Submitted:
01 April 2025
Posted:
02 April 2025
You are already at the latest version
Abstract
Traumatic brain injury (TBI) represents a significant medical concern typically resulting from severe or repetitive mild head impacts. It often results in vision deterioration and can cause sudden death after hospitalization as a result of pathologies. The objective identification options are limited or too invasive to be routinely applied; therefore, we need new tools to identify the severity of the disease. The retina is the only part of the central nervous system that is directly approachable by light and easy to access. In this review, we used an aimed database search, together with a blinded human and artificial intelligence (AI) based scoring system to narrow down the number of articles, based on relevance to the topic. The selected articles went through thorough processing to identify the relevant links in them. Based on this, we hypothesize that the retina can be used as a direct indicator of brain health, including the use of microglia to indicate traumatic brain injury. As an outcome, we concluded that recent developments in technology now allow the imaging of the retinal surface and cells directly to allow retinal cellular and inflammatory assessment and translation of the findings into medical practice.
Keywords:
traumatic brain injury
; retina
; microglia
; visual system
; imaging
; disease
1. Introduction
The phrase "Eyes are the windows to the soul" states that one can infer significant insight about a person's thoughts and feelings by looking into the eyes. While this phrase is often used in literature in our context, we use it metaphorically to explore the potential of the retina showing in revealing traumatic brain injury (TBI) as well as to utilize it to assess the severity of brain injuries.
There is no perfect marker to identify TBI severity to this date, not to mention the need for noninvasive techniques that improve the assessment of disease severity.
Traumatic brain injury (TBI) has been increasingly recognized as a significant contributor to retinal dysfunction, which is not surprising given the shared developmental and physiological characteristics between the retina and the central nervous system (CNS). The impact of TBI on retinal integrity manifests through inflammatory responses, neurodegenerative processes, oxidative stress, and functional impairments, which collectively suggest the retina as a potential biomarker for assessing TBI severity and progression. Here, we aim to sum up the recent literature about brain-retina relations in TBI to identify novel details for identifying TBI severity.
2. Results
Brain traumas (TBI) are often caused by an impact on the skull, but due to the stochastic divergence of non-dissipated pressure changes, the effects can vary significantly ranging from changes in the level of molecular markers to the manifestation of distinct symptoms.
2.1. Manifestations Traumatic Brain Injury in Patients
Mouse models have demonstrated various behavioral manifestations of TBI, including increased light aversion, reduced contrast sensitivity in the optokinetic reflex test, and elevated pupil contraction in the pupillary light response test [1], symptoms alike to those observed following ocular blast injury [2]. Photophobia, increased sensitivity to light, has been reported after TBI and was hypothesized to result from increased sensitivity of intrinsically photosensitive retinal ganglion cells (ipRGCs) due to the injury [3,4]. However, the same study reported no significant differences in pupillary constriction and re-dilation amplitudes in response to blue or red light between photophobic participants with mild TBI and controls, suggesting no increased ipRGC light sensitivity [4]. Interestingly, however, TBI participants exhibited greater variability in their pupil responses to blue light compared to controls, indicating that some alteration in ipRGCs distinct from light sensitivity may underlie TBI-induced photophobia [4].
As revealed before, a significant number of blast-induced TBI patients suffer from visual impairments, with prevalence rates varying based on injury severity and the specific visual function [5]. Among them hemianopia following TBI is relatively common that occurs due to retrochiasmatic damage. Homonymous hemianopia may result from a lesion affecting the contralateral optic tract (OT), lateral geniculate nucleus (LGN), optic radiation (OR), or primary visual cortex (V1) [6].
Beyond the immediate effects, multiple studies indicate a link between a history of TBI and a higher risk of Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, cognitive impairment, and multiple sclerosis, suggesting ongoing neurodegeneration over time following primary and secondary injuries [7]. In the United States, only 3% (~51,000) of all TBIs cases (1.7 million annually) result in death [8], but nearly one-third of injury-related deaths originate from TBI, with an even higher prevalence (up to 85%) of vision impairment among all cases. Among military personnel, blast-mediated TBI (bTBI) injury is the most prevalent, although it is considered a distinct type of injury caused by a pressure wave [9].
TBI symptoms can include double vision or diplopia, asthenopia with blurred vision, reading difficulties such as slower reading speed and loss of place, restricted peripheral vision, increased photosensitivity, and altered color perception. The severity of TBI can differentiate the outcome, with severe TBI (sTBI) bearing the most injuries with multiple lesions and more pronounced inflammatory effects, while moderate and mild TBI can still cause visual dysfunctions. Repetitive mild injuries can lead to the loss of RGCs in animal models, but human-related studies are lacking. Some studies differentiate traumatic optic neuropathy (TON) related to TBI if direct deterioration and accumulated visual symptoms appear, although the diagnosis is quite challenging [10]. TBI includes primary and secondary injuries in the CNS. Primary injury is due to physical force impacting the brain, while secondary injury can occur later, beginning in a few minutes following the impact and lasting for several weeks. It comprises edema, BBB breakdown, ROS release, calcium ion imbalances, and inflammation. Silver staining indicates the maximal peak in tissue degeneration 48 hours after injury, continuing up to 7 days , however, these numbers are just rough approximations due to the post-mortem applicability of the approach [11]. The damage caused by neutrophils also peaks around 12-48 hours after experimental TBI [12].
Even in mild TBI, patients exhibit long-term cognitive disability, neuropsychiatric symptoms, and post-traumatic stress disorder [8]. In patients, the occurrence of post-chiasmal field defects ranged from 3.2% to 39% in TBI [8]. CT and MRI scans are the most widely used brain imaging technologies for the diagnosis of TBI but have obvious limitations, including low sensitivity, resolution, and invasive nature or limited access to equipment [13].
The strong connection between visual deficits and TBI can be easily assessed using the Brain Injury Vision Symptom Survey (BIVSS), a short symptom questionnaire to document vision deficits and distinguish between TBI severity levels [13]. A retrospective analysis of 100 patients with TBI reported that approximately 50% of patients had visual symptoms. One case-control study demonstrated that the prevalence of photophobia in patients with TBI was about four times higher than in healthy controls. Another case-control study showed that patients with TBI reported significantly stronger light-induced headaches than controls [14].
Both single and repeated injury protocols led to the loss of retinal ganglion cells and degeneration of the optic nerve, with these effects being linked to the impact load and the number/frequency of injuries. No phosphorylated tau immunoreactivity was observed in the brains of animals exposed to repetitive mild traumatic brain injury (mTBI). Therefore, the above-listed evidence indicates that repetitive mTBI induced traumatic axonal injury (TAI) affecting specific CNS tracts may serve as new diagnostic tool, particularly those affecting the visual system and/or cerebellum [15].
