
Submitted:
21 March 2025
Posted:
24 March 2025
You are already at the latest version
Abstract
β-Lactamase/transpeptidase-like superfamily proteins are serine proteases that use the Ser-Lys catalytic dyad to carry out their biological functions. Here, we investigate the three known families of β-lactamase/transpeptidase-like superfamily proteins, the β-lactamase/D-Ala carboxypeptidase, glutaminase and Dac-like, and describe the structural catalytic cores that govern the catalytic residues in these proteins. We show that the structural catalytic core of these proteins is a combination of three zones, the mutual three-dimensional arrangement of which correspondingly determines their belonging to one of 7 and 24 established groups and subgroups.
Keywords:
β-lactamase
; DD-carboxypeptidase
; glutaminase
; 3D structure
; catalytic core
; classification
1. Introduction
Serine peptidases/hydrolases are found in all living organisms. These diverse enzymes have been subject of a significant number of structural studies for many years, reflected in creation of the MEROPS and ESTHER databases [1,2]. In most cases, these enzymes carry out catalytic activity through the “classical” catalytic triad: Ser(nucleophile) – His(base) - Asp(acid) [3,4].
The sheer amount of different three-dimensional (3D) structures of serine peptidases/hydrolases that have been submitted to the Protein Data Bank (PDB) [5] necessitated the search for common structural elements that could be used as a basis to cluster and further classify many unrelated structures that belong to the same structural superfamilies. For example, one common structural element is the Structural Catalytic Core (SCC) around the Ser-His-Asp catalytic triad, which has been used to compare representative 3D structures from trypsin-like serine proteases, α/β-hydrolases, SGNH hydrolase-like and subtilisin-like superfamily proteins [6,7,8,9]. The functional idea behind the definition of the SCC was that it consisted of a collection of small closed substructures, called “zones”, where each zone (1) included short segments that were repeatedly found in groups of different enzymes, (2) incorporated “key” residues that fulfilled equivalent functional roles, and (3) were interconnected by various atomic interactions that were also similar within those groups of proteins.
In our previous investigation of the SCC, there remained one uncharacterized superfamily of serine proteases with the key representative enzyme of D-alanyl-D-alanine carboxypeptidase that was purposely excluded. This enzyme is an unconventional serine protease, in which the classical Ser-His-Asp triad of the active site instead contains either a Ser-Lys or a Ser-Tyr catalytic dyad. Indeed, two separate 3D structures of this protein, PDB ID: 3PTE [10] and PDB ID: 1MPL [11], were used in two separate studies of enzyme function, where in one study, the role of the general base was shown to be played by Lys [12], and in the other, by Tyr [13].
According to the Structural Classification of Proteins (SCOP) database [14], D-alanyl-D-alanine carboxypeptidase belongs to the β-lactamase/D-Ala carboxypeptidase structural family and β-lactamase/transpeptidase-like structural superfamily. Three small characteristic motifs: (1) the Ser-Xaa-Xaa-Lys motif, which is also known as the “S-X-X-K” motif; (2) the (Ser/Tyr)-Xaa-(Asn/Cys) motif, which is also known as the “S-X-N” motif; and (3) the (Lys/His)-(Thr/Ser)-Gly motif, which is also known as the “K-T/S-G” motif; and an additional structural element called the omega(Ω)-loop [15] are present at the active site of all β-lactamase/D-Ala carboxypeptidase family enzymes [16,17,18,19]. The role of the Ω-loop in the catalytic activity of different β-lactamases had been studied in detail [20,21,22].
Due to the unconventional composition and variation of the catalytic triad in D-alanyl-D-alanine carboxypeptidase and presence of the unique Ω-loop involved in the formation of the active site, the SCC of the β-lactamase/transpeptidase-like superfamily enzymes had been excluded from our previous studies; which we now address here.
In summary, members of the β-lactamase/D-Ala carboxypeptidase family have three standalone structural elements of their catalytic machinery. One, there is a catalytic dyad, Ser-Lys/Ser-Tyr described above. Two, there is a set of atoms of variable residues called the “oxyanion hole”, which are commonly found in serine peptidases. Three, there is the unique Ω-loop, which is only found in the β-lactamase/D-Ala carboxypeptidase family. Following the SCC approach, these three structural elements of catalytic machinery could be incorporated into their respective structural zones, and if so, the zones could be used as a tool to compare active sites of functionally unrelated “conventional” and “unconventional” triad-type and dyad-type enzymes within the same fold [9].
2. Results and Discussion
As described above, the β-lactamase/D-Ala carboxypeptidase family proteins have three standalone structural elements of the catalytic machinery: (1) the catalytic dyad; (2) the oxyanion hole; and (3) the Ω-loop. We also know that these enzymes have three known sequence motifs, Ser – Xaa – Xaa – Lys (or S-X-X-K), (Ser/Tyr) – Xaa – (Asn/Cys) (or S-X-N) and (Lys/His) – (Thr/Ser) – Gly (or K-T/S-G). These data will be taken as the basis for identification of additional β-lactamase catalytic residue SCC zones. Furthermore, by comparing the respective SCCs in different β-lactamase/transpeptidase-like superfamily proteins, we will group them based on the differences in interactions between key catalytic amino acids and the other structural elements.
2.1. Creating a Dataset of the β-Lactamase/Transpeptidase-Like Representative Structures
In the SCOP database, the β-lactamase/transpeptidase-like superfamily incorporates three families: (1) the β-lactamase/D-Ala carboxypeptidase family (201 different proteins), (2) the glutaminase family (five different proteins) and (3) the Dac-like family (five different proteins) [14]. For each of these 211 different proteins, one representative PDB 3D structure with the best resolution has been selected, thus making a set of 211 representative 3D structures.
2.2. SCC Identification with the Example of β-Lactamase CTX-M-14
2.2.1. Conserved Local Motifs at the Basis of the SCC Identification
Based on the selected representative structure of the β-lactamase/D-Ala carboxypeptidase protein family, we will describe all known conserved structural elements, around which the SCC will be built. Taking into consideration all the structural criteria for creating the dataset of representative structures described above, the 3D structure of β-lactamase CTX-M-14 (PDB ID: 4UA6; 0.79 Å resolution) [23] can be selected to represent the β-lactamase/transpeptidase-like superfamily. This enzyme is a hydrolase and belongs to the class A β-lactamases (http://bldb.eu/S-BLDB.php [24,25,26]). Typical for all β-lactamases of this class, the active site of CTX-M-14 contains the following characteristic motifs: (1) the S-X-X-K motif is Ser70-Xaa-Xaa-Lys73; (2) the S-X-N motif is Ser130-Xaa-Asn132; and (3) the K-T/S-G motif is Lys234-Thr/Ser-Gly236; and the Ω-loop is Asp163-Arg178 (Figure 1) [17,20].
For comparison purposes, we will use the sequence and structure of β-lactamase CTX-M-14 as the reference. In the Ser70-Xaa-Xaa-Lys73 motif, Ser70 and Lys73 are the catalytic nucleophile (Nuc) and general base (Base), respectively (Figure 1). Together with Lys73, Glu166 located in the Ω-loop has also been proposed as a potential general base (Figure 1). The amino acids Ser70, Lys73, Ser130, Glu166, and Asn170 are involved in the enzymatic catalytic activity (Figure 1) [27]. Hydrogen bonds between Ser130 and side-chain groups of Ser70 and Lys234 were also found to be important for protein function [28]. Mutation of Asn132 to alanine showed involvement of this residue in the functional transition-state stabilization [29]. By analogy with the Ω-loop, we will refer to the Ser130-Xaa-Asn132 motif (the S-X-N motif in Figure 1) located in the A-domain of β-lactamase [30] as the A(Alpha)-tripeptide (Figure 1). Finally, the oxyanion hole is formed by the main-chain nitrogen atoms of two amino acids: the catalytic nucleophile, Ser70, and the “Oxy” residue, Ser237, which directly follows Lys234-Thr-Gly236 [30].
2.2.2. The “NucBase-Oxy” Zone
Let us consider interactions between the local substructures, which govern the key motifs: Ser70-Xaa-Xaa-Lys73 (the S-X-X-K motif), Ser130-Xaa-Asn132 (the S-X-N motif), Lys234-Thr/Ser-Gly236 (the K-T/S-G motif) and the Ω-loop described above. The hexapeptide Met68-Lys73, which incorporates the catalytic nucleophile Ser70 and the catalytic base Lys73, is a unique structural segment, which not only contains a catalytic dyad, but also forms two separate local mini-networks of hydrogen bonds and weak interactions with the oxyanion hole and the Ω-loop, respectively. We will designate the Met68-Lys73 hexapeptide as the “NucBase” hexapeptide. The NucBase hexapeptide and the Lys234-Ser237 tetrapeptide, which incorporates the Oxy residue Ser237, form a local interconnected substructure, which incorporates the catalytic residues and the oxyanion hole, and it is internally bound by a mini-network of hydrogen bonds and weak interactions (Table 1 and Table S1). We will refer to this standalone substructure as the “NucBase-Oxy zone” (Figure 2A). The contacts marked as I (between Ser70 and Lys73), III (between Lys234 and Thr235) and V (between Met68 and Thr71; all defined in Table S1 and shown in Figure 2A) are the canonical hydrogen bonds, while the contacts marked as II (between Ser70 and Lys234) and IV (between Met68 and Gly236) are a weak hydrogen bond (Derewenda [31] has written a comprehensive review on weak hydrogen bonds in the 3D structures of proteins and nucleic acids) and a van der Waals interaction, respectively. In Table S1, it is easy to separate standard hydrogen bonds from weak ones. The cut-off distance for a canonical hydrogen bond is ≤ 3.3 Å (Table S1, row 1), while for a weak C–H•O hydrogen bond, the cut-off distances are usually slightly larger (≤ 4.0 Å) with the C–H•O angle strictly ≥ 130° [32].
