
Submitted:
22 March 2025
Posted:
24 March 2025
You are already at the latest version
Abstract
Antiphospholipid syndrome (APS) can affect the kidneys, leading to renal artery and vein thrombosis, allograft loss following transplantation, and microvascular damage referred to as aPL-nephropathy (aPL-N). APL-N is a complex and frequently underdiagnosed condition characterized by an incomplete understanding of its etiopathogenesis and associated with unfavorable renal outcomes. The 2023 ACR/EULAR classification criteria for APS included aPL-N within the microvascular domain. The gold standard for aPL-N is the renal biopsy, revealing lesions associated with acute thrombotic microangiopathy and chronic vascular changes. Nevertheless, reluctance for biopsies due to anticoagulation and thrombocytopenia underscores the need for noninvasive diagnostics. SCommon clinical features include hypertension, microscopic hematuria, proteinuria, and renal insufficiency. Antiphospholipid antibodies seem crucial to kidney damage through thrombotic and inflammatory processes. Studies and experimental models of thrombotic microangiopathy lesions suggest the involvement of the complement cascade, tissue factor, and mammalian target of the rapamycin complex activation pathway. Currently, the management of aPL-N is based mainly on expert opinion, with limited evidence supporting the use of anticoagulants, leading to controversy in their application. Treatment may include heparin, intravenous immunoglobulin, plasma exchange, and targeted therapies tailored to aPL-N mechanisms. Future multicenter studies are essential to clarify their roles.
Keywords:
antiphospholipid antibodies
; lupus anticoagulant
; aPL-nephropathy
; thrombotic angiopathy
; thrombosis
1. Introduction
Antiphospholipid syndrome (APS) is a thrombo-inflammatory disorder with a complex and not fully understood pathogenesis. APS is marked by recurrent thromboembolic occurrences and pregnancy complications linked to the presence of circulating antiphospholipid antibodies (aPL). The aPL included in the recent ACR/EULAR 2023 classification criteria are anti-cardiolipin (aCL), anti-β2-glycoprotein I (anti-β2GPI), and lupus anticoagulant (LAC) [1,2]. With an estimated incidence of 50 patients per 10,000 person-years, APS predominantly affects young female adults aged 15 to 50 years, conferring significant morbidity [3]. The condition includes arterial or venous thrombosis, microvascular thrombosis, or recurrent thrombosis resistant to anticoagulants, potentially causing severe complications [4,5,6]. APS can affect various organs, including the kidneys, potentially leading to a significantly higher rate of damage accrual and disability burden in this young population [7]. As recently reported, about one-third of aPL-positive patients develop kidney failure [8]. The entire kidney vasculature may be involved in APS, such as renal arteries and veins, intrarenal arteries and arterioles, and glomerular capillaries [9]. Renal involvement in APS, particularly microvascular, is often underestimated due to the high-risk nature of renal biopsy amid anticoagulant therapy and potential thrombocytopenia. Indeed, in aPL-positive systemic lupus erythematosus (SLE) patients, kidney biopsies are significantly delayed compared to aPL-negative patients (134.4±60.6 vs. 42.6±60.1 months from SLE diagnosis without differences between aPL-positive SLE patients with or without APS criteria), risking misdiagnosis or delayed diagnosis [10,11]. Recently, the American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) 2023 APS classification criteria included aPL-nephropathy (aPL-N) in the microvascular domain [1]. This review explores the clinical phenotypes associated with renal involvement in APS and the potential pathogenetic mechanisms that underlie these phenomena.
2. Epidemiology of Renal Involvement in APS
The prevalence of kidney involvement in APS remains poorly defined. Among the 1000 patients in the Euro-phospholipid registry, the prevalence of renal disease was 3% [12], of which 2% had glomerular thrombosis, 1% renal infarction, 0.5% renal arterial thrombosis, and 0.2% renal vein thrombosis. However, given the reluctance to perform biopsies, the true prevalence of APS-N is unknown to date. In fact, in some cohorts that specifically evaluated aPL-N lesions, the prevalence ranged from 6% to 45% [10,13,14]. A recent systematic review of the literature on biopsy-proven aPL-N revealed a prevalence of 28.6% [8]. In CAPS, acute renal involvement reaches 71%, as reported in the CAPS-Registry [15].
3. Pathophysiology of Renal Involvement in APS
The pathogenesis of renal APS involvement is complex and not fully understood. The strong link between aPL and aPL-N suggests these antibodies play a crucial role in renal injury. Clinical presentations of macrovascular and microvascular involvement, with acute and chronic lesions, indicate various underlying mechanisms (Figure 1). Macrovascular involvement follows the "two-hit model of APS," where anti-β2GPI initiates a prothrombotic state, further worsened by stasis or inflammation systems [2]. APL interacts with various cell types, including platelets, granulocytes, and endothelial cells, promoting intravascular coagulation by cross-reacting with plasma proteins like tissue factor pathway inhibitors. This triggers monocytes’ tissue factor (TF) release, leading to factor Xa release and thrombin formation generation [16]. Anti-β2GPI antibodies bind targets, aggregating and forming immune complexes on platelets and endothelial cells, enhancing prothrombotic activity by activating the Apolipoprotein E receptor 2 (ApoER2) [16,17]. This mechanism is recognized as an essential trigger in murine models of APS [17]. Interestingly, mice that have anti-β2GPI antibodies and are deficient in the ApoER2 (-/-) gene showed reduced thrombotic events compared to wild-type mice. Immune complexes attach to endothelial cells, prompting the expression of adhesion molecules like selectins and integrins. This mechanism activates endothelial cells and enhances TF expression [2,16,17]. Molecular activations from immune complexes activate the complement system and promote neutrophil extracellular trap release (NETs) [2]. Additionally, the complement-mediated TF seems to contribute to the development of TMA in APS [18]. The activation of the complement cascade is seen as a key factor in aPL-mediated thrombosis, both ex-vivo and in vitro studies [19,20,21]. Research by Seshan et al. showed that both mouse-derived IgG aPL and human aPL can induce glomerular lesions typical of TMA in mice [22]. They observed increased fibrin, TF, and complement C3 deposits in the glomeruli of mice given both mouse and human aPL, highlighting these factors’ role in TMA pathogenesis [22]. Furthermore, neutralizing CD59 in a renal-TMA mouse model resulted in more C5b-9 formation in glomeruli, accompanied by increased platelet and fibrin deposition, more severe endothelial injury, and reduced renal function compared with controls [23]. A clinical study of 42 renal tissue samples confirmed TMA due to various conditions (8 SLE/APS). It found C4d and C5b-9 in 88.1% and 78.6% of TMA cases, respectively, noting distinct staining patterns in the renal vasculature across conditions [24]. C4d staining and microthrombi coexist in aPL-positive SLE biopsy samples patients [23,24,25].
Chronic vascular lesions display different histopathological changes, with FIH being the most common. While the exact cause of these lesions remains unclear, it is probably not just due to thrombotic injury. Canaud et al. suggested that chronic activation of the mammalian target of the rapamycin complex (mTOR) pathway might play a role in these patients, which is associated with the emergence of chronic vasculopathy lesions. Their research showed no vascular lesion recurrence and reduced proliferation in the renal transplant of aPL-N patients treated with sirolimus, confirmed by follow-up assessment biopsies.26 They also observed vascular activation of the mTOR pathway in autopsy specimens from CAPS patients [26].
4. Kidney Involvement in APS
APS renal involvement includes three groups based on location: renal artery lesions, intrarenal vascular lesions, and renal vein thrombosis, as shown in Figure 2. Renal artery lesions and vein thrombosis represent macrovascular, acknowledged in the Sapporo classification criteria, while intrarenal lesions indicating microvascular involvement were recently added to the 2023 ACR/EULAR classification criteria [1].
4.1. Macrovascular Involvement
4.1.1. Renal Arterial Stenosis
Though uncommon in APS, thrombosis of the renal artery and branches leads to renal infarction. First described in the early 90s [28], it can present as unilateral or bilateral thrombosis from in situ thrombosis or arterial embolism, possibly from cardiac chambers or proximal arteries. Renal artery disease may manifest as renal infarction, ischemic acute renal failure, or slowly progressive chronic renal issues failure [29,30,31]. Key clinical signs of renal artery stenosis include severe new-onset hypertension or worsening of existing hypertension, often accompanied by flank pain and macrohematuria [30,31,32]. Sangle et al. studied 77 patients with aPL and uncontrolled hypertension, 91 hypertension clinic attendees, and 92 potential kidney donors. MRI renal angiography diagnosed renal artery stenosis in 26%, notably higher than in other groups [32]. Consequently, APS patients with uncontrolled hypertension should raise suspicions of renal artery thrombosis, and young patients with unexplained renal artery thrombosis should undergo testing for aPL.
Renal artery Doppler ultrasound is preferred for screening renal artery stenosis. Although renal angiography has the highest accuracy, contrast-enhanced CT and MRI are less invasive alternatives with similar diagnostic value performance [33].
Renal artery involvement in APS has two key radiological traits that set it apart from fibrodysplasia and atherosclerosis: (i) it primarily affects the proximal segment of renal arteries, and (ii) it presents as a smooth, well-defined lesion, usually resulting in non-critical stenosis [32]. Growing interest exists in contrast-enhanced ultrasound for assessing perfusion abnormalities in patients with renal impairment infarction [34].
4.1.2. Renal Vein Thrombosis
Renal vein thrombosis can occur unilaterally or bilaterally in individuals with APS, being more common in those also diagnosed with SLE. The main clinical sign linked to renal vein thrombosis is nephrotic-range proteinuria, and in some cases, bilateral involvement may lead to renal failure thrombosis [35,36,37,38]. Acute thrombosis may present with flank pain and hematuria less frequently. In APS patients, consider renal vein thrombosis with sudden nephrotic-range proteinuria, as it may complicate renal transplantation and affect outcomes. Doppler ultrasonography is the preferred diagnostic tool for showing an edematous kidney with reduced echogenicity, parenchymal disruption, and renal vein thrombus. Computed tomography venography may also be performed. Be aware that intravenous line size, injection rate, hydration levels, and cardiac output significantly impact results. Renal vein thrombosis is a filling defect in post-contrast venous phase imaging administration [39].
