Submitted:
01 April 2025
Posted:
02 April 2025
You are already at the latest version
Abstract
The human bone is a dynamic, highly vascularized tissue composed of 60-70% minerals, which include mainly calcium phosphate (CaP) in the form of hydroxyapatite (HA) crystals, 30% organic matrix composed of type I collagen fibers and less than 5% of water and lipids. The crystals are formed inside the matrix vesicles (MVs) and are then released in the organic collagen based fibrous matrix. Extracellular matrix (ECM) formation and mineralization processes, named osteogenesis, are associated with human mesenchymal stem cells (hMSCs) undergoing differentiation into osteoblasts (osteoblastogenesis). Osteogenesis is regulated by multiple intracellular signaling and genetic pathways and by environmental factors. Calcium flow is finely regulated and plays a key role in both osteoblastogenesis and osteogenesis. The formation and accumulation of CaP, the biogenesis of MVs, their secretion and the deposition of HA crystals to fill the organic bone matrix are the fundamental events in the biomineralization process. Here, I will describe and discuss the recent findings and hypothesis on the molecular mechanism regulating this process.
Keywords:
osteoblastogenesis
; osteogenesis
; calcium signaling
; matrix vesicles
; mineralization
1. Cellular, Molecular and Genetic Aspects of Bone Formation
Osteoblasts, osteocytes and osteoclasts are the three cell types which are fundamental for the development, growth, repair and remodeling of bones and contribute to bone homeostasis. Osteoblasts are defined as bone-forming cells and are responsible for bone matrix formation and mineralization, osteocytes are mature bone cells while the cells that break down bone are named osteoclasts [1].
Bone formation is a multi-step process which occurs through the differentiation of mesenchymal stem cells (MSCs) into osteoblasts (osteoblastogenesis), their modulation by other cell types, including immune cells, the production of extracellular matrix (ECM), the biogenesis and release of matrix vesicles (MVs) and ECM mineralization [2].
Osteoblastogenesis is a growth and differentiation program which is divided into four stages characterized by different markers: lineage commitment into osteoprogenitors, proliferation of osteoblast precursors, differentiation in early osteoblasts which are responsible for extracellular matrix maturation, and differentiation in mature osteoblasts which are linked to matrix mineralization [3,4]. Osteoblast differentiation is regulated by three transcription factors, Runt-related transcription factor 2 (also known as Runx2, coded by RUNX2 gene), osterix (coded by SP7/OSX gene) and ATF-4 (coded by ATF4 gene), and is controlled at multiple levels such as transcriptional co-factors, inhibitors, osteo-enhancing and -suppressing miRNAs, epigenetics, systemic factors, hormonal controls and environmental cues such as light–dark cycle (Figure 1) [5].
Runx2 is fundamental for osteoblast differentiation and chondrocyte maturation. In relation to osteoblastogenesis, RUNX2 is expressed in uncommitted MSCs, upregulated in osteoblast precursors, highly upregulated in immature osteoblasts and down-regulated in mature osteoblasts [5,6]. The RUNX2 gene expression is regulated by transcriptional factors, among which MSX-2, DLX-3, DLX-5, SATB2 (Special AT-rich sequence binding protein 2), FoxO1 (Forkhead box protein O1), Bapx1, PPARγ2 (regulator of adipocyte differentiation peroxisome proliferator-activated receptor γ 2) and Hox-A2 [5,7]. SATB2 mediates osteoblast proliferation in the early steps of osteoblastogenesis and contributes to the regulation of differentiation at later stages [5]. Moreover, Ihh/Gli2 signaling favors MSCs differentiation in osteoblast precursor by regulating the expression and stimulating Runx2 osteoblastogenic function [6].
Furthermore, Runx2 is subjected to the control of Wnt and transforming growth factor-β (TGF-β)/bone morphogenic protein (BMP) signaling pathways [8,9]. Interestingly, Wnt signaling activates the upregulation of RUNX2, OSX and DLX5 through the action of β-catenin in the canonical pathway and nuclear factor of activated T cells (NFAT) and phospho-c-JUN in the non-canonical pathway [8].
Similarly, both TGF-β and BMP signaling activate the canonical Smad-dependent pathway, with the phosphorylation of specific Smad proteins which translocate into the nucleus to promote transcription, and the non-canonical Smad-independent p38 MAPK (mitogen-activated protein kinase) signaling pathway. Both pathways converge at the RUNX2 gene in order to trigger the expression of osteoblast specific genes [8,9].
Runx2 is the “master regulator of osteogenesis” as it is the main transcription factor that determines the commitment of mesenchymal progenitors towards the osteogenic lineage [10]. Runx2 promotes proliferation of mesenchymal cells, their commitment into osteoblast lineage cells and their differentiation into osteoblasts through the reciprocal regulation of hedgehog, Fgf, Wnt and Pthlh signaling pathway genes and Dlx5. Runx2 enhances the proliferation of osteoblast precursors by modulating Fgfr2 and Fgfr3 [6].
Runx2 is responsible for the transactivation of major bone matrix protein genes via the osteoblast-specific cis-acting element (OSE) [5]. It forms heterodimers with the co-transcription factor core binding factor β (CBFβ) and regulates the expression of the osteoblast gene markers COL1A1 (coding for collagen α-1(I) chain), SPP1 (also known as OPN, coding for osteopontin), BGLAP (named also OCN, coding for osteocalcin), IBSP (also known as BSP, coding for integrin-binding sialoprotein), OSX and ALPL (also known as TNAP, coding for TNS-ALP, tissue-nonspecific alkaline phosphatase) [3,6,11,12,13,14,15,16].
Numerous transcription factors interact with Runx2 providing with costimulatory signals or repressing its function by affecting its DNA binding activity and/or transactivation potential [17]. Among these, Twist-related protein 1 and Twist-related protein 2 regulate Runx2 at the protein level by physically interacting with it and inhibiting its binding to DNA [18]. Also FoxO1 can interact with Runx2 and cooperates in the transcriptional regulation of osteoblastic markers [7]. Moreover, ATF4 is involved in indirect interactions with Runx2 during cell maturation to increase OCN expression, a marker of terminal osteoblast differentiation [19,20,21].
Runx2 is modulated by several post-translational modifications, such as acetylation, methylation, phosphorylation, SUMOylation, glycosylation and ubiquitination which affect the binding properties to genomic DNA [9,22,23]. Interestingly, the dynamic equilibrium among Runx2 acetylation, deacetylation, and ubiquitination is maintained by transmembrane and coiled-coil domains 1 (TMCO1)-mediated Ca2+ signaling [24]. Recent studies on Runx2 association with cis-regulatory elements (CREs) proved the association of Runx2 with the distal regulatory regions targeting skeletal genes and bone development and with the proximal regions (transcription start sites) of genes related to general cell activities, such as metabolic processes [23]. In line with this, Runx2 is associated with other genes which regulate the energy supply during osteogenesis [5]. Interestingly, Runx2 actively regulates the genes involved in glucose metabolism and energy homeostasis in mesenchymal cells [25]. Runx2 promotes glucose transporter 1 (GLUT1) gene expression, a glucose transporter which is responsible for passive transport of glucose. Its uptake favors osteoblast differentiation and bone formation by inhibiting AMP-activated protein kinase (AMPK), therefore suppressing the AMPK-dependent proteasomal degradation of Runx2, and enhancing the activity of mTORC1 pathway and, hence, protein synthesis, among which collagen, respectively [26]. Recently, a link between the odontogenic differentiation of human dental pulp stem cells (hDPSCs) and GLUT1-mTORC1 axis has been discovered [27]. Interestingly, the reciprocal regulation of Runx2 and GLUT1 contributes to an amplification mechanism allowing proper bone formation [26].
Furthermore, a strong correlation between RUNX family and NFAT family has been found out in several cell types as a RUNX binding sequence has been discovered on the NFAT cytoplasmatic 1-3 (NFATC1-3) promoter, therefore directly regulating its transcription. The inhibition of the RUNX family determined the suppression of NFATC2 family at the transcriptional level and a reduction of the amount of the total NFATc family, determining a lower activation of downstream genes. Therefore, the RUNX-NFAT axis can be targeted to reduce cell growth and other functions [28].
Osterix (Osx) is localized to the nucleus and induces the expression of some mature osteoblast genes, such as COL1A1, SPP1, BGLAP, IBSP, SPARC (also known as secreted protein acidic and cysteine rich, ON, osteonectin), IL10 (coding for interleukin-10), MMP13 (coding for collagenase 3 or matrix metalloproteinase-13), fibromodulin, DKK1 (Dickkopf WNT signaling pathway inhibitor 1) and ZBTB16 (Zinc finger and BTB domain containing 16). Osx can also interact with Runx2 to synergistically regulate gene transcription. OSX gene expression is regulated by two main pathways: one Runx2-dependent and one Runx2-independent [29]. Interestingly, Osx can form a complex with NFATc1 which promotes COL1A1 gene expression [29,30,31].
OSX gene expression is regulated by miRNAs and long non-coding RNAs (lncRNAs) in an epigenetic manner, whereas the protein undergoes post-translational modifications which modulate the DNA binding and transcriptional activity of the transcription factor [29].
ATF4 regulates both the differentiation of osteoblasts and the activation of osteogenic genes by modulating BGLAP, BSP and OSX. Interestingly, it is also involved in the amino acid import in osteoblasts and therefore in the regulation of protein diet and skeletogenesis [5].
The most important mediators of osteoblastogenesis and osteogenesis produced by immune cells are cytokines. Regulatory effects on osteoblastic genes are exerted by neutrophils, T cells, and macrophages which mainly affect the BMPs and Wnt signaling [32,33].
Interestingly, calcium signaling influences both the cellular transcription activity and mineralization of ECM.
2. Calcium Signaling and Cellular Transcription Activity
Oscillations of calcium concentration can activate many processes in cells through the Ca2+/calcineurin (CaN)/NFAT signaling pathway (Figure 2). It regulates growth and development of many types of cells, and, in particular, of osteoblasts, and bone development and regeneration by influencing osteoblasts gene expression [1]. The intracellular calcium oscillations can be induced by important events such as the activation of a variety of signaling pathways, among which the non-canonical Wnt signaling pathway. Interestingly, the activation of surface receptors determines the activation of phosphoinositide phospholipase C (PLC) which is responsible for the formation of inositol-1,4,5-trisphosphate (IP3). This binds to its receptor IP3R on the surface of the endoplasmic reticulum (ER), leading to the efflux of Ca2+ from it. Similarly, cADPR (cADP ribose), a cyclic derivative of ADP, can activate the ryanodine receptor (RyR) and promote Ca2+ efflux. The store calcium depletion determines the activation, oligomeration and translocation of the calcium sensor proteins, stromal interacting proteins (STIM1 or STIM2), on the ER to the ER-plasma membrane (PM) junctions, where they interact with the olfactory receptor class A related 1 (Orai1), determining the formation of the complex contained in the calcium-released activated channel (CRAC) and inducing a sustained influx of Ca2+. Calreticulin (CRT) is a Ca2+-binding multifunctional molecular chaperone in the ER which takes part to the IP3-mediated Ca2+ release [34,35].
Moreover, the Ca2+/CaN/NFAT signaling pathway is activated also by the environmental sensor calcium-sensing receptor (CaSR) which is a unique G protein-coupled receptor (GPCR), can sense the extracellular Ca2+ concentration and can mobilize rapidly the intracellular Ca2+ flux, and by the voltage gated Ca2+ channel (VGCC), the ligand-gated Ca2+ channel and the Na+/Ca2+ exchanger (NCX) which can regulate Ca2+ flowing into the cell [1,36].
Cytoplasmic Ca2+ can bind calmodulin (CaM) which undergoes a conformational change and can therefore bind to specific targets such as calmodulin kinase II (CaMKII) and calcineurin (CaN). CAMKII is released from its autoinhibitory status, maximally activated and subjected to an intraholoenzyme autophosphorylation reaction. Phosphorylated CAMKII activates the CREB/ATF and ERK signaling pathways, which are involved in osteogenesis. The CaMKII/HDAC4 pathway is mediated by TMCO1 and induces the enhancement of RUNX2 stability and promote bone formation. TMCO1 is an ER Ca2+ permeable channel that responds to ER Ca2+ overload, prevents intracellular Ca2+ stores from overfilling and maintains calcium homeostasis in the ER through Ca2+ leakage from it [24]. CaN is activated by interacting with CaM and, in turn, promotes the dephosphorylation of NFAT transcription factors and their nuclear translocation in order to regulate gene expression [1].
Interestingly, it has been observed that a high extracellular concentration of 10 mM Ca2+ determined the nuclear translocation of NFATn [37,38]. Similarly, a concentration of 40 mM Ca2+ at the bone resorption site induced the increment of intracellular Ca2+ concentration [39]. The Ca2+/CaN/NFAT signaling pathway has been closely associated with the physiological activities of osteoblasts and compounds which inhibit this signaling pathway at the same time affect osteoblastogenesis [1].
NFATs are a family of inducible transcriptional regulators identified in T cells over 35 years ago [40]. NFAT gene family contains four classic members, NFATc1 (also named NFATc and NFAT2), NFATc2 (NFATp, NFAT1), NFATc3 (NFAT4) and NFATc4 (NFAT3), which are uniquely expressed only in vertebrates. The termination of the surface signaling and, in turn, a lower Ca2+ influx, leads to rephosphorylation of NFAT proteins by several kinases, among which GSK3, PKA, MEKK, and its export in the cytoplasm in an inactive state [36].
CaN-NFAT signaling pathway plays a fundamental role in bone homeostasis. Osterix-, Runx2- and Smad-dependent osteoblastic genes were upregulated by the overexpression of NFAT [30,36]. Interestingly, NFATc1 cooperates with osterix and Fos/Jun to activate target genes, such as Wnt4, Frizzled9 (Fz), CCL8 and osteocalcin [41]. Moreover, it has also been demonstrated that the induction of NFATc1 nuclear translocation may be associated to the increase of the expression of Wnt3a, Wnt5a and β-catenin [36,42]. Furthermore, it negatively regulates the expression of the Wnt inhibitory genes secreted frizzled-related protein 2 (sfrp2) and Dickkopf 2 (DKK2) [41].
Nevertheless, it has been proved a role of NFATc1 in suppressing the promoter of osteocalcin during osteoblast differentiation possibly mediated by histone deacetylase (HDAC) 3 interaction with Runx2 at OCN promoter and therefore repressing osteoblast maturation [36,43]. Indeed, it has been demonstrated that NFATc1 increased translocation and localization to the nucleus caused osteopetrosis, a rare disorder that causes bones to grow abnormally and with higher density and probability of bone breakage, by regulating chemokines and the Wnt pathways [41].