The interaction between O/Met68 and CA/Gly236 (the interaction marked as IV in Table S1 and Figure 2A) locks the ends of the “circular” structure of the NucBase-Oxy zone, and also affects the relative arrangement of the functionally important nodes of the oxyanion hole, N/Ser70 and N/Ser237, which should keep their position intact during catalysis (Figure 2A). Indeed, according to the PDBsum database [33] and the Ligplot tool [34], in the ligand-bound structures of β-lactamase CTX-M-14 (PDB ID: 4UA9) the two atoms of the oxyanion hole do interact with the ligand, and neither their position nor the interaction between O/Met68 and CA/Gly236 (interaction IV in Table S1) changes when comparing the ligand-free (PDB ID: 4UA6) and ligand-bound (PDB ID: 4UA9) forms of the enzyme. Thus, the observed distance between O/Met68 and CA/Gly236 does show local structural conservation of the NucBase-Oxy zone conformation but also reflects the requirements of protein function.
The rigid main-chain based dual interaction between Gly236 and Asn245 (N/Gly236-O/Asn245 and O/Gly236-N/Asn245; interaction VI in Table S1 and Figure 2A) is the only dual-bond interaction that the residues of the NucBase-Oxy zone form with the rest of the protein and not among residues of the zone or the protein ligand; residue Asn245 is not part of the NucBase-Oxy zone. However, Gly236 and Asn245 are located on two adjacent antiparallel β-strands of the β-sheet, of which one β-strand containing the residue Gly236 is positioned on the edge of the β-sheet (Figure 1), thus linking the NucBase-Oxy zone to the overall protein fold. The Gly236 – Asn245 dual-bond interaction is typical of rigid secondary structures, such as β-sheets. As a result, the positions of the functionally important atoms of N/Ser237 and O/Thr235 (the Oxy atom and interaction III in β-lactamase CTX-M-14, respectively) is stabilized by this dual-bond interaction VI (Figure 2A).
2.2.3. The NucBase-Omega Zone and its Omega (Ω) Subzone
In β-lactamase CTX-M-14, the NucBase hexapeptide Met68-Lys73 and three segments of the Ω-loop, Glu166, Leu169-Asn170, and Asp179 form the NucBase-Omega zone (Figure 2B; Tables 1 and S2). Similar to the NucBase-Oxy zone, the NucBase-Omega zone is a standalone structure – internally interlocked conserved substructure – which creates and supports the connection between the catalytic residues and the Ω-loop. Thus, while the NucBase-Oxy zone extends from the catalytic dyad to the oxyanion hole, the NucBase-Omega zone extends to the Ω-loop. The NucBase-Omega zone also contains a water molecule-“mediator” (HOH2167 in Figure 2B), which forms three hydrogen bonds with the constituent structural elements of the zone, one hydrogen bond per element (Table S2, column V). Figure 2B shows all interactions that interlock the NucBase-Omega zone. These interactions are designated by numbers I through VII and are repeatedly found in proteins of the β-lactamase/transpeptidase-like superfamily (Table S2). In the NucBase-Omega zone, contacts I, IV and VI are typically canonical hydrogen bonds, contact II is a weak hydrogen bond, and contact V consists of two strong hydrogen bonds connecting two key amino acids that in the majority of cases is an antiparallel main-chain O-N + N-O link (Table S2).
Knowledge of the NucBase-Omega zone allowed us to separately pinpoint the Ω-subzone, the “pocket” of NucBase-Omega zone, which incorporates the Ω-loop. This zone-based structural approach further allowed us to identify scaffolds supporting the Ω-loop and the other key functional elements without regard to their sequence length. In a β-lactamase, CTX-M-14, however, the Arg161-Thr180 localization of the Ω-subzone does not differ much from the Asp163-Arg178 localization of the Ω-loop [29], but this is not always the case.
2.2.4. SCC as a Structural Association of the NucBase-Oxy and NucBase-Omega Zones and the A-Tripeptide Link
The combination of bound NucBase-Oxy and NucBase-Omega zones of β-lactamase CTX-M-14 is shown in Figure 3A. The amino acid chain direction of the NucBase segment Met68-Lys73 is antiparallel to the direction of the Oxy segment Lys234-Asp240 and the Ω-subzone, whose chain directions coincide.
Moreover, at one end of the arrangement, shown at the bottom of Figure 3A, the NucBase segment, Oxy segment and the Ω-subzone structurally converge, while at the opposite end they diverge (Figure 3A). Structurally, the divergent part of the NucBase-Omega-Oxy arrangement (upper left part of Figure 3A) is connected by the A-tripeptide Ser130-Asn132. As it was shown in Figure 1, the A(Alpha)-tripeptide in β-lactamase CTX-M-14 is the S-X-N motif, but in the other representative structures of the same superfamily its amino acid composition can vary (shown as “Alpha” in Table 1).
Nevertheless, we will show that the structural role of the A-tripeptide remains the same. In the β-lactamase CTX-M-14, two side-chain oxygen atoms of the terminal residues of the A-tripeptide form hydrogen bonds with the NZ atoms of the Lys73 (catalytic base) and Lys234 (Table S2, interaction VIII), and thus, the A-tripeptide forms a conserved link, the A-tripeptide link, between these two positions. Taken together, the combination of the NucBase-Oxy zone, NucBase-Omega zone and the A-tripeptide link form the Structural Catalytic Core (SCC) of β-lactamase CTX-M-14 and the rest of the β-lactamase/D-Ala carboxypeptidase family. It consists of 19 amino acids and a water molecule mediator, which is incorporated into the NucBase-Omega zone (Table 1; Figure 3A).
2.3. SCC in Proteins of the β-Lactamase/D-Ala Carboxypeptidase Family: Groups, Subgroups and Classes
After examining the β-lactamase CTX-M-14, the remaining 200 representative 3D structures from the β-lactamase/D-Ala carboxypeptidase family of the β-lactamase/transpeptidase-like superfamily proteins were similarly analyzed for the SCCs formed by four main structural elements and incorporating seven key functional amino acids. The results are summarized in Table 1, Table S1 and Table S2.
All representative structures in Table 1, Table S1 and Table S2 are structurally aligned with respect to the position of equivalent key functional amino acids and equivalent amino acid segments in the protein structure. Unlike a standard sequence alignment, a structural alignment shows the alignment of equivalent positions within the protein structure, which may or may not contain similar amino acids. Subsequent identification of similar amino acids in the key positions of a structural alignment can identify key amino acids that can serve as a good basis for structure classification. We have analyzed structural alignments in Table 1, Table S1 and Table S2 and found three such key structural positions that can serve as the basis for classifying and naming SCC groups in all proteins of the β-lactamase/transpeptidase-like superfamily. Consequently, 199 representative structures of this family were divided into six groups and 23 subgroups as described below.
The three key structural positions that were chosen for the naming of groups of the SCC are those equivalent to β-lactamase CTX-M-14 positions 130 (Ser130 in CTX-M-14), 132 (Asn132 in CTX-M-14) and 170 (Asn170 in CTX-M-14). Positions 130 and 132 represent the beginning and the end of the A-tripeptide link, while position 170 is the key interacting amino acid from the Ω-loop (Figure 1; Table 1 and Table S1). In two representative structures, it was not possible to fully identify some of these positions: (1) In β-lactamase SPH-1 (PDB ID: 4EWF), it was not possible to identify a residue of the Ω-loop located at a position equivalent to position 170 of CTX-M-14; and (2) in penicillin binding protein 1A from S. Pneumoniae (PDB ID: 2V2F), it was not possible to determine the beginning and end of the Ω-subzone according to our accepted procedure (Section 2.2.3).
2.3.1. The N-Like Group and its Subgroups
According to the three amino acids described above, the β-lactamase CTX-M-14 is a representative member of “SNN” subgroup (S130-N132-N170). In addition to the structural and functional properties described in Section 2.2.1, Ser130 and Asn132 are directly involved in ligand binding [33.34]. The conserved asparagine at position 170, Asn170, is one of the most important residues involved in the interaction of the Ω-subzone with both the Met68-Cys69 dipeptide and the Ser237-Asp240 tripeptide.
Together with the β-lactamase CTX-M-14, there are 67 representative 3D structures of β-lactamase/D-Ala carboxypeptidase family proteins that belong to the SNN subgroup (shown in round brackets in Table 1; also shown in column “Sum” in Table S1). All 67 proteins have almost identical SCCs and seven key functional amino acids are identical at positions 70, 73, 130, 132, 166, 170, and 234 (Figure 1). Additionally, structurally analogous SCCs are observed in four more subgroups: SSN, SGN, SNS, and SNG, which have undergone single amino acid mutations at positions 132 or 170 to serine or glycine, but otherwise are the same as the SNN subgroup (Table 1 and Table S1). These five subgroups together contain 79 representative structures that belong to class A β-lactamases and together constitute the “N-like” group. The “N-like” name comes from the fact that even though residues at positions 132 or 170 can be mutated to serine or glycine in some subgroups, the structure of the SCC in all members of this group remains similar to SNN.
Finally, some proteins of the N-like group contain a disulfide bond between the cysteine preceding the nucleophile position and the central cysteine of the Ser237-Cys238-Asp240 tripeptide. For example, such a disulfide bond is observed in carbapenemase GES-1 (Table 1, row 3).
