Submitted:
17 March 2025
Posted:
18 March 2025
You are already at the latest version
Abstract
Approximately 70–80% of breast cancers are ER+ and 65% of them also are ER+PR+. In most cases of ER+ advanced disease, endocrine therapy (ET) is the initial treatment with different drugs that act by inhibiting the ER signaling, mainly tamoxifen, a selective estrogen receptor modulator (SERM), or fulvestrant, a selective estrogen receptor degrader (SERD), or by impeding the estrogen formation as aromatase inhibitors (AIs). However, hormone resistance intrinsic or acquired, eventually develops, making disease progression unavoidable. An increased progression-free survival (PFS) and, sometimes, overall survival (OS) recently occurred after the association of ET with the cyclin-dependent kinases 4 and 6 inhibitors (CDK4/6is) that block cell-division cycle in the G1 phase and halt DNA production thus synergizing with ET. This review focuses on the main mechanisms of resistance to ET, whether used alone or in combination with biological agents, and on new drugs/strategies currently in use or under investigation to overcome it. Overcoming resistance to ET is a “work in progress” and early in the next it is expected to better select patients for different therapeutic strategies based on more specific biologic and/or genetic markers. Particularly, liquid biopsy may provide a real-time portrait of the disease state including mechanisms responsible for ET independence and cancer proliferation.
Keywords:
advanced ER+/HER2- breast cancer
; endocrine therapy
; CDK4/6 inhibitors
; endocrine resistance
1. Introduction
Approximately 70–80% of breast cancers are ER+ [1], and 65% of them also are ER+PR+ [2]. In most cases of ER+ advanced disease, endocrine therapy (ET) is the initial treatment option [3,4]. ET includes a variety of drugs that act by inhibiting the ER signaling, mainly tamoxifen, a selective estrogen receptor modulator (SERM), or fulvestrant, a selective estrogen receptor degrader (SERD)ì, or by impeding the estrogen formation as aromatase inhibitors (AIs) [3,4]. However, hormone resistance eventually develops, making disease progression unavoidable [5]. Disease relapse within 24 months from beginning of adjuvant ET or progression during the initial 6 months of 1-line ET for advanced or relapsed breast cancer respectively is called primary endocrine resistance. Disease relapse following the first 24 months of adjuvant ET, or within 12 months following the end of adjuvant ET, or progression more than 6 months from starting ET for metastatic disease is called secondary or acquired resistance [6]. An increased progression-free survival (PFS) and, sometimes, overall survival (OS) of invasive ER+ breast cancer recently occurred after the association of ET with the cyclin-dependent kinases 4 and 6 inhibitors (CDK4/6is) that block cell-division cycle in the G1 phase and halt DNA production thus synergizing with ET [7]. This review focuses on the mechanisms of resistance to ET, whether used alone or in combination with biological agents, and on new drugs/strategies currently in use or under investigation to overcome it.
2. Mechanisms of Resistance to ET
A few various modalities for arising of lack of response to ET have been described [8]. They comprehend genetic aberrations (mutations, fusions, amplifications) in the ligand-binding domain of ESR1 gene which encodes ER-alpha, mechanisms that refer to regulators of the ER pathway, the PI3K/AKT/mTOR or other signaling cascades, metabolic re-programming and further processes.
2.1. ESR1 Genetic Alterations
ESR1 mutations (ESR1m) are among the most common genetic alterations leading to ET failure [9]. ER+ breast cancer patients undergoing ET can develop particular mutations, such as Y537S in ERα that are responsible of unrestrained cell growth and endocrine-resistant metastatic disease [10]. A proteomic analysis in breast cancer cell lines with Y537N and Y537S ER mutations showed a significant increase in immune-related pathways along with proliferation signaling and the associated kinases, mainly mTOR and CDKs. So, ESR1 mutations are associated with an enhanced cyclin-dependent kinase signaling [11]. The ESR1 gene chromosomal abnormalities identified in ET-resistant breast cancer comprehend gene fusions within the same chromosome or with oncogenes located on different chromosomes [12,13]. Furthermore, an enhanced ESR1 copy number with ER-alpha overexpression levels has been observed in 1% to 37% of endocrine-refractory breast cancers [14,15,16,17]. However, the link between ESR1 amplifications and ET resistance, mainly reduced tamoxifen sensitivity, remains controversial [17,18,19].
2.2. Regulators of the ERα Pathway
Co-factors, chromatin modifiers, or miRNAs alterations can be involved. In a study carried out in luminal breast cancers, infiltration of cancer-associated fibroblasts (CAFs) near malignant cells was linked to reduced ER-α expression and function. CAFs promoted estrogen-independent tumor proliferation by sustaining the expression of particular genes associated with no sensitivity to treatment, basal-like differentiation and diffusion while most estrogen-responsive genes were suppressed. Genes down-regulated in cancer cells by CAFs also correlated with low sensitivity to ET while the transforming growth factor-β (TGF-β) and Janus kinase signaling were identified as potential targets to inhibit CAF-promoted changes through ER-α action [20]. MYSM1, a deubiquitinase highly expressed in breast cancer samples, was found to govern ERα activity and its depletion decreased in xenograft models the proliferation of breast cancer-derived cells and improved their sensitivity to anti-estrogen therapies [21]. A bioinformatics analysis in primary N+ breast tumors, validated in a mouse model, identified MNX1 as a possible transcription factor regulating the level of hormone receptors (HR) along with HER2. Accordingly, macrophages emerged as key cells in the tumor microenvironment (TME), driving down-regulation of HR and up-regulation of HER2, likely through the action of MNX1 [22]. SMAD4, by a genome-wide CRISPR analysis, was identified to be relevant in controlling 4-hydroxytamoxifen (OHT) sensitivity in T47D cells. Based on clinical findings it was found that the down-regulation of SMAD4, induced by ET, contributed to the arising of acquired resistance by cooperation of ER with ERBB signaling [23].
2.3. The PI3K/AKT/mTOR and Other Signaling Pathways
Mutations or amplifications in the ERBB2 gene occur in approximately 5% and 2%, respectively, of ER+/HER2− relapsed breast cancer population [24] while roughly half of them show increased activity of the PI3K/Akt pathway [25]. ErbB2/HER2/Neu amplification decreases the therapeutic activity of anti-estrogens, primarily through an enhanced PI3K/Akt and MAPK signaling that account for estrogen-independent ER phosphorylation and activation [26]. Furthermore, mutations in other receptor tyrosine kinase (RTK)-MAPK pathway, such as BRAF, RAS, and MAP2K1, have been identified in metastatic breast cancer not responding to AI [27]. Mutations in the PIK3CA gene occur in about 40% of cases and often occur in combination with endocrine resistance [28,29,30]; besides mutations in AKT1 and PTEN are frequently reported [31]. FGFR, EGFR, and other cell surface RTKs via upstream signaling can promote the hyper-activation of the PI3K/Akt pathway. Additionally, the PI3K/Akt signaling is downstream of several growth factors, insulin-like growth factor-I (IGF-I) and heregulin among them, which use this pathway to activate ER and promote estrogen-independent growth [32]. The increased activity of PI3K signaling accounted for the arising of acquired resistance after prolonged endocrine deprivation in experimental models of ER+ breast cancer cell lines, with proteomic analisys revealing enhanced phosphorylation of mTOR and Akt [27]. Accordingly, experimental and clinical evidence suggest to combine treatments targeting ER and the PI3K/Akt signaling to efficaciously counteract acquired endocrine resistance [33,34]. Everolimus is an mTOR inhibitor with antitumor activity independent of PIK3CA mutation and approved in association with exemestane for treating ER+ metastatic breast cancer [35]. The mechanism of endocrine resistance involving the PI3K/Akt pathway is ligand-independent ER activation. Particularly, S6 kinase, a substrate of mTOR complex 1, phosphorylates the activation function domain 1 (AF1) of ER [36,37]. Everolimus allows to overcome resistance to mTORC1 by binding and allosterically inhibiting it [38]. In patients with ESR1 mutations progressing on prior AI therapy and possible resistance to everolimus, fulvestrant may be a more suitable antiestrogen partner and the National Comprehensive Cancer Network (NCCN) guidelines include fulvestrant or tamoxifen as alternative partners for ET with everolimus [3,4]. Alpelisib, a PI3K inhibitor, in association with fulvestrant received FDA approval for treating PIK3CA-mutated ER+ metastatic breast cancer [3,4,34] without prior exposure to CDK4/6, PI3K, Akt, or mTOR inhibitors [28]. Inhibitors of the PI3K/Akt/mTOR pathway synergize with anti-estrogen therapy due to the significant cross-talk between these pathways and ER [39]. In endocrine-resistant breast cancer cells, the cGAS-STING signaling is diminished compared to endocrine-sensitive cells. This reduction appears to be driven by hyperactivation of the AKT1 kinase, and the inactivation of cGAS-STING signaling creates a positive feedback loop with hyperactivated AKT1, promoting endocrine resistance. Disrupting this feedback loop by using an AKT1 inhibitor alongside a STING agonist reduces the proliferation of endocrine-resistant tumors in mouse models [40]. Amplifications of RTKs such as EGFR (epidermal growth factor receptor) and FGFR1 (fibroblast growth factor receptor 1) are commonly found in breast cancer without response to ET [8]. FGFR1 overexpression often occurs in aggressive Luminal B-like tumors and correlates with poor outcome and resistance to therapy. The amplification of FGFR1 concomitant with other oncogenes suggests it is not a sole oncogenic driver. Although FGFR2 amplification is more rare, it is crucial for regulating hormone receptors, promoting growth and resistance to therapy. FGFR3 and FGFR4 also can be responsible for endocrine resistance by different modalities including by stimulating PI3K/AKT/mTOR and RAS/RAF/MEK/ERK signaling [41]. Neurofibromatosis type 1 (NF1) is a tumor suppressor gene and 27% of breast cancers suffer from harmful NF1 aberrations. Consequently, the absence or loss of function of NF1 account for both the hyper-activation of RAS-dependent signal and the de-repression of cyclin D1, thus favoring lack of response to ET and increased cell growth [42,43,44]. Breast cancer is marked by a highly inflammatory TME. The NF-κB pathway is relevant in linking inflammation to cancer, and also in promoting tumor proliferation and resistance to treatment. Chemotherapy, targeted therapy, ET, and radiotherapy may favor resistance in breast cancer via NF-κB. NF-κB inhibitors in association with tamoxifen importantly improved the breast cancer cells response to tamoxifen [45].
2.4. Metabolic Reprogramming
In some researches, metabolic reprogramming has been found to be associated with endocrinotherapy. Nine metabolic proteins were identified, with phosphomannose mutase 2 (PMM2) emerging as more promising candidate and depletion of PMM2 led to the deterioration of ERα Y537S mutant cells, inhibited cell growth, and reduced ERα signaling. In particular, lowering PMM2 again sensitized ERα Y537S-expressing cells to some drugs given for endocrine treatment and CDK4/CDK6is. Besides, PMM2 decrease lowered FOXA1, a protein that is relevant in regulating ERα. Overall, this suggests that PMM2 in relapsed breast cancer with the ERα Y537S variant is a suitable target for treatment [10]. In ER+ breast cancer cells that were maintained for prolonged time without estrogens (LTED cells), a model commonly used to assess resistance to AI, an increased intra-cellular lipids accumulation was found. This metabolic reprogramming was driven by acetyl-CoA carboxylase-1 (ACC1), and by inhibiting ACC1 the survival of LTED cells significantly decreased. Despite this, the introduction of branched and very long-chain fatty acids counteracted the effects of ACC1 inhibition, a process confirmed in patient-derived samples treated with AIs. Ultimately, ACC1 is a potential target to counteract the development of estrogen independence in ER+ breast cancers [46]. In a study focused on NF1-deficient ER+ breast cancer, NF1 deficiency drove metabolic reprogramming showing constraints on oxidative ATP production, increased glutamine inserted into the TCA cycle, and expansion of lipid pools. Authors concluded that these metabolic alterations favor new synergies between metabolic and targeted inhibitors [47].
2.5. Further Mechanisms
CYP19A1, the gene encoding the aromatase enzyme which produces estrogen is often over-expressed in endocrine-resistant breast cancer that progresses following therapy with AI [48]. Moreover, deletions or mutations in the histone H3K4 methyl-transferase KMT2C (MLL3) have been found in ER+ metastatic breast cancer after AIs therapy and were associated with poor PFS [49]. Long non-coding RNA LINC00152 contributes to tamoxifen resistance by inhibiting ferroptosis, a type of programmed cell death dependent on iron and favored by tamoxifen. Notably, high levels of LINC00152 expression are significantly associated with increased PDE4D, decreased ferroptosis, and poorer clinical outcome highlighting that LINC00152 and its downstream effectors can be promising targets for increasing survival rate in refractory ER+ breast cancer [50]. High expression of collagen type XI alpha 1 (COL11A1) has been associated with therapeutic resistance and poor outcome in breast cancer subjects undergoing tamoxifen. A research evaluated MCF-7/COL11A1 and T47D/COL11A1 cell lines that unlike the parental MCF-7 and T47D cell lines, exhibited greater resistance to growth inhibition induced by 4-OHT while the removal of COL11A1 significantly made these cells sensitive in vitro and in vivo to 4-OHT. Particularly, in tamoxifen refractory cells Erα over-expression occurred, likely because of elevated COL11A1 levels. Additionally, the COL11A1 removal led to ERα and its downstream target genes reduced expression [51]. In breast cancer a meta-analysis validated the combination of breast cancer resistance 4 (BCAR4) gene with negative results along with the resistance to ET. Importantly, BCAR4 expression was clinically relevant in luminal A and B subtypes and a correlation of BCAR4 with lack of response to AIs was identified [52]. In a study, F-box protein 22 (Fbxo22) negativity was associated with resistance to ET. Among all patients treated with SERMs, those without Fbxo22 had poorer outcomes, with 10-year OS rates of 81.3% compared to 92.3% (P = 0.032) suggesting that Fbxo22 negativity significantly affects survival in breast cancer patients with both invasive ductal (IDC) and lobular (ILC) carcinomas, with particularly pronounced disadvantages among postmenopausal women with ILC or those treated with SERMs [53]. Another study showed that RFC3 is relevant for the cell cycle and RFC3 over-expression was found in breast cancer-resistant cells unlike parental cells suggesting that RFC3 favors the lack of response to ET in breast cancer through the cell cycle [54].
3. CDK4/6is in Association with Endocrine-Therapy
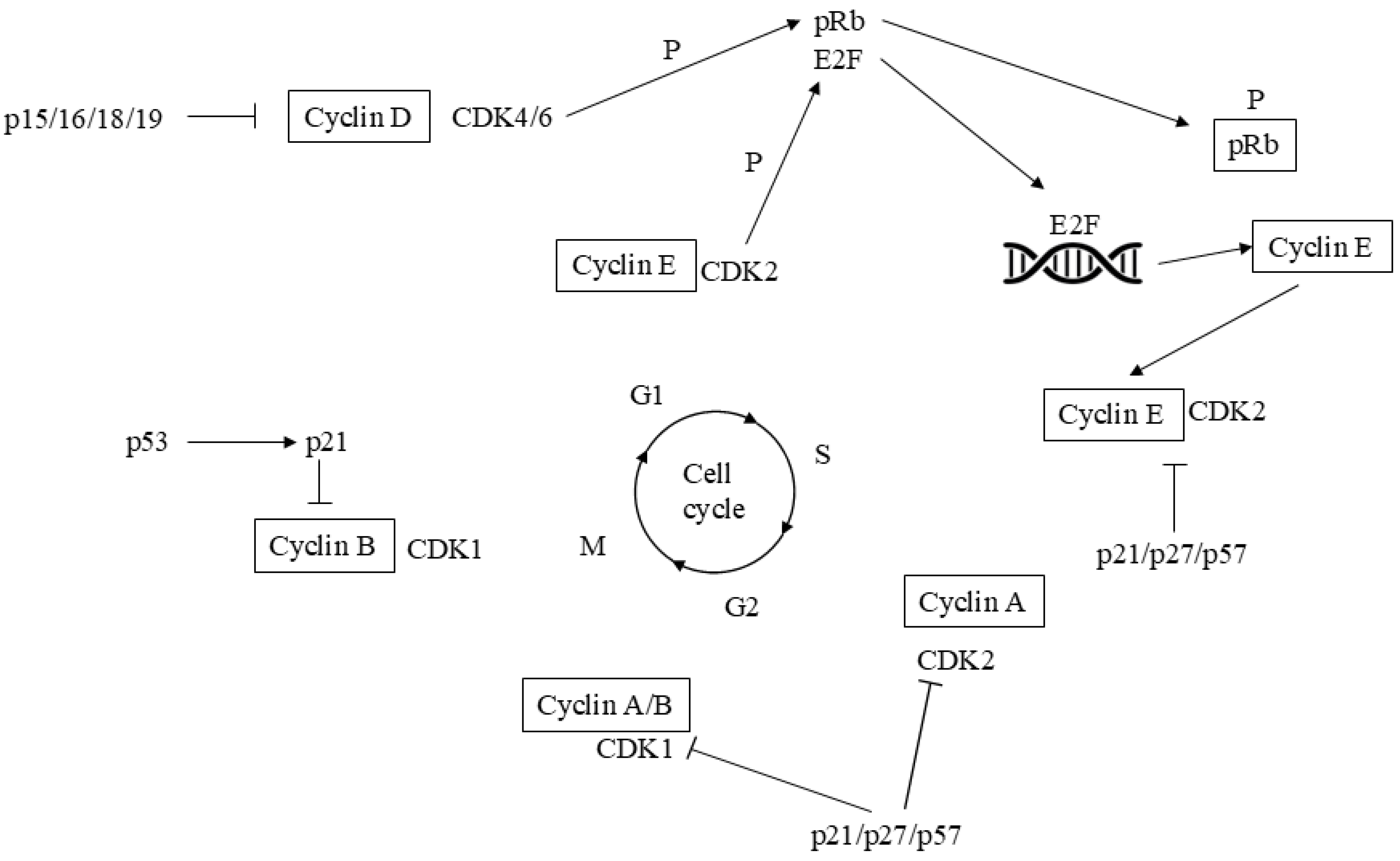
Cell-division cycle is controlled through various cyclins and CDKs (Figure 1). Cyclin D1 interacts with CDK4/6 to regulate advancement to the G1 phase of the cell-division cycle and is known as a target of the PI3K/AKT/mTOR downward signaling [55]. Since tumor development often depends on unregulated cell-division cycle, the cyclin D1/CDK4/6 pathway has become a significant target in cancer treatment, including breast cancer [56,57,58]. Additionally, CDK4/6 have been implicated in cross-talk with the ER signaling pathway [59]. Early research identified a strong correlation between ER expression and the response to CDK4/6is [60]. In the PALOMA-2, MONALEESA-2, MONARCH-3, and MONALEESA-7 phase III trials, palbociclib, ribociclib, and abemaciclib CDK4/6is plus ET improved PFS when they were given as 1-line in pre- and post-menopausal breast cancer populations [61,62,63,64]. Despite this, a signifcant prolonged OS up to-day only occurred in the two MONALEESA-7 and -2 trials with ribociclib plus anti-estrogens in pre- and post-menopausal breast cancer patients [65,66]. So, current guidelines [3,4] recommend CDK4/6is combined with ET as the common 1-line therapy in HR+/HER2- advanced/relapsed breast cancer. In a comprehensive search of PubMed and Embase, that identified peer-reviewed studies from February 2015 to March 2022 reporting cost-effectiveness in metastatic breast cancer treatment, none of the three CDK4/6is had positive incremental net benefits (INB) compared to AIs alone [67]. Furthermore, often the PFS/OS benefit is not significant when the CDK4/6is are given as second-line therapy and chemotherapy is preferred for rapid disease control if the patient has an approaching organ failure or a life-threatening visceral crisis. However, CDK 4/6is given in addition to fulvestrant also were efficacious as second-line ET after relapse or progression on an AI [68,69,70,71].
Figure 1.
Complexity of the molecular pathways governing the function of the cyclin-dependent kinases (CDKs) throught cell-divisio cycle. Cell-division cycle starts depending on tangled interplay of CDK4 and CDK6 complexes which involve D-type cyclins and release E2F transcription fectors by pRb phosphorylation. They regulate the transcription of genes that play a key role in S-phase advance. The S phase progression then is regulated by co-operation of CDK2-cyclin E/cyclin A complexes. CDK function arrangement is promoted by CDKis particularly addressing CDK4/6 or CDK2. The progression from G2 phase to M phase is governed by the collaborating A and B cyclins along with CDK1. CDK: cyclin dependent kinase; p: protein; Rb: retinoblastoma; P: phosphorylation. Also see text.
Figure 1.
Complexity of the molecular pathways governing the function of the cyclin-dependent kinases (CDKs) throught cell-divisio cycle. Cell-division cycle starts depending on tangled interplay of CDK4 and CDK6 complexes which involve D-type cyclins and release E2F transcription fectors by pRb phosphorylation. They regulate the transcription of genes that play a key role in S-phase advance. The S phase progression then is regulated by co-operation of CDK2-cyclin E/cyclin A complexes. CDK function arrangement is promoted by CDKis particularly addressing CDK4/6 or CDK2. The progression from G2 phase to M phase is governed by the collaborating A and B cyclins along with CDK1. CDK: cyclin dependent kinase; p: protein; Rb: retinoblastoma; P: phosphorylation. Also see text.
4. Resistance to ET and/or CDK4/6is
So far, various procedures accounting for no or impaired response to ET and/or CDK4/6is have been identified. They include genetic aberrations involving cell-division cycle regulation, triggering of different molecular pathways, modifications in transcriptional and epigenetic regulators [72,73], acquired CDK6 amplification, and oncogene c-Myc alteration. Immunological alterations in tumor microenvironment (TME) and proliferation despite CDK suppression are further potential mechanisms.
