
Submitted:
20 April 2025
Posted:
21 April 2025
You are already at the latest version
Abstract
Ellipticine, a nitrogen containing compound of plant origin, possesses potent anticancer properties. Mechanism of intercalation into DNA helices and/or topoisomerase II inhibition is ascribed for its pharmaceutical aspects. Multiple biological activities of ellipticine have generated curiosity among researchers belonging to varied disciplines, primarily focussed on unraveling out its mode of action. Employing DFT based B3LYP and functional blended with 6-311 G (d, p) basis function from Gaussian 16 program, electronic parameters and global descriptors of the drug have been examined. A comparative analysis of calculated vibrational assignments of the drug molecule vis-à-vis experimental data from literature has been performed. Again, molecular docking analysis of ellipticine with isomerase transcriptases (PDB ID: 1did, 2ypi and 1xig) has been carried out to understand inhibition activity, binding sites etc.
Keywords:
ellipticine
; DFT
; MEP
; HOMO
; LUMO
; molecular docking
1. Introduction
Natural products play very important role in promoting healthcare and disease preven tion all over the world [1]. Extracted from Ochrosia elliptica leaves firstly in 1959, ellipticine and its derivatives exhibit pronounced anticancer activities [2,3]. The planer polycyclic chromophore consisting of protonatable ring nitrogen shows strong interaction with DNA via intercalation. Having simplest structure ellipticine and many of its derivative compounds are efficiently used in the treatment of brain tumours, acute myeloblastic leukaemia, kidney and breast cancer, HIV etc [4,5,6]. The antitumour property of ellipticine is caused to be due to its ability to intercalate into DNA and inhibit topoisomerases, enzymes that play very important role in the functions of DNA. Interfering replication and transcription processes, ellipticine can cause DNA damage leading to cell death in cancer cells. Despite its potential in cancer treatment, ellipticine has yet to be approved for clinical use due to concerns about its toxicity and poor solubility in water. However, researchers are exploring ways to modify the structure of ellipticine to improve its pharmacokinetics and reduce its side effects, and several derivatives of the compound are currently in development as potential anticancer agents [7].
Binding mechanism of small molecules to nucleic acid helices takes place in many ways such as major groove binding, minor groove binding, intercalation into successive base pairs etc. Aromatic polycyclic molecules having planar structure get inserted into two consecutive DNA base pairs and thereby elicit their pharmacological properties. Such intercalating molecules have drawn special attention of many researchers owing to their active role in antitumor chemotherapy. The strength of noncovalent stacking of most of the intercalating drugs inside DNA base pairs is usually attributed to the biological activity of the molecules. Oftenly intercalators are stabilized inside the nucleic acid helices by forming hydrogen and/or covalent bonds engaging side chain of the drug molecule [8,9,10]. Several earlier studies have been devoted to correlate the drug activity and intercalatibility; however, establishing definite correlation between intercalative mechanism and pharmacological activity of drug molecules is still to be understood [9,10,11].
In this paper in silico investigation of the physicochemical and electronic parameters, MEP surface, HOMO, LUMO energy of ellipticine have been investigated by DFT method [12,13,14]. Further, infrared spectrum of drug molecule has been computed and juxtaposed to experimental data. Elucidation of binding mechanism of ellipticine with three isomerase transcriptases taken from protein data bank with Id: 1did, 2ypi and 1xig has been accomplished for understanding biological aspects of the drug [15,16].
2. Computational Methods
Becke 3 Lee Yang Parr functional of density functional theory and 6-311G (d, p) basis function has been utilised. For this Gaussian 16 program has been used [17]. Visualization has been done by GaussView 6.0 software. Optimized geometry of ellipticine has been subjected to calculate molecular geometrical parameters, frontier molecular orbitals, molecular electrostatic potential surface as well as global reactivity descriptors [18]. Vibrational spectra has also been computed. Further, Swiss Dock web server has been used to simulate molecular docking of ellipticine drug with three protein receptors chosen from protein data bank. UCSF Chimera has been used for visulisation [19,20,21,22,23].
3. Results and Discussion
3.1. Molecular Structure and Global Reactivity Descriptors Analysis 3.1.1. Subsubsection
The optimized geometry of ellipticine is shown in Figure 1. The C-C, C-N and C-H bond lengths of ellipticine ranges from 1.370 to 1.509Å, 1.314 to 1.385Å and 1.081 to 1.095Å respectively; which are in conformity with experimental data4. The computed electronic parameters along with global reactivity descriptors of ellipticine drug are given in Table 1.
The MEP surface of ellipticine signifies its molecular shape and size as depicted in Figure 3. Shading in colours elaborates varied potential zones like positive, neutral, and negative regions and is also very useful in investigating biomolecular interactions etc. The most electronegative region occurs due to nitrogen atom in MEP map. Therefore, it is a suitable site of electrophilic attack. HOMO and LUMO energies characterise molecule’s ability to donate and accept electron. The difference between these orbital energies estimates molecule’s chemical reactivity. For the ellipticine, LUMO-HOMO energy difference is 3.821 eV. This shows that ellipticine is chemically moderate and indicates the charge transfer interactions within the ellipticine drug occurring eventually [24,25]. The HOMO and LUMO surfaces of ellipticine drug are shown in Figure 2. Global parameters have been calculated utilising HOMO and LUMO energies [25,26,27,28,29,30,31]. The chemical reactivity is governed by the structural arrangement of the compound.