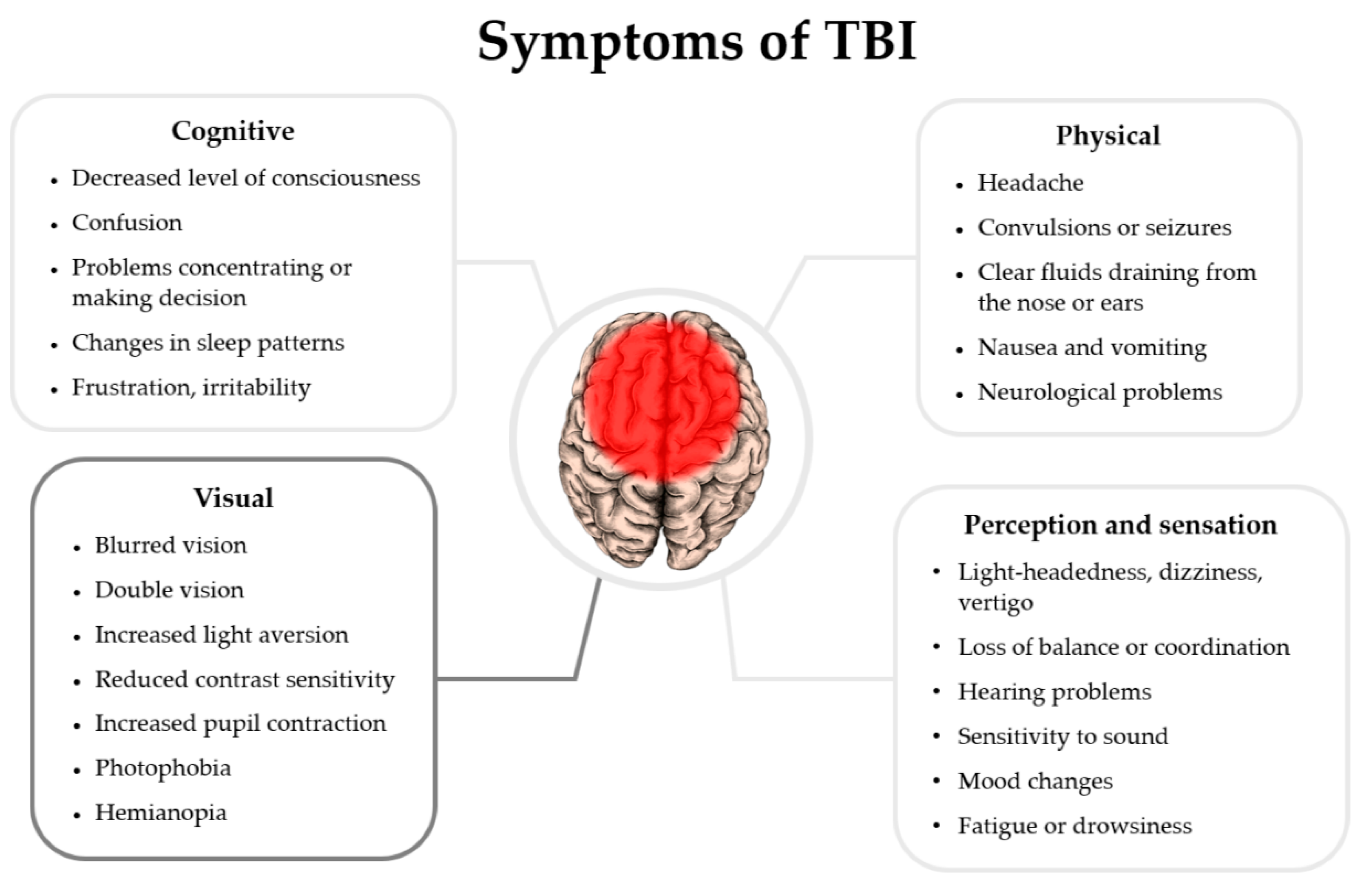
Figure 1.
Symptoms of Traumatic brain injury in patients with related references appear in this review article. The list of symptoms is based on NIH health information data: [16].
Figure 1.
Symptoms of Traumatic brain injury in patients with related references appear in this review article. The list of symptoms is based on NIH health information data: [16].
2.2. Effects of TBI on the Visual System
The retina, as part of the central nervous system, is particularly vulnerable to damage including those induced by traumatic brain injury (TBI). Studies show that blast-induced trauma leads to decreased thickness in the outer nuclear layer of the retina due to a reduction in photoreceptor cell nuclei [2,17]. Additionally, blast-induced TBI and repeated ocular blast injuries cause optic nerve axon loss, correlating with reduced contrast sensitivity [1,2].
In a central fluid percussion injury model of TBI, researchers observed diffuse traumatic axonal injury (TAI) in the optic nerve. Interestingly, however, this TAI did not result in the loss of retinal ganglion cells (RGCs). Instead, the distal optic nerve segments showed degeneration, marked by increased microglia and macrophage activity, while proximal axonal segments remained intact, preserving RGC integrity [18]. Intrinsically photosensitive RGCs (ipRGCs) were also affected, showing decreased soma size and increased melanopsin immunolabeling following blast-induced TBI [1]. Conversely, the density of displaced amacrine cells remained unaffected [19] indicating that the observed changes were conveyed to ganglion cells via their axons.
Functional impacts on the visual system were evident in electroretinogram (ERG) measurements, which revealed decreased A- and B-wave peak amplitudes following blast-induced TBI [1]. Pattern ERG (PERG) assessments of RGC function showed an initial decrease in response amplitude one-week post-injury, a temporary recovery at four weeks, and a subsequent decline by 16 weeks [19,20]. However, under conditions of elevated ocular pressure, a decreased voltage change amplitude was detected at four weeks, aligning with reduced RGC layer thickness and increased RGC spontaneous activity throughout the testing period [19].
Human TBI studies showed some important changes in the retina. Henle’s fiber layer (HFL) is made up of Muller cells and photoreceptor axons coursing obliquely from their nuclei towards their synapse with inner nuclear cells. Because of their directional reflectivity they can be distinguished from the optical outer nuclear and outer plexiform layers close to the fovea. In individuals with a history of TBI, the Henle fiber layer (HFL), the was found to be thicker than in controls, potentially due to changes in the deep capillary plexus structure or increased Müller cell volume [21]. However, global HFL phase retardation remained unchanged [21]. Additionally, ERG A-wave amplitude was reduced, indicating impaired photoreceptor function [21].
Early diffusion tensor imaging (DTI) can identify optic tract lesions causing homonymous hemianopia. DTI also detects diffuse changes in functionally intact structures that resolve over time, likely due to minor diffuse axonal injury recovery. Optical coherence tomography (OCT) reveals retinal thinning consistent with Wallerian degeneration of the optic tract. However, since optic tract atrophy can also stem from lateral geniculate nucleus or primary visual cortex lesions, OCT lacks precise localization value [6].
In another study, OCT revealed a temporal peripapillary retinal nerve fiber layer (pRNFL) thinning associated with traumatic optic neuropathy in patients with chronic mTBI [22].
Chronic decline in optomotor response was observed five months post-blast TBI [20]. Mild TBI leads to widespread axonal damage and microglial activation, with injured axons releasing damage-associated molecular patterns (DAMPs), such as ATP and S100b. These molecules bind to microglial receptors, triggering a pro-inflammatory response that exacerbates damage [23].
Traumatic optic neuropathy (TON) and retinal thinning are distinct forms of injury that appear to contribute to TBI effects in the retina. TON from blunt force trauma affects 0.5–5% of TBI cases, resulting in irreversible vision loss [11]. In studies on mice and Olympic boxers, retinal nerve fiber layer thinning correlated with optic nerve thinning [8]. Ultrasonographic optic nerve sheath diameter (ONSD) measurements can assess increased intracranial pressure (ICP) in TBI patients, showing a near-linear correlation with invasive methods [24].
