Submitted:
07 April 2025
Posted:
08 April 2025
You are already at the latest version
Abstract
(1) Background: Early-onset atrial fibrillation (AF) exhibits distinct clinical and genetic profiles compared to AF in older adults. The increasing detection of AF among younger patients—often in the absence of traditional risk factors—has raised interest in the genetic determinants underlying the condition; this review aims to synthesize current evidence on the genetic architecture of early-onset AF, assess the clinical utility of genetic testing, and discuss future directions for integrating genetic insights into personalized management strategies (2) Methods: We conducted a comprehensive analysis of recent studies, including genome-wide association studies and targeted sequencing efforts, that examined rare pathogenic variants and polygenic risk scores in early-onset AF. The review also considers emerging data on atrial cardiomyopathy and evaluates current guideline recommendations for genetic testing; (3) Results: Data indicate that rare variants, particularly in genes such as TTN, LMNA, and KCNQ1, play a significant role in early-onset AF, with evidence suggesting an association between these mutations and adverse clinical outcomes. Polygenic risk scores further complement traditional risk factors, providing a more nuanced risk stratification. Despite these advances, challenges remain in the interpretation of variants of uncertain significance, cost-effectiveness, and the need for interdisciplinary collaboration in clinical implementation; (4) Conclusions: Integrating genetic evaluation into the diagnostic and management framework of early-onset AF holds promise for improved risk stratification and personalized therapy. Future large-scale, multi-ethnic studies and ongoing refinement of genetic risk models are essential to overcome current limitations and enhance the clinical applicability of genetic testing in this rapidly evolving field.
Keywords:
Early-onset atrial fibrillation
; genetic testing
; atrial cardiomyopathy
; polygenic risk scores
; genome-wide association studies (GWAS)
; inherited arrhythmia syndromes
; cardiac genetics
; risk stratification
; precision medicine in cardiology
1. Introduction
Atrial fibrillation (AF) and atrial flutter (AFL) are common tachyarrhythmias associated with severe complications such as heart failure and stroke, significantly affecting patients' health and quality of life. Their etiology is multifactorial, involving risk factors like hypertension, smoking, obesity, and socio-demographic variables [1,2]. The global prevalence of AF has doubled from 1990 to 2019, with cases expected to rise further, particularly in aging populations [3]. Economic disparities influence disease burden, with lower-resource regions facing higher morbidity and mortality, while developed countries benefit from better diagnostic and treatment measures. [4] Roselli et al. (2018) conducted a multi-ethnic GWAS meta-analysis for atrial fibrillation, analyzing data from over 500,000 individuals—including 65,446 AF cases—and identified 97 AF-associated loci, 67 of which were novel. The study integrated transcriptomic and eQTL data from relevant cardiac tissues to implicate pathways in cardiac development, electrophysiology, and structure, thereby deepening our understanding of AF’s genetic architecture and suggesting new avenues for therapeutic intervention [5]. Additionally, the recent studies [6,7] underscore the complex genetic landscape of AF, involving both rare and common variants, paving the way for improved risk assessment and personalized management strategies. Compared to older adults, young individuals are generally less prone to AF and its related strokes [8]. However, the increasing use of wearable devices has led to earlier detection of AF among the young. In patients under 30, about 25% exhibit reentrant supraventricular tachycardias—specifically, atrioventricular nodal or atrioventricular reentrant tachycardia—which, when treated with targeted ablation, often results in the resolution of AF episodes. Nonetheless, additional considerations are warranted in this age group. Emerging evidence suggests that young patients with AF may also carry a genetic susceptibility to inherited ion channel or cardiomyopathic disorders—even when standard echocardiograms appear normal [9]. Therefore, alongside routine evaluation for newly diagnosed AF, incorporating genetic testing for rare pathogenic variants, advanced imaging, and regular screening could help uncover occult cardiomyopathy. [10] In a prospective observational cohort of patients diagnosed with AF before age 66, sequencing using ion channelopathy and cardiomyopathy gene panels identified disease-associated variants in 10% of the 1293 patients, with most variants found in cardiomyopathy-related genes. [11] Similarly, among 23 unrelated patients with AF onset before 45—who had normal echocardiograms and no other apparent causes—24% carried a pathogenic or likely pathogenic variant, again predominantly in genes related to cardiomyopathy. [12] Although mounting evidence underscores the significant role of genetic factors in AF—particularly in younger patients—the optimal integration of these insights into clinical practice remains an ongoing challenge. In this review, we critically assess current genetic testing strategies and examine their clinical implications for personalized patient management.