Ionization potential represents the energy required to eject an electron from molecule. Chemical inertness or high stability is dictated by high value of ionization potential and vice- versa. As shown in Table 1, ellipticine has ionization potential of 5.459 eV. This shows that ellipticine is a good donor. Adding electron to the neutral molecule leads to release of some energy, which is taken as electron affinity of the molecule. In case of ellipticine its value is 1.639 eV, indicating that drug is moderately reactive (Table 1). The chemical potential is used to estimate the electron’s escaping tendency and also the electronegativity of the molecular system. Further electrophilicity index gives idea about stabilization of the molecule due to surrounding electrons approaching to saturate the system (atom/molecule). If a molecule or molecular system carries lower electrophilicity index then it is considered as highly reactive, nucleophile and better system. The electrophilicity index of ellipticine is 3.296 eV, which is noticeably lower, indicating that ellipticine is a moderate nucleophile.
3.2. Infrared Spectroscopic Analysis
The vibrational spectra of ellipticine drug has been analysed. Considering the characteristic vibrational modes of ethyl, methyl and other groups comprising of different modes, taking into account the stretching modes of CH in CH3 groups, CH3 deformations with HCC bending and NH stretching vibrations assignments, have been carried out [32,33,34,35]. Calculated infrared vibrational frequencies of ellipticine are juxtaposed to the standard data in the Table 2. The vibrational spectrum is given in Figure 4. Ellipticine has 33 atoms and hence associated with 93 normal vibration modes among which 78 modes lie in the IR region. A good correspondence is established between calculated and standard data. Only the basic modes are briefly described here (Table 2).
3.2.1. C-H Vibration
Usually, the CH stretching modes are reported in the region from 2800 to 3200 cm−1. In case of ellipticine, the CH of CH3 group yields calculated vibrational modes at 3021 and 3153 cm−1; which is in accordance with the standard data (Table 2).
3.2.2. N–H Vibration
Normally the N–H modes of stretching ranges from 3300 to 3600 cm−1. For ellipticine, this mode is calculated at 3680.55 cm−1; which is slightly higher than the standard value (Table 2).
3.2.3. Other Modes of Vibration
