Submitted:
02 April 2025
Posted:
02 April 2025
You are already at the latest version
Abstract
Multiple immune cells and stromal components comprise the tumor microenvironment (TME). In this complex TME, tumor-associated macrophages (TAMs) are spatially heterogeneous. Tumor heterogeneity affects tissue remodeling, immune response, angiogenesis, and metastatic potential. This review highlights the roles of macrophages in TME, immunoregulation, and immunotherapy. Also, intricate relationships between TAMs and various immune cells in the TME and immune evasion have been explored comprehensively. TAM heterogeneity and complex interactions with immune cell types may lead to potential therapeutic approaches. Similarly, classic immune checkpoint inhibitors like anti-PD-L-1 and anti-PD-1 offer promising tumor therapies. TAMs regulate PD-1/PD-L1 immunosuppression by suppressing T cell recruitment and function using cytokines, superficial immune checkpoint ligands, and exosomes. TAM’s functions and mechanisms in PD-1/PD-L1 blocker resistance are described in detail. The insights into intricate relationships of TAMs with the immune cells within TME and the role of immune checkpoint inhibitors will further help explore potential future therapeutic advances.
Keywords:
Macrophage
; Polarization
; TAM
; Immune regulation
; Immune checkpoint
; Cancer
; Immunotherapy
1. Introduction
Cancer ranks high among global public health concerns and is leading to high fatalities in multiple countries. It is anticipated that 2,041,910 new cases of cancer and 618,120 cancer-related deaths are projected to occur in the US in 2025 [1]. The high cost of treating cancer patients and their families, as well as the whole economy, has been a significant drag on healthcare investment. Accordingly, to further decrease the mortality and morbidity rates, tumor prevention and treatment are of utmost urgency. Despite the widespread use of chemotherapy, radiation therapy, and surgery as therapies for cancer, some patients have had unsuccessful outcomes [2,3].
In animals, macrophages penetrate various tissues, where they co-develop with the organs they occupy. Until their environment changes, these macrophages generally keep their numbers and morphologies the same. There are three main types of macrophages found in almost every tissue: those that originate in the blood or bone marrow, those that dwell in the tissue itself, and those that arise in the bone marrow but do not produce any inhibitory cells [4]. Due to their phenotypic and functional diversity, macrophages are essential to innate and adaptive immunity. Embryonic hematopoietic stem cells become lung alveolar macrophages, bone osteoclasts, liver, and Kupffer cells. Postnatal bone marrow precursors, specifically monocytes, may become macrophages in a tissue inflammation or steady-state [5].
Immunology, tissue and systemic inflammation, and regeneration are all processes in which macrophages participate. They perform phagocytosis, antigen presentation, microbial cytotoxicity defense, cytokine and complement components, and other tasks [6]. Likewise, immunological tolerance, tissue repair, and inflammation suppression are all achieved by macrophages [7]. Moreover, TAMs help establish the TME. Many cancers include TAMs and increase tumor development, invasion, metastasis, and treatment resistance [8]. Furthermore, macrophage’s functional differences are linked to plasticity, and TME chemicals modulate their functional phenotype [9].
The fourth therapeutic method to develop after conventional therapy is chimeric antigen receptor (CAR)-T cell therapy and immune-checkpoint inhibitors. Some advanced-stage tumors have demonstrated encouraging results when treated with immunotherapies such as CAR-T cell therapy and programmed death receptor 1 (PD-1) inhibitors, which have recently gained clinical approval [10,11,12,13]. Still, there is room for improvement in the effectiveness of PD-1 inhibitors since they were only partially effective in a subset of cancer patients [14].
Regarding tumor development, metastasis, and therapeutic response, the TME is a major player. [15,16]. Investigating the TME to increase the response rate and create novel cancer immunotherapy techniques is essential. Research indicates that macrophages are among the most important immune cells found in the TME [16]. Macrophages, through their phagocytosis function, can initially clear tumor cells. However, when stimulated by TME factors, they eventually change into TAMs with the M2 phenotype, which increases metastasis and tumor growth by suppressing immunity, triggering angiogenesis, and bolstering cancer stem cells [17]. Studying macrophages in the TME is crucial, and future anti-tumor efforts may benefit from targeting macrophages.
2. Macrophage Origin and Polarization
Monocytes in the bloodstream are the progenitors of macrophages, and there is a great deal of diversity among these immune cells [18]. They may originate from tissue macrophages or blood monocytes drawn to chemokines like CCL2 or CSF-1 [19]. Classically activated macrophages (M1) and alternatively activated macrophages (M2) are two types of macrophages that their phenotype and function may identify. [20].
Macrophage Polarization and Differentiation in TME
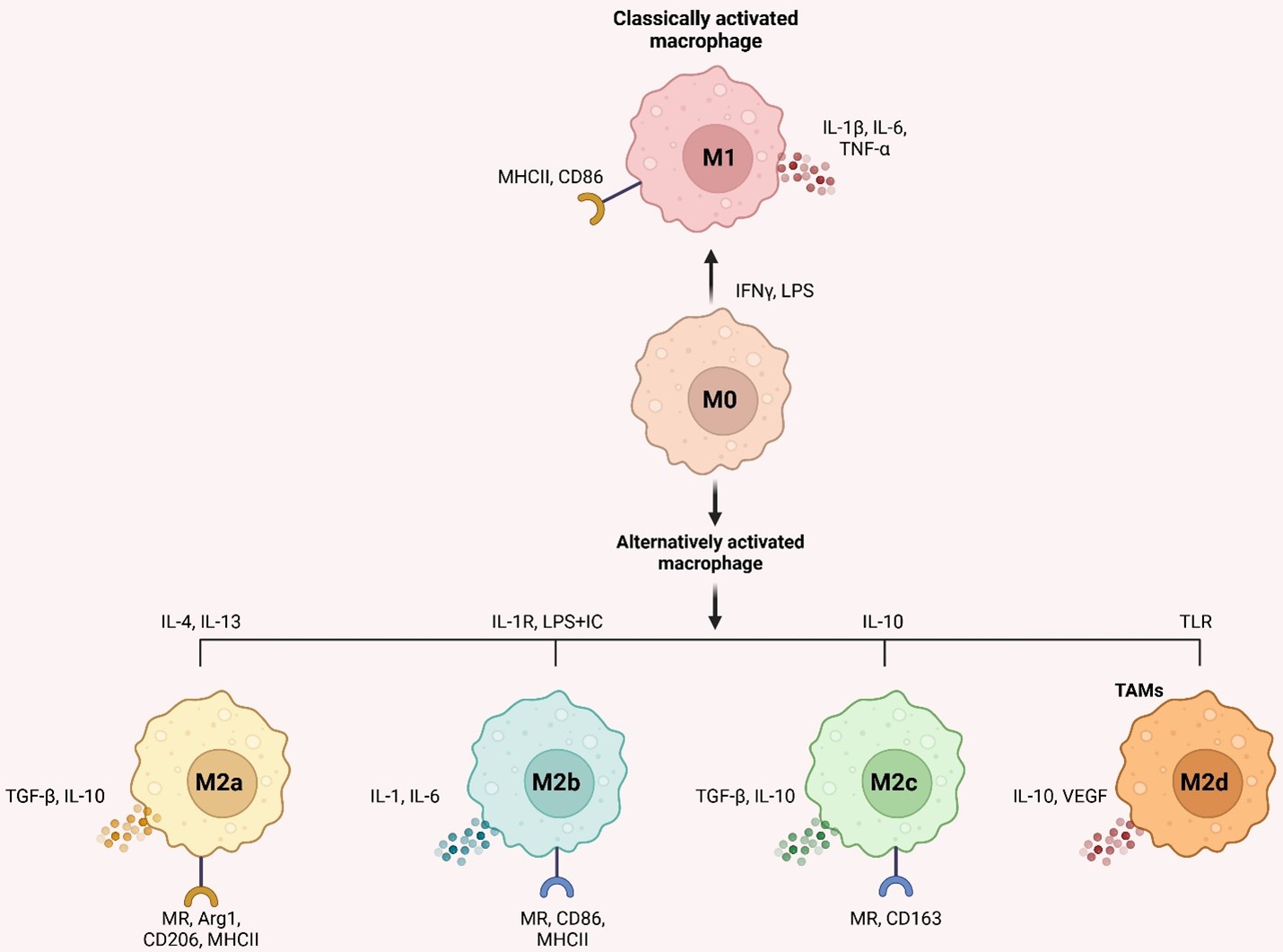
Environmental signals cause macrophages to differentiate into distinct phenotypes for host defense, wound healing, immunological control, and cancer [21]. The malleability of undifferentiated macrophages (M0) allows them to be polarized into two types of activation: conventional (M1) and alternative (M2) [22]. IFN-γ and LPS from bacteria activate M1 macrophages, which release inflammatory molecules such as IL-1β, IL-6, and TNF-α and have pro-inflammatory and anti-tumor characteristics. When tumors grow, spread, and evade the immune system, M2 macrophages are vital, influenced by IL-4 and IL-13 and released by T-helper 2 (Th2) cells. In addition, they release anti-inflammatory components such as IL-10 and TGF-β [23].
Furthermore, abnormalities in the IKKβ/NF-κB pathway may cause TAM polarization [24]. IL-4 binding to its receptor may phosphorylate STAT6, causing M2-like macrophage polarization via the JAK/STAT6 signaling pathway. To induce polarization, phosphorylated STAT6 binds to KLF4 and PPAR-γ simultaneously [25]. M2-like macrophages become M1-like when IL-4's receptor connection is blocked [26]. Multiple mediators, including as IL-4, TGF-β, IL-10, and BMP-7, activate M2 polarization via the PI3K/Akt pathway [27]. Non-coding RNA (lncRNA)-Xist knockdown or miR-101 overexpression in M1-like macrophages suppresses C/EBPα, and KLF6 synthesis leads to M1-to-M2 polarization [28]. Antitumor therapies that regulate M1/M2 polarization are promising owing to TAM’s higher adaptability.
It's important to note that macrophages are not merely M1 and M2 immune cells. IL-4/IL-13, LPS/IL-1 receptor, and IL-10 promote activated M2 macrophages into M2a, M2b, and M2c, respectively [29]. According to Mantovani et al. [30]. M2a and M2b macrophages modulate immunological response and enhance the response of Th2 cells, whereas M2c suppresses immune response and remodels tissue. M2d macrophages (TAMs) activated by Toll-like receptors and expressing VEGF and IL-10 were also hypothesized [31]. M2d macrophages promote tumor growth and angiogenesis [32]. Figure 1 depicts inducers, surface indicators, and cytokines. Targeting TAMs in the TME is becoming more critical since macrophage activation may control tumor growth and inflammation.
3. Macrophages Inside the TME Accelerate Tumor Development
At various points during tumor formation, macrophages play an essential role. Through the first steps of tumor growth, immune cells such as macrophages are drawn to the tumor stroma by cytokines and exosomes released by tumor cells. After they arrive, macrophages aid migration, metastasis, and tumor growth [17]. Macrophages play an essential role in the TME. They can induce tumor necrosis by assertive phagocytosis [33], but there is evidence that TAMs play a substantial role in further tumor advancement. In cancers, TAM increases cancer cell expansion and proliferation, angiogenesis, and lymph angiogenesis and suppresses the effector T cell immune response [34].
At the outset of lung cancer, TAM exhibits characteristics that are thought to be pro-inflammatory and anti-tumor (M1 type). Still, as the disease advances, TAM begins to show characteristics thought to be pro-inflammatory and tumor-promoting [17]. TAM can potentially regulate the immune system and non-immune processes to promote tumor formation [35,36,37]. One example is TAM secretion of many pro-angiogenic factors, which aid in tumor angiogenesis and metastasis [38].
Half of the TME's cells are macrophages, mostly M2 phenotypes. A negative correlation has been observed between tumor micro-vessels, the number of macrophages in the TME, and survival results in patients with non-small cell lung cancer (NSCLC) [39,40]. Recently, it has seen an explosion of research using next-gen and single-cell sequencing technologies that have unveiled the TAM's complex management of the ever-changing cancer environment [20,40]. The primary objective of this section is to deliver an overview of TAM and its role in tumors.
3.1. Implications of M1 Type TAM on Regression of Tumors
It was shown that TAM's primary deleterious process is suppressing anti-tumor immunity. One immunostimulatory cytokine that TAM may be able to lower is IL-12 production. This cytokine can stimulate NK and cytotoxic CD4+ T cells, potentially eradicating tumors [41]. In addition, TAM's production of immunosuppressive elements, including IL-10, transforming growth factor-β, and prostaglandin E2, might have a role in cancer formation [17,42,43].
Arginase 1 (ARG1) is a hydrolase that regulates the breakdown of L-arginine; it is one of the enzymes that TAM may directly block T cell activity via. IL-4, IL-10, and hypoxia are the mediators of many signaling pathways that generate ARG1, which impacts T-cell function by reducing the activity of the semi-essential amino acid L-arginine [43]. Additionally, by blocking the expression of programmed cell death ligand 1 (PD-L1) and B7 homolog 1 on T cells, TAM may enhance T-cell apoptosis [20,43].
3.2. M2 Type TAM's Role in Driving Tumor Growth
Proinflammatory cytokines, such as TNF-α, IL-6, and IL-11, mediate the tumor-promoting function of M2 macrophages in cancer cells by activating the nuclear factor-κB (NF-κB) and signal transduction and activator of transcription 3 (STAT3) pathways [17,20,36,43,44]. Furthermore, M2 TAM accelerated tumor growth via elevated VEFG-A and VEFG-C expression, which enhanced angiogenesis and lymph angiogenesis [36,38,43].
4. Crosstalk Between TAMs and Tumor Cells
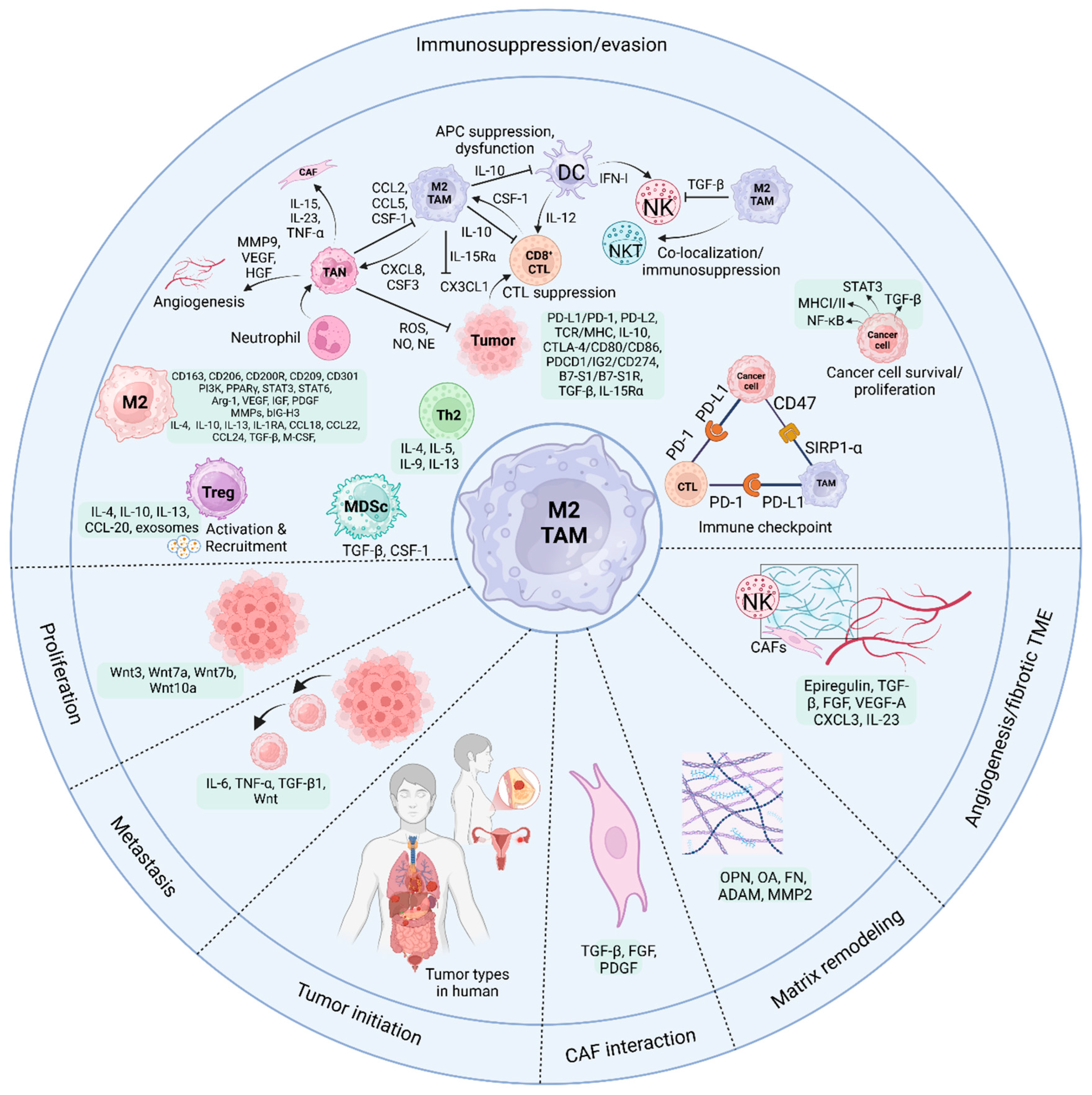
Dendritic cells (DCs), natural killer (NK) cells, mast cells, tumor-associated neutrophils (TANs), recruited macrophages, and cancer cells are among the several cell types seen in the TME [45].
A complicated interaction between several cellular components and environmental factors causes TAM migration to the TME. Cytokines and the production of exosomes are essential components of the juxtacrine and paracrine signaling pathways that are crucial to this process. The TAM colony-stimulating factor (GM-CSF) is a chemokine that tumors secrete and is essential for recruiting TAMs. Research has shown that this factor activates DCs within tumors, demonstrating anti-tumorigenic actions at low levels in the blood. On the other hand, it shifts to recruiting TAMs and promoting oncogenesis in advanced cancer stages when GM-CSF levels are high. Breast, colon, and cholangiocarcinoma cancers have all been linked to increased recruitment and infiltration of TAMs to the tumor site due to upregulation of this cytokine and CSF [46,47].
Furthermore, it has been shown that CSF may stimulate colon cancer cells to produce IL-8 from TAMs. Later, the colon cancer cell’s protein kinase C signaling pathway is activated by IL-8, which causes the tumor to produce more CSF and attract more TAMs [22]. Figure 2 shows the interaction between TAMs and cancer cells. Lung cancer, osteosarcoma, and breast cancer are associated with producing IL-17, IL-34, and CSF-2, three other notable tumor-released cytokines that increase TAM recruitment [48,49]. Not only do cytokines play a crucial role in TAM recruitment, but so do specific chemokines generated by tumors. Examples include C-C motif chemokine ligand 2 (CCL2), CCL5, CCL20, CXCL4, and CXCL12. These ligands are linked to several cancers, including bladder, colon, breast, and NSCLC [50,51]. In the end, the reliance on these substances generated by tumors highlights how vital an inflammatory TME is for attracting TAMs, which aid in tumor development.