4.1. Genetic Alterations Involving Cell Cycle Regulation
Mutational signatures linked to apolipoprotein B mRNA-editing enzyme, catalytic subunit 3 (APOBEC3) enzymes are over-expressed in HR+ cancers following therapy compared to cases without therapy. These APOBEC3 mutations independently joined with shorter PFS in patients receiving a combination of anti-estrogen therapy and CDK4/6is for HR+ relapsed breast cancer. Whole genome sequencing (WGS) of experimental breast cancer and chosen samples from primary and relapsed cancers revealed that active APOBEC3 mutations accounted for resistance to endocrine and targeted therapies, primarily through RB1 mutations inducing its impaired function [74]. In a study, the capability of CDK4/6is in relapsed breast cancer with ESR1m identified through the collection of circulating tumor DNA (ctDNA) was assessed. The authors concluded that ESR1 variants are not linked to pan-CDK4/6 inhibitor resistance, supporting the hypothesis that combining CDK4/6 blockade with a SERD may be an effective strategy for managing ESR1m metastatic breast cancer [75]. Overexpression of the CCND1 gene, which encodes cyclin D1 (the primary partner of CDK4/6), was commonly observed in breast cancer that do not respond to CDK4/6is [76]. Similarly, overexpression of the CCNE1 gene, encoding cyclin E1 (a crucial cofactor for CDK2 essential to induce RB hyper-phosphorylation), joined with lower responses to palbociclib in the PALOMA-3 trial [77]. Activating mutations in crucial sites as the ATP-binding pocket of CDK4/6 even were thought to be implicated in resistance [73]. Furthermore, impairing mutations or loss of the RB1 gene have been reported in breast cancer subjects with low response to CDK4/6is [78,79]. Tumors with germline (g) BRCA2 mutations or with homologous recombination deficiency (HRD) linked to gBRCA2, as well as baseline loss of heterozygosity (LOH) for RB1 exhibited a higher prevalence of RB1 loss-of-function mutations and demonstrated poor outcomes when treated with front-line combinations of CDK4/6is suggesting that using PARP inhibitors before initiating CDK4/6i therapy could help to intercept harmful RB1-loss pathways and mitigate the development of CDK4/6is resistance [80]. In another study, the copy number abnormality (CNA) profiles of early breast cancers from several cohorts were compared to the CNAs of metastatic breast cancers. In ER+/HER2- metastatic breast cancers, among the 21 genes more often with genetic aberration, focal amplification of TERT was linked to worse outcomes. For patients with available data on CDK4/6is prior to biopsy collection, seven genes were identified on post-treatment biopsies. Notably, CDK4 was over-expressed in 9.8% of the pre-treated ER+/HER2- relapsed breast cancer subjects, unlike only 1.5% in the untreated ER+/HER2- metastatic cohort (P = 2.82E-04), as well as in the three early breast cancer population. Moreover, CDK4 over-expression joined with poor outcomes in ER+/HER2- early breast cancers [81]. In proliferating cells, the E3 ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C) is relevant in preventing premature entry into the S phase. A study found that the APC/C inhibitor, EMI1, is essential for allowing to APC/C function to prevent S phase entry in cells blocked by Palbociclib inhibition. This suggests that over-expressing EMI1 cancers are likely to avoid CDK4/6 inhibition, leading to premature and under licensed S phase entry, which may result in increased genome instability [82].
4.2. Activation of Alternative Signaling Pathways
In a study conducted in ER+ HER2 relapsed breast cancer, subjects undergoing ET combined with palbociclib, a panel of miRNAs was examined. Seven miRNAs demonstrated a significant inverse correlation with PFS. Specifically, when PFS was less than six months, some miRNAs joined with poor outcomes. The multivariate analysis confirmed that miR-378e, miR-99b-5p and miR-877-5p, had a significant and independent effect on PFS. Based on the literature and bioinformatics, in most instances the PI3K/AKT/mTOR signaling, cell-division cycle regulators, cyclin D1 and CDKN1B among them as well as autophagy were primarily implicated [83]. Resistance to PI3K inhibitors is indicated by continued RB phosphorylation, which can be effectively addressed by combining a CDK inhibitor with a PI3K inhibitor [59]. Regarding activation of alternative pathways, aberrations in the AKT1, AURKA, and KRAS genes have been identified in HR+/HER2- breast cancer resistant to CDK4/6is. Specifically, functioning genetic aberrations or over-expression in the AKT1 and AKT3 genes are linked to decreased sensitivity to CDK4/6is [78]. Additionally, alterations in all three RAS family members, namely the oncogenic aberrations KRASG12D, HRASK117R, or NRAS amplification, occurred in HR+/HER2− breast cancer that showed poor responses to CDK4/6is [78]. Over-activation of the FGFR signaling pathway was observed in CDK4/6is-resistant breast cancer, with FGFR1/2 overexpression or amplification correlating with reduced sensitivity [84]. Moreover, lack or inactivating aberrations in the FAT1 tumor suppressor gene, which inhibits the Hippo molecular signaling, induced the enrichment of YAP/TAZ transcription factors on the CDK6 promoter, leading to amplification and CDK4/6is resistance in ER+ BC [85]. In a study, the role of the G protein-coupled estrogen receptor (GPER) in the resistance of breast cancer cells to palbociclib was investigated. It was found an ER-alpha under expression associated with a GPER over-expression promoted by the EGFR interacting with the promoter region of GPER. It was also noticed that palbociclib activates pro-inflammatory transcriptional events through GPER signaling in CAFs and co-culture assays confirmed its role in reducing sensitivity to palbociclib and that it also facilitates the functional interaction between breast cancer cells and CAFs [86]. In a separate study, breast cancer cells termed MCF7-FAR and T47D-FAR, refractory to fulvestrant and abemaciclib association were developed. Both MCF7-FAR and T47D-FAR cells exhibited hyperactivation of EGFR, HER2, and AKT signaling and cetuximab recovered tumor response to fulvestrant and abemaciclib in FAR and EGFR-over-expressing breast cancer spheroids and xenografts [87]. A proteogenomic analysis of 22 patient-derived xenografts (PDXs) from ER+ breast cancer revealed that PKMYT1 is regulated by estradiol (E2) and that an intrinsic expression occurs in cases where proliferation is E2-independent. In samples of cancer patients, PKMYT1 mRNA over-expression was linked to resistance against ET and CDK4/6 inhibition. The PKMYT1 antagonist lunresertib (RP-6306) sinergyzed with gemcitabine and particularly reduced the viability of ER+ breast cancer cells refractory to endocrine therapy and palbociclib, even in the lack of activated p53 [88]. In a further study, tumor together with blood samples were collected before the start of combined CDK4/6is and ET, as well as at the disease progression. The most commonly acquired alterations included mutations in PIK3CA and TP53, along with amplification of the pyruvate dehydrogenase lipoamide kinase enzyme 1 (PDK1). Notably, rising levels of PIK3CA-mutated ctDNA were identified 4 to 17 months prior to any imaging results [89]. In another research the circHIAT1/miR-19a-3p/CADM2 axis affected epithelial to mesenchymal transition (EMT) and resistance to palbociclib with circHIAT1 and CADM2 down-regulation in breast cancer tissues and cell lines and miR-19a-3p up-regulation. Particularly, palbociclib-resistant breast cancer cells showed similar trends and overexpressing circHIAT1 in cells restored their sensitivity to palbociclib. Also, the bioactive flavonoid quercetin re-sensitized breast cancer cells to palbociclib. The anti-tumor effects of quercetin appeared to stem from its capability to govern the circHIAT1/miR-19a-3p/CADM2 axis, mainly by the up-regulation of circHIAT1 [90].
4.3. Modifications in Transcriptional and Epigenetic Regulators
In breast cancer, enhanced function of pro-aggressive transcription factors like NF-κB, AP-1, and E2F was reported to join with resistance to CDK4/6is [73]. In luminal A breast cancer subtype, a research revealed that CDK4/6is decrease the expression of the cystine carrier SLC7A11 by counteracting SP1 binding to the SLC7A11 promoter region. In experimental and clinical studies genetic and pharmacological impediment of SP1 or SLC7A1 led to improved response to CDK4/6is and a synergistic reduction in cancer proliferation [91]. In another research, the microphthalmia-associated transcription factor (MITF) became active through O-GlcNAcylation by O-GlcNAc transferase (OGT) in breast cancer cells refractory to palbociclib. Inhibiting either MITF or its O-GlcNAcylation recovered sensitivity of the refractory cells to palbociclib. Additionally, in samples from cancer patients either resistant or undergoing palbociclib, MITF activation was observed [92]. Elevated histone deacetylase (HDAC) activity joined with CDK4/6is tolerance, likely by p21-mediated cell cycle arrest and survival pathways stimulation [93,94]. Additionally, a study found that miR-432 could increase CDK6 by inhibiting TGF-β signaling through SMAD4 decrease therefore, reducing the effectiveness of CDK4/6is [95]. In a research, the lncRNA TROJAN led to CDK2 upregulation by linking to NKRF and blocking its inhibition on RELA/p65, so contributing to decreased response of ER+ breast cancer cells to CDK4/6is [96].
4.4. Acquired CDK6 Amplification
CDK6 hyper-expression can also account for reduced sensitivity to CDK4/6is in breast cancer [97] although it is not clear if this depends on only partial pharmacological inhibition of CDK6 or other kinase-independent activities of CDK6 [98]. Up-regulation of CDK6 may follow treatment with CDK4/6is, and in pre-clinical studies response can be restored through subsequent suppression of CDK6. Recently in a clinical investigation it was demonstrated a negative correlation of CDK6 over-expression and PFS in ER+ breast cancer subjects undergoing CDK4/6is [99]. Even if rare, loss-of-function mutations in the cadherin superfamily member FAT1 [100] join with insensitivity to CDK4/6is, possibly by CDK6 over-expression in ER+ breast cancer patients [85]. Notably, loss of function or removal of FAT1 activates the Hippo pathway [101] that clearly governs apoptosis and cell proliferation [102]. As a result, FAT1 loss promotes increased activity of the Yap/Taz transcription factors, which favours CDK6 amplification. Patients where biallelic FAT1 is not active have a PFS of 2.4 months, in contrast to ER+ breast cancer patients with FAT1 missense mutations showing PFS of 10.1 months that is a slightly shorter than 11.3 months for patients with wild-type FAT1 [85]. The exosomal miRNA-432–5p contributes to the arising of lack of response to CDK4/6is by CDK6 amplification, SMAD4 decvrease, and ultimately lowering G1/S cell-division cycle arrest [95]. The interruption of palbociclib reduces CDK6, cyclin D1-encoding CCND and miRNA-432–5p, while concurrently up-regulates Rb. Thus, ER+ breast cancer cells refractory to palbociclib can be regulated in vitro and in vivo experimental models [95], this may explain why some ER+ breast cancer subjects who progress during a specific CDK4/6i then might become sensitive to another CDK4/6i.
4.5. Oncogene c-Myc Alteration
The c-Myc gene takes part of the MYC family and is highly expressed in breast cancer [103,104]. CDK2, CDK4, and CDK6 activate Myc and its levels are elevated in preclinical models exhibiting resistance to CDK4/6is [105]. Accordingly, the nextMONARCH-1 clinical trial showed an increase in Myc genomic alterations following abemaciclib given alone or in association with a nonsteroidal AI [106]. Furthermore, activation of the S6K1 kinase has been identified in over 10% of ER+ breast cancer subjects, and several researches indicate that high S6K1 levels can promote insensitivity to palbociclib by triggering c-Myc signaling in both experimental models and samples from breast cancer patients [107].
4.6. Immunological Alterations in Tumor Microenvironment (TME)
Experimental and clinical findings suggests that CDK4/6is promote several immune effects that at least in part account for their efficacy [108,109]. By this also it can be inferred that in HR+HER2- breast cancer subjects, not yet well-defined immunological mechanisms may be responsible of low sensitivity to CDK4/6is. Here some immunological alterations more often observed in association with CDK4/6is are considered along with others more likely related to the arising of CDK4/6is resistance. In a research [110], it has been found that PD-L1 amount depends on cyclin D-CDK4 and the cullin 3-SPOP E3 ligase via proteasome-mediated deterioration. CDK4/6is in vivo enhances PD-L1 by inhibiting cyclin D-CDK4-mediated phosphorylation of speckle-type POZ protein (SPOP) therefore favoring SPOP deterioration by the anaphase-promoting complex activator FZR1. Accordingly, mutations rendering SPOP inactive impair ubiquitination-mediated PD-L1 deterioration therefore accounting for high PD-L1 levels and decrease of tumour-infiltrating lymphocytes (TILs) in mouse tumours and in primary samples from prostate cancer patients. Particularly, therapy with CDK4/6is given with anti-PD-1 increased tumour sensitivity and importantly improved OS rates in mouse tumour models. Otherwise, it has been similarly reported that CDK4/6is may sinergyze with the immune checkpoint blockade agents and may have inhibiting effects on TILs thus supporting the clinical use of CDK4/6is plus anti PD-1 or anti CTLA-4 antibodies [111]. Another experimental study [112] was conducted based on parental breast cancer cells and their derivatives that were insensitive to endocrine therapy (EndoR). Parental and estrogen deprivation-refractory MCF7 and T47D cells were used to get derivative breast cancer cells that had become insensitive to palbociclib (PalboR). By a transcriptomic evaluation of these cell lines an "IFN-related palbociclib-resistance Signature" (IRPS) was identified. In two clinical trials where CDK4/6is plus ET were given in the neoadjuvant setting, IRPS and other IFN-related signatures were over-expressed in a cancer population who was intrinsecally insensitive to CDK4/6is. In primary ER+/HER2- tumors, the IRPS score was significantly increased in lum B more than in lumA subtype and showed a positive correlation with amplification of immune checkpoints genes, lack of response to endocrine therapy and poor clinical outcome. In a further experimental study [113] the immunological processes accounting for the arising of insensitivity to CDK4/6is were deeply investigated. An in silico evaluation of the Cancer Genome Atlas (TCGA) was carried out. Moreover, 3 different cohorts of HR+HER2- breast cancer subjects including tumor samples collected over time were examined. An increase in interleukin 17 (IL17) producing gamma/delta T cells occurred in mouse HR+HER2- mammary tumors following CDK4/6 inhibition. In these tumors, circulating IL17 levels joined with poor clinical outcome while inhibiting the gamma/delta TCR, neutralizing IL17 or CCL2 similarly improved the response to CDK4/6is. These findings were confirmed in patients from the TCGA. In fact, patients with an active IL17 signature showed poor clinical outcome along with immunosuppression in the TME. In HR+HER2- breast cancer patients, gamma/delta T cell infiltration in tumor tissue samples correlated with tumor grade, and gamma/delta T cells were placed close to PD-L1+ tumor cells and macrophages.
4.7. Proliferation Mechanisms Despite CDK Suppression
The inherent plasticity of cell-division machinery allows breast cancer cells to divide even when all CDKs involved in interphase are inhibited. Moreover, mice without several CDKs can survive by relying on CDK1. Therefore, it was explored whether breast cells exposed to a combination of CDK2is and CDK4/6is might adopt this specific mechanism [114]. While cells undergoing CDK2i PF3600 [115] can proliferate potentially by utilizing CDK1, combining CDK2i and CDK4/6i impedes that this occurs [116]. In brief, this potential process could be addressed by inhibiting CDK7, which serves two critical functions: 1) as a cycle dependent-activating kinase that phosphorylates CDK1, CDK2, CDK4, and CDK6 and 2) acting as an in-between molecule in RNA polymerase II-mediated transcription [117]. Accordingly, specific CDK7is like samuraciclib have shown therapeutic efficacy in HR+/HER2- advanced breast cancer population resistant to CDK4/6is [118]. Furthermore, enhanced cellular growth alone dictates the response to a CDK7i which helps to explain why there are some cancers sensitive to CDK inhibition more than normally proliferating cells [119]. The main mechanisms of resistance to ET and/or CDK4/6is in advanced ER+/HER2- breast cancer are shown in Figure 2.
5. Common Therapeutic Strategies to Overcome Resistance to ET and/or CDK4/6is
After progression on endocrine plus CDK4/6is therapy, there is no established standard systemic treatment. In patients who maintain an ER dependent signaling, viable options include: a) switching to a different endocrine monotherapy; b) using ET combined with everolimus (an mTOR inhibitor). Additionally, for patients with somatic PIK3CA mutations, ET can be paired with either alpelisib (a PI3K inhibitor) or capivasertib (an AKT inhibitor). In patients who have lost ER dependent signaling, cytotoxic chemotherapy is an alternative option [3,4,5], while continuing CDK4/6is beyond progression, although experimental, is a further choice. In the principal MONALEESA-2/7, MONARCH-3, and PALOMA-1/2 randomized trials, subjects progressing on 1st-line CDK4/6is plus ET were given endocrine monotherapy, chemotherapy or a different CDK4/6is on average in 65%,44% or 18% respectively of cases, and ET plus mTOR inhibitors in 17% of cases [120]. Anti-estrogen therapy alone obtained a short PFS (less than 3 months) [121].
5.1. Fulvestrant, a SERD and Novel Oral SERDs
SERDs are non-steroidal compounds that function as both competitive ER antagonists and inducers of ER degradation via the proteasome. Fulvestrant, the first SERD approved for ER+ metastatic breast cancer, is administered monthly via im. injection [122]. Fulvestrant as second-line ET combined with other different drugs like CDK 4/6is, alpelisib, and everolimus showed to be efficacious in a few randomized trials [28,68,69,71,123,124]. In phase III trials, fulvestrant, given alone after progression on CDK4/6is, was evaluated as a control arm with a limited median PFS (1.9 to 4.7 months, range) [125,126]. ESR1 mutations occur in 25%-40% of patients who were given an AI. In the phase III PADA-1 study, fulvestrant replaced an AI when ctDNA-detected ESR1 mutations occurred before radiological progression; in patients treated with fulvestrant plus CDK4/6is and with AI plus CDK4/6is PFS was 11.9 and 5.7 months respectively [127]. Elacestrant is an oral hybrid SERM/SERD. In EMERALD phase III trial, participants, who previously had undergone 1 or 2 lines of ET plus a CDK4/6i and up to one prior line of chemotherapy, randomly received either elacestrant or standard of care (SOC) with fulvestrant or an AI. Elacestrant compared with SOC significantly increased PFS in subjects with ESR1 mutations and in the overall population [128], suggesting oral SERD for those maintaining some hormone responsiveness following ET plus CDK4/6is. An updating indicated a greater PFS benefit (5.45 and 3.29 months) for patients who had a longer exposure to CDK4/6is, particularly among those with ESR1 mutations [129]. An interim OS analysis favored elacestrant in the ESR1-mutant population unlike non-mutant subgroup. Based on this, elacestrant has obtained FDA approval in post-menopausal women or adult men with ER+HER2- ESR1-mutated advanced or relapsed breast cancer progressing after at least one line of ET. Elacestrant is under evaluation in combination with other drugs (CDK4/6is, everolimus, alpelisib, samuraciclib) in some clinical trials (NCT05963997, NCT06062498, NCT06382948, NCT05563220). The ELEVATE (NCT05563220) trial is a phase Ib/II umbrella study where the clinical performance of elacestrant joined with either alpelisib, capivasertib, everolimus, palbociclib, abemaciclib, or ribociclib is assessed in ER+/HER2- advanced/relapsed breast cancer [130]. ADELA (NCT06382948) is a phase III trial where elacestrant combined with everolimus is compared to elacestrant alone in ER+/HER2-advanced breast cancer population with ESR1-mutated tumors that have progressed on ET and CDK4/6i. Patients must have received at least one and no more than two lines of ET for advanced breast cancer. Patients receiving a CDK4/6i in the adjuvant setting are eligible only if disease progression is diagnosed after ≥ 12 months of treatment but <12 months following CDK4/6i treatment completion. Camizestrant (AZD9833) is another oral SERD, pure antagonist of ER-alpha. Camizestrant versus fulvestrant was assessed in the SERENA-2 phase II trial. In contrast to the EMERALD trial, no patient received more than one line of ET; only 50% of them received CDK4/6is, and no patient received a prior treatment with fulvestrant. Camizestrant significantly prolonged PFS with the 75 mg (7.2 compared to 3.7 months) and the 150 mg dose (7.7 versus 3.7 months) also in ESR1-mutated patients. Moreover, camizestrant at both doses decreased ESR1 mutant ctDNA to not measurable or close to not measurable levels [131]. The phase III SERENA-6 trial evaluates a switch to camizestrant plus the same CDK4/6i versus the continuation of a non-steroidal AI plus CDK4/6i (palbociclib or abemaciclib) in HR+/HER2- advanced or relapsed breast cancer population who have measurable ESR1 mutations in ctDNA during 1-line treatment, before clinical/radiological progresssion (NCT04964934) [132]. Imlunestrant is a SERD which showed 6.5 months as median PFS (3.6–8.3 months, range) when tested as second-line treatment after ET plus CDK4/6i. The EMBER-3 study is a phase III trial where imlunestrant alone is compared with imlunestrant in association with abemaciclib and with an investigator’s choice hormone therapy as 2nd-line therapy after AI or AI plus a CDK4/6i. Imlunestrant alone obtained a significantly longer PFS than conventional therapy in a population with ESR1 mutations but not in the entire sample size. Imlunestrant in combination with abemaciclib unlike imlunestrant alone significantly prolonged PFS irrespective of ESR1 status [133]. Giredestrant (GDC-9545) is a SERD active either as monotherapy or in association with a CDK4/6i in ESR1 mutant or wild-type tumor models [134]. The phase II acelERA Breast Cancer Study evaluated ER+ HER2- relapsed breast cancer patients progressing after 1-2 lines of systemic therapy, including ET for at least 6 months and a targeted agent; one previous line of chemotherapy and fulvestrant were allowed; patients were randomly allocated to giredestrant or fulvestrant/AI (physician’s choice of hormone monotherapy, PCET) until disease progression/unacceptable toxicity. After 7.9 months as median follow-up, in the 303 recruited patients median PFS was 5.6 months in the giredestrant arm vs 5.4 months in the PCET arm. At 6 months 46.8% and 39.6% were the PFS rates in the giredestrant and PCET arms, respectively. Among the 90 ESR1 mutated patients, median PFS was 5.3 months versus 3.5 months in the giredestrant and PCET arms, respectively. Despite the primary endpoint did not attain statistical significance, authors concluded that giredestrant had showed therapeutic efficacy in most subgroups and a trend toward a benefit in the population having ESR1 mutations [135]. Trials evaluating giredestrant alone or in combination with other drugs are ongoing (NCT04802759).
