Submitted:
11 April 2025
Posted:
14 April 2025
You are already at the latest version
Abstract
Tumour-associated angiogenesis plays a key role at all stages of cancer development and progression by providing nutrient supply, promoting the creation of protective niches for therapy-resistant cancer stem cells, and supporting the metastatic cascade. Therapeutic strategies aimed at vascular targeting, including vessel disruption and/or normalization, have yielded promising but inconsistent results, pointing to the need to set up reliable models dissecting the steps of the angiogenic process, as well as the ways to interfere with them, to improve patients’ outcomes while limiting side effects. Murine models have successfully contributed to both translational and pre-clinical cancer research, but they are time-consuming, expensive, and cannot recapitulate the genetic heterogeneity of cancer inside its native microenvironment. Non-animal technologies (NATs) are rapidly emerging as invaluable human-centric tools to reproduce the complex and dynamic tumour ecosystem, particularly tumour-associated vasculature. In the present Review, we will resume currently available NATs able to mimic vascular structure and functions with progressively increasing complexity, starting from two-dimensional static cultures to the more sophisticated tri- dimensional dynamic ones, patient-derived cultures, perfused engineered microvasculature and in silico models. We highlight the added value of a “one health” approach to cancer research including studies on spontaneously occurring tumours in companion animals devoid of the ethical concerns associated with traditional animal studies. Limitations of the present tools to a broader use in pre-clinical oncology, and their translational potential in terms of new target identification, drug development, and personalised therapy, are also discussed.
Keywords:
tumour microenvironment
; tumour associated microvasculature
; cell cultures
; organoids
; microphysiologic systems
; compuattaional pathology
; multiscale modelling
1. Introduction
Cancer emerges when mutated cells grow uncontrollably within a permissive tissue environment [1]. Initially, natural safeguards inhibit neoplastic formation, however, prolonged exposure to carcinogenic factors favours the accumulation of further mutations in cancer cells until they become able to evade these defences. Concurrently, growing tumours alter local and systemic environments, enabling cancer progression and spread [2].
Cancers are complex dynamic ecosystems composed of tumour cells and different non-cancerous cells all embedded in an extracellular matrix (ECM) exhibiting distinctive and unique physical, biochemical, and mechanical properties [3] (Figure 1).
The tumour microenvironment (TME) includes immune cells, cancer-associated fibroblasts, endothelial cells, pericytes, and tissue-resident cell types, all playing critical roles in each stage of cancer progression [3,4] (Figure 1). Tumour-TME crosstalk, mediated by cellular interactions, soluble factors and metabolites availability [5] affects cancer cell survival, proliferation, response to anti-cancer drugs and immune evasion, and is emerging as an attractive therapeutic target.
Typically, all cancers elicit a certain degree of local tissue inflammation. Inflammation initially combats cancer development, but, at the same time, it exercises a selective pressure on cancer cells [6,7,8] (Figure 1 B, C). Those cells that survive, become tolerant to inflammation, and ultimately shape the immune landscape both in the TME [2] and systemically. In solid tumours, these initial malignant lesions are called carcinoma in-situ (Figure 1 B), whose prognosis is favourable if they are promptly detected [9,10].
Tumour associated angiogenesis (TAA), and vascular alterations are hallmarks of the TME, and, following the intuition of J. Folkman [11,12], microvascular involvement is included among the criteria defining invasive versus non-invasive lesions (Figure 1 B vs C). A hypo vascular, hypoxic TME can limit the efficacy of chemo-/radiotherapy by hampering delivery of oxygen and drugs [13,14]. On the other hand, uneven intratumor angiogenesis and angiocrine signalling further promote cancer cells phenotypic heterogeneity [2] (Figure 1 D).
Overall, the TME behaves like a chronic, never-healing wound [15] because the sources of tissue damage and inflammation are not removed. The cellular and molecular dynamics in such systems is complex to predict as it depends on many contextual signals which vary widely over time, different genetic background, across different organs, and even within the same tissue. Moreover, by means of systemic signalling, mediated by soluble factors like cyto/chemokines and extracellular vesicles [16], cancer may induce changes in distant tissues including vascular leakiness, angiogenesis, and immunosuppression. All this forms the pre-metastatic niche [17]. Pre-metastatic niches promote the seeding of metastases and are tumour specific (both within the same cancer type and across different types) dictating preferences for specific target organs (seed and soil theory [18]).
Notably, the Tumour associated micro vasculature (TAMV, Figure 1 C, D) plays a key role at all stages of cancer development and progression, including the “angiogenic switch”, the creation of protective niches for therapy-resistant cancer stem cells (CSC), the creation of pre-metastatic niches, and metastasis.
1.1. Tumour Associated Micro Vasculature
Malignant cancer cells have a high metabolic demand to sustain their abnormal growth, and rapid cell growth promotes intra-tumoral hypoxia and chronic inflammation [19]. All these features contribute to the “angiogenic switch”, i.e., the sustained and uncontrolled induction of pathological angiogenesis leading to the formation of aberrant TAMV (Figure 1 D). Hypoxia activates the cellular oxygen-sensing machinery (Hypoxia-Inducible Factor, HIF, pathway) [20] in endothelial, immune, stromal and cancer cells directly regulating the transcription of pro-angiogenic genes like Vascular Endothelial Growth Factor (VEGF-A), Stromal Derived Factor (SDF-1), and Angiopoietin-2 [19]. However, due to the unbalanced signalling in the TME, the TAMV does not fully mature, is morphologically and functionally abnormal, and provides suboptimal diffusion of oxygen and nutrients, contributing to a self-sustaining hypoxia-inflammatory loop. In such environments, cancer cells can acquire distinct phenotypes, including fast-growing clones, slow growing CSC with self-renewal potential, or migratory clones with high metastatic potential (Figure 1 D).
Endothelial Cells (EC) are the main cellular components of the microvasculature. EC are physiologically heterogeneous across different organs, and within the same organ reflecting a variety of different functions [21]. EC composing the TAMV are phenotypically distinct from their healthy counterparts [22], have distinct metabolism [23], and angiocrine signalling [24].
Dysregulated angiocrine signalling to immune cells contributes to an immunosuppressive TME which hampers natural antitumour responses [25,26,27]. Concurrently the leaky TAMV favour intravasation and dissemination of metastases initiating cells which travel as circulating tumour cells to distant organs along blood or lymphatic streams [28].
In breast cancer, EC, tumour associated perivascular macrophages (TEMs), and specific cancer cells form a functional triad, the tumour microenvironment of metastasis (TMEM) doorways. The number of TMEM doorways in the primary tumours has been found to correlate with the propensity to metastasis [29] (Figure 1 D). It is tempting to assume that similar doorways might exist within other cancer types, possibly with distinct molecular signatures, however, this has not been demonstrated so far.
1.2. Therapeutic Implications
Given the central role of the TAMV in tumour development, vascular targeting has been explored as a therapeutic option in the past 20 years.
Anti-angiogenic therapy (AAT), i.e., attempting to prune the abnormal TAMV via interference with pro-angiogenic signalling, especially the VEGF pathway, has been widely studied and tested experimentally and clinically [32]. Despite the initial promise and some clinical success, AAT has yielded inconsistent clinical results. Not all cancer types are sensitive to AAT, and in those sensitive types, AATs like the monoclonal antibody against VEGF-A, Bevacizumab, causes a host of escape mechanisms like the “angiogenic rebound”, i.e., compensatory up-regulation of VEGF or other angiogenic molecules. Additionally, interference with the VEGF axis causes vessel disruption locally and systemically, producing severe side effects and increasing the propensity to metastasis [22,32].
More recently, vascular normalisation therapy (VNT), i.e., attempting to modulate rather than prune the TMV via fine-tuning of AATs, has been proposed as a promising new route. Animal studies have shown that VNT could alleviate intratumor hypoxia, facilitate delivery of drugs, increase the oxygen-dependent effects of radiotherapy, and favourably modulate the immune TME [13,34,35,36].
Clinical testing of VNT is currently ongoing [36], however previous studies have already highlighted that clinical success of VNT will depend on precisely identifying and measuring the time frame of their therapeutic activity (TAMV normalisation window).
In summary, it is now clear that VNTs are potent tools to modulate the TAMV with huge therapeutic potential. However, their clinical efficacy is currently limited by our capacity to measure and understand the dynamics crosstalk between cancer, the TME, and therapeutic agents.
For example, we do not know how to timely and precisely deliver therapeutic agents to maximise their beneficial effects while minimising their side-effects.
Thanks to intensive research and powerful technologies like OMICS, we are rapidly learning which cell types and molecules are involved in these processes. However, we do not fully understand how these elements crosstalk before, during, and after therapy in different tissues and organs.
Characterising the cellular and molecular dynamics driving TAMV functions is thus a central and unmet clinical need as TAMV associated features, like metastases, still represent the leading cause of cancer-associated death [30,33].
Deciphering the dynamic interplay between cancer cells and the TAMV may offer the opportunity to identify and target pathways common to most cancers. To this purpose, reliable models of TAMV are urgently needed.
Murine models are a cornerstone of biological research and have been successfully used in both translational and pre-clinical research [37]. However, preclinical animal models are typically complex, time-consuming, expensive, and difficult to standardise [38]. More importantly, they do not fully capture the complexity and genetic heterogeneity of human cancers, and they cannot recapitulate the molecular signalling and cancer-TME cellular crosstalk, limiting their translation potential. Indeed, US Food and Drugs Administration agency (FDA) data show that over 90% of drugs successful in pre-clinical animal models, fails to demonstrate safety or effectiveness in humans [39].
We need to develop affordable research tools able to increase our mechanistic understanding of the TME, to facilitate the development of new therapeutic strategies, and to customise treatment to individual patients. Non-Animal Technologies (NATs) able to reproduce selected aspects of human biology are rapidly emerging to fill these gaps.
In the following sections of this review, will discuss advances in modelling the TME, the TAMV, and their dynamic crosstalk, in vitro, ex-vivo, and in silico, focusing on those promising to advance cancer research, therapy, and care.
2. Non-Animal Technologies
2.1. Static Cultures
Cancer research and drug development have long relied upon experiments performed using in vitro culture of cell lines and primary tumour cells grown in two-dimensional (2D) static cultures [40,41,42]. While these culture systems allowed to elucidate basic molecular signatures of cancer, and still are a gold standard in primary drug screenings [43], they fail to properly reproduce spatial and functional complexity of TME, and hence to predict the impact of drugs in individual patients. The pioneering work of Bissell and colleagues [44,45], has clearly demonstrated that normal and tumour cells grown in 2D culture significantly differ from those kept in 3D in terms of morphology, biological behaviour, gene expression profile and response to drugs [46,47].
Experimental cancer models should incorporate elements of the surrounding milieu, to recapitulate tissue specific multi-cellularity and architecture, biochemical and mechanical signals, cell–cell and cell-ECM interactions [40,41,42]. Moreover, in the perspective of patient-specific cancer therapy, the need for personalized tumour models is rapidly emerging [48].