In addition to tumor cells, other stromal cellular components of the TME are also involved in TAM recruitment. Hepatocellular carcinoma progresses because tumor-derived CXCL5 influences TAN recruitment to the tumor site, where they produce CCL2 and CCL17. These, in turn, attract TAMs, which further the cancer's spread [46]. The production of CCL2 and other adipokines by breast cancer cells and cancer-associated adipocytes, such as lauric acid and leptin, invites TAMs to the TME [52]. Moreover, CAFs and mesenchymal stromal cells contribute to an inflammatory TME favorable to the recruitment of TAMs in ways that rely on C-C chemokine receptor type 2 (CCR2) and IL-8.
A protein known as cluster of differentiation 47 (CD47) is involved in another biological pathway that regulates the interaction between tumor cells and TAMs. Tumor cells are among the many cell types that have it on their membrane surfaces. The cells that produce signal regulatory protein alpha (SIRPα), a membrane protein that binds to CD47, are the most prevalent in the bone marrow and TAMs (Figure 2). The usual immunoreceptor tyrosine-based inhibitory motif (ITIM) is formed when these cells come together. The cytosolic tyrosine phosphatases SHP-1 or SHP-2 may be recruited and activated via the contact between the NH2 terminal domain of the ITIM motif and the single domain of CD47. This connection regulates downstream signaling cascades and dephosphorylates many substrates, which limits TAM phagocytosis of cancer cells. The "do-not-eat-me" signal is, hence, another name for CD47 [53].
Macrophage-mediated programmed cell removal (PrCR) is essential to detect and eliminate tumors. Immune responses involving macrophage toll-like receptors (TLRs) initiate the Btk signaling pathway, which in turn triggers phosphorylation of the endoplasmic reticulum and dissociation of the cell surface calreticulin (CRT) [46]. Macrophages express dissociated CRT, forming the CRT/CD91/C1q complexes, allowing them to phagocytose cancer cells [54]. By attaching to SIRPα on macrophages, "Do-not-eat-me" signals prevent tumor cell PrCR production, inhibiting phagocytosis. However, "do-not-eat-me" signals may be blocked by inhibiting CD47 on tumor cells. Thus, inhibiting CD47 on tumor cells and activating the TLR signaling pathway in macrophages may enhance PrCR. Research has shown tumor cells may resist macrophage phagocytosis even after CD47 inhibition. To be more precise, Weissman et al. [55] reported that tumor cells evade macrophage phagocytosis by using a different identification process between the two types of cells. To enhance phagocytosis and eliminate tumor cells in vivo, it is possible to block or downregulate the signaling protein β2-microglobulin, which is present on the tumor cell surface as part of major histocompatibility complex-I (MHC-I). Research has shown that this therapy may significantly extend the longevity of animals with tumors by as much as 70% (Figure 2).
Evidence also suggests a robust relationship between TAM status and cancer stage and prognosis. In most cases, anti-tumor TAMs are often seen in early-stage tumors, while pro-tumor TAMs are more commonly found in advanced-stage tumors. Hence, the TAM polarization index, which measures the ratio of pro-tumor to anti-tumor TAMs, has been suggested to predict cancer outcomes and target potential treatments [56,57]. Upon analyzing the polarization spectra of TAMs in 931 colorectal carcinomas, Väyrynen and colleagues found that a higher M1:M2 density ratio in the tumor stroma was linked to improved cancer-specific survival probability [58]. Nevertheless, it has been shown that the location of these cells is just as important, if not more so, for prognosis than confirming TAMs in the TME [59].
Studying TAMs inside the TME in a specific setting and using a particular approach might lead to substantially different percentages [60]. Several cancers, including melanoma, breast, ovarian, pancreatic, and lung cancers, are associated with a poor prognosis when these markers are present [61]. Pancreatic cancer prognosis, vascularization, and disease progression correlate with TAM levels, particularly CD163+ and CD204+ TAMs [62]. A poor prognosis and an increased risk of recurrence are linked to high levels of TAM infiltration in breast cancer [63]. Based on their research, Jeong and colleagues found that invasive breast cancer (IBC) patients had bigger tumors and a worse prognosis when CD163+ M2-like macrophages were infiltrated into tumor nests. This discovery also functioned as a separate indicator of prognosis for decreased disease-free survival (DFS) and overall survival (OS) [64]. This is mainly because, as previously shown in research [65], TAMs secrete matrix metalloproteinases (MMPs) and other proteases, which aid tumor invasion and metastasis. In contrast, individuals with IBC had an improved prognosis for OS and DFS when CD11c+ M1-like macrophages were seen infiltrating the tumor stroma [66]. Mei et al. similarly discovered in their meta-analyses and systematic review that NSCLC patients' OS improved when the tumor islet included a high density of M1-like TAMs. Still, it worsened when the tumor stroma contained a high density of M2-like TAMs [67]. The patient’s overall survival was unrelated to the total CD68+ TAMs in the tumor islet or stroma, which is intriguing [67]. These results prove that the location of M1- and M2-like TAMs, rather than just the existence of these phenotypes in the TME, is a better indicator of disease outcome.
Because TAM activities are context-dependent and highly changeable, it is essential to consider the unique TAM phenotype and its interactions with the TME when targeting TAMs for cancer treatment [68]. Cancer researchers are investigating potential ways to use TAMs as a therapeutic target. These include reprogramming TAMs to be less tumor-promoting, preventing their recruitment to tumor sites, and improving the immune response against tumors [69]. However, further research is needed to comprehend the complex interaction between macrophages and the TME fully [68,70].
5. Macrophage in Immunoregulation
In TME, the balance between M1 pro- and M2 anti-inflammatory phenotypes in TAM determines the immune response [71]. Figure 3 shows that M2-like TAMs decrease immune surveillance and encourage tumor development, in contrast to M1-like TAMs, which enhance anti-tumor immunological responses. In comparison to M2-like TAMs, which may block tumor-killing cytotoxic T cells, M1-like TAMs attract regulatory T cells and other immunosuppressive cells to help construct an immunosuppressive TME [72].
Another method TAMs contribute to immunosuppression and tumor development is by releasing immunomodulatory chemicals such as PGE2, IL-8, IL-10, and TGF-β [73]. In times of infection or inflammation, IL-10 plays an essential role in preserving tissue homeostasis by enhancing innate immunity, reducing excessive inflammatory responses, and facilitating processes for tissue repair [74]. When they are present in cancer, they suppress the functions of other immune cells, for instance, NK and T cells, which help tumor cells survive by avoiding immune surveillance [75]. Recent research has shown that TAMs influence NK cell cytolytic activity in two ways. In a contact-dependent mechanism mainly mediated by TGF-β, TAMs reduce the cytolytic activity of NK cells. The restoration of NK cell cytotoxicity was seen with suppression of TGF-β. Conversely, TAMs encourage the CD27lowCD11high NK cell phenotype, characterized by reduced activation and tumor-killing capabilities, adding to NK cell exhaustion [76]. By activating Foxp3, TAMs convert Th cells into regulatory T cells (Tregs) in response to the release of TGF-β [77,78]. Tregs are activated by TAMs via the release of TGF-γ and IL-10, and they hinder the activity of CD8+ effector T cells. Last, TAMs secrete chemokines such as CCL5, CCL17, CCL20, and CCL22, which attract CCR4+ Tregs and help them enter the TME [47,79]. The immune suppression of CD8+ cytotoxic T cells is intensified as a result.
In addition, the beneficial interaction between TAMs and Tregs has been extensively studied. By aiding in the formation of an immunosuppressive TME, Tregs contribute to immune evasion in cancer. Research has shown that TAMs may activate Tregs, encouraging monocytes to differentiate into M2 phenotype cells [80]. Clinical trial results show that M2-like immunosuppressive TAM infiltration at metastases suppresses clinically significant immune responses in the metastatic TME (M-TME). Patients without neoadjuvant treatment for high-grade serous carcinomas (HGSCs) had their transcriptome profile examined across 24 matched original and metastatic tumor tissues. The study found that M2-like TAMs may cause T-cell exhaustion in the M-TME because they regulate the expression of cytokine/chemokine signals, such as IL-10 and CCL22. Metastatic site inhibition of immune responses was associated with poor prognoses in HGSC patients with robust M-TME infiltration by M2-like TAMs. Immune effector cell infiltration did not significantly affect patient survival, and 1468 genes showed differential expression between the primary-TME and M-TME of HGSCs [81].
In addition, it has been shown that DCs are impacted by TGF-β and IL-10 produced by TAMs. Evidence shows that these anti-inflammatory cytokines cause antigen-presenting cells to die off, reducing the number of DCs that can infiltrate tumor metastasized sites and migrate to lymph nodes, weakening adaptive immune responses mediated by T cells [82,83].
Another way that TAMs might limit the immune response is via their interactions with other immune cells. A critical component of immunosuppression is programmed cell death protein (PD-1), which belongs to the CD28 superfamily. When regulating the immune system for objectives including cancer prevention, infection control, autoimmune disease treatment, and organ transplant recipient survival, PD-1 consideration is essential [84,85]. Programmed cell death-ligand 1 (PD-L1) is a PD-1 ligand produced by antigen-presenting cells. Collaborating with PD-1 on T cells stops T cells from targeting antigen-presenting cells [84,85]. The PD-1/L1 signaling pathway can restrict the actions of T effector cells, DCs, and NK cells. As shown by the reduction of phagocytosis in TAMs and the suppression of effects on T cell activation, proliferation, and cytokine production, this limitation increases the possibility of tumor immune escape (Figure 3) [86,87].
To specifically decrease the immune response, TAMs may increase the expression of additional surface proteins, including CD80/CD86 or death receptor ligands like Fas-L or TRAIL [88]. These ligands bind to and activate inhibitory receptors on immune effector cells, namely CTLA-4, FAS, and TRAIL-RI/-RII. Stimulating the PD-1 and CTLA-4 receptors reduces cytokine and protein production, which aids cell survival by inhibiting the T cell receptor (TCR) signaling pathway. On top of that, TAMs produce the enzyme Arg-1, which is responsible for L-arginine degradation. Several processes rely on L-arginine, including developing immunological memory, lymphocyte proliferation, TCR complex expression, and the antitumor response mediated by T cells [46,89,90]. Therefore, TAMs reduce adaptive immunity against tumors by reducing immune responses pleiotropically.
Anergic and unresponsive T cells, as opposed to activated ones, might result from defective immunological synapses during the presentation of antigen fragments by macrophages to T cells [91]. The M2-induced Treg population maintains this T cell anergy condition, most noticeable in the lymph nodes that drain tumors [92]. Moreover, Kersten and colleagues show that CD8+ T cells are prepared to saturate following extended, antigen-specific engagement with TAMs, and this saturation is amplified in a hypoxic setting, such as that seen in the TME [93]. This immune-excluded TME pattern is further enhanced by persistent contact with TAMs inhibiting CD8+ T cell tumor invasion. By combining CSF-1R blockage with anti-PD-1 treatment, Peranzoni and colleagues demonstrated that CD8+ T cell tumor infiltration was improved, and tumor growth was delayed [94].
Figure 3.
The TAMs regulate tumors and polarize M1/M2 cells. The tumor stage, tissue, microenvironment, and paracrine signals affect them. Macrophages recruit the support of T and NK cells to fight against cancers. Cancer and tissue repair are two functions of macrophages that produce APCs like M1. Malignant M2 macrophages cause tumors to develop. Macrophages in the hypoxic TME use immunosuppressive mediators to fuel tumor development, angiogenesis, invasion, and metastasis. The same holds true for inflammation, cancer stem cell (CTC) self-renewal, tissue remodeling, and the epithelial-mesenchymal transition (EMT). Moreover, to coordinate CD8+ T-cell responses, TAMs activate immunological checkpoints, downregulate antigen presentation, and release regulatory factors. Additionally, TAMs inhibit dendritic cell antigen presentation and invasion. TAMs release TGF-β and CSF-1, which boost myeloid-derived suppressor cell (MDSC) proliferation. TAMs attract immune-suppressive Treg cells and impair NKT cell activity, suppressing. Furthermore, PD-L1 on TAMs and tumor cells interacts with PD-1 on CTLs to reduce antitumor immunity. CD47-SIRPa interaction between TAMs and cancer cells helps tumors avoid phagocytosis. Moreover, chemokines such as CCL1, CCL17, CCL18, CCL22, and CCL24 are abundantly expressed by M2-like macrophages, along with CD163, CD206, CD200R, CD209, and CD301. A variety of anti-inflammatory factors, such as TGF-β, IL-4, IL-13, IL-10, and IL-1RA, are released by macrophages. Furthermore, M2-like TAMs have reduced amounts of the inflammatory cytokines IL-6, IL-12, IL-23, and TNF-α. In addition to matrix metalloproteinases (MMPs), M2-like macrophages secrete autocrine extracellular matrix components, including fibronectin, beating-h3 (BIG-H3), enzymes that cross-link ECM, transglutaminase, and proteins that bridge gaps in bone [95]. M2-like macrophages increase Arg-1 and VEGF expression, aiding proline and polyamine production. Proline promotes ECM formation, and polyamines enhance cell growth. PDGF and IGF, which enhance cell proliferation, are also released by M2-like macrophages and implicated in angiogenesis [96,97]. Created by BioRender.
Figure 3.
The TAMs regulate tumors and polarize M1/M2 cells. The tumor stage, tissue, microenvironment, and paracrine signals affect them. Macrophages recruit the support of T and NK cells to fight against cancers. Cancer and tissue repair are two functions of macrophages that produce APCs like M1. Malignant M2 macrophages cause tumors to develop. Macrophages in the hypoxic TME use immunosuppressive mediators to fuel tumor development, angiogenesis, invasion, and metastasis. The same holds true for inflammation, cancer stem cell (CTC) self-renewal, tissue remodeling, and the epithelial-mesenchymal transition (EMT). Moreover, to coordinate CD8+ T-cell responses, TAMs activate immunological checkpoints, downregulate antigen presentation, and release regulatory factors. Additionally, TAMs inhibit dendritic cell antigen presentation and invasion. TAMs release TGF-β and CSF-1, which boost myeloid-derived suppressor cell (MDSC) proliferation. TAMs attract immune-suppressive Treg cells and impair NKT cell activity, suppressing. Furthermore, PD-L1 on TAMs and tumor cells interacts with PD-1 on CTLs to reduce antitumor immunity. CD47-SIRPa interaction between TAMs and cancer cells helps tumors avoid phagocytosis. Moreover, chemokines such as CCL1, CCL17, CCL18, CCL22, and CCL24 are abundantly expressed by M2-like macrophages, along with CD163, CD206, CD200R, CD209, and CD301. A variety of anti-inflammatory factors, such as TGF-β, IL-4, IL-13, IL-10, and IL-1RA, are released by macrophages. Furthermore, M2-like TAMs have reduced amounts of the inflammatory cytokines IL-6, IL-12, IL-23, and TNF-α. In addition to matrix metalloproteinases (MMPs), M2-like macrophages secrete autocrine extracellular matrix components, including fibronectin, beating-h3 (BIG-H3), enzymes that cross-link ECM, transglutaminase, and proteins that bridge gaps in bone [95]. M2-like macrophages increase Arg-1 and VEGF expression, aiding proline and polyamine production. Proline promotes ECM formation, and polyamines enhance cell growth. PDGF and IGF, which enhance cell proliferation, are also released by M2-like macrophages and implicated in angiogenesis [96,97]. Created by BioRender.
5.1. TAMs and CD8+ CTLs
Among the many immunosuppressive components of the TME, TAMs have a role in tumor initiation, progression, and immunotherapy resistance. Cancer development is accompanied by fibrosis, which reduces antitumor immune infiltration. A TME, defined by nutritional depletion, hypoxia, acidity, and metabolite buildup, develops due to the Warburg effect and the dysregulated bioenergetic machinery of tumor cells. These factors reduce the antitumor responses of CD8+ T lymphocytes by lowering their effector activity [98].
A recent study reported that in response to the solid fibrotic TME, TAMs initiate collagen production via TGF-β. Collagen-synthesizing macrophages absorb ambient arginine, manufacture proline, and release ornithine, which impairs CD8+ T cell activity in female breast cancer. Thus, a solid and fibrotic TME may inhibit antitumor immunity by physically excluding CD8+ T cells and mechano-metabolic programming TAMs, generating an unfavorable metabolic milieu for anticancer immunotherapies [99].
Few TAM-targeting drugs work preclinically or clinically. To maintain CSF-1 receptor inhibition and high anticancer immunity, the novel reagent FF-10101 formed a covalent link and decreased immunosuppressive TAMs in the TME. Preclinical animal studies showed that FF-10101 decreased immunosuppressive TAMs and boosted TME anticancer TAMs. CD8+ T lymphocytes that target tumor antigens also increased, inhibiting tumor development. FF-10101 with an anti-PD-1 antibody destroyed tumors better than either alone. As in animal models, FF-10101 reduced TAM PD-L1 expression in human cancer tissues. By reducing immunosuppressive TAMs and enhancing tumor antigen-specific T-cell responses, FF-10101 enhances TME. FF-10101 may work with PD-1/PD-L1 immune checkpoint inhibitors in cancer immunotherapy [100].
Xie et al. [101] found that E twenty-six variant transcription factor 4 (ETV4) elevated PD-L1 and chemokine CCL2 production in hepatocellular carcinoma (HCC) cells. This, in turn, led to the accumulation of TAM and myeloid-derived suppressor cells (MDSC) and the suppression of CD8+ T-cells, which facilitated the spread of HCC [102]. In another study, Luan et al. found aberrant complement 5a receptor (C5aR) expression in human ovarian cancer (OC) and elevated C5aR expression on TAMs, which activated TAMs to become immunosuppressive. C5aR deletion or inhibitor therapy mitigated tumor development and restored TAM antitumor response. Mechanistically, C5aR deficiency reprogrammed macrophages from protumor to antitumor, upregulating immune response and stimulation pathways, which enhanced CTL’s antitumor response in a manner dependent on CXCL9. Drugs that inhibit C5aR also enhance immune checkpoint blockage. C5aR expression was linked to CXCL9 production and CD8+ T cell infiltration, and a high C5aR level indicated worse clinical outcomes and lower anti-PD-1 treatment benefits. Their findings illuminate how the C5a-C5aR axis modulates TAM antitumor immune response and suggests therapeutic uses for targeting C5aR.
