Submitted:
27 March 2025
Posted:
28 March 2025
You are already at the latest version
Abstract
Antimicrobial peptides (AMPs) are bioactive molecules that can effectively destabilize microbes cells. AMPs can reduce the side effects of drugs and other bioactive mole-cules, ensure greater bioavailability of the peptide/bioactive molecule combination, and eventu-ally act synergistically in these combinations. Several AMPs such as linear gramicidins and melittin, which are among the oldest AMPs reported, are considered promising anti-cancer weapons among other applications alone or in combinations. By improving drug bioavailability, targeting, and reducing drug resistance, AMP-based formulations offer a promising avenue for future cancer pharmacology.
Keywords:
antimicrobial peptides
; gramicidin D
; self-assembly of AMPs
; nanoparticles
; combinations of AMPs and other bioactive molecules
; synergism
; anti-cancer activity for AMPs
1. Antimicrobial Peptides, Their Assemblies and Biomedical Applications
Natural or synthetic antimicrobial peptides (AMPs) have been explored to create a whole new set of applications in drug delivery, materials science, molecular biology and nanotechnology [1]. For example, antimicrobial peptides can assemble as scaffolds acting with dual functions: bactericidal agents and carriers for antibiotics delivery. In combinatorial therapeutics they provide a valuable approach; inter- and intra-molecular interactions for peptides drive the formation of nanospheres, nano-fibers, nets, sheets and tubes, or even, hydrogels [2]. Some architectures can be obtained from peptide self-assembly such as gels for 3D scaffolds useful in tissue engineering, fibrils, nanospheres used as drug delivery systems, and nanotubes, used in hybrid materials and biomineralization [3].
Cyclic peptides functionalized through conjugation to polymers generated peptide–-polymer nanotubes able to insert in membranes, act as ion channels, deliver anticancer drugs or genetic materials, or provide applications as antivirals [4]. Sophisticated techniques such as super-resolution microscopy with modern image analysis, small-angle neutron scattering, and solid-state nuclear magnetic resonance have allowed the characterization of nanostructures from short peptides, which have strategic applications in biomedicine and nanotechnology. They can function as antimicrobials, anticancer agents, vehicles for controlled drug release, and responsive cell culture materials [5].
In water solution peptides adopt a conformation of minimum free energy under equilibrium conditions. Intermolecular interactions between peptides often drive their self-assembly in solution. They are hydrogen-bonding, π-π, cation-π, electrostatic, hydrophobic, and van der Waals interaction forces. The thermodynamics and kinetics of the self-assembly process for short peptides were comprehensively reviewed [6]. These physical interactions acting cooperatively can yield a variety of nanostructures such as nanotubes, nanospheres, and nanofibers that are finding a myriad of functional applications; forces such as hydrogen bonding, cation-π, and anion-π interactions can be reasonably long-ranged, and able to co-operatively yield energies equivalent to a weak covalent bond [7].
The self-assembly of peptides can provide interesting novel materials such as hydrogels. Hydrogels are classically made from high molecular weight natural polymers such as proteins (e.g., gelatin). Using amino acids or peptides as building blocks for gels brings both biodegradability and biocompatibility to these soft materials. Peptide-based hydrogels are used as scaffolds for wound healing, drug and biomolecule release, cell culture, and tissue engineering [8]. The many applications of peptide fibers assembling as gels have been the subject of excellent reviews [8,9,10]. Bioactive hydrogels based on peptides have also been used for the delivery of antibiotics, antimicrobial peptides, cationic polymers, photosensitizers, and nanomaterials for photodynamic or photothermal therapy. Multiple wound-repairing effects could be obtained from hydrogels such as hemostasis, adhesion, wound contraction and closure, anti-microbial effect, anti-oxidative effect, and others [11].
Lysine-based dendritic hydrogels able to manage severe trauma or intraoperative bleeding have been synthesized [12]. Oxidized carboxymethylcellulose (OCMC) and third-generation lysine peptide dendrimers (OCMC/G3KP) showed inherent hemostatic ability, antibacterial properties, and high adhesiveness. In vivo, this hydrogel promoted superior wound healing in contrast to conventional sutures. The oxidation of hydroxyl groups on carboxymethylcellulose (CMC) yielded aldehydes on the OCMC polymer chain that could covalently bind to amino moieties on the tissue yielding the observed strong adhesiveness whereas dendritic poly (lysine) accounted for the high bactericidal efficiency of the hydrogel [13].
An interesting development has been the hydrogel-based delivery of growth factors, which has been an important therapeutic agent for tissue engineering and regenerative medicine in the past decades. Growth factor requires protection for in vivo delivery due to side effects and rapid degradation. These limitations have been circumvented by using hydrogels for growth factor delivery [14]. For example, the IKVAV sequence is a biologically recognized peptide epitope from laminin, a protein from the extracellular matrix in the central nervous system. This isoleucine-lysine-valine-alanine-valine sequence was added aspartic acid to the N-terminus of the peptide adjusting the apparent pKa of the peptide molecule to ensure that the pH-driven self-assembly would take place at physiological pH. This self-assembled peptide (DIKVAV) formed nanofibers (∼10 nm in diameter) and the resulting hydrogel was used to stabilize and deliver growth factors such as the neurotrophic growth factor glial cell-line derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF) [15].
Peptide self-assembly directly depends on the intervening medium. For example, in toluene, short phenylalanine dipeptides made of only two phenylalanine residues self-assemble as gels where the nanofibrils are interconnected by hydrogen bonding. On the other hand, in ethanol, the π-π stacking of phenyl moieties between chains favored the formation of microcrystals [16]. In the gel, the molecules adopted antiparallel β-sheet structures, whereas in the crystals, parallel β-sheets occurred [16].
Natural antimicrobial peptides such as gramicidin D and melittin also interact in different microenvironments to yield a variety of assemblies [17,18]. Both Gramicidin D and melittin are lytic peptides able to interact with the plasma membranes of eukaryotic cells. For example, their interaction with the membranes of erythrocytes changes permeability to allow passage of ions. As a consequence, osmotic lysis from cell swelling follows and hemolysis takes place. A similar mechanism of action was reported for staphylococcal delta-toxin in which toxin molecules embed themselves in the membrane where they assemble as transmembrane channels [17,19].
Membrane damage in erythrocytes by melittin involved the accumulation of the peptide molecules predominantly in the outer half of the bilayer. This accumulation induced deformation of the erythrocyte into crenature destabilizing the membrane structure and releasing membrane fragments; this mechanism of action was similar to the one of surfactants [20]. At the very low lipid-to-peptide ratio of 50, melittin was found to form pores of 2.5-3.0 nm diameter in palmitoyl oleoyl phosphatidylcholine (POPC) vesicles [21]. More recently, the therapeutic potential of melittin for treating cancer and overcoming its toxicity has been explored thanks to optimized assemblies this peptide can yield with biocompatible carriers. Melittin in stable supramolecular complexes of ≈60 nm with the photosensitizer chlorin e6 (Ce6) coated with hyaluronic acid was safe and displayed reduced hemolysis and selective cytotoxicity against cancer cells. There was a synergism of action for melittin lytic effect and photosensitization of chlorine e6 leading to increased penetration of the complexes in the tumor [22].
Over the last decades, in our laboratory, novel formulations for gramicidin D have been described based on its self-assembly as nanospheres in water dispersions [23], formation of dimeric channels in cationic bilayers [24,25,26,27,28] or synergistic antimicrobial action with the cationic polymer poly (diallyl dimethylammonium chloride) (PDDA) [22,28,29,30,31,32,33,34,35,36,37,38,39]. Gramicidin D nanoparticles and the antimicrobial polymer PDDA displayed convenient synergistic action against bacteria and fungus both in water dispersions[23,39] and in coatings [40]. Successful formulations for antimicrobial peptides involve not only their self-assembly property but also their combination with other bioactive molecules such as cationic lipids, surfactants, and polymers [18].
Similarly to other peptides, conformation, and self-assembly of gramicidin D (Gr) are highly dependent on its microenvironment as illustrated in Figure 2 [24]. Gr medium determined not only its conformation but also its self-assembly. Gr intrinsic fluorescence, circular dichroism (CD) spectroscopy, dynamic light scattering, and zeta potential analysis are essential for characterizing Gr conformations and self-assembled structures in different media. Additionally, bilayer phase transition analysis using extrinsic fluorescence and colloidal stability evaluation over time and varying NaCl concentrations provide further insights into Gr dispersion behavior.
The Gr interaction with dipalmitoyl phosphatidylcholine (DPPC)/dioctadecyl dimethylammonium bromide (DODAB) 1:1 large unilamellar vesicles (LVs) [41] revealed that the Gr dimeric channel in LVs yielded CD and intrinsic fluorescence spectra similar to those in trifluoroethanol (TFE). Furthermore, the Gr channels were functional allowing KCl or glucose permeation through the bilayer [24].
For Gr in the DPPC/DODAB bilayer fragments (BFs), the intertwined dimeric conformation was evidenced by CD and intrinsic fluorescence spectra similar to those in ethanol. Both LVs and BFs protected Gr tryptophan fluorescence against quenching by acrylamide. However, the Stern–Volmer quenching constant was slightly higher for Gr in BFs showing that Gr was more exposed to water in BFs than in LVs [23] (Figure 1).
In solvents such as trifluoroethanol (TFE) or ethanol, gramicidin D assemblies were dimers of Gr molecules in the channel conformation or intertwined assemblies, respectively. In the cationic bilayers, they inserted as dimeric channels in LV or remained as intertwined molecules at the borders of cationic bilayer fragments (Figure 1) [24].
Figure 2.
Turbidity kinetics for DPPC/DODAB large vesicles (LVs) due to the establishment of KCl (●, ○) or glucose osmotic gradients through the LV bilayer (▲, ∆) in the absence (●, ▲) or in the presence of 10% gramicidin at 1.0 mM lipid and 25 °C (○, ∆). Reprinted from [24] with permission Copyright 2020 Elsevier.
Figure 2.
Turbidity kinetics for DPPC/DODAB large vesicles (LVs) due to the establishment of KCl (●, ○) or glucose osmotic gradients through the LV bilayer (▲, ∆) in the absence (●, ▲) or in the presence of 10% gramicidin at 1.0 mM lipid and 25 °C (○, ∆). Reprinted from [24] with permission Copyright 2020 Elsevier.
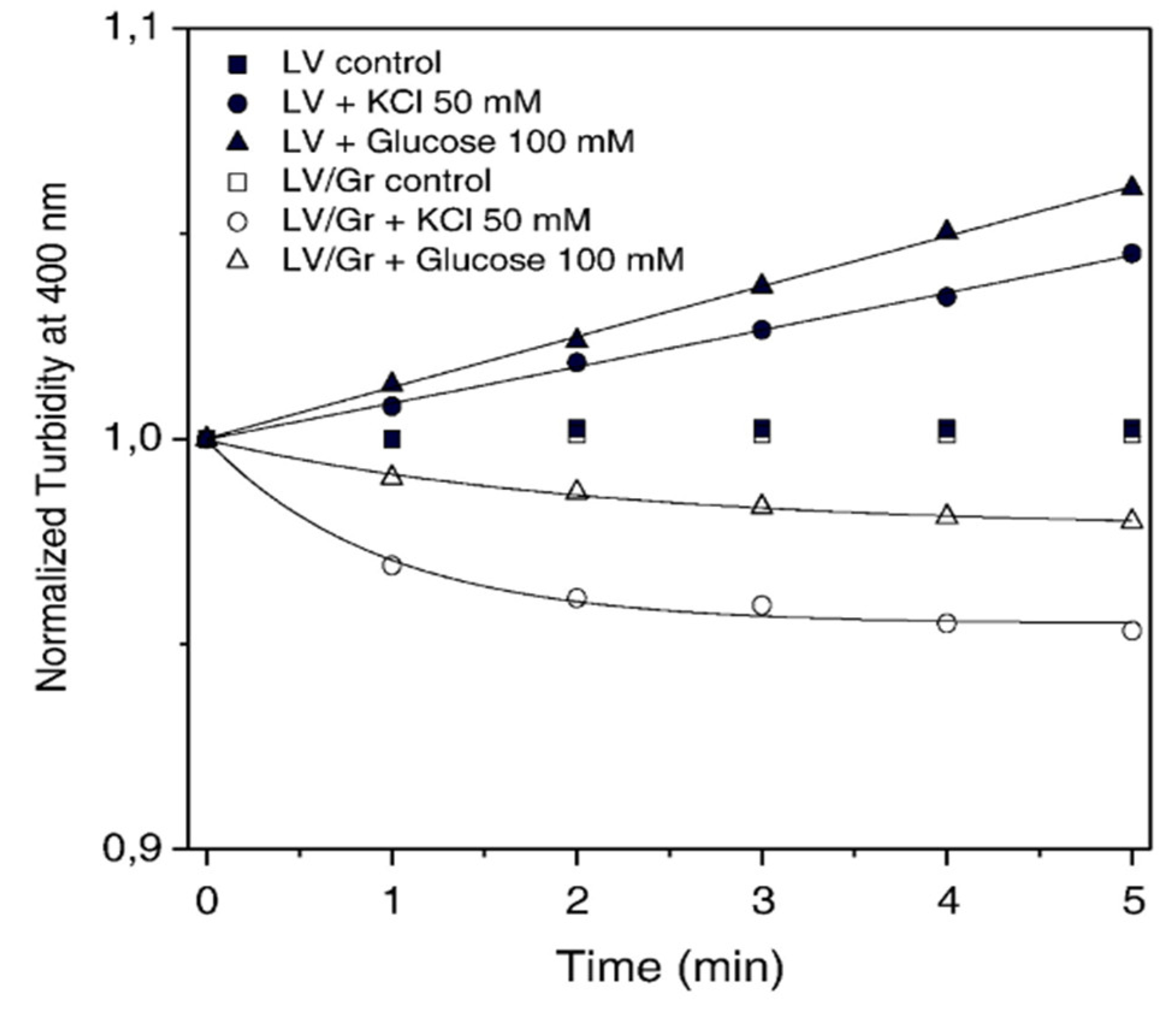
Gramicidin’s effects on the water/solute permeation through the DPPC/DODAB LV bilayer were evaluated from turbidity kinetics as a function of time. For the DPPC/DODAB 1:1 LV dispersions submitted to a final external concentration of 50 mM KCl or 100 mM glucose, the turbidity at 400 nm increased as a function of time (Figure 2) [24]. This resulted from LV shrinkage due to water efflux along the solute gradient. It was previously reported for similar cationic LVs, that shrinkage corresponds to an increase in turbidity as a function of time [42]. Therefore, the DPPC/DODAB LV dispersion responded as expected to the hypertonic external solutions. On the other hand, in the presence of 10% Gr, there was a slight decrease in normalized turbidity at 400 nm as a function of time, suggesting a small swelling of LVs/Gr (Figure 2). Gr channel inserted in the bilayer completely changes its normal permeability profile. The usually low permeability of cations and solutes through the DPPC/DODAB LV bilayer increases substantially in 10% Gr. In Figure 2, KCl or glucose entered from the outer to the inner vesicle compartment carrying water and dissipating the solute concentration gradient to the point of changing shrinkage to swelling.
In synthetic cationic vesicles or bilayer fragments made of dioctadecyl dimethylammonium bromide (DODAB) only, insertion of gramicidin in the DODAB LV bilayer vesicles (1:10 gramicidin: DODAB molar ratio) led similar behavior as the previous one in DPPC: DODAB LV [25]. Figure 3 (a) shows the leakage of phosphorylated intracellular compounds induced by these formulations as a function of DODAB concentration. Consistently, the morphology of bacterial cells seen in Figure 3(b) points out the occurrence of major changes and the appearance of cellular debris indicative of lysis.
Microorganisms often synthesize a wide range of biosurfactants (BS), which have been finding applications in pharmaceutics, food processing, biodegradation, bioremediation, cosmetics, and pest control management [42]. Recent findings reported that BS from pathogenic microorganisms facilitated the host infection. Glycolipid BS could modulate humoral and cellular immune responses, and cyclic lipopeptide surfactin down-regulated expression of several surface molecules such as CD40, CD54, CD80, and major histocompatibility complex class II (MHC-II). They also find applications as immunosuppressive compounds for treating autoimmune diseases [42]. BS interactions with cell membranes lead to antimicrobial, antiviral, and anticancer properties for these molecules [43].
Among the BS, cyclic lipopeptides (CLP) are synthesized and secreted by Bacillus and Pseudomonas. CLP identified in the genus Bacillus are the surfactins, the iturins, the lichenysins, and the fengycins [46,47]. As surfactants, the BSs reduce the surface tension at the air/water interface and the interfacial tension at the oil/water interface. For example, the self-assembly of a cyclic lipopeptides mixture secreted by a Bacillus megaterium strain yielded negatively charged and large aggregates (300-800 nm of mean hydrodynamic radius) [45,46]. Interestingly, these aggregates were not affected by changes in the osmolarity of the outer medium suggesting the absence of an internal aqueous compartment. However, increasing pH and the resulting negative charge promoted disaggregation, enhancing activity against Bacillus cereus [45]. The interaction between CLP and Bacillus cereus cells induced leakage of intracellular phosphorylated compounds from the cells showing their lytic power. In addition, removing the cyclic character of the CLP by hydrolysis of the lactone or lactam rings did not remove the detergency power but abolished the antibiotic activity.
The minimal inhibitory concentration (MIC) of 12 mg/L for the CLP secreted by B. megaterium was well below the critical micelle concentration (CMC) of 100 mg/L. Therefore, B. cereus lysis was not due to a detergency effect only but also to the insertion of individual CLP molecules in the cell membranes unleashing the lytic process which kills the B. cereus cells [46]. Given the natural functions of lipopeptides from Bacillus and Pseudomonas, they have been recognized as more than surfactants and antibiotics [48]. They are a kind of primeval defense of a given microbe against competitors in the same niche.
Based on artificial molecular designs protein folds unseen in nature have been created [49]. Interestingly, two-dimensional giant nanosheets were obtained from the self-assembly of d/l-alternating cyclic peptides [50]. These giant nanosheets represent one of the largest 2D supramolecular materials ever made. Figure 4 illustrates the self-assembly of cyclic peptide nanotubes to yield giant nanosheets; the model for the sequential 1D-to-2D self-assembly of cyclic peptide CPx involved the secondary parallel β-sheet conformation between CPx backbones within nanotubes stabilized via eight hydrogen bonds and the tertiary 1D self-assembly of CPx as amphiphilic peptide nanotubes with hydrophobic (Leu–Trp–Leu in yellow-gray) and hydrophilic (Glu–His–Gln in red-blue-green) surfaces. The 2D self-assembly of CPx into nanosheets comprising nanotube bilayers with hydrophobic cores, composed of Leu zippers (yellow) and Trp stacks (gray), and polar residues on their surface can be seen in Figure 4 [50].
The design of supramolecular assemblies driven by the supramolecular interactions between cyclic peptides was comprehensively reviewed [4].
Some AMPs are part of the innate immune system in mammals ensuring protection against infection; these AMPs are usually called defensins. In pigs, Staphylococcus epidermidis colonization in wounds increases when AMP activation is inhibited [51]. In mice, Salmonella virulence correlates with a natural resistance to AMP action [52]. In humans, Shigella infections correlate with downregulation of enteric cathelicidin and β-defensin-1 expression [53]. In transgenic mice, overexpression of a human AMP gene improves lung clearance of Pseudomonas aeruginosa [54]. Many mammalian antimicrobial or host defense peptides stimulate the host’s immune cellular response aiding in the clearance of invading pathogens [55].
The potential use of AMPs in biomedical applications goes beyond their direct antimicrobial activities for the treatment of antibiotic-resistant infections [56]. Peptides in novel supramolecular assemblies may become essential in vaccine design, antimicrobial chemotherapy, cancer immunotherapy, food preservation, organ transplants, design of novel materials for dentistry, formulations against diabetes, and other important applications; often their therapeutic index can be improved by protecting their activity and increasing their bioavailability [57]. Combinations of peptides with lipids, liposomes, nanoparticles, polymers, micelles, etc., within the limits of nanotechnology may also provide novel applications going beyond antimicrobial therapy for infectious diseases [58].
Interestingly, the self-assembly of the cationic and anticancer killerFLIP peptide [59] yielded improved selectivity against cancer cells [60]. Figure 5 illustrates the behavior of monomers and aggregates of the killerFLIP peptide. Reducing the exposure of peptide hydrophobic moieties in the peptide aggregates to normal cells diminished its affinity toward eukaryotic cell membranes but improved the electrostatic attraction of the aggregates toward the cancer cells [60].