Furthermore, it has been found out that NFATc2 may have a role in the regulation of bone homeostasis. In fact, Nell-1 is a growth factor which stimulates the expression of ECM proteins required for bone formation, is a target gene of Runx2 and can regulate NFATc2 [36,44,45,46,47,48]. Both NFATc1 and NFATc3 genes were upregulated when the extracellular calcium level was increased. NFATc3 may regulate bone homeostasis by leading to higher RANKL expression [37]. Activated osteoblastic RANKL promotes osteoblast differentiation through RANKL reverse signaling and the activation of RUNX2 [49]. Moreover, the NFATc3 expression is induced by the expression of NFATc1 [37].
This data suggests that the involvement of CaN-NFAT pathway in the cellular transcription activity is finely regulated and different isoforms of calcineurin and NFAT, and different NFAT proteins exert distinct functions [36]. Therefore, calcium oscillations can influence gene expression and bone homeostasis.
Moreover, immunomodulation of MSCs by immune cells can promote osteogenesis by affecting NFAT. It has been demonstrated that T cells can promote proliferation and survival of bone marrow stromal cells (SCs) through the binding of CD40 ligand (CD40L), a surface molecule on T cells, to its receptor CD40 on SCs plasma membrane. CD40 signaling is activated and RANKL is produced [50]. CD40 lacks the kinase domain and, upon activation, recruits TNF-alpha Receptor Associated Factors (TRAFs) at the cytoplasmic domain. Both TRAFs-dependent and TRAFs-independent signaling activate phosphatidylinositol 3-phosphate (PI3K), Raf, Lyn, Syk, p38, Erk, and Protein kinase C (PKCs). CD40 can also activate NF-kB pathways, ATF2, ELK1, BLIMP-1, BATF and NFAT, therefore modulating differential gene expression [51]. Interestingly, overexpression of NFATc1 and NFATc3 induced RANKL expression. Moreover, high extracellular Ca2+ determined activation of the CaN/NFAT pathway and the expression of RANKL [37,52,53].
Furthermore, NFAT signaling in osteoblasts induces the expression of chemokines, such as CCL8, that attract monocytic osteoclast precursors. Given the role of NFATc1 in osteoclastogenesis and in light of the above-mentioned studies, bone formation and resorption are integrated processes regulated by NFAT signaling [54].
3. Regulation of Calcium Homeostasis
Ca2+ exerts a key role in the process of osteogenic differentiation. The concentration of cytoplasmic Ca2+ is normally around 0,1-0,2 µM. Calcium channels and transporters are necessary for calcium oscillations. Interestingly, mitochondria, lysosomes and ER are the organelles responsible for intracellular Ca2+ storage. There are crosstalks between organelles and plasma membrane Ca2+ channels in order to regulate cytosolic Ca2+ signals. For example, Ca2+ can modulate the proliferation and differentiation of osteoblasts through the mutual adjustment between CaSR, VGCC and SOCE (store-operated calcium entry, a ubiquitous Ca2+ signaling pathway mediated by CRAC) [1,55]. Furthermore, Ca2+-permeable channels transient receptor potential (TRP), purinergic (P) (P2X and P2Y) receptors and Piezo channels on the PM can increase intracellular Ca2+ concentration. Moreover, activation of K+ channels inducing membrane hyperpolarization indirectly promotes Ca2+ signaling in osteoblasts linage cells [35].
Furthermore, IP3R, RyR, TMCO1 and pannexin3 (Panx3) Ca2+ channel in the ER transmembrane, Na+/Ca2+ exchanger (NCX), Na+-Li+/Ca2+ exchanger (NCLX), Na+-independent Ca2+ exchanger (H+/Ca2+ exchanger, HCX) and the Permeability Transition Pore (PTP) in mitochondria and the transient receptor potential mucolipin subfamily 1 (TRPML1 or ML1) and two-pore channels (TPCs) in lysosomes can favor the increment of Ca2+ in the cytosol [24,35,56,57,58,59,60,61].
The extracellular Ca2+ concentration is normally around 1-2 mM. Its concentration is 0,5-1 mM in the ER, 0,1 µM in the mitochondria and 2 µM in the lysosomes [62]. It has been demonstrated that extracellular Ca2+ concentration of 1,8-7,8 mM induced the increase in cell size and proliferation. Higher extracellular values of Ca2+ concentration of 10-15 mM Ca2+ determined the activation of downstream MAPK signaling pathways mediated by this ion, which promoted the expression of osteogenic differentiation related genes. Nevertheless, Ca2+ concentration of 50 mM hindered the normal adhesion of cells [1].
The increase in calcium flow can generate subplasmalemmal high Ca2+ microdomains (HCMDs). Depending on the cell type and relative position of organelles, channels and other molecules, physiological responses, such as exocytosis, contraction or cell growth, can be induced or Ca2+ can be taken up by mitochondria through the mitochondrial Ca2+ uniporter (MCU) or by ER in order to avoid progression of the Ca2+ wave towards the cell core [63]. Mitochondrial Ca2+ accumulation is usually transient as this organelle is normally empty but it can accumulate large amounts of Ca2+ when its concentration is above 1 µM, nevertheless its overload (over 3 mM/mg protein) can determine damages to the organelle and trigger, in turn, apoptotic mechanisms [63,64,65]. Besides MCU, the leucine zipper, EF-hand containing transmembrane protein 1 (LETM1) allows the transport of calcium ion inside the mitochondria and of proton outside the mitochondrial matrix [66]. The voltage-dependent anion channel (VDAC) is located on the outer membrane of the mitochondria and is responsible for Ca2+ ions and ATP transport [67].
An important crosstalk between mitochondria and ER is due to dynamin-related mitofusins, which are part of the tethering mechanism between these two organelles, and determines the release of Ca2+ from the ER through IP3Rs coupled to the mitochondrial uptake through MCU and the mitochondrial voltage-dependent anion channel 1 (VDAC1) on the internal and outside mitochondrial membranes, respectively (Figure 3) [63]. The MAM (mitochondrial-associated endoplasmic reticulum membranes) regions contain the ER proteins mitofusin 2 (MFN2) and vesicle-associated membrane protein (VAMP)-associated protein B (VAPB) which interact with the mitochondrial mitofusin 1 (MFN1) or MFN2 protein and the protein tyrosine phosphatase-interacting protein 51 (PTPIP51) [68,69,70]. The cytosolic glucose regulated protein 75 (GRP75) is part of the physical tethering of ER and mitochondria and mediated the interaction between VDAC and IP3R [71].
Intracellular Ca2+ is also decreased by NCX at the PM and by Ca2+ extruding ATP-driven enzymatic pumps such as PMCA at the PM, SERCA at ER, and SPCA at the Golgi [72]. It is likely that a mechanism to transport Ca2+ from the cytosol inside lysosomes such as Ca2+ exchangers or Ca2+ ATPases might exist and that a lysosomal-ER crosstalk might be a most critical Ca2+ source [73,74].
Interestingly, the ER drives calcium refilling of lysosomes upon contact with this organelle. In contrast with what was previously suggested, lysosomal acidification mediated by the V-ATPase H+ pump was not associated with Ca2+ refilling, whereas pharmacological depletion or chelation of ER Ca2+ or antagonists of ER IP3Rs were able to prevent Ca2+ refilling of lysosomes. Besides Ca2+ uptake through endocytosis, most of lysosomal Ca2+ requires the activity of lysosomal Ca2+ channels and it has been hypothesized that a functional interaction between ER and lysosomes could be the direct source of Ca2+ to lysosomes through the formation of nanojunctions composed of IP3R on the ER, which is responsible for producing a local high Ca2+ concentration, an unknown low-affinity Ca2+ transport mechanism on the lysosome and unidentified tether proteins which link the ER and lysosomal membrane proteins [73,75].
It has been recently demonstrated that additional mechanisms of mitochondrial disposal and mitochondrial quality control (MQC) recycle whole damaged mitochondria or mitochondrial fragments through the production of mitochondria-derived vesicles (MDVs). These organelles are generated directly through the budding from the mitochondrial membranes to extrude damaged content and can fuse to vesicles deriving from the endosomal-lysosomal system, such as multivesicular bodies (MVBs) and lysosomes. It has been hypothesized that MDVs can contribute to regulate calcium homeostasis by containing Ca2+ ions and fusing with lysosomes [76,77,78,79]. Moreover, direct interaction between mitochondria and lysosomes via non-degradative process has been identified and may favour inter-organelle transfer of calcium. Interestingly, lysosomal guanosine triphosphate (GTP)-bound Rab7 promotes contact formation and tethering to the mitochondria which could be mediated by Rab7 effector proteins. The recruitment of TBC1D15, a Rab7 GTPase-Activating Protein (GAP), to mitochondria mediated by the outer mitochondrial membrane protein Fis1 leads to untethering [80].
The cytosolic calcium ion concentration is controlled by the activation of membrane pumps and channels, the calcium ion capture by subcellular organelles and the action of calcium-binding proteins in the cytosol. Annexins are a group of calcium-binding proteins which have an important role in the mineralization process by working as nucleating centers for extracellular mineral deposition and by providing critical sites for intracellular calcium ion trafficking and cluster formation. Annexins are localized on intracellular organelles, the PM and MVs [81]. Proper regulation of intracellular concentration of Ca2+ or extracellular Ca2+ can enhance osteogenic differentiation and mineralization [1]. The involvement of mitochondria and ER is important in regulating Ca2+ transport and accumulation and the initiation of the mineralization process [82].
4. Formation of the Prebone and Bone Matrix
Osteoid, which is an unmineralized precursor of bone, is secreted at the sites of new bone formation and is normally surrounded and controlled by an organized epithelioid structure made of osteoblasts. The prebone (osteoid) ECM is an organic matrix made of structurals proteins (collagens), which compose dense layers that alternate parallel and orthogonal to the axis of stress loading, non-collagenous matrix proteins (osteonectin, osteocalcin, osteopontin, sialoprotein), proteoglycans (PG), such as decorin, lumican, biglycan and others, and glycosaminoglycans such as chondroitin sulfate and hyaluronan which are macromolecules that provide the framework for collagen formation. The collagen-proteoglycan matrix is able to bind calcium salts. Some of the non-collagenous proteins play important regulatory and structural roles in order to regulate bone formation and regeneration [54,83,84].
Osteoid undergoes mineralization to form mature bone. This process is promoted by vesicles named MVs released by osteoblasts which contain molecules, such as annexin, TNAP and CaP (Figure 4). The calcification process starts with the formation of amorphous calcium phosphate (ACP) due to an aggregation process of Ca2+ and inorganic phosphate (Pi) ions which transform to more stable crystalline phases, such as octacalcium phosphate (OCP, Ca8(HPO4)2(PO4)4·5H2O), calcium-deficient apatite (CDHA, Ca9(HPO4)(PO4)5(OH)) and hydroxyapatite (HA, Ca10(PO4)6(OH)2). HA is the major component of vertebral tooth and bone tissue. As the matrix matures, HA microcrystals are organized into more complex structures. The crystallization process occurs within vesicles, followed by the formation of calcified nodules [83,85,86]. In fact, after the release of these vesicles, needle-like crystals pierce the membrane of MVs and further accumulation of ACP and HA crystals in the extracellular matrix is induced due to the presence of free ion complexes [87]. Both the HA crystals and the free ion complexes serve as foci for mineral formation [1,6,35,55,72]. Further step of mineralization, which determines the formation of the bone (mineralized) ECM, is the formation of mineralized nodules and calcifying globules with deposition within intrafibrillar and interfibrillar spaces of collagen. Glycoproteins, growth factors (GFs) and other non-collagenous proteins can influence mineralization [87]. Calcification of the matrix occurs experimentally only at mildly alkaline levels of pH [83,86]. Formation of prebone and bone matrix occur at the apical surface of osteoblasts [83].
Cellular influx of Pi ions occurs through multimeric membrane exchanger/transporter proteins, including family 20 member A1/sodium-dependent phosphate transporter 1 (SLC20A1/PiT1), family 20 member A2/sodium-phosphate transporter 2 (SLC20A2/PiT2), family 34 member A1/sodium-phosphate co-transporter IIa (SLC34A1/NaPi-IIa) and family 34 member A2/sodium-phosphate co-transporter IIb (SLC34A2/NaPi-IIb) [83]. Efflux of Pi from the cells occurs through the XPR1 transporter protein. Ionic homeostasis of the cytosol is regulated also by other types of exchanger proteins, such as sodium/hydrogen and sodium/potassium membrane pumps [83,88,89,90,91]. Pi are present intracellularly at a higher concentration compared to the plasma [92]. Pi can be localized in the cytoplasm, in the ER and in mitochondria [93,94].
TNAP, phosphoethanolamine/phosphocholine phosphatase 1 (PHOSPHO1) and Ectonucleotide pyrophosphatase/phosphodiesterase I (ENPP1) are responsible for the Pi production. Interestingly, ENPP1 is a membrane-bound glycoprotein that can catalyze ATP and generate PPi and is coupled with TNAP, a glycosylphosphatidylinositol anchor enzyme, which can hydrolyze PPi into Pi to promote mineralization. TNAP is localized in large amounts on the PM and on the vesicles membrane. ClC3 and ClC5 can alter its normal activity [83,87]. Differently, PHOSPHO1 can hydrolyze PPi into Pi inside MVs. Moreover, ANK, which is encoded by the progressive ankylosis gene (Ank), is a non-enzymatic PPi channel that can enable PPi exit from MVs, whereas Pi can be internalized into MVs by sodium-inorganic Pi co-transporters Pit1 and Pit2 [87]. Moreover, TNAP might be able of recruiting calcium ions. It was also found in and around the calcified nodules. The Ca2+ and Pi ions were localized differently, at proteoglycan sites and collagen fibril structures, respectively, in the uncalcified matrix, therefore limiting the CaP production while Ca2+/Pi colocalization was observed in and around the calcified nodules. The activity of TNAP is fundamental for the ECM mineralization [87,95].
The process of mineral deposition produces acid and requires removal of protons for driving mineral precipitation and formation of an extremely dense mineral. This occurs thanks to Cl-/H+ exchangers that move acid into osteoblasts whereas acid extrusion at the basolateral membranes is mediated by sodium-hydrogen exchangers 1 and 6. This process and the osteoblast Pi transport are regulated by the sodium-hydrogen exchanger regulatory factor-1. The process of mineral deposition might require also K+ channels, which might correlate with ClC exchangers and other ion transporters, such as an outward pump of net sodium at the basolateral surface and the reuptake of chloride at the apical surface. Besides acid uptake from bone matrix, water is removed from the dense collagen hydrogel forming the osteoid. Tissue arrays suggest that osteoblasts express aquaporins [83].