Bader et al [103] reported that obesity increased TAM PD-1. When exposed to type I inflammatory cytokines and obesity-related substances such as IFN-γ, TNF, leptin, insulin, and palmitate, macrophages express PD-1 via mTORC1 and glycolysis-dependent approach. PD-1 negative feedback reduced TAM glycolysis, phagocytosis, and T-cell activation. PD-1 inhibition increased macrophage glycolysis, which is necessary for TAM CD86, MHC I and II, and T cell activation. Myeloid cell PD-1 deficiency reduced tumor growth, increased TAM glycolysis and antigen presentation, and exhausted CD8+ T cells. Inflammatory stresses and obesity-associated rise in metabolic signaling TAM PD-1 expression, creating a TAM-specific feedback loop that suppresses tumor immune surveillance. It may boost PD-1 therapy but increase obesity-related cancer risk.
In another study, lentiviral vectors (LVs) generated liver macrophages, including Kupffer cells and TAMs, that transport IFNα to liver metastases. IFNα gene therapy reduces liver metastases in colorectal and pancreatic ductal adenocarcinoma mice. TAM immunological activation, MHC-II-restricted antigen presentation, and reduced CD8+ T cell exhaustion result from IFNα response. IL-10 signaling, Eomes CD4+ T cell proliferation, Tr1 cell traits, and CTLA-4 expression increase with treatment resistance. CTLA-4 immune checkpoint inhibition and IFNα LV infusion boost tumor-reactive T cells, leading to complete response in most animals. These data suggest an unmet medical requirement for therapy [104].
5.2. TAMs and NK Cells
The innate and adaptive immune systems interact via macrophages, phagocytic and antigen-presenting cells. There are two kinds of TAM: TAM1 and TAM2. TAM1 has anti-tumor action, whereas TAM2 promotes tumor growth. IFNγ and LPS trigger TAM1 induction, producing cytokines including TNF-α, IL-1β, IL-6, IL-12, nitric oxide synthase (iNOS), and reactive oxygen species (ROS). In response to cytokines like IL-4, IL-10 and IL-13, TAM2 is activated, generating cytokines like IL-10, TGF-β, and VEFG [105,106].
Initially, TAM1 enhances the expression of CD69, an activation marker, degranulation, and IFNγ secretion of resting autologous NK cells by both soluble factor secretion and direct interaction [106,107]. When TAM produces IFN-γ, it triggers the expression of IL-15Rα on NK cells and subsequent production of IL-15 [108,109]. NK cells produce more IFN-γ due to this cis-presentation. The release of IL-1β, IL-23, and IFN-β, as well as the up-regulation of NKG2D and NKp44 expression, are mechanisms via which TAM1 enhances the cytolytic efficacy of NK cells [106]. Furthermore, the cytotoxicity of murine splenic CD49b+ NK cells and their increased NKG2D expression against several tumor cell lines are both enhanced by macrophages treated with polyI:C. The inhibitory receptor NKG2A identifies Qa-1, which these murine macrophages express, allowing them to evade NK cell death [107,110]. IL-12 family cytokines are produced by monocyte-derived macrophages cultured with IFN-λ/LPS, which stimulates the production of IFN-γ by human NK cells [111].
In one investigation, researchers examined how DCs and NK cells work together to eliminate tumors. One receptor that macrophages and DCs produce, Dectin-1, can recognize N-glycan complexes in tumor cells. This triggers the activation of IRF5, leading to gene transcription downstream. Macrophages and DCs can trigger NK cell’s anti-tumor activity against tumor cells that produce N-glycan due to these processes [112].
On the other hand, TGF-β produced from TAM hinders NK cell’s abilities by reducing cytokine production and degranulation in humans [113], and cytotoxicity in mice [114,115]. TAM also promotes the exhausted phenotype CD27low CD11bhigh in mice [115]. Targeting VEFG-A in myeloid cells enhances chemotherapy transport to the tumor nest and subsequent NK cell recruitment, as shown in a mouse model by Klose et al. [116].
NK cells can impair macrophage’s ability to fight tumors. An exhausted phenotype, defined by a rise in PD-1 and TIM-3 expression and a reduction in NKG2D, is seen, for instance, in prostate cancer patient’s peripheral blood NK cells [117]. Two inhibitory surface molecules worth mentioning are PD-1 and TIM-3. When PD-1 and its ligands, PD-L1 and PD-L2, are overexpressed in some malignancies, they decrease the anti-tumor activity of NK [117,118]. Patients with prostate cancer consistently have NK cells with a significantly reduced degranulation capability [119]. Figure 3 shows that these NK cells release soluble mediators such as CCL2 and IL-10, which play a role in monocyte recruitment and M2-polarization [120].
5.3. TAMs and NKT Cells
TME NKT cell investigations have increased in recent years. Natural killer T cells (NKTs) with decreased CD1d expression may be classified into many types, including Th1-like, Th2-like, Th17-like, Treg-like, and T-follicle-assisted (TFH)-like [121]. NKTs attack tumor cells like NK cells but flip between inflammatory and immunosuppressive characteristics. The central subgroup explored is iNKT (invariant natural killer T). iNKT cells possess inherent characteristics and secrete several cytokines from the Th1 and Th2 groups, such as IFN-γ, IL-4, and GM-CSF, which may control activated APCs [122]. High cytolytic activity, the release of cytotoxic particles including perforin and granzymes, and the activation of death receptor pathways involved in the Fas-FasL-TRAIL-DR5 connection were additional features of iNKT cells, which were unique from other cell types [123].
Recently published research has shown that crosstalk between iNKTs and macrophages helps better understand immune cell interaction in TME. While CD68+ macrophages enhanced periampullary malignant adenoma survival, co-localization of NKp46+ NKT cells with CD163+ macrophages reduced it [124,125]. While NKp46+ NKT cells are abundant in the tumor region close to tumor cells, CD68+ macrophages are primarily found in the stroma, far from tumor cells, in malignant tumor tissues; this limits their capacity to interact [125]. Immunosuppressive macrophage co-localization reduces NKT function (Figure 3). A current study shows that iNKT cells control TAMs. Using CD1d and Fas-FasL, iNKT cells kill M2 TAMs, whereas M1 TAMs with CD40 are protected [126]. A mechanism involving microsomal prostaglandin E synthase-1 (mPGES-1) and 5-lipoxygenase (5-LOX) is proposed by iNKT cells to inhibit M2 TAMs in the pancreatic cancer model [127]. However, iNKT cells improved TAMS M2 polarization, FoxP3 protein expression, Treg frequency, tumor growth, and intestinal adenomatous polyps in colon cancer transgenic mice [121,128]. Few studies have examined the interaction between NKT cells and TAMs, which may vary significantly amongst tumors. More research on the process is needed.
A potential cell platform for CAR treatment in solid malignancies is human NKT cells. Li et al. [129] recently generated universal CAR-engineered NKT (UCAR-NKT) cells that selectively deplete immunosuppressive TAMs to change the TME. UCAR-NKT cells also have fewer challenges with cytokine release syndrome and graft-versus-host disease. To pave the way for the translational and clinical development of UCAR-NKT cell products, preclinical investigations prove their viability and therapeutic potential.
In another study, researchers Zhou et al. [130] discovered that CAR-NKT cells can eliminate M2-like macrophages that express CD1d. Additionally, CAR-NKT cells stimulate endogenous T cells to combat neoantigens linked with tumors and spread epitopes. The findings assist the clinical development of CAR-NKT treatments by showing their multimodal action in solid tumors.
5.4. TAMs and CD4 T Cells
Recent research has focused on CD4+ T cell function in tumor immune evasion and anti-tumor immunity. CD4+ T cells vary like macrophages. Effective CD4+ T cells are regulatory (iTreg and nTreg) or helper (Th1, Th2, Th17, Th9, Th22) in the TME [131,132]. Heterogeneity casts doubt on cancer immunity. CD4+ T cells cannot recognize most cancer cells because they lack MHC class II molecules (HLA-DR). Necrotic tumor cells or cancer cell vesicles eaten by tumor stromal cells usually cross-present tumor antigens via the classical MHCII (HLA-DR) processing pathway [133,134]. MHCII-positive monocytes/macrophages are found in most solid tumors; therefore, CD4+ T cell immunological recognition relies on them. TAMs increase MHCII genes and transmit antigens to CD4+ T cells in early NSCLC lesions [135]. Curiously, one research discovered that CXCL13+ CD4+ T cells are encouraged to invade by activated macrophages via IL15, CXCL9, CXCL10, and CXCL11. Only in the microenvironment does this percentage of T cells boost melanoma patient survival [78]. The elimination of MHC deletion and non-responsive cancer cells is triggered by inflammatory cell death, which is initiated by CD4+ effector T cells and activated iNOS-expressing tumor-killer monocytes and macrophages [136]. There is a distinct opportunity for targeted treatment targeting this small subset of CD4+ effector T cells.
Macrophage-CD4+ T cell interactions may not always kill tumor cells. In cirrhosis, macrophage-induced CD4+ T cells may become TREGs, leading to immunosuppressive TME and HCC [137]. Additionally, TAMs increased Treg cell proliferation and the Treg/CD8+ T cell ratio [138]. In addition to expressing PD-L1 on the cell membrane, TAM in the tumor’s core may recruit invasive Tregs from the tumor’s periphery into the TME, therefore suppressing the immune response (Figure 3) [139,140]. Around tumor cells in stomach cancer models, you can find CD4+FoxP3− T cells, CTLA-4 T cells, and PD-L1 T cells. However, to successfully transfer antigens, CD68+CD163−HLA-DR+ (M1) macrophages are too far away [141]. Patients with head and neck squamous cell carcinoma with PD-1+ helper T cells and CD163+ TAMs outlived those in other subpopulations [142].
Regarding tumor development, immunological homeostasis, and regulating other immune cell functions, macrophages and Tregs play a pivotal role. New research shows that metabolic alterations in Tregs and macrophages regulate signaling cascades and epigenetic reprogramming, affecting their pro- and anticancer activities. So, they are being acknowledged as potential targets for cancer immunotherapy more and more. Some metabolites in the TME may influence their pro- or anti-tumor activities via metabolic machinery interference [143]. Zhang et al. found that colorectal cancer included two main TAM subgroups: C1QC+ and SPP1+. The first subtype is associated with tumor angiogenesis and vascular markers, whereas the second subtype upregulates genes that are involved in complement activation and antigen presentation [144]. A further study found that GITR+CD25+ effector Tregs, which express the scavenger receptor CD36, are the most suppressive subgroup of Tregs in human melanoma tumors [145].
5.5. TAMs and DCs
Standard dendritic cells are cDC and pDC [146]. The cDC1s preferred MHC-I cross-presentation on CD8+ T cells due to its particular antigen-capturing capabilities, whereas the cDC2s opted for MHC-II on CD4+ T cells [147]. TLRs activate pDC, a DC subgroup, to release vast amounts of type I IFNs, which fight cancer. Correctly activated pDC stimulates T cells in vitro [148]. In vivo, studies demonstrate that pDC kills cancer [149]. According to specific research, pDC is associated with poor clinical outcomes caused by tumor suppression resistance and has unfavorable immunomodulatory effects on the TME [150]. Regulatory pDC lacked type I IFN; costimulatory molecules were downregulated, while IDO and PD-L1 were upregulated [151].
The extra-tumor stroma is where PD-L1+ DC and TAMs are most often seen in esophageal cancer patients, and their prognosis is dismal, according to studies [152,153]. Macrophage-DC co-localization may inhibit DC invasion or antigen presentation. In the B78ChOVA melanoma model, TAMs were more invasive and CD103+ cDC1 and CD11b+ cDC2 decreased [121]. Preventing antigen presentation may inhibit CD4+ and CD8+ T cell activation. TAM suppresses CD103+ DC IL-12 production and T cell activation in breast cancer by secreting IL-10 [154] (Figure 3). Unfortunately, previous studies have revealed that the TAM-DC connection does not improve tumor prognosis. Little is known about this interaction, so the mechanism is uncertain.
5.6. TAMs and Neutrophils
Neutrophils first combat infection and inflammation. Vascular chemotaxis transports them into tissues to fight infections. TME neutrophil activation and function vary by numerous parameters. These neutrophils become tumor-linked. Like TAM, TANs have two main polarizing types: N1TANs, which fight cancer, and N2TANs, which promote it. Many studies have studied TAN’s anticancer effects. TANs penetrate cancer cells, expressing co-stimulatory receptors such as 4-1BBL, OX40L, and CD86, activating active T cells and secreting IFN-γ for antitumor activity [121]. Interestingly, IFN-γ activates NK cells by causing TANs to generate IL-18 [155,156]. Furthermore, DC and CD8+ T lymphocytes are stimulated by TAN’s production of TNF-α [157]. TANs may kill cancer cells by secreting ROS, NO, and NE, reducing tumor growth and metastasis [158]. TANs can potentially enhance tumor development by increasing cancer cell proliferation, invasion, angiogenesis, and immunosuppression. The Akt/p38 pathway is activated by TANs, which turn MSCs into TAFs and promote tumor cell proliferation and metastasis via the production of cytokines such as IL-17, IL-23, and TNF-α [159,160]. Like macrophages, TANs increase angiogenesis and cancer cell aggressiveness by producing VEFG, HGF, and MMP9 [161,162]. After G-CSF and TGF-β stimulation, TANs release arginine-1, reactive oxygen species, and nitric oxide, which limit T cell activation [163,164].
In intrahepatic cholangiocarcinoma, most CD66b+ TANs were near CD68+ TAMs and formed small cell clusters in two-thirds of the samples [165]. Tans-Tams clusters boosted HuCCT1, RBE, and SG231 cell proliferation, invasion, colony formation, and lung metastatic tumors compared to TANs or TAMs alone [166,167]. TANs preferentially create OSM (oncostatin M), and TAMs preferentially express IL-11, which promotes STAT3 signaling in ICC cells and tumor development [121,165]. TANs released CCL2, CCL5, and CSF1, which may recruit macrophages to TAMs [168]. TAMs release CXCL8 and CSF3, TAN-related chemokines [168]. A positive feedback loop between TANs and TAMs boosted CSF1 and CXCL8 secretion after co-culture, substantially overlapping geographically and synergistically (Figure 3).
Recently, it was found that CD89hiCD32loCD64lo peripheral blood neutrophils (PBN) and TAN supported tumor cell development in the presence of cetuximab. In contrast, IgA anti-EGFR Abs produced PBN tumoricidal and removed TAN's stimulatory action. This work illuminates how myeloid effectors kill or resist tumor cells during tAb treatment [169]. Lei et al. [170] showed CXCL5-induced neutrophil NETosis. CD8+ T cells treated with neutrophil extracellular traps (NETs) upregulated exhaustion-related and cytosolic DNA sensing pathways and downregulated effector genes. However, A2AR inhibition dramatically decreased CXCL5 expression and neutrophil infiltration, improving CD8+ T cell dysfunction. According to the results, the complicated connection between tumor and immune cells may be a therapeutic target. Schmidt et al. [171] identified poor prognoses in central tumor samples with high stromal and intraepithelial CD66b+ TAN density. Higher neutrophil density in lymph nodes and adjacent normal breast tissue decreased disease-free survival. They previously linked TAN density to CD163+ M2-like TAM density. Low M1/M2 TAM ratios did not negatively prognosticate TANs, whereas high ratios did. TAM polarization state was the only independent predictor in a multivariate TAM and TAN density study. In single-marker analysis of early-stage luminal breast cancer, CD66b+ neutrophils were adverse. Future studies may require TAM analysis to estimate their predictive impact correctly. Malignancies that cannot attract and polarize TAMs may compensate for immunoevasion and disease progression by recruiting TANs. Pan et al. [172] showed that TAMs lacking VSIG4 produced less lactate and histone H3 lysine 18 lactylation, which lowered STAT3-mediated transcription of SPP1, disrupting TAM-neutrophil cell-cell connections. VSIG4 and SPP1 inhibition synergistically increased anti-tumor action. According to the study, VSIG4's epigenetic regulatory function allows TAMs to construct the immunosuppressive TME and hinder antigen-specific immunity against aggressive tumors, in addition to their checkpoint role.
5.7. TAMs and MDSCs
Bone marrow MDSCs produce DC, macrophages, and granulocytes. To create the immunosuppressive tumor myeloid microenvironment, CCL2 and CCL5 draw it to the tumor center [140]. The positive feedback impact of TAM secreting TGF-β has been shown in recent research. The amplification of MDSCs might be enhanced by continuous exposure to TGF-β and CSF-1 [76]. TAMs and MDSCs co-localize at colorectal cancer's aggressive border [173]. HCC also demonstrated TAM and MDSC-induced CD8+ T cell suppression along the tumor border [174,175].
Furthermore, the TRAMP/MICB spontaneous prostate tumor model's flow cytometry revealed a high association between MDSCs in the tumor-infiltrating region and serum soluble MHCI chain-related molecules (SMIC), an NKG2D ligand that activates STAT3, increases MDSCs and polarizes TAMs M2 [176]. Hypoxia improved sialic acid transport and CD45 binding when recruited MDSCs moved to tumors, activated CD45 protein tyrosine phosphorylase, dephosphorylated, and down-regulated STAT3. It accelerated TAM differentiation without hypoxia-inducible factor 1 (HIF-1) [177,178]. Though paradoxical, tumor tissues may up and down-regulate myeloid cell STAT3 activity due to time and space. The above method may upregulate STAT3 to enhance MDSCs from blood arteries entering tumor tissue. When recruited and amplified MDSCs penetrated tumor tissue in the deep vascular deficit area, hypoxia downregulated STAT3 and expedited MDSC differentiation into TAMs. This positive space-time feedback may assist macrophages and MDSC in growing tumors.
Xie et al [102] showed in HCC cells that overexpressing ETV4 stimulated PD-L1 and CCL2 expression, leading to an upregulation of TAM and MDSC infiltration and a downregulation of CD8+ T cell accumulation. Lentivirus- or CCR2-inhibitor CCX872-mediated CCL2 knockdown reduced ETV4-induced TAM, MDSC infiltration, and HCC metastasis. In addition, the ERK1/2 pathway was used by both FGF19/FGFR4 and HGF/c-MET to enhance ETV4 expression. Moreover, a positive feedback loop was established between FGF19, ETV4, and FGFR4 when ETV4 increased FGFR4 expression and FGFR4 downregulation reduced ETV4-enhanced HCC metastasis. Finally, anti-PD-L1, in conjunction with either the FGFR4 inhibitor BLU-554 or the MAPK inhibitor trametinib, significantly reduced the spread of HCC produced by the FGF19-ETV4 signaling pathway. The authors conclude that an effective strategy to suppress HCC metastasis might be to use anti-PD-L1 paired with the FGFR4 inhibitor BLU-554 or the MAPK inhibitor trametinib since ETV4 is a predictive biomarker.
