
Submitted:
14 April 2025
Posted:
15 April 2025
You are already at the latest version
Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disorder primarily characterized by memory loss and cognitive decline, which significantly impacts patients' quality of life and imposes substantial emotional, practical, and economic burdens on their families. As the most common cause of senile dementia, AD currently affects approximately 50 million people worldwide, with projections indicating a threefold increase by 2050 due to rising life expectancy and an aging global population. Diagnosis of AD remains challenging. Neuroimaging techniques reveal atrophy in critical brain regions, particularly in the cortex, hippocampus, and limbic system, which are essential substrates for memory, personality changes, and other cognitive functions. The hallmark molecular changes associated with AD include the accumulation of β-amyloid plaques and the formation of tau protein tangles. Several underlying mechanisms contribute to neuron loss, such as oxidative stress, neuroinflammation, microbial dysbiosis and insulin resistance. In this context, exosomes − small extracellular vesicles that facilitate cell communication − transport proteins, DNA, mRNA, and non-coding RNA (ncRNA), all of which play a significant role in the neurobiology of AD. Furthermore, emerging research indicates that exosomal ncRNAs may serve as promising biomarkers for AD, offering the possibility of improved diagnostic precision. This review explores the potential of exossomal ncRNAs as non-invasive biomarkers for AD, focusing on recent advances and future directions in translational studies.
Keywords:
Alzheimer's disease
; biomarkers
; β-amyloid
; Tau protein
; miRNAs
; circRNAs
1. Introduction
Dementia has increasingly become a focal point in scientific research and public health initiatives. This attention is driven by the condition’s rising prevalence among the aging global population, as well as its profound impact on both affected individuals and their families. Efforts are being made in the realms of prevention, early diagnosis, and clinical intervention to address this growing concern [1]. The increase in life expectancy has contributed to a rise in cases of dementia as well as other neurodegenerative disorders among the elderly. Currently, approximately 50 million individuals worldwide are diagnosed with dementia, a figure projected to triple by 2050 [2]. Alzheimer’s disease is the most common form of dementia globally. This chronic neurological disease was first described by Alois Alzheimer in 1906 and is primarily characterized by progressive memory loss and impairment across multiple cognitive functions [3]. Despite considerable progress in understanding the brain pathology associated with AD, diagnosing and treating this complex disorder remains challenging [1,4].
The incidence of AD is influenced by non-modifiable factors such as advancing age, female sex, lower educational attainment, and genetic predisposition, particularly the presence of the APOE ε4 allele, while ε2 offers some protection. Modifiable factors include cardiovascular conditions (hypertension, diabetes, hypercholesterolemia), lifestyle elements (Mediterranean diet, physical activity, cognitive engagement), and environmental exposures (e.g., air pollution). Depression, obesity, subjective cognitive decline, neurodegenerative markers on imaging, and co-pathologies also contribute to AD risk [5,6,7,8,9,10,11,12,13].
The pathological features of AD encompass the accumulation of β-amyloid (Aβ) plaques and neurofibrillary tangles (NFTs) composed of tau protein within the brain. Additional changes of note include an increase in reactive oxygen species (ROS), the proliferation of glial cells, impaired insulin sensitivity, and modifications in the microbiome [14]. These characteristics have been extensively studied due to their critical importance for both diagnostic and potential therapeutic applications [15].
Exosomes are extracellular vesicles (EVs) that have gained significant prominence in AD research in recent years. These small lipid vesicles carry a variety of molecules, including proteins, DNA, mRNA, and non-coding RNA (ncRNA) from their cells of origin. Notably, ncRNAs are regulators of gene expression and can influence multiple signaling pathways [16,17,18]. AD-related exosomes have been shown to disseminate amyloidogenic peptides, suggesting their involvement in the underlying mechanisms of brain pathology [19].
Research has also investigated blood-derived exosomes as potential biomarkers for AD, focusing on levels of Aβ, amyloid precursor protein (APP) fragments, tau, and phosphorylated tau (p-tau) [16,17,20]. Furthermore, exosomes isolated from AD patients exhibit aberrant contents of synaptic proteins, inflammatory mediators, growth factors, and lysosomal proteins [20,21,22,23]
Exosomal non-coding RNAs (ncRNAs), particularly microRNAs (miRNAs), have emerged as key regulators of APP and tau proteins, both of which are critical in AD pathology [24]. In addition to miRNAs, exosomes transport a variety of other ncRNAs, including long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs), which facilitate cell-to-cell communication and reciprocal influence [25,26]. The expression patterns of these exosomal ncRNAs can vary depending on cell type and disease state, suggesting their potential involvement in AD and other pathologies [27,28]. Recent studies have further clarified the roles of exosomal mRNAs, circRNAs, and miRNAs in AD progression, particularly their regulatory effects on APP and tau proteins [24,29,30]
Regarding the diagnosis of AD, it is crucial to develop simple and effective tests with high specificity to improve diagnostic [15,31,32]. Exosomes represent a promising source of accessible and cost-effective biomarkers for disease monitoring, as their lipid bilayer protects the cargo from enzymatic degradation in the bloodstream [33,34]. This review examines the potential of ncRNAs derived from exosomes as viable biomarkers for AD.
Clinical Diagnosis of AD
Alzheimer’s Disease is clinically defined as a major or mild neurocognitive disorder (NCD), as outlined in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5). The DSM-5 emphasizes a decline in cognitive functioning that is not attributable to delirium or another mental disorder. Key criteria include significant cognitive decline in one or more cognitive domains (e.g., executive function, learning and memory, complex attention, language, social cognition, or perceptual-motor skills) based on concerns from the individual, informants, or clinicians, and preferably documented through standardized neuropsychological testing. For major NCD, these deficits must interfere with independence in daily activities, while for mild NCD, independence remains intact but may require compensatory strategies. A diagnosis of probable AD requires genetic evidence or specific patterns of cognitive decline and progression, whereas possible AD is diagnosed in the absence of such evidence but with clinical support [35,36,37] .
Associated with this clinical presentation, AD is characterized neuropathologically by the presence of amyloid plaques and NFTs, alongside other pathological changes such as inflammatory responses, astrocyte and microglial activation, and synaptic and neuronal loss. Amyloid plaques are extracellular deposits of Aβ peptides that follow a predictable regional progression, starting in the neocortex and advancing to other brain areas, as outlined in Thal phases [38,39,40,41,42] . NFTs, on the other hand, consist of intracellular aggregates of p-tau, which progress in a stereotyped pattern from the transentorhinal region to the limbic system and eventually the neocortex, as described in Braak stages [43]. The National Institute on Aging-Alzheimer’s Association (NIA-AA) guidelines recommend a comprehensive neuropathological assessment using the ABC scoring system, which integrates Thal phases for amyloid plaques, Braak stages for NFTs, and the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria for neuritic plaque density. This approach allows for a detailed classification of AD-related changes, while also accounting for co-pathologies, such as cerebral amyloid angiopathy and other proteinopathies, that may influence clinical presentation and disease progression [38,39,40,41,42].
Historically, the diagnosis of AD relied primarily on clinical assessment, making accurate diagnosis both challenging and time-consuming. Physicians needed to exclude other potential causes of dementia through a rigorous, multi-step process. This typically involved a comprehensive review of medical history, physical examinations, laboratory tests to exclude other conditions (e.g., vitamin B-12, syphilis serology and thyroid hormone levels), imaging studies (e.g., CT scans), and the discontinuation of any medications that might impair cognition. Only after systematically ruling out other causes of cognitive decline could a probable AD diagnosis be established, with confirmation gained by observing disease progression and specific cognitive patterns associated with AD [44] .When clinical findings are inconclusive, re-evaluation is often necessary [45] Moreover, early diagnosis of AD is frequently impeded by social and emotional barriers, including denial, fear of the diagnosis, limited family support, and difficulties in accessing healthcare [46].
Neuropathological studies in individuals diagnosed with AD during their lifetime have significantly advanced our understanding of the disease's complexity and heterogeneity. While the hallmark pathological features—amyloid plaques composed Aβ deposits and NFTs of p-tau proteins—are central to AD diagnosis, not all individuals clinically diagnosed with AD exhibit these findings. Approximately 14% of patients with mild-to-moderate AD lack sufficient neuropathological changes to meet classical diagnostic thresholds, as highlighted in postmortem brain studies. Among clinically diagnosed AD cases, 81% show high or intermediate levels of typical AD pathology, yet only 41% of dementia cases are attributed exclusively to AD pathology. Polymorbidity is common, with frequent co-occurrence of vascular lesions, TDP-43 proteinopathy, and Lewy bodies, which can complicate clinical presentations. This heterogeneity is also reflected in atrophy-based subtypes, such as limbic-predominant and hippocampal-sparing patterns, which correlate with variations in amyloid and tau pathology. The predictable progression of AD pathology described in Thal and Braak staging is often accompanied by comorbidities that may modify disease trajectories and explain variability in treatment outcomes. These findings underscore the importance of integrating neuropathological, clinical, and imaging data to provide a comprehensive understanding of AD and related dementias [41,47,48,49,50].
The diagnosis of AD has undergone significant evolution, with recent developments highlighting the integration of biological and clinical criteria. In 2024, the Alzheimer's Association (AA) workgroup, led by Clifford Jack, proposed revised criteria emphasizing the biological definition of AD, centered on the ATN model, which incorporates biomarkers for Aβ, tau, and neurodegeneration. These criteria distinguish between Core 1 biomarkers - including amyloid PET, CSF measures (specifically CSF Aβ42/40, CSF p-tau181/Aβ42, CSF t-tau/Aβ42), and plasma markers like phosphorylated tau 217 (requiring at least 90% accuracy for diagnostic validity) - and Core 2 biomarkers, comprising tau PET and specific fluid T2 biomarkers such as MTBR-tau243 and other phosphorylated tau forms [51] Core 1 biomarkers define the earliest in vivo detectable stage of AD and can identify disease presence in both symptomatic and asymptomatic individuals. In contrast, Core 2 biomarkers become abnormal later in disease evolution and are more closely linked to symptom onset, serving primarily for biological staging and prognosis rather than diagnosis. Under this framework, an abnormal Core 1 biomarker is sufficient for an AD diagnosis, even in the absence of clinical symptoms.
However, this approach has sparked debate in the scientific community. The conceptualization of AD as a clinical-biological construct has been a cornerstone of the International Working Group’s (IWG) approach, emphasizing the necessity of integrating specific clinical phenotypes with pathophysiological biomarkers. According to the IWG, the diagnosis of AD should rest on the presence of distinct clinical syndromes—such as the amnestic variant or other characteristic phenotypes—supported by positive biomarkers, ensuring that these biological findings are interpreted within a clinical context. This approach contrasts with the AA framework, which allows for the diagnosis of AD in cognitively normal individuals based solely on biomarker positivity [52].
A major point of contention lies in the classification of cognitively normal individuals with positive biomarkers. The AA criteria consider these individuals as having AD, while the IWG advocates for alternative terminology, categorizing them as “asymptomatic at risk for AD” or, in specific scenarios with deterministic biomarker profiles, as having “presymptomatic AD.” [52,53]. The IWG highlights the potential risks of labeling cognitively normal individuals as having AD based solely on biomarkers, including psychological distress, social consequences, and the possibility of overdiagnosis. Moreover, they emphasize that many biomarker-positive individuals may never develop clinical symptoms, underscoring the importance of distinguishing risk states from active disease to avoid unnecessary interventions and anxiety [52,53,54].