Due to matrix growth, osteoblasts undergo either apoptosis or terminal differentiation to form osteocytes which are incorporated into the matrix and communicate with each other and with the cells surrounding the bone matrix through cell processes within canaliculi in the matrix [83,96].
The alkaline nature of bone formation is counteracted by the essential acidic environment of bone degradation where addition of protons is necessary for recovery of ionic phosphate and calcium during bone resorption. This balance is modulated by osteoblasts and osteoclasts, respectively [83].
5. Biogenesis, Trafficking and Release of MVs
Besides limited amounts of calcium ions which diffuse through the membrane and specific transport systems which regulate the cation passage into the cell, specific cellular pathways are responsible for Ca2+ accumulation, trafficking and discharge from intracellular organelles such as vesicles. Mineralization occurs through mineral delivery and deposition as packages of ACP nanospheres, which transform into structures of crystalline apatite (HA) within the collagen matrix [97].
Several findings on the physiological process of biomineralization have been recently published, however a unique process of MVs biogenesis, CaP formation and secretion of vesicles has not been associated with it. Indeed, it has been demonstrated that MVs could form from vesicles, likely lysosomes, that get in contact with mitochondria in order to internalize Ca2+ before being released, from lysosomes that fuse with autophagosomes containing mitochondria which present ACP in the mitochondrial matrix, from multivesicular bodies (MVBs) and from vesicles at later steps of intracellular trafficking [64,77,78,79,80,82,87] (Figure 5).
In line with these works, ACP/HA could be released in MVs secreted from MVBs similarly to exosomes or through ACP/HA-containing MVs that interacts with the PM and are secreted in the ECM [98,99]. Further release of Ca2+, Pi ions and ACP can occur through exocytosis or through polarized budding and pinching-off processes with the release of MVs, similarly to ectosomes (Figure 6) [77,78,100,101,102,103].
5.1. Inter-Organelle Communication and Physicochemical Structure Development of CaP
The formation of ACP precursors supports the process of intracellular initiation of biomineralization in organelles and subsequently the release of vesicles in the ECM. It has been observed that both ER and mitochondria contain both Ca2+ and Pi ions [93,94,101]. It has been hypothesized that the initial process of formation of minerals occurs in the ER and a first contact between ER and mitochondria is required for the formation and accumulation of CaP in mitochondria. Mitochondria-ER contact sites (MERCs) could have an important role in the mineralization process as Ca2+ is accumulated in the bulk cytosol microdomains which are involved in the ions and molecular exchange between the two organelles at a tenfold higher concentration than normal leading to the ER promoting the entry of Ca2+ into mitochondria, which in turn can modulate the Ca2+ signaling. Pi can be transported independently through MERCs. In particular, the transport of Pi could occur through the glucose-6-phosphatase (G6Pase) transport system in the ER which hydrolyzes glucose-6-phosphate to glucose and Pi. Pi can, in turn, be transported through the transport system termed T2 on the ER to the cytoplasm and pass effortlessly through the outer mitochondrial membrane (OMM) due to its high permeability. Subsequently, the phosphate carrier (PIC) on the inner mitochondrial membrane (IMM) barrier is responsible for the uptake of Pi. Due to the transport of Ca2+ and Pi from ER to mitochondria, ACP can be formed in the mitochondrial matrix and in the cytoplasm after the first part of the transport [87].
Furthermore, a second inter-organelle communication is necessary between mitochondria and vesicles in order to have CaP accumulated in vesicles and the nucleation and maturation of minerals. In 1960s insoluble CaP electron-dense granules reported as ACP precursors have been detected in mitochondria [104]. In the same years it has been discovered that the earliest mineral deposits were in contact with membrane-bounded TNAP-positive vesicles initiated from intracellular structures [87,105,106]. The transport of Ca2+ and Pi ions or ACP from mitochondria to vesicles has been observed and could occur through mitophagy, direct contact between mitochondria and lysosomes or other vesicles and formation of MDVs and subsequent contact with other vesicles [64,77,78,79].
Mitophagy is a cytoprotective mechanism and a type of macroautophagy which removes dysfunctional or superfluous mitochondria by transporting them to lysosomes for degradation [107]. The first step of this process is the engulfment of mitochondria in a cup-shaped sequestering compartment named phagophore which matures into the double-membraned vesicle named autophagosome. ACP precursors localized in the mitochondrial matrix are therefore transported to autophagosomes, which are conveyed to lysosomes. The outer membrane of the autophagosome fuses with the lysosome membrane to form an autolysosome, an acidic degradative hybrid organelle, while the inner membrane and the content of the autolysosome are degraded by the lysosomal enzymes and recycled. In these organelles, ACP precursors coalescence to form larger intra-vesicular granules [64].
The transport of Ca2+ and Pi ions or ACP from mitochondria to vesicles may also occur through MDVs which are a set of intracellular vesicles of 70-150 nm in diameter of mitochondrial origin. They are constitutively produced under basal conditions and can be highly produced under specific conditions. MDVs shuttle mitochondrial cargoes to peroxisomes, lysosomes and MVBs and are single- or double-membrane organelles, budding from the OMM or shedding from both OMM and IMM, respectively. The double-membrane MDVs contain portions of mitochondrial matrix and could contain ACP. Both subtypes of MDVs are directed to lysosomes for degradation or lysosomal exocytosis or to MVBs for the exocytosis of MVs [76,108].
Scanning electron-assisted dieletric microscopy and super-resolution microscopy have been used to evaluate the intracellular MVs transport and secretion at a nanolevel resolution. Interestingly, lysotracker-positive vesicles were able to fuse with calcein-positive vesicles adjacent to mitochondria and the newly formed vesicles moved towards the extracellular space. Calcein is a membrane-impermeable calcium binding fluorescent probe and mitochondria have been proposed to be the source of these static calcium-containing vacuoles adjacent to them. The fusion of these vesicles with lysosomes, characterized by the cationic dye lysotracker that preferentially accumulates in acidic lysosome and by the lysosomal resident marker protein 1 (LAMP1) might determine the formation of MVs containing ACP which are directed to the periphery in order to be secreted. When the cells were treated with a lysosomal proton adenosine triphosphatase inhibitor, bafilomycin A1 (BafA), lysosomal acidification was completely abolished and the formation of the hypothesized MVs was also negatively affected. Nevertheless, when the cells were treated with an exocytosis inhibitor, vacuolin-1 (Vac-1) or with the inhibitor of lysosomal exocytosis endosidin 2 (ES2), which targets the exocyst complex subunit, intracellular accumulation of vesicles and calcein occurred and mineralization was blocked [78].
Ca2+- or ACP-containing lysosomes or lysosome-related organelle may fuse with other vesicles, such as the TNAP-positive vesicles originated in the secretion pathway, in order to acquire the typical markers and the specific molecules of MVs. Furthermore, acidocalcidomes, acidic calcium stores, which are another form of the lysosome, have been identified in human cells besides bacteria and can take part to the process of MVs biogenesis. These organelles are rich in phosphorus compounds and their acidity is maintained by proton pumps [109].
Interestingly, the interaction between ER and lysosomes or other vesicles can promote the formation of MVs thanks to the transport of Ca2+ from the ER through IP3R and its internalization by the vesicles through Ca2+ importers [73,75].
Furthermore, it has been hypothesized that MVs can origin from autophagosomes which fuse with endosomes leading to the formation of amphisomes or from other vesicles, besides MDVs, that can provide MVBs with Ca2+ and Pi ions and ACP [110,111,112]. In fact, ion loading by endosomes could occur thanks to specific proteins, such as nucleobindins, calbindins and calreticulins located in the cytoplasm which interact with Ca2+ and favour its transit to endosomes [113,114,115]. Moreover, endosomes could accumulate ions from other sources, such as from ER, mitochondria or from the cytoplasma through channels [111].
Another mechanism through which ACP precursors are transported from mitochondria through vesicles to the ECM is overloaded Ca2+ influx into mitochondria which triggers the opening of mPTP, leading to the release of pro-apoptotic molecules and activation of downstream CASPs, induces ROS generation and apoptosis. Indeed, mitochondrial disfunction leads to apoptotic cell death, the formation of apoptotic bodies which are heterogeneous vesicles containing ACP/HA and promotion of calcification [98,99].
In light of what previously described, it has been hypothesized that MVs have a mixed origin and an overlap of pathways can occur. Moreover, besides the MVs associated with activities of the cellular endosomal-autophagic system [64,78,87,110,112], several works report MVs as derived from protrusions, blebs or buds from the PM [106,116,117,118,119].
5.2. Regulation of MVs Biogenesis and Intracellular Trafficking
Ras-associated binding (Rab) proteins are guanine nucleotide-binding proteins which regulate intracellular membrane trafficking by orchestrating the biogenesis, transport, tethering, and fusion of vesicles with a target membrane. Rabs cycle between two states, an active membrane-bound (GTP-loaded) form and an inactive unbound (GDP-loaded) form. Their cycling is regulated by GTPase-activating proteins (GAPs) for GTP hydrolysis and GTP exchange factor proteins (GEFs) for nucleotide exchange. More than 60 human Rab proteins have been identified. They decorate the surface of specific organelles and recruit effector proteins in order to modulate the membrane trafficking [120].
Other molecules are involved in the regulation of the intracellular trafficking, such as SNARE (Soluble NSF Attachment Protein REceptor) proteins which are fundamental for membrane fusion [121].
It has been demonstrated that Rab4, Rab5, Rab11, Rab14, Rab18, and Rab21 were present in matrix vesicles isolated from SAOS osteoblast-like cells [82,122].
Interestingly, it has been proved that Rab11 is a fundamental molecule for regulating the trafficking of sortilin, a key regulator of smooth muscle cell (SMC) calcification via its recruitment to MVs. Sortilin mediates the loading of TNAP from the Golgi apparatus to MVs. TNAP is able to induce high mineralization competence in the ECM and therefore determining the calcification [123]. Rab11 is also involved in the regulation of the fusion of lysosomes and late endosomes (LE) with Rab11-positive vesicles in order to promote lysosomal exocytosis. Interestingly, both Rab11a and Rab11b have been identified as regulators of Ca2+-induced lysosome exocytosis. The transient interaction occurs at the cell periphery and it is mediated by the exocyst subunit Sec15, a Rab11 effector. It has been hypothesized that Sec15 might function independently of the exocyst complex. Therefore, it could have an important role in mediating the transport of Rab11-positive vesicles towards the periphery and late endocytic compartments along the actin cytoskeleton by interacting with myosin V. Afterwards, Rab11 binds Rab3a and GRAB, a Rab3a GEF. In particular Rab3a GEF activates Rab3a which, in turn, binds to the LE and lysosomes and recruits the non-muscle myosin heavy chain IIA (NMIIA, encoded by MYH9) and synaptotagmin-like protein 4a (Slp-4a, encoded by Sytl4) in order to determine the positioning and exocytosis of lysosomes. It has been demonstrated that this process is triggered by Ca2+ as treatment with the Ca2+ ionophore ionomycin impairs lysosome exocytosis by hindering the interaction between Rab11-positive vesicles with lysosomes and LE [124].
Furthermore, it has been demonstrated that the active form of Rab7 promotes contact formation and tethering between Rab7-positive lysosomes and mitochondria in order to promote inter-organelle transfer of metabolites, including lipids, calcium, or iron whereas the mitochondrial TBC1D15, a Rab7 GAP, determines Rab7 inactivation and contact untethering [80].
Interestingly, Rab32 and phosphofurin acidic cluster protein 2 (PACS2) can influence the localization of calnexin (CNX) and, therefore, the transport of Ca2+ from ER to mitochondria. This occurs through the opening of the IP3Rs, which is regulated by a set of proteins present at or recruited to MAMs, including calnexin (CNX), the Sigma non-opioid intracellular receptor 1 (Sigma1R), presenilin 1 and 2 (PS1 and PS2) [125].
Further studies are necessary to identify other Rab proteins and relatives SNAREs involved in the biogenesis, ion loading and trafficking of MVs.
6. Discussion
The initiation of osteogenesis is related to the differentiation of mesenchymal stem cells into osteoblasts. This process is based on a gene expression program associated to the bone matrix production. Interestingly, the gene expression of important transcription factors is coupled to the oscillations of calcium flow and to the cell signal transduction. In fact, besides various essential cell signaling pathways, including Wnt, BMP and TGF-β, which are activated by stimuli on the PM and promote directly the upregulation of key transcriptional factors in the differentiation process, oscillations of intracellular Ca2+ can influence the expression level of specific genes, such as RUNX2 and OSX [30,45,83]. Osteoblast are cuboidal cells which display large amounts of rough ER and mitochondria and secrete bone organic matrix [126]. Similarly to what has been found out in invertebrates, the biomineralization process is due to the formation of vesicles containing Ca2+ and Pi, the production of ACP in these compartments and their shuttle to the PM. The secretion of MVs containing high concentrations of Ca2+ and Pi and ACP are responsible for the formation of HA crystal deposition in order to fill the organic bone matrix [6,127,128]. Bone formation is characterized by the release of MVs at the apical surface of osteoblasts and mineralization is driven by both active and passive transport and pH control [83]. Therefore, both osteoblast differentiation and bone matrix synthesis are coupled with the regulation of calcium flow. While there are several studies proving the influence of Ca2+ oscillations on gene expression and osteblastogenesis, several aspects of the biomineralization process are not clarified yet.
MVs are 30 to several hundreds of nanometers in diameter [129]. They are characterized by the presence of TNAP, PHOSPHO1, ENPP1, annexins, non-collagenous calcium-binding proteins, accumulation of Ca2+ and Pi ions, ACP and a specific spectrum of phospholipids [130]. Moreover, MVs contain several membrane transporters on their membranes which could promote further Ca2+ and Pi internalization [83]. Similarly to other vesicles, MVs displayed the exosomal markers CD63, CD81 and CD9, and the endosomal markers ALIX, TSG101, and HSP70. Nevertheless, MG63 exosomes exhibited lower enrichment in alkaline phosphatase-specific activity compared to MG63-derived MVs. It has been hypothesized that both MVs and EVs could present similar biogenesis pathway, size, and morphology, but they exhibit different roles [131].