2. Materials and Methods
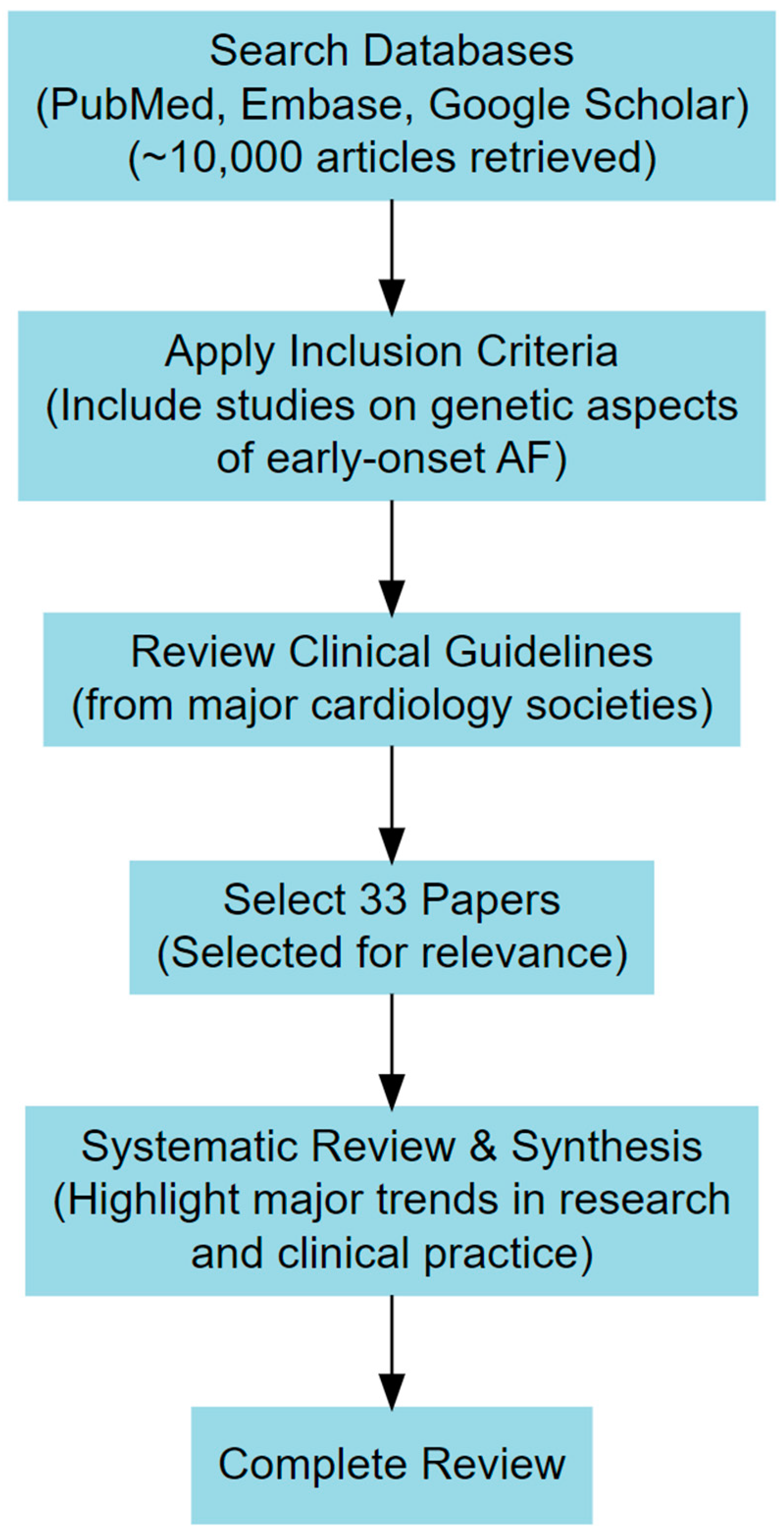
This review was conducted through a comprehensive analysis of the published literature, including genome-wide association studies (GWAS), whole-exome and whole-genome sequencing studies, clinical cohort analyses, and current guideline recommendations. Relevant articles were identified using databases such as PubMed, Embase, and Google Scholar with search terms including "early-onset atrial fibrillation," "genetic testing in AF," "atrial cardiomyopathy," and "polygenic risk scores in AF." Studies were included if they focused on the genetic contributions to early-onset atrial fibrillation, investigated monogenic or polygenic risk factors, evaluated the role of genetic testing in clinical management and risk stratification, or included large-scale population-based genetic studies or clinically relevant cohort studies. Studies were excluded if they primarily addressed atrial fibrillation in older adults (>65 years) without genetic considerations, were limited to case reports or small-scale studies lacking statistical power, or did not include genetic analysis or relevant clinical outcomes. (Figure 1)
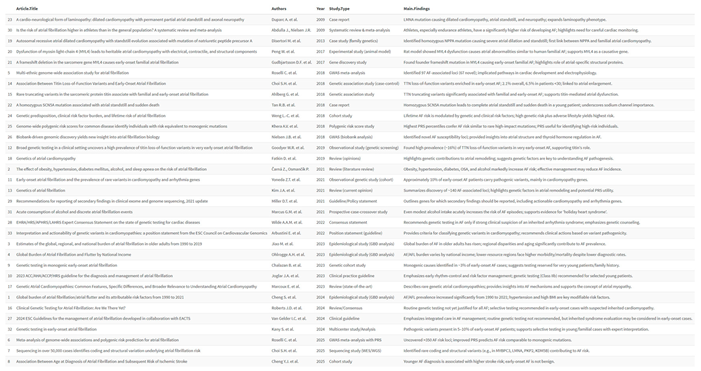
The genetic data analysis incorporated findings from sequencing efforts such as targeted gene panels, exome sequencing, and genome-wide association studies. Emphasis was placed on genes implicated in cardiomyopathies and inherited arrhythmia syndromes, including TTN, LMNA, KCNQ1, and MYH7. Current guidelines from major cardiology societies, including the ESC and AHA/ACC, were reviewed to evaluate recommendations regarding genetic testing in early-onset AF. All selected studies were systematically reviewed, and findings were synthesized to highlight major trends in genetic predisposition, the clinical implications of genetic testing, and future directions in personalized medicine for AF management. (Table 1)
3. Clinical and Genetic Landscape of Early-Onset Atrial Fibrillation
The independent link between AF and increased mortality is well documented in older populations. However, less is known about younger patients with AF despite the absence of traditional risk factors. Early observations linking a family history of AF to increased risk provided initial evidence of a heritable component to the disease. In families with a clear hereditary pattern, Mendelian inheritance enabled linkage analyses to uncover rare variants. The first mutation identified via this approach was a gain-of-function (p.Ser140Gly) variant in the KCNQ1 gene—which encodes a subunit of the cardiac IKs channel—resulting