Submitted:
31 March 2025
Posted:
01 April 2025
You are already at the latest version
Abstract
A new series of 6-arylaminoflavones was synthesized via the Buchwald-Hartwig cross-coupling reaction, aiming to functionalize the flavone core efficiently. Reaction optimization revealed that Pd₂(dba)₃/XantPhos with Cs₂CO₃ in toluene provided the best yields, with isolated yields ranging from 8% to 95%, depending on the arylamine structure. Steric hindrance and electron-withdrawing groups at the arylamine ring negatively impacted the reaction. Cytotoxicity assays indicated that specific substituents at the 6-position influenced biological activity, with trifluoromethyl- (13c) and chlorine-containing (13g) derivatives showing higher selectivity towards prostate cancer cells (PC3). These findings provide insights into the structure-activity relationship of 6-arylaminoflavones while contributing to the development of synthetic methodologies for functionalized flavones.

Keywords:
Aminoflavones
; Buchwald-Hartwig amination
; Drug design
; Cytotoxicity assays
1. Introduction
Flavonoids represent a diverse group of heterocyclic molecules featuring oxygen in their structure. They are categorized into six main subclasses and are commonly found in plant-based foods and beverages including fruits, vegetables, cocoa, cereals, tea, and wine [1]. Among these, flavones consist of two aromatic rings (A and B) and a heterocyclic pyranone ring (C), forming the framework of 2-phenyl-4H-chromen-4-one (Figure 1) [2,3]. In particular, aminoflavones are flavone derivatives that have been modified to include one or more amino groups (-NH2) attached to various positions on the flavone core structure. These amino groups can also be further substituted, enhancing the diversity and potential biological activity of the compounds. We also observed that often the 3’and 4’ positions are mono or di-substituted by heteroatoms such as fluoride or methoxy groups [4].
The 5-aminoflavones 1-2 (Figure 1) showed antiproliferative activity against human breast cancer (MCF-7), with potencies of 7.2 and 1.2 nM, respectively. Compound 2 also suppressed tumor growth in mice, highlighting its potential as a chemotherapeutic agent [5,6]. Compound 3 (AFP464), a synthetic lysyl prodrug of 2, has demonstrated efficacy against breast and renal cancer cell lines [7,8]. Compound 3 has progressed to phase II clinical trials against solid tumors, however its development was discontinued due to pulmonary toxicity [9]. The 3-aminoflavones 4-5 (Figure 1), have demonstrated considerable cytotoxicity against the murine lymphocytic leukemia cell line L1210, with IC50 values of 22 µM and 10 µM, respectively [10]. The 2’-aminoflavone 6 (PD98059) (Figure 1), is a selective MEK1 (IC50 = 2-7 µM) and MEK2 (IC50 = 50 µM) inhibitor that blocks the MAPK/ERK pathway [11], with antiproliferative effects at 50 µM in breast cancer (MCF-7 and MDA-MB-231) [12]. The 6-benzylaminoflavone 7 (Figure 1) exhibited anticancer activity against MCF-7 (IC50 = 9.4 µM), and inhibited topoisomerase II (IC50 = 12 µM) [13]. Despite significant advancements in the synthesis of aminoflavones, the selective functionalization of positions 6 and 7, are particularly underexplored relative to other positions. This highlights an opportunity to explore the 6-position with different amines and observe the impact on tumor cell proliferation.
In this context, the present study aims to synthesize 6-arylaminoflavone derivatives and evaluate their cytotoxic activities against different cancer cell lines. The introduction of various arylamino groups at the 6-position of flavones was explored to understand the influence of these modifications on the biological activity of the compounds. This approach is expected not only to reveal new candidates for anticancer agents but also to provide valuable insights into the structure-activity relationship (SAR) of these flavone derivatives.
2. Material & Methods
2.1. General Information
Reactions were monitored by thin layer chromatography (TLC) with silica gel 60 GF254 (Merck®), 0.20 mm thick. The revelations were carried out by inspection in a chamber with emission of ultraviolet light (UV) with a length of λ = 254 and 366 nm, with subsequent use of a developing chamber with iodine and vanillin solution. Silica gel 60 (230-400 mesh) was used for column chromatography. Solvents were purified by distillation, except for HPLC quality products. Anhydrous solvents were conditioned in the presence of a 3Å molecular sieves. Reactions sensitive to humidity and air were carried out in tubes under an inert atmosphere (N2). FTIR data were obtained using an Agilent Technologies Cary 630 and melting points were measured using a Buchi B-545 melting point apparatus. Structural analyses of 1H (300 MHz), 13C (75 MHz), and 19F (282 MHz) Nuclear Magnetic Resonance (NMR) spectra were performed in a Brucker, Advanced DPX-300 model. Chemical shifts (δ) were expressed in parts per million (ppm) being referenced when compared to DMSO-d6 at 2.55 ppm (1H) and 39.52 ppm (13C), and at 77.16 ppm (13C) when using CDCl3; the coupling constant (J) is expressed in Hertz (Hz). The purity of molecules was analyzed by High-Performance Liquid Chromatography (HPLC), using Shimadzu Proeminence® chromatograph, with a Zorbax SB-Phenyl® analytical column (5μm, 150x4.6 mm). Samples were diluted to 0.25 mg/mL. Detection was conducted by ultraviolet (UV) light at 264 nm. The injection volume was 7 μL. Deionized water and acetonitrile (ACN) was used as the eluent system with 0.1% trifluoroacetic acid (TFA), with a flow rate of 1 mL/minute. The chromatographic run started with 5% of the ACN/TFA mixture, after 10 minutes it became 100% of the mixture. After 15 minutes, the ACN/TFA concentration was reduced to 50%, in 20 minutes it dropped to 25%, ending in 25 minutes with 0%. The measurement of the mass/charge ratios of the 6-arylaminoflavones was obtained using Bruker Daltonics® microOTOF QII / ESI-TOF. They were analyzed using high-performance liquid chromatography (Prominence liquid chromatography, Shimadzu®) coupled to a Q-TOF mass spectrometer, a compact model from Bruker Daltonics GmbH®, with an electrospray ionization interface. The compounds were dissolved in DMSO, separated using Luna C18 column (3 μm, 100 Å, 150×3 mm, Phenomenex® brand) using 0.1% formic acid in deionized water (A) and 0.1% of formic acid in acetonitrile (B) with a flow rate of 0.4 mL/min. The gradient program was: 30% B at the start, 100% B between 3-6 min., returning to 30% B in 6.01 min., and finishing the race with a total time of 11 min. The injection volume was 20 μL, and the column temperature was maintained at 40 °C. The Q-TOF/MS instrument was operated in positive mode using the following parameters: ionic gas source temperature (N2) of 200 ºC; nebulizer pressure of 45 psi; and capillary voltage of 2800 V. The mass spectrometer was operated in MS scan mode. Sodium formate was used as the internal mass calibration.
2.2. Synthetic Methods
2.2.1. General Procedure A: Claisen-Schmidt Condensation
A solution of 5′-bromo-2′-hydroxyacetophenone 8 (1 mmol, 1 eq.), aldehydes 9, 14 or 15 (1.1 mmol, 1.1 eq.), KOH (2 mmol, 2 eq.) in ethanol (6 mL) was heated at reflux for 2.5 h. The reaction mixture was quenched with water and extracted with AcOEt (3x 30 mL). The combined organic phases were collected, dried over anhydrous MgSO4, filtered, and concentred under reduced pressure. The crude product was purified by recrystallization with methanol.
(E)-1-(5-bromo-2-hydroxyphenyl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one (10). The chalcone was obtained as a dark yellow solid (286 mg, 85 %). mp. 127.2-127.9 °C. 1H NMR (300 MHz, CDCl3) δ 12.87 (s, 1H, OH), 8.02 (s, 1H), 7.92 (d, J = 15 Hz, 1H, C=C), 7.57 (d, J = 9 Hz, 1H), 7.41 (d, J = 15 Hz, 1H, C=C), 7.30 (d, J = 9 Hz, 1H), 7.19 (s, 1H), 6.94 (d, J = 9 Hz, 2H), 4.00 (s, 3H, O-CH3) 3.97 (s, 3H, O-CH3). 13C NMR (75 MHz, CDCl3) δ 192.7 (C=O), 162.6, 152.3, 149.6, 147.0, 138.8, 131.8, 127.5, 124.1, 121.5, 120.7, 117.2, 111.4, 110.7, 110.4, 56.2 (2C; O-CH3).