4.2. Microvascular Involvement
4.2.1. APL-Nephropathy
Identified in 1999, aPL-N is a small-vessel vasculopathy characterized by acute and chronic renal lesions. A seminal study by Nochy et al. [40] assessed renal biopsies of sixteen APS patients, revealing histological changes that clarify aPL-N diagnosis: TMA, fibrous intimal hyperplasia, recanalizing thrombi, fibrous arterial occlusion, tubular pseudo-thyroidization, and focal cortical atrophy. TMA is a key histological finding in the acute form of aPL-N (Figure 3A), characterized by fibrin thrombi with fragmented blood cells in small vessels and glomeruli, along with subendothelial oedema. The glomerular basement membrane may show a double-layered, wrinkled appearance in advanced stages due to endothelial detachment and mesangial cell infiltration. Cellular inflammation and immune deposits seen in immunofluorescence are usually absent in primary cases of APS [40]. TMA lesions are more common in CAPS [41].
Fibrous intimal hyperplasia (FIH) is the most common chronic lesion linked to APS, alongside focal cortical atrophy (FCA) and thyroidization (Figure 3B). FIH involves significant myofibroblastic proliferation in the intimal layer, sometimes displaying an "onion skin" appearance. Lumens may become obstructed by fibrous tissue, leading to recanalization and the development of endothelialized channels. FCA, resulting from prolonged reduced blood flow, affects the renal cortex beneath the capsule, causing contour depression and glomerular tissue retraction. Tubular thyroidization shows extensive atrophic tubules containing eosinophilic casts resembling thyroid tissue.
APL-N has been described in both primary and secondary forms of APS. In 2002, Daugas et al. evaluated renal biopsies from a substantial French cohort of patients with SLE, specifically investigating vascular lesions consistent with aPL-N [42]. Their findings showed aPL-N in 63% of aPL-positive patients, 78% of those had a previous thrombosis or pregnancy issues. Other studies supported these results. Tektonidou et al. found APL-N prevalence at 40% in aPL-positive SLE biopsy samples versus 4% in aPL-negative cohorts’ prevalence [43]. A strong association between aPL-N and arterial thrombosis APS and triple-aPL positivity APS phenotypes has been described [8,14,43]. Furthermore, a notable link between aPL-N and livedo reticularis was reported [43].
Noteworthy, patients with lupus nephritis and renal TMA have the worst outcomes among vascular damage manifestations, necessitating thorough evaluation assessment [44,45]. This observation is consistent with a longitudinal study involving 111 individuals diagnosed with lupus nephritis and monitored over 15 years, which revealed that patients who tested positive for aPL encountered the most detrimental renal outcomes [46]. An international multicenter study recently evaluated 123 kidney biopsies from patients who were aPL-positive [14]. Cluster analysis showed that renal TMA, acute or chronic, occurred more frequently in patients with thrombotic APS and triple aPL-positivity. In contrast, other changes, like FIH, were less correlated with systemic thrombotic events. Additionally, the renal TMA cluster was linked to the worst renal prognosis.
aPL-N can present with clinical signs like high blood pressure, microscopic hematuria, and varying levels of proteinuria, which may become severe, reaching the nephrotic range, alongside acute kidney injury or a gradual decline in chronic kidney disease. A recent meta-analysis investigated risk factors associated with kidney failure in biopsy-proven aPL-N, identifying hypertension and triple aPL as strong predictors of adverse renal outcomes in aPL-N [8]. Hypertension is the most common symptom of aPL-N. In a recent systematic review, over 70% of patients with biopsy-confirmed aPL-N were found affected with arterial hypertension, compared to 48% in patients without aPL-N [8]. In the Nochy study, over 90% of participants experienced hypertension, attributed to excess renin production caused by vascular damage [40]. Since hypertension often occurs in chronic kidney disease (CKD) patients, both as a cause and effect, the clinical presentation of aPL-N tends to be nonspecific. Other renal manifestations include mild proteinuria and a gradual decrease in kidney function. The combination of slowly worsening kidney function and mild proteinuria may lead clinicians to hesitate in recommending a kidney biopsy, particularly given the ongoing anticoagulant therapy and/or thrombocytopenia. However, these pathological changes, particularly TMA, are not pathognomonic of APS, making it challenging for clinicians to diagnose aPL-N. Consequently, although APS-N is rare, it may go unrecognized and underdiagnosed.
As outlined in Table 1 and Table 2, the groundbreaking 2023 ACR/EULAR classification criteria include aPL-N within the microvascular domain, considering both suspected and definitive aPL-N based on the presence or absence of kidney biopsy [1]. Furthermore, a clear definition was provided to homogenize the histopathological terminology, including the distinction of acute from chronic aPL-N lesions, as well as the distinction of thrombotic lesions in glomeruli from those in arteries/arterioles, and the separation of fibrous intimal hyperplasia from organized thrombi as reported in Table 3 [47]. The goal is that the new 2023 ACR/EULAR APS classification criteria aPL-N definition, in conjunction with advancing technologies such as digital pathology, machine learning algorithms, spatial transcriptomics, and proteomics, will facilitate precise diagnosis. Furthermore, noninvasive techniques such as intravoxel incoherent motion diffusion-weighted imaging MRI, used to assess renal function in patients with immunoglobulin A nephropathy, may aid in diagnosing aPL-N in the future [49].
4.2.2. Glomerular Lesions
In addition to the acute and chronic vascular lesions discussed earlier, various glomerular lesions have been identified in aPL-N. In 2003, Fakhouri et al. reported that 31% of the 29 patients studied exhibited ‘atypical’ histological forms of aPL-N [49]. Among these patients, they documented different glomerular lesions, including membranous nephropathy (2 cases), minimal change disease/focal segmental glomerulosclerosis (3 cases), mesangial C3 nephropathy (3 cases), and pauci-immune crescentic glomerulonephritis (1 case). Sinico et al., in a multicenter cohort study of 160 PAPS patients, evaluated the kidney biopsy of 10 patients with signs of renal injury; four patients displayed features indicative of aPL-N, four had membranous nephropathy, and two showed proliferative glomerulonephritis [13]. A recent systematic literature review revealed that among 238 biopsy-proven aPL-N cases, 62.2% had diffuse proliferative glomerulonephritis, while 16.2% had membranous glomerulonephritis [8]. It is still uncertain whether a pathophysiological connection exists between these glomerulonephritis and aPL-N or if these lesions occurred independently and are mutually exclusive. There are ongoing studies to clarify this data.
5. Renal Transplantation in APS
Patients with APS-N who undergo renal transplants face a high risk of thromboembolic events in the post-transplant period [50,51,52,53,54]. This risk is most significant in the first week, with renal vein thrombosis being the most common occurrence; intra- and extra-graft thrombosis can also happen [53,54,55]. APS progression in kidney transplant recipients is unpredictable, causing allograft thrombosis and potential loss. Complications include macrovascular thrombosis of renal vessels and TMA. A study of 19 adult kidney recipients with APS revealed a significantly lower 15-year renal allograft survival rate compared to controls (p= 0.0009), especially in those diagnosed with APS later transplant [55].
The estimated prevalence of aPLs in renal transplant recipients is around 3% [51]. Positive aPL test results before transplantation lead to higher chronic vascular scores and faster mGFR decline after one year compared to aPL-negative patients (mean 49.1 ± 18.4 vs 54.4 ± 19.4, p=0.04); LA is the most common aPL antibody. Emerging evidence about non-criteria aPL is being reported. Serrano et al. found renal transplant recipients with IgA anti-B2GP1 face higher thrombosis risk if they develop IgA immunocomplexes with β2-glycoprotein I [56]. Currently, no literature is available on anti-phosphatidylserine/prothrombin antibodies (aPS/PT), which have been associated with thrombosis and LA in APS patients [57,58]. Within our cohort, four out of five APS patients who underwent renal transplants tested positive for aPS/PT antibodies (personal communication).
6. Management of Renal Involvement in APS
Patients with macrovascular involvement who meet the 2023 ACR/EULAR criteria should use vitamin K antagonists, targeting an INR of 2-3 for venous thrombosis or 3–4 for arterial or recurrent thrombosis, as shown in Figure 4. Patients with renal artery stenosis should receive prompt evaluation for angioplasty. Sangle et al. studied 14 patients who underwent angioplasty, noting restenosis in those inadequately anticoagulated (mean INR < 2.5) [59].
There is no consensus on managing aPL-N due to insufficient robust studies [29]. Therapeutic anticoagulation with vitamin K antagonists is recommended for patients meeting aPL-N histological criteria and 2023 ACR/EULAR classification criteria [60]. Although the evidence for combining antiplatelets and vitamin K antagonists (VKA) is limited, clinical observations indicate that this approach may improve renal outcomes (personal experience). Preliminary data from our cohort of patients with microvascular involvement showed that in 4/4 (100%) patients with timely diagnosis of aPL-N, early treatment with combined anti-platelet drugs and VKA led to improvement in renal function (per sonal data). Management strategy for aPL-N in those not meeting classification criteria is unclear. EULAR guidelines recommend low-dose aspirin (100 mg daily) for high-risk patients with triple-aPL positivity who do not meet APS classification criteria [60]. For patients with concurrent lupus nephritis, the use of hydroxychloroquine and immunosuppressive therapies is recommended [61,62]. Since hypertension and proteinuria often occur in aPL-N, the standard treatment includes administering angiotensin system inhibitors [61,62]. Although direct oral anticoagulants (DOACs) have not been studied specifically for aPL-N, their use in APS—especially for high-risk triple-positive APS patients or those experiencing arterial or recurrent thrombotic events, as alarming findings from the TRAPS trial indicate [63,64] —is not advised. A recent meta-analysis of four open-label randomized clinical trials involving 472 patients with APS (median time of control arm in therapeutic range: 60%) found a 5-fold increased risk of subsequent arterial thrombosis (OR 5.43, 95% CI 1.87-15.79), while no difference was observed with regarding the risk of venous thrombosis in the DOACs group compared to the warfarin one [65]. Therefore, given the strong association between arterial thrombosis and aPL-N, it can be assumed that DOACs are not effective in aPL-N. No powered studies exist on anticoagulation’s effects on renal outcomes. Heparin, known for inhibiting the classical complement activation pathway and intravenous immunoglobulin and/or plasma exchange, ought to be regarded as a potential alternative treatment for severe, refractory, and catastrophic APS [66,67].
