KD values reach the analyte concentration level of 1 μM. The latter estimate is slightly lower than that derived from the BLI experiments, which can be rationalized by the same factors.
The KD estimates for Aβ-Sema/Lira complexes (Table 1 and Table 2) are comparable to those for Aβ binding to its natural depot, human serum albumin (HSA) (~0.1 μM [58]), as well as for Aβ complexes with fragments of the receptor for advanced glycation end products, which exhibit neuroprotective activity in both in vitro and in vivo models [59]. Similarly, the KD values for Aβ-Sema/Lira complexes are close to the KD estimate for binding of 125I-labelled Lira to GLP-1 receptor, 1.3×10-7 M [60]. Moreover, these values are close to the peak plasma concentrations of Sema/Lira (20-120 nM [61,62]), indicating that Sema/Lira interaction with Aβ (0.5 nM in plasma [63]) may occur in the circulation.
2.2. Concentration-Dependent Changes in Quaternary Structure of the GLP-1RAs
Since GLP-1RAs are prone to oligomerization and fibrillation [64,65,66], we studied the quaternary structure of GLP-1 and its analogues using dynamic light scattering (DLS) spectroscopy in a buffer with salt conditions close to physiological ones and similar to the BLI experiments (Table 3). Decrease in DLS sensitivity at protein concentrations of 0.05–0.02 mg/mL prevented the measurements at the GLP-1RAs concentrations below 6–12 μM, depending on the peptide.
The main light scattering peak of 5-83 μM GLP-1(7-37) corresponds to particles with a hydrodynamic radius (Rh) exceeding 92 nm, which indicates strong oligomerization of the peptide and explains the inability to describe analytically the BLI data on its interaction with Aβ40/Aβ42 (Figure 1A, Figure 1E).
The DLS data for 6-105 μM Lira show an increase in its degree of multimerization, MWRh/MWm, with protein concentration from 6.5–8.2 to 15.6, consistent with the other reports [66,67,68]. This transition in oligomeric state of Lira correlates with a tendency to changes in the KD values for its interaction with Aβ40/Aβ42 (Table 1).
Similarly to Lira, Exen demonstrates an increase in degree of multimerization with protein concentration (15-234 μM) from 1.6 to 5.9, in agreement with the literature data [69].
The Rh estimates for Sema (12 μM, 47 μM) are consistent with its monomer and dimer, which is below the previous estimates for the formulation buffer composition [66]. Hence, the KD values for Sema interaction with Aβ40/Aβ42 estimated using BLI (Table 1) are close to the corresponding thermodynamic constants. In contrast, in the other cases complicated by oligomerization of the GLP-1RAs, the estimates shown in Table 1 represent only apparent constants.
Overall, the DLS data indicate that the GLP-1Ras, at the concentrations used in the BLI experiments, exist as mixture of oligomers with varying degree of multimerization. However, since degree of their multimerization decreases with decreasing protein concentration, at plasma concentrations of 1-119 pM for GLP-1/Exen) [70,71,72] and 20-120 nM for Sema/Lira [61,62], the GLP-1RAs predominantly exist in monomeric form. In this state, hydrophobic residues and fatty acid moieties are more accessible for interaction with Aβ, which likely facilitates binding.
2.3. Effect of the GLP-1RAs on Aβ fibrillation
The influence of the GLP-1RAs on Aβ40 fibril formation at 30°C was studied using ThT fluorescence assay at ThT and GLP-1RA concentrations of 10 μM for both components (Figure 4). While Lira had no significant effect on fibrillation (Figure 4A), the other GLP-1RAs demonstrate drastically different behavior (Figure 4B). GLP-1(7-37) and Exen both suppress Aβ40 fibrillation, whereas in the presence of Sema there is a clear tendency to stimulate the fibrillation process.
To explore structural features of the grown fibrils, we examined them using negative-staining transmission electron microscopy, TEM (Figure 5). The Aβ40 sample and samples with the addition of Lira/Sema reveal dense clusters of the intertwined mature fibrils up to 2 µm long (Figure 5 A,B,C). When analyzing the samples with the addition of Exen, only scattered fibrils, shorter fibrils compared to the others, were visible (Figure 5D). In addition, in the presence of Exen, large clusters of fibrils, which were characteristic of the other samples, did not form (Figure 5 D). When analyzing the samples with the addition of GLP-1 sporadic small fibril clusters were still observed (Figure 5E,F). In summary, the microscopic data support the finding from the ThT fluorescence assay.
Apparently, the ability of a particular GLP-1RA to affect Aβ fibrillation in vivo depends not only on its in vitro activity, but also on its ability to penetrate the CNS and distribute across the brain regions. Since Exen and GLP-1 readily cross the BBB [24,73], and GLP-1 can be expressed by some population of neurons [22,23], these GLP-1RAs have the potential to suppress Aβ fibrillation also in the brain tissue. On the contrary, Sema does not cross the BBB [74], indicating that its ability to stimulate Aβ fibrillation in vitro (Figure 4B, Figure 5C) is unlikely to be of physiological significance.
Our in vitro data for Exen are consistent with data showing reduced Aβ accumulation in the AD models [40,41]. Interestingly, Lira and Sema are also able to reduce Aβ deposits in AD mouse models [47,48,49,55]. These GLP-1 analogues did not inhibit the process of fibrillation (Figure 4, Figure 5), but in this case other mechanisms are probably involved, such as influence on APP level [47], on insulin signaling and insulin secretion level [75,76,77], as well as reduction of chronic inflammation level [12,49].
2.4. Structural Modeling of the Complexes Between Aβ40 or Its Protofibril and GLP-1(7-37)/Exen
To identify structural patterns in the formation of the GLP-1RA‒Aβ complexes, tertiary structures of the complexes between GLP-1(7-37)/Exen and Aβ40 monomer were modeled using ClusPro docking server [78] (Figure 6A, Figure 6B). Additionally, we investigated the interaction of GLP-1(7-37)/Exen with the protofibrillar form of Aβ40, since this type of interaction (as well as interaction with monomeric Aβ40) may underlie the inhibitory effects of GLP-1(7-37)/Exen on Aβ40 fibril formation that we observed.
The modeling of the complex between GLP-1(7-37) and Aβ40 monomer predicts (Figure 6A) that Aβ40 binds GLP-1(7-37) via N-terminal residues R5, D7, G9, Y10, and Q15 from the α-helix. The predicted Aβ40-binding site of GLP-1(7-37) includes residues L20, E27, F28, W31, L32, K34, and G35. The modeling of structure of the Exen complex with Aβ40 monomer predicts (Figure 6B) that Aβ40 binds Exen via residue L17 (α-helix) and C-terminal residues L34 and V39. The predicted Aβ40-binding site of Exen includes N-terminal H1, residues F6, L10, Q13, M14, E17, L21 (α-helix), and C-terminal residues P38 and S39. Thus, the predicted Aβ40-binding sites of GLP-1(7-37)/Exen are located in the region of residues 5-21 а.а. Meanwhile, the predicted contact residues of the Aβ40 molecule differ significantly for GLP-1(7-37) and Exen, which may reflect limitations of the rigid-body approximation employed in the docking algorithm.
The modeling of the complex between GLP-1(7-37) and Aβ40 protofibril predicts (Figure 6C) that GLP-1(7-37) interacts with chains А and C of the protofibril via residues T13, S14, S18, Y19, E21, Q23, A24, A25, E27, F28, W31, L32, K34, and G35. The chains A and C are predicted to bind GLP-1(7-37) via residues Q15, K16, V18, F19, F20 and V39. Note that the same residues of GLP-1(7-37) (L20, E27, F28, W31, L32, K34, and G35) participate in the binding of both the Aβ40 monomer and Aβ40 protofibril.
The analogous modeling of the complex between Exen and Aβ40 protofibril predicts (Figure 6D) that Exen interacts with chains А and C of the protofibril via residues H1, G2, E3, G4, T5, F6, S8, L10, Q13, M14, E15, E17, R20, and S39. The chains A and C are predicted to bind Exen via residues Q15, V18 and V36. The residues H1, F6, L10, Q13, M14, E17, are common for the binding sites of Exen with Aβ40 monomer and Aβ40 protofibril
2.5. Effect of the GLP-1RAs on Aβ Cytotoxicity to Human Neuroblastoma Cells
Since the deleterious effects of Aβ on neuronal cells are thought to be mediated by its oligomeric forms with increased cytotoxicity [9,79,80], we compared cytotoxicity of Aβ40/Aβ42 alone and in the presence of GLP-1(7-37) or its analogues against human neuroblastoma SH-SY5Y cells using the MTT assay. GLP-1RAs were premixed with Aβ40/Aβ42 at an equimolar ratio in the serum-free medium and added to the SH-SY5Y cells cultured in the same medium to a final concentration of the both components of 10 µM. Staining by the MTT was performed after incubation of the cells for 48 h.
In the absence of Aβ, Lira, Sema and Exen have no effect on survival of SH-SY5Y cells, whereas GLP-1(7-37) enhances cell viability by 33% (Figure 7A). The addition of Aβ40 or Aβ42 alone decreases the cell survival by 22% and 37%, respectively (Figure 7B, Figure 7C). The addition of Lira, Sema or Exen with Aβ40/Aβ42 abolishes this effect. Meanwhile, the addition of GLP-1(7-37) increases the cytotoxicity of Aβ40 (Figure 7B), but does not affect the cytotoxic effect of Aβ42 (Figure 7C).
The most pronounced increase in viability of SH-SY5Y cells was observed for Lira, Sema and Exen upon treatment of the cells with Aβ42 (Figure 7C). Similarly, Sema reversed the effect of Aβ(25-35) on SH-SY5Y cells after their pretreatment with the latter [81]. Pretreatment of neuronal cells with Lira or Exen also protected them from Aβ(25-35) and Aβ42, respectively [30,37,82].
The protective effect of Exen on the Aβ-treated neuroblastoma cells is consistent with results of the Aβ fibrillation experiments (Figure 4 and Figure 5) and rescuing memory deficits in AD mice [40]. However, such benefits have not been replicated in clinical trials [43]. Despite its higher affinity for monomeric Aβ, Lira exhibits a similar set of the properties, except for the lack of significant effect on Aβ40 fibrillation (Table 4). In contrast, both Sema and GLP-1(7-37) show conflicting results in the Aβ fibrillation and Aβ cytotoxicity tests (Table 4), which may reflect differences in the cytotoxic properties of the multimeric forms of Aβ formed in their presence. In the case of Sema, the rapid fibrillation of Aβ40 (Figure 4B) may prevent the accumulation of the more cytotoxic Aβ40 oligomers [9,79,80]. The excess of the latter in the case of GLP-1(7-37) appears to favor its cytotoxicity, despite the suppression of Aβ40 fibrillation in its presence (Table 4).
3. Materials and Methods
3.1. Materials
Lira (Victoza, 6 mg/mL) and Sema (Ozempic, 1.34 mg/mL) were bought from Novo Nordisk (Bagsværd, Denmark). Exen (Byetta, 250 µg/mL) was from Astra Zeneca (Cambridge, UK) and Acmec Biochemical Technology Co., Ltd. (Shanghai, China). GLP-1(7-37) was purchased from Merck KGaA (Darmstadt, Germany), cat. #G9416, and Aladdin (Riverside, USA), cat. #G-118964. Lira, Sema and Exen were dialyzed three times against 1,000-fold excess of deionized water and then dialyzed twice against 50 mM Tris-HCl, 280 mM NaCl, 9.8 mM KCl, 5 mM CaCl2, 2 mM MgCl2, pH 7.4 (buffer A) for all experiments except for BLI and SPR.