Many studies reported that CaP within intracellular MVs was mainly amorphous, nevertheless it has been also found that extracellular MVs contained crystallized CaP, suggesting that CaP forms the more stable form HA in secreted MVs whose membrane is disrupted by the mineralization process determining the release of HA in the ECM [79].
Calcium phosphate can crystallize to form HA in neutral to basic pH. It has been demonstrated that OCP forms preferentially at pH 5.5 or higher whereas HA is formed at pH above 7 (preferentially around pH 7.4-8) [132]. Interestingly, the pH values of the eukaryotic compartments and organelles has been studied and intracellular cytosolic pH is approximately 7.2. This value varies significantly between the different organelles. The pH is generally 6.3 in early endosomes, 5.5 in MVBs and late endosomes and 4.7 in lysosomes. Like endocytic organelles, recycling endosomes have a pH of 6.5, while secretory granules of 5.5. In contrast the pH value is 7.2 in the ER and 8 in mitochondria. pH values are maintained by a balance between ion pumps, leaks and internal ionic balance. Interestingly, endosomal pH can be modulated by affecting the membrane potential. Compared to the cytosolic pH, the pH of ECM is slightly alkaline with a value of 7.3-7.4 [133].
ACP has been detected in cytosol, mitochondria and vesicles, included MVs [79,111,134]. ACP might also be formed in intraluminal vesicles (ILVs, vesicles located inside MVBs), however it has not been clarified yet how MVs acquire a higher pH and other specific markers when they origin from acidic organelles such as lysosomes, lysosome-related organelles and acidocalcisomes and whether TNAP is necessary for promoting ACP formation and accumulation. It can be hypothesized that acidic vesicles could interact with other compartments, such as more alkaline compartments, MDVs, and TNAP-positive secretory granules. In support of this, Rab11 could mediate the transport of TNAP from the secretory pathway to Rab3a-positive LE, lysosomes or lysosomes-related vesicles before exocytosis [123,124]. The acquisition of TNAP on MVs might occur also during the secretion step by transferring TNAP from the PM to the released vesicle.
In support of the relationship between autophagy, and relative lysosome-related vesicles which are produced, and bone mineralization, it has been proved a marked increase in autophagy during osteoblast differentiation. In fact, it has been demonstrated an association between genes involved with the regulation of the autophagic pathway and bone mineral density and a change in the lipidated form of LC3-II protein at the onset of mineralization [135,136]. Furthermore, autophagosomes containing Ca2+ and Pi determined the formation of calcified MVs and promoted the extracellular mineralization [137].
Another interesting hypothesis is that MDVs could constitute the main source of MVs. Nevertheless, specific membrane composition and cargo selection have been reported for single- and double-membrane MDVs and further studies may clarify the correlation between these two subsets of MDVs and MVs biogenesis, the trafficking and further interaction of MDVs with other organelles prior to the unloading into the ECM [108].
It has been demonstrated that Ca2+ load in lysosomes is not pH-dependent while MVs formation is affected by lysosomal pH [73,75,78]. In light of these findings, it can be hypothesized that H+ flux is important for ion homeostasis in lysosomes.
Similarly to the vesicles in the endolysosomal-autophagic system, regulatory proteins such as Rabs and their effectors could regulate membrane fusion between MVs and other compartments, the expression of pumps and channels and the presence of specific enzymes in MVs.
The process of mineral formation begins in the cell and terminates in the ECM. It is a multistep process which includes the biogenesis, ion loading, trafficking and release of MVs. This process is highly regulated and involves several molecules responsible for the maintenance of intracellular ions homeostasis, vesicle trafficking and pH control, those modulating the actin fiber distribution in correlation with the fluidity of the cell membrane and many other cell components [138]. Nevertheless, oscillations of calcium flow have been highlighted as a key player in the mechanism of biomineralization as Ca2+ signaling mediates gene expression and Ca2+ transport affects CaP formation, MV biogenesis and release and the deposition and mineralization of collagen fibers.
Funding
This research received no funding.
Acknowledgments
Figures were created with Biorender.com.
Conflicts of Interest
The author declares no conflict of interest.
References
- Ren, R.; Guo, J.; Chen, Y.; Zhang, Y.; Chen, L.; Xiong, W. The role of Ca. Cell Prolif 2021, 54 (11), e13122. DOI: 10.1111/cpr.13122. [CrossRef]
- Bartold, M.; Gronthos, S.; Haynes, D.; Ivanovski, S. Mesenchymal stem cells and biologic factors leading to bone formation. J Clin Periodontol 2019, 46 Suppl 21, 12-32. DOI: 10.1111/jcpe.13053. [CrossRef]
- Ariffin, N. S. RUNX1 as a Novel Molecular Target for Breast Cancer. Clin Breast Cancer 2022, 22 (6), 499-506. DOI: 10.1016/j.clbc.2022.04.006. [CrossRef]
- Krasnova, O.; Neganova, I. Assembling the Puzzle Pieces. Insights for in Vitro Bone Remodeling. Stem Cell Rev Rep 2023, 19 (6), 1635-1658. DOI: 10.1007/s12015-023-10558-6. [CrossRef]
- Chan, W. C. W.; Tan, Z.; To, M. K. T.; Chan, D. Regulation and Role of Transcription Factors in Osteogenesis. Int J Mol Sci 2021, 22 (11). DOI: 10.3390/ijms22115445. [CrossRef]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int J Mol Sci 2019, 20 (7). DOI: 10.3390/ijms20071694. [CrossRef]
- Teixeira, C. C.; Liu, Y.; Thant, L. M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a novel regulator of osteoblast differentiation and skeletogenesis. J Biol Chem 2010, 285 (40), 31055-31065. DOI: 10.1074/jbc.M109.079962. [CrossRef]
- Khotib, J.; Marhaeny, H. D.; Miatmoko, A.; Budiatin, A. S.; Ardianto, C.; Rahmadi, M.; Pratama, Y. A.; Tahir, M. Differentiation of osteoblasts: the links between essential transcription factors. J Biomol Struct Dyn 2023, 41 (19), 10257-10276. DOI: 10.1080/07391102.2022.2148749. [CrossRef]
- Chen, G.; Deng, C.; Li, Y. P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 2012, 8 (2), 272-288. DOI: 10.7150/ijbs.2929. [CrossRef]
- Otto, F.; Thornell, A. P.; Crompton, T.; Denzel, A.; Gilmour, K. C.; Rosewell, I. R.; Stamp, G. W.; Beddington, R. S.; Mundlos, S.; Olsen, B. R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89 (5), 765-771. DOI: 10.1016/s0092-8674(00)80259-7. [CrossRef]
- Yoshida, C. A.; Furuichi, T.; Fujita, T.; Fukuyama, R.; Kanatani, N.; Kobayashi, S.; Satake, M.; Takada, K.; Komori, T. Core-binding factor beta interacts with Runx2 and is required for skeletal development. Nat Genet 2002, 32 (4), 633-638. DOI: 10.1038/ng1015. [CrossRef]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res 2010, 339 (1), 189-195. DOI: 10.1007/s00441-009-0832-8. [CrossRef]
- Meyer, M. B.; Benkusky, N. A.; Pike, J. W. The RUNX2 cistrome in osteoblasts: characterization, down-regulation following differentiation, and relationship to gene expression. J Biol Chem 2014, 289 (23), 16016-16031. DOI: 10.1074/jbc.M114.552216. [CrossRef]
- Wu, H.; Whitfield, T. W.; Gordon, J. A.; Dobson, J. R.; Tai, P. W.; van Wijnen, A. J.; Stein, J. L.; Stein, G. S.; Lian, J. B. Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome Biol 2014, 15 (3), R52. DOI: 10.1186/gb-2014-15-3-r52. [CrossRef]
- Weng, J. J.; Su, Y. Nuclear matrix-targeting of the osteogenic factor Runx2 is essential for its recognition and activation of the alkaline phosphatase gene. Biochim Biophys Acta 2013, 1830 (3), 2839-2852. DOI: 10.1016/j.bbagen.2012.12.021. [CrossRef]
- Hojo, H.; Saito, T.; He, X.; Guo, Q.; Onodera, S.; Azuma, T.; Koebis, M.; Nakao, K.; Aiba, A.; Seki, M.; et al. Runx2 regulates chromatin accessibility to direct the osteoblast program at neonatal stages. Cell Rep 2022, 40 (10), 111315. DOI: 10.1016/j.celrep.2022.111315. [CrossRef]
- Schroeder, T. M.; Jensen, E. D.; Westendorf, J. J. Runx2: a master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Res C Embryo Today 2005, 75 (3), 213-225. DOI: 10.1002/bdrc.20043. [CrossRef]
- Franco, H. L.; Casasnovas, J.; Rodríguez-Medina, J. R.; Cadilla, C. L. Redundant or separate entities?--roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res 2011, 39 (4), 1177-1186. DOI: 10.1093/nar/gkq890. [CrossRef]
- Makowski, A. J.; Uppuganti, S.; Wadeer, S. A.; Whitehead, J. M.; Rowland, B. J.; Granke, M.; Mahadevan-Jansen, A.; Yang, X.; Nyman, J. S. The loss of activating transcription factor 4 (ATF4) reduces bone toughness and fracture toughness. Bone 2014, 62, 1-9. DOI: 10.1016/j.bone.2014.01.021. [CrossRef]
- Xiao, G.; Jiang, D.; Ge, C.; Zhao, Z.; Lai, Y.; Boules, H.; Phimphilai, M.; Yang, X.; Karsenty, G.; Franceschi, R. T. Cooperative interactions between activating transcription factor 4 and Runx2/Cbfa1 stimulate osteoblast-specific osteocalcin gene expression. J Biol Chem 2005, 280 (35), 30689-30696. DOI: 10.1074/jbc.M500750200. [CrossRef]
- Yang, X.; Matsuda, K.; Bialek, P.; Jacquot, S.; Masuoka, H. C.; Schinke, T.; Li, L.; Brancorsini, S.; Sassone-Corsi, P.; Townes, T. M.; et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 2004, 117 (3), 387-398. DOI: 10.1016/s0092-8674(04)00344-7. [CrossRef]
- Gomathi, K.; Akshaya, N.; Srinaath, N.; Moorthi, A.; Selvamurugan, N. Regulation of Runx2 by post-translational modifications in osteoblast differentiation. Life Sci 2020, 245, 117389. DOI: 10.1016/j.lfs.2020.117389. [CrossRef]
- Hojo, H. Emerging RUNX2-Mediated Gene Regulatory Mechanisms Consisting of Multi-Layered Regulatory Networks in Skeletal Development. Int J Mol Sci 2023, 24 (3). DOI: 10.3390/ijms24032979. [CrossRef]
- Li, J.; Liu, C.; Li, Y.; Zheng, Q.; Xu, Y.; Liu, B.; Sun, W.; Ji, S.; Liu, M.; Zhang, J.; et al. TMCO1-mediated Ca. Nat Commun 2019, 10 (1), 1589. DOI: 10.1038/s41467-019-09653-5. [CrossRef]
- Adhami, M.; Ghori-Javed, F. Y.; Chen, H.; Gutierrez, S. E.; Javed, A. Runx2 regulates the gene network associated with insulin signaling and energy homeostasis. Cells Tissues Organs 2011, 194 (2-4), 232-237. DOI: 10.1159/000324763. [CrossRef]
- Wei, J.; Shimazu, J.; Makinistoglu, M. P.; Maurizi, A.; Kajimura, D.; Zong, H.; Takarada, T.; Lezaki, T.; Pessin, J. E.; Hinoi, E.; et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 2015, 161 (7), 1576-1591. DOI: 10.1016/j.cell.2015.05.029. [CrossRef]
- Liu, L.; Xie, H.; Zhao, S.; Huang, X. The GLUT1-mTORC1 axis affects odontogenic differentiation of human dental pulp stem cells. Tissue Cell 2022, 76, 101766. DOI: 10.1016/j.tice.2022.101766. [CrossRef]
- Masuda, T.; Kubota, H.; Sakuramoto, N.; Hada, A.; Horiuchi, A.; Sasaki, A.; Takeda, K.; Takeda, M.; Matsuo, H.; Sugiyama, H.; et al. RUNX-NFAT Axis As a Novel Therapeutic Target for AML and T Cell Immunity. Blood 2020, 136 (Supplement 1), 25-26. DOI: 10.1182/blood-2020-143458 (accessed 2/6/2025). [CrossRef]
- Liu, Q.; Li, M.; Wang, S.; Xiao, Z.; Xiong, Y.; Wang, G. Recent Advances of Osterix Transcription Factor in Osteoblast Differentiation and Bone Formation. Front Cell Dev Biol 2020, 8, 601224. DOI: 10.3389/fcell.2020.601224. [CrossRef]
- Koga, T.; Matsui, Y.; Asagiri, M.; Kodama, T.; de Crombrugghe, B.; Nakashima, K.; Takayanagi, H. NFAT and Osterix cooperatively regulate bone formation. Nat Med 2005, 11 (8), 880-885. DOI: 10.1038/nm1270. [CrossRef]
- Canalis, E.; Schilling, L.; Eller, T.; Yu, J. Nuclear factor of activated T cells 1 and 2 are required for vertebral homeostasis. J Cell Physiol 2020, 235 (11), 8520-8532. DOI: 10.1002/jcp.29696. [CrossRef]
- Amarasekara, D. S.; Kim, S.; Rho, J. Regulation of Osteoblast Differentiation by Cytokine Networks. Int J Mol Sci 2021, 22 (6). DOI: 10.3390/ijms22062851. [CrossRef]
- Chen, R.; Hao, Z.; Wang, Y.; Zhu, H.; Hu, Y.; Chen, T.; Zhang, P.; Li, J. Mesenchymal Stem Cell-Immune Cell Interaction and Related Modulations for Bone Tissue Engineering. Stem Cells Int 2022, 2022, 7153584. DOI: 10.1155/2022/7153584. [CrossRef]
- Hao, Y.; Yang, N.; Sun, M.; Yang, S.; Chen, X. The role of calcium channels in osteoporosis and their therapeutic potential. Front Endocrinol (Lausanne) 2024, 15, 1450328. DOI: 10.3389/fendo.2024.1450328. [CrossRef]
- Kito, H.; Ohya, S. Role of K. Int J Mol Sci 2021, 22 (19). DOI: 10.3390/ijms221910459. [CrossRef]
- S, C.; M, P. NFAT Signaling and Bone Homeostasis. J Hematol Thromb Dis: 2013; Vol. 1, p 102.
