
Submitted:
25 October 2024
Posted:
28 October 2024
You are already at the latest version
Abstract
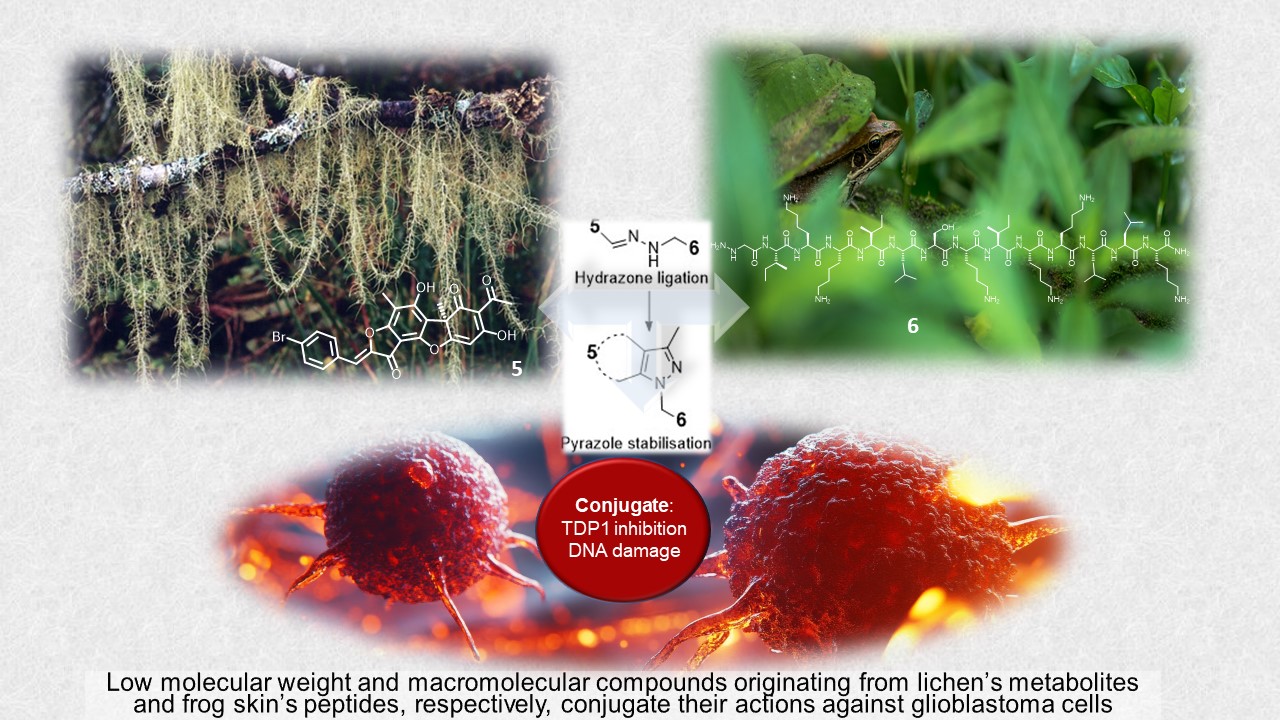
Cationic Antimicrobial Peptides (AMPs), also called Host Defence Peptides have established antimicrobial and anticancer activities. Conjugation of an AMP to a bioactive molecule with complementary activity can address some of the clinical limitations of the peptide candidate. This approach has been particularly applied in antimicrobial applications of AMPs, but remains relatively less explored in the generation of anticancer candidates. Herein, two usnic acid derivatives, based on hydrazinothiazole and benzylidenefuranone pharmacophore moieties, respectively, have been conjugated to L-K6, a lysine/leucine rich AMP, through a new pyrazole ligation intrinsically driven by the cargo molecule. Both components, the usnic acid derivative and the peptide, are selectively active against cancer cells, by targeting the human DNA repair enzyme tyrosyl-DNA phosphodiesterase 1 (TDP1) and through DNA damage, respectively. The two conjugates, based on a hydrazone linkage, exhibited pleiotropic effects, ranging from, overall, reduction of the activity from the parent drugs to their conservation or even enhancement. Notably, the conjugates retain some anti-TDP1 activity and display intermediate, or even higher, cytotoxicities against glioblastoma cells, compared to their individual components.
Keywords:
Antimicrobial peptides
; secondary metabolites
; peptide-drug conjugates
; usnic acid
; anticancer
1. Introduction
Cancer is recognised by the World Health Organisation (WHO) as the second leading cause of death worldwide with 1 in 6 deaths a consequence of malignancy.[1] In 2020, an estimated 10 million people died from cancer and approximately 19.3 million new cases were diagnosed; nonetheless, cancer mortality has been reduced in recent years, given the improvements in treatments available and their accessibility.[2] There is a steady development of new anticancer drugs and forms of chemotherapy to combat the high incidences of cancer. However, with chemotherapy as the main form of treatment, drug resistance complicates the matter significantly.[3] Described as the innate (primary) and/or adaptive (secondary) ability of cancer cells to avoid the effects of drugs through a number of mechanisms, it is a major obstacle in cancer therapy.[4] It is one of the reasons warranting the search for novel anticancer agents and/or treatment modalities.
Small molecules represent the most abundant class of drugs in treating a diverse range of diseases.[5] As chemical compounds of low-molecular weight, they possess the ability to modulate biochemical processes in order to diagnose, treat, or prevent diseases through a wide range of mechanisms.[6] In recent years there has been a shift in research from small-molecules as broad-spectrum cytotoxic drugs to more targeted cytotoxic agents, which are often of high potency and low toxicity.[7] Often kinases [8], epigenetic regulatory proteins [9], proteasomes [10], and DNA damage repair enzymes[11] are targeted as a result.
Tyrosyl-DNA-phosphodiesterase 1 (TDP1) is a critical enzyme in the repair of DNA lesions caused by antitumour drugs in the form of topoisomerase 1 (TOP1) poisons such as camptothecin and its derivatives.[12] Thus, the application of TDP1 inhibitors in cancer treatment would increase the toxicity of anticancer therapeutics targeted at topoisomerases.[13,14,15] Some natural products have been identified as promising platforms for developing effective TDP1 inhibitors [16,17,18], such as the secondary metabolite usnic acid 1 of lichen origin, with broad biological activity.[19] In recent years, the synthesis of usnic acid derivatives generated various polycyclic structures with inhibitory activities against TDP1[20], such as compounds 2 [21], 3 [22], 4 [23], and 5 [19] (Figure 1), the synergistic action of which, when combined with the TOP1 inhibitor topotecan, was confirmed in experiments in cell culture and in animal models.[23,24] In particular, two usnic acid derivatives (4 and 5) active in the low micromolar-nanomolar range were identified as promising candidates.