Personalized 3D cell cultures include tumour spheroids, organoids, and explants. Spheroids are clusters of cells, aggregated under static conditions via hanging drop techniques or culture on non-adhesive substrates [49,50,51]. Spheroids derived from tumour cell lines or primary tumour cells recapitulate main features of human solid tumours like their multi-layered structure where peripheral proliferative cells surround a necrotic core, and hypoxia and nutrient gradients, making them suitable for drug screening [52,53]. More complex approaches that incorporate the ECM take advantage of natural or synthetic polymeric substrates with tuneable composition and stiffness. Such substrates can be formed as hydrogels or solid scaffolds, which provide mechanical support and biomimetic physical and chemical environment [54]. Scaffolds allow tumour and stromal cells to attach, and proliferate, mimicking cell-to-cell and cell-to-ECM signals [54].
A better approximation of tumour multi-cellular complexity is represented by patients-derived organoids, which are 3D cell aggregates generated by culturing pluripotent stem cells (PSC) or cells derived from patients’ tumour tissues (tumoroids) under appropriate conditions [48,55]. Compared to tumour spheroids, which are typically monocultures [50,51], organoids more closely mimic TME heterogeneity and composition, and the structure of the tumour they originate from. However, they still don’t incorporate key elements of the TME like the microvasculature. Moreover, current protocols to generate patient-derived tumour organoids have inconsistent yield, are complex and lengthy to implement, and are scarcely standardised across laboratories [52,55].
Finally, 3D bioprinting technology is emerging as a promising strategy to manufacture tissue-engineered constructs with well-defined 3D geometry [56]. 3D bioprinting can be used to build tumour constructs via sequential layering of living cells (both tumour and stroma) within functionalized biomaterials (bioinks) [56,57].
2.2. Ex Vivo Culture of Tumour Explants
Patient-derived preclinical models that can preserve tumour features and predict outcomes in a personalized manner are greatly solicited in the field of cancer drug discovery and development. Compared to tumour-derived organoids, tumour explants obtained from resected tumour/metastasis or biopsy specimens fully incorporate the native genomic, histological and functional profile of cancer cells within their TME [58,59]. For example, tissue slices of human gliomas have been successfully cultured and used to detect metabolically active EC-associated “hot-spots” in otherwise low-grade lesions [59].
Tumour explants cultured under static conditions have a limited lifespan in culture (less than 3 days) limiting their applicability. This limitation has been addressed through the application of dynamic culture technologies, including the Rotary Cell Culture System (RCCS) bioreactor.
2.3. Dynamic Cultures
The 3D models described above have a common limitation in that they do not allow appropriate transport of nutrients, solutes, and metabolic waste in and out of the cultured tissue, all pre-requisites to sustain long-term cell viability and prevent cell necrosis. Thus, complex cancer cultures typically have short life span in culture.
Seminal discoveries and inventions in recent years have fostered the transition from static to dynamic cultures. Dynamic cultures can better reproduce the changes occurring in a growing tumour by providing efficient mass transfer, i.e., nutrients’ delivery and metabolic waste removal, and thus preserving cell viability within 3D cell/tissue masses [60]. These culture conditions can be obtained using dynamic bioreactors [50,61], including roller bottles, spinner flasks, gyratory shakes, perfusion [62,63], and microgravity bioreactors [48,59,60,61].
Among these systems, the microgravity-based RCCS bioreactor stands out as suitable for culturing functional 3D tissue-like bio-constructs and explants of various origins. RCCS bioreactors were primarily used to facilitate the self-assembly and culture of scaffold-free spheroids [64], and then further tuned to tumour-TME co-cultures on scaffolds [65], and tissue explants to study molecular mechanisms involved in cancer development and biology, and for drug testing in a patient-specific context [66,67,68]. Culture in RCCS bioreactor allows to perform downstream analyses, including “Omics” approaches, to dissect metabolic and functional pathways operating within TMEs [69].
In recent years advances in microfluidics technology, i.e., the study of fluid flow within small (<100 micrometres in diameter) conduits and development of affordable devices operating in such conditions, has paved the way towards developing more biomimetic systems.
3D cell culture based on microfluidic devices (often called lab-on-chip, or micro-physiologic-systems, MPS) allows studying key processes like cancer migration, invasion, and angiogenesis, and drug responses in a miniaturized, yet precisely defined culture environment [70].
3. Modelling Tumour Vasculature and Angiogenesis
Micro-vessels, i.e., blood vessels, or capillaries with a calibre smaller than 100 microns, sit at the interface between blood and all tissues in the body. They mediate oxygen and nutrients diffusion, they regulate tissue development and homeostasis via angiocrine signalling, and they are the gatekeepers of immunity and iflammation [22,32].
Comprehensively modelling such functions in vitro is challenging and many of the currently available models [71,72] typically focus on aspects of only one specific function.
3.1. EC Cultures and PCS
The first step towards modelling the microvasculature in vitro is the ability to culture EC and vascular cells like pericytes, under physiologic conditions to retain their original phenotype and functions. After over 20 years of research, it is now possible to robustly isolate and subculture vascular and microvascular EC and pericytes from human samples (primary cells). Primary EC 2D cultures have been extensively characterised, standardised, and validated to model several functions like the endothelial barrier and angiogenesis (sections below).
A new perspective in culturing human EC comes from pluripotent stem cells (PSC) technology, whereby PSC can be differentiated into EC (PSC-EC) via directed differentiation [73,74] or by inducing transcriptional regulators of the EC fate via transgenic strategies [75]. PSC-EC can be generated in large quantities, they can be genetically edited via CRISPR-Cas9 (impossible with primary cells), and thus they are amenable to genetic/pharmacologic screening for drug development.
PSC-EC are also ideal tools to study developmental vasculogenesis/angiogenesis in vitro, to study tissue-specific vascularisation, and to create vascularised organoids. However, the current protocols to generate PSC-EC are still imperfect and until yet, PSC-EC are more expensive, complicate to culture, and less robust in culture than primary EC.
3.2. In Vitro Tools to Study Vascular Permeability and Trans-Endothelial Cells Migration
As discussed, leaky TAMV mediates aberrant extravasation of solutes and immune cells in the TME while offering an entry/exit point for metastatic cancer cells (Figure 1 D).
Transit of solutes and trans-endothelial migration of cells have been studied in vitro leveraging EC monolayer cultures (Figure 2 A, upper panels) upon semi-permeable membranes [76]. Using such assays, it is possible to observe and measure the effects of experimental perturbations (including patients derived samples [77]) on endothelial integrity, its permeability to macromolecules, and its propensity to allow cells’ transmigration. Trans-endothelial migration assays are amenable to high throughput screenings which, in turn, can be efficiently analysed with dedicated image analysis tools [78,79] to achieve detailed molecular insights at cellular and sub-cellular levels.
Despite their utility, initial trans-endothelial migration assays cannot fully reproduce haemodynamic forces, which are key determinants of the above functions. To overcome this challenge more advanced experimental platforms like MPS are required. In recent years several seminal works have established the utility of MPS to model endothelial barriers in different organs like the lung [80], liver [81] and brain [82], under physiologic perfusion. Recent work has also demonstrated how multiple MPS could be joined together to study systemic crosstalk between different tissue via vascular transport [83,84].
3.3. In Vitro Tools to Study Angiogenesis
The microvasculature, especially the TAMV, constantly remodels in response to homeostatic or pathologic stimuli. As discussed above, the angiogenic switch is a hallmark of cancer with therapeutic potential, and assays to study angiogenesis in vitro have been developed since the early 2000s.
Historically, beyond 2D EC monolayers, EC self-organization in tubule-like structures has been appraised by culturing on extracellular matrices containing appropriate growth factors (Matrigel, Figure 2 A middle panel). However, the Matrigel assay can only recapitulate early phases of vascular assembly, and it is inadequate to study sprouting angiogenesis.
Hydrogel-based angiogenesis models like the fibrin gel assay [85] (Figure 2 A middle panel) offer a more developed alternative to study the initial phases of sprouting angiogenesis. The fibrin gel assay leverages the robust self-assembly of EC into patent microvascular networks within fibrin hydrogels and it has been used to study endothelial signalling during sprouting angiogenesis in different contexts [86,87]. However, microvascular networks grown in fibrin gels cannot mature into well-formed microvasculature mainly because such hydrogels do not possess appropriate mechanical properties.
Developmental and reparative angiogenesis mostly occurs within cell assemblies including matrix-producing stromal cells which offer appropriate biomechanical support and signalling to nascent micro vessels. This fact has inspired the development of a more biomimetic assay, the organotypic vasculo-/angio-genesis assays (OVAA) able to create patent capillaries within layers of matrix-producing fibroblasts or stromal cells [88]. The OVAA generates an interconnected network of patent and functional micro vessels resembling in vivo microvasculature. The OVAA is amenable to high throughput and timelapse imaging and can be used to observe and measure fine cellular and sub-cellular details during sprouting angiogenesis like tip cells, endothelial filopodia, and associated signalling (Figure 2 A bottom panels).
3.4. Vascularised Organoids and 3D Dynamic Cultures
Organoids are multicellular assemblies created by culturing adult or pluripotent stem cells (ASC, PSC respectively) under appropriate in vitro conditions which promote their self-organisation into organ like microtissues able to recapitulate selected tissue functions. Organoids are promising tools for applications like disease modelling and drug screenings. Organoids lifespan and viability in culture are limited due to inappropriate vascular supply, and novel strategies are being developed to overcome this limitation [89,90].
Vascularised organoids can be created by forming organoids in presence of primary or PSC-derived EC and perivascular cells like pericytes or smooth muscle cells [91,92]. Using these strategies several authors have reported successful creation of organoids embedding patent capillary-like structures [89,90]. Such systems can be used to study intratumor angiogenesis and angiocrine signalling, however, they cannot reproduce perfusion-dependent functions like mass transfer unless bioreactors like the RCCS described above (Figure 2 B, C) are used to facilitate it. Notably, the RCCS technology also allows the long-term evaluation of native tumour-associated angiogenic vessels inside cultured tumour explants, including their histochemical identification, quantification and functions [93], (Figure 2 B), and may in perspective be exploited to unveil and validate new molecular targets, thus improving efficacy of anti-angiogenic therapies.
3.5. Microphysiologic Systems to Perfuse Engineered Microvasculature
The research in the past years has yielded robust and flexible assays to create biomimetic capillaries which are amenable to high-throughput screenings and thus can be leveraged in drug discovery. However, until recently, perfusing remodelling capillaries like that in fibrin gels or the OVAA, aiming to enable physiologic mass transfer, has been very challenging.
The work of R Kamm and other groups has pioneered the development of perfusable self-assembling microvasculature in vitro leveraging microfluidics chips and the fibrin gel system described in previous sections. These works have provided many proof-of-principle applications to demonstrate their potential and utility in microvascular and cancer research [94,95,96,97,98]. For example, vascularised MPS can be used to reproduce aspects of the TAMV like its development, and its responses to anticancer drugs [98,99].