Human Aβ40/Aβ42 was expressed in E. coli and purified as described earlier [83]. Briefly, chimera of Aβ with ubiquitin was purified using Ni-NTA affinity chromatography and cleaved with Usp2-cc protease (prepared mainly as described in ref. [84]), followed by purification using Ni-NTA and C18 columns. Quality of the Aβ samples was controlled by SDS-PAGE and electrospray ionization mass spectrometry.
Protein concentrations were measured spectrophotometrically using molar extinction coefficients at 280 nm calculated according to ref. [85]: 6,990 M-1cm-1 for Sema and Lira, 5,500 M-1cm-1 for Exen, and 1,490 M-1cm-1 for Aβ40/Aβ42 at pH 7.4-8.0.
Ethylenediaminetetraacetic acid (EDTA), magnesium chloride, Thioflavin T (ThT), ethanolamine and polyethylene glycol sorbitan monolaurate (TWEEN®) 20 were from Merck KGaA (Darmstadt, Germany). 2-mercaptoethanol (2-ME) was from Amresco® LLC (Vienna, Austria). Urea, imidazole, sodium hydroxide, sodium dodecyl sulfate (SDS) and glycerol were purchased from PanReac AppliChem (Barcelona, Spain). Calcium/magnesium chloride were from Honeywell Fluka (Charlotte, NC, USA). AbiFlow 100 Ni-NTA Agarose was from Abisense (Sirius, Russia). Hydrochloric acid was from Sigma Teс LLC (Khimki, Russia). Ultra-grade Tris, HEPES, sodium chloride and dimethyl sulfoxide (DMSO) were from Helicon (Moscow, Russia). Trifluoroacetic acid (TFA) was purchased from Fisher Scientific Inc. (Waltham, USA). Potassium chloride, Coomassie Brilliant Blue R-250, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and sodium azide were from Dia-M (Moscow, Russia). Acetic acid and ammonium hydroxide were from Chimmed (Moscow, Russia) and Component-reaktiv (Moscow, Russia), respectively. Dulbecco’s Modified Eagle Medium (DMEM), Fetal Bovine Serum (FBS) and penicillin-streptomycin-glutamine were from Gibco (New York, USA). Ampicillin was bought from neoFroxx (Einhausen, Germany). F12 was from PanEco (Moscow, Russia).Stock solution of ThT (0.6 mg/mL) was prepared in deionized water. ThT concentration was measured spectrophotometrically using the molar extinction coefficient at 412 nm of 36,000 M-1cm-1 [86].
Neuroblastoma SH-SY5Y cells were from Prof. Valery P. Zinchenko (Institute of Cell Biophysics of the RAS, Pushchino, Russia).
3.2. BLI Measurements
GLP-1RAs were dialyzed three times against 1,000-fold excess of deionized water and then dialyzed twice against 20 mM Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, pH 7.4 buffer for Exen and Lira or 20 mM HEPES-KOH, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, pH 7.4 for Sema. GLP-1(7-37) was dissolved in the last buffer. The Aβ samples were pretreated by TFA and dissolved in DMSO (2 mg/mL) as described in ref. [58], and stored at –20°C.
Affinity of Aβ40/Aβ42 (ligand) for Exen (4–15 μM), Lira (5–20 μM), Sema (17–38 μM) or GLP-1(7-37) (25–50 μM) (analyte) at 25°C was measured by BLI using a ForteBio Octet® QKe System, 96-well microplates with shaking at 1,000 rpm. Aβ40/Aβ42 (0.05 mg/mL in 10 mM sodium acetate, pH 4.5 buffer) was immobilized on five amino-reactive biosensors while one reference sensor was loaded with 1 M ethanolamine solution through 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride/N-hydroxysulfosuccinimide (EDAC/sulfo-NHS) reaction until the Aβ40/Aβ42 loading level of 3.5 nm was reached, . The rest of the activated amine groups on the biosensors was blocked by 1 M ethanolamine solution. The non-covalently bound Aβ40/Aβ42 molecules were washed off with 0.5% SDS and then with an assay buffer (20 mM HEPES-KOH/Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, pH 7.4). The loading level after the washing was 1.5 nm. Baseline collection time was 300 s, association with an analyte in the assay buffer was recorded for 600 s, and dissociation phase for 600 s or 1,200 s. The ligand was regenerated by triple immersion in 0.1 % SDS water solution for 5 sec, followed by a 30 sec rinsing with the assay buffer. The BLI signal was corrected for baseline drift and non-specific binding by subtraction of the signal from the reference sensor, and fit to the single binding site model (A, analyte; L, ligand):
or the heterogeneous ligand scheme:
where ka and kd are kinetic association and dissociation constants, respectively, and KD are equilibrium dissociation constants. The constants were evaluated for each analyte concentration using ForteBio Data Analysis software v.12.0 (Fremont, CA, USA); standard deviations are indicated.
| A + L |
ka ↔ |
AL | (1) |
|
kd KD |
| A + L1 |
ka1 ↔ |
AL1 | A + L2 |
ka2 ↔ |
AL2 | (2) | |
|
kd1 KD1 |
kd2 KD2 |
3.3. SPR Measurements
SPR studies of Exen, Sema and Lira interaction with monomeric Aβ40/Aβ42 were performed at 25°C using a Bio-Rad ProteOn™ XPR36 instrument mainly according to ref. [87]. Lira and Sema were exhaustively dialyzed against 10 mM sodium phosphate, 50 mM NaCl, pH 7.0 buffer. Exen was exhaustively dialyzed against buffer A. Concentrations of stock solutions were 70-243 µM for Sema, 1,4-2,0 mM for Lira and 27 µM for Exen. The Aβ samples were pretreated by TFA and dissolved in DMSO (2 mg/mL) as described in ref. [58], and stored at –20°C.
Ligand (50 µg/mL Aβ40/Aβ42) was immobilized on a ProteOn GLH sensor chip surface by amine coupling using EDAC/sulfo-NHS, with subsequent blocking of the remaining activated amine groups on the chip surface by 1 M ethanolamine solution. The noncovalently bound Aβ40/Aβ42 molecules were washed off the chip surface with a 0.5% SDS water solution. Analyte (0.0625–2 µM Sema, 1–8 µM Lira and 0.5–6 µM Exen) in the running buffer (10 mM HEPES-NaOH, 150 mM NaCl, 0.05% Tween 20, pH 7.4) was passed over the sensor at a rate of 30 µL/min for 300 s (association phase), followed by flushing the chip with the running buffer for 900 s (dissociation phase). The ligand was regenerated by passage of 10 mM glycine, pH 3.3 buffer, for 50 s. The kinetic SPR data were corrected for baseline drift and non-specific binding, and described using a heterogeneous ligand model (2). The ka, kd, and KD values were estimated using Bio-Rad ProteOn Manager™ v.3.1 software (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The estimates were performed for each data set globally, followed by their averaging; standard deviations are indicated.
3.4. Dynamic Light Scattering Measurements
DLS measurements were carried out using a Zetasizer Nano ZS system (Malvern Instruments Ltd., Malvern, UK). The backscattered light from a 4 mW He-Ne laser 632.8 nm was collected at an angle of 173°. Lira (6-105 µM), Exen (15-234 µM), Sema (12-47 µM) and GLP-1(7-37) (5-83 µM) solutions in 25 mM Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, pH 7.4 buffer were incubated at 25°C for 3 min. The acquisition time for a single autocorrelation function was 100 s. The resulting autocorrelation functions are averaged values from three measurements. The volume-weighted size distributions were calculated using the following parameters for the buffer: refractive index of 1.334 measured with RL3 refractometer (PZO, Warszawa, Poland); the viscosity value η = 0.95 mPa∙s measured using micro-rheology method with a water suspension of standard latex nanoparticles (NIST 3060A, Thermo Fisher Scientific, USA). Molecular mass and its standard deviation corresponding to the volume-weighted hydrodynamic radius MWRh distribution was calculated in approximation of a globular protein according to the equations from ref. [88]. Degree of multimerization was calculated as a MWRh/MWm ratio, where MWm is a molecular mass of monomeric GLP-1RAs calculated from its molecular structure.
3.5. Structural Modeling
The unrelaxed structure of Sema was built in PyMOL v.2.0 software (Schrödinger, Inc., New York, USA) based on the structure of Sema- and taspoglutide-bound GLP-1 receptor in complex with Gs protein (PDB ID: 7KI0, EM, chain E) by combination with a linker and C18 di-acid chain (Figure 1B). Structures of the linker and C18 di-acid chain were built using ChemDraw v.22 (Boston, USA) and minimized in the MM2 field (ChemDraw v.22).
The tertiary structures of Aβ40 (PDB ID 2LFM, NMR, model 1), Aβ40 fibril (PDB ID 2LMN, NMR, model 1), Exen (PDB ID 1JRJ, NMR, chain A, model 1) and GLP-1(7-37) (PDB ID 3IOL, X-ray, chain B) were taken from PDB (www.rcsb.org [89]). Models of tertiary structures of Aβ40/protofibril complexes with Exen or GLP-1(7-37) were built using ClusPro docking server [78]. The resulting complexes were visualized and analyzed using PyMOL v.2 (https://pymol.org). The contact residues in the docking models were calculated using a PyMOL script. The numbering of the contact residues is according to the PDB entries.
3.6. ThT Fluorescence Assay
ThT fluorescence emission measurements were carried out mainly as described in ref. [83] using a BioTek Synergy H1 microplate reader, emission wavelength of 485 nm and excitation at 440 nm. Aβ40 sample was prepared as described in the ref. [83] with some modifications (~ 5 mM NaOH at pH 11.8, 0.5 mg/mL). 20 µM Aβ40 was incubated at 30°C in the absence/presence of 10 µM Sema, Lira, Exen or GLP-1(7-37). The control curves (without Aβ40; Figure S1) were subtracted from the corresponding kinetic curves of Aβ40 samples with/without GLP-1RAs. Each measurement was performed in 2-10 repetitions. The mean fluorescence signal values for each experimental sample were normalized to the average fluorescence signal corresponding to the saturation phase of Aβ40 fibril formation without additives. Data are presented as mean ± standard deviation.
3.7. Transmission Electron Microscopy
A copper grid (300-mesh) coated with a 0.2% formvar film was placed on a 10 µL drop of the sample. After incubating the sample (following the ThT fluorescence assay) for 15 minutes to allow adsorption, the grid was stained with a 1% (w/v) aqueous solution of uranyl acetate for 2 minutes. Excess stain was removed using filter paper, and the grid was rinsed in deionized water for 1 minute. The samples were analyzed using a JEM-1400Plus (HC) transmission electron microscope (JEOL, Ltd., Tokyo, Japan) at an accelerating voltage of 80 keV.
3.8. Cell Viability Assay
Human neuroblastoma SH-SY5Y cells were cultured in DMEM-F12 medium supplemented with 1% penicillin-streptomycin-glutamine and 10% fetal bovine serum at 37°C for 24 h in a humidified atmosphere with 5% CO2. Upon reaching 80% confluence, the cells were harvested and seeded into 96-well plates at a density of 15×105 cells per well in the serum-free DMEM/F12+PSG medium.
The Aβ40/Aβ42 samples were dissolved in fresh 1% NH4OH at a concentration of 0.5 mg/mL, followed by freeze-drying. The dried Aβ40/Aβ42 samples were dissolved in serum-free DMEM medium at a concentration of 40-50 µM (0.17-0.23 mg/mL), followed by mixing with the GLP-1RA (Sema, Lira, Exen or GLP-1) stock solutions in buffer A and the same medium to a final concentration of the both components of 20 µM.
