Submitted:
02 April 2025
Posted:
02 April 2025
You are already at the latest version
Abstract
Thoracic Aortic Aneurysms are life-threatening vascular conditions linked to inherited disorders such as Marfan syndrome, Loeys-Dietz syndrome, vascular Ehlers-Danlos syndrome, and Familial Thoracic Aortic Aneurysms and Dissections. While traditionally associated with extracellular matrix and contractile defects in vascular smooth muscle cells, emerging evidence suggests a key role for mitochondrial dysfunction. Here, we show that overexpression of ACTA2R179H and TGFBR2G357W in murine aortic VSMCs reduces Mitochondrial Transcription Factor A (Tfam) expression, decreases mitochondrial DNA (mtDNA) content, and impairs oxidative phosphorylation, shifting metabolism toward glycolysis. Notably, nicotinamide riboside, a NAD+ precursor, restores mitochondrial respiration, increases Tfam and mtDNA levels, and promotes a contractile phenotype by enhancing actin polymerization and reducing matrix metalloproteinase activity. These findings identify mitochondrial dysfunction as a shared feature in hereditary Thoracic Aortic Aneurysm, not only in Marfan syndrome but also in other genetic forms and highlight mitochondrial boosters as a potential therapeutic strategies.
Keywords:
Mitochondria
; aneurysm
; Marfan Syndrome
; Loeys-Dietz syndrome
; Familial Thoracic Aortic Aneurysm
; Nicotinamide Riboside
1. Introduction
Different inherited disorders significantly affect the structural integrity of the aortic wall, resulting in thoracic aortic aneurysms (TAAs) [1]. This vascular condition is defined by the permanent dilation of the thoracic aorta, which, if not addressed, can lead to life-threatening complications such as aortic rupture or dissection, potentially resulting in fatal hemorrhaging [2]. Many of these disorders are associated with mutations in genes related to the extracellular matrix (ECM) or the contractile apparatus of vascular smooth muscle cells (VSMCs). Among these, Marfan syndrome (MFS) is one of the most prevalent connective tissue disorders, caused by mutations in the FBN1 gene that encodes the ECM protein Fibrillin-1. Patients with MFS typically present elongated bones, ectopia lentis, and reduced life expectancy, largely due to the progression of TAAs [3]. Other genetic conditions impacting the aorta include Loeys–Dietz syndrome, which arises from mutations in genes involved in TGF-β signaling and shares certain skeletal characteristics with MFS. Vascular Ehlers–Danlos syndrome is mainly associated with mutations in the COL3A1 gene (which encodes collagen type III), leading to joint and skin hyperelasticity, short stature, and an increased risk of aortic dissection [5]. In familial nonsyndromic thoracic aortic aneurysms and dissections (FTAAD), while most causative genes remain unidentified, around 20% of cases are associated with mutations in genes that support the contractile apparatus of smooth muscle cells, including smooth muscle actin and myosin (ACTA2 and MYH11 respectively). The primary complication in all these inherited connective tissue disorders is the heightened risk of developing TAAs. Current management strategies for aortic aneurysms largely depend on surgical prophylactic repair [6]. Therefore, it is crucial to identify novel molecular mediators involved in the pathophysiology of TAAs to inform the development of new pharmacologic approaches.
VSMCs found in the medial layer of arteries, are essential for the contraction of the vascular wall and the regulation of vessel diameter. These cells can transition from a quiescent, contractile phenotype to a secretory phenotype, which is linked to increased proliferation, extracellular matrix (ECM) accumulation, and subsequent medial degeneration, all of which contribute to aneurysm formation [7]. Recent studies have begun to investigate the connections between cellular adhesion, cytoskeletal reorganization, and cellular metabolism, highlighting the critical role of ECM composition and stiffness in regulating metabolism. Our previous research has demonstrated that mitochondrial metabolism is a key regulator of the VSMC phenotype during aortic remodeling in Marfan syndrome and is finely tuned by ECM composition [8,9]. Furthermore, we have shown the significance of mitochondrial metabolism in aneurysm development in a conditional mouse model with specific mitochondrial dysfunction in VSMCs through the depletion of Tfam (mitochondrial transcription factor A) [9].