Consideration should be given to targeted therapies such as B-cell-directed treatments, complement inhibitors, tissue factor inhibitors, and mTOR pathway inhibitors. Nonetheless, comprehensive prospective multicenter studies are crucial to understand their effectiveness. A pilot open-label phase II trial evaluated the potential advantages of rituximab in managing non-criteria manifestations of APS. This trial showed that two patients with aPL-N achieved partial responses after receiving two doses of 1,000 mg rituximab on days 1 and 15 [68]. Additionally, case reports highlighted successful anti-CD20 treatment in patients with aPL-N and/or other non-criteria manifestations, such as severe thrombocytopenia, hemolytic anemia, and skin ulcers [69,70]. Belimumab, a BAFF antagonist, was given in two primary APS cases, one with recurrent alveolar hemorrhage and the other with skin ulcers [71]. Both patients showed clinical improvement and successfully discontinued corticosteroids. A phase II trial is evaluating belimumab therapy’s efficacy and safety for refractory or non-criteria antiphospholipid syndrome manifestations, and we are hopeful for its treatment effectiveness in APS patients [72].
Eculizumab—a humanized recombinant monoclonal antibody—binds to C5, thus preventing its cleavage into C5a and C5b. It has been effectively used in refractory catastrophic APS [73], post-kidney transplants-TMA [74,75] and in patients with lupus nephritis accompanied by TMA [76]. Data from the CAPS registry reported 39 cases of CAPS patients treated with eculizumab [77]. Among these, 29 (74.4%) recovered from the CAPS; 25 (64.1%) achieved complete remission, while 4 (10.3%) attained partial remission.
Inhibiting the mTOR pathway offers a promising therapy for aPL-N. This approach reduces vascular proliferation in renal biopsies and lowers the recurrence of vascular lesions in kidney transplant recipients with aPL-N. Research by Canaud et al. showed that 7 of 10 patients (70%) on mTOR inhibition had a functioning allograft 10 years post-transplant, compared to only 3 of 27 untreated patients (11%) [26]. Importantly, the treatment’s efficacy was independent of anticoagulation therapy.
Recent findings have identified a type I interferon signature in primary APS [78,79,80]. Research shows that endothelial progenitor cells from APS patients have decreased differentiation into endothelial cells. Improvement was noted with type I interferon receptor-neutralizing treatment antibody [78]. Transcriptomic analysis of whole blood shows elevated IFN-pathway gene expression in thrombotic PAPS cohorts, underscoring their key role in thrombotic events PAPS [80]. Recently, a murine model of aPL-N revealed increased IFN signature expression levels in APS mice’s kidneys [81]. The novel tyrosine kinase 2 inhibitor reversed pathological vascular changes in mice kidneys and decreased fibrin and C3 deposition. These findings suggest exploring anti-interferon antibodies as a new therapeutic strategy to reduce vascular damage associated with APS
The importance of statin use in APS has gained attention. Experimental models show that reduced TF expression alleviates glomerular injury in both mouse aPL- and human aPL-treated mice [22]. Pravastatin administration down-regulates glomerular TF synthesis and prevents aPL-induced TMA in mice, indicating that targeting TF may be a promising therapeutic strategy for patients TMA [22].
The field of nephrology has greatly benefited from the use of sodium-glucose cotransporter inhibitors (iSGLT-2), which have revolutionized the field and drastically improved outcomes regarding the progression of CKD [82,83]. However, patients with autoimmune diseases such as SLE with lupus nephritis were excluded from the DAPA-CKD trial, and higher doses of glucocorticoids or intravenous use of immunosuppression within 3 months were exclusion criteria of the EMPA-KIDNEY trial. Currently, there are no randomized controlled trials evaluating the efficacy of iSGLT2 in reducing the progression of aPL-N. However, in addition to their nephroprotective effects, iSGLT2 have shown in vitro and in vivo in animal models to influence the mTOR pathway. Therefore, it is plausible that iSGLT2 may confer renal protection also in the context of aPL-N. However, due to the predominantly thrombotic pathogenesis, driven by endothelial damage specific to aPL-N, further clinical studies specifically targeting this patient population are needed to justify the use of iSGLT2 in aPL-N.
7. Unmet Needs and Future Strategies in Antiphospholipid Antibodies Nephropathy
Despite progress in understanding APS, significant unmet needs remain in diagnosing and managing aPL-N. The 2023 ACR/EULAR classification criteria Renal Pathology Subcommittee’s efforts in defining aPL-N have resulted in only minor changes compared to two decades ago [48]. Diagnosing aPL-N depends on histological changes not exclusive to APS. However, this initiative provides a detailed list of histological variations to standardize pathological reporting aPL-N [48]. Advanced technologies like digital pathology and machine learning can identify early endothelial cell injury in glomeruli and blood vessels, improving diagnoses and differentiating TMA causes. Although renal biopsy is the gold standard, performing renal biopsy in APS patients is challenging due to increased thrombotic risk, anticoagulant use, and frequent thrombocytopenia. We lack universally accepted guidelines for anticoagulation management, or platelet count thresholds specifically for aPL-N biopsy. Nonetheless, extrapolating from existing expert consensus and KDIGO and ASH guidelines [84,85,86] for high-risk biopsy situations, a platelet count above 50.000/µL is generally advised as a safe threshold to proceed with percutaneous renal biopsy. Anticoagulation therapy (particularly VKA and DOACs) should be withheld with bridging to low-molecular-weight heparin, when possible, restarting anticoagulation 48–72 hours after biopsy if no bleeding complications occur. When significant thrombocytopenia (platelets <50.000/µL) or high bleeding risk precludes standard percutaneous biopsy, alternative approaches such as transjugular renal biopsy may be considered. Indeed, transjugular renal biopsy provides a safer alternative, minimizing bleeding risk, allowing for prompt anticoagulation restart, and should thus be considered strongly in patients with severe thrombocytopenia or who cannot safely interrupt anticoagulation. Multidisciplinary discussion (nephrologist, rheumatologist, haematologist, vascular medicine physician and radiologist) is recommended for individualized decisions. Challenges in performing kidney biopsies highlight the urgent need for noninvasive diagnostics alternatives [8,10]. Recent advancements in noninvasive renal assessment, such as diffusion-weighted imaging, hold promise for patients with immunoglobulin A and diabetic nephropathy, paving the way for innovative diagnostics approaches [87,88].
Currently, we do not have biomarkers capable of identifying aPL-N in its early stages, particularly for differentiating it from other kidney issues such as lupus nephritis or hypertensive nephropathy. Recent studies of kidney tissue transcriptome data show increased expression of complement, interferon, and NETs genes in APS kidneys, indicating potential therapeutic targets biomarkers [89].
Current guidelines focus on APS broadly, neglecting nephropathy as a distinct condition [61]. Uncertainty persists regarding effective treatment for aPL-N, including anticoagulants, immunosuppressants, and combination therapies. Patient responses vary significantly, highlighting the need for more research on factors influencing treatment outcomes. Urgent long-term studies are needed to assess aPL-N progression and treatment effectiveness. Identifying risk factors for CKD or end-stage renal disease in aPL-N patients is a critical unmet need.
8. Conclusions
APL-N is a complex, often underdiagnosed condition with unclear etiopathogenesis and poor renal outcomes. Improving our understanding of its mechanisms, identifying risk factors, and using noninvasive diagnostic methods could lead to earlier diagnoses and new treatment options, enhancing patient prognosis.
Author Contributions
AH contributed to the study’s conception and design. AH wrote the first draft of the paper. AH and DDP provided the data analyses and directly accessed and verified the underlying data reported in the manuscript. All authors contributed to interpreting the data and critically revised the paper. AH, DDP, IC, ML, FKM, FN, and PS had full access to all the data in the study. All authors had final responsibility for the decision to submit for publication.
Funding
This research received no specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability statement
All data generated or analyzed during this study are included in respective cited articles.
Declaration of Competing Interest
The authors disclosed no conflict of interest.