in reduced action potential duration and refractory period, thereby predisposing affected individuals to persistent AF. Notably, KCNQ1 mutations have also been implicated in long QT and short QT syndromes. Since this discovery, nearly 40 additional genes harboring rare mutations associated with familial and early-onset AF have been reported. Although many of these genes encode ion channel components, an increasing number involve non-ion channel proteins such as transcription factors, myocardial structural elements, and signaling molecules. Experimental models have further revealed that such mutations may disrupt sarcomere structure, impair calcium handling, and reduce conduction velocity—mechanisms that contribute to AF pathogenesis. More recently, rare variants in genes traditionally linked to dilated cardiomyopathy have been associated with early-onset AF even in the absence of overt structural abnormalities. [8] For instance, loss-of-function (LOF) variants in TTN, which encodes the sarcomere protein titin, were observed in 2.1% of early-onset AF cases versus 1.1% of controls, with an even higher prevalence (6.5%) among individuals diagnosed before age 30. Whole-genome sequencing was performed on cohort of 7,740 European-ancestry participants (2,781 early-onset AF cases and 4,959 controls) with a mean sequencing depth of 37.8× and over 98 million variants identified. Association testing of 8,248,975 common variants confirmed genome-wide significance at six known AF loci (e.g., PITX2, PRRX1, NEURL1, ZFHX3, KCNN3, SOX5) and at a novel locus in NAV2 (rs2625322; OR, 1.32; P = 1.46 × 10⁻⁸), with replication in independent cohorts. Analysis of rare loss-of-function variants across candidate genes demonstrated a significant association between TTN LOF variants and early-onset AF (OR, 2.16; P = 1.55 × 10⁻³). In a sensitivity analysis excluding participants with pre-existing heart failure or cardiomyopathy, TTN LOF variants were present in 2.1% of cases versus 1.1% of controls (OR, 1.76; P = 3.42 × 10⁻²). Notably, among individuals with AF onset before age 30, 6.5% carried a TTN LOF variant (OR, 5.94; P = 1.65 × 10⁻⁵), and carriers experienced AF approximately 5.3 years earlier than noncarriers. Restricting the analysis to exons with high cardiac expression further strengthened this association (OR, 4.41; P = 7.34 × 10⁻⁴). These findings were independently validated in an external dataset from the MyCode Community Health Initiative (OR, 2.16; P = 0.01), and a meta-analysis confirmed that TTN LOF variants are significantly associated with early-onset AF (OR, 2.74; P = 6.03 × 10⁻⁵). [9].