In the ellipticine, modes of deformation of ring lie at 1423.39 and 1490.09 cm-1; which are in close proximity with standard data. Torsion and wagging modes occupy lower frequency range. It is therefore apparent that there exists a gross similarity between the calculated infrared spectra and standard data [36,37].
3.3. Molecular Docking
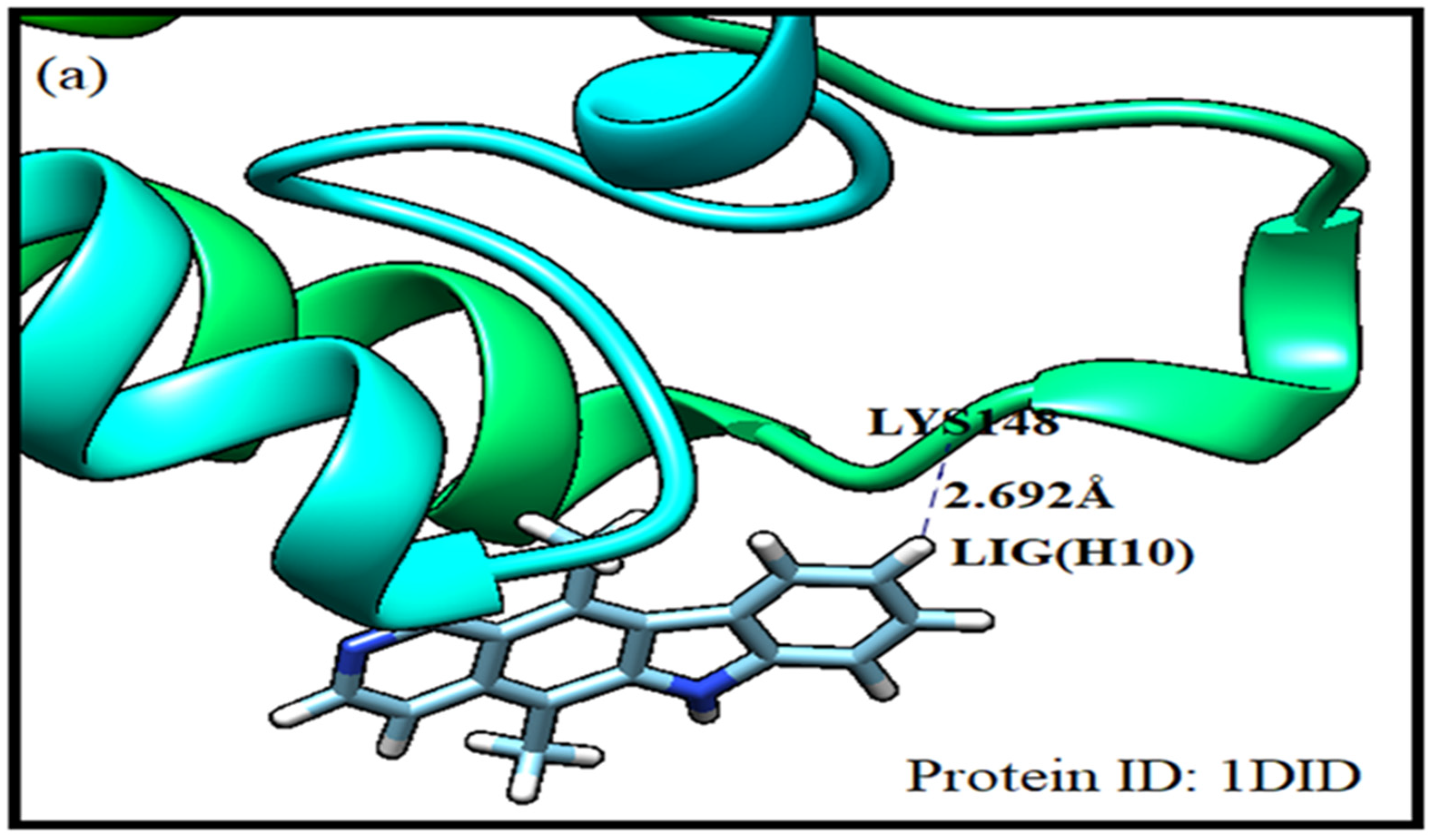
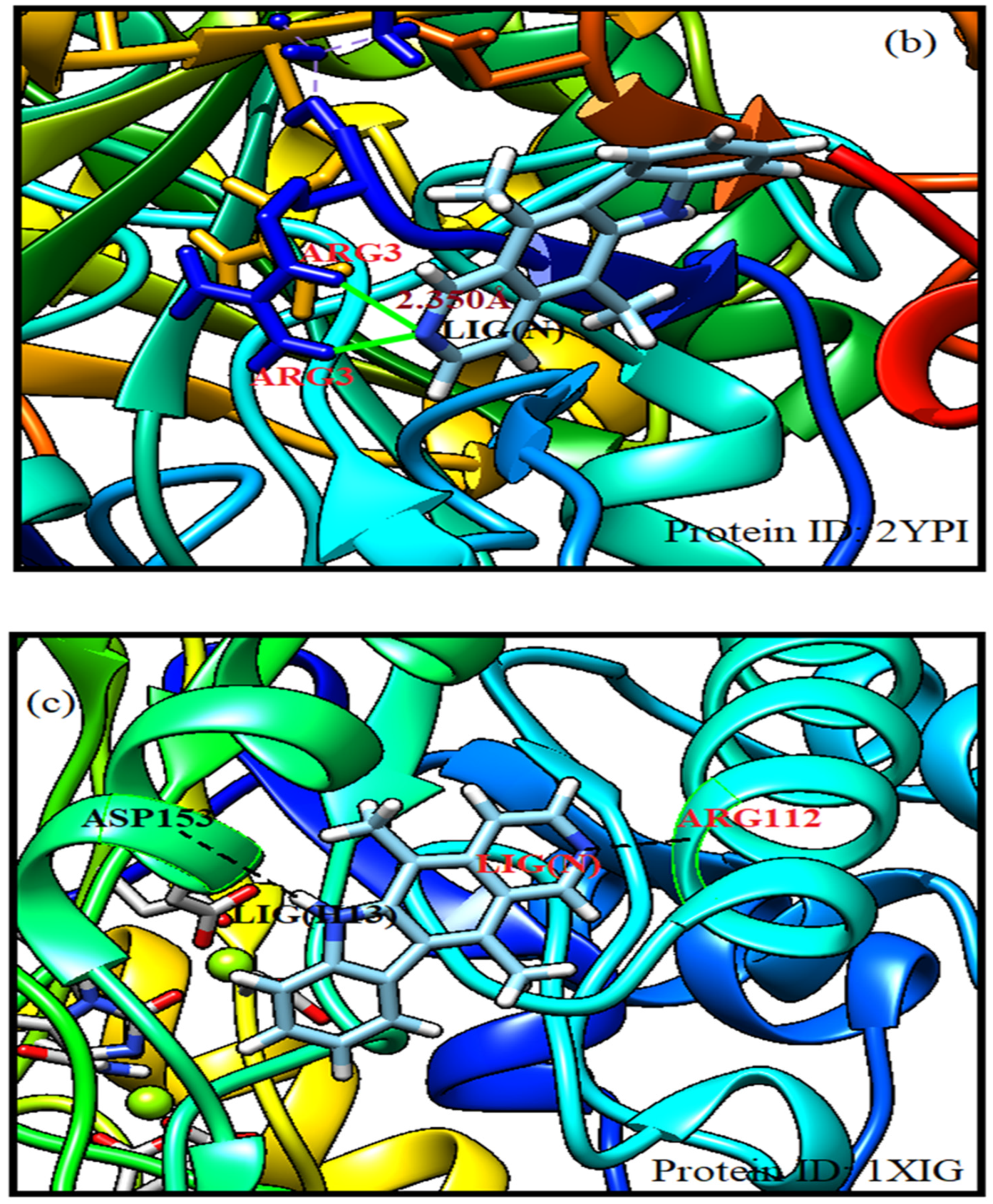
Molecular docking has got enormous application in studying drug-receptor interaction and estimating the drug’s efficacy. Hence it is inevitably used in drug design. Herein energy of each possible configuration of ligand-receptor association is calculated. According to full fitness score binding modalities of ligand-receptor complexes are arranged. SwissDock has been used in the present investigation. Sampling bias has been avoided by covering the whole protein. The protein receptors identified from protein data bank as 1did, 2ypi and 1xig are selected for docking. The receptor 1did shows high binding affinity for ellipticine drug. The receptor residue LYS (148) complexed with ellipticine LIG(H10) having energy of -8.01 kcal/mol at a separation of 2.692 Å with FF score of -1922.49 kcal/mol corresponds to best pose. Another preferred site with energy of -7.69 kcal/mol and FF score of -1920.75 kcal/mol is revealed between GLU (388) and LIG(H13) at a distance of 1.865Å. The 2ypi receptor elicits its several stable complexes with the ellipticine drug. The residue ARG (3) forms with LIG(N) a stable complex with energy of -8.70 kcal/mol. The corresponding FF score is -2401.14 kcal/mol at a drug-receptor separation of 2.350Å. Another preferred binding pose is found between LYS (221) and LIG(N) at a distance of 2.457Å with energy of -8.76 kcal/mol and FF score of -2403.35 kcal/mol. Receptor protein 1xig possesses stronger binding affinity with the ligand as compared to other protein receptors. Herein the ligand is simultaneously complexed with two sites of the receptor namely ARG (112) with LIG (N) and ASP (153) with LIG (H13) having ligand-receptor separated by 2.046 Å and 1.794 Å respectively. The drug/ligand complexed at two sites of the protein receptor 1xig is the most stabilised having energy of -8.80 kcal/mol with FF score of -2172.25 kcal/mol. Evidently ellipticine complexed with 1xig receptor is better stabilised in comparison to its complexes with 1did and 2ypi protein receptors [38].