Based on these findings, we hypothesize that mitochondrial dysfunction in VSMCs is a key factor in aortic remodeling in Marfan syndrome and other type of hereditable-TAAs, therefore, restoring mitochondrial function could prevent aneurysm progression and aortic dissection. Additionally, enhancing mitochondrial function with nicotinamide riboside (NR), a precursor of NAD+, presents a promising therapeutic strategy for managing aortic aneurysms associated with Marfan syndrome, with the potential to prevent aortic dissection.
2. Material and Methods
2.1. Cell Procedures
The isolation and culture of primary vascular smooth muscle cells (VSMCs) from mice were carried out following the protocol described in [16]. Tissue digestion was performed using a solution containing collagenase (1 mg/ml) and elastase (0.5 mg/ml) from Worthington Biochemical until a single-cell suspension was achieved. All experiments using primary VSMCs were conducted between passages 2 and 4. Lentiviral transduction was performed over 5 hours at a multiplicity of infection (MOI) of 3. Subsequently, the medium was replaced with fresh DMEM supplemented with 10% FBS, and the cells were cultured for an additional seven days. Cells were then treated with NR for three more days and serum-starved for 16 hours. HEK-293T (CRL-1573) and Jurkat (Clone E6-1, TIB-152) cell lines were employed for the production of high-titer lentivirus and for titration, respectively. All cell lines were verified to be mycoplasma-free. For cell immunostaining, cells were fixed with 4% paraformaldehyde for 10 minutes and permeabilized with 0.3% Triton X-100 in PBS for 10 minutes. Samples were incubated with 647-Phalloidin (1/500 dilution, Millipore) and DAPI for 15 minutes. Images were acquired using a Zeiss-LSM-800 microscope with a 40x oil immersion objective and the ZEN acquisition software.
2.2. Lentivirus Production and Infection
Lentiviruses expressing shRNA targeting murine shFbn1, and control shRNA were purchased from Sigma-Aldrich. Lentiviruses expressing ACTA2R178H and TGFBR2G317W mutations, along with control viruses, were generously provided by Mark Lindsay [4]. Pseudotyped lentiviruses were generated via transient calcium phosphate transfection of HEK-293T cells and concentrated from the culture supernatant through ultracentrifugation (2 hours at 128,000xg; Ultraclear Tubes; SW28 rotor and Optima L-100 XP Ultracentrifuge; Beckman). The viral particles were suspended in cold, sterile PBS and titrated by transducing Jurkat cells for 48 hours. Transduction efficiency (measured as GFP-positive cells) and cell viability (using propidium iodide staining) were assessed by flow cytometry.
2.3. Extracellular Flux Analysis and Metabolic Assays
Oxygen consumption rates (OCRs) were measured using the XF-96 Extracellular Flux Analyzer (Seahorse Bioscience). A total of 25,000 mouse aortic VSMCs were seeded in unbuffered DMEM medium containing 25 mM glucose and 1 mM CaCl₂. Baseline measurements were recorded three times, followed by sequential addition of oligomycin (1 mM), fluoro-carbonyl cyanide phenylhydrazone (FCCP; 1.5 mM), and a combination of rotenone (100 nM) and antimycin A (1 mM). Extracellular lactate levels were determined using the Accutrend® Plus system (Roche), analyzing 20 μL of conditioned medium collected after 48 hours of activation. Lactate concentrations were normalized to protein content in cell extracts.
2.4. Gelatin Zymography
Supernatants form cell culture were prepared as described [16]. Extracts (15 μg) were fractioned under nonreducing conditions on 10% SDS–polyacrylamide gels containing 1% gelatin (Sigma). Gels were washed three times in 2.5% Triton X-100 for 2 hours at room temperature, incubated overnight at 37 °C in 50 mM Tris-HCl pH 7.5, 10 mM CaCl2 and 200 mM NaCl, and stained with Coomasie Blue. The areas of gelatinolytic or MMP activity were visualized as transparent bands. Images were analyzed with Quantity One software (Bio-Rad).