(E)-1-(5-bromo-2-hydroxyphenyl)-3-phenylprop-2-en-1-one (16). The chalcone was obtained as a yellow solid (151 mg, 53 %). mp. 103.1-104.2 °C. 1H NMR (300 MHz, CDCl3) δ 12.75 (s, 1H, OH), 8.03 (s, 1H), 7.97 (d, J = 15 Hz, 1H, C=C), 7.70 (s, 1H), 7.58 (m, 1H), 7.52 (d, J = 15 Hz, 1H, C=C), 6.96 (d, J = 9 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 192.9 (C=O), 162.7, 146.7, 139.1, 134.5, 132.0, 131.4, 129.2, 129.0, 127.7, 121.4, 120.8, 119.6, 110.6.
(E)-1-(5-bromo-2-hydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (17). The chalcone was obtained as a yellow solid (166 mg, 50 %). mp. 112.0-112.6 °C. 1H NMR (300 MHz, CDCl3) δ 12.88 (s, 1H, OH), 8.02 (s, 1H), 7.94 (d, J = 15 Hz, 1H, C=C), 7.67 (d, J = 9 Hz, 2H), 7.57 (d, J = 9 Hz, 1H), 7.45 (d, J = 15 Hz, 1H, C=C), 7.00-6.93 (m, 3H), 3.90 (s, 3H, O-CH3). 13C NMR (75 MHz, CDCl3) δ 192.8 (C=O), 162.6, 162.5, 146.7, 138.8, 131.9, 131.0, 127.3, 121.6, 120.7, 117.0, 114.8, 110.5, 55.6 (O-CH3).
2.2.2. General Procedure B: Oxidative Cyclization
In a round bottom flask, chalcone 10, 16 or 17 (1 mmol), iodine I2 (10 mol %), and anhydrous DMSO (5 ml) were added. The reaction was kept under constant stirring and in an inert atmosphere for 1.5 h at a temperature between 110-130 °C. After the completion of the reaction, the mixture was extracted with saturated Na2S2O3 and EtOAc (3x 30 mL), dried over anhydrous MgSO4, filtered, and concentred under reduced pressure. The crude product was purified by flash column chromatography (eluent: ethyl acetate/hexane 8:2).
6-Bromo-2-(3,4-dimethoxyphenyl)-4H-chromen-4-one (11). The flavone was obtained as a light-yellow solid (248 mg, 69 %). mp. 199.4-200.4 °C. 1H NMR (300 MHz, CDCl3) δ 8.25 (s, 1H), 7.67 (dd, J = 9 Hz, 2 Hz, 1H), 7.45 (dd, J = 9 Hz, 2 Hz, 1H), 7.37 (d, J = 9 Hz, 1H), 7.28 (s, 1H), 6.90 (d, J = 9 Hz, 1H), 6.66 (s, 1H), 3.90 (s, 3H, O-CH3), 3.89 (s, 3H, O-CH3). 13C NMR (75 MHz, CDCl3) δ 178.3 (C=O), 163.1, 152.0, 151.0, 149.3, 142.4, 141.1, 124.9, 124.5, 124.1, 122.0, 120.0, 119.1, 118.5, 111.2, 110.8, 108.8, 105.9, 56.2 (2C; O-CH3).
6-bromo-2-phenyl-4H-chromen-4-one (18). The flavone was obtained as a white solid (126 mg, 42 %). mp. 195.4-195.6 °C. 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.96 (d, J = 6 Hz, 2H), 7.83 (d, J = 9 Hz, 1H), 7.60 - 7.58 (m, 3H), 7.52 (d, J = 9 Hz, 1H), 6.90 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 177.1 (C=O), 163.9, 155.1, 136.9, 132.0, 131.5, 129.3, 128.5, 126.5, 125.4, 120.2, 118.8, 107.6.
6-Bromo-2-(4-methoxyphenyl)-4H-chromen-4-one (19). The flavone was obtained as a white solid (1 mmol, 74 %). mp. 195.4-195.6 °C. 1H NMR (300 MHz, CDCl3) δ 8.25 (d, J = 3 Hz, 1H), 7.77 (d, J = 9 Hz, 2H), 7.66 (dd, J = 9 Hz, 2.4 Hz, 1H), 7.35 (d, J = 9 Hz, 1H), 6.93 (d, J = 9 Hz, 2H), 6.64 (s, 1H), 3.81 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 177.0 (C=O), 163.8, 162.8, 155.0, 136.6, 128.5, 128.2, 125.4, 123.7, 120.0, 118.6, 114.7, 106.2, 55.7 (O-CH3).
2.2.3. General Procedure C: Buchwald-Hartwig Amination
A flame-dried pressure tube was charged with flavone 11, 18 or 19 (1 mmol), Pd₂(dba)₃ (10 mol%), XantPhos (5 mol%), Cs₂CO₃ (3 equiv.) and correspondent amine* (1.1 mmol). The tube was capped with septum, evacuated for 1 hour with vaccum, heated at approximately 40-50 ºC and then filled with nitrogen twice. Subsequently, anhydrous toluene (5 mL) was added using a syringe, and the mixture was stirred at 110 ºC for 20 h. The resulting solids were removed by vacuum filtration and the filtrate was then concentrated under reduced pressure. The crude material was purified by by flash column chromatography (eluent: hexane/dichloromethane (9:1), to 100% dichloromethane, and concluding with dichloromethane/methanol (99:1)). *Liquid amines were previous degassing and added with the solvent.
2-(3,4-dimethoxyphenyl)-6-((4-methoxyphenyl)amino)-4H-chromen-4-one (13a). The product was obtained as a light-yellow solid (147 mg, 77%). mp. 204.6-205 °C. 1H NMR (300 MHz, CDCl3) δ 7.63 (s, 1H), 7.55 (d, J = 6 Hz, 1H), 7.45 - 7.39 (m, 2H), 7.29 - 7.14 (m, 4H), 6.98 (d, J = 9 Hz, 1H), 6.90 (d, J = 6 Hz, 2H), 6.75 (s, 1H), 3.99 (s, 3H, O-CH3), 3.97 (s, 3H, O-CH3), 3.82 (s, 3H, O-CH3). 13C NMR (75 MHz, CDCl3) δ 178.3 (C=O), 163.2, 152.1, 150.5, 149.4, 124.8, 124.6, 123.0, 122.6, 120.1, 119.1, 115.0, 111.3, 109.1, 108.4, 105.7, 56.21 (O-CH3), 56.20 (O-CH3), 55.7 (O-CH3). IR (ν, cm−1): 1566 e 1544 (C=O), 2902 (Ar-CH), 3205 (N-H). HRMS (ESI-TOF) m/z calcd for C24H21NO5 404.1420 [M-H]+, found 404.1483. Analytical HPLC retention time: 12.1 min; purity: 98.2%.
2-(3,4-dimethoxyphenyl)-6-(phenylamino)-4H-chromen-4-one (13b). The product was obtained as a yellow solid (188 mg, 50%). mp. 197.2-197.5 °C. 1H NMR (300 MHz, CDCl3) δ 7.83 (s, 1H), 7.56 (d, J = 6 Hz, 1H), 7.48 (t, J = 9 Hz, 2H), 7.39 (s, 1H), 7.35 - 7.30 (m, 2H), 7.15 (d, J = 6 Hz, 2H), 7.03 - 6.98 (m, 2H), 6.74 (s, 1H), 6.10 (s, 1H), 4.00 (s, 3H, O-CH3), 3.98 (s, 3H, O-CH3). 13C NMR (75 MHz, CDCl3) δ 178.3 (C=O), 163.1, 152, 151, 149.3, 142.4, 141.1, 129.7, 124.9, 124.5, 124.1, 122, 120, 119.1, 118.5, 111.2, 110.8, 108.8, 105.9, 56.2 (O-CH3). IR (ν, cm−1): 1564 e 1546 (C=O), 3019 (Ar-CH), 3203 (N-H). HRMS (ESI-TOF) m/z calcd for C23H19NO4 374.13143 [M-H]+, found 374.1393. Analytical HPLC retention time: 12.2 min; purity: 99.1%.