Freshly prepared Aβ40/Aβ42, GLP-1RAs, or their mixtures were added (100 µL per well) to the cultures 24 h after seeding to a final concentration of the both components of 10 µM. The final volume of medium in the well was 200 µL. The MTT assay, designed to assess cellular metabolic activity, was performed after incubation of the cells for 48 h. 0.005 mg/mL MTT was added and the cells were incubated for 3 h, followed by solubilization of the cells using DMSO. Absorbance at 550 nm was measured using an BioTek Synergy H1 microplate reader (Agilent Technologies, Inc., Santa Clara, CA, USA). The resulting values were normalized relative to the control group of the untreated cells (100%). Data are presented as mean ± standard deviation (n=5-10).
4. Conclusions
The risk of AD development in patients with diabetes increases by approximately 65% [8], since these diseases share some pathological features [12]. Therefore, antidiabetic drugs, including GLP-1RAs, are now being repurposed for treatment of AD [14]. Although both animal studies and clinical trials have reported beneficial effects of GLP-1RAs on the course of AD [29,36,85], the molecular mechanisms underlying these effects remain poorly understood. Here we demonstrated direct interaction of GLP-1RAs such as GLP-1(7-37), Lira, Sema and Exen with monomeric forms of Aβ40 and Aβ42 under the in vitro conditions mimicking physiological conditions. Comparison of the KD estimates for Aβ-Sema/Lira complexes with peak plasma concentrations of Sema/Lira indicates potential physiological significance of these interactions. This suggestion is supported by the marked effect of Sema on Aβ40 fibrillation in vitro and the effect of Sema/Lyra on Aβ-induced cytotoxicity towards SH-SY5Y cells. Similarly, Exen and GLP-1(7-37) affect Aβ40 fibrillation and cytotoxicity of Aβ against SH-SY5Y cells. Notably, these effects largely depend on the specific GLP-1RA and not necessarily correlate with results of animal and clinical studies (Table 4). The latter also depends on the ability of GLP-1RA to cross the BBB, which is only possessed by Exen, Lira and GLP-1(7-37), but not by Sema.
Our findings indicate that, despite certain structural similarities, individual GLP-1RAs exhibit distinct behaviors in vitro in terms of their affinity for Aβ, their influence on Aβ fibril formation, and their modulation of Aβ-associated cytotoxicity (Table 4). Further clinical trials of GLP-1-based drugs are needed to rule out the possibility of neuronal damage that does not necessarily lead to progression of AD. Our findings not only suggest a new mechanism for the influence of GLP-1RAs on Aβ metabolism in vivo, but also provide a basis for the development of GLP-1RA drugs with more pronounced anti-AD effects.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: The change in ThT fluorescence in solutions with and without Sema (10 µM), Lira (10 µM), GLP-1 (10 µM), Exen (10 µM) over time. Buffer: 25 mM Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 0.05% NaN3, pH 7.4.
Author Contributions
Conceptualization, E.L., M.Sh., E.N.; methodology, E.D., E.L., M.Sh., A.Ch., V.R.; software, E.D., A.M., V.D.; validation, E.L., M.Sh., E.D., A.Ch., A.V.; formal analysis, M.Sh., E.D., E.L., E.N.; investigation, E.L., M.Sh., V.R., A.V., A.M., E.D., V.A., A. Ch.; resources, M.P., V.A., A.N.; data curation, E.L., M.Sh., V.R., S.P.; writing—original draft preparation, E.L., E.D., M.Sh., A.Ch., V.R.; writing—review and editing, S.P.,E.N.; visualization, E.L., M.Sh., E.D., V.R., A.Ch., A.M.; supervision, E.L., E.N.; project administration, E.L.; funding acquisition, E.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 20-74-10072, https://rscf.ru/project/20-74-10072/. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and supplementary materials.
Acknowledgments
We are grateful to Dr. Roman Fadeev (ITEB RAS, Pushchino, Russia) for preliminary data on influence of the GLP-1RAs on Aβ cytotoxicity. The authors are grateful to Vadim V. Rogachevskii (Institute of Cell Biophysics, Russian Academy of Sciences, Pushchino, Moscow Region, Russia) for providing access to the electron microscope of the Shared Core Facilities of the Pushchino Scientific Center for Biological Research (http://www.ckp-rf.ru/ckp/670266/; accessed on 25 March 2025).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| 2-ME | 2-mercaptoethanol |
| Aβ | amyloid-β peptide |
| Aβ40/Aβ42 | amyloid-β peptide, residues 1-40/42 |
| AD | Alzheimer’s disease |
| APP | amyloid precursor protein |
| BBB | blood-brain barrier |
| CNS | central nervous system |
| DM | diabetes mellitus |
| DM2 | type 2 diabetes mellitus |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | dimethyl sulfoxide |
| DPP-4 | dipeptidyl peptidase-4 |
| EDAC/sulfo-NHS | 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride/N-hydroxysulfosuccinimide |
| EDTA EM |
ethylenediaminetetraacetic acid Electron microscope |
| Exen | Exendin-4/Exenatide |
| GLP-1 | glucagon-like peptide 1 |
| GLP-1(7-36), GLP-1(7-37) | N-terminally truncated forms of glucagon-like peptide 1, residues 7-36 or 7-37 |
| GLP-1R | glucagon-like peptide 1 receptor |
| GLP-1RA | glucagon-like peptide 1 receptor agonist |
| HSA | human serum albumin |
| Lira | Liraglutide |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MWm | molecular mass calculated from the amino acid sequence |
| MWRh | molecular mass calculated from the hydrodynamic radius |
| NMR | nuclear magnetic resonance |
| PA | palmitic acid |
| PDB | Protein Data Bank |
| Sema | Semaglutide |
| SDS | sodium dodecyl sulfate |
| TEM | transmission electron microscopy |
| TFA | trifluoroacetic acid |
| ThT | Thioflavin T |
| Tris | tris(hydroxymethyl)aminomethane |
| TWEEN | polyethylene glycol sorbitan monolaurate |
References
- Gustavsson, A.; Norton, N.; Fast, T.; Frölich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P.M.; Ferretti, M.T.; et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 2023, 19, 658–670. [Google Scholar] [CrossRef]
- Harris, E. FDA Green-Lights Second Alzheimer Drug, Donanemab. JAMA 2024, 332, 524–524. [Google Scholar] [CrossRef] [PubMed]
- 2024 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 2024, 20, 3708–3821. [CrossRef]
- Ameen, T.B.; Kashif, S.N.; Abbas, S.M.I.; Babar, K.; Ali, S.M.S.; Raheem, A. Unraveling Alzheimer’s: the promise of aducanumab, lecanemab, and donanemab. The Egyptian Journal of Neurology, Psychiatry and Neurosurgery 2024, 60, 72. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-beta: a crucial factor in Alzheimer’s disease. Med Princ Pract 2015, 24, 1–10. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984, 120, 885-890, doi:S0006-291X(84)80190-4 [pii]10.1016/s0006-291x(84)80190-4. [CrossRef]
- 8. Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 2017, 38, 1205-1235, doi:aps201728 [pii]10.1038/aps.2017.28. [CrossRef]
- Vander Zanden, C.M.; Wampler, L.; Bowers, I.; Watkins, E.B.; Majewski, J.; Chi, E.Y. Fibrillar and Nonfibrillar Amyloid Beta Structures Drive Two Modes of Membrane-Mediated Toxicity. Langmuir 2019, 35, 16024–16036. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Archives of neurology 2004, 61, 661–666. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; Hofman, A.; van Harskamp, F.; Grobbee, D.E.; Breteler, M.M. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 1996, 39, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Han, W.N.; Chai, S.F.; Li, Y.; Fu, C.J.; Wang, C.F.; Cai, H.Y.; Li, X.Y.; Wang, X.; Hölscher, C.; et al. Semaglutide promotes the transition of microglia from M1 to M2 type to reduce brain inflammation in APP/PS1/tau mice. Neuroscience 2024, 563, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Overview: Type 2 diabetes. In InformedHealth.org [Internet]. ; Institute for Quality and Efficiency in Health Care (IQWiG): Cologne, Germany, 2006.
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s Disease as Type 3 Diabetes: Common Pathophysiological Mechanisms between Alzheimer’s Disease and Type 2 Diabetes. Int J Mol Sci 2022, 23, doi:ijms23052687 [pii]ijms-23-02687 [pii]10.3390/ijms23052687. [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim Biophys Acta Mol Basis Dis 2017, 1863, 1078-1089, doi:S0925-4439(16)30215-0 [pii]10.1016/j.bbadis.2016.08.018. [CrossRef] [PubMed]
- Michailidis M; Tata DA; Moraitou D; Kavvadas D; Karachrysafi S; Papamitsou T; Vareltzis P; V., P. Antidiabetic Drugs in the Treatment of Alzheimer’s Disease. Int J Mol Sci 2022, 23.
- Mudaliar, S.; Henry, R.R. The incretin hormones: from scientific discovery to practical therapeutics. Diabetologia 2012, 55, 1865–1868. [Google Scholar] [CrossRef]
- Cobble, M. Differentiating among incretin-based therapies in the management of patients with type 2 diabetes mellitus. Diabetology & Metabolic Syndrome 2012, 4, 8. [Google Scholar] [CrossRef]
- Meier, J. The role of incretin-based therapies in the management of type 2 diabetes mellitus: perspectives on the past, present and future; 2019; Volume 22.
- Lim, G.E.; Brubaker, P.L. Glucagon-Like Peptide 1 Secretion by the L-Cell: The View From Within. Diabetes 2006, 55, S70–S77. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab 2018, 27, 740-756, doi:S1550-4131(18)30179-7 [pii]10.1016/j.cmet.2018.03.001. [CrossRef]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Molecular metabolism 2019, 30, 72–130. [Google Scholar] [CrossRef]
- Holt, M.K.; Richards, J.E.; Cook, D.R.; Brierley, D.I.; Williams, D.L.; Reimann, F.; Gribble, F.M.; Trapp, S. Preproglucagon Neurons in the Nucleus of the Solitary Tract Are the Main Source of Brain GLP-1, Mediate Stress-Induced Hypophagia, and Limit Unusually Large Intakes of Food. Diabetes 2019, 68, 21–33. [Google Scholar] [CrossRef]
- Kastin, A.J.; Akerstrom, V.; Pan, W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci 2002, 18, 7-14, doi:JMN:18:1-2:07 [pii]10.1385/JMN:18:1-2:07. [CrossRef]
- Muscogiuri, G.; DeFronzo, R.A.; Gastaldelli, A.; Holst, J.J. Glucagon-like Peptide-1 and the Central/Peripheral Nervous System: Crosstalk in Diabetes. Trends Endocrinol Metab 2017, 28, 88-103, doi:S1043-2760(16)30126-6 [pii]10.1016/j.tem.2016.10.001. [CrossRef] [PubMed]
- Monney, M.; Jornayvaz, F.R.; Gariani, K. GLP-1 receptor agonists effect on cognitive function in patients with and without type 2 diabetes. Diabetes Metab 2023, 49, 101470, doi:S1262-3636(23)00052-6 [pii]10.1016/j.diabet.2023.101470. [CrossRef] [PubMed]
- Holscher, C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology 2018, 136, 251-259, doi:S0028-3908(18)30040-6 [pii]10.1016/j.neuropharm.2018.01.040. [CrossRef] [PubMed]
- Abbas, T.; Faivre, E.; Holscher, C. Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: Interaction between type 2 diabetes and Alzheimer’s disease. Behav Brain Res 2009, 205, 265-271, doi:S0166-4328(09)00397-0 [pii]10.1016/j.bbr.2009.06.035. [CrossRef]
- During, M.J.; Cao, L.; Zuzga, D.S.; Francis, J.S.; Fitzsimons, H.L.; Jiao, X.; Bland, R.J.; Klugmann, M.; Banks, W.A.; Drucker, D.J.; et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med 2003, 9, 1173-1179, doi:nm919 [pii]10.1038/nm919. [CrossRef]
- Perry, T.; Lahiri, D.K.; Sambamurti, K.; Chen, D.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. Journal of neuroscience research 2003, 72, 603–612. [Google Scholar] [CrossRef]
- Qin, Z.; Sun, Z.; Huang, J.; Hu, Y.; Wu, Z.; Mei, B. Mutated recombinant human glucagon-like peptide-1 protects SH-SY5Y cells from apoptosis induced by amyloid-beta peptide (1-42). Neurosci Lett 2008, 444, 217-221, doi:S0304-3940(08)01164-6 [pii]10.1016/j.neulet.2008.08.047. [CrossRef]
- Ussher, J.R.; Drucker, D.J. Cardiovascular biology of the incretin system. Endocr Rev 2012, 33, 187-215, doi:er.2011-1052 [pii]10.1210/er.2011-1052. [CrossRef]
- Hui, H.; Farilla, L.; Merkel, P.; Perfetti, R. The short half-life of glucagon-like peptide-1 in plasma does not reflect its long-lasting beneficial effects. European journal of endocrinology 2002, 146, 863–869. [Google Scholar] [CrossRef]
- Plamboeck, A.; Holst, J.J.; Carr, R.D.; Deacon, C.F. Neutral Endopeptidase 24.11 and Dipeptidyl Peptidase IV are Both Involved in Regulating the Metabolic Stability of Glucagon-like Peptide-1 in vivo. In Dipeptidyl Aminopeptidases in Health and Disease, Back, N., Cohen, I.R., Kritchevsky, D., Lajtha, A., Paoletti, R., Eds.; Springer US: Boston, MA, 2003; pp. 303-312.