Both frameworks acknowledge the importance of staging disease progression and recognize factors such as cognitive reserve and comorbidities in shaping clinical outcomes. However, differences persist in how these elements are applied in clinical practice. The growing availability of blood-based biomarkers and new therapeutics targeting the core pathology of AD has heightened the urgency of these discussions, particularly regarding the implications for treatment eligibility and healthcare accessibility. These evolving diagnostic frameworks underscore broader challenges in the field, including the standardization of biomarker testing, the need to account for population diversity, and the ethical considerations surrounding early detection. As AD therapeutics and research continue to advance, these criteria will likely be refined, reflecting the dynamic interplay between biological innovations and clinical application [51].
As an example of the clinical application of this recent knowledge, the Brazilian Academy of Neurology (BAN) underscores a clinically grounded approach to the use of AD biomarkers, tailored to the realities of the healthcare system. According to BAN, biomarker testing should be reserved for symptomatic individuals with objectively verified cognitive deficits and must be conducted under the guidance of qualified specialists in Geriatrics, Neurology, or Psychiatry. The Academy advises against the use of biomarkers in asymptomatic individuals or those with subjective cognitive decline, emphasizing that their main clinical usefulness lies in supporting diagnosis for symptomatic cases [55]. This is also particularly relevant for early-onset dementia, rapidly progressive dementias, atypical clinical presentations, or when disease-modifying treatments are being considered.
BAN supports a structured diagnostic algorithm that prioritizes CSF biomarker analysis over amyloid PET, highlighting the greater accessibility and cost-effectiveness of CSF testing in Brazil. BAN also recognizes the potential of plasma biomarkers, provided they undergo rigorous validation to be effectively integrated into clinical practice. It emphasizes that biomarker results must be interpreted within the broader clinical context, as positive AD biomarker findings do not definitively attribute symptoms to AD pathology alone, given the high prevalence of comorbid neuropathologies in older adults [55] .
Progress in Diagnostic Technologies for AD
Significant advancements in understanding the natural history of AD over the past two decades have been driven by technological breakthroughs, including the development of molecular markers for neuroimaging, biochemical analyses of biological fluids, and the implementation of bioinformatics and machine learning [56,57].
The most extensively studied biomarkers for AD are Aβ and tau proteins (Table 1). Key amyloid biomarkers include decreased Aβ42/Aβ40 ratio in CSF, as increased amyloid deposition in brain parenchyma detected in PET imaging. Additionally, elevated levels of p-tau in CSF and tau deposition in brain parenchyma in PET scans serve as effective indicators for AD pathology[58,59]. While the Aβ42/40 ratio is a validated CSF biomarker [60], it poses challenges for blood-based testing due to detection limitations [61,62]. In contrast, recent p-tau blood assays demonstrate higher diagnostic accuracy and disease specificity, making them promising candidates for routine clinical testing [63]. The p-tau217 has been demonstrated to be a superior marker of cognitive progression to dementia in patients with mild cognitive impairment (MCI) compared to p-tau181 [64]. Conversely, p-tau205 showed changes at a later stage and was more strongly associated with tau PET imaging than with amyloid PET imaging. Similar to total tau (t-tau), increased concentrations of neurofilament light chain (NfL) are not specific to AD and can also be elevated in other neurodegenerative disorders, such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD).
In a cohort of Chinese participants, changes in CSF biomarker concentrations were observed over the 20 years preceding clinical diagnosis of sporadic AD, with specific timelines as follows: Aβ42 levels changed 18 years prior, Aβ42/Aβ40 ratio 14 years prior, p-tau181 11 years prior, t-tau 10 years prior, and NfL 9 years prior to diagnosis. As cognitive impairment progressed, the alterations in CSF biomarker levels in the AD group initially accelerated before subsequently slowing down [65].

Table 1.
Overview of diagnostic methodologies and biomarkers for AD.
| Methodology | Biomarker | Advantages | Limitations | Reference |
|---|---|---|---|---|
| Immunoassay from Biological Fluids | ||||
| CSF | Aβ42, t-tau and p-tau, NfL | Widely used in several systematic studies | Requires the presence of cognitive decline that impacts daily activities for the diagnosis of AD | [5] |
| Plasma | Aβ42 and Aβ40, p-tau181, p-tau217, p-tau231, NfL, GFAP | High diagnostic accuracy for identifying AD compared to standard clinical evaluation | More research is needed | [66,67] |
| Serum | Aβ, tau, NfL, miRNA | Ability to accurately diagnose early stages of AD and identify individuals at high risk of cognitive decline in older adults | Diagnosis is based on clinical and pathological criteria | [68] |
| Neuroimaging | ||||
| CT | Brain atrophy and vascular changes | This method also identifies other causes of neurodegeneration | While offering ease of access, there may be less accuracy in the results | [69] |
| PET | Cerebral metabolism involving glycolysis or Aβ protein deposition | Identifies lower glycolysis uptake in medial temporal regions and the cingulum | High cost | [70] |
| MRI | Cerebral atrophy and ventricular dilation | Better visualization of atrophy in the entorhinal cortex, middle temporal lobe, and hippocampus | High cost | [69] |
| MRS | Measures different metabolites: NAA, mI, Chol, Glu, Gln, and GABA | Provides metabolic information that may aid in understanding AD | Difficult access and high cost of use | [71] |
| DTI | Description of white matter microstructure through its tensor model | Identifies potential biomarkers in the early stages of AD | Limitations regarding interpretation | [72] |
| FW | Isolates and quantifies changes in extracellular water | Detects subtle changes in brain tissue that may indicate early stages of DA | Introduces an additional layer of complexity in the analysis and interpretation of data | [73] |
Abbreviations: NfL, Neurofilament light chain; GFAP, Glial fibrillary acidic protein; miRNA, MicroRNA; Ccl2, Chemokine; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; DTI, diffusion tensor imaging; FW, free-water imaging; NAA, N-acetylaspartate; mI, myo-inositol; Chol, cholesterol; Glu, glutamate; Gln, glutamine, and GABA, gamma-aminobutyric acid.
Modern molecular techniques enable the detection of Aβ40 and Aβ42 in both CSF and blood samples, with blood testing offering a less invasive alternative. Commercially available blood immunoassays for p-tau217 have demonstrated comparable accuracy to CSF biomarkers for identifying AD [67]. In a cohort study involving 786 individuals, Ashton et al. (2024) found that the p-tau217 immunoassay exhibited accuracy similar to that of cerebrospinal fluid biomarkers in identifying Aβ and tau pathologies. Plasma p-tau217 results from the ALZpath pTau217 assay were compared with magnetic resonance imaging, β-amyloid PET, tau PET, and CSF biomarkers (Aβ42/40 and p-tau immunoassays). The method was able to detect the Aβ protein across three distinct ranges in three patient cohorts. Additionally, changes were observed over an eight-year period as the disease progressed, with the most significant variation in p-tau217 noted in individuals who were positive for both Aβ and tau.
A study evaluated a two-step diagnostic workflow to screen for Aβ positivity in patients with MCI using plasma p-tau217 as a biomarker [74]. In the first step, a model combining plasma p-tau217 levels, age, and APOE ε4 status stratified patients into low-, intermediate-, and high-risk categories for Aβ-PET positivity. Confirmatory CSF testing was conducted only for intermediate-risk individuals. The workflow demonstrated high accuracy (88.2–92.0%) while reducing the need for CSF or PET confirmatory testing by 61.2–85.9%, depending on the thresholds used. The findings suggest that plasma p-tau217 can effectively aid in stratifying patients for Aβ status, potentially reducing the reliance on more invasive or costly diagnostic methods.
Advances in detection technologies, such as Single-Molecule Array (SIMOA), have enabled precise quantification of low-abundance proteins in biofluids, including Aβ peptides and p-tau, biomarkers relevant to AD. SIMOA has been employed to measure plasma Aβ42/Aβ40 ratios, providing a less invasive and more accessible method for assessing amyloid status compared to PET imaging or CSF sampling [75]. It has also been used for plasma p-tau quantification, supporting its application in early-stage AD detection [76] The method relies on digital bead-based ELISA, where paramagnetic beads coated with capture antibodies isolate single protein molecules. Fluorescence signals are measured in femtoliter-sized wells, allowing for detection of proteins at very low concentrations [77]
Neuroimaging has proven clinically relevant for monitoring AD progression and identifying early-stage indicators. Imaging modalities such as PET, computed tomography (CT), magnetic resonance imaging (MRI), magnetic resonance spectroscopy (MRS), diffusion tensor imaging (DTI), and free-water imaging (FW) enable both structural and functional assessments that aid in predicting AD progression [78,79,80]. PET imaging is considered the gold standard for non-invasive AD diagnostics, playing a critical role in disease monitoring and differential diagnosis.
The development of PET radiotracers for AD has progressed through distinct phases targeting both amyloid and tau pathologies. The amyloid-targeting timeline began in 2003 with [11C]PiB, the first successful selective radioligand for Aβ, which led to the development and FDA approval of three 18F-labeled tracers: [18F]florbetaben (Neuraceq), [18F]florbetapir (Amyvid), and [18F]flutemetamol (Vizamyl)[81,82]. The second generation of amyloid tracers, including [18F]AZD4694 ([18F]NAV4694), addressed limitations such as white matter retention (Rowe et al., 2013).Parallel developments in tau imaging emerged with first-generation tracers such as [18F]THK5117 and [11C]PBB3, followed by [18F]AV-1451 (flortaucipir) which demonstrated significant binding to neurofibrillary tangles. Second-generation tau tracers, including [18F]MK-6240 and [18F]PI-2620, showed improved selectivity and reduced off-target binding to monoamine oxidases [84,85]. This dual-pathway development of both amyloid and tau radiotracers has enabled in vivo visualization of the two primary pathological hallmarks of AD, facilitating early diagnosis, disease progression monitoring, and therapeutic evaluation [86,87]. The evolution of these imaging agents has particularly enhanced understanding of the temporal relationship between amyloid and tau accumulation in the disease process. These advancements prompted to propose a temporal model of the AD pathological cascade, thereby enhancing our capacity to visualize and quantify AD progression at both the molecular and cellular levels [88].
Additional neuroimaging techniques contribute to a comprehensive assessment in AD diagnostics. While CT imaging is less detailed than MRI, it offers a cost-effective option for initial evaluations. MRI with T1-weighted sequences provides high-resolution images, facilitating an in-depth examination of brain structures affected by AD, such as the hippocampus and entorhinal cortex [69]. MRS has demonstrated utility in detecting metabolic changes associated with AD; however, it is used less frequently due to accessibility and cost constraints [89]. DTI, a specialized form of MRI, allows for the examination of white matter microstructure, aiding in the early detection of AD by assessing neural connectivity [90] In parallel, FW offers valuable insights by specifically detecting changes in extracellular water, highlighting early degenerative processes in the brain that conventional imaging techniques might miss [91,92].
Finally, machine learning and regression analysis represent promising tools for early AD diagnosis and predicting disease progression. Techniques such as least squares regression, support vector machines, and regression trees have been utilized to predict neuropsychological scores based on neuroimaging biomarkers [93]. Machine learning models trained on features from multiple imaging modalities have shown potential in accurately predicting cognitive outcomes, offering a compelling approach for precise diagnosis [77,79].
Are exosomes reliable sources of biomarkers for AD diagnosis?