As discussed above, fibrin gel systems cannot support microvascular remodelling and maturation, yielding EC-lined sinusoidal structures rather than well-formed capillaries (Figure 2 A middle panel). Large tubule-like structures have low hydraulic resistance, and they can be easily perfused using gravity driven flow without the need of creating secure microfluidics connections. All this greatly simplifies chip manufacture and creating perfusable vasculature, which allowed all the seminal advances outlined above.
Building upon this research, we addressed two critical components that remained unresolved until recently: the need for continuous perfusion and appropriate mechanical stimulation of engineered capillaries, both chief modulators of microvascular architecture and functions. Via the OVAA co-culture model (Figure 2 A bottom panels) it is possible to provide appropriate extravascular signalling and mechanical stimulation to nascent vessels. To perfuse OVAA capillaries and thus provide intravascular flow-dependent mechanical forces, we developed the Vasculature on Chip platform (VoC) [100]. The VoC is based on optimised cell culture substrates, a system to create secure microfluidic connections, a fluidic design allowing medium recirculation, and a compact flow driver fitting standard cell culture incubators (Figure 2 C right panel).
Through the VoC-OVAA, we successfully demonstrated the creation of perfusable microvascular networks reminiscent of those observed in vivo and capable of remodelling over weeks under continuous flow, and a balance of intravascular and extravascular biomechanical forces (Figure 2 D) [100]. The VoC-OVAA allows long-term culture of perfused micro vessels and can be combined with other tissue culture techniques including organoids representing a promising platform to study angiogenic/angiocrine signalling in tissue microenvironments under physiologic conditions including mass transfer.
These technical advances have been made possible by the recent development of affordable microfluidic devices which enable creating appropriate interfaces between microfluidic channels and biologic systems of interest. Despite these advances, MPS technology is still in its infancy, and the future research will need to address key issues like improving biocompatibility, robustness, standardisation, and parallelisation of MPS platforms.
3.6. Spontaneous Tumour Models in Companion Animals
Spontaneous companion animal cancers (SCAC) [101,102,103] are emerging as an alternative to traditional laboratory animal models (LAMs) for cancer research [104]. Although SCAC involve animals, they don’t pose ethical concerns (as LAMs do instead) because they are spontaneously occurring due to similar risk factors than humans, they are treated with curative intent, and they do not involve any additional procedure on the “patient”. Inspired by the concepts of “one health” and “one medicine”, SCAC can be included in “clinical trials” and help us to shed new light on the fundamental mechanisms of oncogenesis which are common to all higher animals, including companion animals and humans.
LAMs typically use an immunocompromised animal, altering the immune response to the tumour while chemically induced tumours do not fully recapitulate the complex multifactorial nature of carcinogenesis [105]. On the other hand, spontaneously occurring cancers in domestic animals, mimic carcinogenesis and the body’s immune response more naturally, and the close relationship between humans and companion animals, results in shared cancer risk factors. For example, second-hand tobacco exposure has been identified as a risk factor for both human and feline oral squamous cell carcinomas [106,107]. The parallel advancements in human and veterinary medical research have dramatically increased companion animal’s life expectancy, without the possibility to evolve tumour suppressive mechanisms, leading to similarities in carcinogenesis in humans and companion animals [108].
As human and veterinary oncology research continues to progress, we are uncovering several prognostic cancer biomarkers common to humans and animals. There is mutual benefit to a comparative oncological approach, as findings from human cancer research can guide investigations in veterinary research and vice versa.
Diagnostic biopsies and tumour resections of spontaneously occurring cancers as part of the surgical management of the cancer contain a wealth of information including information on the tumour vasculature. For example, micro vessel density (MVD) has been investigated in both human and canine breast cancers and carry a similar prognostic significance [109,110]. In the case of prostate cancer, studies have identified numerous cancer-related genes that are altered in both humans and dogs [104,111]. In addition, VEGFR-2 and VEGF-A are evolutionarily conserved, and both carry similar prognostic trends in the two species [104,112].
Furthermore, certain cancers are rare in humans but relatively common in companion animals. For example, oral melanomas are extremely rare in humans, (1-2% of all oral cancers [113]). However, in canines, oral melanomas are the most common oral malignancy [114]. Dogs have been identified as a promising model to study human oral melanomas, and their high incidence provides ample data to study an otherwise very rare cancer in humans.
Combined with advances in computational pathology highlighted in following sections, cancer tissues can be further interrogated to identify biologically relevant prognostic biomarkers from the tumour microenvironment and associated vasculature.
There is a potential utility in performing veterinary clinical trials in spontaneously occurring tumours in companion animal models to form the evidence-base for subsequent human clinical trials. This allows drugs to be tested with curative and therapeutic intent in a system that more closely mimics the disease in humans. Such clinical trials would also provide researchers with the opportunity to observe longer term follow-ups and quality of life metrics from owner-reported questionnaires.
3.7. Computational Pathology and Artificial Intelligence
Human cancers are highly heterogeneous, morphologically, genotypically, and phenotypically, and this represents a main obstacle to understand their biology and deliver appropriate treatment.
Bioptic and excisional samples are an invaluable resource to better understand tumour heterogeneity, because they encapsulate much of the key information regarding each patient’s specific cancer. In parallel with ex-vivo culture systems described above, histopathologic analysis of diagnostic or cultured samples allows detecting and measuring architectural and cytomorphological features of the TME at the single cell level. Analysis of resulting data via univariate or multivariate statistics offers valuable diagnostic/prognostic information [67,115,116].
Systematic quantification of features of interest in bioptic samples is a daunting task if performed manually weighting on dedicated professionals outside their diagnostic routine and strongly limited by human capabilities and biases. Recent computational pathology and image analysis techniques including AI are very promising to automate and standardise tissue histomorphometry. Automation can remove observer’s biases, thus helping compilation of high-quality, large, potentially multicentric datasets [117].
For example, we have previously used image analysis to automatically measure MVD in cancerous, healthy, or irradiated tissue [115,118]. Translationally, this analysis allowed us to correlate MVD and irradiation dose in jaw bones, with direct clinical implications for oral rehabilitation of cancer patients receiving radiation therapy in the head and neck region [115].
However, standard image analysis requires homogeneous samples in terms of tissue preparation, staining quality and intensity, which is a limitation to reliably compare different experiments or samples from different sources.
Previous work has reported a performance decrease of AI models in segmenting heterogeneous Hematoxylin and Eosin (H&E) images without stain normalisation prior to analyses [119]. We have encountered similar challenges in our own work [120] using archival H&E slides.
For example, the images shown in Figure 3 A have different staining intensities as evident from the corresponding intensity distribution R, G, B histograms. Standard segmentation techniques and even a Convolutional Neural Network (CNN) based segmentator (Stardist [121]) might fail to appropriately segment heterogeneous data (Figure 3 B). To overcome this issue a variety of image restoration techniques can be applied. Figure 3 C show an example workflow where the images are enhanced with a Random Forest based probability mapper prior to segmentation, and these predictions are further refined with Stardist to accurately identify all nuclei including correctly segmenting overlapping objects.
Using this strategy, we have demonstrated how single cells analysis in HPV positive oropharyngeal squamous cells carcinoma patients, can help predicting therapy outcome solely via analysis of diagnostic histological slides [120]. Thus, proper image pre-processing and accurate segmentation of biologically relevant structures in H&E images are the first step in extracting the rich data encoded within the tumour islands, the TME, and the TAMV.
AI, especially CNN [122], is rapidly reshaping cancer research and personalized clinical care through a wide range of applications, encompassing detection, staging and classification of cancer, and molecular characterization of tumours and their microenvironment [123]. Specifically, machine learning and AI have been successfully used to detect and measure micro vessels in cancer patients’ histopathological samples [124]. Advances in this field, together with the integration of additional data types, including genomics, epigenomics, and proteomics, are likely to yield even more accurate models to automatically detect and measure many features of interest to diagnose and prognosticate patient’s response to therapy or clinical outcomes.
The rapid growth of AI technology has also inspired criticisms and concerns. For example, most CNNs developed for image segmentation and classification do not allow experimenters to backtrack the features or weights that led to specific decisions.This poses ethical and legal concerns to the use of such models in the clinical practice where decisions must be always justified. Current research is concentrating on optimising AI algorithms and on developing robust validation strategies for current NNs and explainable AI technologies to overcome these limitations [125].
One method to introduce a degree of interpretability in “black box” deep learning models is to use class activation mapping which highlights areas of the images that contributed strongly to the model’s predictions. While this provides some insights regarding the most important image features, it does not provide clear explanations regarding how the overall decisions are taken [126]. Furthermore, in some instances, categories mapping areas do not correspond to biologically relevant features the pathologist may expect. Another method to improve the explainability of AI models is by utilising biologically significant features established in the literature as model inputs. Indeed, in our previous work [120] we quantified several pathologically known prognostic biomarkers such as the number of tumour-infiltrating lymphocytes, and tumour morphology as inputs to train an AI model to predict patient outcomes. This can help clinicians to build confidence in AI models trained on evidence-based and explainable features.
3.8. Mechanistic Modelling
As discussed above, we need to understand the dynamic evolution of cancer within its TME to treat it with maximal efficacy and minimal side effects. Biologic experimentation, including the in vitro systems discussed in previous paragraphs are often designed and interpreted via statistical methods providing a qualitative understanding of the underlying phenomena. Achieving comprehensive mechanistic understanding is more challenging.
Mathematical biology, i.e., the study of biological processes defined as mathematical rules, offers an exciting perspective to address this problem. By defining, calibrating, and using suitable set of rules (mathematic equations) we can model complex biologic systems and predict their evolution via computer simulations. For example, Physiologically Based Pharmacokinetics modelling, which allows predicting the adsorption, distribution, metabolism and excretion of drugs, are now widely used to investigate new cancer targeting drugs [127].
Physiologically Based Pharmacokinetics models are ideal to study molecules biodistribution and activity at the organs and systems scale, however they are not suitable to model cell-cell interactions at the tissue, cellular, and subcellular scales. Powerful tools developed in the past 20 years now allow modelling complex cell assemblies, and their signalling.
For example, biochemical network modelling (e.g., Boolean networks, ODE systems) which captures the dynamics of molecular reactions (e.g., enzyme kinetics) can help investigating intracellular signalling qualitatively and quantitatively [128,129]. On the other hand, spatial modelling (e.g., agent-based, cellular automata) can help studying cell-cell and cell-environment interactions dependent on relative cell adhesiveness or intrinsic motility [130]. Finally, multiscale simulations combine the advantages of spatial and non-spatial models to recreate virtual tissues composed of different cell types where the behaviour of each cell can be regulated by cell-specific signalling, juxtacrine, paracrine signalling among cells in the simulation, or interactions with the environment. Typically, multiscale tools offer facilities to simulate molecular transport within tissues including cell secretion and uptake allowing to model paracrine and long-range signalling or delivery of therapeutics.