- Lee, H. L.; Bae, O. Y.; Baek, K. H.; Kwon, A.; Hwang, H. R.; Qadir, A. S.; Park, H. J.; Woo, K. M.; Ryoo, H. M.; Baek, J. H. High extracellular calcium-induced NFATc3 regulates the expression of receptor activator of NF-κB ligand in osteoblasts. Bone 2011, 49 (2), 242-249. DOI: 10.1016/j.bone.2011.04.006. [CrossRef]
- Hu, F.; Pan, L.; Zhang, K.; Xing, F.; Wang, X.; Lee, I.; Zhang, X.; Xu, J. Elevation of extracellular Ca2+ induces store-operated calcium entry via calcium-sensing receptors: a pathway contributes to the proliferation of osteoblasts. PLoS One 2014, 9 (9), e107217. DOI: 10.1371/journal.pone.0107217. [CrossRef]
- Zayzafoon, M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J Cell Biochem 2006, 97 (1), 56-70. DOI: 10.1002/jcb.20675. [CrossRef]
- Shaw, J. P.; Utz, P. J.; Durand, D. B.; Toole, J. J.; Emmel, E. A.; Crabtree, G. R. Identification of a putative regulator of early T cell activation genes. Science 1988, 241 (4862), 202-205. DOI: 10.1126/science.3260404. [CrossRef]
- Winslow, M. M.; Pan, M.; Starbuck, M.; Gallo, E. M.; Deng, L.; Karsenty, G.; Crabtree, G. R. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell 2006, 10 (6), 771-782. DOI: 10.1016/j.devcel.2006.04.006. [CrossRef]
- Fromigué, O.; Haÿ, E.; Barbara, A.; Marie, P. J. Essential role of nuclear factor of activated T cells (NFAT)-mediated Wnt signaling in osteoblast differentiation induced by strontium ranelate. J Biol Chem 2010, 285 (33), 25251-25258. DOI: 10.1074/jbc.M110.110502. [CrossRef]
- Choo, M. K.; Yeo, H.; Zayzafoon, M. NFATc1 mediates HDAC-dependent transcriptional repression of osteocalcin expression during osteoblast differentiation. Bone 2009, 45 (3), 579-589. DOI: 10.1016/j.bone.2009.05.009. [CrossRef]
- Chen, W.; Zhang, X.; Siu, R. K.; Chen, F.; Shen, J.; Zara, J. N.; Culiat, C. T.; Tetradis, S.; Ting, K.; Soo, C. Nfatc2 is a primary response gene of Nell-1 regulating chondrogenesis in ATDC5 cells. J Bone Miner Res 2011, 26 (6), 1230-1241. DOI: 10.1002/jbmr.314. [CrossRef]
- Chen, S.; Pan, M. NFAT Signaling and Bone Homeostasis. Journal of Hematology & o Thromboembolic Diseases: 2013; Vol. 1.
- Pan, M. G.; Xiong, Y.; Chen, F. NFAT gene family in inflammation and cancer. Curr Mol Med 2013, 13 (4), 543-554. DOI: 10.2174/1566524011313040007. [CrossRef]
- Matsuo, K.; Galson, D. L.; Zhao, C.; Peng, L.; Laplace, C.; Wang, K. Z.; Bachler, M. A.; Amano, H.; Aburatani, H.; Ishikawa, H.; et al. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J Biol Chem 2004, 279 (25), 26475-26480. DOI: 10.1074/jbc.M313973200. [CrossRef]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E. F.; Mak, T. W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med 2005, 202 (9), 1261-1269. DOI: 10.1084/jem.20051150. [CrossRef]
- Cao, X. RANKL-RANK signaling regulates osteoblast differentiation and bone formation. Bone Res 2018, 6, 35. DOI: 10.1038/s41413-018-0040-9. [CrossRef]
- Gao, Y.; Wu, X.; Terauchi, M.; Li, J. Y.; Grassi, F.; Galley, S.; Yang, X.; Weitzmann, M. N.; Pacifici, R. T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab 2008, 8 (2), 132-145. DOI: 10.1016/j.cmet.2008.07.001. [CrossRef]
- Sarode, A. Y.; Jha, M. K.; Zutshi, S.; Ghosh, S. K.; Mahor, H.; Sarma, U.; Saha, B. Residue-Specific Message Encoding in CD40-Ligand. iScience 2020, 23 (9), 101441. DOI: 10.1016/j.isci.2020.101441. [CrossRef]
- Bai, H.; Zhu, H.; Yan, Q.; Shen, X.; Lu, X.; Wang, J.; Li, J.; Chen, L. TRPV2-induced Ca. Cell Commun Signal 2018, 16 (1), 68. DOI: 10.1186/s12964-018-0280-8. [CrossRef]
- Park, H. J.; Baek, K.; Baek, J. H.; Kim, H. R. TNFα Increases RANKL Expression via PGE₂-Induced Activation of NFATc1. Int J Mol Sci 2017, 18 (3). DOI: 10.3390/ijms18030495. [CrossRef]
- Sitara, D.; Aliprantis, A. O. Transcriptional regulation of bone and joint remodeling by NFAT. Immunol Rev 2010, 233 (1), 286-300. DOI: 10.1111/j.0105-2896.2009.00849.x. [CrossRef]
- Okada, H.; Okabe, K.; Tanaka, S. Finely-Tuned Calcium Oscillations in Osteoclast Differentiation and Bone Resorption. Int J Mol Sci 2020, 22 (1). DOI: 10.3390/ijms22010180. [CrossRef]
- Parekh, A. B.; Putney, J. W. Store-operated calcium channels. Physiol Rev 2005, 85 (2), 757-810. DOI: 10.1152/physrev.00057.2003. [CrossRef]
- Ishikawa, M.; Williams, G.; Forcinito, P.; Petrie, R. J.; Saito, K.; Fukumoto, S.; Yamada, Y. Pannexin 3 ER Ca. Sci Rep 2019, 9 (1), 18759. DOI: 10.1038/s41598-019-55371-9. [CrossRef]
- Jin, X.; Zhang, Y.; Alharbi, A.; Hanbashi, A.; Alhoshani, A.; Parrington, J. Targeting Two-Pore Channels: Current Progress and Future Challenges. Trends Pharmacol Sci 2020, 41 (8), 582-594. DOI: 10.1016/j.tips.2020.06.002. [CrossRef]
- Wang, X.; Zhang, X.; Dong, X. P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151 (2), 372-383. DOI: 10.1016/j.cell.2012.08.036. [CrossRef]
- Boyman, L.; Williams, G. S.; Khananshvili, D.; Sekler, I.; Lederer, W. J. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol 2013, 59, 205-213. DOI: 10.1016/j.yjmcc.2013.03.012. [CrossRef]
- Carraro, M.; Bernardi, P. The mitochondrial permeability transition pore in Ca. Cell Calcium 2023, 111, 102719. DOI: 10.1016/j.ceca.2023.102719. [CrossRef]
- Bischof, H.; Burgstaller, S.; Waldeck-Weiermair, M.; Rauter, T.; Schinagl, M.; Ramadani-Muja, J.; Graier, W. F.; Malli, R. Live-Cell Imaging of Physiologically Relevant Metal Ions Using Genetically Encoded FRET-Based Probes. Cells 2019, 8 (5). DOI: 10.3390/cells8050492. [CrossRef]
- García-Sancho, J. The coupling of plasma membrane calcium entry to calcium uptake by endoplasmic reticulum and mitochondria. J Physiol 2014, 592 (2), 261-268. DOI: 10.1113/jphysiol.2013.255661. [CrossRef]
- Pei, D. D.; Sun, J. L.; Zhu, C. H.; Tian, F. C.; Jiao, K.; Anderson, M. R.; Yiu, C.; Huang, C.; Jin, C. X.; Bergeron, B. E.; et al. Contribution of Mitophagy to Cell-Mediated Mineralization: Revisiting a 50-Year-Old Conundrum. Adv Sci (Weinh) 2018, 5 (10), 1800873. DOI: 10.1002/advs.201800873. [CrossRef]
- Reith, E. J. The binding of calcium within the Golgi saccules of the rat odontoblast. Am J Anat 1976, 147 (3), 267-270. DOI: 10.1002/aja.1001470302. [CrossRef]
- Jiang, D.; Zhao, L.; Clapham, D. E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326 (5949), 144-147. DOI: 10.1126/science.1175145. [CrossRef]
- Báthori, G.; Csordás, G.; Garcia-Perez, C.; Davies, E.; Hajnóczky, G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J Biol Chem 2006, 281 (25), 17347-17358. DOI: 10.1074/jbc.M600906200. [CrossRef]
- de Brito, O. M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456 (7222), 605-610. DOI: 10.1038/nature07534. [CrossRef]
- Stenzinger, A.; Schreiner, D.; Koch, P.; Hofer, H. W.; Wimmer, M. Cell and molecular biology of the novel protein tyrosine-phosphatase-interacting protein 51. Int Rev Cell Mol Biol 2009, 275, 183-246. DOI: 10.1016/S1937-6448(09)75006-3. [CrossRef]
- De Vos, K. J.; Mórotz, G. M.; Stoica, R.; Tudor, E. L.; Lau, K. F.; Ackerley, S.; Warley, A.; Shaw, C. E.; Miller, C. C. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 2012, 21 (6), 1299-1311. DOI: 10.1093/hmg/ddr559. [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M. R.; Cavagna, D.; Nagy, A. I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 2006, 175 (6), 901-911. DOI: 10.1083/jcb.200608073. [CrossRef]
- Vandecaetsbeek, I.; Vangheluwe, P.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb Perspect Biol 2011, 3 (5). DOI: 10.1101/cshperspect.a004184. [CrossRef]
- Garrity, A. G.; Wang, W.; Collier, C. M.; Levey, S. A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. Elife 2016, 5. DOI: 10.7554/eLife.15887. [CrossRef]
- Lloyd-Evans, E.; Waller-Evans, H. Lysosomal Ca. Cold Spring Harb Perspect Biol 2020, 12 (6). DOI: 10.1101/cshperspect.a035311. [CrossRef]
- Zheng, S.; Wang, X.; Zhao, D.; Liu, H.; Hu, Y. Calcium homeostasis and cancer: insights from endoplasmic reticulum-centered organelle communications. Trends Cell Biol 2023, 33 (4), 312-323. DOI: 10.1016/j.tcb.2022.07.004. [CrossRef]
- Ferrucci, L.; Guerra, F.; Bucci, C.; Marzetti, E.; Picca, A. Mitochondria break free: Mitochondria-derived vesicles in aging and associated conditions. Ageing Res Rev 2024, 102, 102549. DOI: 10.1016/j.arr.2024.102549. [CrossRef]
- Iwayama, T.; Bhongsatiern, P.; Takedachi, M.; Murakami, S. Matrix Vesicle-Mediated Mineralization and Potential Applications. J Dent Res 2022, 101 (13), 1554-1562. DOI: 10.1177/00220345221103145. [CrossRef]
- Iwayama, T.; Okada, T.; Ueda, T.; Tomita, K.; Matsumoto, S.; Takedachi, M.; Wakisaka, S.; Noda, T.; Ogura, T.; Okano, T.; et al. Osteoblastic lysosome plays a central role in mineralization. Sci Adv 2019, 5 (7), eaax0672. DOI: 10.1126/sciadv.aax0672. [CrossRef]
- Boonrungsiman, S.; Gentleman, E.; Carzaniga, R.; Evans, N. D.; McComb, D. W.; Porter, A. E.; Stevens, M. M. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc Natl Acad Sci U S A 2012, 109 (35), 14170-14175. DOI: 10.1073/pnas.1208916109. [CrossRef]
- Wong, Y. C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol 2019, 29 (6), 500-513. DOI: 10.1016/j.tcb.2019.02.004. [CrossRef]
- Kirsch, T.; Harrison, G.; Golub, E. E.; Nah, H. D. The roles of annexins and types II and X collagen in matrix vesicle-mediated mineralization of growth plate cartilage. J Biol Chem 2000, 275 (45), 35577-35583. DOI: 10.1074/jbc.M005648200. [CrossRef]
- Shapiro, I. M.; Landis, W. J.; Risbud, M. V. Matrix vesicles: Are they anchored exosomes? Bone 2015, 79, 29-36. DOI: 10.1016/j.bone.2015.05.013. [CrossRef]
- Blair, H. C.; Larrouture, Q. C.; Li, Y.; Lin, H.; Beer-Stoltz, D.; Liu, L.; Tuan, R. S.; Robinson, L. J.; Schlesinger, P. H.; Nelson, D. J. Osteoblast Differentiation and Bone Matrix Formation In Vivo and In Vitro. Tissue Eng Part B Rev 2017, 23 (3), 268-280. DOI: 10.1089/ten.TEB.2016.0454. [CrossRef]
- Cui, J.; Dean, D.; Hornicek, F. J.; Chen, Z.; Duan, Z. The role of extracelluar matrix in osteosarcoma progression and metastasis. J Exp Clin Cancer Res 2020, 39 (1), 178. DOI: 10.1186/s13046-020-01685-w. [CrossRef]
- Bhadada, S. K.; Rao, S. D. Role of Phosphate in Biomineralization. Calcif Tissue Int 2021, 108 (1), 32-40. DOI: 10.1007/s00223-020-00729-9. [CrossRef]
- The Chemical Dynamics of Bone Mineral., William F. Newman and Margaret W. Newman. Chicago, The University of Chicago Press, 1958. xi + 209 pp. $5.00. Arthritis & Rheumatism 1958, 1 (5), 473-474. DOI: https://doi.org/10.1002/art.1780010510. [CrossRef]
- Yan, X.; Zhang, Q.; Ma, X.; Zhong, Y.; Tang, H.; Mai, S. The mechanism of biomineralization: Progress in mineralization from intracellular generation to extracellular deposition. Jpn Dent Sci Rev 2023, 59, 181-190. DOI: 10.1016/j.jdsr.2023.06.005. [CrossRef]
- Merametdjian, L.; Beck-Cormier, S.; Bon, N.; Couasnay, G.; Sourice, S.; Guicheux, J.; Gaucher, C.; Beck, L. Expression of Phosphate Transporters during Dental Mineralization. J Dent Res 2018, 97 (2), 209-217. DOI: 10.1177/0022034517729811. [CrossRef]
- Adams, C. S.; Mansfield, K.; Perlot, R. L.; Shapiro, I. M. Matrix regulation of skeletal cell apoptosis. Role of calcium and phosphate ions. J Biol Chem 2001, 276 (23), 20316-20322. DOI: 10.1074/jbc.M006492200. [CrossRef]
- Hu, P.; Lacruz, R. S.; Smith, C. E.; Smith, S. M.; Kurtz, I.; Paine, M. L. Expression of the sodium/calcium/potassium exchanger, NCKX4, in ameloblasts. Cells Tissues Organs 2012, 196 (6), 501-509. DOI: 10.1159/000337493. [CrossRef]
- Parry, D. A.; Poulter, J. A.; Logan, C. V.; Brookes, S. J.; Jafri, H.; Ferguson, C. H.; Anwari, B. M.; Rashid, Y.; Zhao, H.; Johnson, C. A.; et al. Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta. Am J Hum Genet 2013, 92 (2), 307-312. DOI: 10.1016/j.ajhg.2013.01.003. [CrossRef]
