
Submitted:
02 April 2025
Posted:
02 April 2025
You are already at the latest version
Abstract
Background/Objectives: Bluetongue virus (BTV) is an emerging arbovirus causing significant economic losses in the ruminant industry. Current vaccines offer limited cross-protection against heterologous serotypes and do not enable differentiation between infected and vaccinated animals (DIVA). Subunit-based vaccines provide a potential DIVA-compatible solution. This study aimed to develop a vaccination protocol expressing BTV structural proteins VP7 or VP2 using antibiotic-resistance-free DNA plasmids and replication-defective adenovirus vectors. Methods: We evaluated homologous DNA prime-boost and heterologous DNA prime-adenovirus boost strategies in a murine model, assessing adaptive immune responses and protection against virulent BTV challenge. Results: The heterologous DNA-adenovirus prime-boost strategy expressing both antigens conferred full protection, preventing viremia, while homologous DNA-DNA prime-boost provided only partial protection. Both VP7 and VP2 elicited cellular and humoral immune responses, but the heterologous strategy significantly enhanced anti-BTV IgG, neutralizing antibody titers, and T cell activation. CD8+ T cell responses showed the strongest correlation with viral load reduction, suggesting that immunity to VP7 could offer cross-protection against multiple BTV serotypes. Conclusion: These findings highlight the potential of heterologous DNA-adenovirus vaccination as an effective DIVA-compatible strategy for BTV control. By inducing strong and protective immune responses, this approach could improve disease surveillance and management, ultimately reducing the impact of BTV on livestock industries.
Keywords:
Orbivirus
; T cell response
; neutralization
; DNA vaccine
; adenovirus vaccine
; protection
1. Introduction
Bluetongue virus (BTV) produces a debilitating ruminant disease of mandatory notification to the world organization for animal health (WOAH) that leads to significant economic losses through loss of productivity. The disease is characterized by pyrexia, apathy, loss of appetite, and edemas of the face and lips. In the most severe cases, it generates respiratory distress and hemorrhages that can result in the characteristic cyanotic tongue that gives its name to the disease, and even lead to the death of animals [1]. It can also produce abortions in pregnant ewes, which further contribute to the loss of productivity [2]. An estimated 3 billion dollars are yearly lost to the industry as a result of BTV infections [3]. Typically sheep are more prone to develop severe clinical signs than other ruminants but some serotypes can also severely affect cattle, such as in the case of the 2006 BTV-8 outbreak in Europe [4].
BTV is the prototypical member of the genus Orbivirus, family Sedoreoviridae, that includes other relevant pathogens in animal health such as african horse sickness virus (AHSV) or epizootic hemorrhagic disease virus (EHDV) [5]. It possesses a genome that consists of 10 segments that encodes for 7 structural proteins (named VP1 to VP7) and 4-5 non-structural proteins (named NS1 to NS5) [6,7]. BTV is principally transmitted by the bite of infected Culicoides spp. to the mammalian host [2,8]. Viremia is characteristically long in BTV infection, which probably facilitates the transmission of the virus back to the Culicoides vector [9,10]. Global warming, which has increased the habitat range of traditional competent vectors, as well as the likely adaptation of the virus to Culicoides species present in more temperate regions has led to the establishment of BTV in Europe [11]. To further complicate disease control, BTV also possesses multiple serotypes (at least 29 have been described to date) that offer little heterologous cross-serotype protection. Although current inactivated vaccines are effective for homologous serotype protection, they only offer little protection against a different serotype. These vaccines cannot differentiate between infected and vaccinated animals, the so-called DIVA approach. Vaccines based on the expression of BTV subunits have the potential to be DIVA vaccines, as they only express parts of the virus. Much effort has been made to develop these subunit vaccines against BTV, either through the expression of recombinant BTV proteins or by expressing the coding sequences of these subunits in recombinant viral vaccine platforms [12]. Some of these approaches have also shown some promising protection results as multiserotype vaccines [13,14,15]. This is highly sought after in the field, as production of a single vaccine formulation could be sufficient to protect against BTV in regions in which multiple serotypes circulate.
Protection against BTV infections necessitates of a humoral and a cellular response. Early depletion and transfer studies demonstrated that solely humoral or cellular responses only offered partial protection against infection [16,17]. Vaccination should therefore aim at eliciting both arms of the adaptive immune response. Cellular immunity is particularly relevant in the case of BTV infections as it can be targeted by subunit vaccines to conserved antigens between serotypes such as VP7 and NS1 [18,19]. Indeed, we have found that expressing VP7 in a recombinant replication-defective adenovirus platform could confer BTV protection in sheep in the absence of neutralizing antibody [20], while cellular responses to an NS1 epitope can also be protective in a murine model [21]. In most cases, cellular protection is unlikely to confer sterile protection against BTV, and potent humoral antigens such as VP2, which contains the majority of neutralizing determinants, should be included in vaccines.
In the present work, we evaluated the immunogenicity and protective potential of a DNA plasmid platform (pPAL) that lacks resistance gene for selection and that the European Medicines Agency recently approved for leishmania vaccination in dogs [22]. Expression of SARS-Cov2 spike and nucleoprotein in this DNA platform was also an effective vaccine in animal models against virulent challenge with SARS-Cov2 [23]. As a proof of principle, we chose to express the highly conserved antigen VP7 and the main determinants for neutralizing antibodies VP2 from BTV-8 in this DNA expression system. We used a prime-boost regime either with two inoculations of the pPAL plasmids, or with a pPAL plasmid priming followed by a recombinant replication-defective adenovirus booster that encoded the same viral antigens. We evaluated the responses to vaccination and virulent BTV-8 challenge using only VP7 or VP2 as antigens, or with a combination of both antigens.
2. Material and Methods
2.1. Ethical Statement
All the animal experiments were carried out in a disease-secure isolation facility (BSL3) at the Centro de Investigación en Sanidad Animal (CISA), in strict accordance with the recommendations in the guidelines of the Code for Methods and Welfare Considerations in Behavioral Research with Animals (Directive 86/609EC; RD1201/2005), and all efforts were made to minimize suffering. Experiments were approved by the Committee on the Ethics of Consejo Superior de Investigaciones Científicas (CSIC) and the National Animal Welfare Committee (PROEX 295.6/21).
2.2. Cell Lines and Viruses
HEK293 (ATCC CRL-1573) cells were grown as described in [24]. BTV-8 (NET2006/04) was grown in BHK-21 cells (ATCC CCL-10) and tittered in Vero cells (ATCC CCL-81) by standard plaque assays as described in [18,25,26]. The construction, preparation and purification of the recombinant replication-defective human adenovirus 5 (Ad) AdVP7, AdVP2 (both antigens from BTV-8), and AdDsRed (control adenovirus that encodes a red fluorescent protein) used in the present work has been described in [20]. BTV-8 was inactivated (iBTV) with binary ethylenimine (BEI) as described in [18,25].
2.3. Generation of pPAL-VP7 and VP2 Plasmids
BTV-8 VP7 (KU569990 to KU569999) and VP2 (AB037932.1) sequences were optimized by the Monte Carlo approach according to relative codon usage frequencies and cloned into the pPAL expression plasmid that lacks an antibiotic resistance gene as described [23]. Plasmid DNA (pPAL-empty, pPAL-VP7 and pPAL-VP2) were obtained by endotoxin-free giga plasmid, (Qiagen, Hilden, Germany).
2.4. Immunofluorescence Study of VP7 and VP2 Expression
HEK293 cells were seeded in 12 well plates containing coverslips and subsequently transfected using transIT-LT1 reagent (Mirus Bio). After 48h, cells were fixed using 4% paraformaldehyde for 20 min, washed 3 times with PBS and used for immunofluorescence studies. Cells were permeabilized with PBS+ 0.1% (v/v) triton X-100 (Sigma) for 10 min at room temperature. Preparations were blocked for 45 min at room temperature with Dako background reducing antibody diluent (Agilent), and subsequently incubated with primary antibodies overnight at 4ºC (anti-VP7 antibody (VMRD) or anti-VP2-8 rabbit sera obtained in-house from adenovirus-vaccinated animals [13]). Cells were washed in PBS and stained with highly cross-adsorbed goat anti-mouse/rabbit Alexa Fluor 488 secondary antibody (Thermofisher) and counterstained with 4’, 6-diamidino-2-phenylindole DAPI (Sigma) to visualize nucleic acids. Coverslips were mounted with Prolong mounting media (Thermofisher) and confocal images acquired using a Zeiss using a LSM 880 confocal microscope (Zeiss). Image analysis was performed with the ImageJ software (http://rsbweb.nih.gov/ij/ US National Institutes ofHealth).
2.5. Mouse Vaccination, Serum Preparation and BTV-8 Challenge
This study was approved by the institution ethical committee (PROEX 295.6/21). All steps were taken to minimize animal suffering and animals were assessed daily once infected for clinical signs and euthanized once they reach the defined humane endpoint. To determine vaccine immunogenicity and protection from viral challenge, the IFN-α/βR0/0 (IFNAR(-/-)) mouse model lacking type I IFN receptor was chosen given their susceptibility to BTV infection [26]. IFNAR(-/-) mice on a C57BL/6 genetic background were kindly provided by Professor R. Zinkernagel (Institute of Experimental Medicine, Zurich) and bred in our animal facility. For priming immunizations, 8- to 10-week-old female IFNAR(-/-) mice were inoculated with 40μg pPAL plasmid intramuscularly followed by an immediate electroporation cycle [23]. For combined VP7 and VP2 plasmid inoculations, 40μg of each pPAL plasmid was administered. After 15 days, mice were boosted with the same antigens, either administered as pPAL plasmids as indicated above, or by intramuscular inoculation of a 109 IU recombinant replication-defective human adenovirus 5 (Ad) that encoded the antigen. For combined VP7 and VP2 adenovirus booster, mice received 109 IU of each adenovirus construct. Mice were randomly separated in 8 groups: 2 control groups that received homologous pPAL-empty (pPAL) prime-boost or heterologous pPAL prime and AdDsRed boost; 2 groups received solely the BTV antigen VP7, administered as pPAL-VP7 prime-boost or as pPAL-VP7-prime and AdVP7-boost; 2 groups received solely the BTV antigen VP2, administered as pPAL-VP2 prime-boost or as pPAL-VP2-prime and AdVP2-boost; the last two groups received a combination of VP7 and VP2 antigens, administered as a combined pPAL-VP7 and pPAL-VP2 plasmids prime-boost or as combined pPAL-VP7 and pPAL-VP2 plasmids prime and combined AdVP7 and AdVP2 boost. Animals were challenged with BTV-8 (103pfu) given intraperitoneally 15 days after the booster vaccination. To obtain serum for humoral immunity assays, mouse blood was collected prior to immunization, booster immunization, challenge, and at the end of the experiment in surviving animals. Blood was allowed to clot at room temperature for 30 min in collection tubes and spun down at 10,000xg to extract serum. Serum was stored at -80ºC until use. To evaluate viremia, a drop of blood was obtained in EDTA containing tubes prior to challenge and at days 3, 5, 7 and 12 post-challenge. EDTA-blood was stored at -80ºC until use.
