Submitted:
17 March 2025
Posted:
18 March 2025
You are already at the latest version
Abstract
Trichloroethylene (TCE) is an organic solvent used in industrial applications worldwide. Despite a recently proposed ban in the US, its usage over the last century produced widespread and long-lasting environmental contamination. TCE has been linked to multiple adverse health outcomes, with evidence growing that is a Parkinson’s disease (PD) risk factor. Exposure to TCE and other solvents in contaminated water at Camp Lejeune, North Carolina is associated with 70% increased PD risk in US veterans who lived on the base. However, little is known about PD risk from TCE in civilian populations, particularly in children who were exposed in early life. Importantly, the developing brain is highly susceptible to toxicant exposure, and previous work shows that exposure to TCE can result in neurodevelopmental deficits that manifest in adolescence and into adulthood. Given the number of individuals who have yet to “age” into idiopathic PD, understanding the mechanisms that underlie neurodegeneration from early life exposure to TCE could help to develop early interventions in at-risk populations. To further examine this, we review the existing literature on environmental exposures to PD-related toxicants during early-life and their long-term consequences. In addition, we discuss the potential for TCE-induced neurotoxic mechanisms to prime the brain for PD risk. Finally, we highlight the need for future studies to evaluate the impact of early-life TCE exposure across the lifespan.
Keywords:
trichloroethylene
; Parkinson’s disease
; developmental exposure
; neurotoxicity
Introduction
Trichloroethylene is a globally used organic solvent with numerous industrial uses, particularly in machine degreasing, as a chemical feedstock, and previously as a common dry-cleaning solvent [1,2]. Due to the widespread use of TCE, vast environmental contamination in the air, soil, and water surrounding industrial sites remains a significant problem within the US and around the world [3]. TCE represents an insidious exposure hazard to humans as it is environmentally persistent, odorless at low concentrations, and poses a vapor intrusion risk that contaminates indoor air in buildings and homes [4]. While water contamination remains a concern in many countries, inhalation is the most common route of exposure. Thus, environmental exposure to TCE may go unknown or unmonitored for years [3,5].
TCE was recently proposed to be prohibited for use by the Environmental Protection Agency (EPA) because of its classification as a human health hazard, mostly due to its carcinogenicity [6]. Epidemiological studies have demonstrated that occupational exposure to TCE increases the risk of kidney cancer, while other studies have shown a potential increased risk of liver cancer, non-Hodgkin lymphoma, and male breast cancer [7,8,9,10]. Experimental studies demonstrated that exposure to TCE increases the incidence of kidney, liver, and testicular tumors in rodents [11]. These findings from epidemiological and experimental studies provided sufficient evidence for the International Agency for Research on Cancer (IARC) to deem that TCE is a Group 1 carcinogen based on evidence in both animals and humans (particularly kidney cancer in humans; [12].
In addition to its carcinogenic nature, TCE also has direct neurotoxic properties and has been associated with increased risk for Parkinson’s disease (PD) [4,13,14,15,16]. Previously, most data showing PD risk from TCE exposure resulted from occupational exposures. However, recent epidemiological studies show that veterans who lived at Camp Lejeune, a United States military base that reported contamination in drinking water with volatile organic compounds (VOCs; TCE and related solvent tetrachloroethylene (perchloroethylene [PCE]) from 1953 to 1987 had 70% increased risk of developing PD compared to veterans who were stationed at Camp Pendleton, CA, a military camp that did not have reported TCE contamination during the same timeframe (odds ratio 1.70; 95% CI, 1.39-2.07) [17]. In a follow-up study of this same cohort, veterans who were exposed to TCE had higher hazard ratios (HR) for symptoms typically related in later stages of PD, including fall (HR 2.64, 95% CI 0.97–7.21), psychosis (HR 2.19, 95% CI 0.99–4.83), and fracture (HR 2.44, 95% CI 0.91–6.55) than in non-exposed veterans, suggesting that exposure to TCE might contribute to faster onset of PD progression or more severe symptom presentation [18]. To further support the biological basis for PD risk from TCE, experimental studies in rodents demonstrate that exposure to TCE causes the significant and selective loss of dopaminergic neurons in the substantia nigra (SN) [16,19,20], as well as other hallmark PD pathology such as alpha-synuclein (αSyn) accumulation, neuroinflammation, endolysosomal dysfunction, and oxidative damage [21,22,23].
To date, both epidemiological and experimental data for PD risk from TCE exposure have focused on adult populations with high exposure levels (e.g., military veterans or adult animal studies); however, given the widespread TCE contamination, exposure may happen at any point across the lifespan. The long prodromal period of PD, which can begin and estimated 10-20 years prior to clinical diagnosis, suggests that the pathology and degenerative processes caused by environmental risk factors are initiated decades before diagnosis [24]. As such, TCE exposures temporally separated from disease onset may contribute to PD risk may be to PD via mechanisms of latent or silent neurotoxicity, where the triggering event that sparks neurotoxicity takes years to manifest as disease. In latent or silent neurotoxicity, the effects of exposures that are insufficient to cause PD alone are unmasked later in life by the cumulative effects of exposures over the lifespan or the effects of aging [25,26,27,28]. This is consistent with the idea that accumulating factors over the lifespan accelerates the normal pace of dopaminergic neuron dysfunction and loss with age, eventually exceeding a threshold for PD development [29].
The developing brain is particularly sensitive to toxicant exposure both in gestation and in the postnatal period [30,31]. The developmental origins of health and disease (DoHAD) hypothesis posits that exposures in early life, from conception through adolescence, can produce long-lasting changes that contribute to the risk of later-life disease [32,33]. Many toxicants, including TCE, can cross the placenta and directly impact the developing fetus, as well as induce placental damage that can indirectly impact fetal development [34]. Existing evidence shows that exposure to TCE during pregnancy causes neural tube and cardiac defects in the developing fetus [35,36]. Furthermore, TCE is present in breast milk samples from communities in Arizona with contaminated drinking water [37]. These studies provide evidence for the premise that exposure to TCE during neurodevelopment may make the brain more susceptible to neurodegenerative diseases, including PD, late in life (Figure 1). As PD is the fastest growing neurologic disorder, and the average lifespan continues to increase, understanding the overall risk and the mechanisms that drive neurotoxicity from exposures across the lifespan will be critical for preventative measures and therapeutic developments.