2.3.2. The W-Group
The second, “W-group” (class D β-lactamases (http://bldb.eu/S-BLDB.php [24,25,26])), included 45 structures where tryptophan occupies position 170 (position numbering according to CTX-M-14; see “Omega” column in Table 1) instead of asparagine. This group can be divided into three subgroups: SVW, SIW, and SLW, where according to our accepted naming position 130 is occupied by serine (S), position 132 is occupied by valine (V), isoleucine (I) or leucine (L), and position 170 is occupied by tryptophan (W). The three subgroups consist of 36, 5, and 4 representative structures, respectively (column “Sum” in Table S1). The appearance of branched hydrophobic residues in the W-group instead of a small or polar residue as in the N-like group is related to the protein function of these enzymes. For example, in β-lactamase OXA-24/40 (PDB ID: 5TG4), the appearance of valine at position 130 (132 in CTX-M-14) is consistent with the need to contact the hydrophobic aromatic ring of the ligand [33,34]. In addition to the 45 structures mentioned above, the W-group also includes 5 additional representative structures within two subgroups, SNW (e.g. methicillin resistance mecR1 protein) and STW (e.g. regulatory protein BlaR1), that are not β-lactamases, and thus are not included in the β-Lactamase database (http://bldb.eu/S-BLDB.php). Here again, position 132 is occupied by a small polar amino acid.
The structure of β-lactamase OXA-143 (PDB ID: 5IY2) can be taken as the representative for the W-group because the crystal structure has the highest resolution (1.15 Å) [35]. Comparing SCCs between CTX-M-14 and OXA-143, we were able to draw conclusions about observed differences of the SCCs between the N-like group and the W-group. The most noticeable differences in their organization of the SCCs are linked to the construction of their NucBase-Omega zones (Figure 3B vs. 3A). The tryptophan side-chain group (shown in green in Figure 3B) is significantly larger in size compared to the asparagine side-chain group (shown in green in Figure 3A) and does not contain an oxygen atom. Therefore, instead of the OD1/Asn170-CA/Cys69 weak hydrogen bond seen in β-lactamase CTX-M-14, the CD1/Trp167-O/Ala80 contact is present in β-lactamase OXA-143 (column II in Table S2). In addition, a hydrogen bond is formed directly between tryptophan and the catalytic base: NE1/Trp167-OQ2/Lys84 (Table S2, column I). Taken together, the need for an analogue of Glu166 disappears. The physical “shift” of the hydrogen bond CD1/Trp167-O/Ala80 towards the catalytic nucleophile in OXA-143 and the rest of the W-group proteins (see CD1/Trp167-O/Ala80 in OXA-143 in Figure 3B versus OD1/Asn170-CA/Cys69 in CTX-M-14 in Figure 3A) is coupled by the appearance of an additional conventional hydrogen bond: O/Trp167-N/Ala80 (OXA-143; Table S2; column III). It is important to note that while in the β-lactamase CTX-M-14 the main-chain oxygen of Asn170 forms a hydrogen bond with N/Asp240 of the Oxy segment, in the β-lactamase OXA-143 the existence of O/Trp167-N/Ala80 eliminates the possibility of contacts between tryptophan and the last two residues of the Oxy segment and effectively removes those residues from the SCC (Table 1). As we will show below, interactions of the amino acid at position 170 (Trp in the W-group) with the residue preceding the catalytic nucleophile, together with the “shortening” of the Oxy segment in the SCC, are specific structural markers of the β-lactamase/transpeptidase-like superfamily proteins.
Another important difference between the SCCs of the two groups of β-lactamases is the replacement of asparagine (Asn132 in CTX-M-14 from the N-like group) of the A-tripeptide with valine (Val130 in OXA-143 from the W-group) (Figure 3B vs. 3A). However, even in the W-group, the A-tripeptide link is formed due to the contact of valine with carboxylic acid, which is covalently linked to the catalytic Lys84 (Figure 3B). Note that in 4 proteins out of 5 belonging to the W-group β-lactamases, asparagine is located instead of valine in the A-tripeptide, and the formation of the BaseAlpha zone occurs in the same way as in class A β-lactamases (Table S2, column VIII). Finally, the SCC of the W-group proteins consists of 17 amino acids instead of 19 seen in the N-like group (class A of the β-lactamases) (Table 1).
In section 2.3.1, it was shown that in some N-like group proteins there is a disulfide bond that stabilizes the conformation of the NucBase-Oxy zone. In particular, the cysteine located before the catalytic nucleophile in the amino acid sequence (position “nuc-1”) takes part in its formation. Thus, the amino acid at position nuc-1 is one of the key residues in the formation of both the NucBase-Oxy and NucBase-Omega zones, and therefore the entire SCC. Further evidence for the important structural role of the nuc-1 residue in formation of the NucBase-Omega zone is seen in W-group proteins. In β-lactamase BSU-2 and methicillin resistance mecR1 protein, instead of the frequently observed alanine, the nuc-1 position is occupied by glutamine or asparagine (Table 1, rows 7 and 9, respectively). In these two proteins, the side-chain group of the nuc-1 residues forms two hydrogen bonds with glutamine/glutamic acid at the C-terminus of the Ω-subzone (Table S2, rows 7 and 9, columns III and V).
2.3.3. The G-Group
The third group of proteins (23 representative structures) has glycine at sequence position 170 (position numbering according to CTX-M-14), thus it is referred to as the “G-group”. The G-group consists of four subgroups with D-alanyl-D-alanine carboxypeptidase as its representative (PDB ID: 1YQS, R = 1.05 Å) [36]. The SCC of D-alanyl-D-alanine carboxypeptidase is shown in Figure 3C.
Based on the naming introduced here, D-alanyl-D-alanine carboxypeptidase belongs to the YNG subgroup (Table 1). Unlike the N-like group and the W-group proteins, D-alanyl-D-alanine carboxypeptidase has two internal weak hydrogen bonds, CA/Gly238-O/Gly61 and O/Gly238-CA/Gly61 (in place of CD1/Trp167-O/Ala80 and O/Trp167-N/Ala80 bonds in β-lactamase OXA-143), which together account for approximately 60% of one standard hydrogen bond. A water molecule mediator is also present in the G-group proteins (Figure 3C; Table S2). Due to the absence of a side chain in Gly238, the main-chain oxygen of Ala237 is involved in the NucBase-Omega zone-forming contacts with the catalytic base (Table S2, column I). The conformation of the side-chain group of the catalytic base is stabilized by its interaction with Asn161 of the A-tripeptide via hydrogen bonding (Table S2, column VIII). An additional zone-forming bond O/Val240-N/Val60 is formed between the terminal amino acids of the NucBase and the Ω-subzone segments (Figure 3C; Table S2, column IV). As a result, the lengths of the Oxy segments in carboxypeptidase and β-lactamase OXA-143 coincide. However, the carboxypeptidase Oxy segment has one noticeable difference compared to the respective segments from the N-like and W groups: the initial amino acid of this segment in the G-group is not necessarily lysine (histidine in D-alanyl-D-alanine carboxypeptidase (Table 1). Otherwise, the NucBase-Oxy zones are very similar in the three groups of proteins (Figure 3A, Figure 3B and Figure 3C; Table S2).
In D-alanyl-D-alanine carboxypeptidase, interaction between the NucBase-Oxy zone and the A-tripeptide link are done through the Tyr159−His298 pair, which is unique for the G-group of proteins. If we consider all four subgroups of the G-group simultaneously, then in two out of the four subgroups, serine at the first position of the A-tripeptide is replaced by tyrosine (Table 1). SCC of the G-group consists of 16 amino acids.
Finally, the G-group also contains a representative protein, 6-aminohexanoate-dimer hydrolase, with methionine at the nuc-1 position (Table 1), which interacts with the Oxy residue, and thus, participates in stabilizing the NucBase-Oxy zone.
2.3.4. The G-Like Group
The G-like group includes 39 proteins whose SCC is nearly the same as the SCC of the G-group, despite the fact that they do not have glycine at position 170 (see Figure 3D vs. 3C; position numbering according to CTX-M-14; Table 1). Proteins of the G-like group are divided into 6 subgroups, of which the YNY subgroup (33 representative proteins) is the largest. β-lactamase TRU-1 (PDB ID: 6FM6, 1.05 Å resolution) [37] from the YNY subgroup was selected as the representative structure of the G-like group. Proteins of the YNY subgroup belong to class C β-lactamases (http://bldb.eu/S-BLDB.php [24,25,26]). The only feature of this group that distinguishes it from the other groups is the presence of tryptophan at the first position of the Oxy segment in some subgroups instead of a lysine (see “Oxy” column in Table 1, lines 17, 18 and 19). In such structures, the NE1 atom of tryptophan substitutes for the NZ atom of lysine in all respective interactions between the A-tripeptide and the Oxy segment (Table S2, column VIII). Tyrosine at the first position of the A-tripeptide is conserved with very few exceptions in the G-like group (see the “Alpha” column in Table 1). Similar to the G-group, the SCC of the G-like group proteins is constructed from 16 amino acids.
2.3.5. The Q-Like Group
Unlike the groups described above, the Q-like group is relatively small. It contains six representative structures from two subgroups (Table 1). The SNQ subgroup is dominant. It includes five representative structures. The last representative structure of this group (PDB ID: 5TFQ) has threonine at position 170 (position numbering according to CTX-M-14; Thr158 in 5TFQ), and thus formally forms the SNT subgroup. Due to the predominance of glutamine at position 170, this group of proteins is referred to as the “Q-like”.
As with the N-like group, the Q-like group proteins are class A β-lactamases (http://bldb.eu/S-BLDB.php). However, there are several fundamental differences between the SCCs of the N- and Q-like groups. Let us compare the SCCs of β-lactamases CTX-M-14 and CPA-1 (PDB ID: 6V4W, 1.29 Å resolution) [38] (see Figure 4A vs. 3A). Firstly, there is predominantly glutamine at position 170 in the Q-like group that is never found in the N-like group. Secondly, the side-chain group of this glutamine (Gln177 in CPA-1) contacts the side-chain group of Gln67, adjacent to the catalytic nucleophile Ser68. An analogous interaction does not exist in any of the groups discussed above. Gln67 and HOH403 act as intermediaries in the contact of Gln177 with the functionally important Glu167, a structural analogue of Glu166 in the β-lactamase CTX-M-14 (see section 2.2.1). Finally, unlike the N-like group proteins, the Oxy segment of the Q-like group contains only 4 residues, similar to some other groups shown in Table 1. The SCC of the Q-like group proteins consists of 16 amino acids in total.