Peptide-based cancer therapy is also among the alternative treatment options currently explored. Malignant cells possess cancer-specific proteins on their membrane, which may be utilised in finding, or designing molecules that can selectively bind to these proteins.[25] For example, some peptides specifically recognise and bind to the membrane proteins of tumour cells with tumoricidal effects. Alternatively, those of cationic nature are attracted to the anionic phospholipids present on the outer membrane of cancer cells. [26,27] This mechanism is exploited by antimicrobial peptides (AMPs), which either are of natural origin or synthetically produced. Of potential use against infectious disease of viral, bacterial, and fungal origins, they have also been shown to possess promising and unique anticancer activities.[28] Structurally, they can be classified as α-helical, β-sheet, extended, or loop, of amphiphatic nature, due to being made-up predominantly of hydrophobic and cationic residues.[27] In terms of selectivity, AMPs with anticancer properties are classifiable into two groups, peptides active against cancer and microbial cells while not damaging normal mammalian cells, and peptides active against all three cell types.[28] However, the mechanism, through which anticancer peptides kill malignant cells can be affected by various factors and is not systemically understood in terms of binding, biofunction, and uptake. Among mechanistically characterised AMPs, L-K6 is a leucine/lysine rich sequence with selective anticancer activity, primarily exerted by DNA binding and nuclear damage.[29]
Cationic AMPs and the closely related cell-penetrating peptides (CPPs), including L-K6, are able to translocate biological membranes and exhibit therefore characteristics of possible delivery vectors for small-molecule drugs, with improved tumour targeting ability, via potential specific membrane interactions and cell-internalisation.[30] By conjugating a small-molecule drug to the peptide, transport of this bioactive cargo into the target cell may be simplified and associated with minimal toxicity, good efficacy, high potency, and potential to circumvent resistance mechanisms such as efflux pumps. Conversely, some limitations often affecting peptides, such as short half-life and low oral bioavailability [31], can potentially be mitigated through conjugation.[32] This approach has been widely investigated in the modification of AMPs for antimicrobial applications,[33,34] but has been comparatively less studied with anticancer candidates,[35] in particular in the conjugation of bioactive molecules beyond established chemotherapeutic drugs.
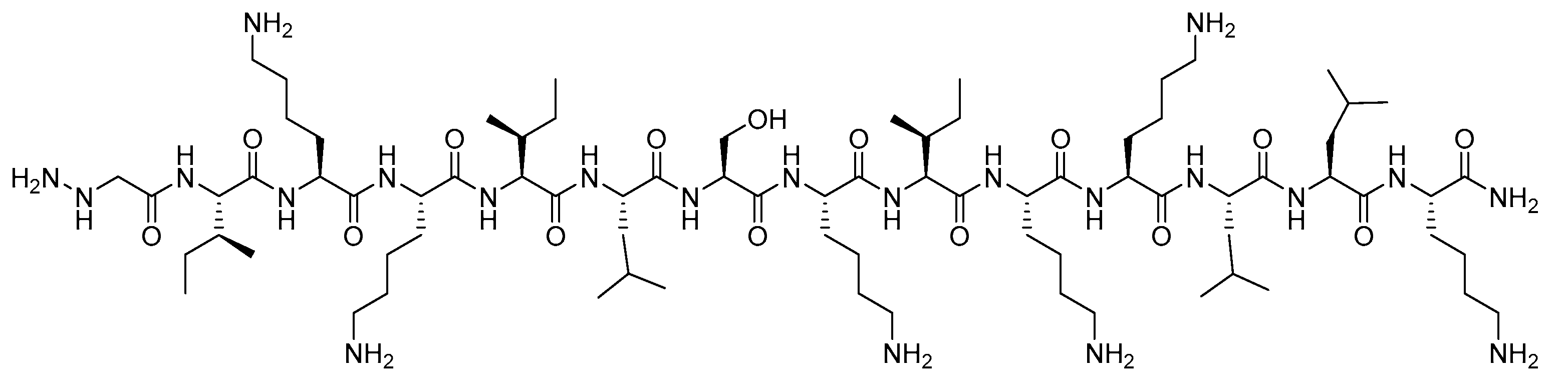
The work depicted in this paper outlines the preparation of an α-hydrazinoacetylated L-K6 peptide (6) (Figure 2) by solid-phase peptide synthesis (SPPS) and its subsequent conjugation with two usnic acid derivatives (4 and 5) by a pyrazole ligation exploiting for the first time an intrinsic structural feature of the latter molecules. These conjugates can mediate the joint and selective (peptide-mediated) delivery to cancer cells of one agent inducing DNA damage, and a second agent preventing repair. This combination can broaden the activity of the peptide, in particular against resistant cells, and possibly extend the inhibitory activities of the two components against insensitive cells. Accordingly, the testing of these conjugates, for inhibitory activity against TDP1 and cytotoxicity against human glioblastoma and adenocarcinoma cell lines, is reported.
2. Results
2.1. Synthesis
Besides being anticancer agents in their own right, the usnic acid derivatives 4 and 5 feature a 1,3-diketone group which can be targeted for effective and selective conjugation to a peptide sequence. It can indeed be exploited in a Click Chemistry approach by imine-based ligation with an α-hydrazinoacetylated peptide. This approach allows the use of fully deprotected sequences, thereby precluding any restriction in their contents and alleviating concerns related to selectivity (with regard to the conjugation site), or the stability of the cargo to the reaction conditions used for peptide deprotection.[36,37] The usnic acid derivatives, 4 and 5, were prepared as previously described.[19,23]
The modified peptide 6 was synthesised using automated SPPS, producing the native sequence with a purity of 95%. N-terminal modification with a hydrazine linker was performed manually by coupling tri-Boc-α-hydrazinoacetic to the resin-bound peptide, followed by standard cleavage and deprotection, yielding 6 with a purity of 93%.
As is known, the reaction of usnic acid with monosubstituted hydrazines proceeds at the C11=O carbonyl group with the intermediate formation of hydrazone and in most cases is accompanied by intramolecular cyclisation with the formation of a pyrazole ring annelated with a C ring.[38] The only exception is the reaction with arylhydrazines containing strong acceptor substituents, which stops at the stage of hydrazone formation.[39] In all cases, only one isomer of the pyrazole derivative is formed with the arrangement of the five-membered ring at the 2,3 positions of the dibenzofuran framework. This reaction mechanism was exploited here to selectively, efficiently and stably conjugate the peptide. Practically, while the conjugation reaction is usually carried out in alcohol at room temperature or under heating, here insufficient solubility of compounds 4 and 5 in anhydrous methanol imposed the use of other solvents. THF, chloroform, and combination thereof also failed to provide homogeneous solutions and reaction at higher temperature (40oC) remained unsuccessful. Conjugation in anhydrous DMF, at room temperature was therefore attempted, providing sufficient solubility of both reaction partners and conversion to the desired products, which were subsequently isolated by precipitation from diethyl ether.
Mass spectrometry analysis of the peptide conjugate confirmed the loss of two water molecules accompanying the formation of the hydrazone and intramolecular cyclisation alike the Knorr pyrazole synthesis. Accordingly, conjugation of 5 was performed in the same solvent and manner as 4 and the same intramolecular cyclisation observed, with yields of 55% and 63% for conjugates 7 and 8, respectively (Scheme 1 and Scheme 2).
2.2. TDP1 Inhibition
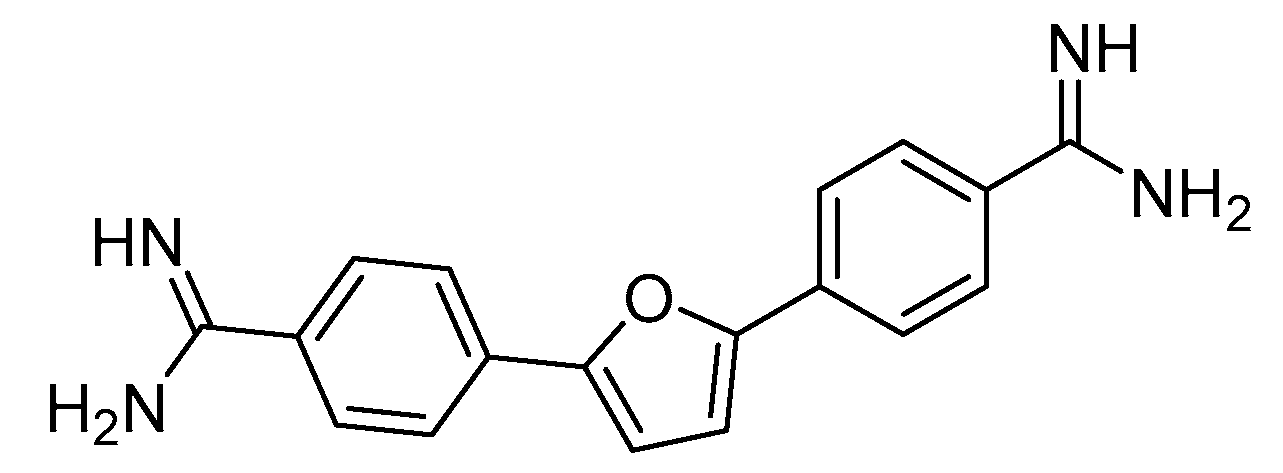
These conjugates and their individual components were tested in a real-time fluorescent assay of TDP1 inhibition.[40] Hydrazinoacetylated L-K6 (6) was able to inhibit TDP1, but with an IC50 value significantly higher than those of its conjugates 7 and 8 (Table 1). The latter show pronounced inhibitory activity against TDP1, but with approximately 6.5 and 2.3 fold increases in their IC50’s, compared to their parent TDP1 inhibitors 4 and 5, respectively. These results are compared in Table 1 to furamidine, the most potent commercial TDP1 inhibitor reported to date, used here as a positive control.[41]
Figure 3.