Biomedical research employs two targeted delivery strategies to fight cancer: passive and active targeting [61]. The active targeting relies on ligand-receptor binding, which improves selective accumulation to targeted sites and thus discriminates between the diseased and healthy tissues. The passive targeting could be achieved by targeting the endothelial cells, tumor cells, or the acidic environment of tumors. The passive targeting agents are mainly nanoparticles (1-1000 nm in size), which accumulate around the tumors with leaky vasculature due to enhanced permeation and retention (EPR) effects in tumors [61].
The peptide derivative FA-EEYSV-NH2 with a folic acid (FA) moiety targeted folate receptors in cancer cells and also self-assembled as nanoparticles or fibers depending on pH. This endowed this peptide with dual targeting (the EPR effect and the active targeting) with improved therapeutic index due to their specific internalization and toxicity towards the cancer cells [62]. The self-assembly of these molecules was pH-dependent yielding nanoparticles at pH 7 or fibers at pH 5.Figure 6 illustrates the pH-dependent assemblies of FA-EEYSV-NH2 both in the normal cell microenvironment as NPs at pH 7.0 and as nanofibers at pH 5.The modification of the self-assemblies from nanoparticles to nanofibers when pH decreased from 7.0 to 5.0 enhanced the retention and therapeutic index of the peptide specifically in the tumor.
Peptides are frequently used as nonviral vectors for nucleic acid delivery, alongside cationic liposomes, polymers, and dendrimers [63]. A variety of peptides can penetrate cells, interact with cell membranes, and be modified to target different compartments of the cell including the cell nucleus. The main driving forces for the interaction between non-viral vectors and nucleic acids in vitro are the electrostatic interaction and the hydrophobic effect. Cationic artificial vectors assemble with oppositely charged nucleic acids functioning as protectors against the biodegradation by nucleases that may take place in vivo. Furthermore, innate immunity against nucleic acid delivery also represents a significant barrier preventing genetic material delivery to cells in vivo [64]. For vaccines, the first step is the introduction of mRNA into mammalian cells. For delivery of genetic material and gene therapy, for example, the immune stimulation triggered in vivo may compromise this non-immunotherapy-related application.
Gene therapy depends on the introduction of DNA into target cells followed by DNA transcription and mRNA translation as expressed proteins. Peptides have been successful in promoting DNA compaction into biocompatible, stable, and protected NPs facilitating delivery into cells. The design of peptides can also be tailored to a high efficiency of gene delivery [64]. Encapsulating DNA in synthetic particles protects it from degradation and enhances cellular uptake. However, the DNA/peptide interaction must be reversible to release the DNA for gene therapy. Figure 7 illustrates how a DNA/peptide assembly may reach the cell nucleus for DNA delivery [64].
Several anti-cancer drugs in noncovalent assemblies with the cyclic peptide cyclo-Histidine-Histidine (Cyclo-HH) were evaluated both theoretically (from molecular dynamics simulations) and experimentally [65]. Most drugs tested required zinc and nitrate for forming the co-assemblies with the peptide, displaying biocompatibility and successful intracellular release except for cisplatin (CIS). Drug encapsulation and release were higher for epirubicin (EPI), doxorubicin (DOX), and methotrexate (MTX), in comparison to mitomycin-C (MIT) and 5-fluorouracil (5FU).
Targeted cancer therapy delivers a toxic payload selectively to target-expressing cells thereby preventing the toxicity of chemotherapy on normal tissues [66]. Recently, as alternatives to targeting tumors using antibodies-drug conjugates, the use of peptide-drug has been explored [67,68]. An exponentially growing research area in cancer therapy involves targeting specific cancer cells using combinations of peptides and drugs where the peptide binds to receptors occurring in specific cancer cells [69]. For example, covalent linkage using a bifunctional linker such as an oxime moiety allowed the binding of peptide and daunomicin and targeted delivery of the conjugate avoiding the drug systemic toxicity [69]. In another instance, peptide-drug conjugates increased the water solubility of paclitaxel, the tumor permeability of paclitaxel, the selective localization of the drug in the tumor, and the overall therapeutic index of the drug [70].
Another interesting approach has been targeting the epidermal growth factor receptor with an antibody linked to an anticancer lytic peptide. For example, anti-EGFR scFv is an antibody directed to epidermal growth factor receptor (EGFR) so that a fusion protein with a cetuximab-derived anti-EGFR scFv and the anticancer lytic peptide (ACLP) ZXR2, connected by a linker (G4S)3 and MMP2 cleavage site; this recombinant protein displayed anticancer properties on EGFR-overexpressed cancer cells [71].
Proteins and drugs have limited entrance into the brain due to the blood-brain barrier (BBB). Interestingly, the transcytosis ability of some peptides called Angiopeps allowed their accumulation in the brain endothelial cells [72]. These peptides are considered vectors capable of drug delivery to the brain [73]. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1). This peptide has three amino groups for covalently linking anti-cancer drugs such as daunomycin; the most cytotoxic conjugate was the one linking the drug to the peptide N-terminus [74]. In addition, tumors such as glioblastomas can bind Angiopeps; from fluorescence labeling of Angiopep-2 with Alexa488, the peptide was found co-localized with the low-density lipoprotein receptor protein 1 (LRP1) in brain endothelial cell monolayers. Angiopep-2 entry across the BBB is, in part, mediated by LRP1.
Peptides have also been used as alternatives to monoclonal antibodies (mAbs) to revert the immunosuppressive effect of cancers on oncolytic immune response by T cells [75]. Instead of using entire mAbs, peptides were used to block the PD-1/PD-L1 pathway and its immune suppressive effect, which occurs associated with cancers. PD-1 means Programmed Death 1 protein and PD-L1 means Programmed Death Ligand 1 represents a very important alternative in immunotherapy against cancer. For example, a series of macrocyclic peptides have been developed that exhibit binding strengths to PD-L1 over a range of sub-micromolar to micromolar concentrations; the macrocyclic PD-L1 targeting peptide pAC65 displayed a potency equivalent to the current FDA-approved mAbs for cancer immunotherapy [75]. Compared to mAbs, peptides are smaller in size, can have their synthesis easily scaled up in the biotechnology industry with reproducible results in different batches, display lower immunogenicity and longer shelf life; the use of peptides in cancer therapy was recently reviewed [76].
Once recognized the joint power of lytic peptides, peptides self-assembly, and nanotechnology, the importance of novel formulations for lytic peptides becomes clear. At this point, a good example refers to the lytic peptide gramicidin D. Gramicidin D (Gr) was first isolated by Dubos from soil samples containing Bacillus brevis [77]. It is a channel-forming polypeptide with 15 L- and D-amino acids residues including a β-helix secondary structure with an internal pore. Gr molecules combine to form homodimers with β-helix pore structures spanning the cell membrane. As a case study, we evaluate this important lytic peptide, its self-assembly, and its perspectives in future pharmacology against infectious diseases and cancer.
As models for membrane proteins, linear gramicidins have been extensively characterized regarding their organization, dynamics, and function. Gr pores in membranes resulting from self-assembled dimeric channels have been described in lipid bilayers, model membranes, and natural membranes [78,79,80,81,82,83]. Upon insertion in membranes, Gr molecules acquire a helix conformation and Gr dimeric channels become pores for single-file passive diffusion of Na+ and K+ cations with Na+ influx and K+ efflux from cells. Gr dimeric channels are cation-selective, form pores in model membranes, and conduct around 107 ions/ second [84]. Natural Gr (gramicidin D) consists mostly of gramicidin A (85%), which has four tryptophan residues at positions 9, 11, 13, and 15 imparting intrinsic fluorescence to the molecule [85]. The hydrophobic amino acid residues in the Gr molecule account for its low solubility in water and drive Gr self-assembly in water dispersions as nanoparticles with about 150 nm of mean hydrodynamic diameter [18,23,39,40]. Gramicidin D displayed a strict dependency on the nano-formulation for activity [18,23,24,25,27,57,86]. In combination with complementary antimicrobials such as the cationic polymer PDDA, synergism of action was observed. This complementary action involved PDDA ability to withdraw biopolymers from the microbial cell wall opening the way for Gr NPs interaction with the inner cell membrane. PDDA disrupts the cell wall and Gr NPs disrupts the cell membrane [18,23,39,40]. Figure 8 illustrates the combined activity of Gr NPs and nanodisks built to display an outermost layer of PDDA.
Other Gr formulations have been proposed involving graphene oxide [87], silver NPs stabilized with dodecane thiol [88] and fluoride for applications against biofilms on teeth [89].
First reported by our lab [23], the Gr NPs may become important not only for improving Gr antimicrobial performance but also as novel anti-cancer assemblies. Dissipation of ion concentration gradients across cell membranes hampers cells from properly regulating their intracellular environment. The potential anticancer properties of channel formers have not been sufficiently explored in pharmacology.
Energy depletion in cancer cells induced by Gr led to cancer cell death in renal carcinoma [90]. The selective fusion of cancer cell membranes with liposomes containing the fusogenic peptide pHLIP® at the low pH of tumors was an interesting strategy applied for Gr delivery [91]. The systemic toxicity of Gr could be circumvented by intra-tumoral administration, which was reported to be both safe and effective in murine xenograft studies in the absence of significant side effects [92].
An excellent review on the pharmacological potential of Gr against cancer pointed out that a comprehensive understanding of the effects of ionophore drugs upon cancer cells vs. normal tissues is still lacking and will be necessary before Gr uses in the clinic can evolve [90].
Several reports emphasize AMPs’ unexplored potential for novel applications, including against cancer. In a very interesting approach, the gramicidin A unimolecular channel was targeted to cancer cells by covalently binding a galactose molecule to the N-terminus of Gr A; thereby, the conjugate containing galactose moiety recognized the asialoglycoprotein on cancer cells, formed a unimolecular transmembrane channel and induced cancer cells death by apoptosis [93].
Cationic and lytic antimicrobial peptides have been blocked by negative charges on cancer cells; these charges are due to heparan sulfate on their surfaces [94]. Gramicidin D has the advantage of being a neutral peptide that inserts in cell membranes due to the hydrophobic effect.
Aiming at synergistic combinations of gramicidin A and doxorubicin against the spheroids from colorectal cancer cells (HT-29), the spheroid evolution, cell viability, and ATP levels were monitored at 24 and 48 h after the applied treatments; there was significant drop in cell viability and cellular ATP levels for all the experimental treatments suggesting that simultaneous use of Gr A and doxorubicin indeed acted synergistically against the spheroids [95].
Acting over a nanomolar range of concentrations against the human breast cancer cell line MCF-7, gramicidin A not only dissipated ion gradients across cancer cells but also became inserted in the inner mitochondrial membrane, reducing the H+ gradient and hampering ATP synthesis; there was a cytostatic action at 1 nanomolar Gr; the mitochondrial dysfunction induced mitophagy and, the lack of ATP caused cell cycle arrest [96]. Gr also inhibited the proliferation of human gastric cancer cells, arrested their cell cycle, and induced apoptosis [97]. Iturin A is a natural biosurfactant secreted by Bacillus which is also able to disrupt the cell membrane due to its cyclic character [45,46]. In combination with Gr A, there was cell membrane disruption, inhibited proliferation, apoptosis, and regulated inflammation in breast cancer cells [98].
In comparison with the general populations of cancer cells, cancer stem cells (CSCs) display a lack of differentiation, self-renewal capability, pluripotency, resistance to chemo- and radiotherapy, and higher tumorigenicity. In pancreatic ductal adenocarcinoma, CSCs contribute to an aggressive prognosis for this cancer and treatment resistance [99]. The effect of Gr A at 0.05 μM on pancreatic cancer stem cells was tumor sphere disintegration and a reduction in cell counting; there was CD47 down-regulation and modulation of macrophage/tumor cell interaction [100]. In another instance, Gr A inhibited the proliferation of both acute promyelocytic and chronic myeloid leukemia cell lines in the absence of hemolytic effects; combinations of GrA with other anticancer drugs were also evaluated revealing down-regulation of oncogenes, such as c-Myc, Eya3, and Axin 2 [101].
Table 1 provides an overview of assemblies involving peptides and their potential biomedical applications.
2. Structure-Function for AMPs and Peptide Mimetics Against Pathogens and Cancer
The discovery of new agents that promote pathogens cell lysis at concentrations that barely affect mammalian cells is of utmost importance to circumvent the problem of multidrug resistance often observed in the clinic for infections by microbial strains. Many natural peptides have evolved as antimicrobial agents (AMPs) in the most diverse types of living organisms [102,103,104]. What guarantees the effectiveness of AMPs is their mechanism of action based on their ability to bind to the microbial cell membrane affecting its functionality and leading to loss of microbial cell viability.
Structure determinants of high antimicrobial activity have been modeled in amphiphilic ternary copolymers and found to have anti-fouling moieties such as poly ethylene glycol, cationic groups, and hydrophobic lateral chains [105]. These functional groups imparted high antimicrobial potency and hemocompatibility. The separation of hydrophilic and hydrophobic domains in di-block copolymers was related to the loss of antimicrobial activity attributed to the stability of core-shell assemblies [105]. Mimicking antimicrobial peptides with polymers has been a powerful approach to create potent antimicrobial polymers promoting bacterial cell rupture and overcoming multidrug resistance of pathogenic strains [33,34,37,106]. The organization of functional subunits within AMPs has a significant influence on their performance. For example, segmentation of hydrophobic and cationic functionalities on antimicrobial polymers affected their selectivity between bacteria and mammalian cells [107].
A novel antimicrobial peptide mimic has been constructed from a combination of reversible addition-fragmentation chain transfer (RAFT) polymerization and ring-opening metathesis polymerization (ROMP); bottle brush copolymers cause lysis in the membrane and destabilize its integrity having the ability to self-assemble, a process that reduces their antimicrobial capacity. By attaching antimicrobial copolymers to a bottle brush structure, antimicrobial activity was improved. In this sense, the construction of several bottlebrush copolymers with varied architectures allowed to establish their structure-antimicrobial function relationship. The structure control in the design of peptides for high bioactivity and low cytotoxicity towards the host has often been reported from determinations of their hemolytic effect [108,109]. The utility of antimicrobial polymers and their assemblies showed how novel polymer structures can be directed to mimic antimicrobial peptides [57,58,106].
Amphiphilic Peptides (APs) are surfactant-like peptides (SLPs) whose sequence of hydrophobic and hydrophilic amino acid residues characterize their amphiphilic nature. Their primary sequence can yield secondary conformations resulting in different self-assembled nanostructures. A variety of shapes (nanotubes, nano rings, nano sheets or nanofibers) from self-assembled peptides can be obtained. Nanofibers, for example, have been observed for peptides with beta-sheets as secondary structures. Aiming at the applications of APs, their useful antimicrobial activity needs to be preserved in vivo to prevent biodegradation by proteolytic enzymes. Acylation of the N-terminus has been utilized for improved stability. The resultant self-assembly driven by the hydrophobic effect between acyl moieties often allows PAs to organize themselves into micelles with protected antibiofilm activity [110].
AMPs against resistant microorganisms typically have between 10 and 100 amino acid residues [111]. Self-assembly of AMPs into nanometer-scale structures can improve their antimicrobial activity, prolong their half-life, and increase their bioavailability. Six different AMPs with nine amino acid residues and central symmetry, containing triple tryptophan (WWW) or double tryptophan (WW) motifs were tested pointing out peptide K6 as the most potent against P. aeruginosa and S. aureus thanks to its ability to self-assemble into micelles. The K6 peptide also had potent action against biofilms formed by P. aeruginosa and S. aureus, revealing an increase in its lytic capacity against bacterial cell membranes. The reason why these peptides have this strong antimicrobial capacity is the fact that the WWW or WW motifs were placed in the middle or at the ends of the peptide to provide hydrophobicity and enhance self-assembly into micelles, facilitating their interaction with bacterial membranes and disruption of its integrity [112].
Another alternative against antimicrobial resistance has been combining antimicrobials, in particular, those with different targets [113]. Dosages could be reduced attenuating collateral effects; synergism or antagonism between antimicrobials could even be theoretically predicted [114]. A variety of natural AMPs with different structure and mode of action (porcine protegrin 1, caprine bactenecin ChBac3.4, human cathelicidin LL-37, human alpha- and beta-defensins such as HNP-1, HNP-4, hBD-2, hBD-3, and egg white lysozyme) have been combined with antibiotics such as gentamicin, ofloxacin, oxacillin, rifampicin or polymyxin B towards selected bacteria, including drug-sensitive and drug-resistant strains. For quantifying synergy, fractional inhibitory concentration indexes obtained from “checkerboard titrations” allowed to infer that synergy in antibacterial action mainly occurs between highly membrane-active AMPs (e.g., protegrin 1, hBD-3) and antibiotics with intracellular targets such as gentamicin or rifampicin [115]. Their synergistic use can cause cytotoxic effects even in strains resistant to conventional medicines.
Novel semi-synthetic biopolymers based on cellulose have been developed [116]. They are water-soluble alternatives to the insoluble cellulose representing a proper scaffold to incorporate various bioactive agents with beneficial therapeutic effects. Applications for these materials in wound dressings, tissue engineering, and drug delivery devices have been reviewed [87]. For example, hydroxypropyl methylcellulose and xyloglucan were crosslinked with citric acid resulting in a transparent and sustainable blend film, which could be loaded with gentamicin sulfate (GS) for providing outstanding antibacterial activity against Staphylococcus aureus and Escherichia coli [117]. In another instance, sodium alginate (Alg) reacted with gentamicin sulfate (GS) in an aqueous solution via carbodiimide crosslinking yielding a broad-spectrum bactericidal hydrogel capable of killing Pseudomonas aeruginosa, Escherichia coli and Staphylococcus aureus [118].
The shrinkage of hydrogels has also been exploited for wound closure and pushing-out of embedded drugs (drug release) as recently reviewed [119].
Bone infections such as chronic osteomyelitis, similarly to cancers, often require excision of the infected area together with some extent of healthy bone and soft tissues [120]. Besides surgery, antibiotics are also required for treatment to avoid recurrence of the infection [121]. Despite its nephrotoxicity and ototoxicity, vancomycin glycopeptide is an antibiotic in clinical use to prevent and treat the infections caused by Gram-positive bacteria, especially drug-resistant strains such as MRSA that occur in chronic osteomyelitis. Vancomycin administration intravenously can cause severe nephrotoxicity and ototoxicity. In contrast, local antibiotic delivery can maintain a relatively high concentration for a longer time improving the bioavailability of vancomycin. A vancomycin-loaded bone-like hydroxyapatite/poly amino acid scaffold was reported as biosafe promoting continuous release of vancomycin at the desired site both in vitro and in vivo. The 4–6 weeks release of vancomycin corresponded to the requirements of antibiotics used to treat chronic osteomyelitis [122].
Although AMPs have been recognized as one of the most promising routes toward developing next-generation antimicrobial compounds, major obstacles have been related to the lack of methodologies for systematic rational design and optimization of new AMPs [123]. In a very interesting study, a potent new AMP was developed using atomic detail molecular dynamics simulations for predicting structures; from a 14-residue poly leucine template, a minimalistic potent new AMP of only four types of amino acids (LDKA) formed large pores in microbial membranes at very low peptide-to-lipid ratios (1:1000) and low micromolar concentrations; there was negligible hemolysis at bactericidal concentrations; despite being too short a sequence to span cellular membranes, there was stacking of channels in each bilayer leaflet [123].
The challenges balancing enhanced antimicrobial efficacy with increased toxicity were faced in a de novo design of d-type β-hairpin AMP able to self-assemble as nanofibers in the bacterial membrane; the W-4 peptide showed high antimicrobial activity and a low hemolytic potency; in vivo, there was a synergistic effect in combination with antibiotics and wound-healing properties [124].
De novo-designed surfactant-like peptides (SLPs) are similar to conventional surfactants, with consecutive hydrophobic amino acid residues along their hydrophobic tail region and one or two charged residues as the hydrophilic head group. SLPs are amphiphilic molecules exhibiting short length, well-defined hydrophobic and hydrophilic regions, and excellent water solubility; importantly they self-assemble in a controllable manner depending on their molecular design and microenvironment. The properties and applications of SLPs were recently reviewed [125]. Multi-spanning integral membrane proteins, including G-protein coupled receptors (GPCR), ion channels, and ion transporters are very important as drug targets but their functional conformation is short-lived and requires stabilization to be studied and characterized in vitro. SLPs have been very successful in membrane protein stabilization and characterization [126]. Expression, stabilization, and purification of membrane proteins have been achieved via diverse protein synthesis systems plus detergents involving cell-free associated with self-assembled SLPs [127]. Cell-free productions associated with self-assembly lipid peptides have become cost-effective and competitive. Selecting the right surfactant is thus crucial because bottlenecks in elucidating the structure and function of membrane proteins involve the difficulty of producing large quantities of functional receptors. Various self-assembly peptide surfactants in commercial E. coli cell-free systems can rapidly produce milligrams of soluble GPCRs.