2.3.6. The Group of “Inactive” β-Lactamases, i.e. Those Unable to Perform Catalysis
All proteins we have considered so far are enzymatically active. However, the β-lactamase/D-ala carboxypeptidase family includes two structures whose proteins are inactive due to amino acid changes at the catalytic nucleophile and base positions, and yet they have the same fold and belong to the same protein family (see “Inactive β-lactamase group” in Table 1). Let us consider how changes at the two catalytically important amino acid positions affect SCC of β-lactamase B. ambifaria MC40-6 (PDB ID: 5IHV, 1.10 Å resolution) [39]. Essentially, the SCC in this protein is similar to the SCC of the N-like group, class A β-lactamases (see Figure 4B vs. 3A). Replacing serine with glycine at the catalytic nucleophile position (Gly47 in Figure 4B) has no effect on the structure of SCC because of the main-chain interactions and the small size of glycine. All contacts lost due to the exchange of the catalytic base to alanine (Ala50 in Figure 4B) are formed instead by the bound small molecule, ethane 1,2-diol (used in the crystallization solution, EDO302), which maintains the local conformation. Finally, substitution of asparagine for lysine at position 132 of the A-tripeptide has very little effect on the conformation of the NucBase-Omega zone and the A-tripeptide link. Like β-lactamase CTX-M-14, the SCC of β-lactamase B. ambifaria MC40-6 consists of 19 amino acids.
2.4. SCC in Proteins of the Glutaminase Family (Example: Glutaminase 1)
The glutaminase family is one of the three families within the β-lactamase/transpeptidase-like superfamily [14]. This family includes 5 representative structures that form only one group according to our classification, the “C-group” (Figure 4C, Table 1).
The glutaminase 1 structure (PDB ID: 1U60, 1.61 Å resolution) [40] is the representative structure of this family and group. In the NucBase-Omega zone, the CA atom of Cys196 from the Ω-subzone (shown in green in Figure 4C) interacts with the main-chain oxygen atom of an amino acid from the NucBase zone (Glu65 in Figure 4C) by means of a weak hydrogen bond, as also seen in proteins of the G- and G-like groups (Table S2, column II). However, the zone-forming contact between the Ω-subzone and Lys69 (the catalytic base in glutaminase 1) is formed differently when compared with the G- and G-like groups (Figure 4C vs. Figure 3C/Figure 3D). In glutaminase 1, the donor of the intermediary atom is several residues away from the rest of the Ω-loop (Tyr192 in Figure 4C).
Another feature of this group of proteins is the presence of a hydrophobic amino acid at position 130 of the A-tripeptide (Leu115 in glutaminase 1; position numbering according to CTX-M-14). At the same time, an intermediary residue Tyr244 is found to interact with the Oxy subzone (Figure 4C; Table S2, column VIII) unlike the G- and G-like groups that have tyrosine at position 130 of the A-tripeptide. The SCC in proteins from the C-group consists of 17 amino acids (Table 1, row 24).
2.5. SCC in Proteins of the Dac-Like Family (Example: D-Alanyl-D-Alanine Carboxypeptidase DacB)
The SCC of D-alanyl-D-alanine carboxypeptidase DacB (PDB ID: 2EX2, R = 1.55 Å) [41] (Figure 4D, Table 1) is the representative structure for five proteins that form the G-group of the Dac-like family proteins (line 25 in Table 1), which is similar to the G-group of D-alanyl-D-alanine carboxypeptidases (line 11 in Table 1). However, there are two differences between the SCCs of the Dac-like and standard D-alanyl-D-alanine carboxypeptidases. The first difference is the presence of a serine instead of tyrosine at the position 130 of the A-tripeptide (Ser306 in Figure 4D; position numbering according to CTX-M-14). The second difference is the extension of the NucBase segment by one residue due to the presence of a proline at the N-terminus. Despite these differences, both Dac-like and standard D-alanyl-D-alanine carboxypeptidases belong to the same group type, G-group.
3. Materials and Methods
The Protein Data Bank (PDB, http://www.rcsb.org/; 10 October 2024 [5]) and the Structural Classification of Proteins database (SCOP, https://www.ebi.ac.uk/pdbe/scop/; 10 October 2024 [14]) were used to retrieve 211 representative structures of proteins from the β-lactamase/transpeptidase-like superfamily (SCOP ID: 3001604).
Structure visualization and structural analysis of interactions (hydrogen bonds, non-polar and the other weak interactions) were done using Maestro software (Schrödinger Release 2023-1: Schrödinger, LLC, New York, NY, USA, 2021; http://www.schrodinger.com/; 10 October 2024). The class of β-lactamases was determined using the Ambler Classification system and the β-lactamase database (BLDT, http://bldb.eu/S-BLDB.php; 10 October 2024) [26]. Identification of protein residues involved in contact with a ligand was carried out using the PDBsum database (https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/; 10 October 2024 [33]) and the Ligplot tool [34].
Weak hydrogen bonds were identified using criteria from [32]. The π-π stacking and the other similar interactions were analyzed using the Residue Interaction Network Generator (RING, https://ring.biocomputingup.it/; 10 October 2024) [42]. Figures were drawn with MOLSCRIPT [43] and the PyMOL molecular graphics software (https://pymol.org/; 10 October 2024).
4. Conclusions
Structural studies of the catalytic sites of 199 proteins from the superfamily of β-lactamase/transpeptidase-like proteins revealed the similarity and differences among the Structural Catalytic Cores (SCCs) in these proteins, which consist of three distinct zones: the NucBase-Oxy zone, the NucBase-Omega zone, and the A-tripeptide link. The NucBase-Oxy zone is formed by the NucBase hexapeptide, containing a catalytic nucleophile and a catalytic base, and the Oxy tripeptide, followed by the amino acid that forms the oxyanion hole (Oxy tetrapeptide). In the process of constructing the NucBase-Omega zone, a structural requirement for localizing the omega(Ω)-subzone was formulated.
There are two ways in which the NucBase-Oxy zone and the NucBase-Omega zone join together to construct the SCCs. Firstly, when the amino acid at position 170 (position numbering according to CTX-M-14) of the Ω-subzone contacts simultaneously the nuc-1 residue from the NucBase hexapeptide and the Oxy hexapeptide. This variant is observed in the N-like group of proteins (class A β-lactamases). Secondly, when the same amino acid at position 170 (position numbering according to CTX-M-14) of the Ω-subzone contacts only the nuc-1 residue of the NucBase hexapeptide and nothing else. The second variant is observed in all of the other representative proteins.
If we consider both the type of amino acid at the position 170 of the Ω-subzone and the nature of its interaction with the NucBase-Oxy zone, then all proteins of the β-lactamase/transpeptidase-like superfamily can be divided into seven different SCC groups, which further can be divided into 24 subgroups according to the key residue of the A-tripeptide. The proposed structural classification of the β-lactamase/transpeptidase-like proteins can be easily expanded to accommodate all new structures by using the approach proposed herein.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.: Table S1. Conserved geometric parameters (distance and angle) of contacts in 25 NucBase-Oxy zones of the beta-lactamase/transpeptidase-like superfamily proteins. Table S2. Conserved geometric parameters (distance and angle) of contacts in 25 NucBase-Omega and BaseAlpha zones of the beta-lactamase/transpeptidase-like superfamily proteins.
Author Contributions
A.I.D.: Study design, Formal analysis, Methodology, Visualization, Writing – Original Draft, Writing – Review & Editing; K.D.: Formal analysis, Methodology, Visualization, Writing – Original Draft, Writing – Review & Editing; M.S.J.: Formal analysis, Methodology, Writing – Original Draft; V.N.U.: Study design, Formal analysis, Methodology, Visualization, Investigation, Writing – Original Draft, Writing – Review & Editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data supporting reported results can be found in the Supplementary Materials.