Furamidine (9).
2.3. Cytotoxicity
Next, the conjugates and their parent agents were evaluated in a cytotoxicity assay. Human glioblastoma (SNB19, T98G) and adenocarcinoma (MCF-7) cell lines were used in this study. At a general level, results showed that the parent TDP1 inhibitors 4 and 5 are more cytotoxic against the adenocarcinoma cell line, than against the glioblastoma ones (Table 2). Interestingly, this difference in cytotoxicity is inversed with the conjugates, 7 and 8, which are, generally, more cytotoxic against the glioblastoma cell lines than the adenocarcinoma one, while the hydrazinoacetylated peptide itself 6 is, as 4 and 5, more potent against the latter cells.
Comparing the conjugates and their individual agents, the usnic acid-based candidates (4 and 5) are in most cases significantly more cytotoxic than the peptide 6 and its conjugates (7 and 8). Against the adenocarcinoma cells, the conjugation is either detrimental, with an activity lower than those of both parent compounds, when 4 is the anti-TDP1 candidate, or neutral, with regard to the activity of the less active agent (6), when 5 is the anti-TDP1 candidate. Against the glioblastoma T98G cells, the conjugation yields activities that are intermediate between those of the usnic acid and peptide components, for both the hydrazinothiazole- and benzylidenefuranone-based candidates (4 and 5, respectively). Interestingly, against SNB19 glioblastoma cells, the results favorably compare to those obtained with the adenocarcinoma ones, as here the conjugation either generally maintains the activity of the more active agent, the usnic acid derivative 4, with conjugate 7, or even significantly improves the activities of both parent compounds, 5 and 6, with conjugate 8. To determine if some specificity can be achieved against these SNB19 cells, compounds 4 - 8 were also tested against a non-malignat (Vero) cell line. Although only conjugate 7 demonstrated some specificity to cancer SNB19 cells in comparison with Vero cells based on GI50 data, for more important GI80 data, both conjugates were more toxic to SNB19 cells than to Vero cells.
3. Discussion
Peptide drug conjugates are an emerging class of therapeutic agents. For example, between 1980 and 2018, the proportion of therapeutic peptides entering clinical development as conjugates increased from 5 to 30%.[42] Conjugation of peptides to small molecules can combine the advantages of peptide-based pharmacology with traditional medicinal chemistry,[32,43] but the synthesis of such conjugates is associated with a number of challenges. Two main strategies are generally implemented for the conjugation, solid-phase or solution-phase synthesis. The former uses one or more selected residue(s) in an otherwise fully protected sequence. It affords complete selectivity of the conjugation site(s), but require subsequent cleavage from the resin and global deprotection, under (strongly acidic) conditions that are often not compatible with the stability of the conjugate and/or the non-peptidic component. Alternatively, conjugation in solution with a protected (cleaved) sequence retains selectivity, but also faces similar stability limitations, as super acid-sensitive moieties exist as linkers for the solid support, but not for all amino acid side-chains protections. A solution phase conjugation with a fully deprotected peptide circumvents the final deprotection step, but at the expense of selectivity. Otherwise, abiotic functional groups placed on the peptide and the low-molecular weight entity, as partners of a biorthogonal reaction can address the latter limitation, but Click Chemistry[44] can require the use of metal catalysts that might be difficult to remove from the peptide, or the use of reactive moieties (e.g., constrained alkyne) which introduce additional structures significantly deviating from those of the peptide and the conjugated agent. On the other hand, hydrazone ligation as implemented here, allows the simple (methodologically and structurally) establishment of a covalent linkage between a peptide modified with a functional group reminiscent of α- or ε-amino groups in peptides and a molecule containing a carbonyl group. Here, the conjugation can be achieved by simply dissolving together the macromolecular and molecular components, does not require any additional reagent or catalyst, is realised with high atom economy, and preserves the integrity of the latter component. [45,46,47,48,49]
A peptide-drug conjugate typically comprises three components, the peptide, commonly used as a targeting ligand, a low-molecular weight agent called cargo and the linker.[50] The latter can possess important functions with regards to stability and circulation time of the conjugate, as well as potential release of the drug in its free form at the target site.[51] In the context of cancer therapy, an appropriate linker between the peptide and a cytotoxic drug provides a specific chemical bridge, potentially allowing the peptide to selectively deliver and release, or not, the drug in (the vicinity of) cancer cells.[52] Hence, linker choice is determined by the conjugation chemistry and the desired biological outcome. Here, the hydrazine-based linker, introduced by amidation of the peptide’s N-terminus provides different advantages. Targeting the N-terminus of peptides is indeed a promising strategy to achieve single-site modification at a unique and conserved peptide’s group, which is moreover conveniently solvent exposed for functionalisation by SPPS.[53] The acid-sensitive hydrazone linkage formed is reported to be stable at neutral pH, therefore in plasma (pH 7.38 - 7.42), healthy tissues and cells, but readily cleaved in the acidic conditions found in endosomes and lysosomes where pH lies between 4.5 and 6.0.[50,54,55] Overall here lies potential for release of these two agents in cancer cells, if delivered through a lysosomal delivery route.[52] On the other hand, when complete stability, even at low pH, of the hydrazone-based conjugate is desirable, the linker can be reduced to the corresponding hydrazine.[56,57] In our case, enhanced stability is directly provided by the intramolecular (pyrazole) cyclisation, following the hydrazone formation. Indeed, this ligation strategy has already been established with hydrazine peptides and 1,3-diketone groups, demonstrating to yield conjugates stable at neutral and acidic (4.5) pH for at least seven days at temperature ≤37 °C.[58] The novelty of the present ligation lies in the use of the 1,3-diketone group imbedded in the structure of the bioactive cargos. In addition to this efficient conjugation of molecules containing a 1,3-diketone system, and beside the independently reported use of C-terminal acyl pyrazoles in native chemical ligation,[59] it could be envisaged, owing to the structural similarity of the pyrazole to the well-known triazole, itself considered a peptide bond isostere,[60] to extend this effective pyrazole ligation to two peptide fragments, namely one hydrazine and one 1,3-diketone functionalized sequence.