- Collins L, C.R. Glucagon-Like Peptide-1 Receptor Agonists. In StatPearls [Internet]; 2025.
- Gupta, V. Glucagon-like peptide-1 analogues: An overview. Indian J Endocrinol Metab 2013, 17, 413-421, doi:IJEM-17-413 [pii]10.4103/2230-8210.111625. [CrossRef]
- Li, Y.; Duffy, K.B.; Ottinger, M.A.; Ray, B.; Bailey, J.A.; Holloway, H.W.; Tweedie, D.; Perry, T.; Mattson, M.P.; Kapogiannis, D.; et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. Journal of Alzheimer’s disease : JAD 2010, 19, 1205–1219. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Jiang, R.; Xu, Y.; Zhao, X.; Li, Y. Exendin-4 antagonizes Aβ1-42-induced attenuation of spatial learning and memory ability. Exp Ther Med 2016, 12, 2885–2892. [Google Scholar] [CrossRef]
- Garabadu, D.; Verma, J. Exendin-4 attenuates brain mitochondrial toxicity through PI3K/Akt-dependent pathway in amyloid beta (1–42)-induced cognitive deficit rats. Neurochemistry International 2019, 128, 39–49. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Xu, Z.; Chen, S.; Yao, W.; Gao, X. GLP-1 receptor agonists downregulate aberrant GnT-III expression in Alzheimer’s disease models through the Akt/GSK-3β/β-catenin signaling. Neuropharmacology 2018, 131, 190–199. [Google Scholar] [CrossRef]
- Song, X.; Sun, Y.; Wang, Z.; Su, Y.; Wang, Y.; Wang, X. Exendin-4 alleviates β-Amyloid peptide toxicity via DAF-16 in a Caenorhabditis elegans model of Alzheimer’s disease. Frontiers in Aging Neuroscience 2022, 14. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Kahan, J.; Ell, P.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J Parkinsons Dis 2014, 4, 337-344, doi:H26015442R458926 [pii]10.3233/JPD-140364. [CrossRef]
- Mullins, R.J.; Mustapic, M.; Chia, C.W.; Carlson, O.; Gulyani, S.; Tran, J.; Li, Y.; Mattson, M.P.; Resnick, S.; Egan, J.M.; et al. A Pilot Study of Exenatide Actions in Alzheimer’s Disease. Curr Alzheimer Res 2019, 16, 741-752, doi:CAR-EPUB-100792 [pii]10.2174/1567205016666190913155950. [CrossRef]
- Kumar, V.; Xin, X.; Ma, J.; Tan, C.; Osna, N.; Mahato, R.I. Therapeutic targets, novel drugs, and delivery systems for diabetes associated NAFLD and liver fibrosis. Adv Drug Deliv Rev 2021, 176, 113888, doi:S0169-409X(21)00280-5 [pii]10.1016/j.addr.2021.113888. [CrossRef]
- Meece, J. Pharmacokinetics and Pharmacodynamics of Liraglutide, a Long-Acting, Potent Glucagon-Like Peptide-1 Analog. Pharmacotherapy 2009, 29, 33S–42S. [Google Scholar] [CrossRef]
- Jantrapirom, S.; Nimlamool, W.; Chattipakorn, N.; Chattipakorn, S.; Temviriyanukul, P.; Inthachat, W.; Govitrapong, P.; Potikanond, S. Liraglutide Suppresses Tau Hyperphosphorylation, Amyloid Beta Accumulation through Regulating Neuronal Insulin Signaling and BACE-1 Activity. Int J Mol Sci 2020, 21, doi:ijms21051725 [pii]ijms-21-01725 [pii]10.3390/ijms21051725. [CrossRef]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 2011, 31, 6587–6594. [Google Scholar] [CrossRef]
- McClean, P.L.; Hölscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 2014, 76 Pt A, 57–67. [Google Scholar] [CrossRef]
- McClean, P.L.; Jalewa, J.; Hölscher, C. Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav Brain Res 2015, 293, 96–106. [Google Scholar] [CrossRef]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Moller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Braendgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front Aging Neurosci 2016, 8, 108. [Google Scholar] [CrossRef]
- Watson, K.T.; Wroolie, T.E.; Tong, G.; Foland-Ross, L.C.; Frangou, S.; Singh, M.; McIntyre, R.S.; Roat-Shumway, S.; Myoraku, A.; Reiss, A.L.; et al. Neural correlates of liraglutide effects in persons at risk for Alzheimer’s disease. Behav Brain Res 2019, 356, 271-278, doi:S0166-4328(18)30643-0 [pii]10.1016/j.bbr.2018.08.006. [CrossRef]
- Lau, J.; Bloch, P.; Schaffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J Med Chem 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Chang, Y.F.; Zhang, D.; Hu, W.M.; Liu, D.X.; Li, L. Semaglutide-mediated protection against Abeta correlated with enhancement of autophagy and inhibition of apotosis. J Clin Neurosci 2020, 81, 234-239, doi:S0967-5868(20)31533-2 [pii]10.1016/j.jocn.2020.09.054. [CrossRef]
- Yang, X.; Feng, P.; Zhang, X.; Li, D.; Wang, R.; Ji, C.; Li, G.; Holscher, C. The diabetes drug semaglutide reduces infarct size, inflammation, and apoptosis, and normalizes neurogenesis in a rat model of stroke. Neuropharmacology 2019, 158, 107748, doi:S0028-3908(19)30307-7 [pii]10.1016/j.neuropharm.2019.107748. [CrossRef]
- Zhang, Y.; Tang, C.; He, Y.; Zhang, Y.; Li, Q.; Zhang, T.; Zhao, B.; Tong, A.; Zhong, Q.; Zhong, Z. Semaglutide ameliorates Alzheimer’s disease and restores oxytocin in APP/PS1 mice and human brain organoid models. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2024, 180, 117540. [Google Scholar] [CrossRef]
- A Research Study Investigating Semaglutide in People With Early Alzheimer’s Disease (EVOKE Plus). 2024.
- Cummings, J.L.; Atri, A.; Feldman, H.H.; Hansson, O.; Sano, M.; Knop, F.K.; Johannsen, P.; León, T.; Scheltens, P. evoke and evoke+: design of two large-scale, double-blind, placebo-controlled, phase 3 studies evaluating efficacy, safety, and tolerability of semaglutide in early-stage symptomatic Alzheimer’s disease. Alzheimer’s research & therapy 2025, 17, 14. [Google Scholar] [CrossRef]
- Deryusheva, E.I.; Shevelyova, M.P.; Rastrygina, V.A.; Nemashkalova, E.L.; Vologzhannikova, A.A.; Machulin, A.V.; Nazipova, A.A.; Permyakova, M.E.; Permyakov, S.E.; Litus, E.A. In Search for Low-Molecular-Weight Ligands of Human Serum Albumin That Affect Its Affinity for Monomeric Amyloid beta Peptide. Int J Mol Sci 2024, 25, doi:ijms25094975 [pii]ijms-25-04975 [pii]10.3390/ijms25094975. [CrossRef]
- Kamynina, A.V.; Esteras, N.; Koroev, D.O.; Bobkova, N.V.; Balasanyants, S.M.; Simonyan, R.A.; Avetisyan, A.V.; Abramov, A.Y.; Volpina, O.M. Synthetic Fragments of Receptor for Advanced Glycation End Products Bind Beta-Amyloid 1–40 and Protect Primary Brain Cells From Beta-Amyloid Toxicity. Frontiers in Neuroscience 2018, 12. [Google Scholar] [CrossRef]
- Lv, J.; Pan, Y.; Li, X.; Cheng, D.; Liu, S.; Shi, H.; Zhang, Y. The Imaging of Insulinomas Using a Radionuclide-Labelled Molecule of the GLP-1 Analogue Liraglutide: A New Application of Liraglutide. PloS one 2014, 9, e96833. [Google Scholar] [CrossRef]
- Jacobsen, L.V.; Flint, A.; Olsen, A.K.; Ingwersen, S.H. Liraglutide in Type 2 Diabetes Mellitus: Clinical Pharmacokinetics and Pharmacodynamics. Clinical pharmacokinetics 2016, 55, 657–672. [Google Scholar] [CrossRef]
- Yang, X.D.; Yang, Y.Y. Clinical Pharmacokinetics of Semaglutide: A Systematic Review. Drug design, development and therapy 2024, 18, 2555–2570. [Google Scholar] [CrossRef]
- Mehta, P.D.; Pirttila, T.; Mehta, S.P.; Sersen, E.A.; Aisen, P.S.; Wisniewski, H.M. Plasma and cerebrospinal fluid levels of amyloid beta proteins 1-40 and 1-42 in Alzheimer disease. Archives of neurology 2000, 57, 100–105. [Google Scholar] [CrossRef]
- Zapadka, K.L.; Becher, F.J.; Uddin, S.; Varley, P.G.; Bishop, S.; Gomes Dos Santos, A.L.; Jackson, S.E. A pH-Induced Switch in Human Glucagon-like Peptide-1 Aggregation Kinetics. J Am Chem Soc 2016, 138, 16259–16265. [Google Scholar] [CrossRef]
- Prada Brichtova, E.; Krupova, M.; Bour, P.; Lindo, V.; Gomes Dos Santos, A.; Jackson, S.E. Glucagon-like peptide 1 aggregates into low-molecular-weight oligomers off-pathway to fibrillation. Biophys J 2023, 122, 2475-2488, doi:S0006-3495(23)00298-9 [pii]10.1016/j.bpj.2023.04.027. [CrossRef]