Extracellular vesicles are lipid-coated structures produced by nearly all cell types and released into the extracellular milieu through membrane budding. They carry a variety of intracellular molecules that serve different roles within the organism, including plasma membrane restoration, intercellular communication, and the elimination of cellular waste [94,95] The cargo and function of EVs likely contribute to their heterogeneity, leading to their classification into exosomes (30 to 150 nm) and microvesicles (50 to 1000 nm). Notably, these structures differ in their biogenesis: microvesicles are formed directly through outward budding, primarily from apoptotic cells, while exosomes are generated during the formation of endosomes (Figure 1). The endocytic pathway also provides the molecular machinery for the autophagy pathway, and both exosome secretion and autophagy play crucial roles in eliminating cellular waste in a coordinated manner [96]
The role of exosomes in AD is complex, as they are implicated in both disease progression and the maintenance of normal brain physiology. Exosomes are known to be neuroprotective, supporting neuronal survival, myelination, neurogenesis, and immune responses following brain injury [97,98] Conversely, exosomes also contribute to AD pathology by facilitating the spread of pathogenic proteins, such as Aβ and p-tau, to the brain and potentially influencing the formation of Aβ plaques (Figure 2) [99,100]
Due to their nanoscale size and lipid composition, exosomes can easily traverse different organs and cells, even crossing the blood-brain barrier to interact with neurons, astrocytes, and microglia. They are delivered to target cells either by fusing with the cell membrane or through endocytosis, releasing their cargo, which may include molecules such as proteins, mRNA, miRNA, and circRNA, into the extracellular milieu. This process can potentially modulate gene expression [101]. The contents of exosomes also reflect the pathophysiological status of the originating cells, resulting in heterogeneity in terms of size, release pathway, content, and function [94]. Non-coding RNAs, such as miRNA and circRNA, present in exosomes are increasingly being explored as potential biomarkers for various neurodegenerative diseases, including AD and chronic cognitive disorders and syndromes.
Exosomes represent a promising source of biomarkers, as they are readily detectable in body fluids, particularly in serum and plasma. Furthermore, they contain specific membrane biomolecules that facilitate the identification of their originating cells. It is now possible to distinguish exosomes from different sources using commercial kits or magnetic beads linked to monoclonal antibodies, such as those for neuronal-enriched exosomes (L1 Cell Adhesion Molecule - L1CAM) and astrocyte-enriched exosomes (Glutamine aspartate transporter - GLAST) [102,103].
Exosomal Proteins
Previous studies have demonstrated that neuronal exosomes carry a variety of misfolded proteins, including Aβ, α-synuclein, full-length APP, and p-tau [100] (Figure 2). Notably, the gene locus Bridging Integrator 1 (BIN1), which is linked to late-onset AD, has been shown to facilitate the transport of tau via exosomes both in vitro and in vivo [104]. The tau protein in exosomes represents a promising strategy for diagnostic purposes, as full-length tau has been found to be more abundant in exosomes than in the free solutions of plasma or cerebrospinal fluid (CSF) of AD patients, whereas healthy individuals do not exhibit full-length tau [105].
Exosomes also carry enzymes such as BACE1, PSEN1, PSEN2, and Adam10 (Figure 2), which are responsible for cleaving APP, generating Aβ peptides, and consequently contributing to the formation of Aβ plaques [106]. Interestingly, when peripheral blood exosomes are injected into the hippocampus of an AD mouse model, they diffuse to the cortex and exhibit affinity with microglia, clustering around Aβ plaques (Figure 2). This suggests that exosomes play a role in amyloid deposition in AD, mediated by microglia ([100]). Consistently, exosomes stimulate the aggregation of Aβ1-42 in vitro. Furthermore, it has been shown that exosomes from AD patients are more neurotoxic than those from healthy individuals, impairing calcium homeostasis and mitochondrial function [107]. Additionally, the presence of exosomes is enriched in the hippocampus of AD patients compared to those with Parkinson’s disease or healthy individuals [108]. The release of exosomes is stimulated by glutamatergic activity in somato-dendritic compartments [109] and by neutral sphingomyelinase 2 (nSMase2), an enzyme that converts sphingomyelin to ceramide. Notably, inhibiting nSMase2 with GW4869 reduces exosome production, accompanied by a decrease in brain ceramide and Aβ plaque deposition [110] .Moreover, amyloid peptides induce the secretion of exosomes containing PAR-4 and C18 ceramide, which activate Caspase 3 and consequently lead to astrocyte apoptosis in vitro [111].
Exosomal microRNAs
miRNAs are small, double-stranded RNAs that modulate gene expression by targeting mRNA for degradation or inhibiting translation, a process known as RNA interference (RNAi) [112,113] Several miRNAs, including miR-9, miR-29a, miR-107, miR-125b, and miR-135b, regulate various aspects of AD pathology, such as Aβ metabolism, tau phosphorylation, and neuroinflammation [114]. These miRNAs function as negative regulators of BACE1, thereby reducing Aβ production and potentially slowing the progression of AD [103].
Exosomal miRNAs are attractive biomarkers due to their stability in bodily fluids and their ability to reflect the origin and function of their originating cells. Studies have shown that miRNAs associated with AD risk genes (e.g., miR-107, miR-135b-5p) are elevated in neuronal exosomes from AD patients, while others, such as miR-320a, are reduced in the CSF of AD patients compared to healthy controls [115].
Exosomal Circular RNAs
CircRNAs, which are abundant in exosomes, play a crucial role in their functionality [111,116]. These stable, single-stranded RNA molecules are formed through back-splicing, resulting in a half-life exceeding 48 hours [117,118,119]. They are classified into three main types: intronic circRNAs (ciRNAs), exonic circRNAs (EcircRNAs), and exon-intron circRNAs (EIciRNAs) [120,121]. EcircRNAs, which comprise over 80% of circRNAs, are derived from pre-mRNA exons and are found in both the cytoplasm and exosomes, whereas ciRNAs originate from introns and EIciRNAs are primarily located in the nucleus [122,123]
CircRNAs regulate gene expression by binding to miRNA response elements (MREs), effectively acting as sponges that sequester miRNAs from their target mRNAs [116] Additionally, circRNAs interact with proteins, influencing cellular functions and highlighting their complex regulatory roles [124,125]
Studies indicate that circRNAs can circulate through serum and are enriched in exosomes, with their levels influenced by associated miRNA variations, allowing them to transfer biological activity to target cells [116,126,127,128] A prominent circRNA, CDR1as, exhibits high expression in the normal brain but shows reduced levels in the hippocampus and cortex of individuals with AD. CDR1-as functions as a sponge for miR-7; its decrease impairs the degradation of BACE1 and APP, leading to an abnormal increase in Aβ levels [129,130,131].
Exosomal miRNAs as Potential Biomarkers for AD Diagnosis
Recent research has focused on the role of miRNAs in AD pathogenesis associated with EVs such as exosomes (Table 2). Duan et al., 2024, examined serum-derived exosomal miRNAs in AD patients using high-throughput sequencing and RT-qPCR [132]. The authors identified hsa-miR-125b-1-3p, hsa-miR-193a-5p, hsa-miR-378a-3p, hsa-miR-378i, and hsa-miR-451a as differentially expressed miRNAs. Notably, hsa-miR-451a exhibited an area under the curve (AUC) of 0.728. Pathway enrichment analysis linked these miRNAs to neuroactive ligand-receptor interactions, PI3K-Akt signaling, and cytokine-cytokine receptor interactions [132]. Previous studies using EVs isolated from plasma samples also showed that miR-451a was significantly deregulated in AD relative to DLB [133] as well as in prodomal AD patients, highlighting its potential for differential diagnosis.
A study involving 42 AD patients and 19 healthy controls (HC) revealed significant alterations in EV membrane antigens, inflammatory cytokine content, and miRNA expression. Specifically, myelin oligodendrocyte glycoprotein (MOG) and axonal glycoprotein CD171 correlate with disease severity. Five key miRNAs (let-7g-5p, miR126-3p, miR142-3p, miR146a-5p, and miR223-3p) showed a significant reduction in AD patients, suggesting a link between disease severity and neuroinflammation [134]. Serum exosomal miR-223 levels were significantly downregulated in dementia patients compared to controls. miR-223 levels correlated with Mini-Mental State Examination (MMSE) scores and inflammatory markers (IL-1β, IL-6, TNF-α). Previous studies showed that its downregulation promotes inflammatory responses in macrophages. ROC curve analysis indicated strong diagnostic potential (AUC = 0.875) [135]. Visconte et al., 2023, found an increased expression of mR-223 in EVs from AD patients, with a positive correlation between miR-223-3p and t-tau levels, suggesting a possible involvement in the cognitive decline and neurodegeneration in AD [136].
The microRNA hsa-miR-126-3p has been identified in two distinct studies on AD. Gámez-Valero et al., 2019, found this miRNA to be significantly reduced in AD patients compared to healthy controls. Aharon et al., 2020, also observed a decrease in this miRNA in patients with severe AD compared to healthy individuals. hsa-miR-126-3p plays crucial roles in neuronal homeostasis and inflammatory response. Its reduced expression may be associated with various pathophysiological mechanisms of AD, such as neuroinflammation and apoptosis [133,134]. miR-126-3p regulates the NF-κB pathway, a key factor in the exacerbated inflammatory response observed in AD [137]. Its downregulation may contribute to a chronic neuroinflammatory environment, leading to sustained microglial activation and progressive neuronal damage.
Furthermore, studies suggest that miR-126-3p plays a fundamental role in maintaining the integrity of the blood-brain barrier (BBB). Its reduced expression may increase BBB permeability, facilitating the infiltration of inflammatory cells and neurotoxic molecules into the central nervous system [138]. The decrease in miR-126-3p may also impact cellular pathways involved in protein degradation, promoting the accumulation of Aβ levels [139]. This alteration is directly linked to the formation of senile plaques and neurofibrillary tangles—hallmark features of AD. Gámez-Valero et al., 2019, suggested that the downregulation of miR-126-3p could help differentiate AD from other dementias, such as DLB. Target gene analysis revealed the regulation of pathways related to phosphorylation enzymes, the proteasome, and apoptosis, reinforcing its involvement in AD pathophysiology. However, further studies with larger cohorts and detailed functional analyses of its molecular interactions are needed to validate the clinical use of miR-126-3p as a biomarker [133].
Cha et al., 2019, observed reduced levels of miR-132 in exosomes derived from the nervous system in the plasma of AD patients compared to controls [140]. miRNA analysis of the brains of AD patients revealed lower miR-132 levels compared to high-risk pathological controls. In plasma, miR-132-3p in exosomes from the nervous system exhibited high sensitivity and specificity for diagnosing AD. miR-132 plays a key role in molecular events leading to Aβ deposition by regulating Aβ metabolism, including tau, MAPK, and Sirtuin1 (SIRT1) pathways [141]. Another study demonstrated that miR-132/212 deficiency in an animal model resulted in exacerbated tau pathology and memory deficits, which could be partially reversed by replacing with miR-132 mimics [142]. Furthermore, reduced miR-132 expression in the hippocampus and medial frontal gyrus of AD patients (Braak scores 4 to 6) compared to non-demented controls (Braak scores 0 to 3) correlated with impairments in neuronal differentiation, mediated through the regulation of p250GAP, a Rho family GTPase enriched in the brain ([143].
Recent studies have highlighted the role of miRNAs in the pathogenesis, particularly in relation to gene regulation and disease-modifying factors such as education and depression.
Wang et al., 2022, identified hsa-miR-20a-5p as a key regulator associated with AD risk in a Chinese cohort. The expression of hsa-miR-20a-5p was reduced in AD patients; however, this reduction was less pronounced in individuals with higher levels of education [144]. Notably, miR-20a-5p directly targets several genes implicated in AD, including APP, whose increased expression enhances Aβ production. The study also highlighted hsa-miR-185-5p as another downregulated miRNA in AD patients. Similar to miR-20a-5p, the decrease in hsa-miR-185-5p expression was partially offset by higher educational attainment. Functional enrichment analysis showed that validated targets of these miRNA are involved in AD-related pathways including Presenilin 1 (PSEN1), an essential component of the γ-secretase complex, which was found to be upregulated in AD patients. Additionally, the downregulation of hsa-miR-185-5p was particularly evident in AD patients with concurrent depressive symptoms. One of its predicted targets, Breast Cancer 1 (BRCA1), plays a role in PSEN1 turnover and was found to be overexpressed in AD. Collectively, these findings suggest that reduced hsa-miR-20a-5p expression contributes to increased APP levels, while the downregulation of hsa-miR-185-5p and hsa-miR-181c-5p leads to upregulation of their respective targets, facilitating Aβ accumulation and plaque formation in AD pathology [144].