2.6. BTV Viremia Detection by RT-qPCR
RNA was obtained from EDTA blood using the IndiSpin pathogen kit (Indical) following the manufacturer’s instructions. RNA was stored at -80ºC until use. BTV viremia was assessed as described in [27]. Data are presented as Ct value of the amplified BTV segment 5 (S5). Detection limit for the assay was Ct <36.
2.7. Splenocyte Isolation
Splenocytes were isolated as described in [25]. Briefly, spleens were obtained from animals sacrificed at day 7 post-boost (5 animals per vaccinated groups and 3 per control groups) and disaggregated using a 50μm cell strainer using complete splenocyte media (RPMI supplemented with 10% FBS (Lonza Biowhittaker), 4 mM L-glutamine, 10 mM HEPES, 1% 100X non-essential aminoacids, 1 mM sodium pyruvate, 100U/mL penicillin/100 μg/ mL streptomycin and 50nM β-mercaptoethanol (all from Gibco, Invitrogen)). Erythrocytes were lysed by incubation 15min at room temperature with lysis buffer (155mM NH4Cl, 12mM NaHCO3, 140μM EDTA, pH 7.4). After two further washes in complete splenocyte media, cells were counted and ready for use in ELISpot assays and flow cytometry-based assays.
2.8. Mouse IFN-γ ELISpot Assay
Sterile ELISpot plates (MSIPS4510 plate, Millipore) were activated with 35% sterile ethanol, washed once with sterile water and twice with sterile PBS prior to coating with anti-mouse IFN-γ capture antibody (Mouse IFN-γ ELISPOT pair, BD Biosciences). Plates were then blocked in complete splenocyte media for 2 hours at room temperature (RT). Freshly-isolated splenocytes (2×105 per well) were then plated and incubated at 37ºC, 5%CO2 for 24 hours with BEI-inactivated BTV-8 (iBTV) (equivalent to 104pfu/well prior to inactivation), a VP7 immunogenic peptide pool (10μg/ml) (VP7(139) GRWFMRAAQAVTAVV; VP7 (324) RPEFAIHGVNPMPGP; VP7 (72) AAGINVGPI; VP7(80) ISPDYTQHM; VP7 (283) TAILNRTTL; VP7 (327) FAIHGVNPM [19]), concanavalin-A (1.25μg/ml) as positive control or left unstimulated as negative control. All cultures were performed in triplicates. Cells were then removed and plates washed with PBS+0.1%Tween (PBS-T) before incubation with biotinylated capture antibody. Plates were washed in PBS-T and incubated with streptavidin-alkaline phosphatase, before being revealed with dissolved SigmaFAST BCIP/NBT tablets (Sigma). Plates were allowed to air dry and spots were counted using an AID ELISpot counter. ELISpot were considered valid when spot counts in unstimulated well were below 30 and standard deviation of triplicates was within 25% of the mean. Data are presented as spots above background (i.e. counts in stimulated wells - count in unstimulated wells) normalized to 1×106 splenocytes.
2.9. Flow Cytometry: Intracellular Cytokine Staining (ICS) and T Cell Activation Marker Staining
Freshly isolated splenocytes (1×106/well) were stimulated with iBTV or left unstimulated as control in flat-bottom 96 well plates. For intracellular cytokine staining (ICS), cells were incubated 2 hours with iBTV prior to addition of Brefeldin-A (10μg/mL), Monensin (4μM), and anti-CD107a antibody (all from Biolegend) for a further 4 hours. Cells were then stained with viability marker LIVE/DEAD Near IR (Thermofisher) and for surface antigens CD3, CD4 and CD8. Cells were then fixed and permeabilized with BD Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s protocol, and stained for intracellular cytokines IFN-γ, TNF-α, and IL-2. For T cell activation markers, cells were incubated with iBTV for 8 hours in presence of Monensin and anti-CD154 antibody (both from iolegend). Cells were then placed on ice stained for viability with LIVE/DEAD Near IR marker and for surface markers CD3, CD4, CD8, and activation markers CD44 and CD137 (all from Biolegend). Cells were then fixed in 4% paraformaldehyde at room temperature for 15min and washed twice in staining buffer. Samples were acquired on a FACSCelesta flow cytometer (Becton-Dickinson). Gating strategy for ICS has been described in [23]. Fluorescence minus-one (FMO) stainings were included to confirm target cell gating. Analysis was performed with FlowJo software.
2.10. Anti-BTV IgG ELISA
ELISA maxisorp plates (Corning) were coated with iBTV (equivalent to 104pfu/well) overnight at 4ºC. Plates were then blocked with PBS+5% skimmed milk, washed extensively with PBS + 0.1% tween (PBS-T), and incubated with heat-inactivated sera at different dilutions in PBS+2% milk for 2 hours at room temperature. As control to evaluate anti-BTV-IgG binding, sera from naïve mice were used. Plates were then extensively washed with PBS-T and incubated for 1h 30 min with horseradish peroxidase-conjugated anti-mouse IgG antibody (Biorad). Plates were washed extensively with PBS-T and ELISA revealed with TMB substrate (Thermofisher). Reactions were stopped with addition of sulfuric acid (1M) and absorbance read at 450nm using a FluoSTAR Omega ELISA plate reader (BMG Labtech). Anti-IgG titers were calculated as the inverse dilution required to obtain twice the background absorbance for each mouse (i.e. absorbance of 1:200 dilution of naïve sera) [13]. The negative dilution values were given a value of 1 for graphical representation.
2.11. BTV Seroneutralization Assays
Heat-inactivated mouse serum dilutions were incubated with 100pfu BTV-8 for 1h at 37ºC in 96 flat-bottom well plates and subsequently Vero cells (2x104 per cells) were plated and incubated for 96 hours. Cultures were then fixed with 2% formaldehyde and stained with crystal violet to evaluate cytopathic effects (CPE). Neutralizing antibody titers were estimated as the serum concentration at which CPE is <50% of wells.
2.12. Statistical Analysis
All statistical analysis was performed using GraphPad Prism 8 software. Statistical tests used for experimental data analysis are indicated in the figure legends. GraphPad Prism software was used to calculate the Pearson correlation coefficient r and their p-value by matching BTV S5 Ct values and immune parameter values.
3. Results
3.1. pPAL-Mediated Expression of BTV Proteins VP7 and VP2 in Transfected Cells
The pPAL plasmid platform has been successfully used as a DNA vaccine to express antigens from SARS-CoV2 [23] and leishmania [22], able to induce protection after challenge. This platform has the added advantage of not containing an antibiotic-resistance gene for selection, thus making it suitable for approval as a vaccine by regulating authorities. We therefore express the BTV-8 antigens VP7 and VP2 that elicit cellular and humoral response in pPAL to assess the potency of the platform as a BTV vaccine. To determine expression of VP7 and VP2 genes, HEK293 cells were transfected with pPAL-VP7, pPAL-VP2 or pPAL-empty as control. After 48 hours, cells were fixed and VP7 and VP2 proteins expression was assessed by immunofluorescence studies (Figure 1). Both VP7 and VP2 expression was detected with specific antibodies, whereas in control transfected cells with pPAL-empty no fluorescence was observed, which demonstrates that BTV-VP7 and -VP2 antigens are expressed in transfected cells. pPAL-VP7 and pPAL-VP2 could therefore be suitable antigen delivery platforms.
3.2. Heterologous pPAL Prime + Adenovirus Booster Vaccination Protects Against BTV Lethal Challenge
We next designed several vaccination regimes to determine the immunogenicity of VP7 and VP2 expressed using the pPAL platform, either as a homologous pPAL prime-boost vaccine or as a heterologous prime-boost vaccine consisting of a pPAL prime followed by a boost with replication-defective adenovirus that expresses the same antigen. We have previously characterized the adenovirus constructs used in the present work and demonstrated their immunogenicity and capacity to protect against virulent BTV challenge [13,20]. We therefore primed IFNAR(-/-) mice by intramuscular inoculation followed by immediate electroporation with pPAL constructs expressing VP7, VP2 (derived from BTV-8), a combination of both plasmids (VP7+VP2), or an empty pPAL construct as control. For booster immunizations, mice either were inoculated with the same pPAL constructs as described above, or received an intramuscular injection of human replication-defective adenovirus 5 (Ad5) construct that expressed the same antigen(s) given for priming. Vaccinated mice were challenged 15 days after booster vaccination with 1000 pfu of BTV-8 and their disease status monitored daily thereafter. The homologous DNA+DNA vaccination with BTV antigens partially protected mice against BTV challenge (Figure 2A). The presence of VP2 in the DNA vaccine regime improved survival when compared to only VP7. Mice that received heterologous vaccination regime consisting in pPAL prime followed by Ad5 booster expressing BTV antigens were fully protected. All control mice that received immunizations that did not include BTV antigens (i.e pPAL+pPAL or pPAL+Ad5DsRed) succumbed to the disease. We also assessed viremia at several timepoints post-challenge by RT-qPCR (Figure 2B-D). In accordance with the survival data, viremia was controlled by vaccination in heterologous pPAL+Ad5 groups that express BTV antigens, which resulted in the survival of all mice. In the heterologous pPAL+Ad5 that received both BTV antigens, we did not detect viremia, indicating that protection could be sterile in this group. Homologous pPAL vaccination only controlled replication in some individuals, which ultimately led to their survival. Viremia data shows that BTV replicated quickly in control groups, which resulted in the death of the animals. The delivery of VP2 and VP7 through a heterologous DNA + Adenovirus regime is therefore sufficient to drastically reduced viral replication and protect IFNAR(-/-) mice from lethal BTV challenge.