- Wang, K.; Chen, K. Direct Assessment of Oligomerization of Chemically Modified Peptides and Proteins in Formulations using DLS and DOSY-NMR. Pharm Res 2023, 40, 1329-1339, doi:10.1007/s11095-022-03468-8 [pii]10.1007/s11095-022-03468-8. [CrossRef] [PubMed]
- Venanzi, M.; Savioli, M.; Cimino, R.; Gatto, E.; Palleschi, A.; Ripani, G.; Cicero, D.; Placidi, E.; Orvieto, F.; Bianchi, E. A spectroscopic and molecular dynamics study on the aggregation process of a long-acting lipidated therapeutic peptide: the case of semaglutide. Soft Matter 2020, 16, 10122–10131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lomakin, A.; Kanai, S.; Alex, R.; Benedek, G.B. Transformation of oligomers of lipidated peptide induced by change in pH. Mol Pharm 2015, 12, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Gast, K.; Evers, A.; Kurz, M.; Pfeiffer-Marek, S.; Schuler, A.; Seckler, R.; Thalhammer, A. A Conserved Hydrophobic Moiety and Helix-Helix Interactions Drive the Self-Assembly of the Incretin Analog Exendin-4. Biomolecules 2021, 11, doi:biom11091305 [pii]biomolecules-11-01305 [pii]10.3390/biom11091305. [CrossRef] [PubMed]
- Calanna, S.; Christensen, M.; Holst, J.; Laferrère, B.; Gluud, L.; Vilsbøll, T.; Knop, F. Secretion of Glucagon-Like Peptide-1 in Patients With Type 2 Diabetes Mellitus - Systematic Review and Meta-Analysis of Clinical Studies. Diabetologia 2013, 56. [Google Scholar] [CrossRef]
- Balks, H.J.; Holst, J.J.; von zur Mühlen, A.; Brabant, G. Rapid Oscillations in Plasma Glucagon-Like Peptide-1 (GLP-1) in Humans: Cholinergic Control of GLP-1 Secretion via Muscarinic Receptors1. The Journal of Clinical Endocrinology & Metabolism 1997, 82, 786–790. [Google Scholar] [CrossRef]
- Fineman, M.; Flanagan, S.; Taylor, K.; Aisporna, M.; Shen, L.Z.; Mace, K.F.; Walsh, B.; Diamant, M.; Cirincione, B.; Kothare, P.; et al. Pharmacokinetics and Pharmacodynamics of Exenatide Extended-Release After Single and Multiple Dosing. Clinical pharmacokinetics 2011, 50, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Kastin, A.J.; Akerstrom, V. Entry of exendin-4 into brain is rapid but may be limited at high doses. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 2003, 27, 313–318. [Google Scholar] [CrossRef]
- Gabery, S.; Salinas, C.G.; Paulsen, S.J.; Ahnfelt-Rønne, J.; Alanentalo, T.; Baquero, A.F.; Buckley, S.T.; Farkas, E.; Fekete, C.; Frederiksen, K.S.; et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI insight 2020, 5. [Google Scholar] [CrossRef]
- Lovshin, J.A.; Drucker, D.J. Incretin-based therapies for type 2 diabetes mellitus. Nature Reviews Endocrinology 2009, 5, 262–269. [Google Scholar] [CrossRef]
- Dejgaard, T.; Frandsen, C.; Kielgast, U.; Størling, J.; Overgaard, A.; Svane, M.; Olsen, M.H.; Thorsteinsson, B.; Andersen, H.; Krarup, T.; et al. Liraglutide enhances insulin secretion and prolongs the remission period in adults with newly diagnosed type 1 diabetes (the NewLira study): A randomized, double-blind, placebo-controlled trial. Diabetes, obesity & metabolism 2024, 26. [Google Scholar] [CrossRef]
- Zheng, Z.; Zong, Y.; Ma, Y.; Tian, Y.; Pang, Y.; Zhang, C.; Gao, J. Glucagon-like peptide-1 receptor: mechanisms and advances in therapy. Signal Transduction and Targeted Therapy 2024, 9, 234. [Google Scholar] [CrossRef]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28, 1071-1081 e1073, doi:S0969-2126(20)30209-4 [pii]10.1016/j.str.2020.06.006. [CrossRef] [PubMed]
- Jarero-Basulto, J.J.; Gasca-Martínez, Y.; Rivera-Cervantes, M.C.; Gasca-Martínez, D.; Carrillo-González, N.J.; Beas-Zárate, C.; Gudiño-Cabrera, G. Cytotoxic Effect of Amyloid-β1-42 Oligomers on Endoplasmic Reticulum and Golgi Apparatus Arrangement in SH-SY5Y Neuroblastoma Cells. NeuroSci 2024, 5, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Vander Zanden, C.M.; Chi, E.Y. Passive Immunotherapies Targeting Amyloid Beta and Tau Oligomers in Alzheimer’s Disease. Journal of pharmaceutical sciences 2020, 109, 68–73. [Google Scholar] [CrossRef]
- Chang, Y.F.; Zhang, D.; Hu, W.M.; Liu, D.X.; Li, L. Semaglutide-mediated protection against Aβ correlated with enhancement of autophagy and inhibition of apotosis. J Clin Neurosci 2020, 81, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Wang, L.X.; Chen, Z.; Liu, L.B. Liraglutide prevents beta-amyloid-induced neurotoxicity in SH-SY5Y cells via a PI3K-dependent signaling pathway. Neurological research 2016, 38, 313–319. [Google Scholar] [CrossRef]
- Litus, E.A.; Kazakov, A.S.; Deryusheva, E.I.; Nemashkalova, E.L.; Shevelyova, M.P.; Machulin, A.V.; Nazipova, A.A.; Permyakova, M.E.; Uversky, V.N.; Permyakov, S.E. Ibuprofen Favors Binding of Amyloid-beta Peptide to Its Depot, Serum Albumin. Int J Mol Sci 2022, 23, doi:ijms23116168 [pii]ijms-23-06168 [pii]10.3390/ijms23116168. [CrossRef]
- Catanzariti, A.M.; Soboleva, T.A.; Jans, D.A.; Board, P.G.; Baker, R.T. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci 2004, 13, 1331-1339, doi:13/5/1331 [pii]0131331 [pii]10.1110/ps.04618904. [CrossRef]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci 1995, 4, 2411–2423. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Mallender, W.D.; Inestrosa, N.C.; Rosenberry, T.L. Thioflavin T is a fluorescent probe of the acetylcholinesterase peripheral site that reveals conformational interactions between the peripheral and acylation sites. J Biol Chem 2001, 276, 23282-23287, doi:S0021-9258(20)78313-4 [pii]10.1074/jbc.M009596200. [CrossRef]
- Deryusheva, E.I.; Shevelyova, M.P.; Rastrygina, V.A.; Nemashkalova, E.L.; Vologzhannikova, A.A.; Machulin, A.V.; Nazipova, A.A.; Permyakova, M.E.; Permyakov, S.E.; Litus, E.A. In Search for Low-Molecular-Weight Ligands of Human Serum Albumin That Affect Its Affinity for Monomeric Amyloid β Peptide. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: a point where biology waits for physics. Protein Sci 2002, 11, 739-756, doi:0110739 [pii]10.1110/ps.4210102. [CrossRef] [PubMed]
- Berman, H.M.; Burley, S.K. Protein Data Bank (PDB): Fifty-three years young and having a transformative impact on science and society. Quarterly reviews of biophysics 2025, 58, e9. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The alignment of amino acid sequences (panel A) and model structures of the GLP-1RAs (B-E): Sema (B: based on PDB entry 7KI0, EM, chain E), Exen PDB ID 1JRJ, NMR, chain A, model 1), GLP-1(7-37) (D: PDB ID 3IOL, X-ray, chain B) and Lira (E: PDB ID 4APD, NMR, chains A, B, model 1). The amino acid residues that differ from those in GLP-1 are marked in orange (Aib, 2-aminoisobutyric acid). The lysine residues of Sema and Lira modified by the linkers with fatty acids are highlighted in blue. The numbering of the residues is according to the PDB entries.
Figure 1.
The alignment of amino acid sequences (panel A) and model structures of the GLP-1RAs (B-E): Sema (B: based on PDB entry 7KI0, EM, chain E), Exen PDB ID 1JRJ, NMR, chain A, model 1), GLP-1(7-37) (D: PDB ID 3IOL, X-ray, chain B) and Lira (E: PDB ID 4APD, NMR, chains A, B, model 1). The amino acid residues that differ from those in GLP-1 are marked in orange (Aib, 2-aminoisobutyric acid). The lysine residues of Sema and Lira modified by the linkers with fatty acids are highlighted in blue. The numbering of the residues is according to the PDB entries.
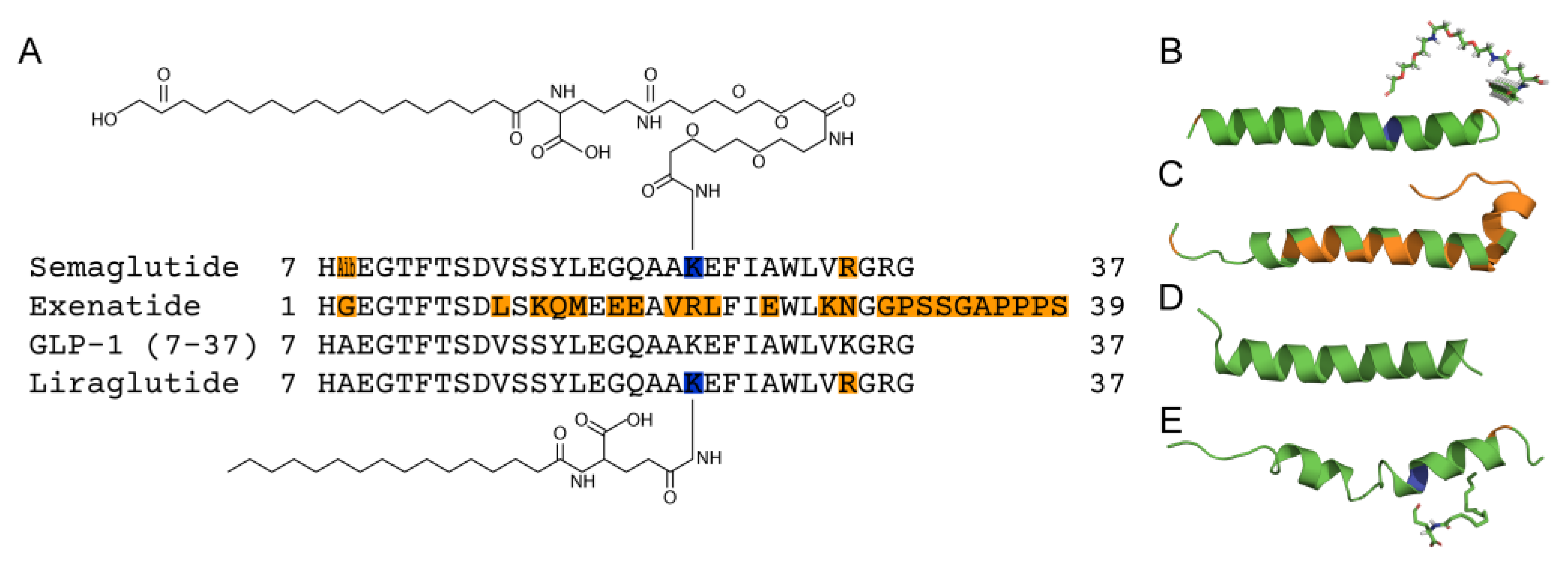
Figure 2.