The studies by Duan et al., 2024, and Kumar et al., 2023, highlight the potential role of miR-125b as a biomarker for AD [132]. hsa-miR-125b-1-3p was found to be differentially expressed in serum-derived exosomes of AD patients, showing an AUC of 0.765, with a sensitivity of 82.1%, and a specificity of 67.7%. Functional enrichment analysis indicated its involvement in key neurobiological pathways, including the PI3K-Akt and Hippo signaling pathways—an evolutionarily conserved signaling network that plays a crucial role in regulating numerous biological processes [132]; [145].
Table 2.
Exosomal miRNAs in biofluids as prospective biomarkers for AD diagnosis.
| EV samples | miRNA's | Biomarkers Characteristics | Reference |
|---|---|---|---|
| Plasma | let-7g; miR126-3p; miR142-3p; miR146a; mir223-3p; mir26b | Low level in severe AD (SAD). KEGG pathway analysis - The analysis revealed 43 KEGG pathways associated with the most significant miRNAs, with a focus on p53, toll-like receptors, MAPK, NF-kappa B, AD, apoptosis, and PI3K-Akt. Increase of endothelial EVs. Elevated levels of EVs expressing the axonal glycoprotein CD171. Increase of inflammatory cytokines and reduction of over 50% growth factors, compared to EVs of HC. SAD score and miRNA expression presented correlation (let-7g r = 0.5223, miR126-3p r = 0.4564, miR142-3p r = 0.4675, miR146a r = 0.5433, mir223-3p r = 0.4779). |
[146] |
| CSF, serum and plasma | miR-193b | Low level in CSF, serum and plasma of AD, and serum and plasma of MCI. Potential target of the 3' UTR of APP. Negative correlation between levels of miR-193b and Aβ42 in the CSF of patients with DAT (r =−0.442), and control group (r =−0.503). |
[147] |
| Plasma | miR-342-3p | Differential expression in AD group and correlated with other miRNAs decreased in AD. | [148] |
| miR-185-5p, hsa-miR-20a-5p, and hsa-miR-497-5p | Related to AD and education level. | ||
| Plasma | hsa-miR-185-5p, hsa-miR-181c-5p, hsa-miR-451a, and hsa-miR-664a-3p | Decreased hsa-miR-185-5p in AD improves the expression of PSEN1 and GSK3β, which further increases Aβ generation. The 3′ UTR of hsa-miR-181c-5p contains a predicted binding site for IL1. In AD patients, IL1 is associated with Aβ generation. hsa-miR-451a correlated with clinical measurements of education (R = 0.477), depression (R = 0.605), and leisure activity (R = 0.411). hsa-miR-664a-3p was upregulated in AD patients, which downregulated CREB1 and BDNF expression levels, thereby leading to a cognitive decline in AD patients. |
[149] |
| Serum | miR-223 | Decreased in patients with dementia. miR-223 level correlated with Mental State Examination (MMSE) scores, Clinical Dementia Rating (CDR) scores, magnetic resonance spectroscopy (MRS) spectral ratios and serum concentrations of IL-1b, IL-6, TNF-α, and CRP. | [150] |
| miR-223 downregulated in the dementia group compared to the control group. Differential expression of miR-223 between AD and Vascular Dementia (VaD) groups. Higher miR-223 levels in AD patients under medical care than those at their first clinical visit. Levels of miR-223 in the blood of dementia patients have a positive correlation with the scores on the MMSE and CDR scales (r = 0.365 and 0.4598, respectively). miR-223 levels in patients with dementia present positive correlation with the scores on the MMSE and CDR scales (r = 0.365 and 0.4598, respectively). Levels of IL-1β, IL-6, TNF-α, and PCR elevated in patients with dementia. Higher in AD compared to VaD. A correlation was found between the levels of miR-223 and the concentrations of IL-1β, IL-6, TNF-α, and PCR (r = -0.5504, -0.4549, -0.5152, -0.4977, respectively). miR-223 present AUC of 0.875 (95% CI: 0.7779–0.9721). |
|||
| Plasma | miR-16-5p, miR-19b-3p, miR-25-3p, miR-30b-5p, miR-92a-3p and miR-451a | Validation analysis confirmed significant upregulation of miR-16-5p, miR-25-3p, miR-92a-3p, and miR-451a in prodromal AD patients, suggesting these dysregulated miRNAs are involved in the early progression of AD. Group of AD patients presented positive correlations between Aβ42 and miR-30b-5p (r = 0.67) and between h-tau and miR-223-3p (r = 0.62). |
[151] |
| Plasma | hsa-miR-451a e hsa-miR-21-5p hsa-miR-23a-3p, hsa-miR-126-3p, hsa-let-7i-5p e hsa-miR-151a-3p |
Down-regulated in AD samples respect to dementia with Lewy bodies (DLB) patients. Decreased in AD respect to controls. |
[152] |
| Cortical gray matter | miR-132 and miR-212 | Levels of miR-132 separated controls from AD-MCI with an AUC of 0.58 (95% CI: 0.38–0.78) and controls from AD dementia with an AUC of 0.77 (95% CI: 0.61–0.93). miR-212 showed better discrimination than miR-132 between AD-MCI and controls, and AD and controls. miR-212 levels separated controls from AD-MCI with an AUC of 0.68 (95% CI: 0.5–0.86) and controls from AD dementia with an AUC of 0.84 (95% CI: 0.72–0.96). miR-212 achieved a sensitivity of 92.2% (95% CI: 68.5–99.6%) and a specificity of 69.0% (95% CI: 50.8–82.7%). | [140] |
| Plasma | miR-502-5p miR-483-5p |
AUC is 0.872, sensitivity 79.2 % and specificity 83.3 %. Area Under the Curve (AUC) is 0.901, sensitivity 79.2 % and specificity 100 %. |
[153] |
| Serum | hsa-miR-125b-1-3p, hsa-miR-193a-5p, hsa-miR-378a-3p, hsa-miR-378i and hsa-miR-451 | hsa-miR-125b-1-3p has an AUC of 0.765 in the AD group compared to the healthy group. Sensitivity (82.1) and specificity (67.7%). | [126,154,155] |
| CSF | miR-455–3p | Elevated levels in AD patients compared to controls (AUC = 0.745). | [156] |
| Plasma (NCAM/ABCA1 dual-labeled exosomal Aβ42/40) | miR-384 | The AUC of NCAM/ABCA1 dual-labeled exosomal Aβ42/40 for diagnosis of SCD was higher than that of Aβ42, T-tau, and P-T181-tau; the AUC of NCAM/ABCA1 dual-labeled exosomal miR-384 for diagnosis of SCD was higher than that of Aβ42, Aβ42/40, T-tau, P-T181-tau, and NfL. miR-384 can downregulate the expression and activity of BACE. |
[157] |
| Plasma (Neurons: EVL1CAM) | miR-29a-5p, miR-125b-5p, and miR-210-3p | MCI, MCI-AD, and AD dementia (AUC = 0.948). | [158] |
| miR-210-3p and miR-132-5p | MCI (AUC = 0.941). | ||
| miR-106-5p |
AD dementia (AUC = 1.000). |
||
| miR-106b-5p |
Negative correlation with cortical thickness in regions prone to age-related dementias as imaged in MRI. | ||
|
Plasma (Astrocytes: sEVGLAST) |
miR-107 |
MCI, MCI-AD, and AD dementia (AUCs = 0.964); AD dementia (AUC = 1.000). |
|
| miR-107 and miR 132-5p | Negative correlation with the cortical thickness. | ||
| and miR-210-3p | MCI (AUCs = 0.941). | ||
| miR-29a-5p and miR-106-5p | Overall cognitive impairment (AUC = 0.925). | ||
|
Plasma (Microglia: sEVTMEM119) |
miR-29a-5p |
MCI (AUC = 0.840). |
|
| miR-132-5p and miR-125b-5p | AD dementia (AUC = 1.000). | ||
| miR-106b-5p and miR-132-5p | Negative correlation with the temporal cortical thickness. | ||
| Plasma (Oligodendrocytes: sEVPDGFRα) | miR-29a-5p | AD dementia (AUC = 1.000). Negative correlation with temporal cortical thickness. | |
| Plasma (Pericytes: sEVPDGFRβ) | miR-9-5p | Overall cognitive impairment (AUC = 0.935), MCI (AUC = 0.931), and AD (AUC = 1.000). | |
| Plasma (Endothelial cells: sEVCD31) | miR-132-5p | Overall impairment and MCI, and prediction of AD (AUC = 1.000). | |
| miR-210-3p | Negative correlation with cortical thickness. | ||
| Plasma (Pericytes: sEVPDGFRβ and Endothelial cells: sEVCD31) | miR-9-5p (sEVPDGFRβ) and miR-132-5p (sEVCD31) | Overall cognitive impairment (AUC = 1.000). | |
| miR-132-5p (sEVCD31) and miR-135b-5p (sEVPDGFRβ) | MCI and AD. |
Abbreviations: EV, extracellular vesicle; SAD, severe AD; KEGG pathway, Kyoto Encyclopedia of Genes and Genomes; HC, healthy control; CSF: cerebrospinal fluid; MCI, mild cognitive impairment; 3′ UTR, 3'-untranslated region; APP, amyloid precursor protein; DAT, dementia of Alzheimer-type; PSEN1, Presenilin 1; GSK3β, Glycogen synthase kinase-3β; IL1, Interleukin-1; CREB1, cAMP-response element binding protein; BDNF, brain-derived neurotrophic factors; MMSE, Mental State Examination scores; CDR, Clinical Dementia Rating; MRS, magnetic resonance spectroscopy; TNF-α, Tumor necrosis factor alpha; VaD ,Vascular Dementia; AUC, area under the curve; DLB, dementia with Lewy bodies; CI, Confidence interval; NCAM/ABCA1, neural cell adhesion molecule (NCAM)/ATP-binding cassette transporter A1 (ABCA1); SCD, subjective cognitive decline; BACE-1, beta-secretase-1.
Concluding Remarks and Prospects
The potential of exosomes and their molecular contents, particularly miRNAs and circRNAs, as biomarkers in the context of AD is an area of significant interest. Exosomes have the capacity to serve as carriers of critical biomarkers linked to disease, and their molecular profiles may provide valuable insights into disease mechanisms and progression.
Future research should prioritize the exploration of exosomal biology to enhance our understanding of how these vesicles and their contents can function as reliable biomarkers for AD diagnosis. Identifying specific exosomal miRNAs and circRNAs associated with AD could lead to the development of novel diagnostic tools. Furthermore, elucidating the mechanisms by which exosomes influence neuronal health may reveal important connections between these biomarkers and the pathophysiology of AD.
In conclusion, exosomal ncRNAs may play a significant role in the diagnosis of AD, alongside well-established biomarkers such as Aβ and tau, thereby helping to refine diagnostic results. The effectiveness of these biomarkers will depend on coordinated changes in the patterns of these molecules, as well as the validation of findings through multicenter studies with independent cohorts.
