
Submitted:
07 April 2025
Posted:
07 April 2025
You are already at the latest version
Abstract
Rhabdoid tumours (RTs) are aggressive neoplasms most often characterised by biallelic loss of the SMARCB1 gene, which encodes a core subunit of the SWI/SNF chromatin remodelling complex. Despite their relative genetic stability, RTs exhibit a highly malignant phenotype and poor prognosis. This review aims to explore the mechanisms underlying SMARCB1 aberrations, their role in driving hallmarks of cancer, and emerging therapeutic strategies for RTs. The loss of SMARCB1 drives multiple cancer hallmarks by disrupting key regulatory pathways. It promotes unchecked cell proliferation through alterations in p16INK4a and Myc signalling. SMARCB1-deficient tumours possess immune evading capabilities via PD-L1 overexpression and immune checkpoint activation. SMARCB1 deficiency also alters cellular energetics by various mechanisms. The nucleotide biosynthesis pathway has been demonstrated to be upregulated in RT organoids, as shown by increased levels of pathway metabolites. Enzymes of the mevalonate pathway such as HMG-CoA reductase and mevalonate kinase are also dysregulated. Targeting glutathione metabolism with eprenetapopt may result in oxidative stress and induce apoptosis in RTs. Widespread epigenetic aberrations, including increased EZH2 activity, are being targeted with inhibitors such as tazemetostat. Future horizons in RT treatment include immunotherapies, epigenetic modifiers, and gene therapies. The synergy and optimal timing of targeted therapy with conventional treatment needs to be better characterised in order to proceed to later stage clinical trials and clinical practice.
Keywords:
rhabdoid tumours
; SMARCB1 deficiency
; hallmarks of cancer
; energy metabolism
; targeted therapy
Introduction
The SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 (SMARCB1) gene is located on chromosome 22q11.2 and encodes a core subunit of the ATP-dependent SWI/SNF complex [1]. It is involved in chromatin remodelling and regulation of gene expression. Loss of the SMARCB1 has been linked to various neoplasms, including rhabdoid tumours (RT), epithelioid sarcoma, and renal medullary carcinoma [2]. RTs are rare neoplasms which typically present in the paediatric population before the age of 3 [3]. They have been described in nearly all anatomical locations, and are divided into those located intracranially, most commonly in the cerebellum [4], or extracranially in organs such as the kidney, gastrointestinal track, or liver [5]. Apart from the frequent biallelic loss of SMARCB1, RTs are considered relatively genetically stable, despite their highly aggressive phenotype [6]. Germline mutations also predispose patients to various malignancies in a condition known as rhabdoid tumour predisposition syndrome [7]. Loss of SMARCB1 promotes cancer through various pathways (Figure 1), which will be described in more detail in this review. This article will also serve to discuss the mechanisms by which aberrations of the SMARCB1 gene occur, their implications in driving the various hallmarks of cancer, and provide an overview of the novel therapeutic approaches in clinical trials for treatment of RT.
The SMARCB1 gene extends over 50 kilobases and its peptide sequence is encoded by nine exons [8]. The encoded protein has a molecular weight of 47kDa with a length of 385 amino acids. It is composed of 4 domains, including 2 highly conserved tandem repeat 1 and 2 domains (RPT1/2), flanked by the N-terminal Winged Helix DNA binding domain and the C-terminal coiled-coil domain (CTD). Data derived from COSMIC v95 revealed 815 alterations in the SMARCB1 gene, largely clustered at the CTD. The majority of mutations (49.33%) were missense, followed by frameshift mutations (20.12%) and nonsense mutations (19.88%). Silent and inframe mutations accounted for approximately 10% of genetic aberrations [2].
Confirmation of the role of SMARCB1 as a tumour suppressor gene (TSG) arose when conditional gene knockout in adult mice resulted in CD8+ mature lymphoma and fully penetrant cancer formation [9]. Later studies were also able to produce RTs by inducing SMARCB1 deletion in earlier stages of embryonic development [10]. While complete loss-of-function of SMARCB1 is the best characterised genetic mutation in RT, some cases are associated with cytoplasmic localisation of the C-terminal truncated version of the gene, compromising its ability to exert its biological functions in the nucleus. Exportin 1 is thought to be responsible for this abnormal localisation through its interaction with the RPT2 domain of SMARCB1. As a result, nuclear export inhibitors such as leptomycin B and selinexor may be effective in these cases [11]. However, clinical efficacy is yet to be established [12].
Driver of the Hallmarks of Cancer
The hallmarks of cancer were introduced by Hanahan and Weinberg in 2000, and were later expanded in 2011 and 2022 [13,14,15]. They describe a set of capabilities acquired by human cells during the carcinogenic process, and provide a systematic structure for studying cancer, designing therapeutic targets, and guiding experimental models. Figure 2 summarises the role of SMARCB1 loss as a driver of the hallmarks of cancer in RTs.
Evading Growth Suppressors and Sustaining Proliferative Signalling
SMARCB1 has been shown to regulate the p16INK4a cyclin-dependent kinase (CDK) inhibitor which prevents the activation of the CDK4/6-cyclin D1 complex, responsible for Rb protein phosphorylation and hence the transcription of genes associated with G1/S phase progression [16]. Weissmiller and colleagues found that SNF5 encoded by SMARCB1 inhibits the DNA-binding ability of Myc, impeding its ability to recognise target genes. Further-supporting these findings, the reintroduction of SNF5 into SMARCB1-null cells mimicked the primary transcriptional effects of MYC inhibition [17]. Myc is a helix-loop-helix transcription factor that is one of the best-described drivers of cell proliferation [18]. As a heterodimer with MAX, it regulates the expression of various genes involved in cell-cycle regulation, apoptosis, angiogenesis and cell metabolism [19,20,21]. OmoMYC (OMO-103) is a drug that targets Myc signalling by binding to c- and n-Myc, Max, and Miz1 and was shown to reduce RT cell viability in in-vitro studies [17].
Another implicated oncogene is the epidermal growth factor receptor (EGFR), where a phosphoproteomic study highlighted its abnormal expression in RT and that its inhibition hinders cell proliferation [22]. Overexpression of Aurora kinase A (AURKA) has been described as a result of SMARCB1 deficiency, which plays a key role in mitotic division and cell survival. Its targeting is also being investigated in clinical trials [23]. CAR-T cell targeting of glypican 3 (GPC3), an over-expressed membrane-bound proteoglycan which regulates cell growth and apoptosis [24], is subject to various ongoing clinical trials (NCT05103631, NCT04715191, NCT04377932) [25,26,27].
The use of small molecules aimed at recovering the lost function of TSGs in malignancy poses significant challenges, and many tumour suppressors are considered “undruggable” for this reason [28]. Synthetic lethal relationships therefore provide a promising approach for the targeting of such genetic abnormalities [29]. Radko-Juettner et al in 2024 showed through a genome-wide CRISPR screen that the DDB1-CUL4-associated factor 5 (DCAF5) gene, which functions as substrate receptor for E3-ubiquitin ligase, is required for the survival of SMARCB1-mutated RTs. In addition, depletion of DCAF5 allowed SWI/SNF to reaccumulate and restored histone marks such as H3K27ac and H3K4me1 at active enhancers, normalising cellular differentiation and reversing the cancer phenotype. These results suggest that SMARCB1 mutation leads to the destabilisation of the SWI/SNF complexes, which are subsequently cleared by the ubiquitin quality control pathway, in order to keep malignant cells viable [30]. As such, targeting of DCAF5 may serve as a potential novel therapeutic approach in the treatment of RTs [31].
Tumour-Promoting Inflammation and Avoiding Immune Destruction
The microenvironment of certain subsets of RTs has been shown to be highly inflammatory and to express a high level of genes involved in CD8+ T cell activation, homing, and tumour infiltration such as CXCL10, PRF1, and CLEC9A. Yet, RTs deploy various survival mechanisms that allow them to thrive in such a hostile environment. Immunohistochemistry of RT revealed overexpression of IL-10, programmed death ligand 1 (PD-L1), PD-L1-expressing CD68+ myeloid cells, as well as other immune checkpoint proteins such as HAVCR2 [32]. One study found that in samples of 30 paediatric RTs, 47% stained positive for PD-L1 expression, and therefore may benefit from immune checkpoint inhibitors [33]. It is therefore no surprise that various clinical trials are currently examining the efficacy of a multitude of immune checkpoint inhibitors in the treatment of RTs (Table 1).
Deregulating Cellular Energetics
An emerging metabolic target in SMARCB1-deficient tumours is the cholesterol biosynthetic pathway (Figure 3). Cholesterol plays a key role in the tumoural immune response by regulating the function of various immune cells, including dendritic cells, CD8⁺ T lymphocytes, NK cells, and γδ T cells [34]. One metabolite in the cholesterol biosynthetic pathway, isopentenyl pyrophosphate (IPP), regulates the activity of glutathione peroxidase 4 (GPX4), which inhibits ferroptosis to promote cancer cell survival [35]. In addition, cholesterol contributes to the maintenance of cancer stem cells by activating downstream of several developmental signalling pathways, including Hedgehog and Notch [36]. In a newly established RT cell line it was found that restoration of SMARCB1 significantly downregulated the expression of enzymes such as HMG-CoA reductase (HMGCR) and mevalonate kinase, and that the frequently prescribed cholesterol-lowering drug simvastatin induced apoptosis in RT cells [37]. A phase I study (NCT02390843) of simvastatin in combination with topotecan and cyclophosphamide in relapsed and/or refractory paediatric patients with solid and central nervous system (CNS) tumours including RTs has since been carried out, and found that the combination was well-tolerated [38].