In an effective, low cost and resilient approach to inhibit infection by the SARS-CoV-2 virus, self-assembling peptides (SAPs) with tailored compositions were shown to bind to the SARS-CoV-2 Spike protein [128]. This antiviral platform has been developed based on a mixture of hetero-peptides self-assembled into functionalized β-sheets capable of specific binding to viral protein complexes; the peptides self-assembled into fibrils which also kept binding to viral proteins and were resilient to viral mutations. The viral infection involves the viral ‘Spike’ protein binding to the angiotensin-converting enzyme 2 (ACE2) receptor of the host, which is ubiquitously expressed in human cells [129]. With SAPs binding to the Spike protein, the virus binding to the ACE2 receptor of the host was hampered as was the virus infective power [128].
Lysosomal proteases and cathepsins degrade proteins and are upregulated during tumor growth and metastasis. In particular, cathepsin B is overexpressed in cancer cells, mainly in lysosomes being considered an important target for treating cancer [47,48]. Recently, a sophisticated peptide amphiphile with a lysosomal protease cathepsin B substrate sequence (Arg–Arg, RR)[130,131] and also other functionalities were designed to target cancer lysosomes [132]. This peptide named NDI-Lyso was obtained by solid-phase peptide synthesis with three motifs: (i) a self-assembling Phe–Phe (FF) sequence, (ii) a second self-assembling sequence hexyl naphthalene diimide alanine (C6-NDI-Ala) with dual function, self-assembly via aromatic–aromatic interactions and fluorescence tracking inside the cells [133], and (iii) a cell-penetrating peptide sequence Arg–Arg–Arg–Arg–Lys (RRRRK) consisting of an arginyl glycyl aspartic acid (RGD) sequence for binding to the extracellular matrix of the cell and cathepsin B substrate sequence (Arg–Arg, RR) for localization of the peptide amphiphiles in the cancer lysosomes [132]. One should notice that cleavage converting NDI-Lyso-RGD to NDI-R3 changes the hydrophobic-hydrophilic character of the molecule producing a more hydrophobic molecule so that the self-assembly further improves driven by increased hydrophobic effect. Thereby, the peptide self-assembly yielding fibers could be observed inside the lysosomes with lysosomal swelling and rupture, cancer cell death by apoptosis, and reduction of tumor growth in a variety of cancerous and drug-resistant tumors. In addition, cytotoxicity evaluated from IC50 determinations occurred at low values of peptide concentration (∼10 μM) in a wide variety of cancerous and drug-resistant cell lines [132]. This work shows the sophisticated NDI-Lyso-RGD peptide targeted to the lysosomes of cancer cells both before and after its cleavage by cathepsin B; the mechanism of action for the NDI-Lyso-RGD peptide involves binding to the extracellular matrix integrin receptors, internalization targeting the lysosome, peptide cleavage by cathepsin; self-assembly of the NDI-R3 peptide, lysosomal disruption and apoptosis [132]. Figure 9 shows a scheme of peptides able to become active in lysosomes, self-assembling thereafter, and producing aggregates that disrupt this organelle.
Bacteriophages evolved to attack microbes with high efficiency, eventually inducing their cell lysis. Recently, bacteriophage-inspired peptides have been developed to fight antibiotic-resistant bacteria. Phage mimetic nanoparticles (PhaNPs) with high antibacterial activity have been designed to avoid the immune responses of humans. For example, core-shell nanoparticles, with a silica core, were conjugated with silver-coated gold nanospheres to which the synthetic antimicrobial peptide Syn-71 was covalently bound (PhaNP@Syn71); infected wounds in mice treated with these nanoparticles and tested for inhibition of bacterial growth showed above 99.99% of microbial killing in absence of toxicity against HaCaT human skin cells, suggesting good biocompatibility[134].
Mycobacteria infections such as tuberculosis remain the leading cause of death, especially in the developing countries [135]. Peptidoglycan (PG), the major component of the cell wall in mycobacteria, is often a prime drug target [136]. PG consists of glycan strands of N-acetyl muramic acid (NAM) and N-acetyl glucosamine (NAG), linked by β-1,4 glycosidic bonds. Several modifications have been described in mycobacterial PG. The presence of a mixture of both the typical NAM and its hydroxylated derivative, N-glycolyl muramic acid (MurGlyc) has been observed with mycobacterial PG endowed with additional hydrogen bonds able to strengthen the mycobacteria cell wall [137]. Given the lack of efficient drugs to hydrolyze the cell wall, drug-resistant strains of Mycobacterium tuberculosis point out the urgent need for alternative therapeutic agents. Viruses that infect bacteria such as the bacteriophages, thanks to their endolysins, can break down bacterial peptidoglycan at the terminal stage of the phage reproduction cycle [138]. However, a better understanding of the regulatory mechanism of these endolysins and their mode of action is required before further developments effective in the clinic can emerge [89,90].
Several issues need to be addressed for combating cancer: the development of drug resistance derived from the activation of drug efflux pumps, the antiapoptotic defense, the enhanced DNA repair ability, and the antioxidant systems [141]. Some strategies can increase the sensitivity of the resistant cancer cells to drugs. For example, synergistic approaches involve combining chemotherapy with photodynamic therapy, gene therapy, or immunotherapy [18]. Several therapeutic agents gathered in one single nanometric system often reveal the power of biomimetic nanotechnology. In this regard, lytic peptides can contribute to cancer cell lysis releasing many antigens from the tumor to elicit an efficient immune response against several antigens of the tumoral cells [86].
Another interesting strategy has been testing the structure-function relationship for several parts of a given lytic protein molecule. Triallysin is a protein found in the salivary glands of Triatoma infestans, the Chagas disease vector. This protein creates pores in the cell membrane of a variety of prokaryotic and eukaryotic cells. Peptides synthesized from the N-terminal region of triallysin showed similar helical secondary structures in solvents or in detergent micelles and exhibited the lytic property not only against the protozoan but also against Escherichia coli over a range of micromolar concentrations. The most active peptide was the peptide P6 [142].
A cyclic peptide PROTAC induced intracellular degradation of palmitoyltransferase and reduced PD-L1 expression in human cervical cancer cells [143]. As a consequence, immune suppressive effects of the PD-L1 pathway were prevented, and enhancement of immunity against the tumor was achieved.
The structure-function relationship for antimicrobial peptides has been systematically reviewed for decades [57,106,144,145,146,147,148]. They performed well regarding their desired and desirable actions such as membrane perturbing [144], lytic [145,146,147,149], oncolytic [147], antibacterial [57,150], and/or biofilm disruptive effects [148,151,152] on a variety of prokaryotic and/or eukaryotic cells. Common drawbacks in vivo such as general cytotoxicity, poor stability due to the proteolytic action of enzymes, and poor bioavailability due to rapid clearance have also been pointed out but considered controllable from the formulation point of view [18,23]. In this regard, there is yet much to be learned and explored regarding the effect of the formulation on peptide action. Another promising area for research refers to the similar lytic behavior of antimicrobial polymers and peptides [106]. In these systems, the lytic effect can also be controlled by the degree of freedom of the antimicrobial in the formulation [37]. Applications of the lytic effect in cancer treatment can be foreseen as promising given appropriate formulations.
3. Peptides and Their Assemblies for Treating Cancers
In this section, the importance of cell-penetrating peptides (CPPs), tumor-targeting, lytic peptides, and biomimetic delivery systems in future pharmacology are discussed.
CPPs can deliver a variety of drugs, despite their short time of action and poor cell specificity [153,154]. Against solid tumors such as breast, glioma, colorectal, and melanoma cancers, peptide-based therapeutics have been reviewed and show promising perspectives [155]. CPPs are efficient in delivering large quantities of drugs but their non-specific activity has been requiring additional strategies such as tumor microenvironment targeting and/or specific targeting to cancer cell receptors [156]. Figure 10 illustrates the diverse mechanisms by which anticancer peptides induce tumor cell death through immune modulation, apoptosis induction, and membrane disruption. Peptides as immune modulators enhance antitumor immunity by activating CD4+ helper T cells and CD8+ cytotoxic T cells. Death receptor-activating peptides engage TNF-R1, Fas, and TRAIL-R2, leading to caspase-8 activation and triggering the extrinsic apoptosis pathway. Mitochondria-disrupting peptides induce mitochondrial membrane destabilization, resulting in caspase-3 activation and intrinsic apoptosis. In addition, membrane-disrupting peptides compromise the integrity of the cancer cell membrane, leading to uncontrolled water efflux and cell shrinkage.
The non-membrane targets of AMPs and their potential to combat antibiotic-resistant infections have been the subject of comprehensive review articles. Their mechanisms of action usually target intracellular components essential for bacterial survival such as transcription, translation, cell wall synthesis, cell division, chaperone proteins, enzyme activity, and channel proteins. All these actions make AMPs promising candidates for novel antimicrobial treatments, with specificity against prokaryotic microorganisms. Non-membrane-targeting AMPs can infiltrate bacterial biofilms effectively but their role in cancer treatments has not been established yet [157,158].
Among the lytic peptides with anticancer activity, two natural peptides should be pointed out: melittin (MLT)[159] and gramicidin D [160].
Melittin (MLT) is a 26 amino acid amphiphilic cationic peptide found in the venom of the European honeybee, which has been described as a promising agent in anticancer treatment since the 1950s [161]. However, MLT indiscriminate lytic cytotoxicity in vivo has been a major drawback impeding its use in the clinic; MLT insertion into the lipid bilayers of membranes leads to disruption of cells [162,163,164], which was reported effective even against solid tumors resulting in cancer cell lysis [165]. MLT was also reported to act via cellular apoptosis due to the lysis of mitochondrial membranes [166].
In tumors, due to the disruption of cancer cell membranes, MLT can induce tumor necrosis or apoptosis with the release of intracellular contents such as whole-tumor antigens and damage-associated molecular patterns [167]. Recently, in nanometric formulations of MLT with lipid nanoparticles targeted to lymph nodes (LN), an important and systemic anti-tumor immune response was achieved; there was whole tumor antigen release in situ and activation of antigen-presenting cells (APCs) in LNs accompanied by improved antigen-specific CD8+ T cell responses; experiments in mice models with tumors, revealed primary and distant tumor growth inhibition of 95% and 92%, respectively [168]. This α-melittin-NPs formulation was an effective LN-targeted whole-cell nanovaccine against the tumor representing a valuable immunotherapeutic tool.
The problem of targeting anticancer peptides and reducing their indiscriminate action against all host cells was addressed using an anticancer polymeric prodrug candidate of AMPs; there was the synthesis of a dual-release prodrug combining an AMP and doxorubicin as the anticancer agent and evaluation of their activities against cancer cells [169]. The synthesis yielded a poly (ethylene glycol) PEG-based dual-release prodrug or two individual pegylated prodrugs susceptible to hydrolysis of cathepsin B-labile peptide linker and an acid-sensitive acyl hydrazone bond for the AMP and doxorubicin prodrugs, respectively; there were differential anti-cancer activities for the polymer conjugates as compared to the free peptide evaluated against ovarian cancer and non-malignant cells. Pegylation was reversible due to protease activity and assured improved therapeutics against cancer. Using a derivative of the P18 peptide, namely, the D-P18 (D-P18 peptide was synthesized from D -amino acids and modified with leucine residue at position 8), yielded a small increase in the therapeutic index to only 2.1 against Hs832 and A2780P cells; this peptide induced the mitochondria-associated pathway of apoptosis in pancreatic carcinoma and cervical cancer cells lines [170]; polymeric prodrugs of the D-P18 also displayed this same mechanism of action[169]. An α-helical peptide derived from magainin 2 was pegylated and had its antimicrobial activity reduced and cytotoxicity against CHO-K1 cells abolished [171]. Oncolytic peptides can also be synthesized from modifications or stapling of other existing peptides such as the Ano-3/3s peptide produced from wasp venom, which is resistant to proteases, is membranolytic and induces prolonged immune responses against melanoma cells [172].
The polymer-peptides conjugates significantly reduced side effects, killing cancer cells and reducing cytotoxicity to normal cells [173,174]. There was selectivity against tumor lineages, absence of a resistance phenotype (a common occurrence when chemotherapy is used), and superior tumor cytotoxicity as compared to doxorubicin alone against breast cancer tumor spheres [173]. Combination therapies of two anticancer agents with different mechanisms of action, despite their complex development, are less prone to drug resistance than treatments with single drugs [174]. The activity of antimicrobial peptides with selective antitumor mechanisms has been reviewed [175].
In an interesting biomimetic anti-cancer approach, a construction based on self-assembled NPs with amphiphilic penetrating Zhein protein from corn and a rabies virus glycoprotein carrying temozolomide (TMZ) was able to treat glioma, the most frequent brain cancer. The self-assembled amphiphilic NPs penetrated the blood-brain barrier, targeted the tumor with the virus glycoprotein, and released TMZ. These NPs were considered ideal for treating brain diseases due to their biocompatible character, BBB penetration, targeting to the brain, and nanometric size [176].
Several lytic peptides with activity against cancer cells have been reported [59,145,177,178]. For example, a short cationic peptide composed of D- and L-leucines, lysines, and arginines displayed selective toxicity toward cancer cells and prevented the formation of lung metastasis in mice with no detectable side effects; the mechanism of action involved membrane depolarization of cancer cells at a few micromolar concentration; the simple peptide structure, high solubility in water and resistance to degradation in vivo made it a good candidate for treating cancer [178]. In general, lytic anti-cancer peptides empowered by self-assembly [179], cyclization [180], or diastereoisomery from the manipulation of the amino acids sequence [181], novel nanoformulations for peptide loading [23,25,27,57,173] and conjugation of peptide drugs to natural or synthetic polymers have been improving their perspectives for use in anti-cancer therapy. The current methods and formulations to enhance the half-life of peptide drugs and improve peptide drug delivery have been reviewed [182].
With the advancement of technology, conventional cancer treatments have become increasingly surmountable. One of the factors that lead to this is the increased resistance of tumor cells and bacteria against existing drugs. Cancer and microbial infections are still diseases that cause high mortality rates worldwide, and the discovery of new antitumor and antimicrobial drugs is still challenging for medicine, especially concerning the consequences of side effects [163,183]. The use of targeting peptides appears as a promising strategy in this context. From this perspective, peptides act by reducing the amount of drug administered, increasing bioavailability, increasing specific drug targeting, and being less cytotoxic in non-tumor cells [155,184].
The LTX-315 is an oncolytic peptide able to change the tumor microenvironment (TME) and induce immune-mediated antitumor action [185]. TME often hampers the penetration and activity of T-cells able to combat the tumor [186]. Combinatorial strategies have been proposed such as oncolytic virotherapy so that genetically modified or naturally occurring viruses can infect and replicate selectively in cancer cells being able to induce not only lysis but also immunogenic cell death [187,188]. Upon oncolytic cell disruption, tumor-associated antigens are released and taken up by dendritic cells, which activate specific anti-tumor T cells; furthermore, the lysis of cancer cells releases factors known as damage-associated molecular patterns that activate the immunogenic cell death. Recently, oncolytic viruses encoding tumor antigens and tumor antigen-decorated adenoviral platforms have been used as cancer vaccines eliciting local and systemic antitumor response in a poorly immunogenic tumor melanoma mice model [189]. Recently, new branched oncolytic peptides BOP7 and BOP9 proved to elicit the release of damage-associated molecular patterns DAMPS, mediators of ICD, in pancreatic cancer cells [190]. These peptides selectively killed tumor cells but not cells of non-tumor origin. This result was interpreted as due to their repeated cationic sequences for multivalent binding to heparan sulfate glycosaminoglycans displaying multiple anionic charges on cancer cells. BOPs triggered the release of DAMPs, particularly HMGB1, IFN-β, and ATP, by dying cells whereas in vivo, nude mice showed 20% inhibition of tumor grafting and growth in pancreatic cancer.
Preventing the development of resistance is the biggest challenge when it comes to the mechanism of attack on tumor cells. In this scenario, membrane-disrupting peptides/peptidomimetics (MDPs) are antitumor drugs that directly attack the physical structure of the membrane. Its isolated or combined action in the form of adjuvants guarantees rapid attack on cancer cells, intensive action, and reduced or absent resistance. Therefore, recent MDP designs promise the development of more effective therapy capable of decreasing resistance and reducing side effects [191].
Table 2 shows the variety of formulations for AMPS against pathogens or cancer.
5. Conclusions
A multidisciplinary approach is needed for fighting drug resistance in infectious diseases and/or cancer. Natural or synthetic AMPs have been intensively and extensively studied for many reasons. They have easily changeable structure-function relationships, often display the property of self-assembly, can be easily endowed with targeting, have unlimited possibilities for combination or conjugation with specific drugs and mechanically disrupt cells via mechanisms difficult to counteract from the microbe or cancer cell point of view. The joint venture of nanotechnology, self-assembly, and combinations with other bioactive compounds has been poorly but promisingly explored and advancements can be foreseen for novel AMPs nanometric formulations in biomedical applications. Synergistic action for components in each formulation has been reducing doses needed for microbial or cancer cell death in many instances. Sinergy represents one of the most promising avenues to follow for achieving efficient treatments against infectious diseases and cancer. The clinical application of oncolytic peptides has not been achieved yet due to structural instability, proteolytic degradation, and undesired toxicity when AMPs are administered systemically. The most prolific trend nowadays is mimicking AMPs with other molecules such as polymers. Oncolytic peptides can be copied by oncolytic polymers that are chemically stable, not susceptible to degradation in vivo, and easier to scale up in the biotechnology industry. Immunogenic cell death triggered by cancer cell lysis is one of the most promising approaches for fighting cancer.
Author Contributions
Conceptualization, A.M.C.-R.; methodology, A.M.C.-R., R. M. S., Y.P.B.; validation, A.M.C.-R., Y.P.B.; formal analysis, A.M.C.-R.; investigation, A.M.C.-R., R.M.S.; resources, A.M.C.-R.; writing—original draft preparation, A.M.C.-R., R.M.S.; writing—review and editing, A.M.C.-R., Y.P.B.; supervision, A.M.C.-R.; project administration, A.M.C.-R.; funding acquisition, A.M.C.-R. All authors have read and agreed to the published version of the manuscript.
Funding
This bibliography research, critical evaluation, discussion, and review writing was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), grants number 304091/2023-5 and 302758/2019-4 to A. M. C.-R. The APC was funded by CNPq grants to A. M. C.-R numbers 304091/2023-5 and 302758/2019-4. R. M. S. was the recipient of a MSc fellowship from CAPES grant 88887.829313/2023-00.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or description of available literature in the review; in the writing of the manuscript; or in the decision to publish the synthesis of the bibliographic search.