The mechanism through which anticancer peptides operate in terms of cellular entry is not systemically understood, but through previous studies conducted with L-K6, it is proposed that targeted cancer cell binding and internalisation are major factors in ultimate cell death.[29] The internalised peptide can cause considerable nucleus damage in MCF-7 cells, without significant cytoskeleton and mitochondria disruption. Such results indicate a nucleus targeting capacity and a potential to serve as a peptidic anticancer drug. N-terminal hydrazinoacetylation of L-K6 does not appear to significantly alter the cytotoxic properties of this peptide (native sequence reported IC50 of 23 μM [29]), although slightly lower activities are observed with the modified sequence (GI50 of 39 μM). On the other hand, the parent TDP1 inhibitors 4 and 5 possess more pronounced cytotoxic activities in the low micromolar range against all tested cell lines compared to 6, except for 5 against SNB19 cells, where neither this compound, nor the modified peptide 6, demonstrate some toxicity in the concentration range tested. Interestingly, in this case, the conjugate 8 has potent cytotoxic activity against these glioblastoma cells (16 μM), while maintaining TDP1 inhibitor activity in the nanomolar range. In conclusion, while this conjugation of an AMP and a secondary metabolite did not provide a general approach to produce anticancer candidates with favorable emergent properties, it nonetheless generated a new lead compound against SNB19 cells.Future investigations will be required to comprehensively evaluate the potential of 8 against these glioblastoma, and possibly other cancer cells, at a mechanistic level and in terms of selectivity, potency, and capacity to overcome resistance.
4. Materials and Methods
4.1. Chemistry
Amino acids were purchased from CEM Corporation, UK. All other reagents and solvents were sourced from Sigma-Aldrich (Merck, Ireland), unless otherwise stated. Chromatographic analysis and purification by reversed-phase high-performance liquid chromatography (RP-HPLC) was performed on a Shimadzu Prominence HPLC (Mason Technology, Ireland) using Phenomenex Gemini columns (5 μm, C18, 110 Å, 4.6 mmD x 250 mmL or 10 mmD x 250 mmL, for the analytical and semi-preparative columns, respectively). The mobile phase, unless otherwise stated, consisted of buffer A: 0.1% trifluoroacetic acid (TFA) in water; mobile phase B: 0.1% TFA in acetonitrile with a gradient of 5 to 65% B in 18 column volumes (analytical) or 5 column volumes (semi-preparative) with a flow rate of 1 mL min-1 (analytical) or 4 mL min-1 (semi-preparative) and wavelength detection between 190-800 nm using a PDA detector. Purified peptides were characterised using an Ultraflextreme Matrix Assisted Laser Desorption Ionisation – Time of Flight (MALDI-TOF) mass spectrometer (Bruker, US). Approximately 1 mg of each sample was dissolved in 20 μL of 50% CH3CN/0.1% TFA. One μL of each sample solution was added to 1 μL of a matrix solution (α-cyano-4-hydroxycinnamic acid, 10 mg in 1 mL of 50% CH3CN/0.1% TFA). Routine analysis for reaction monitoring was performed using an Advion Expression Compact Mass Spectrometer (CMS) (Advion, US). Compounds 4 and 5 were prepared according to published procedures.[19,23]
4.1.1. Peptide Synthesis and Purification
All peptide sequences were assembled by automated SPPS on a CEM Liberty Blue™ Automated Microwave Peptide Synthesiser (CEM Corporation, UK) from 9-fluorenylmethyloxycarbonyl (Fmoc)-protected L-amino acids, according to the Fmoc-tBu strategy with N,N’-diisopropylcarbodiimide/ ethyl cyanohydroxyiminoacetate (Oxyma Pure) coupling chemistry, from a Rink Amide MBHA resin (0.78 mmol/g) on a scale of 0.1-0.25 mM and with final Fmoc deprotection. Single coupling cycles using an excess of Fmoc-amino acids (4 equivalents) were employed unless otherwise stated. The addition of the tri-Boc-α-hydrazinoacetic acid linker was carried out manually using a syringe fitted with a Teflon frit. Removal of Fmoc was carried out using 20% v/v piperidine in N,N-dimethylformamide (DMF) and the reaction was monitored by the UV absorbance at 301 nm of the dibenzofulvene-piperidine adduct in the run-off.
After assembly, the peptide was released from the resin by treatment with a cleavage cocktail consisting of 840 μL of trifluoroacetic acid, 100 μL thioanisole, 50 μL water, 10 μL triisopropylsilane, added to the resin and left stirring for 120 minutes. The peptide was then precipitated with diethyl ether as a white solid, collected by centrifugation and washed twice with diethyl ether.
Synthesis of hydrazinoacetylated L-K6 (H2NHNCOC)Ile-Lys-Lys-Ile-Leu-Ser-Lys-Ile-Lys-Lys-Leu-Leu-Lys-NH2 (6): Following washing of the resin-bound (L-K6 native) peptide with CH2Cl2 and DMF, (Boc)2N-N(Boc)CH2COOH (195 mg, 0.5 mmol) was coupled using HATU (190 mg, 0.5 mmol) and DIPEA (1.25 mL, 1.0 mmol) activation (120 min at rt with agitation), for each cycle of a double coupling procedure. The resin was then successively washed with DMF and CH2Cl2 and dried under vacuum. An aliquot cleavage of the peptide from the resin was performed for mass spectrometry analysis. The cleavage, isolation and purification of the modified peptide was performed as described above. Following a freeze-drying step, the peptide was obtained as a white powder (298 mg, 37%). Analytical HPLC (C18): tR= 28.687 min, 97% purity. ESI MS (m/z) Calcd for C77H150N22O15: 1624.149 Found: 812.6 [M + 2H]2+ MALDI-TOF (m/z) Calcd for C77H150N22O15: 1624.149 Found: 1624.202 [M+H]+.
Synthesis of L-K6-4 conjugate (4-NHNCOC-Ile-Lys-Lys-Ile-Leu-Ser-Lys-Ile-Lys-Lys-Leu-Leu-Lys-NH2, 7):6 (40.7 mg, 0.025 mmol) was added to a solution of 4 (14.6 mg, 0.025 mmol) in anhydrous DMF (1 mL) and left stirring for 24 hours under argon. Precipitation using Et2O (10 mL), centrifugation and washes x2 with Et2O followed. The pellet was dissolved in distilled water and lyophilised. Upon drying a small amount of product was dissolved in water and prepared for mass spectrometry (MALDI-TOF) analysis. Purification of the final peptide was performed by RP-HPLC following standard procedure. Following a freeze-drying step, the peptide was obtained as a pale-yellow powder (30 mg, 55%). Analytical HPLC (C18): tR= 37.593 min, 86% purity. ESI MS (m/z) Calcd for C103H166BrN25O19S: 2170.58 Found: 724.4 [M + 3H]3+. MALDI-TOF (m/z) Calcd for C103H166BrN25O19S: 2170.58 Found: 2170.244 [M+].
Synthesis of L-K6-5 conjugate (5-NHNCOC-Ile-Lys-Lys-Ile-Leu-Ser-Lys-Ile-Lys-Lys-Leu-Leu-Lys-NH2, 8): performed as described above for 7. Following a freeze-drying step, the peptide was obtained as a pale-yellow powder (33 mg, 63%). Analytical HPLC (C18): tR= 36.357 min, 94% purity. ESI MS (m/z) Calcd for C102H163BrN22O20: 2095.16 Found: 699.8 [M + 3H]3+. MALDI-TOF (m/z) Calcd for C102H163BrN22O20: 2095.16 Found: 2096.349 [M+].