2-(3,4-dimethoxyphenyl)-6-((4-(trifluoromethyl)phenyl)amino)-4H-chromen-4-one (13c). The product was obtained as a yellow solid (203 mg, 46%). mp. 221.4-221.9 °C. 1H NMR (300 MHz, CDCl3) δ 7.84 (s, 1H), 7.50 - 7.42 (m, 5H), 7.32 (s, 1H), 7.19 (s, 1H), 7.04 (d, J = 9 Hz, 2H), 6.92 (d, J = 9 Hz, 1H), 6.72 (s, 1H), 3.91 (s, 3H, O-CH3), 3.90 (s, 3H, O-CH3). 13C NMR (75 MHz, CDCl3) δ 178 (C=O), 163.8, 152.5, 152.1, 149.6, 146.2, 139.3, 127 (q, J = 7,5 Hz, C-F), 126.1, 124.9, 124.3, 120.3, 119.5, 116.1 (2C), 113.9, 111.4, 109.1, 106.0, 56.3 (2C; O-CH3). 19F NMR (282 MHz, CDCl3) δ - 60.6. IR (ν, cm−1): 1559 e 1546 (C=O), 3024 (Ar-CH), 3173 (N-H). HRMS (ESI-TOF) m/z calcd for C24H18F3NO4 442.1188 [M-H]+, found 442.1264. Analytical HPLC retention time: 12.7 min; purity: 99.1%.
2-(3,4-dimethoxyphenyl)-6-((2-(trifluoromethyl)phenyl)amino)-4H-chromen-4-one (13d). The product was obtained as a yellow solid (22 mg, 10%). mp. 214.8-215.2 °C. 1H NMR (300 MHz, CDCl3) δ 7.79 (s, 1H), 7.55-7.46 (m, 3H), 7.36 - 7.28 (m, 4H), 6.99 - 6.90 (m, 2H), 6.77 (s, 1H), 3.91 (s, 3H, O-CH3), 3.89 (s, 3H, O-CH3). 13C NMR (75 MHz, CDCl3) δ 177.9 (C=O), 163.9, 152.5, 152.0, 149.5, 141.2, 140.0, 133.2, 127.2 (q, J = 7.5 Hz, C-F), 126.2, 124.7, 124.3, 121.4, 120.4, 119.5, 119.3, 119.1, 118.9, 113.5, 111.4, 109.1, 105.8, 56.3 (2C; O-CH3). 19F NMR (282 MHz, CDCl3) δ - 60.6. IR (ν, cm−1): 1564 e 1547 (C=O), 2969 (Ar-CH), 3183 (N-H). HRMS (ESI-TOF) m/z calcd for C24H18F3NO4 442.1188 [M-H]+, found 442.1261. Analytical HPLC retention time: 12.6 min; purity: 98.8%.
2-(3,4-dimethoxyphenyl)-6-((4-fluorophenyl)amino)-4H-chromen-4-one (13e). The product was obtained as a yellow solid (50 mg, 13 %). mp. 190.7-191.6 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.41 (s, 1H), 7.65 (d, J = 6 Hz, 2H), 7.55 (d, J = 9 Hz, 2H), 7.41 (d, J = 6 Hz, 1H), 7.17 - 7.09 (m, 5H), 6.93 (s, 1H), 3.89 (s, 3H, O-CH3), 3.84 (s, 3H, O-CH3). 13C NMR (75 MHz, DMSO-d6) δ 176.7 (C=O), 162.1, 157.0 (d, J = 237.3 Hz, C-F), 151.7, 149.5, 149.0, 141.9, 138.9, 138.9, 124.0, 123.6, 123.2, 120.0 (d, J = 7.9 Hz, C-F), 119.5 (d, J = 9.6 Hz, C-F), 115.9 (d, J = 22.4 Hz, C-F), 111.7, 109.3, 106.6, 104.9, 55.8 (O-CH3), 55.6 (O-CH3). 19F NMR (282 MHz, DMSO-d6) δ -120.5. IR (ν, cm−1): 1566 e 1534 (C=O), 3020 (Ar-CH), 3192 (N-H). HRMS (ESI-TOF) m/z calcd for C23H18FNO4 392.1220 [M-H]+, found 392.1279. Analytical HPLC retention time: 12.3 min; purity: 98.1%.
2-(3,4-dimethoxyphenyl)-6-((2-fluorophenyl)amino)-4H-chromen-4-one (13f). The product was obtained as a dark brown solid (196 mg, 50%). mp.: 172.9-173.5 °C. 1H NMR (300 MHz, CDCl3) δ 7.88 (s, 1H), 7.58 - 7.51 (m, 2H), 7.44 - 7.39 (m, 2H), 7.16 - 6.93 (m, 5H), 6.75 (s, 1H), 6.05 (s, 1H), 4.00 (s, 3H, O-CH3), 3.98 (s, 3H, O-CH3). 13C NMR (75 MHz, CDCl3) δ 178.2 (C=O), 163.2, 153.5 (d, J = 241.8 Hz, C-F), 152.1, 151.4, 149.3, 140, 130.8 (d, J = 11.1 Hz, C-F), 124.8, 124.7, 124.7, 124.4, 121.9 (d, J = 7.3 Hz, C-F), 120.0, 119.3, 118.2, 118.2, 115.8 (d, J = 19.2 Hz, C-F), 111.4 (d, J = 29.8 Hz, C-F), 108.8, 106.0, 56.2 (2C; O-CH3). 19F NMR (282 MHz, CDCl3) δ - 129.6. IR (ν, cm−1): 1583 e 1551 (C=O), 2935 (Ar-CH), 3164 (N-H). HRMS (ESI-TOF) m/z calcd for C23H18FNO4 392.1220 [M-H]+, found 392.1282. Analytical HPLC retention time: 12.2 min.; purity: 98.9%.
6-((4-chlorophenyl)amino)-2-(3,4-dimethoxyphenyl)-4H-chromen-4-one (13g). The product was obtained as a yellow solid (163 mg, 39%). mp. 201.3-201.8 °C. 1H NMR (300 MHz, CDCl3) δ 7.80 (s, 1H), 7.58 - 7.50 (m, 2H), 7.40 (m, 2H), 7.27 (d, J = 9 Hz, 2H), 7.07 (d, J = 6 Hz, 2H), 7.00 (d, J = 9 Hz, 1H), 6.74 (s, 1H), 6.09 (s, 1H), 4.01 (s, 3H, O-CH3), 3.99 (s, 3H, O-CH3). 13C NMR (75 MHz, CDCl3) δ 178.2 (C=O), 163.3, 152.1, 151.2, 149.4, 141.2, 140.7, 129.6 (2C), 126.6, 124.9, 124.4, 124.3, 120.1, 119.6, 119.3 (2C), 111.3, 111.2, 108.8, 105.9, 56.2 (O-CH3). IR (ν, cm−1): 1574 e 1553 (C=O), 3015 (Ar-CH), 3181 (N-H). HRMS (ESI-TOF) m/z calcd for C23H18ClNO4 408.09245 [M-H]+, found 408.1022. Analytical HPLC retention time: 12.7 min; purity: 99.7%.