3.3. Heterologous pPAL Prime + Adenovirus Booster Vaccination Induces Potent Humoral Responses
To identify the correlates of protection with adaptive immunity elicited by vaccination, we first assessed humoral responses triggered by the different vaccine regimes. We measured the presence of anti-BTV IgG in the serum of immunized mice after booster vaccination (Figure 3A). All vaccination regimes, which included VP7 and/or VP2, led to the generation of anti-BTV IgG when compared to controls that did not received BTV antigens. Booster immunization with adenovirus improved antibody titers for the groups vaccinated with VP7 alone and the group that received VP7+VP2 as antigens. No significant differences in antibody titers were detected in the VP2 alone vaccinated groups. We also determined the presence of neutralizing antibodies (NAb) to BTV, since their induction is important for disease control [17]. As predicted, neutralizing antibodies were detected in groups immunized with the VP2 antigen (VP2 alone and VP2+VP7) since the majority of neutralizing determinants are directed to this antigen (Figure 3B). Adenovirus booster vaccination nonetheless significantly improved anti-BTV NAb titer when compared to homologous pPAL vaccination. Overall, these data indicate that adenovirus booster vaccination improves the quality of the antibody response to BTV as shown by the increase in anti-BTV NAb titers.
3.4. Heterologous pPAL Prime + Adenovirus Booster Vaccination Induces Potent Anti-BTV Cellular Responses
We employed several strategies to study the cellular responses elicited by vaccination. We first analyzed T cell activation in response to iBTV stimulation by assessing by flow cytometry the expression of the antigen-specific activation markers CD154 on activated CD44+ CD4+ T cells and CD137 on CD44high CD8+ T cells (Figure 4A). These markers were chosen as they are transiently expressed on T cells in response to antigen stimulation [28,29]. Upon iBTV in vitro stimulation, we detected a significant increase in CD154+ cells in activated CD4+T cells for all vaccination regimes, which indicates that immunization elicited BTV-specific CD4+ T cell (Figure 4B). We did not detect a significant increase in CD137+ cells in activated CD8+ T cells in the pPAL-VP7 + pPAL-VP7 and in the pPAL-VP7+VP2 + pPAL-VP7+VP2 groups (Figure 4C). The presence of these activated CD8+ T cells significantly increased for the other vaccination regimes stimulated with iBTV. Importantly, the presence of CD154+ CD44+ CD4+ T cells and CD137+ CD44high CD8+ T cells was significantly higher in heterologous prime boost groups when compared to homologous DNA groups. Overall, our data indicate that heterologous immunization with pPAL and adenovirus increased the number of BTV-specific T cells when compared to homologous pPAL vaccination.
We also analyzed the cellular responses elicited by the different vaccination regimes using IFN-γ ELISpot assays by measuring the anti-BTV cellular response using iBTV or a pool of immunogenic VP7 peptides [19] in splenocytes 7 days after booster immunization (Figure 5A and B). All vaccination regimes that included BTV antigens induced cellular responses to iBTV, although responses in the two groups that only received VP7 did not reach significance. Booster vaccination with AdVP2 significantly improved cellular responses to iBTV. When responses were assessed with VP7 peptide pool, we could only detect potent induction of anti-VP7 cellular immunity in the group that used AdVP7 as booster immunization. Adenovirus-based booster vaccination thus significantly improved cellular responses to VP7 when compared to homologous pPAL strategy. We next used intracellular cytokine staining (ICS) for IFN-γ, TNF-α, and IL-2, and staining of CD107a as surrogate marker for cytotoxicity and flow cytometry analysis to determine which T cell population were activated by immunization (Figure 5C and D). We evaluated in CD4+ and CD8+ T cells the expression of at least one functional marker (IFN-γ, TNF-α, IL-2, or CD107a) in response to iBTV stimulation. We found that all vaccination regimes induced CD4+ and CD8+ T cells that could specifically recognize iBTV. Adenovirus booster immunization significantly increased CD4+ and CD8+ T cell responses to iBTV when compared to homologous pPAL immunization independently of the given antigen. In line with the T cell activation data (Figure 4), we found in ELISpot and ICS studies that heterologous immunization with pPAL and adenovirus increased the number of T cells responding to BTV stimulation when compared to homologous pPAL vaccination.
Finally, we analyzed the polyfunctionality of anti-BTV CD4+ and CD8+ T cells by analyzing the concomitant production of IFN-γ, TNF-α, and IL-2, and the expression of the degranulation marker CD107a in flow cytometry (Figure 6). In CD4+ T cells, we found that homologous pPAL immunization could elicit potentially cytotoxic CD4+ T cells (CD107a+) (Figure 6A). We also detected CD4+ T cells expressing TNF-α in most animals but low levels of IL-2 and IFN-γ production with homologous pPAL BTV vaccination. Heterologous immunization increased the percentage of CD4+ T cells producing these cytokines to iBTV, but did not elicit significant levels of cytotoxic CD4+ T cells as measured by CD107a expression. Importantly, we detected IFN-γ-producing CD4+ T cells to iBTV with heterologous vaccination regimes. In CD8+ T cells, homologous pPAL immunization only elicited low levels of IFN-γ, TNF-α, and IL-2 producing cells (except for two mice in the VP2 immunized group) (Figure 6B). Heterologous immunization with pPAL and adenovirus increased the percentage of BTV-specific cytotoxic CD8+ T cells as well as that of cytokine producing cells responding to iBTV when compared to homologous pPAL vaccination. We also evaluated the capacity of CD4+ and CD8+ T cells to express simultaneously multiple cytokine/degranulation marker as a measure of the T cell polyfunctionality elicited by the vaccine (Figure 6C and D). Most vaccination regimes elicited limited polyfunctional T CD4+ or CD8+ T cells, but inclusion of VP7 delivery through adenovirus booster (pPAL-VP7 + AdVP7 and pPAL-VP2+VP7 + AdVP7+VP2 groups) increased the percentage of polyfunctional T cells when compared to homologous DNA boost. Overall, our study indicates that heterologous pPAL + Adenovirus vaccination with BTV antigen VP7 and VP2 improves T cells responses to the virus, not only quantitatively, but also qualitatively.
3.5. Vaccine-Induced Adaptive Immune Response Parameters Correlate with Protection
We wanted to determine which parameters of the adaptive immune responses elicited by vaccination would correlate with protection as analyzed by others [30]. We therefore calculated the Pearson correlation coefficients r between adaptive immune measurements and viremia at day 3 and 5 pi (Table 1). We found a significant positive correlation (r>0.5; p<10-3) between all immune parameters and Ct values for BTV-S5, i.e. immune parameters correlated with a reduction in viral load. This confirms that induction of adaptive immune responses to BTV by the vaccine associates with a reduction in viremia at day 3 and 5pi that ultimately leads to recovery from the disease. It is worth noting that activation of CD8+ T cells (as measured by cytokine secretion and degranulation) displayed the highest correlation with reduced disease burden. The presence of neutralizing antibodies to BTV was also a higher correlate of reduced disease than anti-BTV IgG. Overall, these data highlight the necessity for BTV vaccines to induce cellular immunity as well as humoral immunity to protect adequately against BTV.
4. Discussion
In the present work, we show that delivery of BTV antigens VP7 and VP2 through a DNA priming followed by an adenovirus booster can fully protect mice against virulent BTV challenge. Heterologous prime-boost strategies have been used in the past and shown promising results in protecting against BTV [14,15,31,32,33]. BTV antigen delivery through DNA prime followed by modified vaccinia virus Ankara (MVA) booster can protect mice from BTV lethal challenge [14,31]. Similarly, antigen delivery through avian reovirus microspheres followed by MVA booster was also successful for protection in murine models [32]. Heterologous strategies involving 2 viral vectors (ChAdOx1 vector and MVA) have also shown some efficacy in mice and sheep [15,33]. Our strategy nonetheless makes use of two delivery systems (pPAL and adenovirus platforms) that have been approved as vaccine by regulatory agencies. We also present an exhaustive assessment of the immune response generated by these heterologous strategies as well as their correlation with protection. We found that in the murine model, induction of anti-BTV CD8+ T cells is crucial as this parameter showed the highest correlation coefficient with protection. We also found that neutralizing antibody induction correlated strongly with protection. Vaccination should therefore aim at stimulating both immune parameters, and vaccine potency is likely to rely on both parameters.
In support of this, we found that viremia was better controlled in groups that received VP2 than in the group that only received VP7. This is likely due to the presence of anti-VP2 neutralizing antibodies to BTV in these animals that quickly limit replication, whereas anti-VP7 antibodies cannot effectively neutralize BTV infection. We nonetheless detected low NAb titers in one mouse immunized solely with VP7 following adenovirus boost. Although antibodies to BTV-VP7 inner core protein are not thought to produce neutralization, reports exist of neutralizing determinants on the VP6 inner core proteins of rotavirus [34,35], another member of the Sedoreoviridae family. Inner core proteins in Sedoreoviridae are mainly exposed intracellularly during infection [6], thus antibodies are unlikely to “classically” neutralized infection by preventing virus entry. It is more probable that neutralization against VP7 determinants would occur through intracellular mechanisms mediated for instance by antibody binding to the intracellular IgG receptor TRIM21 [36]. The observation that some neutralizing antibodies can be elicited against BTV-VP7 suggests that neutralizing determinant could also exist on this conserved BTV protein. Further work will be required to elucidate this observation.
Another important aspect that is critical for BTV protection is the choice of antigen to include in the vaccine formulation. Our rationale was to focus on structural proteins as antigens, since they are present on the viral particles and this should allow for quicker recognition and elimination of virus and infected cells when compared to non-structural proteins, which require viral replication to occur for expression to begin. We chose VP7 and VP2 as antigens, as they are abundant proteins on the virion (260 and 60 copies, respectively [6]) and therefore their availability for presentation to the immune system as viral antigen should be high during infection. Moreover, they are also known to contain cellular and humoral determinants [19,37]. We have found that VP7 could represent the basis for cross-serotype protection and that this is likely due to cellular immunity since we detected protection in mice and sheep in the absence of neutralizing antibodies [13,20]. Other studies also confirmed that induction of immunity to VP7 participate in protection [31,32]. The non-structural proteins NS1 and NS2 have also been used as immunogens in other studies to trigger cellular immunity. These non-structural proteins are conserved between serotypes and can therefore be used to elicit serotype cross-reactive T cells [15,18,21]. Vaccination with these antigens can lead to protection in animal models, but protection was limited in sheep, possibly because only targeting these antigens is insufficient to control viral replication in the early stages of infection [15,33]. As previously discussed, VP2 improved protection against virulent BTV challenge in our experiment, and this is likely due to the induction of antibodies that can rapidly neutralize BTV infection. A recent study has corroborated this, showing that inclusion of VP2 in a recombinant MVA with NS1 and a truncated NS2 could offer protection in sheep against BTV challenge, whereas concomitant expression of VP7 with NS1 and the truncated NS2 only offered partial protection [38]. This further indicates that the inclusion of cellular and humoral immune targets of BTV is a requisite to design effective BTV vaccines.