- Goretti Penido, M.; Alon, U. S. Phosphate homeostasis and its role in bone health. Pediatr Nephrol 2012, 27 (11), 2039-2048. DOI: 10.1007/s00467-012-2175-z. [CrossRef]
- Landis, W. J.; Glimcher, M. J. Electron optical and analytical observations of rat growth plate cartilage prepared by ultracryomicrotomy: the failure to detect a mineral phase in matrix vesicles and the identification of heterodispersed particles as the initial solid phase of calcium phosphate deposited in the extracellular matrix. J Ultrastruct Res 1982, 78 (3), 227-268. DOI: 10.1016/s0022-5320(82)80001-4. [CrossRef]
- Appleton, J.; Lyon, R.; Swindin, K. J.; Chesters, J. Ultrastructure and energy-dispersive x-ray microanalysis of cartilage after rapid freezing, low temperature freeze drying, and embedding in Spurr's resin. J Histochem Cytochem 1985, 33 (10), 1073-1079. DOI: 10.1177/33.10.3900194. [CrossRef]
- Hsu, H. H.; Morris, D. C.; Davis, L.; Moylan, P.; Anderson, C. H. In vitro Ca deposition by rat matrix vesicles: is the membrane association of alkaline phosphatase essential for matrix vesicle-mediated calcium deposition? Int J Biochem 1993, 25 (12), 1737-1742. DOI: 10.1016/0020-711x(88)90301-1. [CrossRef]
- Mackie, E. J. Osteoblasts: novel roles in orchestration of skeletal architecture. Int J Biochem Cell Biol 2003, 35 (9), 1301-1305. DOI: 10.1016/s1357-2725(03)00107-9. [CrossRef]
- Mahamid, J.; Aichmayer, B.; Shimoni, E.; Ziblat, R.; Li, C.; Siegel, S.; Paris, O.; Fratzl, P.; Weiner, S.; Addadi, L. Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. Proc Natl Acad Sci U S A 2010, 107 (14), 6316-6321. DOI: 10.1073/pnas.0914218107. [CrossRef]
- Boraldi, F.; Lofaro, F. D.; Quaglino, D. Apoptosis in the Extraosseous Calcification Process. Cells 2021, 10 (1). DOI: 10.3390/cells10010131. [CrossRef]
- Brookes, P. S.; Yoon, Y.; Robotham, J. L.; Anders, M. W.; Sheu, S. S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004, 287 (4), C817-833. DOI: 10.1152/ajpcell.00139.2004. [CrossRef]
- Rohde, M.; Mayer, H. Exocytotic process as a novel model for mineralization by osteoblasts in vitro and in vivo determined by electron microscopic analysis. Calcif Tissue Int 2007, 80 (5), 323-336. DOI: 10.1007/s00223-007-9013-5. [CrossRef]
- Tang, C.; Wei, Y.; Gu, L.; Zhang, Q.; Li, M.; Yuan, G.; He, Y.; Huang, L.; Liu, Y.; Zhang, Y. Biomineral Precursor Formation Is Initiated by Transporting Calcium and Phosphorus Clusters from the Endoplasmic Reticulum to Mitochondria. Adv Sci (Weinh) 2020, 7 (8), 1902536. DOI: 10.1002/advs.201902536. [CrossRef]
- Akisaka, T.; Kawaguchi, H.; Subita, G. P.; Shigenaga, Y.; Gay, C. V. Ultrastructure of matrix vesicles in chick growth plate as revealed by quick freezing and freeze substitution. Calcif Tissue Int 1988, 42 (6), 383-393. DOI: 10.1007/BF02556357. [CrossRef]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int J Mol Sci 2020, 21 (7). DOI: 10.3390/ijms21072576. [CrossRef]
- GREENAWALT, J. W.; ROSSI, C. S.; LEHNINGER, A. L. EFFECT OF ACTIVE ACCUMULATION OF CALCIUM AND PHOSPHATE IONS ON THE STRUCTURE OF RAT LIVER MITOCHONDRIA. J Cell Biol 1964, 23 (1), 21-38. DOI: 10.1083/jcb.23.1.21. [CrossRef]
- Anderson, H. C. Electron microscopic studies of induced cartilage development and calcification. J Cell Biol 1967, 35 (1), 81-101. DOI: 10.1083/jcb.35.1.81. [CrossRef]
- Bonucci, E. Fine structure of early cartilage calcification. J Ultrastruct Res 1967, 20 (1), 33-50. DOI: 10.1016/s0022-5320(67)80034-0. [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D. W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct Target Ther 2023, 8 (1), 304. DOI: 10.1038/s41392-023-01503-7. [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H. J.; Landi, F.; Bucci, C.; Marzetti, E. Mitochondrial-Derived Vesicles: The Good, the Bad, and the Ugly. Int J Mol Sci 2023, 24 (18). DOI: 10.3390/ijms241813835. [CrossRef]
- Docampo, R. The origin and evolution of the acidocalcisome and its interactions with other organelles. Mol Biochem Parasitol 2016, 209 (1-2), 3-9. DOI: 10.1016/j.molbiopara.2015.10.003. [CrossRef]
- Liou, W.; Geuze, H. J.; Geelen, M. J.; Slot, J. W. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J Cell Biol 1997, 136 (1), 61-70. DOI: 10.1083/jcb.136.1.61. [CrossRef]
- Mahamid, J.; Sharir, A.; Gur, D.; Zelzer, E.; Addadi, L.; Weiner, S. Bone mineralization proceeds through intracellular calcium phosphate loaded vesicles: a cryo-electron microscopy study. J Struct Biol 2011, 174 (3), 527-535. DOI: 10.1016/j.jsb.2011.03.014. [CrossRef]
- Ponpuak, M.; Mandell, M. A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr Opin Cell Biol 2015, 35, 106-116. DOI: 10.1016/j.ceb.2015.04.016. [CrossRef]
- Somogyi, E.; Petersson, U.; Hultenby, K.; Wendel, M. Calreticulin--an endoplasmic reticulum protein with calcium-binding activity is also found in the extracellular matrix. Matrix Biol 2003, 22 (2), 179-191. DOI: 10.1016/s0945-053x(02)00117-8. [CrossRef]
- Petersson, U.; Somogyi, E.; Reinholt, F. P.; Karlsson, T.; Sugars, R. V.; Wendel, M. Nucleobindin is produced by bone cells and secreted into the osteoid, with a potential role as a modulator of matrix maturation. Bone 2004, 34 (6), 949-960. DOI: 10.1016/j.bone.2004.01.019. [CrossRef]
- Lavoie, C.; Meerloo, T.; Lin, P.; Farquhar, M. G. Calnuc, an EF-hand Ca(2+)-binding protein, is stored and processed in the Golgi and secreted by the constitutive-like pathway in AtT20 cells. Mol Endocrinol 2002, 16 (11), 2462-2474. DOI: 10.1210/me.2002-0079. [CrossRef]
- Bonucci, E. Fine structure and histochemistry of "calcifying globules" in epiphyseal cartilage. Z Zellforsch Mikrosk Anat 1970, 103 (2), 192-217. DOI: 10.1007/BF00337312. [CrossRef]
- Ali, S. Y.; Sajdera, S. W.; Anderson, H. C. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc Natl Acad Sci U S A 1970, 67 (3), 1513-1520. DOI: 10.1073/pnas.67.3.1513. [CrossRef]
- Anderson, H. C.; Matsuzawa, T.; Sajdera, S. W.; Ali, S. Y. Membranous particles in calcifying cartilage matrix. Trans N Y Acad Sci 1970, 32 (5), 619-630. DOI: 10.1111/j.2164-0947.1970.tb02737.x. [CrossRef]
- Anderson, H. C. Molecular biology of matrix vesicles. Clin Orthop Relat Res 1995, (314), 266-280.
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab family of small GTPases: an updated view on their regulation and functions. FEBS J 2021, 288 (1), 36-55. DOI: 10.1111/febs.15453. [CrossRef]
- Han, J.; Pluhackova, K.; Böckmann, R. A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front Physiol 2017, 8, 5. DOI: 10.3389/fphys.2017.00005. [CrossRef]
- Rosenthal, A. K.; Gohr, C. M.; Ninomiya, J.; Wakim, B. T. Proteomic analysis of articular cartilage vesicles from normal and osteoarthritic cartilage. Arthritis Rheum 2011, 63 (2), 401-411. DOI: 10.1002/art.30120. [CrossRef]
- Goettsch, C.; Hutcheson, J. D.; Aikawa, M.; Iwata, H.; Pham, T.; Nykjaer, A.; Kjolby, M.; Rogers, M.; Michel, T.; Shibasaki, M.; et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest 2016, 126 (4), 1323-1336. DOI: 10.1172/JCI80851. [CrossRef]
- Escrevente, C.; Bento-Lopes, L.; Ramalho, J. S.; Barral, D. C. Rab11 is required for lysosome exocytosis through the interaction with Rab3a, Sec15 and GRAB. J Cell Sci 2021, 134 (11). DOI: 10.1242/jcs.246694. [CrossRef]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol 2016, 131 (4), 505-523. DOI: 10.1007/s00401-015-1528-7. [CrossRef]
- Komarova, S. V.; Ataullakhanov, F. I.; Globus, R. K. Bioenergetics and mitochondrial transmembrane potential during differentiation of cultured osteoblasts. Am J Physiol Cell Physiol 2000, 279 (4), C1220-1229. DOI: 10.1152/ajpcell.2000.279.4.C1220. [CrossRef]
- Vidavsky, N.; Masic, A.; Schertel, A.; Weiner, S.; Addadi, L. Mineral-bearing vesicle transport in sea urchin embryos. J Struct Biol 2015, 192 (3), 358-365. DOI: 10.1016/j.jsb.2015.09.017. [CrossRef]
- Marsh, M. E. Biomineralization in coccolithophores. Gravit Space Biol Bull 1999, 12 (2), 5-14.
- Hasegawa, T. Ultrastructure and biological function of matrix vesicles in bone mineralization. Histochem Cell Biol 2018, 149 (4), 289-304. DOI: 10.1007/s00418-018-1646-0. [CrossRef]
- Wuthier, R. E.; Lipscomb, G. F. Matrix vesicles: structure, composition, formation and function in calcification. Front Biosci (Landmark Ed) 2011, 16 (8), 2812-2902. DOI: 10.2741/3887. [CrossRef]
- Skelton, A. M.; Cohen, D. J.; Boyan, B. D.; Schwartz, Z. Osteoblast-Derived Matrix Vesicles Exhibit Exosomal Traits and a Unique Subset of microRNA: Their Caveolae-Dependent Endocytosis Results in Reduced Osteogenic Differentiation. Int J Mol Sci 2023, 24 (16). DOI: 10.3390/ijms241612770. [CrossRef]
- De Rooij, J. F.; Heughebaert, J. C.; Nancollas, G. H. A ph study of calcium phosphate seeded precipitation. Journal of Colloid and Interface Science 1984, 100 (2), 350-358. DOI: https://doi.org/10.1016/0021-9797(84)90440-5. [CrossRef]
- Casey, J. R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 2010, 11 (1), 50-61. DOI: 10.1038/nrm2820. [CrossRef]
- Kerschnitzki, M.; Akiva, A.; Ben Shoham, A.; Asscher, Y.; Wagermaier, W.; Fratzl, P.; Addadi, L.; Weiner, S. Bone mineralization pathways during the rapid growth of embryonic chicken long bones. J Struct Biol 2016, 195 (1), 82-92. DOI: 10.1016/j.jsb.2016.04.011. [CrossRef]
- Liu, F.; Fang, F.; Yuan, H.; Yang, D.; Chen, Y.; Williams, L.; Goldstein, S. A.; Krebsbach, P. H.; Guan, J. L. Suppression of autophagy by FIP200 deletion leads to osteopenia in mice through the inhibition of osteoblast terminal differentiation. J Bone Miner Res 2013, 28 (11), 2414-2430. DOI: 10.1002/jbmr.1971. [CrossRef]
- Nollet, M.; Santucci-Darmanin, S.; Breuil, V.; Al-Sahlanee, R.; Cros, C.; Topi, M.; Momier, D.; Samson, M.; Pagnotta, S.; Cailleteau, L.; et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 2014, 10 (11), 1965-1977. DOI: 10.4161/auto.36182. [CrossRef]
- Yan, J.; Shen, M.; Sui, B.; Lu, W.; Han, X.; Wan, Q.; Liu, Y.; Kang, J.; Qin, W.; Zhang, Z.; et al. Autophagic LC3. Sci Adv 2022, 8 (19), eabn1556. DOI: 10.1126/sciadv.abn1556. [CrossRef]
- Hale, J. E.; Wuthier, R. E. The mechanism of matrix vesicle formation. Studies on the composition of chondrocyte microvilli and on the effects of microfilament-perturbing agents on cellular vesiculation. J Biol Chem 1987, 262 (4), 1916-1925.
Figure 1.
Transcriptional regulation during osteoblastogenesis and osteogenesis. Runx2 is the first transcription factor required for the differentiation of MSCs into osteoblasts. It induces the commitment of the osteoblast lineage, the differentiation of MSCs into early osteoblasts promoting osteoid formation and triggers the expression of osteogenic genes. Osx further regulate at later steps the differentiation of MSCs into mature osteoblasts. The TNAP, Col1 and BSP osteogenic genes are upregulated in early osteoblasts, while SPARC, OCN and OPN are upregulated in mature osteoblasts.
Figure 1.
Transcriptional regulation during osteoblastogenesis and osteogenesis. Runx2 is the first transcription factor required for the differentiation of MSCs into osteoblasts. It induces the commitment of the osteoblast lineage, the differentiation of MSCs into early osteoblasts promoting osteoid formation and triggers the expression of osteogenic genes. Osx further regulate at later steps the differentiation of MSCs into mature osteoblasts. The TNAP, Col1 and BSP osteogenic genes are upregulated in early osteoblasts, while SPARC, OCN and OPN are upregulated in mature osteoblasts.
