
Submitted:
07 April 2025
Posted:
08 April 2025
You are already at the latest version
Abstract
Autism spectrum disorder (ASD) is a pervasive condition of neurodevelopmental origin with an increasing burden on society. Idiopathic ASD is notorious for its heterogenous behavioural manifestations, and despite substantial efforts, its etiopathology is still unclear. An increasing amount of data point at the causative role of critical developmental alterations in the first year of life, although the contribution of fetal, environmental, and genetic factors cannot be clearly distinguished. This review attempts to propose a narrative starting from neuropathological findings in ASD, involving insulin-like growth factor 1 (IGF-1) as a key modulator, and demonstrates how the most consistent gestational risk factors of ASD – maternal insulin resistance and fetal growth insufficiency – converge at the perinatal dysregulation of offspring anabolism in the critical period of early development. A unifying hypothesis is derived, stating that co-occurence of these gestational conditions leads to postnatal biphasic dysregulation of IGF-1 tone in the offspring, leading first to insulin-dependent accelerated development, then to subsequent arrest of growth and brain maturation in ASD as an etiologic process. This hypothesis is tested for its explanation of various widely reported risk factors and observations of idiopathic ASD, including early postnatal growth abnormalities, the pervasive spectrum of symptoms, familial predisposition, and male susceptibility. Finally, further directions of research are outlined.
Keywords:
autism
; brain overgrowth
; insulin-like growth factor 1
; intrauterine growth restriction
; insulin resistance
1. Introduction
1.1. Background
Autism spectrum disorder (ASD) is a group of pervasive developmental and behavioural disabilities [1,2] with a steadily increasing burden on society and families [3]. ASD is diagnosed based on behavioural symptoms, but its manifestations are notoriously heterogeneous [2,4]. Despite high prevalence and increasing awareness, little is known about the etiology of ASD [5,6] and although numerous genetically defined syndromes display high comorbidity with autism, idiopathic ASD represents the majority of cases [7,8]. Despite clearly demonstrated familial risk and high heritability of idiopathic ASD [9,10], increasingly large-scale genetic studies with a concomitantly increasing number of candidate genes have so far failed to find mono- or oligogenic causes for ASD [8,11].
The disorder is typically diagnosed in toddlers or in early childhood [12], although first manifestations seem to occur around 6-12 months of age [13,14]. The diagnosis of autism is rather stable throughout childhood [15,16] and adolescence [17,18] suggesting a critical role for early brain maldevelopment in the emergence of autism symptoms [19,20]. Therefore, in addition to the heterogeneity in symptoms, studies of ASD are also hampered by timing of symptom onset, namely incidence during a period of intense brain development and maturation of the nervous system [21,22], requiring longitudinal observations and analysis often in at-risk populations placed in a developmental framework [19,23].
Recognizing the proposed early origin of the disorder, environmental, familial, and gestational risk factors have been investigated extensively, and several risk factors of idiopathic ASD have been identified [1,24]. Among familial and environmental factors, an affected sibling [9,25] or male sex [26,27] are widely documented to substantially increase the risk for autism. Furthermore, increased maternal and paternal age have also been demonstrated as risk factors [28,29]. Among gestational and maternal conditions, the most robust and consistently identified risk factors for ASD are conditions related to maternal hypertension, namely preeclampsia and gestational or chronic hypertension [24,30,31]; maternal use of antidepressants, in particular selective serotonin reuptake inhibitors (SSRIs) [24,32,33] and symptoms related to maternal metabolic disturbances like gestational diabetes [34,35,36,37] and pre-gestational overweight or obesity [24,38,39]. For perinatal factors, signs of fetal growth insufficiency, like preterm birth [35,40] or low or very low birth weight [35,41] are associated with ASD risk, but, interestingly, macrosomia (fetal overgrowth) also bears a slight risk for later ASD diagnosis [41].
Although ASD is typically not diagnosed before the age of 18 months, at the population level several growth and developmental deviations have been described to precede or correlate with later ASD diagnosis. Accelerated early growth has been demonstrated for body length [42,43], total brain volume [20,44,45,46], cortical surface area [47] and excess extra-axial fluid volume [48]. Importantly, following a period of overgrowth in toddlers, a slowing or even arrested growth has been reported for many of these parameters in early childhood [46,47,49,50].
In addition to that observed in anthropometric measures, a biphasic developmental trajectory was also found for brain structural connectivity [51]. In particular, two highly valuable longitudinal imaging studies, comparing at-risk infants later diagnosed with ASD to non-affected peers [52] or diagnosed toddlers to typical controls [53]), reported structural hyperconnectivity between brain regions at earlier scans, in contrast to hypoconnectivity at the later time points (24- ca. 36 months, depending on the study). In studies of head circumference direct comparisons found early overgrowth in ASD (and in pervasive developmental disorder not otherwise specified, in earlier studies), which was absent in patients with attention deficit hyperactivity disorder (ADHD) [54,55] or generalized or other developmental delays [43,56].
Despite robust data on well-defined prenatal risk factors and postnatal developmental alterations with substantial comorbidity with ASD, to date no theoretical framework exists for the etiology of this disruptive disorder which convincingly matches these observations. In recognition of the pervasive heterogeneity of the disorder, recently multiple etiologies have been considered to account for the diverse manifestations of the dysfunctions [57]. However, it has yet to be demonstrated that symptoms group according to suggested pathomechanisms or risk factors [4]. Moreover, multiple disjunct etiologies or genetic causation has to account for the increasing prevalence of ASD, with the disorder retaining its heterogeneity of manifestations, stable pattern of risk factors and characteristic developmental trajectory. Alternatively, instead of multiple etiologies or risk genes, a central pathomechanism could be considered in idiopathic ASD, a process that could inherently account for the heterogeneity of symptoms and be associated with known ASD risk factors. In the latter sections such a hypothesis of idiopathic ASD etiology is proposed and discussed in light of scientific observations on ASD.
1.2. Neuropathological Findings in ASD and a Neurodevelopmental Narrative
Despite occasional and specific peripheral comorbidities of ASD it is reasonable to assume that the brain is the most-affected organ in the disorder. Neuropathology studies of autism patients report a diverse variety of microscopic brain alterations [58,59,60,61]. The most consistent microscopic observation, demonstrated in about 75% of cases, is the decreased number of Purkinje cells (PCs) in the cerebellum [58,60] – typically in the form of dispersed and partial loss [62,63]. The origin of this cell loss was believed to be prenatal, suggested by the lack of retrograde cell loss in the inferior olive [64], however, the presence of excess cerebellar basket and stellar cells [65] and the lack of hypoplastic folia [60] suggests a relatively late timing of this event, and therefore precise time of atrophy could not be convincingly established yet (discussed in [60]). Late postnatal loss of PCs in autism has not been considered in the literature, due to the putative dependence of inferior olivary neuron survival on their PC targets [64]. However, two findings support the possibility of postnatal atrophy. First, the dependence of inferior olive neurons on their target PCs seems to weaken during postnatal development [66,67]. Namely, although retrograde inferior olivary numeric atrophy has been demonstrated in various mutant mouse strains with early PC loss, in strains with the latest postnatal onset of PC death (around postanal days 30 and 20 in leaner and nervous mice, respectively), little or no loss of inferior olivary neurons is reported [67,68]. Secondly, although retrograde atrophy of the inferior olive has been observed in human patients following acute cerebellar lesions [69], gradual and partial loss of PCs, in contrast, does not necessarily lead to detectable inferior olivary atrophy in adults [69,70]. Intriguingly, neuropathologic analysis of the youngest ASD specimens (3 to 4 years) revealed no PC loss [58,63,71], in line with the possibility of late postnatal atrophy. In summary, PC loss in ASD in latter neonatal development cannot be excluded as a possibility and might be even in line with pathological findings [65]. If PCs were present throughout regular development of the cerebellum, their loss might report on later changes in internal environment, like sudden loss of neurotrophic or survival factors, as PCs are known for their particular vulnerability to pathophysiological insults [72,73,74].
When searching for survival-promoting agents of PCs, insulin-like growth factor 1 (IGF-1) has been identified as a hormone having a pronounced and reproducible positive effect on PC survival [75,76] and development [77] both in vitro and in vivo [78,79,80]. The marked survival-promoting effect of IGF-1 on PCs seems specific in comparison to numerous other neurotrophic factors like nerve growth factor, brain-derived neurotrophic factor [81,82,83], ciliary neurotrophic factor [84], basic fibroblast growth factor, insulin and insulin-like growth factor 2 [75], or to contradictory effects of neurotrophin-3 [81,83] or glial cell line-derived neurotrophic factor [79,85]. Thus, although other survival-promoting factors might also play a role, PC loss in ASD might be a result of late postnatal attenuation in the neurotrophic tone of IGF-1. Strikingly, in two highly valuable clinical datasets, liquor IGF-1 level of younger ASD patients, ages 1.9 years to 5 years, was found to be extremely low and lower than that of age-matched nonaffected patients [86] or neurotypical controls [87], a difference not found for nerve growth factor [88], insulin-like growth factor 2 or IGF-1 in ASD patients above the age of 5 [87]. Several authors have even suggested the loss of IGF-1 tone as an underlying mechanism in ASD [89,90,91] and treatment with IGF-1 or analogues has been proposed for treatment of ASD or related disorders [91,92,93]. However, the hypothesis of IGF-1 deficit, unfortunately, does not account for the most consistent characteristics and risk factors of idiopathic ASD, such as early growth and connectivity anomalies, or various perinatal or familial risk factors. Furthermore, although IGF-1 regulates PC survival and development, IGF-1 knockout mice are not characterized by PC loss [94], which suggests a more complex dysregulation of IGF-1 in ASD.
1.3. IGF-1 and Its Dysregulation in Perinatal Complications
IGF-1 is a trophic factor, a mediator of growth via its major molecular target, the IGF-1 receptor, ubiquitously expressed in most tissues. Circulating IGF-1 is predominantly secreted from the liver but can be taken up by the brain, enabling modulation of central levels by circulating IGF-1 of liver origin [95,96]. IGF-1 is widely expressed also in other tissues [97] including the brain [76] and while paracrine action contributes to postnatal growth [98], liver-produced systemic IGF-1 also exerts substantial somatic growth-promoting effects [99,100], indicating that multiple sources of IGF-1 act jointly on tissue development and that endocrine IGF-1 also affects growth. Secretion of IGF-1 from the liver is predominantly regulated via the growth hormone releasing hormone (GHRH) – growth hormone (GH) – IGF-1 axis [101] but peculiarly, perinatal regulation of endocrine IGF-1 is different, creating a vulnerability through a specific constellation of perinatal factors associated with later ASD risk. Finally, unlike the closely related insulin, IGF-1 is bound to a family of carrier proteins (insulin-like growth factor-binding proteins, IGFBP), which can regulate IGF-1 availability and thus exert further control on IGF-1 function in vivo.