Effects of Early Life Exposure to TCE
A handful of experimental studies have evaluated the effects of early life exposure to TCE on developmental processes. These studies have focused on modulation of the immune system from consumption of TCE via contaminated drinking water. For example, continuous exposure to 0.1 mg/mL TCE from gestation through early life caused a significant elevation in thymocyte number at PND 20 with a return to baseline at PND 20 and 42 in autoimmune-susceptible MRL+/+ mice [38]. In comparison, when MRL +/+ mice were exposed to higher doses of TCE (0.5 or 2.5 mg/mL) throughout gestation and early life through drinking water, there was no significant change in thymic cellularity at 7-8 weeks of age [39]. Similarly, MRL +/+ mice exposed to either 0.01 or 0.1 mg/mL of TCE either only in gestation or beginning in lactation did not have overall differences in thymic cell numbers [40]. Additionally, MRL +/+ mice showed reductions in splenic cellularity at PND 49 when exposed to 0.01 or 0.1mg/mL from lactation to PND 49 through drinking water [40]. Interestingly, total splenic cell numbers were reduced in MRL +/+ mice exposed to 0.1 mg/mL only in gestation compared to mice not exposed to TCE [40]. Splenic cellularity was not significantly different from non-exposed controls at 7-8 weeks of age in MRL+/+ mice exposed to either 0.5 or 2.5 mg/mL of TCE from gestation to euthanasia [39]. Interestingly, these studies have not reported changes to splenic or thymic cellularity when exposure to TCE began during lactation instead of gestation [40]. Further, peripheral CD4+ T cells from MRL+/+ mice produce greater levels of classic pro-inflammatory cytokine interferon-γ (IFN-γ) in mice treated with 2.5 mg/mL of TCE from 4-6 weeks of age, with a return to baseline at 7-8 weeks [39]. Additionally, at a lower dose of 0.1 mg/mL, splenic CD4+ T cells from MRL+/+ mice exposed from gestation until euthanasia had increased production of IFN-γ and tumor necrosis factor ɑ (TNF-α) [38].
In addition, experimental evidence also suggests that early life exposure causes dysregulation of the immune cell population in the brain during mid-adulthood. Mice exposed to either 0.05 or 500μg/mL of TCE from gestation through PND 154 had elevations in total splenic cell numbers, although there were no changes in percentages of these cells at PND 259 [41]. Furthermore, these mice had significant liver pathology scores, even after over 100 days of consuming noncontaminated water, showing that TCE-induced liver toxicity is persistent [41]. A separate study reported that early-life exposure to 500 μg/mL of TCE from gestation through PND 259 produced altered DNA modifications in effector memory CD4+ splenic T cells in mice [42]. This study also showed that mice treated with TCE through PND 259 displayed 113 hypermethylated regulatory elements and 16 hypomethylated elements, while mice treated with TCE for a shorter early life duration had only 4 hypermethylated elements [42]. This study suggests that epigenetic changes resulting from TCE exposure might be dynamic.
In relation to neurotoxicity, there is experimental evidence that hippocampal and cerebellar development is impacted by early life exposure to TCE. In particular, mice exposed to 0.1 mg/kg of TCE beginning in either gestation or lactation displayed reduced levels of the antioxidant glutathione and increased levels of oxidative stress markers 3-nitrotyrosine (3-NT) in the cerebellum by PND 42 [38,43,44]. Similar results were reported in the hippocampus, showing reductions in neurotrophic factors by PND 42 [45]. TCE-exposed offspring also exhibited hyperactivity and novelty-seeking behavior at the same time point, suggesting that developmental exposure to TCE caused functional behavioral differences [43,44]. While these studies did not specifically measure neurotoxicity in the dopaminergic system or the midbrain basal ganglia, a reduction in glutathione with elevated oxidative stress in the SN is a key feature of PD [46,47,48], suggesting that TCE-induced impairment of the glutathione system could drive future neurodegeneration or render the brain more vulnerable to additional stress.
Overall, the long-term effects of TCE exposure during development on the risk for neurodegeneration remain understudied and relatively unknown. While the studies described above provide evidence that early life exposure to TCE can promote oxidative stress in developing brain regions, there is a lack of information on whether these insults are persistent and contribute to neurodegenerative processes as the brain ages. While findings from long-term exposure studies in mid-adulthood suggest that epigenetic modifications might be dynamic, increased numbers of immune cell populations are still sustained even after 3 months of no exposure, suggesting that at least some insults are persistent [49]. However, it is important to note that these exposure paradigms expose the mice to TCE for extended periods even after the onset of maturity. Thus, research is needed on the long-term effects of exposure occurring along the entire developmental timeline.