Currently, there are several software to build and execute multiscale simulations with different focus. Notable examples like Morpheus [131] and Compucell 3D [132] offer a multiscale approach to virtual tissues development including the ability to create complex models of angiogenesis and cancer microenvironment [133,134,135].
For example, Figure 4 A and B shows results obtained by reimplementing the simulation developed by R Merks and colleagues [135] in Compucell 3D. The simulation allows exploring the role of contact inhibited EC chemotaxis and cell adhesion in self-organising networks like that observed in the in vitro Matrigel assay. The simulation reproduces experimental scenarios paralleling the results of perturbation experiments. However, due to its simplicity, and similarly to its in vitro counterpart, this simulation cannot reproduce more complex scenarios like sprouting angiogenesis.
More complex TME simulations can be created, for example, Figure 4 C shows qualitative results of a CC3D model reproducing cancer induced sprouting angiogenesis promoted by tumour-secreted VEGF which induces migratory tip cells and proliferating stalk cells selected by a lateral inhibition mechanism (adaptation after Shirinifard et al. [133]).
This kind of simulations can be invaluable to better interpret experimental results or clinical data, to optimally design new experiments, and, in the future, even to predict disease evolution, anticipate response to therapy, and design personalised therapeutic programs for each patient [136].
The promise is huge, however, selecting and calibrating appropriate rules is a formidable challenge as shown by substantial literature in the field. For example, it is established that simple models of cancer growth limited by immune response can generate complex and difficult to predict dynamic behaviours [137,138]. Much has been done to address these and other problems like appropriately leverage Omics “big data” [139]. However, there is still an urgent need to standardise in silico experiments across different platforms, and to establish suitable metrics to enable meaningful comparisons with experimental data, and cross-validation.
Purposedly designed dynamic cell culture systems described throughout the review can help experimentally measuring the dynamics of multicellular environments like the TME. Co-developing in vitro and in silico models, including suitable metrics common to both, will enable cross-validating simulations against experimental evidence increasing their predictive potential. Validated simulations will then allow inexpensively formulating and testing new hypotheses, and to plan optimally informative experiments.
4. Conclusions/Perspectives
The recent shift in the US FDA's stance towards non-animal pre-clinical testing highlights the growing need for new tools in biomedical research and drug development [39]. While achieving true biomimicry of human tissue in vitro is undoubtedly an ambitious goal, new NATs like patient-derived organoids, tumoroids, ex vivo cultures, and MPS are emerging as promising starting points. Specifically, human-based models of tumour vasculature and angiogenesis are actively pursued and expected to result in a better definition of the molecular mechanisms operating in the TME, and thus in the development of novel therapeutic approaches to manipulate and target cancer progression in individual patients.
Intra-tumour hypoxia and biochemical imbalance, along with resulting phenotypic heterogeneity and co-option of physiologic functions towards metastasis, are still key unresolved issue in cancer therapy. The TAMV is central to all these processes and despite inconsistent results of the first generation of VTTs observed in clinical trials, it is still one of the most promising targets for novel cancer therapies.
Despite significant recent progress, numerous challenges still need to be addressed to bring vascularized tumour models closer to clinical use, including the implementation and standardization of suitable in vitro techniques, as well as the development of widely used analysis tools and metrics for quantification [140].
We envisage that novel microfluidics technologies, bioreactor systems, and biomaterials will help the development of increasingly biomimetic MPS, where engineered or patients-derived tissue, like tissue slices [59], can be robustly cultured for extended time.
Mechanistically, new complex assays and MPS are very promising to replace animal experimentation in both basic and pre-clinical research. Such systems leverage the simplicity of in vitro experimental manipulation, including transgenic approaches, the possibility to appreciate variability in human cells phenotype, and the ability to model complex functions. Furthermore, like 2D culture systems are a gold standard in primary drug/toxicology screenings [43], we envisage that more complex but robust in vitro screening will take the lead in modelling functions like angiogenesis and microvascular homeostasis, reducing the need for animal testing in such contexts.
Tissue, systems, and organism level interactions have been very challenging to model in vitro until recently, leaving animal experimentation as the only viable option. New technologies like ex vivo cultures [68] offer an exciting translational perspective to model and study tissue-level functions, and new multiorgan MPS are being developed to achieve a system level perspective [83]. We envisage that by fostering tight collaboration between clinician, biologist and engineers, such technologies will mature until eventually surpassing in vivo experiments in delivering research or prognostic value.
Capturing, studying, and understanding the variability of cancer phenotypes and manifestations among different patients, with different genetic background, lifestyles, and exposure to risk factors, is a key unmet need that eludes the capabilities of typical animal experiments. Organoids technology can aid unravelling these aspects in vitro [141], and AI-powered digital pathology and OMICS are powerful tools to tackle this challenge, by providing tissue level insights. These technologies are revealing the complexity of hFuman cancer tissue composition [142], and via appropriate statistic modelling they are highlighting useful biomarkers for patient’s stratification and therapy optimisation [120,143]. In addition, taking inspiration form the “one health” concept (https://www.who.int/health-topics/one-health), there is a clear indication to investigate cancers and their development in domesticated animals [104]. This way we can bypass the ethical concerns weighting on traditional animal models, mine an existing and potentially huge source of new data, and promote an integrated health care for humans and companion animals.
The need to mine large data generated via high throughput screenings, OMICS and computational pathology has sparked intense research in bioinformatic yielding numerous tools to infer mechanistic knowledge from large data. Further than this, mechanistic mathematical modelling is offering exciting perspectives to model and simulate multiscale functions, from genes, through cells and tissue, to organs and systems in silico. Despite the promises, creating, calibrating, validating, and standardising these models, is still a huge and only partially addressed challenge.
As discussed above in vitro and in silico technologies can complement and empower each other to create NATs for research which can be further developed to pre-clinical or point-of-care precision medicine applications [144].We believe that all of this will ultimately yield a better understanding of cancer, the TME, and their dynamic responses to therapies to the benefit of patients.
In conclusion, NATs can help us tackle unmet research and clinical needs emphasizing sustainability and translation: To understand the dynamic evolution of the TAMV and TME, to devise new therapeutic strategies, to aid drug discovery, to expedite the diagnostic process, and to personalise therapy.
Acknowledgments
The authors wish to acknowledge the work Dr F Pedica, and Dr F Chesnais which have contributed with fruitful discussions and experimental work.
References
- Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000). [CrossRef]
- de Visser, K. E. & Joyce, J. A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 41, 374–403 (2023). [CrossRef]
- Mantovani, A., Allavena, P., Marchesi, F. & Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov 21, 799–820 (2022). [CrossRef]
- Chen, Y., McAndrews, K. M. & Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol 18, 792–804 (2021). [CrossRef]
- Arner, E. N. & Rathmell, J. C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 41, 421–433 (2023). [CrossRef]
- Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008). [CrossRef]
- Colotta, F., Allavena, P., Sica, A., Garlanda, C. & Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30, 1073–1081 (2009). [CrossRef]
- Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [CrossRef]
- van Seijen, M. et al. Ductal carcinoma in situ: to treat or not to treat, that is the question. Br J Cancer 121, 285–292 (2019). [CrossRef]
- Knoblauch, M., Kühn, F., von Ehrlich-Treuenstätt, V., Werner, J. & Renz, B. W. Diagnostic and Therapeutic Management of Early Colorectal Cancer. Visc Med 39, 10–16 (2023). [CrossRef]
- Folkman, J., Merler, E., Abernathy, C. & Williams, G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med 133, 275–288 (1971). [CrossRef]
- Folkman, J. Tumor angiogenesis: therapeutic implications. N Engl J Med 285, 1182–1186 (1971). [CrossRef]
- Leroi, N., Lallemand, F., Coucke, P., Noel, A. & Martinive, P. Impacts of Ionizing Radiation on the Different Compartments of the Tumor Microenvironment. Front Pharmacol 7, 78 (2016). [CrossRef]
- Minchinton, A. I. & Tannock, I. F. Drug penetration in solid tumours. Nat Rev Cancer 6, 583–592 (2006). [CrossRef]
- Deyell, M., Garris, C. S. & Laughney, A. M. Cancer metastasis as a non-healing wound. Br J Cancer 124, 1491–1502 (2021). [CrossRef]
- Whiteside, T. L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv Clin Chem 74, 103–141 (2016). [CrossRef]
- Patras, L., Shaashua, L., Matei, I. & Lyden, D. Immune determinants of the pre-metastatic niche. Cancer Cell 41, 546–572 (2023). [CrossRef]
- Fidler, I. J. & Nicolson, G. L. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J Natl Cancer Inst 57, 1199–1202 (1976). [CrossRef]
- Wicks, E. E. & Semenza, G. L. Hypoxia-inducible factors: cancer progression and clinical translation. J Clin Invest 132, e159839 (2022). [CrossRef]
- Mole, D. R. & Ratcliffe, P. J. Cellular oxygen sensing in health and disease. Pediatr Nephrol 23, 681–694 (2008). [CrossRef]
- Kalucka, J. et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 180, 764-779.e20 (2020). [CrossRef]
- Eelen, G., Treps, L., Li, X. & Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis Updated. Circ Res 127, 310–329 (2020). [CrossRef]