In the fetus, IGF-1 release from the liver is regulated by circulating insulin with little effect of GH [102,103,104], also mirrored in the close to normal birth weight of GH-deficient infants [105]. In newborns, circulating IGF-1 is a determinant of somatic growth [106,107,108] and is associated with insulin level [109,110] with no or negative correlation to GH or GH binding protein [111,112,113], in line with typically no signs of growth deficit in congenital GH deficiency prior to 6-12 months [105]. Regulation of circulating IGF-1 in infants thus gradually changes from insulin-dependent to GH-dependent throughout the first year of life [113,114].
As IGF-1 is a major regulator of fetal growth, two groups of gestational maternal complications associated with metabolic and growth alterations in the offspring can lead to its dysregulation. Fetal growth restriction or intrauterine growth restriction (IUGR) often leads to a neonate with birth weight lower than 10th percentile. These small for gestational age (SGA) infants are characterized by short stature, decreased serum or cord IGF-1 level [112,115,116], and elevated insulin sensitivity at birth [116,117]. Their majority undergo a period of compensatory accelerated early postnatal growth (catch-up growth) [118,119] and reach a more insulin-resistant metabolic state [117,120,121]. An opposite alteration from typical perinatal growth trajectory and metabolism is observed after macrosomia or fetal overgrowth. Macrosomic, or large for gestational age, newborns display hyperinsulinemia [122,123] and higher IGF-1 levels [124,125] in a more insulin resistant state [123,126,127] and often suffer decelerated early postnatal growth – catch-down growth [128,129,130]. Catch-up and catch-down growth take place mainly within the first 6-9 months [128,129], the time window of insulin-dependent IGF-1 secretion in the newborn. SGA infants experiencing catch-up growth have elevated circulating IGF-1 levels compared to those remaining short or light [131,132].
These specific perinatal growth anomalies are highlighted because their maternal risk factors show a striking overlap with those of idiopathic ASD discussed earlier. In one group of conditions, SGA offspring and low birth weight are associated with maternal SSRI and antidepressant use [133,134], and IUGR is highly comorbid with preeclampsia [135,136] and gestational hypertension [137,138]. Conversely, in a second group of gestational conditions, macrosomia is strongly associated with maternal metabolic disturbances like gestational diabetes [139], obesity [140] or higher than recommended gestational weight gain [141], driven by insulin resistance [142]. Importantly, as discussed above, both groups of maternal conditions are strongly connected with ASD risk in the offspring [24,31,37,39].
2. Hypothesis
2.1. A Possible Etiological Role of IGF-1 Dysregulation in Idiopathic ASD
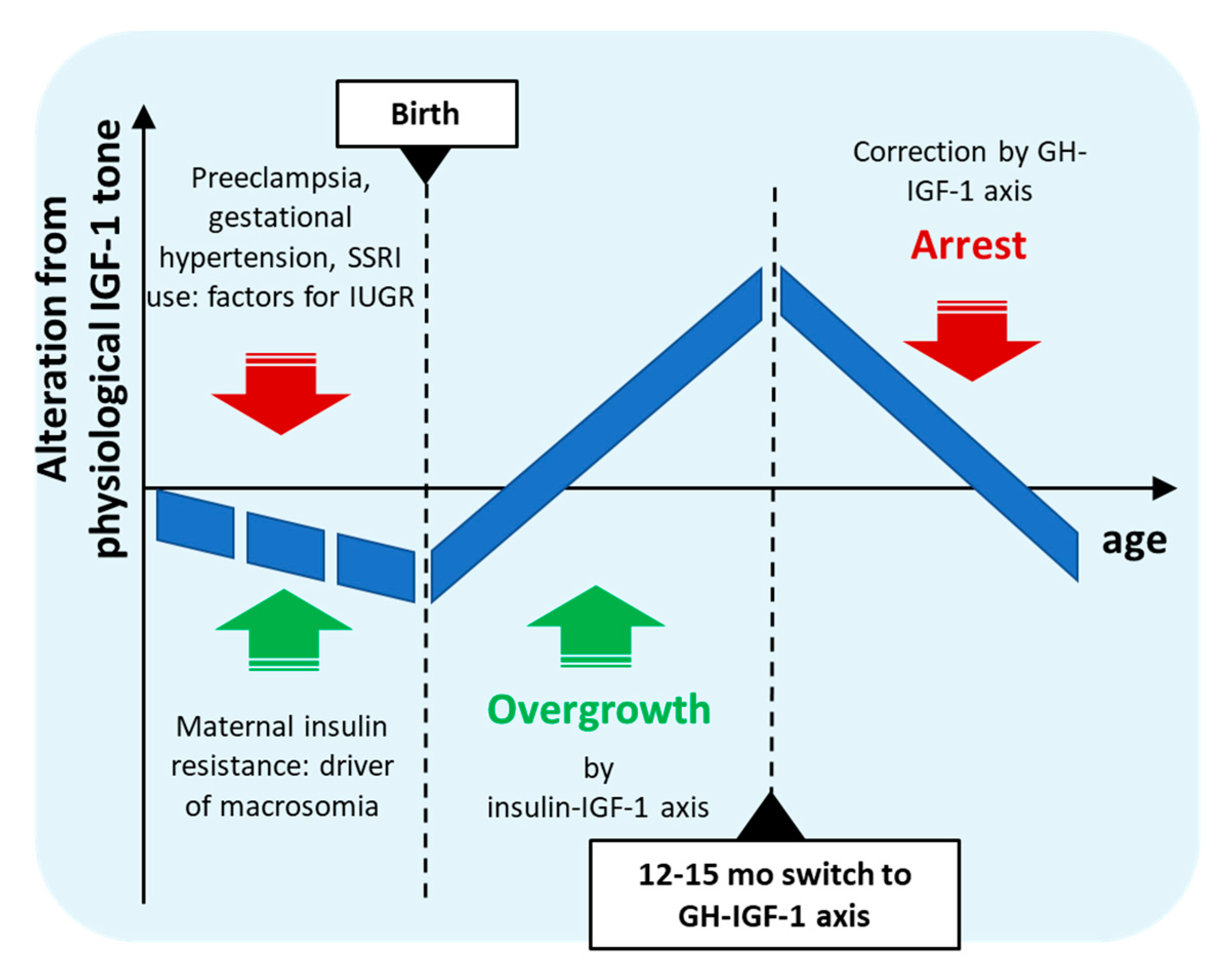
The gestational risk factors of ASD described above thus form two groups associated with opposing dysregulation of fetal growth, neonatal insulin sensitivity, circulating neonatal or cord IGF-1 level, and infant growth trajectory. More importantly, timing of their postanatal consequences also overlap with the earliest developmental alterations preceding ASD in infancy. Based on these overlaps the novel hypothesis for idiopathic ASD presented here is that simultaneous occurrence of these counteracting conditions during gestation could lead to a complex dysregulation of perinatal insulin homeostasis and insulin-mediated IGF-1 action in the affected offspring. Prenatally, these co-occurring factors might partially neutralize each other despite misalignment of fetal insulin sensitivity, however, after birth, isolated from maternal circulation, asynchronous compensatory processes could lead to biphasic dysregulation of neonatal insulin sensitivity and IGF-1 tone. In the first phase, separated from the placental unit, prevailing elevated insulin action in tissues could result in higher IGF-1 secretion, leading to accelerated growth in a sensitive period of brain development in infancy. Next, by the age of 12-15 months, maturation of the GHRH-GH-IGF-1 axis and sharp negative feedback due to elevated IGF-1 levels could lead to an abrupt drop in GHRH-GH-IGF-1 tone, and concomitantly to arrest of IGF-1-dependent developmental processes and slowed maturation of the central nervous system. According to this hypothesis, thus, prenatal gestational anabolic disturbances result in a complexly dysregulated neonatal insulin-IGF-1 axis, leading first to elevated and later to decreased IGF-1 levels in affected infants leading to somatic and brain overgrowth and subsequent growth arrest and delayed maturation (summarized in Figure 1). This biphasic postnatal growth differs from that of catch-up growth in SGA or low birth weight infants in that the latter is characterized by in utero growth restriction or insufficiency leading to delayed growth up to birth, while cooccurrence of maternal insulin resistance would partly compensate IUGR, resulting in a generally well-developed newborn with high capacity for overdevelopment in a postnatal compensatory growth spurt.
Since somatic growth anomalies, observed only in a subset of ASD patients, supposedly do not form the etiologic factors of ASD, core symptoms should depend on developmental alterations in affected brain regions. Brain development and connectivity of ASD brains seem to show peculiar dynamics with early age [19,20,51]. Early connectivity abnormalities – mixed functional over- and underconnectivity – at 6 months were used to predict later ASD diagnosis with 100% specificity [143] and the alterations seem specific to ASD [144]. Additionally, brain overgrowth predominantly in cortical surface area by 6 and 12 months identified affected infants with high specificity (95%) [20]. As these developmental trajectories are distinct from those in global developmental delay [145] and ADHD [146] and as diagnosis stability of ASD is relatively high, it can be reasoned that this early period of brain overgrowth and probably overconnectivity is specific to autism and might form the pathomechanistic basis of idiopathic ASD [147].
A potential role of IGF-1 in ASD has been suggested in the literature and even IGF-1 administration has been considered as a treatment of ASD [89,90,91,148,149], but the hypothesis of early postnatal elevated central IGF-1 tone or its biphasic dysregulation has not been raised before. The above hypothesis thus represents a novel viewpoint of the possible neurodevelopmental dysregulation by IGF-1 in the appearence of idiopathic ASD.
2.2. Explanation of Observations and Risk Factors of ASD
IGF-1 is a pleiotropic modulator of growth [97] and brain development [150], therefore, dysregulation of IGF-1 tone affects a broad range of neurodevelopmental processes and therefore could account for the most pervasive observation in ASD: the puzzling diversity of brain developmental, pathophysiological, and pathological findings, as well as the resulting variety of behavioural or neurological symptoms. According to this view, it is not a single site, but most of the organism and the central nervous system affected by abnormal biphasic early overgrowth, and genetic and other factors could further shape the appearance of ASD. Since the highly arbored PCs of the cerebellum are known to display elevated vulnerability to pathophysiological conditions in vivo [72,74,151], their survival and development could be the most affected by altered central IGF-1 availability. Unprogrammed postnatal elevation of IGF-1 tone followed by an abrupt drop could lead to late and selective postnatal PC loss, without substantial atrophy of cerebellar basket cells, cerebellar stellar cells, and inferior olivary neurons. As cited above, the lack of PC loss in the youngest ASD cases subjected to neuropathological analysis is noteworthy, not contradicting the supposed PC loss throughout early childhood. This assumption is in line with the extremely low liquor levels of IGF-1 between 1.9 to 5 years in ASD patients [86,87]. Similarly, although peripheral and central IGF-1 do not necessarily correlate, lower serum level of IGF-1 was found in ASD patients of 2-3 years age compared to age-matched controls [152], and lower urinary secretion of IGF-1 has been reported in ASD in a very similar age range (2-5 years) [153]. Interestingly, in older pediatric subjects cerebrospinal fluid (CSF) levels of IGF-1 did not differ between affected and control subjects [87], while serum levels were reported varied in older children with ASD [152,154,155,156].