A recent study by Kes and colleagues used patient-derived organoids to investigate the metabolic vulnerabilities of RT [39]. Gene expression analysis and liquid chromatography-mass spectrometry revealed that de novo nucleotide biosynthesis is significantly upregulated in RT, as demonstrated by the increased levels of metabolites such as uridine monophosphate (UMP) and inosine monophosphate (IMP). Pharmacological inhibition (Figure 4) using methotrexate and BAY-2402234, targeting purine and pyrimidine synthesis pathways respectively, resulted in significant cytotoxic effects in RT tumoroids while sparing normal kidney organoids. Further in vivo validation in RT xenograft mouse models demonstrated that methotrexate treatment delayed tumour progression [39]. While the direct link between these metabolic aberrations and SMARCB1 loss has not been confirmed, SMARCB1 loss is likely to be indirectly or directly causal given that it is the defining driver of RTs. Further research into the precise role of SMARCB1 deficiency as a driver of nucleotide synthesis may yield novel therapeutic targets and enhance the understanding of the pathogenesis of the disease.
Non-Mutational Epigenetic Reprogramming
Widespread transcriptional dysregulation and aberrant chromatin-remodelling is known to occur as a result of SMARCB1 loss in RT [40]. In mouse embryonic fibroblasts, SMARCB1 deletion resulted in decreased levels of active histone markers H3K27ac and H3Kme1 at enhancers [41]. The action of the polycomb repressive complex 2 (PRC2), whose catalytic subunit EZH2 contains histone methyltransferase activity, is opposed by the SWI/SNF complex. As a result, SMARCB1 mutation is associated with uncontrolled PRC2-mediated inhibition of BAF target genes. Tazemetostat, an inhibitor of EZH2, has received FDA approval for treatment of SMARCB1-deficient epithelioid sarcoma [42], and trials for other malignancies including RT are ongoing. In the absence of SMARCB1, RT cells upregulate GBAF expression, a SWI/SNF subcomplex which does not contain the SMARCB1 subunit. GBAF’s subunits BRD9 and GLTSCR1 are proposed drug targets, and their degradation or inhibition has been shown to reduce viability of RT cell lines [43]. SMARCB1-deficiency also prevents the recruitment of cBAF and PBAF to the promoter and enhancer regions of SLC7A11, which plays a crucial role in glutathione metabolism by supplying cysteine for glutathione synthesis. Treatment with eprenetapopt has been shown to further-decrease glutathione levels, resulting in oxidative stress and apoptosis [44].
It has recently been demonstrated that in SMARCB1-deficient cells, CBP/p300-mediated H3K27 acetylation promotes the expression of Kringle containing transmembrane protein 2 (KREMEN2) [45], encoding a protein which complexes with KREMEN1 to regulate the Wnt/β catenin pathway [46] and inhibit apoptosis [47]. SMARCB1-deficient cells subsequently become dependent on KREMEN2 for survival. Dual inhibitors of CBP/p300 have been shown to suppress growth and induce apoptosis in cell lines and xenograft models of SMARCB1-deficient cells but not SMARCB1-expressing cells [45]. This serves as another example of synthetic lethal vulnerabilities in SMARCB-1 deficient RTs, which are highlighted in Figure 5.
Ongoing Trials and Future Direction
Immunotherapies
Immune checkpoint proteins (ICPs) are molecules which control the activation of T lymphocytes, a mechanism employed by the body in order to limit the indiscriminate killing of healthy tissues [48]. Various ICPs have been discovered to date, the most notable being PD-1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). PD-1 on T cells binds to PD-L1 expressed on tumour cells and antigen-presenting cells to suppress T-cell proliferation, activity, and cytokine production [49]. CTLA-4 competes with CD28 for the binding of co-stimulatory molecules CD80/86 [50]. A more recently discovered ICP is T cell immunoreceptor with Ig and ITIM domains (TIGIT) which inhibits T and NK cell activation through interaction with CD155 [51].
Immune checkpoint inhibitors (ICIs) have been hailed as exciting anti-cancer therapeutics in the last few decades. Single-agent inhibition of PD-1 or PD-L1 in adults has demonstrated promising activity in various solid tumours. However, in the paediatric population, efficacy with mono-blockade has been limited to just a few tumours, particularly lymphomas [52]. Various ongoing clinical trials are therefore investigating the efficacy of combinations of ICIs in the treatment of SMARCB-1 deficient RTs. A phase II trial (NCT04416568) is currently recruiting patients to investigate the combination of nivolumab (anti-PD1) with ipilimumab (anti-CTLA4) in children and young adults with SMARCB1-deficient tumours, including RTs, epithelioid sarcomas, and chordomas [53]. In mouse models of solid tumours, co-blockade of both PD-L1 and TIGIT has been shown to promote the clonal expansion of multipotent anti-tumoural T lymphocytes [54]. Inhibition of PD-L1 and TIGIT through atezolizumab and tiragolumab respectively is subject to a phase I/II clinical trial for the treatment of relapsed or refractory SMARCB1- or SMARCA4-deficient tumours [55]. Another proposed mechanism of enhancing ICI efficacy is through simultaneous inhibition of EZH2, a histone methyltransferase which has been shown to suppress MHC-I presentation and upregulate PD-L1 [56]. TAZNI (NCT05407441) is a phase I/II study recruiting to evaluate the combination of an EZH2 inhibitor, tazemetostat, with nivolumab and ipilimumab for SMARCA4/SMARCB-1 deficient paediatric malignancies [57].
Chimeric antigen receptor T (CAR-T) cells are genetically engineered T lymphocytes that express synthetic receptors designed to specifically recognise and bind target antigens on tumour cells [58]. This results in the activation of intracellular signalling domains such as CD3ζ, 4-1BB, or CD28 to elicit cancer cytotoxicity [59]. Multiple CAR-T cell strategies are being investigated for RTs (Table 2). CAR-T cells are often targeted against over-expressed tumour antigens, such as GPC3 in the case of solid tumours including RTs. These trials (NCT05103631, NCT04715191, NCT04377932) are investigating the use of IL-15/21 to enhance CAR-T cell efficacy and persistence [25,26,27]. CAR-T cells may also serve as ICIs. For example, several trials (NCT05835687, NCT04897321) are exploring the targeting of B7-H3, a transmembrane immunoregulatory protein, by CAR-T cells for the treatment of solid.

Table 1.
Active clinical trials utilising immune checkpoint inhibitors to target RTs. At the top of the list are the latest trials to be entered to clinicaltrials.gov..
Table 1.
Active clinical trials utilising immune checkpoint inhibitors to target RTs. At the top of the list are the latest trials to be entered to clinicaltrials.gov..
| Trial number | Phase | SMARCB-1 targeting therapies | Disorder | Target | Status | Sites | Primary outcome measures |
|---|---|---|---|---|---|---|---|
| NCT06622941 [60] | II | Nivolumab (ONO-4538) | RT | PD-1 | Not yet recruiting | Osaka and Tokyo, Japan. | Objective response rate |
| NCT05407441 [57] | I/II | Tazemetostat, nivolumab, ipilimumab. | SMARCB1 negative or SMARCA4- deficient tumours including RT | EZH2, PD-1, CTLA-4 | Recruiting | Boston, USA. | Toxicity and dosing parameters |
| NCT05286801 [55] | I/II | Tiragolumab and Atezolizumab | SMARCB1 or SMARCA4 deficient tumours including RT | TIGIT, PD-L1 | Recruiting | USA, Canada, and Australia. | Objective response rate and dose-limiting toxicities |
| NCT04416568 [53] | II | Nivolumab and Ipilimumab | SMARCB1-negative tumours | PD-1, CTLA-4 | Recruiting | Texas, USA. | Objective overall response rate |
Table 2.
Active clinical trials utilising CAR-T cells and cytotoxic lymphocytes against RTs..