Persistent Consequences of Early Life and Developmental Exposure to Other PD-Related Toxicants
Dieldrin
Dieldrin is an organochlorine pesticide that, like TCE, has been associated with increased risk of PD [50]. The literature on late-life neurotoxic effects of early-life exposure to dieldrin is more extensive than TCE. Mouse studies show that both adult and developmental dieldrin exposures disrupt the expression of PD-related proteins, cause oxidative stress, and produce an increase in susceptibility in adult male offspring to secondary toxicants [51,52,53,54,55]. In this paradigm, dams are exposed via dieldrin-contaminated food (0.3 mg/kg) starting 4 weeks prior to mating and continuing through gestation and lactation; exposure to pups end at weaning (PND21) [52,53,55]. At 12 weeks of age, male offspring show increased toxicity induced by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and by synucleinopathy in the ɑSyn preformed fibril (αSyn PFF) model [52,53,55]. Specifically, mice exposed to dieldrin during development had higher levels of ɑSyn and greater loss of striatal dopamine induced by MPTP injections than mice not exposed [52]. In addition, using the same exposure paradigm, male offspring exposure to dieldrin demonstrated exacerbated motor dysfunction and striatal dopaminergic turnover 6 months after unilateral injection of ɑSyn PFFs into the striatum at 12 weeks of age [53]. Furthermore, these offspring display sex-specific and age-specific alterations to DNA modifications at PND0, 6 weeks, 12 weeks, and 9 months of age in the midbrain in pathways related to dopaminergic neuron differentiation, synaptogenesis, synaptic plasticity, and glial-neuron interactions [54,56]. Finally, dieldrin exposure led to sex-specific differences in the expression of neuroinflammatory genes at 12 weeks of age [53]. This series of studies support the idea that dieldrin-induced changes persist after the exposure period and disrupt critical neurodevelopmental and neuroinflammatory pathways, impacting the risk of late-life disease.
Paraquat
Paraquat is a widely used herbicide that is associated with increased Parkinson’s risk in individuals who worked with paraquat (odds ratio 2.15, 95% CI 1.46, 3.19) [57]. Data suggests that developmental paraquat exposure can affect the developing fetus. A study of newborns born to women who live in agricultural areas of Thailand that use the herbicide found higher concentrations of paraquat in the meconium if their mothers had used herbicides while pregnant, lived around farms that used paraquat, or consumed water from community wells near agricultural areas [58]. Another study of paraquat exposure in pregnant women found significantly increased infant mortality rates, underscoring the overt toxicity of paraquat to developing systems [59].
Experimental studies evaluating motor behavior and dopaminergic neuron density in adulthood following early life paraquat exposure suggest that exposure to this toxicant during neurodevelopment can have latent effects on the dopaminergic system. One study found that Swiss mice treated with 20 mg/kg of paraquat during gestation reduced tyrosine hydroxylase (TH) immunoreactivity at PND 60 [60]. In addition, early-life paraquat exposure has often been evaluated alongside a secondary insult in adulthood. For example, mice administered 0.8 mg/kg of paraquat via intraperitoneal injection from PND 5-19 and additional doses of 0.8 mg/kg of paraquat at 8 months of age demonstrated significant deficits in working memory compared to both controls treated with saline at both time points, and greater deficits than mice given paraquat at only one of the time points [61]. Similar findings were reported in a study of Wistar rats injected with either paraquat or zinc from PND 5-19 and later given additional doses of either paraquat or 2 mg/kg of zinc 2 months later [62]. Administration of 0.5 mg/kg of paraquat from PND 5-19 increased the susceptibility of the SN to challenge of the same dose of paraquat or 2 mg/kg of zinc for 12 weeks in adulthood, with rats showing a significant loss of TH immunoreactivity [62]. Finally, there is evidence that co-administration of 0.3 mg/kg of paraquat and 1 mg/kg of the fungicide maneb from PND 5-19 and also at 3 months of age had the largest reduction of TH immunoreactivity in the SN compared to mice treated with saline or paraquat and maneb at only one of the timepoints [63]. Collectively, these findings suggest that while paraquat exposure during early life can produce motor deficits and loss of dopaminergic neurons in the SN similar to PD pathology, this exposure might also make the brain more susceptible to secondary insults.
Tetrachloroethylene
Tetrachloroethylene, also called perchloroethylene (PCE), is an organic solvent closely related to TCE and also a contaminant at Camp Lejeune [17]. Like TCE, there is very little published data on neurodegeneration after exposure to PCE during development. However, existing literature in humans shows an increased prevalence of various disorders in individuals whose mothers were exposed to PCE. For example, in Cape Cod, Massachusetts, an area that had a PCE-contaminated water supply from the 1960s-1980s [64,65,66], individuals who were exposed to PCE during gestation were at higher risk of developing cancer and epilepsy in adulthood [66]. Developmental PCE exposure is associated with increased risk ratio (RR) for psychiatric disorders, including bipolar disorder (RR 1.8, 95% CI 0.9-1.4) and post-traumatic stress disorder (PTSD, RR 1.5, 95% CI 0.9-2.5), alongside an increased risk of demonstrating risky behaviors such as drinking (RR 1.3, 95% CI 1.0-1.7) or illicit drug use in adulthood (RR 1.5, 95% CI 1.2-1.9) [64,65]. While these studies do demonstrate the long-term effects of PCE exposure in early life, there are currently no reports of neurodegenerative diseases after this exposure, likely due to the relatively young age of the individuals in these specific reports. Additionally, there is a lack of experimental data on early life PCE exposure in general.
Rotenone
Rotenone is a pesticide that can act as a mitochondrial complex I inhibitor (OR 2.5, 95% CI 1.3-4.7) [67]. Some rotenone studies have explored the effect of inflammation during development on rotenone exposure later in life. In these models, lipopolysaccharide (LPS), a bacterial endotoxin known to cause activation of the innate immune system and trigger inflammatory responses, is used to induce inflammation [68]. Rats exposed to 1 mg/kg of LPS via intraperitoneal injection in the postnatal period causes long-lasting increases in oxidative stress markers and microglia in the cerebellum of offspring at 3 months old [69]. Rats administered 1000 EU/g LPS via intracerebral injection on PND 5 followed by secondary insult with 1.25 mg/kg of rotenone via subcutaneous minipump infusion from PND 70-84 showed reduced TH and neuronal nuclei (NeuN) positive cells in the SN on PND 98 [70]. Interestingly, this loss, while present in mice administered only rotenone or LPS, was significantly exacerbated by the combined exposure [70].