Curiously, we observed that homologous pPAL immunization could potentially elicit anti-BTV cytotoxic CD107a+ CD4+ T cells. We have previously observed a similar phenotype using pPAL vaccination against SARS-CoV2 [23]. DNA vaccines are known to potentiate cellular responses [39], although this phenotype has not been described before to the best of our knowledge. Whether this observation is specific to the pPAL expression system or it can be extended to other DNA vaccine platforms remains to be determined. The exact contribution of these cytotoxic CD4+ T cells to infection resolution is not fully elucidated [40]. Some studies indicate that they participate in virus clearance [41,42], however their presence could also lead to the loss of B cell germinal center observed in patients that succumbed to SARS-Cov2 infection [43]. Our data nonetheless indicate that BTV is better controlled in this murine model by CD8+ T cell activation, and as such vaccination should aim at triggering this cell population.
Although our data highlights the importance of cellular immunity for protection against BTV in IFNAR(-/-) mice, the immune mechanisms that lead to protection could be different in the natural host. Indeed, a recent study has indicated that CD8+ T cells could be detrimental for bluetongue disease outcome in sheep possibly through immune-mediated pathogenicity [44]. We have however found in several experimental infections with BTV in sheep that CD8+ T cells expand at day 10-15pi [27,45,46] and this typically coincides with reduced viremia, which points at a role for these T cells in the removal of BTV-infected cells. We nonetheless detected that during the peak of viral replication T cell responses are acutely suppressed and this coincided with the overexpression of genes and molecules involved in the immunoregulatory PD1/PD-L1 pathway [27]. In this context, and given the putative role of CD8+ T cells in bluetongue immunopathology, it could be hypothesized that in the natural host these T cells would become activated during acute viral infection to clear infected cells but their prolonged activation could lead to immunopathology, hence the triggering of immune checkpoints to tightly regulate the activity of the population.
It is also important to note that, although IFNAR(-/-) mice are excellent models to evaluate vaccine candidates against BTV, these mice are partially immunocompromised. As such, this model has some limitations when evaluating immune responses to vaccines [47,48]. We found that IFN-γ production was quite limited in these experiments when compared to previous work with immunocompetent transgenic mice susceptible to SARS-CoV2 [23]. This could be due to the IFN deficiency of this murine model that could result in impaired IFN type II responses. In comparison, TNF-α responses appear enhanced when contrasted to previous studies in fully immunocompetent mice, which suggest that IFNAR(-/-) mice may compensate their IFN-I deficiency by promoting pro-inflammatory cytokines production such as TNF-α. This is an important consideration when using this model to evaluate immune responses to vaccine candidates, given the importance of IFN-I in modulating adaptive immunity [49]. As a result, IFN-I deficiency in these animals could limit and bias the adaptive immunity we observe. In spite of this, we found that IFNAR(-/-) mice mounted T cell responses to a similar repertoire of VP7 epitopes than wild-type mice with the same genetic background [19], and as many vaccination studies attest immune responses in these mice are sufficient to control viral replication. It would be nonetheless interesting to perform in future work a parallel vaccination study with immunocompetent mice to further evaluate the capacity of the IFNAR(-/-) murine model to mount fully effective adaptive immune responses. These studies could help in the interpretation of immune results in IFNAR(-/-) mice.
Finally, our data show that heterologous prime-boost improves the titers of neutralizing antibodies, when compared to the homologous DNA vaccination strategy. We also found in a previous study that homologous adenovirus expressing VP2 elicited minimal titers of neutralizing antibodies [13]. Although this could be due to the antigen, as we have been able to obtain adequate neutralizing antibody titers to other viral antigens such as the hemagglutinin from peste des petits ruminants virus with recombinant adenovirus vaccines when using homologous prime-boost vaccination [50]. Adequate neutralizing antibody titers against SARS-CoV2 can also be reached with homologous recombinant adenovirus vaccines or with pPAL [23,51]. Nonetheless, one of the main advantages of heterologous prime-boost vaccination over homologous vaccination is that it improves neutralization antibody generation. For instance, in immunocompromised patients, adenovirus prime followed by mRNA vaccine boost against COVID’19 was shown to improve neutralizing antibody titers [52]. Similarly, we found that DNA prime-adenovirus boost with BTV antigens improved cellular responses when compared to homologous DNA vaccination. Adenovirus-based VP7 delivery improved the polyfunctionality of anti-BTV T cells, a feature associated to more effective immune responses to pathogens [53]. Evidence exists that heterologous prime-boost strategies also enhance cellular immunity by increasing the number of responding cells and promoting effector memory differentiation [52,54,55,56]. Heterologous prime-boost strategies can therefore be advantageous as they potentiate both arms of adaptive immunity. Although in the context of veterinary medicine the licensing of two vaccine reagents could be costly, it could still be a useful strategy to explore to produce BTV vaccines that induce long-lasting immunity, particularly if they allow for multiserotype protection.
We herein report a promising strategy for BTV vaccination that consists in a heterologous prime boost strategy with DNA and adenovirus vectors that express VP7 and VP2 proteins from BTV. Expression of both antigens induced potent cellular and humoral immunity that led to protection. Importantly, we could not detect viremia in the vaccinated group that received both antigens, which indicates that this vaccine could produce sterile protection. It will be important in future studies to evaluate the protective efficacy of this heterologous prime-boost regime in a natural host of the disease. Heterologous DNA-adenovirus prime-boost vaccinations are therefore a promising strategy to explore for the development of DIVA BTV vaccines.
5. Conclusions
This study demonstrates that a heterologous DNA-adenovirus prime-boost vaccination strategy expressing BTV-VP7 and -VP2 provides full protection against virulent BTV challenge in a murine model. This approach enhances both cellular and humoral immune responses, with CD8+ T cell activation and neutralizing antibody titers strongly correlating with viral clearance. VP2 contributed significantly to protection by inducing neutralizing antibodies, while VP7 may also have unrecognized neutralizing determinants. The inclusion of both antigens ensures rapid immune recognition and effective protection. Heterologous prime-boost vaccination proved superior to homologous strategies by enhancing adaptive immunity, making it a promising approach despite potential regulatory challenges. Although IFNAR(-/-) mice are useful for vaccine evaluation, further studies in natural hosts are needed. Overall, this strategy represents a strong candidate for DIVA-compatible BTV vaccination.
Author Contributions
P.M-A., L.G-M., A.B-C., A.L-L. J.L., P.A., A.A. and V.M., performed experimental work and analyzed and interpreted data. V.L. and V.M. provided expert input and reviewed the manuscript. J.R. wrote the manuscript. J.R. and N.S. conceptualized the study, designed the experiments, analyzed and interpreted the data and accountable for accuracy and integrity of the work. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by grant PID2021-124872OB-100 from Ministerio de Ciencia, Innovación y Universidades (Spain) and co-funded by the European Union’s Horizon Europe Project 101136346 EUPAHW.
Conflicts of Interest
The authors do not have any conflict of interest.
References
- Rojas, J.M. , et al., Diagnosing bluetongue virus in domestic ruminants: current perspectives. Vet Med (Auckl), 2019. 10: p. 17-27.
- Rojas, J.M., V. Martín, and N. Sevilla, Vaccination as a Strategy to Prevent Bluetongue Virus Vertical Transmission. Pathogens, 2021. 10(11).
- Rushton, J. and N. Lyons, Economic impact of Bluetongue: a review of the effects on production. Vet Ital, 2015. 51(4): p. 401-6.
- Santman-Berends, I.M.G.A. , et al., Bluetongue virus serotype 8 (BTV-8) infection reduces fertility of Dutch dairy cattle and is vertically transmitted to offspring. Theriogenology, 2010. 74(8): p. 1377-1384.
- Attoui, H. , et al., Orbiviruses☆, in Reference Module in Biomedical Sciences. 2016, Elsevier.
- Roy, P. , Bluetongue virus assembly and exit pathways. Adv Virus Res, 2020. 108: p. 249-273.
- Stewart, M. , et al., Characterization of a second open reading frame in genome segment 10 of bluetongue virus. J Gen Virol, 2015. 96(11): p. 3280-3293.
- Baylis, M., L. O'Connell, and P.S. Mellor, Rates of bluetongue virus transmission between Culicoides sonorensis and sheep. Med Vet Entomol, 2008. 22(3): p. 228-37.
- Barratt-Boyes, S.M. and N.J. MacLachlan, Dynamics of viral spread in bluetongue virus infected calves. Vet Microbiol, 1994. 40(3-4): p. 361-71.
- Takamatsu, H. , et al., A possible overwintering mechanism for bluetongue virus in the absence of the insect vector. J Gen Virol, 2003. 84(Pt 1): p. 227-235.
- Goffredo, M. , et al., Vector species of Culicoides midges implicated in the 2012-2014 Bluetongue epidemics in Italy. Vet Ital, 2015. 51(2): p. 131-8.
- van Rijn, P.A. , Prospects of Next-Generation Vaccines for Bluetongue. Frontiers in Veterinary Science, 2019. 6(407).
- Rojas, J.M. , et al., Vaccination With Recombinant Adenoviruses Expressing the Bluetongue Virus Subunits VP7 and VP2 Provides Protection Against Heterologous Virus Challenge. Frontiers in Veterinary Science, 2021. 8(158).
- Calvo-Pinilla, E. , et al., Multiserotype protection elicited by a combinatorial prime-boost vaccination strategy against bluetongue virus. PLoS One, 2012. 7(4): p. e34735.
- Utrilla-Trigo, S. , et al., The Combined Expression of the Nonstructural Protein NS1 and the N-Terminal Half of NS2 (NS2(1-180)) by ChAdOx1 and MVA Confers Protection against Clinical Disease in Sheep upon Bluetongue Virus Challenge. J Virol, 2022. 96(3): p. e0161421.
- Jeggo, M.H., R. C. Wardley, and J. Brownlie, A study of the role of cell-mediated immunity in bluetongue virus infection in sheep, using cellular adoptive transfer techniques. Immunology, 1984. 52(3): p. 403-10.