A subsequent smaller study reported TTN nonsense variants in 16% of patients with early-onset AF (<45 years) without significant comorbidities. [12] Despite these advances, only a subset of the identified genetic variants has been robustly linked to AF causation. More recent studies have revealed that early-onset AF is frequently associated with rare genetic variants in genes implicated in inherited cardiomyopathy and arrhythmia syndromes, suggesting that in some cases, AF may represent the initial manifestation of a more severe underlying genetic disorder. [13] For example, sequencing of a 145-gene panel for cardiomyopathy and arrhythmia identified pathogenic or likely pathogenic variants in 10.1% of patients diagnosed with AF before age 66 and in 16.8% of those diagnosed before age 30. [14] Despite these findings, the impact of rare genetic variants on long-term clinical outcomes in early-onset AF remains undefined. In conditions such as hypertrophic and dilated cardiomyopathy, Brugada syndrome, and long QT syndrome, genotype-positive patients consistently exhibit worse outcomes, including higher rates of malignant ventricular arrhythmias, progression to end-stage heart failure, and increased mortality. Based on these observations, it has been hypothesized that disease-associated rare variants in patients with early-onset AF would correlate with a higher risk of mortality and that there might be an interaction between genotype-positive status and younger age at diagnosis regarding mortality risk. In a cohort of 1293 early-onset atrial fibrillation patients (72% male, median age 56 years, all diagnosed before age 66) followed for a median of 9.9 years (IQR 6.9–13.2 years), Yoneda et al reported that 219 patients (16.9%) died during the follow-up period. There were 73 cardiomyopathy-related deaths, 40 sudden deaths, and 10 stroke-related deaths. Whole-genome sequencing identified disease-associated rare variants in 10.1% of the cohort (131 patients). Of these, the majority were found in cardiomyopathy (CM) genes—with 93 carriers in Dilated CM (DCM) genes, 43 in Hypertrophic CM (HCM) genes, and 37 in Arrhythmogenic CM (AC/ARVC) genes—while only 15 patients carried variants associated with channelopathies, respectively 2 (0.2%) variants for Brugada syndrome, 12 (0.9%) for LQTS, and 1 (0.1%) for CPVT. When the analysis was restricted to major disease genes, the numbers were 69 (5.3%) for DCM, 27 (2.1%) for HCM, 5 (0.4%) for AC/ARVC, 2 (0.2%) for Brugada syndrome, 11 (0.9%) for LQTS, and 1 (0.1%) for CPVT. [4,8] Notably, patients with a disease-associated variant had a higher mortality rate (24% vs 16% in those without) and multivariable analysis confirmed that variant status significantly increased the risk of all-cause mortality, particularly among those diagnosed with AF at a younger age. When stratified by specific genes, the most prevalent variants were observed in TTN (26% mortality among 38 carriers), MYH7 (33% mortality among 18 carriers), LMNA (22% mortality among 9 carriers), with no deaths observed among carriers of MYH6 (0/10) or KCNQ1 (0/8). These findings highlight the prognostic significance of rare, primarily CM-related genetic variants in early-onset AF. [15] These observations underscore the potential importance of incorporating genetic testing into the clinical assessment of early-onset AF to improve risk stratification and guide personalized management strategies.
4. Atrial Cardiomyopathy and Early Onset Atrial Fibrillation
Unlike ventricular cardiomyopathies such as dilated, hypertrophic, and arrhythmogenic right ventricular cardiomyopathy, which are diagnosed based on well-defined structural abnormalities, the recent consensus on atrial cardiomyopathy (AtCM) proposes that a diagnosis may be established with a broad array of alterations in atrial electrical properties, chamber size, or contractile function. These changes can be detected using noninvasive methods, including ECG tracings (with or without Holter monitoring or invasive electrophysiological studies), transthoracic echocardiography, or cardiac MRI. [16]Primary atrial cardiomyopathy arises from variants in genes that are functionally active in the atria and play key roles in atrial development and the maintenance of electrical, structural, and metabolic properties. For instance, variants in the NPPA gene, which encodes atrial natriuretic peptide A, have been linked to a distinctive phenotype characterized by massive bi-atrial dilation, early supraventricular arrhythmias, and progressive contractile dysfunction leading to atrial standstill. [17] Similarly, mutations in the MYL4 gene, which encodes the atrial-selective essential myosin light chain, have been associated with both electrical and mechanical defects [18,19].