Acknowledgments
We thank the Biocenter Finland Bioinformatics Network (Jukka Lehtonen) and CSC IT Center for Science for computational support for the project. The Structural Bioinformatics Laboratory is part of the Solution for Health strategic area of Åbo Akademi University and within the InFLAMES Flagship program on inflammation and infection, Åbo Akademi University and the University of Turku, funded by the Academy of Finland.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rawlings ND, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2000; 28(1):323-325. [CrossRef]
- Chatonnet A, Perochon M, Velluet E, Marchot P. The ESTHER database on alpha/beta hydrolase fold proteins - An overview of recent developments. Chem Biol Interact. 2023; 383:110671. [CrossRef]
- Dodson G, Wlodawer A. Catalytic triads and their relatives. Trends Biochem Sci. 1998; 23(9):347-352. [CrossRef]
- Polgár L. The catalytic triad of serine peptidases. Cell Mol Life Sci. 2005; 62(19-20):2161-2172. [CrossRef]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000; 28(1):235-242. [CrossRef]
- Denesyuk AI, Johnson MS, Salo-Ahen OMH, Uversky VN, Denessiouk K. NBCZone: Universal three-dimensional construction of eleven amino acids near the catalytic nucleophile and base in the superfamily of (chymo)trypsin-like serine fold proteases. Int J Biol Macromol. 2020; 153:399-411. [CrossRef]
- Denesyuk A, Dimitriou PS, Johnson MS, Nakayama T, Denessiouk K. The acid-base-nucleophile catalytic triad in ABH-fold enzymes is coordinated by a set of structural elements. PLoS One. 2020; 15(2):e0229376. [CrossRef]
- Denessiouk K, Denesyuk AI, Permyakov SE, Permyakov EA, Johnson MS, Uversky VN. The active site of the SGNH hydrolase-like fold proteins: Nucleophile-oxyanion (Nuc-Oxy) and Acid-Base zones. Curr Res Struct Biol. 2023; 7:100123. [CrossRef]
- Denesyuk AI, Denessiouk K, Johnson MS, Uversky VN. Structural Catalytic Core in Subtilisin-like Proteins and Its Comparison to Trypsin-like Serine Proteases and Alpha/Beta-Hydrolases. Int J Mol Sci. 2024; 25(22):11858. [CrossRef]
- Kelly JA, Kuzin AP. The refined crystallographic structure of a DD-peptidase penicillin-target enzyme at 1.6 Å resolution. J Mol Biol. 1995; 254(2):223-236. [CrossRef]
- Silvaggi NR, Anderson JW, Brinsmade SR, Pratt RF, Kelly JA. The crystal structure of phosphonate-inhibited D-Ala-D-Ala peptidase reveals an analogue of a tetrahedral transition state. Biochemistry. 2003; 42(5):1199-1208. [CrossRef]
- Ekici OD, Paetzel M, Dalbey RE. Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008; 17(12):2023-2037. [CrossRef]
- Buller AR, Townsend CA. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc Natl Acad Sci U S A. 2013; 110(8):E653-E661. [CrossRef]
- Andreeva A, Kulesha E, Gough J, Murzin AG. The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 2020; 48(D1):D376-D382. [CrossRef]
- Leszczynski JF, Rose GD. Loops in globular proteins: a novel category of secondary structure. Science. 1986; 234(4778):849-855. [CrossRef]
- Goffin C, Ghuysen JM. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol Mol Biol Rev. 1998; 62(4):1079-1093. [CrossRef]
- Majiduddin FK, Materon IC, Palzkill TG. Molecular analysis of beta-lactamase structure and function. Int J Med Microbiol. 2002; 292(2):127-137. [CrossRef]
- Tooke CL, Hinchliffe P, Bragginton EC, Colenso CK, Hirvonen VHA, Takebayashi Y, Spencer J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J Mol Biol. 2019; 431(18):3472-3500. [CrossRef]
- Kaderabkova N, Bharathwaj M, Furniss RCD, Gonzalez D, Palmer T, Mavridou DAI. The biogenesis of β-lactamase enzymes. Microbiology (Reading). 2022; 168(8):001217. [CrossRef]
- Banerjee S, Pieper U, Kapadia G, Pannell LK, Herzberg O. Role of the omega-loop in the activity, substrate specificity, and structure of class A beta-lactamase. Biochemistry. 1998; 37(10):3286-3296. [CrossRef]
- Szarecka A, Lesnock KR, Ramirez-Mondragon CA, Nicholas HB Jr, Wymore T. The Class D beta-lactamase family: residues governing the maintenance and diversity of function. Protein Eng Des Sel. 2011; 24(10):801-809. [CrossRef]
- Egorov A, Rubtsova M, Grigorenko V, Uporov I, Veselovsky A. The Role of the Ω-Loop in Regulation of the Catalytic Activity of TEM-Type β-Lactamases. Biomolecules. 2019; 9(12):854. [CrossRef]
- Nichols DA, Hargis JC, Sanishvili R, Jaishankar P, Defrees K, Smith EW, Wang KK, Prati F, Renslo AR, Woodcock HL, Chen Y. Ligand-Induced Proton Transfer and Low-Barrier HB Revealed by X-ray Crystallography. J Am Chem Soc. 2015; 137(25):8086-8095. [CrossRef]
- Ambler RP. The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci. 1980; 289(1036):321-331. [CrossRef]
- Hall BG, Barlow M. Structure-based phylogenies of the serine beta-lactamases. J Mol Evol. 2003; 57(3):255-260. [CrossRef]
- Naas T, Oueslati S, Bonnin RA, Dabos ML, Zavala A, Dortet L, Retailleau P, Iorga BI. Beta-lactamase database (BLDB) - structure and function. J Enzyme Inhib Med Chem. 2017; 32(1):917-919. [CrossRef]
- Agarwal V, Yadav TC, Tiwari A, Varadwaj P. Detailed investigation of catalytically important residues of class A β-lactamase. J Biomol Struct Dyn. 2023; 41(5):2046-2073. [CrossRef]
- Sun T, Bethel CR, Bonomo RA, Knox JR. Inhibitor-resistant class A beta-lactamases: consequences of the Ser130-to-Gly mutation seen in Apo and tazobactam structures of the SHV-1 variant. Biochemistry. 2004; 43(44):14111-14117. [CrossRef]
- Jacob F, Joris B, Lepage S, Dusart J, Frère JM. Role of the conserved amino acids of the 'SDN' loop (Ser130, Asp131 and Asn132) in a class A beta-lactamase studied by site-directed mutagenesis. Biochem J. 1990; 271(2):399-406. [CrossRef]
- Herzberg O. Refined crystal structure of beta-lactamase from Staphylococcus aureus PC1 at 2.0 Å resolution. J Mol Biol. 1991; 217(4):701-719. [CrossRef]
- Derewenda ZS. C-H Groups as Donors in Hydrogen Bonds: A Historical Overview and Occurrence in Proteins and Nucleic Acids. Int J Mol Sci. 2023; 24(17):13165. [CrossRef]
- Derewenda ZS, Derewenda U, Kobos PM. (His)C epsilon-H...O=C < hydrogen bond in the active sites of serine hydrolases. J Mol Biol. 1994; 241(1):83-93. [CrossRef]
- Laskowski RA, Hutchinson EG, Michie AD, Wallace AC, Jones ML, Thornton JM. PDBsum: a Web-based database of summaries and analyses of all PDB structures. Trends Biochem Sci. 1997; 22(12):488-490. [CrossRef]
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995; 8(2):127-134. [CrossRef]
- Toth M, Smith CA, Antunes NT, Stewart NK, Maltz L, Vakulenko SB. The role of conserved surface hydrophobic residues in the carbapenemase activity of the class D β-lactamases. Acta Crystallogr D Struct Biol. 2017; 73(Pt 8):692-701. [CrossRef]
- Llinás A, Ahmed N, Cordaro M, Laws AP, Frère JM, Delmarcelle M, Silvaggi NR, Kelly JA, Page MI. Inactivation of bacterial DD-peptidase by beta-sultams. Biochemistry. 2005; 44(21):7738-7746. [CrossRef]
- Pozzi C, Di Pisa F, De Luca F, Benvenuti M, Docquier JD, Mangani S. Atomic-Resolution Structure of a Class C β-Lactamase and Its Complex with Avibactam. ChemMedChem. 2018; 13(14):1437-1446. [CrossRef]
- Tan K, Welk L, Endres M, Joachimiak A. The crystal structure of a beta-lactamase from Chitinophaga pinensis DSM 2588. 2019. [CrossRef]
- Mayclin SJ, Abendroth J, Lorimer DD, Edwards TE. Crystal structure of a beta-lactamase from Burkholderia ambifaria. 2016. [CrossRef]
- Brown G, Singer A, Proudfoot M, Skarina T, Kim Y, Chang C, Dementieva I, Kuznetsova E, Gonzalez CF, Joachimiak A, Savchenko A, Yakunin AF. Functional and structural characterization of four glutaminases from Escherichia coli and Bacillus subtilis. Biochemistry. 2008; 47(21):5724-5735. [CrossRef]
- Kishida H, Unzai S, Roper DI, Lloyd A, Park SY, Tame JR. Crystal structure of penicillin binding protein 4 (dacB) from Escherichia coli, both in the native form and covalently linked to various antibiotics. Biochemistry. 2006; 45(3):783-792. [CrossRef]
- Clementel D, Del Conte A, Monzon AM, Camagni GF, Minervini G, Piovesan D, Tosatto SCE. RING 3.0: fast generation of probabilistic residue interaction networks from structural ensembles. Nucleic Acids Res. 2022; 50(W1):W651-W656. [CrossRef]
- Kraulis, P.J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991; 24:946-950. [CrossRef]
- Smith CA, Nossoni Z, Toth M, Stewart NK, Frase H, Vakulenko SB. Role of the Conserved Disulfide Bridge in Class A Carbapenemases. J Biol Chem. 2016; 291(42):22196-22206. [CrossRef]
- Smith CA, Caccamo M, Kantardjieff KA, Vakulenko S. Structure of GES-1 at atomic resolution: insights into the evolution of carbapenamase activity in the class A extended-spectrum beta-lactamases. Acta Crystallogr D Biol Crystallogr. 2007; 63(Pt 9):982-992. [CrossRef]
- Nukaga M, Becka SA, Zeiser ET, Hoshino Y, LiPuma JJ, Papp-Wallace KM. Frameshift mutations in genes encoding PBP3 and PBP4 trigger an unusual, extreme beta-lactam resistance phenotype in Burkholderia multivorans. 2021. [CrossRef]
- Elings W, Tassoni R, van der Schoot SA, Luu W, Kynast JP, Dai L, Blok AJ, Timmer M, Florea BI, Pannu NS, Ubbink M. Phosphate Promotes the Recovery of Mycobacterium tuberculosis β-Lactamase from Clavulanic Acid Inhibition. Biochemistry. 2017; 56(47):6257-6267. [CrossRef]
- Stewart NK, Bhattacharya M, Toth M, Smith CA, Vakulenko SB. A surface loop modulates activity of the Bacillus class D β-lactamases. J Struct Biol. 2020; 211(2):107544. [CrossRef]
- Michalska K, Tesar C, Endres M, Joachimiak A, Satchell KJ. Crystal structure of class D beta-lactamase from Sebaldella termitidis ATCC 33386. 2018. [CrossRef]
- Marrero A, Mallorquí-Fernández G, Guevara T, García-Castellanos R, Gomis-Rüth FX. Unbound and acylated structures of the MecR1 extracellular antibiotic-sensor domain provide insights into the signal-transduction system that triggers methicillin resistance. J Mol Biol. 2006; 361(3):506-521. [CrossRef]
- Kerff F, Charlier P, Colombo ML, Sauvage E, Brans A, Frère JM, Joris B, Fonzé E. Crystal structure of the sensor domain of the BlaR penicillin receptor from Bacillus licheniformis. Biochemistry. 2003; 42(44):12835-12843. [CrossRef]
- Jeong JH, Cha HJ, Kim YG. Crystal Structures of Penicillin-Binding Protein D2 from Listeria monocytogenes and Structural Basis for Antibiotic Specificity. Antimicrob Agents Chemother. 2018; 62(9):e00796-18. [CrossRef]
- Fonze E, Rhazi N, Nguyen-Disteche M, Charlier P. DD-transpeptidase. 2000. [CrossRef]
- Negoro S, Ohki T, Shibata N, Mizuno N, Wakitani Y, Tsurukame J, Matsumoto K, Kawamoto I, Takeo M, Higuchi Y. Structure of 6-aminohexanoate-dimer hydrolase. 2005. [CrossRef]
- Bompard-Gilles C, Remaut H, Villeret V, Prangé T, Fanuel L, Delmarcelle M, Joris B, Frère J, Van Beeumen J. Crystal structure of a D-aminopeptidase from Ochrobactrum anthropi, a new member of the 'penicillin-recognizing enzyme' family. Structure. 2000; 8(9):971-980. [CrossRef]
- Wagner UG, Petersen EI, Schwab H, Kratky C. EstB from Burkholderia gladioli: a novel esterase with a beta-lactamase fold reveals steric factors to discriminate between esterolytic and beta-lactam cleaving activity. Protein Sci. 2002; 11(3):467-478. [CrossRef]
- Cha SS, An YJ, Jeong CS, Kim MK, Jeon JH, Lee CM, Lee HS, Kang SG, Lee JH. Structural basis for the β-lactamase activity of EstU1, a family VIII carboxylesterase. Proteins. 2013; 81(11):2045-2051. [CrossRef]
- Liang Y, Lu X. Structural insights into the catalytic mechanism of lovastatin hydrolase. J Biol Chem. 2020; 295(4):1047-1055. [CrossRef]
- Macheboeuf P, Di Guilmi AM, Job V, Vernet T, Dideberg O, Dessen A. Active site restructuring regulates ligand recognition in class A penicillin-binding proteins. Proc Natl Acad Sci U S A. 2005; 102(3):577-582. [CrossRef]
- Nocek B, Hatzos-Skintges C, Babnigg G, Joachimiak A. Crystal structure of a representative of class A beta-lactamase from Bacteroides cellulosilyticus DSM 14838. 2016. [CrossRef]
Figure 1.