The timing of the suggested biphasic alteration in circulating IGF-1 level coincides well with the ASD-specific switch in growth rate compared to controls [51,157] in light of the observations that neonatal somatic growth correlated to circulating IGF-1 levels [107,158]. In infants with later ASD diagnosis, excess extraaxial volume [48] and brain volume [47] seem to stabilize after 12-18 months. Similarly, although imaging studies reported structural hyperconnectivity prior to 20 months, hypoconnectivity was observed in ASD patients by the age of 2-3 years [52,53]. Additionally, a particularly interesting analysis has found that white matter overgrowth in ASD is specific to myelinization processes starting within the first 4 months after birth [159]. Based on the well-described effect of IGF-1 on brain development [150], the timing of these brain myelination periods overlaps well with the proposed timing of early postnatal developmental dysregulations in ASD.
The most consistent non-gestational risk factors of ASD could also be explained by the concept of perinatal IGF-1 dysregulation. Boys bear approximately 3-fold risk of ASD incidence compared to girls [27]. Male infants are known to be born larger [160,161], have higher brain volume [162,163] and undergo slightly faster postnatal brain growth than females [21,164]. The IGF-1 level of newborn boys, in contrast, is reported as lower or similar to that of girls, both in serum [107,165,166,167] and CSF [168], and female infants are more insulin-resistant than boys [161,169]. Therefore, male infants, since they display higher growth rate with lower or similar IGF-1 levels and concomitant higher insulin sensitivity than females, might be more sensitive to insulin-regulated hypertrophy mediated by excess IGF-1 and its abrupt drop, and this amplification of IGF-1-mediated effects might be the cause of their susceptibility to ASD.
The single highest risk for idiopathic ASD is an affected sibling [1,25] with clear familial connection [10] despite lack of high penetrance single risk gene variants [170]. Genetic architecture clearly modifies the penetrance of neurodevelopmental disturbances and thus definitely exerts a contribution to appearence of ASD that should not be underestimated. Still, according to the hypothesis above, combined incidence of two groups of gestational conditions could be the specific etiologic trigger for idiopathic ASD, and therefore careful separation of genetic influence from the influence of the intrauterine environment is required. Contribution of genetic and environmental factors is most often assessed by analyzing concordance in monozygotic and dizygotic twins [10]. However as intrauterine growth restriction, as presented above, can be suspected in the etiology of ASD, such a heritability analysis has to take into account the difference in placental organization of dizygotic and monozygotic twins. Dizygotic twins namely are obligatory dichorionic, they possess separate placentas, while the majority of monozygotic twin pairs are monochorionic, sharing the same placenta. Placental dysfunction is a major contributor to fetal growth insufficiency and IUGR [171,172], therefore, monochorionicity might lead to overestimation of heritability as impaired placental function might not be separated from genetic overlap. It has to be noted that preeclampsia, the strongest maternal risk factor of ASD and a risk factor for IUGR, parallels idiopathic ASD in its familial aggregation with yet unclear genetic etiology [173].
The steeply increasing prevalence of ASD can be partly related to the increased awereness and diagnostic tolerance of the behavioral spectrum, however, a true increase in prevalence is nevertheless suggested [174,175]. According to the etiologic hypothesis presented, cooccurrence of maternal insulin resistance and risk factors of IUGR like chronic or gestational hypertonia are prerequisites for biphasic neonatal metabolic dysregulation. While prevalence trends in gestational hypertensive disorders in the last decades are reported as varied [176,177,178], prevalence of pregestational obesity and pregestational/gestational diabetes has been rising significantly since decades [179,180,181,182] in line with the steadily increasing ASD prevalence.
Finally, increasing maternal age, too, is unambiguously a risk factor for metabolic syndrome – by multiple definitions – in women of childbearing age in developed countries [183,184], and also for its various manifestations like gestational diabetes [179,185], overweight, or obesity [186]. Increasing maternal age as a risk factor for ASD could therefore indirectly transmit the effect of these gestational conditions.
2.3. Prospective Testing of the Hypothesis
The etiologic hypothesis presented here attempts to add points for consideration in order to improve our understanding of this pervasive disorder. Numerous epidemiologic studies have investigated the contribution of environmental, gestational, and maternal conditions to ASD risk. The principle proposed above is that of a combination of gestational conditions leading to opposing postnatal growth dysregulation (e.g. maternal metabolic disturbances in combination with preeclampsia, gestational hypertension or SSRI use). Therefore, investigation of combinations of these specific conditions in regard to ASD incidence risk would be interesting, suggested in general in [30]. Additionally, as signs of maternal metabolic syndrome and insulin resistance are risk factors of ASD [37] and as gestational diabetes bears a significant risk of later type 2 diabetes of the mother, the apparent risk posed by having an ASD offspring on later maternal type 2 diabetes is worth investigating. Moreover, as discussed above, heritability studies on ASD prevalence in monozygotic twins taking chorionicity into account could improve dissection of genetic and intrauterine environmental effects, further increasing our understanding of role of genetic influence. Higher concordance in monochorionic compared dichorionic twins would support a role of placental dysfunction in the latter incidence of ASD.
Finally, and perhaps most importantly, CSF IGF-1 level determination could be extended to at-risk neonates 3-6 months old in order to test the hypothesis of early elevated level of this neurotrophic factor as such an analysis has not been reported yet. The lower concentration of arginine vasopressin, but not oxytocin, in the CSF of 0-3 months old neonates in high concordance with idiopathic ASD diagnosis in a quasi-prospective re-evaluated random population sample [187] supports that neonatal liquor composition could show clear alteration in ASD from typical development at such an early age.
Limitations of the above hypothesis lie in the lack in incorporation of the paracrine effects of IGF-1 or the complex contribution of IGFBPs, and the role of genetic predisposition in affecting the incidence and the phenotype of emerging ASD, factors that could serve as subjects of further research.
3. Concluding Remarks
The most consistent maternal and gestational risk factors of idiopathic ASD overlap with two groups of gestational conditions affecting fetal growth through the insulin-IGF-1 axis. Asynchronous termination of these drivers after birth would lead to a biphasic postnatal dysregulation of IGF-1 tone in the infant, with the early insulin resistance-driven overgrowth in the first life year coinciding with the well-documented and specific accelerated development and growth in ASD. This excess IGF-1 spurt might lead to irreversible structural connectivity abnormalities specific to ASD followed by slowed maturation of neurocircuits, resulting in lasting network dysfunction. If this concept wins confirmation in at-risk infants, in addition to providing new research avenues, it could support effective early prevention initiatives for this burdensome condition.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The helpful suggestions and comments by Dr. Nóra Hartvig and Dr. András Boros and careful reading of the manuscript by Dr. Klára Felsővályi are acknowledged.
Conflicts of Interest
A.V. is employee of Gedeon Richter Plc.
Abbreviations
The following abbreviations are used in this manuscript:
| ADHD | Attention deficit hyperactivity disorder |
| ASD | Autism spectrum disorder |
| CSF | Cerebrospinal fluid |
| GH | Growth hormone |
| GHRH | Growth hormone releasing hormone |
| IGF-1 | Insulin-like growth factor 1 |
| IGFBP | Insulin-like growth factor-binding protein |
| IUGR | Intrauterine growth restriction |
| PC | Purkinje cell |
| SGA | Small for gestational age |
| SSRI | Selective serotonin reuptake inhibitor |
References
- Lord, C.; Brugha, T.S.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.J.H.; Jones, R.M.; Pickles, A.; State, M.W.; et al. Autism Spectrum Disorder. Nat Rev Dis Primers 2020, 6, 5. [Google Scholar] [CrossRef]
- Masi, A.; DeMayo, M.M.; Glozier, N.; Guastella, A.J. An Overview of Autism Spectrum Disorder, Heterogeneity and Treatment Options. Neurosci Bull 2017, 33, 183–193. [Google Scholar] [CrossRef]