5.2. PI3K/AKT/mTOR Pathway Inhibitors
PIK3CA somatic mutations occur in about 30%–50% of relapsed HR+/HER2− breast cancer population [136]. In the phase III SOLAR-1 trial, alpelisib, a PI3Kalpha-specific inhibitor, was evaluated in association with fulvestrant in patients resistant to prior ET. In patients with PIK3CA mutations, the combination, unlike fulvestrant alone, significantly prolonged PFS but not OS (11.0 vs. 5.7 months respectively) [137]. However, in this trial only 6% of subjects were previously given a CDK4/6i. The phase II BYLieve trial confirmed that adding alpelisib to ET is effective for relapsed HR+/HER2− breast cancer subjects carrying a PIK3CA mutation and progressing on CDK4/6is and ET [138,139]. The phase II FAKTION trial recruited women with HR+/HER2− relapsed breast cancer progressing after or during AI and with no prior CDK4/6i; patients received the AKT inhibitor capivasertib plus fulvestrant, and a significant PFS and OS benefit occurred in those carrying mutations in PTEN, AKT1 or PIK3CA genes [140,141]. The phase III CAPItello-291 trial enrolled patients progressing on AI plus a CDK4/6i; subjects could have received 1-2 lines of ET, and no more than one line of chemotherapy in the advanced/relapsed setting; patients who received a prior treatment with fulvestrant, AKT, PI3K and mTOR inhibitors were excluded; the enrolled subjects were or were not carriers of mutations in the genes of the PIK3CA-AKT-PTEN pathway. The capivasertib–fulvestrant association doubled the median PFS against the placebo given with fulvestrant (median PFS 7.2 vs. 3.6 months in the overall population; 7.3 months vs 3.1 months in the AKT pathway mutated population) [142]. Inavolisib is a PIK3CA inhibitor that also induces the deterioration of the alpha isoform of the p110 catalytic subunit of PIK3CA. Inavolisib showed to synergize with palbociclib and fulvestrant in experimental tumor models. INAVO-120 (NCT04191499) is a phase III randomized study where the association of inavolisib with palbociclib and fulvestrant was compared with that of placebo plus palbociclib and fulvestrant as 1st line treatment in subjects with PIK3CA-mutated, ER+/HER2- advanced or relapsed breast cancer who had recurred during or within one year after adjuvant ET. The median PFS was 15.0 months and 7.3 months in the inavolisib arm and in the placebo arm respectively [143]. INAVO 121 will compare inavolisib plus fulvestrant with alpelisib plus fulvestrant in a population with PIK3CA-mutated, ER+/HER2- advanced or relapsed breast cancer progressing after ET plus CDK4/6is (NCT05646862). Other AKT inhibitors such as ipatasertib are under evaluation in trials following the occurrence of resistance to CDK4/6is [144] (NCT04650581). The BOLERO-2 evaluated the mTOR inhibitor everolimus in patients progressing on an AI [35]. Everolimus administered in association with exemestane prolonged the median PFS (10.6 vs. 4.1 months), but not OS [145]. Although no patient in this study had been previously given a CDK4/6i, the use of ET combined with everolimus after progression on CDK4/6is is suggested by several researches [146,147]. Dual PI3K and mTOR inhibitors are also under evaluation. Particularly, a phase III trial (VIKTORIA-1, NCT05501886) is evaluating gedatolisib, an intravenously administered pan-PI3K/mTOR inhibitor [148], in combination with fulvestrant with or without palbociclib in advanced or metastatic HR+/HER2- breast cancer progressing on or after aromatase inhibitor plus CDK4/6i.
5.3. Antibody-Drug Conjugates (ADCs)
ADCs are composed of antibodies that target tumor antigens conjugated to a chemotherapeutic agent [149,150]. Upon delivery of the cytotoxic agent to breast cancer cells, the cleavable linker provokes a bystander killing, as it affects neighboring cancer cells, even if they do not express the target antigen [151]. The HER2-targeted ADC trastuzumab deruxtecan is engineered by linking a humanized anti-HER2 monoclonal antibody to a topoisomerase I inhibitor payload via a tetrapeptide cleavable linker. In the DESTINY-Breast04 phase III trial, it significantly improved outcomes in HR+ HER2 low (1+ or 2+ score by immuno-histochemistry [IHC] and negative in situ hybridization), advanced or relapsed breast cancer subjects progressing after up two different regimens of chemotherapy. The trial reported a PFS of 10 and an OS of 23.9 months compared to 5.4 and 17.5 months respectively with physician’s choice chemotherapy. Notably, patients previously treated or not with CDK4/6is experienced a comparable PFS benefit [152]. Therefore, the drug received FDA approval for this population. DESTINY-Breast-06 is a phase III trial involving ER+/HER2 low or ultra-low (IHC 0 with membrane staining) metastatic breast cancer patients progressing after one or more lines of ET with no previous chemotherapy for metastatic disease. Most of these patients received prior CDK4/6is. Patients were given trastuzumab deruxtecan or one among capecitabine, or paclitaxel, or nab-paclitaxel at the physician’s choice. Seventy hundred thirteen of the 866 enrolled were HER2-low and 153 were HER2-ultra-low subjects. Considering HER2 low population, the median PFS was 13.2 months in the trastuzumab deruxtecan arm and 8.1 months in the chemotherapy arm [153]; similar findings were described in HER2-ultra-low patients [154]. Sacituzumab govitecan, another ADC, is an antibody against the transmembrane glycoprotein trophoblast cell-surface antigen 2 (Trop-2) linked to SN-38, a topoisomerase I inhibitor [155]. Trop-2 amplification occurs in various malignancies and in more than 90% of ER+/HER2- breast cancer cells [156]. The TROPiCS-02 phase III trial recruited ER+/HER2- breast cancer patients progressing after three different lines of systemic therapy, a CDK4/6i among them; in this setting, a significant prolonging of PFS to 5.5 months occurred with sacituzumab govitecan, versus 4 months with the physician’s choice chemotherapy; the clinical benefit was independent of Trop-2 expression. Following these results sacituzumab govitecan received approval by the FDA for this ER+ patient cohort [157,158]. In the recent TROPION-PanTumor01 phase I trial, datopotamab deruxtecan, an ADC similar to trastuzumab deruxtecan, showed promising activity and low toxicity in heavily pretreated ER+/HER2- breast cancer subjects, most of them treated with a CDK4/6i [159]. The TROPION-Breast-01 phase III trial evaluated the clinical performance of datopotamab deruxtecan versus one among eribulin or vinorelbine or capecitabine or gemcitabine at investigator’s choice in advanced breast cancer subjects progressed on ET and after 1-2 prior lines of chemotherapy; those who were given datopotamab deruxtecan showed statistically significant prolonged median PFS (6.9 vs 4.9 months) and a good safety profile [160,161].
6. Other Therapeutic Strategies
6.1. Continuing CDK4/6is
In the MAINTAIN phase II trial, HR+/HER2- relapsed breast cancer population progressing during ET plus CDK4/6i changed the ET (to fulvestrant or exemestane) and were randomly allocated to treatment with ribociclib or placebo. Of the 119 recruited patients, 103 (86.5%) had been previously given palbociclib and 14 (11.7%) ribociclib. Median PFS was 5.9 months in patients allocated to switched ET plus ribociclib arm, versus 2.76 months in whom allocated to switched ET plus placebo arm. The benefit was mainly seen in the ESR1 wild-type subset; however, a small sample size were ESR1 mutant patients, with a higher frequency of CCND1 and/or FGFR1 gene amplifications [125]. In contrast, in the phase II PACE trial only a slightly prolonged PFS occurred in patients carrying ESR1 or PIK3CA mutations when palbociclib with fulvestrant were continued after progression [162]. Likewise, in the PALMIRA trial, continuing palbociclib beyond progression in combination with a 2nd–line ET did not prolong PFS in comparison with only ET as 2nd line [163]. Perhaps, switching to a different CDK4/6i is to be preferred rather than maintaining the same agent; different mechanisms of action and resistance among different CDK4/6is could account for the differing outcomes across these trials. The phase III post-MONARCH trial is conducted on ER+/HER2- advanced or relapsed breast cancer population progressing on ET plus CDK4/6i as first line treatment. Subjects received abemaciclib + fulvestrant or placebo + fulvestrant. Nearly all enrolled subjects had been mainly treated with palbociclib and ribociclib in the advanced setting. Following a median follow-up of 13 months, the median PFS was 6.0 months in subjects receiving abemaciclib + fulvestrant and 5.3 months in those who were given fulvestrant + placebo. The PFS benefit was irrespective of ESR1 or PIK3CA mutations [164]
6.2. Next-Generation Endocrine Agents
Complete estrogen receptor antagonists (CERANs), SERMs and SERM/SERD Hybrids (SSHs), selective estrogen receptor covalent antagonists (SERCAs), selective human ER partial agonists (ShERPAs) and PROteolysis Targeting Chimeras (PROTACs) are new anti-estrogens [165]. Selective androgen receptor modulators (SARMs) also are novel endocrine agents. Most of them are still under clinical development.
6.2.1. CERANs
CERANs circumvent endocrine resistance in breast tumors by inactivating the two distinct transcriptional activation domains of the ER, the activation function 1 and 2 (AF1 and AF2). AF1 is activated through various pathways like mTOR, PI3K, and MAPK, while AF2 activation occurs through the estrogen ligand itself. While SERDs and SERMs mainly target AF2, CERANs target both AF1 and AF2 [166]. Palazestrant (OP-1250) is an oral CERAN that induced decrease of both wild-type and mutant ER breast tumors in xenograft models [167]. Currently, it is on evaluation in phase I/II trials (NCT05266105, NCT06016738).
6.2.2. SSHs
SERMs act as ER antagonists by inhibiting AF2, while also showing agonist effects through AF1 depending on cell type and involving diverse co-activators and co-repressors. In an experimental model of AI-resistant breast cancer, lasofoxifene alone or in combination with palbociclib inhibited tumor growth more than fulvestrant, independent of ESR1 mutation [168]. The phase II ELAINE-1 trial was conducted in HR+/HER2− relapsed breast cancer, 40% of subjects having the Y537S ESR1 mutation, and progressing on prior AI and CDK4/6is; lasofoxifene given as single agent against fulvestrant did not significantly prolong PFS [169]. The ELAINE-2 trial examined lasofoxifene plus abemaciclib in pre-treated subjects with ESR1-mutated metastatic breast cancer. The clinical benefit rate (CBR), ORR and median PFS were 65.5%,55.6% and about 13 months respectively [170]. The phase III ELAINE-3 study is assessing lasofoxifene + abemaciclib versus fulvestrant + abemaciclib in metastatic subjects with ESR1 mutation progressing during AI plus palbociclib or ribociclib (NCT05696626) [171] The SERM/SERD bazedoxifene showed clinical efficacy as anticancer agent in HR+ endocrine-resistant breast cancer models, particularly in Y537S ESR1 mutation carriers [172]. A phase Ib/II trial reported a CBR of 33.3% for bazedoxifene and palbociclib in these subjects regardless of ESR1 mutation [173].
6.2.3. SERCAs
SERCAs neutralises ER by targeting a specific cysteine residue and favoring morphological changes. In experimental models, H3B-6545 antagonized both the wild-type and mutant ER and showed greater anti-cancer activity than fulvestrant [174]. A phase I/II trial enrolled HER2− relapsed breast cancer subjects who had already received CDK4/6is (87%), fulvestrant (71%), or chemotherapy (54%), 58% of them carrying ESR1 mutations. An ORR of 16.4% and a median PFS of 3.8 months occurred in the preliminary analysis with a manageable safety profile [175].
6.2.4. ShERPAs
ShERPAs mimic the action of beta-estradiol binding to ER within the nucleus and prompting its extranuclear translocation thus inhibiting the growth of ER+ tumor cells. ShERPAs showed promising efficacy in tamoxifen-refractory breast cancer cells and in xenograft models [176]. TTC-352 showed anti-tumor activity in a phase I study in subjects progressing following two or more lines of ET including one line with a CDK4/6i [177].
6.2.5. PROTACs
PROTACs are bifunctional molecules designed to bind a particular target protein, like the ER while concurrently recruiting an E3 ubiquitin ligase. A PROTAC, by bringing the target protein and E3 ligase together, enables the ubiquitylation of the target, which then leads to its deterioration via the ubiquitin-proteasome system. Following this, it initiates another degradation cycle [178]. Vepdegestrant (ARV-471) has shown to be an anticancer drug superior to fulvestrant in xenograft models [179]. The phase I/II VERITAC trial enrolled patients who had previously received anti-estrogens and CDK 4/6is; vepdegestrant showed a good clinical activity in the whole studied population and in subjects carrying ESR1 mutations (CBR up to 38.9 and 54.5% respectively) [180,181]. In pretreated HR+ HER2- patients, vepdegestrant plus palbociclib showed a good activity (CBR 63% in ITT subjects and 72.4% in ESR1 mutants) [182]. The phase III VERITAC-2 trial (NCT05654623) is evaluating the clinical performance of vepdegestrant compared with fulvestrant in subjects progressing following first-line CDK4/6 inhibitor and ET. Other trials are evaluating vepdegestrant in combination with various drugs, including CDK4/6is (NCT06125522, NCT06206837, NCT05548127, NCT05909397, NCT05654623, NCT05573555, NCT04072952). AC682 is a chimeric compound that given alone or plus CDK4/6is or PI3K/mTOR pathway inhibitors showed anti-cancer activity in experimental ER+ breast cancer models, including those with ESR1 mutations, [183]. A phase I trial is currently underway (NCT05080842).
6.2.6. SARMs
The androgen receptor (AR) is a steroid nuclear receptor commonly expressed in HR+HER2− breast cancer [184] and SARMs act as either AR agonists or antagonists. AR may have different roles in ER+ compared to ER-negative breast cancers. AR in HR+ breast cancer correlates with a favorable prognosis and AR agonism inhibits the progression of both endocrine-responsive and -refractory breast cancers [185,186]. Enzalutamide, a nonsteroidal anti-androgen, in combination with exemestane versus exemestane alone was tested in a randomized phase II trial but did not prolong PFS in subjects previously receiving ET [187]. Recently, it was found that the AR to ER ratio in breast cancer influences the sensitivity to AR-targeted treatments, suggesting the potential utility of enzalutamide in ER+ tumors with a low AR/ER ratio, and AR agonists like RAD140 in those with a high AR/ER ratio [188]. In a phase II trial, enobosarm, a novel oral selective AR activator, achieved a CBR of 32% and 29%, depending on the dose used. Furthermore, the ORR was 48% and 0% in patients with over or less than 40% AR staining respectively [189]. Randomized phase III trials are ongoing (NCT05065411) [190].
6.3. Agents Targeting CDK7 and CDK2
CDK 7 is a component of the CDK-activating kinase (CAK) complex and plays dual role in transcription and cell-division cycle advance [191]. The anti-cancer efficacy of CDK7 inhibition in ER+ breast tumor are partially dependent on p53 and involve both cell cycle arrest and c-Myc suppression. Unlike the cytostatic effects seen with ET and CDK4/6is, CDK7 inhibition appears to exert cytotoxic effects. It also reduces ER phosphorylation at S118, although extended CDK7 inhibition can lead to increased ER signaling. Elevated c-Myc activity and intact p53 may serve as potential predictors of sensitivity to treatments based on CDK7is [192]. CDK7is showed significant anti-cancer activity especially in TNBC and HR+ breast cancers [191]. The CAK complex activates p53, which is involved in DNA repair, therefore CDK7is may be effective against aggressive luminal tumors with TP53 alterations [193]. Samuraciclib (ICEC0942), a selective CDK7i, blocked the cell cycle and promoted apoptosis in experimental studies [194]. In HR+/HER2− relapsed breast cancer population progressing after an AI plus a CDK4/6i, samuraciclib plus fulvestrant was responsible of 36% CBR [118]. Samuraciclib is under evaluation, alone or in combination, in various clinical trials (NCT05963984, NCT05963997, NCT06125522). Cyclin E/CDK2 has an important role in CDK4/6is resistance [105,111] and targeting CDK2 either with or without responsiveness to CDK4/6is is under evaluation. In pre-clinical models, dinaciclib, a non-selective CDK2i, combined with palbociclib and letrozole, was more efficacious than palbociclib and/or letrozole only [105]. Other CDK2is are under investigation in phase I/II trials in ER+/HER2- breast cancer patients (NCT04553133, NCT05252416). In the VELA phase I trial (NCT05252416) the CDK2i BLU-222 administered alone in HR+/HER2- breast and further different advanced solid cancers showed clinical efficacy and low toxicity [195]. Likewise in another phase I/II trial (NCT04553133) the novel CDK2i PF-07104091 showed clinical efficacy in previously largely treated advanced/relapsed breast cancer population progressing during ET plus CDK4/6is [196]. Similarly, an ongoing phase 1/ 2 trial (NCT03519178) is evaluating the CDK2/CDK4/CDK6 inhibitor PF-06873600 alone or with letrozole or fulvestrant in the same population progressing after CDK4/6is, ET and ≤2 lines of chemotherapy.
6.4. Immune Therapy (IT)
IT includes ESRmut vaccines, beta-interferon-interleukin-2 sequence given in association with ET or treatment with macrophages inhibitors. Mutations in the ESR1 gene can account for neo-epitopes that could be targeted through IT. In in vitro cytotoxicity assays demonstrated that expanded antigen-specific CTLs could effectively lyse peptide-pulsed targets and breast cancer cells expressing five peptides as the most immunogenic candidates, derived from D538G, Y537S, and E380Q that is the three most prevalent ESR1 mutations [197]. Accordingly, in a study using high performance liquid chromatography (HPLC), the presentation of ESR1 and ESR1mut peptides on human MHC was shown in an ER+ breast cancer subject along with the presence of human T cells reactive to ESR1mut epitopes [198]. This observation supported the development of ESR1mut vaccines. Since early nineties, our research group discovered a synergism of beta-interferon-interleukin-2 sequence when administered in association with conventional ET in a ER+ endocrine dependent relapsed breast cancer population. Since then, a few patients were recruited in a pilot study where they received this IT first combined with tamoxifen which at that time was the 1-line standard ET and thereafter with AIs in some others. The initial findings were reported more times [199,200,201] and recently [202] the final results were discussed with 95 controls and 42 cases overall retrospectively compared. The 95 controls were ER+/HER2− relapsed breast cancer subjects who had undergone first-line ET with AIs or fulvestrant. Twenty-eight of them (28.9%) also were given biological drugs, CDK4/6is among them. The 42 cases were ER+ endocrine dependent relapsed subjects who were given beeta-interferon-interleukin-2 sequence plus 1st-line ET. In particular, 39 (92.9%) subjects received SERMs/SERDs and in the 3 remaining AIs were administered. Median PFS and OS were significantly prolonged in the 42 studied patients compared with the 95 controls (median time 33 vs. 18 months, p = 0.002, and 81 vs. 62 months, p = 0.019 respectively) (Figure 3). We hypothesized that “in responsive metastatic disease a stable or decreased tumor burden and a lower genetic instability due to the quiescent state (G0-G1 state) of tumor cells induced by antiestrogens is also likely to reduce the immune evasion and immune inhibition. This favors the immune attack stimulated by the concomitant immune therapy” [200]. This mechanistic rationale is widely different from that of CDK4/6is therefore the proposed association can be an alternative to CDK4/6is or a reasonable choice for patients who maintain ER signaling after they had progressed with ET and CDK4/6is. Table 1 summarizes the main characteristics of clinical studies carried out with current and experimental therapies in endocrine-CDK4/6is resistant advanced ER+/HER2- breast cancer.
6.5. Further Drugs and Targets
They comprehend FGFR inhibitors, MITF or MYSM1 inhibition, quercetin and targeting CXCR1/2 receptor.
FGFR inhibitors erdafitinib, lucitanib, and dovitinib have reported conflicting results, reflecting the complication of FGFR signaling in breast cancer. Thus, in the next studies defining biomarkers and combined treatments to improve the effectiveness of FGFR-targeted therapies should be prioritized [41]. Inhibition of MITF or its O-GlcNAcylation restores sensitivity to palbociclib in resistant cells. Therefore, targeting MITF is a potential strategy for managing CDK4/6 inhibitor-resistant breast cancer [98]. MYSM1, a deubiquitinase, is an epigenetic regulator of ERα activity and relevant in tumor growth. Silencing MYSM1 inhibits the increase of breast cancer-derived cells in xenograft models and increases their sensitivity to antiestrogens. A virtual screening identified Imatinib as a small molecule capable of interacting with the catalytic MPN domain of MYSM1, this effectively inhibited breast cancer cell growth highlighting the MYSM1-ERα axis as a promising target to overcome endocrine independence in breast cancer [21]. In-silico analyses likely indicate quercetin as CDK2 inhibitor in ER+ breast cancer [203] and that quercetin can re-sensitize breast cancer cells to palbociclib by governing circHIAT1/miR-19a-3p/CADM2 axis. So, quercetin can revert lack of sensitivity to palbociclib and can hinder EMT [96]. Endocrine-resistant breast cancer (ERBC) cells secrete CXCL1, which links with the CXCR1/2 on fibroblasts, activating the ERK/MAPK signaling and inducing CXCL1 amplification. CXCL1, in turn, interacts with CXCR1/2 on ERBC cells, further activating the ERK/MAPK pathway and promoting ERBC cell proliferation and spread thus highlighting CXCL1 as a key element in ERBC proliferation. Accordingly, reparixin in association with CDK4/6is is promising to counteract endocrine-refractory breast cancer progression and diffusion [204].