References
- Levin, A.; Hakala, T.A.; Schnaider, L.; Bernardes, G.J.L.; Gazit, E.; Knowles, T.P.J. Biomimetic Peptide Self-Assembly for Functional Materials. Nat Rev Chem 2020, 4, 615–634. [Google Scholar] [CrossRef] [PubMed]
- Simonson, A.W.; Aronson, M.R.; Medina, S.H. Supramolecular Peptide Assemblies as Antimicrobial Scaffolds. Molecules 2020, 25, 2751. [Google Scholar] [CrossRef] [PubMed]
- Juković, M.; Ratkaj, I.; Kalafatovic, D.; Bradshaw, N.J. Amyloids, Amorphous Aggregates and Assemblies of Peptides – Assessing Aggregation. Biophysical Chemistry 2024, 308, 107202. [Google Scholar] [CrossRef]
- Song, Q.; Cheng, Z.; Kariuki, M.; Hall, S.C.L.; Hill, S.K.; Rho, J.Y.; Perrier, S. Molecular Self-Assembly and Supramolecular Chemistry of Cyclic Peptides. Chem. Rev. 2021, 121, 13936–13995. [Google Scholar] [CrossRef]
- Hu, X.; Liao, M.; Gong, H.; Zhang, L.; Cox, H.; Waigh, T.A.; Lu, J.R. Recent Advances in Short Peptide Self-Assembly: From Rational Design to Novel Applications. Current Opinion in Colloid & Interface Science 2020, 45, 1–13. [Google Scholar] [CrossRef]
- Wang, J.; Liu, K.; Xing, R.; Yan, X. Peptide Self-Assembly: Thermodynamics and Kinetics. Chem. Soc. Rev. 2016, 45, 5589–5604. [Google Scholar] [CrossRef]
- Mahadevi, A.S.; Sastry, G.N. Cooperativity in Noncovalent Interactions. Chem. Rev. 2016, 116, 2775–2825. [Google Scholar] [CrossRef]
- Dasgupta, A.; Mondal, J.H.; Das, D. Peptide Hydrogels. RSC Adv. 2013, 3, 9117. [Google Scholar] [CrossRef]
- Yan, C.; Pochan, D.J. Rheological Properties of Peptide-Based Hydrogels for Biomedical and Other Applications. Chem. Soc. Rev. 2010, 39, 3528. [Google Scholar] [CrossRef]
- Tomasini, C.; Castellucci, N. Peptides and Peptidomimetics That Behave as Low Molecular Weight Gelators. Chem. Soc. Rev. 2013, 42, 156–172. [Google Scholar] [CrossRef]
- Wu, L.; He, Y.; Mao, H.; Gu, Z. Bioactive Hydrogels Based on Polysaccharides and Peptides for Soft Tissue Wound Management. J. Mater. Chem. B 2022, 10, 7148–7160. [Google Scholar] [CrossRef]
- Villa-Camacho, J.C.; Ghobril, C.; Anez-Bustillos, L.; Grinstaff, M.W.; Rodríguez, E.K.; Nazarian, A. The Efficacy of a Lysine-Based Dendritic Hydrogel Does Not Differ from Those of Commercially Available Tissue Sealants and Adhesives: An Ex Vivo Study. BMC Musculoskelet Disord 2015, 16, 116. [Google Scholar] [CrossRef]
- Zhu, H.; Mei, X.; He, Y.; Mao, H.; Tang, W.; Liu, R.; Yang, J.; Luo, K.; Gu, Z.; Zhou, L. Fast and High Strength Soft Tissue Bioadhesives Based on a Peptide Dendrimer with Antimicrobial Properties and Hemostatic Ability. ACS Appl. Mater. Interfaces 2020, 12, 4241–4253. [Google Scholar] [CrossRef] [PubMed]
- Shan, B.; Wu, F. Hydrogel-Based Growth Factor Delivery Platforms: Strategies and Recent Advances. Advanced Materials 2023, 2210707. [Google Scholar] [CrossRef]
- Bruggeman, K.F.; Rodriguez, A.L.; Parish, C.L.; Williams, R.J.; Nisbet, D.R. Temporally Controlled Release of Multiple Growth Factors from a Self-Assembling Peptide Hydrogel. Nanotechnology 2016, 27, 385102. [Google Scholar] [CrossRef]
- Zhu, P.; Yan, X.; Su, Y.; Yang, Y.; Li, J. Solvent-Induced Structural Transition of Self-Assembled Dipeptide: From Organogels to Microcrystals. Chemistry A European J 2010, 16, 3176–3183. [Google Scholar] [CrossRef]
- Bernheimer, A.W.; Rudy, B. Interactions between Membranes and Cytolytic Peptides. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes 1986, 864, 123–141. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Antimicrobial Peptides and Their Assemblies. Future Pharmacology 2023, 3, 763–788. [Google Scholar] [CrossRef]
- Portlock, S.H.; Clague, M.J.; Cherry, R.J. Leakage of Internal Markers from Erythrocytes and Lipid Vesicles Induced by Melittin, Gramicidin S and Alamethicin: A Comparative Study. Biochimica et Biophysica Acta (BBA) - Biomembranes 1990, 1030, 1–10. [Google Scholar] [CrossRef]
- Katsu, T.; Kuroko, M.; Morikawa, T.; Sanchika, K.; Fujita, Y.; Yamamura, H.; Uda, M. Mechanism of Membrane Damage Induced by the Amphipathic Peptides Gramicidin S and Melittin. Biochimica et Biophysica Acta (BBA) - Biomembranes 1989, 983, 135–141. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; Selsted, M.E.; White, S.H. Sizing Membrane Pores in Lipid Vesicles by Leakage of Co-Encapsulated Markers: Pore Formation by Melittin. Biophysical Journal 1997, 72, 1762–1766. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Zhu, Y.; Xu, K.; Wu, F. Turning Toxicants into Safe Therapeutic Drugs: Cytolytic Peptide−Photosensitizer Assemblies for Optimized In Vivo Delivery of Melittin. Adv Healthcare Materials 2018, 7, 1800380. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Betancourt, Y.; Zaia, R.; Evangelista, M.F.; Ribeiro, R.T.; Roncoleta, B.M.; Mathiazzi, B.I.; Carmona-Ribeiro, A.M. Characterization and Differential Cytotoxicity of Gramicidin Nanoparticles Combined with Cationic Polymer or Lipid Bilayer. Pharmaceutics 2022, 14, 2053. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.A.; Olivares-Ortega, C.; Soto-Arriaza, M.A.; Carmona-Ribeiro, A.M. Interaction of Gramicidin with DPPC/DODAB Bilayer Fragments. Biochim Biophys Acta 2012, 1818, 3064–3071. [Google Scholar] [CrossRef]
- Ragioto, D.A.; Carrasco, L.D.; Carmona-Ribeiro, A.M. Novel Gramicidin Formulations in Cationic Lipid as Broad-Spectrum Microbicidal Agents. Int J Nanomedicine 2014, 9, 3183–3192. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; Midmore, B.R. Synthetic Bilayer Adsorption onto Polystyrene Microspheres. Langmuir 1992, 8, 801–806. [Google Scholar] [CrossRef]
- Xavier, G.R.S.; Carmona-Ribeiro, A.M. Cationic Biomimetic Particles of Polystyrene/Cationic Bilayer/Gramicidin for Optimal Bactericidal Activity. Nanomaterials 2017, 7. [Google Scholar] [CrossRef]
- Lincopan, N.; Espíndola, N.; Vaz, A.; Carmonaribeiro, A. Cationic Supported Lipid Bilayers for Antigen Presentation. International journal of pharmaceutics 2007, 340, 216–222. [Google Scholar] [CrossRef]
- Martins, L.M.S.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Cationic Vesicles as Bactericides. Langmuir 1997, 13, 5583–5587. [Google Scholar] [CrossRef]
- Vieira, D.B.; Carmona-Ribeiro, A.M. Cationic Lipids and Surfactants as Antifungal Agents: Mode of Action. J Antimicrob Chemother 2006, 58, 760–767. [Google Scholar] [CrossRef]
- Melo, L.D.; Mamizuka, E.M.; Carmona-Ribeiro, A.M. Antimicrobial Particles from Cationic Lipid and Polyelectrolytes. Langmuir 2010, 26, 12300–12306. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.D.; Palombo, R.R.; Petri, D.F.S.; Bruns, M.; Pereira, E.M.A.; Carmona-Ribeiro, A.M. Structure–Activity Relationship for Quaternary Ammonium Compounds Hybridized with Poly(Methyl Methacrylate). ACS Appl. Mater. Interfaces 2011, 3, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- de Melo Carrasco, L.D.; Sampaio, J.L.M.; Carmona-Ribeiro, A.M. Supramolecular Cationic Assemblies against Multidrug-Resistant Microorganisms: Activity and Mechanism of Action. Int J Mol Sci 2015, 16, 6337–6352. [Google Scholar] [CrossRef]
- Carrasco, L.D. de M.; Bertolucci, R.J.; Ribeiro, R.T.; Sampaio, J.L.M.; Carmona-Ribeiro, A.M. Cationic Nanostructures against Foodborne Pathogens. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Lipid Bilayer Fragments and Disks in Drug Delivery. Curr Med Chem 2006, 13, 1359–1370. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Bilayer-Forming Synthetic Lipids: Drugs or Carriers? Current Medicinal Chemistry 2003, 10, 2425–2446. [Google Scholar] [CrossRef]
- Sanches, L.M.; Petri, D.F.S.; de Melo Carrasco, L.D.; Carmona-Ribeiro, A.M. The Antimicrobial Activity of Free and Immobilized Poly (Diallyldimethylammonium) Chloride in Nanoparticles of Poly (Methylmethacrylate). Journal of Nanobiotechnology 2015, 13, 58. [Google Scholar] [CrossRef]
- Galvão, C.N.; Sanches, L.M.; Mathiazzi, B.I.; Ribeiro, R.T.; Petri, D.F.S.; Carmona-Ribeiro, A.M. Antimicrobial Coatings from Hybrid Nanoparticles of Biocompatible and Antimicrobial Polymers. International Journal of Molecular Sciences 2018, 19, 2965. [Google Scholar] [CrossRef]
- Zaia, R.; Quinto, G.M.; Camargo, L.C.S.; Ribeiro, R.T.; Carmona-Ribeiro, A.M. Transient Coatings from Nanoparticles Achieving Broad-Spectrum and High Antimicrobial Performance. Pharmaceuticals 2023, 16, 816. [Google Scholar] [CrossRef]
- Camargo, L.C.D.S.; Bazan, B.R.; Ribeiro, R.T.; Quinto, G.M.; Muniz, A.C.B.; Carmona-Ribeiro, A.M. Antimicrobial Coatings from Gramicidin D Nanoparticles and Polymers. RSC Pharm. 2024. 10.1039.D4PM00124A. [Google Scholar] [CrossRef]
- Sobral, C.N.C.; Soto, M.A.; Carmona-Ribeiro, A.M. Characterization of DODAB/DPPC Vesicles. Chemistry and Physics of Lipids 2008, 152, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Carmona Ribeiro, A.M.; Chaimovich, H. Preparation and Characterization of Large Dioctadecyldimethylammonium Chloride Liposomes and Comparison with Small Sonicated Vesicles. Biochimica et Biophysica Acta (BBA) - Biomembranes 1983, 733, 172–179. [Google Scholar] [CrossRef]
- Sajid, M.; Ahmad Khan, M.S.; Singh Cameotra, S.; Safar Al-Thubiani, A. Biosurfactants: Potential Applications as Immunomodulator Drugs. Immunology Letters 2020, 223, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Ceresa, C.; Fracchia, L.; Sansotera, A.C.; De Rienzo, M.A.D.; Banat, I.M. Harnessing the Potential of Biosurfactants for Biomedical and Pharmaceutical Applications. Pharmaceutics 2023, 15, 2156. [Google Scholar] [CrossRef]
- Pueyo, M.T.; Mutafci, B.A.; Soto-Arriaza, M.A.; Di Mascio, P.; Carmona-Ribeiro, A.M. The Self-Assembly of a Cyclic Lipopeptides Mixture Secreted by a B. Megaterium Strain and Its Implications on Activity against a Sensitive Bacillus Species. PLoS One 2014, 9, e97261. [Google Scholar] [CrossRef]
- Pueyo, M.T.; Bloch, C.; Carmona-Ribeiro, A.M.; di Mascio, P. Lipopeptides Produced by a Soil Bacillus Megaterium Strain. Microb Ecol 2009, 57, 367–378. [Google Scholar] [CrossRef]
- Banat, I.M.; Franzetti, A.; Gandolfi, I.; Bestetti, G.; Martinotti, M.G.; Fracchia, L.; Smyth, T.J.; Marchant, R. Microbial Biosurfactants Production, Applications and Future Potential. Appl Microbiol Biotechnol 2010, 87, 427–444. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; De Bruijn, I.; Nybroe, O.; Ongena, M. Natural Functions of Lipopeptides from Bacillus and Pseudomonas: More than Surfactants and Antibiotics. FEMS Microbiol Rev 2010, 34, 1037–1062. [Google Scholar] [CrossRef]
- Ljubetič, A.; Gradišar, H.; Jerala, R. Advances in Design of Protein Folds and Assemblies. Current Opinion in Chemical Biology 2017, 40, 65–71. [Google Scholar] [CrossRef]
- Insua, I.; Montenegro, J. 1D to 2D Self Assembly of Cyclic Peptides. J. Am. Chem. Soc. 2020, 142, 300–307. [Google Scholar] [CrossRef]
- Cole, A.M.; Shi, J.; Ceccarelli, A.; Kim, Y.-H.; Park, A.; Ganz, T. Inhibition of Neutrophil Elastase Prevents Cathelicidin Activation and Impairs Clearance of Bacteria from Wounds. Blood 2001, 97, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Groisman, E.A.; Parra-Lopez, C.; Salcedo, M.; Lipps, C.J.; Heffron, F. Resistance to Host Antimicrobial Peptides Is Necessary for Salmonella Virulence. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 11939–11943. [Google Scholar] [CrossRef] [PubMed]
- Islam, D.; Bandholtz, L.; Nilsson, J.; Wigzell, H.; Christensson, B.; Agerberth, B.; Gudmundsson, G.H. Downregulation of Bactericidal Peptides in Enteric Infections: A Novel Immune Escape Mechanism with Bacterial DNA as a Potential Regulator. Nat Med 2001, 7, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Bals, R.; Weiner, D.J.; Meegalla, R.L.; Wilson, J.M. Transfer of a Cathelicidin Peptide Antibiotic Gene Restores Bacterial Killing in a Cystic Fibrosis Xenograft Model. J. Clin. Invest. 1999, 103, 1113–1117. [Google Scholar] [CrossRef]
- Bowdish, D.M.E.; Davidson, D.J.; Lau, Y.E.; Lee, K.; Scott, M.G.; Hancock, R.E.W. Impact of LL-37 on Anti-Infective Immunity. Journal of Leukocyte Biology 2004, 77, 451–459. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E.W. Immune Modulation by Multifaceted Cationic Host Defense (Antimicrobial) Peptides. Nat Chem Biol 2013, 9, 761–768. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; De Melo Carrasco, L.D. Novel Formulations for Antimicrobial Peptides. International Journal of Molecular Sciences 2014, 15, 18040–18083. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Self-Assembled Antimicrobial Nanomaterials. IJERPH 2018, 15, 1408. [Google Scholar] [CrossRef]
- Pennarun, B.; Gaidos, G.; Bucur, O.; Tinari, A.; Rupasinghe, C.; Jin, T.; Dewar, R.; Song, K.; Santos, M.T.; Malorni, W.; et al. killerFLIP: A Novel Lytic Peptide Specifically Inducing Cancer Cell Death. Cell Death Dis 2013, 4, e894–e894. [Google Scholar] [CrossRef]
- Vaezi, Z.; Bortolotti, A.; Luca, V.; Perilli, G.; Mangoni, M.L.; Khosravi-Far, R.; Bobone, S.; Stella, L. Aggregation Determines the Selectivity of Membrane-Active Anticancer and Antimicrobial Peptides: The Case of killerFLIP. Biochimica et Biophysica Acta (BBA) - Biomembranes 2020, 1862, 183107. [Google Scholar] [CrossRef]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An Overview of Active and Passive Targeting Strategies to Improve the Nanocarriers Efficiency to Tumour Sites. Journal of Pharmacy and Pharmacology 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fan, Z.; Zhang, X.; Li, H.; Sun, Y.; Cao, M.; Wei, G.; Wang, J. pH-Responsive Self-Assemblies from the Designed Folic Acid-Modified Peptide Drug for Dual-Targeting Delivery. Langmuir 2021, 37, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Z.; Ma, H.; Cao, M. Application of Peptides in Construction of Nonviral Vectors for Gene Delivery. Nanomaterials 2022, 12, 4076. [Google Scholar] [CrossRef] [PubMed]
- Urello, M.; Hsu, W.-H.; Christie, R.J. Peptides as a Material Platform for Gene Delivery: Emerging Concepts and Converging Technologies. Acta Biomaterialia 2020, 117, 40–59. [Google Scholar] [CrossRef]
- Vlachou, A.; Kumar, V.B.; Tiwari, O.S.; Rencus-Lazar, S.; Chen, Y.; Ozguney, B.; Gazit, E.; Tamamis, P. Co-Assembly of Cancer Drugs with Cyclo-HH Peptides: Insights from Simulations and Experiments. ACS Appl. Bio Mater. 2024, 7, 2309–2324. [Google Scholar] [CrossRef]
- Pryyma, A.; Matinkhoo, K.; Bu, Y.J.; Merkens, H.; Zhang, Z.; Bénard, F.; Perrin, D.M. Synthesis and Preliminary Evaluation of Octreotate Conjugates of Bioactive Synthetic Amatoxins for Targeting Somatostatin Receptor (Sstr2) Expressing Cells. RSC Chem. Biol. 2022, 3, 69–78. [Google Scholar] [CrossRef]
- Kim, H.; Hwang, D.; Choi, M.; Lee, S.; Kang, S.; Lee, Y.; Kim, S.; Chung, J.; Jon, S. Antibody-Assisted Delivery of a Peptide–Drug Conjugate for Targeted Cancer Therapy. Mol. Pharmaceutics 2019, 16, 165–172. [Google Scholar] [CrossRef]
- Wang, M.; Liu, J.; Xia, M.; Yin, L.; Zhang, L.; Liu, X.; Cheng, Y. Peptide-Drug Conjugates: A New Paradigm for Targeted Cancer Therapy. European Journal of Medicinal Chemistry 2024, 265, 116119. [Google Scholar] [CrossRef]
- Mező, G.; Gomena, J.; Ranđelović, I.; Dókus, E.; Kiss, K.; Pethő, L.; Schuster, S.; Vári, B.; Vári-Mező, D.; Lajkó, E.; et al. Oxime-Linked Peptide–Daunomycin Conjugates as Good Tools for Selection of Suitable Homing Devices in Targeted Tumor Therapy: An Overview. IJMS 2024, 25, 1864. [Google Scholar] [CrossRef]
- Wang, L.; Chen, H.; Wang, F.; Zhang, X. The Development of Peptide-Drug Conjugates (PDCs) Strategies for Paclitaxel. Expert Opinion on Drug Delivery 2022, 19, 147–161. [Google Scholar] [CrossRef]
- Zhang, R.; Pei, P.; Wang, Y.; Guo, Q.; Luo, S.; Chen, L. A Single-chain Variable Fragment-anticancer Lytic Peptide (scFv-ACLP) Fusion Protein for Targeted Cancer Treatment. Chem Biol Drug Des 2023, 101, 1406–1415. [Google Scholar] [CrossRef]
- Demeule, M.; Currie, J.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.; Béliveau, R. Involvement of the Low-density Lipoprotein Receptor-related Protein in the Transcytosis of the Brain Delivery Vector Angiopep. Journal of Neurochemistry 2008, 106, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Régina, A.; Ché, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Identification and Design of Peptides as a New Drug Delivery System for the Brain. J Pharmacol Exp Ther 2008, 324, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Pethő, L.; Oláh-Szabó, R.; Mező, G. Influence of the Drug Position on Bioactivity in Angiopep-2—Daunomycin Conjugates. IJMS 2023, 24, 3106. [Google Scholar] [CrossRef]
- Rodriguez, I.; Kocik-Krol, J.; Skalniak, L.; Musielak, B.; Wisniewska, A.; Ciesiołkiewicz, A.; Berlicki, Ł.; Plewka, J.; Grudnik, P.; Stec, M.; et al. Structural and Biological Characterization of pAC65, a Macrocyclic Peptide That Blocks PD-L1 with Equivalent Potency to the FDA-Approved Antibodies. Mol Cancer 2023, 22, 150. [Google Scholar] [CrossRef]
- Chen, X.; Wang, D.; Jiang, Y.-B.; Jiang, T. Design of Cell-Specific Targeting Peptides for Cancer Therapy. Targets 2024, 2, 186–201. [Google Scholar] [CrossRef]
- Van Epps, H.L. René Dubos: Unearthing Antibiotics. The Journal of Experimental Medicine 2006, 203, 259–259. [Google Scholar] [CrossRef]
- Kelkar, D.A.; Chattopadhyay, A. The Gramicidin Ion Channel: A Model Membrane Protein. Biochim Biophys Acta 2007, 1768, 2011–2025. [Google Scholar] [CrossRef]
- Koeppe, R.E.; Anderson, O.S. Engineering the Gramicidin Channel. Annual Review of Biophysics and Biomolecular Structure 1996, 25, 231–258. [Google Scholar] [CrossRef]
- Greathouse, D.V.; Koeppe, R.E.; Providence, L.L.; Shobana, S.; Andersen, O.S. [28] Design and Characterization of Gramicidin Channels. In Methods in Enzymology; Elsevier, 1999; Vol. 294, pp. 525–550 ISBN 978-0-12-182195-1.