- Li, X., Sun, X. & Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 30, 414–433 (2019). [CrossRef]
- Butler, J. M., Kobayashi, H. & Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer 10, 138–146 (2010). [CrossRef]
- Gajewski, T. F., Schreiber, H. & Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14, 1014–1022 (2013). [CrossRef]
- Whiteside, T. L., Demaria, S., Rodriguez-Ruiz, M. E., Zarour, H. M. & Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin Cancer Res 22, 1845–1855 (2016). [CrossRef]
- Mantovani, A., Marchesi, F., Jaillon, S., Garlanda, C. & Allavena, P. Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell Mol Immunol 18, 566–578 (2021). [CrossRef]
- Kang, Y. & Pantel, K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 23, 573–581 (2013). [CrossRef]
- Coste, A. et al. Hematogenous Dissemination of Breast Cancer Cells From Lymph Nodes Is Mediated by Tumor MicroEnvironment of Metastasis Doorways. Front Oncol 10, 571100 (2020). [CrossRef]
- Lambert, A. W., Pattabiraman, D. R. & Weinberg, R. A. Emerging Biological Principles of Metastasis. Cell 168, 670–691 (2017). [CrossRef]
- Massagué, J. & Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov 11, 971–994 (2021). [CrossRef]
- Potente, M., Gerhardt, H. & Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887 (2011). [CrossRef]
- Ganesh, K. & Massagué, J. Targeting metastatic cancer. Nat Med 27, 34–44 (2021). [CrossRef]
- Fan, P. et al. Alleviating hypoxia to improve cancer immunotherapy. Oncogene (2023) doi:10.1038/s41388-023-02869-2. [CrossRef]
- Jain, R. K. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622 (2014). [CrossRef]
- Huang, Y. et al. Improving immune-vascular crosstalk for cancer immunotherapy. Nat Rev Immunol 18, 195–203 (2018). [CrossRef]
- Zitvogel, L., Pitt, J. M., Daillère, R., Smyth, M. J. & Kroemer, G. Mouse models in oncoimmunology. Nat Rev Cancer 16, 759–773 (2016). [CrossRef]
- Bédard, P. et al. Innovative Human Three-Dimensional Tissue-Engineered Models as an Alternative to Animal Testing. Bioengineering (Basel) 7, 115 (2020). [CrossRef]
- Moutinho, S. Researchers and regulators plan for a future without lab animals. Nat Med 29, 2151–2154 (2023). [CrossRef]
- Pampaloni, F., Reynaud, E. G. & Stelzer, E. H. K. The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol 8, 839–845 (2007). [CrossRef]
- Kapałczyńska, M. et al. 2D and 3D cell cultures – a comparison of different types of cancer cell cultures. Arch Med Sci 14, 910–919 (2018). [CrossRef]
- Zimmer, J., Castriconi, R. & Scaglione, S. Editorial: Recent 3D Tumor Models for Testing Immune-Mediated Therapies. Front Immunol 12, 798493 (2021). [CrossRef]
- Allemang, A. et al. Assessing the genotoxicity and carcinogenicity of 2-chloroethanol through structure activity relationships and in vitro testing approaches. Food Chem Toxicol 168, 113290 (2022). [CrossRef]
- Lee, G. Y., Kenny, P. A., Lee, E. H. & Bissell, M. J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods 4, 359–365 (2007). [CrossRef]
- Roskelley, C. D., Desprez, P. Y. & Bissell, M. J. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci U S A 91, 12378–12382 (1994). [CrossRef]
- Chang, T. T. & Hughes-Fulford, M. Monolayer and spheroid culture of human liver hepatocellular carcinoma cell line cells demonstrate distinct global gene expression patterns and functional phenotypes. Tissue Eng Part A 15, 559–567 (2009). [CrossRef]
- Riedl, A. et al. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J Cell Sci 130, 203–218 (2017). [CrossRef]
- van Renterghem, A. W. J., van de Haar, J. & Voest, E. E. Functional precision oncology using patient-derived assays: bridging genotype and phenotype. Nat Rev Clin Oncol 20, 305–317 (2023). [CrossRef]
- Weiswald, L.-B., Bellet, D. & Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 17, 1–15 (2015). [CrossRef]
- Bray, L. J., Hutmacher, D. W. & Bock, N. Addressing Patient Specificity in the Engineering of Tumor Models. Front Bioeng Biotechnol 7, 217 (2019). [CrossRef]
- Gunti, S., Hoke, A. T. K., Vu, K. P. & London, N. R. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers (Basel) 13, 874 (2021). [CrossRef]
- Lee, K.-H. & Kim, T.-H. Recent Advances in Multicellular Tumor Spheroid Generation for Drug Screening. Biosensors (Basel) 11, 445 (2021). [CrossRef]
- Nunes, A. S., Barros, A. S., Costa, E. C., Moreira, A. F. & Correia, I. J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol Bioeng 116, 206–226 (2019). [CrossRef]
- Carletti, E., Motta, A. & Migliaresi, C. Scaffolds for tissue engineering and 3D cell culture. Methods Mol Biol 695, 17–39 (2011). [CrossRef]
- Drost, J. & Clevers, H. Organoids in cancer research. Nat Rev Cancer 18, 407–418 (2018). [CrossRef]
- Datta, P., Dey, M., Ataie, Z., Unutmaz, D. & Ozbolat, I. T. 3D bioprinting for reconstituting the cancer microenvironment. NPJ Precis Oncol 4, 18 (2020). [CrossRef]
- Augustine, R. et al. 3D Bioprinted cancer models: Revolutionizing personalized cancer therapy. Transl Oncol 14, 101015 (2021). [CrossRef]
- Powley, I. R. et al. Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br J Cancer 122, 735–744 (2020). [CrossRef]
- Kirby, A. J. et al. Multicellular ‘hotspots’ harbor high-grade potential in lower-grade gliomas. Neurooncol Adv 3, vdab026 (2021). [CrossRef]
- Navran, S. The application of low shear modeled microgravity to 3-D cell biology and tissue engineering. Biotechnol Annu Rev 14, 275–296 (2008). [CrossRef]
- Selden, C. & Fuller, B. Role of Bioreactor Technology in Tissue Engineering for Clinical Use and Therapeutic Target Design. Bioengineering (Basel) 5, 32 (2018). [CrossRef]
- Muraro, M. G. et al. Ex-vivo assessment of drug response on breast cancer primary tissue with preserved microenvironments. Oncoimmunology 6, e1331798 (2017). [CrossRef]
- Manfredonia, C. et al. Maintenance of Primary Human Colorectal Cancer Microenvironment Using a Perfusion Bioreactor-Based 3D Culture System. Adv Biosyst 3, e1800300 (2019). [CrossRef]
- Ferreira, L. P., Gaspar, V. M. & Mano, J. F. Design of spherically structured 3D in vitro tumor models -Advances and prospects. Acta Biomater 75, 11–34 (2018). [CrossRef]
- Belloni, D. et al. Modeling multiple myeloma-bone marrow interactions and response to drugs in a 3D surrogate microenvironment. Haematologica 103, 707–716 (2018).
- Grimm, D. et al. Growing tissues in real and simulated microgravity: new methods for tissue engineering. Tissue Eng Part B Rev 20, 555–566 (2014). [CrossRef]
- Guzzeloni, V., Veschini, L., Pedica, F., Ferrero, E. & Ferrarini, M. 3D Models as a Tool to Assess the Anti-Tumor Efficacy of Therapeutic Antibodies: Advantages and Limitations. Antibodies (Basel) 11, 46 (2022). [CrossRef]
- Ferrarini, M. et al. 3D-Dynamic Culture Models of Multiple Myeloma. Methods Mol Biol 1612, 177–190 (2017). [CrossRef]
- Ferrero, E. et al. Immunometabolic activation of macrophages leads to cytokine production in the pathogenesis of KRAS-mutated histiocytosis. Rheumatology (Oxford) 61, e93–e96 (2022). [CrossRef]
- Holton, A. B. et al. Microfluidic Biopsy Trapping Device for the Real-Time Monitoring of Tumor Microenvironment. PLoS One 12, e0169797 (2017). [CrossRef]
- Marei, I. et al. 3D Tissue-Engineered Vascular Drug Screening Platforms: Promise and Considerations. Front Cardiovasc Med 9, 847554 (2022). [CrossRef]
- Simons, M. et al. State-of-the-Art Methods for Evaluation of Angiogenesis and Tissue Vascularization: A Scientific Statement From the American Heart Association. Circ. Res. 116, 99–132 (2015). [CrossRef]
- Oh, J. E., Jung, C. & Yoon, Y.-S. Human Induced Pluripotent Stem Cell-Derived Vascular Cells: Recent Progress and Future Directions. J Cardiovasc Dev Dis 8, 148 (2021). [CrossRef]
- Palpant, N. J. et al. Generating high-purity cardiac and endothelial derivatives from patterned mesoderm using human pluripotent stem cells. Nature Protocols 12, 15–31 (2016). [CrossRef]
- Kumar, M. et al. A fully defined matrix to support a pluripotent stem cell derived multi-cell-liver steatohepatitis and fibrosis model. Biomaterials 276, 121006 (2021). [CrossRef]
- Ferrero, E. et al. CD14+ CD34+ peripheral blood mononuclear cells migrate across endothelium and give rise to immunostimulatory dendritic cells. J Immunol 160, 2675–2683 (1998).
- Langheim, S. et al. Increased expression and secretion of resistin in epicardial adipose tissue of patients with acute coronary syndrome. Am. J. Physiol. Heart Circ. Physiol. 298, H746-753 (2010). [CrossRef]
- Chesnais, F. et al. High-content image analysis to study phenotypic heterogeneity in endothelial cell monolayers. J Cell Sci 135, jcs259104 (2022). [CrossRef]
- Veschini, L. et al. High-Content Imaging to Phenotype Human Primary and iPSC-Derived Cells. Methods Mol Biol 2185, 423–445 (2021). [CrossRef]
- Kerns, S. J. et al. Human immunocompetent Organ-on-Chip platforms allow safety profiling of tumor-targeted T-cell bispecific antibodies. eLife 10, e67106 (2021). [CrossRef]
- Ewart, L. et al. Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology. Commun Med 2, 1–16 (2022). [CrossRef]
- Pediaditakis, I. et al. A microengineered Brain-Chip to model neuroinflammation in humans. iScience 25, (2022). [CrossRef]
- Ronaldson-Bouchard, K. et al. Engineering complexity in human tissue models of cancer. Advanced Drug Delivery Reviews 184, 114181 (2022). [CrossRef]
- Ronaldson-Bouchard, K. et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat Biomed Eng 6, 351–371 (2022). [CrossRef]
- Nakatsu, M. N., Davis, J. & Hughes, C. C. W. Optimized fibrin gel bead assay for the study of angiogenesis. J Vis Exp 186 (2007). [CrossRef]
- Jauhiainen, S. et al. ErbB signaling is a potential therapeutic target for vascular lesions with fibrous component. Elife 12, e82543 (2023). [CrossRef]
- Francis, C. R., Kincross, H. & Kushner, E. J. Rab35 governs apicobasal polarity through regulation of actin dynamics during sprouting angiogenesis. Nat Commun 13, 5276 (2022). [CrossRef]
- Hetheridge, C., Mavria, G. & Mellor, H. Uses of the in vitro endothelial-fibroblast organotypic co-culture assay in angiogenesis research. Biochem. Soc. Trans. 39, 1597–1600 (2011). [CrossRef]
- Strobel, H. A., Moss, S. M. & Hoying, J. B. Vascularized Tissue Organoids. Bioengineering (Basel) 10, 124 (2023). [CrossRef]
- Yu, J. Vascularized Organoids: A More Complete Model. Int J Stem Cells 14, 127–137 (2021). [CrossRef]
- Orlova, V. V. et al. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat Protoc 9, 1514–1531 (2014). [CrossRef]
- Kumar, A. et al. Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Rep 19, 1902–1916 (2017). [CrossRef]
- Ferrarini, M. et al. Ex-vivo dynamic 3-D culture of human tissues in the RCCSTM bioreactor allows the study of Multiple Myeloma biology and response to therapy. PLoS One 8, e71613 (2013). [CrossRef]
- Osaki, T., Sivathanu, V. & Kamm, R. D. Vascularized microfluidic organ-chips for drug screening, disease models and tissue engineering. Curr. Opin. Biotechnol. 52, 116–123 (2018). [CrossRef]
- Chen, M. B. et al. On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat Protoc 12, 865–880 (2017).