Author Contributions
Conceptualization, S.S.T.A and R.T.A.; writing—S.S.T.A. original draft preparation, S.S.T.A., R.T.A. C.L.F. M.V.R., R.T.A.; writing—review and editing, S.S.T.A., R.T.A. C.L.F. M.V.R., L.D.S.S., P.R.P.B; D.D.C.B., F.V.G., and R.T.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; et al. Alzheimer disease. Nat Rev Dis Primers. 2021, 7, 33. [Google Scholar] [PubMed]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022, 7, e105–e125. [Google Scholar] [CrossRef] [PubMed]
- Grøntvedt, G.R.; Schröder, T.N.; Sando, S.B.; White, L.; Bråthen, G.; Doeller, C.F. Alzheimer’s disease. Current Biology. 2018, 28, R645–R649. [Google Scholar] [CrossRef] [PubMed]
- Madnani, R.S. Alzheimer’s disease: a mini-review for the clinician. Front Neurol. 2023, 14, 1178588. [Google Scholar] [CrossRef]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; et al. Clinical diagnosis of Alzheimer’s disease: recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar]
- Zhao, N.; Ren, Y.; Yamazaki, Y.; Qiao, W.; Li, F.; Felton, L.M.; et al. Alzheimer’s Risk Factors Age, APOE Genotype, and Sex Drive Distinct Molecular Pathways. Neuron. 2020, 106, 727–742e6. [Google Scholar] [CrossRef]
- Grant, W.B. A Brief History of the Progress in Our Understanding of Genetics and Lifestyle, Especially Diet, in the Risk of Alzheimer’s Disease. Journal of Alzheimer’s Disease. 2024, 100, S165–78. [Google Scholar] [CrossRef]
- Nordestgaard, L.T.; Christoffersen, M.; Frikke-Schmidt, R. Shared Risk Factors between Dementia and Atherosclerotic Cardiovascular Disease. Int J Mol Sci. 2022, 23, 9777. [Google Scholar]
- Xu, W.; Tan, L.; Wang, H.F.; Jiang, T.; Tan, M.S.; Tan, L.; et al. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2015, 86, 1299–1306. [Google Scholar] [CrossRef]
- Li, J.Q.; Tan, L.; Wang, H.F.; Tan, M.S.; Tan, L.; Xu, W.; et al. Risk factors for predicting progression from mild cognitive impairment to Alzheimer’s disease: a systematic review and meta-analysis of cohort studies. J Neurol Neurosurg Psychiatry. 2016, 87, 476–484. [Google Scholar] [CrossRef]
- Arenaza-Urquijo, E.M.; Vemuri, P. Resistance vs resilience to Alzheimer disease. Neurology. 2018, 90, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Monsell, S.E.; Mock, C.; Fardo, D.W.; Bertelsen, S.; Cairns, N.J.; Roe, C.M.; et al. Genetic Comparison of Symptomatic and Asymptomatic Persons With Alzheimer Disease Neuropathology. Alzheimer Dis Assoc Disord. 2017, 31, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Prpar Mihevc, S.; Majdič, G. Canine Cognitive Dysfunction and Alzheimer’s Disease – Two Facets of the Same Disease? Front Neurosci. 2019, 13, 604. [Google Scholar] [CrossRef]
- Panek, W.K.; Murdoch, D.M.; Gruen, M.E.; Mowat, F.M.; Marek, R.D.; Olby, N.J. Plasma Amyloid Beta Concentrations in Aged and Cognitively Impaired Pet Dogs. Mol Neurobiol. 2021, 58, 483–489. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimer’s & Dementia. 2015, 11, 600. [Google Scholar]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; et al. Association of Extracellular Vesicle Biomarkers With Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2019, 76, 1340. [Google Scholar] [CrossRef]
- Bhat, A.A.; Younes, S.N.; Raza, S.S.; Zarif, L.; Nisar, S.; Ahmed, I.; et al. Role of non-coding RNA networks in leukemia progression, metastasis and drug resistance. Mol Cancer. 2020, 19, 57. [Google Scholar] [CrossRef]
- Soares Martins, T.; Trindade, D.; Vaz, M.; Campelo, I.; Almeida, M.; Trigo, G.; et al. Diagnostic and therapeutic potential of exosomes in Alzheimer’s disease. J Neurochem. 2021, 156, 162–181. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. The FASEB Journal. 2016, 30, 4141–4148. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Winston, C.N.; Goetzl, E.J.; Schwartz, J.B.; Elahi, F.M.; Rissman, R.A. Complement protein levels in plasma astrocyte-derived exosomes are abnormal in conversion from mild cognitive impairment to Alzheimer’s disease dementia. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2019, 11, 61–66. [Google Scholar]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2016, 3, 63–72. [Google Scholar]
- Chen Jjiao Yang, G.; Yan Qqing Zhao, J.; Li, S. Exosome-encapsulated microRNAs as promising biomarkers for Alzheimer’s disease. Rev Neurosci. 2019, 31, 77–87. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics. 2013, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Van Balkom, B.W.M.; Eisele, A.S.; Michiel Pegtel, D.; Bervoets, S.; Verhaar, M.C. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J Extracell Vesicles. 2015, 4, 26760. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.A.; Ludwig, R.G.; Garcia-Martin, R.; Brandão, B.B.; Kahn, C.R. Extracellular miRNAs: From Biomarkers to Mediators of Physiology and Disease. Cell Metab. 2019, 30, 656–673. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; et al. Exosome and Exosomal MicroRNA: Trafficking, Sorting, and Function. Genomics Proteomics Bioinformatics. 2015, 13, 17–24. [Google Scholar] [CrossRef]
- Shobeiri, P.; Alilou, S.; Jaberinezhad, M.; Zare, F.; Karimi, N.; Maleki, S.; et al. Circulating long non-coding RNAs as novel diagnostic biomarkers for Alzheimer’s disease (AD): A systematic review and meta-analysis. PLoS One. 2023, 18, e0281784. [Google Scholar] [CrossRef]
- Tang, Z.; Cheng, X.; Su, X.; Wu, L.; Cai, Q.; Wu, H. Treponema denticola Induces Alzheimer-Like Tau Hyperphosphorylation by Activating Hippocampal Neuroinflammation in Mice. J Dent Res. 2022, 101, 992–1001. [Google Scholar] [CrossRef]
- Graff-Radford, J.; Yong, K.X.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; et al. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021, 20, 222–234. [Google Scholar] [CrossRef]
- Phochantachinda, S.; Chatchaisak, D.; Temviriyanukul, P.; Chansawang, A.; Pitchakarn, P.; Chantong, B. Ethanolic Fruit Extract of Emblica officinalis Suppresses Neuroinflammation in Microglia and Promotes Neurite Outgrowth in Neuro2a Cells. Evidence-Based Complementary and Alternative Medicine. 2021, 2021, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.; Wildman, D.E. Extracellular Vesicles and the Promise of Continuous Liquid Biopsies. J Pathol Transl Med. 2018, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kanninen, K.M.; Bister, N.; Koistinaho, J.; Malm, T. Exosomes as new diagnostic tools in CNS diseases. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2016, 1862, 403–410. [Google Scholar] [CrossRef]
- Apostolova, L. Early-Stage Alzheimer Primer. J Clin Psychiatry. 2021, 82, 32781. [Google Scholar] [CrossRef]
- Eramudugolla, R.; Mortby, M.E.; Sachdev, P.; Meslin, C.; Kumar, R.; Anstey, K.J. Evaluation of a research diagnostic algorithm for DSM-5 neurocognitive disorders in a population-based cohort of older adults. Alzheimers Res Ther. 2017, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, P.S.; Blacker, D.; Blazer, D.G.; Ganguli, M.; Jeste, D.V.; Paulsen, J.S.; et al. Classifying neurocognitive disorders: the DSM-5 approach. Nat Rev Neurol. 2014, 10, 634–642. [Google Scholar] [CrossRef]
- Thal, D.R.; Capetillo-Zarate, E.; Del Tredici, K.; Braak, H. The Development of Amyloid β Protein Deposits in the Aged Brain. Science of Aging Knowledge Environment. 2006, 2006, re1. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s & Dementia. 2012, 8, 1–13. [Google Scholar]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics. 2022, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019, 14, 32. [Google Scholar]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Schilling, L.P.; Balthazar, M.L.F.; Radanovic, M.; Forlenza, O.V.; Silagi, M.L.; Smid, J.; et al. Diagnóstico da doença de Alzheimer: recomendações do Departamento Científico de Neurologia Cognitiva e do Envelhecimento da Academia Brasileira de Neurologia. Dement Neuropsychol. 2022, 16 (Suppl. 1), 25–39. [Google Scholar] [CrossRef]
- Murden, R.A. The Diagnosis of Alzheimer’s Disease. In 1990. p. 59–64.