- Leigh, J.P.; Du, J. Brief Report: Forecasting the Economic Burden of Autism in 2015 and 2025 in the United States. J Autism Dev Disord 2015, 45, 4135–4139. [Google Scholar] [CrossRef]
- Nordahl, C.W.; Andrews, D.S.; Dwyer, P.; Waizbard-Bartov, E.; Restrepo, B.; Lee, J.K.; Heath, B.; Saron, C.; Rivera, S.M.; Solomon, M.; et al. The Autism Phenome Project: Toward Identifying Clinically Meaningful Subgroups of Autism. Front Neurosci 2022, 15, 786220. [Google Scholar] [CrossRef] [PubMed]
- Amaral, D.G. Examining the Causes of Autism. Cerebrum 2017, 2017, 1–12. [Google Scholar]
- Hertz-Picciotto, I.; Schmidt, R.J.; Krakowiak, P. Understanding Environmental Contributions to Autism: Causal Concepts and the State of Science. Autism Research 2018, 11, 554–586. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.F.; Casanova, E.L.; Frye, R.E.; Baeza-Velasco, C.; LaSalle, J.M.; Hagerman, R.J.; Scherer, S.W.; Natowicz, M.R. Editorial: Secondary vs. Idiopathic Autism. Front Psychiatry 2020, 11, 297. [Google Scholar] [CrossRef]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and Epigenetics of Autism Spectrum Disorder—Current Evidence in the Field. J Appl Genet 2019, 60, 37–47. [Google Scholar] [CrossRef]
- Hansen, S.N.; Schendel, D.E.; Francis, R.W.; Windham, G.C.; Bresnahan, M.; Levine, S.Z.; Reichenberg, A.; Gissler, M.; Kodesh, A.; Bai, D.; et al. Recurrence Risk of Autism in Siblings and Cousins: A Multinational, Population-Based Study. J Am Acad Child Adolesc Psychiatry 2019, 58, 866–875. [Google Scholar] [CrossRef]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of Autism Spectrum Disorders: A Meta-Analysis of Twin Studies. J Child Psychol Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef]
- Manoli, D.S.; State, M.W. Autism Spectrum Disorder Genetics and the Search for Pathological Mechanisms. Am J Psychiatr 2021, 178, 30–38. [Google Scholar] [CrossRef] [PubMed]
- van’t Hof, M.; Tisseur, C.; van Berckelear-Onnes, I.; van Nieuwenhuyzen, A.; Daniels, A.M.; Deen, M.; Hoek, H.W.; Ester, W.A. Age at Autism Spectrum Disorder Diagnosis: A Systematic Review and Meta-Analysis from 2012 to 2019. Autism 2021, 25, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Ozonoff, S.; Iosif, A.-M.; Baguio, F.; Cook, I.C.; Moore Hill, M.; Hutman, T.; Rogers, S.J.; Rozga, A.; Sangha, S.; Sigman, M.; et al. A Prospective Study of the Emergence of Early Behavioral Signs of Autism. J Am Acad Child Adolesc Psychiatry 2010, 49, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.J.H.; Venema, K.; Earl, R.; Lowy, R.; Barnes, K.; Estes, A.; Dawson, G.; Webb, S.J. Reduced Engagement with Social Stimuli in 6-Month-Old Infants with Later Autism Spectrum Disorder: A Longitudinal Prospective Study of Infants at High Familial Risk. J Neurodev Disord 2016, 8, 7. [Google Scholar] [CrossRef]
- Gotham, K.; Pickles, A.; Lord, C. Trajectories of Autism Severity in Children Using Standardized ADOS Scores. Pediatrics 2012, 130, e1278. [Google Scholar] [CrossRef]
- Brian, J.; Bryson, S.E.; Smith, I.M.; Roberts, W.; Roncadin, C.; Szatmari, P.; Zwaigenbaum, L. Stability and Change in Autism Spectrum Disorder Diagnosis from Age 3 to Middle Childhood in a High-Risk Sibling Cohort. Autism 2016, 20, 888–892. [Google Scholar] [CrossRef]
- Kočovská, E.; Billstedt, E.; Ellefsen, A.; Kampmann, H.; Gillberg, I.C.; Biskupstø, R.; Andorsdóttir, G.; Stóra, T.; Minnis, H.; Gillberg, C. Autism in the Faroe Islands: Diagnostic Stability from Childhood to Early Adult Life. Scient World J 2013, 592371. [Google Scholar] [CrossRef]
- Orm, S.; Andersen, P.N.; Fossum, I.N.; Øie, M.G.; Skogli, E.W. Brief Report: Autism Spectrum Disorder Diagnostic Persistence in a 10-Year Longitudinal Study. Res Autism Spectr Disord 2022, 97, 102007. [Google Scholar] [CrossRef]
- Piven, J.; Elison, J.T.; Zylka, M.J. Toward a Conceptual Framework for Early Brain and Behavior Development in Autism. Mol Psychiatry 2017, 22, 1385–1394. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Gu, H.; Munsell, B.C.; Kim, S.H.; Styner, M.; Wolff, J.J.; Elison, J.T.; Swanson, M.R.; Zhu, H.; Botteron, K.N.; et al. Early Brain Development in Infants at High Risk for Autism Spectrum Disorder. Nature 2017, 542, 348–351. [Google Scholar] [CrossRef]
- Holland, D.; Chang, L.; Ernst, T.M.; Curran, M.; Buchthal, S.D.; Alicata, D.; Skranes, J.; Johansen, H.; Hernandez, A.; Yamakawa, R.; et al. Structural Growth Trajectories and Rates of Change in the First 3 Months of Infant Brain Development. JAMA Neurol 2014, 71, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Knickmeyer, R.C.; Gouttard, S.; Kang, C.; Evans, D.; Wilber, K.; Smith, J.K.; Hamer, R.M.; Lin, W.; Gerig, G.; Gilmore, J.H. A Structural MRI Study of Human Brain Development from Birth to 2 Years. J Neurosci 2008, 28, 12176–12182. [Google Scholar] [CrossRef] [PubMed]
- Amaral, D.G. The Promise and the Pitfalls of Autism Research: An Introductory Note for New Autism Researchers. Brain Res 2011, 1380, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Son, M.J.; Son, C.Y.; Radua, J.; Eisenhut, M.; Gressier, F.; Koyanagi, A.; Carvalho, A.F.; Stubbs, B.; Solmi, M.; et al. Environmental Risk Factors and Biomarkers for Autism Spectrum Disorder: An Umbrella Review of the Evidence. Lancet Psychiatry 2019, 6, 590–600. [Google Scholar] [CrossRef]
- Ozonoff, S.; Young, G.S.; Carter, A.; Messinger, D.; Yirmiya, N.; Zwaigenbaum, L.; Bryson, S.; Carver, L.J.; Constantino, J.N.; Dobkins, K.; et al. Recurrence Risk for Autism Spectrum Disorders: A Baby Siblings Research Consortium Study. Pediatrics 2011, 128, e488. [Google Scholar] [CrossRef]
- Maenner, M.J.; Shaw, K.A.; Bakian, A. V.; Bilder, D.A.; Durkin, M.S.; Esler, A.; Furnier, S.M.; Hallas, L.; Hall-Lande, J.; Hudson, A.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years — Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2018. MMWR. Surveill Summ 2021, 70, 1–16. [Google Scholar] [CrossRef]
- Loomes, R.; Hull, L.; Polmear Locke Mandy, W. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J Am Acad Child Adolesc Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef]
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Advanced Parental Age and Autism Risk in Children: A Systematic Review and Meta-Analysis. Acta Psychiatr Scand 2017, 135, 29–41. [Google Scholar] [CrossRef]
- Sandin, S.; Schendel, D.; Magnusson, P.; Hultman, C.; Surén, P.; Susser, E.; GrØnborg, T.; Gissler, M.; Gunnes, N.; Gross, R.; et al. Autism Risk Associated with Parental Age and with Increasing Difference in Age between the Parents. Mol Psychiatry 2016, 21, 693–700. [Google Scholar] [CrossRef]
- Katz, J.; Reichenberg, A.; Kolevzon, A. Prenatal and Perinatal Metabolic Risk Factors for Autism: A Review and Integration of Findings from Population-Based Studies. Curr Opin Psychiatry 2021, 34, 94–104. [Google Scholar] [CrossRef]
- Dachew, B.A.; Mamun, A.; Maravilla, J.C.; Alati, R. Pre-Eclampsia and the Risk of Autism-Spectrum Disorder in Offspring: Meta-Analysis. Br J Psychiatr 2018, 212, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, Y.C.; Keskin-Arslan, E.; Acar, S.; Sozmen, K. Prenatal Selective Serotonin Reuptake Inhibitor Use and the Risk of Autism Spectrum Disorder in Children: A Systematic Review and Meta-Analysis. Reprod Toxicol 2016, 66, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Leshem, R.; Bar-Oz, B.; Diav-Citrin, O.; Gbaly, S.; Soliman, J.; Renoux, C.; Matok, I. Selective Serotonin Reuptake Inhibitors (SSRIs) and Serotonin Norepinephrine Reuptake Inhibitors (SNRIs) During Pregnancy and the Risk for Autism Spectrum Disorder (ASD) and Attention Deficit Hyperactivity Disorder (ADHD) in the Offspring: A True Effect or a Bias? A Systematic Review & Meta-Analysis. Curr Neuropharmacol 2021, 19, 896–906. [Google Scholar] [CrossRef]
- Xu, G.; Jing, J.; Bowers, K.; Liu, B.; Bao, W. Maternal Diabetes and the Risk of Autism Spectrum Disorders in the Offspring: A Systematic Review and Meta-Analysis. J Autism Dev Disord 2014, 44, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Geng, H.; Liu, W.; Zhang, G. Prenatal, Perinatal, and Postnatal Factors Associated with Autism: A Meta-Analysis. Medicine 2017, 96, e6696. [Google Scholar] [CrossRef]
- Rowland, J.; Wilson, C.A. The Association between Gestational Diabetes and ASD and ADHD: A Systematic Review and Meta-Analysis. Sci Rep 2021, 11, 5136. [Google Scholar] [CrossRef]
- Abraham, D.A.; Rajanandh, M.G. Does Metabolic Syndrome during Pregnancy Really a Risk to Autism Spectrum Disorder? Diab Metabol Syndr: Clin Res Rev 2020, 14, 1591–1592. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, S.; Xu, S.; Weng, S.; Liu, Z. Maternal Body Mass Index and Risk of Autism Spectrum Disorders in Offspring: A Meta-Analysis. Sci Rep 2016, 6, 34248. [Google Scholar] [CrossRef]
- Modabbernia, A.; Velthorst, E.; Reichenberg, A. Environmental Risk Factors for Autism: An Evidence-Based Review of Systematic Reviews and Meta-Analyses. Mol Autism 2017, 8, 13. [Google Scholar] [CrossRef]
- Agrawal, S.; Rao, S.C.; Bulsara, M.K.; Patole, S.K. Prevalence of Autism Spectrum Disorder in Preterm Infants: A Meta-Analysis. Pediatrics 2018, 142, e20180134. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, J.; Su, Y.; Lu, H.; Li, J.; Wang, L.; Shang, S.; Yue, W. Association of Birth Weight with Risk of Autism: A Systematic Review and Meta-Analysis. Res Autism Spectr Disord 2022, 92, 101934. [Google Scholar] [CrossRef]