Notably, genes such as SCN5A and LMNA, despite their widespread expression, can also contribute to atrial standstill, although they are more commonly associated with extra-atrial manifestations. [20,21] Ahlberg et al. observed a significant enrichment of titin-truncating variants (TTNtv) in familial and early-onset lone AF, with validation in independent cohorts indicating that TTNtv are a major genetic contributor to this condition. Experimental studies in a CRISPR/Cas9-modified zebrafish model further demonstrated that TTN truncation leads to disrupted sarcomere structure, increased atrial fibrosis, and electrophysiological abnormalities, such as prolonged PR intervals. [22] Considering the potential development of atrial cardiomyopathy in diverse clinical contexts is essential, and a high index of suspicion should prompt targeted electrical and imaging investigations. Recognizing atrial cardiomyopathy is practically relevant, especially for predicting AF recurrence after interventions such as cardioversion or ablation and for guiding thromboembolic prophylaxis decisions. Although the genetics of atrial cardiomyopathy is still in its early stages—with causative rare variants proving useful in Mendelian forms of disease, the role of genetic testing in predicting individual risk remains largely unestablished. [16] Advances in polygenic risk scores derived from common SNPs have shown incremental value in AF risk stratification, sometimes equaling the impact of single rare pathogenic variants [23,24]. However, cohort studies indicate that known genes and loci account for only a small fraction of AF heritability [25], which currently limits the utility of routine genetic testing. A deeper understanding of the genetic basis of atrial cardiomyopathy may help explain the missing heritability of AF and yield novel risk markers for AF, heart failure, and stroke. Finally, emerging evidence suggests that atrial fibrillation in younger patients may represent the initial presentation of an underlying atrial cardiomyopathy driven by rare variants in cardiomyopathy or arrhythmia syndrome genes. Despite data indicating the potential utility of genetic testing for atrial cardiomyopathy (AtCM), current guidelines from major scientific societies do not yet endorse its routine use. [26]
5. Current Guideline Recommendation on Genetic Testing in Atrial Fibrillation
European Society of Cardiology’s Guidelines do not provide a recommendation for genetic testing in early onset Atrial Fibrillation, although an increasing number of common genetic polymorphisms and variations discovered through sequencing have been linked to AF. These genetic factors can be aggregated into instruments, which yield cumulative risk estimates that exceed those associated with single, monogenic AF mutations. Polygenic risk scores, which combine this genetic information, enhance the prediction of AF by providing additional insights beyond traditional risk factors and will likely improve further as more AF-associated variants are identified. Ultimately, polygenic and genome-wide risk scores could help pinpoint individuals at the highest risk for targeted AF screening. However, because such genetic data are not yet widely available, their clinical applicability and cost-effectiveness still need to be demonstrated. [18] On the other hand, the 2023 ACC/AHA guidelines assign a Class IIb recommendation for genetic testing in patients with atrial fibrillation under 45 years of age. Additionally, for those under 30 with unexplained AF, may be considered an electrophysiological study to evaluate and treat supraventricular tachycardia which is a potential trigger for AF, also under a Class IIb recommendation. [10] Finally, the consensus document from European, US, Latin American, and Asian-Pacific societies recommends considering genetic testing for individuals with familial AF who are under 60 years of age. [27]
6. Clinical Implications of Genetic Testing
Advances in our understanding of the genetic underpinnings of AF emphasize the importance of integrating genetic testing for early-onset AF into clinical practice. Genetic testing should be performed by an interdisciplinary team, comprising cardiologists specializing in electrophysiology and cardiomyopathies, cardiovascular geneticists, and genetic counselors. The ACMG currently advises the reporting of secondary findings for many genes implicated in early-onset AF (e.g., TTN, LMNA, MYBPC3, KCNQ1, PKP2) regardless of the primary indication for testing. [28] When genetic testing is pursued specifically for early-onset AF, a comprehensive evaluation of supporting and opposing factors is essential. Favorable indicators include very early disease onset (<45 years) or a family history of AF or cardiomyopathy in individuals under 65, as well as imaging evidence suggestive of ventricular cardiomyopathy, such as increased chamber volumes or subtly reduced contractile function. Additionally, clinical features pointing to a specific monogenic defect—such as early-onset AF with conduction disease or ventricular arrhythmias (as seen with LMNA mutations) or with QT abnormalities (as observed with KCNQ1 mutations)—should raise suspicion of a genetic cause. The ECG can