- Parker, T.D.; Slattery, C.F.; Zhang, J.; Nicholas, J.M.; Paterson, R.W.; Foulkes, A.J.M.; et al. Cortical microstructure in young onset Alzheimer’s disease using neurite orientation dispersion and density imaging. Hum Brain Mapp. 2018, 39, 3005–3017. [Google Scholar]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb Perspect Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Alafuzoff, I.; Libard, S. Ageing-Related Neurodegeneration and Cognitive Decline. Int J Mol Sci. 2024, 25, 4065. [Google Scholar] [CrossRef]
- Boyle, P.A.; Yu, L.; Leurgans, S.E.; Wilson, R.S.; Brookmeyer, R.; Schneider, J.A.; et al. Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Ann Neurol. 2019, 85, 114–124. [Google Scholar] [CrossRef]
- White, L.R.; Corrada, M.M.; Kawas, C.H.; Cholerton, B.A.; Edland, S.E.; Flanagan, M.E.; et al. Neuropathologic Changes of Alzheimer’s Disease and Related Dementias: Relevance to Future Prevention. Journal of Alzheimer’s Disease. 2023, 95, 307–316. [Google Scholar]
- Jack, C.R.; Andrews, J.S.; Beach, T.G.; Buracchio, T.; Dunn, B.; Graf, A.; et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimer’s & Dementia. 2024, 20, 5143–5169. [Google Scholar]
- Dubois, B.; Villain, N.; Schneider, L.; Fox, N.; Campbell, N.; Galasko, D.; et al. Alzheimer Disease as a Clinical-Biological Construct—An International Working Group Recommendation. JAMA Neurol. 2024, 81, 1304. [Google Scholar] [PubMed]
- Jack, C.R.; Andrews, J.S.; Beach, T.G.; Buracchio, T.; Dunn, B.; Graf, A.; et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimer’s & Dementia. 2024, 20, 5143–5169. [Google Scholar]
- McKenna, M.C.; O’Philibin, L.; Foley, T.; Dolan, C.; Kennelly, S.; O’Dowd, S.; et al. The Use of Biomarkers in Diagnosing Alzheimer’s Disease: Recommendations of the Irish Working Group on Biological Approaches to the Diagnosis of Dementia. Ir Med J. 2024, 117, 1062. [Google Scholar] [PubMed]
- Studart Neto, A.; Barbosa, B.J.A.P.; Coutinho, A.M.; Souza, L.C.; de Schilling, L.P.; da Silva, M.N.M.; et al. Guidelines for the use and interpretation of Alzheimer’s disease biomarkers in clinical practice in Brazil: recommendations from the Scientific Department of Cognitive Neurology and Aging of the Brazilian Academy of Neurology. Dement Neuropsychol. 2024, 18, e2024C001. [Google Scholar] [PubMed]
- Okazawa, H.; Nogami, M.; Ishida, S.; Makino, A.; Mori, T.; Kiyono, Y.; et al. PET/MRI multimodality imaging to evaluate changes in glymphatic system function and biomarkers of Alzheimer’s disease. Sci Rep. 2024, 14, 12310. [Google Scholar]
- Jack, C.R. Advances in Alzheimer’s disease research over the past two decades. Lancet Neurol. 2022, 21, 866–869. [Google Scholar]
- Ashton, N.J.; Janelidze, S.; Al Khleifat, A.; Leuzy, A.; van der Ende, E.L.; Karikari, T.K.; et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat Commun. 2021, 12, 3400. [Google Scholar]
- Benedet, A.L.; Milà-Alomà, M.; Vrillon, A.; Ashton, N.J.; Pascoal, T.A.; Lussier, F.; et al. Differences Between Plasma and Cerebrospinal Fluid Glial Fibrillary Acidic Protein Levels Across the Alzheimer Disease Continuum. JAMA Neurol. 2021, 78, 1471. [Google Scholar]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol Psychiatry. 2021, 26, 5481–5503. [Google Scholar]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019, 93, e1647–e1659. [Google Scholar]
- Karikari, T.K.; Ashton, N.J.; Brinkmalm, G.; Brum, W.S.; Benedet, A.L.; Montoliu-Gaya, L.; et al. Blood phospho-tau in Alzheimer disease: analysis, interpretation, and clinical utility. Nat Rev Neurol. 2022, 18, 400–418. [Google Scholar] [CrossRef]
- Ashton, N.J.; Puig-Pijoan, A.; Milà-Alomà, M.; Fernández-Lebrero, A.; García-Escobar, G.; González-Ortiz, F.; et al. Plasma and CSF biomarkers in a memory clinic: Head-to-head comparison of phosphorylated tau immunoassays. Alzheimer’s & Dementia. 2023, 19, 1913–1924. [Google Scholar]
- Mielke, M.M.; Aakre, J.A.; Algeciras-Schimnich, A.; Proctor, N.K.; Machulda, M.M.; Eichenlaub, U.; et al. Comparison of CSF phosphorylated tau 181 and 217 for cognitive decline. Alzheimer’s & Dementia. 2022, 18, 602–611. [Google Scholar]
- Jia, J.; Ning, Y.; Chen, M.; Wang, S.; Yang, H.; Li, F.; et al. Biomarker Changes during 20 Years Preceding Alzheimer’s Disease. New England Journal of Medicine. 2024, 390, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Tideman, P.; Mattsson-Carlgren, N.; Schindler, S.E.; Smith, R.; Ossenkoppele, R.; et al. Blood Biomarkers to Detect Alzheimer Disease in Primary Care and Secondary Care. JAMA. 2024, 332, 1245. [Google Scholar]
- Ashton, N.J.; Brum, W.S.; Di Molfetta, G.; Benedet, A.L.; Arslan, B.; Jonaitis, E.; et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurol. 2024, 81, 255. [Google Scholar] [PubMed]
- Farvadi, F.; Hashemi, F.; Amini, A.; Vakilinezhad, M.A.; Raee, M.J. Early Diagnosis of Alzheimer’s Disease with Blood Test; Tempting but Challenging. Int J Mol Cell Med. 2023, 12, 172. [Google Scholar]
- Livingston, G.; Huntley, J.; Liu, K.Y.; Costafreda, S.G.; Selbæk, G.; Alladi, S.; et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. The Lancet. 2024, 404, 572–628. [Google Scholar]
- Petrie, E.C.; Cross, D.J.; Galasko, D.; Schellenberg, G.D.; Raskind, M.A.; Peskind, E.R.; et al. Preclinical Evidence of Alzheimer Changes. Arch Neurol. 2009, 66, 632–637. [Google Scholar] [CrossRef]
- Sheikh-Bahaei, N.; Chen, M.; Pappas, I. Magnetic Resonance Spectroscopy (MRS) in Alzheimer’s Disease. In 2024. p. 115–42.
- Chen, X.; Firulyova, M.; Manis, M.; Herz, J.; Smirnov, I.; Aladyeva, E.; et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023, 615, 668–677. [Google Scholar] [CrossRef]
- Kamagata, K.; Andica, C.; Takabayashi, K.; Saito, Y.; Taoka, T.; Nozaki, H.; et al. Associação de índices de ressonância magnética do sistema glinfático com deposição amiloide e cognição em comprometimento cognitivo leve e doença de Alzheimer. Neurology. 2022, 2648–2660. [Google Scholar]
- Brum, W.S.; Cullen, N.C.; Janelidze, S.; Ashton, N.J.; Zimmer, E.R.; Therriault, J.; et al. A two-step workflow based on plasma p-tau217 to screen for amyloid β positivity with further confirmatory testing only in uncertain cases. Nat Aging. 2023, 3, 1079–1090. [Google Scholar] [CrossRef]
- Colmant, L.; Boyer, E.; Gerard, T.; Sleegers, K.; Lhommel, R.; Ivanoiu, A.; et al. Definition of a Threshold for the Plasma Aβ42/Aβ40 Ratio Measured by Single-Molecule Array to Predict the Amyloid Status of Individuals without Dementia. Int J Mol Sci. 2024, 25, 1173. [Google Scholar] [CrossRef]
- Ding, X.L.; Tuo, Q.Z.; Lei, P. An Introduction to Ultrasensitive Assays for Plasma Tau Detection. Journal of Alzheimer’s Disease. 2021, 80, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.T.; Rocchi, L.; Hammond, P.; Hardy, C.J.D.; Warren, J.D.; Ridha, B.H.; et al. Effect of donepezil on transcranial magnetic stimulation parameters in Alzheimer’s disease. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2018, 4, 103–107. [Google Scholar]
- Kamagata, K.; Andica, C.; Hatano, T.; Ogawa, T.; Takeshige-Amano, H.; Ogaki, K.; et al. Advanced diffusion magnetic resonance imaging in patients with Alzheimer’s and Parkinson’s diseases. Neural Regen Res. 2020, 15, 1590. [Google Scholar] [CrossRef] [PubMed]
- Tabarestani, S.; Eslami, M.; Cabrerizo, M.; Curiel, R.E.; Barreto, A.; Rishe, N.; et al. A Tensorized Multitask Deep Learning Network for Progression Prediction of Alzheimer’s Disease. Front Aging Neurosci. 2022, 14, 810873. [Google Scholar] [CrossRef]
- Wen, J.; Thibeau-Sutre, E.; Diaz-Melo, M.; Samper-González, J.; Routier, A.; Bottani, S.; et al. Convolutional neural networks for classification of Alzheimer’s disease: Overview and reproducible evaluation. Med Image Anal. 2020, 63, 101694. [Google Scholar] [CrossRef]
- Bischof, G.N.; Bartenstein, P.; Barthel, H.; van Berckel, B.; Doré, V.; van Eimeren, T.; et al. Toward a Universal Readout for 18 F-Labeled Amyloid Tracers: The CAPTAINs Study. Journal of Nuclear Medicine. 2021, 62, 999–1005. [Google Scholar] [CrossRef]
- Filippi, L.; Chiaravalloti, A.; Bagni, O.; Schillaci, O. 18F-labeled radiopharmaceuticals for the molecular neuroimaging of amyloid plaques in Alzheimer’s disease. Am J Nucl Med Mol Imaging. 2018, 8, 268–281. [Google Scholar]
- Rowe, C.C.; Pejoska, S.; Mulligan, R.S.; Jones, G.; Chan, J.G.; Svensson, S.; et al. Head-to-Head Comparison of 11 C-PiB and 18 F-AZD4694 (NAV4694) for β-Amyloid Imaging in Aging and Dementia. Journal of Nuclear Medicine. 2013, 54, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.A.; Chiotis, K.; Rodriguez-Vieitez, E.; Lemoine, L.; Ågren, H.; Nordberg, A. Cross-interaction of tau PET tracers with monoamine oxidase B: evidence from in silico modelling and in vivo imaging. Eur J Nucl Med Mol Imaging. 2019, 46, 1369–1382. [Google Scholar] [CrossRef]
- Groot, C.; Villeneuve, S.; Smith, R.; Hansson, O.; Ossenkoppele, R. Tau PET Imaging in Neurodegenerative Disorders. Journal of Nuclear Medicine. 2022, 63 (Supplement 1), 20S–26S. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef]
- Xia, C.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; et al. [18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s & Dementia. 2013, 9, 666–676. [Google Scholar]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Wittens, M.M.J.; Allemeersch, G.J.; Sima, D.M.; Naeyaert, M.; Vanderhasselt, T.; Vanbinst, A.M.; et al. Inter- and Intra-Scanner Variability of Automated Brain Volumetry on Three Magnetic Resonance Imaging Systems in Alzheimer’s Disease and Controls. Front Aging Neurosci. 2021, 13, 746982. [Google Scholar] [CrossRef]
- Chen, X.; Firulyova, M.; Manis, M.; Herz, J.; Smirnov, I.; Aladyeva, E.; et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023, 615, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.; Hillmer, L.J.; Lu, H.; Arfanakis, K.; Gold, B.T.; Bauer, C.E.; et al. MRI free water as a biomarker for cognitive performance: Validation in the MarkVCID consortium. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2022, 14, e12362. [Google Scholar]
- Sathe, A.; Yang, Y.; Schilling, K.G.; Shashikumar, N.; Moore, E.; Dumitrescu, L.; et al. Free-water: A promising structural biomarker for cognitive decline in aging and mild cognitive impairment. Imaging Neuroscience. 2024, 2, 1–16. [Google Scholar] [CrossRef]
- Moradi, E.; Hallikainen, I.; Hänninen, T.; Tohka, J. Rey’s Auditory Verbal Learning Test scores can be predicted from whole brain MRI in Alzheimer’s disease. Neuroimage Clin. 2017, 13, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology , function , and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Ciferri, M.C.; Quarto, R.; Tasso, R. Extracellular Vesicles as Biomarkers and Therapeutic Tools: From Pre-Clinical to Clinical Applications. Biology 2021, 10, 359. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Baixauli, F.; Mittelbrunn, M.; Fernández-Delgado, I.; Torralba, D.; Moreno-Gonzalo, O.; et al. ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nat Commun. 2016, 7, 13588. [Google Scholar] [CrossRef] [PubMed]
- Krämer-Albers, E.; Bretz, N.; Tenzer, S.; Winterstein, C.; Möbius, W.; Berger, H.; et al. Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: Trophic support for axons? Proteomics Clin Appl. 2007, 1, 1446–1461. [Google Scholar] [CrossRef]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; et al. Neurotransmitter-Triggered Transfer of Exosomes Mediates Oligodendrocyte–Neuron Communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef] [PubMed]
- Drago, F.; Lombardi, M.; Prada, I.; Gabrielli, M.; Joshi, P.; Cojoc, D.; et al. ATP Modifies the Proteome of Extracellular Vesicles Released by Microglia and Influences Their Action on Astrocytes. Front Pharmacol. 2017, 8, 910. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, H.; Zhu, Z.; Minhas, J.K.; Jin, Y. Enrichment of selective miRNAs in exosomes and delivery of exosomal miRNAs in vitro and in vivo. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2017, 312, L110–21. [Google Scholar] [CrossRef]