4.2. Real-time Detection of TDP1 Activity
The oligonucleotide biosensor 5'-FAM-AAC GTC AGG GTC TTC C- BHQ1-3' was used for TDP1 enzyme activity real-time fluorescence detection that is a 16-mer single-stranded oligonucleotide with a 5'-fluorophore (FAM), and a 3'-quencher (BHQ1) [40]. Recombinant protein TDP1 was expressed in E. coli (pET 16B plasmid containing TDP1 cDNA was kindly provided by Dr. K.W. Caldecott, University of Sussex, United Kingdom) and isolated as described [61]. The final volume (200 μL) of reaction mixture contained TDP1 reaction buffer (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 7 mM β-mercaptoethanol), 50 nM oligonucleotide substrate, varied concentrations of the tested compounds. Purified TDP1 was added in a final concentration of 1.5 nM. The TDP1 reaction mixtures were incubated at a constant temperature of 26°C in a POLARstar OPTIMA fluorimeter (BMG LABTECH, GmbH). Fluorescence intensity was measured (Ex. 485/Em. 520 nm) every 1 min for 10 minutes. The efficiency of TDP1 inhibition was evaluated by comparing the rate of increase in fluorescence of biosensor in the presence of compound to that of DMSO (1.5%) in control wells. IC50 values were determined using an eleven-point concentration response curve by MARS Data Analysis 2.0 (BMG LABTECH) and the slope during the linear phase was calculated. The IC50 measurements were carried out in at least three independent experiments.
4.3. Cell Culture and Cytotoxicity Assay
The human cancer cells of the SNB19, T98G (cells of human glioblastoma) MCF-7 (adenocarcinoma) and the cells derived from the kidney of an African green monkey (Vero) were used in this study. These cell lines were obtained from a collection of cells of State Research Center for Virology and Biotechnology "Vector" of Rospotrebnadzor, Koltsovo, Novosibirsk Region. They were cultured in the DMEM/F12 medium that contained 10% fetal bovine serum, L-glutamine (2 mmol/L), gentamicin (80 µg/ml) in a CO2 incubator at 37oC. The tested compounds were dissolved in DMSO and added to the cellular culture at the required concentrations. Three wells were used for each concentration. The cells which were incubated without the compounds were used as a control. Cells were placed on 96-well microplates and cultivated at 37 oС in 5% CO2/95% air for 48 h. The cell viability was assessed through an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-phenyl-2H-tetrazolium bromide] conversion assay. 1% MTT was added to each well. Four hours later the medium was removed, leaving the formazan crystals, and isopropanol was added and mixed for 15 min. Optical density of the samples was measured on a TECAN Sunrise multi-well spectrophotometer at the wavelength of 570 nm with reference 670 nm. The 50% cytotoxic dose (GI50) of each compound (i.e., the compound concentration that lowers the amount of cells to 50% in a culture, or decreases the optical density twice as compared to the control wells) was calculated from the data obtained. Statistical processing of the results was performed using the Microsoft Excel-2007, STATISTICA 6.0, and GraphPad Prism 5.0 programs. The results are given as an average value ± a deviation from the average.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org: chromatograms and mass spectra of the parent L-K6 peptide, its hydrazinoacetylated derivative (6) and of the two conjugates (7 and 8).
Author Contributions
Conceptualization, A.G.P, O.A.L., M.D. and K.P.V.; methodology, M.D., and K.P.V.; validation, S.O’F.; formal analysis, S.O’F. and J.L.; investigation, S.O’F.,Y.K., O.A.L., N.S.D., A.L.Z., M.A.P., N.F.S.; resources, A.G.P, M.D., O.I.L. and N.F.S.; writing—original draft preparation, S.O’F.; writing—review and editing, S.O’F., M.D., O.A.L., A.L.Z., O.I.L., N.F.S. and K.P.V.; supervision, M.D., M.V. and K.P.V; project administration, M.D. and K.P.V.; funding acquisition, A.G.P. and M.D. All authors have read and agreed to the published version of the manuscript.
Funding
This publication has emanated from research conducted with the financial support of Science Foundation Ireland under Grant number 12/RC/2275_P2. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Acknowledgments
The authors acknowledge the Erasmus+ Programme of the European Union for the mobility of researchers between RCSI and Novosibirsk State University funded under Key Action 1 International Credit Mobility Programme, grant numbers 2015-2-IE02-KA107-000423 and 2018-1-IE02-KA107-000606, and between RCSI and the University of Lorraine under the Lifelong Learning Programme and Key Action 103.
Conflicts of Interest
“The authors declare no conflict of interest.”
References
- WHO Health Topics: Cancer. https://www.who.int/health-topics/cancer#tab=tab_1 12/07/2021.
- H Sung, J Ferlay, R L Siegel, M Laversanne, I Soerjomataram, A Jemal and F Bray, Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers and 185 countries, CA Cancer Clin, 2020, 71, 209-249. [CrossRef]
- K O Alfarouk, C-M Stock, S Taylor, M Walsh, A K Muddathir, D Verduzco, A H H Bashir, O Y Mohammed, G O Elhassan, S Harguindey, S J Reshkin, M E Ibrahim and C Rauch, Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp, Cancer Cell Int, 2015, 15, 71. [CrossRef]
- J-C Marine, S-J Dawson and M. A. Dawson, Non-genetic mechanisms of therapeutic resistance in cancer, Nat Rev Cancer, 2020, 20, 743-756. [CrossRef]
- M Chhabra in Translational Biotechnology: A Journey from laboratory to clinics, (Ed. Y Hasija), Academic Press, United States, 2021, pp. 137-164.
- H X Ngo and S Garneau-Tsodikova, What are the drugs of the future?, Med Chem Commun, 2018, 9, 757-758. [CrossRef]
- L Zhong, Y Li, L Xiong, W Wang, M Wu, T Yuan, W Yang, C Tian, Z Miao, T Wang and S Yang, Small molecules in targeted cancer therapy: advances, challenges, and future perspectives, Sig Transduct Target Ther, 2021, 6, 201. [CrossRef]
- K S Bhullar, N Orrego Lagarón, E M McGowan, I Parmar, A Jha, B P Hubbard and H P Vasantha Rupasinghe, Kinase-targeted cancer therapies: progress, challenges and future directions, Mol Cancer, 2018, 17, 48. [CrossRef]
- J W Park and J W Han, Targeting epigenetics for cancer therapy, Arch Pharm Res, 2019, 42, 159-170. [CrossRef]
- E Manasanch and R Orlowski, Proteasome inhibitors in cancer therapy, Nat Rev Clin Oncol, 2017, 14, 417-433. [CrossRef]
- M R Kelley and M L Fishel, DNA Repair Proteins as Molecular Targets for Cancer Therapeutics, Anticancer Agents Med Chem, 2008, 8, 4, 417-425. [CrossRef]
- J Murai, S N Huang, B B Das, T S Dexheimer, S Takeda and Y Pommier, Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Repairs DNA Damage Induced by Topoisomerases I and II and Base Alkylation in Vertebrate Cells, J Biol Chem, 2012, 287, 16, 12848-12857. [CrossRef]