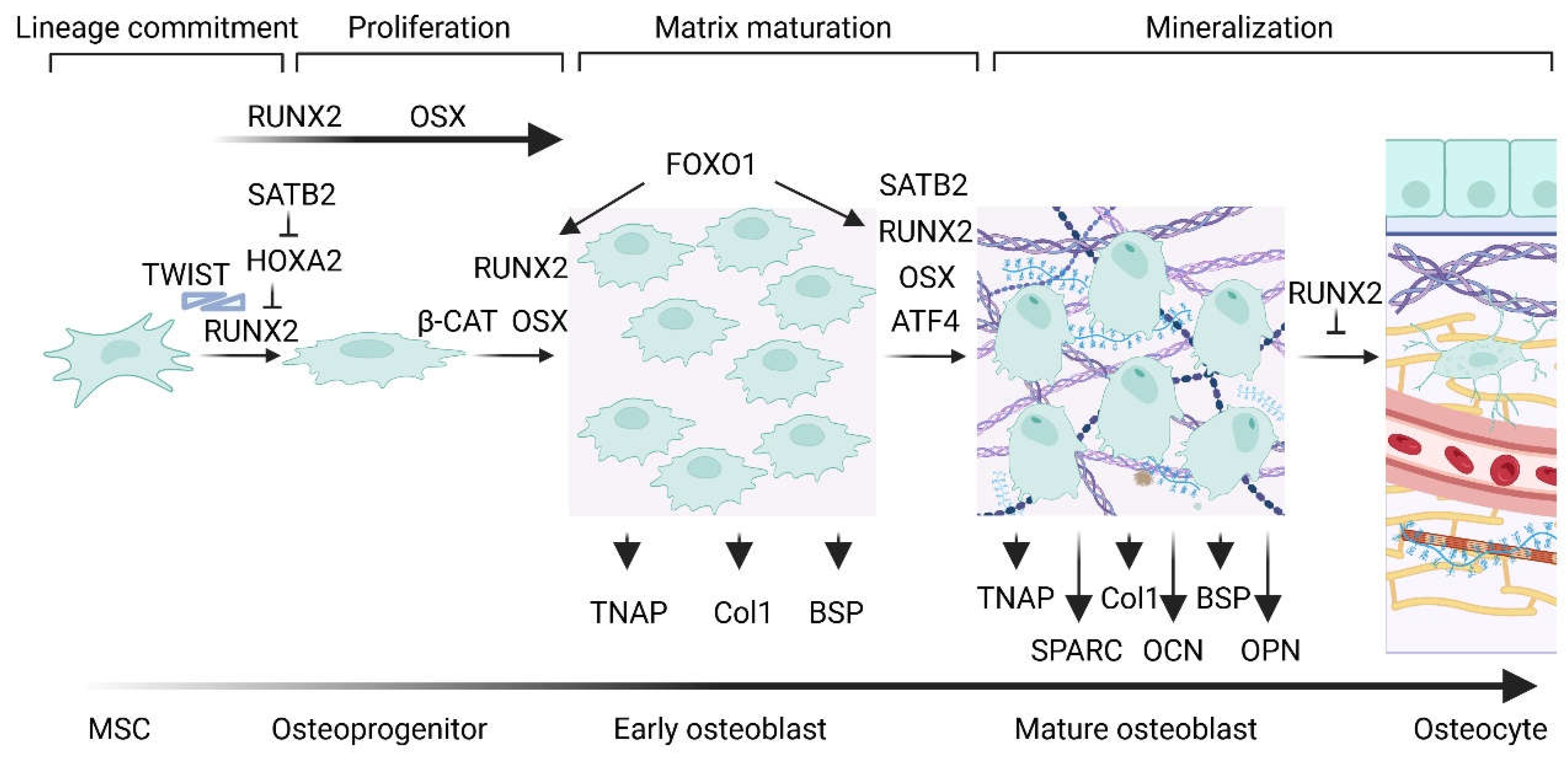
Figure 2.
Schematic view of membrane channels, Ca2+ and inorganic phosphate (Pi) ion trafficking and amorphous calcium phosphate (ACP)/HA formation. Calcium transporters located at the PM, ER, mitochondria and lysosomes (Lys) work coordinately to regulate specific functions which include calcium sensing, calcium homeostasis, signal transduction and organelle calcium loading. The Ca2+/CaN/NFAT signaling pathway promotes the process of osteoblast differentiation mediated by the nuclear translocation of NFAT.
Figure 2.
Schematic view of membrane channels, Ca2+ and inorganic phosphate (Pi) ion trafficking and amorphous calcium phosphate (ACP)/HA formation. Calcium transporters located at the PM, ER, mitochondria and lysosomes (Lys) work coordinately to regulate specific functions which include calcium sensing, calcium homeostasis, signal transduction and organelle calcium loading. The Ca2+/CaN/NFAT signaling pathway promotes the process of osteoblast differentiation mediated by the nuclear translocation of NFAT.
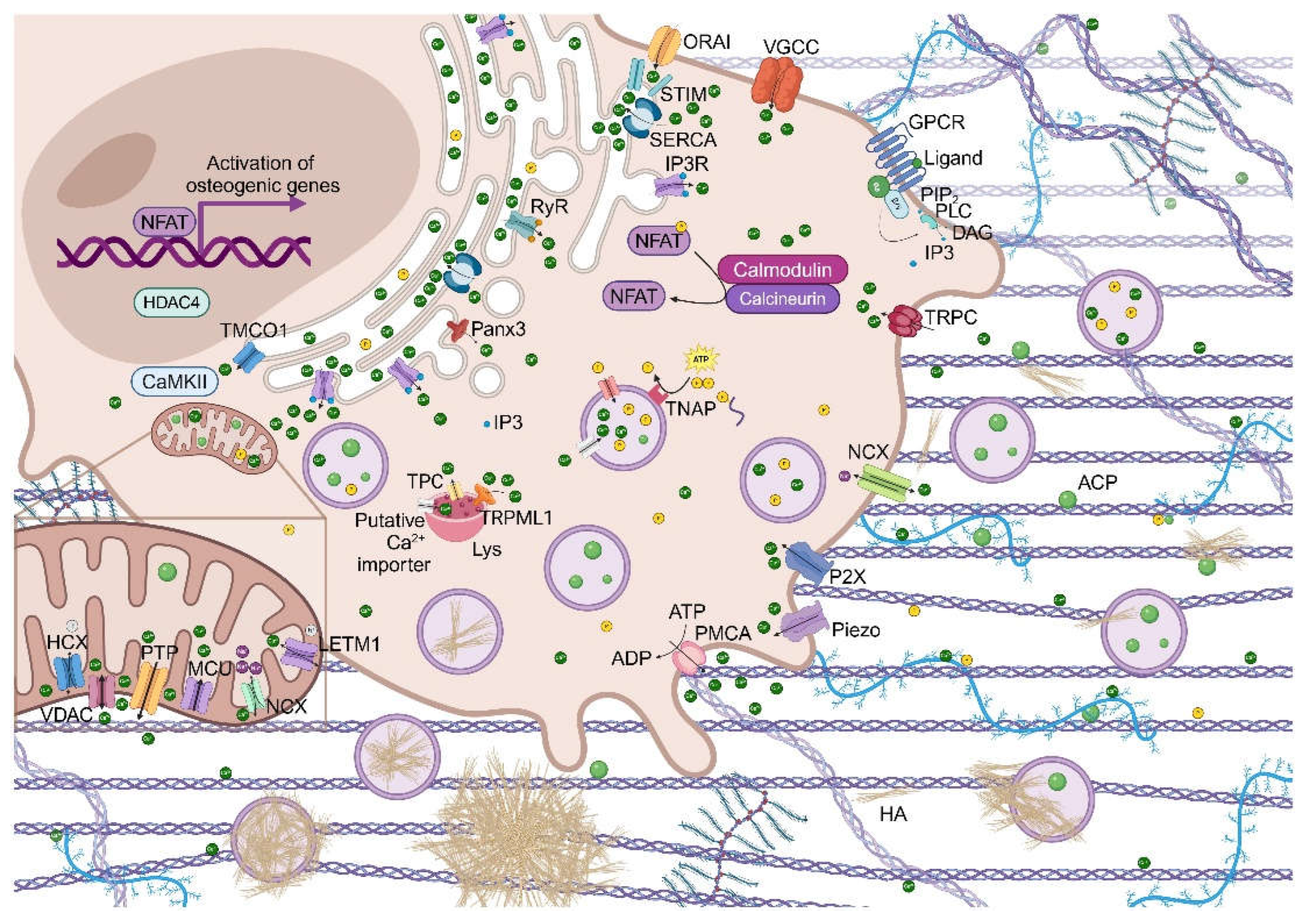
Figure 3.
Coordination of Ca2+ homeostasis in ER and other intracellular organelles, such as mitochondria and lysosomes. Ca2+ ions transfer can occur from ER, which can interact both with mitochondria and lysosomes, or from mitochondria to lysososomes/vesicles through direct interaction or MDVs (brown square). Green squares: a) Ca2+ is released from the ER through IP3R and it is internalized by mitochondria through VDAC on the outer membrane and MCU on the internal mitochondrial membrane. The interaction between the IP3R and VDAC is mediated by GRP75. The interaction between the two organelles is mediated by MFN2 on the ER and MFN1 or MFN2 on the mitochondria and by VAP8 on the ER and PTPIP51 on the mitochondria. b) Ca2+ is released from the ER through IP3R and it is internalized by the lysosome through Ca2+ importers. c) Mitochondria releases MDVs, small vesicles that shuttle mitochondrial constituents to other organelles, which contain Ca2+ ions. MDVs deliver Ca2+ ions to lysosomes. d) Inter-organelle transfer of Ca2+ from mitochondria to lysosomes is mediated by Rab7. Untethering of the two organelles occurs due to Fis1 located on the mitochondria which recruits TBC1D15, a Rab7 GAP.
Figure 3.
Coordination of Ca2+ homeostasis in ER and other intracellular organelles, such as mitochondria and lysosomes. Ca2+ ions transfer can occur from ER, which can interact both with mitochondria and lysosomes, or from mitochondria to lysososomes/vesicles through direct interaction or MDVs (brown square). Green squares: a) Ca2+ is released from the ER through IP3R and it is internalized by mitochondria through VDAC on the outer membrane and MCU on the internal mitochondrial membrane. The interaction between the IP3R and VDAC is mediated by GRP75. The interaction between the two organelles is mediated by MFN2 on the ER and MFN1 or MFN2 on the mitochondria and by VAP8 on the ER and PTPIP51 on the mitochondria. b) Ca2+ is released from the ER through IP3R and it is internalized by the lysosome through Ca2+ importers. c) Mitochondria releases MDVs, small vesicles that shuttle mitochondrial constituents to other organelles, which contain Ca2+ ions. MDVs deliver Ca2+ ions to lysosomes. d) Inter-organelle transfer of Ca2+ from mitochondria to lysosomes is mediated by Rab7. Untethering of the two organelles occurs due to Fis1 located on the mitochondria which recruits TBC1D15, a Rab7 GAP.
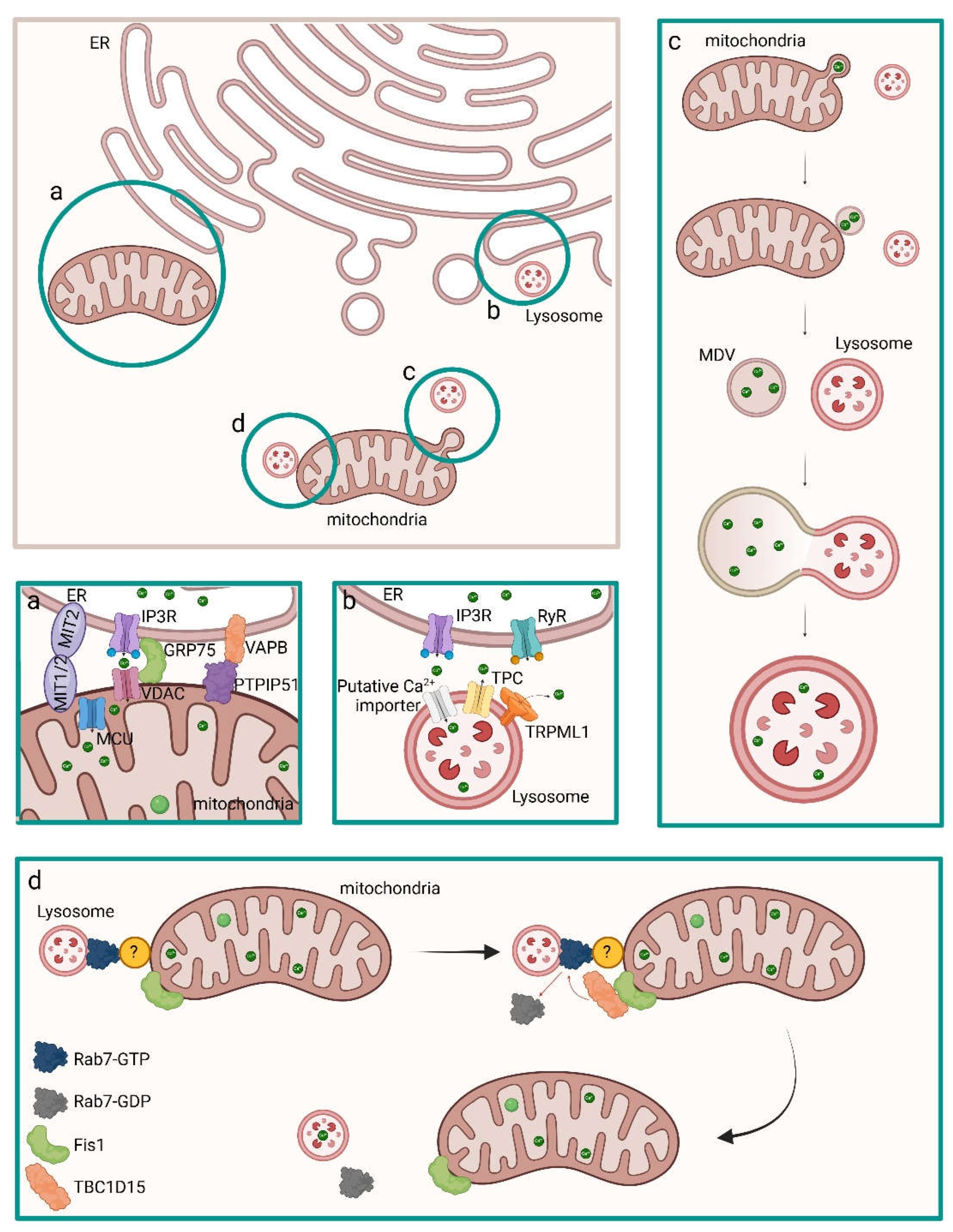
Figure 4.