References
- Barbhaiya, M.; Zuily, S.; Naden, R.; Hendry, A.; Manneville, F.; Amigo, M.-C.; Amoura, Z.; Andrade, D.; Andreoli, L.; Artim-Esen, B.; et al. 2023 ACR/EULAR antiphospholipid syndrome classification criteria. Ann. Rheum. Dis. 2023, 82, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Branch, D.W.; Ortel, T.L. Antiphospholipid syndrome: advances in diagnosis, pathogenesis, and management. BMJ 2023, 380, e069717. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.; Sciascia, S.; de Groot, P.G.; Devreese, K.; Jacobsen, S.; Ruiz-Irastorza, G.; Salmon, J.E.; Shoenfeld, Y.; Shovman, O.; Hunt, B.J. Antiphospholipid syndrome. Nat. Rev. Dis. Prim. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.W.M.; De Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef]
- Asherson, R.A. The catastrophic antiphospholipid (Asherson's) syndrome. Autoimmun. Rev. 2006, 6, 64–67. [Google Scholar] [CrossRef]
- Pengo, V.; Ruffatti, A.; Legnani, C.; Gresele, P.; Barcellona, D.; Erba, N.; Testa, S.; Marongiu, F.; Bison, E.; Denas, G.; et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J. Thromb. Haemost. 2010, 8, 237–242. [Google Scholar] [CrossRef]
- Hoxha, A.; Perin, N.; Lovisotto, M.; Calligaro, A.; Del Ross, T.; Favaro, M.; Tonello, M.; Doria, A.; Simioni, P. Risk factors for damage accrual in primary antiphospholipid syndrome: A retrospective single-center cohort study. J. Autoimmun. 2024, 144, 103180. [Google Scholar] [CrossRef]
- Hoxha, A.; Lovisotto, M.; Perin, N.; Nalesso, F.; Del Prete, D.; Simioni, P. Anti-phospholipid antibodies nephropathy is associated with an increased risk of kidney failure: a systematic literature review and meta-analysis. Clin. Kidney J. 2024, 17, sfae302. [Google Scholar] [CrossRef]
- Scheen, M.; Adedjouma, A.; Esteve, E.; Buob, D.; Abisror, N.; Planche, V.; Fain, O.; Boffa, J.J.; De Seigneux, S.; Mekinian, A.; et al. Kidney disease in antiphospholipid antibody syndrome: Risk factors, pathophysiology and management. Autoimmun. Rev. 2022, 21, 103072. [Google Scholar] [CrossRef]
- Silvariño, R.; Sant, F.; Espinosa, G.; Pons-Estel, G.; Solé, M.; Cervera, R.; Arrizabalaga, P. Nephropathy associated with antiphospholipid antibodies in patients with systemic lupus erythematosus. Lupus 2011, 20, 721–729. [Google Scholar] [CrossRef]
- Ruffatti, A.; Tonello, M.; Calligaro, A.; Del Ross, T.; Favaro, M.; Zen, M.; Hoxha, A.; Alaibac, M. Prevalence and adverse consequences of delayed diagnosis and misdiagnosis in thrombotic antiphospholipid syndrome. An observational cohort study and a review of the literature. Clin. Rheumatol. 2023, 42, 3007–3019. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Piette, J.; Font, J.; Khamashta, M.A.; Shoenfeld, Y.; Camps, M.T.; Jacobsen, S.; Lakos, G.; Tincani, A.; Kontopoulou-Griva, I.; et al. Antiphospholipid syndrome: Clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients: Clinical and immunologic manifestations of APS. Arthritis Rheum. 2002, 46, 1019–1027. [Google Scholar] [CrossRef]
- Sinico, R.A.; Cavazzana, I.; Nuzzo, M.; Vianelli, M.; Napodano, P.; Scaini, P.; Tincani, A. Renal Involvement in Primary Antiphospholipid Syndrome: Retrospective analysis of 160 patients. Clin. J. Am. Soc. Nephrol. 2010, 5, 1211–1217. [Google Scholar] [CrossRef]
- Sciascia, S.; Yazdany, J.; Moroni, G.; Becker, J.U.; Seshan, S.V.; Andrade, D.; Emmi, G.; Cuadrado, M.J.; Radin, M.; Cecchi, I.; et al. Clinical-Pathological Characteristics of Renal Injuries Identify Different Clusters in Patients With Antiphospholipid Antibodies. Kidney Int. Rep. 2023, 8, 754–763. [Google Scholar] [CrossRef]
- Cervera, R.; Bucciarelli, S.; Plasín, M.A.; Gómez-Puerta, J.A.; Plaza, J.; Pons-Estel, G.; Shoenfeld, Y.; Ingelmo, M.; Espinos, G.; Catastrophic Antiphospholipid Syndrome (CAPS) Registry Project Group (European Forum On Antiphospholipid Antibodies). Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of a series of 280 patients from the “CAPS Registry”. J. Autoimmun. 2009, 32, 240–245. [Google Scholar] [CrossRef]
- Müller-Calleja, N.; Hollerbach, A.; Ritter, S.; Pedrosa, D.G.; Strand, D.; Graf, C.; Reinhardt, C.; Strand, S.; Poncelet, P.; Griffin, J.H.; et al. Tissue factor pathway inhibitor primes monocytes for antiphospholipid antibody-induced thrombosis. Blood 2019, 134, 1119–1131. [Google Scholar] [CrossRef]
- Romay-Penabad, Z.; Aguilar-Valenzuela, R.; Urbanus, R.T.; Derksen, R.H.W.M.; Pennings, M.T.T.; Papalardo, E.; Shilagard, T.; Vargas, G.; Hwang, Y.; de Groot, P.G.; et al. Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood 2011, 117, 1408–1414. [Google Scholar] [CrossRef]
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J.D. A Novel C5a Receptor-Tissue Factor Cross-Talk in Neutrophils Links Innate Immunity to Coagulation Pathways. J. Immunol. 2006, 177, 4794–4802. [Google Scholar] [CrossRef]
- Pierangeli, S.S.; Girardi, G.; Vega-Ostertag, M.; Liu, X.; Espinola, R.G.; Salmon, J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody–mediated thrombophilia. Arthritis Rheum. 2005, 52, 2120–2124. [Google Scholar] [CrossRef]
- Romay-Penabad, Z.; X, X.L.; Montiel-Manzano, G.; De Martínez, E.P.; Pierangeli, S.S. C5a Receptor-Deficient Mice Are Protected from Thrombophilia and Endothelial Cell Activation Induced by Some Antiphospholipid Antibodies. Ann. New York Acad. Sci. 2007, 1108, 554–566. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Braunstein, E.M.; Yuan, X.; Yu, J.; Alexander, A.; Chen, H.; Gavriilaki, E.; Alluri, R.; Streiff, M.B.; Petri, M.; et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 2020, 135, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Seshan, S.V.; Franzke, C.-W.; Redecha, P.; Monestier, M.; Mackman, N.; Girardi, G. Role of tissue factor in a mouse model of thrombotic microangiopathy induced by antiphospholipid antibodies. Blood 2009, 114, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; E Alpers, C.; Pippin, J.; Shankland, S.J.; Kurokawa, K.; Adler, S.; Morgan, B.P.; Johnson, R.J.; Couser, W.G. CD59 protects glomerular endothelial cells from immune-mediated thrombotic microangiopathy in rats. J. Am. Soc. Nephrol. 1998, 9, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.S.; Baelde, H.J.; Zandbergen, M.; Wilhelmus, S.; van Es, L.A.; de Fijter, J.W.; Bruijn, J.A.; Bajema, I.M.; Cohen, D. Complement Factor C4d Is a Common Denominator in Thrombotic Microangiopathy. J. Am. Soc. Nephrol. 2015, 26, 2239–2247. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, X.-W.; Sun, C.-Y.; Dai, M.; Yan, Y.-C.; Yang, C.-D. Association between anti-β2 glycoprotein I antibodies and renal glomerular C4d deposition in lupus nephritis patients with glomerular microthrombosis: a prospective study of 155 cases. Lupus 2010, 19, 1195–1203. [Google Scholar] [CrossRef]
- Canaud, G.; Bienaimé, F.; Tabarin, F.; Bataillon, G.; Seilhean, D.; Noël, L.-H.; Dragon-Durey, M.-A.; Snanoudj, R.; Friedlander, G.; Halbwachs-Mecarelli, L.; et al. Inhibition of the mTORC Pathway in the Antiphospholipid Syndrome. New Engl. J. Med. 2014, 371, 303–312. [Google Scholar] [CrossRef]
- Wilson, W.A.; Gharavi, A.E.; Koike, T.; Lockshin, M.D.; Branch, D.W.; Piette, J.-C.; Brey, R.; Derksen, R.; Harris, E.N.; Hughes, G.R.V.; et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: Report of an International workshop. Arthritis Rheum. 1999, 42, 1309–1311. [Google Scholar] [CrossRef]
- Ostuni, P.; Lazzarin, P.; Pengo, V.; Ruffatti, A.; Schiavon, F.; Gambari, P. Renal artery thrombosis and hypertension in a 13 year old girl with antiphospholipid syndrome. Ann. Rheum. Dis. 1990, 49, 184–187. [Google Scholar] [CrossRef]
- Tektonidou, M.G. Antiphospholipid Syndrome Nephropathy: From Pathogenesis to Treatment. Front. Immunol. 2018, 9, 1181. [Google Scholar] [CrossRef]
- Amigo, M.C.; Garcia-Torres, R.; Robles, M.; Bochicchio, T.; A Reyes, P. Renal involvement in primary antiphospholipid syndrome. J Rheumatol. 1992, 19, 1181–5. [Google Scholar]
- Ames, P.R.; Cianciaruso, B.; Bellizzi, V.; Balletta, M.; Lubrano, E.; Scarpa, R.; Brancaccio, V. Bilateral renal artery occlusion in a patient with primary antiphospholipid antibody syndrome: thrombosis, vasculitis or both? J. Rheumatol. 1992, 19, 1802–6. [Google Scholar] [PubMed]
- Sangle, S.R.; D'Cruz, D.P.; Jan, W.; Karim, M.Y.; A Khamashta, M.; Abbs, I.C.; Hughes, G.R.V. Renal artery stenosis in the antiphospholipid (Hughes) syndrome and hypertension. Ann. Rheum. Dis. 2003, 62, 999–1002. [Google Scholar] [CrossRef]
- O'Neill, W.C.; Bardelli, M.; Yevzlin, A.S. Imaging For Renovascular Disease. Semin. Nephrol. 2011, 31, 272–282. [Google Scholar] [CrossRef]
- Girometti, R.; Stocca, T.; Serena, E.; Granata, A.; Bertolotto, M. Impact of contrast-enhanced ultrasound in patients with renal function impairment. World J. Radiol. 2017, 9, 10–16. [Google Scholar] [CrossRef]
- Ko, W.-S.; Lim, P.-S.; Sung, Y.-P. Renal vein thrombosis as first clinical manifestation of the primary antiphospholipid syndrome. Nephrol. Dial. Transplant. 1995, 10, 1929–1931. [Google Scholar] [CrossRef]
- Morgan, R.; Feneley, R. Renal vein thrombosis caused by primary antiphospholipid syndrome. Br. J. Urol. 1994, 74, 807–808. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Brandao, L.; Geary, D.; Licht, C. Primary antiphospholipid syndrome presenting as renal vein thrombosis and membranous nephropathy. Pediatr. Nephrol. 2011, 26, 979–985. [Google Scholar] [CrossRef]
- Oliveira, C.; D`oliveira, I.; Bacchiega, A.; Klumb, E.; Albuquerque, E.; Souza, E.; Suassuna, J.; Ribeiro, F. Renal transplantation in lupus nephritis: a Brazilian cohort. Lupus 2011, 21, 570–574. [Google Scholar] [CrossRef]
- Glazer G, Francis I, Gross B, Amendola M Glazer GM, Francis IR, Gross BH, Amendola MA. Computed tomography of renal vein thrombosis. J Comput Assist Tomogr. 1984;8(2):288-93.