Using scRNA-seq analysis, Tang et al. [179] identified two states of macrophage cells: MDSC-like (expressing CD300E, VCAN, EGFR, FCN1, and CCL20) and TAM-like (expressing C1QA, APOE, CD163, MRC1, and FOLR2). Additionally, they discovered that macrophages resembling MDSCs and TAMs increased levels of proteins that inhibit the immune system and promote cancer development, including SPP1, CCL20, and TIMP1. Additionally, increased MDSC- and TAM-like macrophages indicate an immunosuppressive milieu that promotes tumor development and immune evasion. Immunosuppressive myeloid cells, including TAMs and MDSCs, and immune-inhibitory factors may reduce the effectiveness of ICB in gastric cancer liver metastasis. Findings from the research highlight the need to investigate the liver microenvironment for tumor cell and immune system interactions to improve liver metastasis therapies, circumvent immune resistance, and improve patient outcomes. Tabachnick-Cherny et al. [180] used a single-cell technique to investigate MCC myeloid signatures. The authors then confirmed markers in tumor samples before PD-L1 blockade using myeloid spatial biology. Researchers identified and characterized MCC's dominant myeloid cells, TAMs, which show M-MDSC traits such as increased S100A8 and S100A9 genes and immunosuppressive cytokines like IL-10 and VEGF.
Zhang et al. [181] showed that C3 of RCC cell-derived EVs aids metastasis by attracting polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) and polarizing TAMs into an immunosuppressive phenotype. From a mechanistic standpoint, EV C3 increased TAM polarization and PMN-MDSC recruitment by inducing lung macrophages to secrete CCL2 and CXCL1. Significantly, in a mouse model of RCC-induced C3-induced lung metastasis, the inhibitors RS504393 and Navarixin, which target the CCL2/CCR2 or CXCL1/CXCR2 axis, respectively, successfully decrease the metastasis. From a clinical standpoint, the prognosis is not suitable for RCC patients who exhibit a high level of C3. They found that RCC metastasis is facilitated by tumor-derived EV C3, which produces an immunosuppressive TME via TAMs.
5.8. TAMs and γδ T Cells
The subgroup of T lymphocytes known as γδ T cells, which contribute to the immune cells that infiltrate tumors, constitutes 1% to 10% of the T cells in the peripheral blood of healthy individuals [182]. These T cells express T-cell receptors (TCRs) with γ and δ chains, distinguishing them from those with α and β chains [182]. While most T cells in the body express αβ TCRs, γδ T cells have recently come to the forefront due to their unique characteristics, which make them promising targets in cancer immunotherapy [183]. γδ T cells deliver antigens without the aid of MHC, making them unique. This is beneficial for immunotherapy since standard T cell-based treatments are hampered by cancer cell’s ability to downregulate MHC-I expression to escape detection [184].
The results demonstrated that lipid formulations increased circulating γδ T cells via MPS cells and suppressed TAMs in a breast cancer mouse model [185]. TAMs may be responsible for the reported anti-tumor activity in mice models lacking γδ T cells by endocytosing n-BPs, which have been proven to deplete these cells [185]. The ratio of M1:M2 TAMs in tumor models suggests that liposomal ZOL may preferentially remove M2 macrophages. However, it may also remove M1 anti-tumoral and M2 pro-tumoral TAMs in hepatocellular carcinoma and triple-negative breast cancer xenograft mice models [186,187,188]. Adoptive immunotherapy using liposomal ALD-activated Vγ9Vδ2 T cells effectively treats mouse epithelial ovarian cancer [189,190]. In laboratory and animal studies, Nanocarriers of lipids have shown excellent efficacy in stimulating γδ T-cell-mediated lysis of cancer cells. Still, toxicity and bioavailability difficulties remain in attaining therapeutic usefulness. The study by Gao et al. [191] demonstrated that BTNL2 stimulates γδT17 cells to release IL-17A and bring in myeloid suppressor cells, leading to a rise in the quantity of TME mesenchymal stem cells, M2 macrophages, and TAMs.
6. Immunotherapy Employing Macrophages and Anti-PD-1/PD-L1
Several immune checkpoint blockade therapies are available, but the two most common are anti-PD-1 and anti-PD-L1 treatments. Cancer immunotherapy that inhibits PD-1 aims to counteract immune suppression rather than boost immunity. Inhibitors of the PD-1/PD-L1 pathway have enhanced the cytotoxicity of T cells and the treating cancer [192]. As a means of immune system evasion and inhibition, TAMs express PD-L1 and PD-1. Inhibiting PD-1/PD-L1 may restore PD-1+ TAM phagocytosis, which in turn reduces tumor burden [193].
6.1. Effects of TAMs on PD-1/PD-L1 Expression
Immunotherapy targeting PD-1/PD-L1 has been used or attempted in the treatment of many solid tumors, such as lung cancer, advanced metastatic melanoma, esophageal cancer, and colorectal cancer. [194,195]. The PD-1/PD-L1 pathway was aberrantly activated in several cancers [13,196]. Nevertheless, PD-1 inhibitors failed to improve outcomes for many patients despite strong PD-L1 expression, and the exact reasons behind this are still poorly understood.
As previously shown in studies, TAMs activate many signaling pathways that influence PD-1/PD-L1 expression, which impacts the effectiveness of PD-1/PD-L1 inhibitors. There is a positive connection between CD163+ TAMs in the TME and PD-L1 expression in many tumor types, including pancreatic and liver tumors. Multiple signaling pathways being activated, such as phosphoinositide 3-kinase (PI3K)/AKT, NF-κB, Janus kinase (JAK)/STAT3, or Extracellular signal-regulated kinase (ERK) 1 and 2, may lead to upregulation of PD-L1 expression by various cytokines generated by TAM, such as IL-6 and TNF-α [197,198]. Moreover, TNF-α may potentially upregulate PD-L1 expression of proteins by post-translational regulation [197].
6.2. TAMs and Resistance to Anti-PD-1
Several factors have been associated with anti-PD-1 resistance, including TME and PD-L1 expression on tumor cells. As mentioned earlier, regulating PD-L1 protein expression by TAM's cytokine is a notable indicator for anti-PD-1/PD-L1 treatment. The TME now contains many immune cells, and the cancer ecology has changed over the years, impacting cancer growth intricately [199,200]. The response to immunotherapy was shown to be associated with the interaction between macrophages and other immune cells [199]. Researchers found that immunotherapy is less effective when FAP+ fibroblasts and SPP1+ macrophages interact, leading to the development of immune-excluded desmoplasic structures and a restriction of T-cells [199]. The therapeutic benefit of checkpoint inhibitors may be predicted in triple-negative breast cancer patients with high numbers of CXCL13+ T cells, which are linked to the pro-inflammatory characteristics of macrophages [200].
A few biological processes in cancer rely on exosomes, which are tiny extracellular vesicles. Metastasis and tumor development are supported by pre-metastatic niches, which may be enhanced by exosomes produced by macrophages, according to recent research. Depending on their source, EVs produced by M2 macrophages may influence the immune cell spectrum in the TME, induce resistance to anti-PD-1/PD-L1 treatment, or increase the expression of drug-resistant genes in tumor cells [201,202]. Therefore, cancer patients may acquire resistance to anti-PD-1 therapy because of the interaction between TAMs and TME. This discovery lends credence to integrating anti-PD-1/PD-L1 treatment with macrophage targeting.
6.3. Macrophage Immune Responses to Anti-PD-1/PD-L1 Therapy
According to prior investigations, the TME is affected by PD-1 inhibitors in several tumors [203]. Researchers used single-cell RNA sequencing to demonstrate that the TME in NSCLC patients treated with neoadjuvant PD-1 blockade and chemotherapy transformed. TAMS shifted from an anti-tumor phenotype to a neutral one [204]. In addition, anti-PD-L1 treatment may inhibit tumor growth by lowering PD-L1 expression and raising the levels of two co-stimulatory molecules, CD86 and MHC-II [205]. Furthermore, anti-PD-L1 treatment improved macrophage phagocytic capacity and immunological function, activating T cells in the TME and eliminating cancer cells [205]. Hence, in specific individuals, anti-PD-L1 treatment has the potential to repolarize macrophages, improve their phagocytic capacity, and alleviate TME.
7. Targeting TAM Immunotherapy
Future research into TAM might provide fruitful results due to its role in tumor immunity and progression. There are now two main approaches to treating macrophages: reducing the amount of TAM or reprogramming it. Combinations of immunotherapy, chemotherapy, and radiation were prevalent in clinical trials aiming to increase the effectiveness of treatment by targeting TAMs (Table 1) [206,207,208,209,210,211,212,213,214,215,216,217].
7.1. Reduced TAM Levels
One potential cancer treatment technique, whether used alone or in conjunction with chemotherapy, is the depletion of macrophages in the TME. When the signal transduction axis of the CSF1/CSF1R receptor is inhibited, macrophages undergo apoptosis. This axis is critical for the survival of macrophages. To begin with, T-cell responses may be enhanced by blocking CSF-1R in conjunction with radiation or chemotherapy. Glioblastoma brain tumors may have their survival time extended by stimulating the CD8+ T-cell response and depleting the immunosuppressive TAM by blocking CSF1R signaling [218]. Some cancers, including orthotopic glioblastoma and localized prostate cancer, are now undergoing clinical studies that combine chemotherapy with CSF1R inhibitors [218,219]. Moreover, specific immunotherapies, such as CD-40 agonists [220] and PD-1 inhibitors [221], maybe more effective by inhibiting CSF1/CSF1R.
Another way to decrease TAM in the TME was to prevent monocyte recruitment from the circulation to the tumor site, as TAM was changed from monocytes. Transduction of signals between C-C motif ligand 2 (CCL2) and CC chemokine receptor 2 (CCR2) is essential for the recruitment of monocytes from bone marrow to the location of tumors [222]. When CCR2 is inhibited, monocytes stay in the bone marrow instead of circulating in the bloodstream. This decreases the number of TAMs, which reduces the recruitment of monocytes to the tumor site and metastatic foci, reducing the tumor and improving survival [223,224,225].
Macrophage recruitment also involves the CXCL12-CXCR4 and the angiopoietin 2 (ANG2)-TIE2 axis, among others [226,227,228]. Consequently, TEM depletion might lead to vascular deterioration, ANG2 neutralization could enhance the response to vascular VEGFA blockage, and TEM recruitment inhibition could decrease tumor development [229].
7.2. TAM Reprogramming
After TAMs are eliminated, the immune-stimulating role of macrophages declines, as they are the primary phagocytes and antigen-presenting cells in the tumor. Therefore, a better way to treat cancer would be to repolarize or reprogram TAM such that it has more anti-tumor functions and less tumor-promoting effects. For instance, in the breast cancer mouse model, blocking IL-10 signal transduction greatly enhances chemotherapy effectiveness because TAM is the primary source of IL-10. To counteract the anti-tumor effect of paclitaxel and carboplatin on CD8+ T cells, TAM secretes IL-10, suppressing the production of IL-12 by APCs [41]. Furthermore, TAM repolarization causes it to produce the proinflammatory cytokine IFN-α in a tumor setting, potentially activating NK cells and T cells and considerably reducing tumor development in a mouse model [230]. Inhibiting histone deacetylase (HDAC) may reprogram macrophage epigenetics and activate T cells for an immunological response [231,232]. A specific class IIa HDAC inhibitor increased the efficacy of chemotherapy and immune checkpoint inhibitors in a breast cancer model by inducing an anti-tumor macrophage phenotype and enhancing the T-cell immunological response [231]. Furthermore, macrophage immunosuppression in TAM may be driven by PI3K signaling activation, while T-cell responses can be improved by reprogramming macrophages to decrease PI3K signaling [233,234].
7.3. Therapy Using Macrophages
In hematological cancers, CAR-T cells are beneficial, but in solid tumors, where T cell infiltration is limited, the effectiveness of CAR-T treatment is still limited [235,236]. Nevertheless, this drawback is circumvented by CAR-macrophages (CAR-M) since circulating monocytes may resupply the macrophages in the TME. Macrophage polarization, anti-cancer activity, higher phagocytic capacity, and antigen-dependent activities, including the production of cytokines, might be improved by CAR expression [237]. CAR-M cells have anti-tumor effects in both primary and metastatic cancers, support phagocytosis, and display M1 capabilities somewhat consistently [238]. The anti-cancer effectiveness of CAR-M in various tumor types is now being investigated in many ongoing or planned clinical studies.
7.4. Integrating Anti-PD-1 Treatment with Macrophage Targeting in Cancer
In vivo and in vitro studies on cancer treatment combining macrophage targeting with anti-PD-1 therapy have been conducted [205,239,240,241]. In hepatocellular carcinoma, repolarization of TAM might enhance the efficiency of anti-PD-1 therapy, as mentioned before, and is a potentially helpful technique for cancer treatment [241]. Radiotherapy and chemotherapy can potentially improve the effectiveness of immunotherapy for cancer by resetting macrophages to an M1 phenotype [240]. To improve the survival result of tumor-based immunotherapy, vinblastine may activate NF-κB, increase CD8+ T-cell populations, and polarize TAMs to the M1 phenotype [240]. By reprogramming immunosuppressive TAMs, bi-target treatments like PD-1-IL-2 cytokine variation (IL2v) boost therapeutic effectiveness [239]. IL2v uses anti-PD-1 as a target moiety fused with an immuno-stimulatory IL2v. Ultimately, it seems that a potential approach to combating medication resistance in cancer patients might include combining anti-PD-1 treatment with macrophage targeting.
8. Conclusions
Macrophages have a role in several cancer-related cellular processes, and tumor formation is linked to the interaction of macrophages with cancer cells or other immune cells. Because of their prominent role in TME, TAMs can potentially be a treatment-promoting target for cancer. It is possible to achieve remarkable anti-tumor efficacy by targeting macrophages alone or in conjunction with radiation, chemotherapy, and immune checkpoint inhibitors. Therapeutic targets include the mechanisms upstream and downstream of macrophage function regulation. The therapeutic utility of genetic engineering in reprogramming macrophages from tumor-promoting TAM to anti-tumor macrophages is very high. While there has been some success in clinical trials and preclinical investigations combining macrophage targeting with anti-PD-1 therapy in cancer, this therapeutic method is still in its early stages and requires further research. The variety and adaptability of TAM-targeted therapy's mononuclear phagocytes are challenges to its development and implementation [70]. A justification for selectively depleting tumor-promoting macrophages and eliminating tumors has been provided by demonstrating the variety of macrophages and their connection with other immune cells via the single-cell level of TME dissection [40,199]. Cancer treatments that target macrophages are in their early stages, and more research and clinical studies are required to prove their effectiveness and safety.
Author Contributions
Writing—original paper preparation, review, editing, illustrative material preparation, A.S.
Funding
This work received no funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L., et al., Cancer statistics, 2025. CA Cancer J Clin. 2025, 75, 10–45.
- Siegel, R.L., et al., Cancer statistics, 2023. CA Cancer J Clin 2023, 73, 17–48.
- Shi, D.; Gao, L.; Wan, X.-C.; Tian, T.; Hu, J.; Zhang, Q.-L.; Su, Y.-F.; Zeng, Y.-P.; Hu, Z.-J.; Yu, B.-H.; et al. Clinicopathologic features and abnormal signaling pathways in plasmablastic lymphoma: a multicenter study in China. BMC Med. 2022, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Zhang, S.; He, G.; Liu, J.; Fan, Q.; et al. Targeting tumor-associated macrophages in hepatocellular carcinoma: biology, strategy, and immunotherapy. Cell Death Discov. 2023, 9, 1–15. [Google Scholar] [CrossRef]
- Guan, F., et al., Tissue macrophages: origin, heterogenity, biological functions, diseases and therapeutic targets. Signal Transduct Target Ther. 2025, 10, 93.
- Zhou, X.; Wu, Y.; Zhu, Z.; Lu, C.; Zhang, C.; Zeng, L.; Xie, F.; Zhang, L.; Zhou, F. Mucosal immune response in biology, disease prevention and treatment. Signal Transduct. Target. Ther. 2025, 10, 1–32. [Google Scholar] [CrossRef]
- Feng, X., et al., IFN-τ Maintains Immune Tolerance by Promoting M2 Macrophage Polarization via Modulation of Bta-miR-30b-5p in Early Uterine Pregnancy in Dairy Cows. Cells 2025, 14.
- He, Y.; Hong, Q.; Chen, S.; Zhou, J.; Qiu, S. Reprogramming tumor-associated macrophages in gastric cancer: a pathway to enhanced immunotherapy. Front. Immunol. 2025, 16, 1558091. [Google Scholar] [CrossRef]
- Wu, Y.; Park, J.; Xu, E.; Kim, D.; Lee, J.; Oh, Y.-K. MicroRNA-induced reprogramming of tumor-associated macrophages for modulation of tumor immune microenvironment. J. Control. Release 2025, 381, 113593. [Google Scholar] [CrossRef]
- Kamdar, M.; Solomon, S.R.; Arnason, J.; Johnston, P.B.; Glass, B.; Bachanova, V.; Ibrahimi, S.; Mielke, S.; Mutsaers, P.; Hernandez-Ilizaliturri, F.; et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet 2022, 399, 2294–2308. [Google Scholar] [CrossRef]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. New Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, Wyrwicz L, Yamaguchi K, Skoczylas T, Campos Bragagnoli A, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet (London, England). 2021, 398, 27–40.