- Migneault, F.; Dieudé, M.; Turgeon, J.; Beillevaire, D.; Hardy, M.P.; Brodeur, A.; et al. Apoptotic exosome-like vesicles regulate endothelial gene expression, inflammatory signaling, and function through the NF-κB signaling pathway. Sci Rep. 2020, 10, 12562. [Google Scholar] [CrossRef]
- Martinez-Valbuena, I.; Visanji, N.P.; Kim, A.; Lau, H.H.C.; So, R.W.L.; Alshimemeri, S.; et al. Alpha-synuclein seeding shows a wide heterogeneity in multiple system atrophy. Transl Neurodegener. 2022, 11, 7. [Google Scholar] [CrossRef]
- Kumar, A.; Su, Y.; Sharma, M.; Singh, S.; Kim, S.; Peavey, J.J.; et al. MicroRNA expression in extracellular vesicles as a novel blood-based biomarker for Alzheimer’s disease. Alzheimer’s & Dementia. 2023, 19, 4952–4966. [Google Scholar]
- Crotti, A.; Sait, H.R.; McAvoy, K.M.; Estrada, K.; Ergun, A.; Szak, S.; et al. BIN1 favors the spreading of Tau via extracellular vesicles. Sci Rep. 2019, 9, 9477. [Google Scholar] [CrossRef]
- Guix, F.; Corbett, G.; Cha, D.; Mustapic, M.; Liu, W.; Mengel, D.; et al. Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles. Int J Mol Sci. 2018, 19, 663. [Google Scholar] [CrossRef] [PubMed]
- Bellingham, S.A.; Guo, B.B.; Coleman, B.M.; Hill, A.F. Exosomes: Vehicles for the Transfer of Toxic Proteins Associated with Neurodegenerative Diseases? Front Physiol. 2012, 3, 124. [Google Scholar] [CrossRef]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; et al. Extracellular vesicle-associated Aβ mediates trans-neuronal bioenergetic and Ca2+-handling deficits in Alzheimer’s disease models. NPJ Aging Mech Dis. 2016, 2, 16019. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; et al. Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proceedings of the National Academy of Sciences. 2006, 103, 11172–11177. [Google Scholar] [CrossRef]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; et al. Neurotransmitter-Triggered Transfer of Exosomes Mediates Oligodendrocyte–Neuron Communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging. 2014, 35, 1792–1800. [Google Scholar] [CrossRef]
- Wang, G.; Dinkins, M.; He, Q.; Zhu, G.; Poirier, C.; Campbell, A.; et al. Astrocytes Secrete Exosomes Enriched with Proapoptotic Ceramide and Prostate Apoptosis Response 4 (PAR-4). Journal of Biological Chemistry. 2012, 287, 21384–21395. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Titze-de-Almeida, R.; David, C.; Titze-de-Almeida, S.S. The Race of 10 Synthetic RNAi-Based Drugs to the Pharmaceutical Market. Pharm Res. 2017, 34, 1339–1363. [Google Scholar] [CrossRef] [PubMed]
- Takousis, P.; Sadlon, A.; Schulz, J.; Wohlers, I.; Dobricic, V.; Middleton, L.; et al. Differential expression of microRNAs in Alzheimer’s disease brain, blood, and cerebrospinal fluid. Alzheimer’s & Dementia. 2019, 15, 1468–1477. [Google Scholar]
- Tan, Y.J.; Wong, B.Y.X.; Vaidyanathan, R.; Sreejith, S.; Chia, S.Y.; Kandiah, N.; et al. Altered Cerebrospinal Fluid Exosomal microRNA Levels in Young-Onset Alzheimer’s Disease and Frontotemporal Dementia. J Alzheimers Dis Rep. 2021, 5, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, Q.; Bao, C.; Li, S.; Guo, W.; Zhao, J.; et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015, 25, 981–984. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; et al. Global quantification of mammalian gene expression control. Nature. 2011, 473, 337–342. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013, 19, 141–157. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Titze-de-Almeida, S.S.; Titze-de-Almeida, R. Progress in circRNA-Targeted Therapy in Experimental Parkinson’s Disease. Pharmaceutics 2023, 15, 2035. [Google Scholar] [CrossRef]
- Isaac, R.; Reis, F.C.G.; Ying, W.; Olefsky, J.M. Exosomes as mediators of intercellular crosstalk in metabolism. Cell Metab. 2021, 33, 1744–1762. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zheng, Y.; Zhang, J.; Chen, S.; Zhao, F. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun. 2016, 7, 12060. [Google Scholar] [CrossRef]
- Isaac, R.; Reis, F.C.G.; Ying, W.; Olefsky, J.M. Exosomes as mediators of intercellular crosstalk in metabolism. Cell Metab. 2021, 33, 1744–1762. [Google Scholar] [PubMed]
- Ulshöfer, C.J.; Pfafenrot, C.; Bindereif, A.; Schneider, T. Methods to study circRNA-protein interactions. Methods. 2021, 196, 36–46. [Google Scholar] [PubMed]
- Aufiero, S.; van den Hoogenhof, M.M.G.; Reckman, Y.J.; Beqqali, A.; van der Made, I.; Kluin, J.; et al. Cardiac circRNAs arise mainly from constitutive exons rather than alternatively spliced exons. RNA. 2018, 24, 815–827. [Google Scholar] [CrossRef]
- Duan, Y.; Wang, Y.; Liu, Y.; Jin, Z.; Liu, C.; Yu, X.; et al. Circular RNAs in Parkinson’s Disease: Reliable Biological Markers and Targets for Rehabilitation. Mol Neurobiol. 2023, 60, 3261–3276. [Google Scholar] [PubMed]
- Preußer, C.; Hung, L.; Schneider, T.; Schreiner, S.; Hardt, M.; Moebus, A.; et al. Selective release of circRNAs in platelet-derived extracellular vesicles. J Extracell Vesicles. 2018, 7, 1424473. [Google Scholar] [CrossRef]
- Han, Q.F.; Li, W.J.; Hu, K.S.; Gao, J.; Zhai, W.L.; Yang, J.H.; et al. Exosome biogenesis: machinery, regulation, and therapeutic implications in cancer. Mol Cancer. 2022, 21, 207. [Google Scholar]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013, 495, 384–388. [Google Scholar]
- Shi, L.; Yan, P.; Liang, Y.; Sun, Y.; Shen, J.; Zhou, S.; et al. Circular RNA expression is suppressed by androgen receptor (AR)-regulated adenosine deaminase that acts on RNA (ADAR1) in human hepatocellular carcinoma. Cell Death Dis. 2017, 8, e3171–e3171. [Google Scholar]
- Zhao, J.; Deng, Y.; Jiang, Z.; Qing, H. G Protein-Coupled Receptors (GPCRs) in Alzheimer’s Disease: A Focus on BACE1 Related GPCRs. Front Aging Neurosci. 2016, 8, 58. [Google Scholar] [CrossRef]
- Duan, X.; Zheng, Q.; Liang, L.; Zhou, L. Serum Exosomal miRNA-125b and miRNA-451a are Potential Diagnostic Biomarker for Alzheimer’s Diseases. Degener Neurol Neuromuscul Dis. 2024, 14, 21–31. [Google Scholar]
- Gámez-Valero, A.; Campdelacreu, J.; Vilas, D.; Ispierto, L.; Reñé, R.; Álvarez, R.; et al. Exploratory study on microRNA profiles from plasma-derived extracellular vesicles in Alzheimer’s disease and dementia with Lewy bodies. Transl Neurodegener. 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Aharon, A.; Spector, P.; Ahmad, R.S.; Horrany, N.; Sabbach, A.; Brenner, B.; et al. Extracellular Vesicles of Alzheimer’s Disease Patients as a Biomarker for Disease Progression. Mol Neurobiol. 2020, 57, 4156–4169. [Google Scholar] [CrossRef]
- Han, P.F.; Wei, L.; Duan, Z.Q.; Zhang, Z.L.; Chen, T.Y.; Lu, J.G.; et al. Contribution of IL-1β, 6 and TNF-α to the form of post-traumatic osteoarthritis induced by “idealized” anterior cruciate ligament reconstruction in a porcine model. Int Immunopharmacol. 2018, 65, 212–220. [Google Scholar]
- Visconte, C.; Fenoglio, C.; Serpente, M.; Muti, P.; Sacconi, A.; Rigoni, M.; et al. Altered Extracellular Vesicle miRNA Profile in Prodromal Alzheimer’s Disease. Int J Mol Sci. 2023, 24, 14749. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wang, L. Inflamma-MicroRNAs in Alzheimer’s Disease: From Disease Pathogenesis to Therapeutic Potentials. Front Cell Neurosci. 2021, 15, 785433. [Google Scholar] [CrossRef] [PubMed]
- Xi, T.; Jin, F.; Zhu, Y.; Wang, J.; Tang, L.; Wang, Y.; et al. MicroRNA-126-3p attenuates blood-brain barrier disruption, cerebral edema and neuronal injury following intracerebral hemorrhage by regulating PIK3R2 and Akt. Biochem Biophys Res Commun. 2017, 494, 144–151. [Google Scholar] [CrossRef]
- Chum, P.P.; Bishara, M.A.; Solis, S.R.; Behringer, E.J. Cerebrovascular miRNAs Track Early Development of Alzheimer’s Disease and Target Molecular Markers of Angiogenesis and Blood Flow Regulation. Journal of Alzheimer’s Disease. 2024, 99, S187–234. [Google Scholar] [CrossRef]
- Cha, D.J.; Mengel, D.; Mustapic, M.; Liu, W.; Selkoe, D.J.; Kapogiannis, D.; et al. miR-212 and miR-132 Are Downregulated in Neurally Derived Plasma Exosomes of Alzheimer’s Patients. Front Neurosci. 2019, 13, 1208. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; et al. Plasma Exosomal miRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PLoS One. 2015, 10, e0139233. [Google Scholar] [CrossRef]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum Mol Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; et al. Identification of miRNA Changes in Alzheimer’s Disease Brain and CSF Yields Putative Biomarkers and Insights into Disease Pathways. Journal of Alzheimer’s Disease. 2008, 14, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhen, H.; Sun, Y.; Rong, S.; Li, B.; Song, Z.; et al. Plasma Exo-miRNAs Correlated with AD-Related Factors of Chinese Individuals Involved in Aβ Accumulation and Cognition Decline. Mol Neurobiol. 2022, 59, 6790–6804. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Hu, Y.; Lan, T.; Guan, K.L.; Luo, T.; Luo, M. The Hippo signalling pathway and its implications in human health and diseases. Signal Transduct. Target. Ther. 2022, 7, 1–20. [Google Scholar] [CrossRef]
- Aharon, A.; Spector, P.; Ahmad, R.S.; Horrany, N.; Sabbach, A.; Brenner, B.; et al. Extracellular Vesicles of Alzheimer’s Disease Patients as a Biomarker for Disease Progression. Mol Neurobiol. 2020, 57, 4156–4169. [Google Scholar]
- LIUCG; SONGJ; ZHANGYQ; WANGPC MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer’s disease. Mol Med Rep. 2014, 10, 2395–2400. [CrossRef] [PubMed]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; et al. Plasma Exosomal miRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PLoS One. 2015, 10, e0139233. [Google Scholar] [CrossRef]
- Wang, L.; Zhen, H.; Sun, Y.; Rong, S.; Li, B.; Song, Z.; et al. Plasma Exo-miRNAs Correlated with AD-Related Factors of Chinese Individuals Involved in Aβ Accumulation and Cognition Decline. Mol Neurobiol. 2022, 59, 6790–6804. [Google Scholar]
- Wei, H.; Xu, Y.; Xu, W.; Zhou, Q.; Chen, Q.; Yang, M.; et al. Serum Exosomal miR-223 Serves as a Potential Diagnostic and Prognostic Biomarker for Dementia. Neuroscience. 2018, 379, 167–176. [Google Scholar] [CrossRef]
- Visconte, C.; Fenoglio, C.; Serpente, M.; Muti, P.; Sacconi, A.; Rigoni, M.; et al. Altered Extracellular Vesicle miRNA Profile in Prodromal Alzheimer’s Disease. Int J Mol Sci. 2023, 24, 14749. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Campdelacreu, J.; Vilas, D.; Ispierto, L.; Reñé, R.; Álvarez, R.; et al. Exploratory study on microRNA profiles from plasma-derived extracellular vesicles in Alzheimer’s disease and dementia with Lewy bodies. Transl Neurodegener. 2019, 8, 31. [Google Scholar] [CrossRef]
- Liu, T.; Li, N.; Pu, J.; Zhang, C.; Xu, K.; Wang, W.; et al. The plasma derived exosomal miRNA-483-5p/502-5p serve as potential MCI biomarkers in aging. Exp Gerontol. 2024, 186, 112355. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Zheng, Q.; Liang, L.; Zhou, L. Serum Exosomal miRNA-125b and miRNA-451a are Potential Diagnostic Biomarker for Alzheimer’s Diseases. Degener Neurol Neuromuscul Dis. 2024, 14, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Doecke, J.D.; Sharples, R.A.; Villemagne, V.L.; Fowler, C.J.; Rembach, A.; et al. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol Psychiatry. 2015, 20, 1188–1196. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, P.H. Elevated levels of MicroRNA-455-3p in the cerebrospinal fluid of Alzheimer’s patients: A potential biomarker for Alzheimer’s disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2021, 1867, 166052. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meng, S.; Di, W.; Xia, M.; Dong, L.; Zhao, Y.; et al. Amyloid-β protein and MicroRNA-384 in NCAM-Labeled exosomes from peripheral blood are potential diagnostic markers for Alzheimer’s disease. CNS Neurosci Ther. 2022, 28, 1093–1107. [Google Scholar] [CrossRef]
- Kumar, A.; Su, Y.; Sharma, M.; Singh, S.; Kim, S.; Peavey, J.J.; et al. MicroRNA expression in extracellular vesicles as a novel blood-based biomarker for Alzheimer’s disease. Alzheimer’s & Dementia. 2023, 19, 4952–4966. [Google Scholar]
Figure 1.