- Nochy, D.; Daugas, E.; Droz, D.; Beaufils, H.; Grünfeld, J.-P.; Piette, J.-C.; Bariety, J.; Hill, G. The Intrarenal Vascular Lesions Associated with Primary Antiphospholipid Syndrome. J. Am. Soc. Nephrol. 1999, 10, 507–518. [Google Scholar] [CrossRef]
- Tektonidou, M.; Sotsiou, F.; Moutsopoulos, H.M. Antiphospholipid syndrome (APS) nephropathy in catastrophic, primary, and systemic lupus erythematosus-related APS. J Rheumatol. 2008, 35, 1983–8. [Google Scholar]
- Daugas, E.; Nochy, D.; Huong, D.L.T.; Duhaut, P.; Beaufils, H.A.A.G.A.; Caudwell, V.A.A.; Bariety, J.; Piette, J.-C.; Hill, G. Antiphospholipid Syndrome Nephropathy in Systemic Lupus Erythematosus. J. Am. Soc. Nephrol. 2002, 13, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Tektonidou, M.G.; Sotsiou, F.; Nakopoulou, L.; Vlachoyiannopoulos, P.G.; Moutsopoulos, H.M. Antiphospholipid syndrome nephropathy in patients with systemic lupus erythematosus and antiphospholipid antibodies: Prevalence, clinical associations, and long-term outcome. Arthritis Rheum. 2004, 50, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Mejía-Vilet, J.M.; Córdova-Sánchez, B.M.; O Uribe-Uribe, N.; Correa-Rotter, R.; E Morales-Buenrostro, L. Prognostic significance of renal vascular pathology in lupus nephritis. Lupus 2017, 26, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Strufaldi, F.L.; Neves, P.D.M.d.M.M.; Dias, C.B.; Yu, L.; Woronik, V.; Cavalcante, L.B.; Malheiros, D.M.A.C.; Jorge, L.B. Renal thrombotic microangiopathy associated to worse renal prognosis in Lupus Nephritis. J. Nephrol. 2021, 34, 1–10. [Google Scholar] [CrossRef]
- Moroni, G.; Ventura, D.; Riva, P.; Panzeri, P.; Quaglini, S.; Banfi, G.; Simonini, P.; Bader, R.; Meroni, P.L.; Ponticelli, C. Antiphospholipid antibodies are associated with an increased risk for chronic renal insufficiency in patients with lupus nephritis. Am. J. Kidney Dis. 2004, 43, 28–36. [Google Scholar] [CrossRef]
- Barbhaiya, M.; Taghavi, M.; Zuily, S.; Domingues, V.; Chock, E.Y.; Tektonidou, M.G.; Erkan, D.; Seshan, S.V.; New APS Classification Criteria Steering Committee and APS ACTION Collaborators. Efforts to Better Characterize “Antiphospholipid Antibody Nephropathy” for the 2023 ACR/EULAR Antiphospholipid Syndrome Classification Criteria: Renal Pathology Subcommittee Report. J. Rheumatol. 2023, 51, 150–159. [Google Scholar] [CrossRef]
- Liang, P.; Yuan, G.; Li, S.; Peng, Y.; Xu, C.; Benkert, T.; Hu, D.; Han, M.; Li, Z. Noninvasive Assessment of the Renal Function, Oxford Classification and Prognostic Risk Stratification of IgAN by Using Intravoxel Incoherent Motion Diffusion-Weighted Imaging and Blood Oxygenation Level-Dependent MRI. J. Magn. Reson. Imaging 2022, 58, 879–891. [Google Scholar] [CrossRef]
- Fakhouri, F.; Noël, L.-H.; Zuber, J.; Beaufils, H.; Martinez, F.; Lebon, P.; Papo, T.; Chauveau, D.; Bletry, O.; Grünfeld, J.-P.; et al. The expanding spectrum of renal diseases associated with antiphospholipid syndrome. Am. J. Kidney Dis. 2003, 41, 1205–1211. [Google Scholar] [CrossRef]
- Stone, J.H.; Amend, W.J.; Criswell, L.A. Antiphospholipid antibody syndrome in renal transplantation: Occurrence of clinical events in 96 consecutive patients with systemic lupus erythematosus. Am. J. Kidney Dis. 1999, 34, 1040–1047. [Google Scholar] [CrossRef]
- Canaud, G.; Bienaimé, F.; Noël, L.; Royal, V.; Alyanakian, M.; Dautzenberg, M.; Rabant, M.; Posson, J.; Thervet, E.; Anglicheau, D.; et al. Severe Vascular Lesions and Poor Functional Outcome in Kidney Transplant Recipients with Lupus Anticoagulant Antibodies. Am. J. Transplant. 2010, 10, 2051–2060. [Google Scholar] [CrossRef]
- Gauthier, M.; Canoui-Poitrine, F.; Guéry, E.; Desvaux, D.; Hue, S.; Canaud, G.; Stehle, T.; Lang, P.; Kofman, T.; Grimbert, P.; et al. Anticardiolipin antibodies and 12-month graft function in kidney transplant recipients: a prognosis cohort survey. Nephrol. Dial. Transplant. 2018, 33, 709–716. [Google Scholar] [CrossRef]
- Vaidya, S. Ten-yr renal allograft survival of patients with antiphospholipid antibody syndrome. Clin. Transplant. 2012, 26, 853–856. [Google Scholar] [CrossRef]
- Wagenknecht, D.; Fastenau, D.; Torry, R.; Carter, C.; Haag, B.; McIntyre, J. Antiphospholipid antibodies are a risk factor for early renal allograft failure: isolation of antiphospholipid antibodies from a thrombosed renal allograft. Transplant. Proc. 1999, 31, 285–288. [Google Scholar] [CrossRef]
- Furmańczyk-Zawiska, A.; Bułło-Piontecka, B.; Komorniczak, M.; Dębska-Ślizień, A.; Augustyniak-Bartosik, H.; Durlik, M. Antiphospholipid Syndrome in Renal Allograft Recipients—A Long-Term Multicenter Analysis. J. Clin. Med. 2023, 12, 667. [Google Scholar] [CrossRef]
- Serrano, M.; Martínez-Flores, J.A.; Pérez, D.; García, F.; Cabrera, O.; Pleguezuelo, D.; Paz-Artal, E.; Morales, J.M.; González, E.; Serrano, A. β 2 -Glycoprotein I/IgA Immune Complexes: A Marker to Predict Thrombosis After Renal Transplantation in Patients With Antiphospholipid Antibodies. Circulation 2017, 135, 1922–1934. [Google Scholar] [CrossRef]
- Hoxha, A.; Ruffatti, A.; Mattia, E.; Meneghel, L.; Tonello, M.; Salvan, E.; Pengo, V.; Punzi, L. Relationship between antiphosphatidylserine/prothrombin and conventional antiphospholipid antibodies in primary antiphospholipid syndrome. cclm 2015, 53, 1265–1270. [Google Scholar] [CrossRef]
- Hoxha, A.; Ruffatti, A.; Tonello, M.; Bontadi, A.; Salvan, E.; Banzato, A.; Pengo, V.; Punzi, L. Antiphosphatidylserine/prothrombin antibodies in primary antiphospholipid syndrome. Lupus 2012, 21, 787–789. [Google Scholar] [CrossRef]
- Sangle, S.R.; D'Cruz, D.P.; Abbs, I.C.; Khamashta, M.A.; Hughes, G.R.V. Renal artery stenosis in hypertensive patients with antiphospholipid (Hughes) syndrome: outcome following anticoagulation. Rheumatology 2005, 44, 372–377. [Google Scholar] [CrossRef]
- Tektonidou, M.G.; Andreoli, L.; Limper, M.; Amoura, Z.; Cervera, R.; Costedoat-Chalumeau, N.; Cuadrado, M.J.; Dörner, T.; Ferrer-Oliveras, R.; Hambly, K.; et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann. Rheum. Dis. 2019, 78, 1296–1304. [Google Scholar] [CrossRef]
- Tektonidou, M. Identification and treatment of APS renal involvement. Lupus 2014, 23, 1276–1278. [Google Scholar] [CrossRef]
- Korkmaz, C.; Kabukcuoglu, S.; Isiksoy, S.; Yalçin, A.U. Renal involvement in primary antiphospholipid syndrome and its response to immunosuppressive therapy. Lupus 2003, 12, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Pengo, V.; Denas, G.; Zoppellaro, G.; Jose, S.P.; Hoxha, A.; Ruffatti, A.; Andreoli, L.; Tincani, A.; Cenci, C.; Prisco, D.; et al. Rivaroxaban vs warfarin in high-risk patients with antiphospholipid syndrome. Blood 2018, 132, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Pengo, V.; Hoxha, A.; Andreoli, L.; Tincani, A.; Silvestri, E.; Prisco, D.; Fierro, T.; Gresele, P.; Cafolla, A.; De Micheli, V.; et al. Trial of Rivaroxaban in AntiPhospholipid Syndrome (TRAPS): Two-year outcomes after the study closure. J. Thromb. Haemost. 2020, 19, 531–535. [Google Scholar] [CrossRef]
- Khairani, C.D.; Bejjani, A.; Piazza, G.; Jimenez, D.; Monreal, M.; Chatterjee, S.; Pengo, V.; Woller, S.C.; Cortes-Hernandez, J.; Connors, J.M.; et al. Direct Oral Anticoagulants vs Vitamin K Antagonists in Patients With Antiphospholipid Syndromes: Meta-Analysis of Randomized Trials. Circ. 2022, 81, 16–30. [Google Scholar] [CrossRef]
- Kronbichler, A.; Brezina, B.; Quintana, L.F.; Jayne, D.R. Efficacy of plasma exchange and immunoadsorption in systemic lupus erythematosus and antiphospholipid syndrome: A systematic review. Autoimmun. Rev. 2016, 15, 38–49. [Google Scholar] [CrossRef]
- Ruffatti, A.; De Silvestro, G.; Marson, P.; Tonello, M.; Calligaro, A.; Favaro, M.; Del Ross, T.; Hoxha, A.; Mattia, E.; Pengo, V. Catastrophic antiphospholipid syndrome: Lessons from 14 cases successfully treated in a single center. A narrative report. J. Autoimmun. 2018, 93, 124–130. [Google Scholar] [CrossRef]
- Erkan, D.; Vega, J.; Ramón, G.; Kozora, E.; Lockshin, M.D. A pilot open-label phase II trial of rituximab for non-criteria manifestations of antiphospholipid syndrome. Arthritis Rheum. 2012, 65, 464–471. [Google Scholar] [CrossRef]
- Sciascia, S.; Naretto, C.; Rossi, D.; Bazzan, M.; Roccatello, D. Treatment-induced downregulation of antiphospholipid antibodies: effect of rituximab alone on clinical and laboratory features of antiphospholipid syndrome. Lupus 2011, 20, 1106–1108. [Google Scholar] [CrossRef]
- Tsagalis, G.; Psimenou, E.; Nakopoulou, L.; Laggouranis, A. Effective treatment of antiphospholipid syndrome with plasmapheresis and rituximab. Hippokratia 2010, 14, 215–6. [Google Scholar]
- Yazici, A.; Yazirli, B.; Erkan, D. Belimumab in primary antiphospholipid syndrome. Lupus 2017, 26, 1123–1124. [Google Scholar] [CrossRef]