- Li, J.-W.; Deng, C.; Zhou, X.-Y.; Deng, R. The biology and treatment of Epstein-Barr virus-positive diffuse large B cell lymphoma, NOS. Heliyon 2023, 10, e23921. [Google Scholar] [CrossRef]
- Goc, J.; Lv, M.; Bessman, N.J.; Flamar, A.-L.; Sahota, S.; Suzuki, H.; Teng, F.; Putzel, G.G.; Eberl, G.; Withers, D.R.; et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell 2021, 184, 5015–5030.e16. [Google Scholar] [CrossRef]
- Scholler, N.; Perbost, R.; Locke, F.L.; Jain, M.D.; Turcan, S.; Danan, C.; Chang, E.C.; Neelapu, S.S.; Miklos, D.B.; Jacobson, C.A.; et al. Tumor immune contexture is a determinant of anti-CD19 CAR T cell efficacy in large B cell lymphoma. Nat. Med. 2022, 28, 1872–1882. [Google Scholar] [CrossRef]
- Hirz, T.; Mei, S.; Sarkar, H.; Kfoury, Y.; Wu, S.; Verhoeven, B.M.; Subtelny, A.O.; Zlatev, D.V.; Wszolek, M.W.; Salari, K.; et al. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single-cell and spatial transcriptomic analyses. Nat. Commun. 2023, 14, 1–20. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer 2023, 23, 238–257. [Google Scholar] [CrossRef]
- Chan, J.M.; Quintanal-Villalonga, Á.; Gao, V.R.; Xie, Y.; Allaj, V.; Chaudhary, O.; Masilionis, I.; Egger, J.; Chow, A.; Walle, T.; et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 2021, 39, 1479–1496.e18. [Google Scholar] [CrossRef]
- Park, M.; Kim, Y.S.; Song, H. Macrophages: a double-edged sword in female reproduction and disorders. Exp. Mol. Med. 2025, 57, 285–297. [Google Scholar] [CrossRef]
- Kloosterman, D.J.; Akkari, L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell 2023, 186, 1627–1651. [Google Scholar] [CrossRef]
- Lösslein, A.K.; Henneke, P. Macrophage Differentiation and Metabolic Adaptation in Mycobacterial Infections. Annu. Rev. Immunol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Verona, F.; Di Bella, S.; Schirano, R.; Manfredi, C.; Angeloro, F.; Bozzari, G.; Todaro, M.; Giannini, G.; Stassi, G.; Veschi, V. Cancer stem cells and tumor-associated macrophages as mates in tumor progression: mechanisms of crosstalk and advanced bioinformatic tools to dissect their phenotypes and interaction. Front. Immunol. 2025, 16, 1529847. [Google Scholar] [CrossRef] [PubMed]
- López-Collazo, E.; Hurtado-Navarro, L.; L, E. Cell fusion as a driver of metastasis: re-evaluating an old hypothesis in the age of cancer heterogeneity. Front. Immunol. 2025, 16, 1524781. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yi, Y.; Han, C.; Shi, B. NF-κB signaling pathway in tumor microenvironment. Front. Immunol. 2024, 15, 1476030. [Google Scholar] [CrossRef]
- Daniel, B., et al., The IL-4/STAT6/PPARγ signaling axis is driving the expansion of the RXR heterodimer cistrome, providing complex ligand responsiveness in macrophages. Nucleic Acids Res 2018, 46, 4425–4439. [CrossRef]
- Liu, H.; Amakye, W.K.; Ren, J. Codonopsis pilosula polysaccharide in synergy with dacarbazine inhibits mouse melanoma by repolarizing M2-like tumor-associated macrophages into M1-like tumor-associated macrophages. Biomed. Pharmacother. 2021, 142, 112016. [Google Scholar] [CrossRef]
- Vergadi, E., et al., Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J Immunol 2017, 198, 1006–1014. [CrossRef]
- Zhao, Y.; Yu, Z.; Ma, R.; Zhang, Y.; Zhao, L.; Yan, Y.; Lv, X.; Zhang, L.; Su, P.; Bi, J.; et al. lncRNA-Xist/miR-101-3p/KLF6/C/EBPα axis promotes TAM polarization to regulate cancer cell proliferation and migration. Mol. Ther. - Nucleic Acids 2021, 23, 536–551. [Google Scholar] [CrossRef]
- Singer, M.; Zhang, Z.; Dayyani, F.; Zhang, Z.; Yaghmai, V.; Choi, A.; Valerin, J.; Imagawa, D.; Abi-Jaoudeh, N. Modulation of Tumor-Associated Macrophages to Overcome Immune Suppression in the Hepatocellular Carcinoma Microenvironment. Cancers 2024, 17, 66. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Snijckers, R.P.M.; Foks, A.C. Adaptive immunity and atherosclerosis: aging at its crossroads. Front. Immunol. 2024, 15, 1350471. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Dong, C.; Li, B. Effects of macrophages in OSCC progression. Front. Immunol. 2025, 15, 1517886. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Davidson, D.; Li, R.; Zhong, M.-C.; Qian, J.; Chen, J.; Veillette, A. Inflammatory macrophages exploit unconventional pro-phagocytic integrins for phagocytosis and anti-tumor immunity. Cell Rep. 2021, 37, 110111. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Jiang, Q.; Han, M.; Ye, F.; Wang, M.; Qiu, Y.; Wang, J.; Gao, M.; Hou, F.; Wang, H. FBXO38 regulates macrophage polarization to control the development of cancer and colitis. Cell. Mol. Immunol. 2023, 20, 1367–1378. [Google Scholar] [CrossRef]
- Zhang, G.; Gao, Z.; Guo, X.; Ma, R.; Wang, X.; Zhou, P.; Li, C.; Tang, Z.; Zhao, R.; Gao, P. CAP2 promotes gastric cancer metastasis by mediating the interaction between tumor cells and tumor-associated macrophages. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 1–35. [Google Scholar] [CrossRef]
- Park, M.D. , et al. , TREM2 macrophages drive NK cell paucity and dysfunction in lung cancer. Nat Immunol 2023, 24, 792–801. [Google Scholar]
- Yu, Y.; Dai, K.; Gao, Z.; Tang, W.; Shen, T.; Yuan, Y.; Wang, J.; Liu, C. Sulfated polysaccharide directs therapeutic angiogenesis via endogenous VEGF secretion of macrophages. Sci. Adv. 2021, 7, eabd8217. [Google Scholar] [CrossRef]
- Casanova-Acebes, M.; Dalla, E.; Leader, A.M.; LeBerichel, J.; Nikolic, J.; Morales, B.M.; Brown, M.; Chang, C.; Troncoso, L.; Chen, S.T.; et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 2021, 595, 578–584. [Google Scholar] [CrossRef]
- Leader, A.M.; Grout, J.A.; Maier, B.B.; Nabet, B.Y.; Park, M.D.; Tabachnikova, A.; Chang, C.; Walker, L.; Lansky, A.; Le Berichel, J.; et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021, 39, 1594–1609.e12. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Michielin, O.; Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 2022, 19, 402–421. [Google Scholar] [CrossRef] [PubMed]
- Aegerter, H.; Lambrecht, B.N.; Jakubzick, C.V. Biology of lung macrophages in health and disease. Immunity 2022, 55, 1564–1580. [Google Scholar] [CrossRef]
- Kerneur, C.; Cano, C.E.; Olive, D. Major pathways involved in macrophage polarization in cancer. Front. Immunol. 2022, 13, 1026954. [Google Scholar] [CrossRef]
- Gao, J., et al., PANoptosis: bridging apoptosis, pyroptosis, and necroptosis in cancer progression and treatment. Cancer Gene Ther 2024, 31, 970–983. [CrossRef]
- Toledo, B.; Chen, L.Z.; Paniagua-Sancho, M.; Marchal, J.A.; Perán, M.; Giovannetti, E. Deciphering the performance of macrophages in tumour microenvironment: a call for precision immunotherapy. J. Hematol. Oncol. 2024, 17, 1–34. [Google Scholar] [CrossRef]
- Basak, U.; Sarkar, T.; Mukherjee, S.; Chakraborty, S.; Dutta, A.; Dutta, S.; Nayak, D.; Kaushik, S.; Das, T.; Sa, G. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front. Immunol. 2023, 14, 1295257. [Google Scholar] [CrossRef]
- Tomassetti, C.; Insinga, G.; Gimigliano, F.; Morrione, A.; Giordano, A.; Giurisato, E. Insights into CSF-1R Expression in the Tumor Microenvironment. Biomedicines 2024, 12, 2381. [Google Scholar] [CrossRef]
- Pan, Y.; Yuan, C.; Zeng, C.; Sun, C.; Xia, L.; Wang, G.; Chen, X.; Zhang, B.; Liu, J.; Ding, Z.-Y. Cancer stem cells and niches: challenges in immunotherapy resistance. Mol. Cancer 2025, 24, 1–22. [Google Scholar] [CrossRef]
- Yao, J.; Ji, L.; Wang, G.; Ding, J. Effect of neutrophils on tumor immunity and immunotherapy resistance with underlying mechanisms. Cancer Commun. 2024, 45, 15–42. [Google Scholar] [CrossRef]
- Luo, D.; Zhou, J.; Ruan, S.; Zhang, B.; Zhu, H.; Que, Y.; Ying, S.; Li, X.; Hu, Y.; Song, Z. Overcoming immunotherapy resistance in gastric cancer: insights into mechanisms and emerging strategies. Cell Death Dis. 2025, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Dong, S.; Huang, R.; Chen, X. Cancer-Associated Adipocytes and Breast Cancer: Intertwining in the Tumor Microenvironment and Challenges for Cancer Therapy. Cancers 2023, 15, 726. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Weng, L.; Wang, Y.; Zhang, J.; Wu, Q.; Zhao, P.; Shi, Y.; Wang, P.; Fang, L. Deciphering the role of CD47 in cancer immunotherapy. J. Adv. Res. 2023, 63, 129–158. [Google Scholar] [CrossRef]
- Brady, R.V.; Thamm, D.H. Tumor-associated macrophages: Prognostic and therapeutic targets for cancer in humans and dogs. Front. Immunol. 2023, 14. [Google Scholar] [CrossRef]
- Barkal, A.A.; Weiskopf, K.; Kao, K.S.; Gordon, S.R.; Rosental, B.; Yiu, Y.Y.; George, B.M.; Markovic, M.; Ring, N.G.; Tsai, J.M.; et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2018, 19, 76–84. [Google Scholar] [CrossRef]
- Xu, H., et al., Immunoglobulin-like transcript 5 polarizes M2-like tumor-associated macrophages for immunosuppression in non-small cell lung cancer. Int J Cancer, 2025.
- Zhang, Y.; Wang, B.; Chen, J.; Li, T. Role of exosomal miRNAs and macrophage polarization in gastric cancer: A novel therapeutic strategy. Eur. J. Pharmacol. 2025, 990, 177268. [Google Scholar] [CrossRef]
- Väyrynen, J.P., et al., The Prognostic Role of Macrophage Polarization in the Colorectal Cancer Microenvironment. Cancer Immunol Res 2021, 9, 8–19. [CrossRef]
- Robinson, S.D.; Filippopoulou, C.; Besta, S.; Samuels, M.; Betrán, A.L.; Abu Ajamieh, M.; Vella, V.; Jones, W.; Giamas, G. Spatial biology – unravelling complexity within the glioblastoma microenvironment. Trends Mol. Med. 2025. [Google Scholar] [CrossRef]
- Gangadaran, P.; Onkar, A.; Rajendran, R.L.; Goenka, A.; Oh, J.M.; Khan, F.; Nagarajan, A.K.; Muthu, S.; Krishnan, A.; Hong, C.M.; et al. Noninvasive in vivo imaging of macrophages: understanding tumor microenvironments and delivery of therapeutics. Biomark. Res. 2025, 13, 1–25. [Google Scholar] [CrossRef]
- Cai, D.X.; Tian, F.; Zhang, D.K.; Tu, J.F.; Wang, Y.H. CRABP2 (Cellular Retinoic Acid Binding Protein 2D): A novel biomarker for the diagnosis and prognosis involved in immune infiltration of lung adenocarcinoma. J. Cancer 2025, 16, 1631–1646. [Google Scholar] [CrossRef]
- Qi, Y.-Q.; Xiong, F.; Chen, Y.-J. The correlation between tumor-associated macrophages and the prognosis of east Asian hepatocellular carcinoma patients: A systematic review and meta-analysis. Pathol. - Res. Pr. 2023, 252, 154919. [Google Scholar] [CrossRef] [PubMed]
- Nedeljković, M.; Vuletić, A.; Martinović, K.M. Divide and Conquer—Targeted Therapy for Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2025, 26, 1396. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Hwang, I.; Kang, S.H.; Shin, H.C.; Kwon, S.Y. Tumor-Associated Macrophages as Potential Prognostic Biomarkers of Invasive Breast Cancer. J. Breast Cancer 2019, 22, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Baghel, K.S.; Tewari, B.N.; Shrivastava, R.; Malik, S.A.; Lone, M.U.-D.; Jain, N.K.; Tripathi, C.; Kanchan, R.K.; Dixit, S.; Singh, K.; et al. Macrophages promote matrix protrusive and invasive function of breast cancer cells via MIP-1β dependent upregulation ofMYO3Agene in breast cancer cells. OncoImmunology 2016, 5, e1196299–e1196299. [Google Scholar] [CrossRef]
- Jahandideh, A.; Yarizadeh, M.; Masjedi, M.N.-K.; Fatehnejad, M.; Jahandideh, R.; Soheili, R.; Eslami, Y.; Zokaei, M.; Ahmadvand, A.; Ghalamkarpour, N.; et al. Macrophage’s role in solid tumors: two edges of a sword. Cancer Cell Int. 2023, 23, 1–25. [Google Scholar] [CrossRef]
- Mei, J.; Xiao, Z.; Guo, C.; Pu, Q.; Ma, L.; Liu, C.; Lin, F.; Liao, H.; You, Z.; Liu, L. Prognostic impact of tumor-associated macrophage infiltration in non-small cell lung cancer: A systemic review and meta-analysis. Oncotarget 2016, 7, 34217–34228. [Google Scholar] [CrossRef]
- Jin, R.; Neufeld, L.; McGaha, T.L. Linking macrophage metabolism to function in the tumor microenvironment. Nat. Cancer 2025, 6, 239–252. [Google Scholar] [CrossRef]
- Ma, M.; Zhang, Y.; Pu, K.; Tang, W. Nanomaterial-enabled metabolic reprogramming strategies for boosting antitumor immunity. Chem. Soc. Rev. 2024, 54, 653–714. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Bao, C.; Ma, Q.; Ying, X.; Wang, F.; Hou, Y.; Wang, D.; Zhu, L.; Huang, J.; He, C. Histone lactylation in macrophage biology and disease: from plasticity regulation to therapeutic implications. EBioMedicine 2024, 111, 105502. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Li, Y.; Gong, Q.; Luo, K. Metformin-based nanomedicines for reprogramming tumor immune microenvironment. Theranostics 2025, 15, 993–1016. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zheng, L.; Qi, C. Myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment and their targeting in cancer therapy. Mol. Cancer 2025, 24, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Kim, B.; Bello, A.B.; Moon, J.J.; Arai, Y.; Lee, S.-H. Regenerative Functions of Regulatory T Cells and Current Strategies Utilizing Mesenchymal Stem Cells in Immunomodulatory Tissue Regeneration. Tissue Eng. Regen. Med. 2025, 22, 167–180. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, H.S.; Kim, W.; Lee, E.H.; Kim, S.; Kim, T.; Shin, E.-A.; Pyo, K.-H.; Lee, H.; Jin, S.H.; et al. Isoxazole-based molecules restore NK cell immune surveillance in hepatocarcinogenesis by targeting TM4SF5 and SLAMF7 linkage. Signal Transduct. Target. Ther. 2025, 10, 1–18. [Google Scholar] [CrossRef]
- Fanijavadi, S.; Thomassen, M.; Jensen, L.H. Targeting Triple NK Cell Suppression Mechanisms: A Comprehensive Review of Biomarkers in Pancreatic Cancer Therapy. Int. J. Mol. Sci. 2025, 26, 515. [Google Scholar] [CrossRef]
- Imani, S.; Kaboli, P.J.; Babaeizad, A.; Maghsoudloo, M. Neoantigen mRNA vaccines and A 2 A receptor antagonism: A strategy to enhance T cell immunity. Hum. Vaccines Immunother. 2025, 21, 2458936. [Google Scholar] [CrossRef]
- Griffiths, J.I.; Cosgrove, P.A.; Medina, E.F.; Nath, A.; Chen, J.; Adler, F.R.; Chang, J.T.; Khan, Q.J.; Bild, A.H. Cellular interactions within the immune microenvironment underpins resistance to cell cycle inhibition in breast cancers. Nat. Commun. 2025, 16, 2132. [Google Scholar] [CrossRef]
- Li, Z.; Duan, D.; Li, L.; Peng, D.; Ming, Y.; Ni, R.; Liu, Y. Tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for hepatocellular carcinoma: recent research progress. Front. Pharmacol. 2024, 15, 1382256. [Google Scholar] [CrossRef]
- Toghraie, F.S.; Bayat, M.; Hosseini, M.S.; Ramezani, A. Tumor-infiltrating myeloid cells; mechanisms, functional significance, and targeting in cancer therapy. Cell. Oncol. 2025, 1–32. [Google Scholar] [CrossRef]
- Hensler, M.; Kasikova, L.; Fiser, K.; Rakova, J.; Skapa, P.; Laco, J.; Lanickova, T.; Pecen, L.; Truxova, I.; Vosahlikova, S.; et al. M2-like macrophages dictate clinically relevant immunosuppression in metastatic ovarian cancer. J. Immunother. Cancer 2020, 8, e000979. [Google Scholar] [CrossRef]
- Trebska-McGowan, K.; Chaib, M.; Alvarez, M.A.; Kansal, R.; Pingili, A.K.; Shibata, D.; Makowski, L.; Glazer, E.S. TGF-β Alters the Proportion of Infiltrating Immune Cells in a Pancreatic Ductal Adenocarcinoma. J. Gastrointest. Surg. 2022, 26, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Mirlekar, B. Tumor promoting roles of IL-10, TGF-β, IL-4, and IL-35: Its implications in cancer immunotherapy. SAGE Open Med. 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wang, S.; Xia, L.; Sun, Z.; Chan, K.M.; Bernards, R.; Qin, W.; Chen, J.; Xia, Q.; Jin, H. Hepatocellular carcinoma: signaling pathways and therapeutic advances. Signal Transduct. Target. Ther. 2025, 10, 1–43. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, J.-B.; Sun, R.; Wang, N.; Weng, X.-Q.; Xu, T.-Y.; Fu, D.; Feng, Y.; Xu, P.-P.; Cheng, S.; et al. Dual targeting PD-L1 and 4-1BB to overcome dendritic cell-mediated lenalidomide resistance in follicular lymphoma. Signal Transduct. Target. Ther. 2025, 10, 1–13. [Google Scholar] [CrossRef]
- Xiong, Z.; Huang, Y.; Cao, S.; Huang, X.; Zhang, H. A new strategy for the treatment of advanced ovarian cancer: utilizing nanotechnology to regulate the tumor microenvironment. Front. Immunol. 2025, 16, 1542326. [Google Scholar] [CrossRef]