7. Discussion and Conclusions
In the last decade, the survival of recurred ER+ breast cancer population has improved and the CDK4/6is introduction into current clinical practice in addition to ET was a main novel therapeutic strategy. However, inevitably resistance develops over time and a better comprehension of the reasons accounting for resistance to anti-estrogens and/or CDK4/6is has paved the way for new drugs and treatment modalities. In patients maintaining an ER dependent signaling, fulvestrant, a SERD, as single agent after progression on CDK4/6is demonstrated a limited efficacy in phase III trials. Better results have been reported with fulvestrant given as second-line ET in addition to CDK4/6is, alpelisib, and everolimus. Namely, alpelisib, a PI3K inhibitor, plus fulvestrant has been approved for ER+/HER2-, PIK3CA mutated advanced breast cancer patients following prior ET. Capivasertib, an AKT inhibitor, plus fulvestrant has shown important improvement in PFS and OS in HR+/HER2− relapsed breast cancer subjects with mutated PTEN, AKT1, or PIK3CA genes and progressing after AI plus CDK4/6i. Other AKT inhibitors are under evaluation after progression during CDK4/6is. Everolimus, an mTOR inhibitor, is currently used in addition to exemestane in HR+/HER2− relapsed breast cancer population progressing after ET, with or without CDK4/6is. Meanwhile other SERDs have been investigated as elacestrant or camizestrant and imlunestrant which are currently under evaluation alone or in combination with different biological drugs. Among more recent SERMs, lasofoxifene in association with abemaciclib is under evaluation in relapsed subjects with ESR1 mutation progressing on an AI plus CDK4/6i. Vepdegestrant, a PROTAC, is currently assessed compared with fulvestrant in subjects progressing following 1st-line therapy with a CDK4/6 inhibitor and ET. Continuing CDK4/6is therapy after progression has shown contrasting results, while replacing it with another CDK4/6i may be more effective. However, the phase III PADA-1 study, conducted in patients submitted to 1st-line therapy with a CDK4/6i and an AI, showed an improvement in PFS in patients who replaced AI with fulvestrant when an early ESR1 mutation detection before radiological progression and continuing CDK4/6i therapy, occurred. Samuraciclib, a CDK7i, given in addition to fulvestrant showed efficacy in subjects with HR+/HER2− relapsed breast cancer progressing during an AI plus a CDK4/6i. Several CDK2is, alone or in combination with ET, are under investigation in phase I/II clinical trials in ER+/HER2- breast cancer patients. As to immunotherapy, ER+ breast cancer is commonly thought to be a “cold” tumor and checkpoint inhibitors did not show a significant activity. Despite this, since early nineties our research group discovered a synergism of beta-interferon-interleukin-2 sequence given in association with conventional ET in ER+ relapsed breast cancer population. The proposed mechanistic rationale of this IT is widely different from that of CDK4/6is, therefore its association with ET can be a reasonable first choice or an alternative for patients progressing after ET and CDK4/6is. In conclusion, overcoming resistance to ET is a “work in progress” and future studies are expected to better select patients for different therapeutic strategies based on more specific biologic and/or genetic markers. Liquid biopsy may provide a real-time portrait of the disease state with insights into disease biology, including processes responsible for ET independence and cancer proliferation. A few different liquid-based biomarkers with prognostic and predictive capabilities in ER+/HER2- breast cancer are going to be available in the current clinical practice [205,206]. Next-generation sequencing (NGS) is an important diagnostic tool, that can provide a comprehensive analysis of PI3K pathway alterations and other actionable markers, such as ESR1 and BRCA. Due to this, NGS of tumor or plasma samples should become the standard of care for patients with HR+/HER2 – MBC [207].
Funding
This paper received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; Perou, C.M.; Regan, M.M.; Rimm, D.L.; Symmans, W.F.; Torlakovic, E.E.; Varella, L.; Viale, G.; Weisberg, T.F.; McShane, L.M.; Wolff, A.C. Estrogen and Progesterone Receptor Testing in Breast Cancer: ASCO/CAP Guideline Update. J Clin Oncol. 2020, 38, 1346–1366. [Google Scholar] [CrossRef]
- Lumachi, F.; Santeufemia, D.A.; Basso, S.M. Current medical treatment of estrogen receptor-positive breast cancer. World J Biol Chem. 2015, 6, 231–239. [Google Scholar] [CrossRef]
- NCCN guidelines. Breast cancer, accessed March15. 2025.
- ESMO Clinical Practice Guidelines: Breast Cancer; accessed March 15; 15 March 2025.
- Ferro, A.; Campora, M.; Caldara, A.; De Lisi, D.; Lorenzi, M.; Monteverdi, S.; Mihai, R.; Bisio, A.; Dipasquale, M.; Caffo, O.; Ciribilli, Y. Novel Treatment Strategies for Hormone Receptor (HR)-Positive, HER2-Negative Metastatic Breast Cancer. J Clin Med. 2024, 13, 3611. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Costa, A.; Norton, L.; Senkus, E.; Aapro, M.; André, F.; Barrios, C.H.; Bergh, J.; Biganzoli, L.; Blackwell, K.L.; Cardoso, M.J.; Cufer, T.; El Saghir, N.; Fallowfield, L.; Fenech, D.; Francis, P.; Gelmon, K.; Giordano, S.H.; Gligorov, J.; Goldhirsch, A.; Harbeck, N.; Houssami, N.; Hudis, C.; Kaufman, B.; Krop, I.; Kyriakides, S.; Lin, U.N.; Mayer, M.; Merjaver, S.D.; Nordström, E.B.; Pagani, O.; Partridge, A.; Penault-Llorca, F.; Piccart, M.J.; Rugo, H.; Sledge, G.; Thomssen, C.; Van't Veer, L.; Vorobiof, D.; Vrieling, C.; West, N.; Xu, B.; European School of Oncology; European Society of Medical Oncology. ESO-ESMO 2nd international consensus guidelines for advanced breast cancer (ABC2). Breast. 2014, 23, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell. 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; Penson, A.; Shen, R.; Pareja, F.; Kundra, R.; Middha, S.; Cheng, M.L.; Zehir, A.; Kandoth, C.; Patel, R.; Huberman, K.; Smyth, L.M.; Jhaveri, K.; Modi, S.; Traina, T.A.; Dang, C.; Zhang, W.; Weigelt, B.; Li, B.T.; Ladanyi, M.; Hyman, D.M.; Schultz, N.; Robson, M.E.; Hudis, C.; Brogi, E.; Viale, A.; Norton, L.; Dickler, M.N.; Berger, M.F.; Iacobuzio-Donahue, C.A.; Chandarlapaty, S.; Scaltriti, M.; Reis-Filho, J.S.; Solit, D.B.; Taylor, B.S.; Baselga, J. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell. 2018, 34, 427–438.e6. [Google Scholar] [CrossRef]
- Cipolletti, M.; Acconcia, F. PMM2 controls ERα levels and cell proliferation in ESR1 Y537S variant expressing breast cancer cells. Mol Cell Endocrinol. 2024, 584, 112160. [Google Scholar] [CrossRef]
- De Marchi, T.; Lai, C.F.; Simmons, G.M.; Goldsbrough, I.; Harrod, A.; Lam, T.; Buluwela, L.; Kjellström, S.; Brueffer, C.; Saal, L.H.; Malmström, J.; Ali, S.; Niméus, E. Proteomic profiling reveals that ESR1 mutations enhance cyclin-dependent kinase signaling. Sci Rep. 2024, 14, 6873. [Google Scholar] [CrossRef]
- Veeraraghavan, J.; Tan, Y.; Cao, X.X.; Kim, J.A.; Wang, X.; Chamness, G.C.; Maiti, S.N.; Cooper, L.J.; Edwards, D.P.; Contreras, A.; Hilsenbeck, S.G.; Chang, E.C.; Schiff, R.; Wang, X.S. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun. 2014 5, 4577. [CrossRef]
- Lei, J.T.; Shao, J.; Zhang, J.; Iglesia, M.; Chan, D.W.; Cao, J.; Anurag, M.; Singh, P.; He, X.; Kosaka, Y.; Matsunuma, R.; Crowder, R.; Hoog, J.; Phommaly, C.; Goncalves, R.; Ramalho, S.; Peres, R.M.R.; Punturi, N.; Schmidt, C.; Bartram, A.; Jou, E.; Devarakonda, V.; Holloway, K.R.; Lai, W.V.; Hampton, O.; Rogers, A.; Tobias, E.; Parikh, P.A.; Davies, S.R.; Li, S.; Ma, C.X.; Suman, V.J.; Hunt, K.K.; Watson, M.A.; Hoadley, K.A.; Thompson, E.A.; Chen, X.; Kavuri, S.M.; Creighton, C.J.; Maher, C.A.; Perou, C.M.; Haricharan, S.; Ellis, M.J. Functional Annotation of ESR1 Gene Fusions in Estrogen Receptor-Positive Breast Cancer. Cell Rep. 2018, 24, 1434–1444.e7. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Hoog, J.; Chin, S.F.; Tao, Y.; Zayed, A.A.; Chin, K.; Teschendorff, A.E.; Quackenbush, J.F.; Marioni, J.C.; Leung, S.; Perou, C.M.; Neilsen, T.O.; Ellis, M.; Gray, J.W.; Bernard, P.S.; Huntsman, D.G.; Caldas, C. ESR1 gene amplification in breast cancer: a common phenomenon? Nat Genet. 2008, 40, 806–807. [Google Scholar] [CrossRef]
- Holst, F.; Moelans, C.B.; Filipits, M.; Singer, C.F.; Simon, R.; van Diest, P.J. On the evidence for ESR1 amplification in breast cancer. Nat Rev Cancer. 2012, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Moelans, C.B.; Holst, F.; Hellwinkel, O.; Simon, R.; van Diest, P.J. ESR1 amplification in breast cancer by optimized RNase FISH: frequent but low-level and heterogeneous. PLoS One 2013, 8, e84189. [Google Scholar] [CrossRef] [PubMed]
- Albertson, D.G. Conflicting evidence on the frequency of ESR1 amplification in breast cancer. Nat Genet. 2008, 40, 821–822. [Google Scholar] [CrossRef]
- Nielsen, K.V.; Ejlertsen, B.; Müller, S.; Møller, S.; Rasmussen, B.B.; Balslev, E.; Lænkholm, A.V.; Christiansen, P.; Mouridsen, H.T. Amplification of ESR1 may predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res Treat. 2011, 127, 345–355. [Google Scholar] [CrossRef]
- Tomita, S.; Zhang, Z.; Nakano, M.; Ibusuki, M.; Kawazoe, T.; Yamamoto, Y.; Iwase, H. Estrogen receptor alpha gene ESR1 amplification may predict endocrine therapy responsiveness in breast cancer patients. Cancer Sci. 2009, 100, 1012–1017. [Google Scholar] [CrossRef]
- Reid, S.E.; Pantaleo, J.; Bolivar, P.; Bocci, M.; Sjölund, J.; Morsing, M.; Cordero, E.; Larsson, S.; Malmberg, M.; Seashore-Ludlow, B.; Pietras, K. Cancer-associated fibroblasts rewire the estrogen receptor response in luminal breast cancer, enabling estrogen independence. Oncogene. 2024, 43, 1113–1126. [Google Scholar] [CrossRef]
- Luan, R.; He, M.; Li, H.; Bai, Y.; Wang, A.; Sun, G.; Zhou, B.; Wang, M.; Wang, C.; Wang, S.; Zeng, K.; Feng, J.; Lin, L.; Wei, Y.; Kato, S.; Zhang, Q.; Zhao, Y. MYSM1 acts as a novel co-activator of ERα to confer antiestrogen resistance in breast cancer. EMBO Mol Med. 2024, 16, 10–39. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, F.; Huang, Z.; Liu, X.; Xia, G.; Huang, J.; Yang, Y.; Li, J.; Huang, J.; Liu, Y.; Zhou, T.; Qi, W.; Gao, G.; Yang, X. Macrophages Promote Subtype Conversion and Endocrine Resistance in Breast Cancer. Cancers (Basel). 2024, 16, 678. [Google Scholar] [CrossRef]
- Li, K.; Shu, D.; Li, H.; Lan, A.; Zhang, W.; Tan, Z.; Huang, M.; Tomasi, M.L.; Jin, A.; Yu, H.; Shen, M.; Liu, S. SMAD4 depletion contributes to endocrine resistance by integrating ER and ERBB signaling in HR + HER2- breast cancer. Cell Death Dis. 2024, 15, 444. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P.; Duffy, M.J. Prognostic and predictive biomarkers in breast cancer: Past, present and future. Semin Cancer Biol. 2018, 2018 52(Pt 1) Pt 1, 56–73. [Google Scholar] [CrossRef]
- Millis, S.Z.; Ikeda, S.; Reddy, S.; Gatalica, Z.; Kurzrock, R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors. JAMA Oncol. 2016, 2, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, H.; Lenferink, A.E.; Simpson, J.F.; Pisacane, P.I.; Sliwkowski, M.X.; Forbes, J.T.; Arteaga, C.L. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000, 60, 5887–5894. [Google Scholar] [PubMed]
- Fribbens, C.; Garcia Murillas, I.; Beaney, M.; Hrebien, S.; O'Leary, B.; Kilburn, L.; Howarth, K.; Epstein, M.; Green, E.; Rosenfeld, N.; Ring, A.; Johnston, S.; Turner, N. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Ann Oncol. 2018, 29, 145–153. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; Yamashita, T.; Lu, Y.S.; Inoue, K.; Takahashi, M.; Pápai, Z.; Longin, A.S.; Mills, D.; Wilke, C.; Hirawat, S.; Juric, D.; SOLAR-1 Study Group. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N Engl J Med. 2018, 379, 2052–2062. [Google Scholar] [CrossRef]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; van Riel, J.M.G.H.; Bloemendal, H.J.; Steeghs, N.; Lolkema, M.P.; Voest, E.E.; van de Werken, H.J.G.; Jager, A.; Cuppen, E.; Sleijfer, S.; Martens, J.W.M. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Stoica, G.E.; Franke, T.F.; Moroni, M.; Mueller, S.; Morgan, E.; Iann, M.C.; Winder, A.D.; Reiter, R.; Wellstein, A.; Martin, M.B.; Stoica, A. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 2003, 22, 7998–8011. [Google Scholar] [CrossRef]
- Sanchez, C.G.; Ma, C.X.; Crowder, R.J.; Guintoli, T.; Phommaly, C.; Gao, F.; Lin, L.; Ellis, M.J. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011, 13, R21. [Google Scholar] [CrossRef]
- Miller, T.W.; Hennessy, B.T.; González-Angulo, A.M.; Fox, E.M.; Mills, G.B.; Chen, H.; Higham, C.; García-Echeverría, C.; Shyr, Y.; Arteaga, C.L. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010, 120, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris HA3rd Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; Beck, J.T.; Ito, Y.; Yardley, D.; Deleu, I.; Perez, A.; Bachelot, T.; Vittori, L.; Xu, Z.; Mukhopadhyay, P.; Lebwohl, D.; Hortobagyi, G.N. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Yamnik, R.L.; Holz, M.K. mTOR/S6K1 and MAPK/RSK signaling pathways coordinately regulate estrogen receptor alpha serine 167 phosphorylation. FEBS Lett. 2010, 584, 124–128. [Google Scholar] [CrossRef]
- Yamnik, R.L.; Digilova, A.; Davis, D.C.; Brodt, Z.N.; Murphy, C.J.; Holz, M.K. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J Biol Chem. 2009, 284, 6361–6369. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Sabatini, D.M. mTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010, 22, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist. 2018, 23, 528–539. [Google Scholar] [CrossRef]
- Zhang, K.M.; Zhao, D.C.; Li, Z.Y.; Wang, Y.; Liu, J.N.; Du, T.; Zhou, L.; Chen, Y.H.; Yu, Q.C.; Chen, Q.S.; Cai, R.Z.; Zhao, Z.X.; Shan, J.L.; Hu, B.X.; Zhang, H.L.; Feng, G.K.; Zhu, X.F.; Tang, J.; Deng, R. Inactivated cGAS-STING Signaling Facilitates Endocrine Resistance by Forming a Positive Feedback Loop with AKT Kinase in ER+HER2- Breast Cancer. Adv Sci (Weinh) 2024, 11, e2403592. [Google Scholar] [CrossRef]
- Marin, A.; Morales, F.; Walbaum, B. Fibroblast growth factor receptor signaling in estrogen receptor-positive breast cancer: mechanisms and role in endocrine resistance. Front Oncol. 2024, 2024 14, 1406951. [Google Scholar] [CrossRef]
- Zheng, Z.Y.; Anurag, M.; Lei, J.T.; Cao, J.; Singh, P.; Peng, J.; Kennedy, H.; Nguyen, N.C.; Chen, Y.; Lavere, P.; Li, J.; Du, X.H.; Cakar, B.; Song, W.; Kim, B.J.; Shi, J.; Seker, S.; Chan, D.W.; Zhao, G.Q.; Chen, X.; Banks, K.C.; Lanman, R.B.; Shafaee, M.N.; Zhang, X.H.; Vasaikar, S.; Zhang, B.; Hilsenbeck, S.G.; Li, W.; Foulds, C.E.; Ellis, M.J.; Chang, E.C. Neurofibromin Is an Estrogen Receptor-α Transcriptional Co-repressor in Breast Cancer. Cancer Cell. 2020, 37, 387–402.e7. [Google Scholar] [CrossRef]
- Pearson, A.; Proszek, P.; Pascual, J.; Fribbens, C.; Shamsher, M.K.; Kingston, B.; O'Leary, B.; Herrera-Abreu, M.T.; Cutts, R.J.; Garcia-Murillas, I.; Bye, H.; Walker, B.A.; Gonzalez De Castro, D.; Yuan, L.; Jamal, S.; Hubank, M.; Lopez-Knowles, E.; Schuster, E.F.; Dowsett, M.; Osin, P.; Nerurkar, A.; Parton, M.; Okines, A.F.C.; Johnston, S.R.D.; Ring, A.; Turner, N.C. Inactivating NF1 Mutations Are Enriched in Advanced Breast Cancer and Contribute to Endocrine Therapy Resistance. Clin Cancer Res. 2020, 26, 608–622. [Google Scholar] [CrossRef]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat Commun. 2021, 12, 1786. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jin, Y.; Lin, M.; Zeng, C.; Zhang, J. NF-κB signaling in therapy resistance of breast cancer: Mechanisms, approaches, and challenges. Life Sci 2024, 348, 122684. [Google Scholar] [CrossRef]
- Bacci, M.; Lorito, N.; Smiriglia, A.; Subbiani, A.; Bonechi, F.; Comito, G.; Morriset, L.; El Botty, R.; Benelli, M.; López-Velazco, J.I.; Caffarel, M.M.; Urruticoechea, A.; Sflomos, G.; Malorni, L.; Corsini, M.; Ippolito, L.; Giannoni, E.; Meattini, I.; Matafora, V.; Havas, K.; Bachi, A.; Chiarugi, P.; Marangoni, E.; Morandi, A. Acetyl-CoA carboxylase 1 controls a lipid droplet-peroxisome axis and is a vulnerability of endocrine-resistant ER+ breast cancer. Sci Transl Med. 2024, 16, eadf9874. [Google Scholar] [CrossRef] [PubMed]
- House, R.R.J.; Tovar, E.A.; Redlon, L.N.; Essenburg, C.J.; Dischinger, P.S.; Ellis, A.E.; Beddows, I.; Sheldon, R.D.; Lien, E.C.; Graveel, C.R.; Steensma, M. NF1 deficiency drives metabolic reprogramming in ER+ breast cancer. Mol Metab. 2024, 80, 101876. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Barozzi, I.; Faronato, M.; Lombardo, Y.; Steel, J.H.; Patel, N.; Darbre, P.; Castellano, L.; Győrffy, B.; Woodley, L.; Meira, A.; Patten, D.K.; Vircillo, V.; Periyasamy, M.; Ali, S.; Frige, G.; Minucci, S.; Coombes, R.C.; Magnani, L. Differential epigenetic reprogramming in response to specific endocrine therapies promotes cholesterol biosynthesis and cellular invasion. Nat Commun. 2015 6, 10044.