- Jeggo, M.H., R. C. Wardley, and W.P. Taylor, Role of neutralising antibody in passive immunity to bluetongue infection. Res Vet Sci, 1984. 36(1): p. 81-6.
- Rojas, J.M. , et al., Ovine and murine T cell epitopes from the non-structural protein 1 (NS1) of bluetongue virus serotype 8 (BTV-8) are shared among viral serotypes. Veterinary research, 2014. 45(1): p. 30-30.
- Rojas, J.M. , et al., T cell responses to bluetongue virus are directed against multiple and identical CD4+ and CD8+ T cell epitopes from the VP7 core protein in mouse and sheep. Vaccine, 2011. 29(40): p. 6848-57.
- Martin, V. , et al., Protective Efficacy in Sheep of Adenovirus-Vectored Vaccines against Bluetongue Virus Is Associated with Specific T Cell Responses. PLoS One, 2015. 10(11): p. e0143273.
- Marín-López, A. , et al., CD8 T Cell Responses to an Immunodominant Epitope within the Nonstructural Protein NS1 Provide Wide Immunoprotection against Bluetongue Virus in IFNAR(-/-) Mice. J Virol, 2018. 92(16).
- Alonso, A. , et al., A non-replicative antibiotic resistance-free DNA vaccine delivered by the intranasal route protects against canine leishmaniasis. Front Immunol, 2023. 14: p. 1213193.
- Alcolea, P.J. , et al., Non-replicative antibiotic resistance-free DNA vaccine encoding S and N proteins induces full protection in mice against SARS-CoV-2. Front Immunol, 2022. 13: p. 1023255.
- Avia, M. , et al., Virus-induced autophagic degradation of STAT2 as a mechanism for interferon signaling blockade. EMBO Rep, 2019. 20(11): p. e48766.
- Rodriguez-Calvo, T. , et al., Type I interferon limits the capacity of bluetongue virus to infect hematopoietic precursors and dendritic cells in vitro and in vivo. J Virol, 2014. 88(2): p. 859-67.
- Calvo-Pinilla, E. , et al., Establishment of a bluetongue virus infection model in mice that are deficient in the alpha/beta interferon receptor. PLoS One, 2009. 4(4): p. e5171.
- Louloudes-Lazaro, A. , et al., Comprehensive immune profiling reveals that Orbivirus infection activates immune checkpoints during acute T cell immunosuppression. Front Immunol, 2023. 14: p. 1255803.
- Frentsch, M. , et al., Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nat Med, 2005. 11(10): p. 1118-24.
- Wölfl, M. , et al., Use of CD137 to study the full repertoire of CD8+ T cells without the need to know epitope specificities. Cytometry A, 2008. 73(11): p. 1043-9.
- Proctor, J. , et al., Heterologous vaccine immunogenicity, efficacy, and immune correlates of protection of a modified-live virus porcine reproductive and respiratory syndrome virus vaccine. Frontiers in Microbiology, 2022. 13.
- Calvo-Pinilla, E. , et al., Heterologous prime boost vaccination with DNA and recombinant modified vaccinia virus Ankara protects IFNAR(-/-) mice against lethal bluetongue infection. Vaccine, 2009. 28(2): p. 437-45.
- Marín-López, A. , et al., Microspheres-prime/rMVA-boost vaccination enhances humoral and cellular immune response in IFNAR(-/-) mice conferring protection against serotypes 1 and 4 of bluetongue virus. Antiviral Res, 2017. 142: p. 55-62.
- Utrilla-Trigo, S. , et al., Heterologous Combination of ChAdOx1 and MVA Vectors Expressing Protein NS1 as Vaccination Strategy to Induce Durable and Cross-Protective CD8+ T Cell Immunity to Bluetongue Virus. Vaccines (Basel), 2020. 8(3).
- Caddy, S.L. , et al., Intracellular neutralisation of rotavirus by VP6-specific IgG. PLoS Pathog, 2020. 16(8): p. e1008732.
- Corthésy, B. , et al., Rotavirus anti-VP6 secretory immunoglobulin A contributes to protection via intracellular neutralization but not via immune exclusion. J Virol, 2006. 80(21): p. 10692-9.
- Li, X. , et al., Multiple Roles of TRIM21 in Virus Infection. Int J Mol Sci, 2023. 24(2).
- White, J.R. and B.T. Eaton, Conformation of the VP2 protein of bluetongue virus (BTV) determines the involvement in virus neutralization of highly conserved epitopes within the BTV serogroup. J Gen Virol, 1990. 71 ( Pt 6): p. 1325-32.
- Jiménez-Cabello, L. , et al., Co-expression of VP2, NS1 and NS2-Nt proteins by an MVA viral vector induces complete protection against bluetongue virus. Frontiers in Immunology, 2024. 15.
- Kozak, M. and J. Hu, DNA Vaccines: Their Formulations, Engineering and Delivery. Vaccines, 2024. 12(1): p. 71.
- Cenerenti, M. , et al., The Era of Cytotoxic CD4 T Cells. Frontiers in Immunology, 2022. 13.
- Weiskopf, D. , et al., Dengue virus infection elicits highly polarized CX3CR1+ cytotoxic CD4+ T cells associated with protective immunity. Proc Natl Acad Sci U S A, 2015. 112(31): p. E4256-63.
- Wilkinson, T.M. , et al., Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med, 2012. 18(2): p. 274-80.
- Kaneko, N. , et al., Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell, 2020. 183(1): p. 143-157.e13.
- Newbrook, K. , et al., Specific T-cell subsets have a role in anti-viral immunity and pathogenesis but not viral dynamics or onwards vector transmission of an important livestock arbovirus. Front Immunol, 2024. 15: p. 1328820.
- Rodriguez-Martin, D. , et al., The Interplay between Bluetongue Virus Infections and Adaptive Immunity. Viruses, 2021. 13(8).
- Rojas, J.M., T. Rodriguez-Calvo, and N. Sevilla, Recall T cell responses to bluetongue virus produce a narrowing of the T cell repertoire. Vet Res, 2017. 48(1): p. 38.
- Wong, G. and X.G. Qiu, Type I interferon receptor knockout mice as models for infection of highly pathogenic viruses with outbreak potential. Zool Res, 2018. 39(1): p. 3-14.
- Marín-Lopez, A. , et al., Modeling Arboviral Infection in Mice Lacking the Interferon Alpha/Beta Receptor. Viruses, 2019. 11(1).
- Rojas, J.M. , et al., Viral pathogen-induced mechanisms to antagonize mammalian interferon (IFN) signaling pathway. Cell Mol Life Sci, 2021. 78(4): p. 1423-1444.
- Rojas, J.M. , et al., Vaccination with recombinant adenovirus expressing peste des petits ruminants virus-F or -H proteins elicits T cell responses to epitopes that arises during PPRV infection. Vet Res, 2017. 48(1): p. 79.
- Liu, X. , et al., Safety and immunogenicity of heterologous versus homologous prime-boost schedules with an adenoviral vectored and mRNA COVID-19 vaccine (Com-COV): a single-blind, randomised, non-inferiority trial. Lancet, 2021. 398(10303): p. 856-869.
- Chu, C. , et al., Immune response of heterologous versus homologous prime-boost regimens with adenoviral vectored and mRNA COVID-19 vaccines in immunocompromised patients. Frontiers in Immunology, 2023. 14.
- Han, Q. , et al., Polyfunctional responses by human T cells result from sequential release of cytokines. Proc Natl Acad Sci U S A, 2012. 109(5): p. 1607-12.
- Ratto-Kim, S. , et al., Heterologous prime-boost regimens using rAd35 and rMVA vectors elicit stronger cellular immune responses to HIV proteins than homologous regimens. PLoS One, 2012. 7(9): p. e45840.
- Siddiqui, A. , et al., Revival of the heterologous prime-boost technique in COVID-19: An outlook from the history of outbreaks. Health Science Reports, 2022. 5(2): p. e531.
- Heinen, N. , et al., In-depth analysis of T cell immunity and antibody responses in heterologous prime-boost-boost vaccine regimens against SARS-CoV-2 and Omicron variant. Front Immunol, 2022. 13: p. 1062210.
Figure 1.
Immunofluorescence studies confirmed VP2 and VP7 expression by pPAL plasmids. HEK-293 cells were transfected with pPAL-VP2 pPAL-VP7, or pPAL-empty as control. After 48h, cells were fixed permeabilized and stained to detect the expression of VP2-8 or VP7, before counterstaining nucleic acids with DAPI. (A) VP2-8 expression in pPAL-empty or pPAL-VP2-8 transfected HEK293 cells. (B) VP7 expression in pPAL-empty or pPAL-VP7 transfected HEK293 cells.
Figure 1.
Immunofluorescence studies confirmed VP2 and VP7 expression by pPAL plasmids. HEK-293 cells were transfected with pPAL-VP2 pPAL-VP7, or pPAL-empty as control. After 48h, cells were fixed permeabilized and stained to detect the expression of VP2-8 or VP7, before counterstaining nucleic acids with DAPI. (A) VP2-8 expression in pPAL-empty or pPAL-VP2-8 transfected HEK293 cells. (B) VP7 expression in pPAL-empty or pPAL-VP7 transfected HEK293 cells.