- Ricci, J.-E. Tumor-induced metabolic immunosuppression: Mechanisms and therapeutic targets. Cell Rep. 2025, 44, 115206. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, Y.; Liu, J.; Guo, L.; Guo, Q.; Liu, W. Bone marrow immune cells and drug resistance in acute myeloid leukemia. Exp. Biol. Med. 2025, 250, 10235. [Google Scholar] [CrossRef]
- E Menjivar, R.; Nwosu, Z.C.; Du, W.; Donahue, K.L.; Hong, H.S.; Espinoza, C.; Brown, K.; Velez-Delgado, A.; Yan, W.; Lima, F.; et al. Arginase 1 is a key driver of immune suppression in pancreatic cancer. eLife 2023, 12. [Google Scholar] [CrossRef]
- Yang, L.; Chu, Z.; Liu, M.; Zou, Q.; Li, J.; Liu, Q.; Wang, Y.; Wang, T.; Xiang, J.; Wang, B. Amino acid metabolism in immune cells: essential regulators of the effector functions, and promising opportunities to enhance cancer immunotherapy. J. Hematol. Oncol. 2023, 16, 1–33. [Google Scholar] [CrossRef]
- Chi, H.; Pepper, M.; Thomas, P.G. Principles and therapeutic applications of adaptive immunity. Cell 2024, 187, 2052–2078. [Google Scholar] [CrossRef]
- Tian, Z.; Chen, H.; Zhao, P. Compliant immune response of silk-based biomaterials broadens application in wound treatment. Front. Pharmacol. 2025, 16, 1548837. [Google Scholar] [CrossRef] [PubMed]
- Kersten, K.; Hu, K.H.; Combes, A.J.; Samad, B.; Harwin, T.; Ray, A.; Rao, A.A.; Cai, E.; Marchuk, K.; Artichoker, J.; et al. Spatiotemporal co-dependency between macrophages and exhausted CD8+ T cells in cancer. Cancer Cell 2022, 40, 624–638.e9. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. npj Precis. Oncol. 2024, 8, 1–19. [Google Scholar] [CrossRef]
- Soriano-Cruz, M.; Vázquez-González, W.G.; Molina-Vargas, P.; Faustino-Trejo, A.; Chávez-Rueda, A.K.; Legorreta-Haquet, M.V.; Aguilar-Ruíz, S.R.; Chávez-Sánchez, L. Exosomes as Regulators of Macrophages in Cardiovascular Diseases. Biomedicines 2024, 12, 2683. [Google Scholar] [CrossRef]
- Shen, L.; Li, Y.; Zhao, H. Fibroblast growth factor signaling in macrophage polarization: impact on health and diseases. Front. Immunol. 2024, 15, 1390453. [Google Scholar] [CrossRef]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Tharp, K.M.; Kersten, K.; Maller, O.; Timblin, G.A.; Stashko, C.; Canale, F.P.; Menjivar, R.E.; Hayward, M.-K.; Berestjuk, I.; Hoeve, J.T.; et al. Tumor-associated macrophages restrict CD8+ T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat. Cancer 2024, 5, 1045–1062. [Google Scholar] [CrossRef]
- Sato, T.; Sugiyama, D.; Koseki, J.; Kojima, Y.; Hattori, S.; Sone, K.; Nishinakamura, H.; Ishikawa, T.; Ishikawa, Y.; Kato, T.; et al. Sustained inhibition of CSF1R signaling augments antitumor immunity through inhibiting tumor-associated macrophages. J. Clin. Investig. 2025, 10. [Google Scholar] [CrossRef]
- Luan, X.; Lei, T.; Fang, J.; Liu, X.; Fu, H.; Li, Y.; Chu, W.; Jiang, P.; Tong, C.; Qi, H.; et al. Blockade of C5a receptor unleashes tumor-associated macrophage antitumor response and enhances CXCL9-dependent CD8+ T cell activity. Mol. Ther. 2023, 32, 469–489. [Google Scholar] [CrossRef]
- Xie, M., et al., FGF19/FGFR4-mediated elevation of ETV4 facilitates hepatocellular carcinoma metastasis by upregulating PD-L1 and CCL2. J Hepatol 2023, 79, 109–125. [CrossRef] [PubMed]
- Bader, J.E.; Wolf, M.M.; Lupica-Tondo, G.L.; Madden, M.Z.; Reinfeld, B.I.; Arner, E.N.; Hathaway, E.S.; Steiner, K.K.; Needle, G.A.; Hatem, Z.; et al. Obesity induces PD-1 on macrophages to suppress anti-tumour immunity. Nature 2024, 630, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Kerzel, T.; Giacca, G.; Beretta, S.; Bresesti, C.; Notaro, M.; Scotti, G.M.; Balestrieri, C.; Canu, T.; Redegalli, M.; Pedica, F.; et al. In vivo macrophage engineering reshapes the tumor microenvironment leading to eradication of liver metastases. Cancer Cell 2023, 41, 1892–1910.e10. [Google Scholar] [CrossRef] [PubMed]
- Muteeb, G.; Khafaga, D.S.; El-Morsy, M.T.; Farhan, M.; Aatif, M.; Hosney, M. Targeting tumor-associated macrophages with nanocarrier-based treatment for breast cancer: A step toward developing innovative anti-cancer therapeutics. Heliyon 2024, 10, e37217. [Google Scholar] [CrossRef]
- Coënon, L.; Geindreau, M.; Ghiringhelli, F.; Villalba, M.; Bruchard, M. Natural Killer cells at the frontline in the fight against cancer. Cell Death Dis. 2024, 15, 1–14. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, H.; Jounaidi, Y. Comprehensive snapshots of natural killer cells functions, signaling, molecular mechanisms and clinical utilization. Signal Transduct. Target. Ther. 2024, 9, 1–39. [Google Scholar] [CrossRef]
- Vidal-Manrique, M.; Nieuwenstein, T.; Hooijmaijers, L.; de Jonge, P.; Djojoatmo, M.; Jansen, J.; van der Waart, A.; Brock, R.; Dolstra, H. IL-15 transpresentation by ovarian cancer cells improves CD34 + progenitor-derived NK cell's anti-tumor functionality. OncoImmunology 2025, 14, 2465010. [Google Scholar] [CrossRef]
- Jin, P.; Bai, M.; Li, J.; Jia, W.; Yu, J.; Meng, X. Synergistic enhancement of radio-immunotherapy efficacy by IL-15 via macrophage activation and memory T cell response. Cancer Lett. 2025, 613, 217511. [Google Scholar] [CrossRef]
- Horta, A.L.; Gigley, J.; Boutet, M.; Lavau, G.; Weiss, L.M.; Huang, H. Memory-like NK Cells Are a Critical Component of Vaccine-Induced Immunity to Trypanosoma cruzi Infection. J. Immunol. 2024, 212, 617–631. [Google Scholar] [CrossRef]
- Guo, R.; Wang, R.; Zhang, W.; Li, Y.; Wang, Y.; Wang, H.; Li, X.; Song, J. Macrophage Polarisation in the Tumour Microenvironment: Recent Research Advances and Therapeutic Potential of Different Macrophage Reprogramming. Cancer Control. 2025, 32. [Google Scholar] [CrossRef]
- Chiba, S.; Ikushima, H.; Ueki, H.; Yanai, H.; Kimura, Y.; Hangai, S.; Nishio, J.; Negishi, H.; Tamura, T.; Saijo, S.; et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. eLife 2014, 3, e04177. [Google Scholar] [CrossRef] [PubMed]
- Aftabi, S. , et al., Therapeutic targeting of TGF-β in lung cancer. The FEBS Journal. n/a(n/a).
- Mathews, J.A.; Borovsky, D.T.; Reid, K.T.; Murphy, J.M.; Colpitts, S.J.; Carreira, A.S.; Moya, T.A.; Chung, D.C.; Novitzky-Basso, I.; Mattsson, J.; et al. Single cell profiling of hematopoietic stem cell transplant recipients reveals TGF-β1 and IL-2 confer immunoregulatory functions to NK cells. iScience 2024, 27, 111416. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.-Y.; Zhu, Y.; Xie, S.-Z.; Qin, L.-X. Immunosuppressive tumor microenvironment and immunotherapy of hepatocellular carcinoma: current status and prospectives. J. Hematol. Oncol. 2024, 17, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.; Krzywinska, E.; Castells, M.; Gotthardt, D.; Putz, E.M.; Kantari-Mimoun, C.; Chikdene, N.; Meinecke, A.-K.; Schrödter, K.; Helfrich, I.; et al. Targeting VEGF-A in myeloid cells enhances natural killer cell responses to chemotherapy and ameliorates cachexia. Nat. Commun. 2016, 7, 12528. [Google Scholar] [CrossRef]
- Jiang, P.; Jing, S.; Sheng, G.; Jia, F. The basic biology of NK cells and its application in tumor immunotherapy. Front. Immunol. 2024, 15, 1420205. [Google Scholar] [CrossRef]
- Lin, X., et al., Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol Cancer 2024, 23, 108. [CrossRef]
- Fanijavadi, S.; Hansen, T.F.; Zedan, A.H. NK Cell-Microbiota Interaction Biomarker Strategy: Advancing Prostate Cancer Management. Biomolecules 2025, 15, 273. [Google Scholar] [CrossRef]
- Tredicine, M.; Mucci, M.; Recchiuti, A.; Mattoscio, D. Immunoregulatory mechanisms of the arachidonic acid pathway in cancer. FEBS Lett. 2025. [CrossRef]
- Ji, S.; Shi, Y.; Yin, B. Macrophage barrier in the tumor microenvironment and potential clinical applications. Cell Commun. Signal. 2024, 22, 1–14. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, G.; Chai, D.; Dang, Y.; Zheng, J.; Li, H. iNKT: A new avenue for CAR-based cancer immunotherapy. Transl. Oncol. 2022, 17, 101342. [Google Scholar] [CrossRef]
- Díaz-Basabe, A.; Strati, F.; Facciotti, F. License to Kill: When iNKT Cells Are Granted the Use of Lethal Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 3909. [Google Scholar] [CrossRef] [PubMed]
- Cruz, S.M.; Sholevar, C.J.; Judge, S.J.; Darrow, M.A.; Iranpur, K.R.; Farley, L.E.; Lammers, M.; Razmara, A.M.; Dunai, C.; Gingrich, A.A.; et al. Intratumoral NKp46+ natural killer cells are spatially distanced from T and MHC-I+ cells with prognostic implications in soft tissue sarcoma. Front. Immunol. 2023, 14, 1230534. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, S.; Micke, P.; Elebro, J.; Heby, M.; Hrynchyk, I.; Nodin, B.; Leandersson, K.; Mezheyeuski, A.; Jirström, K. Topographical Distribution and Spatial Interactions of Innate and Semi-Innate Immune Cells in Pancreatic and Other Periampullary Adenocarcinoma. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Delfanti, G.; Dellabona, P.; Casorati, G.; Fedeli, M. Adoptive Immunotherapy With Engineered iNKT Cells to Target Cancer Cells and the Suppressive Microenvironment. Front. Med. 2022, 9, 897750. [Google Scholar] [CrossRef]
- Janakiram, N.B., et al., Loss of natural killer T cells promotes pancreatic cancer in LSL-Kras(G12D/+) mice. Immunology 2017, 152, 36–51. [CrossRef]
- Cruz, M.S.; Loureiro, J.P.; Oliveira, M.J.; Macedo, M.F. The iNKT Cell–Macrophage Axis in Homeostasis and Disease. Int. J. Mol. Sci. 2022, 23, 1640. [Google Scholar] [CrossRef]
- Li, Y.-R.; Zhou, Y.; Yu, J.; Zhu, Y.; Lee, D.; Zhu, E.; Li, Z.; Kim, Y.J.; Zhou, K.; Fang, Y.; et al. Engineering allorejection-resistant CAR-NKT cells from hematopoietic stem cells for off-the-shelf cancer immunotherapy. Mol. Ther. 2024, 32, 1849–1874. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Dou, Z.; Delfanti, G.; Tsahouridis, O.; Pellegry, C.M.; Zingarelli, M.; Atassi, G.; Woodcock, M.G.; Casorati, G.; et al. CAR-redirected natural killer T cells demonstrate superior antitumor activity to CAR-T cells through multimodal CD1d-dependent mechanisms. Nat. Cancer 2024, 5, 1607–1621. [Google Scholar] [CrossRef]
- Kostic, M., et al., Dissecting the immune response of CD4+ T cells in Alzheimer’s disease. Reviews in the Neurosciences 2025, 36, 139–168. [CrossRef]
- Khalaf, K.; Chamieh, M.; Welc, N.; Singh, C.; Kaouk, J.L.; Kaouk, A.; Mackiewicz, A.; Kaczmarek, M.; Perek, B. Cellular aspects of immunity involved in the development of atherosclerosis. Front. Immunol. 2025, 16, 1461535. [Google Scholar] [CrossRef]
- Martinenaite, E.; Lecoq, I.; Aaboe-Jørgensen, M.; Ahmad, S.M.; Perez-Penco, M.; Glöckner, H.J.; Chapellier, M.; de la Torre, L.L.; Olsen, L.R.; Rømer, A.M.A.; et al. Arginase-1-specific T cells target and modulate tumor-associated macrophages. J. Immunother. Cancer 2025, 13, e009930. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Nelde, A.; Ramirez, B.C.; Hatin, I.; Arbes, H.; François, P.; Demais, S.; Labaronne, E.; Decimo, D.; Guiguettaz, L.; et al. Unveiling conserved HIV-1 open reading frames encoding T cell antigens using ribosome profiling. Nat. Commun. 2025, 16, 1–18. [Google Scholar] [CrossRef]
- Chandra, D.J.; Alber, B.; Saultz, J.N. The Immune Resistance Signature of Acute Myeloid Leukemia and Current Immunotherapy Strategies. Cancers 2024, 16, 2615. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Sun, L.; Lin, H.; Liao, Y.; Yang, H.; Mao, Y. Harnessing innate immune pathways for therapeutic advancement in cancer. Signal Transduct. Target. Ther. 2024, 9, 1–59. [Google Scholar] [CrossRef]
- Tsomidis, I.; Voumvouraki, A.; Kouroumalis, E. Immune Checkpoints and the Immunology of Liver Fibrosis. Livers 2025, 5, 5. [Google Scholar] [CrossRef]
- Xu, H., et al., Immunoglobulin-like transcript 5 polarizes M2-like tumor-associated macrophages for immunosuppression in non-small cell lung cancer. International Journal of Cancer. n/a(n/a).
- Ge, Y., et al., Utilizing Nanoparticles to Overcome Anti-PD-1/PD-L1 Immunotherapy Resistance in Non-Small Cell Lung cancer: A Potential Strategy. Int J Nanomedicine 2025, 20: p. 2371-2394.
- Xia, Y.; Huang, C.; Zhong, M.; Zhong, H.; Ruan, R.; Xiong, J.; Yao, Y.; Zhou, J.; Deng, J. Targeting HGF/c-MET signaling to regulate the tumor microenvironment: Implications for counteracting tumor immune evasion. Cell Commun. Signal. 2025, 23, 1–18. [Google Scholar] [CrossRef]
- Bos, J.; Schooten, T.G.-V.; Brugman, C.; Jamaludin, F.; van Laarhoven, H.; Derks, S. The tumor immune composition of mismatch repair deficient and Epstein-Barr virus-positive gastric cancer: A systematic review. Cancer Treat. Rev. 2024, 127, 102737. [Google Scholar] [CrossRef]
- Jumaniyazova, E.; Lokhonina, A.; Dzhalilova, D.; Miroshnichenko, E.; Kosyreva, A.; Fatkhudinov, T. The Role of Macrophages in Various Types of Tumors and the Possibility of Their Use as Targets for Antitumor Therapy. Cancers 2025, 17, 342. [Google Scholar] [CrossRef]
- Vilbois, S.; Xu, Y.; Ho, P.-C. Metabolic interplay: tumor macrophages and regulatory T cells. Trends Cancer 2023, 10, 242–255. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Greene, T.T.; Jo, Y.; Chiale, C.; Macal, M.; Fang, Z.; Khatri, F.S.; Codrington, A.L.; Kazane, K.R.; Akbulut, E.; Swaminathan, S.; et al. Metabolic deficiencies underlie reduced plasmacytoid dendritic cell IFN-I production following viral infection. Nat. Commun. 2025, 16, 1–19. [Google Scholar] [CrossRef]
- Niemetz, L.; Bodmer, B.S.; Olal, C.; Escudero-Pérez, B.; Hoehn, K.; Bencsik, A.; A Vickers, M.; Rodríguez, E.; Oestereich, L.; Hoenen, T.; et al. Ebola Virus Infection of Flt3-Dependent, Conventional Dendritic Cells and Antigen Cross-presentation Leads to High Levels of T-Cell Activation. J. Infect. Dis. 2024, 231, 501–511. [Google Scholar] [CrossRef]
- Vafaeian, A.; Rajabi, F.; Rezaei, N. Toll-like receptors in atopic dermatitis: pathogenesis and therapeutic implications. Heliyon 2025, 11, e42226. [Google Scholar] [CrossRef]
- Chen, M.Y.; Zhang, F.; Goedegebuure, S.P.; Gillanders, W.E. Dendritic cell subsets and implications for cancer immunotherapy. Front. Immunol. 2024, 15, 1393451. [Google Scholar] [CrossRef]
- Luo, D.; Zhou, J.; Ruan, S.; Zhang, B.; Zhu, H.; Que, Y.; Ying, S.; Li, X.; Hu, Y.; Song, Z. Overcoming immunotherapy resistance in gastric cancer: insights into mechanisms and emerging strategies. Cell Death Dis. 2025, 16, 1–18. [Google Scholar] [CrossRef]
- Niveau, C.; Cettour-Cave, M.; Mouret, S.; Cuevas, E.S.; Pezet, M.; Roubinet, B.; Gil, H.; De Fraipont, F.; Landemarre, L.; Charles, J.; et al. MCT1 lactate transporter blockade re-invigorates anti-tumor immunity through metabolic rewiring of dendritic cells in melanoma. Nat. Commun. 2025, 16, 1–24. [Google Scholar] [CrossRef]
- Guo, F.; Kong, W.; Li, D.; Zhao, G.; Anwar, M.; Xia, F.; Zhang, Y.; Ma, C.; Ma, X. M2-type tumor-associated macrophages upregulated PD-L1 expression in cervical cancer via the PI3K/AKT pathway. Eur. J. Med Res. 2024, 29, 357. [Google Scholar] [CrossRef]
- Ma, X.; Guo, Z.; Wei, X.; Zhao, G.; Han, D.; Zhang, T.; Chen, X.; Cao, F.; Dong, J.; Zhao, L.; et al. Spatial Distribution and Predictive Significance of Dendritic Cells and Macrophages in Esophageal Cancer Treated With Combined Chemoradiotherapy and PD-1 Blockade. Front. Immunol. 2022, 12, 786429. [Google Scholar] [CrossRef]
- Xie, D.; Lu, G.; Mai, G.; Guo, Q.; Xu, G. Tissue-resident memory T cells in diseases and therapeutic strategies. Medcomm 2025, 6, e70053. [Google Scholar] [CrossRef]
- Li, Y., et al., DNMT1 inhibition improves the activity of memory-like natural killer cells by enhancing the level of autophagy. Mol Biol Rep 2024, 52, 68.
- Li, L.; Xu, T.; Qi, X. Balanced regulation of ROS production and inflammasome activation in preventing early development of colorectal cancer. Immunol. Rev. 2024, 329. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Tu, C.; Xiong, H.; Xu, Y.; Shi, X.; Zhang, X.; Yang, R.; Zhang, N.; Lin, B.; Liu, M.; et al. GITRL enhances cytotoxicity and persistence of CAR-T cells in cancer therapy. Mol. Ther. 2025. [Google Scholar] [CrossRef] [PubMed]
- Luyang, H.; Zeng, F.; Lei, Y.; He, Q.; Zhou, Y.; Xu, J. Bidirectional role of neutrophils in tumor development. Mol. Cancer 2025, 24, 1–16. [Google Scholar] [CrossRef]
- Schmitt, H., M.F. Neurath, and R. Atreya, Role of the IL23/IL17 Pathway in Crohn's Disease. Front Immunol 2021, 12: p. 622934.