- Gala, K.; Li, Q.; Sinha, A.; Razavi, P.; Dorso, M.; Sanchez-Vega, F.; Chung, Y.R.; Hendrickson, R.; Hsieh, J.J.; Berger, M.; Schultz, N.; Pastore, A.; Abdel-Wahab, O.; Chandarlapaty, S. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERα enhancer function. Oncogene. 2018, 37, 4692–4710. [Google Scholar] [CrossRef]
- Saatci, O.; Alam, R.; Huynh-Dam, K.T.; Isik, A.; Uner, M.; Belder, N.; Ersan, P.G.; Tokat, U.M.; Ulukan, B.; Cetin, M.; Calisir, K.; Gedik, M.E.; Bal, H.; Sener Sahin, O.; Riazalhosseini, Y.; Thieffry, D.; Gautheret, D.; Ogretmen, B.; Aksoy, S.; Uner, A.; Akyol, A.; Sahin, O. Targeting LINC00152 activates cAMP/Ca2+/ferroptosis axis and overcomes tamoxifen resistance in ER+ breast cancer. Cell Death Dis. 2024, 15, 418. [Google Scholar] [CrossRef]
- Fu, C.; Duan, S.; Zhou, X.; Meng, Y.; Chen, X. Overexpression of COL11A1 confers tamoxifen resistance in breast cancer. NPJ Breast Cancer. 2024, 10, 38. [Google Scholar] [CrossRef]
- Liao, M.; Webster, J.; Coonrod, E.M.; Weilbaecher, K.N.; Maher, C.A.; White, N.M. BCAR4 Expression as a Predictive Biomarker for Endocrine Therapy Resistance in Breast Cancer. Clin Breast Cancer. 2024, 24, 368–375.e2. [Google Scholar] [CrossRef]
- Nakagawa, S.; Miyashita, M.; Maeda, I.; Goda, A.; Tada, H.; Amari, M.; Kojima, Y.; Tsugawa, K.; Ohi, Y.; Sagara, Y.; Sato, M.; Ebata, A.; Harada-Shoji, N.; Suzuki, T.; Nakanishi, M.; Ohta, T.; Ishida, T. Potential role of Fbxo22 in resistance to endocrine therapy in breast cancer with invasive lobular carcinoma. Breast Cancer Res Treat. 2024, 204, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Ye, L.; Sun, S.; Yuan, J.; Huang, J.; Zeng, Z. Involvement of RFC3 in tamoxifen resistance in ER-positive breast cancer through the cell cycle. Aging (Albany NY). 2023, 15, 13738–13752. [Google Scholar] [CrossRef] [PubMed]
- Muise-Helmericks, R.C.; Grimes, H.L.; Bellacosa, A.; Malstrom, S.E.; Tsichlis, P.N.; Rosen, N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem. 1998, 273, 29864–29872. [Google Scholar] [CrossRef]
- Kenny, F.S.; Hui, R.; Musgrove, E.A.; Gee, J.M.; Blamey, R.W.; Nicholson, R.I.; Sutherland, R.L.; Robertson, J.F. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res. 1999, 5, 2069–2076. [Google Scholar] [PubMed]
- Kilker, R.L.; Planas-Silva, M.D. Cyclin D1 is necessary for tamoxifen-induced cell cycle progression in human breast cancer cells. Cancer Res. 2006, 66, 11478–11484. [Google Scholar] [CrossRef]
- Wilcken, N.R.; Prall, O.W.; Musgrove, E.A.; Sutherland, R.L. Inducible overexpression of cyclin D1 in breast cancer cells reverses the growth-inhibitory effects of antiestrogens. Clin Cancer Res. 1997, 3, 849–854. [Google Scholar]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; Lehar, J.; Wiesmann, M.; Wartmann, M.; Chen, Y.; Cao, Z.A.; Pinzon-Ortiz, M.; Kim, S.; Schlegel, R.; Huang, A.; Engelman, J.A. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014, 26, 136–149. [Google Scholar] [CrossRef]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 17. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.L.; Winer, E.P.; Janni, W.; Verma, S.; Conte, P.; Arteaga, C.L.; Cameron, D.A.; Mondal, S.; Su, F.; Miller, M.; Elmeliegy, M.; Germa, C.; O'Shaughnessy, J. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol. 2018, 29, 1541–1547, Erratum in: Ann Oncol. 2019, 30, 1842. [Google Scholar] [CrossRef]
- Im, S.A.; Lu, Y.S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.S.; Campos-Gomez, S.; Villanueva-Vazquez, R.; Jung, K.H.; Chakravartty, A.; Hughes, G.; Gounaris, I.; Rodriguez-Lorenc, K.; Taran, T.; Hurvitz, S.; Tripathy, D. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N Engl J Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S.; Toi, M.; Iwata, H.; Goetz, M.P. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer. 2019, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Finn, R.S.; Diéras, V.; Ettl, J.; Lipatov, O.; Joy, A.A.; Harbeck, N.; Castrellon, A.; Iyer, S.; Lu, D.R.; Mori, A.; Gauthier, E.R.; Bartlett, C.H.; Gelmon, K.A.; Slamon, D.J. Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast Cancer Res Treat. 2019, 174, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W.; Conte, P.; Cameron, D.A.; André, F.; Arteaga, C.L.; Zarate, J.P.; Chakravartty, A.; Taran, T.; Le Gac, F.; Serra, P.; O'Shaughnessy, J. Overall Survival with Ribociclib plus Letrozole in Advanced Breast Cancer. N Engl J Med. 2022, 386, 942–950. [Google Scholar] [CrossRef]
- Lu, Y.S.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Cardoso, F.; Harbeck, N.; Hurvitz, S.; Chow, L.; Sohn, J.; Lee, K.S.; Campos-Gomez, S.; Villanueva Vazquez, R.; Jung, K.H.; Babu, K.G.; Wheatley-Price, P.; De Laurentiis, M.; Im, Y.H.; Kuemmel, S.; El-Saghir, N.; O'Regan, R.; Gasch, C.; Solovieff, N.; Wang, C.; Wang, Y.; Chakravartty, A.; Ji, Y.; Tripathy, D. Updated Overall Survival of Ribociclib plus Endocrine Therapy versus Endocrine Therapy Alone in Pre- and Perimenopausal Patients with HR+/HER2- Advanced Breast Cancer in MONALEESA-7: A Phase III Randomized Clinical Trial. Clin Cancer Res. 2022, 28, 851–859. [Google Scholar] [CrossRef]
- Masurkar, P.P.; Prajapati, P.; Canedo, J.; Goswami, S.; Earl, S.; Bhattacharya, K. Cost-effectiveness of CDK4/6 inhibitors in HR+/HER2- metastatic breast cancer: a systematic review and meta-analysis. Curr Med Res Opin. 2024, 40, 1753–1767. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; André, F.; Puyana Theall, K.; Huang, X.; Giorgetti, C.; Huang Bartlett, C.; Cristofanilli, M. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N Engl J Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; Nusch, A.; Sonke, G.S.; De la Cruz-Merino, L.; Beck, J.T.; Pivot, X.; Sondhi, M.; Wang, Y.; Chakravartty, A.; Rodriguez-Lorenc, K.; Taran, T.; Jerusalem, G. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N Engl J Med. 2020, 382, 514–524. [Google Scholar] [CrossRef]
- Neven, P.; Fasching, P.A.; Chia, S.; Jerusalem, G.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Martín, M.; Nusch, A.; Sonke, G.S.; De la Cruz-Merino, L.; Beck, J.T.; Zarate, J.P.; Wang, Y.; Chakravartty, A.; Wang, C.; Slamon, D.J. Updated overall survival from the MONALEESA-3 trial in postmenopausal women with HR+/HER2- advanced breast cancer receiving first-line ribociclib plus fulvestrant. Breast Cancer Res. 2023, 25, 103. [Google Scholar] [CrossRef]
- Sledge GWJr Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; Koh, H.; Grischke, E.M.; Conte, P.; Lu, Y.; Barriga, S.; Hurt, K.; Frenzel, M.; Johnston, S.; Llombart-Cussac, A. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 116–124. [Google Scholar] [CrossRef]
- Álvarez-Fernández, M.; Malumbres, M. Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell. 2020, 37, 514–529. [Google Scholar] [CrossRef]
- Lee, J.S.; Hackbart, H.; Cui, X.; Yuan, Y. CDK4/6 Inhibitor Resistance in Hormone Receptor-Positive Metastatic Breast Cancer: Translational Research, Clinical Trials, and Future Directions. Int J Mol Sci. 2023, 24, 11791. [Google Scholar] [CrossRef]
- Gupta, A.; Gazzo, A.; Selenica, P.; Safonov, A.; Pareja, F.; da Silva, E.M.; Brown, D.N.; Zhu, Y.; Patel, J.; Blanco-Heredia, J.; Stefanovska, B.; Carpenter, M.A.; Pei, X.; Frosina, D.; Jungbluth, A.A.; Ladanyi, M.; Curigliano, G.; Weigelt, B.; Riaz, N.; Powell, S.N.; Razavi, P.; Harris, R.S.; Reis-Filho, J.S.; Marra, A.; Chandarlapaty, S. APOBEC3 mutagenesis drives therapy resistance in breast cancer. bioRxiv 2024, 591453. [Google Scholar]
- Lloyd, M.R.; Brett, J.O.; Carmeli, A.; Weipert, C.M.; Zhang, N.; Yu, J.; Bucheit, L.; Medford, A.J.; Wagle, N.; Bardia, A.; Wander, S.A. CDK4/6 Inhibitor Efficacy in ESR1-Mutant Metastatic Breast Cancer. NEJM Evid. 2024, 3. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; Boehnke, K.; Mur, C.; Marugán, C.; Baquero, C.; Yu, C.; Bray, S.M.; Wulur, I.H.; Bi, C.; Chu, S.; Qian, H.R.; Iversen, P.W.; Merzoug, F.F.; Ye, X.S.; Reinhard, C.; De Dios, A.; Du, J.; Caldwell, C.W.; Lallena, M.J.; Beckmann, R.P.; Buchanan, S.G. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell. 2017, 32, 761–776.e6. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; André, F.; Bayar, M.A.; Michiels, S.; Zhang, Z.; Giorgetti, C.; Arnedos, M.; Huang Bartlett, C.; Cristofanilli, M. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J Clin Oncol. 2019, 37, 1169–1178. [Google Scholar] [CrossRef]
- Wander, S.A.; Cohen, O.; Gong, X.; Johnson, G.N.; Buendia-Buendia, J.E.; Lloyd, M.R.; Kim, D.; Luo, F.; Mao, P.; Helvie, K.; Kowalski, K.J.; Nayar, U.; Waks, A.G.; Parsons, S.H.; Martinez, R.; Litchfield, L.M.; Ye, X.S.; Yu, C.; Jansen, V.M.; Stille, J.R.; Smith, P.S.; Oakley, G.J.; Chu, Q.S.; Batist, G.; Hughes, M.E.; Kremer, J.D.; Garraway, L.A.; Winer, E.P.; Tolaney, S.M.; Lin, N.U.; Buchanan, S.G.; Wagle, N. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov. 2020, 10, 1174–1193. [Google Scholar] [CrossRef]
- Palafox, M.; Monserrat, L.; Bellet, M.; Villacampa, G.; Gonzalez-Perez, A.; Oliveira, M.; Brasó-Maristany, F.; Ibrahimi, N.; Kannan, S.; Mina, L.; Herrera-Abreu, M.T.; Òdena, A.; Sánchez-Guixé, M.; Capelán, M.; Azaro, A.; Bruna, A.; Rodríguez, O.; Guzmán, M.; Grueso, J.; Viaplana, C.; Hernández, J.; Su, F.; Lin, K.; Clarke, R.B.; Caldas, C.; Arribas, J.; Michiels, S.; García-Sanz, A.; Turner, N.C.; Prat, A.; Nuciforo, P.; Dienstmann, R.; Verma, C.S.; Lopez-Bigas, N.; Scaltriti, M.; Arnedos, M.; Saura, C.; Serra, V. High p16 expression and heterozygous RB1 loss are biomarkers for CDK4/6 inhibitor resistance in ER+ breast cancer. Nat Commun. 2022, 13, 5258. [Google Scholar] [CrossRef]
- Safonov, A.; Marra, A.; Bandlamudi, C.; O'Leary, B.; Wubbenhorst, B.; Ferraro, E.; Moiso, E.; Lee, M.; An, J.; Donoghue, M.T.A.; Will, M.; Pareja, F.; Nizialek, E.; Lukashchuk, N.; Sofianopoulou, E.; Liu, Y.; Huang, X.; Ahmed, M.; Mehine, M.M.; Ross, D.; Mandelker, D.; Ladanyi, M.; Schultz, N.; Berger, M.F.; Scaltriti, M.; Reis-Filho, J.S.; Li, B.T.; Offit, K.; Norton, L.; Shen, R.; Shah, S.; Maxwell, K.N.; Couch, F.; Domchek, S.M.; Solit, D.B.; Nathanson, K.L.; Robson, M.E.; Turner, N.C.; Chandarlapaty, S.; Razavi, P. Tumor suppressor heterozygosity and homologous recombination deficiency mediate resistance to front-line therapy in breast cancer. bioRxiv 2024 02.05.57 8934.
- Sablin, M.P.; Gestraud, P.; Jonas, S.F.; Lamy, C.; Lacroix-Triki, M.; Bachelot, T.; Filleron, T.; Lacroix, L.; Tran-Dien, A.; Jézéquel, P.; Mauduit, M.; Barros Monteiro, J.; Jimenez, M.; Michiels, S.; Attignon, V.; Soubeyran, I.; Driouch, K.; Servant, N.; Le Tourneau, C.; Kamal, M.; André, F.; Bièche, I. Copy number alterations in metastatic and early breast tumours: prognostic and acquired biomarkers of resistance to CDK4/6 inhibitors. Br J Cancer. 2024, 131, 1060–1067. [Google Scholar] [CrossRef]
- Mouery, B.L.; Baker, E.M.; Mei, L.; Wolff, S.C.; Mills, C.A.; Fleifel, D.; Mulugeta, N.; Herring, L.E.; Cook, J.G. APC/C prevents a noncanonical order of cyclin/CDK activity to maintain CDK4/6 inhibitor-induced arrest. Proc Natl Acad Sci U S A. 2024, 121, e2319574121. [Google Scholar] [CrossRef]
- Torrisi, R.; Vaira, V.; Giordano, L.; Fernandes, B.; Saltalamacchia, G.; Palumbo, R.; Carnaghi, C.; Basilico, V.; Gentile, F.; Masci, G.; De Sanctis, R.; Santoro, A. Identification of a Panel of miRNAs Associated with Resistance to Palbociclib and Endocrine Therapy. Int J Mol Sci. 2024, 25, 1498. [Google Scholar] [CrossRef]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; Ericsson, P.G.; Lee, K.M.; Nixon, M.J.; Schwarz, L.J.; Sanders, M.E.; Dugger, T.C.; Cruz, M.R.; Behdad, A.; Cristofanilli, M.; Bardia, A.; O'Shaughnessy, J.; Nagy, R.J.; Lanman, R.B.; Solovieff, N.; He, W.; Miller, M.; Su, F.; Shyr, Y.; Mayer, I.A.; Balko, J.M.; Arteaga, C.L. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; Morris, L.; Selenica, P.; Eichenberger, E.; Shen, R.; Schultz, N.; Rosen, N.; Scaltriti, M.; Brogi, E.; Baselga, J.; Reis-Filho, J.S.; Chandarlapaty, S. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell. 2018, 34, 893–905.e8. [Google Scholar] [CrossRef] [PubMed]
- Talia, M.; Cirillo, F.; Scordamaglia, D.; Di Dio, M.; Zicarelli, A.; De Rosis, S.; Miglietta, A.M.; Capalbo, C.; De Francesco, E.M.; Belfiore, A.; Grande, F.; Rizzuti, B.; Occhiuzzi, M.A.; Fortino, G.; Guzzo, A.; Greco, G.; Maggiolini, M.; Lappano, R. The G Protein Estrogen Receptor (GPER) is involved in the resistance to the CDK4/6 inhibitor palbociclib in breast cancer. J Exp Clin Cancer Res. 2024, 43, 171. [Google Scholar] [CrossRef] [PubMed]
- Belli, S.; Esposito, D.; Ascione, C.M.; Messina, F.; Napolitano, F.; Servetto, A.; De Angelis, C.; Bianco, R.; Formisano, L. EGFR and HER2 hyper-activation mediates resistance to endocrine therapy and CDK4/6 inhibitors in ER+ breast cancer. Cancer Lett. 2024, 593, 216968. [Google Scholar] [CrossRef]
- Chen, A.; Kim, B.J.; Mitra, A.; Vollert, C.T.; Lei, J.T.; Fandino, D.; Anurag, M.; Holt, M.V.; Gou, X.; Pilcher, J.B.; Goetz, M.P.; Northfelt, D.W.; Hilsenbeck, S.G.; Marshall, C.G.; Hyer, M.L.; Papp, R.; Yin, S.Y.; De Angelis, C.; Schiff, R.; Fuqua, S.A.W.; Ma, C.X.; Foulds, C.E.; Ellis, M.J. PKMYT1 Is a Marker of Treatment Response and a Therapeutic Target for CDK4/6 Inhibitor-Resistance in ER+ Breast Cancer. Mol Cancer Ther. 2024, 23, 1494–1510. [Google Scholar] [CrossRef]
- Kindt, C.K.; Alves, C.L.; Ehmsen, S.; Kragh, A.; Reinert, T.; Vogsen, M.; Kodahl, A.R.; Rønlev, J.D.; Ardik, D.; Sørensen, A.L.; Evald, K.; Clemmensen, M.L.; Staaf, J.; Ditzel, H.J. Genomic alterations associated with resistance and circulating tumor DNA dynamics for early detection of progression on CDK4/6 inhibitor in advanced breast cancer. Int J Cancer. 2024, 155, 2211–2222. [Google Scholar] [CrossRef]
- Li, X.; Niu, C.; Yi, G.; Zhang, Y.; Jin, W.; Zhang, Z.; Zhang, W.; Li, B. Quercetin inhibits the epithelial-mesenchymal transition and reverses CDK4/6 inhibitor resistance in breast cancer by regulating circHIAT1/miR-19a-3p/CADM2 axis. PLoS One. 2024, 19, e0305612. [Google Scholar] [CrossRef]
- Cui, Y.; Li, Y.; Xu, Y.; Liu, X.; Kang, X.; Zhu, J.; Long, S.; Han, Y.; Xue, C.; Sun, Z.; Du, Y.; Hu, J.; Pan, L.; Zhou, F.; Xu, X.; Li, X. SLC7A11 protects luminal A breast cancer cells against ferroptosis induced by CDK4/6 inhibitors. Redox Biol. 2024, 76, 103304. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, S.; Kai, Y.; Zhang, Y.Q.; Peng, C.; Li, Z.; Mughal, M.J.; Julie, B.; Zheng, X.; Ma, J.; Ma, C.X.; Shen, M.; Hall, M.D.; Li, S.; Zhu, W. O-GlcNAcylation of MITF regulates its activity and CDK4/6 inhibitor resistance in breast cancer. Nat Commun. 2024, 15, 5597. [Google Scholar] [CrossRef]
- Lee, J.; Lim, B.; Pearson, T.; Tripathy, D.; Ordentlich, P.; Ueno, N.T. The synergistic antitumor activity of entinostat (MS-275) in combination with palbociclib (PD 0332991) in estrogen receptor-positive and triple-negative breast cancer. Cancer Res 2018, 78 (4_Supplement), P5-21-15. [Google Scholar] [CrossRef]
- Papadimitriou, M.C.; Pazaiti, A.; Iliakopoulos, K.; Markouli, M.; Michalaki, V.; Papadimitriou, C.A. Resistance to CDK4/6 inhibition: Mechanisms and strategies to overcome a therapeutic problem in the treatment of hormone receptor-positive metastatic breast cancer. Biochim Biophys Acta Mol Cell Res. 2022, 1869, 119346. [Google Scholar] [CrossRef]
- Cornell, L.; Wander, S.A.; Visal, T.; Wagle, N.; Shapiro, G.I. MicroRNA-Mediated Suppression of the TGF-β Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance. Cell Rep. 2019, 26, 2667–2680.e7. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ge, L.P.; Li, D.Q.; Shao, Z.M.; Di, G.H.; Xu, X.E.; Jiang, Y.Z. LncRNA TROJAN promotes proliferation and resistance to CDK4/6 inhibitor via CDK2 transcriptional activation in ER+ breast cancer. Mol Cancer. 2020, 19, 87. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017, 36, 2255–2264. [Google Scholar] [CrossRef]
- Wu, X.; Yang, X.; Xiong, Y.; Li, R.; Ito, T.; Ahmed, T.A.; Karoulia, Z.; Adamopoulos, C.; Wang, H.; Wang, L.; Xie, L.; Liu, J.; Ueberheide, B.; Aaronson, S.A.; Chen, X.; Buchanan, S.G.; Sellers, W.R.; Jin, J.; Poulikakos, P.I. Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat Cancer. 2021, 2, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Al-Qasem, A.J.; Alves, C.L.; Ehmsen, S.; Tuttolomondo, M.; Terp, M.G.; Johansen, L.E.; Vever, H.; Hoeg, L.V.A.; Elias, D.; Bak, M.; Ditzel, H.J. Co-targeting CDK2 and CDK4/6 overcomes resistance to aromatase and CDK4/6 inhibitors in ER+ breast cancer. NPJ Precis Oncol. 2022, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.G.; Saba, N.F.; Teng, Y. The diverse functions of FAT1 in cancer progression: good, bad, or ugly? J Exp Clin Cancer Res. 2022, 41, 248. [Google Scholar] [CrossRef]
- Martin, D.; Degese, M.S.; Vitale-Cross, L.; Iglesias-Bartolome, R.; Valera, J.L.C.; Wang, Z.; Feng, X.; Yeerna, H.; Vadmal, V.; Moroishi, T.; Thorne, R.F.; Zaida, M.; Siegele, B.; Cheong, S.C.; Molinolo, A.A.; Samuels, Y.; Tamayo, P.; Guan, K.L.; Lippman, S.M.; Lyons, J.G.; Gutkind, J.S. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat Commun. 2018, 9, 2372. [Google Scholar] [CrossRef]
- Steinhardt, A.A.; Gayyed, M.F.; Klein, A.P.; Dong, J.; Maitra, A.; Pan, D.; Montgomery, E.A.; Anders, R.A. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008, 39, 1582–1589. [Google Scholar] [CrossRef]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules. 2017, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.Y.; Li, X.T.; Xu, K.; Wang, R.T.; Guan, X.X. c-MYC mediates the crosstalk between breast cancer cells and tumor microenvironment. Cell Commun Signal. 2023, 21, 28. [Google Scholar] [CrossRef]
- Pandey, K.; Park, N.; Park, K.S.; Hur, J.; Cho, Y.B.; Kang, M.; An, H.J.; Kim, S.; Hwang, S.; Moon, Y.W. Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers (Basel). 2020, 12, 3566. [Google Scholar] [CrossRef]
- Goetz, M.P.; Hamilton, E.P.; Campone, M.; Hurvitz, S.A.; Cortes, J.; Johnston, S.R.D.; Jerusalem, G.H.M.; Graham, H.; Wang, H.; Litchfield, L.; Jansen, V.M.; Martin, M. Acquired genomic alterations in circulating tumor DNA from patients receiving abemaciclib alone or in combination with endocrine therapy. J Clin Oncol 2020, 38 (suppl), 15. [Google Scholar] [CrossRef]
- Mo, H.; Liu, X.; Xue, Y.; Chen, H.; Guo, S.; Li, Z.; Wang, S.; Li, C.; Han, J.; Fu, M.; Song, Y.; Li, D.; Ma, F. S6K1 amplification confers innate resistance to CDK4/6 inhibitors through activating c-Myc pathway in patients with estrogen receptor-positive breast cancer. Mol Cancer. 2022, 21, 171. [Google Scholar] [CrossRef]
- Petroni, G.; Buqué, A.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Immunomodulation by targeted anticancer agents. Cancer Cell 2021, 39, 310–345. [Google Scholar] [CrossRef] [PubMed]
- Petroni, G.; Formenti, S.C.; Chen-Kiang, S.; Galluzzi, L. Immunomodulation by anticancer cell cycle inhibitors. Nat Rev Immunol 2020, 20, 669–679. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 2018 553, 91–95. [Google Scholar] [CrossRef]
- Teh, J.L.F. Arrested developments: CDK4/6 inhibitor resistance and alterations in the tumor immune microenvironment. Clin. Cancer Res 2019, 25(3), 921–927. [Google Scholar] [CrossRef]
- De Angelis, C. Activation of the IFN signaling pathway is associated with resistance to CDK4/6 inhibitors and immune checkpoint activation in ER-positive breast cancer. Clin. Cancer Res. 2021, 27(17), 4870–4882. [Google Scholar] [CrossRef]
- Petroni, G. IL17-Producing γδ T Cells Promote Resistance to CDK4/6 Inhibitors in HR+HER2-Breast cancer. BMJ Specialist Journals, 2022. [Google Scholar]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Freeman-Cook, K.D.; Hoffman, R.L.; Behenna, D.C.; Boras, B.; Carelli, J.; Diehl, W.; Ferre, R.A.; He, Y.A.; Hui, A.; Huang, B.; Huser, N.; Jones, R.; Kephart, S.E.; Lapek, J.; McTigue, M.; Miller, N.; Murray, B.W.; Nagata, A.; Nguyen, L.; Niessen, S.; Ninkovic, S.; O'Doherty, I.; Ornelas, M.A.; Solowiej, J.; Sutton, S.C.; Tran, K.; Tseng, E.; Visswanathan, R.; Xu, M.; Zehnder, L.; Zhang, Q.; Zhang, C.; Dann, S. Discovery of PF-06873600, a CDK2/4/6 Inhibitor for the Treatment of Cancer. J Med Chem. 2021, 64, 9056–9077. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Moser, J.; Hoffman, T.E.; Watts, L.P.; Min, M.; Musteanu, M.; Rong, Y.; Ill, C.R.; Nangia, V.; Schneider, J.; Sanclemente, M.; Lapek, J.; Nguyen, L.; Niessen, S.; Dann, S.; VanArsdale, T.; Barbacid, M.; Miller, N.; Spencer, S.L. Rapid adaptation to CDK2 inhibition exposes intrinsic cell-cycle plasticity. Cell. 2023, 186, 2628–2643.e21. [Google Scholar] [CrossRef]
- Glover-Cutter, K.; Larochelle, S.; Erickson, B.; Zhang, C.; Shokat, K.; Fisher, R.P.; Bentley, D.L. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol. 2009, 29, 5455–5464. [Google Scholar] [CrossRef] [PubMed]
- Coombes, R.C.; Howell, S.; Lord, S.R.; Kenny, L.; Mansi, J.; Mitri, Z.; Palmieri, C.; Chap, L.I.; Richards, P.; Gradishar, W.; Sardesai, S.; Melear, J.; O'Shaughnessy, J.; Ward, P.; Chalasani, P.; Arkenau, T.; Baird, R.D.; Jeselsohn, R.; Ali, S.; Clack, G.; Bahl, A.; McIntosh, S.; Krebs, M.G. Dose escalation and expansion cohorts in patients with advanced breast cancer in a Phase I study of the CDK7-inhibitor samuraciclib. Nat Commun. 2023, 14, 4444, Erratum in: Nat Commun. 2023, 14, 4741. [Google Scholar] [CrossRef]
- Wilson, G.A.; Vuina, K.; Sava, G.; Huard, C.; Meneguello, L.; Coulombe-Huntington, J.; Bertomeu, T.; Maizels, R.J.; Lauring, J.; Kriston-Vizi, J.; Tyers, M.; Ali, S.; Bertoli, C.; de Bruin, R.A.M. Active growth signaling promotes senescence and cancer cell sensitivity to CDK7 inhibition. Mol Cell. 2023, 83, 4078–4092.e6. [Google Scholar] [CrossRef]
- Munzone, E.; Pagan, E.; Bagnardi, V.; Montagna, E.; Cancello, G.; Dellapasqua, S.; Iorfida, M.; Mazza, M.; Colleoni, M. Systematic review and meta-analysis of post-progression outcomes in ER+/HER2- metastatic breast cancer after CDK4/6 inhibitors within randomized clinical trials. ESMO Open. 2021, 6, 100332. [Google Scholar] [CrossRef]
- Gennari A, ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 2021, 2021 32, 1475–1495.