- Yamamoto, K. et al. Construction of Continuous Capillary Networks Stabilized by Pericyte-like Perivascular Cells. Tissue Eng Part A 25, 499–510 (2019). [CrossRef]
- Boussommier-Calleja, A. et al. The effects of monocytes on tumor cell extravasation in a 3D vascularized microfluidic model. Biomaterials 198, 180–193 (2019). [CrossRef]
- Sobrino, A. et al. 3D microtumors in vitro supported by perfused vascular networks. Sci Rep 6, 31589 (2016). [CrossRef]
- Hachey, S. J. et al. A human vascularized microtumor model of patient-derived colorectal cancer recapitulates clinical disease. Transl Res 255, 97–108 (2023). [CrossRef]
- Chesnais, F. et al. Continuously perfusable, customisable, and matrix-free vasculature on a chip platform. Lab Chip 23, 761–772 (2023). [CrossRef]
- Razavirad, A., Rismanchi, S., Mortazavi, P. & Muhammadnejad, A. Canine Mammary Tumors as a Potential Model for Human Breast Cancer in Comparative Oncology. Veterinary Medicine International 2024, 9319651 (2024). [CrossRef]
- Palma, S. D., McConnell, A., Verganti, S. & Starkey, M. Review on Canine Oral Melanoma: An Undervalued Authentic Genetic Model of Human Oral Melanoma? Vet Pathol 58, 881–889 (2021). [CrossRef]
- Wong, K. et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat Commun 10, 353 (2019). [CrossRef]
- Oh, J. H. & Cho, J.-Y. Comparative oncology: overcoming human cancer through companion animal studies. Exp Mol Med 55, 725–734 (2023). [CrossRef]
- Onaciu, A. et al. Spontaneous and Induced Animal Models for Cancer Research. Diagnostics (Basel) 10, 660 (2020). [CrossRef]
- Mariano, L. C., Warnakulasuriya, S., Straif, K. & Monteiro, L. Secondhand smoke exposure and oral cancer risk: a systematic review and meta-analysis. Tob Control 31, 597–607 (2022). [CrossRef]
- Zaccone, R. et al. Environmental risk factors for the development of oral squamous cell carcinoma in cats. J Vet Intern Med 36, 1398–1408 (2022). [CrossRef]
- Sarver, A. L., Makielski, K. M., DePauw, T. A., Schulte, A. J. & Modiano, J. F. Increased risk of cancer in dogs and humans: a consequence of recent extension of lifespan beyond evolutionarily-determined limitations? Aging Cancer 3, 3–19 (2022). [CrossRef]
- Carvalho, M. I. et al. EGFR and microvessel density in canine malignant mammary tumours. Res Vet Sci 95, 1094–1099 (2013). [CrossRef]
- Queiroga, F. L., Pires, I., Parente, M., Gregório, H. & Lopes, C. S. COX-2 over-expression correlates with VEGF and tumour angiogenesis in canine mammary cancer. Vet J 189, 77–82 (2011). [CrossRef]
- Laufer-Amorim, R. et al. Comprehensive Genomic Profiling of Androgen-Receptor-Negative Canine Prostate Cancer. Int J Mol Sci 20, 1555 (2019). [CrossRef]
- Nordby, Y. et al. Stromal expression of VEGF-A and VEGFR-2 in prostate tissue is associated with biochemical and clinical recurrence after radical prostatectomy. Prostate 75, 1682–1693 (2015). [CrossRef]
- Warszawik-Hendzel, O., Słowińska, M., Olszewska, M. & Rudnicka, L. Melanoma of the oral cavity: pathogenesis, dermoscopy, clinical features, staging and management. J Dermatol Case Rep 8, 60–66 (2014). [CrossRef]
- Bergman, P. J. Canine oral melanoma. Clin Tech Small Anim Pract 22, 55–60 (2007). [CrossRef]
- Patel, V. et al. The impact of intensity-modulated radiation treatment on dento-alveolar microvasculature in pharyngeal cancer implant patients. J Oral Rehabil 47, 1411–1421 (2020). [CrossRef]
- Nafe, R. & Schlote, W. Histomorphometry of brain tumours. Neuropathol Appl Neurobiol 30, 315–328 (2004). [CrossRef]
- Mahmood, H. et al. Use of artificial intelligence in diagnosis of head and neck precancerous and cancerous lesions: A systematic review. Oral Oncol 110, 104885 (2020). [CrossRef]
- Veschini, L. et al. The vasostatin-1 fragment of chromogranin A preserves a quiescent phenotype in hypoxia-driven endothelial cells and regulates tumor neovascularization. FASEB J. 25, 3906–3914 (2011). [CrossRef]
- Madusanka, N., Jayalath, P., Fernando, D., Yasakethu, L. & Lee, B.-I. Impact of H&E Stain Normalization on Deep Learning Models in Cancer Image Classification: Performance, Complexity, and Trade-Offs. Cancers (Basel) 15, 4144 (2023). [CrossRef]
- Hue, J., Valinciute, Z., Thavaraj, S. & Veschini, L. Multifactorial estimation of clinical outcome in HPV-associated oropharyngeal squamous cell carcinoma via automated image analysis of routine diagnostic H&E slides and neural network modelling. Oral Oncol 141, 106399 (2023). [CrossRef]
- Graham, S. et al. CoNIC Challenge: Pushing the frontiers of nuclear detection, segmentation, classification and counting. Med Image Anal 92, 103047 (2024). [CrossRef]
- Graham, S. et al. Hover-Net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images. Medical Image Analysis 58, 101563 (2019). [CrossRef]
- Cabral, B. P., Braga, L. A. M., Syed-Abdul, S. & Mota, F. B. Future of Artificial Intelligence Applications in Cancer Care: A Global Cross-Sectional Survey of Researchers. Curr Oncol 30, 3432–3446 (2023). [CrossRef]
- Timakova, A. et al. Artificial Intelligence Assists in the Detection of Blood Vessels in Whole Slide Images: Practical Benefits for Oncological Pathology. Biomolecules 13, 1327 (2023). [CrossRef]
- Gniadek, T., Kang, J., Theparee, T. & Krive, J. Framework for Classifying Explainable Artificial Intelligence (XAI) Algorithms in Clinical Medicine. Online J Public Health Inform 15, e50934 (2023). [CrossRef]
- Ali, S. et al. The enlightening role of explainable artificial intelligence in medical & healthcare domains: A systematic literature review. Comput Biol Med 166, 107555 (2023). [CrossRef]
- Wang, X. et al. Application of physiologically based pharmacokinetics modeling in the research of small-molecule targeted anti-cancer drugs. Cancer Chemother Pharmacol 92, 253–270 (2023). [CrossRef]
- Calzone, L., Noël, V., Barillot, E., Kroemer, G. & Stoll, G. Modeling signaling pathways in biology with MaBoSS: From one single cell to a dynamic population of heterogeneous interacting cells. Comput Struct Biotechnol J 20, 5661–5671 (2022). [CrossRef]
- Choi, K. et al. Tellurium: An extensible python-based modeling environment for systems and synthetic biology. Biosystems 171, 74–79 (2018). [CrossRef]
- Graner, F. & Glazier, J. A. Simulation of biological cell sorting using a two-dimensional extended Potts model. Phys Rev Lett 69, 2013–2016 (1992). [CrossRef]
- Starruß, J., de Back, W., Brusch, L. & Deutsch, A. Morpheus: a user-friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 30, 1331–1332 (2014). [CrossRef]
- Swat, M. H. et al. Multi-scale modeling of tissues using CompuCell3D. Methods Cell Biol 110, 325–366 (2012). [CrossRef]
- Shirinifard, A. et al. 3D multi-cell simulation of tumor growth and angiogenesis. PLoS One 4, e7190 (2009). [CrossRef]
- Merks, R. M. H. & Glazier, J. A. Dynamic mechanisms of blood vessel growth. Nonlinearity 19, C1–C10 (2006). [CrossRef]
- Merks, R. M. H., Perryn, E. D., Shirinifard, A. & Glazier, J. A. Contact-Inhibited Chemotaxis in De Novo and Sprouting Blood-Vessel Growth. PLOS Computational Biology 4, e1000163 (2008). [CrossRef]
- Niarakis, A. et al. Immune digital twins for complex human pathologies: applications, limitations, and challenges. NPJ Syst Biol Appl 10, 141 (2024). [CrossRef]
- Yang, H. M. Mathematical modeling of solid cancer growth with angiogenesis. Theor Biol Med Model 9, 2 (2012). [CrossRef]
- Moffett, A. S., Deng, Y. & Levine, H. Modeling the Role of Immune Cell Conversion in the Tumor-Immune Microenvironment. Bull Math Biol 85, 93 (2023). [CrossRef]
- Uatay, A. et al. Physiological Indirect Response Model to Omics-Powered Quantitative Systems Pharmacology Model. J Pharm Sci S0022-3549(23)00438–0 (2023). [CrossRef]
- Pereira, M. et al. A Comprehensive Look at In Vitro Angiogenesis Image Analysis Software. Int J Mol Sci 24, 17625 (2023). [CrossRef]
- Yan, H. H. N. et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 23, 882-897.e11 (2018). [CrossRef]
- Oyoshi, H. et al. Comprehensive single-cell analysis demonstrates radiotherapy-induced infiltration of macrophages expressing immunosuppressive genes into tumor in esophageal squamous cell carcinoma. Sci Adv 9, eadh9069 (2023). [CrossRef]
- Shen, Y., Ni, S., Li, S. & Lv, B. Role of stemness-related genes TIMP1, PGF, and SNAI1 in the prognosis of colorectal cancer through single-cell RNA-seq. Cancer Med 12, 11611–11623 (2023). [CrossRef]
- Laubenbacher, R. et al. Building digital twins of the human immune system: toward a roadmap. npj Digit. Med. 5, 1–5 (2022). [CrossRef]
Figure 1.
Schematic representation of cancer development: A) healthy stratified epithelial tissue encompassing sub epithelial connective tissue and epithelium separated by a continuous basal membrane. B) Carcinoma in situ eliciting tissue inflammation. C) Invasive carcinoma composed of distinct cancer clones (CEpC) with differential properties. Invasive CEpC subvert the microenvironment generating cancer associated fibroblasts (CAF) and altered ECM D) Close-up view of invasive TME and its relations with the TAMV. Inflammation can control cancer growth in some but not all CEpC clones. Hypoxia and CEpC mutations favour the “angiogenic switch”, and CEpC-EC interactions can generate further phenotypic heterogeneity. Cancer cells which can co-opt TME components like EC and Macrophages (MΦ) to intravasate and then disseminate to distant organs. In breast cancer a pro-metastatic Tie2-expressing-MΦ /EC/Cancer cell triad, the tumour microenvironment of metastasis doorway (TMEM doorway), has been identified.
Figure 1.