2.5. Quantitative PCR
Total RNA was extracted with TRIzol (Life Technologies). To quantify mitochondrial DNA (mtDNA), total DNA from cells was isolated using the SurePrep kit (Fisher Scientific) following the manufacturer’s protocols. DNA was amplified by real-time PCR using primers targeting cytochrome c oxidase subunit 1 (mt-Co1) and mitochondrial 16S rRNA and normalized to nuclear-encoded control genes B2M and H2K. Quantitative PCR (qPCR) reactions were performed in triplicate using SYBR Master Mix (Promega), following the manufacturer's instructions. Post-amplification melting curve analysis was conducted to verify probe specificity, ensuring a single melting temperature (Tm) peak per reaction. Relative mRNA quantification was carried out using the 2−ΔCT method, with B2M, YWHAZ, and PP1A serving as normalization controls. Expression fold changes were calculated relative to control mRNA levels. qPCR was performed with the following primers:
Tfam CAGGAGGCAAAGGATGATTC; CCAAGACTTCATTTCATTGTCG
Ppargc1a GGCACGCAGCCCTATTCA; CGACACGGAGAGTTAAAGGAAGA
mt-Co1 CTCGCCTAATTTATTCCACTTCA; GGGGCTAGGGGTAGGGTTAT
Mmp2 CAAGTTCCCCGGCGATGTC; TTCTGGTCAAGGTCACCTGTC
Mmp9 CACCACAGCCAACTATGACCA; CAGGAAGACGAAGGGGAAGAC
Spp1 ATGAGATTGGCAGTGATTTG; CATCCTTTTCTTCAGAGGAC
Col1a1 GCTCCTCTTAGGGGCCACT; CCACGTCTCACCATTGGGG
16s Mt-rRNA CCGCAAGGGAAAGATGAAAGAC; TCGTTTGGTTTCGGGGTTTC
Hk2 nDNA GCCAGCCTCTCCTGATTTTAGTGT; GGGAACACAAAAGACCTCTTCTGG
B2m TACATACGCCTGCAGAGTTAAGCA; TGATCACATGTCTCGATCCCAG
Pp1a ACGCCACTGTCGCTTTTC; GCAAACAGCTCGAAGGAGAC
Ywhaz TTACTTGGCCGAGGTTGCT; TGCTGTGACTGGTCCACAAT
3. Results
To investigate whether mitochondrial metabolism is affected in other thoracic aortic aneurysms and dissections (TAADs), we modeled familial thoracic aortic aneurysm and dissections (FTAAD) and Loeys-Dietz syndrome (LDS) by using lentivectors to overexpress human ACTA2R179H and TGFBR2G357W in primary murine aortic vascular smooth muscle cells (VSMCs). These constructs were generously provided by Mark Lindsay [4]. Overexpression of ACTA2R179H and TGFBR2G357W resulted in reduced levels of Tfam mtDNA and decreased mRNA expression of mitochondrial regulators (Figure 1A). Flux analysis of the oxygen consumption rate (OCR), an indicator of mitochondrial oxidative phosphorylation, demonstrated diminished mitochondrial respiration in TAA-VSMCs (Figure 1B). This decline in mitochondrial function was accompanied by increased extracellular lactate production, suggesting a shift toward glycolysis as the primary source of cellular energy in the presence of TAAD mutations (Figure 1B). Notably, incubation with nicotinamide riboside (NR) reversed the impact of both mutations on these metabolic parameters (Figure 1A,B).
Furthermore, NR enhanced VSMCs-contractile phenotype, as indicated by increased F-actin polymerization in FBN1-silenced cells modeling Marfan syndrome, as well as in ACTA2R179H and TGFBR2G357W -VSMCs (Figure 2A,B), while also reducing the expression and activity of matrix metalloproteinases and other secretory markers (Figure 2C,D).