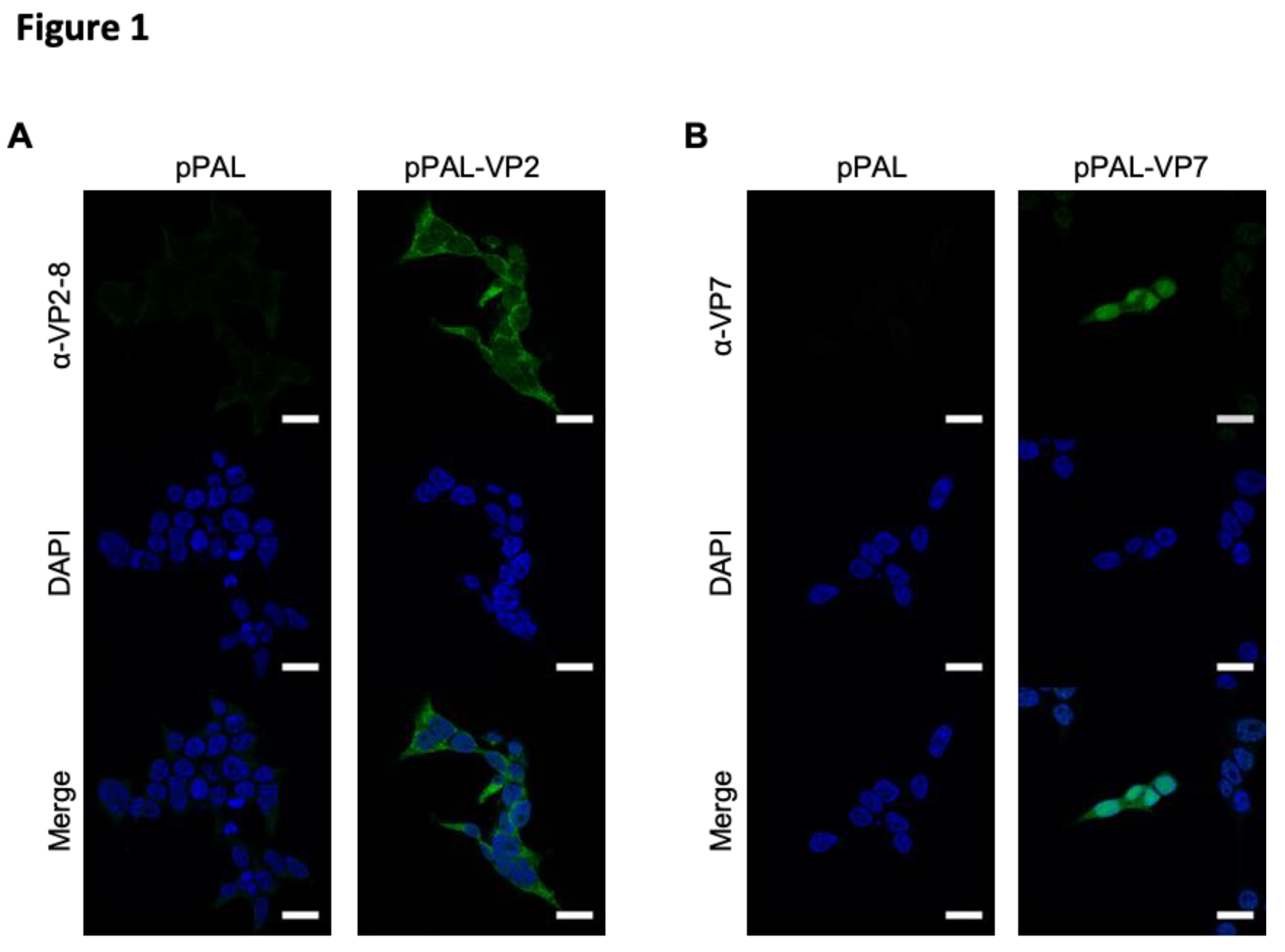
Figure 2.
pPAL and adenovirus vaccination protects against BTV-8 challenge and controls viral replication. (A) IFNAR(-/-) mice were immunized with VP7 and/or VP2 delivered as homologous pPAL or heterologous pPAL+adenovirus immunizations. Mice were challenged with 1000pfu BTV-8, and monitored daily for appearance of clinical signs of disease. Survival curve for each vaccinated group is shown. (B-D) Blood samples were obtained from infected mice at day 0 (pre-challenge) and at days 3, 5, 7 and 12 post-infection (DPI), and RNA extracted. RT-qPCR was performed to detect BTV RNA presence in these samples. (B) Ct mean values for BTV segment 5 fragment (BTV S5) are plotted for all timepoints assessed for each treatment group. Ct individual values at (C) day 3 post-infection (D3PI) and (D) day 5pi (D5PI) are plotted for each vaccinated group. * p<0.05; **p<0.01; *** p<0.001 One-way ANOVA with Fisher’s LSD post-test. Detection limit of RT-qPCR for BTV S5 fragment (Ct<36) is indicated.
Figure 2.
pPAL and adenovirus vaccination protects against BTV-8 challenge and controls viral replication. (A) IFNAR(-/-) mice were immunized with VP7 and/or VP2 delivered as homologous pPAL or heterologous pPAL+adenovirus immunizations. Mice were challenged with 1000pfu BTV-8, and monitored daily for appearance of clinical signs of disease. Survival curve for each vaccinated group is shown. (B-D) Blood samples were obtained from infected mice at day 0 (pre-challenge) and at days 3, 5, 7 and 12 post-infection (DPI), and RNA extracted. RT-qPCR was performed to detect BTV RNA presence in these samples. (B) Ct mean values for BTV segment 5 fragment (BTV S5) are plotted for all timepoints assessed for each treatment group. Ct individual values at (C) day 3 post-infection (D3PI) and (D) day 5pi (D5PI) are plotted for each vaccinated group. * p<0.05; **p<0.01; *** p<0.001 One-way ANOVA with Fisher’s LSD post-test. Detection limit of RT-qPCR for BTV S5 fragment (Ct<36) is indicated.
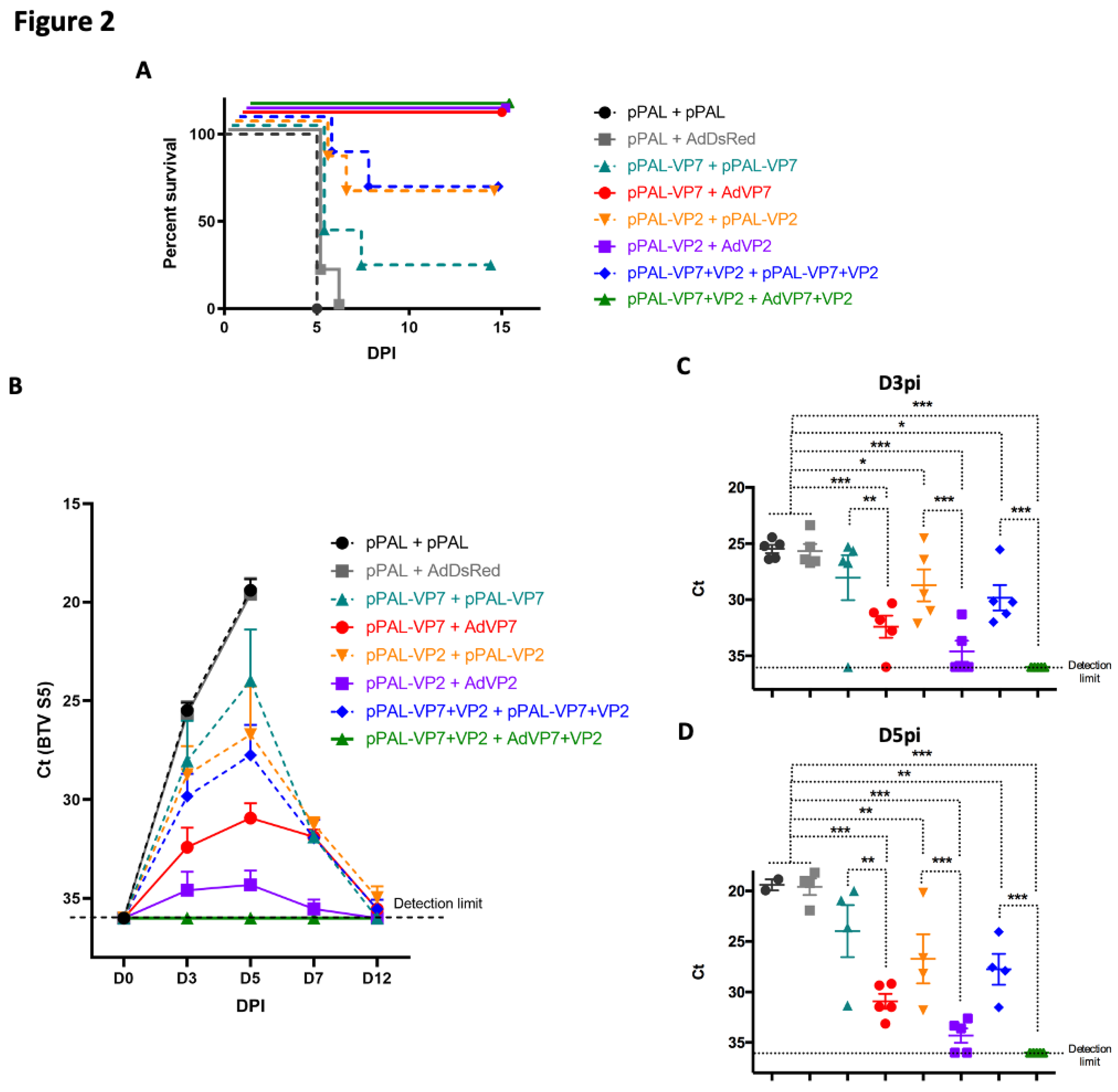
Figure 3.
pPAL and adenovirus immunization induces anti-BTV humoral immune responses. Sera from immunized mice were collected 14 days post-booster vaccination and analyzed for the presence of (A) anti-BTV IgG and (B) neutralizing antibodies (NAb) to BTV-8. (A) Anti-BTV IgG titers (1/) (mean ± SEM) were assessed by ELISA in immunized animals for each vaccination regime. (B) NAb titers to BTV-8 (1/) (mean ± SEM) were measured by seroneutralization assays in Vero cells. * p<0.05; **p<0.01; ***p<0.001 One-way ANOVA with Fisher’s LSD post-test.
Figure 3.
pPAL and adenovirus immunization induces anti-BTV humoral immune responses. Sera from immunized mice were collected 14 days post-booster vaccination and analyzed for the presence of (A) anti-BTV IgG and (B) neutralizing antibodies (NAb) to BTV-8. (A) Anti-BTV IgG titers (1/) (mean ± SEM) were assessed by ELISA in immunized animals for each vaccination regime. (B) NAb titers to BTV-8 (1/) (mean ± SEM) were measured by seroneutralization assays in Vero cells. * p<0.05; **p<0.01; ***p<0.001 One-way ANOVA with Fisher’s LSD post-test.