- Campbell, D.J.; Chang, J.; Chawarska, K. Early Generalized Overgrowth in Autism Spectrum Disorder: Prevalence Rates, Gender Effects, and Clinical Outcomes. J Am Acad Child Adolesc Psychiatry 2014, 53, 1063–1073.e5. [Google Scholar] [CrossRef] [PubMed]
- Chawarska, K.; Campbell, D.; Chen, L.; Shic, F.; Klin, A.; Chang, J. Early Generalized Overgrowth in Boys With Autism. Arch Gen Psychiatry 2011, 68, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas-de-la-Parra, A.; Lewis, J.D.; Fonov, V.S.; Botteron, K.N.; McKinstry, R.C.; Gerig, G.; Pruett, J.R.; Dager, S.R.; Elison, J.T.; Styner, M.A.; et al. A Voxel-Wise Assessment of Growth Differences in Infants Developing Autism Spectrum Disorder. Neuroimage Clin 2021, 29, 102551. [Google Scholar] [CrossRef]
- Sacco, R.; Gabriele, S.; Persico, A.M. Head Circumference and Brain Size in Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Psychiatry Res Neuroimaging 2015, 234, 239–251. [Google Scholar] [CrossRef]
- Courchesne, E.; Carper, R.; Akshoomoff, N. Evidence of Brain Overgrowth in the First Year of Life in Autism. J Am Med Assoc 2003, 290, 337–344. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Poe, M.D.; Gerig, G.; Styner, M.; Chappell, C.; Smith, R.G.; Vachet, C.; Piven, J. Early Brain Overgrowth in Autism Associated With an Increase in Cortical Surface Area Before Age 2 Years. Arch Gen Psychiatry 2011, 68, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.D.; Nordahl, C.W.; Li, D.D.; Lee, A.; Angkustsiri, K.; Emerson, R.W.; Rogers, S.J.; Ozonoff, S.; Amaral, D.G. Extra-Axial Cerebrospinal Fluid in High-Risk and Normal-Risk Children with Autism Aged 2–4 Years: A Case-Control Study. Lancet Psychiatry 2018, 5, 895–904. [Google Scholar] [CrossRef]
- Surén, P.; Stoltenberg, C.; Bresnahan, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; Schjølberg, S.; Susser, E.; et al. Early Growth Patterns in Children with Autism. Epidemiol 2013, 24, 660–670. [Google Scholar] [CrossRef]
- Lange, N.; Travers, B.G.; Bigler, E.D.; Prigge, M.B.D.; Froehlich, A.L.; Nielsen, J.A.; Cariello, A.N.; Zielinski, B.A.; Anderson, J.S.; Fletcher, P.T.; et al. Longitudinal Volumetric Brain Changes in Autism Spectrum Disorder Ages 6-35 Years. Autism Res 2015, 8, 82–93. [Google Scholar] [CrossRef]
- Conti, E.; Calderoni, S.; Marchi, V.; Muratori, F.; Cioni, G.; Guzzetta, A. The First 1000 Days of the Autistic Brain: A Systematic Review of Diffusion Imaging Studies. Front Hum Neurosci 2015, 9, 159. [Google Scholar] [CrossRef]
- Wolff, J.J.; Gu, H.; Gerig, G.; Elison, J.T.; Styner, M.; Gouttard, S.; Botteron, K.N.; Dager, S.R.; Dawson, G.; Estes, A.M.; et al. Differences in White Matter Fiber Tract Development Present From 6 to 24 Months in Infants With Autism. Am J Psychiatr 2012, 169, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Solso, S.; Xu, R.; Proudfoot, J.; Hagler, D.J.; Campbell, K.; Venkatraman, V.; Carter Barnes, C.; Ahrens-Barbeau, C.; Pierce, K.; Dale, A.; et al. Diffusion Tensor Imaging Provides Evidence of Possible Axonal Overconnectivity in Frontal Lobes in Autism Spectrum Disorder Toddlers. Biol Psychiatry 2016, 79, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, K.; Räikkönen, K.; Pesonen, A.K.; Andersson, S.; Kajantie, E.; Eriksson, J.G.; Vartia, T.; Wolke, D.; Lano, A. Trajectories of Growth and Symptoms of Attention-Deficit/Hyperactivity Disorder in Children: A Longitudinal Study. BMC Pediatr 2011, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; García-Esteban, R.; Iñiguez, C.; Costa, O.; Fernández-Somoano, A.; Rodríguez-Delhi, C.; Ibarluzea, J.; Lertxundi, A.; Tonne, C.; Sunyer, J.; et al. Head Circumference and Child ADHD Symptoms and Cognitive Functioning: Results from a Large Population-Based Cohort Study. Eur Child Adolesc Psychiatry 2019, 28, 377–388. [Google Scholar] [CrossRef]
- Xiao, Z.; Qiu, T.; Ke, X.; Xiao, X.; Xiao, T.; Liang, F.; Zou, B.; Huang, H.; Fang, H.; Chu, K.; et al. Autism Spectrum Disorder as Early Neurodevelopmental Disorder: Evidence from the Brain Imaging Abnormalities in 2-3 Years Old Toddlers. J Autism Dev Disord 2014, 44, 1633–1640. [Google Scholar] [CrossRef]
- Constantino, J.N.; Charman, T. Diagnosis of Autism Spectrum Disorder: Reconciling the Syndrome, Its Diverse Origins, and Variation in Expression. Lancet Neurol 2016, 15, 279–291. [Google Scholar] [CrossRef]
- Bailey, A.; Luthert, P.; Dean, A.; Harding, B.; Janota, I.; Montgomery, M.; Rutter, M.; Lantos, P. A Clinicopathological Study of Autism. Brain 1998, 121, 889–905. [Google Scholar] [CrossRef]
- Casanova, M.F.; Buxhoeveden, D.P.; Switala, A.E.; Roy, E.; Switala, A. Minicolumnar Pathology in Autism. Neurology 2002, 58, 428–432. [Google Scholar] [CrossRef]
- Palmen, S.J.M.C.; Van Engeland, H.; Hof, P.R.; Schmitz, C. Neuropathological Findings in Autism. Brain 2004, 127, 2572–2583. [Google Scholar] [CrossRef]
- Donovan, A.P.A.; Basson, M.A. The Neuroanatomy of Autism – a Developmental Perspective. J Anat 2017, 230, 4–15. [Google Scholar] [CrossRef]
- Whitney, E.R.; Kemper, T.L.; Bauman, M.L.; Rosene, D.L.; Blatt, G.J. Cerebellar Purkinje Cells Are Reduced in a Subpopulation of Autistic Brains: A Stereological Experiment Using Calbindin-D28k. Cerebellum 2008, 7, 406–416. [Google Scholar] [CrossRef]
- Wegiel, J.; Flory, M.; Kuchna, I.; Nowicki, K.; Ma, S.Y.; Imaki, H.; Wegiel, J.; Cohen, I.L.; London, E.; Wisniewski, T.; et al. Stereological Study of the Neuronal Number and Volume of 38 Brain Subdivisions of Subjects Diagnosed with Autism Reveals Significant Alterations Restricted to the Striatum, Amygdala and Cerebellum. Acta Neuropathol Commun 2014, 2, 141. [Google Scholar] [CrossRef]
- Kemper, T.L.; Bauman, M.L. The Contribution of Neuropathologic Studies to the Understanding of Autism. Neurology Clinics 1993, 11, 175–187. [Google Scholar] [CrossRef]
- Whitney, E.R.; Kemper, T.L.; Rosene, D.L.; Bauman, M.L.; Blatt, G.J. Density of Cerebellar Basket and Stellate Cells in Autism: Evidence for a Late Developmental Loss of Purkinje Cells. J Neurosci Res 2009, 87, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Triarhou, L.C.; Ghetti, B. Stabilisation of Neurone Number in the Inferior Olivary Complex of Aged “Purkinje Cell Degeneration” Mutant Mice. Acta Neuropathol 1991, 81, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Zanjani, H.; Herrup, K.; Mariani, J. Cell Number in the Inferior Olive of Nervous and Leaner Mutant Mice. J Neurogenet 2004, 18, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Heckroth, J.A.; Abbott, L.C. Purkinje Cell Loss from Alternating Sagittal Zones in the Cerebellum of Leaner Mutant Mice. Brain Res 1994, 658, 93–104. [Google Scholar] [CrossRef]
- Holmes, G. On the Connections of the Inferior Olives with the Cerebellum in Man. Brain 1908, 31, 125–137. [Google Scholar] [CrossRef]
- Louis, E.D.; Babij, R.; Cortés, E.; Vonsattel, J.P.G.; Faust, P.L. The Inferior Olivary Nucleus: A Postmortem Study of Essential Tremor Cases versus Controls. Mov Disord 2013, 28, 779–786. [Google Scholar] [CrossRef]
- Williams, R.S.; Hauser, S.L.; Purpura, D.P.; Delong,; G Robert; Swisher, C. N. Autism and Mental Retardation Neuropathologic Studies Performed in Four Retarded Persons With Autistic Behavior. Arch Neurol 1980, 34, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Mavroudis, I.; Petridis, F.; Kazis, D.; Njau, S.N.; Costa, V.; Baloyannis, S.J. Purkinje Cells Pathology in Alzheimer’s Disease. Am J Alzheimers Dis Other Demen 2019, 34, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, K.; Wang, L.; Kruse, J.; Winters, A.; Sumien, N.; Shetty, R.; Prah, J.; Liu, R.; Shi, J.; Forster, M.; et al. Early Loss of Cerebellar Purkinje Cells in Human and a Transgenic Mouse Model of Alzheimer’s Disease. Neurol Res 2021, 43, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Mcknight, S.; Ai, J.; Baker, A.J. Purkinje Cell Vulnerability to Mild and Severe Forebrain Head Trauma. J Neuropathol Exp Neurol 2006, 65, 226–234. [Google Scholar] [CrossRef]
- Torres-Aleman, I.; Pons, S.; Santos-Benito, F.F. Survival of Purkinje Cells in Cerebellar Cultures Is Increased by Insulin-like Growth Factor I. Eur J Neurosci 1992, 4, 864–869. [Google Scholar] [CrossRef]
- Croci, L.; Barili, V.; Chia, D.; Massimino, L.; Van Vugt, R.; Masserdotti, G.; Longhi, R.; Rotwein, P.; Consalez, G.G. Local Insulin-like Growth Factor I Expression Is Essential for Purkinje Neuron Survival at Birth. Cell Death Differ 2011, 18, 48–59. [Google Scholar] [CrossRef]
- Fukudome, Y.; Tabata, T.; Miyoshi, T.; Haruki, S.; Araishi, K.; Sawada, S.; Kano, M. Insulin-like Growth Factor-I as a Promoting Factor for Cerebellar Purkinje Cell Development. Eur J Neurosci 2003, 17, 2006–2016. [Google Scholar] [CrossRef]
- Ye, P.; Xing, Y.; Dai, Z.; D’Ercole, J.A. In Vivo Actions of Insulin-like Growth Factor-I (IGF-1) on Cerebellum Development in Transgenic Mice: Evidence That IGF-I Increases Proliferation of Granule Cell Progenitors. Dev Brain Res 1996, 95, 44–54. [Google Scholar] [CrossRef]
- Tolbert, D.L.; Clark, B.R. GDNF and IGF-I Trophic Factors Delay Hereditary Purkinje Cell Degeneration and the Progression of Gait Ataxia. Exp Neurol 2003, 183, 205–219. [Google Scholar] [CrossRef]