- Brownd, M.; McKay, M.J.; Greathouse, D.V.; Andersen, O.S.; Koeppe, R.E. Gramicidin Subunits That Cross Membranes and Form Ion Channels. Biophysical Journal 2018, 114, 454a. [Google Scholar] [CrossRef]
- Steinem, C.; Janshoff, A.; Ulrich, W.P.; Sieber, M.; Galla, H.J. Impedance Analysis of Supported Lipid Bilayer Membranes: A Scrutiny of Different Preparation Techniques. Biochim Biophys Acta 1996, 1279, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.K.; Weichbrodt, C.; Steinem, C. Impedance Analysis of Gramicidin D in Pore-Suspending Membranes. Soft Matter 2009, 5, 3347. [Google Scholar] [CrossRef]
- Urban, B.W.; Hladky, S.B.; Haydon, D.A. Ion Movements in Gramicidin Pores. An Example of Single-File Transport. Biochimica et Biophysica Acta (BBA) - Biomembranes 1980, 602, 331–354. [Google Scholar] [CrossRef]
- Mukherjee, S.; Chattopadhyay, A. Motionally Restricted Tryptophan Environments at the Peptide-Lipid Interface of Gramicidin Channels. Biochemistry 1994, 33, 5089–5097. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M. Chapter 1 - Immunoadjuvants for Cancer Immunotherapy. In Nanomedicine in Cancer Immunotherapy; Kesharwani, P., Ed.; Academic Press, 2024; pp. 1–36 ISBN 978-0-443-18770-4.
- Abdelhamid, H.N.; Khan, M.S.; Wu, H.-F. Graphene Oxide as a Nanocarrier for Gramicidin (GOGD) for High Antibacterial Performance. RSC Adv. 2014, 4, 50035–50046. [Google Scholar] [CrossRef]
- Gambucci, M.; Gentili, P.L.; Sassi, P.; Latterini, L. A Multi-Spectroscopic Approach to Investigate the Interactions between Gramicidin A and Silver Nanoparticles. Soft Matter 2019, 15, 6571–6580. [Google Scholar] [CrossRef]
- Nelson, J.W.; Zhou, Z.; Breaker, R.R. Gramicidin D Enhances the Antibacterial Activity of Fluoride. Bioorganic & Medicinal Chemistry Letters 2014, 24, 2969–2971. [Google Scholar] [CrossRef]
- David, J.M.; Rajasekaran, A.K. Gramicidin A: A New Mission for an Old Antibiotic. J Kidney Cancer VHL 2015, 2, 15–24. [Google Scholar] [CrossRef]
- Wijesinghe, D.; Arachchige, M.C.M.; Lu, A.; Reshetnyak, Y.K.; Andreev, O.A. pH Dependent Transfer of Nano-Pores into Membrane of Cancer Cells to Induce Apoptosis. Sci Rep 2013, 3, 3560. [Google Scholar] [CrossRef]
- David, J.M.; Owens, T.A.; Barwe, S.P.; Rajasekaran, A.K. Gramicidin A Induces Metabolic Dysfunction and Energy Depletion Leading to Cell Death in Renal Cell Carcinoma Cells. Molecular Cancer Therapeutics 2013, 12, 2296–2307. [Google Scholar] [CrossRef]
- Haoyang, W.-W.; Xiao, Q.; Ye, Z.; Fu, Y.; Zhang, D.-W.; Li, J.; Xiao, L.; Li, Z.-T.; Hou, J.-L. Gramicidin A-Based Unimolecular Channel: Cancer Cell-Targeting Behavior and Ion Transport-Induced Apoptosis. Chem. Commun. 2021, 57, 1097–1100. [Google Scholar] [CrossRef]
- Fadnes, B.; Rekdal, Ø.; Uhlin-Hansen, L. The Anticancer Activity of Lytic Peptides Is Inhibited by Heparan Sulfate on the Surface of the Tumor Cells. BMC Cancer 2009, 9, 183. [Google Scholar] [CrossRef]
- Raileanu, M.; Popescu, A.; Bacalum, M. Antimicrobial Peptides as New Combination Agents in Cancer Therapeutics: A Promising Protocol against HT-29 Tumoral Spheroids. Int J Mol Sci 2020, 21, 6964. [Google Scholar] [CrossRef]
- Xue, Y.-W.; Itoh, H.; Dan, S.; Inoue, M. Gramicidin A Accumulates in Mitochondria, Reduces ATP Levels, Induces Mitophagy, and Inhibits Cancer Cell Growth. Chem Sci 2022, 13, 7482–7491. [Google Scholar] [CrossRef]
- Chen, T.; Wang, Y.; Yang, Y.; Yu, K.; Cao, X.; Su, F.; Xu, H.; Peng, Y.; Hu, Y.; Qian, F.; et al. Gramicidin Inhibits Human Gastric Cancer Cell Proliferation, Cell Cycle and Induced Apoptosis. Biol Res 2019, 52, 57. [Google Scholar] [CrossRef]
- Altin-Celik, P.; Eken, A.; Derya-Andeden, M.; Eciroglu, H.; Uzen, R.; Donmez-Altuntas, H. Iturin A and Gramicidin A Inhibit Proliferation, Trigger Apoptosis, and Regulate Inflammation in Breast Cancer Cells. Journal of Drug Delivery Science and Technology 2024, 100, 106121. [Google Scholar] [CrossRef]
- Bubin, R.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences 2023, 24, 7030. [Google Scholar] [CrossRef]
- Wang, R.-Q.; Geng, J.; Sheng, W.-J.; Liu, X.-J.; Jiang, M.; Zhen, Y.-S. The Ionophore Antibiotic Gramicidin A Inhibits Pancreatic Cancer Stem Cells Associated with CD47 Down-Regulation. Cancer Cell Int 2019, 19, 145. [Google Scholar] [CrossRef]
- Maham, S.; Awan, S.N.; Adnan, F.; Kakar, S.J.; Mian, A.; Khan, D. PB1837: ANTIBIOTICS; A POSSIBLE ALTERNATE TREATMENT OPTION FOR MYELOID LEUKEMIA. Hemasphere 2023, 7, e1313111. [Google Scholar] [CrossRef]
- Bush, K.; Courvalin, P.; Dantas, G.; Davies, J.; Eisenstein, B.; Huovinen, P.; Jacoby, G.A.; Kishony, R.; Kreiswirth, B.N.; Kutter, E.; et al. Tackling Antibiotic Resistance. Nat Rev Microbiol 2011, 9, 894–896. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6. [Google Scholar] [CrossRef]
- Yang, B.; Li, W.; Mao, Y.; Zhao, Y.; Xue, Y.; Xu, X.; Zhao, Y.; Liu, K. Study on Antimicrobial Activity of Sturgeon Skin Mucus Polypeptides (Rational Design, Self-Assembly and Application). Food Chemistry: X 2024, 21, 101236. [Google Scholar] [CrossRef] [PubMed]
- Namivandi-Zangeneh, R.; Kwan, R.J.; Nguyen, T.-K.; Yeow, J.; Byrne, F.L.; Oehlers, S.H.; Wong, E.H.H.; Boyer, C. The Effects of Polymer Topology and Chain Length on the Antimicrobial Activity and Hemocompatibility of Amphiphilic Ternary Copolymers. Polym. Chem. 2018, 9, 1735–1744. [Google Scholar] [CrossRef]
- Carmona-Ribeiro, A.M.; de Melo Carrasco, L.D. Cationic Antimicrobial Polymers and Their Assemblies. Int J Mol Sci 2013, 14, 9906–9946. [Google Scholar] [CrossRef]
- Kuroki, A.; Sangwan, P.; Qu, Y.; Peltier, R.; Sanchez-Cano, C.; Moat, J.; Dowson, C.G.; Williams, E.G.L.; Locock, K.E.S.; Hartlieb, M.; et al. Sequence Control as a Powerful Tool for Improving the Selectivity of Antimicrobial Polymers. ACS Appl. Mater. Interfaces 2017, 9, 40117–40126. [Google Scholar] [CrossRef]
- Laroque, S.; Reifarth, M.; Sperling, M.; Kersting, S.; Klöpzig, S.; Budach, P.; Storsberg, J.; Hartlieb, M. Impact of Multivalence and Self-Assembly in the Design of Polymeric Antimicrobial Peptide Mimics. ACS Appl. Mater. Interfaces 2020, 12, 30052–30065. [Google Scholar] [CrossRef]
- Afoshin, A.S.; Kudryakova, I.V.; Borovikova, A.O.; Suzina, N.E.; Toropygin, I.Yu.; Shishkova, N.A.; Vasilyeva, N.V. Lytic Potential of Lysobacter Capsici VKM B-2533T: Bacteriolytic Enzymes and Outer Membrane Vesicles. Sci Rep 2020, 10, 9944. [Google Scholar] [CrossRef]
- Adak, A.; Castelletto, V.; De Sousa, A.; Karatzas, K.-A.; Wilkinson, C.; Khunti, N.; Seitsonen, J.; Hamley, I.W. Self-Assembly and Antimicrobial Activity of Lipopeptides Containing Lysine-Rich Tripeptides. Biomacromolecules 2024, 25, 1205–1213. [Google Scholar] [CrossRef]
- Jhong, J.-H.; Chi, Y.-H.; Li, W.-C.; Lin, T.-H.; Huang, K.-Y.; Lee, T.-Y. dbAMP: An Integrated Resource for Exploring Antimicrobial Peptides with Functional Activities and Physicochemical Properties on Transcriptome and Proteome Data. Nucleic Acids Research 2019, 47, D285–D297. [Google Scholar] [CrossRef]
- Chou, S.; Guo, H.; Zingl, F.G.; Zhang, S.; Toska, J.; Xu, B.; Chen, Y.; Chen, P.; Waldor, M.K.; Zhao, W.; et al. Synthetic Peptides That Form Nanostructured Micelles Have Potent Antibiotic and Antibiofilm Activity against Polymicrobial Infections. Proc. Natl. Acad. Sci. U.S.A. 2023, 120, e2219679120. [Google Scholar] [CrossRef]
- Zimmermann, G.R.; Lehár, J.; Keith, C.T. Multi-Target Therapeutics: When the Whole Is Greater than the Sum of the Parts. Drug Discovery Today 2007, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol Rev 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Zharkova, M.S.; Orlov, D.S.; Golubeva, O.Yu.; Chakchir, O.B.; Eliseev, I.E.; Grinchuk, T.M.; Shamova, O.V. Application of Antimicrobial Peptides of the Innate Immune System in Combination With Conventional Antibiotics—A Novel Way to Combat Antibiotic Resistance? Front. Cell. Infect. Microbiol. 2019, 9, 128. [Google Scholar] [CrossRef]
- Tudoroiu, E.-E.; Dinu-Pîrvu, C.-E.; Albu Kaya, M.G.; Popa, L.; Anuța, V.; Prisada, R.M.; Ghica, M.V. An Overview of Cellulose Derivatives-Based Dressings for Wound-Healing Management. Pharmaceuticals 2021, 14, 1215. [Google Scholar] [CrossRef] [PubMed]
- Kondaveeti, S.; Damato, T.C.; Carmona-Ribeiro, A.M.; Sierakowski, M.R.; Petri, D.F.S. Sustainable Hydroxypropyl Methylcellulose/Xyloglucan/Gentamicin Films with Antimicrobial Properties. Carbohydr Polym 2017, 165, 285–293. [Google Scholar] [CrossRef]
- Kondaveeti, S.; Bueno, P.V. de A.; Carmona-Ribeiro, A.M.; Esposito, F.; Lincopan, N.; Sierakowski, M.R.; Petri, D.F.S. Microbicidal Gentamicin-Alginate Hydrogels. Carbohydrate Polymers 2018, 186, 159–167. [Google Scholar] [CrossRef]
- Feng, W.; Wang, Z. Tailoring the Swelling-Shrinkable Behavior of Hydrogels for Biomedical Applications. Advanced Science 2023, 10, 2303326. [Google Scholar] [CrossRef]
- Valour, F.; Bouaziz, A.; Karsenty, J.; Ader, F.; Lustig, S.; Laurent, F.; Chidiac, C.; Ferry, T. Determinants of Methicillin-Susceptible Staphylococcus Aureusnative Bone and Joint Infection Treatment Failure: A Retrospective Cohort Study. BMC Infect Dis 2014, 14, 443. [Google Scholar] [CrossRef]
- Fleiter, N.; Walter, G.; Bösebeck, H.; Vogt, S.; Büchner, H.; Hirschberger, W.; Hoffmann, R. Clinical Use and Safety of a Novel Gentamicin-Releasing Resorbable Bone Graft Substitute in the Treatment of Osteomyelitis/Osteitis. Bone & Joint Research 2014, 3, 223–229. [Google Scholar] [CrossRef]
- Cao, Z.; Jiang, D.; Yan, L.; Wu, J. In Vitro and in Vivo Drug Release and Antibacterial Properties of the Novel Vancomycin-Loaded Bone-like Hydroxyapatite/Poly Amino Acid Scaffold. IJN 2017, Volume 12, 1841–1851. [Google Scholar] [CrossRef]
- Chen, C.H.; Starr, C.G.; Troendle, E.; Wiedman, G.; Wimley, W.C.; Ulmschneider, J.P.; Ulmschneider, M.B. Simulation-Guided Rational de Novo Design of a Small Pore-Forming Antimicrobial Peptide. J. Am. Chem. Soc. 2019, 141, 4839–4848. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ouyang, X.; Liu, Y.; Ba, Z.; Yang, Y.; Zhang, J.; Yang, P.; Yang, T.; Wang, Y.; Zhao, Y.; et al. Novel β-Hairpin Antimicrobial Peptide Containing the β-Turn Sequence of -NG- and the Tryptophan Zippers Facilitate Self-Assembly into Nanofibers, Exhibiting Excellent Antimicrobial Performance. J. Med. Chem. 2024, 67, 6365–6383. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Zhao, Y.; Zhou, P.; Carter, J.; Li, Z.; Waigh, T.A.; Lu, J.R.; Xu, H. Surfactant-like Peptides: From Molecular Design to Controllable Self-Assembly with Applications. Coordination Chemistry Reviews 2020, 421, 213418. [Google Scholar] [CrossRef]
- Koutsopoulos, S.; Kaiser, L.; Eriksson, H.M.; Zhang, S. Designer Peptidesurfactants Stabilize Diverse Functional Membrane Proteins. Chem. Soc. Rev. 2012, 41, 1721–1728. [Google Scholar] [CrossRef]
- Zheng, X.; Dong, S.; Zheng, J.; Li, D.; Li, F.; Luo, Z. Expression, Stabilization and Purification of Membrane Proteins via Diverse Protein Synthesis Systems and Detergents Involving Cell-Free Associated with Self-Assembly Peptide Surfactants. Biotechnology Advances 2014, 32, 564–574. [Google Scholar] [CrossRef]
- Dodd-o, J.; Roy, A.; Siddiqui, Z.; Jafari, R.; Coppola, F.; Ramasamy, S.; Kolloli, A.; Kumar, D.; Kaundal, S.; Zhao, B.; et al. Antiviral Fibrils of Self-Assembled Peptides with Tunable Compositions. Nat Commun 2024, 15, 1142. [Google Scholar] [CrossRef]
- Beyerstedt, S.; Casaro, E.B.; Rangel, É.B. COVID-19: Angiotensin-Converting Enzyme 2 (ACE2) Expression and Tissue Susceptibility to SARS-CoV-2 Infection. Eur J Clin Microbiol Infect Dis 2021, 40, 905–919. [Google Scholar] [CrossRef]
- Shim, M.K.; Park, J.; Yoon, H.Y.; Lee, S.; Um, W.; Kim, J.-H.; Kang, S.-W.; Seo, J.-W.; Hyun, S.-W.; Park, J.H.; et al. Carrier-Free Nanoparticles of Cathepsin B-Cleavable Peptide-Conjugated Doxorubicin Prodrug for Cancer Targeting Therapy. Journal of Controlled Release 2019, 294, 376–389. [Google Scholar] [CrossRef]
- Gao, W.; Cao, W.; Zhang, H.; Li, P.; Xu, K.; Tang, B. Targeting Lysosomal Membrane Permeabilization to Induce and Image Apoptosis in Cancer Cells by Multifunctional Au–ZnO Hybrid Nanoparticles. Chem. Commun. 2014, 50, 8117. [Google Scholar] [CrossRef]
- Jana, B.; Jin, S.; Go, E.M.; Cho, Y.; Kim, D.; Kim, S.; Kwak, S.K.; Ryu, J.-H. Intra-Lysosomal Peptide Assembly for the High Selectivity Index against Cancer. J. Am. Chem. Soc. 2023, 145, 18414–18431. [Google Scholar] [CrossRef]
- Singha, N.; Gupta, P.; Pramanik, B.; Ahmed, S.; Dasgupta, A.; Ukil, A.; Das, D. Hydrogelation of a Naphthalene Diimide Appended Peptide Amphiphile and Its Application in Cell Imaging and Intracellular pH Sensing. Biomacromolecules 2017, 18, 3630–3641. [Google Scholar] [CrossRef] [PubMed]
- Olesk, J.; Donahue, D.; Ross, J.; Sheehan, C.; Bennett, Z.; Armknecht, K.; Kudary, C.; Hopf, J.; Ploplis, V.A.; Castellino, F.J.; et al. Antimicrobial Peptide-Conjugated Phage-Mimicking Nanoparticles Exhibit Potent Bactericidal Action against Streptococcus Pyogenes in Murine Wound Infection Models. Nanoscale Adv. 2024, 6, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The Challenge of New Drug Discovery for Tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for Bad Bugs: Confronting the Challenges of Antibacterial Discovery. Nat Rev Drug Discov 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Batt, S.M.; Burke, C.E.; Moorey, A.R.; Besra, G.S. Antibiotics and Resistance: The Two-Sided Coin of the Mycobacterial Cell Wall. Cell Surf 2020, 6, 100044. [Google Scholar] [CrossRef]
- Loessner, M.J. Bacteriophage Endolysins — Current State of Research and Applications. Current Opinion in Microbiology 2005, 8, 480–487. [Google Scholar] [CrossRef]
- Zamora-Caballero, S.; Chmielowska, C.; Quiles-Puchalt, N.; Brady, A.; Del Sol, F.G.; Mancheño-Bonillo, J.; Felipe-Ruíz, A.; Meijer, W.J.J.; Penadés, J.R.; Marina, A. Antagonistic Interactions between Phage and Host Factors Control Arbitrium Lysis–Lysogeny Decision. Nat Microbiol 2024, 9, 161–172. [Google Scholar] [CrossRef]
- Nair, G.; Jain, V. An Intramolecular Cross-Talk in D29 Mycobacteriophage Endolysin Governs the Lytic Cycle and Phage-Host Population Dynamics. Sci. Adv. 2024, 10, eadh9812. [Google Scholar] [CrossRef]
- Zhu, Y.; Jia, H.; Duan, Q.; Wu, F. Nanomedicines for Combating Multidrug Resistance of Cancer. WIREs Nanomed Nanobiotechnol 2021, 13, e1715. [Google Scholar] [CrossRef]