- Di Leo, A.; Jerusalem, G.; Petruzelka, L.; Torres, R.; Bondarenko, I.N.; Khasanov, R.; Verhoeven, D.; Pedrini, J.L.; Smirnova, I.; Lichinitser, M.R.; Pendergrass, K.; Malorni, L.; Garnett, S.; Rukazenkov, Y.; Martin, M. Final overall survival: fulvestrant 500 mg vs 250 mg in the randomized CONFIRM trial. J Natl Cancer Inst. 2014, 106, djt337. [Google Scholar] [CrossRef]
- Kornblum, N.; Zhao, F.; Manola, J.; Klein, P.; Ramaswamy, B.; Brufsky, A.; Stella, P.J.; Burnette, B.; Telli, M.; Makower, D.F.; Cheema, P.; Truica, C.I.; Wolff, A.C.; Soori, G.S.; Haley, B.; Wassenaar, T.R.; Goldstein, L.J.; Miller, K.D.; Sparano, J.A. Randomized Phase II Trial of Fulvestrant Plus Everolimus or Placebo in Postmenopausal Women With Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer Resistant to Aromatase Inhibitor Therapy: Results of PrE0102. J Clin Oncol. 2018, 36, 1556–1563. [Google Scholar] [PubMed]
- Schmid, P.; Zaiss, M.; Harper-Wynne, C.; Ferreira, M.; Dubey, S.; Chan, S.; Makris, A.; Nemsadze, G.; Brunt, A.M.; Kuemmel, S.; Ruiz, I.; Perelló, A.; Kendall, A.; Brown, J.; Kristeleit, H.; Conibear, J.; Saura, C.; Grenier, J.; Máhr, K.; Schenker, M.; Sohn, J.; Lee, K.S.; Shepherd, C.J.; Oelmann, E.; Sarker, S.J.; Prendergast, A.; Marosics, P.; Moosa, A.; Lawrence, C.; Coetzee, C.; Mousa, K.; Cortés, J. Fulvestrant Plus Vistusertib vs Fulvestrant Plus Everolimus vs Fulvestrant Alone for Women With Hormone Receptor-Positive Metastatic Breast Cancer: The MANTA Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Kalinsky, K.; Accordino, M.K.; Chiuzan, C.; Mundi, P.S.; Sakach, E.; Sathe, C.; Ahn, H.; Trivedi, M.S.; Novik, Y.; Tiersten, A.; Raptis, G.; Baer, L.N.; Oh, S.Y.; Zelnak, A.B.; Wisinski, K.B.; Andreopoulou, E.; Gradishar, W.J.; Stringer-Reasor, E.; Reid, S.A.; O'Dea, A.; O'Regan, R.; Crew, K.D.; Hershman, D.L. Randomized Phase II Trial of Endocrine Therapy With or Without Ribociclib After Progression on Cyclin-Dependent Kinase 4/6 Inhibition in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer: MAINTAIN Trial. J Clin Oncol. 2023, 41, 4004–4013. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, G.J.; Fernando, T.M.; Bowen, R.; Jerzak, K.J.; Song, X.; Decker, T.; Boyle, F.; McCune, S.; Armstrong, A.; Shannon, C.; Bertelli, G.; Chang, C.W.; Desai, R.; Gupta, K.; Wilson, T.R.; Flechais, A.; Bardia, A. VERONICA: Randomized Phase II Study of Fulvestrant and Venetoclax in ER-Positive Metastatic Breast Cancer Post-CDK4/6 Inhibitors - Efficacy, Safety, and Biomarker Results. Clin Cancer Res. 2022, 28, 3256–3267. [Google Scholar] [CrossRef]
- Bidard, F.C.; Hardy-Bessard, A.C.; Dalenc, F.; Bachelot, T.; Pierga, J.Y.; de la Motte Rouge, T.; Sabatier, R.; Dubot, C.; Frenel, J.S.; Ferrero, J.M.; Ladoire, S.; Levy, C.; Mouret-Reynier, M.A.; Lortholary, A.; Grenier, J.; Chakiba, C.; Stefani, L.; Plaza, J.E.; Clatot, F.; Teixeira, L.; D'Hondt, V.; Vegas, H.; Derbel, O.; Garnier-Tixidre, C.; Canon, J.L.; Pistilli, B.; André, F.; Arnould, L.; Pradines, A.; Bièche, I.; Callens, C.; Lemonnier, J.; Berger, F.; Delaloge, S.; PADA-1 investigators. Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising ESR1 mutation during aromatase inhibitor and palbociclib therapy (PADA-1): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2022, 23, 1367–1377. [Google Scholar] [CrossRef]
- Bidard, F.C.; Kaklamani, V.G.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; Khong, H.; Kocsis, J.; Dalenc, F.; Dillon, P.M.; Babu, S.; Waters, S.; Deleu, I.; García Sáenz, J.A.; Bria, E.; Cazzaniga, M.; Lu, J.; Aftimos, P.; Cortés, J.; Liu, S.; Tonini, G.; Laurent, D.; Habboubi, N.; Conlan, M.G.; Bardia, A. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From the Randomized Phase III EMERALD Trial. J Clin Oncol. 2022, 40, 3246–3256, Erratum in: J Clin Oncol. 2023, 41, 3962. [Google Scholar] [CrossRef]
- Bardia, A.; Cortés, J.; Bidard, F.C.; Neven, P.; Garcia-Sáenz, J.; Aftimos, P.; O'Shaughnessy, J.; Lu, J.; Tonini, G.; Scartoni, S.; Paoli, A.; Binaschi, M.; Wasserman, T.; Kaklamani, V. Elacestrant in ER+, HER2- Metastatic Breast Cancer with ESR1-Mutated Tumors: Subgroup Analyses from the Phase III EMERALD Trial by Prior Duration of Endocrine Therapy plus CDK4/6 Inhibitor and in Clinical Subgroups. Clin Cancer Res. 2024, 30, 4299–4309. [Google Scholar] [CrossRef]
- Rugo, H.S.; O'Shaughnessy, J.; Cortes, J.; et al. Elacestrant in various combinations in patients (pts) with estrogen receptor-positive (ER+), HER2-negative (HER2-) locally advanced or metastatic breast cancer (adv/mBC): Preliminary data from ELEVATE, a phase 1b/2, open-label, umbrella study. J Clin Oncol. 2024, 42 (Suppl 16), 1069. [Google Scholar] [CrossRef]
- Oliveira, M.; Pominchuk, D.; Nowecki, Z.; Hamilton, E.; Kulyaba, Y.; Andabekov, T.; Hotko, Y.; Melkadze, T.; Nemsadze, G.; Neven, P.; Vladimirov, V.; Zamagni, C.; Denys, H.; Forget, F.; Horvath, Z.; Nesterova, A.; Ajimi, M.; Kirova, B.; Klinowska, T.; Lindemann, J.P.O.; Lissa, D.; Mathewson, A.; Morrow, C.J.; Traugottova, Z.; van Zyl, R.; Arkania, E. Camizestrant, a next-generation oral SERD, versus fulvestrant in post-menopausal women with oestrogen receptor-positive, HER2-negative advanced breast cancer (SERENA-2): a multi-dose, open-label, randomised, phase 2 trial. Lancet Oncol. 2024, 25, 1424–1439. [Google Scholar] [CrossRef]
- Turner, N.; Huang-Bartlett, C.; Kalinsky, K.; Cristofanilli, M.; Bianchini, G.; Chia, S.; Iwata, H.; Janni, W.; Ma, C.X.; Mayer, E.L.; Park, Y.H.; Fox, S.; Liu, X.; McClain, S.; Bidard, F.C. Design of SERENA-6, a phase III switching trial of camizestrant in ESR1-mutant breast cancer during first-line treatment. Future Oncol. 2023, 19, 559–573. [Google Scholar] [CrossRef]
- Jhaveri, K.L.; Neven, P.; Casalnuovo, M.L.; Kim, S.B.; Tokunaga, E.; Aftimos, P.; Saura, C.; O'Shaughnessy, J.; Harbeck, N.; Carey, L.A.; Curigliano, G.; Llombart-Cussac, A.; Lim, E.; García Tinoco, M.L.; Sohn, J.; Mattar, A.; Zhang, Q.; Huang, C.S.; Hung, C.C.; Martinez Rodriguez, J.L.; Ruíz Borrego, M.; Nakamura, R.; Pradhan, K.R.; Cramer von Laue, C.; Barrett, E.; Cao, S.; Wang, X.A.; Smyth, L.M.; Bidard, F.C.; EMBER-3 Study Group. Imlunestrant with or without Abemaciclib in Advanced Breast Cancer. N Engl J Med. 2024. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zbieg, J.R.; Blake, R.A.; Chang, J.H.; Daly, S.; DiPasquale, A.G.; Friedman, L.S.; Gelzleichter, T.; Gill, M.; Giltnane, J.M.; Goodacre, S.; Guan, J.; Hartman, S.J.; Ingalla, E.R.; Kategaya, L.; Kiefer, J.R.; Kleinheinz, T.; Labadie, S.S.; Lai, T.; Li, J.; Liao, J.; Liu, Z.; Mody, V.; McLean, N.; Metcalfe, C.; Nannini, M.A.; Oeh, J.; O'Rourke, M.G.; Ortwine, D.F.; Ran, Y.; Ray, N.C.; Roussel, F.; Sambrone, A.; Sampath, D.; Schutt, L.K.; Vinogradova, M.; Wai, J.; Wang, T.; Wertz, I.E.; White, J.R.; Yeap, S.K.; Young, A.; Zhang, B.; Zheng, X.; Zhou, W.; Zhong, Y.; Wang, X. GDC-9545 (Giredestrant): A Potent and Orally Bioavailable Selective Estrogen Receptor Antagonist and Degrader with an Exceptional Preclinical Profile for ER+ Breast Cancer. J Med Chem. 2021, 64, 11841–11856. [Google Scholar] [CrossRef]
- Martín, M.; Lim, E.; Chavez-MacGregor, M.; Bardia, A.; Wu, J.; Zhang, Q.; Nowecki, Z.; Cruz, F.M.; Safin, R.; Kim, S.B.; Schem, C.; Montero, A.J.; Khan, S.; Bandyopadhyay, R.; Moore, H.M.; Shivhare, M.; Patre, M.; Martinalbo, J.; Roncoroni, L.; Pérez-Moreno, P.D.; Sohn, J.; acelERA Breast Cancer Study Investigators. Giredestrant for Estrogen Receptor-Positive, HER2-Negative, Previously Treated Advanced Breast Cancer: Results From the Randomized, Phase II acelERA Breast Cancer Study. J Clin Oncol. 2024, 42, 2149–2160. [Google Scholar] [CrossRef]
- Miron, A.; Varadi, M.; Carrasco, D.; Li, H.; Luongo, L.; Kim, H.J.; Park, S.Y.; Cho, E.Y.; Lewis, G.; Kehoe, S.; Iglehart, J.D.; Dillon, D.; Allred, D.C.; Macconaill, L.; Gelman, R.; Polyak, K. PIK3CA mutations in in situ and invasive breast carcinomas. Cancer Res. 2010, 70, 5674–5678. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; Inoue, K.; Pápai, Z.; Takahashi, M.; Ghaznawi, F.; Mills, D.; Kaper, M.; Miller, M.; Conte, P.F.; Iwata, H.; Rugo, H.S. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; Turner, N.; Sophos, N.; Zarate, J.P.; Arce, C.; Shen, Y.M.; Turner, S.; Kanakamedala, H.; Hsu, W.C.; Chia, S. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): one cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021, 22, 489–498, Erratum in: Lancet Oncol. 2021, 22, e184. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): one cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef]
- Tokunaga, E.; Kataoka, A.; Kimura, Y.; Oki, E.; Mashino, K.; Nishida, K.; Koga, T.; Morita, M.; Kakeji, Y.; Baba, H.; Ohno, S.; Maehara, Y. The association between Akt activation and resistance to hormone therapy in metastatic breast cancer. Eur J Cancer. 2006, 42, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.J.; Casbard, A.; Carucci, M.; Ingarfield, K.; Butler, R.; Morgan, S.; Meissner, M.; Bale, C.; Bezecny, P.; Moon, S.; Twelves, C.; Venkitaraman, R.; Waters, S.; de Bruin, E.C.; Schiavon, G.; Foxley, A.; Jones, R.H. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive, HER2-negative breast cancer (FAKTION): overall survival, updated progression-free survival, and expanded biomarker analysis from a randomised, phase 2 trial. Lancet Oncol. 2022, 23, 851–864. [Google Scholar]
- Turner, N.C.; Oliveira, M.; Howell, S.J.; Dalenc, F.; Cortes, J.; Gomez Moreno, H.L.; Hu, X.; Jhaveri, K.; Krivorotko, P.; Loibl, S.; Morales Murillo, S.; Okera, M.; Park, Y.H.; Sohn, J.; Toi, M.; Tokunaga, E.; Yousef, S.; Zhukova, L.; de Bruin, E.C.; Grinsted, L.; Schiavon, G.; Foxley, A.; Rugo, H.S.; CAPItello-291 Study Group. Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2023, 388, 2058–2070. [Google Scholar] [CrossRef]
- Turner, N.C.; Im, S.A.; Saura, C.; Juric, D.; Loibl, S.; Kalinsky, K.; Schmid, P.; Loi, S.; Sunpaweravong, P.; Musolino, A.; Li, H.; Zhang, Q.; Nowecki, Z.; Leung, R.; Thanopoulou, E.; Shankar, N.; Lei, G.; Stout, T.J.; Hutchinson, K.E.; Schutzman, J.L.; Song, C.; Jhaveri, K.L. Inavolisib-Based Therapy in PIK3CA-Mutated Advanced Breast Cancer. N Engl J Med. 2024, 391, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Bardia, A.; Kim, S.B.N.; et al. Ipatasertib in combination with palbociclib (palbo) and fulvestrant (fulv) in patients with hormone receptor-positive HER2-negative advanced breast cancer. Cancer Res 2022, 82 (4 Suppl), P5-16-11. [Google Scholar] [CrossRef]
- Piccart, M.; Hortobagyi, G.N.; Campone, M.; Pritchard, K.I.; Lebrun, F.; Ito, Y.; Noguchi, S.; Perez, A.; Rugo, H.S.; Deleu, I.; Burris HA3rd Provencher, L.; Neven, P.; Gnant, M.; Shtivelband, M.; Wu, C.; Fan, J.; Feng, W.; Taran, T.; Baselga, J. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2. Ann Oncol. 2014, 25, 2357–2362. [Google Scholar] [CrossRef]
- Dhakal, A.; Antony Thomas, R.; Levine, E.G.; Brufsky, A.; Takabe, K.; Hanna, M.G.; Attwood, K.; Miller, A.; Khoury, T.; Early, A.P.; Soniwala, S.; O'Connor, T.; Opyrchal, M. Outcome of Everolimus-Based Therapy in Hormone-Receptor-Positive Metastatic Breast Cancer Patients After Progression on Palbociclib. Breast Cancer (Auckl). 2020, 2020 14, 1178223420944864. [Google Scholar] [CrossRef]
- Cook, M.M.; Al Rabadi, L.; Kaempf, A.J.; Saraceni, M.M.; Savin, M.A.; Mitri, Z.I. Everolimus Plus Exemestane Treatment in Patients with Metastatic Hormone Receptor-Positive Breast Cancer Previously Treated with CDK4/6 Inhibitor Therapy. Oncologist. 2021, 26, 101–106. [Google Scholar] [CrossRef]
- Layman, R.M.; Han, H.S.; Rugo, H.S.; Stringer-Reasor, E.M.; Specht, J.M.; Dees, E.C.; Kabos, P.; Suzuki, S.; Mutka, S.C.; Sullivan, B.F.; Gorbatchevsky, I.; Wesolowski, R. Gedatolisib in combination with palbociclib and endocrine therapy in women with hormone receptor-positive, HER2-negative advanced breast cancer: results from the dose expansion groups of an open-label, phase 1b study. Lancet Oncol. 2024, 25, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Reichert, J.M.; Senter, P.D.; Lambert, J.M.; Beck, A. Antibody-drug conjugates come of age in oncology. Nat Rev Drug Discov. 2023, 22, 641–661. [Google Scholar] [CrossRef]
- Grinda, T.; Rassy, E.; Pistilli, B. Antibody-Drug Conjugate Revolution in Breast Cancer: The Road Ahead. Curr Treat Options Oncol. 2023, 24, 442–465. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer. 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; Lee, K.S.; Niikura, N.; Park, Y.H.; Xu, B.; Wang, X.; Gil-Gil, M.; Li, W.; Pierga, J.Y.; Im, S.A.; Moore, H.C.F.; Rugo, H.S.; Yerushalmi, R.; Zagouri, F.; Gombos, A.; Kim, S.B.; Liu, Q.; Luo, T.; Saura, C.; Schmid, P.; Sun, T.; Gambhire, D.; Yung, L.; Wang, Y.; Singh, J.; Vitazka, P.; Meinhardt, G.; Harbeck, N.; Cameron, D.A.; DESTINY-Breast04 Trial Investigators. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N Engl J Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Bardia, A.; Hu, X.; Dent, R.; Yonemori, K.; Barrios, C.H.; O'Shaughnessy, J.A.; Wildiers, H.; Pierga, J.Y.; Zhang, Q.; Saura, C.; Biganzoli, L.; Sohn, J.; Im, S.A.; Lévy, C.; Jacot, W.; Begbie, N.; Ke, J.; Patel, G.; Curigliano, G.; DESTINY-Breast06 Trial Investigators. Trastuzumab Deruxtecan after Endocrine Therapy in Metastatic Breast Cancer. N Engl J Med. 2024, 391, 2110–2122. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Hu, T.X.; Dent, R.A.; Yonemori, K.; Barrios CHSr O'Shaughnessy, J.; et al. Trastuzumab deruxtecan (T-DXd) vs physician’s choice of chemotherapy (TPC) in patients (pts) with hormone receptor-positive (HR+), human epidermal growth factor receptor 2 (HER2)-low or HER2-ultralow metastatic breast cancer (mBC) with prior endocrine therapy (ET): Primary results from DESTINY-Breast06 (DB-06). J Clin Oncol 2024, 42 (suppl 17), abstr LBA1000. [Google Scholar]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs. 2020, 80, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Vidula, N.; Yau, C.; Rugo, H. Trophoblast Cell Surface Antigen 2 gene (TACSTD2) expression in primary breast cancer. Breast Cancer Res Treat. 2022, 194, 569–575. [Google Scholar] [CrossRef]
- Rugo, H.S.; Bardia, A.; Marmé, F.; Cortes, J.; Schmid, P.; Loirat, D.; Trédan, O.; Ciruelos, E.; Dalenc, F.; Pardo, P.G.; Jhaveri, K.L.; Delaney, R.; Fu, O.; Lin, L.; Verret, W.; Tolaney, S.M. Sacituzumab Govitecan in Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer. J Clin Oncol. 2022, 40, 3365–3376. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.; Bardia, A.; Marmé, F.; Cortés, J.; Schmid, P.; Loirat, D.; et al. Sacituzumab Govitecan (SG) vs Treatment of Physician’s Choice (TPC): Efficacy by Trop-2 Expression in the TROPiCS-02 Study of Patients (Pts) With HR+/HER2– Metastatic Breast Cancer (mBC). Cancer Res 2023, 83 (5_Supplement), GS1-11. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Krop, I.; Juric, D.; Kogawa, T.; Hamilton, E.; Spira, A.I.; et al. Phase 1 TROPION-PanTumor01 Study Evaluating Datopotamab Deruxtecan (Dato-DXd) in Unresectable or Metastatic Hormone Receptor–Positive/HER2–Negative Breast Cancer (BC). Cancer Res 2023, 83 (5_Supplement), PD13-08. [Google Scholar] [CrossRef]
- Bardia, A.; Jhaveri, K.; Im, S.A.; De Laurentiis, M.; Xu, B.; Pernas, S.; et al. Randomized phase 3 study of datopotamab deruxtecan vs chemotherapy for patients with previously-treated inoperable or metastatic hormone receptor-positive, HER2-negative breast cancer: Results from TROPION-Breast01. Cancer Res 2024, 84 (9_Supplement), GS02-01. [Google Scholar] [CrossRef]
- Bardia, A.; Jhaveri, K.; Im, S.A.; Pernas, S.; De Laurentiis, M.; Wang, S.; Martínez Jañez, N.; Borges, G.; Cescon, D.W.; Hattori, M.; Lu, Y.S.; Hamilton, E.; Zhang, Q.; Tsurutani, J.; Kalinsky, K.; Rubini Liedke, P.E.; Xu, L.; Fairhurst, R.M.; Khan, S.; Denduluri, N.; Rugo, H.S.; Xu, B.; Pistilli, B.; TROPION-Breast01 Investigators. Datopotamab Deruxtecan Versus Chemotherapy in Previously Treated Inoperable/Metastatic Hormone Receptor-Positive Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer: Primary Results From TROPION-Breast01. J Clin Oncol. 2024, JCO2400920. [Google Scholar] [CrossRef]