| Trial number | Phase | SMARCB-1 targeting therapies | Disorder | Target | Status | Sites | Primary outcome measures |
|---|---|---|---|---|---|---|---|
| NCT06193759 [61] | I | Cytotoxic T lymphocytes directed against proteogenomically determined tumour-specific antigens | Paediatric brain tumours, including RT. | Various tumour-specific antigens | Recruiting | Washington, USA. | Various adverse event and toxicity parameters |
| NCT05835687 [62] | I | Locoregional autologous B7-H3-CAR T cells | Primary CNS neoplasms including RT | B7-H3-positive tumours | Recruiting | Tennessee, USA. | Maximum tolerated dose |
| NCT05103631 [25] | I | GPC3-CART cells and IL-15 | Solid tumours including RT | GPC3-positive tumours | Recruiting | Texas, USA. | Dose-limiting toxicities |
| NCT04897321 [63] | I | Autologous B7-H3-CAR T cells | Solid tumours including RT | B7-H3-positive tumours | Recruiting | Tennessee, USA. | Maximum tolerated dose |
| NCT04715191 [26] | I | GPC3-CART cells and IL-15/21 | Paediatric solid tumours including RT | GPC3-positive tumours | Recruiting | Texas, USA. | Dose-limiting toxicities |
| NCT04377932 [27] | I | GPC3-CART cells and IL-15 | Paediatric solid tumours including RT | GPC3-positive tumours | Recruiting | Texas, USA. | Dose-limiting toxicities |
| NCT04185038 [64] | I | Locoregional autologous B7-H3-CAR T cells | Paediatric CNS tumours including RTs | B7-H3-positive tumours | Recruiting | Washington, USA. | Feasibility and adverse event parameters |
| NCT03618381 [65] | I | EGFR806 CAR T Cell Immunotherapy | Recurrent/refractory solid tumours in children and young adults including RT | EGFR | Recruiting | Washington, USA. | Maximum tolerated dose, feasibility, and adverse event parameters. |
| NCT04483778 [66] | I | B7-H3-CAR T cells | Recurrent/refractory solid tumours in children and young adults including RT | B7-H3-positive tumours | Active, not recruiting | Washington, USA. | Various safety, tolerability, toxicity, and feasibility parameters |
Targeting Epigenetic Aberrations
The role of epigenetics in cancer development and progression has been spotlighted as a key area of interest in recent years. In 2024, an article by Esteller and colleagues proposed the concept of six potentially targetable epigenetic hallmarks of cancer [67]. Epigenetic therapies in cancer aim to modify aberrant epigenetic patterns, such as DNA methylation, histone modifications, and chromatin remodelling [68]. The phase I study of tazemetostat, which targets the PRC2 catalytic subunit EZH2, (NCT02601937) included paediatric patients with relapsed or refractory RTs, other SMARCB1-deficient tumours, and synovial sarcoma [69]. Objective responses were seen in 14% of the dose expansion cohort population, with a 24% response in intracerebral RT patients [70].
Vorinostat is an agent which inhibits the activity of histone deacetylases [71]. A phase I trial (NCT01076530) investigated the combination of vorinostat with temozolomide for the treatment of relapsed/refractory paediatric CNS tumours [72]. 19 patients were recruited including two with rhabdoid tumours. Clinical efficacy was modest, with only three patients exhibiting stable disease and one having a partial response. Nevertheless, the combination was well-tolerated and myelosuppression was identified as the dose-limiting toxicity. It was also noted that accumulation of acetylated H3 histone in peripheral blood mononuclear cells occurred following administration of vorinostat [73]. While clinical utility with epigenetic targeting appears limited thus far, it remains a relatively novel field of cancer research. Future studies focused on identifying predictive biomarkers and exploring synergistic combinations may improve outcomes.
Targeting Auxiliary Oncogenic Aberrations
AURKA plays a pivotal role in mitotic spindle formation and its overexpression has implicated in various malignancies including RTs [74,75]. A phase II clinical trial evaluated alisertib, an AURKA inhibitor, as monotherapy in patients under 22 years with recurrent or progressive atypical teratoid rhabdoid tumours (ATRTs). The study did not meet its primary efficacy endpoint of ≥10 patients progression-free at 12 weeks. However, alisertib was found to be well-tolerated with a 6-month progression-free survival (PFS) of 31%±8.2%. A third of patients also demonstrated disease stabilisation beyond 6 months [76]. Mutations leading to truncated SMARCB1 can result in its exportin-1-mediated mislocalisation in the cytoplasm and hence loss of tumour suppressive capacity. A Phase I trial for paediatric patients with recurrent/refractory solid and CNS tumours established the maximal tolerated dose and recommended a phase II (NCT05985161) starting dose of 35 mg/m² for patients with recurrent/progressive ATRTs [76,77]. The proteins CDK4/6 are crucial for cell cycle progression. A phase I trial of CDK4/6 inhibitor ribociclib in paediatric patients with RTs, neuroblastoma, and other solid tumours showed acceptable safety and prolonged stable disease in a subset of patients [78]. A phase II study is currently recruiting to assess the efficacy of ribociclib with topotecan and temozolomide in paediatric patients with relapsed or refractory neuroblastoma and solid tumours including RTs [79]. Table 3 provides an overview of the active clinical trials targeting aberrations in the cell cycle (CDK4/6) and the dysregulated nuclear export of SMARCB1 (exportin-1). Future efforts should attempt to identify predictive biomarkers to stratify patients most likely to benefit from inhibitors of these auxiliary oncogenic aberrations.
Next Frontiers in Rhabdoid Tumour Research and Treatment
Gene therapy holds promise as a potential approach to treating cancers through the modification or restoration of gene expression [82]. A study published by Kim et al in 2024 explored the use of scL-SMARCB1, a nanomedicine used to deliver wild type SMARCB1 into ATRT cells via transferrin receptor-mediated endocytosis [83]. scL-SMARCB1 therapy was found to upregulate the expression of MYC-repressed genes such as CDKN1C and CDKN2A, while down-regulating MYC-activated genes. AURKA expression was also significantly suppressed. It was also found that transfection with SMARCB1 potentiated cytotoxicity induced by cisplatin and radiation therapy [83]. This highlights how genetic therapy may not only restore biological markers in rhabdoid tumours, but can also enhance their sensitivity to conventional chemoradiation. This may have potential to increase therapeutic efficacy and limit toxicity to healthy tissue.
Artificial intelligence (AI) may serve as a powerful tool for analysing complex genomic data in order to identify new therapeutic indications for drugs. One study has described the application of the Response Algorithm for Drug Positioning and Rescue (RADR®) AI platform, combined with the CellMiner Cross Database, in uncovering novel therapeutic insights for the acylfulvene derivative anti-cancer drugs LP-100 and LP-184 [84]. Gene set enrichment analysis (GSEA) revealed a significant negative correlation between LP-184 sensitivity and SMARCB1 expression. These computational findings were then validated in SMARCB1-deficient ATRT cell lines and through in vivo experiments [84]. This demonstrates the utility of AI-driven platforms in rapidly identifying novel therapeutic indications for drugs. Such approach may especially be useful for rarer cancers such as SMARCB1-deficient rhabdoid malignancies, which tend to have relatively limited specific research focus.
The discussed trials and future innovations highlight how personalised therapies based on genomic, metabolomic, and immunological profiling have the potential to be the mainstay of RT treatment and disease monitoring. Emerging evidence also highlights the importance of metabolic reprogramming in RTs, with pathways such as cholesterol biosynthesis and nucleotide metabolism presenting novel therapeutic vulnerabilities. Integrating metabolomic profiling into future studies may uncover additional biomarkers and enable the development of metabolism-targeted therapies in SMARCB1-deficient tumours. Identifying subsets of patients who are most likely to respond to the various treatments will be crucial. The effects, optimal timing, and integration of combination of SMARCB1 targeted treatments with conventional therapies must be better characterised in order to enhance patient outcomes and allow incorporation into later phase clinical trials and clinical practice. Additional research and genomic analysis is required to further-elucidate the role of SMARCB1 in tumour-suppression and to identify additional molecular targets.
Author Contributions
Abdul L Shakerdi: writing original draft, figure design. Graham P Pidgeon: conceptualisation, writing original draft, correcting drafts.
Conflicts of interest
The authors declare that they have no conflicts of interest regarding the publication of this review.
References
- Pawel, B. R., SMARCB1-deficient Tumors of Childhood: A Practical Guide. Pediatric and Developmental Pathology 2017, 21 (1), 6-28. [CrossRef]
- Cooper, G. W.; Hong, A. L., SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers (Basel) 2022, 14 (15). [CrossRef]
- Gupta, N. K.; Godbole, N.; Sanmugananthan, P.; Gunda, S.; Kasula, V.; Baggett, M.; Gajjar, A.; Kouam, R. W.; D'Amico, R.; Rodgers, S., Management of Atypical Teratoid/Rhabdoid Tumors in the Pediatric Population: A Systematic Review and Meta-Analysis. World Neurosurg 2024, 181, e504-e515. [CrossRef]
- Nemes, K.; Bens, S.; Bourdeaut, F.; Johann, P.; Kordes, U.; Siebert, R.; Frühwald, M. C., Rhabdoid Tumor Predisposition Syndrome. In GeneReviews(®), Adam, M. P.; Feldman, J.; Mirzaa, G. M.; Pagon, R. A.; Wallace, S. E.; Amemiya, A., Eds. University of Washington, Seattle. Copyright © 1993-2025, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA), 1993.
- Sultan, I.; Qaddoumi, I.; Rodríguez-Galindo, C.; Nassan, A. A.; Ghandour, K.; Al-Hussaini, M., Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatric Blood & Cancer 2010, 54 (1), 35-40. [CrossRef]
- Lee, R. S.; Stewart, C.; Carter, S. L.; Ambrogio, L.; Cibulskis, K.; Sougnez, C.; Lawrence, M. S.; Auclair, D.; Mora, J.; Golub, T. R.; Biegel, J. A.; Getz, G.; Roberts, C. W. M., A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. The Journal of Clinical Investigation 2012, 122 (8), 2983-2988. [CrossRef]
- Nemes, K.; Fruhwald, M. C., Emerging therapeutic targets for the treatment of malignant rhabdoid tumors. Expert Opin Ther Targets 2018, 22 (4), 365-379. [CrossRef]
- Kalimuthu, S. N.; Chetty, R., Gene of the month: SMARCB1. J Clin Pathol 2016, 69 (6), 484-9. [CrossRef]
- Roberts, C. W.; Leroux, M. M.; Fleming, M. D.; Orkin, S. H., Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2002, 2 (5), 415-25. [CrossRef]
- Han, Z. Y.; Richer, W.; Freneaux, P.; Chauvin, C.; Lucchesi, C.; Guillemot, D.; Grison, C.; Lequin, D.; Pierron, G.; Masliah-Planchon, J.; Nicolas, A.; Ranchere-Vince, D.; Varlet, P.; Puget, S.; Janoueix-Lerosey, I.; Ayrault, O.; Surdez, D.; Delattre, O.; Bourdeaut, F., The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat Commun 2016, 7, 10421. [CrossRef]
- Pathak, R.; Zin, F.; Thomas, C.; Bens, S.; Gayden, T.; Karamchandani, J.; Dudley, R. W.; Nemes, K.; Johann, P. D.; Oyen, F.; Kordes, U.; Jabado, N.; Siebert, R.; Paulus, W.; Kool, M.; Frühwald, M. C.; Albrecht, S.; Kalpana, G. V.; Hasselblatt, M., Inhibition of nuclear export restores nuclear localization and residual tumor suppressor function of truncated SMARCB1/INI1 protein in a molecular subset of atypical teratoid/rhabdoid tumors. Acta Neuropathol 2021, 142 (2), 361-374. [CrossRef]
- Cooper, G. W.; Hong, A. L., SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14 (15), 3645.