- Sciascia, S.; Radin, M.; Cecchi, I.; Barinotti, A.; Rubini, E.; Rossi, D.; Fenoglio, R.; Vaccarino, A.; Menegatti, E.; Roccatello, D. Open-label, prospective, phase II descriptive pilot trial of belimumab therapy for refractory and/or non-criteria manifestations of antiphospholipid syndrome: study protocol. Clin. Exp. Rheumatol. 2022, 41, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Kronbichler, A.; Frank, R.; Kirschfink, M.; Szilágyi, Á.; Csuka, D.; Prohászka, Z.; Schratzberger, P.; Lhotta, K.; Mayer, G. Efficacy of Eculizumab in a Patient With Immunoadsorption-Dependent Catastrophic Antiphospholipid Syndrome: a case report. Medicine 2014, 93, e143–e143. [Google Scholar] [CrossRef]
- Lonze, B.E.; Zachary, A.A.; Magro, C.M.; Desai, N.M.; Orandi, B.J.; Dagher, N.N.; Singer, A.L.; Carter-Monroe, N.; Nazarian, S.M.; Segev, D.L.; et al. Eculizumab Prevents Recurrent Antiphospholipid Antibody Syndrome and Enables Successful Renal Transplantation. Am. J. Transp. 2014, 14, 459–465. [Google Scholar] [CrossRef]
- Ávila, A.; Gavela, E.; Sancho, A. Thrombotic Microangiopathy After Kidney Transplantation: An Underdiagnosed and Potentially Reversible Entity. Front. Med. 2021, 8. [Google Scholar] [CrossRef]
- Sciascia, S.; Radin, M.; Yazdany, J.; Tektonidou, M.; Cecchi, I.; Roccatello, D.; Dall’era, M. Expanding the therapeutic options for renal involvement in lupus: eculizumab, available evidence. Rheumatol. Int. 2017, 37, 1249–1255. [Google Scholar] [CrossRef]
- López-Benjume, B.; Rodríguez-Pintó, I.; Amigo, M.C.; Erkan, D.; Shoenfeld, Y.; Cervera, R.; Espinosa, G. Eculizumab use in catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis from the “CAPS Registry”. Autoimmun. Rev. 2022, 21, 103055. [Google Scholar] [CrossRef]
- Grenn, R.C.; Yalavarthi, S.; A Gandhi, A.; Kazzaz, N.M.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Cabral, A.R.; McCune, W.J.; Bockenstedt, P.L.; Knight, J.S. Endothelial progenitor dysfunction associates with a type I interferon signature in primary antiphospholipid syndrome. Ann. Rheum. Dis. 2017, 76, 450–457. [Google Scholar] [CrossRef]
- Cecchi, I.; Radin, M.; Barinotti, A.; Foddai, S.G.; Menegatti, E.; Roccatello, D.; Suárez, A.; Sciascia, S.; Rodríguez-Carrio, J. Type I interferon pathway activation across the antiphospholipid syndrome spectrum: associations with disease subsets and systemic antiphospholipid syndrome presentation. Front. Immunol. 2024, 15, 1351446. [Google Scholar] [CrossRef]
- Verrou, K.-M.; Sfikakis, P.P.; Tektonidou, M.G. Whole blood transcriptome identifies interferon-regulated genes as key drivers in thrombotic primary antiphospholipid syndrome. J. Autoimmun. 2022, 134, 102978. [Google Scholar] [CrossRef]
- Tang, K.-T.; Chen, Y.-S.; Chen, T.-T.; Chao, Y.-H.; Kung, S.-P.; Chen, D.-Y.; Lin, C.-C. Inhibiting Tyrosine Kinase 2 Ameliorates Antiphospholipid Syndrome Nephropathy. Mediat. Inflamm. 2024, 2024, 5568822. [Google Scholar] [CrossRef]
- Heerspink HJL, Stef’ansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, Mann JFE, McMurray JJV, Lindberg M, Rossing P,; DAPA-CKD Trial Committees and Investigators. Dapagliflozin in Patients with Chronic Kidney Disease. N Engl J Med. 2020; 8;383:1436-1446. [CrossRef]
- The EMPA-KIDNEY Collaborative Group; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. New Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Beck, L.H.; Ayoub, I.; Caster, D.; Choi, M.J.; Cobb, J.; Geetha, D.; Rheault, M.N.; Wadhwani, S.; Yau, T.; Whittier, W.L. KDOQI US Commentary on the 2021 KDIGO Clinical Practice Guideline for the Management of Glomerular Diseases. Am. J. Kidney Dis. 2023, 82, 121–175. [Google Scholar] [CrossRef]
- Floege, J.; Floege, J.; Jayne, D.R.; Jayne, D.R.; Sanders, J.-S.F.; Sanders, J.-S.F.; Tesar, V.; Tesar, V.; Balk, E.M.; Balk, E.M.; et al. Executive summary of the KDIGO 2024 Clinical Practice Guideline for the Management of ANCA–Associated Vasculitis. Kidney Int. 2024, 105, 447–449. [Google Scholar] [CrossRef]
- Middeldorp, S.; Nieuwlaat, R.; Kreuziger, L.B.; Coppens, M.; Houghton, D.; James, A.H.; Lang, E.; Moll, S.; Myers, T.; Bhatt, M.; et al. American Society of Hematology 2023 guidelines for management of venous thromboembolism: thrombophilia testing. Blood Adv. 2023, 7, 7101–7138. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, A.; Gao, J.; Zou, M.; Du, J.; Wu, P.-Y.; Zhang, J.; Mao, Y.; Song, Y.; Chen, M. Diffusion-weighted, intravoxel incoherent motion, and diffusion kurtosis tensor MR imaging in chronic kidney diseases: Correlations with histology. Magn. Reson. Imaging 2024, 106, 1–7. [Google Scholar] [CrossRef]
- Sułkowska, K.; Palczewski, P.; Furmańczyk-Zawiska, A.; Perkowska-Ptasińska, A.; Wójcik, D.; Szeszkowski, W.; Durlik, M.; Gołębiowski, M.; Małkowski, P. Diffusion Weighted Magnetic Resonance Imaging in the Assessment of Renal Function and Parenchymal Changes in Chronic Kidney Disease: A Preliminary Study. Ann. Transplant. 2020, 25, e920232–e920232. [Google Scholar] [CrossRef]
- Tektonidou, M.G.; Verrou, K.-M.; Gakiopoulou, H.; Manoloukos, M.; Lembessis, P.; Hatzis, P.; Sfikakis, P.P. Kidney whole-transcriptome profiling in primary antiphospholipid syndrome reveals complement, interferons and NETs-related gene expression. Rheumatology 2024, 63, 3184–3190. [Google Scholar] [CrossRef]
Figure 1.
Potential pathogenesis of antiphospholipid antibodies nephropathy and possible pathway inhibitors. A) Kidney vasculature. B) The pathophysiology of APS is explained by the -"two-hits hypothesis,"- where aPL, particularly anti-β2GPI, acts as the initial catalyst in creating a prothrombotic state. However, this alone is insufficient to cause thrombosis. A second hit, such as stasis or inflammation, results in endothelial cell damage that disrupts natural anticoagulant systems. The endothelial cell dysfunction mediated by aPL activates endothelial cells, platelets, monocytes, and neutrophils promoting the release of neutrophil extracellular traps and tissue factor expression as well as complement cascade activation. C) The complement-mediated tissue factor plays a critical role in the pathogenesis of thrombotic microangiopathy in APS; thus, treatment with C5a inhibitors, alongside anticoagulation and/or anti-platelet therapies, may reduce kidney injury. D) The activation of neutrophils by aPL leads to the release of neutrophil extracellular traps and tissue factor expression, fostering thrombus development and vascular injury. Therefore, anticoagulation and/or anti-platelet therapy may sufficiently address vascular injury. Sometimes, treatment with immunosuppressants, such as rituximab, belimumab, hydroxychloroquine, or statins that inhibit tissue factor expression, could be beneficial. E) The aPL antibodies interact with endothelial cells via the mTOR pathway, whereby the activation of the mTOR complex stimulates the growth and proliferation of endothelial cells, contributing to the chronic vasculopathy seen in chronic APL-N lesions. Consequently, treatment with mTOR inhibitors combined with anticoagulation and/or anti-platelet therapy may help alleviate kidney injury. APS: antiphospholipid syndrome; aPL: antiphospholipid antibodies; mammalian target of rapamycin: mTOR. Created with https://BioRender.com.
Figure 1.
Potential pathogenesis of antiphospholipid antibodies nephropathy and possible pathway inhibitors. A) Kidney vasculature. B) The pathophysiology of APS is explained by the -"two-hits hypothesis,"- where aPL, particularly anti-β2GPI, acts as the initial catalyst in creating a prothrombotic state. However, this alone is insufficient to cause thrombosis. A second hit, such as stasis or inflammation, results in endothelial cell damage that disrupts natural anticoagulant systems. The endothelial cell dysfunction mediated by aPL activates endothelial cells, platelets, monocytes, and neutrophils promoting the release of neutrophil extracellular traps and tissue factor expression as well as complement cascade activation. C) The complement-mediated tissue factor plays a critical role in the pathogenesis of thrombotic microangiopathy in APS; thus, treatment with C5a inhibitors, alongside anticoagulation and/or anti-platelet therapies, may reduce kidney injury. D) The activation of neutrophils by aPL leads to the release of neutrophil extracellular traps and tissue factor expression, fostering thrombus development and vascular injury. Therefore, anticoagulation and/or anti-platelet therapy may sufficiently address vascular injury. Sometimes, treatment with immunosuppressants, such as rituximab, belimumab, hydroxychloroquine, or statins that inhibit tissue factor expression, could be beneficial. E) The aPL antibodies interact with endothelial cells via the mTOR pathway, whereby the activation of the mTOR complex stimulates the growth and proliferation of endothelial cells, contributing to the chronic vasculopathy seen in chronic APL-N lesions. Consequently, treatment with mTOR inhibitors combined with anticoagulation and/or anti-platelet therapy may help alleviate kidney injury. APS: antiphospholipid syndrome; aPL: antiphospholipid antibodies; mammalian target of rapamycin: mTOR. Created with https://BioRender.com.