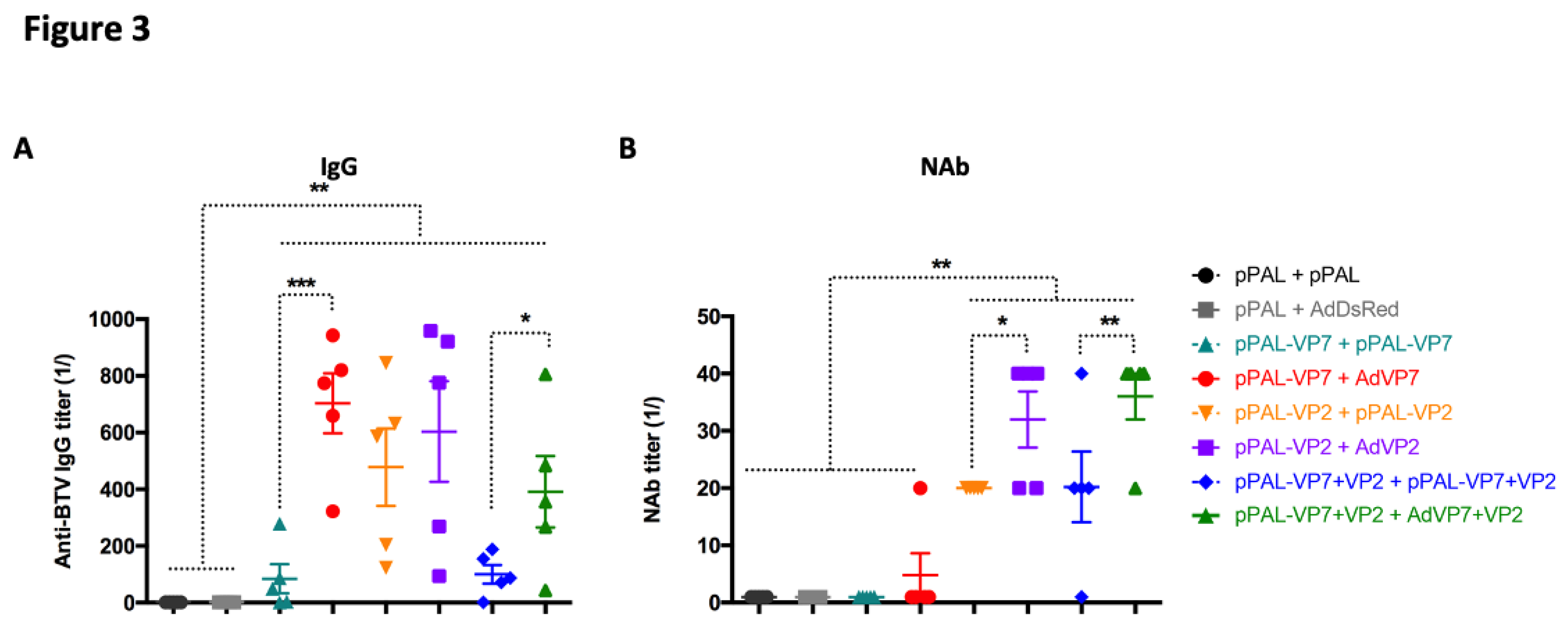
Figure 4.
Activation marker induction in CD4+ and CD8+ T cells stimulated with iBTV. Splenocytes were stimulated with iBTV (or left unstimulated as control) for 8h and stained for CD3, CD4, CD8, CD44, CD137 and CD154 expression. (A) Gating strategy for CD4+ CD44+ CD154+ and CD8+ CD44high CD137+ cells is shown. CD44+ CD154+ events for CD4+ T cells and CD44high CD137+ events for CD8+ T cells were considered activated antigen-specific cells. FSC-A/SSC-A dot-plots were used for splenocyte gating, followed by doublet and dead cell exclusion. CD3/CD4 and CD3/CD8 dot-plots were used for CD4+ T and CD8+ T cell gating respectively. Activated antigen-specific cell gating was established using unstimulated splenocytes for each culture as shown. (B) Percentage of CD4+ CD44+ CD154+ cells in iBTV cultures of vaccinated or control mice. (C) Percentage of CD8+ CD44high CD137+ cells in iBTV cultures of vaccinated or control mice. *p<0.05; **p<0.01; ***p<0.001 One-way ANOVA with Fisher’s LSD post-test.
Figure 4.
Activation marker induction in CD4+ and CD8+ T cells stimulated with iBTV. Splenocytes were stimulated with iBTV (or left unstimulated as control) for 8h and stained for CD3, CD4, CD8, CD44, CD137 and CD154 expression. (A) Gating strategy for CD4+ CD44+ CD154+ and CD8+ CD44high CD137+ cells is shown. CD44+ CD154+ events for CD4+ T cells and CD44high CD137+ events for CD8+ T cells were considered activated antigen-specific cells. FSC-A/SSC-A dot-plots were used for splenocyte gating, followed by doublet and dead cell exclusion. CD3/CD4 and CD3/CD8 dot-plots were used for CD4+ T and CD8+ T cell gating respectively. Activated antigen-specific cell gating was established using unstimulated splenocytes for each culture as shown. (B) Percentage of CD4+ CD44+ CD154+ cells in iBTV cultures of vaccinated or control mice. (C) Percentage of CD8+ CD44high CD137+ cells in iBTV cultures of vaccinated or control mice. *p<0.05; **p<0.01; ***p<0.001 One-way ANOVA with Fisher’s LSD post-test.
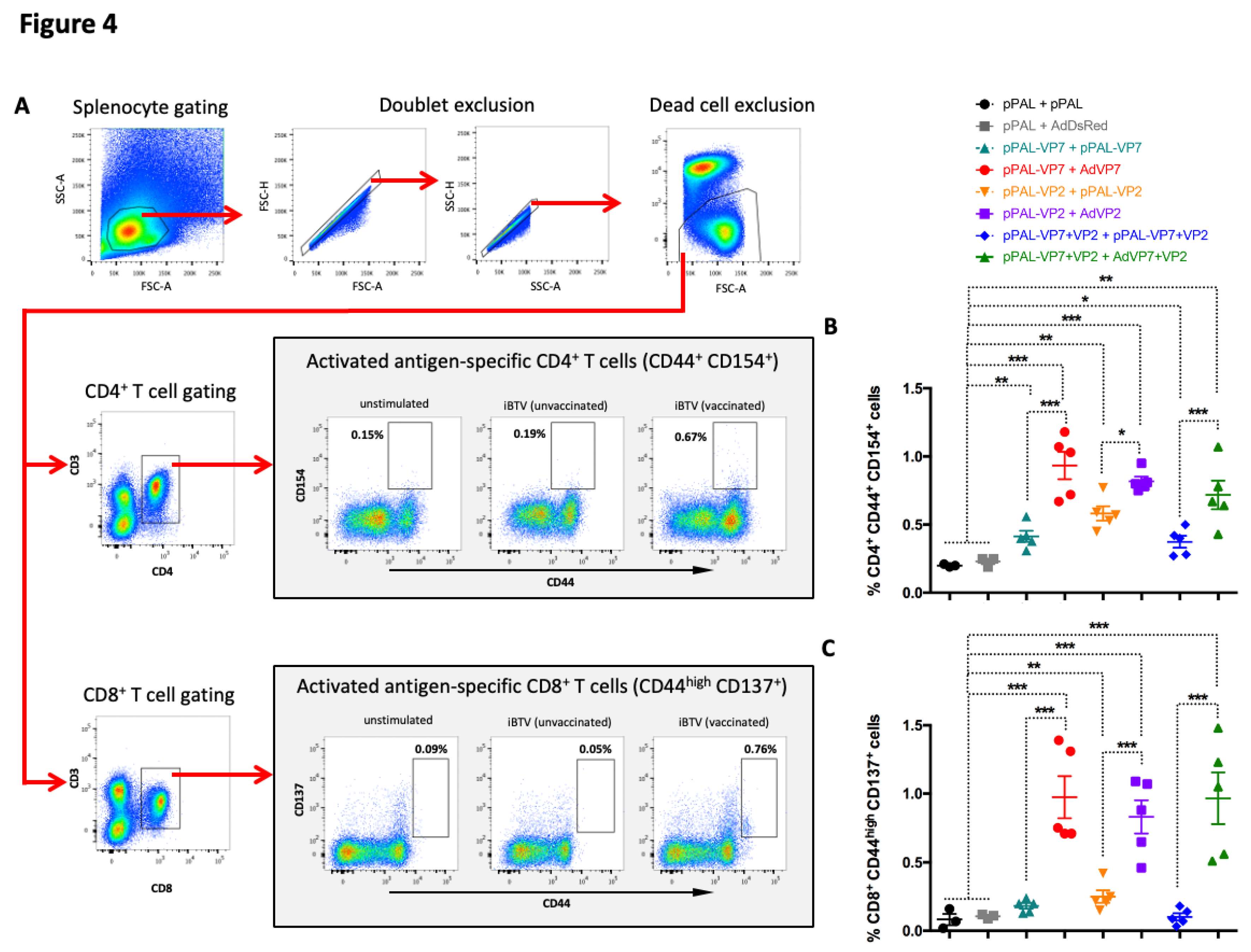
Figure 5.
Heterologous prime-boost improves T cell responses to BTV. (A and B) Splenocytes were seeded in anti-IFN-γ coated ELISPOT plates and stimulated overnight with (A) iBTV or (B) a VP7 immunogenic peptide pool. Splenocytes were discarded and ELISPOT plates revealed for IFN-γ secretion. (C and D) Intracellular cytokine staining (ICS) for CD107a, IFN-γ, IL-2, and TNF-α was performed on iBTV stimulated splenocytes and marker expression analyzed by flow cytometry in CD4+ and CD8+ T cells. Total percentage of (C) CD4+ or (D) CD8+ T cells responding to iBTV stimulation (i.e. expressing at least one stimulation marker). * p<0.05; **p<0.01; *** p<0.001. One-way ANOVA with Fisher’s LSD post-test.
Figure 5.
Heterologous prime-boost improves T cell responses to BTV. (A and B) Splenocytes were seeded in anti-IFN-γ coated ELISPOT plates and stimulated overnight with (A) iBTV or (B) a VP7 immunogenic peptide pool. Splenocytes were discarded and ELISPOT plates revealed for IFN-γ secretion. (C and D) Intracellular cytokine staining (ICS) for CD107a, IFN-γ, IL-2, and TNF-α was performed on iBTV stimulated splenocytes and marker expression analyzed by flow cytometry in CD4+ and CD8+ T cells. Total percentage of (C) CD4+ or (D) CD8+ T cells responding to iBTV stimulation (i.e. expressing at least one stimulation marker). * p<0.05; **p<0.01; *** p<0.001. One-way ANOVA with Fisher’s LSD post-test.