- Hanahan, D.; Weinberg, R. A., Hallmarks of cancer: the next generation. Cell 2011, 144 (5), 646-74. [CrossRef]
- Hanahan, D., Hallmarks of Cancer: New Dimensions. Cancer Discovery 2022, 12 (1), 31-46. [CrossRef]
- Hanahan, D.; Weinberg, R. A., The hallmarks of cancer. Cell 2000, 100 (1), 57-70. [CrossRef]
- Betz, B. L.; Strobeck, M. W.; Reisman, D. N.; Knudsen, E. S.; Weissman, B. E., Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 2002, 21 (34), 5193-203. [CrossRef]
- Weissmiller, A. M.; Wang, J.; Lorey, S. L.; Howard, G. C.; Martinez, E.; Liu, Q.; Tansey, W. P., Inhibition of MYC by the SMARCB1 tumor suppressor. Nat Commun 2019, 10 (1), 2014. [CrossRef]
- García-Gutiérrez, L.; Delgado, M. D.; León, J., MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes (Basel) 2019, 10 (3). [CrossRef]
- Amati, B.; Land, H., Myc—Max—Mad: a transcription factor network controlling cell cycle progression, differentiation and death. Current Opinion in Genetics & Development 1994, 4 (1), 102-108. [CrossRef]
- Baudino, T. A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J. A.; Maclean, K. H.; White, E. L.; Davis, A. C.; Ihle, J. N.; Cleveland, J. L., c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 2002, 16 (19), 2530-43. [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G., Regulation of cancer cell metabolism: oncogenic MYC in the driver’s seat. Signal Transduction and Targeted Therapy 2020, 5 (1), 124. [CrossRef]
- Darr, J.; Klochendler, A.; Isaac, S.; Geiger, T.; Eden, A., Phosphoproteomic analysis reveals Smarcb1 dependent EGFR signaling in Malignant Rhabdoid tumor cells. Mol Cancer 2015, 14, 167. [CrossRef]
- Lee, S.; Cimica, V.; Ramachandra, N.; Zagzag, D.; Kalpana, G. V., Aurora A is a repressed effector target of the chromatin remodeling protein INI1/hSNF5 required for rhabdoid tumor cell survival. Cancer Res 2011, 71 (9), 3225-35. [CrossRef]
- Kohashi, K.; Nakatsura, T.; Kinoshita, Y.; Yamamoto, H.; Yamada, Y.; Tajiri, T.; Taguchi, T.; Iwamoto, Y.; Oda, Y., Glypican 3 expression in tumors with loss of SMARCB1/INI1 protein expression. Hum Pathol 2013, 44 (4), 526-33. [CrossRef]
- Interleukin-15 Armored Glypican-3-specific Chimeric Antigen Receptor Expressing Autologous T Cells As Immunotherapy for Patients with SOLID TUMORS (CATCH). Center for, C.; Gene Therapy, B. C. o. M.; The Methodist Hospital Research, I., Eds. 2021.
- Interleukin-15 and -21 Armored Glypican-3-specific Chimeric Antigen Receptor Expressing Autologous T Cells as an Immunotherapy for Children With Solid Tumors (CARE). Center for, C.; Gene Therapy, B. C. o. M., Eds. 2021.
- Interleukin-15 Armored Glypican-3-specific Chimeric Antigen Receptor Expressing Autologous T Cells as Immunotherapy for Children With Solid Tumors. Center for, C.; Gene Therapy, B. C. o. M., Eds. 2020.
- Gregory, G. L.; Copple, I. M., Modulating the expression of tumor suppressor genes using activating oligonucleotide technologies as a therapeutic approach in cancer. Mol Ther Nucleic Acids 2023, 31, 211-223. [CrossRef]
- Ge, M.; Luo, J.; Wu, Y.; Shen, G.; Kuang, X., The biological essence of synthetic lethality: Bringing new opportunities for cancer therapy. MedComm – Oncology 2024, 3 (1), e70. [CrossRef]
- Radko-Juettner, S.; Yue, H.; Myers, J. A.; Carter, R. D.; Robertson, A. N.; Mittal, P.; Zhu, Z.; Hansen, B. S.; Donovan, K. A.; Hunkeler, M.; Rosikiewicz, W.; Wu, Z.; McReynolds, M. G.; Roy Burman, S. S.; Schmoker, A. M.; Mageed, N.; Brown, S. A.; Mobley, R. J.; Partridge, J. F.; Stewart, E. A.; Pruett-Miller, S. M.; Nabet, B.; Peng, J.; Gray, N. S.; Fischer, E. S.; Roberts, C. W. M., Targeting DCAF5 suppresses SMARCB1-mutant cancer by stabilizing SWI/SNF. Nature 2024, 628 (8007), 442-449. [CrossRef]
- Dedes, K. J.; Wilkerson, P. M.; Wetterskog, D.; Weigelt, B.; Ashworth, A.; Reis-Filho, J. S., Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle 2011, 10 (8), 1192-9. [CrossRef]
- Chun, H. E.; Johann, P. D.; Milne, K.; Zapatka, M.; Buellesbach, A.; Ishaque, N.; Iskar, M.; Erkek, S.; Wei, L.; Tessier-Cloutier, B.; Lever, J.; Titmuss, E.; Topham, J. T.; Bowlby, R.; Chuah, E.; Mungall, K. L.; Ma, Y.; Mungall, A. J.; Moore, R. A.; Taylor, M. D.; Gerhard, D. S.; Jones, S. J. M.; Korshunov, A.; Gessler, M.; Kerl, K.; Hasselblatt, M.; Fruhwald, M. C.; Perlman, E. J.; Nelson, B. H.; Pfister, S. M.; Marra, M. A.; Kool, M., Identification and Analyses of Extra-Cranial and Cranial Rhabdoid Tumor Molecular Subgroups Reveal Tumors with Cytotoxic T Cell Infiltration. Cell Rep 2019, 29 (8), 2338-2354 e7. [CrossRef]
- Forrest, S. J.; Al-Ibraheemi, A.; Doan, D.; Ward, A.; Clinton, C. M.; Putra, J.; Pinches, R. S.; Kadoch, C.; Chi, S. N.; DuBois, S. G.; Leavey, P. J.; LeBoeuf, N. R.; Mullen, E.; Collins, N.; Church, A. J.; Janeway, K. A., Genomic and Immunologic Characterization of INI1-Deficient Pediatric Cancers. Clin Cancer Res 2020, 26 (12), 2882-2890. [CrossRef]
- Pecci, F.; Cognigni, V.; Giudice, G. C.; Paoloni, F.; Cantini, L.; Saini, K. S.; Abushukair, H. M.; Naqash, A. R.; Cortellini, A.; Mazzaschi, G.; Alia, S.; Membrino, V.; Araldi, E.; Tiseo, M.; Buti, S.; Vignini, A.; Berardi, R., Unraveling the link between cholesterol and immune system in cancer: From biological mechanistic insights to clinical evidence. A narrative review. Critical Reviews in Oncology/Hematology 2025, 209, 104654. [CrossRef]
- Ye, L.; Wen, X.; Qin, J.; Zhang, X.; Wang, Y.; Wang, Z.; Zhou, T.; Di, Y.; He, W., Metabolism-regulated ferroptosis in cancer progression and therapy. Cell Death & Disease 2024, 15 (3), 196. [CrossRef]
- Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Li, J.; Xu, H.; Zhao, Y.; Yu, X.; Shi, S., Functional significance of cholesterol metabolism in cancer: from threat to treatment. Experimental & Molecular Medicine 2023, 55 (9), 1982-1995. [CrossRef]
- Matsumoto, F.; Yokogami, K.; Yamada, A.; Moritake, H.; Watanabe, T.; Yamashita, S.; Sato, Y.; Takeshima, H., Targeting cholesterol biosynthesis for AT/RT: comprehensive expression analysis and validation in newly established AT/RT cell line. Hum Cell 2024, 37 (2), 523-530. [CrossRef]
- A Phase 1 Study Using Simvastatin in Combination With Topotecan and Cyclophosphamide in Relapsed and/or Refractory Pediatric Solid and CNS Tumors. Children's Healthcare of, A., Ed. 2015.