4. Discussion
Our findings suggest that reduced Tfam expression and mtDNA levels induce a metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis in VSMCs affected by familial TAAs. This metabolic reprogramming is increasingly recognized as a hallmark of aneurysm pathogenesis, contributing to vascular remodeling and disease progression [9,10,11]. Mitochondrial dysfunction alters bioenergetics and cellular processes such as apoptosis and matrix remodeling, exacerbating aneurysm development [9].
Improving mitochondrial fitness in vitro restored the contractile phenotype of these cells, supporting the idea that mitochondrial dysfunction plays a causative role in TAA pathogenesis. This aligns with prior studies showing that mitochondrial-targeted therapies can improve vascular smooth muscle function and reduce maladaptive phenotypic switching [9,12,13,14].
Notably, nicotinamide riboside (NR) increased Tfam expression, mtDNA levels, and mitochondrial metabolism while reducing the secretory phenotype in TAA VSMCs. NR, a precursor of NAD+, is known to promote mitochondrial biogenesis and improve metabolic function in vascular diseases [9]. These findings suggest that NR may counteract the glycolytic shift observed in TAA VSMCs, potentially reducing disease progression.
Our results reinforce the idea that metabolic remodeling is a key driver of hereditary TAA progression. Given the limited therapeutic options, targeting mitochondrial metabolism, particularly with NAD+ boosters, could provide a promising strategy for treating genetic aortopathies [15]. Further studies are needed to explore the long-term efficacy of mitochondrial-targeted therapies in aneurysm prevention and treatment.
Author Contributions
JO. and N.M-B. conceptualized and did part of the experiments; A.R-O; D.M-R. did some part of the experi-ments and wrote parts of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
A.R-O is supported by the Conchita-Rábago Foundation 2024 grant. N M-B is supported by Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (ISCiii/FEDER, PI21/01126 and PI24/00180, CP19/00151 Miguel Servet contract), Sociedad Española de Arteriosclerosis, and CIBERCV, Spain.. J.O. is supported by a Ramón y Cajal contract (RYC2021-033343-I), and grant from Spanish Science Ministry (PID2022-1377300A-100) and Marfan Spanish association (SIMA, www.Marfan.es).
Institutional Review Board Statement
The experiment was approved by IIS-FJD Ethics Committee and the Madrid regional authorities, conformed to EU Directive 2010/63/EU and Recommendation 2007/526/EC (Approval Code: PROEX 74.7/23), date of approval: 5th September 2023
Data Availability Statement
The data materials and methods supporting the findings of this study are available from the corresponding author on request.
References
- Quintana, R.A., and Taylor, W.R. (2019). Cellular Mechanisms of Aortic Aneurysm Formation. Circ Res 124, 607–618. [CrossRef]
- Rylski, B., Schilling, O., and Czerny, M. (2023). Acute aortic dissection: evidence, uncertainties, and future therapies. Eur Heart J 44, 813–821. [CrossRef]
- Zeigler, S.M. , Sloan, B., and Jones, J.A. (2021). Pathophysiology and Pathogenesis of Marfan Syndrome. In, pp. 185–206. [CrossRef]
- Lino Cardenas, C.L., Kessinger, C.W., Cheng, Y., MacDonald, C., MacGillivray, T., Ghoshhajra, B., Huleihel, L., Nuri, S., Yeri, A.S., Jaffer, F.A., et al. (2018). An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat Commun 9, 1009. [CrossRef]