Repetitive mild TBI led to more profound retinal nerve fiber layer (RNFL) thinning compared to single-blast injury, likely due to the short interval (48 hours) between impacts, which prevented optic nerve edema from subsiding [25]. In mice subjected to blast-induced TBI, vitreous detachment, hemorrhage, subretinal bleeding, and photoreceptor degeneration were observed [26]. Complement C3 deposits appeared in the retinogeniculate synapses of the dorsal lateral geniculate nucleus (dLGN) three days post-TBI, with microglial changes persisting up to 49 days. Complement inhibitor CR2-Crry countered these effects, promoting neuron survival [27]. Repetitive mTBI caused visual pathway dysfunction distal to the retina, as evidenced by the near-complete erosion of visual-evoked potential (VEP) without notable ERG changes. This suggests traumatic optic neuropathy and widespread axonal injury as contributors to visual dysfunction [28]. Extensive microglial activation was noted in TAI sites, especially the optic tract, superior colliculus, and corpus callosum [15]. Patients with migraine-related photosensitivity exhibited light-triggered migraine attacks via the ipRGC pathway [29]. In mTBI, cone response reduction and rod pathway overactivation at high light levels may explain photalgia. This phenomenon likely results from the loss of cone-generated inhibition, leading to ERG shifts toward rod-like properties [30].
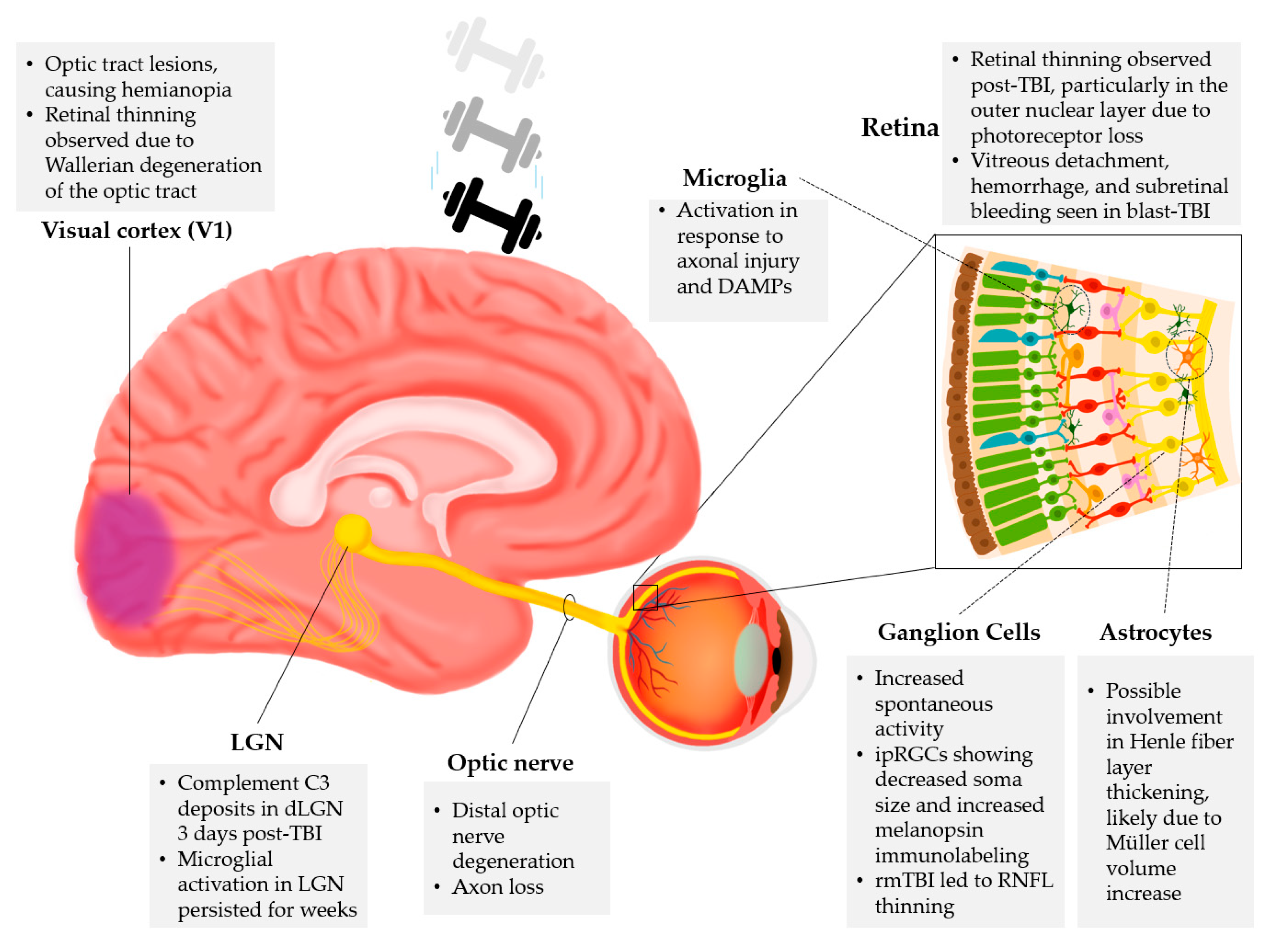
Figure 2.
Effects of TBI in the visual system. Based on the processed literature, different levels and centers are affected by traumatic brain injury in the visual system, including Visual cortex (V1), Lateral geniculate nucleus (LGN), Optic nerve, Retina -including cell types: Microglia, Ganglion cells, and Astrocytes.
Figure 2.
Effects of TBI in the visual system. Based on the processed literature, different levels and centers are affected by traumatic brain injury in the visual system, including Visual cortex (V1), Lateral geniculate nucleus (LGN), Optic nerve, Retina -including cell types: Microglia, Ganglion cells, and Astrocytes.
2.3. Cellular and Molecular Markers of TBI in the Retina
TBI-induced pathologies extend beyond the location of the primary insult in the central nervous system (CNS) and manifest as distinct morphological and functional changes in the retina. These changes are accompanied by alterations in key retinal cellular markers, whose comprehensive list is provided in the paragraphs below.
Glial activation and intermediate filament (IF) remodeling happen in the retina as part of TBI. The upregulation of GFAP immunoreactivity in the mouse retina post-TBI or acoustic blast overpressure (ABO) signals the activation of Müller glia [17,31,32]. However, GFAP is not the sole IF affected; other type III IFs like vimentin and desmin, and type VI IFs such as nestin and synemin, are also remodeled or induced in the rat retina following ABO exposure [32]. The resulting glial phenotype depends on the specific retinal region [32]. Similarly, increased IBA1 immunoreactivity reflects the activation of microglia and a TBI-induced inflammatory response [1,17,31]. Pro-inflammatory microglial activation is further evidenced by elevated CD68 expression, indicating phagocytic activity [17].
There is a strong link in Tau phosphorylation and neurodegeneration. Phosphorylated tau, a hallmark of neurofibrillary tangles in neurodegenerative diseases, is elevated in retinal horizontal cells and Müller glia post-TBI [17]. Here, the release of lysophosphatidic acid (LPA) from activated microglia, astrocytes, and platelets can induce further inflammatory processes and tau phosphorylation [33].
Cytokines and oxidative stress walk hand in hand to alter cellular survival. Increased retinal expression of cytokines, including interleukin (IL)-1B, IL-1a, IL-6, and tumor necrosis factor, occurs post-blast-induced TBI [31]. Blocking the IL-1 pathway with the IL-1 receptor antagonist anakinra reduces glial activation, preserves retinal ganglion cell (RGC) signaling, and protects the structural integrity of both RGCs and the optic nerve [31].
TBI is also associated with an overexpression of reactive oxygen species (ROS), which may contribute to retinal pathologies [34]. ROS overexpression in RGCs triggers the upregulation of Kruppel-like factor 4 (KLF4), activating pro-apoptotic p53 signaling while inhibiting pro-survival STAT3 signaling [34].
TBI leads to blood-brain barrier (BBB) and blood-retina barrier (BRB) disruption, allowing peripheral immune cell infiltration [11]. Infiltrating cells, such as macrophages and neutrophils, exacerbate inflammation and contribute to retinal nerve fiber layer thinning [11].