- Zhao, J.; Lu, Q.; Liu, Y.; Shi, Z.; Hu, L.; Zeng, Z.; Tu, Y.; Xiao, Z.; Xu, Q. Th17 Cells in Inflammatory Bowel Disease: Cytokines, Plasticity, and Therapies. J. Immunol. Res. 2021, 2021, 1–14. [Google Scholar] [CrossRef]
- Surman, M.; Przybyło, M.; Wilczak, M. Melanoma-derived extracellular vesicles transfer proangiogenic factors. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2025, 33, 245–262. [Google Scholar] [CrossRef]
- Mehdikhani, F.; Hajimehdipoor, H.; Tansaz, M.; Maresca, M.; Rajabi, S. Sesquiterpene Lactones as Promising Phytochemicals to Cease Metastatic Propagation of Cancer. Biomolecules 2025, 15, 268. [Google Scholar] [CrossRef]
- García-Navas, R.; Gajate, C.; Mollinedo, F. Neutrophils drive endoplasmic reticulum stress-mediated apoptosis in cancer cells through arginase-1 release. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Yang, Q.; Cui, M.; Wang, J.; Zhao, Y.; Yin, W.; Liao, Z.; Liang, Y.; Jiang, Z.; Li, Y.; Guo, J.; et al. Circulating mitochondrial DNA promotes M2 polarization of tumor associated macrophages and HCC resistance to sorafenib. Cell Death Dis. 2025, 16, 1–14. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, P.; Sun, R.; Li, J.; Hu, Z.; Xin, H.; Luo, C.; Zhou, J.; Fan, J.; Zhou, S. Tumor-associated neutrophils and macrophages interaction contributes to intrahepatic cholangiocarcinoma progression by activating STAT3. J. Immunother. Cancer 2021, 9, e001946. [Google Scholar] [CrossRef]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, A.; Sudarskikh, T.; Kovalev, O.; Kzhyshkowska, J.; Larionova, I. Interaction of tumor-associated macrophages with stromal and immune components in solid tumors: Research progress (Review). Int. J. Oncol. 2023, 62, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. npj Precis. Oncol. 2024, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Rao, A.S.; Stadanlick, J.; Bruns, K.; Sullivan, N.T.; Bermudez, A.; Honig-Frand, A.; Krouse, R.; Arambepola, S.; Guo, E.; et al. Human Tumor–Associated Macrophages and Neutrophils Regulate Antitumor Antibody Efficacy through Lethal and Sublethal Trogocytosis. Cancer Res. 2024, 84, 1029–1047. [Google Scholar] [CrossRef]
- Lei, Q.; Zhen, S.; Zhang, L.; Zhao, Q.; Yang, L.; Zhang, Y. A2AR-mediated CXCL5 upregulation on macrophages promotes NSCLC progression via NETosis. Cancer Immunol. Immunother. 2024, 73, 1–16. [Google Scholar] [CrossRef]
- Schmidt, E.; Distel, L.; Erber, R.; Büttner-Herold, M.; Rosahl, M.-C.; Ott, O.J.; Strnad, V.; Hack, C.C.; Hartmann, A.; Hecht, M.; et al. Tumor-Associated Neutrophils Are a Negative Prognostic Factor in Early Luminal Breast Cancers Lacking Immunosuppressive Macrophage Recruitment. Cancers 2024, 16, 3160. [Google Scholar] [CrossRef]
- Pan, Z.; Chen, J.; Xu, T.; Cai, A.; Han, B.; Li, Y.; Fang, Z.; Yu, D.; Wang, S.; Zhou, J.; et al. VSIG4+ tumor-associated macrophages mediate neutrophil infiltration and impair antigen-specific immunity in aggressive cancers through epigenetic regulation of SPP1. J. Exp. Clin. Cancer Res. 2025, 44, 1–19. [Google Scholar] [CrossRef]
- Chu, X.; Tian, Y.; Lv, C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol. Cancer 2024, 23, 1–24. [Google Scholar] [CrossRef]
- Werner, W.; Kuzminskaya, M.; Lurje, I.; Tacke, F.; Hammerich, L. Overcoming Resistance to Immune Checkpoint Blockade in Liver Cancer with Combination Therapy: Stronger Together? Semin. Liver Dis. 2024, 44, 159–179. [Google Scholar] [CrossRef]
- Yin, Y.; Feng, W.; Chen, J.; Chen, X.; Wang, G.; Wang, S.; Xu, X.; Nie, Y.; Fan, D.; Wu, K.; et al. Immunosuppressive tumor microenvironment in the progression, metastasis, and therapy of hepatocellular carcinoma: from bench to bedside. Exp. Hematol. Oncol. 2024, 13, 1–37. [Google Scholar] [CrossRef]
- Xiao, G.; Wang, X.; Sheng, J.; Lu, S.; Yu, X.; Wu, J.D. Soluble NKG2D ligand promotes MDSC expansion and skews macrophage to the alternatively activated phenotype. J. Hematol. Oncol. 2015, 8, 13–13. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Kumar, A.; Amdare, N.P.; Pathak, R. Current Landscape of Cancer Immunotherapy: Harnessing the Immune Arsenal to Overcome Immune Evasion. Biology 2024, 13, 307. [Google Scholar] [CrossRef]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef]
- Tang, X.; Gao, L.; Jiang, X.; Hou, Z.; Wang, Y.; Hou, S.; Qu, H. Single-cell profiling reveals altered immune landscape and impaired NK cell function in gastric cancer liver metastasis. Oncogene 2024, 43, 2635–2646. [Google Scholar] [CrossRef]
- Tabachnick-Cherny, S.; Pulliam, T.; Rodriguez, H.J.; Fan, X.; Hippe, D.S.; Jones, D.C.; Moshiri, A.S.; Smythe, K.S.; Kulikauskas, R.M.; Zaba, L.C.; et al. Characterization of Immunosuppressive Myeloid Cells in Merkel Cell Carcinoma: Correlation with Resistance to PD-1 Pathway Blockade. Clin. Cancer Res. 2023, 30, 1189–1199. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Gu, Y.; Liu, T.; Zhao, X.; Cheng, S.; Duan, L.; Huang, C.; Wu, S.; Gao, S. Complement C3 of tumor-derived extracellular vesicles promotes metastasis of RCC via recruitment of immunosuppressive myeloid cells. Proc. Natl. Acad. Sci. 2025, 122. [Google Scholar] [CrossRef]
- Guo, F.; Song, Y.; Dong, S.; Wei, J.; Li, B.; Xu, T.; Wang, H. Characterization and anti-tuberculosis effects of γδ T cells expanded and activated by Mycobacterium tuberculosis heat-resistant antigen. Virulence 2025, 16, 2462092. [Google Scholar] [CrossRef]
- Fang, Y.; Chen, Y.; Niu, S.; Lyu, Z.; Tian, Y.; Shen, X.; Li, Y.-R.; Yang, L. Biological functions and therapeutic applications of human mucosal-associated invariant T cells. J. Biomed. Sci. 2025, 32, 1–17. [Google Scholar] [CrossRef]
- Hu, Y., et al., γδ T cells: origin and fate, subsets, diseases and immunotherapy. Signal Transduct Target Ther 2023, 8, 434. [CrossRef]
- Revesz, I.A., et al., Effective γδ T-cell clinical therapies: current limitations and future perspectives for cancer immunotherapy. Clin Transl Immunology 2024, 13, e1492. [CrossRef]
- Petruk, N.; Sousa, S.; Croset, M.; Polari, L.; Zlatev, H.; Selander, K.; Mönkkönen, J.; Clézardin, P.; Määttä, J. Liposome-encapsulated zoledronate increases inflammatory macrophage population in TNBC tumours. Eur. J. Pharm. Sci. 2023, 190, 106571. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lin, X.; Cui, X. Effect of Liposome-Encapsulated Zoledronic Acid on Microenvironment of Hepatocellular Carcinoma May Depend on the Ratio Between M1 and M2 Polarized Macrophages. Bull. Exp. Biol. Med. 2020, 170, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Wendong, Y.; Hengwu, X.; Yanhong, C.; Yingying, X.; Feng, Z.; Zeng, W.; Xinjun, C. Mannose modified co-loaded zoledronic liposomes deplete M2-tumor-associated macrophages to enhance anti-tumor effect of doxorubicin on TNBC. J. Drug Deliv. Sci. Technol. 2022, 74. [Google Scholar] [CrossRef]
- Man, F.; Lim, L.; Volpe, A.; Gabizon, A.; Shmeeda, H.; Draper, B.; Parente-Pereira, A.C.; Maher, J.; Blower, P.J.; Fruhwirth, G.O.; et al. In Vivo PET Tracking of 89Zr-Labeled Vγ9Vδ2 T Cells to Mouse Xenograft Breast Tumors Activated with Liposomal Alendronate. Mol. Ther. 2019, 27, 219–229. [Google Scholar] [CrossRef]
- Parente-Pereira, A.C., et al., Adoptive immunotherapy of epithelial ovarian cancer with Vγ9Vδ2 T cells, potentiated by liposomal alendronic acid. J Immunol 2014, 193, 5557–66. [CrossRef]
- Gao, Z.; Bai, Y.; Lin, A.; Jiang, A.; Zhou, C.; Cheng, Q.; Liu, Z.; Chen, X.; Zhang, J.; Luo, P. Gamma delta T-cell-based immune checkpoint therapy: attractive candidate for antitumor treatment. Mol. Cancer 2023, 22, 31. [Google Scholar] [CrossRef]
- Shiravand, Y., et al., Immune Checkpoint Inhibitors in Cancer Therapy. Curr Oncol 2022, 29, 3044–3060. [CrossRef]
- Liu, Y.; Tan, H.; Dai, J.; Lin, J.; Zhao, K.; Hu, H.; Zhong, C. Targeting macrophages in cancer immunotherapy: Frontiers and challenges. J. Adv. Res. 2025. [CrossRef]
- Janjigian, Y.Y.; Kawazoe, A.; Bai, Y.; Xu, J.; Lonardi, S.; Metges, J.P.; Yanez, P.; Wyrwicz, L.S.; Shen, L.; Ostapenko, Y.; et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2-positive gastric or gastro-oesophageal junction adenocarcinoma: interim analyses from the phase 3 KEYNOTE-811 randomised placebo-controlled trial. Lancet 2023, 402, 2197–2208. [Google Scholar] [CrossRef]
- Brahmer, J.R., et al., Phase I Study of Single-Agent Anti-Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J Clin Oncol 2023, 41, 715–723. [CrossRef]
- Li, J.-W.; Shi, D.; Wan, X.-C.; Hu, J.; Su, Y.-F.; Zeng, Y.-P.; Hu, Z.-J.; Yu, B.-H.; Zhang, Q.-L.; Wei, P.; et al. Universal extracellular vesicles and PD-L1+ extracellular vesicles detected by single molecule array technology as circulating biomarkers for diffuse large B cell lymphoma. OncoImmunology 2021, 10, 1995166. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Ji, Q. Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front. Immunol. 2022, 13, 874589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H., et al., Roles of tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer 2023, 22, 58. [CrossRef] [PubMed]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat. Commun. 2022, 13, 1–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1578–1593.e8. [Google Scholar] [CrossRef]
- Ning, J.; Hou, X.; Hao, J.; Zhang, W.; Shi, Y.; Huang, Y.; Ruan, X.; Zheng, X.; Gao, M. METTL3 inhibition induced by M2 macrophage-derived extracellular vesicles drives anti-PD-1 therapy resistance via M6A-CD70-mediated immune suppression in thyroid cancer. Cell Death Differ. 2023, 30, 2265–2279. [Google Scholar] [CrossRef]
- You, Q.; Wang, F.; Du, R.; Pi, J.; Wang, H.; Huo, Y.; Liu, J.; Wang, C.; Yu, J.; Yang, Y.; et al. m6A Reader YTHDF1-Targeting Engineered Small Extracellular Vesicles for Gastric Cancer Therapy via Epigenetic and Immune Regulation. Adv. Mater. 2022, 35, e2204910. [Google Scholar] [CrossRef]
- Li, J.; Wu, C.; Hu, H.; Qin, G.; Wu, X.; Bai, F.; Zhang, J.; Cai, Y.; Huang, Y.; Wang, C.; et al. Remodeling of the immune and stromal cell compartment by PD-1 blockade in mismatch repair-deficient colorectal cancer. Cancer Cell 2023, 41, 1152–1169.e7. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, L.; Xia, H.; Yan, Y.; Zhu, X.; Sun, F.; Sun, L.; Li, S.; Li, D.; Wang, J.; et al. Tumor microenvironment remodeling after neoadjuvant immunotherapy in non-small cell lung cancer revealed by single-cell RNA sequencing. Genome Med. 2023, 15, 1–25. [Google Scholar] [CrossRef]
- Song, C.-H.; Kim, N.; Nam, R.H.; Choi, S.I.; Jang, J.Y.; Kim, J.W.; Na, H.Y.; Lee, H.-N. Combination treatment with 17β-estradiol and anti-PD-L1 suppresses MC38 tumor growth by reducing PD-L1 expression and enhancing M1 macrophage population in MC38 colon tumor model. Cancer Lett. 2022, 543, 215780. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Kluger, H.; George, S.; Tykodi, S.S.; Kuzel, T.M.; Perets, R.; Nair, S.; Procopio, G.; A Carducci, M.; Castonguay, V.; et al. FRACTION-RCC: nivolumab plus ipilimumab for advanced renal cell carcinoma after progression on immuno-oncology therapy. J. Immunother. Cancer 2022, 10, e005780. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M., et al., Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 2016, 17, 651–62.
- Haag, G.M.; Springfeld, C.; Grün, B.; Apostolidis, L.; Zschäbitz, S.; Dietrich, M.; Berger, A.-K.; Weber, T.F.; Zoernig, I.; Schaaf, M.; et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer – The PICCASSO phase I trial. Eur. J. Cancer 2022, 167, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Cassier, P.A.; Italiano, A.; Gomez-Roca, C.; Le Tourneau, C.; Toulmonde, M.; D'Angelo, S.P.; Weber, K.; Loirat, D.; Jacob, W.; Jegg, A.-M.; et al. Long-term clinical activity, safety and patient-reported quality of life for emactuzumab-treated patients with diffuse-type tenosynovial giant-cell tumour. Eur. J. Cancer 2020, 141, 162–170. [Google Scholar] [CrossRef]
- Gomez-Roca, C.; Cassier, P.; Zamarin, D.; Machiels, J.-P.; Gracia, J.L.P.; Hodi, F.S.; Taus, A.; Garcia, M.M.; Boni, V.; Eder, J.P.; et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naïve or experienced for immune checkpoint blockade. J. Immunother. Cancer 2022, 10, e004076. [Google Scholar] [CrossRef]
- Machiels, J.-P.; Gomez-Roca, C.; Michot, J.-M.; Zamarin, D.; Mitchell, T.; Catala, G.; Eberst, L.; Jacob, W.; Jegg, A.-M.; A Cannarile, M.; et al. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J. Immunother. Cancer 2020, 8, e001153. [Google Scholar] [CrossRef]
- Razak, A.R.; Cleary, J.M.; Moreno, V.; Boyer, M.; Aller, E.C.; Edenfield, W.; Tie, J.; Harvey, R.D.; Rutten, A.; A Shah, M.; et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J. Immunother. Cancer 2020, 8, e001006. [Google Scholar] [CrossRef]
- Johnson, M.; Dudek, A.Z.; Sukari, A.; Call, J.; Kunk, P.R.; Lewis, K.; Gainor, J.F.; Sarantopoulos, J.; Lee, P.; Golden, A.; et al. ARRY-382 in Combination with Pembrolizumab in Patients with Advanced Solid Tumors: Results from a Phase 1b/2 Study. Clin. Cancer Res. 2022, 28, 2517–2526. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Burg, J.M.; Mick, R.; Trosko, J.A.; Li, D.; Shaik, M.N.; Tolcher, A.W.; Hamid, O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. OncoImmunology 2013, 2, e23033. [Google Scholar] [CrossRef]
- Bauer, C.; Kühnemuth, B.; Duewell, P.; Ormanns, S.; Gress, T.; Schnurr, M. Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer. Cancer Lett. 2016, 381, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, X.; Dong, S.; Guo, Y.; Luo, Z.; Zhuang, S.-M.; Liu, J.; Liu, T.; Liao, J.; Wen, W. Modulating tumor-associated macrophages through CSF1R inhibition: a potential therapeutic strategy for HNSCC. J. Transl. Med. 2025, 23, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liaw, K.; Reddy, R.; Sharma, A.; Li, J.; Chang, M.; Sharma, R.; Salazar, S.; Kannan, S.; Kannan, R.M. Targeted systemic dendrimer delivery of CSF-1R inhibitor to tumor-associated macrophages improves outcomes in orthotopic glioblastoma. Bioeng. Transl. Med. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- A Siddiqui, B.; Chapin, B.F.; Jindal, S.; Duan, F.; Basu, S.; Yadav, S.S.; Gu, A.-D.; Espejo, A.B.; Kinder, M.; A Pettaway, C.; et al. Immune and pathologic responses in patients with localized prostate cancer who received daratumumab (anti-CD38) or edicotinib (CSF-1R inhibitor). J. Immunother. Cancer 2023, 11, e006262. [Google Scholar] [CrossRef]
- Wiehagen, K.R.; Girgis, N.M.; Yamada, D.H.; Smith, A.A.; Chan, S.R.; Grewal, I.S.; Quigley, M.; Verona, R.I. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 1109–1121. [Google Scholar] [CrossRef]
- Omstead, A.N.; Paskewicz, M.; Gorbunova, A.; Zheng, P.; Salvitti, M.S.; Mansoor, R.; Reed, P.; Ballengee, S.; Wagner, P.L.; A Jobe, B.; et al. CSF-1R inhibitor, pexidartinib, sensitizes esophageal adenocarcinoma to PD-1 immune checkpoint blockade in a rat model. Carcinog. 2022, 43, 842–850. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41. [Google Scholar] [CrossRef]
- Kim, D.; An, L.; Moon, J.; Maymi, V.I.; McGurk, A.I.; Rudd, B.D.; Fowell, D.J.; White, A.C. Ccr2+ Monocyte-Derived Macrophages Influence Trajectories of Acquired Therapy Resistance in Braf-Mutant Melanoma. Cancer Res. 2023, 83, 2328–2344. [Google Scholar] [CrossRef]
- Miyamoto, T.; Murakami, R.; Hamanishi, J.; Tanigaki, K.; Hosoe, Y.; Mise, N.; Takamatsu, S.; Mise, Y.; Ukita, M.; Taki, M.; et al. B7-H3 Suppresses Antitumor Immunity via the CCL2–CCR2–M2 Macrophage Axis and Contributes to Ovarian Cancer Progression. Cancer Immunol. Res. 2021, 10, 56–69. [Google Scholar] [CrossRef]
- Trac, N.; Chen, L.-Y.; Zhang, A.; Liao, C.-P.; Poon, C.; Wang, J.; Ando, Y.; Joo, J.; Garri, C.; Shen, K.; et al. CCR2-targeted micelles for anti-cancer peptide delivery and immune stimulation. J. Control. Release 2020, 329, 614–623. [Google Scholar] [CrossRef]
- Wang, D. , et al., Exosome-encapsulated miRNAs contribute to CXCL12/CXCR4-induced liver metastasis of colorectal cancer by enhancing M2 polarization of macrophages. Cancer Lett 2020, 474: p. 36-52.