Figure 5.
Molecular docking simulation of ellipticine with protein receptors.
4. Conclusions
In silico bond lengths, bond angles and vibrational modes of ellipticine nearly correspond to standard data. Frontier orbitals and molecular electrostatic potential surface indicate chemical reactivity and charge transfer within ellipticine drug. The prominent vibrational modes of the ellipticine drug are in gross agreement with standard data. As evident from molecular docking simulation, among three protein isomerases chosen from the protein data bank, binding of ellipticine with the 1xig receptor gives rise to the most stable complex.
Author Contributions
Conceptualization, methodology, investigation, writing—review and editing: Abhinav Mishra. Software, validation and formal analysis: Dipendra Sharma. Data curation: Priti Dubey. Supervision: S. N. Tiwari.
Funding
This research received no external funding.
Institutional Review Board Statement
Not Applicable.
Data Availability Statement
Data is unavailable due to privacy or ethical restrictions.
Acknowledgments
D. Sharma expresses gratitude to UGC, New Delhi, India, for providing the Start-Up grant [F.30-505/2020(BSR)].
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| HOMO | Highest Unoccupied Molecular Orbitals |
| LUMO | Lowest Unoccupied Molecular Orbitals |
| DNA | Deoxyribonucleic Acid |
| ELP | Ellipticine |
| DFT | Density Functional Theory |
| MEP | Molecular Electrostatic Potential |
References
- Goodwin, S.; Smith, A. F.; Horning, E. C. Alkaloids of Ochrosia elliptica Labill. J. Am. Chem. Soc. 1959, 81, 1903–1908. [Google Scholar] [CrossRef]
- Auclair, C. Multimodal action of antitumor agents on DNA: the ellipticine series. Arch. Biochem. 1987, 259, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pecq, J.B.L.; Xuong, N. D.; Gosse, C.; Paoletti, C. Peroxidase-catalyzed covalent binding of the antitumor drug N2-methyl-9-hydroxyellipticinium to DNA in vitro. Proc. Nat. Acad. Sci. USA. 1974, 71, 5078–5082. [Google Scholar] [PubMed]
- Jain, S. C.; Bhandary, K. K.; Sobell, H. M. Drug—nucleic acid interaction: X-ray crystallographic determination of an ethidium—dinucleoside monophosphate crystalline complex, ethidium: 5-iodouridylyl (3′-5′) adenosine. J. Molec. Biol. 1979, 135, 813–840. [Google Scholar] [CrossRef]
- Dethe, D. H.; Murhade, G. M. European. Diversity-Oriented Synthesis of Calothrixins and Ellipticines. J. Org. Chem. 2014, 31, 6953–6962. [Google Scholar]
- Garbett, N. C.; Graves, D. E. Extending nature’s leads: the anticancer agent ellipticine. Curr. Med. Chem. Anticancer. Agents. 2004, 4, 149–172. [Google Scholar] [CrossRef]
- Miller, C. M.; Sullivan, E. C.; McCarthy, F. O. Novel 11-Substituted Ellipticines as Potent Anticancer Agents with Divergent Activity against Cancer Cells. Pharmacouticals. 2019, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, K.; Magistrato, A. Modeling anticancer drug–DNA interactions via mixed QM/MM molecular dynamics simulations. Org. Biomol. Chem. 2006, 4, 2507–2517. [Google Scholar] [CrossRef]
- Sanyal, N. K.; Roychoudhury, M.; Tiwari, S. N. Quantum mechanical studies on the activity of anticancerous drug—Ellipticine. J. Biosci. 1985, 83, 713–720. [Google Scholar] [CrossRef]
- Sanyal, N. K.; Roychoudhury, M.; Tiwari, S. N.; Indian Journal of Biochemistry & Biophysics, 1990, 27, 222-227.
- Tiwari, G.; Sharma, D. Stacking interactions between demethylated ellipticines and DNA base pairs--a quantum mechanical study. Mater. Today. Proc. 2019, 29, 844–849. [Google Scholar] [CrossRef]
- Tiwari, G.; Kumar, A.; Dwivedi, K. K.; Sharma, D. In Silico Investigation of Electronic Structure, Binding Patterns and Molecular Docking of Nevirapine: An anti-HIV Type-1 Drug. Polycycl. Aromat. Compd. 2022, 42, 2789–2804. [Google Scholar] [CrossRef]
- Tiwari, G.; Chauhan, M. S.; Sharma, D. Estimation of Binding Sites of Efavirenz with 3EO9 Receptor: In Silico Molecular Docking and Molecular Dynamics Studies. Polycycl. Aromat. Compd. 2022, 1–16. [Google Scholar] [CrossRef]
- Tiwari, G.; Sharma, D.; Dwivedi, K. K.; Dwivedi, M. K. Electronic structure and pair potential energy analysis of 4-n-methoxy-4′-cyanobiphenyl: A nematic liquid crystal. AIP conference Proceedings 2016, 1728, 020206. [Google Scholar]
- Tiwari, G.; Kumar, A.; Sharma, D. Molecular structure, spectroscopic (IR, Raman, NMR, UV–vis) and molecular docking studies of an anticancer drug: isoDC81. Materials Today: Proceedings 2021, 47, 1707–1713. [Google Scholar] [CrossRef]
- Tiwari, G.; Sharma, D.; Singh, N. B. Electronic Structure and Molecular Docking Studies of an anti-HIV Drug: Stavudine. J. Sci. Ind. Res. 2020, 337–339. [Google Scholar]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al.; Gaussian 16, Revision C.01, Gaussian. Inc. Wallingford. CT, 2016.