In a similar exposure paradigm, rats treated with 2 μg/g of LPS via intraperitoneal injection on PND 5 and administered 1.25 mg/kg of rotenone via minipump infusion from PND 70-84 also demonstrated reduced TH and NeuN positive cells [71]. Additionally, rats administered LPS and rotenone had significant motor deficits by PND 98 [70,71]. Rats administered 1 mg/kg of LPS via intracerebral injection on PND 5 and 1.25 mg/kg of rotenone from PND 70-84 had significantly more ɑSyn aggregates in the cytoplasmic compartment of the SN compared to rats given only LPS or rotenone alone [72]. These rats co-exposed to LPS and rotenone also demonstrated elevated dopamine transporter (DAT) levels in the cytoplasmic compartment of the SN compared to those given only LPS or rotenone [72]. Further, co-exposure of LPS and rotenone reduced mitochondrial complex I activity in the striatum more than in rats exposed to only one toxicant [72]. This activity was also significantly lower in the co-exposed rats compared to those given only rotenone [72]. Furthermore, rats administered with 10,000 EU/mL of LPS in gestation and 1.25 mg/kg of rotenone via intrajugular infusion for 14 days at 16 months of age had the greatest loss of TH immunoreactivity in the SN compared to groups treated with only LPS or rotenone [73]. These studies demonstrate that the administration of LPS in early life results in nigrostriatal degeneration and increases susceptibility to secondary exposures like rotenone later in life.
Next Steps for PD Prevention
The prohibition of TCE for most industrial uses by the EPA in 2024 represents a milestone for the PD community. However, whether the ban will be enacted remains unclear, TCE is still used worldwide, and it remains a significant and persistent environmental risk. Much of the human research that associated PD risk with TCE exposure has investigated risk in individuals who experienced occupational exposure to TCE or those living in known drinking water contamination areas. Due to the widespread contamination, it is likely that exposures in the general population across the lifespan also pose a risk. However, there are significant challenges to studying this in human populations. Because TCE is predominantly inhaled, studies focused solely on drinking water levels of TCE may be missing significant populations with high exposure. Additionally, we lack sensitive tools to track ambient TCE and other volatile exposures in epidemiological studies, relying instead on emissions data that can be extremely difficult to quantify over the neurodevelopmental period. In this context, experimental studies can close the gap by measuring mechanisms involved in neurotoxicity that prime the brain for neurodegeneration later in life. Given the existing data on early-life exposure to TCE and other PD-related toxicants, further research into the potential effects of early-life TCE exposures is warranted. As PD incidence continues to rise, studies of the mechanisms that drive neurotoxicity across the lifespan will be a critical part of developing preventative measures and therapeutic advancements.
Acknowledgements
This work is supported by the National Institutes of health (1R01ES034846 to BRD).
Conflicts of Interest
Dr. De Miranda is a consultant for Bell Legal and The Miller Firm.
References
- Doherty, R.E. A history of the production and use of carbon tetrachloride, tetrachloroethylene, trichloroethylene and 1, 1, 1-trichloroethane in the United States: Part 1—Historical background; carbon tetrachloride and tetrachloroethylene. Environmental forensics. 2000, 1, 69–81. [Google Scholar]
- Bakke, B.; Stewart, P.A.; Waters, M.A. Uses of and exposure to trichloroethylene in U.S. industry: A systematic literature review. J Occup Environ Hyg. 2007, 4, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Schaum, J. Exposure assessment of trichloroethylene. Environmental health perspectives 2000, 108 (Suppl 2), 359–363. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Zafar, M.; Lettenberger, S.E.; Pawlik, M.E.; Kinel, D.; Frissen, M.; Schneider, R.B.; Kieburtz, K.; Tanner, C.M.; De Miranda, B.R.; Goldman, S.M.; Bloem, B.R. Trichloroethylene: An Invisible Cause of Parkinson's Disease? J Parkinsons Dis. 2023, 13, 203–218. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Todd, G.D.; Ruiz, P.; Mumtaz, M.; Wohlers, D.; Klotzbach, J.M.; Diamond, G.L.; Coley, C.; Citra, M.J. Toxicological profile for trichloroethylene (TCE)2019.
- Trichloroethylene (TCE); Regulation Under the Toxic Substances Control Act (TSCA), (2024).