- Martins, R.M.; Sforça, M.L.; Amino, R.; Juliano, M.A.; Oyama, S.; Juliano, L.; Pertinhez, T.A.; Spisni, A.; Schenkman, S. Lytic Activity and Structural Differences of Amphipathic Peptides Derived from Trialysin , Biochemistry 2006, 45, 1765–1774. [Google Scholar] [CrossRef]
- Shi, Y.-Y.; Dong, D.-R.; Fan, G.; Dai, M.-Y.; Liu, M. A Cyclic Peptide-Based PROTAC Induces Intracellular Degradation of Palmitoyltransferase and Potently Decreases PD-L1 Expression in Human Cervical Cancer Cells. Front. Immunol. 2023, 14, 1237964. [Google Scholar] [CrossRef]
- Saberwal, G.; Nagaraj, R. Cell-Lytic and Antibacterial Peptides That Act by Perturbing the Barrier Function of Membranes: Facets of Their Conformational Features, Structure-Function Correlations and Membrane-Perturbing Abilities. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes 1994, 1197, 109–131. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, C.; Hansel, W. Membrane Disrupting Lytic Peptides for Cancer Treatments. CPD 2004, 10, 2299–2310. [Google Scholar] [CrossRef]
- Bechinger, B. Structure and Function of Membrane-Lytic Peptides. Critical Reviews in Plant Sciences 2004, 23, 271–292. [Google Scholar] [CrossRef]
- Liu, H.; Shen, W.; Liu, W.; Yang, Z.; Yin, D.; Xiao, C. From Oncolytic Peptides to Oncolytic Polymers: A New Paradigm for Oncotherapy. Bioactive Materials 2024, 31, 206–230. [Google Scholar] [CrossRef]
- Vieira-da-Silva, B.; Castanho, M.A.R.B. The Structure and Matrix Dynamics of Bacterial Biofilms as Revealed by Antimicrobial Peptides’ Diffusion. Journal of Peptide Science 2023, 29, e3470. [Google Scholar] [CrossRef]
- Hansel, W.; Enright, F.; Leuschner, C. Destruction of Breast Cancers and Their Metastases by Lytic Peptide Conjugates in Vitro and in Vivo. Molecular and Cellular Endocrinology 2007, 260–262, 183–189. [Google Scholar] [CrossRef]
- Ciulla, M.G.; Gelain, F. Structure–Activity Relationships of Antibacterial Peptides. Microbial Biotechnology 2023, 16, 757–777. [Google Scholar] [CrossRef]
- Yasir, M.; Willcox, M.D.P.; Dutta, D. Action of Antimicrobial Peptides against Bacterial Biofilms. Materials (Basel) 2018, 11, 2468. [Google Scholar] [CrossRef]
- Pontes, J.T.C.D.; Toledo Borges, A.B.; Roque-Borda, C.A.; Pavan, F.R. Antimicrobial Peptides as an Alternative for the Eradication of Bacterial Biofilms of Multi-Drug Resistant Bacteria. Pharmaceutics 2022, 14, 642. [Google Scholar] [CrossRef]
- Raucher, D.; Ryu, J.S. Cell-Penetrating Peptides: Strategies for Anticancer Treatment. Trends in Molecular Medicine 2015, 21, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular Uptake of the Tat Protein from Human Immunodeficiency Virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Karami Fath, M.; Babakhaniyan, K.; Zokaei, M.; Yaghoubian, A.; Akbari, S.; Khorsandi, M.; Soofi, A.; Nabi-Afjadi, M.; Zalpoor, H.; Jalalifar, F.; et al. Anti-Cancer Peptide-Based Therapeutic Strategies in Solid Tumors. Cell Mol Biol Lett 2022, 27, 33. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, N.; Wang, J.; Liu, Y.; Wang, K.; Zhang, J.; Pan, X. The Recent Advance of Cell-Penetrating and Tumor-Targeting Peptides as Drug Delivery Systems Based on Tumor Microenvironment. Mol. Pharmaceutics 2023, 20, 789–809. [Google Scholar] [CrossRef]
- Cardoso, M.H.; Meneguetti, B.T.; Costa, B.O.; Buccini, D.F.; Oshiro, K.G.N.; Preza, S.L.E.; Carvalho, C.M.E.; Migliolo, L.; Franco, O.L. Non-Lytic Antibacterial Peptides That Translocate Through Bacterial Membranes to Act on Intracellular Targets. IJMS 2019, 20, 4877. [Google Scholar] [CrossRef]
- K R, G.; Balenahalli Narasingappa, R.; Vishnu Vyas, G. Unveiling Mechanisms of Antimicrobial Peptide: Actions beyond the Membranes Disruption. Heliyon 2024, 10, e38079. [Google Scholar] [CrossRef]
- Zhang, H.-Q.; Sun, C.; Xu, N.; Liu, W. The Current Landscape of the Antimicrobial Peptide Melittin and Its Therapeutic Potential. Front. Immunol. 2024, 15, 1326033. [Google Scholar] [CrossRef]
- Choi, M.S.; Lee, C.Y.; Kim, J.H.; Lee, Y.M.; Lee, S.; Kim, H.J.; Heo, K. Gramicidin, a Bactericidal Antibiotic, Is an Antiproliferative Agent for Ovarian Cancer Cells. Medicina 2023, 59, 2059. [Google Scholar] [CrossRef]
- Havas, L.J. Effect of Bee Venom on Colchicine-Induced Tumours. Nature 1950, 166, 567–568. [Google Scholar] [CrossRef]
- Mufson, R.A.; Laskin, J.D.; Fisher, P.B.; Weinstein, I.B. Melittin Shares Certain Cellular Effects with Phorbol Ester Tumour Promoters. Nature 1979, 280, 72–74. [Google Scholar] [CrossRef]
- Parchebafi, A.; Tamanaee, F.; Ehteram, H.; Ahmad, E.; Nikzad, H.; Haddad Kashani, H. The Dual Interaction of Antimicrobial Peptides on Bacteria and Cancer Cells; Mechanism of Action and Therapeutic Strategies of Nanostructures. Microb Cell Fact 2022, 21, 118. [Google Scholar] [CrossRef] [PubMed]
- Gajski, G.; Garaj-Vrhovac, V. Melittin: A Lytic Peptide with Anticancer Properties. Environmental Toxicology and Pharmacology 2013, 36, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, C.; Tenstad, O.; Baumann, A.; Martinez, A.; Myklebust, R.; Bjerkvig, R.; Prestegarden, L. Identification of a Novel Lytic Peptide for the Treatment of Solid Tumours. Genes Cancer 2014, 5, 186–200. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. Host Defense Peptides as New Weapons in Cancer Treatment. CMLS, Cell. Mol. Life Sci. 2005, 62, 784–790. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma Membrane Changes during Programmed Cell Deaths. Cell Res 2018, 28, 9–21. [Google Scholar] [CrossRef]
- Yu, X.; Dai, Y.; Zhao, Y.; Qi, S.; Liu, L.; Lu, L.; Luo, Q.; Zhang, Z. Melittin-Lipid Nanoparticles Target to Lymph Nodes and Elicit a Systemic Anti-Tumor Immune Response. Nat Commun 2020, 11, 1110. [Google Scholar] [CrossRef]
- Kelly, G.J.; Kia, A.F.-A.; Hassan, F.; O’Grady, S.; Morgan, M.P.; Creaven, B.S.; McClean, S.; Harmey, J.H.; Devocelle, M. Polymeric Prodrug Combination to Exploit the Therapeutic Potential of Antimicrobial Peptides against Cancer Cells. Org. Biomol. Chem. 2016, 14, 9278–9286. [Google Scholar] [CrossRef]
- O’Connor, S.; Szwej, E.; Nikodinovic-Runic, J.; O’Connor, A.; Byrne, A.T.; Devocelle, M.; O’Donovan, N.; Gallagher, W.M.; Babu, R.; Kenny, S.T.; et al. The Anti-Cancer Activity of a Cationic Anti-Microbial Peptide Derived from Monomers of Polyhydroxyalkanoate. Biomaterials 2013, 34, 2710–2718. [Google Scholar] [CrossRef]
- Imura, Y.; Nishida, M.; Matsuzaki, K. Action Mechanism of PEGylated Magainin 2 Analogue Peptide. Biochimica et Biophysica Acta (BBA) - Biomembranes 2007, 1768, 2578–2585. [Google Scholar] [CrossRef]
- Wu, Y.; Lu, D.; Jiang, Y.; Jin, J.; Liu, S.; Chen, L.; Zhang, H.; Zhou, Y.; Chen, H.; Nagle, D.G.; et al. Stapled Wasp Venom-Derived Oncolytic Peptides with Side Chains Induce Rapid Membrane Lysis and Prolonged Immune Responses in Melanoma. J. Med. Chem. 2021, 64, 5802–5815. [Google Scholar] [CrossRef]
- Chen, C.H.; Liu, Y.; Eskandari, A.; Ghimire, J.; Lin, L.C.; Fang, Z.; Wimley, W.C.; Ulmschneider, J.P.; Suntharalingam, K.; Hu, C.J.; et al. Integrated Design of a Membrane-Lytic Peptide-Based Intravenous Nanotherapeutic Suppresses Triple-Negative Breast Cancer. Advanced Science 2022, 9, 2105506. [Google Scholar] [CrossRef] [PubMed]
- Dancey, J.E.; Chen, H.X. Strategies for Optimizing Combinations of Molecularly Targeted Anticancer Agents. Nat Rev Drug Discov 2006, 5, 649–659. [Google Scholar] [CrossRef]
- Deslouches, B.; Di, Y.P. Antimicrobial Peptides with Selective Antitumor Mechanisms: Prospect for Anticancer Applications. Oncotarget 2017, 8, 46635–46651. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Y.; Wang, H.; Zhang, K.; Liu, Y.; Li, Q.; Li, C.; Wen, Z.; Chen, Z. Biomimetic Nanocarriers Loaded with Temozolomide by Cloaking Brain-Targeting Peptides for Targeting Drug Delivery System to Promote Anticancer Effects in Glioblastoma Cells. Heliyon 2024, 10, e28256. [Google Scholar] [CrossRef]
- Leuschner, C.; Enright, F.M.; Gawronska, B.; Hansel, W. Membrane Disrupting Lytic Peptide Conjugates Destroy Hormone Dependent and Independent Breast Cancer Cells in Vitro and in Vivo. Breast Cancer Res Treat 2003, 78, 17–27. [Google Scholar] [CrossRef]
- Papo, N.; Shahar, M.; Eisenbach, L.; Shai, Y. A Novel Lytic Peptide Composed of Dl-Amino Acids Selectively Kills Cancer Cells in Culture and in Mice. Journal of Biological Chemistry 2003, 278, 21018–21023. [Google Scholar] [CrossRef]
- Tu, Z.; Hao, J.; Kharidia, R.; Meng, X.G.; Liang, J.F. Improved Stability and Selectivity of Lytic Peptides through Self-Assembly. Biochemical and Biophysical Research Communications 2007, 361, 712–717. [Google Scholar] [CrossRef]
- Zhong, J.; Chau, Y. Antitumor Activity of a Membrane Lytic Peptide Cyclized with a Linker Sensitive to Membrane Type 1-Matrix Metalloproteinase. Molecular Cancer Therapeutics 2008, 7, 2933–2940. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. New Lytic Peptides Based on the d, l -Amphipathic Helix Motif Preferentially Kill Tumor Cells Compared to Normal Cells. Biochemistry 2003, 42, 9346–9354. [Google Scholar] [CrossRef]
- Li, C.M.; Haratipour, P.; Lingeman, R.G.; Perry, J.J.P.; Gu, L.; Hickey, R.J.; Malkas, L.H. Novel Peptide Therapeutic Approaches for Cancer Treatment. Cells 2021, 10, 2908. [Google Scholar] [CrossRef]
- Pan, X.; Xu, J.; Jia, X. Research Progress Evaluating the Function and Mechanism of Anti-Tumor Peptides. CMAR 2020, Volume 12, 397–409. [Google Scholar] [CrossRef]
- Conibear, A.C.; Schmid, A.; Kamalov, M.; Becker, C.F.W.; Bello, C. Recent Advances in Peptide-Based Approaches for Cancer Treatment. CMC 2020, 27, 1174–1205. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.; Marabelle, A.; Baurain, J.-F.; Jebsen, N.L.; Jøssang, D.E.; Awada, A.; Kristeleit, R.; Loirat, D.; Lazaridis, G.; Jungels, C.; et al. Safety, Antitumor Activity, and T-Cell Responses in a Dose-Ranging Phase I Trial of the Oncolytic Peptide LTX-315 in Patients with Solid Tumors. Clinical Cancer Research 2021, 27, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving Cancer Immunotherapy Using Nanomedicines: Progress, Opportunities and Challenges. Nat Rev Clin Oncol 2020, 17, 251–266. [Google Scholar] [CrossRef]
- Ylösmäki, E.; Cerullo, V. Design and Application of Oncolytic Viruses for Cancer Immunotherapy. Curr Opin Biotechnol 2020, 65, 25–36. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning Cold Tumors into Hot Tumors by Improving T-Cell Infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Feola, S.; Russo, S.; Martins, B.; Lopes, A.; Vandermeulen, G.; Fluhler, V.; De Giorgi, C.; Fusciello, M.; Pesonen, S.; Ylösmäki, E.; et al. Peptides-Coated Oncolytic Vaccines for Cancer Personalized Medicine. Front. Immunol. 2022, 13, 826164. [Google Scholar] [CrossRef]
- Rencinai, A.; Tollapi, E.; Marianantoni, G.; Brunetti, J.; Henriquez, T.; Pini, A.; Bracci, L.; Falciani, C. Branched Oncolytic Peptides Target HSPGs, Inhibit Metastasis, and Trigger the Release of Molecular Determinants of Immunogenic Cell Death in Pancreatic Cancer. Front. Mol. Biosci. 2024, 11, 1429163. [Google Scholar] [CrossRef]
- Lin, L.; Chi, J.; Yan, Y.; Luo, R.; Feng, X.; Zheng, Y.; Xian, D.; Li, X.; Quan, G.; Liu, D.; et al. Membrane-Disruptive Peptides/Peptidomimetics-Based Therapeutics: Promising Systems to Combat Bacteria and Cancer in the Drug-Resistant Era. Acta Pharmaceutica Sinica B 2021, 11, 2609–2644. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Borrelli, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Antimicrobial Peptides as Anticancer Agents: Functional Properties and Biological Activities. Molecules 2020, 25, 2850. [Google Scholar] [CrossRef]
- Leite, M.L.; da Cunha, N.B.; Costa, F.F. Antimicrobial Peptides, Nanotechnology, and Natural Metabolites as Novel Approaches for Cancer Treatment. Pharmacology & Therapeutics 2018, 183, 160–176. [Google Scholar] [CrossRef]
Figure 1.
Gramicidin D molecules in different microenvironments such as lipids large vesicles (LV) or lipid bilayer fragments (BF).
Figure 1.
Gramicidin D molecules in different microenvironments such as lipids large vesicles (LV) or lipid bilayer fragments (BF).
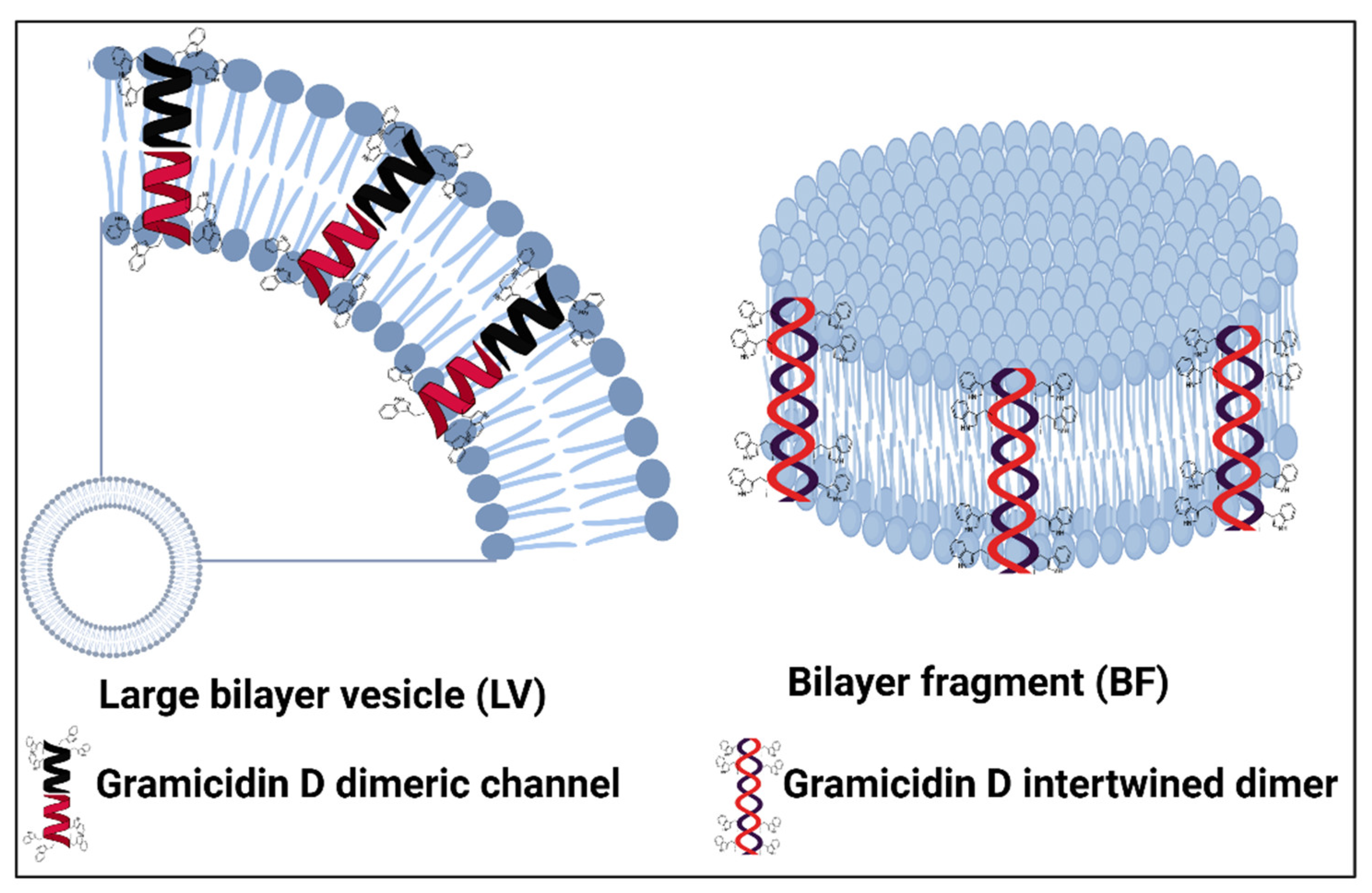
Figure 3.
(a) Leakage of phosphorylated compounds (%) from bacteria for different dispersions at 3.7×108 to 4.8×109 CFU/mL (Escherichia coli) or 5.2×109 to 5.1×1010 CFU/mL (Staphylococcus aureus). The dispersions were tested over a range of DODAB or DODAB/Gr concentrations after one hour of interaction with both types of bacteria. As a control, over a range of Gr concentrations (10−2 to 10−4 mM), no leakage was detected from bacteria after the same interaction time. (b) Micrographs of Escherichia coli (A–C) or Staphylococcus aureus (D–F) cells untreated (A, D) or treated with DODAB BF/Gr (B, E) or DODAB LV/Gr dispersions (C, F), obtained by scanning electron microscopy. Cells appear enlarged by 10,000× (E. coli) or 20,000× (S. aureus). Against E. coli cells, in (B), DODAB =0.005 mM (DODAB BF/Gr) and, in (C), DODAB =0.01 mM (DODAB LV/Gr). Against S. aureus, in (E), DODAB =0.01 mM (DODAB BF/Gr) and, in (F), DODAB =0.05 mM (DODAB LV/Gr). Abbreviations: DODAB, dioctadecyl dimethylammonium bromide; LV, closed bilayers; BF, bilayer disks; Gr, Gramicidin. Reproduced from reference [25].
Figure 3.
(a) Leakage of phosphorylated compounds (%) from bacteria for different dispersions at 3.7×108 to 4.8×109 CFU/mL (Escherichia coli) or 5.2×109 to 5.1×1010 CFU/mL (Staphylococcus aureus). The dispersions were tested over a range of DODAB or DODAB/Gr concentrations after one hour of interaction with both types of bacteria. As a control, over a range of Gr concentrations (10−2 to 10−4 mM), no leakage was detected from bacteria after the same interaction time. (b) Micrographs of Escherichia coli (A–C) or Staphylococcus aureus (D–F) cells untreated (A, D) or treated with DODAB BF/Gr (B, E) or DODAB LV/Gr dispersions (C, F), obtained by scanning electron microscopy. Cells appear enlarged by 10,000× (E. coli) or 20,000× (S. aureus). Against E. coli cells, in (B), DODAB =0.005 mM (DODAB BF/Gr) and, in (C), DODAB =0.01 mM (DODAB LV/Gr). Against S. aureus, in (E), DODAB =0.01 mM (DODAB BF/Gr) and, in (F), DODAB =0.05 mM (DODAB LV/Gr). Abbreviations: DODAB, dioctadecyl dimethylammonium bromide; LV, closed bilayers; BF, bilayer disks; Gr, Gramicidin. Reproduced from reference [25].