- Kes, M. M. G.; Morales-Rodriguez, F.; Zaal, E. A.; de Souza, T.; Proost, N.; van de Ven, M.; van den Heuvel-Eibrink, M. M.; Jansen, J. W. A.; Berkers, C. R.; Drost, J., Metabolic profiling of patient-derived organoids reveals nucleotide synthesis as a metabolic vulnerability in malignant rhabdoid tumors. Cell Reports Medicine 2025, 6 (1), 101878. [CrossRef]
- Kenny, C.; O'Meara, E.; Ulas, M.; Hokamp, K.; O'Sullivan, M. J., Global Chromatin Changes Resulting from Single-Gene Inactivation-The Role of SMARCB1 in Malignant Rhabdoid Tumor. Cancers (Basel) 2021, 13 (11). [CrossRef]
- Wang, X.; Lee, R. S.; Alver, B. H.; Haswell, J. R.; Wang, S.; Mieczkowski, J.; Drier, Y.; Gillespie, S. M.; Archer, T. C.; Wu, J. N.; Tzvetkov, E. P.; Troisi, E. C.; Pomeroy, S. L.; Biegel, J. A.; Tolstorukov, M. Y.; Bernstein, B. E.; Park, P. J.; Roberts, C. W., SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet 2017, 49 (2), 289-295. [CrossRef]
- Hoy, S. M., Tazemetostat: First Approval. Drugs 2020, 80 (5), 513-521. [CrossRef]
- Wang, X.; Wang, S.; Troisi, E. C.; Howard, T. P.; Haswell, J. R.; Wolf, B. K.; Hawk, W. H.; Ramos, P.; Oberlick, E. M.; Tzvetkov, E. P.; Ross, A.; Vazquez, F.; Hahn, W. C.; Park, P. J.; Roberts, C. W. M., BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat Commun 2019, 10 (1), 1881. [CrossRef]
- Sasaki, M.; Ogiwara, H., Efficacy of glutathione inhibitor eprenetapopt against the vulnerability of glutathione metabolism in SMARCA4-, SMARCB1- and PBRM1-deficient cancer cells. Scientific Reports 2024, 14 (1), 31321. [CrossRef]
- Sasaki, M.; Kato, D.; Murakami, K.; Yoshida, H.; Takase, S.; Otsubo, T.; Ogiwara, H., Targeting dependency on a paralog pair of CBP/p300 against de-repression of KREMEN2 in SMARCB1-deficient cancers. Nature Communications 2024, 15 (1), 4770. [CrossRef]
- Mao, B.; Wu, W.; Davidson, G.; Marhold, J.; Li, M.; Mechler, B. M.; Delius, H.; Hoppe, D.; Stannek, P.; Walter, C.; Glinka, A.; Niehrs, C., Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature 2002, 417 (6889), 664-7. [CrossRef]
- Sumia, I.; Pierani, A.; Causeret, F., Kremen1-induced cell death is regulated by homo- and heterodimerization. Cell Death Discov 2019, 5, 91. [CrossRef]
- Marhelava, K.; Pilch, Z.; Bajor, M.; Graczyk-Jarzynka, A.; Zagozdzon, R., Targeting Negative and Positive Immune Checkpoints with Monoclonal Antibodies in Therapy of Cancer. Cancers (Basel) 2019, 11 (11). [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y., PD-1 and PD-L1 in cancer immunotherapy: clinical implications and future considerations. Hum Vaccin Immunother 2019, 15 (5), 1111-1122. [CrossRef]
- Liu, P. C.; Ssu, C. T.; Tsao, Y. P.; Liou, T. L.; Tsai, C. Y.; Chou, C. T.; Chen, M. H.; Leu, C. M., Cytotoxic T lymphocyte-associated antigen-4-Ig (CTLA-4-Ig) suppresses Staphylococcus aureus-induced CD80, CD86, and pro-inflammatory cytokine expression in human B cells. Arthritis Res Ther 2020, 22 (1), 64. [CrossRef]
- Bolm, L.; Petruch, N.; Sivakumar, S.; Annels, N. E.; Frampton, A. E., Gene of the month: T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT). J Clin Pathol 2022, 75 (4), 217-221. [CrossRef]
- Davis, K. L.; Fox, E.; Isikwei, E.; Reid, J. M.; Liu, X.; Minard, C. G.; Voss, S.; Berg, S. L.; Weigel, B. J.; Mackall, C. L., A Phase I/II Trial of Nivolumab plus Ipilimumab in Children and Young Adults with Relapsed/Refractory Solid Tumors: A Children's Oncology Group Study ADVL1412. Clin Cancer Res 2022, 28 (23), 5088-5097. [CrossRef]
- Phase 2 Proof of Concept Study of Nivolumab and Ipilimumab in Children and Young Adults With Relapsed or Refractory INI1-negative Cancers. Gateway for Cancer, R., Ed. 2020.
- Nutsch, K.; Banta, K. L.; Wu, T. D.; Tran, C. W.; Mittman, S.; Duong, E.; Nabet, B. Y.; Qu, Y.; Williams, K.; Muller, S.; Patil, N. S.; Chiang, E. Y.; Mellman, I., TIGIT and PD-L1 co-blockade promotes clonal expansion of multipotent, non-exhausted antitumor T cells by facilitating co-stimulation. Nat Cancer 2024, 5 (12), 1834-1851. [CrossRef]
- A Phase 1/2 Study of Tiragolumab (NSC# 827799) and Atezolizumab (NSC# 783608) in Patients With Relapsed or Refractory SMARCB1 or SMARCA4 Deficient Tumors. 2022.
- Kang, N.; Eccleston, M.; Clermont, P. L.; Latarani, M.; Male, D. K.; Wang, Y.; Crea, F., EZH2 inhibition: a promising strategy to prevent cancer immune editing. Epigenomics 2020, 12 (16), 1457-1476. [CrossRef]
- TAZNI: a Phase I/II Combination Trial of Tazemetostat with Nivolumab and Ipilimumab for Children with INI1-Negative or SMARCA4-Deficient Tumors. Bristol-Myers, S.; Epizyme, I., Eds. 2022.
- Sterner, R. C.; Sterner, R. M., CAR-T cell therapy: current limitations and potential strategies. Blood Cancer Journal 2021, 11 (4), 69. [CrossRef]
- Dabas, P.; Danda, A., Revolutionizing cancer treatment: a comprehensive review of CAR-T cell therapy. Med Oncol 2023, 40 (9), 275. [CrossRef]
- A Multicenter, Open-label, Uncontrolled Phase II Study to Investigate Efficacy and Safety of ONO4538 in Patients With Rhabdoid Tumor. 2024.
- Immunotherapy for Malignant Pediatric Brain Tumors Employing Adoptive Cellular Therapy (IMPACT). 2023.
- Loc3CAR: Locoregional Delivery of B7-H3-specific Chimeric Antigen Receptor Autologous T Cells for Pediatric Patients With Primary CNS Tumors. 2023.
- B7-H3-Specific Chimeric Antigen Receptor Autologous T-Cell Therapy for Pediatric Patients With Solid Tumors (3CAR). 2021.
- Phase 1 Study of B7-H3-Specific CAR T Cell Locoregional Immunotherapy for Diffuse Intrinsic Pontine Glioma/Diffuse Midline Glioma and Recurrent or Refractory Pediatric Central Nervous System Tumors. 2019.
- Phase I Study of EGFR806 CAR T Cell Immunotherapy for Recurrent/Refractory Solid Tumors in Children and Young Adults. 2018.
- Phase I Study of B7H3 CAR T Cell Immunotherapy for Recurrent/Refractory Solid Tumors in Children and Young Adults. 2020.
- Esteller, M.; Dawson, M. A.; Kadoch, C.; Rassool, F. V.; Jones, P. A.; Baylin, S. B., The Epigenetic Hallmarks of Cancer. Cancer Discov 2024, 14 (10), 1783-1809. [CrossRef]
- Sadida, H. Q.; Abdulla, A.; Marzooqi, S. A.; Hashem, S.; Macha, M. A.; Akil, A. S. A.; Bhat, A. A., Epigenetic modifications: Key players in cancer heterogeneity and drug resistance. Transl Oncol 2024, 39, 101821. [CrossRef]
- A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma. 2015.
- Chi, S. N.; Bourdeaut, F.; Casanova, M.; Kilburn, L. B.; Hargrave, D. R.; McCowage, G. B.; Pinto, N. R.; Yang, J.; Chadha, R.; Kahali, B.; Tapia, C.; Nysom, K., Update on phase 1 study of tazemetostat, an enhancer of zeste homolog 2 inhibitor, in pediatric patients with relapsed or refractory integrase interactor 1–negative tumors. Journal of Clinical Oncology 40 (16_suppl), 10040-10040. [CrossRef]
- Ge, Z.; Da, Y.; Xue, Z.; Zhang, K.; Zhuang, H.; Peng, M.; Li, Y.; Li, W.; Simard, A.; Hao, J.; Yao, Z.; Zhang, R., Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp Neurol 2013, 241, 56-66. [CrossRef]
- A Phase I Study of SAHA and Temozolomide in Children With Relapsed or Refractory Primary Brain or Spinal Cord Tumors. 2010.