2-(3,4-dimethoxyphenyl)-6-(p-tolylamino)-4H-chromen-4-one (13h). The product was obtained as a pale-yellow solid (210 mg, 95%). mp. 187.1-187.4 °C. 1H NMR (300 MHz, CDCl3) δ 7.63 (s, 1H), 7.41 (d, J = 9 Hz, 1H), 7.30-7.26 (m, 2H), 7.02 - 6.94 (m, 5H), 6.85 (d, J = 9 Hz, 1H), 6.60 (s, 1H), 3.87 (s, 3H, O-CH3), 3.85 (s, 3H, O-CH3), 2.21 (s, 3H, Ar-CH3). 13C NMR (75 MHz, CDCl3) δ 178.2 (C=O), 163.0, 152.0, 150.6, 149.3, 142.0, 139.7, 131.8, 130.1 (2C), 124.7, 124.5, 123.4, 119.9, 119.5 (2C), 118.9, 111.2, 109.6, 108.9, 105.7, 56.11 (O-CH3), 56.10 (O-CH3), 20.8 (Ar-CH3). IR (ν, cm−1): 1564 e 1544 (C=O), 2987 (Ar-CH), 3211 (N-H). HRMS (ESI-TOF) m/z calcd for C24H21NO4 388.14708 [M-H]+, found 388.1520. Analytical HPLC retention time: 12.5 min; purity: 98.4%.
6-((4-methoxyphenyl)amino)-2-phenyl-4H-chromen-4-one (20a) The product was obtained as an orange solid (40 mg, 23%). mp.166.9-167.4 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.20 (s, 1H), 8.07 (m, 2H), 7.65 - 7.58 (m, 4H), 7.43 (s, 1H), 7.38-7.35 (m, 1H), 7.12 (d, J = 9 Hz, 2H), 6.95-6.92 (m, 3H), 3.75 (s, 3H, O-CH3). 13C NMR (75 MHz, DMSO-d6) δ 176.9 (C=O), 161.9, 154.6, 149.0, 143.5, 135.2, 131.5, 131.4 (2C), 129.0 (2C), 126.1, 124.1, 122.6, 121.6 (2C), 119.4, 114.7 (2C), 105.9, 104.9, 55.2 (O-CH3). IR (ν, cm−1): 1568 e 1553 (C=O), 3023 (Ar-CH), 3179 (N-H). HRMS (ESI-TOF) m/z calcd for C22H17NO3 343.12086 [M-H]+, found 344.1280. Analytical HPLC retention time: 12.5 min; purity: 99.7%.
2-phenyl-6-(phenylamino)-4H-chromen-4-one (20b). The product was obtained as a yellow solid (120 mg, 55%). mp. 193.1-193.9 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.00 (s, 1H), 8.10 (m, 2H), 7.78 - 7.74 (m, 2H), 7.60 - 7.58 (m, 7H), 7.23 (d, J = 6 Hz, 2H), 6.99 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 176.9 (C=O), 162.4, 150.8, 146.8, 139.6, 131.8, 131.3, 129.2 (2C), 126.74 (2C), 126.70 (2C), 126.3, 125.9, 124.11, 119.9, 115.5 (2C), 110.8, 106.3. IR (ν, cm−1): 1566 e 1540 (C=O), 3019 (Ar-CH), 3175 (N-H). HRMS (ESI-TOF) m/z calcd for C21H15NO2 314.11029 [M-H]+, found 314.1170. Analytical HPLC retention time: 12.5 min; purity: 99.1%.
2-phenyl-6-((4-(trifluoromethyl)phenyl)amino)-4H-chromen-4-one(20c). The product was obtained as a golden yellow solid (32 mg, 8.4%). mp. 241.3-241.7 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.00 (s, 1H), 8.10 (m, 2H), 7.77- 7.75 (m, 2H), 7.60 – 758 (m, 6H), 7.23 (d, J = 6 Hz, 2H), 7.00 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 176.7 (C=O), 162.3, 150.7, 146.7, 139.5, 131.7, 131.2, 129.1 (2C), 126.6 (q, J = 7.5 Hz, C-F), 126.2 (2C), 125.8, 124.0, 123.0, 122.9, 119.8, 119.7, 119.3, 115.4 (2C), 110.7, 106.2. 19F NMR (282 MHz, DMSO-d6) δ -58.2. IR (ν, cm−1): 1559 e 1544 (C=O), 3021 (Ar-CH), 3166 (N-H). HRMS (ESI-TOF) m/z calcd for C22H14F3NO2 382.09766 [M-H]+, found 382.1058. Analytical HPLC retention time: 12.9 min; purity: 98.1%.
2-(4-methoxyphenyl)-6-((4-methoxyphenyl)amino)-4H-chromen-4-one (21a). The product was obtained as a yellow solid (152 mg, 41%). mp. 196.4-196.8 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.79 (m, 2H), 7.53 (s, 1H), 7.34 (d, J = 9 Hz, 1H), 7.19 (s, 1H), 7.05 - 7.03 (m, 1H), 6.94 (d, J = 9 Hz, 2H), 6.81 (d, J = 6 Hz, 2H), 6.65 (s, 1H), 3.81 (s, 3H, O-CH3), 3.73 (s, 3H, O-CH3). 13C NMR (75 MHz, DMSO-d6) δ 178.3 (C=O), 163.2, 162.4, 156.1, 150.5, 143.2, 135.1, 128.1 (2C), 124.9, 124.5, 123.0 (2C), 122.6, 119.1, 115.1 (2C), 114.6 (2C), 108.5, 105.5, 55.7 (O-CH3), 55.6 (O-CH3). IR (ν, cm−1): 1562 e 1542 (C=O), 3017 (Ar-CH), 3183 (N-H). HRMS (ESI-TOF) m/z calcd for C23H19NO4 374.13143 [M-H]+, found 374.1361. Analytical HPLC retention time: 12.4 min; purity: 99.6%.
2-(4-methoxyphenyl)-6-(phenylamino)-4H-chromen-4-one (21b). The product was obtained as a yellow solid (165 mg, 69%). mp. 176.7-177.1 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.47 (s, 1H), 8.03 (d, J = 9 Hz, 2H), 7.67 - 7.62 (m, 3H), 7.48 (d, J = 9 Hz, 1H), 7.30 (t, J = 9 Hz, 1H), 7.16 - 7.10 (m, 4H), 6.92 (t, J = 6 Hz, 1H), 6.86 (s, 1H), 3.86 (s, 3H, O-CH3). 13C NMR (75 MHz, DMSO-d6) δ 176.7 (C=O), 162.2, 161.9, 149.6, 142.6, 141.4, 129.3 (2C), 128.0 (2C), 124.0, 123.8, 123.5, 120.7, 119.4, 117.6 (2C), 114.5 (2C), 107.5, 104.6, 55.5 (O-CH3). IR (ν, cm−1): 1560 e 1546 (C=O), 3033 (Ar-CH), 3175 (N-H). HRMS (ESI-TOF) m/z calcd for C22H17NO3 344.12086 [M-H]+, found 344.1276. Analytical HPLC retention time: 12.6 min; purity: 96.8%.
2-(4-methoxyphenyl)-6-((4-(trifluoromethyl)phenyl)amino)-4H-chromen-4-one (21c). The product was obtained as a yellow solid (25 mg, 0.7 mmol, 9%). mp. 200.3-201.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.95 (s, 1H), 8.00 (d, J = 9 Hz, 1H), 7.72 - 7.67 (m, 2H), 7.56 (m, 3H), 7.21 (d, J = 6 Hz, 2H), 7.08 (d, J = 9 Hz, 2H), 6.86 (s, 1H), 3.83 (s, 3H, O-CH3). 13C NMR (75 MHz, DMSO-d6) δ 176.5 (C=O), 162.3, 162, 150.6, 146.8, 139.2, 128.0 (2C), 126.6 (d, J = 7.5 Hz, C-F), 125.5, 124, 123.3, 123, 119.6, 119.2, 115.2 (2C), 114.4 (2C), 110.9, 104.8, 55.4 (O-CH3). 19F NMR (282 MHz, DMSO-d6) δ -58.2. IR (ν, cm−1): 1553 e 1542 (C=O), 3091 (Ar-CH), 3192 (N-H). HRMS (ESI-TOF) m/z calcd for C23H16F3NO3 412.10823 [M-H]+, found 412.1147. Analytical HPLC retention time: 13.9 min; purity: 95.4%.