Kinetics of GLP-1(7-37) (blue), Exen (red), Lira (violet) and Sema (green) interaction with monomeric Aβ40 (panels A-D) or Aβ42 (E-H) immobilized on sensor surface by amine coupling, monitored using BLI at 25°C (20 mM HEPES-KOH/Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, pH 7.4). The analyte concentrations are indicated nearby the sensograms. The black curves are theoretical, calculated according to the single binding site scheme (1) or heterogeneous ligand model (2) (see Table 1 for the fitting parameters).
Figure 2.
Kinetics of GLP-1(7-37) (blue), Exen (red), Lira (violet) and Sema (green) interaction with monomeric Aβ40 (panels A-D) or Aβ42 (E-H) immobilized on sensor surface by amine coupling, monitored using BLI at 25°C (20 mM HEPES-KOH/Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, pH 7.4). The analyte concentrations are indicated nearby the sensograms. The black curves are theoretical, calculated according to the single binding site scheme (1) or heterogeneous ligand model (2) (see Table 1 for the fitting parameters).
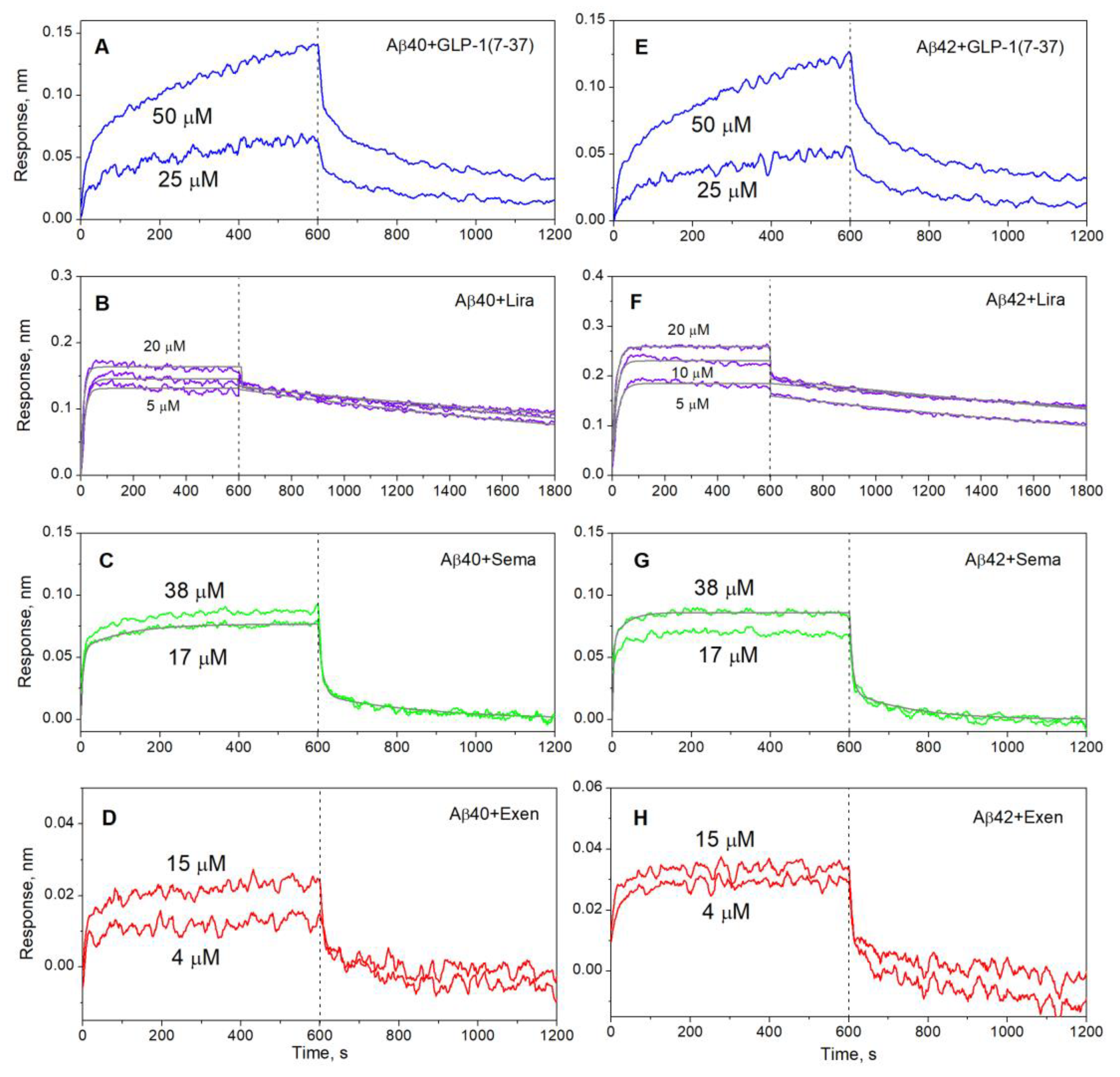
Figure 3.
Kinetics of interaction between monomeric Aβ40 (panels A-C) or Aβ42 (D-F) and Lira (violet), Sema (green) or Exen (red) at 25°C, monitored using SPR (10 mM HEPES-NaOH, 150 mM NaCl, 0.05% Tween 20, pH 7.4). The analyte concentrations are indicated nearby the sensograms. The black curves are theoretical, calculated according to the heterogeneous ligand model (2) (see Table 2 for the fitting parameters).
Figure 3.
Kinetics of interaction between monomeric Aβ40 (panels A-C) or Aβ42 (D-F) and Lira (violet), Sema (green) or Exen (red) at 25°C, monitored using SPR (10 mM HEPES-NaOH, 150 mM NaCl, 0.05% Tween 20, pH 7.4). The analyte concentrations are indicated nearby the sensograms. The black curves are theoretical, calculated according to the heterogeneous ligand model (2) (see Table 2 for the fitting parameters).
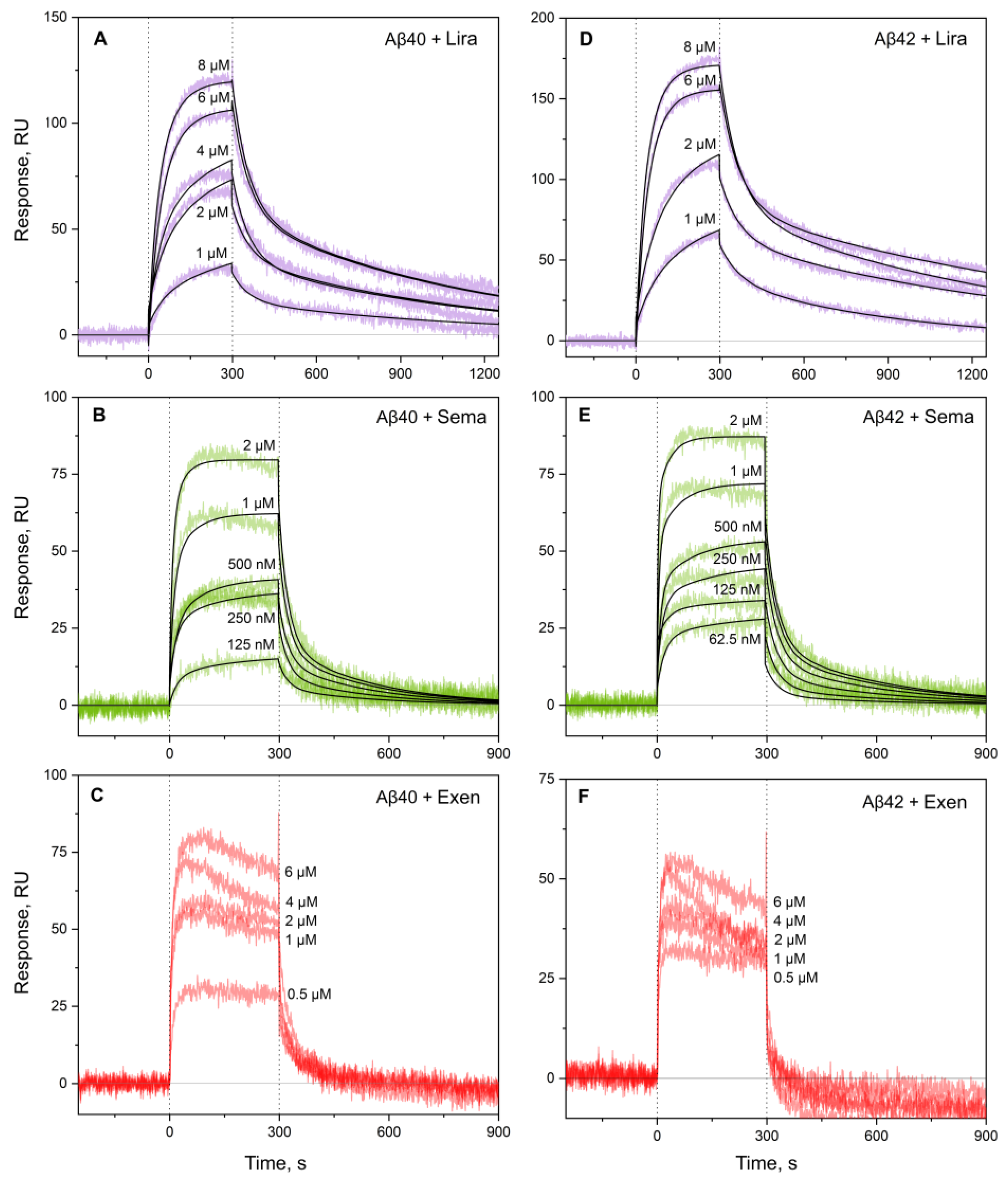
Figure 4.
Kinetics of fluorescence intensity at 485 nm of 10 μM ThT added to 20 µM Aβ40 in the absence or in the presence of 10 µM Lira (panel A), Sema, Exen, or GLP-1(7-37) (B) at 30°C (25 mM Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 0.05% NaN3, pH 7.4). Standard deviations of the fluorescence signals are indicated. Excitation wavelength 440 nm.
Figure 4.
Kinetics of fluorescence intensity at 485 nm of 10 μM ThT added to 20 µM Aβ40 in the absence or in the presence of 10 µM Lira (panel A), Sema, Exen, or GLP-1(7-37) (B) at 30°C (25 mM Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 0.05% NaN3, pH 7.4). Standard deviations of the fluorescence signals are indicated. Excitation wavelength 440 nm.