- Guha, N.; Loomis, D.; Grosse, Y.; Lauby-Secretan, B.; Ghissassi, F.E.; Bouvard, V.; Benbrahim-Tallaa, L.; Baan, R.; Mattock, H.; Straif, K. Carcinogenicity of trichloroethylene, tetrachloroethylene, some other chlorinated solvents, and their metabolites. The Lancet Oncology. 2012, 13, 1192–1193. [Google Scholar] [CrossRef]
- Karami, S.; Lan, Q.; Rothman, N.; Stewart, P.A.; Lee, K.-M.; Vermeulen, R.; Moore, L.E. Occupational trichloroethylene exposure and kidney cancer risk: A meta-analysis. Occupational and Environmental Medicine. 2012, 69, 858. [Google Scholar] [CrossRef]
- Siegel Scott, C.; Jinot, J. Trichloroethylene and Cancer: Systematic and Quantitative Review of Epidemiologic Evidence for Identifying Hazards. International Journal of Environmental Research and Public Health [Internet]. 2011, 8, 4238–4272. [Google Scholar] [CrossRef]
- Ruckart, P.Z.; Bove, F.J.; Shanley, E.; Maslia, M. Evaluation of contaminated drinking water and male breast cancer at Marine Corps Base Camp Lejeune, North Carolina: A case control study. Environmental Health. 2015, 14, 74. [Google Scholar] [CrossRef]
- Maltoni, C.; Lefemine, G.; Cotti, G.; Perino, G. Long-term carcinogenicity bioassays on trichloroethylene administered by inhalation to Sprague-Dawley rats and Swiss and B6C3F1 mice. Annals of the New York Academy of Sciences. 1988, 534, 316–42. [Google Scholar]
- Rusyn, I.; Chiu, W.A.; Lash, L.H.; Kromhout, H.; Hansen, J.; Guyton, K.Z. Trichloroethylene: Mechanistic, epidemiologic and other supporting evidence of carcinogenic hazard. Pharmacology & Therapeutics. 2014, 141, 55–68. [Google Scholar] [CrossRef]
- Bove, F.J.; Ruckart, P.Z.; Maslia, M.; Larson, T.C. Mortality study of civilian employees exposed to contaminated drinking water at USMC Base Camp Lejeune: A retrospective cohort study. Environ Health. 2014, 13, 68. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guehl, D.; Bezard, E.; Dovero, S.; Boraud, T.; Bioulac, B.; Gross, C. Trichloroethylene and parkinsonism: A human and experimental observation. European journal of neurology. 1999, 6, 609–611. [Google Scholar] [CrossRef]
- Goldman, S.M.; Quinlan, P.J.; Ross, G.W.; Marras, C.; Meng, C.; Bhudhikanok, G.S.; Comyns, K.; Korell, M.; Chade, A.R.; Kasten, M.; Priestley, B.; Chou, K.L.; Fernandez, H.H.; Cambi, F.; Langston, J.W.; Tanner, C.M. Solvent exposures and Parkinson disease risk in twins. Ann Neurol. 2012, 71, 776–784. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gash, D.M.; Rutland, K.; Hudson, N.L.; Sullivan, P.G.; Bing, G.; Cass, W.A.; Pandya, J.D.; Liu, M.; Choi, D.-Y.; Hunter, R.L.; Gerhardt, G.A.; Smith, C.D.; Slevin, J.T.; Prince, T.S. Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Annals of Neurology. 2008, 63, 184–192. [Google Scholar] [CrossRef]
- Goldman, S.M.; Weaver, F.M.; Stroupe, K.T.; Cao, L.; Gonzalez, B.; Colletta, K.; Brown, E.G.; Tanner, C.M. Risk of Parkinson Disease Among Service Members at Marine Corps Base Camp Lejeune. JAMA Neurol. 2023, 80, 673–681. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goldman, S.M.; Weaver, F.M.; Gonzalez, B.; Stroupe, K.T.; Cao, L.; Colletta, K.; Brown, E.G.; Tanner, C.M. Parkinson's Disease Progression and Exposure to Contaminated Water at Camp Lejeune. Movement Disorders. 2024, 39, 1732–1739. [Google Scholar] [CrossRef]
- Liu, M.; Choi, D.Y.; Hunter, R.L.; Pandya, J.D.; Cass, W.A.; Sullivan, P.G.; Kim, H.C.; Gash, D.M.; Bing, G. Trichloroethylene induces dopaminergic neurodegeneration in Fisher 344 rats. J Neurochem. 2010, 112, 773–783. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, M.; Shin, E.-J.; Dang, D.-K.; Jin, C.-H.; Lee, P.H.; Jeong, J.H.; Park, S.-J.; Kim, Y.-S.; Xing, B.; Xin, T.; Bing, G.; Kim, H.-C. Trichloroethylene and Parkinson’s Disease: Risk Assessment. Molecular Neurobiology. 2018, 55, 6201–6214. [Google Scholar] [CrossRef]
- Adamson, A.; Ilieva, N.; Stone, W.J.; De Miranda, B.R. Low-dose inhalation exposure to trichloroethylene induces dopaminergic neurodegeneration in rodents. Toxicol Sci. 2023. [CrossRef] [PubMed]
- De Miranda, B.R.; Castro, S.L.; Rocha, E.M.; Bodle, C.R.; Johnson, K.E.; Greenamyre, J.T. The industrial solvent trichloroethylene induces LRRK2 kinase activity and dopaminergic neurodegeneration in a rat model of Parkinson's disease. Neurobiol Dis. 2021, 153, 105312. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, N.M.; Hoffman, E.K.; Ghalib, M.A.; Greenamyre, J.T.; De Miranda, B.R. LRRK2 kinase inhibition protects against Parkinson's disease-associated environmental toxicants. Neurobiol Dis. 2024, 196, 106522. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, H.; Ritz, B. The Search for Environmental Causes of Parkinson's Disease: Moving Forward. J Parkinsons Dis. [CrossRef] [PubMed] [PubMed Central]