- Hummel, T. R.; Wagner, L.; Ahern, C.; Fouladi, M.; Reid, J. M.; McGovern, R. M.; Ames, M. M.; Gilbertson, R. J.; Horton, T.; Ingle, A. M.; Weigel, B.; Blaney, S. M., A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: a Children's Oncology Group phase 1 consortium study. Pediatr Blood Cancer 2013, 60 (9), 1452-7. [CrossRef]
- Nemes, K.; Johann, P. D.; Tüchert, S.; Melchior, P.; Vokuhl, C.; Siebert, R.; Furtwängler, R.; Frühwald, M. C., Current and Emerging Therapeutic Approaches for Extracranial Malignant Rhabdoid Tumors. Cancer Manag Res 2022, 14, 479-498. [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z., Targeting AURKA in Cancer: molecular mechanisms and opportunities for Cancer therapy. Molecular Cancer 2021, 20 (1), 15. [CrossRef]
- Upadhyaya, S.; Campagne, O.; Robinson, G. W.; Onar-Thomas, A.; Orr, B.; Billups, C. A.; Tatevossian, R. G.; Broniscer, A.; Kilburn, L. B.; Baxter, P. A.; Smith, A. A.; Crawford, J.; Partap, S.; Ellison, D. W.; Stewart, C. F.; Patay, Z.; Gajjar, A. J., Phase II study of alisertib as a single agent in recurrent or progressive atypical teratoid rhabdoid tumors. Journal of Clinical Oncology 38 (15_suppl), 10542-10542. [CrossRef]
- Green, A. L.; Minard, C. G.; Liu, X.; Safgren, S. L.; Pinkney, K.; Harris, L.; Link, G.; DeSisto, J.; Voss, S.; Nelson, M. D.; Reid, J. M.; Fox, E.; Weigel, B. J.; Glade Bender, J., Phase 1 trial of selinexor in pediatric recurrent/refractory solid and CNS tumors (ADVL1414): A Children's Oncology Group Phase 1 Consortium Trial. Clin Cancer Res 2025. [CrossRef]
- Geoerger, B.; Bourdeaut, F.; DuBois, S. G.; Fischer, M.; Geller, J. I.; Gottardo, N. G.; Marabelle, A.; Pearson, A. D. J.; Modak, S.; Cash, T.; Robinson, G. W.; Motta, M.; Matano, A.; Bhansali, S. G.; Dobson, J. R.; Parasuraman, S.; Chi, S. N., A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors. Clin Cancer Res 2017, 23 (10), 2433-2441. [CrossRef]
- Phase I/II Multicenter Study to Assess Efficacy and Safety of Ribociclib (LEE011) in Combination With Topotecan and Temozolomide (TOTEM) in Pediatric Patients With Relapsed or Refractory Neuroblastoma and Other Solid Tumors. Innovative Therapies For Children with Cancer, C., Ed. 2022.
- A Multi-Center Phase II Study of Selinexor in Treating Recurrent or Refractory Wilms Tumor and Other Pediatric Solid Tumors. 2023.
- PHASE 1/2 STUDY TO EVALUATE PALBOCICLIB (IBRANCE®) IN COMBINATION WITH IRINOTECAN AND TEMOZOLOMIDE OR IN COMBINATION WITH TOPOTECAN AND CYCLOPHOSPHAMIDE IN PEDIATRIC PATIENTS WITH RECURRENT OR REFRACTORY SOLID TUMORS. Children's Oncology, G., Ed. 2018.
- Cesur-Ergün, B.; Demir-Dora, D., Gene therapy in cancer. J Gene Med 2023, 25 (11), e3550. [CrossRef]
- Kim, S. S.; Moghe, M.; Rait, A.; Donaldson, K.; Harford, J. B.; Chang, E. H., SMARCB1 Gene Therapy Using a Novel Tumor-Targeted Nanomedicine Enhances Anti-Cancer Efficacy in a Mouse Model of Atypical Teratoid Rhabdoid Tumors. Int J Nanomedicine 2024, 19, 5973-5993. [CrossRef]
- McDermott, J.; Sturtevant, D.; Kathad, U.; Varma, S.; Zhou, J.; Kulkarni, A.; Biyani, N.; Schimke, C.; Reinhold, W. C.; Elloumi, F.; Carr, P.; Pommier, Y.; Bhatia, K., Artificial intelligence platform, RADR®, aids in the discovery of DNA damaging agent for the ultra-rare cancer Atypical Teratoid Rhabdoid Tumors. Frontiers in Drug Discovery 2022, 2. [CrossRef]
Figure 1.
Aberrant pathways induced by SMARCB1 deficiency and corresponding therapeutic targets. ‘PRC2’ denotes polycomb repressive complex 2, ‘AURKA’ Aurora Kinase A, ‘EGFR’ epidermal growth factor receptor, ‘BRD9’ bromodomain-containing protein 9, ‘GLTSCR1’ glioma tumour suppressor candidate region gene 1, ‘Rb’ retinoblastoma, ‘CDK4/6’ cyclin-dependent kinase 4/6, ‘HMGCR’ HMG-CoA reductase. Figure created using BioRender.
Figure 1.
Aberrant pathways induced by SMARCB1 deficiency and corresponding therapeutic targets. ‘PRC2’ denotes polycomb repressive complex 2, ‘AURKA’ Aurora Kinase A, ‘EGFR’ epidermal growth factor receptor, ‘BRD9’ bromodomain-containing protein 9, ‘GLTSCR1’ glioma tumour suppressor candidate region gene 1, ‘Rb’ retinoblastoma, ‘CDK4/6’ cyclin-dependent kinase 4/6, ‘HMGCR’ HMG-CoA reductase. Figure created using BioRender.
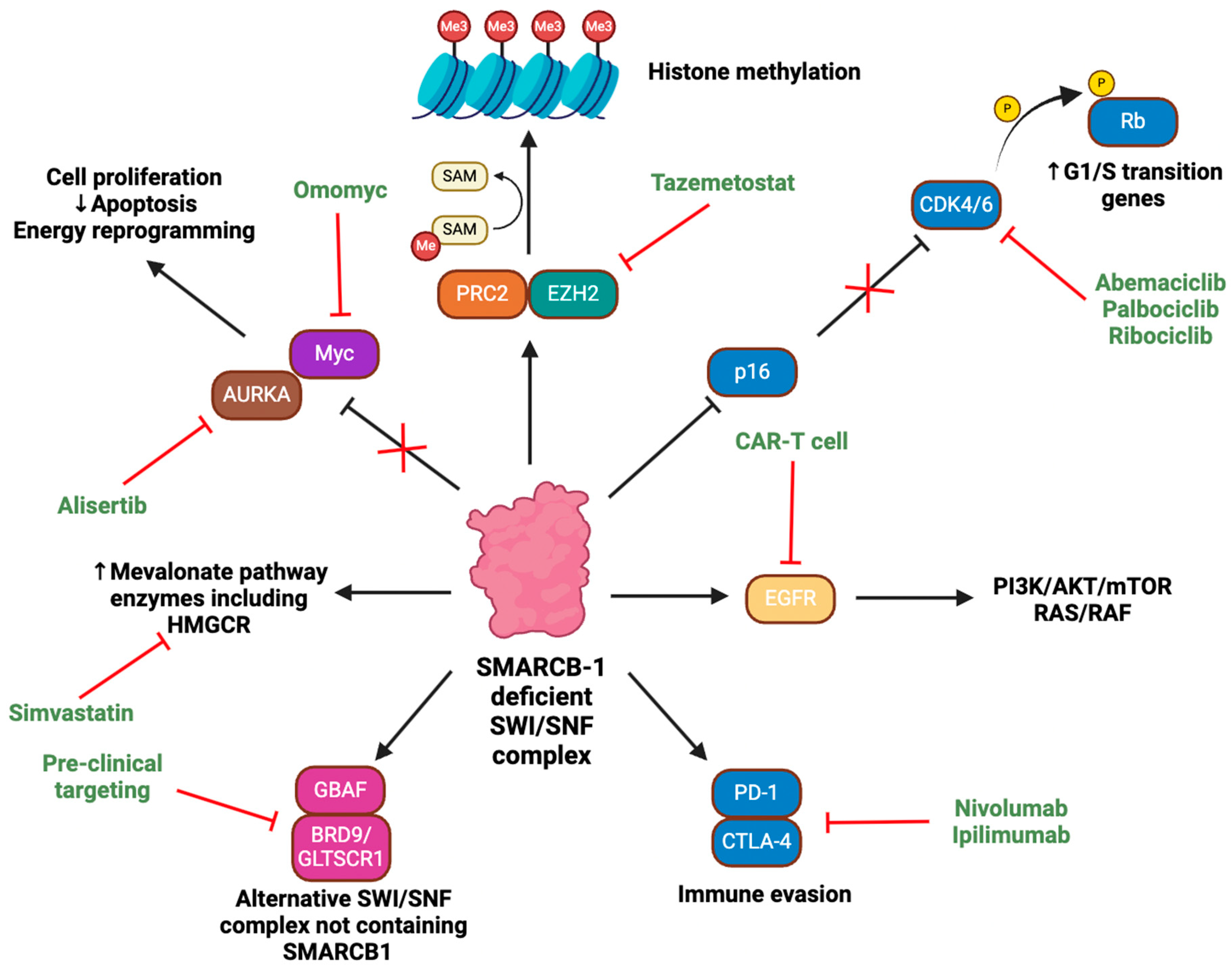
Figure 2.
A summary of the role of SMARCB1 deficiency as a driver of the hallmarks of cancer in RT. The upregulation of nucleotide synthesis has yet to be confirmed as a causal outcome of SMARCB1 deficiency. Figure created using BioRender.
Figure 2.
A summary of the role of SMARCB1 deficiency as a driver of the hallmarks of cancer in RT. The upregulation of nucleotide synthesis has yet to be confirmed as a causal outcome of SMARCB1 deficiency. Figure created using BioRender.