Schematic representation of the origin and function of exosomes in AD. Exosomes (30–150 nm) originate from multivesicular bodies, whereas microvesicles (50–1000 nm) bud directly from the plasma membrane. Once released into circulation, exosomes can cross the blood-brain barrier, as shown. Two main roles of exosomes in AD are depicted: (1) Neuroprotection, contributing to processes such as myelination, neurogenesis, immune activation, and energy metabolism; and (2) Progression of AD, mediating the spread of pathological proteins and apoptosis.
Figure 1.
Schematic representation of the origin and function of exosomes in AD. Exosomes (30–150 nm) originate from multivesicular bodies, whereas microvesicles (50–1000 nm) bud directly from the plasma membrane. Once released into circulation, exosomes can cross the blood-brain barrier, as shown. Two main roles of exosomes in AD are depicted: (1) Neuroprotection, contributing to processes such as myelination, neurogenesis, immune activation, and energy metabolism; and (2) Progression of AD, mediating the spread of pathological proteins and apoptosis.
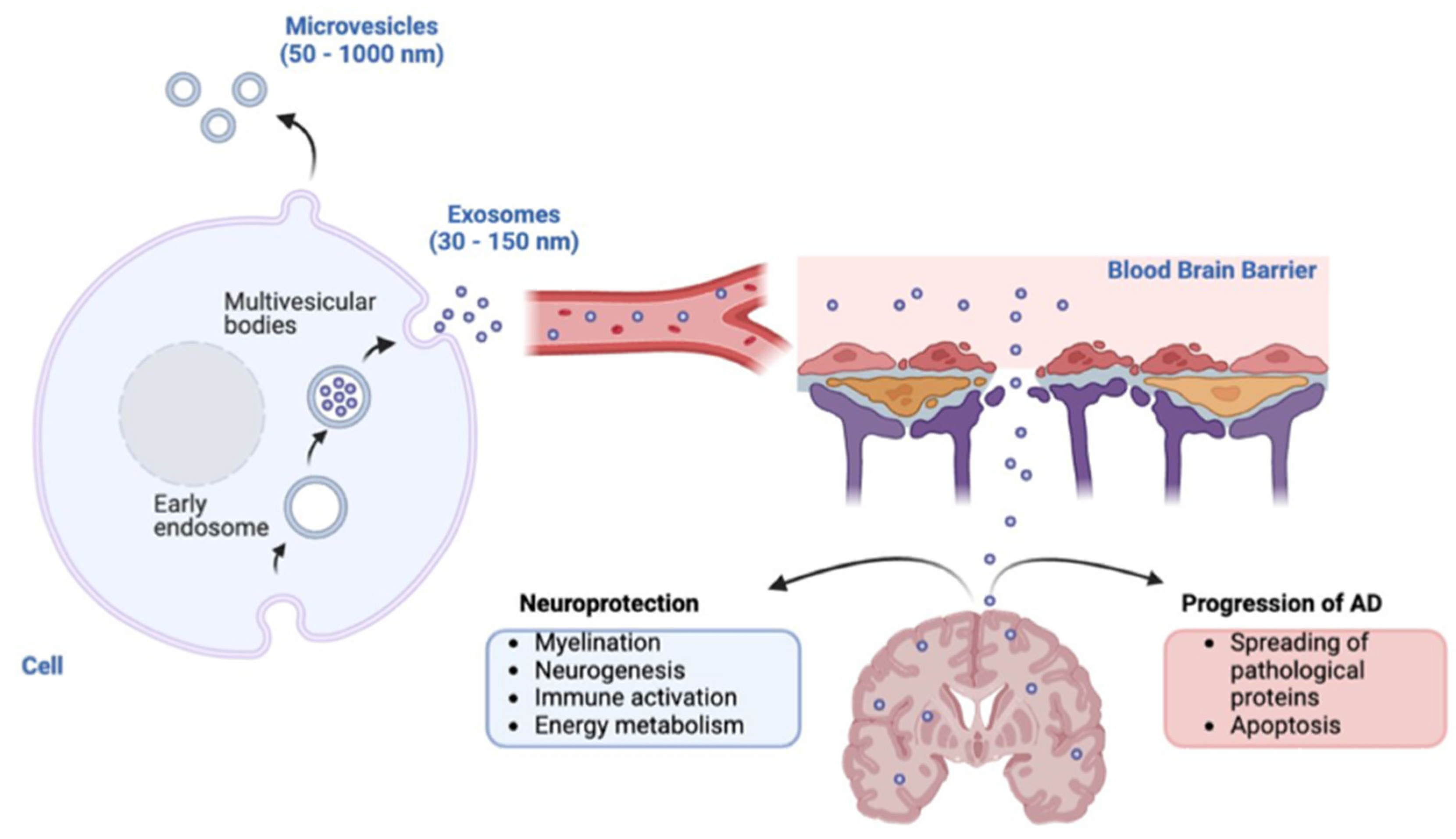
Figure 2.
Exosomes and AD neuropathology. The figure depicts the formation of exosomes from multivesicular endosomes (MVEs) in cells and their role in AD. Exosomes carry key molecules, including amyloid precursor protein (APP), Aβ peptides, full-length tau, α-synuclein, BIN1, miRNAs, and tetraspanins, which contribute to AD pathology. These vesicles may transport molecules that participate in the cleavage of APP into Aβ peptides and, also possibly, in the deposition of tau and Aβ plaques in the brain. The bottom section highlights the distribution of tau and Aβ aggregates, alongside exosomes, in the brain of AD patients.
Figure 2.
Exosomes and AD neuropathology. The figure depicts the formation of exosomes from multivesicular endosomes (MVEs) in cells and their role in AD. Exosomes carry key molecules, including amyloid precursor protein (APP), Aβ peptides, full-length tau, α-synuclein, BIN1, miRNAs, and tetraspanins, which contribute to AD pathology. These vesicles may transport molecules that participate in the cleavage of APP into Aβ peptides and, also possibly, in the deposition of tau and Aβ plaques in the brain. The bottom section highlights the distribution of tau and Aβ aggregates, alongside exosomes, in the brain of AD patients.
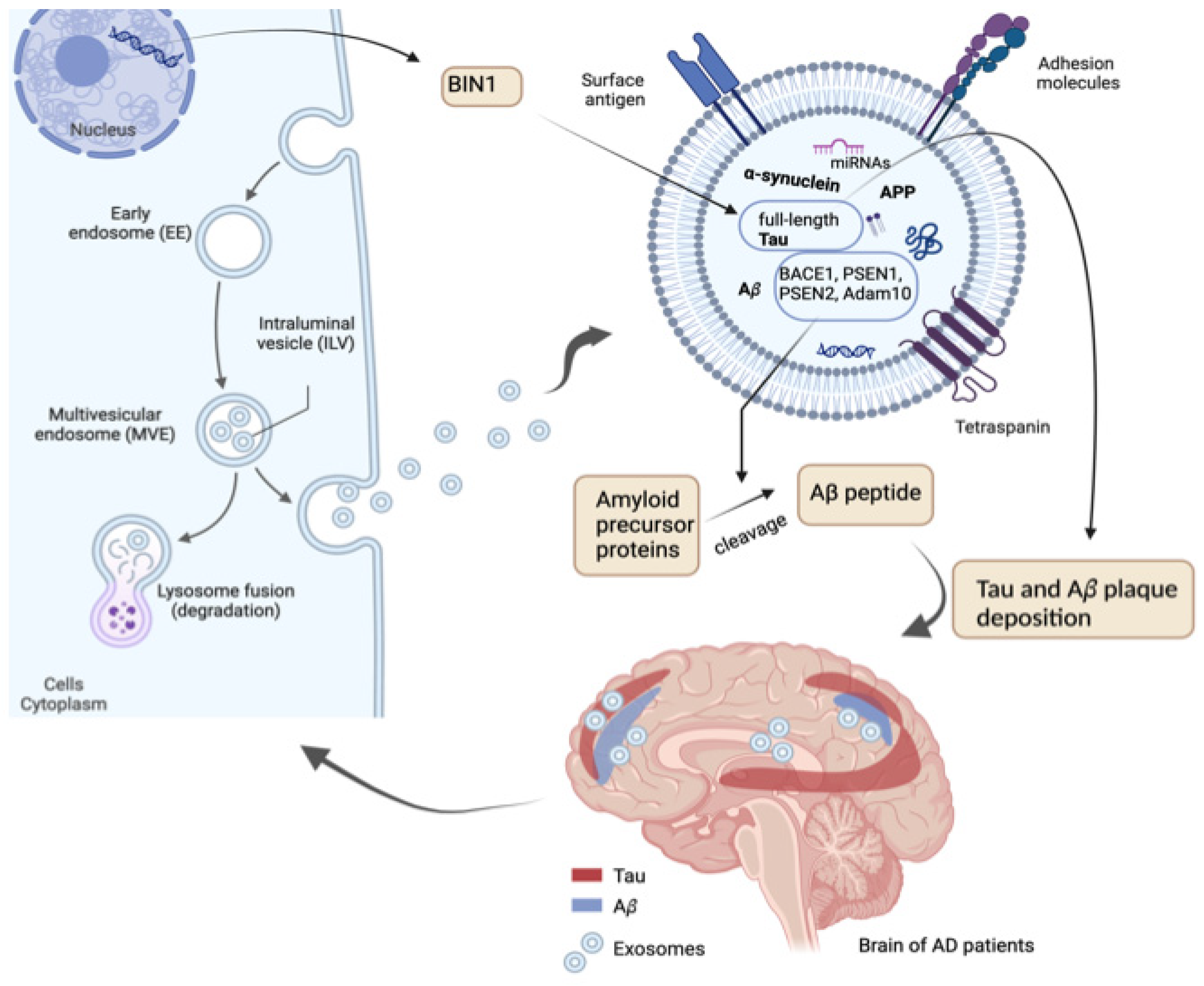
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