- Dennington, R.; Keith, T. A.; GaussView. Version 6.1, Semichem. Inc. Shawnee Mission, KS, 2016.
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic. Acids. Res. 2011, 39. [Google Scholar] [CrossRef]
- Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Pandey A K, Sharma A K, Shukla S, Mishra A, Singh V, Dwivedi A. Molecular docking, QTAIM analysis, UV-Vis spectra, vibrational analysis, and electronic properties of Lantadenes C and D—A complete comparative study. Int. J. Comput. Mater. Sci. Eng. 2023, 13, 2350028. [CrossRef]
- Mishra, A.; Sharma, D.; Tiwari, S. N. First Principle Studies of Electronic Properties, Global Reactivity Descriptors, and Molecular Docking of Olivacine Drug. Macromol. Symp. 2024, 413, 2400075. [Google Scholar] [CrossRef]
- Tiwari, A.; Fernades, R.S.; Dey, N.; Kanungo, S. Comparative analysis of the hydrazine interaction with arylene diimide derivatives: complementary approach using first principles calculation and experimental confirmation. Langmuir 2024, 40, 10966–10979. [Google Scholar] [CrossRef]
- Fleming, J.; Frontier orbitals and organic chemical reactions. 249 S., John Wiley u. Sons LTD., London, in: H. G. O. Becker (Ed.), J. Für Prakt. Chemie, John Wiley & Sons, Ltd., 1978, 879–880.
- Murray, J. S.; Sen, K. D.; eds. Molecular electrostatic potentials: concepts and applications. 1st ed. Elsevier. 1996.
- Koopmans, T. Physica 1934, 1, 104–113.
- Parr, R. G.; Pearson, R. G. Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R. G.; Szentpály, L. V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Parr, R. G.; Donnelly, R. A.; Levy, M.; Palke, W. E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1977, 68, 3801–3807. [Google Scholar] [CrossRef]
- Pauling, L.; The Nature of Chemical Bond. 3rd ed. Cornell University Press, Ithaca, New York 1960.
- Mishra, A.; Sharma, D.; Tiwari, S. N. DFT and Molecular Docking Studies of an Antiviral Drug: Molnupiravir. Indian J. Pure Appl. Phys. 2023, 61, 810–812. [Google Scholar]
- Snyder, R. G.; Scherer, J. R. Band structure in the C–H stretching region of the Raman spectrum of the extended polymethylene chain: Influence of Fermi resonance. J. Chem. Phys. 1979, 71, 3221. [Google Scholar] [CrossRef]
- Snyder, R. G.; Maroncelli, M.; Strauss, H. L.; Elliger, C. A.; Cameron, D. G.; Casal, H. L.; Mantsch, H. H. Distribution of Gauche Bonds in Crystalline n-C₂₁H₄₄ in Phase II. J. Am. Chem. Soc. 1983, 105, 133–134. [Google Scholar] [CrossRef]
- Snyder, R. G.; Srivatsavoy, V. J. P.; Cates, D. A.; Strauss, H. L.; White, J. W.; Dorset, D. L. Conformational Disorder in the Rotator Phases of n-Alkanes: A Raman Spectroscopic Study. J. Phys. Chem. 1994, 98, 674–684. [Google Scholar] [CrossRef]
- Maroncelli, M.; Qi, S. P.; Strauss, H. L.; Snyder, R. G. Nonplanar Conformers and the Phase Behavior of Solid n-Alkanes. J. Am. Chem. Soc. 1982, 104, 6237–6247. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman characteristic group frequencies: tables and charts, 3rd ed. New York: John Wiley, 2004.
- Mishra, A.; Sharma, D.; Tiwari, S. N. International Conference on Nanotechnology: Opportunities and Challenges, 2022, 117-123.
- https://www.rcsb.org/structure/1DID; https://www.rcsb.org/structure/2YPI; https://www.rcsb.org/structure/1XIG.
Figure 1.
Optimized molecular geometry of ellipticine drug.