- Kuivaniemi, H., and Tromp, G. (2019). Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene 707, 151–171. [CrossRef]
- Kuzmik, G.A., Sang, A.X., and Elefteriades, J.A. (2012). Natural history of thoracic aortic aneurysms. J Vasc Surg 56, 565–571. [CrossRef]
- Benke, K., Ágg, B., Szilveszter, B., Tarr, F., Nagy, Z.B., Pólos, M., Daróczi, L., Merkely, B., and Szabolcs, Z. (2013). The role of transforming growth factor-beta in Marfan syndrome. Cardiol J 20, 227–234. [CrossRef]
- Oller, J., Gabandé-Rodríguez, E., Roldan-Montero, R., Ruiz-Rodríguez, M.J., Redondo, J.M., Martín-Ventura, J.L., and Mittelbrunn, M. (2022). Rewiring Vascular Metabolism Prevents Sudden Death due to Aortic Ruptures—Brief Report. Arterioscler Thromb Vasc Biol 42, 462–469. [CrossRef]
- Oller, J., Gabandé-Rodríguez, E., Ruiz-Rodríguez, M.J., Desdín-Micó, G., Aranda, J.F., Rodrigues-Diez, R., Ballesteros-Martínez, C., Blanco, E.M., Roldan-Montero, R., Acuña, P., et al. (2021). Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm. Circulation 143, 2091–2109. [CrossRef]
- Nguyen, T.A.V., Lino, C.A., Hang, H.T., Alves, J.V., Thang, B.Q., Shin, S.J., Sugiyama, K., Matsunaga, H., Takeyama, H., Yamashiro, Y., et al. (2023). Protective Role of Endothelial Fibulin-4 in Valvulo-Arterial Integrity. J Am Heart Assoc 12. [CrossRef]
- Zong, Y., Li, H., Liao, P., Chen, L., Pan, Y., Zheng, Y., Zhang, C., Liu, D., Zheng, M., and Gao, J. (2024). Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther 9, 124. [CrossRef]
- van der Pluijm, I., Burger, J., van Heijningen, P.M., IJpma, A., van Vliet, N., Milanese, C., Schoonderwoerd, K., Sluiter, W., Ringuette, L.-J., Dekkers, D.H.W., et al. (2018). Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc Res 114, 1776–1793. [CrossRef]
- Zhang, X., Zhang, Z., Wan, S., Qi, J., Hao, Y., An, P., Luo, Y., and Luo, J. (2024). Ameliorative Effect of Coenzyme Q10 on Phenotypic Transformation in Human Smooth Muscle Cells with FBN1 Knockdown. Int J Mol Sci 25, 2662. [CrossRef]
- Hibender, S., Franken, R., Van Roomen, C., Ter Braake, A., Van Der Made, I., Schermer, E.E., Gunst, Q., Van Den Hoff, M.J., Lutgens, E., Pinto, Y.M., et al. (2016). Resveratrol Inhibits Aortic Root Dilatation in the Fbn1 C1039G/+ Marfan Mouse Model. Arterioscler Thromb Vasc Biol 36, 1618–1626. [CrossRef]
- Katsyuba, E., Romani, M., Hofer, D., and Auwerx, J. (2020). NAD+ homeostasis in health and disease. Preprint at Nature Research. [CrossRef]
- Oller, J., Méndez-Barbero, N., Ruiz, E.J., Villahoz, S., Renard, M., Canelas, L.I., Briones, A.M., Alberca, R., Lozano-Vidal, N., Hurlé, M.A., et al. (2017). Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med 23, 200–212. [CrossRef]
Figure 1.
Nicotidamide Riboside treatment improves mitochondrial function in VSMCs with Thoracic Aortic Aneurysm mutations. Effect of NR on mouse primary VSMCs overexpressing ACTA2R179 or TGFBR2G357W (to model FTAAD and LDS, respectively). VSMCs was insolated and transduced with lentivectors. After 2 days of transduction, VSMCs were treated with NR (0.25 mg/ml) for 5 days. (A) qPCR analysis of relative mtDNA content and RT–qPCR analysis of Tfam, Mt-Co1 and Ppargc1a mRNA expression. (B) OCR measured by SeahorseTm at basal respiration and after addition of oligomycin (I) and FCCP (II) to measure maximal respiration, followed by a combination of rotenone and antimycin A (III). The right-most panel shows extracellular lactate levels. Data are mean ± s.e.m. Statistical significance was assessed by one-way ANOVA: *P < 0.05, **P < 0.01 vs. Control; # P < 0.05, ## P < 0.01 vs. NR treatment.