2.3. Cytotoxicity Assays
To assess the cytotoxic effects of the 6-arylaminoflavones, a viability assay was conducted using four different human cell lines: HEK293T (human embryonic kidney), PC3 (prostate adenocarcinoma), A172 (glioblastoma), and MDA-MB-231 (breast adenocarcinoma). All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, adjusted to pH 7.2, and maintained at 37 °C in a 5% CO2 atmosphere. Cells were seeded in 96-well plates at a density of 1 × 10⁵ cells per well in 100 µL of culture medium. Untreated control cells received only medium and solvent (0.1% DMSO as the final concentration). The cells were incubated with compounds (100 µM) for 48 hours. Then, 10 µL of a freshly prepared MTT ([3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]) solution (5 mg/mL, in PBS) was added to each well, followed by incubation for 3 hours at 37 °C. Subsequently, 100 µL of DMSO was added to each well to solubilize the formazan, and the absorbance was measured in a plate reader at 540 nm. Cell viability was calculated using the following formula: Viability (%) = [100 × (sample absorbance)/(control absorbance)]. The numerical data was obtained from one experiment performed in quadruplicates.
3. Results and Discussion
Motivated by the scarcity of 6-arylaminoflavone derivatives reported in the literature and their promising antitumor potential, we designed a novel series of 6-arylaminoflavones featuring 3,4-dimethoxy substitutions on the aromatic ring B, along with diverse substituents at the R3 position of the aniline ring. This study aimed to evaluate the influence of these substituents on biological activity, thereby contributing to the elucidation of the structure-activity relationship (SAR).
The synthetic pathway was accomplished in three key steps: the first two stages were dedicated to the construction of the flavone core, while the final step focused on the introduction of diverse arylamino groups at the 6-position of this ring. For the synthesis of flavone core, firstly, a Claisen-Schmidt condensation [14] was performed between 5′-bromo-2′-hydroxyacetophenone 8 and 3,4-dimethoxybenzaldehyde 9 (Scheme 1). This reaction was carried out under reflux conditions in ethanol, using potassium hydroxide (KOH) as the base, yielding the corresponding α,β-unsaturated chalcone 10 with an 85% yield. Subsequently, an oxidative cyclization step was conducted using dimethyl sulfoxide (DMSO) and iodine (I₂) as reagents [15]. In this step, DMSO was a dual function agent - a solvent and oxidizing agent, while iodine acted as an electrophilic species [16], enabling the efficient formation of the 6-bromoflavone intermediate 11 with a 69% yield.
The final step was dedicated to the synthesis of 6-arylaminoflavone derivatives employing a Buchwald-Hartwig coupling reaction. To optimize the reaction conditions, strategic exploratory experiments were performed using classical conditions [17], employing flavone 11 and 4-methoxyaniline 12a as model substrate. The influence of the palladium source, phosphine ligand, base, and solvent were evaluated at 110°C to assess their impact on the reaction outcome (Table 1).
The optimization of reaction conditions for the synthesis of 6-arylaminoflavone via Buchwald-Hartwig coupling reactions revealed significant differences in the efficiency of the employed conditions [18], consistent with the existing literature on these methodologies [19]. Initially, the coupling reaction was conducted using Pd₂(dba)₃ as the catalyst, XantPhos as the ligand, and toluene as the solvent, while evaluating four distinct bases (entries 1-4). In the first experiment, CsF was tested as the base, but no reaction was observed (entry 1). Upon employing a stronger base, NaOtBu, only trace amounts of the product were detected, likely due to the significant steric hindrance of the terc-Butyl group. However, the desired compound 13a was successfully obtained when alkali metal carbonates (K₂CO₃, Cs₂CO₃) were utilized (entries 3-4). The optimal result was achieved with Cs₂CO₃, which afforded a good yield (77%; entry 4). The Cs2CO3 proved to be superior to other bases tested, reinforcing the need for moderately strong bases to efficiently activate the nucleophilic amine and subsequently form the C-N bond [20]. Subsequently, an alternative ligand -DavePhos - was employed (entry 5); however, it yielded results comparable to those obtained with XantPhos. Nevertheless, XantPhos, being a bidentate ligand, likely offers enhanced chelation and steric stabilization, resulting in a marginally superior yield [21]. Other palladium catalyst, such as PdCl2(PPh3)2, were also evaluated; however, the reactions did not yield superior results compared to those obtained with Pd2(dba)3 (20%; entry 6). Finally, replacing toluene with THF as the solvent (entry 7) resulted in reduced yields of the coupling product. This decrease in efficiency can be attributed to the coordination of polar solvents, such as THF, with the palladium center, which likely disrupts the catalytic cycle and compromises the reaction’s overall performance [22]. With optimized conditions established, which included flavone 11 (1.0 equiv), amine (1.2 equiv), Pd2(dba)3 (10 mol %), XantPhos (5 mol %), Cs2CO3 (3.0 equiv), and toluene at 110 °C for 20 h (entry 4), we investigated the scope of the Buchwald-Hartwig coupling reactions (Scheme 2).
As illustrated in Scheme 2, the formation of 6-arylaminoflavones is markedly influenced by the substituents on the arylamine ring, with electronic and steric factors playing a critical role in determining the reaction yields. Better yields were obtained when anilines substituted with electron-donating groups was employed (13a and 13h; 77% and 95%). When an aniline containing an electron-neutral substituent was employed, the desired product 13b was obtained in only moderate yield (50%). The electron withdrawing groups such as trifluoromethyl and fluorine, afforded the 6-arylaminoflavones products 13d-g in a poor to a moderate yield. The presence of strongly electron-withdrawing groups, such as cyano, nitro, or carboxyl, resulted in no reaction 13j-13l, likely due to their ability to hinder the effective formation of the C–N bond under the conditions of the Buchwald-Hartwig coupling [23,24]. Regarding compounds 13m-n, which are not arylamines, these reactions were designed with the aim of improving the solubility of the 6-arylaminoflavones. However, no product was obtained, likely due to the nature of the groups involved, which were not suitable for the desired reaction pathway.
To assess the cytotoxic effects of the compounds 13a-h, we initially conducted an MTT assay using four tumorigenic cell lines (A172, HEK293T, MDA-MB-231, and PC3) [25]. The first screening was performed at a concentration of 100 µM to identify compounds exhibiting cytotoxicity at a high concentration (Figure 2). We observed that substituents in the 6-arylamine group strongly impact cellular viability when compared to the vehicle control (DMSO). Comparing 13a with 13b, we found that 4-methoxy substituent appears to reduce the basal toxic effects, as 13a was non-toxic to the tested cell lines. Of note, 13c (4-trifluoromethyl) exhibited preferential toxicity towards PC-3 (prostate adenocarcinoma) cells compared to the other cell lines, whereas, shifting the trifluoromethyl from the para-position in 13c to the ortho-position in 13d, almost abolished the toxic effect to tumor cells. On the other hand, this effect was inverted when the 4-fluoro group (13e) was replaced by a 2-fluoro (13f). Furthermore, switching the 4-fluoro (13e) to a 4-chloride (13g) regained toxicity towards A172, MDA-MB-231, and especially for PC-3 cells as observed in 13c, whereas a 4-methyl (13h) substitution abolished this effect.
Based on these findings, we sought to further explore the influence of B-ring modifications on cytotoxicity by synthesizing six new analogues of 13a-c under the same previously described conditions (Scheme 3). The synthesis followed a three-step approach, as detailed below.
The Claisen-Schmidt condensation was performed with 8 and benzaldehyde 14 or 4-methoxybenzaldehyde 15 to afford the corresponding α,β-unsaturated chalcone 16-17 with 53% and 50% yield, respectively. The oxidative cyclization gave the 6-bromoflavone intermediates 18-19 with 42-74% yield. Finally, the previous optimized condition for the synthesis of 13a was used to achieve the final compounds. Noteworthy, this procedure proved less efficient for 6-arylaminoflavone bearing a benzene (20a-c) or 4-methoxybenzene (21a-c) substituents in the B-ring, particularly when the 6-substitution was a 4-trifluoromethyl group. Nonetheless, we successfully obtained the desired derivatives of 13a-c bearing the modification at the B-ring for cytotoxic evaluation.