Schematic representation of cancer development: A) healthy stratified epithelial tissue encompassing sub epithelial connective tissue and epithelium separated by a continuous basal membrane. B) Carcinoma in situ eliciting tissue inflammation. C) Invasive carcinoma composed of distinct cancer clones (CEpC) with differential properties. Invasive CEpC subvert the microenvironment generating cancer associated fibroblasts (CAF) and altered ECM D) Close-up view of invasive TME and its relations with the TAMV. Inflammation can control cancer growth in some but not all CEpC clones. Hypoxia and CEpC mutations favour the “angiogenic switch”, and CEpC-EC interactions can generate further phenotypic heterogeneity. Cancer cells which can co-opt TME components like EC and Macrophages (MΦ) to intravasate and then disseminate to distant organs. In breast cancer a pro-metastatic Tie2-expressing-MΦ /EC/Cancer cell triad, the tumour microenvironment of metastasis doorway (TMEM doorway), has been identified.
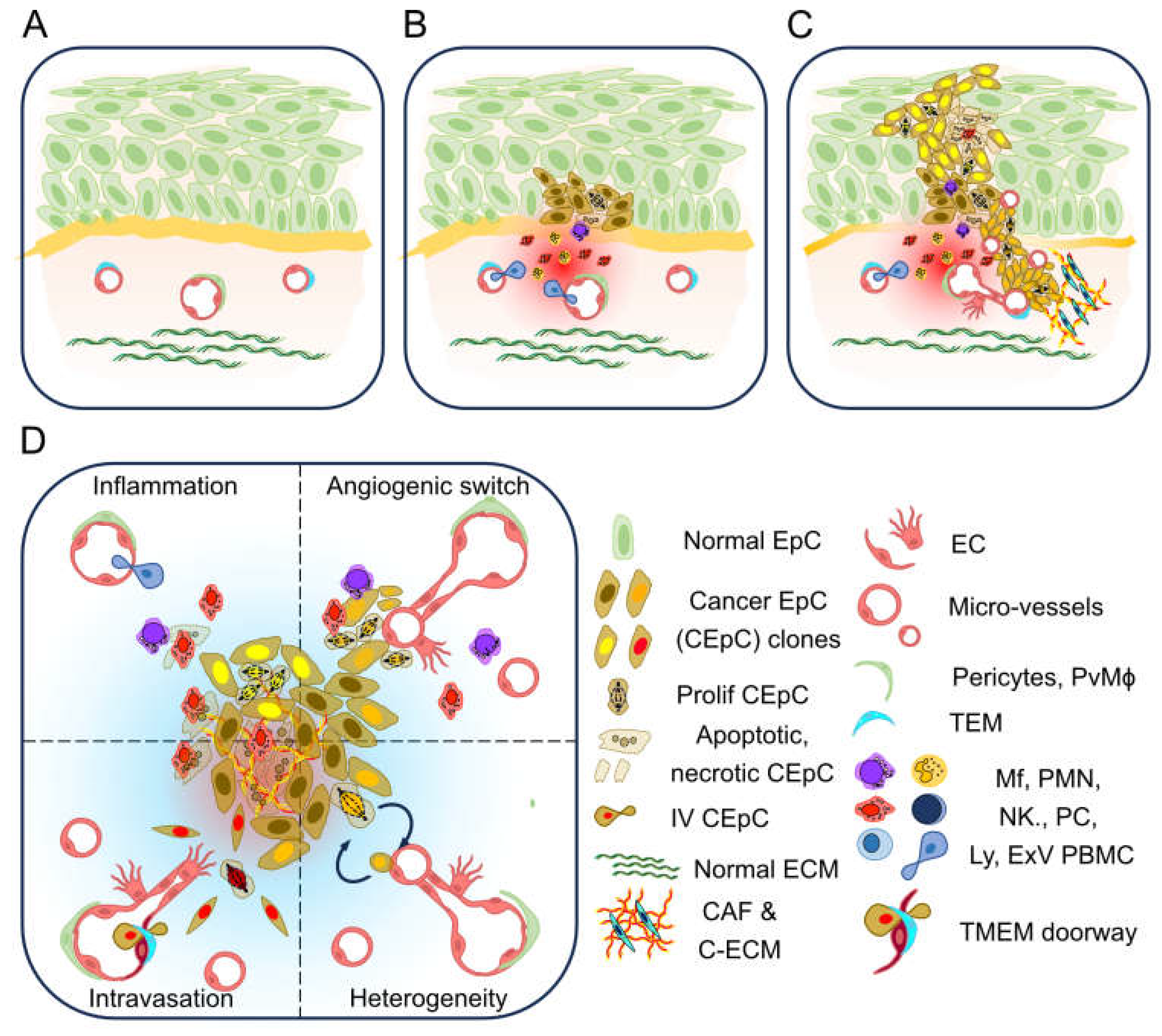
Figure 2.
Overview of in vitro NATs to study the TME and TAMV: A) EC cultures in monolayer, Matrigel, fibrin gel, or in the OVAA system. The OVAA can be imaged at high magnification allowing to appreciate details like individual tip cells (red arrow) and their filopodia. B) Immuno-stained slides of RCCS cultured human hepatocarcinoma at diagnosis, or upon 3 days of RCCS culture in presence or absence of Sorafenib (SF). CD31 marks microvessels. C) Photographs of the RCCS and VoC systems within standard tissue incubators. D) RFP labelled EC in the VoC-OVAA system upon 10 days of perfusion.
Figure 2.
Overview of in vitro NATs to study the TME and TAMV: A) EC cultures in monolayer, Matrigel, fibrin gel, or in the OVAA system. The OVAA can be imaged at high magnification allowing to appreciate details like individual tip cells (red arrow) and their filopodia. B) Immuno-stained slides of RCCS cultured human hepatocarcinoma at diagnosis, or upon 3 days of RCCS culture in presence or absence of Sorafenib (SF). CD31 marks microvessels. C) Photographs of the RCCS and VoC systems within standard tissue incubators. D) RFP labelled EC in the VoC-OVAA system upon 10 days of perfusion.
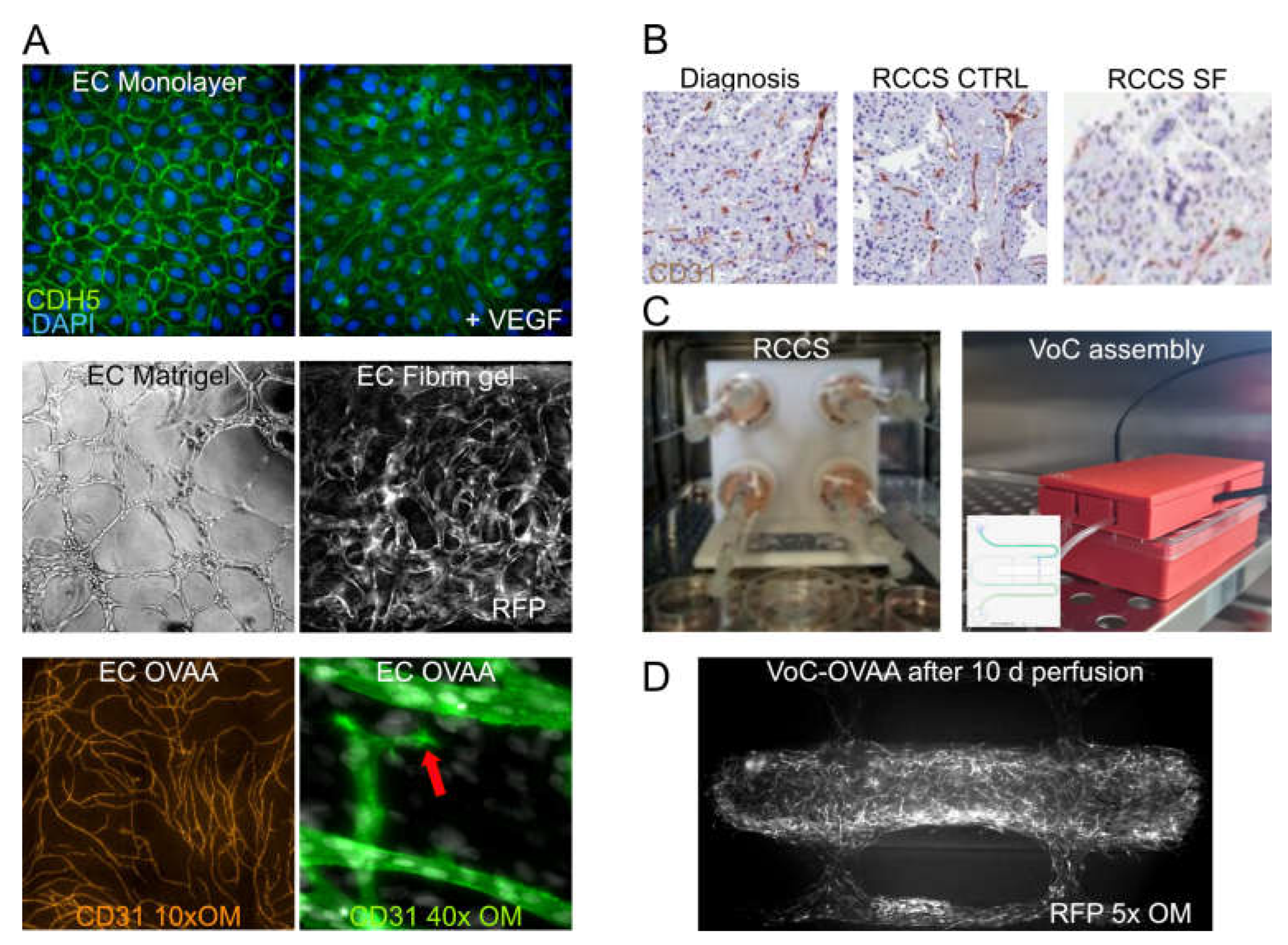
Figure 3.
Strategies to improve pixel level segmentation of H&E images. A) Two examples of images with different staining intensities (faded and dark) and relative RGB intensity histograms. Solid line for faded images and dashed line for dark ones B) Nuclei segmentations using either Stardist (CNN based), or CellProfiler (intensity based). Stardist (default weights) under-segments faded images and over-segments dark ones. A cell profiler pipeline tailored on darker images, fails to segment faded images. C) A sequential workflow to segment both dark stained and faded Hematoxylin and Eosin (H&E) images employing a random forest pixel classifier (as implemented in QuPAth), followed by refinement with Stardist, and the results plugged into CellProfiler for object analysis and classification. Scale bars = 100μm.
Figure 3.
Strategies to improve pixel level segmentation of H&E images. A) Two examples of images with different staining intensities (faded and dark) and relative RGB intensity histograms. Solid line for faded images and dashed line for dark ones B) Nuclei segmentations using either Stardist (CNN based), or CellProfiler (intensity based). Stardist (default weights) under-segments faded images and over-segments dark ones. A cell profiler pipeline tailored on darker images, fails to segment faded images. C) A sequential workflow to segment both dark stained and faded Hematoxylin and Eosin (H&E) images employing a random forest pixel classifier (as implemented in QuPAth), followed by refinement with Stardist, and the results plugged into CellProfiler for object analysis and classification. Scale bars = 100μm.