The 3D structure of the active site in beta-lactamase CTX-M-14. Three short amino acid segments: Ser70-Xaa-Xaa-Lys73 (violet), Ser130-Xaa-Asn132 (grey), Lys234-Thr/Ser-Gly236 (orange), show the location of the corresponding sequence motifs. Amino acids names are given as single-letter designations. The letter “X” denotes the possibility of the presence of any amino acid at a given position of the motif. The designation “Oxyanion hole” shows the location of the corresponding functional site formed by atoms N/Ser70 and N/Ser237. The Omega-loop is shown in green, with the initial (Asp163) and final (Arg178) amino acids and two functionally important residues (Glu166 and Asn170) are indicated.
Figure 1.
The 3D structure of the active site in beta-lactamase CTX-M-14. Three short amino acid segments: Ser70-Xaa-Xaa-Lys73 (violet), Ser130-Xaa-Asn132 (grey), Lys234-Thr/Ser-Gly236 (orange), show the location of the corresponding sequence motifs. Amino acids names are given as single-letter designations. The letter “X” denotes the possibility of the presence of any amino acid at a given position of the motif. The designation “Oxyanion hole” shows the location of the corresponding functional site formed by atoms N/Ser70 and N/Ser237. The Omega-loop is shown in green, with the initial (Asp163) and final (Arg178) amino acids and two functionally important residues (Glu166 and Asn170) are indicated.
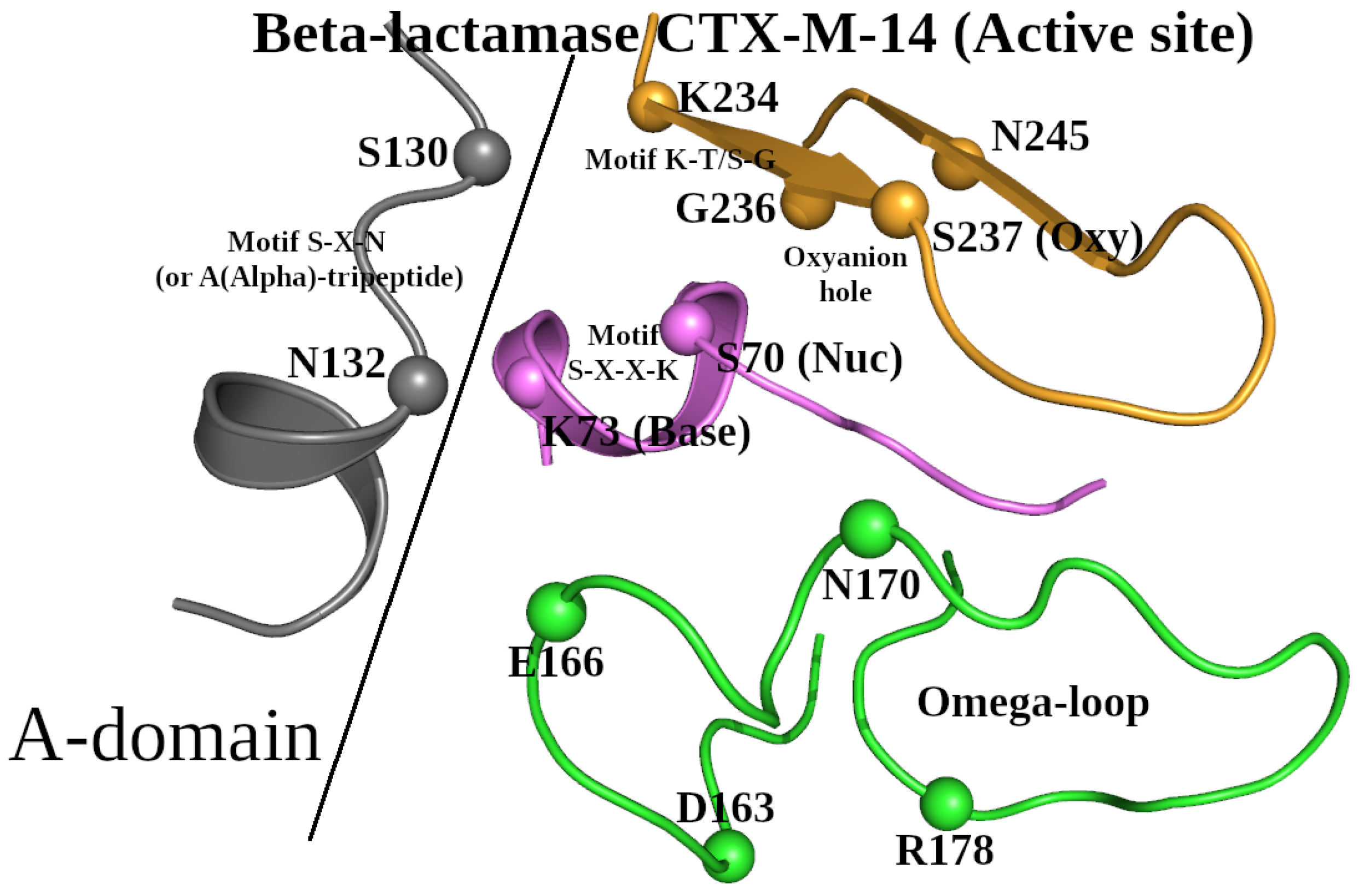
Figure 2.
Formation of NucBase-Oxy (A) and NucBase-Omega (B) zones by peptide segments Met68-Lys73 (violet), Lys234-Ser237 (orange) and residues Glu166, Leu169-Asn170, Asp179 of the Ω-subzone (green) in beta-lactamase CTX-M-14. The terms “Nuc”, “Base”, and “Oxy” are used to denote the functional characteristics of the catalytic nucleophile Ser70 and base Lys73, and the atom N/Ser237 respectively involved in formation of the oxyanion hole. The dashed lines show the canonic and weak hydrogen bonds of two types: zone-forming and internally stabilizing. Water HOH2167 is shown to stabilize the conformation of the NucBase-Omega zone.
Figure 2.
Formation of NucBase-Oxy (A) and NucBase-Omega (B) zones by peptide segments Met68-Lys73 (violet), Lys234-Ser237 (orange) and residues Glu166, Leu169-Asn170, Asp179 of the Ω-subzone (green) in beta-lactamase CTX-M-14. The terms “Nuc”, “Base”, and “Oxy” are used to denote the functional characteristics of the catalytic nucleophile Ser70 and base Lys73, and the atom N/Ser237 respectively involved in formation of the oxyanion hole. The dashed lines show the canonic and weak hydrogen bonds of two types: zone-forming and internally stabilizing. Water HOH2167 is shown to stabilize the conformation of the NucBase-Omega zone.