- Bitoun, E.; Finelli, M.J.; Oliver, P.L.; Lee, S.; Davies, D.K.E. AF4 Is a Critical Regulator of the IGF-1 Signaling Pathway during Purkinje Cell Development. J Neurosci 2009, 29, 15366–15374. [Google Scholar] [CrossRef]
- Mount, H.T.J.; Dreyfus, C.F.; Back, I.B. Neurotrophin-3 Selectively Increases Cultured Purkinje Cell Survival. Neuroreport 1994, 5, 2497–2500. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Cory, S.; Dreyfus, C.F.; Black, I.B. NGF and Excitatory Neurotransmitters Regulate Survival and Morphogenesis of Cultured Cerebellar Purkinje Cells. J Neurosci 1991, 11, 462–471. [Google Scholar] [CrossRef]
- Ghoumari, A.M.; Wehrlé, R.; De Zeeuw, C.I.; Sotelo, C.; Dusart, I. Inhibition of Protein Kinase C Prevents Purkinje Cell Death but Does Not Affect Axonal Regeneration. J Neurosci 2002, 22, 3531–3542. [Google Scholar] [CrossRef]
- Morrison, M.E.; Mason, C.A. Granule Neuron Regulation of Purkinje Cell Development: Striking a Balance Between Neurotrophin and Glutamate Signaling. J Neurosci 1998, 18, 3563–3573. [Google Scholar] [CrossRef] [PubMed]
- Mount, H.T.J.; Dean, D.O.; Alberch, J.; Dreyfus, C.F.; Black, I.B. Glial Cell Line-Derived Neurotrophic Factor Promotes the Survival and Morphologic Differentiation of Purkinje Cells. Proc Natl Acad Sci U S A 1995, 92, 9092–9096. [Google Scholar] [CrossRef]
- Vanhala, R.; Turpeinen, U.; Riikonen, R. Low Levels of Insulin-like Growth Factor-I in Cerebrospinal Fluid in Children with Autism. Dev Med Child Neurol 2001, 43, 614–616. [Google Scholar] [CrossRef]
- Riikonen, R.; Makkonen, I.; Vanhala, R.; Turpeinen, U.; Kuikka, J.; Kokki, H. Cerebrospinal Fluid Insulin-like Growth Factors IGF-1 and IGF-2 in Infantile Autism. Dev Med Child Neurol 2006, 48, 751. [Google Scholar] [CrossRef]
- Riikonen, R.; Vanhala, R. Levels of Cerebrospinal Fluid Nerve-Growth Factor Differ in Infantile Autism and Rett Syndrome. Dev Med Child Neurol 1999, 41, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, R. Insulin-like Growth Factors in the Pathogenesis of Neurological Diseases in Children. Int J Mol Sci 2017, 18, 2056. [Google Scholar] [CrossRef]
- Bou Khalil, R. Insulin-Growth-Factor-1 (IGF-1): Just a Few Steps behind the Evidence in Treating Schizophrenia and/or Autism. CNS Spectr 2019, 24, 277–278. [Google Scholar] [CrossRef]
- Steinman, G. IGF – Autism Prevention/Amelioration. Med Hypotheses 2019, 122, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, R. Treatment of Autistic Spectrum Disorder with Insulin-like Growth Factors. Eur J Paediatr Neurol 2016, 20, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Costales, J.; Kolevzon, A. The Therapeutic Potential of Insulin-like Growth Factor-1 in Central Nervous System Disorders. Neurosci Biobehav Rev 2016, 63, 207–222. [Google Scholar] [CrossRef]
- Cheng, C.M.; Joncas, G.; Reinhardt, R.R.; Farrer, R.; Quarles, R.; Janssen, J.; Mcdonald, M.P.; Crawley, J.N.; Powell-Braxton, L.; Bondy, C.A. Biochemical and Morphometric Analyses Show That Myelination in the Insulin-like Growth Factor 1 Null Brain Is Proportionate to Its Neuronal Composition. J Neurosci 1998, 18, 5673–5681. [Google Scholar] [CrossRef]
- Pulford, B.E.; Ishii, D.N. Uptake of Circulating Insulin-Like Growth Factors (IGFs) into Cerebrospinal Fluid Appears to Be Independent of the IGF Receptors as Well as IGF-Binding Proteins. Endocrinology 2001, 142, 213–220. [Google Scholar] [CrossRef]
- Pan, W.; Kastin, A.J. Interactions of IGF-1 with the Blood-Brain Barrier in Vivo and in Situ. Neuroendocrinology 2000, 72, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.I.; Clemmons, D.R. Insulin-Like Growth Factors and Their Binding Proteins: Biological Actions. Endocr Rev 1995, 16, 1–35. [Google Scholar] [CrossRef]
- Yakar, S.; Liu, J.-L.; Stannard, B.; Butler, A.; Accili, D.; Sauer, B.; Leroith, D. Normal Growth and Development in the Absence of Hepatic Insulin-like Growth Factor I. Proc Natl Acad Sci U S A 1999, 96, 7324–7329. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, H.; Yakar, S.; LeRoith, D. Elevated Levels of Insulin-like Growth Factor (IGF)-I in Serum Rescue the Severe Growth Retardation of IGF-I Null Mice. Endocrinology 2009, 150, 4395–4403. [Google Scholar] [CrossRef]
- Stratikopoulos, E.; Szabolcs, M.; Dragatsis, I.; Klinakis, A.; Efstratiadis, A. The Hormonal Action of IGF1 in Postnatal Mouse Growth. Proc Natl Acad Sci U S A 2008, 105, 19378–19383. [Google Scholar] [CrossRef]
- Murray, P.G.; Clayton, P.E. Endocrine Control of Growth. Am J Med Genet C Semin Med Genet 2013, 163, 76–85. [Google Scholar] [CrossRef]
- Oliver, M.H.; Harding, J.E.; Breier, B.H.; Gluckman, P.D. Fetal Insulin-like Growth Factor (IGF)-I and IGF-II Are Regulated Differently by Glucose or Insulin in the Sheep Fetus. Reprod Fertil Dev 1996, 8, 167–172. [Google Scholar] [CrossRef]
- Fowden, A.L. The Insulin-like Growth Factors and Feto-Placental Growth. Placenta 2003, 24, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Hindmarsh, P.C.; Stanhope, R.G.; Turton, J.P.G.; Cole, T.J.; Preece, M.A.; Dattani, M.T. The Role of Growth Hormone in Determining Birth Size and Early Postnatal Growth, Using Congenital Growth Hormone Deficiency (GHD) as a Model. Clin Endocrinol (Oxf) 2005, 63, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Dattani, M.T.; Malhotra, N. A Review of Growth Hormone Deficiency. Paediatr Child Health 2019, 29, 285–292. [Google Scholar] [CrossRef]
- Ong, K.K.; Langkamp, M.; Ranke, M.B.; Whitehead, K.; Hughes, I.A.; Acerini, C.L.; Dunger, D.B. Insulin-like Growth Factor I Concentrations in Infancy Predict Differential Gains in Body Length and Adiposity: The Cambridge Baby Growth Study. Am J Clin Nutr 2009, 90, 156–161. [Google Scholar] [CrossRef]
- Wang, X.; Xing, K.H.; Qi, J.; Guan, Y.; Zhang, J. Analysis of the Relationship of Insulin-like Growth Factor-1 to the Growth Velocity and Feeding of Healthy Infants. Growth Hormone & IGF Research 2013, 23, 215–219. [Google Scholar] [CrossRef]
- Skalkidou, A.; Petridou, E.; Papathoma, E.; Salvanos, H.; Trichopoulos, D. Growth Velocity during the First Postnatal Week of Life Is Linked to a Spurt of IGF-I Effect. Paediatr Perinat Epidemiol 2003, 17, 281–286. [Google Scholar] [CrossRef]
- Iñiguez, G.; Ong, K.; Bazaes, R.; Avila, A.; Salazar, T.; Dunger, D.; Mericq, V. Longitudinal Changes in Insulin-like Growth Factor-I, Insulin Sensitivity, and Secretion from Birth to Age Three Years in Small-for-Gestational-Age Children. J Clin Endocrin Metabol 2006, 91, 4645–4649. [Google Scholar] [CrossRef]
- Simmons, D. Interrelation between Umbilical Cord Serum Sex Hormones, Sex Hormone-Binding Globulin, Insulin-like Growth Factor I, and Insulin in Neonates from Normal Pregnancies and Pregnancies Complicated by Diabetes. J Clin Endocrinol Metab 1995, 80, 2217–2221. [Google Scholar] [CrossRef]
- Leger, J.; Noel, M.; Limal, J.M.; Czernichow, P. Growth Factors and Intrauterine Growth Retardation. II. Serum Growth Hormone, Insulin-like Growth Factor (IGF) I, and IGF-Binding Protein 3 Levels in Children with Intrauterine Growth Retardation Compared with Normal Control Subjects: Prospective Study from Birth to Two Years of Age. Study Group of IUGR. Pediatr Res 1996, 40, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Ogilvy-Stuart, A.L.; Hands, S.J.; Adcock, C.J.; Holly, J.M.P.; Matthews, D.R.; Mohamed-Ali, V.; Yudkin, J.S.; Wilkinson, A.R.; Dunger, D.B. Insulin, Insulin-Like Growth Factor I (IGF-I), IGF-Binding Protein-1, Growth Hormone, and Feeding in the Newborn. J Clin Endocrin Metabol 1998, 83, 3550–3557. [Google Scholar] [CrossRef] [PubMed]
- Low, L.C.K.; Tam, S.Y.M.; Kwan, E.Y.W.; Tsang, A.M.C.; Karlberg, J. Onset of Significant GH Dependence of Serum IGF-I and IGF-Binding Protein 3 Concentrations in Early Life. Pediatr Res 2001, 50, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Michaelsen, K. Effect of Protein Intake from 6 to 24 Months On-like Growth Factor 1 (IGF-1) Levels, Body, Linear Growth Velocity, and Linear Growth: What Are the Implications for stunting and Wasting? Food Nutr Bull 2013, 34, 268–271. [Google Scholar] [CrossRef]
- Ibáñez, L.; Sebastiani, G.; Lopez-Bermejo, A.; Díaz, M.; Gómez-Roig, M.D.; De Zegher, F. Gender Specificity of Body Adiposity and Circulating Adiponectin, Visfatin, Insulin, and Insulin Growth Factor-I at Term Birth: Relation to Prenatal Growth. J Clin Endocrin Metabol 2008, 93, 2774–2778. [Google Scholar] [CrossRef]
- Beltrand, J.; Nicolescu, R.; Kaguelidou, F.; Verkauskiene, R.; Sibony, O.; Chevenne, D.; Claris, O.; Lévy-Marchal, C. Catch-up Growth Following Fetal Growth Restriction Promotes Rapid Restoration of Fat Mass but without Metabolic Consequences at One Year of Age. PLoS One 2009, 4, e5343. [Google Scholar] [CrossRef]
- Mericq, V.; Ong, K.K.; Bazaes, R.; Peña, V.; Avila, A.; Salazar, T.; Soto, N.; Iñiguez, G.; Dunger, D.B. Longitudinal Changes in Insulin Sensitivity and Secretion from Birth to Age Three Years in Small- and Appropriate-for-Gestational-Age Children. Diabetologia 2005, 48, 2609–2614. [Google Scholar] [CrossRef]