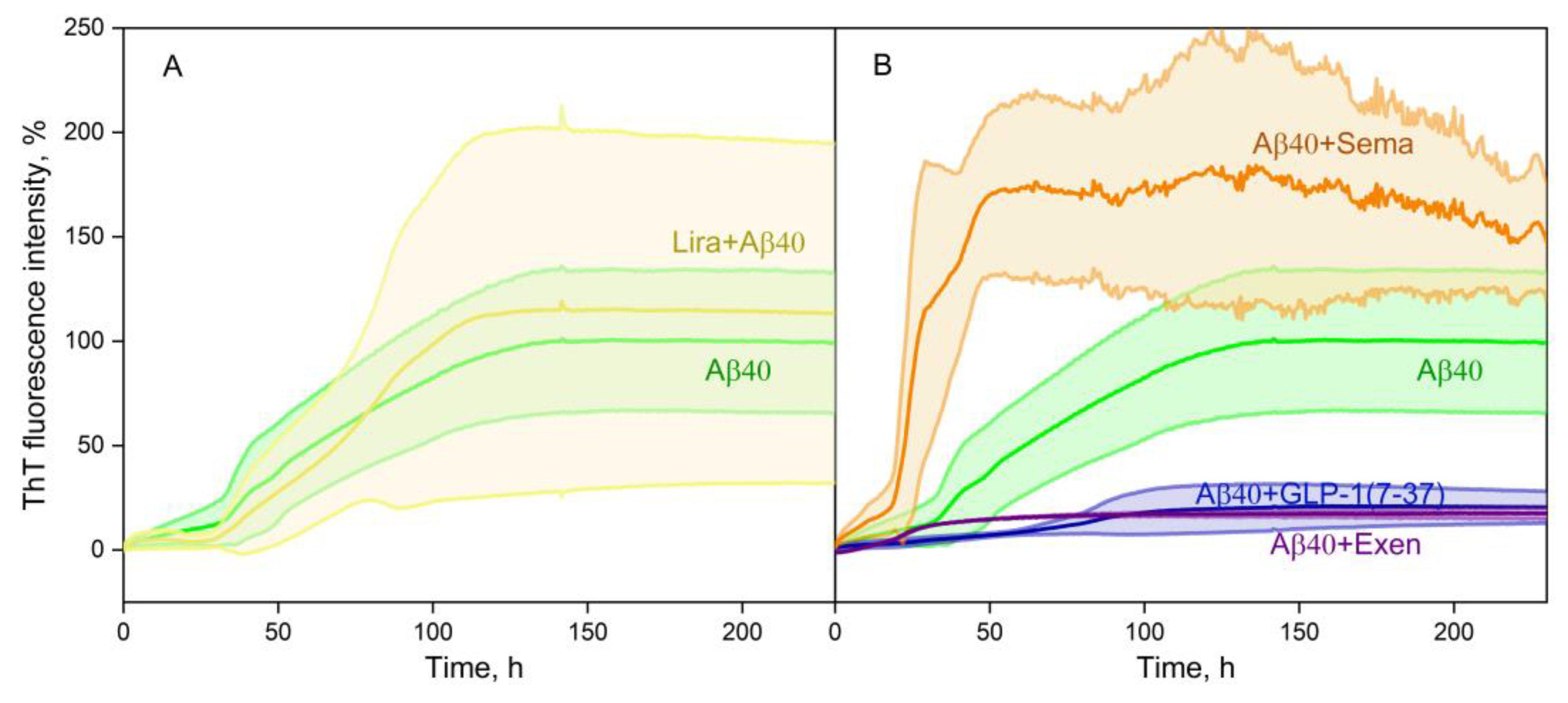
Figure 5.
Negative-staining TEM images of the Aβ40 fibers grown in the course of the ThT fluorescence assay shown in Figure 4 in the absence (panel A) or in the presence of 10 μM Lira (B), 10 μM Sema (C), 10 μM Exen (D), GLP-1(7-37) (E, F). The scale bars represent 1 μm .
Figure 5.
Negative-staining TEM images of the Aβ40 fibers grown in the course of the ThT fluorescence assay shown in Figure 4 in the absence (panel A) or in the presence of 10 μM Lira (B), 10 μM Sema (C), 10 μM Exen (D), GLP-1(7-37) (E, F). The scale bars represent 1 μm .
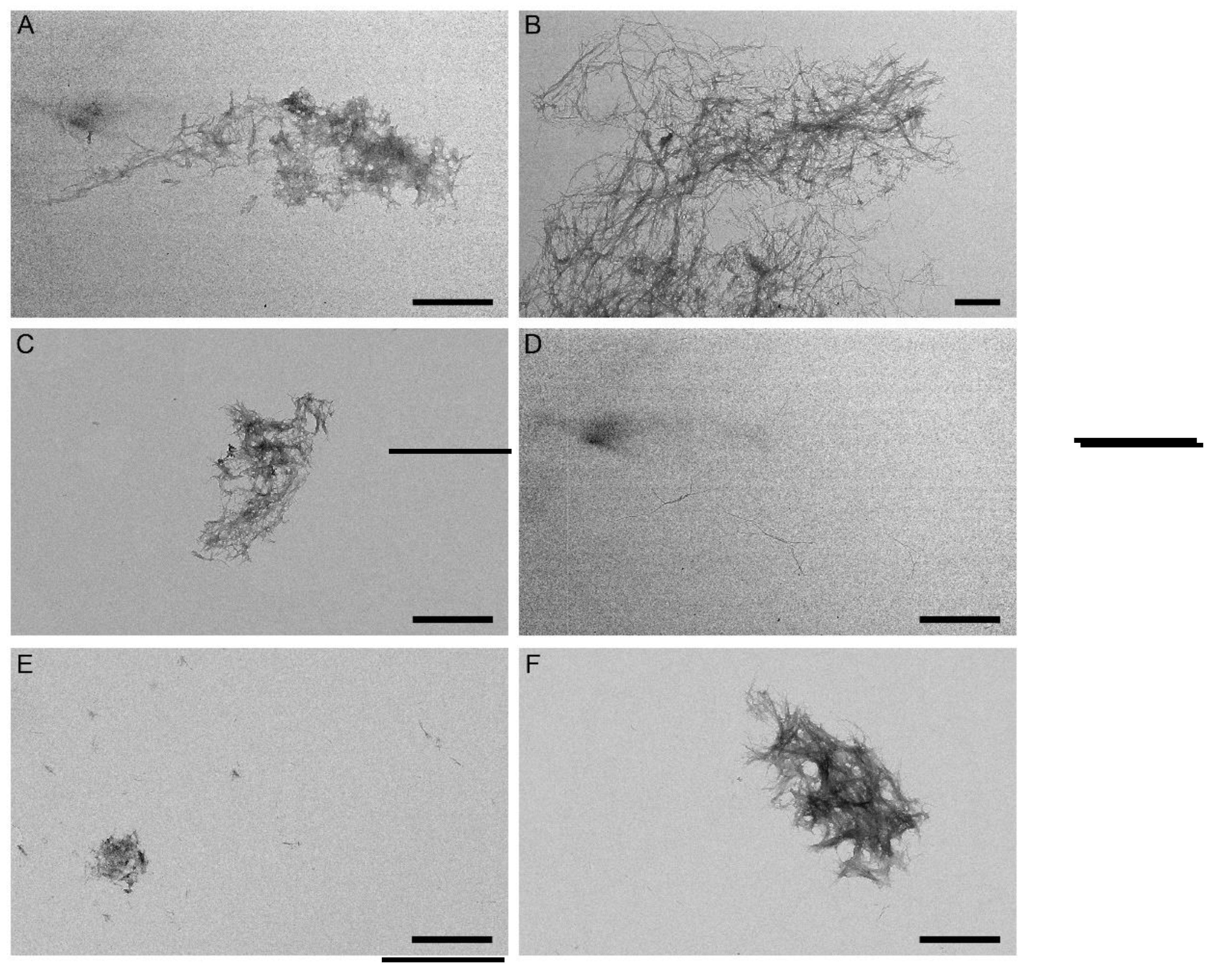
Figure 6.
The models of tertiary structures of the complexes between monomeric Aβ40 (taken from PDB entry 2LFM, model 1) or Aβ40 protofibril (PDB entry 2LMN, model 1) (shown in green) and GLP-1(7-37) (PDB entry 3IOL, chain B) (panels A, C) or Exen (PDB entry 1JRJ, chain A) (B, D) (colored cyan) built using ClusPro docking server. The contact residues are shown in orange. The numbering of the residues is according to the PDB entries.
Figure 6.
The models of tertiary structures of the complexes between monomeric Aβ40 (taken from PDB entry 2LFM, model 1) or Aβ40 protofibril (PDB entry 2LMN, model 1) (shown in green) and GLP-1(7-37) (PDB entry 3IOL, chain B) (panels A, C) or Exen (PDB entry 1JRJ, chain A) (B, D) (colored cyan) built using ClusPro docking server. The contact residues are shown in orange. The numbering of the residues is according to the PDB entries.
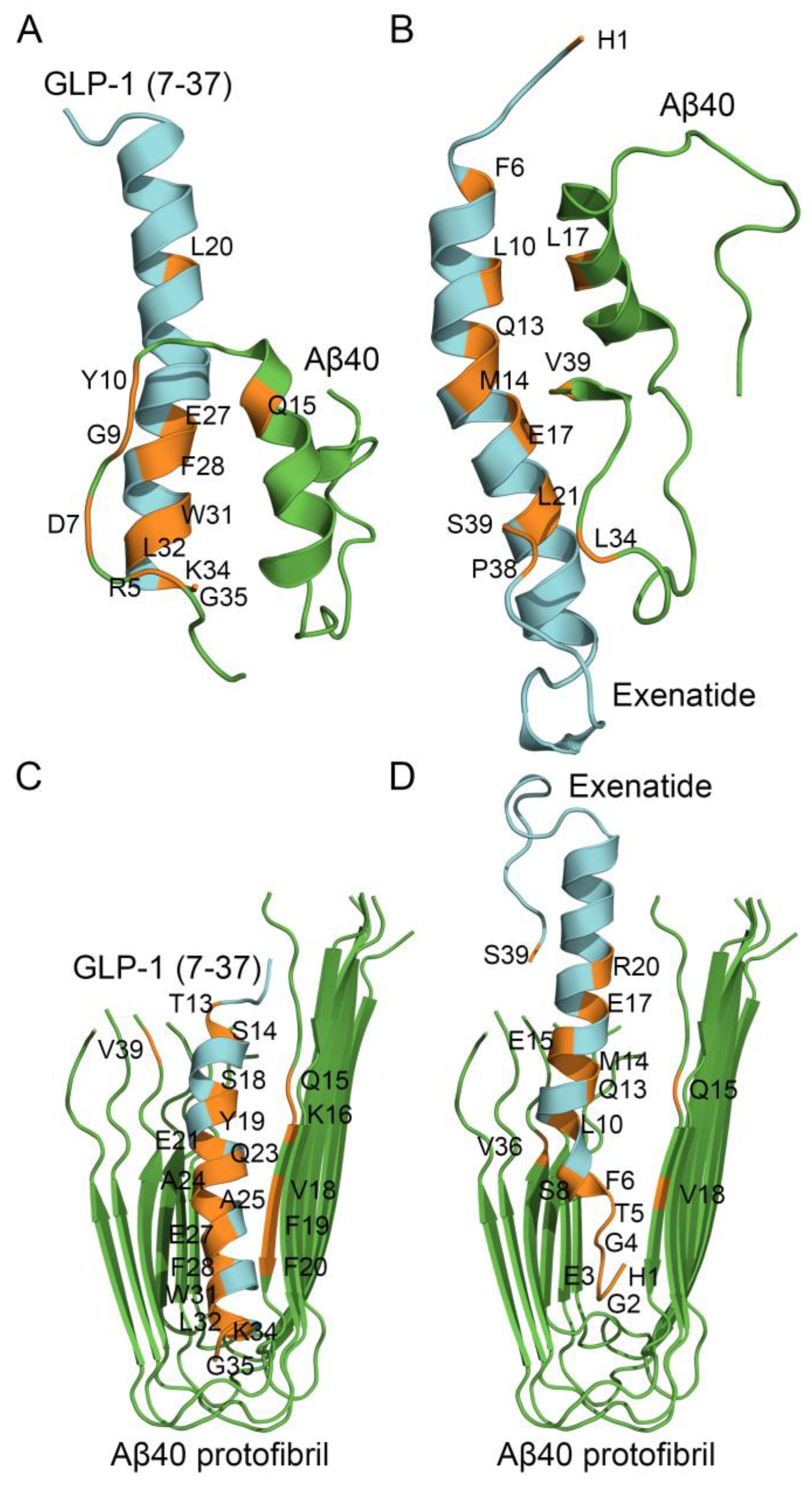
Figure 7.