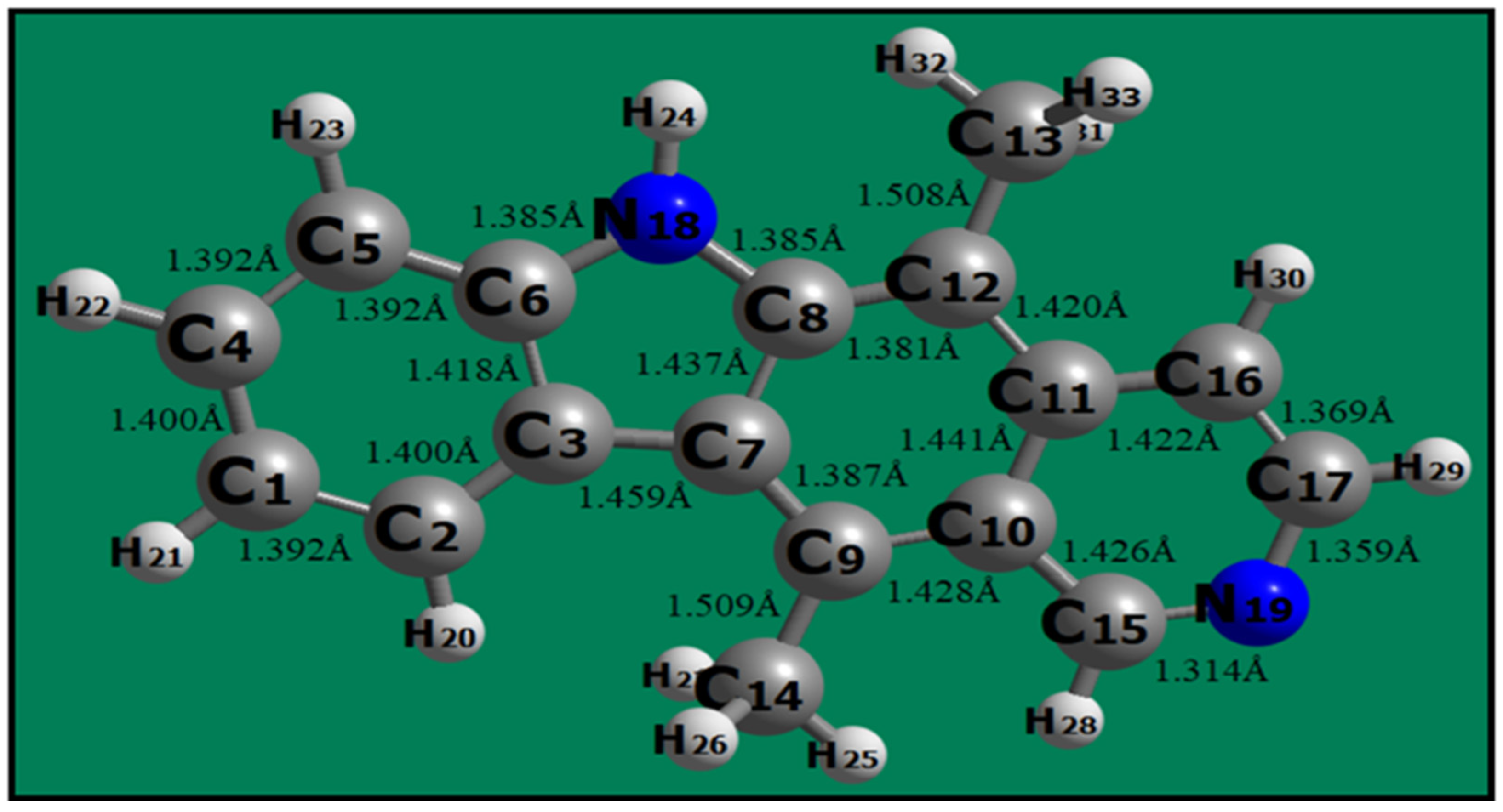
Figure 2.
Molecular electrostatic potential surface of ellipticine drug.
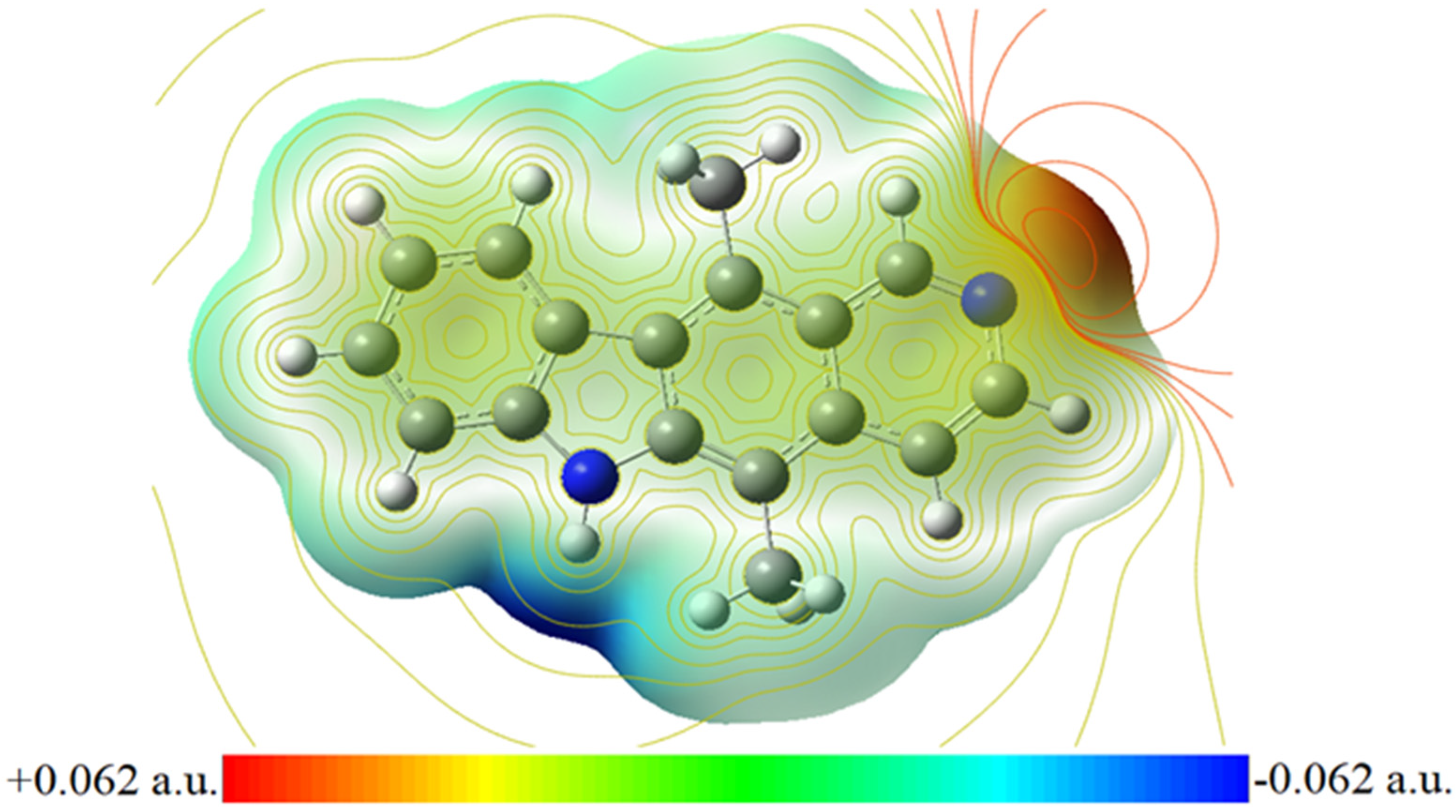
Figure 3.