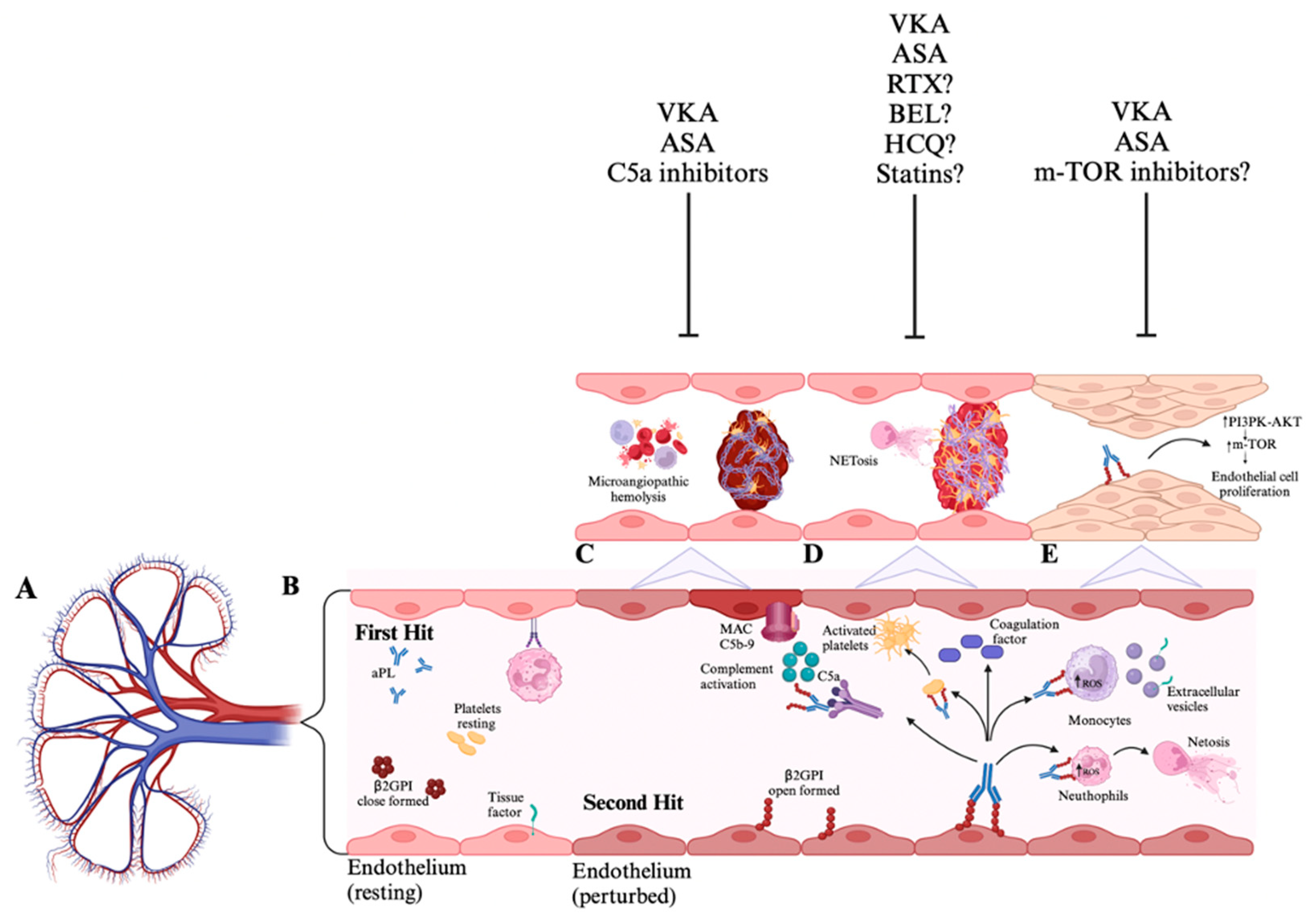
Figure 2.
Involvement of kidney vasculature showing arterial, vein thrombosis, and microvascular lesions characterized by glomerular and arteriolar thrombosis. Created with https://BioRender.com.
Figure 2.
Involvement of kidney vasculature showing arterial, vein thrombosis, and microvascular lesions characterized by glomerular and arteriolar thrombosis. Created with https://BioRender.com.
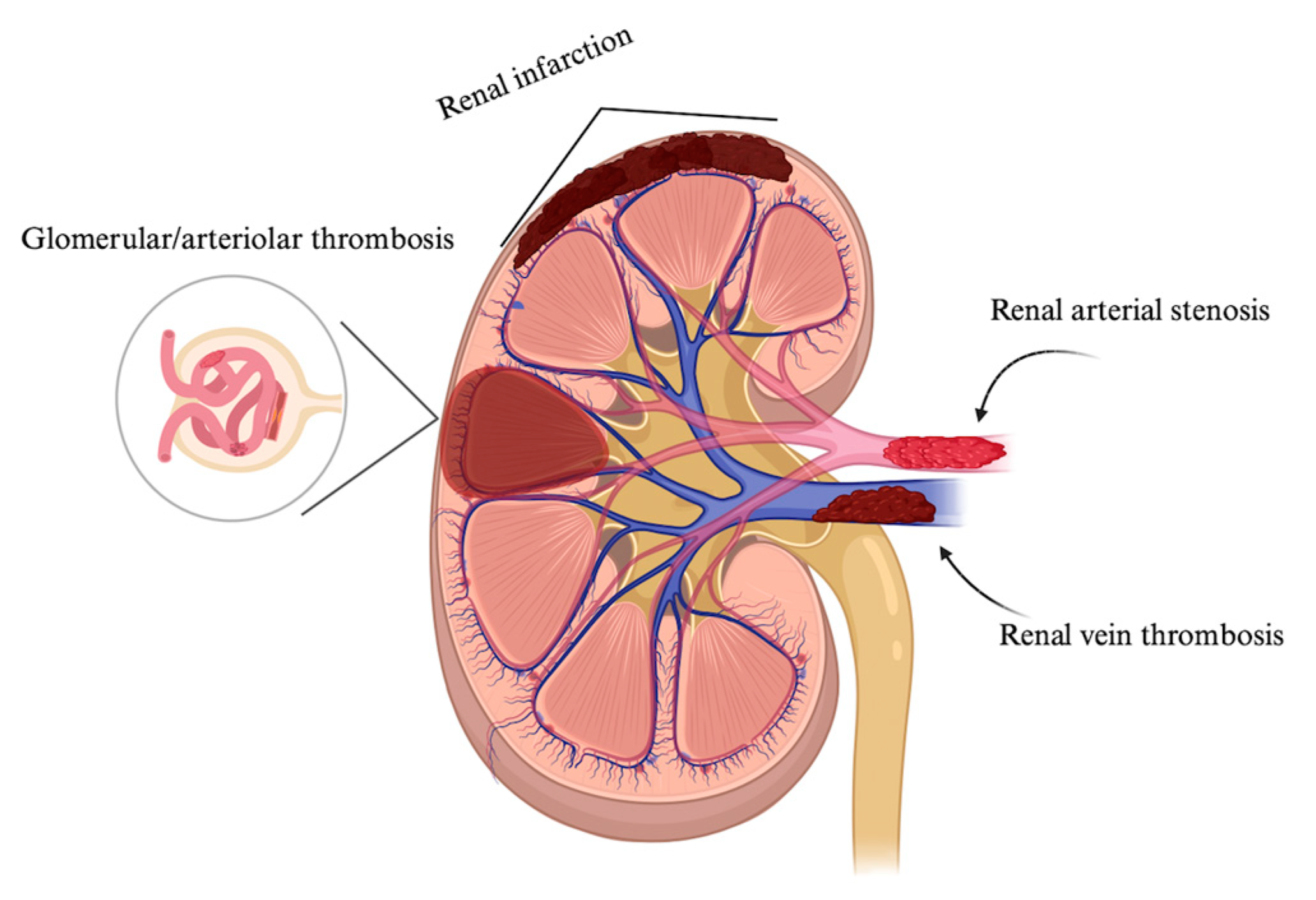
Figure 3.
Antiphospholipid antibodies nephropathy histology. A) (Masson’s Trichrome stain 400X, scale bar 10 μm) The glomerular loops appear large and fill the urinary space. In some areas, the glomerular basement membranes have a fluffy appearance. Fragmented red blood cells are present in the lumen. The mesangial spaces appear expanded with pale-staining material (mesangiolysis). B) (Masson’s Trichrome stain 200X, scale bar 10 μm) Tubulointerstitial compartment with sclerotic lesions, interstitial fibrosis, and tubular atrophy.
Figure 3.
Antiphospholipid antibodies nephropathy histology. A) (Masson’s Trichrome stain 400X, scale bar 10 μm) The glomerular loops appear large and fill the urinary space. In some areas, the glomerular basement membranes have a fluffy appearance. Fragmented red blood cells are present in the lumen. The mesangial spaces appear expanded with pale-staining material (mesangiolysis). B) (Masson’s Trichrome stain 200X, scale bar 10 μm) Tubulointerstitial compartment with sclerotic lesions, interstitial fibrosis, and tubular atrophy.
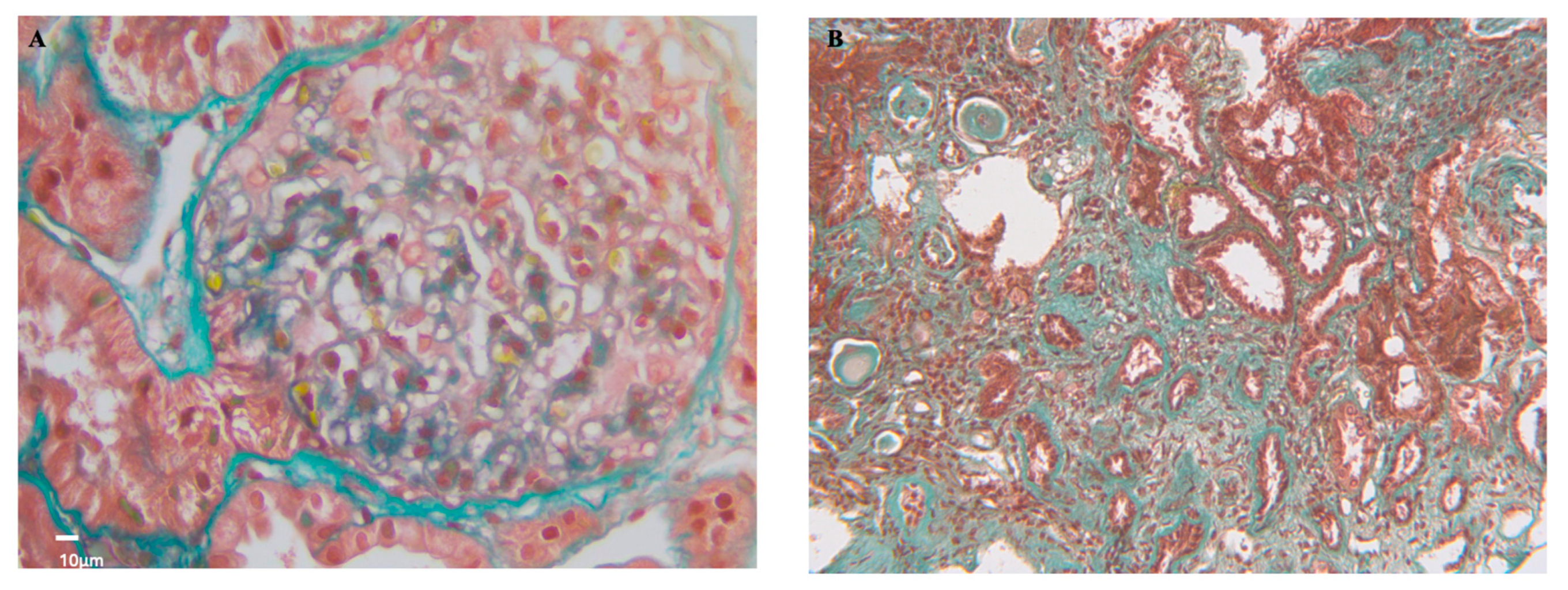
Figure 4.