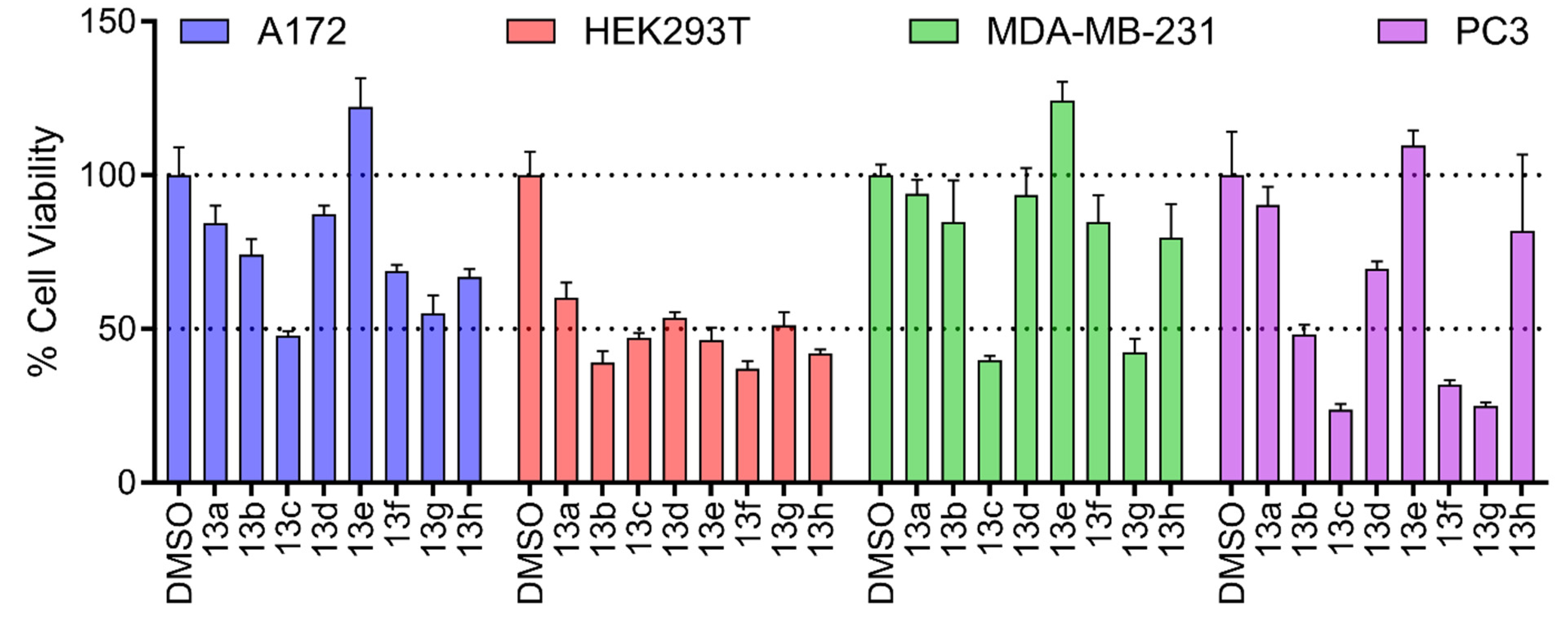
Overall, the new arylaminoflavone derivatives were less toxic to the tumorigenic cells when compared to their parent compounds (Figure 3). This effect was particularly pronounced for compounds 21a-c, which exhibited cell viability above 50%, and was most evident for compound 20c, whose toxicity was comparable to the vehicle control. By analyzing the matched pairs (for instance, 20a and 21a) we observed that all compounds were less toxic than their parent compound (21a), independently of the substitutions at the 6-position. This highlights that most of the cytotoxicity is largely influenced by the most commonly explored region (Figure 1) at the ring- B.
4. Conclusions
This study highlights the significant potential of 6-arylaminoflavone in medicinal chemistry, owing to their diverse biological activities and structural versatility. The research successfully optimized synthetic methods, particularly using palladium-catalyzed Buchwald-Hartwig coupling reaction to improve yields and efficiency in producing 6-arylaminoflavone derivatives. The cytotoxicity assays demonstrated that certain structural modifications are crucial for cytotoxicity of these compounds. Specifically, compounds with a 3,4-dimethoxybenzene motif in ring-B and a 4-trifluoromethyl (13c) or 4-chloride (13g) substituents at the 6-arylamino ring showed higher cytotoxicity to the prostate and breast adenocarcinoma cells. Conversely, whenever the B-ring is modified the cytotoxicity is abrogated. These findings provide valuable insights into the structure-activity relationships of 6-arylaminoflavone, suggesting that careful design and modification of these compounds can enhance their therapeutic potential.
Author Contributions
K.E.P. and M.R.C. contributed equally to this work. K.E.P. and M.R.C. performed the main experiments and data analysis. G.A.M. conducted the biological studies. N.H.M.A. contributed to the analysis of compounds. K.B.W. supervised the biological studies. M.F.Z.J.T. and R.P.-F. assisted in writing and reviewing the final manuscript. R.P.-F. also conceived and supervised the study.
Acknowledgments
The authors are grateful to the Faculty of Pharmacy of the University of Sao Paulo, which allowed the development of this work, and to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and São Paulo Research Foundation (FAPESP), grant numbers: 114897/2022-0, 2024/07723-2, 2021/08260-8, 2021/04853-4, 2014/50897-0, and 465651/2014-3 for financial support.
Conflicts of Interest
The authors declare no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Beecher, G. R. Overview of Dietary Flavonoids: Nomenclature, Occurrence and Intake. The Journal of Nutrition 2003, 133, 3248S–3254S. [Google Scholar] [CrossRef] [PubMed]
- Boniface, P. K.; Elizabeth, F. I. Flavones as a Privileged Scaffold in Drug Discovery: Current Developments. COS 2019, 16, 968–1001. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, M.; Silakari, O. Flavones: An Important Scaffold for Medicinal Chemistry. European Journal of Medicinal Chemistry 2014, 84, 206–239. [Google Scholar] [CrossRef]
- Wang, X.; Cao, Y.; Chen, S.; Lin, J.; Bian, J.; Huang, D. Anti-Inflammation Activity of Flavones and Their Structure–Activity Relationship. J. Agric. Food Chem. 2021, 69, 7285–7302. [Google Scholar] [CrossRef] [PubMed]
- Akama, T.; Shida, Y.; Sugaya, T.; Ishida, H.; Gomi, K.; Kasai, M. Novel 5-Aminoflavone Derivatives as Specific Antitumor Agents in Breast Cancer. J. Med. Chem. 1996, 39, 3461–3469. [Google Scholar] [CrossRef]
- Akama, T.; Ishida, H.; Shida, Y.; Kimura, U.; Gomi, K.; Saito, H.; Fuse, E.; Kobayashi, S.; Yoda, N.; Kasai, M. Design and Synthesis of Potent Antitumor 5,4‘-Diaminoflavone Derivatives Based on Metabolic Considerations. J. Med. Chem. 1997, 40, 1894–1900. [Google Scholar] [CrossRef]
- Callero, M. A.; Rodriguez, C. E.; Sólimo, A.; Bal De Kier Joffé, E.; Loaiza Perez, A. I. The Immune System As a New Possible Cell Target for AFP 464 in a Spontaneous Mammary Cancer Mouse Model: AFP 464 T ARGETS T HE I MMUNE S YSTEM. J. Cell. Biochem. 2017, 118, 2841–2849. [Google Scholar] [CrossRef]
- Luzzani, G. A.; Callero, M. A.; Kuruppu, A. I.; Trapani, V.; Flumian, C.; Todaro, L.; Bradshaw, T. D.; Loaiza Perez, A. I. In Vitro Antitumor Effects of AHR Ligands Aminoflavone (AFP 464) and Benzothiazole (5F 203) in Human Renal Carcinoma Cells. J of Cellular Biochemistry 2017, 118, 4526–4535. [Google Scholar] [CrossRef]