Figure 1.
Nicotidamide Riboside treatment improves mitochondrial function in VSMCs with Thoracic Aortic Aneurysm mutations. Effect of NR on mouse primary VSMCs overexpressing ACTA2R179 or TGFBR2G357W (to model FTAAD and LDS, respectively). VSMCs was insolated and transduced with lentivectors. After 2 days of transduction, VSMCs were treated with NR (0.25 mg/ml) for 5 days. (A) qPCR analysis of relative mtDNA content and RT–qPCR analysis of Tfam, Mt-Co1 and Ppargc1a mRNA expression. (B) OCR measured by SeahorseTm at basal respiration and after addition of oligomycin (I) and FCCP (II) to measure maximal respiration, followed by a combination of rotenone and antimycin A (III). The right-most panel shows extracellular lactate levels. Data are mean ± s.e.m. Statistical significance was assessed by one-way ANOVA: *P < 0.05, **P < 0.01 vs. Control; # P < 0.05, ## P < 0.01 vs. NR treatment.
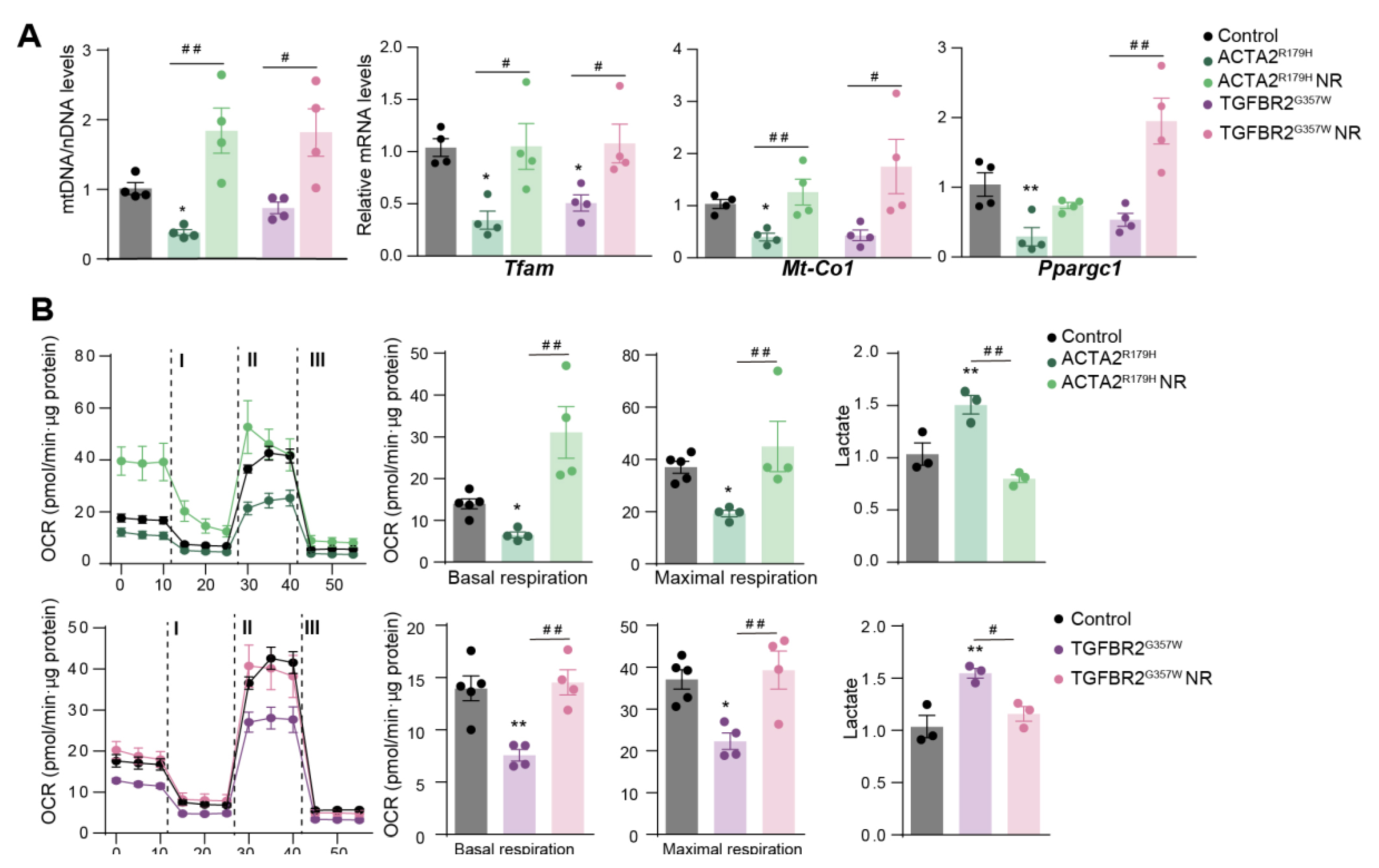
Figure 2.