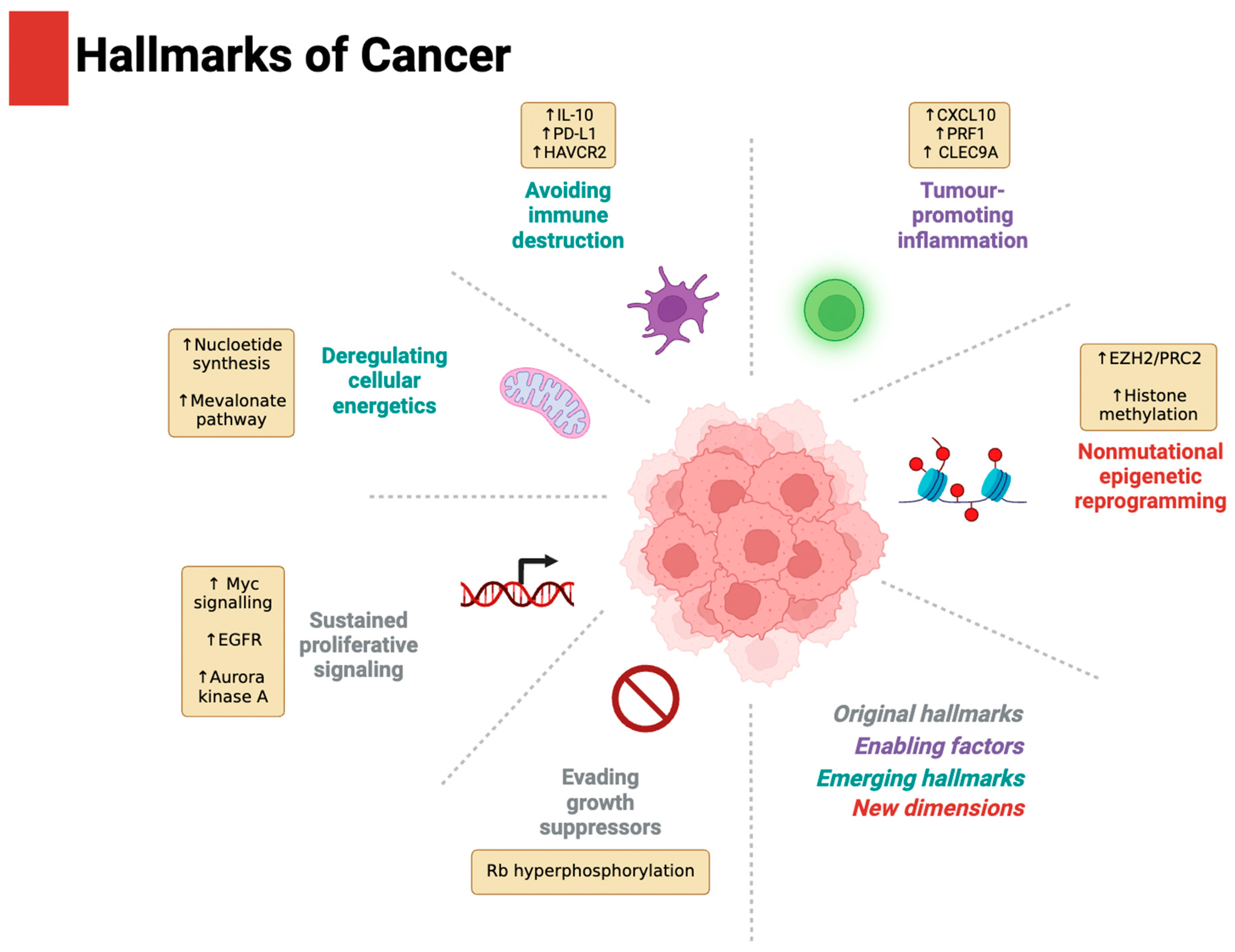
Figure 3.
The mevalonate pathway, highlighting the inhibition of HMG-CoA reductase using simvastatin. ‘HMG-CoA’ denotes β-Hydroxy β-methylglutaryl-CoA. Figure created using BioRender.
Figure 3.
The mevalonate pathway, highlighting the inhibition of HMG-CoA reductase using simvastatin. ‘HMG-CoA’ denotes β-Hydroxy β-methylglutaryl-CoA. Figure created using BioRender.
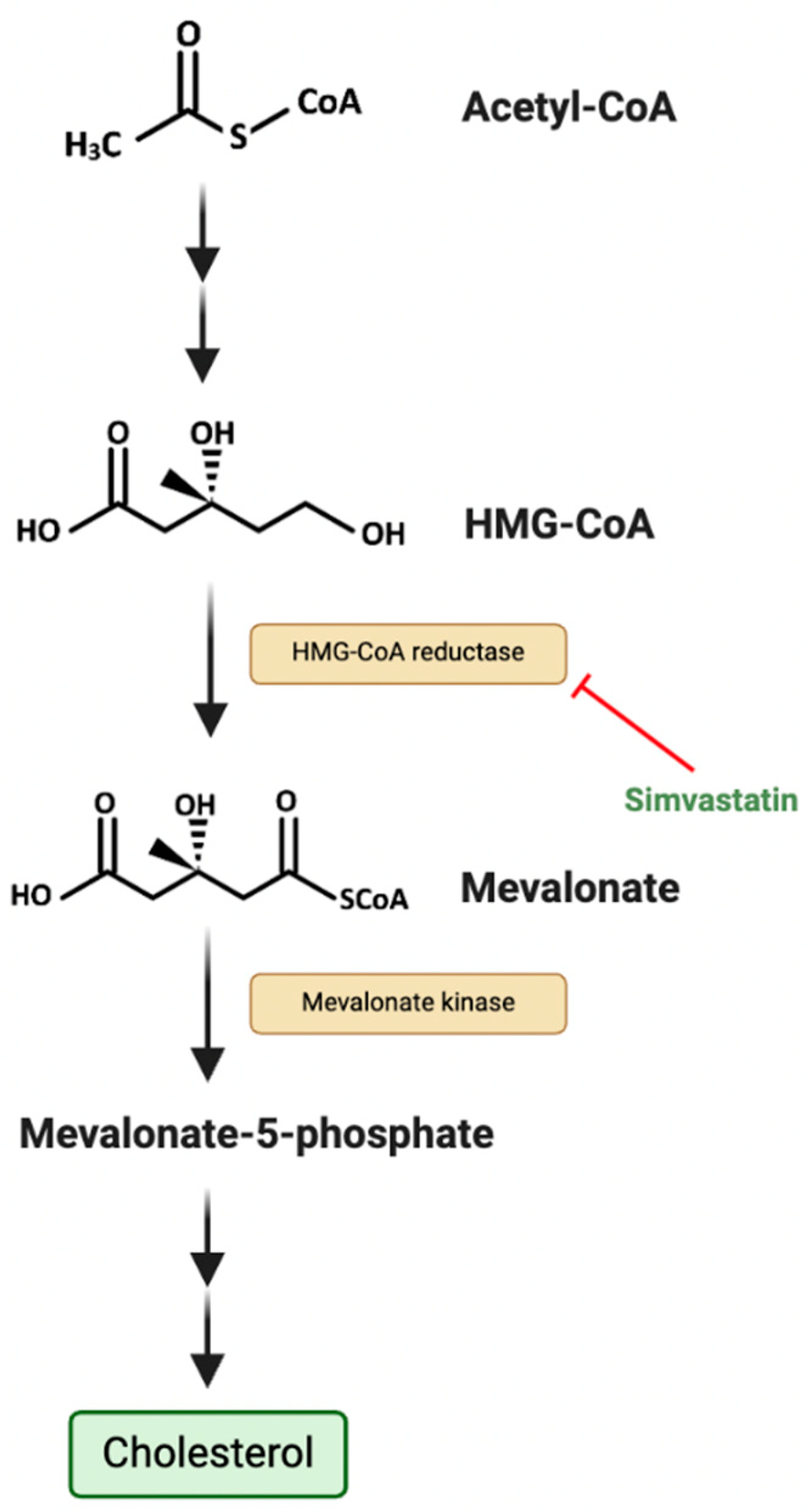
Figure 4.
The nucleotide biosynthetic pathway as a potential metabolic vulnerability in RT. ‘AMP’ denotes adenosine monophosphate, ‘GMP’ guanine monophosphate, ‘UMP’ uridine monophosphate, ‘CTP’ cytidine triphosphate. Figure created using BioRender.
Figure 4.
The nucleotide biosynthetic pathway as a potential metabolic vulnerability in RT. ‘AMP’ denotes adenosine monophosphate, ‘GMP’ guanine monophosphate, ‘UMP’ uridine monophosphate, ‘CTP’ cytidine triphosphate. Figure created using BioRender.