The activation of inflammatory pathways and the disruption of barriers (BBB and BRB) push the CNS beyond the limit and induce secondary processes. The CCL20/CCR6 axis, known for modulating inflammation, plays a critical role in neurodegeneration post-TBI, spinal cord injury, and cerebral ischemia. Elevated CCL20 levels are detected in human plasma within 24 hours of severe TBI. [35]
Importantly, apoptosis and microglial activation together with an accelerated immune response can be detected widely in the retina and in the brain. In a severe and repetitive mild TBI model, Caspase-3 activation signals apoptotic events and axonal deterioration [36]. Interestingly, while microglia avoid apoptotic induction, astrocytes express activated Caspase-3, an early marker of retinal damage [36].
Complement C3 deposits appear in retinogeniculate synapses post-TBI, where CR2-Crry complement inhibition shows a neuroprotective effect [27]. Additionally, dendritic cells from Rag1-/- mice may present RGC antigens, eliciting an immune response in wild-type mice [20].
Traumatic axonopathy is associated with a robust phagocytic response, observed through CD68 immunohistochemistry. However, in Sarm1 knockout mice, the suppression of CD68 activation suggests a potential direct role of SARM1 in microglial activation [37].
Increased β-amyloid and lipid peroxidation product 4-hydroxy-trans-2-nonenal (4HNE) levels are detected in the retina acutely post-blast injury, consistent with elevated oxidative stress markers in human plasma [38].
In repetitive diffuse mTBI models, tauopathy is accelerated in genetically susceptible mice [39]. In α RGCs, downregulating phosphatase and tensin homolog (PTEN) promote regeneration, further enhanced by osteopontin and insulin-like growth factor 1 [40].

Table 1.
Cellular and Molecular Retinal Markers of TBI.
| Marker | Function | Study |
|---|---|---|
| GFAP (Glial Fibrillary Acidic Protein) | Indicates Müller glia activation in the retina post-TBI or acoustic blast overpressure | [17,31,32] |
| IBA1 (Ionized calcium-binding adapter molecule 1) | Reflects microglial activation and inflammation post-TBI | [1,17,31] |
| CD68 (Cluster of Differentiation 68) | Marker of pro-inflammatory microglial activation and indicates phagocytic response in traumatic axonopathy | [17,37] |
| Phosphorylated Tau | Indicates neurodegeneration; associated with neurofibrillary tangles | [17] |
| LPA (Lysophosphatidic acid) | Induces inflammatory processes, astrocyte proliferation, and tau phosphorylation | [33,41,42] |
| IL-1B, IL-1a, IL-6, TNF (Interleukin; Tumor Necrosis Factor) | Cytokines involved in inflammation and oxidative stress post-TBI | [31] |
| KLF4 (Kruppel-like factor 4) | Triggers pro-apoptotic p53 signaling and inhibits pro-survival STAT3 signaling in RGCs | [34] |
| CCL20 ( Chemokine (C-C motif) ligand 20) | Involved in neurodegeneration and inflammation post-TBI | [35] |
| Caspase-3 | Marker of apoptosis, expressed in astrocytes in retinal damage | [36] |
| Complement C3 | Deposits in retinogeniculate synapses post-TBI; inhibition is neuroprotective | [27] |
| β-amyloid and 4HNE (4-hydroxy-trans-2-nonenal) | Oxidative stress markers post-TBI | [38] |
| PTEN ( phosphatase and tensin homolog) | Downregulation promotes regeneration of α RGCs | [40] |
| Osteopontin and IGF-1 ( insulin-like growth factor 1) | Enhances RGC regeneration | [40] |
2.4. Possible Translational Implementation of Retinal Markers in TBI
The effects of traumatic brain injury (TBI) can be detected in the retina, even in cases where the injury's impact is insufficient to produce observable alterations in the brain.. For example, even in cases when blast-induced trauma failed to increase GFAP, IBA1 and phosphorylated tau immunoreactivity in the prefrontal cortex, levels of these damage and inflammatory markers were elevated in the retina [17]. This suggests that the retina may be more sensitive to TBI and therefore can serve as a useful indicator of injury following blast exposure [17]. In many life situations where TBI is common, including the military and sports, participants would greatly benefit from the ability to detect TBI early via non-invasive assessments of retinal structure and/or visual function. Furthermore, the reliable detection of TBI through assessment of the visual system would also have implications within the legal framework, particularly in cases of suspected abusive head trauma (AHT) in children [43]. For example, retinal hemorrhages involving multiple layers of the retina and extending to the ora serrata have been observed to occur more frequently in cases of abusive head trauma (AHT) compared to non-abusive head trauma [43].
To effectively use visual system symptoms as indicators of TBI, it is imperative to develop reliable and non-invasive tests for the early detection of visual deficits. Pattern electroretinography (PERG) may be a useful tool as it can assess RGC function non-invasively. In fact, it has previously shown that marked PERG signal changes occur in response amplitude in mice following blast-induced TBI [19]. It appears that performing the PERG measurements under conditions of increased ocular pressure may help reveal differences in retinal function that go undetected in standard PERG measurements [19].
In terms of possible interventions to reduce TBI-induced retinal pathologies, the intravenous administration of an anti-LPA antibody 1 hour after blast exposure improved rat visual acuity, improved retinal signaling function, and preserved neuronal cell integrity following blast exposure [33]. Furthermore, in contrast to control antibody-treated rats, the anti-LPA antibody-treated counterparts did not show an increase in GFAP-positive activated Müller cells eight days after blast exposure [33]. Ultimately, the administration of anti-LPA antibody immediately post-blast exposure seems effective at reducing the pro-inflammatory effects of LPA described earlier. Another possible therapeutic target to reduce TBI-induced retinal pathologies is KLF4. KLF4 knockdown has been found to significantly enhance ciliary neurotrophic factor-induced RGC axon regeneration following optic nerve crush [34]. This finding suggests that blocking the action of KLF4 following TBI-induced tissue trauma may serve to enhance tissue repair and thus attenuate retinal pathologies. Alternatively, since blocking the interleukin pathway with the IL-1 receptor antagonist anakinra was found to reduce glial activation and RGC and optic nerve damage following blast-induced TBI, this suggests that retinal inflammatory pathways may be effective therapeutic targets to attenuate TBI-induced retinal pathologies [31]. There is also evidence that cannabinoid receptor type 2 (CB2) reverse agonist treatment up to 2 days following TBI can also help mitigate visual system deficits [1]. Specifically, intraperitoneal raloxifene injections up to 48 hours post-blast-induced TBI appear to return contrast sensitivity, visual acuity, light aversion, ERG A- and B-wave peak amplitudes, pupil light reflex, optic nerve axon abundance, ipRGC soma size and ipRGC melanopsin immunoreactivity to sham blast levels [1]. Similar results were obtained after raloxifene treatment post-repeated ocular blast injury [2]. Raloxifene also seemed to shift microglia away from the pro-inflammatory M1 state towards the protective M2 state following TBI or repeated ocular blast injury [1,2]. What makes raloxifene an especially promising therapeutic agent is that it has already been FDA-approved for the treatment of osteoporosis, leading to the possibility of simply using it for the treatment of TBI-induced visual deficits as well [1]. Results suggest that greater doses of raloxifene may have a therapeutic effect on the optic nerve, as it reduces inflammation by improving phagocytosis of axonal debris, thereby saving fewer injured axons [23]. Another pharmacological intervention being explored is the treatment of TBI with P7C3-S243, a neuroprotective agent [19]. A study in a mouse model of blast-induced TBI found intraperitoneal injections of P7C3-S243, starting 5 minutes post-blast and continuing twice daily, to be effective in maintaining pre-blast PERG responses, suggesting maintained visual function, instead of the reduced PERG amplitudes seen in a vehicle treatment group [19].