also offer clues, including bundle branch block, atrioventricular block, T-wave inversions, or QRS abnormalities, indicative of underlying cardiomyopathy. Conversely, AF typically associated with common risk factors—such as obesity, diabetes, hypertension, sedentary lifestyle, sleep apnea, or smoking—predominantly affects older individuals, making age the strongest marker for a non-genetic etiology. Nevertheless, some risk factors, such as height, are age-independent, and others, like endurance sports, are prevalent in younger populations. Moreover, toxic exposures (alcohol, drugs) and hormonal imbalances (hyperthyroidism) can also trigger AF in younger patients. [29,30] The primary objective of genetic testing in early-onset AF is to identify carriers of pathogenic or likely pathogenic (P/LP) variants that underlie inherited cardiomyopathies and arrhythmia syndromes. Consequently, gene panels focusing on cardiomyopathy and arrhythmia should be considered, with particular emphasis on those genes with robust evidence linking them to these conditions especially those for which P/LP variants are deemed actionable by the ACMG. Furthermore, the identification of a cardiomyopathy variant should prompt rigorous clinical and instrumental follow-up to monitor for the eventual emergence of a phenotype, inform sudden cardiac death risk stratification, and guide decisions regarding potential implantable cardioverter-defibrillator (ICD) implantation. Finally, identifying a rare cardiomyopathy or arrhythmia syndrome variant allows for cascade screening to detect at-risk family members for heart failure and sudden cardiac death. [31] Huang et al using the UK Biobank exome sequencing cohort confirmed the association between TTN truncating variants (TTN vs) and an increased risk of atrial fibrillation (AF) and dilated cardiomyopathy (DCM). Notably, TTN vs and polygenic risk scores (PRS) showed additive effects with traditional AF risk factors such as hypertension, diabetes, obesity, and smoking, indicating that genetic factors contribute to AF risk beyond modifiable clinical factors. Even after excluding participants with DCM, TTN vs remained associated with a higher risk of AF, suggesting a potential independent role in AF development, possibly through subclinical structural remodeling. In participants who developed AF, TTN vs were also linked to a higher risk of DCM, underscoring the importance of long-term cardiac monitoring for TTN vs carriers and their at-risk relatives. Additionally, the study highlighted the importance of risk factor management, even in those with high genetic risk. Individuals with TTN vs or elevated PRS but no clinical risk factors had a lower AF risk than those with low genetic risk and multiple risk factors. These findings suggest that genetic data could enhance AF risk stratification, guide early prevention strategies, and support personalized management in at-risk individuals through targeted lifestyle modifications and medical interventions. [32]
7. Challenges, Limitations, and Future Directions
One of the biggest challenges is related to the interpretation and management of variants of uncertain significance (VUS). Yoneda et al highlighted that most patients with early onset AF would carry a VUS. [11] Variants initially classified as VUS or pathogenic may be reclassified over time as new evidence emerges, including validated biomarkers, additional cases or family studies confirming or refuting pathogenicity, functional research, or the discovery of novel causative genes. Conversely, it is rare for a variant initially deemed benign to be later classified as pathogenic, though exceptions may occur with rare synonymous variants that introduce cryptic splice sites identified through functional studies. Regular reassessment of genetic data, incorporating familial segregation and functional evidence, is essential to reduce clinical uncertainty. While a VUS remains clinically inactionable, its reclassification as pathogenic can have significant clinical implications, including enhanced monitoring of carriers, early therapeutic interventions, evaluation of concealed arrhythmogenic risk, and consideration of prenatal or pre-implantation genetic diagnosis.[33] Among the limitations, the primary concern is the cost of genetic testing and data analysis. Cost-effectiveness studies are necessary, and careful patient selection is essential to minimize healthcare expenditures and avoid inconclusive outcomes. A thorough family history assessment and the exclusion of secondary causes of AF in otherwise healthy individuals are crucial to prevent unnecessary testing. Genetic testing in early-onset AF represents a promising avenue for improved prognostic stratification and to enhance clinical and cardiovascular imaging surveillance for carriers of pathogenic or likely pathogenic (P/LP) genetic variants. Additionally, it offers the opportunity for cascade testing in at-risk family members. To ensure accurate interpretation of results, especially in cases of VUS or inconclusive findings that may cause unnecessary anxiety, collaboration between cardiologists with genetic expertise and clinical geneticists is essential for effective personalized medicine.