Frontier orbitals of ellipticine drug molecule.
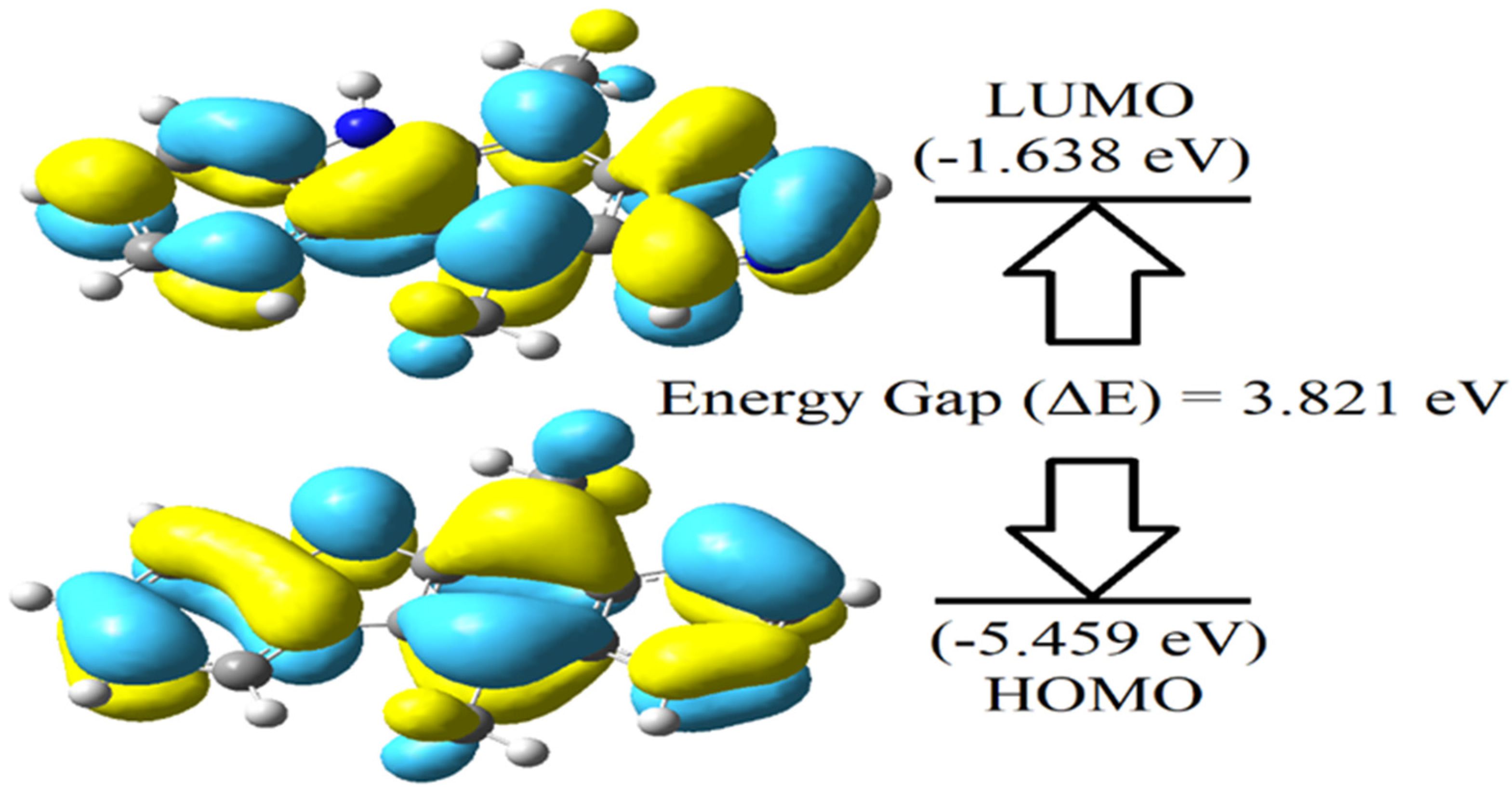
Figure 4.
Infrared spectrum of ellipticine molecule.