- Reuhl, K. Delayed expression of neurotoxicity: The problem of silent damage. Neurotoxicology. 1991, 12, 341–346. [Google Scholar]
- Kraft, A.D.; Aschner, M.; Cory-Slechta, D.A.; Bilbo, S.D.; Caudle, W.M.; Makris, S.L. Unmasking silent neurotoxicity following developmental exposure to environmental toxicants. Neurotoxicology and Teratology. 2016, 55, 38–44. [Google Scholar] [CrossRef]
- Cory-Slechta, D.A.; Thiruchelvam, M.; Barlow, B.K.; Richfield, E.K. Developmental pesticide models of the Parkinson disease phenotype. Environmental health perspectives. 2005, 113, 1263–1270. [Google Scholar] [PubMed]
- Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends in neurosciences. 2007, 30, 244–250. [Google Scholar]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a primary risk factor for Parkinson's disease: Evidence from studies of non-human primates. Nature Reviews Neuroscience. 2011, 12, 359–366. [Google Scholar] [PubMed]
- Rodier, P.M. Developing brain as a target of toxicity. Environmental Health Perspectives. 1995; 103, (suppl 6), 73–76. [Google Scholar] [CrossRef]
- Lanphear, B.P. The Impact of Toxins on the Developing Brain. Annual Review of Public Health. 2015; 36, 211–230. [Google Scholar] [CrossRef]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.C.; Boileau, P.; Le Bouc, Y.; Deal, C.L.; Lillycrop, K.; Scharfmann, R.; Sheppard, A.; Skinner, M.; Szyf, M.; Waterland, R.A.; Waxman, D.J.; Whitelaw, E.; Ong, K.; Albertsson-Wikland, K. Child Health, Developmental Plasticity, and Epigenetic Programming. Endocrine Reviews. 2011, 32, 159–224. [Google Scholar] [CrossRef]
- Heindel, J.J.; Vandenberg, L.N. Developmental origins of health and disease: A paradigm for understanding disease cause and prevention. Current opinion in pediatrics. 2015, 27, 248–253. [Google Scholar]
- Ghantous, H.; Danielsson, B.R.; Dencker, L.; Gorczak, J.; Vesterberg, O. Trichloroacetic acid accumulates in murine amniotic fluid after tri- and tetrachloroethylene inhalation. Acta Pharmacol Toxicol (Copenh). 1986, 58, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Forand, S.P.; Lewis-Michl, E.L.; Gomez, M.I. Adverse birth outcomes and maternal exposure to trichloroethylene and tetrachloroethylene through soil vapor intrusion in New York State. Environ Health Perspect. 2012, 120, 616–621. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goldberg, S.J.; Lebowitz, M.D.; Graver, E.J.; Hicks, S. An association of human congenital cardiac malformations and drinking water contaminants. Journal of the American College of Cardiology. 1990, 16, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Beamer, P.I.; Luik, C.E.; Abrell, L.; Campos, S.; Martínez, M.E.; Sáez, A.E. Concentration of trichloroethylene in breast milk and household water from Nogales, Arizona. Environmental science & technology. 2012, 46, 9055–9061. [Google Scholar]
- Blossom, S.J.; Doss, J.C.; Hennings, L.J.; Jernigan, S.; Melnyk, S.; James, S.J. Developmental exposure to trichloroethylene promotes CD4+ T cell differentiation and hyperactivity in association with oxidative stress and neurobehavioral deficits in MRL+/+ mice. Toxicology and Applied Pharmacology. 2008, 231, 344–353. [Google Scholar] [CrossRef]
- Blossom, S.J.; Doss, J.C. Trichloroethylene Alters Central and Peripheral Immune Function in Autoimmune-Prone MRL+/+ Mice Following Continuous Developmental and Early Life Exposure. Journal of Immunotoxicology. 2007, 4, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, K.M.; Woodruff, W.; Blossom, S.J. Differential immunotoxicity induced by two different windows of developmental trichloroethylene exposure. Autoimmune Dis. 2014, 2014, 982073. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gilbert, K.M.; Bai, S.; Barnette, D.; Blossom, S.J. Exposure Cessation During Adulthood Did Not Prevent Immunotoxicity Caused by Developmental Exposure to Low-Level Trichloroethylene in Drinking Water. Toxicological Sciences. 2017, 157, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Byrum, S.D.; Washam, C.L.; Patterson, J.D.; Vyas, K.K.; Gilbert, K.M.; Blossom, S.J. Continuous Developmental and Early Life Trichloroethylene Exposure Promoted DNA Methylation Alterations in Polycomb Protein Binding Sites in Effector/Memory CD4+ T Cells. Frontiers in Immunology. 2019, 10. [Google Scholar] [CrossRef]
- Blossom, S.J.; Cooney, C.A.; Melnyk, S.B.; Rau, J.L.; Swearingen, C.J.; Wessinger, W.D. Metabolic changes and DNA hypomethylation in cerebellum are associated with behavioral alterations in mice exposed to trichloroethylene postnatally. Toxicology and Applied Pharmacology. 2013, 269, 263–269. [Google Scholar] [CrossRef]