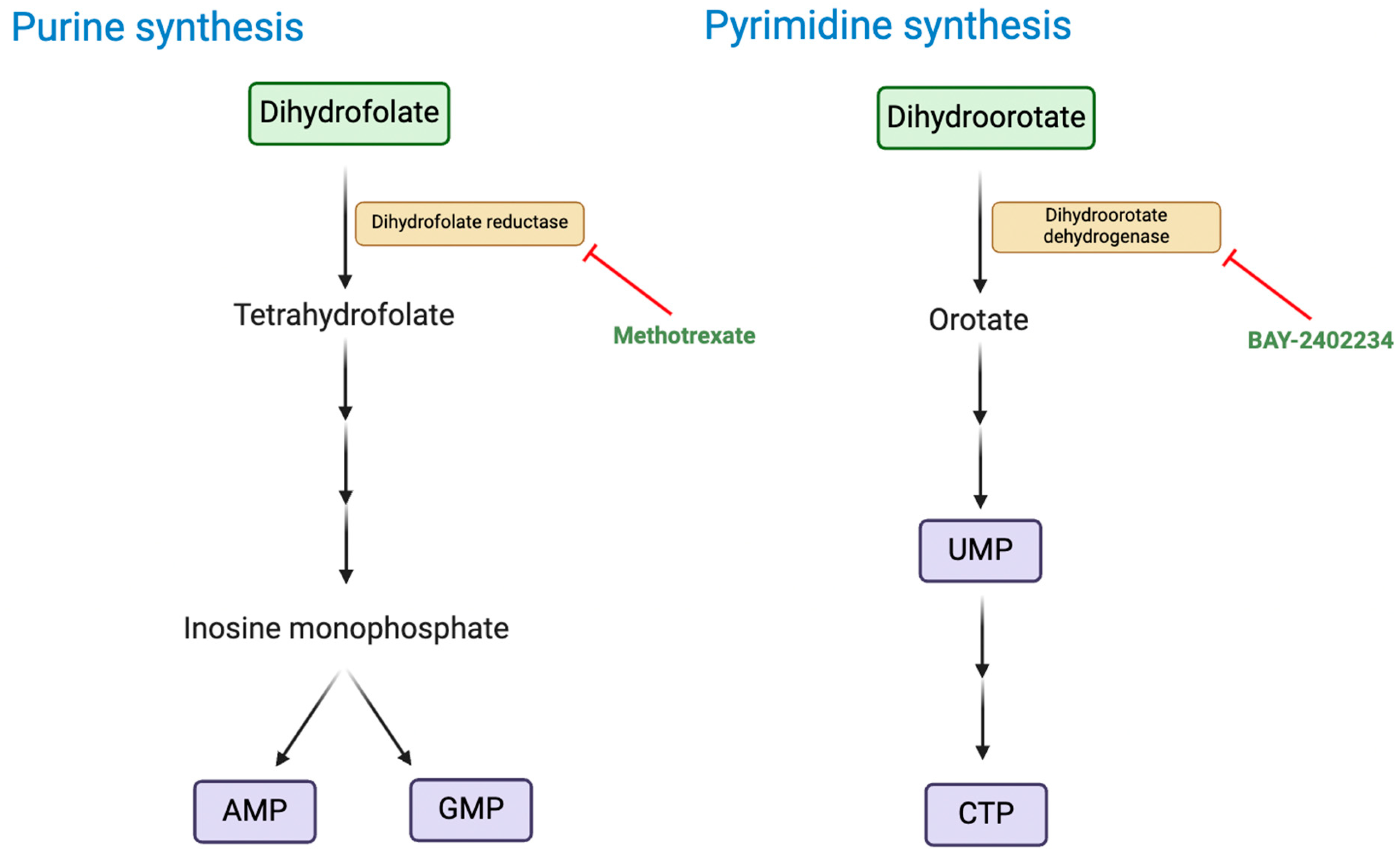
Figure 5.
Synthetic lethal vulnerabilities in RTs. In SMARCB1-deficient cells, SMARCB1-deficient SWI/SNF complexes are targeted for ubiquitination and proteasomal degradation by the DCAF5/CUL4 E3 ligase complex. Inhibition of DCAF5 stabilises these defective SWI/SNF complexes, leading to toxic accumulation and loss of cell viability. In addition, CBP/p300-mediated H3K27 acetylation upregulates KREMEN2 expression, which suppresses apoptosis and promotes cell survival. Inhibition of CBP/p300 reduces KREMEN2 levels, promoting apoptosis. Figure created using BioRender.
Figure 5.
Synthetic lethal vulnerabilities in RTs. In SMARCB1-deficient cells, SMARCB1-deficient SWI/SNF complexes are targeted for ubiquitination and proteasomal degradation by the DCAF5/CUL4 E3 ligase complex. Inhibition of DCAF5 stabilises these defective SWI/SNF complexes, leading to toxic accumulation and loss of cell viability. In addition, CBP/p300-mediated H3K27 acetylation upregulates KREMEN2 expression, which suppresses apoptosis and promotes cell survival. Inhibition of CBP/p300 reduces KREMEN2 levels, promoting apoptosis. Figure created using BioRender.
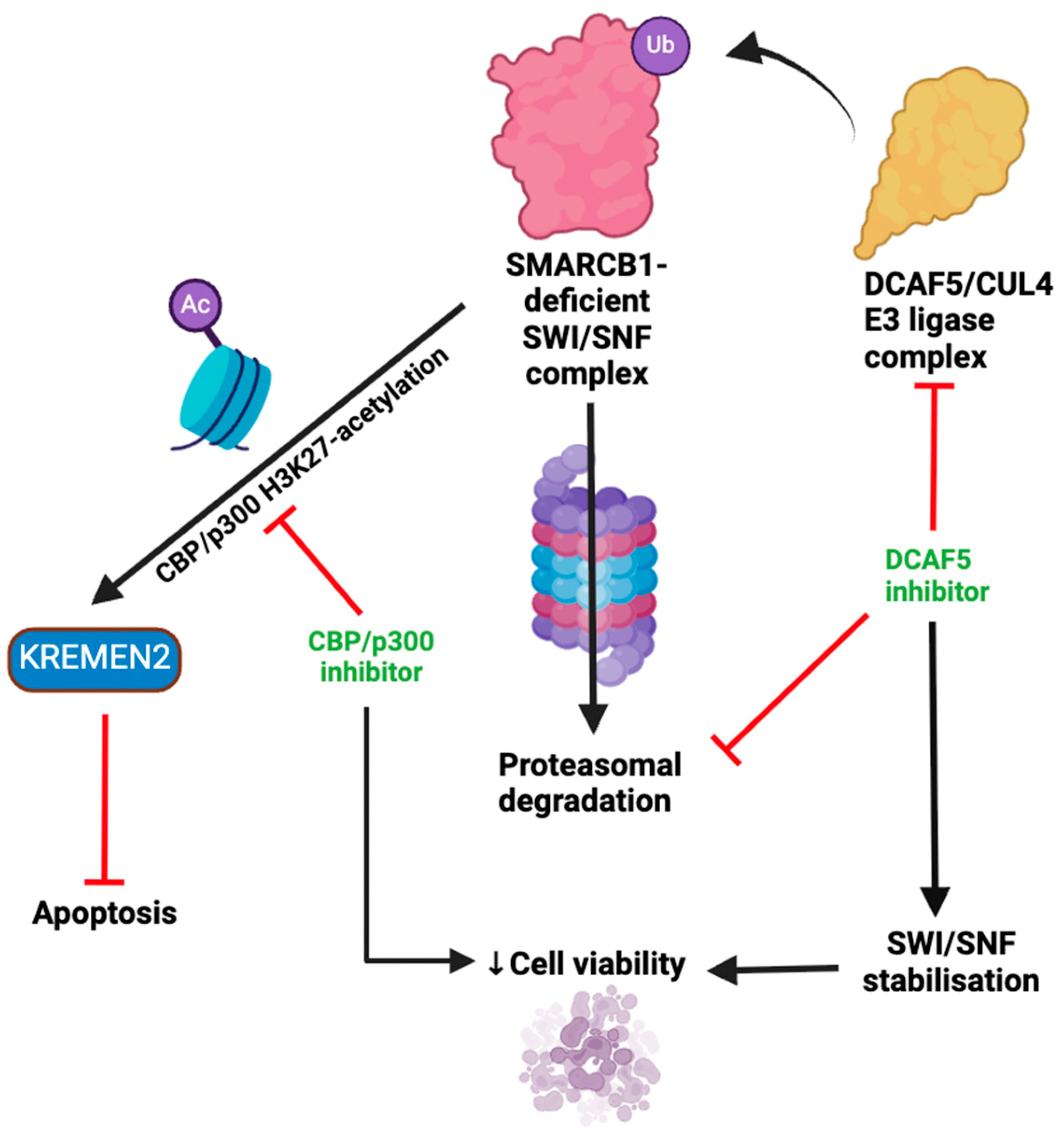
Table 3.
Clinical trials targeting the cell cycle checkpoint control, the dysregulated nuclear export of SMARCB1, and Aurora kinase A.
Table 3.
Clinical trials targeting the cell cycle checkpoint control, the dysregulated nuclear export of SMARCB1, and Aurora kinase A.
| Trial number | Phase | SMARCB-1 targeting therapies | Disorder | Target | Status | Sites | Primary outcome measures |
|---|---|---|---|---|---|---|---|
| NCT05985161 [80] | II | Selinexor | Solid tumours including RT | Exportin-1 | Recruiting | Various USA sites. | Complete and partial response |
| NCT05429502 [79] | I/II | Ribociclib | Solid tumours including RT | CDK4/6 | Recruiting | USA, UK, Spain, Singapore, Italy, Germany, France, and Australia. | Overall response rate and dose-limiting toxicities |
| NCT03709680 [81] | I/II | Palbociclib | Recurrent/refractory solid tumours including RT | CDK4/6 | Active, not recruiting | > 10 countries including USA, UK, Brazil, and Korea. | Event-free survival, first cycle dose-limiting toxicities, frequency of adverse events, complete response or partial response |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