8. Conclusions
In summary, the integration of genetic testing into the clinical management of early-onset atrial fibrillation (AF) offers significant potential for improving risk stratification, guiding therapeutic decisions, and enabling the early detection of inherited cardiomyopathies and arrhythmia syndromes. Advances in genetic research have highlighted the importance of rare pathogenic variants, particularly in genes like TTN, LMNA, and KCNQ1, while polygenic risk scores provide additional insights into the complex interplay of genetic and clinical risk factors. Although current guidelines offer limited recommendations for genetic testing, particularly in younger patients with AF, the expanding evidence base underscores the value of tailored genetic evaluation. Additionally, cascade testing of at-risk relatives can facilitate early diagnosis and preventive care, further reducing the burden of adverse cardiovascular outcomes. Nevertheless, challenges remain, including the interpretation VUS, ensuring equitable access to testing, and the need for interdisciplinary collaboration to optimize the clinical application of genetic data. Future large-scale, multi-ethnic studies and ongoing re-evaluation of genetic variants are essential to refine classification frameworks and enhance predictive accuracy. Ultimately, a personalized, genetics-informed approach to early-onset AF management could significantly improve patient outcomes, underscoring the need for continued research, clinical integration, and guideline adaptation in this rapidly evolving field.
Author Contributions
A.L: Conceptualization, Methodology, Formal analysis, Investigation, Resources, Data Curation, Writing - Original Draft, Writing – Review & Editing, Supervision; T.F: Conceptualization, Methodology, Formal analysis, Data Curation, Writing, Review & Editing, Supervision; G.S: Conceptualization, Methodology, Formal analysis, Data Curation Writing – Review & Editing, Supervision, Project administration; G.C.: Conceptualization, Methodology, Formal analysis, Investigation, Resources, Data Curation Writing – Review & Editing, Supervision, Project administration; Funding: This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
AF – Atrial Fibrillation
AFL – Atrial Flutter
GWAS – Genome-Wide Association Studye
QTL – Expression Quantitative Trait Locus
LOF – Loss of Function
TTN – Titin
DCM – Dilated Cardiomyopathy
HCM – Hypertrophic Cardiomyopathy
ACM/ARVC – Arrhythmogenic Cardiomyopathy / Arrhythmogenic Right Ventricular Cardiomyopathy
CPVT – Catecholaminergic Polymorphic Ventricular Tachycardia
LQTS – Long QT Syndrome
AtCM – Atrial Cardiomyopathy
ECG – Electrocardiogram
MRI – Magnetic Resonance Imaging
TTNtv – Titin-Truncating Variants
PRS – Polygenic Risk Score
ICD – Implantable Cardioverter-Defibrillator
ACMG – American College of Medical Genetics
VUS – Variant of Uncertain Significance
ACC/AHA – American College of Cardiology / American Heart Association
References
- Shantsila E, Choi EK, Lane DA, Joung B, Lip GYH. Atrial fibrillation: comorbidities, lifestyle, and patient factors. Lancet Reg Health Eur. 2024;37:100784. Published 2024 Feb 1. [CrossRef]
- Čarná Z, Osmančík P. The effect of obesity, hypertension, diabetes mellitus, alcohol, and sleep apnea on the risk of atrial fibrillation. Physiol Res. 2021 Dec 30;70(Suppl4):S511-S525. [CrossRef] [PubMed] [PubMed Central]
- Jiao M, Liu C, Liu Y, Wang Y, Gao Q, Ma A. Estimates of the global, regional, and national burden of atrial fibrillation in older adults from 1990 to 2019: insights from the Global Burden of Disease study 2019. Front Public Health. 2023;11:1137230. Published 2023 Jun 12. [CrossRef]
- Ohlrogge AH, Brederecke J, Schnabel RB. Global Burden of Atrial Fibrillation and Flutter by National Income: Results From the Global Burden of Disease 2019 Database. J Am Heart Assoc. 2023 Sep 5;12(17):e030438. [CrossRef]
- 5. Roselli C, Chaffin MD, Weng LC, Aeschbacher S, Ahlberg G, Albert CM, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–33. [CrossRef]
- Roselli C, Surakka I, Olesen MS, Sveinbjornsson G, Marston NA, Choi SH, et al. Meta-analysis of genome-wide associations and polygenic risk prediction for atrial fibrillation in more than 180,000 cases. Nat Genet. 2025 Mar;57(3):539-547. [CrossRef]
- Choi, S.H., Jurgens, S.J., Xiao, L. et al. Sequencing in over 50,000 cases identifies coding and structural variation underlying atrial fibrillation risk. Nat Genet 57, 548–562 (2025). [CrossRef]
- Yun-Jiu Cheng, Hai Deng, Hui-Qiang Wei, Wei-Dong Lin, Zhuomin Liang, Yili Chen et al. Association Between Age at Diagnosis of Atrial Fibrillation and Subsequent Risk of Ischemic Stroke. Journal of the American Heart Association. 2025;14(4). [CrossRef]
- Chalazan, B. , Freeth, E., Mohajeri, A. et al. Genetic testing in monogenic early-onset atrial fibrillation. Eur J Hum Genet 31, 769–775 (2023). [CrossRef]
- Joglar JA, Chung MK, Armbruster AL, Benjamin EJ, Chyou JY, Cronin EM, et al. 2023 ACC/AHA/ACCP/HRS guideline for the diagnosis and management of atrial fibrillation: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines. Circulation. 2023;149:e1–156. [CrossRef]
- Yoneda ZT, Anderson KC, Quintana JA, et al. Early-onset atrial fibrillation and the prevalence of rare variants in cardiomyopathy and arrhythmia genes. JAMA Cardiol. 2021;6:1371–1379.