Management algorithm of kidney damage in antiphospholipid syndrome. * New onset/refractory hypertension, glomerular proteinuria/hematuria, acute kidney failure, chronic or end-stage-renal-disease of unknown origin, history of SLE. ^ Medium-high titer anticardiolipin antibodies and/or anti-β2glycoprotein I IgG/IgM isotype and/or lupus anticoagulant confirmed ≥12 weeks apart. ^^ Pulmonary edema, uncontrolled hypertension, ischemic nephropathy, progressive renal failure. # allograft, acute kidney injury, single kidney. § Unexplained persistent: (a) new-onset/deterioration hypertension; (b) proteinuria≥0.5 gm in 24-hour urine specimen or protein: creatinine ratio ≥0.5 mg/mg (50 mg/mmoles); (c) acute renal failure; or (d) glomerular microscopic hematuria. ** INR 2-3 if associated with venous thrombosis, INR 3-4 if associated with arterial thrombosis. Created with https://BioRender.com.
Figure 4.
Management algorithm of kidney damage in antiphospholipid syndrome. * New onset/refractory hypertension, glomerular proteinuria/hematuria, acute kidney failure, chronic or end-stage-renal-disease of unknown origin, history of SLE. ^ Medium-high titer anticardiolipin antibodies and/or anti-β2glycoprotein I IgG/IgM isotype and/or lupus anticoagulant confirmed ≥12 weeks apart. ^^ Pulmonary edema, uncontrolled hypertension, ischemic nephropathy, progressive renal failure. # allograft, acute kidney injury, single kidney. § Unexplained persistent: (a) new-onset/deterioration hypertension; (b) proteinuria≥0.5 gm in 24-hour urine specimen or protein: creatinine ratio ≥0.5 mg/mg (50 mg/mmoles); (c) acute renal failure; or (d) glomerular microscopic hematuria. ** INR 2-3 if associated with venous thrombosis, INR 3-4 if associated with arterial thrombosis. Created with https://BioRender.com.
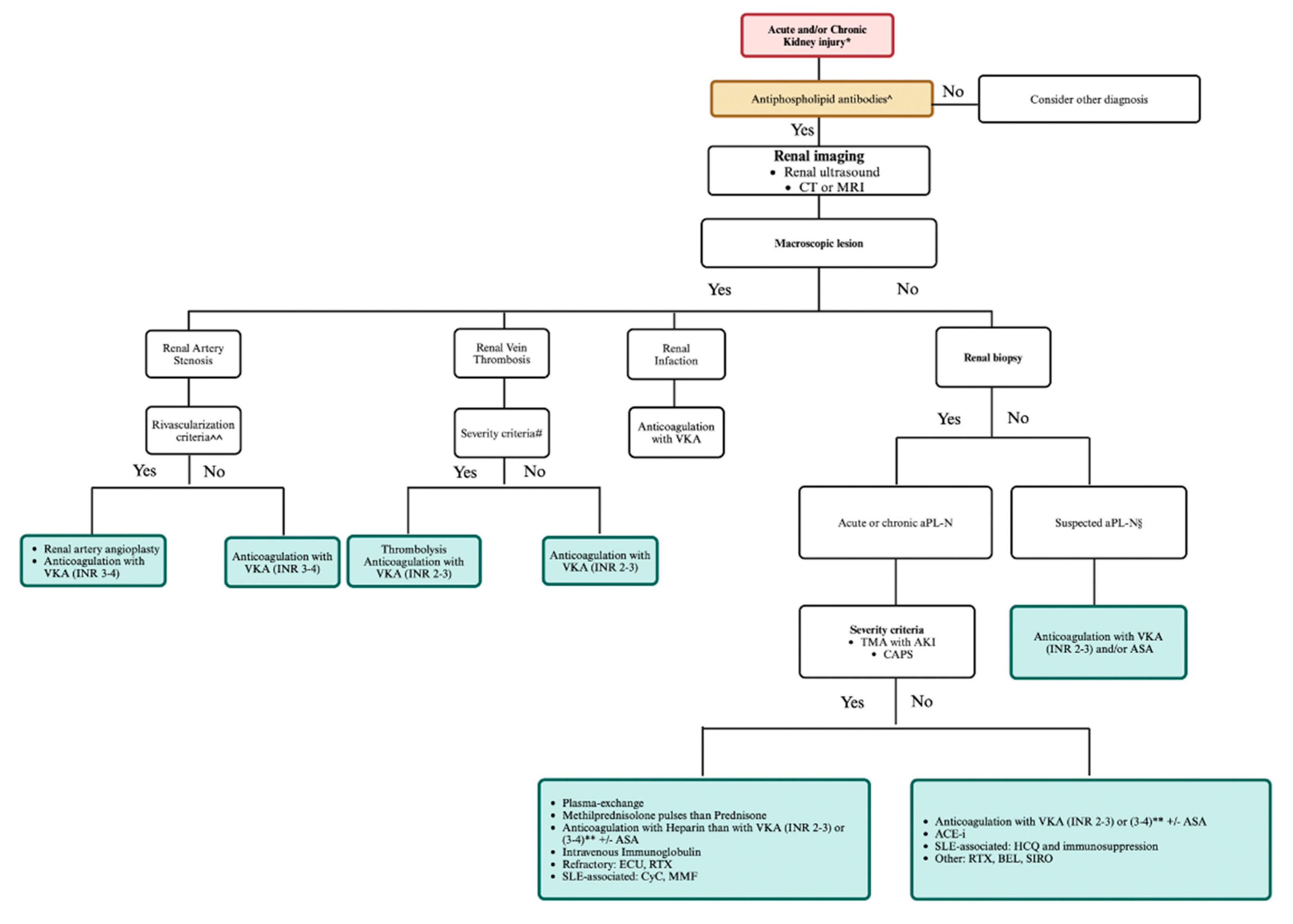

Table 1.
2023 ACR/EULAR definition for antiphospholipid syndrome.
| Entry criteria | |||
|---|---|---|---|
| ≥ 1 clinical criterion plus ≥ 1 aPL test | |||
| (aPL positive within three years from the clinical criterion) | |||
| Clinical Domain | Score | ||
| Domain 1 | Venous Thromboembolism | ||
| with VTE high-risk profile | 1 | ||
| without VTE high-risk profile | 3 | ||
| Domain 2 | Arterial Thrombosis | ||
| with AT high-risk profile | 2 | ||
| without AT high-risk profile | 4 | ||
| Domain 3 | Microvascular | ||
| Suspected* | 2 | ||
| Livedo racemose | |||
| Livedoid vasculopathy | |||
| Acute/chronic aPL-N | |||
| Pulmonary hemorrhage | |||
| Established § | 5 | ||
| Livedoid vasculopathy | |||
| Acute/chronic aPL-N | |||
| Pulmonary hemorrhage | |||
| Myocardial disease | |||
| Adrenal hemorrhage | |||
| Domain 4 | Obstetric | ||
| ≥ 3 consecutive pre-fetal (<10WG) and/or early fetal (< 16WG) | 1 | ||
| Fetal death (≥ 16 WG) without PEC/PI | 1 | ||
| Severe PEC or Severe PI (<34 WG) | 3 | ||
| Severe PEC and Severe PI (<34 WG) | 4 | ||
| Domain 5 | Cardiac Valve | ||
| Thickening | 2 | ||
| Vegetation | 4 | ||
| Domain 6 | Hematology | ||
| Thrombocytopenia (20-130x109/L) | 2 | ||
| Laboratory Domain | |||
| Domain 7 | Lupus anticoagulant test | ||
| One-time positive | 1 | ||
| Persistent positive | 5 | ||
| Domain 8 | Anti-cardiolipin and anti-β2-glycoprotein I ^ | ||
| Moderate-high positive IgM aCL and/or anti-β2GPI | 1 | ||
| Moderate positive IgG aCL and/or anti-β2GPI | 4 | ||
| High positive IgG aCL or anti-β2GPI | 5 | ||
| High positive IgG aCL and anti-β-2GPI | 7 | ||
* Diagnosed by exam and/or laboratory test and/or imaging. § Diagnosed by imaging and/or pathology. ^ Solid phase assay. aPL: antiphospholipid antibodies; VTE: venous thromboembolism; AT: arterial thrombosis; aPL-N: antiphospholipid antibodies-nephropathy; WG: week of gestation; PEC: pre-eclampsia; PI: placental insufficiency; aCL:anti-cardiolipin antibodies; anti-β2GPI: anti-β2 glycoprotein I antibodies; IgM: immunoglobulin M; IgG: immunoglobulin G.
Table 2.
2023 ACR/EULAR definition for Antiphospholipid Antibody Nephropathy.
| aPL-N | Definition |
|---|---|
| Suspected aPL-N * | New-onset hypertension or deterioration of previously well-controlled hypertension |
| Proteinuria≥0.5 gm in 24-hour urine specimen or protein: creatinine ratio ≥0.5 mg/mg (50 mg/mmoles) | |
| Acute renal failure | |
| Glomerular microscopic hematuria | |
| Established aPL-N^ | Acute renal vascular or glomerular thrombotic microangiopathy |
| Chronic renal vascular or glomerular lesions |
aPL-N: antiphospholipid antibody nephropathy; gm: gramme; mg: milligrams; mmloes: micromoles. * By physical examination or laboratory tests. ^ By pathology.
Table 3.
2023 ACR/EULAR histology definition for Antiphospholipid Antibody Nephropathy.
| Type of lesion | Definition | ||||||
| Acute aPL-N | Fibrin thrombi in arterioles or glomeruli without inflammatory cells or immune complexes | ||||||
| Chronic aPL-N | Organized arterial or arteriolar microthrombi with or without recanalization | ||||||
| Fibrous and fibrocellular arterial or arteriolar occlusions | |||||||
| Cortical atrophy with or without thyroidization | |||||||
| Fibrous intimal hyperplasia | |||||||
| Organized glomerular thrombi | |||||||
aPL-N: antiphospholipid antibodies-nephropathy.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