A study shows that taurine, which is found in free form in neurons and serves as an important factor in brain growth and development, may also play a role in neuroprotection after TBI by preserving neuronal ultrastructure, enhancing mitochondrial function, and modulating apoptosis-related protein expression [44].
It is known that for individuals with a genetic predisposition to amyloidosis in Alzheimer’s disease (AD), exposure to blast TBI accelerates damage to retinal ganglion cells and the optic nerve. This damage is linked to a modest but detectable increase in cerebral cortical Aβ pathology. These findings suggest that genetic risk factors for AD may heighten the retina’s vulnerability to blast-induced injury. A study demonstrates the long-term effects of blast injury on both retinal function and structure, as well as amyloid accumulation, future translational biomarker research could focus on integrating retinal function assessments with brain amyloidosis imaging using positron emission tomography (PET). This approach is particularly valuable for aging veterans with a history of blast TBI [45].
The US Centers for Disease Control and Prevention (CDC) reports that individuals with a history of moderate TBI are 2.3 times more likely to develop Alzheimer’s disease (AD), attributing this risk to the persistent neuroinflammation following TBI. This is particularly significant as research suggests that TBI may accelerate the onset of AD pathology by 4 to 10 years. Given that AD pathogenesis involves similar disruptions in cellular homeostasis, microglial and astrocyte activation, and inflammatory processes, the impact of trauma on the central nervous system is a compelling area of study [46]. Nerve sheath diameter is linked to retinal health through its ganglion cell origin. As RGCs are affected by either direct eye traumas or TBI, deterioration of the axons can be measured with ultrasound imaging [24]. Another important study investigating TBI-induced retinal nerve fiber layer (RNFL) thinning found 30% of eyes to have significant RNFL thinning and 40% to have visual function defects 6 months following mTBI, at which point the structural changes and visual function impairment peaked. Scatter visual field defects were among the most frequently documented deficits with a strong, significant association with RNFL thinning [47]. Due to the strong connection between cerebral blood flow and retinal blood flow, non-invasive ocular imaging of retinal blood flow can be used to pinpoint TBI severity. Related to blood flow, retinal oximetry can actively determine the oxyhemoglobin/deoxyhemoglobin ratio and oxygen utilization in the retina [8].
Subtle spectral changes were detected with Raman-spectroscopy, likely linked to a decrease in cardiolipin and indicating metabolic disruption from the brain and retina following injury [48].
The differences in healing abilities in the retina may come from the fact that certain RGC subtypes appear to regenerate better than others. Alpha RGCs (αRGCs) intrinsically have high rapamycin target (mTOR) levels and also selectively express osteopontin and receptors for the insulin-like growth factor 1. Among surviving RGCs after optic nerve crush, αRGCs account for nearly all RGC regeneration [40].
As we described multiple times, microglia show great promise in detecting the early signs of TBI in a multitude of models [26], but this ability has never been linked to the option to use them as an additional marker for categorising TBI severity.
Automated perimetry measurements in veterans with blast-related TBI have proven to be a practical and effective tool for detecting visual field deficits when certain conditions are met, including testing during a time of maximal alertness and the absence of sensorineural medications. 15% of participants showed significant visual field defects, such as hemianopia or quadrantanopia, while an additional 36% showed abnormal global visual field indices. These results suggest that there is a high prevalence of visual field deficits among individuals with blast-related TBI [5].
There is another method, called multifocal pupillographic objective perimetry (mfPOP), which can also provide reliable results. Athletes who recently suffered mild TBI were studied and their visual response amplitude showed a significant decrease compared to control subjects. Furthermore, a significant negative correlation was observed between retinal thickness and the latency of the mfPOP response in athletes with acute mTBI. This indicates that structural changes in the retina after injury may be associated with functional visual impairment. The results here should also be treated with caution, considering that various antidepressants, decongestants and other systemic diseases can affect pupil size [49].
That said, microglia show great promise in detecting the early signs of TBI in a multitude of models [26], but this ability has never been linked to the option to use them as an additional marker for categorising TBI severity.
3. Discussion
There are many post-mortem studies available that can help us determine the time scale of TBI. We now know that most of the tissue degeneration happens 48 hours after injury and continues for up to 7 days or even for weeks [11].
The early signs of TBI cannot be assessed with ultrasonographic optic nerve sheath diameter (ONSD), as a certain post-injury period is needed for nerve thinning to occur. However, the literature does not provide us a well-outlined post-injury time scale; therefore, we are unable to tell whether a method exists for both the early and late detection of TBI. Of course, early assessment is the most important. Further studies are needed to determine its usability but it gives us a potential method for detailed assessment of TBI.
Utilizing non-invasive ocular imaging of retinal blood flow, oxygen levels or cells to assess TBI severity cannot be emphasized enough for early detection, as cerebral blood flow and metabolism are closely associated with retinal blood flow [8]. Live microglial cells can be visualized in the human eye label-free by employing adaptive optics [50]. This method bears the greatest potential in the identification of TBI severity without excessive harm and costs since studies suggest that microglia can be used as the first line of markers in TBI due to their obvious role in protection, including the induction of inflammation and elimination of cellular debris [36]. The other glial element in the retina, astrocytes, is the key player in retinal homeostasis, which is strongly affected by TBI. Avoiding the elimination of astrocytes possibly helps preventing the accumulation of pathological symptoms in the CNS [36], while astrocytes can also be imaged in the eye together with ganglion cells in the GCL [51].
TBI intervention relies on corticosteroids and/or surgical decompression with limited success [11].
Treating the effects of TBI with antioxidant therapies might be limited due to the oversuppression of beneficial ROS. However, controlled ROS inhibition can help, like microglial inhibition with Minocycline, which can lower ROS by suppressing the amount of active microglia. Also, ROS inhibition may have an efficacy time window that lasts only 3-6 hours after the head trauma [11]. On the other hand, as indicated, arginase can compete with NOS also limiting oxidative stress [52].
Given the retina’s accessibility and shared pathology with the CNS, ophthalmic imaging techniques such as OCT and pupillometry have emerged as potential non-invasive diagnostic tools for assessing TBI severity. Studies indicate that alterations in RNFL thickness, retinal ganglion cell layer integrity, vascular abnormalities, and microglial activation due to inflammation may serve as quantifiable biomarkers for TBI-related neurodegeneration. These advancements underscore the potential for integrating retinal assessments into clinical protocols for TBI evaluation and management.
4. Conclusions
TBI exerts significant effects on the retina, characterized by neuroinflammatory responses, oxidative stress, structural degeneration, and functional impairments. The similarity between retinal and CNS pathophysiology highlights the potential applicability of retinal biomarkers in diagnosing and monitoring TBI progression. Further interdisciplinary research is necessary to elucidate the mechanisms underlying TBI-induced retinal pathology and to develop targeted therapeutic strategies aimed at mitigating neurodegenerative sequelae.
To conclude, the effects of TBI on the visual system are multifaceted, ranging from structural degeneration to functional deficits. The degree of damage depends on the type and severity of the injury. Both early and delayed imaging methods, such as DTI and OCT, can aid in the diagnosis and monitoring of TBI-related visual impairments and TBI itself. More recently, because of the strong relationship between TBI and the caused visual impairments, we hypothesize that direct imaging of the eye with adaptive optics can be used to assess the level of TBI, based on the latest literature.