- Goodyer WR, Dunn K, Caleshu C, et al. Broad genetic testing in a clinical setting uncovers a high prevalence of titin loss-of-function variants in very early onset atrial fibrillation. Circ Genom Precis Med. 2019;12:e002713.
- Kim JA, Chelu MG, Li N. Genetics of atrial fibrillation. Curr Opin Cardiol. 2021;36(3):281–287. [CrossRef]
- Choi SH, Weng LC, Roselli C, et al. Association Between Titin Loss-of-Function Variants and Early-Onset Atrial Fibrillation. JAMA. 2018;320(22):2354–2364.
- Yoneda ZT, Anderson KC, Ye F, Quintana JA, O'Neill MJ, Sims RA, et al. Mortality Among Patients With Early-Onset Atrial Fibrillation and Rare Variants in Cardiomyopathy and Arrhythmia Genes. JAMA Cardiol. 2022;7(7):733–741. [CrossRef]
- Fatkin D, Huttner IG, Johnson R. Genetics of atrial cardiomyopathy. Curr Opin Cardiol. 2019;34:275–81.
- Disertori M, Quintarelli S, Grasso M, et al. Autosomal recessive atrial dilated cardiomyopathy with standstill evolution associated with mutation of natriuretic peptide precursor A. Circ Cardiovasc Genet. 2013;6:27–36.
- Peng W, Li M, Li H, et al. Dysfunction of myosin light-chain 4 (MYL4) leads to heritable atrial cardiomyopathy with electrical, contractile, and structural components: evidence from genetically-engineered rats. J Am Heart Assoc. 2017;6:e007030.
- Gudbjartsson DF, Holm H, Sulem P, et al. A frameshift deletion in the sarcomere gene MYL4 causes early-onset familial atrial fibrillation. Eur Heart J. 2017;38:27–34.
- Tan RB, Gando I, Bu L, et al. A homozygous SCN5A mutation associated with atrial standstill and sudden death. Pacing Clin Electrophysiol. 2018. [CrossRef]
- Duparc A, Cintas P, Somody E, et al. A cardio-neurological form of laminopathy: dilated cardiomyopathy with permanent partial atrial standstill and axonal neuropathy. Pacing Clin Electrophysiol. 2009;32:410–415.
- Ahlberg G, Refsgaard L, Lundegaard PR, et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun. 2018;9:4316. [CrossRef]
- Weng LC, Preis SR, Hulme OL, et al. Genetic predisposition, clinical risk factor burden, and lifetime risk of atrial fibrillation. Circulation. 2018;137:1027–1038.
- Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic risk scores for common disease identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224.
- Nielsen JB, Thorolfsdottir RB, Fritsche LG, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50:1234–1239.
- Van Gelder IC, Rienstra M, Bunting KV, et al. 2024 ESC Guidelines for the management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J. 2024;45(36):3314–3414. [CrossRef]
- Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace. 2022;24:1307–67. [CrossRef]
- Miller DT, Lee K, Gordon AS, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23:1391–1398. [CrossRef]
- Abdulla J, Nielsen JR. Is the risk of atrial fibrillation higher in athletes than in the general population? A systematic review and meta-analysis. Europace. 2009;11:1156–1159. [CrossRef]
- Marcus GM, Vittinghoff E, Whitman IR, et al. Acute consumption of alcohol and discrete atrial fibrillation events. Ann Intern Med. 2021;174:1503–1509. [CrossRef]
- Kany S, Jurgens SJ, Rämö JT, et al. Genetic testing in early-onset atrial fibrillation. Eur Heart J. 2024;45(34):3111–3123. [CrossRef]
- Huang K, Trinder M, Roston TM, et al. The Interplay Between Titin, Polygenic Risk, and Modifiable Cardiovascular Risk Factors in Atrial Fibrillation. Can J Cardiol. 2021;37(6):848–856. [CrossRef]
- Arbustini E, Behr ER, Carrier L, et al. Interpretation and actionability of genetic variants in cardiomyopathies: a position statement from the European Society of Cardiology Council on cardiovascular genomics. Eur Heart J. 2022;43:1901–1916. [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