- N I Rechkunova, N A Lebedeva and O I Lavrik, Tyrosyl-DNA Phosphodiesterase 1 is a new player in repair of apurinic/apyrimidinic sites, Russ J Bioorg Chem, 2015, 41, 474-480. [CrossRef]
- G L Beretta, G Cossa, L Gatti, F Zunino and P Perego, Tyrosyl-DNA phosphodiesterase 1 targeting for modulation of camptothecin-based treatment, Curr Med Chem, 2010, 17, 15, 1500-1508. [CrossRef]
- E J. Brettrager and R C.A.M. van Waardenburg, Targeting Tyrosyl-DNA phosphodiesterase I to enhance toxicity of phosphodiester linked DNA-adducts, Cancer Drug Resist, 2019, 2, 4, 1153-1163. [CrossRef]
- T B. Smallwood, Lauren R. H. Krumpe, C D. Payne, V G. Klein, B R. O'Keefe, R J. Clark, C I. Schroeder and K. J. Rosengren, Picking the tyrosine-lock: chemical synthesis of the tyrosyl-DNA phosphodiesterase I inhibitor recifin A and analogues, Chem Sci, 2024, 15, 13227-13233. [CrossRef]
- A L. Zakharenko, O A. Luzina, A A. Chepanova, N S. Dyrkheeva, N F. Salakhutdinov and O I. Lavrik, Natural products and their derivatives as inhibitors of the DNA repair enzyme Tyrosyl-DNA Phosphodiesterase 1, Int J Mol Sci, 2023, 24, 6. [CrossRef]
- A Bermingham, E Price, C Marchand, A Chergui, A Naumova, E L. Whitson, L R. H Krumpe, E I. Goncharova, J R. Evans, T C. McKee, C J. Henrich, Y Pommier and B R. O'Keefe, Identification of natural products that inhibit the catalytic function of human Tyrosyl-DNA Phosphodiesterase (TDP1), SLAS Discov, 2017, 22, 9, 1093-1105. [CrossRef]
- Zakharova, O A. Luzina, A Zakharenko, D Sokolov, A Filimonov, N Dyrkheeva, A Chepanova, E Ilina, A Ilyina, K Klabenkova, B Chelobanov, D Stetsenko, A Zafar, C Eurtivong, J Reynisson, K P. Volcho, N F. Salakhutdinov and O Lavrik, Synthesis and evaluation of aryliden- and hetarylidenfuranone derivatives of usnic acid as highly potent Tdp1 inhibitors, Bioorg Med Chem, 2018, 26, 15, 4470-4480. [CrossRef]
- A S Filimonov, A A Chepanova, O A Luzina, A L Zakharenko, O D Zakharova, E S Ilina, N S Dyrkheeva, M S Kuprushkin, A V Kolotaev, D S Khachatryan, J Patel, I K H Leung, R Chand, D M Ayine-Tora, J Reynisson, K P Volcho, N F Salakhutdinov and O I Lavrik, New hydrazinothiazole derivatives of usnic acid as potent Tdp1 inhibitors, Molecules, 2019, 20, 24, 3711. [CrossRef]
- A Zakharenko, O Luzina, O Koval, D Nilov, I Gushchina, N Dyrkheeva, V Švedas, N Salakhutdinov and O Lavrik, Tyrosyl-DNA Phosphodiesterase 1 Inhibitors: Usnic acid enamines enhance the cytotoxic effect of camptothecin, J Nat Prod, 2016, 79, 206, 2961-2967. [CrossRef]
- A L Zakharenko, O A Luzina, D N Sokolov, O D Zakharova, M E Rakhmanova, A A Chepanova, N S Dyrkheeva, O I Lavrik and N F Salakhutdinov, Usnic acid derivatives are effective inhibitors of tyrosyl-DNA phosphodiesterase 1, Russ J Bioorg Chem, 2017, 43, 84-90. [CrossRef]
- A L. Zakharenko, O A. Luzina, K P. Volcho, N F. Salakhutdinov, O I. Lavrik, D N. Sokolov, V I. Kaledin, V P. Nikolin, N A. Popova, J Patel, O D. Zakharova, A A. Chepanova, A Zafar, J Reynisson, E Leung and I K. H. Leung, Novel tyrosyl-DNA phosphodiesterase 1 inhibitors enhance the therapeutic impact of topoteсan on in vivo tumor models, Eur J Med Chem, 2019, 161, 581-593. [CrossRef]
- T E Kornienko, A A Chepanova, A L Zakharenko, A S Filimonov, O A Luzina, N S Dyrkheeva, V P Nikolin, N A Popova, N F Salakhutdinov and O I Lavrik, Enhancement of the antitumor and antimetastatic effect of Topotecan and normalization of blood counts in mice with lewis carcinoma by Tdp1 Inhibitors - New usnic acid derivatives, Int J Mol Sci, 2024, 25, 2. [CrossRef]
- K R Kampen, Membrane proteins: The key players of a cancer cell, J Membrane Biol, 2011, 242, 69-74. [CrossRef]
- D Wua, Y Gao, Y Qi, L Chen, Y Maa and Y Li, Peptide-based cancer therapy: Opportunity and challenge, Cancer Lett, 2014, 351, 1, 13-22. [CrossRef]
- A Reinhardt and I. Neundorf, Design and application of antimicrobial peptide conjugates, Int J Mol Sci, 2016, 17, 701. [CrossRef]
- D Gaspar, A S Veiga and M A R BCastanho, From antimicrobial to anticancer peptides: A review, Front Microbiol, 2013, 4, 294. [CrossRef]
- C Wang, S Dong, L Zhang, Y Zhao, L Huang, X Gong, H Wang and D Shang, Cell surface binding, uptaking and anticancer activity of L-K6, a lysine/leucine-rich peptide, on human breast cancer MCF-7 cells, Sci Rep, 2017, 7, 8293. [CrossRef]
- M Kalmouni, S Al-Hosani and M. Magzoub, Cancer-targeting peptides, Cell Mol Life Sci, 2019, 76, 2171-2183. [CrossRef]
- C Recio, F Maione, A J Iqbal, N Mascolo and V De Feo, The potential therapeutic application of peptides and peptidomimetics in cardiovascular disease, Front Pharmacol, 2017, 7, 526. [CrossRef]
- B M Cooper, J Legre, D H O' Donovan, M Ölwegård Halvarsson and D R Spring, Peptides as a platform for targeted therapeutics for cancer: peptide-drug conjugates (PDCs), Chem Soc Rev, 2021, 3, 50, 1480-1494. [CrossRef]
- W Li, F Separovic, N M O'Brien-Simpson and J D Wade, Chemically modified and conjugated antimicrobial peptides against superbugs, Chem Soc Rev, 2021, 50, 8, 4932-4973. [CrossRef]
- S Prasad Selvaraj and J-Y Chen, Conjugation of antimicrobial peptides to enhance therapeutic efficacy, Eur J Med Chem, 2023, 259, 115680. [CrossRef]
- S F. A. Rizvi, H Zhang and Q Fang, Engineering peptide drug therapeutics through chemical conjugation and implication in clinics, Med Res Rev, 2024. [CrossRef]
- D Bonnet, C Grandjean, P Rousselot-Pailley, P Joly, L Bourel-Bonnet, V Santraine, H Gras-Masse and O Melnyk, Solid-phase functionalization of peptides by an a-Hydrazinoacetyl group, J Org Chem, 2003, 68, 7033-7040. [CrossRef]
- L Guy, J Vidal and A Collet, Design and synthesis of hydrazinopeptides and their evaluation as human eukocyte elastase inhibitors, J Med Chem, 1998, 41, 4833-4843. [CrossRef]
- D N. Sokolov, O A. Luzina and N F. Salakhutdinov, Usnic acid: preparation, structure, properties and chemical transformations, Russ Chem Rev, 2012, 81, 8, 747-768. [CrossRef]
- Luzina, M P. Polovinka, N F. Salakhutdinov and G. A. Tolstikov, Chemical modification of usnic acid: III.* Reaction of (+)-usnic acid with substituted phenylhydrazines, Russ J Org Chem, 2009, 45, 1783-1789. [CrossRef]
- A Zakharenko, T Khomenko, S Zhukova, O Koval, O Zakharova, R Anarbaev, N Lebedeva, D Korchagina, N Komarova, V Vasiliev, J Reynisson, K Volcho, N Salakhutdinov and O Lavrik, Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety, Bioorg Med Chem, 2015, 9, 23, 2044-2052. [CrossRef]
- S Antony, C Marchand, A G Stephen, L Thibaut, K K Agama, R J Fisher and Y. Pommier, Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1, Nucleic Acids Res, 2007, 13, 35, 4474-4484. [CrossRef]
- J L. Lau and M K. Dunn, Therapeutic peptides: Historical perspectives, current development trends and future directions, Bioorganic Med Chem Lett, 2018, 26, 2700-2707,.