- Albertsson-Wikland, K.; Karlberg, J. Natural Growth in Children Born Small for Gestational Age with and without Catch-up Growth. Acta Paediatr 1994, 83, 64–70. [Google Scholar] [CrossRef]
- Hokken-Koelega, A.C.S.; De Ridder, M.A.J.; Lemmen, R.J.; Hartog, H. Den; De Muinck Keizer-Schrama, S.M.P.F.; Drop, S.L.S. Children Born Small for Gestational Age: Do They Catch Up? Pediatr Res 1995, 38, 267–271. [Google Scholar] [CrossRef]
- Ibáñez, L.; López-Bermejo, A.; Díaz, M.; Marcos, M.V.; Casano, P.; De Zegher, F. Abdominal Fat Partitioning and High-Molecular-Weight Adiponectin in Short Children Born Small for Gestational Age. J Clin Endocrin Metabol 2009, 94, 1049–1052. [Google Scholar] [CrossRef]
- Geremia, C.; Cianfarani, S. Insulin Sensitivity in Children Born Small for Gestational Age (SGA). Rev Diab Stud 2004, 1, 58–58. [Google Scholar] [CrossRef]
- Chiesa, C.; Osborn, J.F.; Haass, C.; Natale, F.; Spinelli, M.; Scapillati, E.; Spinelli, A.; Pacifico, L. Ghrelin, Leptin, IGF-1, IGFBP-3, and Insulin Concentrations at Birth: Is There a Relationship with Fetal Growth and Neonatal Anthropometry? Clin Chem 2008, 54, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.J.; Ebenbichler, C.F.; Huter, O.; Bodner, J.; Lechleitner, M.; Föger, B.; Patsch, J.R.; Desoye, G. Fetal Leptin and Insulin Levels Only Correlate in Large-for-Gestational Age Infants. Eur J Endocrinol 2000, 142, 623–629. [Google Scholar] [CrossRef]
- Yalinbas, E.; Binay, C.; Simsek, E.; Aksit, M. The Role of Umbilical Cord Blood Concentration of IGF-I, IGF-II, Leptin, Adiponectin, Ghrelin, Resistin, and Visfatin in Fetal Growth. Am J Perinatol 2019, 36, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Christou, H.; Connors, J.M.; Ziotopoulou, M.; Hatzidakis, V.; Papathanassoglou, E.; Ringer, S.A.; Mantzoros, C.S. Cord Blood Leptin and Insulin-like Growth Factor Levels Are Independent Predictors of Fetal Growth. J Clin Endocrin Metabol 2001, 86, 935–938. [Google Scholar] [CrossRef]
- Simental-Mendía, L.E.; Castañeda-Chacón, A.; Rodríguez-Morán, M.; Guerrero-Romero, F. Birth-Weight, Insulin Levels, and HOMA-IR in Newborns at Term. BMC Pediatr 2012, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Cekmez, F.; Canpolat, F.E.; Pirgon, O.; Çetinkaya, M.; Aydinoz, S.; Suleymanoglu, S.; Ipcioglu, O.M.; Sarici, S.U. Apelin, Vaspin, Visfatin and Adiponectin in Large for Gestational Age Infants with Insulin Resistance. Cytokine 2011, 56, 387–391. [Google Scholar] [CrossRef]
- Chiavaroli, V.; Cutfield, W.S.; Derraik, J.G.B.; Pan, Z.; Ngo, S.; Sheppard, A.; Craigie, S.; Stone, P.; Sadler, L.; Ahlsson, F. Infants Born Large-for-Gestational-Age Display Slower Growth in Early Infancy, but No Epigenetic Changes at Birth. Sci Rep 2015, 5, 14540. [Google Scholar] [CrossRef]
- Taal, H.R.; Vd Heijden, A.J.; Steegers, E.A.P.; Hofman, A.; Jaddoe, V.W.V. Small and Large Size for Gestational Age at Birth, Infant Growth, and Childhood Overweight. Obesity 2013, 21, 1261–1268. [Google Scholar] [CrossRef]
- Dunn, R.K.; Uhing, M.; Goday, P.S. Catch-down Growth in Infants Born Large for Gestational Age. Nutrition in Clinical Practice 2021, 36, 1215–1219. [Google Scholar] [CrossRef]
- Ohkawa, N.; Shoji, H.; Ikeda, N.; Suganuma, H.; Shimizu, T. Relationship between Insulin-like Growth Factor 1, Leptin and Ghrelin Levels and Catch-up Growth in Small for Gestational Age Infants of 27–31 Weeks during Neonatal Intensive Care Unit Admission. J Paediatr Child Health 2017, 53, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Özkan, H.; Aydın, A.; Demir, N.; Erci, T.; Büyükgebiz, A. Associations of IGF-I, IGFBP-1 and IGFBP-3 on Intrauterine Growth and Early Catch-Up Growth. Biol Neonate 1999, 76, 274–282. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Q.; Cao, S.; Pang, J.; Zhang, H.; Feng, T.; Deng, Y.; Yao, J.; Li, H. A Meta-Analysis of Selective Serotonin Reuptake Inhibitors (SSRIs) Use during Prenatal Depression and Risk of Low Birth Weight and Small for Gestational Age. J Affect Disord 2018, 241, 563–570. [Google Scholar] [CrossRef]
- Toh, S.; Mitchell, A.A.; Louik, C.; Werler, M.M.; Chambers, C.D.; Hernández-Díaz, S. Antidepressant Use during Pregnancy and the Risk of Preterm Delivery and Fetal Growth Restriction. J Clin Psychopharmacol 2009, 29, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Makino, S.; Oguma, K.; Imai, H.; Takamizu, A.; Koizumi, A.; Yoshida, K. Fetal Growth Restriction as the Initial Finding of Preeclampsia Is a Clinical Predictor of Maternal and Neonatal Prognoses: A Single-Center Retrospective Study. BMC Pregnancy Childbirth 2021, 21, 678. [Google Scholar] [CrossRef]
- Srinivas, S.K.; Edlow, A.G.; Neff, P.M.; Sammel, M.D.; Andrela, C.M.; Elovitz, M.A. Rethinking IUGR in Preeclampsia: Dependent or Independent of Maternal Hypertension? J Perinatol 2009, 29, 680–684. [Google Scholar] [CrossRef]
- Zhang, H.-G.; Yang, L.; Qiao, Z.-X.; Guo, W. Effect of Gestational Hypertension on Fetal Growth Restriction, Endocrine and Cardiovascular Disorders. Asian J Surg 2022, 45, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- Sehested, L.T.; Pedersen, P. Prognosis and Risk Factors for intrauterine Growth Retardation. Dan Med J 2014, 61, A4826. [Google Scholar]
- Ehrenberg, H.M.; Mercer, B.M.; Catalano, P.M. The Influence of Obesity and Diabetes on the Prevalence of Macrosomia. Am J Obstet Gynecol 2004, 191, 964–968. [Google Scholar] [CrossRef]
- Dai, R. xue; He, X.J.; Hu, C.L. Maternal Pre-Pregnancy Obesity and the Risk of Macrosomia: A Meta-Analysis. Arch Gynecol Obstet 2018, 297, 139–145. [Google Scholar] [CrossRef]
- Goldstein, R.F.; Abell, S.K.; Ranasinha, S.; Misso, M.; Boyle, J.A.; Black, M.H.; Li, N.; Hu, G.; Corrado, F.; Rode, L.; et al. Association of Gestational Weight Gain with Maternal and Infant Outcomes: A Systematic Review and Meta-Analysis. J Am Med Assoc 2017, 317, 2207–2225. [Google Scholar] [CrossRef] [PubMed]
- Ahlsson, F.; Diderholm, B.; Jonsson, B.; Nordén-Lindberg, S.; Olsson, R.; Ewald, U.; Forslund, A.; Stridsberg, M.; Gustafsson, J. Insulin Resistance, a Link between Maternal Overweight and Fetal Macrosomia in Nondiabetic Pregnancies. Horm Res Paediatr 2010, 74, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Emerson, R.W.; Adams, C.; Nishino, T.; Hazlett, H.C.; Wolff, J.J.; Zwaigenbaum, L.; Constantino, J.N.; Shen, M.D.; Swanson, M.R.; Elison, J.T.; et al. Functional Neuroimaging of High-Risk 6-Month-Old Infants Predicts a Diagnosis of Autism at 24 Months of Age. Sci Transl Med 2017, 9, eaag2882. [Google Scholar] [CrossRef] [PubMed]
- Conti, E.; Calderoni, S.; Marchi, V.; Muratori, F.; Cioni, G.; Guzzetta, A. Network Over-Connectivity Differentiates Autism Spectrum Disorder from Other Developmental Disorders in Toddlers: A Diffusion MRI Study. Hum Brain Mapp 2017, 38, 2333–2344. [Google Scholar] [CrossRef]
- Sun, H.; Li, Q.; Xiao, R.; Zhang, Z.; Yang, X.; Yang, J.; Jin, B.; Wen, J.; Wu, Y.; Yang, H.; et al. A Structural MRI Study of Global Developmental Delay in Infants (<2 Years Old). Front Neurol 2022, 13, 952405. [Google Scholar] [CrossRef]
- Soldateli, B.; Silveira, R.C.; Procianoy, R.S.; Belfort, M.; Caye, A.; Leffa, D.; Franz, A.P.; Barros, F.C.; Santos, I.S.; Matijasevich, A.; et al. Association between Preterm Infant Size at 1 Year and ADHD Later in Life: Data from 1993 and 2004 Pelotas Birth Cohorts. Eur Child Adolesc Psychiatry 2023, 32, 1589–1597. [Google Scholar] [CrossRef]
- Wolff, J.J.; Swanson, M.R.; Elison, J.T.; Gerig, G.; Pruett, J.R.; Styner, M.A.; Vachet, C.; Botteron, K.N.; Dager, S.R.; Estes, A.M.; et al. Neural Circuitry at Age 6 Months Associated with Later Repetitive Behavior and Sensory Responsiveness in Autism. Mol Autism 2017, 8, 8. [Google Scholar] [CrossRef]
- Shen, J.; Liu, L.; Yang, Y.; Zhou, M.; Xu, S.; Zhang, W.; Zhang, C. Insulin-Like Growth Factor 1 Has the Potential to Be Used as a Diagnostic Tool and Treatment Target for Autism Spectrum Disorders. Cureus 2024, 16, e65393. [Google Scholar] [CrossRef]
- Dönmez, B.; Erbakan, K.; Erbaş, O. The Role of Insulin-like Growth Factor on Autism Spectrum Disorder. J Exp Basic Med Sci 2021, 430–435. [Google Scholar] [CrossRef]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The Role of Insulin-Like Growth Factor 1 (IGF-1) in Brain Development, Maturation and Neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef]
- Welsh, J.P.; Yuen, G.; Placantonakis, D.G.; Vu, T.; Haiss, F.; O’Hearn, E.; Molliver, M.E.; Aicher, S.A. Why Do Purkinje Cells Die so Easily after Global Brain Ischemia? Aldolase C, EAAT4, and the Cerebellar Contribution to Posthypoxic Myoclonus. Adv Neurol 2002, 89, 331–359. [Google Scholar]
- Li, Z.; Xiao, G.-Y.; He, C.-Y.; Liu, X.; Fan, X.; Zhao, Y.; Wang, N.-R. Serum Levels of Insulin-like Growth Factor-1 and Insulin-like Growth Factor Binding Protein-3 in Children with Autism Spectrum Disorder. Chin J Contempor Pediatr 2022, 24, 186–191. [Google Scholar]
- Anlar, B.; Oktem, F.; Bakkaloglu, B.; Haliloglu, M.; Oguz, H.; Unal, F.; Pehlivanturk, B.; Gokler, B.; Ozbesler, C.; Yordam, N. Urinary Epidermal and Insulin-Like Growth Factor Excretion in Autistic Children. Neuropediatrics 2007, 38, 151–153. [Google Scholar] [CrossRef]