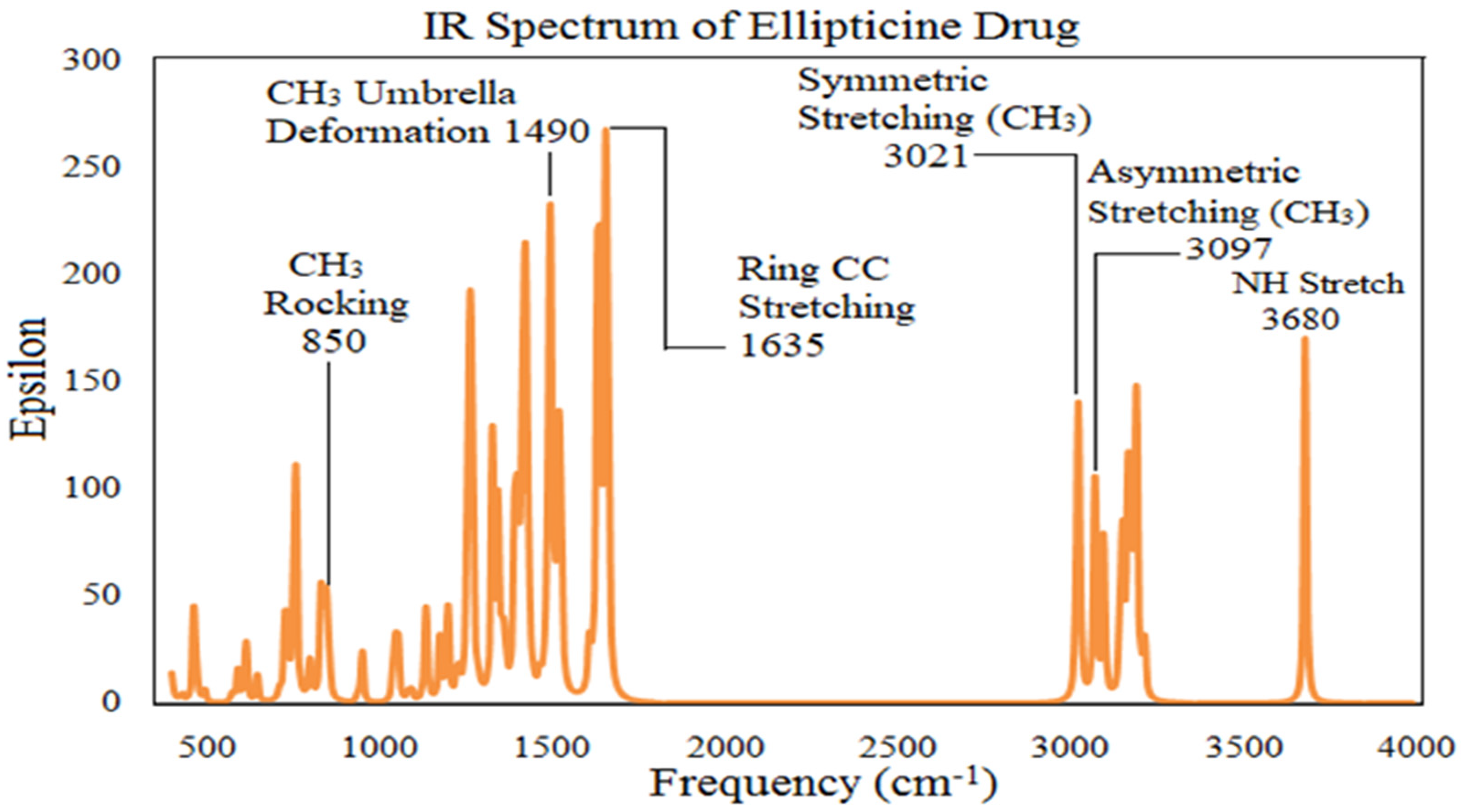

Table 1.
The electronic and global reactivity descriptors of ellipticine.
| Total energy | -20842.791 eV |
| Dipole moment | 4.153 debye |
| HOMO energy (ELUMO) | -5.459 eV |
| LUMO energy (EHOMO) | -1.638 eV |
|
Frontier orbital energy gap ΔELUMO−HOMO = ELUMO − EHOMO |
3.821 eV |
| Chemical potential (µ = −χ) | -3.549 eV |
| Electronegativity (χ = (I + EA)/2) | 3.549 eV |
| Electron affinity (EA= −ELUMO) | 1.638 eV |
| Ionization potential (I= −EHOMO) | 5.459 eV |
| Global hardness (η= (I − A)/2) | 1.911 eV |
| Global softness (S=1/2η) | 0.262 eV-1 |
| Global electrophilicity (ω=μ2/2η) | 3.296 eV |
Table 2.
Significant vibrational assignments of ellipticine drug molecule.
| Standard Frequency (cm-1) |
Unscaled Calculated Frequency (cm-1) |
Calculated Intensity | Vibrational Assignments |
| 3500 | 3680.55 | 50.18 | NH Stretching |
| - | 3192.89 | 23.48 | |
| - | 3188.93 | 28.41 | |
| - | 3169.36 | 28.96 | |
| - | 3165.38 | 1.47 | |
| 2964 | 3153.59 | 23.37 | Asymmetric Stretching (CH3) |
| - | 3097.31 | 23.83 | |
| 2866 | 3021.88 | 39.76 | Symmetric Stretching (CH3) |
| 1522 | 1635.94 | 119.79 | Ring CC Stretching |
| 1523.19 | 57.76 | ||
| 1456 | 1490.09 | 62.06 | CH3 Umbrella Deformation |
| 1374 | 1428.08 | 35.80 | |
| - | 1423.39 | 37.00 | |
| - | 1415.24 | 33.27 | |
| - | 1395.96 | 48.29 | |
| - | 1345.74 | 29.72 | |
| - | 1328.86 | 36.58 | |
| - | 1268.87 | 41.57 | |
| - | 1260.65 | 65.53 | |
| 824 | 850.29 | 24.99 | CH3 Rocking |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