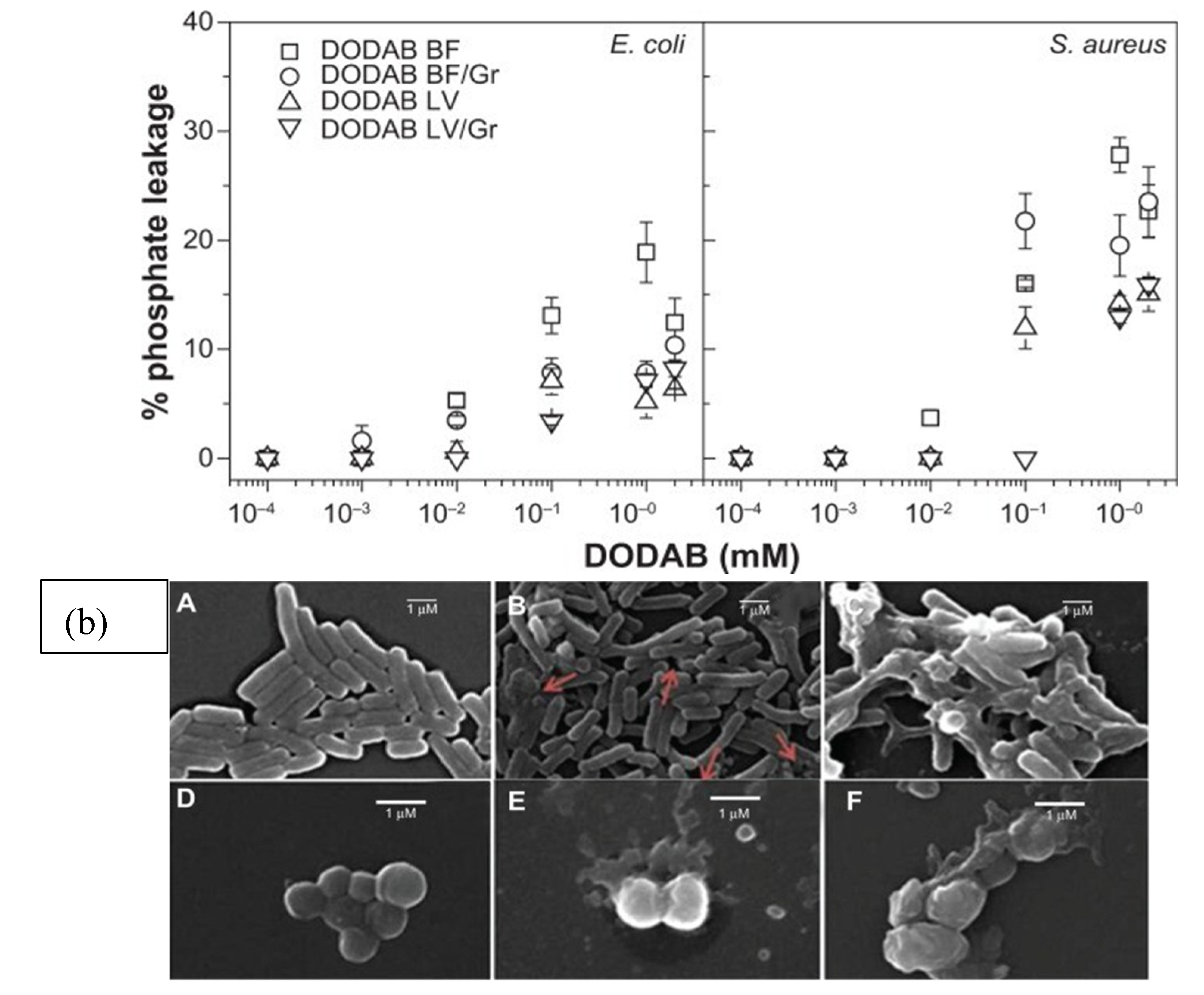
Figure 4.
Nanosheets assembled from nanotubes of the cyclic peptide CPx. The chemical structure and self-assembly of the cyclic peptide CPx in water (a). Formation of amphiphilic nanotubes based on the multiple hydrogen-bonding interactions between the peptide rings (b). Formation of 2D nanosheet assembly by the amphiphilic nanotubes (c). Reprinted from [50] with permission. Copyright 2020 American Chemical Society.
Figure 4.
Nanosheets assembled from nanotubes of the cyclic peptide CPx. The chemical structure and self-assembly of the cyclic peptide CPx in water (a). Formation of amphiphilic nanotubes based on the multiple hydrogen-bonding interactions between the peptide rings (b). Formation of 2D nanosheet assembly by the amphiphilic nanotubes (c). Reprinted from [50] with permission. Copyright 2020 American Chemical Society.
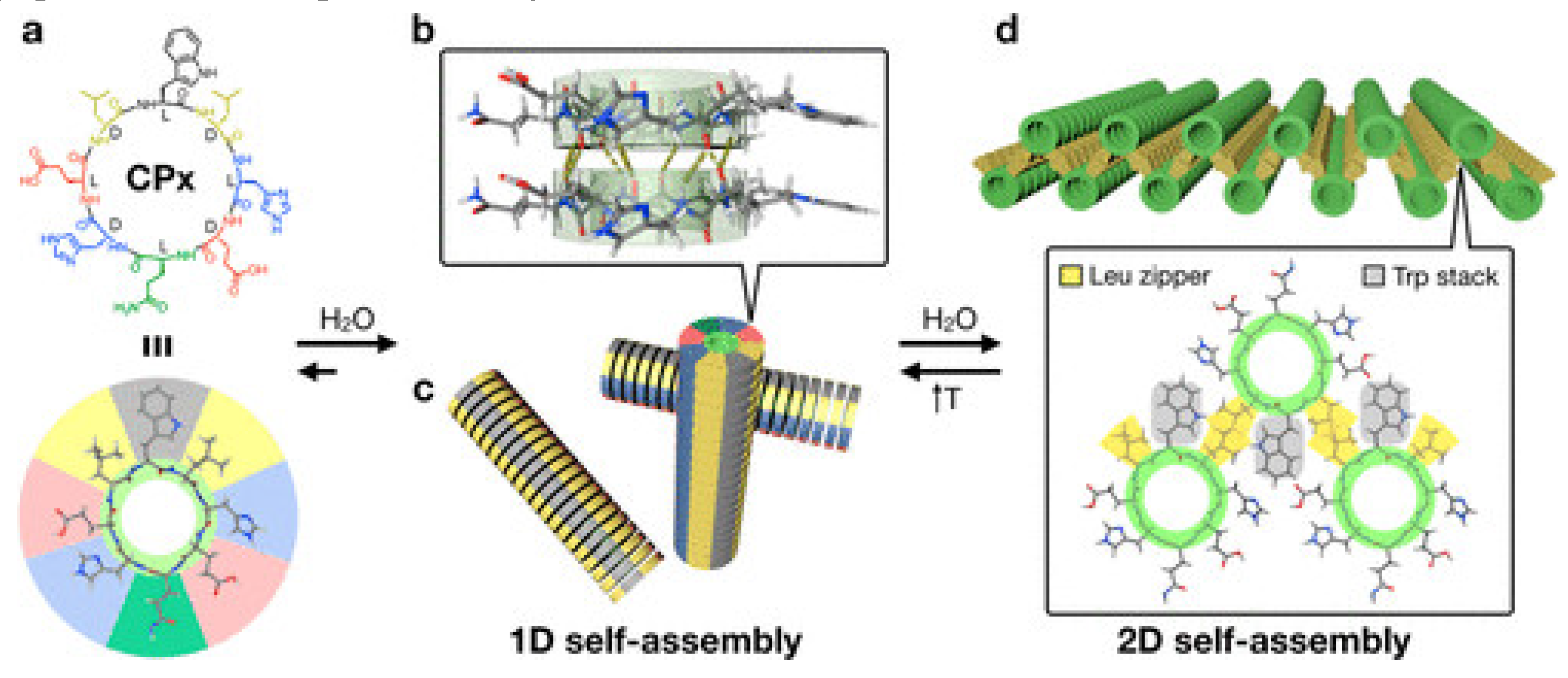
Figure 5.
On top is the sequence for the amphiphilic killerFLIP peptide with hydrophobic residues in green, cationic residues in blue, and polar residues in black. The self-assembly of the killerFLIP peptide reduces its selectivity towards normal eukaryotic cell membranes but improves its binding to cancer cells. The effect is explained by considering two facts: cancer cells bear a much higher negative charge than the normal cells which are practically neutral; the hydrophobic attraction between the peptide and the normal cells is reduced due to the inner localization of the hydrophobic moieties in the aggregate. Reprinted from [60] with permission Copyright 2020 Elsevier.
Figure 5.
On top is the sequence for the amphiphilic killerFLIP peptide with hydrophobic residues in green, cationic residues in blue, and polar residues in black. The self-assembly of the killerFLIP peptide reduces its selectivity towards normal eukaryotic cell membranes but improves its binding to cancer cells. The effect is explained by considering two facts: cancer cells bear a much higher negative charge than the normal cells which are practically neutral; the hydrophobic attraction between the peptide and the normal cells is reduced due to the inner localization of the hydrophobic moieties in the aggregate. Reprinted from [60] with permission Copyright 2020 Elsevier.
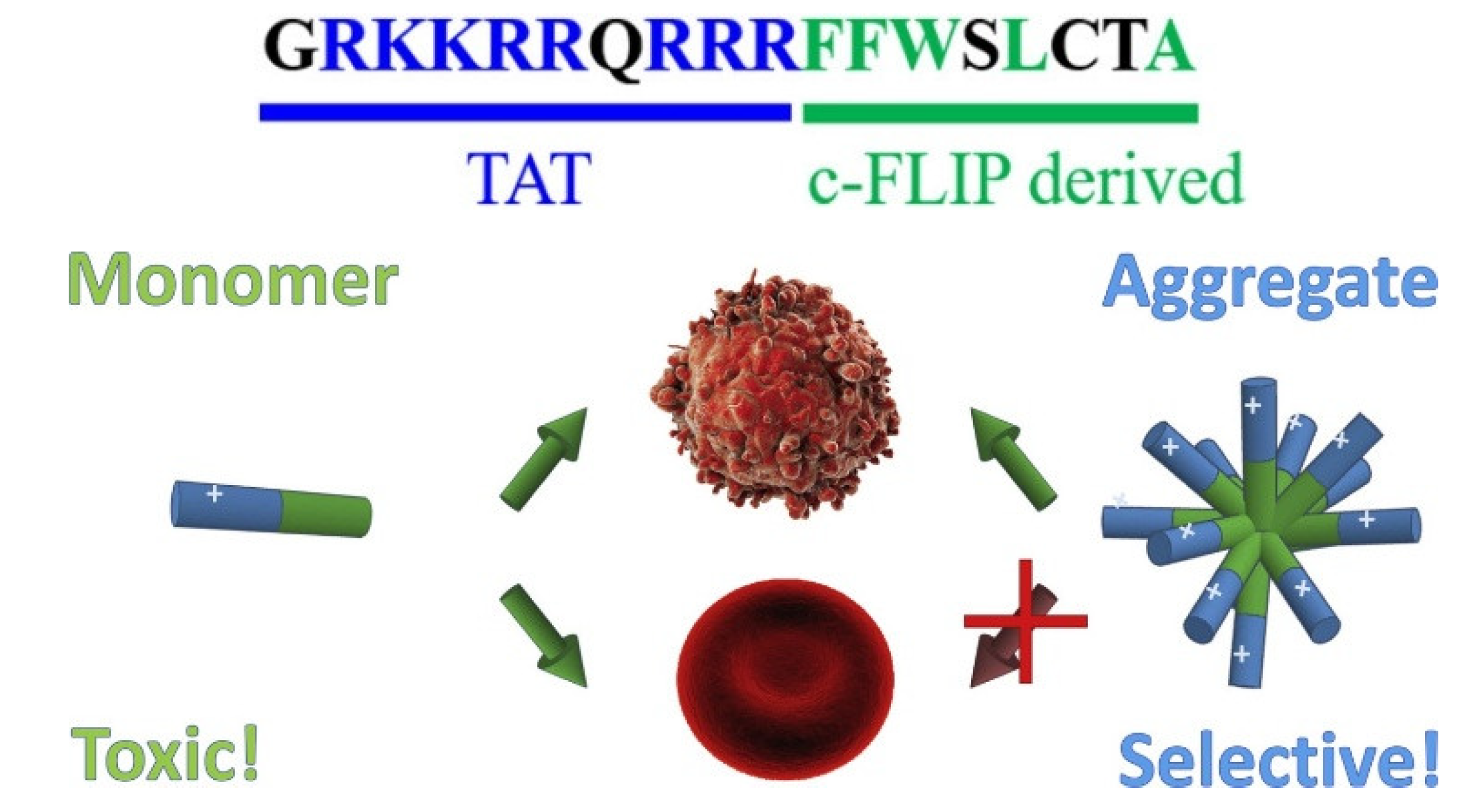
Figure 6.
pH-responsive self-assemblies from the folic acid (FA)-modified peptide derivative FA-EEYSV-NH2. In the acidic pH of the tumor, there was improved passive targeting of the peptide due to higher retention of the fibers than the one of the nanoparticles. Reprinted from [62] with permission Copyright 2021 American Chemical Society.
Figure 6.
pH-responsive self-assemblies from the folic acid (FA)-modified peptide derivative FA-EEYSV-NH2. In the acidic pH of the tumor, there was improved passive targeting of the peptide due to higher retention of the fibers than the one of the nanoparticles. Reprinted from [62] with permission Copyright 2021 American Chemical Society.
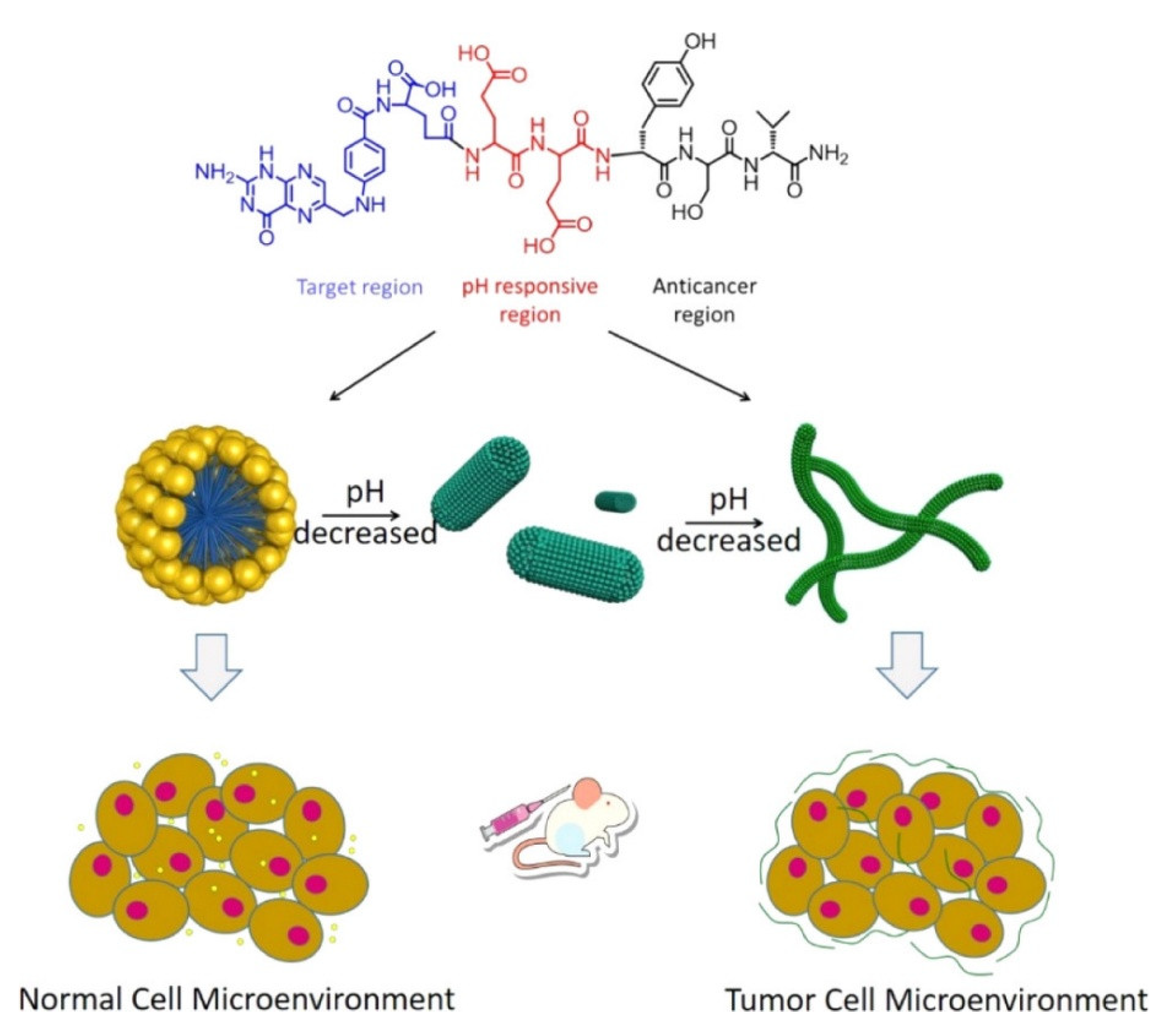
Figure 7.
Peptide/DNA nanoparticle formation, uptake by endocytosis, endosomal escape to the cytosol, and DNA entry into the nucleus aiming at high gene expression with minimal associated toxicity. Reprinted from [64] with permission Copyright (2020) Elsevier.
Figure 7.
Peptide/DNA nanoparticle formation, uptake by endocytosis, endosomal escape to the cytosol, and DNA entry into the nucleus aiming at high gene expression with minimal associated toxicity. Reprinted from [64] with permission Copyright (2020) Elsevier.
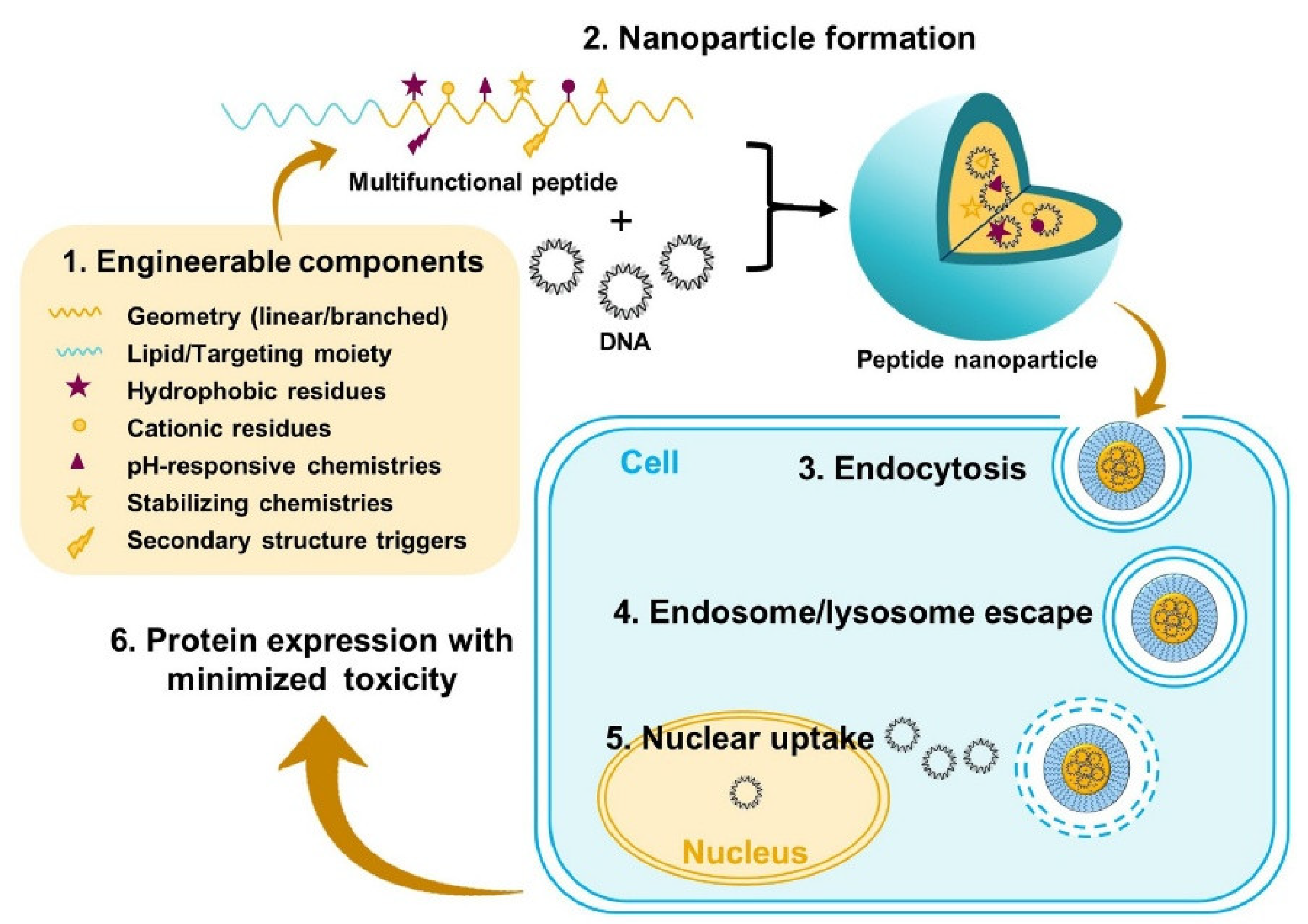
Figure 8.