Effect of 10 µM Aβ40/Aβ42 and/or 10 µM Lira/Sema/Exen/GLP-1(7-37) on viability of SH-SY5Y cells assessed by an MTT assay (serum-free medium, 5% CO2, 37°C, incubation for 48 h). A, effect of the GLP-1RAs on the cell viability. B, effect of Aβ40 and the GLP-1RAs in the presence of Aβ40 on the cell viability. C, effect of Aβ42 and the GLP-1RAs in the presence of Aβ42 on the cell viability. Mean values and standard deviations are indicated.
Figure 7.
Effect of 10 µM Aβ40/Aβ42 and/or 10 µM Lira/Sema/Exen/GLP-1(7-37) on viability of SH-SY5Y cells assessed by an MTT assay (serum-free medium, 5% CO2, 37°C, incubation for 48 h). A, effect of the GLP-1RAs on the cell viability. B, effect of Aβ40 and the GLP-1RAs in the presence of Aβ40 on the cell viability. C, effect of Aβ42 and the GLP-1RAs in the presence of Aβ42 on the cell viability. Mean values and standard deviations are indicated.
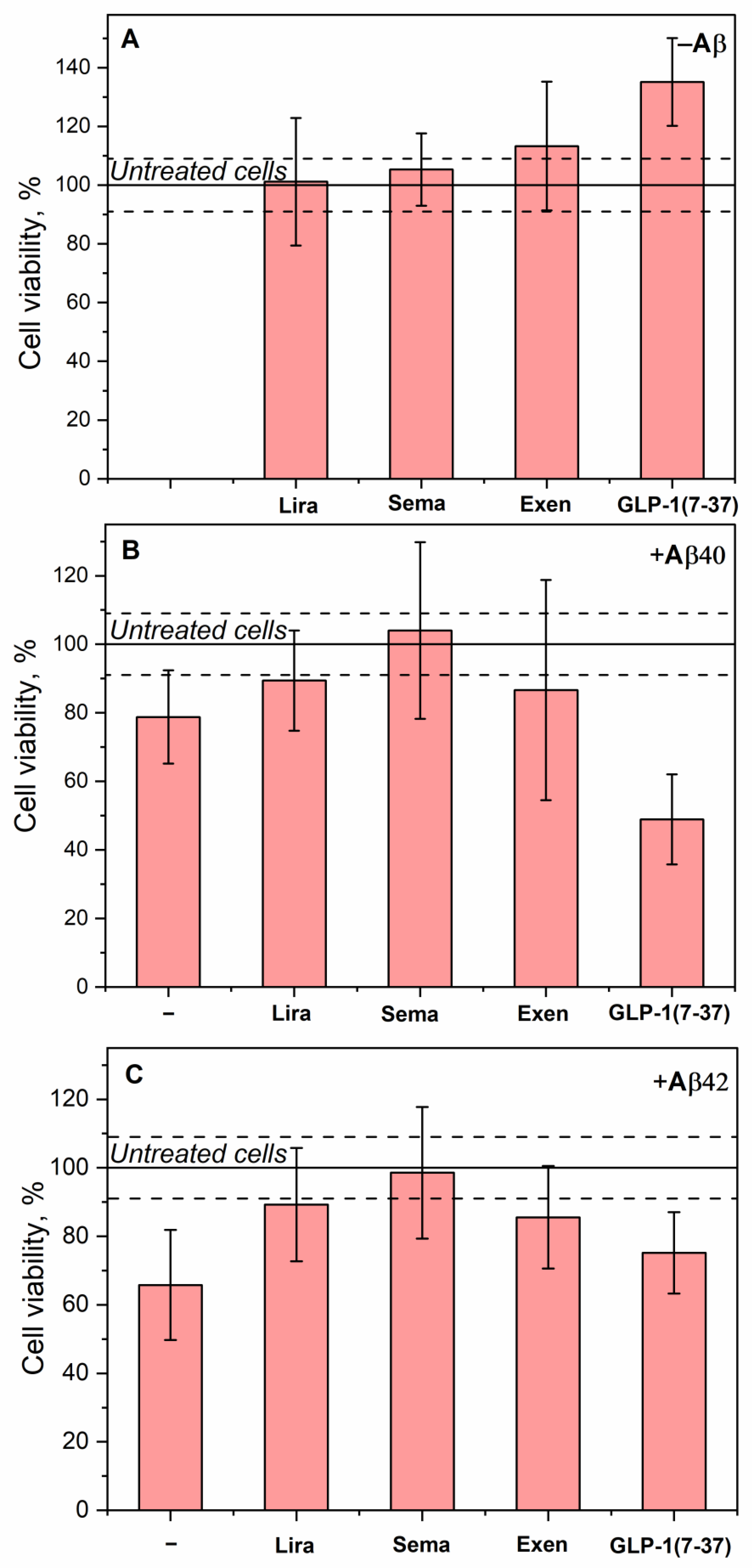

Table 1.
Parameters of the interaction between monomeric Aβ40/Aβ42 and the GLP-1RAs at 25°C, estimated from the BLI data shown in Figure 2 using either single binding site model (1) or heterogeneous ligand scheme (2).
Table 1.
Parameters of the interaction between monomeric Aβ40/Aβ42 and the GLP-1RAs at 25°C, estimated from the BLI data shown in Figure 2 using either single binding site model (1) or heterogeneous ligand scheme (2).
| [Lira], µM | ka, M-1s-1 | kd, s-1 | KD, M | ka, M-1s-1 | kd, s-1 | KD, M | |
| Lira | Aβ40 | Aβ42 | |||||
| 20 | (8.4±2.8)×103 | (9.0±0.4)×10-4 | (1.1±0.4)×10-7 | (5.7±1.1)×103 | (6.0±0.2)×10-4 | (1.1±0.2)×10-7 | |
| 10 | (7.3±0.4)×103 | (3.46±0.07)×10-4 | (4.8±0.3)×10-8 | (8.0±0.7)×103 | (4.82±0.12)×10-4 | (6.0±0.2)×10-8 | |
| 5 | (1.34±0.09)×104 | (5.56±0.11)×10-4 | (4.2±0.3)×10-8 | (1.21±0.08)×104 | (5.40±0.12)×10-4 | (4.5±0.3)×10-8 | |
| [Sema], µM | ka1, M-1s-1 | kd1, s-1 | KD1, M | ka2, M-1s-1 | kd2, s-1 | KD2, M | |
| Sema | Aβ40 | ||||||
| 17 | 310±52 | (3.7±0.2)×10-3 | (1.2±0.2)×10-5 | (5.7±1.1)×103 | (9.1±0.6)×10-2 | (1.6±0.3)×10-5 | |
| Aβ42 | |||||||
| 38 | 582±104 | (6.4±0.6)×10-3 | (1.1±0.2)×10-5 | (6.1±2.7)×103 | (1.34±0.15)×10-1 | (2.2±1.0)×10-5 | |
Table 2.
Parameters of the interaction between monomeric Aβ40/Aβ42 and the GLP-1RAs at 25°C, estimated from the SPR data shown in Figure 3 using the heterogeneous ligand model (2).
Table 2.
Parameters of the interaction between monomeric Aβ40/Aβ42 and the GLP-1RAs at 25°C, estimated from the SPR data shown in Figure 3 using the heterogeneous ligand model (2).
| [GLP-1RA], μM | ka1, M-1s-1 | kd1, s-1 | KD1, M | ka2, M-1s-1 | kd2, s-1 | KD2, M | |
| Aβ40 | |||||||
| Sema | 0.06-2 | (9.6±2.4)×103 | (3.2±0.9)×10-3 | (3.4±0.5)×10-7 | (3.90±1.12)×104 | (4.32±0.12)×10-2 | (1.2±0.4)×10-6 |
| Lira | 1-8 | (1.44±0.05)×103 | (1.19±0.13)×10-3 | (9.5±0.7)×10-7 | (1.9±0.3)×103 | (1.70±0.10)×10-2 | (9.1±1.6)×10-6 |
| Aβ42 | |||||||
| Sema | 0.06-2 | (1.26±0.11)×104 | (4.1±1.4)×10-3 | (3.4±1.4)×10-7 | (1.34±0.18)×105 | (3.8±0.7)×10-2 | (3.0±0.9)×10-7 |
| Lira | 1-8 | (2.24±0.18)×103 | (1.14±0.10)×10-3 | (5.2±0.8)×10-7 | (2.9±0.04)×103 | (1.64±0.14)×10-2 | (5.6±0.3)×10-6 |
Table 3.
Concentration dependence of hydrodynamic radius (Rh), molecular mass (MWRh) and degree of multimerization (MWRh/MWm) for the GLP-1RAs at 25oC, determined by DLS (25 mM Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, pH 7.4).
Table 3.
Concentration dependence of hydrodynamic radius (Rh), molecular mass (MWRh) and degree of multimerization (MWRh/MWm) for the GLP-1RAs at 25oC, determined by DLS (25 mM Tris-HCl, 140 mM NaCl, 4.9 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, pH 7.4).
| GLP-1RA | [GLP-1RA], µM | Rh, nm | MWRh, kDa | MWRh/MWm |
|---|---|---|---|---|
| GLP-1(7-37) | 5-83 | >92 | >7×105 | >210 |
| Lira | 105 | 3.08±0.15 | 54.7±7.8 | 15.6±2.2 |
| 52 | 3.13±0.05 | 57.1±2.6 | 16.3±0.7 | |
| 13 | 2.25±0.12 | 22.7±6.1 | 6.5±1.7 | |
| 6 | 2.45±0.16 | 28.8±3.6 | 8.2±1.0 | |
| Exen | 234 | 2.20±0.01 | 24.58±0.02 | 5.88±0.04 |
| 115 | 2.27±0.16 | 26.7±5.7 | 6.4±1.4 | |
| 29 | 1.53±0.06 | 8.8±0.9 | 2.1±0.2 | |
| 15 | 1.39±0.07 | 6.7±0.9 | 1.6±0.2 | |
| Sema | 47 | 1.22±0.04 | 4.1±0.4 | 1.2±0.1 |
| 12 | 1.42±0.15 | 6.2±2.1 | 1.9±0.6 |
Table 4.
Summary of the properties of the GLP1-RAs obtained in this work and described in the literature. The counteracting effects are highlighted in bold.
Table 4.
Summary of the properties of the GLP1-RAs obtained in this work and described in the literature. The counteracting effects are highlighted in bold.
| Minimal KD for Aβ Binding According to BLI | Effect on Aβ40 Fibrillation (Figure 4 and Figure 5) | Effect on Aβ Cytotoxicity to SH-SY5Y Cells (Figure 7) | Ability to Cross the BBB | AD Animal Data | Clinical Data, AD | |
|---|---|---|---|---|---|---|
| Lira | 4.2×10-8 M | No effect | Protection | + [47] | Prevents memory loss, reduces Aβ amyloid deposits [47,48,49] | No effect [50] |
| Sema | 1.1×10-5 M | Stimulation | Protection | − [74] | Positive effects on cognitive function, reduction of Aβ amyloid deposits [55] | Phase 3 clinical trials (NCT04777396 and NCT04777409) |
| Exen | ~(0.4–1.5)×10-5 M | Inhibition | Protection | + [73] | Positive effects on learning and memory ability, reduces Aβ deposition [38,39,40,41] | No effect [43] |
| GLP-1(7-37) | ~(2.5–5.0)×10-5 M | Inhibition | Increases Aβ40 cytotoxicity | + [24] | Positive effects on learning and memory [29] | No data |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