- Mayer, E.L.; Ren, Y.; Wagle, N.; Mahtani, R.; Ma, C.; DeMichele, A.; Cristofanilli, M.; Meisel, J.; Miller, K.D.; Abdou, Y.; Riley, E.C.; Qamar, R.; Sharma, P.; Reid, S.; Sinclair, N.; Faggen, M.; Block, C.C.; Ko, N.; Partridge, A.H.; Chen, W.Y.; DeMeo, M.; Attaya, V.; Okpoebo, A.; Alberti, J.; Liu, Y.; Gauthier, E.; Burstein, H.J.; Regan, M.M.; Tolaney, S.M. PACE: A Randomized Phase II Study of Fulvestrant, Palbociclib, and Avelumab After Progression on Cyclin-Dependent Kinase 4/6 Inhibitor and Aromatase Inhibitor for Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor-Negative Metastatic Breast Cancer. J Clin Oncol. 2024, 42, 2050–2060. [Google Scholar]
- Llombart Cussac, A.; Harper-Wynne, C.; Perello, A.; Hennequin, A.; Fernandez, A.; Colleoni, M. Second-line endocrine therapy (ET) with or without palbociclib (P) maintenance in patients (pts) with hormone receptor-positive (HR[+])/human epidermal growth factor receptor 2-negative (HER2[-]) advanced breast cancer (ABC): PALMIRA trial. J Clin Oncol 2023, 41 (suppl 16), abstr 1001. [Google Scholar] [CrossRef]
- Kalinski, K.; Bianchini, G.; Hamilton, E.P.; Graff, S.L.; Park, K.H.; Jeselsohn, R.; Demirci, U.; Martin, M.; Layman, R.M.; Hurvitz, S.A.; Sammons, S.L.; Kaufman, P.A.; Muñoz, M.; Tseng, L.M.; Knoderer, H.; Nguyen, B.; Zhou, Y.; Ravenberg, E.; Litchfield, L.M.; Wander, S.A. Abemaciclib plus fulvestrant vs fulvestrant alone for HR+, HER2- advanced breast cancer following progression on a prior CDK4/6 inhibitor plus endocrine therapy: Primary outcome of the phase 3 postMONARCH trial. J Clin Oncol 1001, 42 (suppl 17), abstr LBA1001. [Google Scholar] [CrossRef]
- Raheem, F.; Karikalan, S.A.; Batalini, F.; El Masry, A.; Mina, L. Metastatic ER+ Breast Cancer: Mechanisms of Resistance and Future Therapeutic Approaches. Int J Mol Sci. 2023, 24, 16198. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Klein, P.; Tiersten, A.; Sparano, J.A. An emerging generation of endocrine therapies in breast cancer: a clinical perspective. NPJ Breast Cancer. 2023, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Parisian, A.D.; Barratt, S.A.; Hodges-Gallagher, L.; Ortega, F.E.; Peña, G.; Sapugay, J.; Robello, B.; Sun, R.; Kulp, D.; Palanisamy, G.S.; Myles, D.C.; Kushner, P.J.; Harmon, C.L. Palazestrant (OP-1250), A Complete Estrogen Receptor Antagonist, Inhibits Wild-type and Mutant ER-positive Breast Cancer Models as Monotherapy and in Combination. Mol Cancer Ther. 2024, 23, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Lainé, M.; Greene, M.E.; Kurleto, J.D.; Bozek, G.; Leng, T.; Huggins, R.J.; Komm, B.S.; Greene, G.L. Lasofoxifene as a potential treatment for aromatase inhibitor-resistant ER-positive breast cancer. Breast Cancer Res. 2024, 26, 95. [Google Scholar] [CrossRef]
- Goetz, M.P.; Bagegni, N.A.; Batist, G.; Brufsky, A.; Cristofanilli, M.A.; Damodaran, S.; Daniel, B.R.; Fleming, G.F.; Gradishar, W.J.; Graff, S.L.; Grosse Perdekamp, M.T.; Hamilton, E.; Lavasani, S.; Moreno-Aspitia, A.; O'Connor, T.; Pluard, T.J.; Rugo, H.S.; Sammons, S.L.; Schwartzberg, L.S.; Stover, D.G.; Vidal, G.A.; Wang, G.; Warner, E.; Yerushalmi, R.; Plourde, P.V.; Portman, D.J.; Gal-Yam, E.N. Lasofoxifene versus fulvestrant for ER+/HER2- metastatic breast cancer with an ESR1 mutation: results from the randomized, phase II ELAINE 1 trial. Ann Oncol. 2023, 34, 1141–1151. [Google Scholar] [CrossRef]
- Damodaran, S.; O'Sullivan, C.C.; Elkhanany, A.; Anderson, I.C.; Barve, M.; Blau, S.; Cherian, M.A.; Peguero, J.A.; Goetz, M.P.; Plourde, P.V.; Portman, D.J.; Moore, H.C.F. Open-label, phase II, multicenter study of lasofoxifene plus abemaciclib for treating women with metastatic ER+/HER2- breast cancer and an ESR1 mutation after disease progression on prior therapies: ELAINE 2. Ann Oncol. 2023, 34, 1131–1140. [Google Scholar] [CrossRef]
- Goetz, M.P.; Wander, S.A.; Bachelot, T.; et al. Open-label, randomized, multicenter, phase 3, ELAINE 3 study of the efficacy and safety of lasofoxifene plus abemaciclib for treating ER+/HER2-, locally advanced or metastatic breast cancer with an ESR1 mutation. J Clin Oncol. 2024, 42 S16, TPS1127. [Google Scholar] [CrossRef]
- Fanning, S.W.; Jeselsohn, R.; Dharmarajan, V.; Mayne, C.G.; Karimi, M.; Buchwalter, G.; Houtman, R.; Toy, W.; Fowler, C.E.; Han, R.; Lainé, M.; Carlson, K.E.; Martin, T.A.; Nowak, J.; Nwachukwu, J.C.; Hosfield, D.J.; Chandarlapaty, S.; Tajkhorshid, E.; Nettles, K.W.; Griffin, P.R.; Shen, Y.; Katzenellenbogen, J.A.; Brown, M.; Greene, G.L. The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. Elife. 2018, 2018 7, e37161. [Google Scholar] [CrossRef]
- Tsuji, J.; Li, T.; Grinshpun, A.; Coorens, T.; Russo, D.; Anderson, L.; Rees, R.; Nardone, A.; Patterson, C.; Lennon, N.J.; Cibulskis, C.; Leshchiner, I.; Tayob, N.; Tolaney, S.M.; Tung, N.; McDonnell, D.P.; Krop, I.E.; Winer, E.P.; Stewart, C.; Getz, G.; Jeselsohn, R. Clinical Efficacy and Whole-Exome Sequencing of Liquid Biopsies in a Phase IB/II Study of Bazedoxifene and Palbociclib in Advanced Hormone Receptor-Positive Breast Cancer. Clin Cancer Res. 2022, 28, 5066–5078. [Google Scholar] [CrossRef] [PubMed]
- Furman, C.; Puyang, X.; Zhang, Z.; Wu, Z.J.; Banka, D.; Aithal, K.B.; Albacker, L.A.; Hao, M.H.; Irwin, S.; Kim, A.; Montesion, M.; Moriarty, A.D.; Murugesan, K.; Nguyen, T.V.; Rimkunas, V.; Sahmoud, T.; Wick, M.J.; Yao, S.; Zhang, X.; Zeng, H.; Vaillancourt, F.H.; Bolduc, D.M.; Larsen, N.; Zheng, G.Z.; Prajapati, S.; Zhu, P.; Korpal, M. Covalent ERα Antagonist H3B-6545 Demonstrates Encouraging Preclinical Activity in Therapy-Resistant Breast Cancer. Mol Cancer Ther. 2022, 21, 890–902. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Wang, J.S.; Pluard, T.; et al. H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer—A phase II study. Cancer Res. 2022, 82, P1-17-1. [Google Scholar] [CrossRef]
- Xiong, R.; Patel, H.K.; Gutgesell, L.M.; Zhao, J.; Delgado-Rivera, L.; Pham, T.N.D.; Zhao, H.; Carlson, K.; Martin, T.; Katzenellenbogen, J.A.; Moore, T.W.; Tonetti, D.A.; Thatcher, G.R.J. Selective Human Estrogen Receptor Partial Agonists (ShERPAs) for Tamoxifen-Resistant Breast Cancer. J Med Chem. 2016, 59, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.Z.; Liu, L.C.; Fischer, J.H.; et al. , Phase 1 study of TTC-352 in patients with metastatic breast cancer progressing on endocrine and CDK4/6 inhibitor therapy. Breast Cancer Res Treat. 2020, 183, 617–627. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; Zinn, N.; Grandi, P.; Shimamura, S.; Bergamini, G.; Faelth-Savitski, M.; Bantscheff, M.; Cox, C.; Gordon, D.A.; Willard, R.R.; Flanagan, J.J.; Casillas, L.N.; Votta, B.J.; den Besten, W.; Famm, K.; Kruidenier, L.; Carter, P.S.; Harling, J.D.; Churcher, I.; Crews, C.M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Snyder, L.B.; Flanagan, J.J.; Qian, Y.; et al. The discovery of ARV-471, an orally bioavailable estrogen receptor degrading PROTAC for the treatment of patients with breast cancer. AACR Meeting 2021. Cancer Res. 2021, 81, 44. [Google Scholar] [CrossRef]
- Hamilton, E.; Vahdat, L.; Han, H.; et al. First-in-human safety and activity of ARV-471, a novel PROTAC® estrogen receptor degrader, in ER+/HER2- locally advanced or metastatic breast cancer. Cancer Res. 2022, 82, PD13-08. [Google Scholar] [CrossRef]
- Schott, A.F.; Hurvitz, S.; Ma, C.; et al. GS3-03 ARV-471, a PROTAC® estrogen receptor (ER) degrader in advanced ER-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer: phase 2 expansion (VERITAC) of a phase 1/2 study. Cancer Res. 2023, 83, GS3-03. [Google Scholar] [CrossRef]
- Hamilton, E.; Jeselsohn, R.; Hurvitz, S.; et al. Hamilton E, Jeselsohn R, Hurvitz S, et al. Vepdegestrant, a PROteolysis TArgeting Chimera (PROTAC) estrogen receptor (ER) degrader, plus palbociclib in ER–positive/human epidermal growth factor receptor 2 (HER2)–negative advanced breast cancer: phase 1b cohort. Cancer Res. 2024, 84 (Suppl. S9), PS15-03. [Google Scholar]
- Gough, S.M.; Flanagan, J.J.; Teh, J.; Andreoli, M.; Rousseau, E.; Pannone, M.; Bookbinder, M.; Willard, R.; Davenport, K.; Bortolon, E.; Cadelina, G.; Gordon, D.; Pizzano, J.; Macaluso, J.; Soto, L.; Corradi, J.; Digianantonio, K.; Drulyte, I.; Morgan, A.; Quinn, C.; Békés, M.; Ferraro, C.; Chen, X.; Wang, G.; Dong, H.; Wang, J.; Langley, D.R.; Houston, J.; Gedrich, R.; Taylor, I.C. Oral Estrogen Receptor PROTAC Vepdegestrant (ARV-471) Is Highly Efficacious as Monotherapy and in Combination with CDK4/6 or PI3K/mTOR Pathway Inhibitors in Preclinical ER+ Breast Cancer Models. Clin Cancer Res. 2024, 30, 3549–3563. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, C.; Bianco-Miotto, T.; Jindal, S.; Butler, L.M.; Leung, S.; McNeil, C.M.; O'Toole, S.A.; Ebrahimie, E.; Millar, E.K.A.; Sakko, A.J.; Ruiz, A.I.; Vowler, S.L.; Huntsman, D.G.; Birrell, S.N.; Sutherland, R.L.; Palmieri, C.; Hickey, T.E.; Tilley, W.D. The Magnitude of Androgen Receptor Positivity in Breast Cancer Is Critical for Reliable Prediction of Disease Outcome. Clin Cancer Res. 2018, 24, 2328–2341. [Google Scholar] [CrossRef]
- Hickey, T.E.; Selth, L.A.; Chia, K.M.; Laven-Law, G.; Milioli, H.H.; Roden, D.; Jindal, S.; Hui, M.; Finlay-Schultz, J.; Ebrahimie, E.; Birrell, S.N.; Stelloo, S.; Iggo, R.; Alexandrou, S.; Caldon, C.E.; Abdel-Fatah, T.M.; Ellis, I.O.; Zwart, W.; Palmieri, C.; Sartorius, C.A.; Swarbrick, A.; Lim, E.; Carroll, J.S.; Tilley, W.D. The androgen receptor is a tumor suppressor in estrogen receptor-positive breast cancer. Nat Med. 2021, 27, 310–320. [Google Scholar] [CrossRef]
- Khan, A.F.; Karami, S.; Peidl, A.S.; Waiters, K.D.; Babajide, M.F.; Bawa-Khalfe, T. Androgen Receptor in Hormone Receptor-Positive Breast Cancer. Int J Mol Sci. 2023, 25, 476. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Abramson, V.; Colleoni, M.; Traina, T.; Holmes, F.; Garcia-Estevez, L.; Hart, L.; Awada, A.; Zamagni, C.; Morris, P.G.; Schwartzberg, L.; Chan, S.; Gucalp, A.; Biganzoli, L.; Steinberg, J.; Sica, L.; Trudeau, M.; Markova, D.; Tarazi, J.; Zhu, Z.; O'Brien, T.; Kelly, C.M.; Winer, E.; Yardley, D.A. A Randomized Placebo Controlled Phase II Trial Evaluating Exemestane with or without Enzalutamide in Patients with Hormone Receptor-Positive Breast Cancer. Clin Cancer Res. 2020, 26, 6149–6157. [Google Scholar] [CrossRef]
- Wei, L.; Gao, H.; Yu, J.; Zhang, H.; Nguyen, T.T.L.; Gu, Y.; Passow, M.R.; Carter, J.M.; Qin, B.; Boughey, J.C.; Goetz, M.P.; Weinshilboum, R.M.; Ingle, J.N.; Wang, L. Pharmacological Targeting of Androgen Receptor Elicits Context-Specific Effects in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2023, 83, 456–470. [Google Scholar] [CrossRef]
- Palmieri, C.; Linden, H.M.; Birrell, S.; et al. Palmieri C, Linden HM, Birrell S, et al. Efficacy of enobosarm, a selective androgen receptor (AR) targeting agent, correlates with the degree of AR positivity in advanced AR+/estrogen receptor (ER)+ breast cancer in an international phase 2 clinical study. J Clin Oncol 2021, 39, 1020. [Google Scholar] [CrossRef]
- Rinn, K.; Linden, H.; Schwartzberg, L.; et al. Design of Active Phase 3 ENABLAR-2 Study Evaluating Enobosarm +/- Abemaciclib in Patients with AR+ER+HER2- 2nd-Line Metastatic Breast Cancer Following Tumor Progression on an Estrogen Blocking Agent Plus Palbociclib or Ribociclib. Cancer Res 2024, 84 (9_Supplement, PO4-27-06. [Google Scholar] [CrossRef]
- Song, X.; Fang, C.; Dai, Y.; Sun, Y.; Qiu, C.; Lin, X.; Xu, R. Cyclin-dependent kinase 7 (CDK7) inhibitors as a novel therapeutic strategy for different molecular types of breast cancer. Br J Cancer. 2024, 130, 1239–1248. [Google Scholar] [CrossRef]
- Guarducci, C.; Nardone, A.; Russo, D.; Nagy, Z.; Heraud, C.; Grinshpun, A.; Zhang, Q.; Freelander, A.; Leventhal, M.J.; Feit, A.; Cohen Feit, G.; Feiglin, A.; Liu, W.; Hermida-Prado, F.; Kesten, N.; Ma, W.; De Angelis, C.; Morlando, A.; O'Donnell, M.; Naumenko, S.; Huang, S.; Nguyen, Q.D.; Huang, Y.; Malorni, L.; Bergholz, J.S.; Zhao, J.J.; Fraenkel, E.; Lim, E.; Schiff, R.; Shapiro, G.I.; Jeselsohn, R. Selective CDK7 Inhibition Suppresses Cell Cycle Progression and MYC Signaling While Enhancing Apoptosis in Therapy-resistant Estrogen Receptor-positive Breast Cancer. Clin Cancer Res. 2024, 30, 1889–1905. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; Stover, D.G.; Lim, E.; Wang, Z.C.; Iglehart, J.D.; Young, R.A.; Gray, N.S.; Zhao, J.J. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015, 163, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Periyasamy, M.; Sava, G.P.; Bondke, A.; Slafer, B.W.; Kroll, S.H.B.; Barbazanges, M.; Starkey, R.; Ottaviani, S.; Harrod, A.; Aboagye, E.O.; Buluwela, L.; Fuchter, M.J.; Barrett, A.G.M.; Coombes, R.C.; Ali, S. ICEC0942, an Orally Bioavailable Selective Inhibitor of CDK7 for Cancer Treatment. Mol Cancer Ther. 2018, 17, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Juric, D.; Henick, B.S.; et al. BLU-222, an oral, potent, and selective CDK2 inhibitor, in patients with advanced solid tumors: Phase 1 monotherapy dose escalation. J Clin Oncol 2023, 41 (suppl 16), abstr 3095. [Google Scholar] [CrossRef]
- Yap, T.A.; Elhaddad, A.M.; Grisham, R.N.; et al. First-in-human phase 1/2a study of a potent and novel CDK2-selective inhibitor PF-07104091 in patients (pts) with advanced solid tumors, enriched for CDK4/6 inhibitor resistant HR+/HER2- breast cancer. J Clin Oncol. 2023, 41 16s, abstr 3010. [Google Scholar] [CrossRef]
- Goldberg, J.; Qiao, N.; Guerriero, J.L.; Gross, B.; Meneksedag, Y.; Lu, Y.F.; Philips, A.V.; Rahman, T.; Meric-Bernstam, F.; Roszik, J.; Chen, K.; Jeselsohn, R.; Tolaney, S.M.; Peoples, G.E.; Alatrash, G.; Mittendorf, E.A. Estrogen Receptor Mutations as Novel Targets for Immunotherapy in Metastatic Estrogen Receptor-positive Breast Cancer. Cancer Res Commun. 2024, 4, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Dailey, G.P.; Rabiola, C.A.; Lei, G.; Wei, J.; Yang, X.Y.; Wang, T.; Liu, C.X.; Gajda, M.; Hobeika, A.C.; Summers, A.; Marek, R.D.; Morse, M.A.; Lyerly, H.K.; Crosby, E.J.; Hartman, Z.C. Vaccines targeting ESR1 activating mutations elicit anti-tumor immune responses and suppress estrogen signaling in therapy resistant ER+ breast cancer. Hum Vaccin Immunother. 2024, 20, 2309693. [Google Scholar] [CrossRef]
- Nicolini, A.; Carpi, A. Beta-interferon and interleukin-2 prolong more than three times the survival of 26 consecutive endocrine dependent breast cancer patients with distant metastases: an exploratory trial. Biomed Pharmacother. 2005, 59, 253–263. [Google Scholar] [CrossRef]
- Nicolini, A.; Carpi, A.; Ferrari, P.; Biava, P.M.; Rossi, G. Immunotherapy and Hormone-therapy in Metastatic Breast Cancer: A Review and an Update. Curr Drug Targets. 2016, 17, 1127–1139. [Google Scholar] [CrossRef]
- Nicolini, A.; Rossi, G.; Ferrari, P.; Morganti, R.; Carpi, A. A new immunotherapy schedule in addition to first-line hormone therapy for metastatic breast cancer patients in a state of clinical benefit during hormone therapy. J Mol Med (Berl). 2020, 98, 375–382. [Google Scholar] [CrossRef]
- Nicolini, A.; Rossi, G.; Ferrari, P.; Morganti, R.; Carpi, A. Final results of a 2:1 control–case observational study using interferon beta and interleukin-2, in addition to first-line hormone therapy, in estrogen receptor-positive, endocrine-responsive metastatic breast cancer patients. J Cancer Metastasis Treat 2022, 8, 13. [Google Scholar] [CrossRef]
- Sultan, R.; Ahmed, A.; Wei, L.; Saeed, H.; Islam, M.; Ishaq, M. The anticancer potential of chemical constituents of Moringa oleifera targeting CDK-2 inhibition in estrogen receptor positive breast cancer using in-silico and in vitro approches. BMC Complement Med Ther. 2023, 23, 396. [Google Scholar] [CrossRef] [PubMed]
- Pandithar, S.; Galke, D.; Akume, A.; Belyakov, A.; Lomonaco, D.; Guerra, A.A.; Park, J.; Reff, O.; Jin, K. The role of CXCL1 in crosstalk between endocrine resistant breast cancer and fibroblast. Mol Biol Rep. 2024, 51, 331. [Google Scholar] [CrossRef] [PubMed]
- Pastò, B.; Vida, R.; Dri, A.; Foffano, L.; Della Rossa, S.; Gerratana, L.; Puglisi, F. Beyond Hormone Receptors: liquid biopsy tools to unveil new clinical meanings and empower therapeutic decision-making in Luminal-like metastatic breast cancer. Breast. 2024, 79, 103859. [Google Scholar] [CrossRef]
- Foffano, L.; Cucciniello, L.; Nicolò, E.; Migliaccio, I.; Noto, C.; Reduzzi, C.; Malorni, L.; Cristofanilli, M.; Gerratana, L.; Puglisi, F. Cyclin-dependent kinase 4 and 6 inhibitors (CDK4/6i): Mechanisms of resistance and where to find them. Breast. 2024 79, 103863. [CrossRef]
- Fusco, N.; Malapelle, U. Next-generation sequencing for PTEN testing in HR+/HER2- metastatic breast cancer. Crit Rev Oncol Hematol. 2025, 207, 104626. [Google Scholar] [CrossRef]
Figure 2.