- Blossom, S.J.; Melnyk, S.B.; Li, M.; Wessinger, W.D.; Cooney, C.A. Inflammatory and oxidative stress-related effects associated with neurotoxicity are maintained after exclusively prenatal trichloroethylene exposure. Neurotoxicology. 2017, 59, 164–74. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blossom, S.J.; Melnyk, S.; Cooney, C.A.; Gilbert, K.M.; James, S.J. Postnatal exposure to trichloroethylene alters glutathione redox homeostasis, methylation potential, and neurotrophin expression in the mouse hippocampus. Neurotoxicology. 2012, 33, 1518–1527. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martin, H.L.; Teismann, P. Glutathione--a review on its role and significance in Parkinson's disease. FASEB J. 2009, 23, 3263–3272. [Google Scholar] [CrossRef] [PubMed]
- Zeevalk, G.D.; Razmpour, R.; Bernard, L.P. Glutathione and Parkinson's disease: Is this the elephant in the room? Biomed Pharmacother. 2008, 62, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, K.M.; Bai, S.; Barnette, D.; Blossom, S.J. Exposure Cessation During Adulthood Did Not Prevent Immunotoxicity Caused by Developmental Exposure to Low-Level Trichloroethylene in Drinking Water. Toxicol Sci. 2017, 157, 429–437. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kanthasamy, A.G.; Kitazawa, M.; Kanthasamy, A.; Anantharam, V. Dieldrin-induced neurotoxicity: Relevance to Parkinson's disease pathogenesis. Neurotoxicology. 2005, 26, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, J.M.; Richardson, J.R.; Guillot, T.S.; McCormack, A.L.; Di Monte, D.A.; Jones, D.P.; Pennell, K.D.; Miller, G.W. Dieldrin exposure induces oxidative damage in the mouse nigrostriatal dopamine system. Exp Neurol. 2007, 204, 619–630. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Richardson, J.R.; Caudle, W.M.; Wang, M.; Dean, E.D.; Pennell, K.D.; Miller, G.W.; Richardson, J.R.; Caudle, W.M.; Wang, M.; Dean, E.D. Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson's disease. The FASEB journal. 2006, 20, 1695–1697. [Google Scholar]
- Gezer, A.O.; Kochmanski, J.; VanOeveren, S.E.; Cole-Strauss, A.; Kemp, C.J.; Patterson, J.R.; Miller, K.M.; Kuhn, N.C.; Herman, D.E.; McIntire, A.; Lipton, J.W.; Luk, K.C.; Fleming, S.M.; Sortwell, C.E.; Bernstein, A.I. Developmental exposure to the organochlorine pesticide dieldrin causes male-specific exacerbation of alpha-synuclein-preformed fibril-induced toxicity and motor deficits. Neurobiol Dis. 2020, 141, 104947. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kochmanski, J.; VanOeveren, S.E.; Patterson, J.R.; Bernstein, A.I. Developmental Dieldrin Exposure Alters DNA Methylation at Genes Related to Dopaminergic Neuron Development and Parkinson’s Disease in Mouse Midbrain. Toxicological Sciences. 2019, 169, 593–607. [Google Scholar] [CrossRef]
- Boyd, S.L.; Kuhn, N.C.; Patterson, J.R.; Stoll, A.C.; Zimmerman, S.A.; Kolanowski, M.R.; Neubecker, J.J.; Luk, K.C.; Ramsson, E.S.; Sortwell, C.E.; Bernstein, A.I. Developmental exposure to the Parkinson’s disease-associated organochlorine pesticide dieldrin alters dopamine neurotransmission in α-synuclein pre-formed fibril (PFF)-injected mice. Toxicological Sciences. 2023, 196, 99–111. [Google Scholar] [CrossRef]
- Kochmanski, J.; Virani, M.; Kuhn, N.C.; Boyd, S.L.; Becker, K.; Adams, M.; Bernstein, A.I. Developmental origins of Parkinson’s disease risk: Perinatal exposure to the organochlorine pesticide dieldrin leads to sex-specific DNA modifications in critical neurodevelopmental pathways in the mouse midbrain. Toxicological Sciences. 2024, 201, 263–281. [Google Scholar] [CrossRef]
- Paul, K.C.; Cockburn, M.; Gong, Y.; Bronstein, J.; Ritz, B. Agricultural paraquat dichloride use and Parkinson's disease in California's Central Valley. Int J Epidemiol. 2024, 53. [Google Scholar] [CrossRef] [PubMed]
- Konthonbut, P.; Kongtip, P.; Nankongnab, N.; Tipayamongkholgul, M.; Yoosook, W.; Woskie, S. Paraquat Exposure of Pregnant Women and Neonates in Agricultural Areas in Thailand. International Journal of Environmental Research and Public Health. 2018, 15, 1163. [Google Scholar] [CrossRef] [PubMed]
- Trakulsrichai, S.; Paisanrodjanarat, B.; Sriapha, C.; Tongpoo, A.; Udomsubpayakul, U.; Wananukul, W. Clinical outcome of paraquat poisoning during pregnancy. Clinical Toxicology. 2019, 57, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Ait-Bali, Y.; Ba-M’hamed, S.; Bennis, M. Prenatal Paraquat exposure induces neurobehavioral and cognitive changes in mice offspring. Environmental Toxicology and Pharmacology. 2016, 48, 53–62. [Google Scholar] [CrossRef]