- Goetz, M. P.; Reid, J. M.; Qi, Y.; Chen, A.; McGovern, R. M.; Kuffel, M. J.; Scanlon, P. D.; Erlichman, C.; Ames, M. M. A Phase I Study of Once-Weekly Aminoflavone Prodrug (AFP464) in Solid Tumor Patients. JCO 2011, 29, 2546–2546. [Google Scholar] [CrossRef]
- Dauzonne, D.; Folléas, B.; Martinez, L.; Chabot, G. Synthesis and in Vitro Cytotoxicity of a Series of 3-Aminoflavones. European Journal of Medicinal Chemistry 1997, 32, 71–82. [Google Scholar] [CrossRef]
- Alessi, D. R.; Cuenda, A.; Cohen, P.; Dudley, D. T.; Saltiel, A. R. PD 098059 Is a Specific Inhibitor of the Activation of Mitogen-Activated Protein Kinase Kinase in Vitro and in Vivo. Journal of Biological Chemistry 1995, 270, 27489–27494. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ge, C.-C.; Wang, J.; Wu, X.-X.; Li, X.-M.; Li, W.; Wang, S.-S.; Liu, T.; Hou, J.-Z.; Sun, H.; Fang, D.; Xie, S.-Q. MEK Inhibitor, PD98059, Promotes Breast Cancer Cell Migration by Inducing β-Catenin Nuclear Accumulation. Oncology Reports 2017, 38, 3055–3063. [Google Scholar] [CrossRef] [PubMed]
- Thorat, N. M.; Sarkate, A. P.; Lokwani, D. K.; Tiwari, S. V.; Azad, R.; Thopate, S. R. N-Benzylation of 6-Aminoflavone by Reductive Amination and Efficient Access to Some Novel Anticancer Agents via Topoisomerase II Inhibition. Mol Divers 2021, 25, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Elkanzi, N. A. A.; Hrichi, H.; Alolayan, R. A.; Derafa, W.; Zahou, F. M.; Bakr, R. B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega 2022, 7, 27769–27786. [Google Scholar] [CrossRef]
- Torres Ribón, D. J.; Roa De La Fuente, L. F.; Ceronio, N. R.; Abreu, O. H.; Rivera, M. R.; Juárez, J. R.; Torralba, R.; Ximello, M. V.; Sauret, Q. T.; Sánchez, C. A. Iodine Promoted One-Pot Synthesis of Flavones. Results in Chemistry 2025, 13, 101968. [Google Scholar] [CrossRef]
- Leonte, D.; Ungureanu, D.; Zaharia, V. Flavones and Related Compounds: Synthesis and Biological Activity. Molecules 2023, 28, 6528. [Google Scholar] [CrossRef]
- Dorel, R.; Grugel, C. P.; Haydl, A. M. The Buchwald–Hartwig Amination After 25 Years. Angew Chem Int Ed 2019, 58, 17118–17129. [Google Scholar] [CrossRef]
- Fitzner, M.; Wuitschik, G.; Koller, R. J.; Adam, J.-M.; Schindler, T.; Reymond, J.-L. What Can Reaction Databases Teach Us about Buchwald–Hartwig Cross-Couplings? Chem. Sci. 2020, 11, 13085–13093. [Google Scholar] [CrossRef]
- Clarke, G. E.; Firth, J. D.; Ledingham, L. A.; Horbaczewskyj, C. S.; Bourne, R. A.; Bray, J. T. W.; Martin, P. L.; Eastwood, J. B.; Campbell, R.; Pagett, A.; MacQuarrie, D. J.; Slattery, J. M.; Lynam, J. M.; Whitwood, A. C.; Milani, J.; Hart, S.; Wilson, J.; Fairlamb, I. J. S. Deciphering Complexity in Pd–Catalyzed Cross-Couplings. Nat Commun 2024, 15, 3968. [Google Scholar] [CrossRef]
- Sunesson, Y.; Limé, E.; Nilsson Lill, S. O.; Meadows, R. E.; Norrby, P.-O. Role of the Base in Buchwald–Hartwig Amination. J. Org. Chem. 2014, 79, 11961–11969. [Google Scholar] [CrossRef]
- Surry, D. S.; Buchwald, S. L. Biaryl Phosphane Ligands in Palladium-Catalyzed Amination. Angew Chem Int Ed 2008, 47, 6338–6361. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, J.; Clark, J. H.; Fairlamb, I. J. S.; Slattery, J. M. Solvent Effects in Palladium Catalysed Cross-Coupling Reactions. Green Chem. 2019, 21, 2164–2213. [Google Scholar] [CrossRef]
- Tian, J.; Wang, G.; Qi, Z.-H.; Ma, J. Ligand Effects of BrettPhos and RuPhos on Rate-Limiting Steps in Buchwald–Hartwig Amination Reaction Due to the Modulation of Steric Hindrance and Electronic Structure. ACS Omega 2020, 5, 21385–21391. [Google Scholar] [CrossRef] [PubMed]
- Seifinoferest, B.; Tanbakouchian, A.; Larijani, B.; Mahdavi, M. Ullmann-Goldberg and Buchwald-Hartwig C−N Cross Couplings: Synthetic Methods to Pharmaceutically Potential N-Heterocycles. Asian J Org Chem 2021, 10, 1319–1344. [Google Scholar] [CrossRef]
- Stepanenko, A. A.; Dmitrenko, V. V. HEK293 in Cell Biology and Cancer Research: Phenotype, Karyotype, Tumorigenicity, and Stress-Induced Genome-Phenotype Evolution. Gene 2015, 569, 182–190. [Google Scholar] [CrossRef]
Figure 1.
Structure of some cytotoxic aminoflavones and their core structure.

Scheme 1.
Synthetic route of the flavone core.

Scheme 2.
Substrate scope of 6-arylaminoflavone derivatives synthesis via Buchwald-Hartwig coupling.
Scheme 2.
Substrate scope of 6-arylaminoflavone derivatives synthesis via Buchwald-Hartwig coupling.
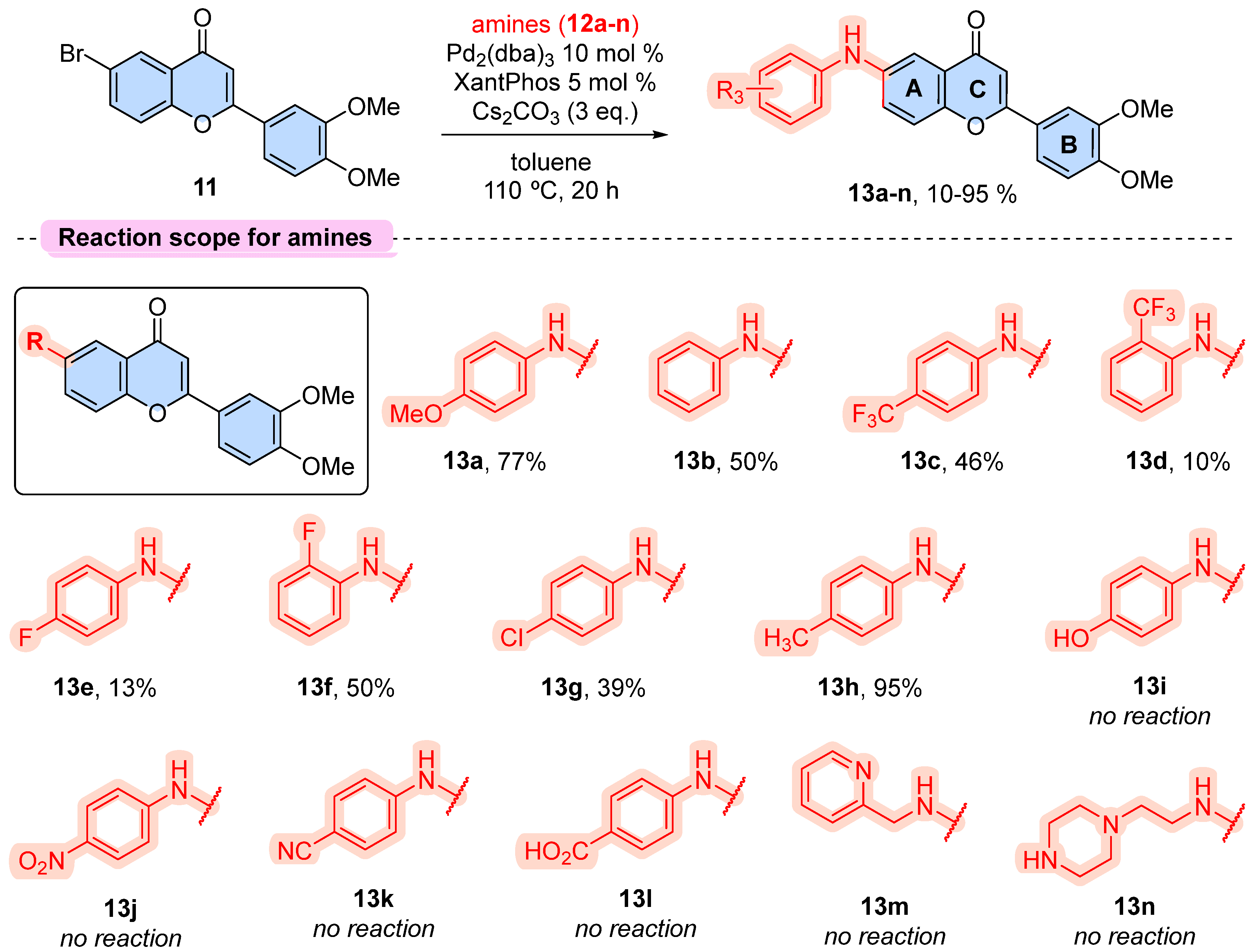
Figure 2.