The mineralization process is regulated by enzymes and transporters. Pi is produced by TNAP on the PM while channels are responsible for Ca2+ and Pi intracellular homeostasis (left). MVs are characterized by the presence of CD9, CD63 and CD81 tetraspannins on the membrane. SMPD3 (sphingomyelin phosphodiesterase 3) catalyzes the hydrolysis of sphingomyelin (SM) to form ceramide and phosphocholine (PC), whereas TNAP, ENPP1 and PHOSPHO1 are responsible for the formation of Pi. PHOSPHO1 is a phosphatase with high activity toward phosphoethanolamine (PEA), PC and pyrophosphate (PP). ANX, Pit1 and Ca2+ importers regulate the transport of ions inside the MVs (center). The mineralization process takes place in MVs which are released by osteoblasts. Ca2+ and Pi are internalized and forms ACP which is transformed into OCP and subsequently into HA. This penetrates through the vesicle membrane thanks to phospholipases leading to the break down of MV membrane. The following nucleation of HA crystals and spreading into the ECM is regulated by extracellular Ca2+, Pi, H+, BSP-1, OCN and OPN.
Figure 4.
The mineralization process is regulated by enzymes and transporters. Pi is produced by TNAP on the PM while channels are responsible for Ca2+ and Pi intracellular homeostasis (left). MVs are characterized by the presence of CD9, CD63 and CD81 tetraspannins on the membrane. SMPD3 (sphingomyelin phosphodiesterase 3) catalyzes the hydrolysis of sphingomyelin (SM) to form ceramide and phosphocholine (PC), whereas TNAP, ENPP1 and PHOSPHO1 are responsible for the formation of Pi. PHOSPHO1 is a phosphatase with high activity toward phosphoethanolamine (PEA), PC and pyrophosphate (PP). ANX, Pit1 and Ca2+ importers regulate the transport of ions inside the MVs (center). The mineralization process takes place in MVs which are released by osteoblasts. Ca2+ and Pi are internalized and forms ACP which is transformed into OCP and subsequently into HA. This penetrates through the vesicle membrane thanks to phospholipases leading to the break down of MV membrane. The following nucleation of HA crystals and spreading into the ECM is regulated by extracellular Ca2+, Pi, H+, BSP-1, OCN and OPN.
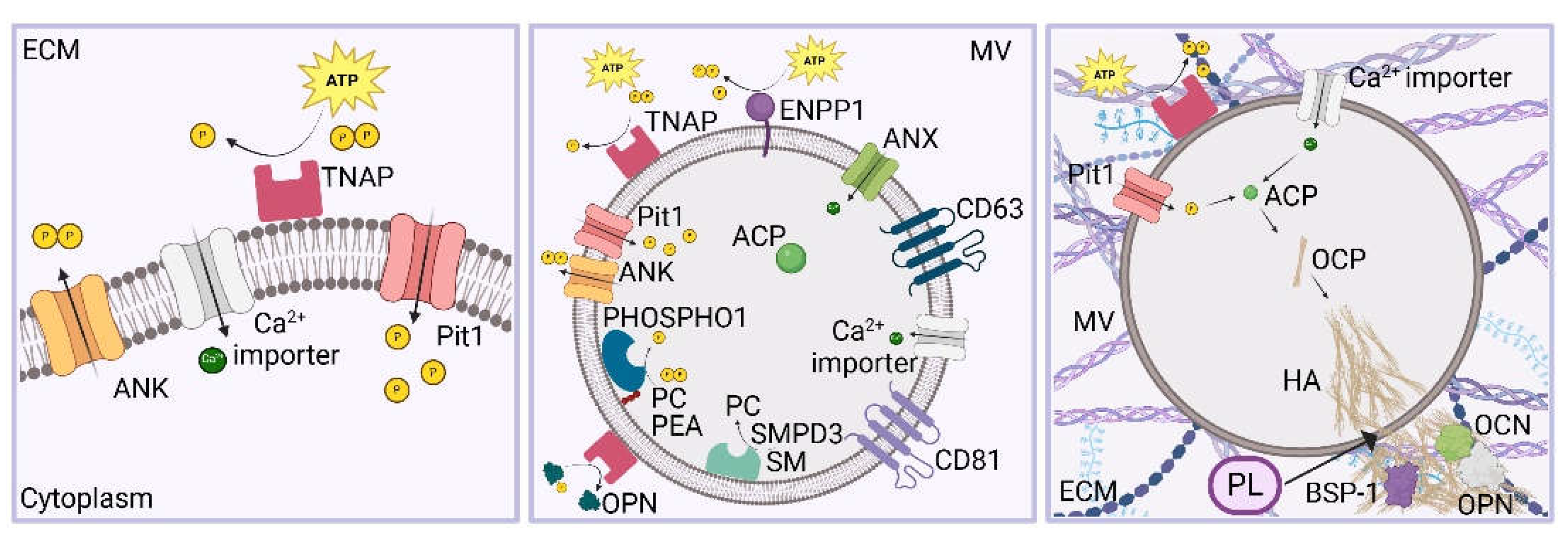
Figure 5.
Biogenesis of MVs and intracellular biomineralization process. Ca2+, Pi and ACP are produced in mitochondria and transferred to intracellular MVs through mitophagy (a), MDVs (b) or interaction with lysosomes (c). MVs are generated also through the transfer of Ca2+ and Pi ions from ER to lysosomes (d). It remains unclear whether autolysosomes and lysosomes undergo further steps in order to acquire the typical characteristics of MVs.
Figure 5.
Biogenesis of MVs and intracellular biomineralization process. Ca2+, Pi and ACP are produced in mitochondria and transferred to intracellular MVs through mitophagy (a), MDVs (b) or interaction with lysosomes (c). MVs are generated also through the transfer of Ca2+ and Pi ions from ER to lysosomes (d). It remains unclear whether autolysosomes and lysosomes undergo further steps in order to acquire the typical characteristics of MVs.
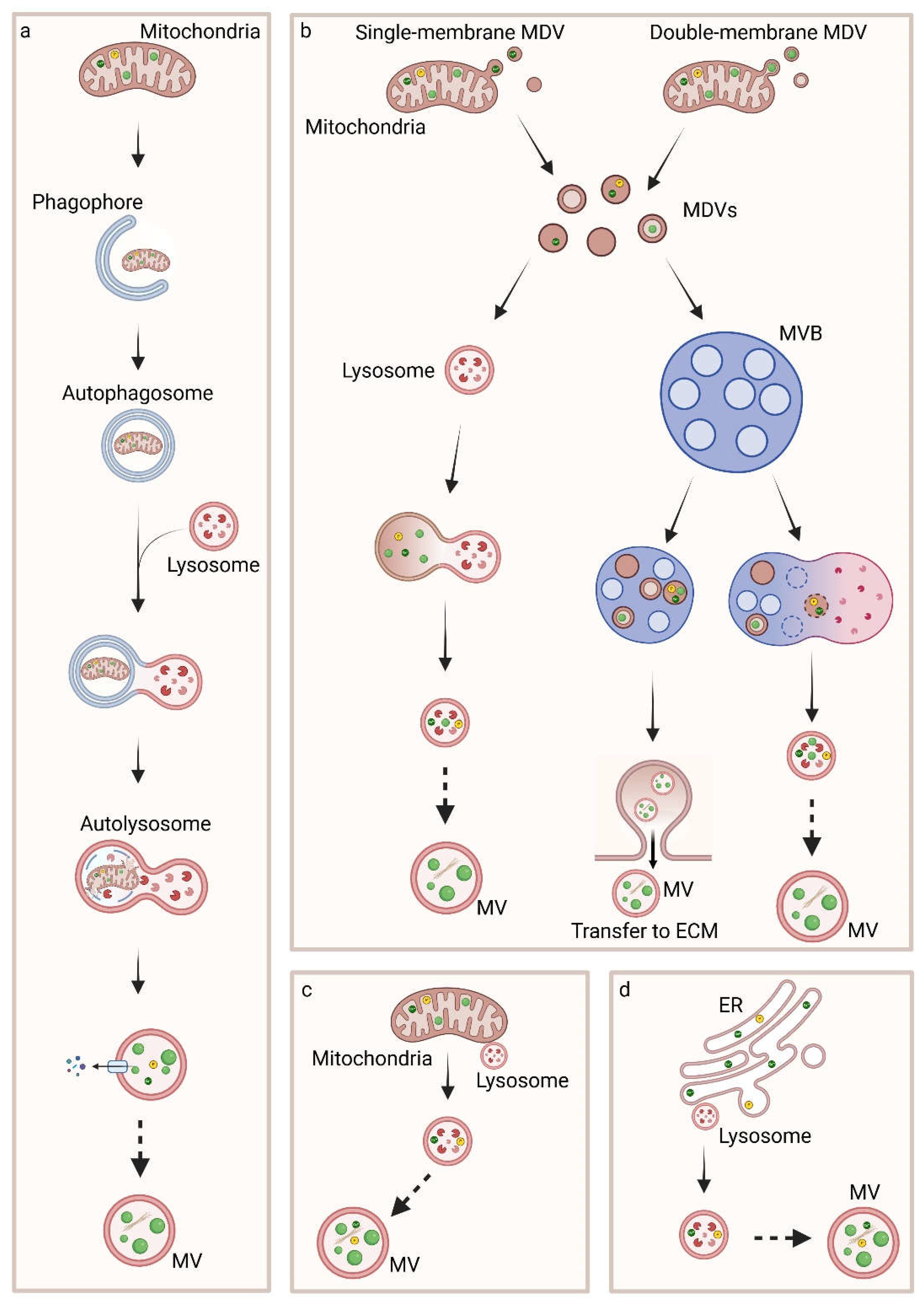
Figure 6.
Release of Ca2+ and Pi ions, ACP and MVs. MVBs (a) or MVs (b) interact with the PM and lead to the release of Ca2+ and Pi ions, ACP and HA in the ECM. Moreover, free Ca2+ and Pi ions and ACP can be released in the ECM as microsomes (c) or through exocytosis (d).
Figure 6.
Release of Ca2+ and Pi ions, ACP and MVs. MVBs (a) or MVs (b) interact with the PM and lead to the release of Ca2+ and Pi ions, ACP and HA in the ECM. Moreover, free Ca2+ and Pi ions and ACP can be released in the ECM as microsomes (c) or through exocytosis (d).
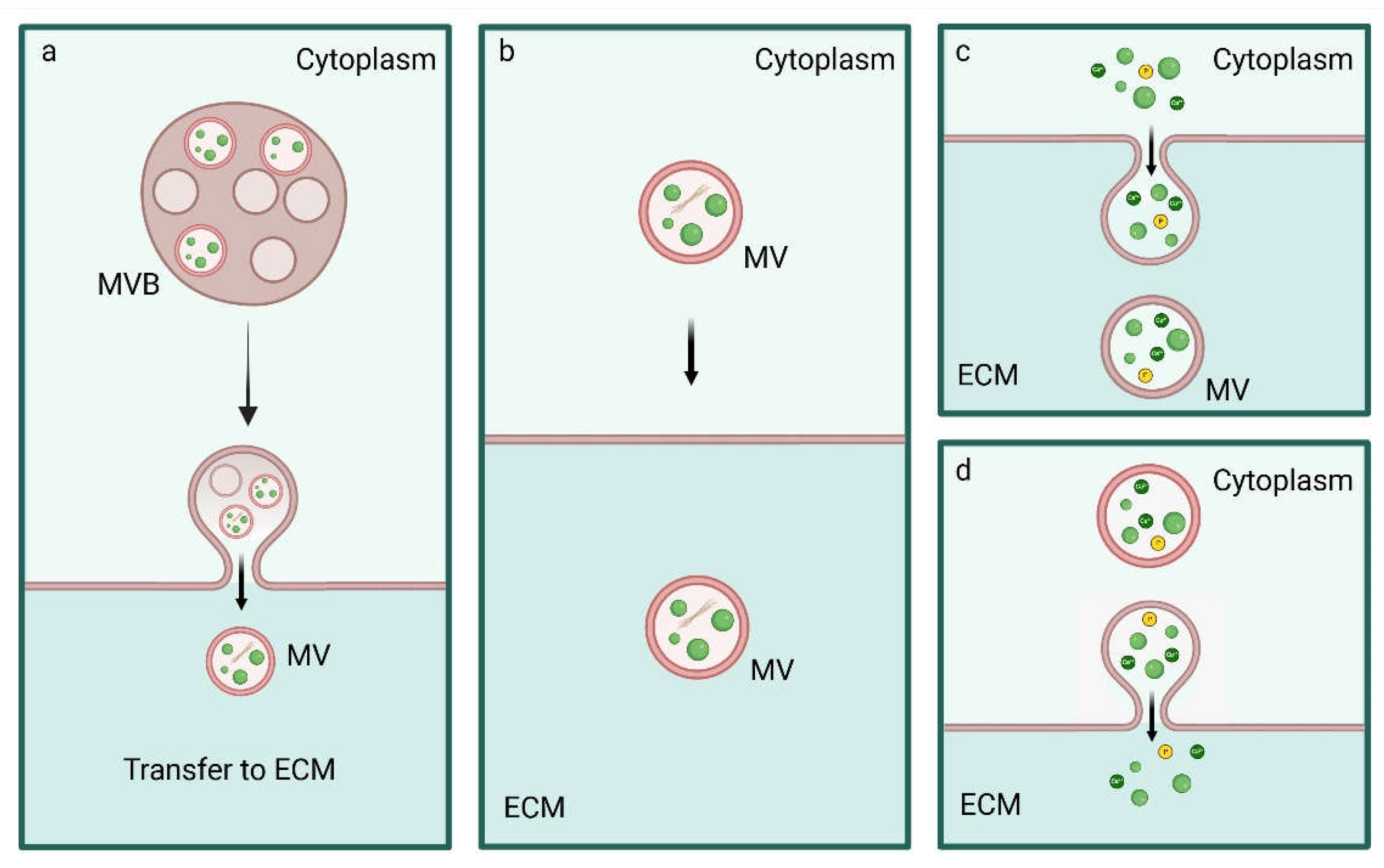
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