- B M. Cooper, J Iegre, D H. O'Donovan, M O. Halvarsson and D R. Spring, Peptides as a platform for targeted therapeutics for cancer: peptide-drug conjugates (PDCs), Chem Soc Rev, 2021, 50, 1480-1494. [CrossRef]
- W Tang and M L. Becker, “Click” reactions: a versatile toolbox for the synthesis of peptide-conjugates, Chem Soc Rev, 2014, 43, 20, 7013-7039. [CrossRef]
- J B. Matson and S I. Stupp, Drug release from hydrazone-containing peptide amphiphiles, Chem Commun, 2011, 47, 7962-7964. [CrossRef]
- G J. Kelly, A Foltyn-Arfa Kia, F Hassan, S O'Grady, M P. Morgan, B S. Creaven, S McClean, J H. Harmey and M Devocelle, Polymeric prodrug combination to exploit the therapeutic potential of antimicrobial peptides against cancer cells, Org Biomol Chem, 2016, 14, 9278-9286. [CrossRef]
- A Dal Pozzo, M-H Ni, E Esposito, S Dallavalle, L Musso, A Bargiotti, C Pisano, L Vesci, F Bucci, M Castorina, R Foderà, G Giannini, C Aulicino and S Penco, Novel tumor-targeted RGD peptide–camptothecin conjugates: Synthesis and biological evaluation, Bioorg Med Chem, 2010, 18, 64-72. [CrossRef]
- R A. Firestone, D Willner, S J. Hofstead, H D. King, T Kaneko, G R. Braslawsky, R S. Greenfield, P A. Trail, S J. Lasch, A J. Henderson, A M. Casazza, I Hellström and K E. Hellström, Synthesis and antitumour activity of the immunoconjugate BR96-Dox, J Control Release, 1996, 39, 2-3, 251-259. [CrossRef]
- A Saghaeidehkordi, S Chen, S Yang and K Kaur, Evaluation of a keratin 1 targeting peptide-doxorubicin conjugate in a mouse model of triple-negative breast cancer, Pharmaceutics, 2021, 13, 5, 661-675. [CrossRef]
- M Alas, A Saghaeidehkordi and K Kaur, Peptide–drug conjugates with different linkers for cancer therapy, J Med Chem, 2021, 64, 1. [CrossRef]
- E Ziaei, A Saghaeidehkordi, C Dill, I Maslennikov, S Chen and K. Kaur, Targeting triple negative breast cancer cells with novel cytotoxic peptide-doxorubicin conjugates, Bioconjug Chem, 2019, 30, 12. [CrossRef]
- P Hoppenz, S Els-Heindl and A G Beck-Sickinger, Peptide-drug conjugates and their targets in advanced cancer therapies, Front Chem, 2020, 8, 571. [CrossRef]
- L De Rosa, R Di Stasi, A Romanelli and L D D’Andrea, Exploiting protein N-Terminus for site-specific bioconjugation, Molecules, 2021, 26, 12, 3521. [CrossRef]
- S-S Li, M Zhang, J-H Wang, F Yang, B Kang, J-J Xu and H-Y Chen, Monitoring the changes of pH in lysosomes during autophagy and apoptosis by plasmon enhanced raman imaging, Anal Chem, 2019, 91, 13, 8398–8405. [CrossRef]
- J R McCombs and S C Owen, Antibody Drug Conjugates: Design and selection of linker, payload and conjugation chemistry, AAPS J, 2015, 17, 2, 339-351. [CrossRef]
- B C. Atkinson and A R. Thomson, Structured cyclic peptide mimics by chemical ligation, Pept Sci, 2022, 114, 5. [CrossRef]
- M I. Meschaninova, N S. Entelis, E L. Chernolovskaya and A G. Venyaminova, A verstaile solid-phase approach to the synthesis of oligonucleotide conjugates with biodegradeable hydrazone linker, Molecules, 2021, 26, 8, 2119-2134. [CrossRef]
- M Mallig, D Hymel and F Liu, Further Exploration of Hydrazine-Mediated Bioconjugation Chemistries, Org Lett, 2020, 22, 16, 6677-6681. [CrossRef]
- P Liao and C He, Azole reagents enabled ligation of peptide acyl pyrazoles for chemical protein synthesis, Chem Sci, 2024, 15, 7965-7974. [CrossRef]
- H C. Kolb and K B. Sharpless, The growing impact of click chemistry on drug discovery, Drug Discov Today, 2003, 8, 24, 1128-1137. [CrossRef]
- N Dyrkheeva, R Anarbaev, N Lebedeva, M Kuprushkin, A Kuznetsova, N Kuznetsov, N Rechkunova and O Lavrik, Human Tyrosyl-DNA Phosphodiesterase 1 Possesses Transphosphooligonucleotidation Activity With Primary Alcohols, Front Cell Dev Biol, 2020, 8. [CrossRef]
Figure 1.
Structures of usnic acid and derivatives with TDP1 inhibitory activities.
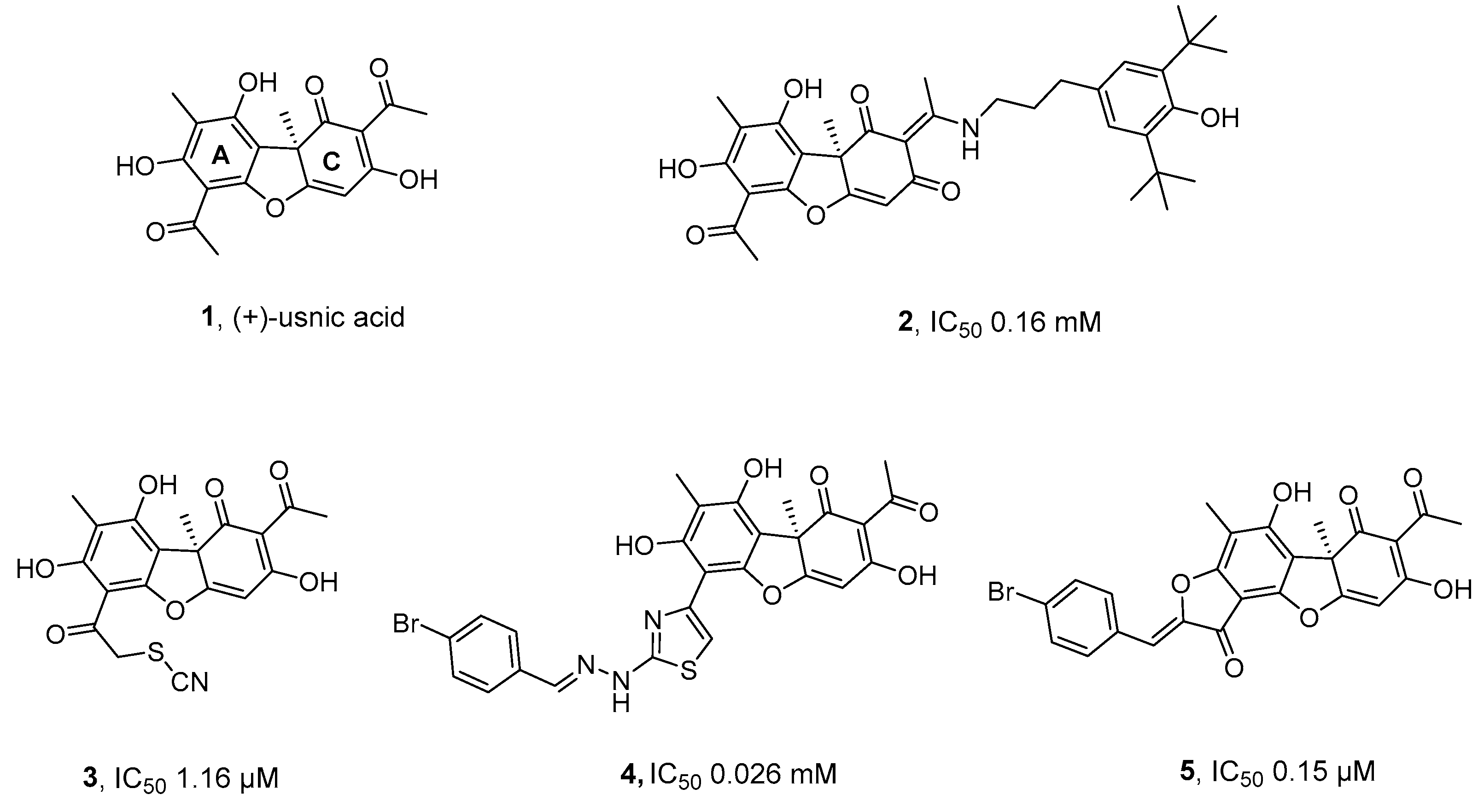
Figure 2.