- Zuo, Z.; Li, J.; Zhang, B.; Hang, A.; Wang, Q.; Xiong, G.; Tang, L.; Zhou, Z.; Chang, X. Early-Life Exposure to Paraquat Aggravates Sex-Specific and Progressive Abnormal Non-Motor Neurobehavior in Aged Mice. Toxics. 2023, 11, 842. [Google Scholar] [CrossRef]
- Mittra, N.; Chauhan, A.K.; Singh, G.; Patel, D.K.; Singh, C. Postnatal zinc or paraquat administration increases paraquat or zinc-induced loss of dopaminergic neurons: Insight into augmented neurodegeneration. Molecular and cellular biochemistry. 2020, 467, 27–43. [Google Scholar]
- Colle, D.; Santos, D.B.; Naime, A.A.; Gonçalves, C.L.; Ghizoni, H.; Hort, M.A.; Farina, M. Early postnatal exposure to paraquat and maneb in mice increases nigrostriatal dopaminergic susceptibility to a Re-challenge with the same pesticides at adulthood: Implications for Parkinson’s disease. Neurotoxicity Research. 2020, 37, 210–26. [Google Scholar] [CrossRef]
- Aschengrau, A.; Janulewicz, P.A.; White, R.F.; Vieira, V.M.; Gallagher, L.G.; Getz, K.D.; Webster, T.F.; Ozonoff, D.M. Long-term Neurotoxic Effects of Early-life Exposure to Tetrachloroethylene-contaminated Drinking Water. Annals of Global Health. 2016, 82, 169–179. [Google Scholar] [CrossRef]
- Aschengrau, A.; Weinberg, J.M.; Janulewicz, P.A.; Romano, M.E.; Gallagher, L.G.; Winter, M.R.; Martin, B.R.; Vieira, V.M.; Webster, T.F.; White, R.F.; Ozonoff, D.M. Occurrence of mental illness following prenatal and early childhood exposure to tetrachloroethylene (PCE)-contaminated drinking water: A retrospective cohort study. Environ Health. 2012, 11, 2. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aschengrau, A.; Winter, M.R.; Vieira, V.M.; Webster, T.F.; Janulewicz, P.A.; Gallagher, L.G.; Weinberg, J.; Ozonoff, D.M. Long-term health effects of early life exposure to tetrachloroethylene (PCE)-contaminated drinking water: A retrospective cohort study. Environmental Health. 2015, 14, 36. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; Comyns, K.; Richards, M.B.; Meng, C.; Priestley, B.; Fernandez, H.H.; Cambi, F.; Umbach, D.M.; Blair, A.; Sandler, D.P.; Langston, J.W. Rotenone, paraquat, and Parkinson's disease. Environ Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Page, M.J.; Kell, D.B.; Pretorius, E. The Role of Lipopolysaccharide-Induced Cell Signalling in Chronic Inflammation. Chronic Stress. 2022, 6, 24705470221076390. [Google Scholar] [CrossRef]
- Pires, J.M.; Foresti, M.L.; Silva, C.S.; Rêgo, D.B.; Calió, M.L.; Mosini, A.C.; Nakamura, T.K.E.; Leslie, A.T.F.; Mello, L.E. Lipopolysaccharide-Induced Systemic Inflammation in the Neonatal Period Increases Microglial Density and Oxidative Stress in the Cerebellum of Adult Rats. Frontiers in Cellular Neuroscience. 2020, 14. [Google Scholar] [CrossRef]
- Fan, L.-W.; Tien, L.-T.; Lin, R.C.; Simpson, K.L.; Rhodes, P.G.; Cai, Z. Neonatal exposure to lipopolysaccharide enhances vulnerability of nigrostriatal dopaminergic neurons to rotenone neurotoxicity in later life. Neurobiology of Disease. 2011, 44, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Fan, L.-W.; Kaizaki, A.; Tien, L.-T.; Ma, T.; Pang, Y.; Lin, S.; Lin, R.C.S.; Simpson, K.L. Neonatal Systemic Exposure to Lipopolysaccharide Enhances Susceptibility of Nigrostriatal Dopaminergic Neurons to Rotenone Neurotoxicity in Later Life. Developmental Neuroscience. 2013, 35, 155–71. [Google Scholar] [CrossRef] [PubMed]
- Tien, L.-T.; Kaizaki, A.; Pang, Y.; Cai, Z.; Bhatt, A.J.; Fan, L.-W. Neonatal exposure to lipopolysaccharide enhances accumulation of α-synuclein aggregation and dopamine transporter protein expression in the substantia nigra in responses to rotenone challenge in later life. Toxicology. 2013, 308, 96–103. [Google Scholar] [CrossRef]
- Ling, Z.; Chang, Q.A.; Tong, C.W.; Leurgans, S.E.; Lipton, J.W.; Carvey, P.M. Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysaccharide prenatally. Experimental Neurology. 2004, 190, 373–383. [Google Scholar] [CrossRef]
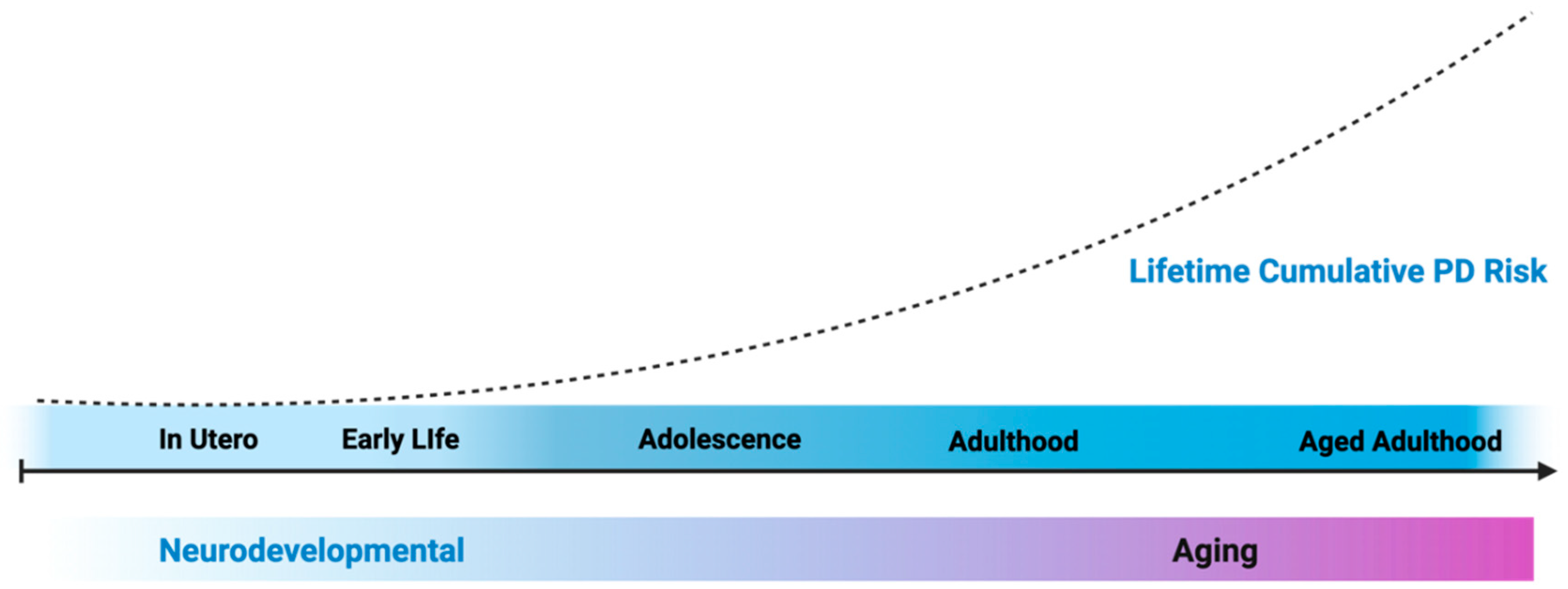
Figure 1.
Exposures can happen at any period across the lifespan, but susceptibility to neurotoxicants associated with PD may be greater during periods of neurodevelopment, driving pathology driving the long prodromal period of PD progression. Figure generated using Biorender.
Figure 1.
Exposures can happen at any period across the lifespan, but susceptibility to neurotoxicants associated with PD may be greater during periods of neurodevelopment, driving pathology driving the long prodromal period of PD progression. Figure generated using Biorender.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