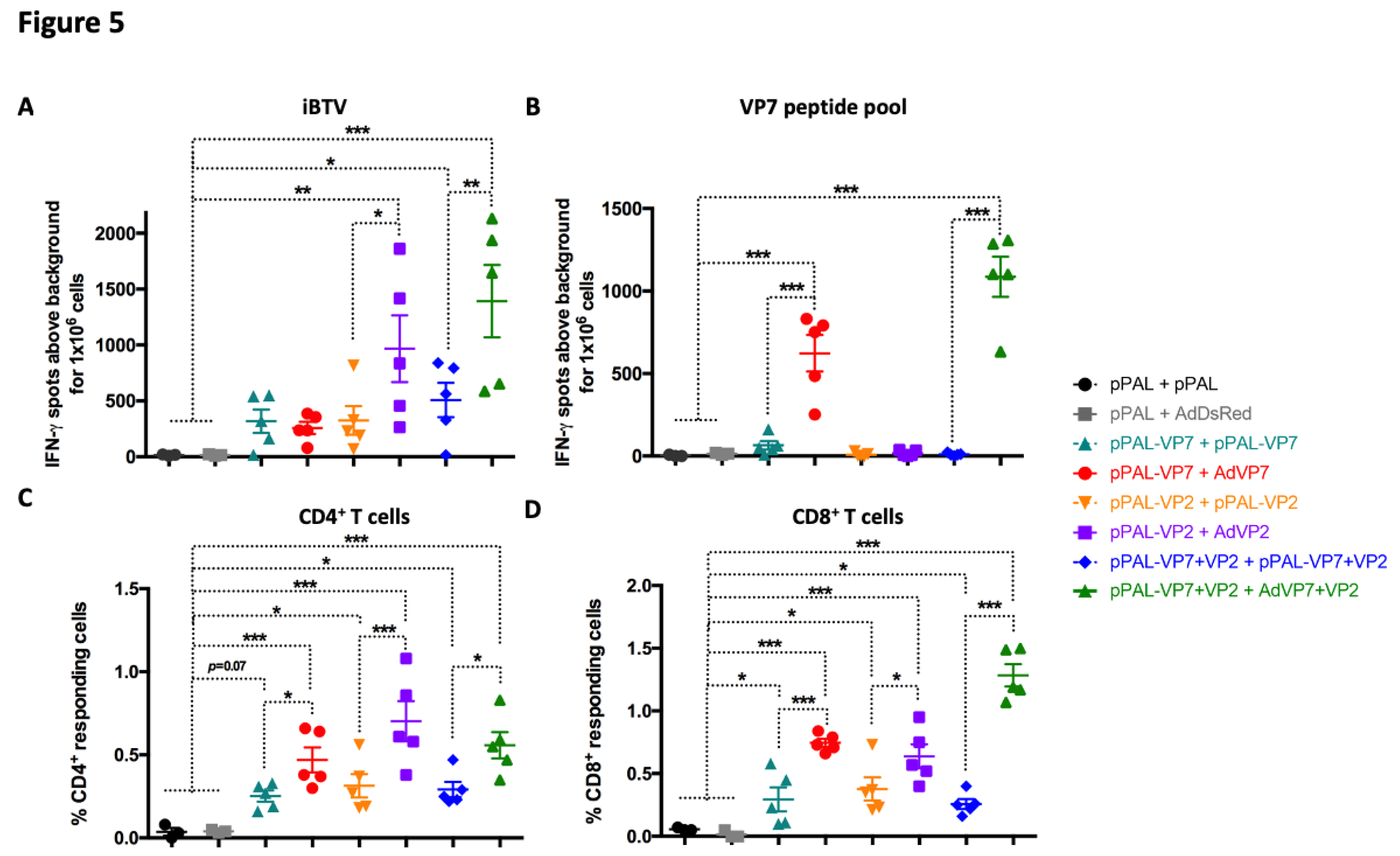
Figure 6.
Heterologous prime-boost with VP7 improves the induction of BTV-specific polyfunctional T cells. Splenocytes were stimulated with iBTV and expression of CD107a, IFN-γ, IL-2, and TNF-α were assessed in CD4+ and CD8+ T cells by intracellular cytokine staining and flow cytometry analysis. (A and B) Expression of CD107a, IFN-γ, IL-2, and TNF-α in (A) CD4+ T cells and (B) CD8+ T cells. * p<0.05; **p>0.01; *** p<0.001 One way ANOVA with Fisher‘s LSD post-test. (C and D) Percentage of simultaneous expression of one, two, three, or four stimulation markers in (C) CD4+ T cells and in (D) CD8+ T cells in response to iBTV for each vaccine regimen.
Figure 6.
Heterologous prime-boost with VP7 improves the induction of BTV-specific polyfunctional T cells. Splenocytes were stimulated with iBTV and expression of CD107a, IFN-γ, IL-2, and TNF-α were assessed in CD4+ and CD8+ T cells by intracellular cytokine staining and flow cytometry analysis. (A and B) Expression of CD107a, IFN-γ, IL-2, and TNF-α in (A) CD4+ T cells and (B) CD8+ T cells. * p<0.05; **p>0.01; *** p<0.001 One way ANOVA with Fisher‘s LSD post-test. (C and D) Percentage of simultaneous expression of one, two, three, or four stimulation markers in (C) CD4+ T cells and in (D) CD8+ T cells in response to iBTV for each vaccine regimen.
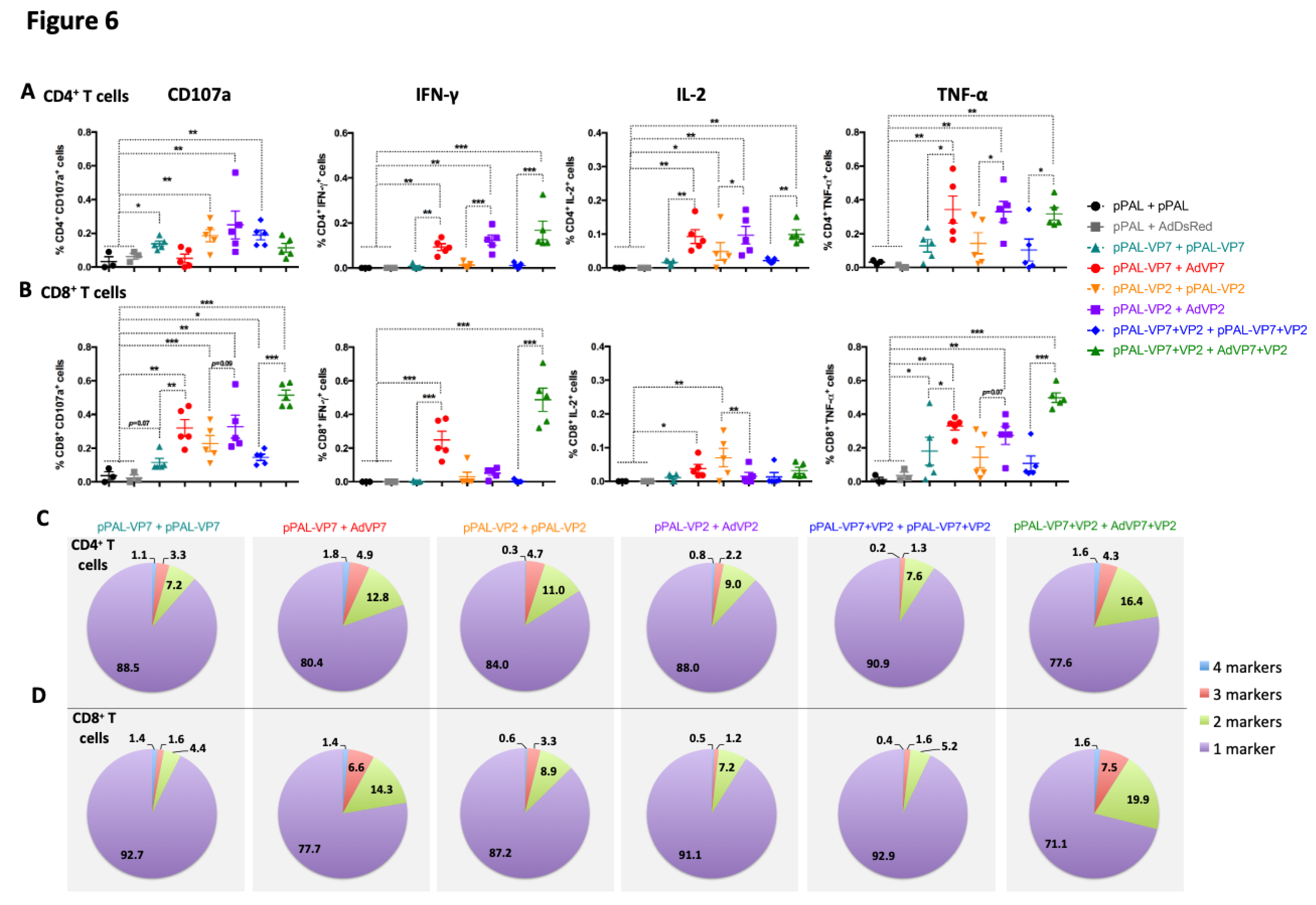

Table 1.
Pearson correlation coefficients (r) between immune parameters induced by vaccination and viral load at day 3 and 5 post-infection. Pearson correlation coefficients r were calculated with GraphPad Prism software using Ct values for BTV S5 and values of immune parameters. All coefficients had significant p values (p<10-3).
Table 1.
Pearson correlation coefficients (r) between immune parameters induced by vaccination and viral load at day 3 and 5 post-infection. Pearson correlation coefficients r were calculated with GraphPad Prism software using Ct values for BTV S5 and values of immune parameters. All coefficients had significant p values (p<10-3).
| Immune parameter | Ct(BTV S5) D3pi | Ct(BTV S5) D5pi | p-value | |||||
| IgG titre | 0.541 | 0.611 | p<10-3 | |||||
| NAb titre | 0.671 | 0.703 | p<10-4 | |||||
| IFN-γ counts (ELISpot) | 0.626 | 0.682 | p<10-5 | |||||
| CD4+CD44+CD154+ cells | 0.600 | 0.653 | p<10-6 | |||||
| CD8+CD44highCD137+ cells | 0.696 | 0.674 | p<10-7 | |||||
| CD4+(CD107a+/ IFN-γ+/ TNF-α+/ IL-2+) cells | 0.694 | 0.768 | ||||||
| CD8+ (CD107a+/ IFN-γ+/ TNF-α+/ IL-2+) cells | 0.791 | 0.814 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