Gramicidin D nanoparticles in aqueous dispersions and coatings. (a) Macroscopic and microscopic features of gramicidin D dispersions in water seen from photos as a turbid dispersion or visualized from scanning electron micrographs (SEM) as reprinted from [23]. (b) Nanodisks prepared from DODAB bilayer fragments (DODAB BF) surrounded by a layer of carboxymethylcellulose (CMC) and an uppermost layer of poly diallyl dimethylammonium bromide (PDDA). Reprinted from reference [33]. (c) SEM images for mixtures of PDDA (1.0 mg/mL) and P. aeruginosa multidrug-resistant (MDR) bacteria (8.9 × 105 cells/mL). Reprinted from [33].(d) Coatings casted on glass cover slips from aqueous dispersions of Gr nanoparticles and DODAB BF/CMC/PDDA nanodisks. Reproduced from reference [39]. (e) Microbicidal activity for the coatings against bacteria and fungus. Reproduced from [39].
Figure 8.
Gramicidin D nanoparticles in aqueous dispersions and coatings. (a) Macroscopic and microscopic features of gramicidin D dispersions in water seen from photos as a turbid dispersion or visualized from scanning electron micrographs (SEM) as reprinted from [23]. (b) Nanodisks prepared from DODAB bilayer fragments (DODAB BF) surrounded by a layer of carboxymethylcellulose (CMC) and an uppermost layer of poly diallyl dimethylammonium bromide (PDDA). Reprinted from reference [33]. (c) SEM images for mixtures of PDDA (1.0 mg/mL) and P. aeruginosa multidrug-resistant (MDR) bacteria (8.9 × 105 cells/mL). Reprinted from [33].(d) Coatings casted on glass cover slips from aqueous dispersions of Gr nanoparticles and DODAB BF/CMC/PDDA nanodisks. Reproduced from reference [39]. (e) Microbicidal activity for the coatings against bacteria and fungus. Reproduced from [39].
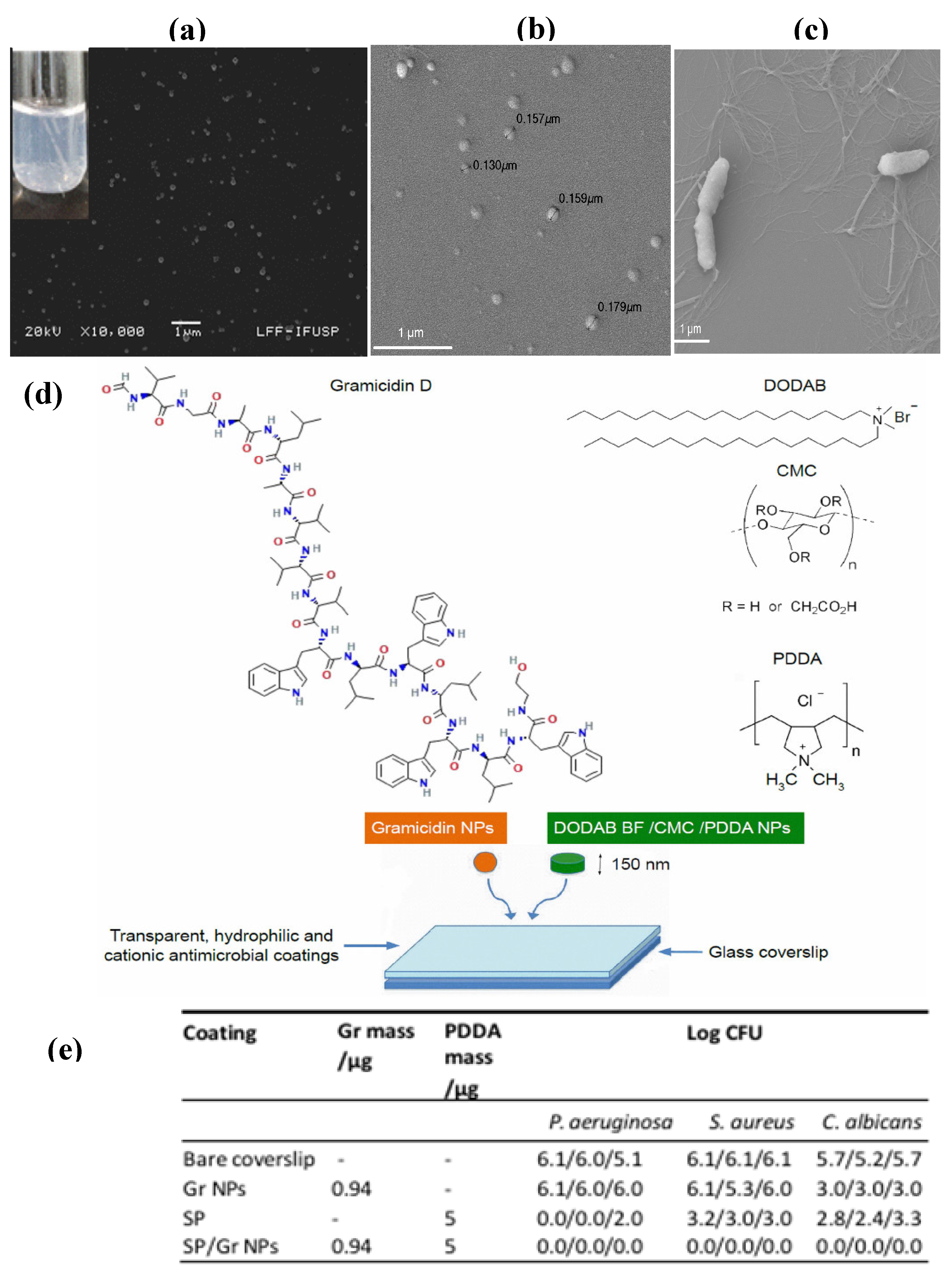
Figure 9.
Schematic of the mechanism of a self-assembling peptide designed for targeted cancer therapy through lysosomal activation and disruption. The peptide is first injected systemically and subsequently binds to specific cell surface receptors on cancer cells, facilitating its uptake via endocytosis. Once inside the lysosome, lysosomal proteases, such as cathepsin B, cleave the peptide, converting it into its active self-assembling form. This transformation leads to the formation of nanofibers, which disrupt the lysosomal membrane, causing lysosomal leakage and subsequent cell damage. The loss of lysosomal integrity ultimately triggers apoptosis, leading to tumor cell death. This approach exploits enzyme-responsive activation and peptide self-assembly as a precise strategy for targeted cancer therapy, offering a promising avenue for treating various drug-resistant tumors.
Figure 9.
Schematic of the mechanism of a self-assembling peptide designed for targeted cancer therapy through lysosomal activation and disruption. The peptide is first injected systemically and subsequently binds to specific cell surface receptors on cancer cells, facilitating its uptake via endocytosis. Once inside the lysosome, lysosomal proteases, such as cathepsin B, cleave the peptide, converting it into its active self-assembling form. This transformation leads to the formation of nanofibers, which disrupt the lysosomal membrane, causing lysosomal leakage and subsequent cell damage. The loss of lysosomal integrity ultimately triggers apoptosis, leading to tumor cell death. This approach exploits enzyme-responsive activation and peptide self-assembly as a precise strategy for targeted cancer therapy, offering a promising avenue for treating various drug-resistant tumors.
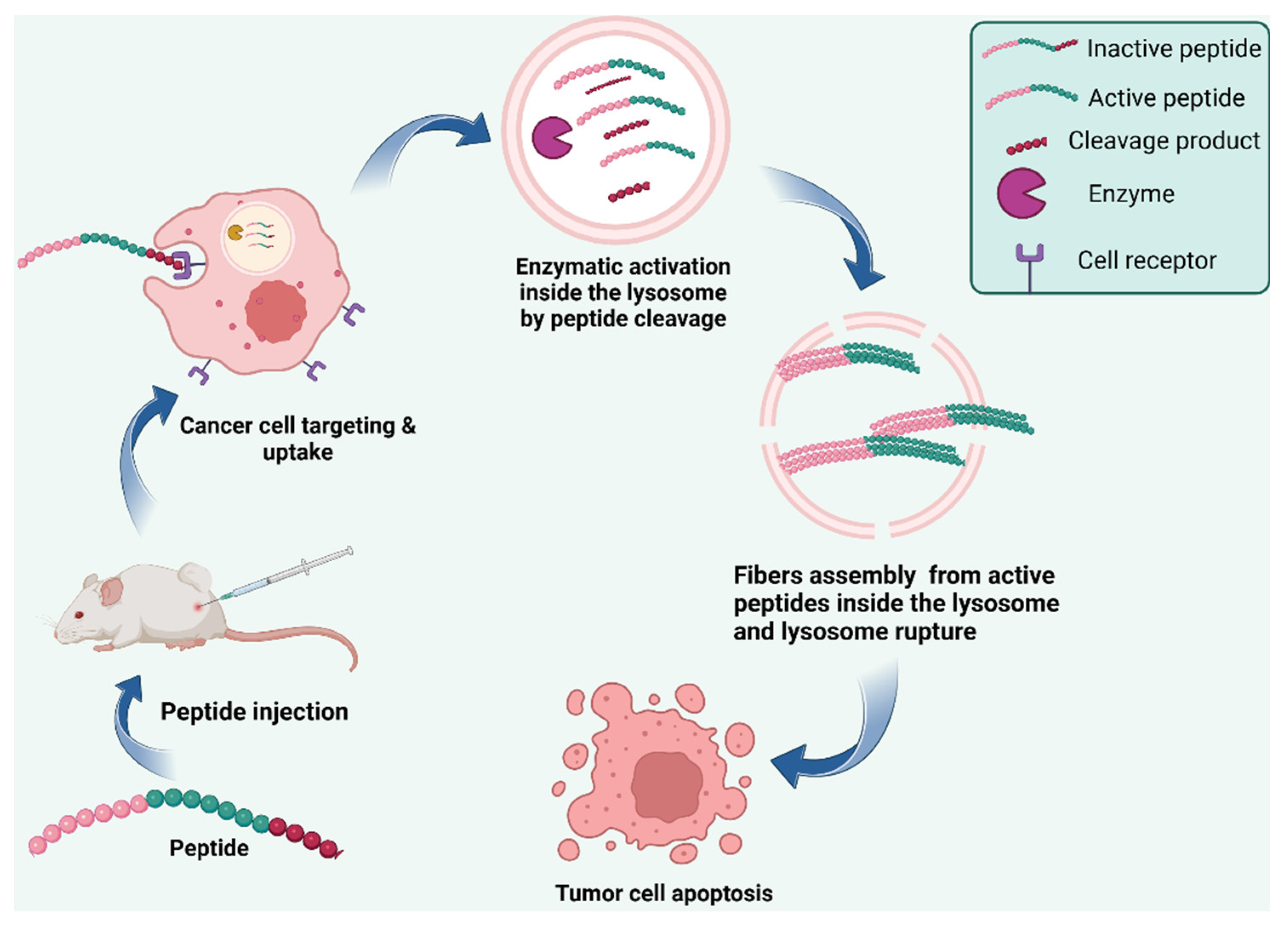
Figure 10.
Mechanisms by which anticancer peptides induce tumor cell death. Immune-modulator peptides enhance T cell responses, promoting CD4+ helper T cells and CD8+ cytotoxic T cells. Death receptor-activating peptides bind to TNF-R1, Fas, and TRAIL-R2, triggering caspase-8 activation and initiating the apoptosis pathway. Mitochondria-disrupting peptides destabilize mitochondrial membranes, leading to caspase-3 activation and apoptosis. Additionally, membrane-disrupting peptides directly permeabilize the cancer cell membrane to ions and other small molecules, causing uncontrolled water efflux and cell shrinkage.
Figure 10.
Mechanisms by which anticancer peptides induce tumor cell death. Immune-modulator peptides enhance T cell responses, promoting CD4+ helper T cells and CD8+ cytotoxic T cells. Death receptor-activating peptides bind to TNF-R1, Fas, and TRAIL-R2, triggering caspase-8 activation and initiating the apoptosis pathway. Mitochondria-disrupting peptides destabilize mitochondrial membranes, leading to caspase-3 activation and apoptosis. Additionally, membrane-disrupting peptides directly permeabilize the cancer cell membrane to ions and other small molecules, causing uncontrolled water efflux and cell shrinkage.
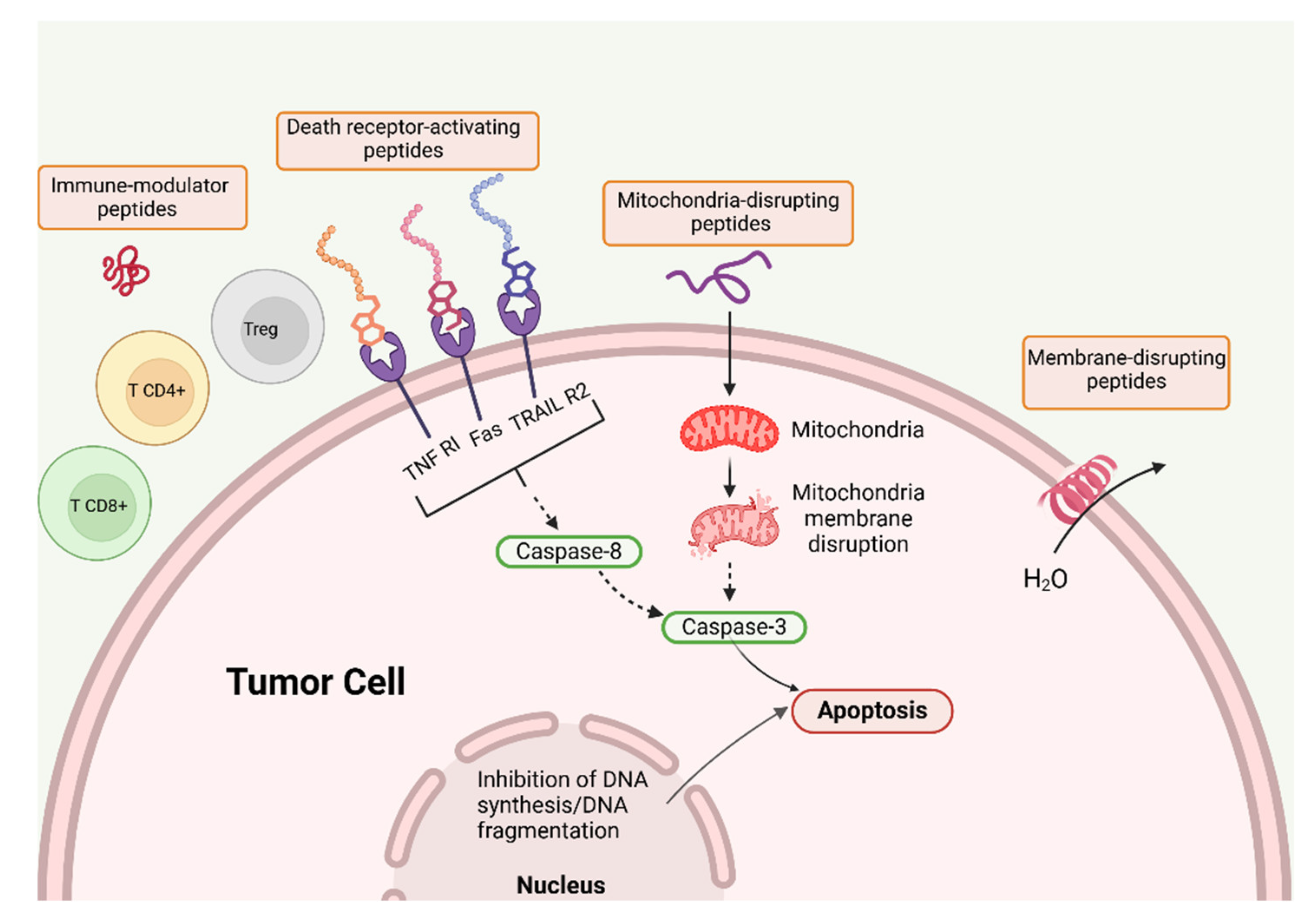

Table 1.
Antimicrobial peptides (AMP), their assemblies, and biomedical applications.
| AMP | Assembly | Biomedical application | Refs. |
|---|---|---|---|
| Poly (lysine) | Dendrimers-based hydrogel | Trauma and intraoperative bleeding | [12] |
| G3KP | OCMC/G3KP hydrogel | Hemostasis, adhesiveness, wound healing, bactericidal effect | [13] |
| DIKVAV | Self-assembled hydrogel | Growth factor delivery | [15] |
| Melittin | Melittin /Chlorin e6 /hyaluronic acid nanoparticles (60 nm in size) | Selective cytotoxicity against cancer cells | [22] |
| Gramicidin D | Self-assembled nanoparticles in water dispersion (150 nm in size) | Microbicidal effect against S. aureus and C. albicans | [23] |
| Gramicidin D and cationic polymer PDDA | Coatings from nanoparticles (self-assembled NPs of Gr D and self-assembled NPs of bilayer fragments/carboxymethylcellulose/PDDA) | Broad spectrum, high performance, synergistic activity against bacteria and fungus | [39] |
| Cyclic lipopeptides from Bacillus megaterium | Aggregates (80-800nm) | Microbicidal/ lytic action against Bacillus cereus | [45,46] |
| Killer FLIP peptide GRKKRRQRRRFFWSLCTA |
Self-assembled aggregates | Selective lysis of cancer cells | [59,60] |
| Folic acid (FA)-modified peptide FA-EEYSV-NH | Fibers | Specific toxicity against cancer cells | [62] |
| Cyclo-HH/anticancer drugs/Zn2+/NO3− | Assemblies | Drug delivery to the brain | [65] |
| Angiopep-2/daunomycin | Conjugates | Drug delivery to the brain | [74] |
| Macrocyclic peptide pAC65 | Immunotherapy against cancer | [75] | |
| Fusogenic peptide pHLIP® /Gr A | pHLIP®/ GrA in liposomes | Acidic pH transfer of Gr A to cancer cells | [89] |
| galactose−Gr A | Galactose conjugated to Gr A N terminus | Targeting galactose−Gr A to asialoglycoprotein in cancer cells | [91] |
| Gr A /doxorubicin | Combination | Synergism against colorectal cancer cells (HT-29) | [92] |
| Gr A / iturin A | Combination | Apoptosis of breast cancer cells | [95] |
| Gr A | Inhibition of pancreatic cancer stem cells proliferation | [97] | |
| Gr A | Alone or in combination with anticancer drugs | Inhibition of the proliferation of acute promyelocytic and chronic myeloid leukemia cell lines in absence of hemolytic effects; downregulation of leukemia oncogenes | [98] |
Table 2.
The variety of antimicrobial peptides (AMPs) formulations and their utilities against pathogens or cancers.
Table 2.
The variety of antimicrobial peptides (AMPs) formulations and their utilities against pathogens or cancers.
| AMP | Formulation | Anti-infective or antitumoral uses | Refs. |
|---|---|---|---|
| Gramicidin D and cationic polymer PDDA | Coatings from gramicidin D nanoparticles and PDDA polymer | Broad spectrum, high performance, synergistic activity against bacteria and fungus | [40] |
| Palmitoylated lipopeptides with 2 lysine residues C16-YKK or C16-WKK | Micelles | Activity against bacteria | [110] |
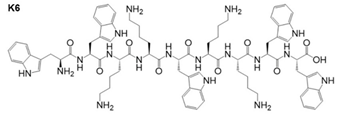 Peptide K6 and similar derivatives |
Micelles | Antibiofilm activity against bacteria | [112] |
| Membrane-active AMPs (e.g., protegrin 1, hBD-3) and antibiotics with intracellular targets (e.g., gentamicin, rifampicin) | Combinations AMPs and antibiotics | Synergy in antibacterial action | [115] |
| Hydroxypropylmethylcellulose/ xyloglucan/ gentamicin | Cross-linked transparent films | Wound healing monitoring/ Antibacterial activity against Staphylococcus aureus and Escherichia coli | [117] |
| Vancomycin-loaded bone-like hydroxyapatite/poly amino acid | Scaffold | Bone tissue engineering preventing infection | [122] |
| Peptide conjugated to short peptides capable of self-assembling into functionalized β-fibrils | Peptide aggregates at the host receptor for virus binding | Antiviral therapeutics | [128] |
| Intra-lysosomal peptide assembly after cathepsin cleavage of amphiphile peptide NDI-Lyso-RGD | Peptide fibrils inside lysosomes | Cancer cell death due to lysosomal bursting in absence of apoptosis | [132] |
| Cyclic peptide PROTAC |
Degradation of palmitoylacyltransferase | Down regulation of PD-L1 expression in human cervical cancer cells. Enhancement of anti-tumor immunity | [143] |
| Oncolytic peptides BOP7 and BOP9 in pancreatic cancer | Release of damage-associated molecular patterns DAMPS, mediators of ICD | Killing of pancreatic cancer cells in vitro. ICD in vivo | [190] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