5. Methods
To identify articles relevant to this review we first used the following search in the NCBI database (NIH, USA): (retina AND (traumatic brain injury[Title/Abstract]). Based on this we got 142 articles in the first round of search. Following this we used a 1-10 scoring system (1-3: non-relevant, 4-6: somewhat relevant, 7-9 very relevant, 10 perfect match to the topic) on the titles and abstracts. Four authors independently scored all articles based on the titles and abstracts. We used the Copilot (Microsoft) as a fifth scorer. The AI model was limited to testing only 13-15 articles after testing scoring reliability based on divergence (error rate > 4 human mean error) to human scores, therefore we introduced all articles in 12 packages. This resulted in an acceptable score with low divergence from the human mean. The average scores (4 humans, 1 AI) of 66 searched papers were above the median score. We included these in our detailed reading and analysis. These articles were equally shared between 5 authors after blind selection and the relevant information was added to the pre-defined paragraphs in an ad-hoc manner. Correct phrasing, information linking and writing were done by all authors, on a shared Google Drive platform, subsequently. Additionally, X articles were added to the Discussion to aid understanding of recent developments and how these can come together in aiding the translation of our findings in the literature search. Figures were done with ibisPaint X and MS PowerPoint based on the literature.
Acknowledgment
This study was supported by the NKFI and the European Union under the action of the ERA-NET COFUND (2019-2.1.7-ERANET-2021-00018; NEURON (NEURON-066 (Rethealthsi) to B.V. The study was also financially supported by the NKFI (OTKA NN128293) (B.V.) from the European Union and the State of Hungary, co-financed by the European Social Fund in the framework of National Excellence Program (B.V.). Project no. TKP2021-EGA-16 has been implemented with the support provided by the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA-16 funding scheme (B.V.). Supported by the EKÖP-24-4-I-PTE-11 (GS) and EKÖP-24-3-I-PTE-129 (BB) New National Excellence Program of the Ministry of Human Capacities
References
References
- Honig, M. G.; Del Mar, N. A.; Henderson, D. L.; Ragsdale, T. D.; Doty, J. B.; Driver, J. H.; Li, C.; Fortugno, A. P.; Mitchell, W. M.; Perry, A. M.; Moore, B. M.; Reiner, A. Amelioration of Visual Deficits and Visual System Pathology after Mild TBI via the Cannabinoid Type-2 Receptor Inverse Agonism of Raloxifene. Exp. Neurol. 2019, 322, 113063. [Google Scholar] [CrossRef] [PubMed]
- Honig, M. G.; Del Mar, N. A.; Henderson, D. L.; O’Neal, D.; Yammanur, M.; Cox, R.; Li, C.; Perry, A. M.; Moore, B. M.; Reiner, A. Raloxifene, a Cannabinoid Type-2 Receptor Inverse Agonist, Mitigates Visual Deficits and Pathology and Modulates Microglia after Ocular Blast. Exp. Eye Res. 2022, 218, 108966. [Google Scholar] [CrossRef]
- Waddell, P. A.; Gronwall, D. M. A. Sensitivity to Light and Sound Following Minor Head Injury. Acta Neurol. Scand. 1984, 69, 270–276. [Google Scholar] [CrossRef]
- Yuhas, P. T.; Shorter, P. D.; McDaniel, C. E.; Earley, M. J.; Hartwick, A. T. E. Blue and Red Light-Evoked Pupil Responses in Photophobic Subjects with TBI. Optom. Vis. Sci. 2017, 94, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Lemke, S.; Cockerham, G. C.; Glynn-Milley, C.; Lin, R.; Cockerham, K. P. Automated Perimetry and Visual Dysfunction in Blast-Related Traumatic Brain Injury. Ophthalmology 2016, 123, 415–424. [Google Scholar] [CrossRef]
- Decramer, T.; Van Keer, K.; Stalmans, P.; Dupont, P.; Sunaert, S.; Theys, T. Tracking Posttraumatic Hemianopia. J. Neurol. 2018, 265, 41–45. [Google Scholar] [CrossRef]
- Lyons, H. S.; Sassani, M.; Hyder, Y.; Mitchell, J. L.; Thaller, M.; Mollan, S. P.; Sinclair, A. J.; mTBI Predict, Consortium; Sinclair, A.; Finch, A.; Hampshire, A.; Sitch, A.; Mazaheri, A.; Bagshaw, A.; Palmer, A.; Strom, A.; Waitt, A.; Yiangou, A.; Abdel-Hay, A.; Bennett, A.; Clark, A.; Hunter, A.; Seemungal, B.; Witton, C.; Dooley, C.; Bird, D.; Fernandez-Espejo, D.; Smith, D.; Ford, D.; Sherwood, D.; Holding, D.; Wilson, D.; Palmer, E.; Golding, J.; Dehghani, H.; Park, H.; Lyons, H.; Smith, H.; Brunger, H.; Ellis, H.; Idrees, I.; Varley, I.; Hubbard, J.; Cao, J.; Deeks, J.; Mitchell, J.; Novak, J.; Pringle, J.; Terry, J.; Rogers, J.; Read, T.; Fildes, J.; Mullinger, K.; Hill, L.; Aurisicchio, M.; Thaller, M.; Wilson, M.; Pearce, M.; Sassani, M.; Brookes, M.; Mahmud, M.; Rayhan, R.; Jenkinson, N.; Karavitaki, N.; Capewell, N.; Grech, O.; Jensen, O.; Hellyer, P.; Woodgate, P.; Coleman, S.; Reynolds, R.; Blanch, R. J.; Morris, K.; Ottridge, R.; Upthegrove, R.; Dardis, R.; Arachchige, R. W.; Berhane, S.; Lucas, S.; Prosser, S.; Sharifi, S.; Dharm-Datta, S.; Mollan, S.; Ellmers, T.; Ghafari, T.; Goldstone, T.; Hawa, W.; Gao, Y.; Blanch, R. J. A Systematic Review of Optical Coherence Tomography Findings in Adults with Mild Traumatic Brain Injury. Eye 2024, 38, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Mufti, O.; Mathew, S.; Harris, A.; Siesky, B.; Burgett, K. M.; Verticchio Vercellin, A. C. Ocular Changes in Traumatic Brain Injury: A Review. Eur. J. Ophthalmol. 2020, 30, 867–873. [Google Scholar] [CrossRef]
- Evans, L. P.; Roghair, A. M.; Gilkes, N. J.; Bassuk, A. G. Visual Outcomes in Experimental Rodent Models of Blast-Mediated Traumatic Brain Injury. Front. Mol. Neurosci. 2021, 14, 659576. [Google Scholar] [CrossRef]
- Das, M.; Tang, X.; Mohapatra, S. S.; Mohapatra, S. Vision Impairment after Traumatic Brain Injury: Present Knowledge and Future Directions. Rev. Neurosci. 2019, 30, 305–315. [Google Scholar] [CrossRef]
- Ryan, A. K.; Rich, W.; Reilly, M. A. Oxidative Stress in the Brain and Retina after Traumatic Injury. Front. Neurosci. 2023, 17, 1021152. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, C. N.; Watson, J. B.; Higgins, E. K.; Quan, N.; Bachstetter, A. D. Inflammatory Regulation of CNS Barriers After Traumatic Brain Injury: A Tale Directed by Interleukin-1. Front. Immunol. 2021, 12, 688254. [Google Scholar] [CrossRef] [PubMed]
- Rauchman, S. H.; Albert, J.; Pinkhasov, A.; Reiss, A. B. Mild-to-Moderate Traumatic Brain Injury: A Review with Focus on the Visual System. Neurol. Int. 2022, 14, 453–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.; Qiu, T.; Xiao, Z. Photophobia in Headache Disorders: Characteristics and Potential Mechanisms. J. Neurol. 2022, 269, 4055–4067. [Google Scholar] [CrossRef]
- Xu, L.; Nguyen, J. V.; Lehar, M.; Menon, A.; Rha, E.; Arena, J.; Ryu, J.; Marsh-Armstrong, N.; Marmarou, C. R.; Koliatsos, V. E. Repetitive Mild Traumatic Brain Injury with Impact Acceleration in the Mouse: Multifocal Axonopathy, Neuroinflammation, and Neurodegeneration in the Visual System. Exp. Neurol. 2016, 275, 436–449. [Google Scholar] [CrossRef]
- National Institue of Neurological Disorders and Stroke. Traumatic Brain Injury (TBI). National Institue of Neurological Disorders and Stroke. https://www.ninds.nih.gov/health-information/disorders/traumatic-brain-injury-tbi#.