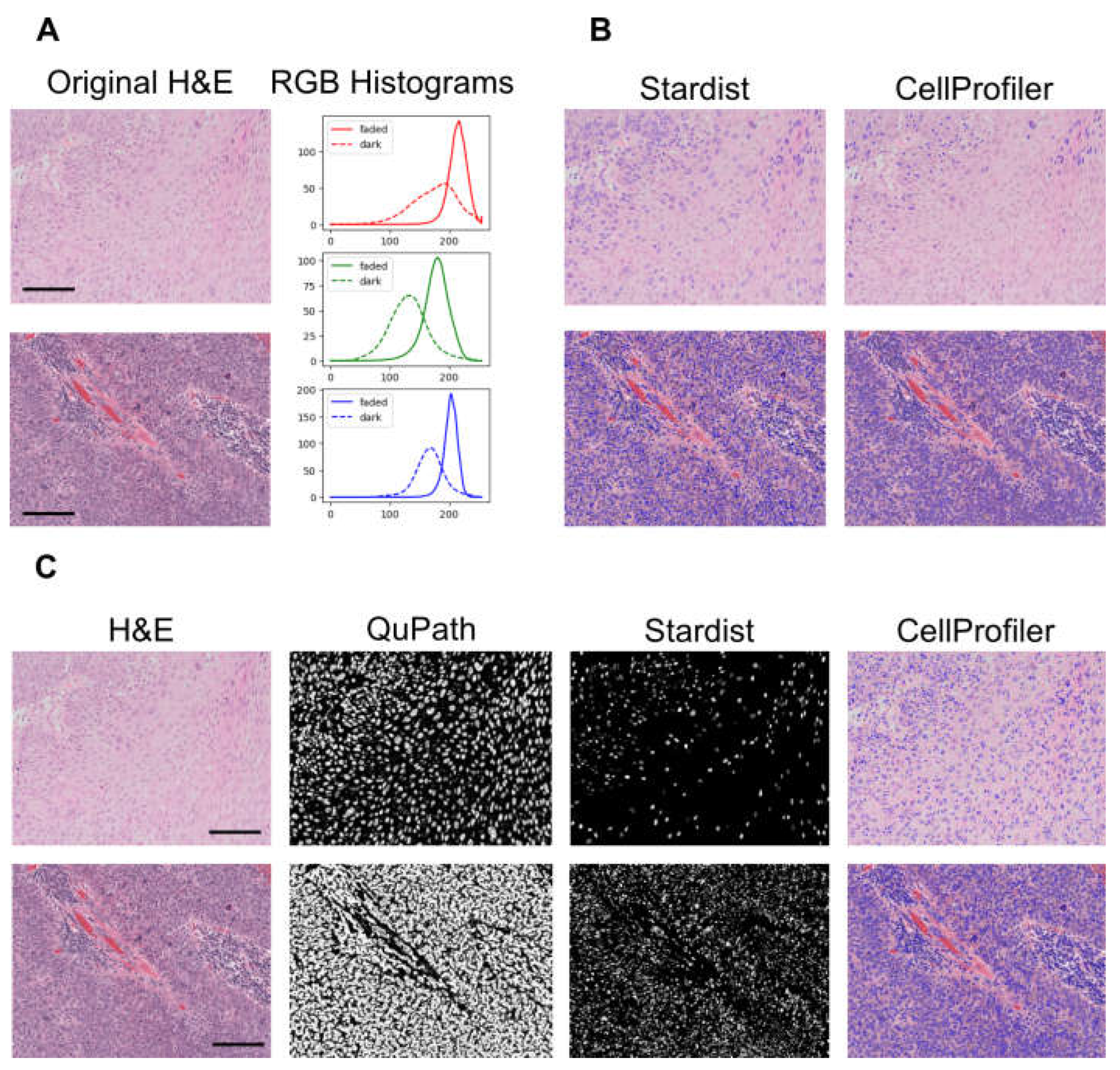
Figure 4.
Agent Based Cell Models of vascular assembly and tumour angiogenesis. A-B) Simulations calibrated to reproduce results of the in vitro tubulogenesis assay on Matrigel (Reimplementation of ref [135]) based on cell adhesion energies and contact-inhibited chemotaxis of EC. A) Time course evolution (600 MCS ~ 12h, 1 MCS ~ 90s) of the “tubulogenesis” simulations with varying initial cell densities. Paralleling in vitro assays, the simulation demonstrates the importance of cell density to achieve structured networks. B) Replicate simulations (n = 5) with varying λ-chemotaxis parameter, i.e., the responsiveness of EC to VEGF chemo-attraction. High values represent standard experimental conditions where networks of tubular structures are formed efficiently. Decreasing values mimic experimental conditions where VEGF signalling is inhibited, for example with anti KDR (human VEGF receptor 2) monoclonal antibodies. The simulation’s results parallel in vitro observation, where interference with VEGF signalling prevents tubules organisation forming blob-like structures. C) 2D reimplementation of a tumour angiogenesis simulation [133]. The simulation recapitulates the crosstalk between tumour cells (TC, brown) and EC (green) mediated by VEGF signalling and oxygen. Hypoxic TC produce long-range diffusing VEGF, promoting angiogenic sprouting which in turn is regulated by VEGF induction and lateral inhibition from neighbouring EC. As new vessels reach the TC, these proliferate and decrease VEGF production. The depicted time course (1 MCS ~2 h) displays the efficient formation of a vascular network, that, via steps of intermittent hypoxia, reach a stable state where most TC are oxygenated and cease to produce VEGF.
Figure 4.
Agent Based Cell Models of vascular assembly and tumour angiogenesis. A-B) Simulations calibrated to reproduce results of the in vitro tubulogenesis assay on Matrigel (Reimplementation of ref [135]) based on cell adhesion energies and contact-inhibited chemotaxis of EC. A) Time course evolution (600 MCS ~ 12h, 1 MCS ~ 90s) of the “tubulogenesis” simulations with varying initial cell densities. Paralleling in vitro assays, the simulation demonstrates the importance of cell density to achieve structured networks. B) Replicate simulations (n = 5) with varying λ-chemotaxis parameter, i.e., the responsiveness of EC to VEGF chemo-attraction. High values represent standard experimental conditions where networks of tubular structures are formed efficiently. Decreasing values mimic experimental conditions where VEGF signalling is inhibited, for example with anti KDR (human VEGF receptor 2) monoclonal antibodies. The simulation’s results parallel in vitro observation, where interference with VEGF signalling prevents tubules organisation forming blob-like structures. C) 2D reimplementation of a tumour angiogenesis simulation [133]. The simulation recapitulates the crosstalk between tumour cells (TC, brown) and EC (green) mediated by VEGF signalling and oxygen. Hypoxic TC produce long-range diffusing VEGF, promoting angiogenic sprouting which in turn is regulated by VEGF induction and lateral inhibition from neighbouring EC. As new vessels reach the TC, these proliferate and decrease VEGF production. The depicted time course (1 MCS ~2 h) displays the efficient formation of a vascular network, that, via steps of intermittent hypoxia, reach a stable state where most TC are oxygenated and cease to produce VEGF.
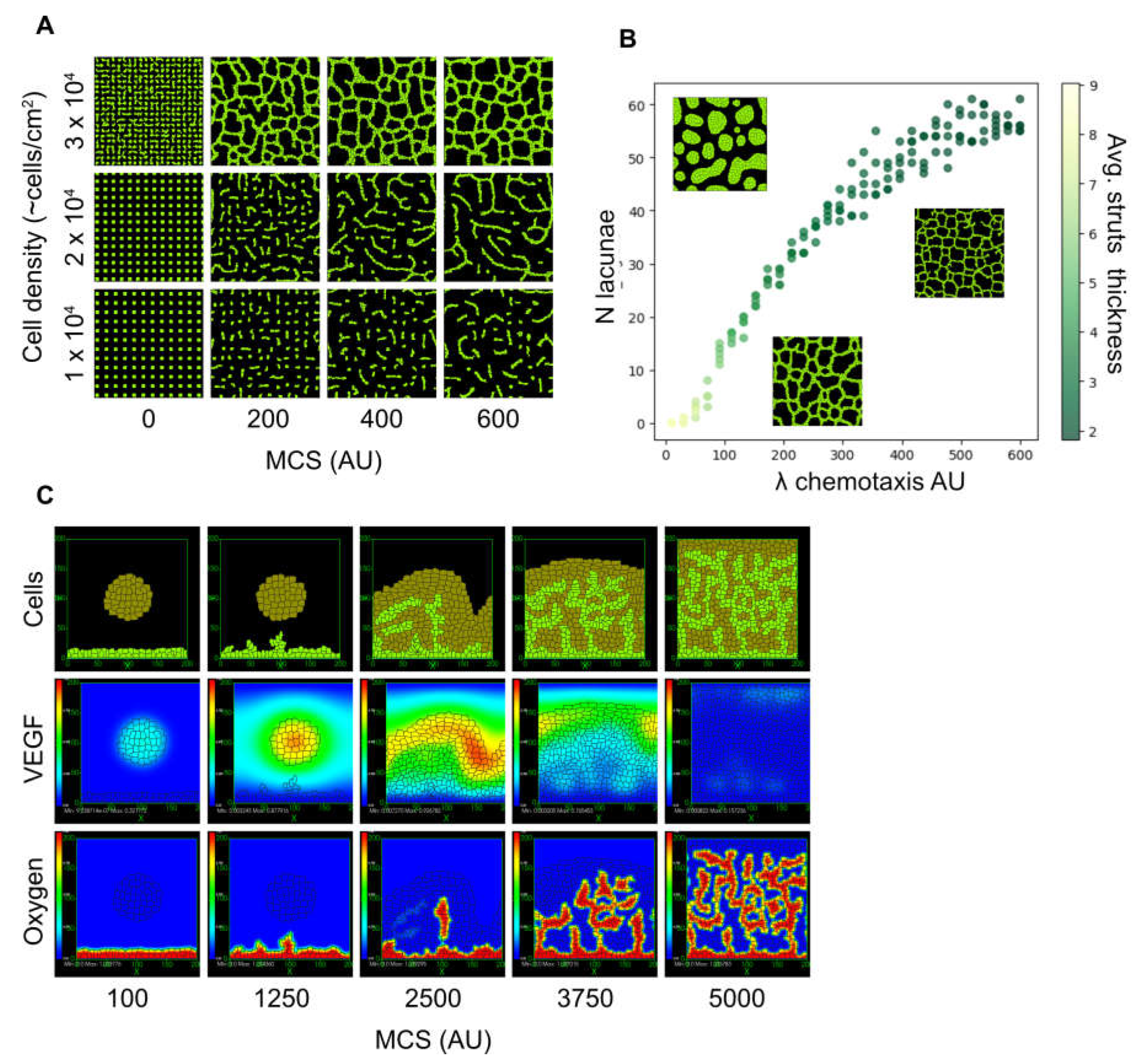
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