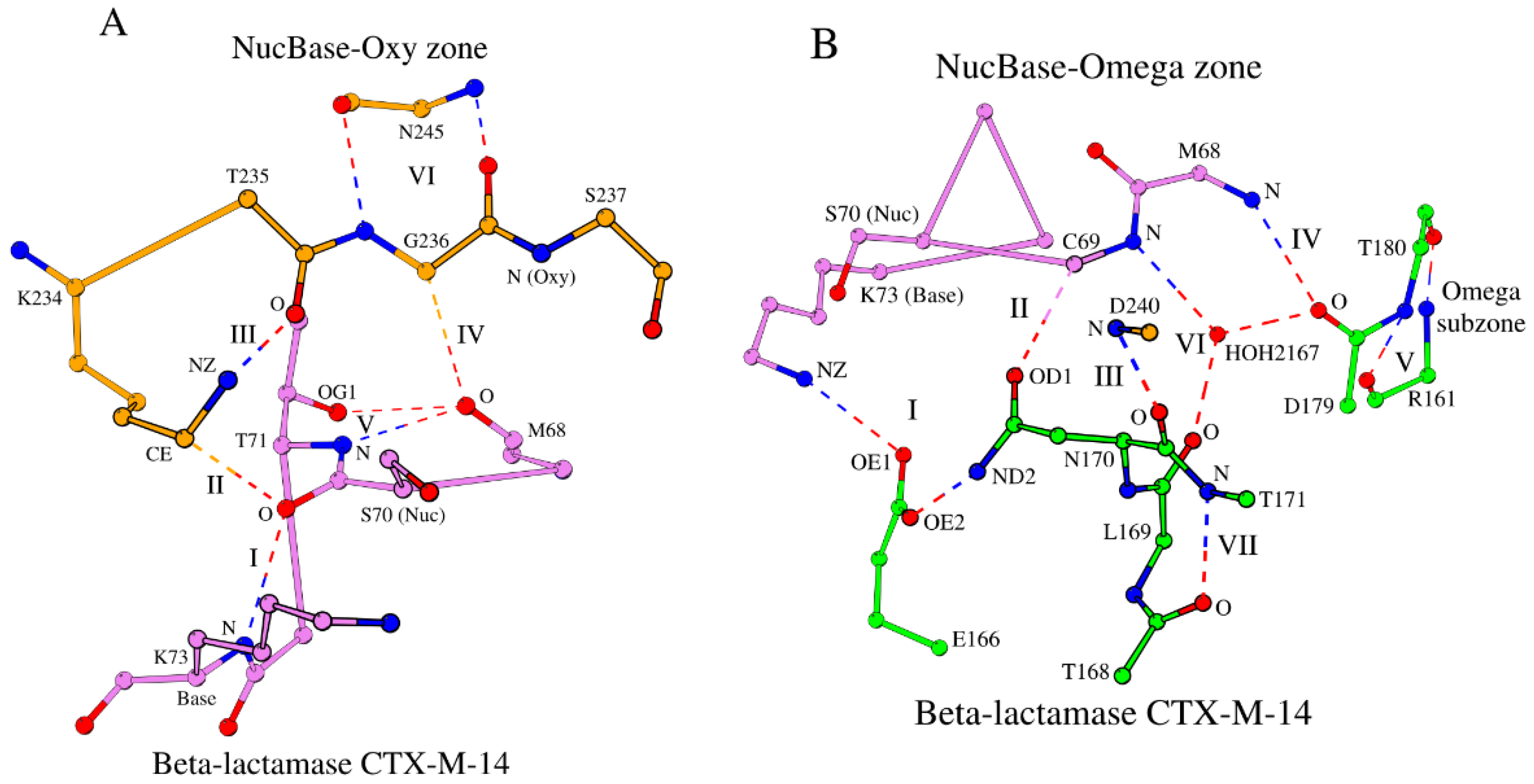
Figure 3.
Formation of the SCCs by combining three zones: NucBase-Oxy (orange and violet), NucBase-Omega (green and violet) and BaseAlpha (grey and violet), in the four most numerous groups: N-like (A), W (B), G (C) and G-like (D), of the beta-lactamase/D-ala carboxypeptidase family. The dashed lines show the canonic and weak hydrogen bonds of two types: zone-forming and internal stabilizing. Conserved water molecules are shown to stabilize the conformation of the NucBase-Omega zones.
Figure 3.
Formation of the SCCs by combining three zones: NucBase-Oxy (orange and violet), NucBase-Omega (green and violet) and BaseAlpha (grey and violet), in the four most numerous groups: N-like (A), W (B), G (C) and G-like (D), of the beta-lactamase/D-ala carboxypeptidase family. The dashed lines show the canonic and weak hydrogen bonds of two types: zone-forming and internal stabilizing. Conserved water molecules are shown to stabilize the conformation of the NucBase-Omega zones.
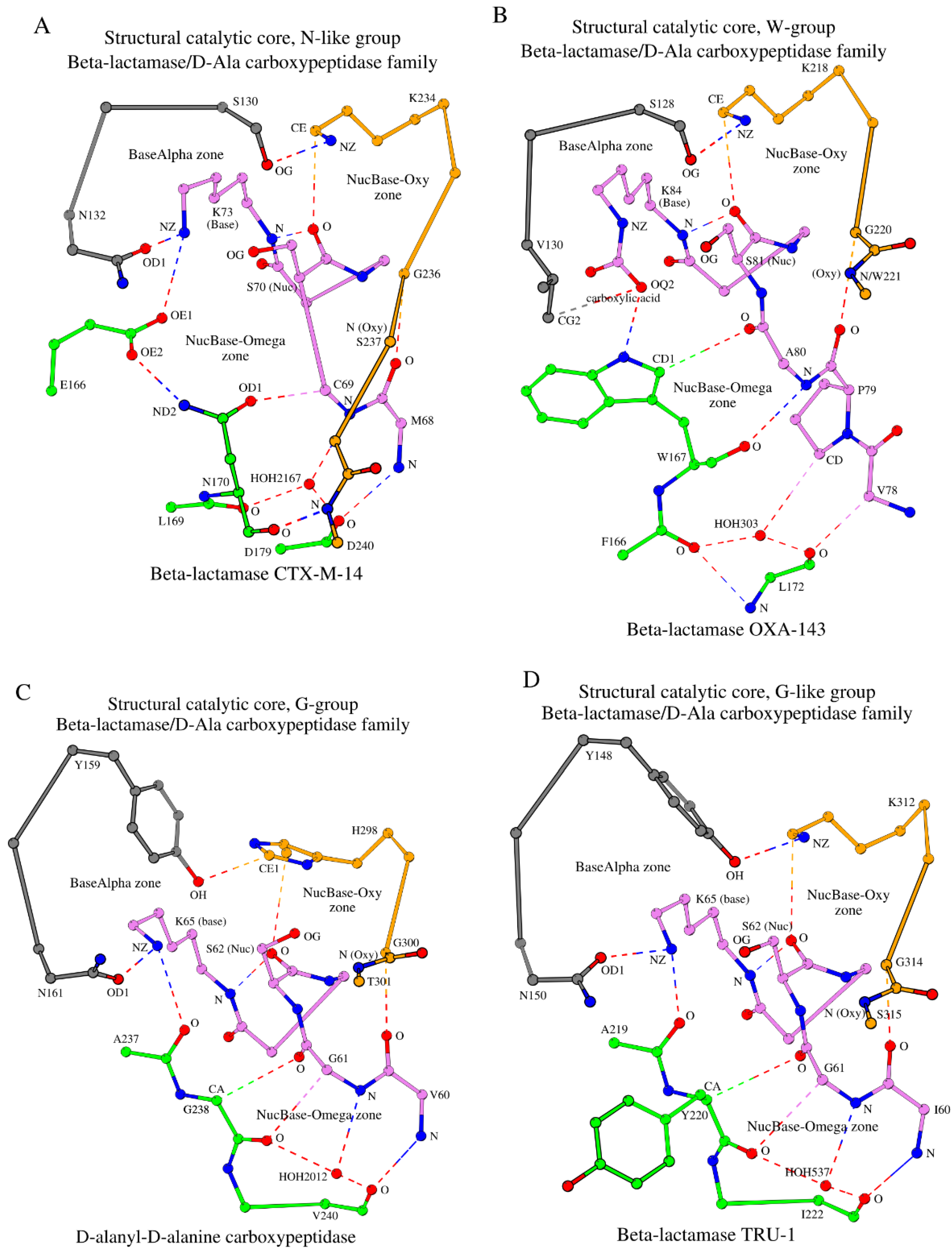
Figure 4.
Formation of the SCCs in two minor groups: Q-like (A) and inactive beta-lactamases (B), of the beta-lactamase/D-ala carboxypeptidase family, as well as in proteins of the glutaminase (C) and Dac-like (D) families. EDO302 is 1,2 ethanediol (B).
Figure 4.
Formation of the SCCs in two minor groups: Q-like (A) and inactive beta-lactamases (B), of the beta-lactamase/D-ala carboxypeptidase family, as well as in proteins of the glutaminase (C) and Dac-like (D) families. EDO302 is 1,2 ethanediol (B).