Nicotinamide Riboside treatment restores the balance between the synthetic and contractile phenotypes in VSMCs with Thoracic Aortic Aneurysm mutations. Effect of NR on mouse primary VSMCs silenced for Fbn1 (ShFbn1), overexpressing ACTA2R179 or TGFBR2G357W (to model MFS, FTAAD and LDS, respectively). VSMCs was isolated and transduced with lentivectors. After 2 days of transduction, VSMCs were treated with NR (0.25 mg/ml) for 5 days. (A) Representative confocal imaging of filamentous actin (F-actin) stained with phalloidin (green); nuclei are stained with Dapi (Blue); (n=4) and quantification in murine VSMCs silenced for Fbn1 (ShFbn1 or control shRNA, shControl). (B) Representative confocal imaging of filamentous actin (F-actin) stained with phalloidin (green); nuclei are stained with Dapi (Blue); (n=4) and quantification in murine VSMCs in VSMCs overexpressing ACTA2R179 or TGFBR2G357W. (C) Representative Gelatin-zymography analysis of Mmp2 and Mmp9 activity in 24h in conditional medium and quantification. (D) RT-qPCR analysis of Mmp9, Mmp2, Spp1, and Col1a1 mRNA expression. Data are mean ± s.e.m. Statistical significance was assessed by one-way ANOVA: **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control, ## P < 0.01, ###P < 0.001, vs. NR treatment.
Figure 2.
Nicotinamide Riboside treatment restores the balance between the synthetic and contractile phenotypes in VSMCs with Thoracic Aortic Aneurysm mutations. Effect of NR on mouse primary VSMCs silenced for Fbn1 (ShFbn1), overexpressing ACTA2R179 or TGFBR2G357W (to model MFS, FTAAD and LDS, respectively). VSMCs was isolated and transduced with lentivectors. After 2 days of transduction, VSMCs were treated with NR (0.25 mg/ml) for 5 days. (A) Representative confocal imaging of filamentous actin (F-actin) stained with phalloidin (green); nuclei are stained with Dapi (Blue); (n=4) and quantification in murine VSMCs silenced for Fbn1 (ShFbn1 or control shRNA, shControl). (B) Representative confocal imaging of filamentous actin (F-actin) stained with phalloidin (green); nuclei are stained with Dapi (Blue); (n=4) and quantification in murine VSMCs in VSMCs overexpressing ACTA2R179 or TGFBR2G357W. (C) Representative Gelatin-zymography analysis of Mmp2 and Mmp9 activity in 24h in conditional medium and quantification. (D) RT-qPCR analysis of Mmp9, Mmp2, Spp1, and Col1a1 mRNA expression. Data are mean ± s.e.m. Statistical significance was assessed by one-way ANOVA: **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control, ## P < 0.01, ###P < 0.001, vs. NR treatment.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