- Mills, J.L.; Hediger, M.L.; Molloy, C.A.; Chrousos, G.P.; Manning-Courtney, P.; Yu, K.F.; Brasington, M.; England, L.J. Elevated Levels of Growth-Related Hormones in Autism and Autism Spectrum Disorder. Clin Endocrinol (Oxf) 2007, 67, 230–237. [Google Scholar] [CrossRef]
- Simsek, F.; Isık, Ü.; Aktepe, E.; Kılıc, F.; Sirin, F.B.; Bozkurt, M. Comparison of Serum VEGF, IGF-1, and HIF-1α Levels in Children with Autism Spectrum Disorder and Healthy Controls. J Autism Dev Disord 2021, 51, 3564–3574. [Google Scholar] [CrossRef] [PubMed]
- Abedini, M.; Mashayekhi, F.; Salehi, Z. Analysis of Insulin-like Growth Factor-1 Serum Levels and Promoter (Rs12579108) Polymorphism in the Children with Autism Spectrum Disorders. J Clin Neurosci 2022, 99, 289–293. [Google Scholar] [CrossRef]
- Courchesne, E.; Campbell, K.; Solso, S. Brain Growth across the Life Span in Autism: Age-Specific Changes in Anatomical Pathology. Brain Res 2011, 1380, 138–145. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.; Cranendonk, A.; Twisk, J.W.R.; Van Weissenbruch, M.M. IGF-I and Relation to Growth in Infancy and Early Childhood in Very-Low-Birth-Weight Infants and Term Born Infants. PLoS One 2017, 12, e0171650. [Google Scholar] [CrossRef]
- Herbert, M.R.; Ziegler, D.A.; Makris, N.; Filipek, P.A.; Kemper, T.L.; Normandin, J.J.; Sanders, H.A.; Kennedy, D.N.; Caviness, V.S. Localization of White Matter Volume Increase in Autism and Developmental Language Disorder. Ann Neurol 2004, 55, 530–540. [Google Scholar] [CrossRef]
- Villar, J.; Ismail, L.C.; Victora, C.G.; Ohuma, E.O.; Bertino, E.; Altman, D.G.; Lambert, A.; Papageorghiou, A.T.; Carvalho, M.; Jaff, Y.A.; et al. International Standards for Newborn Weight, Length, and Head Circumference by Gestational Age and Sex: The Newborn Cross-Sectional Study of the INTERGROWTH-21 St Project. Lancet 2014, 384, 857–868. [Google Scholar] [CrossRef]
- Wilkin, T.J.; Murphy, M.J. The Gender Insulin Hypothesis: Why Girls Are Born Lighter than Boys, and the Implications for Insulin Resistance. Int J Obes 2006, 30, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Durston, S.; Hulshoff Pol, H.E.; Casey, B.J.; Giedd, J.N.; Buitelaar, J.K.; Van Engeland, H. Anatomical MRI of the Developing Human Brain: What Have We Learned? J Am Acad Child Adolesc Psychiatry 2001, 40, 1012–1020. [Google Scholar] [CrossRef]
- Paus, T. Sex Differences in the Human Brain: A Developmental Perspective. Prog Brain Res 2010, 186, 13–28. [Google Scholar] [CrossRef]
- Bethlehem, R.A.I.; Seidlitz, J.; White, S.R.; Vogel, J.W.; Anderson, K.M.; Adamson, C.; Adler, S.; Alexopoulos, G.S.; Anagnostou, E.; Areces-Gonzalez, A.; et al. Brain Charts for the Human Lifespan. Nature 2022, 604, 525–533. [Google Scholar] [CrossRef]
- Geary, M.P.P.; Pringle, P.J.; Rodeck, C.H.; Kingdom, J.C.P.; Hindmarsh, P.C. Sexual Dimorphism in the Growth Hormone and Insulin-like Growth Factor Axis at Birth. J Clin Endocrin Metabol 2003, 88, 3708–3714. [Google Scholar] [CrossRef] [PubMed]
- Yüksel, B.; Özbek, M.N.; Mungan, N.Ö.; Darendeliler, F.; Budan, B.; Bideci, A.; Çetinkaya, E.; Berberoǧlu, M.; Evliyaoǧlu, O.; Yeflilkaya, E.; et al. Serum IGF-1 and IGFBP-3 Levels in Healthy Children between 0 and 6 Years of Age. J Clin Res Pediatr Endocrinol 2011, 3, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Chellakooty, M.; Juul, A.; Boisen, K.A.; Damgaard, I.N.; Kai, C.M.; Schmidt, I.M.; Petersen, J.H.; Skakkebæk, N.E.; Main, K.M. A Prospective Study of Serum Insulin-like Growth Factor I (IGF-I) and IGF-Binding Protein-3 in 942 Healthy Infants: Associations with Birth Weight, Gender, Growth Velocity, and Breastfeeding. J Clin Endocrin Metabol 2006, 91, 820–826. [Google Scholar] [CrossRef]
- Bunn, R.C.; King, W.D.; Winkler, M.K.; Fowlkes, J.L. Early Developmental Changes in IGF-I, IGF-II, IGF Binding Protein-1, and IGF Binding Protein-3 Concentration in the Cerebrospinal Fluid of Children. Pediatr Res 2005, 58, 89–93. [Google Scholar] [CrossRef]
- Shields, B.M.; Knight, B.; Hopper, H.; Hill, A.; Powell, R.J.; Hattersley, A.T.; Clark, P.M. Measurement of Cord Insulin and Insulin-Related Peptides Suggests That Girls Are More Insulin Resistant than Boys at Birth. Diabetes Care 2007, 30, 2661–2666. [Google Scholar] [CrossRef]
- Qiu, S.; Qiu, Y.; Li, Y.; Cong, X. Genetics of Autism Spectrum Disorder: An Umbrella Review of Systematic Reviews and Meta-Analyses. Transl Psychiatry 2022, 12, 249. [Google Scholar] [CrossRef]
- Zur, R.L.; Kingdom, J.C.; Parks, W.T.; Hobson, S.R. The Placental Basis of Fetal Growth Restriction. Obstetr Gynecol Clin 2020, 47, 81–98. [Google Scholar] [CrossRef]
- Surico, D.; Bordino, V.; Cantaluppi, V.; Mary, D.; Gentilli, S.; Oldani, A.; Farruggio, S.; Melluzza, C.; Raina, G.; Grossini, E. Preeclampsia and Intrauterine Growth Restriction: Role of Human Umbilical Cord Mesenchymal Stem Cells-Trophoblast Crosstalk. PLoS One 2019, 14, e0218437. [Google Scholar] [CrossRef]
- Steinthorsdottir, V.; McGinnis, R.; Williams, N.O.; Stefansdottir, L.; Thorleifsson, G.; Shooter, S.; Fadista, J.; Sigurdsson, J.K.; Auro, K.M.; Berezina, G.; et al. Genetic Predisposition to Hypertension Is Associated with Preeclampsia in European and Central Asian Women. Nat Commun 2020, 11, 5976. [Google Scholar] [CrossRef] [PubMed]
- Chiarotti, F.; Venerosi, A. Epidemiology of Autism Spectrum Disorders: A Review of Worldwide Prevalence Estimates since 2014. Brain Sci 2020, 10, 274. [Google Scholar] [CrossRef]
- Depastas, C.; Kalaitzaki, A. The Epidemiology of Autism Spectrum Disorder and Factors Contributing to the Increase in Its Prevalence. Arch Hellen Med 2022, 39, 308–312. [Google Scholar]
- Wang, W.; Xie, X.; Yuan, T.; Wang, Y.; Zhao, F.; Zhou, Z.; Zhang, H. Epidemiological Trends of Maternal Hypertensive Disorders of Pregnancy at the Global, Regional, and National Levels: A Population-based Study. BMC Preg Childbirth 2021, 21, 364. [Google Scholar] [CrossRef] [PubMed]
- Horgan, R.; Monteith, C.; McSweeney, L.; Ritchie, R.; Dicker, P.; EL-Khuffash, A.; Malone, F.D.; Kent, E. The Emergence of a Change in the Prevalence of Preeclampsia in a Tertiary Maternity Unit (2004–2016). J Matern Fetal Neonat Med 2022, 35, 3129–3134. [Google Scholar] [CrossRef]
- Cameron, N.A.; Everitt, I.; Seegmiller, L.E.; Yee, L.M.; Grobman, W.A.; Khan, S.S. Trends in the Incidence of New-Onset Hypertensive Disorders of Pregnancy Among Rural and Urban Areas in the United States, 2007 to 2019. J Am Heart Assoc 2022, 11, e023791. [Google Scholar] [CrossRef]
- Venkatesh, K.K.; Harrington, K.; Cameron, N.A.; Petito, L.C.; Powe, C.E.; Landon, M.B.; Grobman, W.A.; Khan, S.S. Trends in Gestational Diabetes Mellitus among Nulliparous Pregnant Individuals with Singleton Live Births in the United States between 2011 to 2019: An Age-Period-Cohort Analysis. Am J Obstet Gynecol MFM 2023, 5, 100785. [Google Scholar] [CrossRef]
- Wang, M.C.; Freaney, P.M.; Perak, A.M.; Greenland, P.; Lloyd-Jones, D.M.; Grobman, W.A.; Khan, S.S. Trends in Prepregnancy Obesity and Association with Adverse Pregnancy Outcomes in the United States, 2013 to 2018. J Am Heart Assoc 2021, 10, e020717. [Google Scholar] [CrossRef]
- Fisher, S.C.; Kim, S.Y.; Sharma, A.J.; Rochat, R.; Morrow, B. Is Obesity Still Increasing among Pregnant Women? Prepregnancy Obesity Trends in 20 States, 2003-2009. Prev Med (Baltim) 2013, 56, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Baraban, E.; McCoy, L.; Simon, P. Increasing Prevalence of Gestational Diabetes and Pregnancy-Related Hypertension in Los Angeles County, California, 1991–2003. Prev Chronic Dis 2008, 5, 1–9. [Google Scholar]
- Vishram, J.K.K.; Borglykke, A.; Andreasen, A.H.; Jeppesen, J.; Ibsen, H.; Jørgensen, T.; Palmieri, L.; Giampaoli, S.; Donfrancesco, C.; Kee, F.; et al. Impact of Age and Gender on the Prevalence and Prognostic Importance of the Metabolic Syndrome and Its Components in Europeans. the MORGAM Prospective Cohort Project. PLoS One 2014, 9, e107294. [Google Scholar] [CrossRef] [PubMed]
- Hildrum, B.; Mykletun, A.; Hole, T.; Midthjell, K.; Dahl, A.A. Age-Specific Prevalence of the Metabolic Syndrome Defined by the International Diabetes Federation and the National Cholesterol Education Program: The Norwegian HUNT 2 Study. BMC Public Health 2007, 7, 220. [Google Scholar] [CrossRef]
- Li, G.; Wei, T.; Ni, W.; Zhang, A.; Zhang, J.; Xing, Y.; Xing, Q. Incidence and Risk Factors of Gestational Diabetes Mellitus: A Prospective Cohort Study in Qingdao, China. Front Endocrinol (Lausanne) 2020, 11, 636. [Google Scholar] [CrossRef]
- Vahratian, A. Prevalence of Overweight and Obesity among Women of Childbearing Age: Results from the 2002 National Survey of Family Growth. Matern Child Health J 2009, 13, 268–273. [Google Scholar] [CrossRef]
- Oztan, O.; Garner, J.P.; Constantino, J.N.; Parker, K.J. Neonatal CSF Vasopressin Concentration Predicts Later Medical Record Diagnoses of Autism Spectrum Disorder. Proc Natl Acad Sci U S A 2020, 117, 10609–10613. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram of age-dependent IGF-1 tone during and following gestational conditions with counteracting effect on fetal growth.
Figure 1.
Schematic diagram of age-dependent IGF-1 tone during and following gestational conditions with counteracting effect on fetal growth.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