Cellular toxicity of 6-arylaminoflavones 13a-h. Results are expressed as viable cells relative to vehicle control (DMSO). Data are expressed as mean ± standard deviation of one experiment performed in quadruplicate (n = 4).
Figure 2.
Cellular toxicity of 6-arylaminoflavones 13a-h. Results are expressed as viable cells relative to vehicle control (DMSO). Data are expressed as mean ± standard deviation of one experiment performed in quadruplicate (n = 4).
Scheme 3.
Synthetic route to obtain 6-arylaminoflavone derivatives (20a-c, 21a-c) with modification in the B-ring.
Scheme 3.
Synthetic route to obtain 6-arylaminoflavone derivatives (20a-c, 21a-c) with modification in the B-ring.

Figure 3.
Cellular toxicity of 6-arylaminoflavones 20a-c and 21a-c. Results are expressed as viable cells relative to vehicle control (DMSO). Data are expressed as mean ± standard deviation of one experiment performed in quadruplicate (n = 4).
Figure 3.
Cellular toxicity of 6-arylaminoflavones 20a-c and 21a-c. Results are expressed as viable cells relative to vehicle control (DMSO). Data are expressed as mean ± standard deviation of one experiment performed in quadruplicate (n = 4).
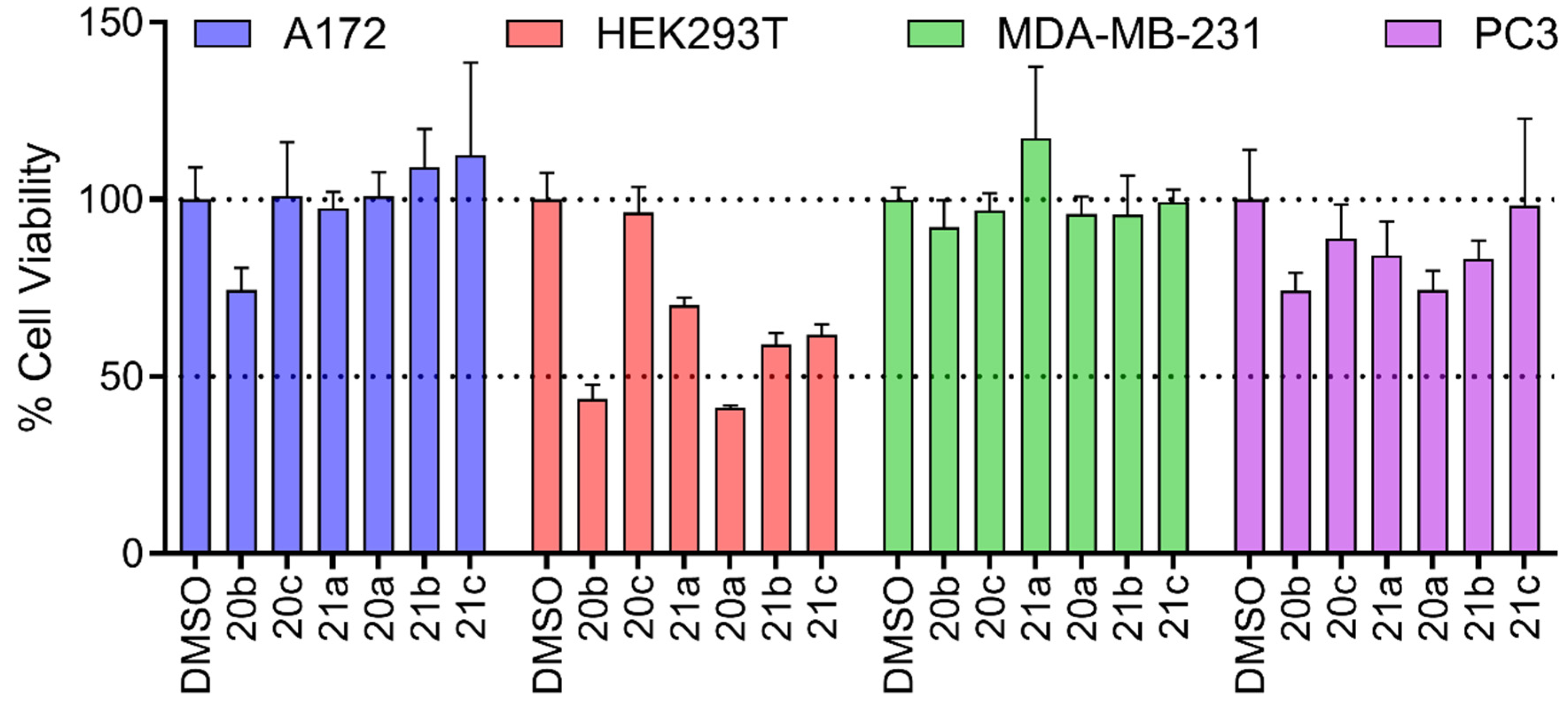

Table 1.
Optimization of the Buchwald-Hartwig reaction conditions.a.
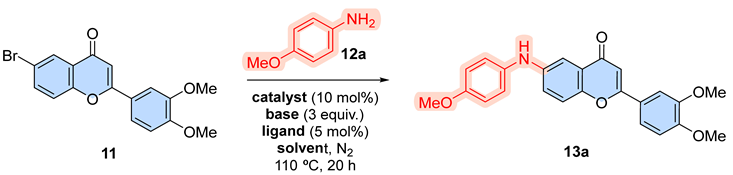 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Solvent | Yield (%) |
| 1 | Pd2(dba)3 | XantPhos | CsF | toluene | No reaction |
| 2 | Pd2(dba)3 | XantPhos | NaOtBu | toluene | trace |
| 3 | Pd2(dba)3 | XantPhos | K2CO3 | toluene | 32 |
| 4 | Pd2(dba)3 | XantPhos | Cs2CO3 | toluene | 77 |
| 5 | Pd2(dba)3 | DavePhos | Cs2CO3 | toluene | 75 |
| 6 | PdCl2(PPh3)2 | XantPhos | Cs2CO3 | toluene | 20 |
| 7 | Pd2(dba)3 | XantPhos | Cs2CO3 | THF | 35 |
aReaction conditions: 11 (1 mmol) and 12a (1.1 mmol) at reflux under sealed tube.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