- Mammadova, N.; Ghaisas, S.; Zenitsky, G.; Sakaguchi, D. S.; Kanthasamy, A. G.; Greenlee, J. J.; West Greenlee, M. H. Lasting Retinal Injury in a Mouse Model of Blast-Induced Trauma. Am. J. Pathol. 2017, 187, 1459–1472. [Google Scholar] [CrossRef]
- Wang, J.; Fox, M. A.; Povlishock, J. T. Diffuse Traumatic Axonal Injury in the Optic Nerve Does Not Elicit Retinal Ganglion Cell Loss. J. Neuropathol. Exp. Neurol. 2013, 72, 768–781. [Google Scholar] [CrossRef]
- Dutca, L. M.; Stasheff, S. F.; Hedberg-Buenz, A.; Rudd, D. S.; Batra, N.; Blodi, F. R.; Yorek, M. S.; Yin, T.; Shankar, M.; Herlein, J. A.; Naidoo, J.; Morlock, L.; Williams, N.; Kardon, R. H.; Anderson, M. G.; Pieper, A. A.; Harper, M. M. Early Detection of Subclinical Visual Damage After Blast-Mediated TBI Enables Prevention of Chronic Visual Deficit by Treatment With P7C3-S243. Invest. Ophthalmol. Vis. Sci. 2014, 55, 8330–8341. [Google Scholar] [CrossRef]
- Harper, M. M.; Gramlich, O. W.; Elwood, B. W.; Boehme, N. A.; Dutca, L. M.; Kuehn, M. H. Immune Responses in Mice after Blast-Mediated Traumatic Brain Injury TBI Autonomously Contribute to Retinal Ganglion Cell Dysfunction and Death. Exp. Eye Res. 2022, 225, 109272. [Google Scholar] [CrossRef]
- Stern-Green, E. A.; Klimo, K. R.; Day, E.; Shelton, E. R.; Robich, M. L.; Jordan, L. A.; Racine, J.; VanNasdale, D. A.; McDaniel, C. E.; Yuhas, P. T. Henle Fiber Layer Thickening and Deficits in Objective Retinal Function in Participants with a History of Multiple Traumatic Brain Injuries. Front. Neurol. 2024, 15, 1330440. [Google Scholar] [CrossRef]
- Chan, J. W.; Hills, N. K.; Bakall, B.; Fernandez, B. Indirect Traumatic Optic Neuropathy in Mild Chronic Traumatic Brain Injury. Investig. Opthalmology Vis. Sci. 2019, 60, 2005. [Google Scholar] [CrossRef] [PubMed]
- Honig, M. G.; Del Mar, N. A.; Henderson, D. L.; O’Neal, D.; Doty, J. B.; Cox, R.; Li, C.; Perry, A. M.; Moore, B. M.; Reiner, A. Raloxifene Modulates Microglia and Rescues Visual Deficits and Pathology After Impact Traumatic Brain Injury. Front. Neurosci. 2021, 15, 701317. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Palacios, K.; Vásquez-García, S.; Fariyike, O. A.; Robba, C.; Rubiano, A. M.; the noninvasive ICP monitoring international consensus, group; Taccone, F. S.; Rasulo, F.; Badenes, R. R.; Menon, D.; Sarwal, A. A.; Cardim, D. D.; Czosnyka, M.; Hirzallah, M.; Geeraerts, T.; Bouzat, P.; Lochner, P. G.; Aries, M.; Wong, Y. L.; Abulhassan, Y.; Sung, G.; Prabhakar, H.; Shrestha, G.; Bustamante, L.; Jibaja, M.; Pinedo, J.; Sanchez, D.; Mendez, J. M.; Vásquez, F.; Shukla, D. P.; Worku, G.; Tirsit, A.; Indiradevi, B.; Shabani, H.; Adeleye, A.; Munusamy, T.; Ain, A.; Paiva, W.; Godoy, D.; Brasil, S.; Robba, C.; Rubiano, A.; Vásquez-García, S. Using Optic Nerve Sheath Diameter for Intracranial Pressure (ICP) Monitoring in Traumatic Brain Injury: A Scoping Review. Neurocrit. Care 2024, 40, 1193–1212. [Google Scholar] [CrossRef]
- Tzekov, R.; Quezada, A.; Gautier, M.; Biggins, D.; Frances, C.; Mouzon, B.; Jamison, J.; Mullan, M.; Crawford, F. Repetitive Mild Traumatic Brain Injury Causes Optic Nerve and Retinal Damage in a Mouse Model: J. Neuropathol. Exp. Neurol. 2014, 73, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Evans, L. P.; Newell, E. A.; Mahajan, M.; Tsang, S. H.; Ferguson, P. J.; Mahoney, J.; Hue, C. D.; Vogel, E. W.; Morrison, B.; Arancio, O.; Nichols, R.; Bassuk, A. G.; Mahajan, V. B. Acute Vitreoretinal Trauma and Inflammation after Traumatic Brain Injury in Mice. Ann. Clin. Transl. Neurol. 2018, 5, 240–251. [Google Scholar] [CrossRef]
- Borucki, D. M.; Rohrer, B.; Tomlinson, S. Complement Propagates Visual System Pathology Following Traumatic Brain Injury. J. Neuroinflammation 2024, 21, 98. [Google Scholar] [CrossRef] [PubMed]
- Morriss, N. J.; Conley, G. M.; Hodgson, N.; Boucher, M.; Ospina-Mora, S.; Fagiolini, M.; Puder, M.; Mejia, L.; Qiu, J.; Meehan, W.; Mannix, R. Visual Dysfunction after Repetitive Mild Traumatic Brain Injury in a Mouse Model and Ramifications on Behavioral Metrics. J. Neurotrauma 2021, 38, 2881–2895. [Google Scholar] [CrossRef]
- Elenberger, J.; Kim, B.; De Castro-Abeger, A.; Rex, T. S. Connections between Intrinsically Photosensitive Retinal Ganglion Cells and TBI Symptoms. Neurology 2020, 95, 826–833. [Google Scholar] [CrossRef]
- Tyler, C. W.; Likova, L. T. Brain Trauma Impacts Retinal Processing: Photoreceptor Pathway Interactions in Traumatic Light Sensitivity. Doc. Ophthalmol. 2022, 144, 179–190. [Google Scholar] [CrossRef]
- Evans, L. P.; Woll, A. W.; Wu, S.; Todd, B. P.; Hehr, N.; Hedberg-Buenz, A.; Anderson, M. G.; Newell, E. A.; Ferguson, P. J.; Mahajan, V. B.; Harper, M. M.; Bassuk, A. G. Modulation of Post-Traumatic Immune Response Using the IL-1 Receptor Antagonist Anakinra for Improved Visual Outcomes. J. Neurotrauma 2020, 37, 1463–1480. [Google Scholar] [CrossRef]
- Skelton, L. A.; Ramachandra Rao, S.; Allen, R. S.; Motz, C. T.; Pardue, M. T.; Fliesler, S. J. Retinal Gliosis and Phenotypic Diversity of Intermediate Filament Induction and Remodeling upon Acoustic Blast Overpressure (ABO) Exposure to the Rat Eye. Exp. Eye Res. 2023, 234, 109585. [Google Scholar] [CrossRef] [PubMed]