- Kocher, F.; Puccini, A.; Untergasser, G.; Martowicz, A.; Zimmer, K.; Pircher, A.; Baca, Y.; Xiu, J.; Haybaeck, J.; Tymoszuk, P.; et al. Multi-omic Characterization of Pancreatic Ductal Adenocarcinoma RelatesCXCR4mRNA Expression Levels to Potential Clinical Targets. Clin. Cancer Res. 2022, 28, 4957–4967. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Dong, C.; Jia, W.; Ma, B. Exosomal miR-655-3p inhibits growth, and invasion and macrophage M2 polarization through targeting CXCR4 in papillary thyroid carcinoma. Acta Biochim. Pol. 2022, 69, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Thapa, K.; Khan, H.; Kaur, G.; Kumar, P.; Singh, T.G. Therapeutic targeting of angiopoietins in tumor angiogenesis and cancer development. Biochem. Biophys. Res. Commun. 2023, 687, 149130. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, X.; Li, J.; Zhang, P.; Li, H.; Chen, G.; Zhang, W.; Wang, T.; Frazer, I.; Ni, G. Caerin 1.1/1.9 Enhances Antitumour Immunity by Activating the IFN-α Response Signalling Pathway of Tumour Macrophages. Cancers 2022, 14, 5785. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Repolarizing macrophages improves breast cancer therapy. Cell Res. 2017, 27, 963–964. [Google Scholar] [CrossRef]
- Goulielmaki, E.; Bermudez-Brito, M.; Andreou, M.; Tzenaki, N.; Tzardi, M.; de Bree, E.; Tsentelierou, E.; Makrigiannakis, A.; Papakonstanti, E.A. Pharmacological inactivation of the PI3K p110δ prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Dis. 2018, 9, 678. [Google Scholar] [CrossRef]
- Cheng, Y.; Bai, F.; Ren, X.; Sun, R.; Guo, X.; Liu, W.; Wang, B.; Yang, Y.; Zhang, X.; Xu, Y.; et al. Phosphoinositide-Binding Protein TIPE1 Promotes Alternative Activation of Macrophages and Tumor Progression via PIP3/Akt/TGFβ Axis. Cancer Res. 2022, 82, 1603–1616. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- Jogalekar, M.P.; Rajendran, R.L.; Khan, F.; Dmello, C.; Gangadaran, P.; Ahn, B.-C. CAR T-Cell-Based gene therapy for cancers: new perspectives, challenges, and clinical developments. Front. Immunol. 2022, 13, 925985. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Tichet, M.; Wullschleger, S.; Chryplewicz, A.; Fournier, N.; Marcone, R.; Kauzlaric, A.; Homicsko, K.; Deak, L.C.; Umaña, P.; Klein, C.; et al. Bispecific PD1-IL2v and anti-PD-L1 break tumor immunity resistance by enhancing stem-like tumor-reactive CD8+ T cells and reprogramming macrophages. Immunity 2023, 56, 162–179.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N., et al., Vinblastine resets tumor-associated macrophages toward M1 phenotype and promotes antitumor immune response. J Immunother Cancer 2023, 11.
- Zhou, C.; Weng, J.; Liu, C.; Liu, S.; Hu, Z.; Xie, X.; Gao, D.; Zhou, Q.; Sun, J.; Xu, R.; et al. Disruption of SLFN11 Deficiency–Induced CCL2 Signaling and Macrophage M2 Polarization Potentiates Anti–PD-1 Therapy Efficacy in Hepatocellular Carcinoma. Gastroenterology 2023, 164, 1261–1278. [Google Scholar] [CrossRef]
Figure 1.
How various environmental stimuli influence the trajectory of macrophage differentiation. Classically activated macrophages are polarized into alternatively activated macrophages under various interferons, LPS, and interleukins. Created by BioRender.
Figure 1.
How various environmental stimuli influence the trajectory of macrophage differentiation. Classically activated macrophages are polarized into alternatively activated macrophages under various interferons, LPS, and interleukins. Created by BioRender.
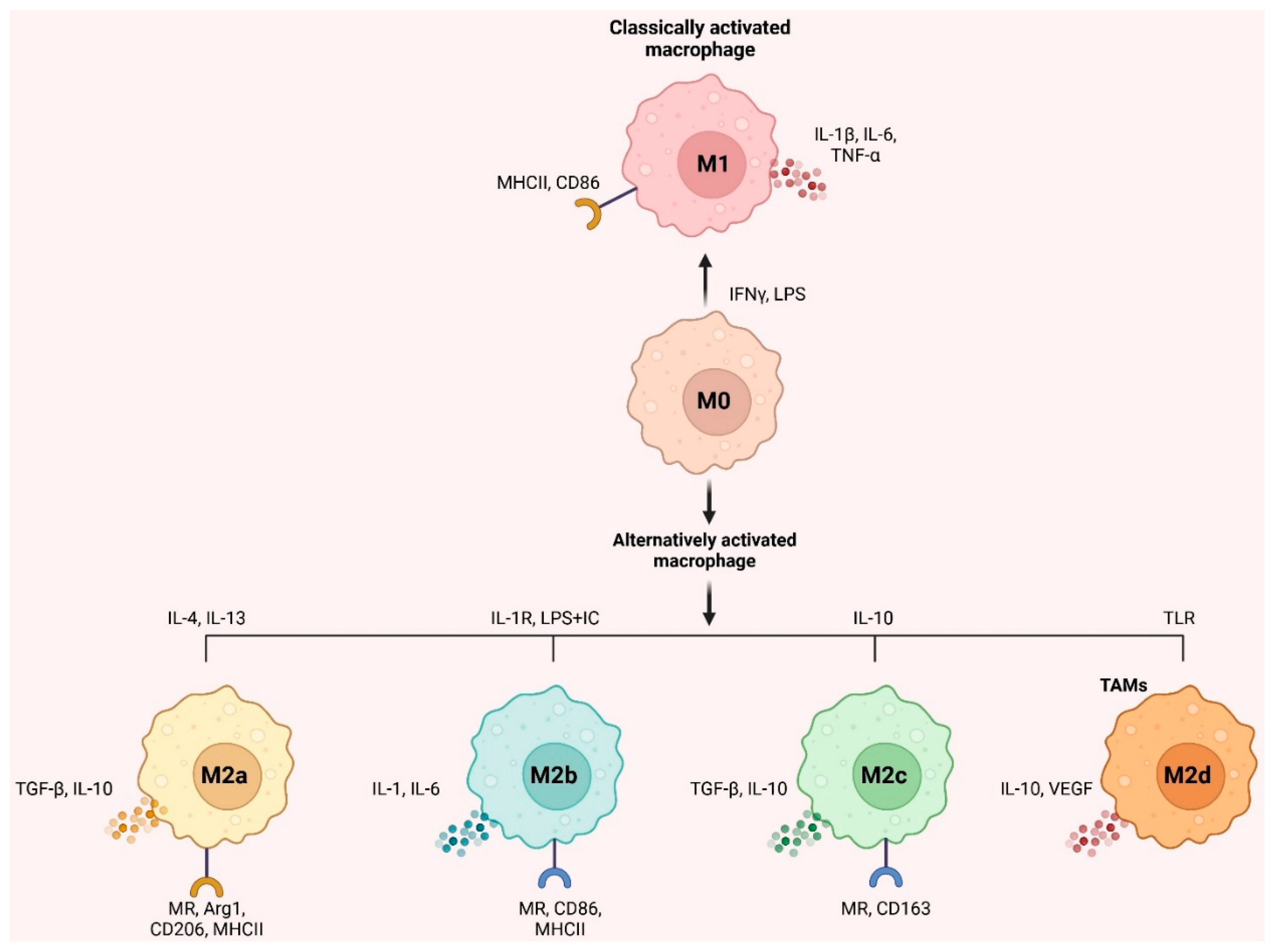
Figure 2.
TAMs produce cancer-promoting IL-6, IL-8, and IL-10. Similarly, they produce T-cell-killing IL-8. Multiple tumor cell-TAM juxtacrine connections restrain immunity. PD-1/L1 inhibits macrophages to hide malignancy from the immune system, and through B7-H3, cancer immune evasion suppresses T cells. The "do-not-eat-me" signal is sent via SIRPα/CD47 and CD24/Siglec-10 pathways. Tumor cells may overexpress CD47 and CD24 to escape macrophages. LILRB1/MHC class I component β2-microglobulin is essential for tumor defense against macrophages. Cells exchange circRNA, miRNA, lncRNA, ICAM-1, REG3b, lipids, proteins, and mRNA via exosomes. TAM associate ApoE and other exosome-delivered chemicals activate PI3K-Akt, causing EMT, cytoskeletal remodeling, and cancer cell migration. Eph44-Ephrin juxtacrine controls immune cell motility, activation, survival, and death. CD44 affects phagocytosis, inflammation, and multidrug resistance and is an effective phagocytic receptor due to HA binding and metabolization. Eph44-ephrin, CD90-CD11b, BTN3N3-L-SECtin, and CD44-HA all have a role in keeping cancer stem cells alive. The paracrine system promotes TAM proliferation, differentiation, and TGF-β production, leading to tumor cell invasion and treatment resistance. The autocrine relationship between IL-1RL1 and IL-33 preserves the tumor cell's stemness. Thus, the complex TME interactions promote crosstalk. ApoE, Apolipoprotein E; CCL, C-C motif chemokine Ligand; EMT, epithelial-mesenchymal transition; GM-CSF, granulocyte-macrophage colony-stimulating factor; HA, hyaluronic acid; IL, interleukin; JAK, Janus kinase; lncRNA, long non-coding RNA; M-CSF, macrophage colony-stimulating factor; MGF-E8, milk fat globule-EGF factor 8; MHC, major histocompatibility complex; miRNA, microRNA; PD-L1, programmed death-ligand 1; Siglec, sialic acid binding Ig-like lectin. Created by BioRender.
Figure 2.
TAMs produce cancer-promoting IL-6, IL-8, and IL-10. Similarly, they produce T-cell-killing IL-8. Multiple tumor cell-TAM juxtacrine connections restrain immunity. PD-1/L1 inhibits macrophages to hide malignancy from the immune system, and through B7-H3, cancer immune evasion suppresses T cells. The "do-not-eat-me" signal is sent via SIRPα/CD47 and CD24/Siglec-10 pathways. Tumor cells may overexpress CD47 and CD24 to escape macrophages. LILRB1/MHC class I component β2-microglobulin is essential for tumor defense against macrophages. Cells exchange circRNA, miRNA, lncRNA, ICAM-1, REG3b, lipids, proteins, and mRNA via exosomes. TAM associate ApoE and other exosome-delivered chemicals activate PI3K-Akt, causing EMT, cytoskeletal remodeling, and cancer cell migration. Eph44-Ephrin juxtacrine controls immune cell motility, activation, survival, and death. CD44 affects phagocytosis, inflammation, and multidrug resistance and is an effective phagocytic receptor due to HA binding and metabolization. Eph44-ephrin, CD90-CD11b, BTN3N3-L-SECtin, and CD44-HA all have a role in keeping cancer stem cells alive. The paracrine system promotes TAM proliferation, differentiation, and TGF-β production, leading to tumor cell invasion and treatment resistance. The autocrine relationship between IL-1RL1 and IL-33 preserves the tumor cell's stemness. Thus, the complex TME interactions promote crosstalk. ApoE, Apolipoprotein E; CCL, C-C motif chemokine Ligand; EMT, epithelial-mesenchymal transition; GM-CSF, granulocyte-macrophage colony-stimulating factor; HA, hyaluronic acid; IL, interleukin; JAK, Janus kinase; lncRNA, long non-coding RNA; M-CSF, macrophage colony-stimulating factor; MGF-E8, milk fat globule-EGF factor 8; MHC, major histocompatibility complex; miRNA, microRNA; PD-L1, programmed death-ligand 1; Siglec, sialic acid binding Ig-like lectin. Created by BioRender.
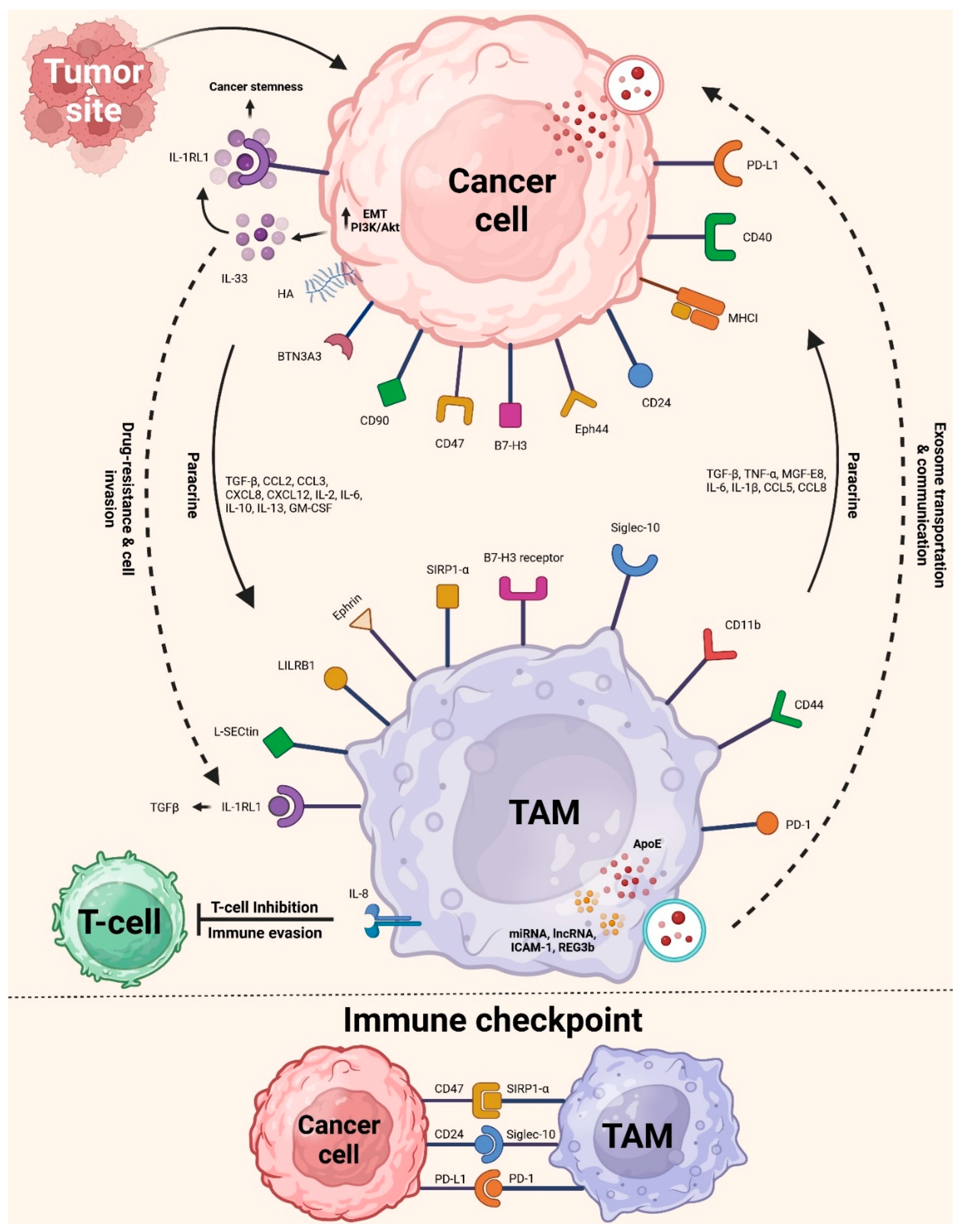

Table 1.
Clinical studies of several drugs that target TAMs.
| Drug | Phase | Cancer type | Combination therapy | NCT identifier |
|---|---|---|---|---|
| Chemokine inhibitors | ||||
| Carlumab (anti-CCL2 antibodies; Centocor) | Phase II (completed) | Prostate cancer | NA | NCT00992186 |
| BMS-813160 (CCR2/CCR5 antagonist; Bristol Myers Squibb) | Phase II (completed) | Renal cell carcinoma | Nivolumab (OPDIVO) in conjunction with ipilimumab (Yervoy) | NCT02996110 |
| Phase I/II (completed) | Pancreatic cancer, colorectal cancer, non-small cell lung cancer | Nab-paclitaxel with nivolumab | NCT03184870 | |
| Phase II (ongoing) | Hepatocellular carcinoma | Nivolumab | NCT04123379 | |
| PF-4136309 (CCR2 antagonist; Pfizer) | Phase II (completed) | PDAC | Nab-paclitaxel, gemcitabine | NCT01413022 |
| CSF1R inhibitors | ||||
| PLX3397 (Plexxikon) | Phase I/II (ongoing) | Tumors of the nerve sheath and sarcoma | Sirolimus (Rapamune) | NCT02584647 |
| Phase I/II (Terminated) | Melanoma and solid tumors, both at advanced stages | Pembrolizumab (Keytruda) | NCT02452424 | |
| Phase I/II (Completed) | Breast carcinoma | Eribulin (Halaven) | NCT01596751 | |
| Phase I/II (completed) | Glioblastoma | Radiotherapy, temozolomide (TMZ) | NCT01790503 | |
| BLZ945 (Novartis) | Phase I/II (Terminated) | Solid tumors | PDR001 (anti- PD1) | NCT02829723 |
| Antibodies targeting CSF1R | ||||
| LY3022855 (Eli Lilly's IMC-C S4) | Phase I/II (completed) | Melanoma | MEK/BRAF inhibitors | NCT03101254 |
| Emactuzumab (RO5509554/RG7155; Roche) | Phase II (Terminated) | Gynecological neoplasms and ovarian cancer | Gynecological neoplasms and ovarian cancer | NCT02923739 |
| Phase I/II (ongoing) | PDAC | Nab- paclitaxel, gemcitabine | NCT03193190 | |
| AMG820 (Amgen) | Phase I/II (completed) | Pancreatic cancer, CRC, NSCLC | Pembrolizumab | NCT02713529 |
| ARRAY-382 (Pfizer) | Phase I/II (terminated) | Solid tumors | Solid tumors | NCT02880371 |
| Agonist anti-CD40 antibodies (cont.) | ||||
| APX005M (Apexigen) | Phase II (completed) | Oesophageal cancer | Radiation, paclitaxel, carboplatin | NCT03214250 |
| Phase I/II (completed) | Pancreatic cancer | Nab- paclitaxel, gemcitabine, nivolumab | NCT03214250 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