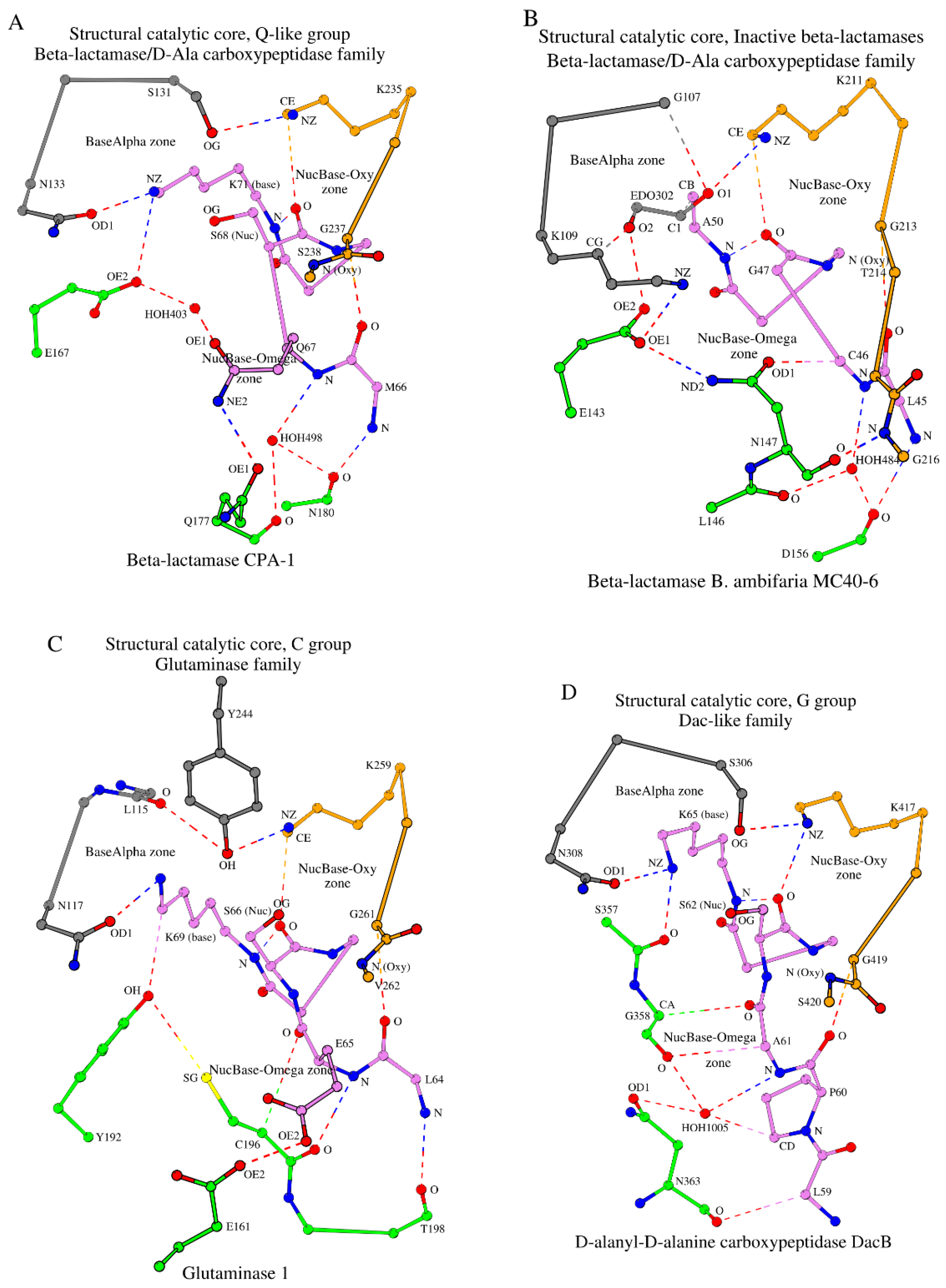

Table 1.
Structural catalytic core (SCC) in 25 β-lactamase/transpeptidase-like superfamily representative proteins.
Table 1.
Structural catalytic core (SCC) in 25 β-lactamase/transpeptidase-like superfamily representative proteins.
| N | PDB ID | R (Å) | Protein | NucBase | Alpha | Omega | Oxy | Mediator | Ref. |
| Superfamily: β-lactamase/transpeptidase-like | |||||||||
| Family: β-lactamase/D-Ala carboxypeptidase | |||||||||
| N-like group (Class A) (79) | |||||||||
| SNN subgroup (67) | |||||||||
| 1 | 4UA6_A | 0.79 | β-lactamase CTX-M-14 | 68 MCSTSK 73 | 130 SDN 132 | E166, 169 LN••D 179 | 234 KTGSGD 240 | HOH2167 | 23 |
| SNS subgroup (5) | |||||||||
| 2 | 5F82_A | 0.96 | Carbapenemase GES-5 | 62 MGSTFK 67 | 125 SDN 127 | E161, 164 MS••D 174 | 229 KTGTCA 234 | HOH498 | 44 |
| SNG subgroup (2) | |||||||||
| 3 | 2QPN_A | 1.10 | Carbapenemase GES-1 | 62 MCSTFK 67 | 125 SDN 127 | E161, 164 MG••D 174 | 229 KTGTCA 234 | HOH338 | 45 |
| SSN subgroup (2) | |||||||||
| 4 | 7DDM_A | 1.20 | β-lactamase PenA39 | 68 FCSTFK 73 | 130 SDS 132 | E166, 169 LN••D 179 | 234 KTGTGD 240 | HOH476 | 46 |
| SGN subgroup (3) | |||||||||
| 5 | 5NJ2_A | 1.19 | β-lactamase BlaC | 68 FCSTFK 73 | 128 SDG 130 | E168, 171 LN••D 181 | 236 KTGTGD 242 | HOH547 | 47 |
| W group (Class D) (45) | |||||||||
| SVW subgroup (36) | |||||||||
| 6 | 5IY2_B | 1.15 | β-lactamase OXA-143 | 78 VPASTFK 84 | 128 SAV 130 | 166 FW••L 172 | 218 KSGW 221 | HOH303 | 35 |
| SIW subgroup (5) | |||||||||
| 7 | 6W5E_A | 1.30 | β-lactamase BSU-2 | 98 TPQSTFK 104 | 149 SAI 151 | 187 FW••L 193 | 239 KTGT 242 | HOH450 | 48 |
| SLW subgroup (4) | |||||||||
| 8 | 6N1N_A | 1.60 | β-lactamase STD-1 | 62 LPASTFK 68 | 113 SAL 115 | 151 FW••L 157 | 203 KTGW 206 | HOH510 | 49 |
| W group (5) | |||||||||
| SNW subgroup (4) | |||||||||
| 9 | 2IWB_A | 1.80 | Methicillin resistance mecR1 protein |
388 SPNSTYK 394 | 439 SVN 441 | 476 YW••L 482 | 528 KTGT 531 | HOH2115 | 50 |
| STW subgroup (1) | |||||||||
| 10 | 1NRF_A | 2.50 | Regulatory protein BlaR1 | 399 APASTYK 405 | 450 STT 452 | 487 YW••L 493 | 539 KTGT542 | HOH738 | 51 |
| G group (23) | |||||||||
| YNG subgroup (4) | |||||||||
| 11 | 1YQS_A | 1.05 | D-alanyl-D-alanine carboxypeptidase | 60 VGSVTK 65 | 159 YSN 161 | 237 AG••V 240 | 298 HTGT 301 | HOH2012 | 36 |
| SNG subgroup (17) | |||||||||
| 12 | 5ZQA_A | 1.55 | Lmo2812 protein | 56 IASLSK 61 | 118 SAN 120 | 158 SG••A 167 | 222 KTGF 225 | HOH515 HOH418 |
52 |
| SCG subgroup (1) | |||||||||
| 13 | 1ES5_A | 1.40 | DD-transpeptidase | 33 TGSTTK 38 | 96 SGC 98 | 143 DG••N 150 | 213 KTGA 216 | HOH479 HOH347 |
53 |
| YSG subgroup (1) | |||||||||
| 14 | 1WYB_A | 1.80 | 6-aminohexanoate-dimer hydrolase | 110 LMSVSK 115 | 215 YCS 217 | 266 HG••V 269 | 342 GIGI 345 | CG/L109 CD1/L109 |
54 |
| G-like group (39) | |||||||||
| YNY subgroup (Class C) (33) | |||||||||
| 15 | 6FM6_A | 1.05 | β-lactamase TRU-1 | 60 IGSVSK 65 | 148 YSN 150 | 219 AY••I 222 | 312 KTGS 315 | HOH537 | 37 |
| YNA subgroup (1) | |||||||||
| 16 | 1EI5_A | 1.90 | D-aminopeptidase | 60 ICSVSK 65 | 153 YCN 153 | 225 DA••I 228 | 287 HGGA 290 | HOH531 | 55 |
| YLA subgroup (2) | |||||||||
| 17 | 1CI9_A | 1.80 | Esterase EstB | 73 LASVTK 78 | 181 YSL 183 | 274 GA••M 277 | 348 WGGV 351 | HOH1050 | 56 |
| YHQ subgroup (1) | |||||||||
| 18 | 4IVK_A | 1.80 | Carboxylesterase | 98 IYSMSK 103 | 218 YGH 220 | 295 GQ••M 298 | 381 WGGA 384 | HOH666 | 57 |
| YPH subgroup (1) | |||||||||
| 19 | 6KJC_A | 2.30 | Lovastatin esterase | 55 LASATK 60 | 170 YGP 172 | 252 GH••L 255 | 344 WGGG 347 | Y54 | 58 |
| SNM subgroup (1) | |||||||||
| 20 | 2BG1_A | 1.90 | Penicillin-binding protein 1b |
457 SPASTTK 463 | 516 SWN 518 | 555 PM••I 560 | 651 KTGT 654 | OG/S457 | 59 |
| Q-like group (Class A) (6) | |||||||||
| SNQ subgroup (5) | |||||||||
| 21 | 6V4W_A | 1.29 | β-lactamase CPA-1 | 66 MQSVFK 71 | 131 SDN 133 | E167, 177 –Q••N 180 | 235 KTGS 238 | HOH498 | 38 |
| SNT subgroup (1) | |||||||||
| 22 | 5TFQ_A | 1.07 | β-lactamase HGB-2 | 46 LLSVFK 51 | 112 SDN 114 | E148, 157 –T••N 160 | 215 KTGS 218 | HOH472 | 60 |
| Inactive beta-lactamase group (2) | |||||||||
| GKN subgroup (2) | |||||||||
| 23 | 5IHV_A | 1.10 | β-lactamase B. ambifaria MC40-6 |
45 LCGTYA 50 | 107 GDK 109 | E143, 146 LN••D 156 |
211 KAGTGG 216 | HOH484 | 39 |
| Family: Glutaminase | |||||||||
| C group (5) | |||||||||
| ONC subgroup (5) | |||||||||
| 24 | 1U60_A | 1.61 | Glutaminase 1 | 64 LESISK 69 | 115 LVN 117 | E161, Y192, 196 –C••T 198 |
259 KSGV 262 | N/A | 40 |
| Family: Dac-like | |||||||||
| G group (5) | |||||||||
| SNG subgroup (5) | |||||||||
| 25 | 2EX2_A | 1.55 | D-alanyl-D-alanine carboxypeptidase DacB | 59 LPASTQK 65 | 306 SDN 308 | 357 SG••N 363 | 417 KTGS 420 | HOH1005 | 41 |
N/A–Not Available.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