- Arun, P.; Rossetti, F.; DeMar, J. C.; Wang, Y.; Batuure, A. B.; Wilder, D. M.; Gist, I. D.; Morris, A. J.; Sabbadini, R. A.; Long, J. B. Antibodies Against Lysophosphatidic Acid Protect Against Blast-Induced Ocular Injuries. Front. Neurol. 2020, 11, 611816. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Zeng, T.; Ren, J.; Wang, K.; Jin, Y.; Zhou, L.; Gao, L. KLF 4 Knockdown Attenuates TBI -Induced Neuronal Damage through P53 and JAK - STAT 3 Signaling. CNS Neurosci. Ther. 2017, 23, 106–118. [Google Scholar] [CrossRef]
- Das, M.; Tang, X.; Han, J. Y.; Mayilsamy, K.; Foran, E.; Biswal, M. R.; Tzekov, R.; Mohapatra, S. S.; Mohapatra, S. CCL20-CCR6 Axis Modulated Traumatic Brain Injury-Induced Visual Pathologies. J. Neuroinflammation 2019, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Kovács-Öller, T.; Zempléni, R.; Balogh, B.; Szarka, G.; Fazekas, B.; Tengölics, Á. J.; Amrein, K.; Czeiter, E.; Hernádi, I.; Büki, A.; Völgyi, B. Traumatic Brain Injury Induces Microglial and Caspase3 Activation in the Retina. Int. J. Mol. Sci. 2023, 24, 4451. [Google Scholar] [CrossRef]
- Alexandris, A. S.; Lee, Y.; Lehar, M.; Alam, Z.; McKenney, J.; Perdomo, D.; Ryu, J.; Welsbie, D.; Zack, D. J.; Koliatsos, V. E. Traumatic Axonal Injury in the Optic Nerve: The Selective Role of SARM1 in the Evolution of Distal Axonopathy. J. Neurotrauma 2023, 40, 1743–1761. [Google Scholar] [CrossRef]
- Mohan, K.; Kecova, H.; Hernandez-Merino, E.; Kardon, R. H.; Harper, M. M. Retinal Ganglion Cell Damage in an Experimental Rodent Model of Blast-Mediated Traumatic Brain Injury. Investig. Opthalmology Vis. Sci. 2013, 54, 3440. [Google Scholar] [CrossRef]
- Xu, L.; Ryu, J.; Nguyen, J. V.; Arena, J.; Rha, E.; Vranis, P.; Hitt, D.; Marsh-Armstrong, N.; Koliatsos, V. E. Evidence for Accelerated Tauopathy in the Retina of Transgenic P301S Tau Mice Exposed to Repetitive Mild Traumatic Brain Injury. Exp. Neurol. 2015, 273, 168–176. [Google Scholar] [CrossRef]
- Crair, M. C.; Mason, C. A. Reconnecting Eye to Brain. J. Neurosci. 2016, 36, 10707–10722. [Google Scholar] [CrossRef]
- Shano, S.; Moriyama, R.; Chun, J.; Fukushima, N. Lysophosphatidic Acid Stimulates Astrocyte Proliferation through LPA1. Neurochem. Int. 2008, 52, 216–220. [Google Scholar] [CrossRef]
- Sayas, C. L.; Ariaens, A.; Ponsioen, B.; Moolenaar, W. H. GSK-3 Is Activated by the Tyrosine Kinase Pyk2 during LPA1 -Mediated Neurite Retraction. Mol. Biol. Cell 2006, 17, 1834–1844. [Google Scholar] [CrossRef]
- Harris, C. K.; Stagner, A. M. The Eyes Have It: How Critical Are Ophthalmic Findings to the Diagnosis of Pediatric Abusive Head Trauma? Semin. Ophthalmol. 2023, 38, 3–8. [Google Scholar] [CrossRef]
- Wang, Q.; Fan, W.; Cai, Y.; Wu, Q.; Mo, L.; Huang, Z.; Huang, H. Protective Effects of Taurine in Traumatic Brain Injury via Mitochondria and Cerebral Blood Flow. Amino Acids 2016, 48, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Harper, M. M.; Hedberg-Buenz, A.; Herlein, J.; Abrahamson, E. E.; Anderson, M. G.; Kuehn, M. H.; Kardon, R. H.; Poolman, P.; Ikonomovic, M. D. Blast-Mediated Traumatic Brain Injury Exacerbates Retinal Damage and Amyloidosis in the APPswePSENd19e Mouse Model of Alzheimer’s Disease. Investig. Opthalmology Vis. Sci. 2019, 60, 2716. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Thamaraikani, T.; Vellapandian, C. A Review of the Retinal Impact of Traumatic Brain Injury and Alzheimer’s Disease: Exploring Inflammasome Complexes and Nerve Fiber Layer Alterations. Cureus 2024. [Google Scholar] [CrossRef] [PubMed]
- Kumar Das, N.; Das, M. Structural Changes in Retina (Retinal Nerve Fiber Layer) Following Mild Traumatic Brain Injury and Its Association with Development of Visual Field Defects. Clin. Neurol. Neurosurg. 2022, 212, 107080. [Google Scholar] [CrossRef]
- Banbury, C.; Styles, I.; Eisenstein, N.; Zanier, E. R.; Vegliante, G.; Belli, A.; Logan, A.; Goldberg Oppenheimer, P. Spectroscopic Detection of Traumatic Brain Injury Severity and Biochemistry from the Retina. Biomed. Opt. Express 2020, 11, 6249. [Google Scholar] [CrossRef]
- Sabeti, F.; Carle, C. F.; Jaros, R. K.; Rohan, E. M. F.; Waddington, G.; Lueck, C. J.; Hughes, D.; Maddess, T. Objective Perimetry in Sporting-Related Mild Traumatic Brain Injury. Ophthalmology 2019, 126, 1053–1055. [Google Scholar] [CrossRef]
- Rui, Y.; Zhang, M.; Lee, D. M. W.; Snyder, V. C.; Raghuraman, R.; Gofas-Salas, E.; Mecê, P.; Yadav, S.; Tiruveedhula, P.; Grieve, K.; Sahel, J.-A.; Errera, M.-H.; Rossi, E. A. Label-Free Imaging of Inflammation at the Level of Single Cells in the Living Human Eye. Ophthalmol. Sci. 2024, 4, 100475. [Google Scholar] [CrossRef]
- Hammer, D. X.; Kovalick, K.; Liu, Z.; Chen, C.; Saeedi, O. J.; Harrison, D. M. Cellular-Level Visualization of Retinal Pathology in Multiple Sclerosis With Adaptive Optics. Investig. Opthalmology Vis. Sci. 2023, 64. [Google Scholar] [CrossRef]
- Fouda, A. Y.; Eldahshan, W.; Narayanan, S. P.; Caldwell, R. W.; Caldwell, R. B. Arginase Pathway in Acute Retina and Brain Injury: Therapeutic Opportunities and Unexplored Avenues. Front. Pharmacol. 2020, 11, 277. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