Chemical structure of hydrazinoacetylated L-K6 peptide, 6.
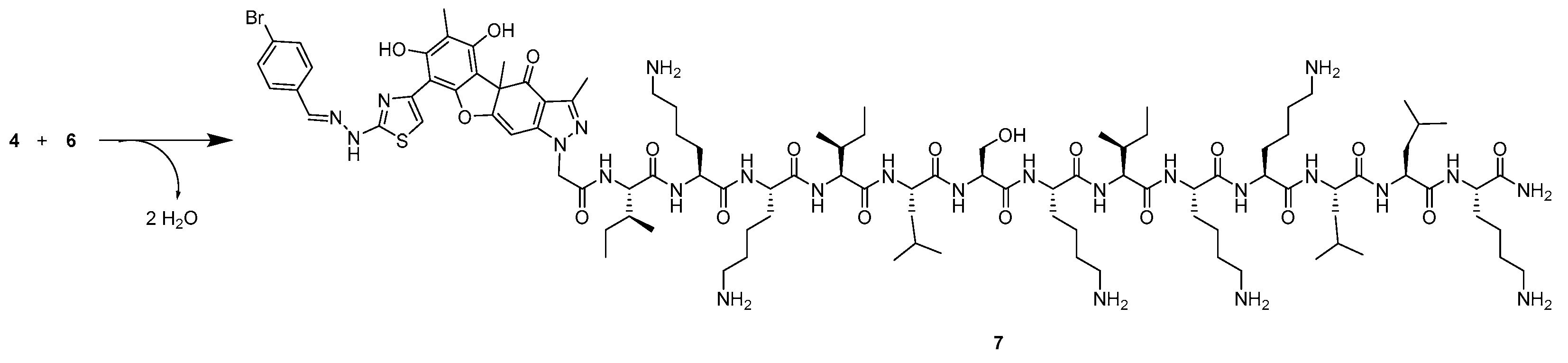
Scheme 1.
Formation of the conjugate 7 from the hydrazinothiazole-based usnic acid derivative 4 and the hydrazinoacetylated peptide 6.
Scheme 1.
Formation of the conjugate 7 from the hydrazinothiazole-based usnic acid derivative 4 and the hydrazinoacetylated peptide 6.
Scheme 2.
Formation of the conjugate 8 from the benzylidenefuranone-based usnic acid derivative 5 and the hydrazinoacetylated peptide 6.
Scheme 2.
Formation of the conjugate 8 from the benzylidenefuranone-based usnic acid derivative 5 and the hydrazinoacetylated peptide 6.
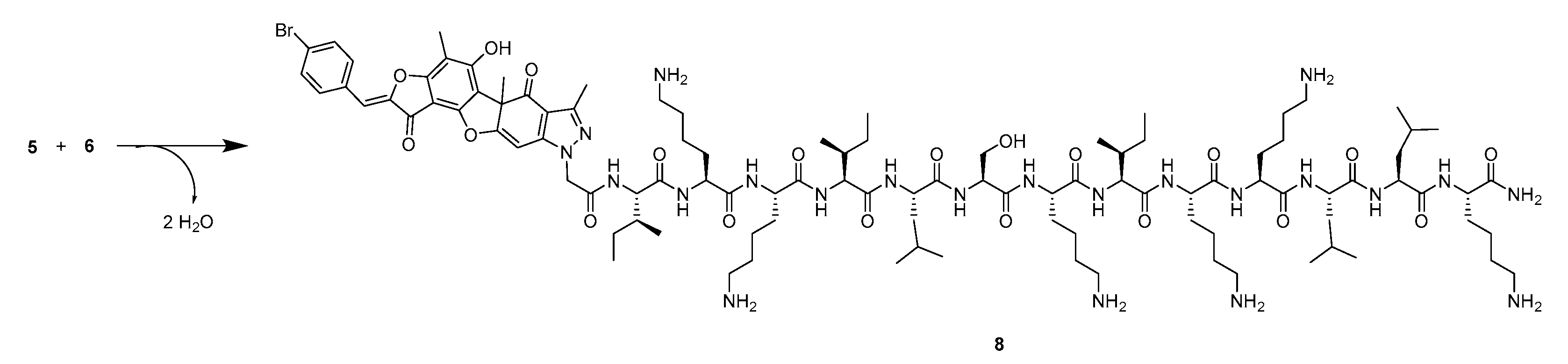

Table 1.
Results of real-time detection of TDP1 activity.
| Compounds | IC50, nM |
|---|---|
| 4 | 26±11 |
| 5 | 150±30 |
| 6 | 1800+600 |
| 7 | 168+23 |
| 8 | 340+34 |
| 9 | 1200±300 |
Table 2.
Cytotoxicity study with adenocarcinoma (MCF-7) and human glioblastoma (SNB19, T98G) cell lines.
Table 2.
Cytotoxicity study with adenocarcinoma (MCF-7) and human glioblastoma (SNB19, T98G) cell lines.
| Cell Line | Cytotoxic dose1 (µM) | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|
| GI50 | 2.8±0.07 | 2.2±0.31 | 39±6.08 | 65±15.29 | 36±1.24 | |
| MCF-7 | GI80 | 4.7±0.21 | 3.9±0.87 | 81±16 | >100 | 72±2.4 |
| GI90 | 5.5±0.41 | 4.7±1.59 | >100 | >100 | >100 | |
| GI50 | 20.5±2.27 | >100 | >100 | 18±0.35 | 16.5±0.35 | |
| SNB19 | GI80 | 61±1.78 | >100 | >100 | 68±3.34 | 56±0.7 |
| GI90 | 83±1.87 | >100 | >100 | >100 | 85±2.32 | |
| GI50 | 10.1±0.67 | 21±2.6 | >100 | 42±3.02 | 38±1.22 | |
| T98G | GI80 | 51±1.08 | >100 | >100 | 82±1.78 | 80±2.86 |
| GI90 | 90±3.94 | >100 | >100 | >100 | >100 | |
| Vero | GI50 | 4,1±1,66 | >100 | 64±13,71 | 33±3,53 | 18±3,18 |
| GI80 | 48±16,76 | >100 | >100 | >100 | >100 | |
| GI90 | >100 | >100 | >100 | >100 | >100 |
1 Cytotoxic dose (GI50) is the concentration that lowers the amount of cells to 50% in a culture.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