Main mechanisms of resistance to endocrine therapy and/or CDK4/6 inhibitors in advanced ER+/HER2-breast cancer. a) they include ERBB2, FGFR1/2, EGFR, IGFR1, NOTCH; b) in Akt1, PI3KCA, PTEN, HRAS, KRAS, NRAS; c) ESR1 Y537S/Y537N mutations; RTK: receptor tyrosine kinase; PI3K: phosphoinositide 3 kinase; PTEN: phosphatase and tensin homolog; Akt: protein kinase B; mTOR: mammalian target of rapamycin; EGFR: epidermal growth factor receptor; ERBB2: receptor tyrosine-protein kinase erbB2; FGFR1/2: fibriblast growth factor receptor 1/2; IGFR1: insulin-like growth factor receptor 1; NOTCH: notch signalling pathway; MAPK: mitogen-activated protein kinase; Rb: retinoblastoma; RAS: rat sarcoma virus protein superfamily; NF1: neurofibromatosis type 1 gene; RAF: rapidly accelerated fibrosarcoma protein kinase; MEK: mitogen-activated protein kinase kinase; ERK: extracellular signal-regulated kinase; FAT1: protein encoded by FAT gene that takes part of the cadherin superfamily proteins; MST1/2: macrophage-stimulating protein 1/2; LATS1/2: large tumor suppressor kinase 1/2; YAP: yes-associated protein-1; TAZ: transcriptional adaptor putative zinc finger; TEAD: TEA domain family member; CDK: cyclin-dependent kinase; p: protein; SMAD4: smad family member 4; PMM2 m.r.; phosphomannomutase 2 metabolic reprogramming; APOBEC3 g.a.: apolipoprotein B mRNA editing enzyme, catalytic subunit 3, genetic alteration; PKMYT: membrane-associated tyrosine and threonine specific cdc 2-inhibitory kinase; MYSM1: Myblike, SWIRM and MNP domain 1; MNX1: motor neuron and pancreas homeobox 1; ACC1 m.r.: acetyl-CoA carboxylase 1 metabolic reprogramming; CYP19A1: member of the cytochrome P450 superfamily; MLL3 g.a.: myeloid lymphoid or mixed lineage leukemia protein 3 genetic alterations; COLL11A1: collagen, type XI, alpha 1; BCAR4: breast cancer anti-estrogen resistance protein 4; RFC3: replication factor C subunit 3; AEs: antiestrogens; ERalpha: estrogen receptor alpha; ERE: estrogen receptor element; TATA: tiamine-adenine sequence; c-myc: family of genes overexpressed in various cancers and homologous with an avian virus; circHIAT1: circular RNA HIAT1; miR-19a-3p: microRNA 19a-3p; CADM2: cell adhesion molecule 2; NF-kB: nuclear factor kappa-light chain enhancer of activated B cells; AP-1: activating protein-1; E2F: transcription factor E2F; t.f.: franscription factor; HDAC: hystone deacetylase; LncRNA: long non coding RNA; AURKA: aurora kinase A; CCNE1/2: genes encoding cyclin E1/E2 proteins; DNMT1: DNA methyltransferase 1; DDR: DNA damage repair; BRCA1/2: breast cancer type 1/2 genes; HRR: homologous recombination repair; HRD: homologous repair deficiency; TP53: tumor protein 53; DDCKPS: DNA damage checkpoints signalling; ATM: part of serine/threonine protein kinase ATM; ATR: ataxia-teleangectasia and RAD3 related; CHEK2: checkpoint kinase 2; PD-L1: programmed death ligand 1; TILs: tumor infiltrating lymphocytes; IRPS: interferon-related palbociclib-resistance signature; IL: interleukin; P: phosphorylation. increase. Decrease. (Also see text).
Figure 2.
Main mechanisms of resistance to endocrine therapy and/or CDK4/6 inhibitors in advanced ER+/HER2-breast cancer. a) they include ERBB2, FGFR1/2, EGFR, IGFR1, NOTCH; b) in Akt1, PI3KCA, PTEN, HRAS, KRAS, NRAS; c) ESR1 Y537S/Y537N mutations; RTK: receptor tyrosine kinase; PI3K: phosphoinositide 3 kinase; PTEN: phosphatase and tensin homolog; Akt: protein kinase B; mTOR: mammalian target of rapamycin; EGFR: epidermal growth factor receptor; ERBB2: receptor tyrosine-protein kinase erbB2; FGFR1/2: fibriblast growth factor receptor 1/2; IGFR1: insulin-like growth factor receptor 1; NOTCH: notch signalling pathway; MAPK: mitogen-activated protein kinase; Rb: retinoblastoma; RAS: rat sarcoma virus protein superfamily; NF1: neurofibromatosis type 1 gene; RAF: rapidly accelerated fibrosarcoma protein kinase; MEK: mitogen-activated protein kinase kinase; ERK: extracellular signal-regulated kinase; FAT1: protein encoded by FAT gene that takes part of the cadherin superfamily proteins; MST1/2: macrophage-stimulating protein 1/2; LATS1/2: large tumor suppressor kinase 1/2; YAP: yes-associated protein-1; TAZ: transcriptional adaptor putative zinc finger; TEAD: TEA domain family member; CDK: cyclin-dependent kinase; p: protein; SMAD4: smad family member 4; PMM2 m.r.; phosphomannomutase 2 metabolic reprogramming; APOBEC3 g.a.: apolipoprotein B mRNA editing enzyme, catalytic subunit 3, genetic alteration; PKMYT: membrane-associated tyrosine and threonine specific cdc 2-inhibitory kinase; MYSM1: Myblike, SWIRM and MNP domain 1; MNX1: motor neuron and pancreas homeobox 1; ACC1 m.r.: acetyl-CoA carboxylase 1 metabolic reprogramming; CYP19A1: member of the cytochrome P450 superfamily; MLL3 g.a.: myeloid lymphoid or mixed lineage leukemia protein 3 genetic alterations; COLL11A1: collagen, type XI, alpha 1; BCAR4: breast cancer anti-estrogen resistance protein 4; RFC3: replication factor C subunit 3; AEs: antiestrogens; ERalpha: estrogen receptor alpha; ERE: estrogen receptor element; TATA: tiamine-adenine sequence; c-myc: family of genes overexpressed in various cancers and homologous with an avian virus; circHIAT1: circular RNA HIAT1; miR-19a-3p: microRNA 19a-3p; CADM2: cell adhesion molecule 2; NF-kB: nuclear factor kappa-light chain enhancer of activated B cells; AP-1: activating protein-1; E2F: transcription factor E2F; t.f.: franscription factor; HDAC: hystone deacetylase; LncRNA: long non coding RNA; AURKA: aurora kinase A; CCNE1/2: genes encoding cyclin E1/E2 proteins; DNMT1: DNA methyltransferase 1; DDR: DNA damage repair; BRCA1/2: breast cancer type 1/2 genes; HRR: homologous recombination repair; HRD: homologous repair deficiency; TP53: tumor protein 53; DDCKPS: DNA damage checkpoints signalling; ATM: part of serine/threonine protein kinase ATM; ATR: ataxia-teleangectasia and RAD3 related; CHEK2: checkpoint kinase 2; PD-L1: programmed death ligand 1; TILs: tumor infiltrating lymphocytes; IRPS: interferon-related palbociclib-resistance signature; IL: interleukin; P: phosphorylation. increase. Decrease. (Also see text).
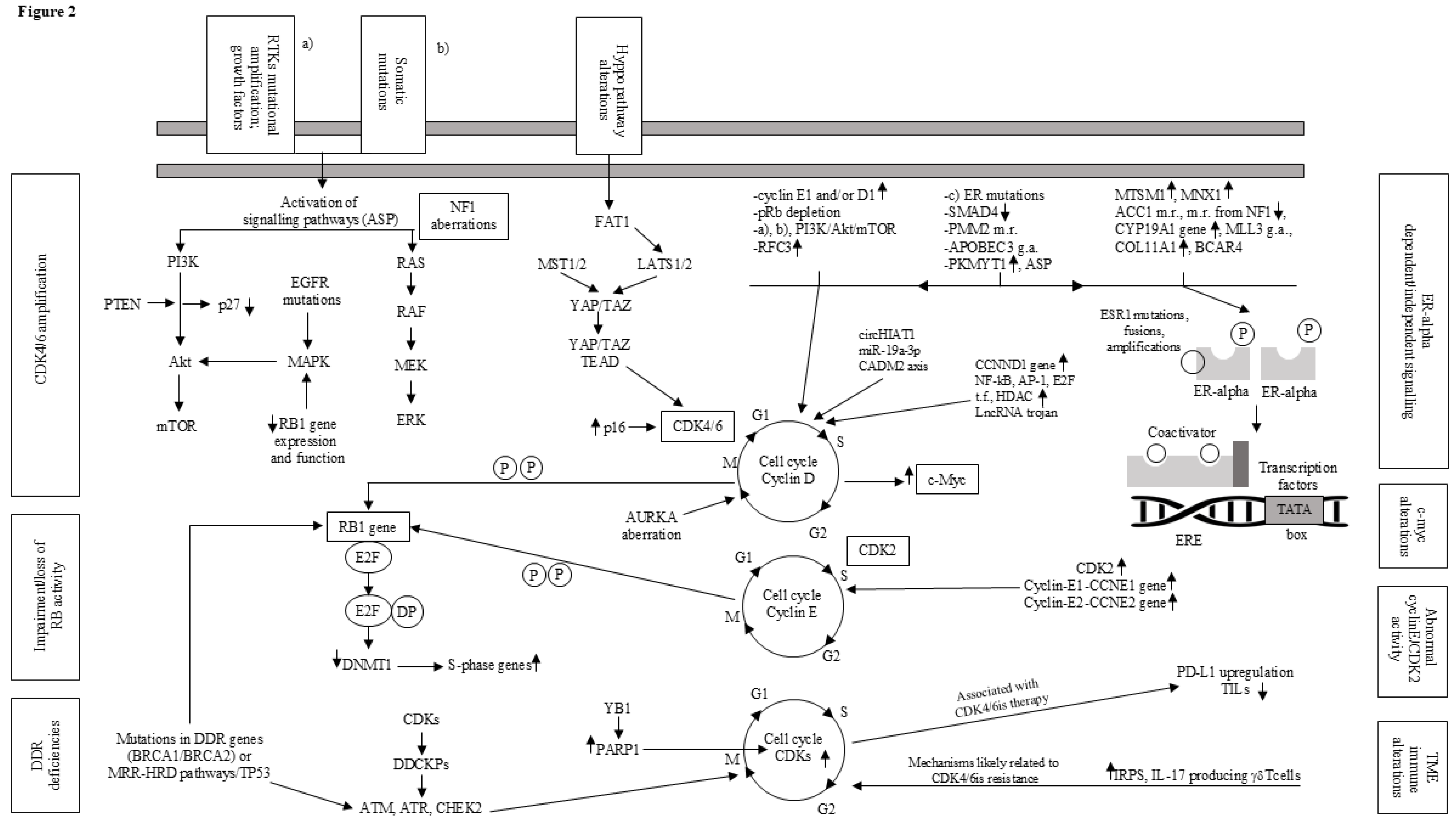
Figure 3.
Progression-free survival correlated to hormonal therapy (survival median time of HIT 33.1 (95% CI 24.5–41.8); survival median time of HT 18 (95% CI 12.1–23.8)). HIT: hormone-immunotherapy; HT: hormone-therapy.
Figure 3.
Progression-free survival correlated to hormonal therapy (survival median time of HIT 33.1 (95% CI 24.5–41.8); survival median time of HT 18 (95% CI 12.1–23.8)). HIT: hormone-immunotherapy; HT: hormone-therapy.
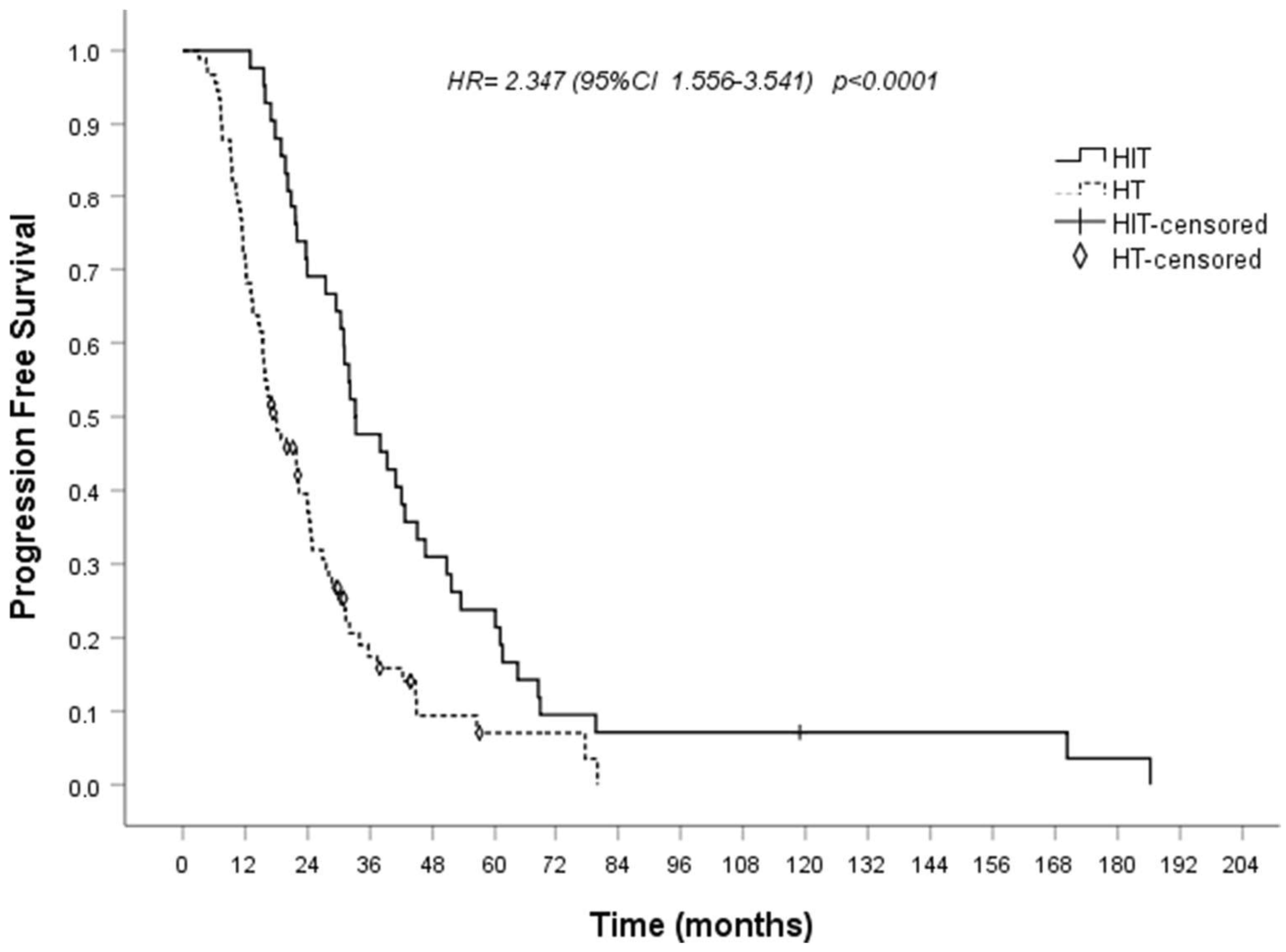

Table 1.
Current and experimental therapies in advanced endocrine-CDK4/6is resistant ER+/HER2- breast cancer.
Table 1.
Current and experimental therapies in advanced endocrine-CDK4/6is resistant ER+/HER2- breast cancer.
| A) continues | ||||||
| Study (phase) | Therapy | Setting | Main endpoints | Reference | ||
| CONFIRM (III) | Fulvestrant 500 mg vs 250 mg | Postmenopausal MBC women progressing on ET | mPFS 6.5 months mOS 26.4 months |
122 | ||
| MAINTAIN (Control arm) | Fulvestrant 500 mg | ABC patients, progressed on ET plus a CDK4/6i | mPFS 2.76 months | 125 | ||
| VERONICA (Control arm) |
Fulvestrant 500 mg | MBC patients, post CDK4/6i progression | m PFS 1.94 months | 126 | ||
| EMERALD (III) | Elacestrant vs standard ET | ABC patients after 1-2 lines of ET including a CDK4/6i, and ≤ 1 chemotherapy | 6-month PFS rate: 34.3% vs 20.4% in all patients; 40.8% vs 19.1%, in patients with ESR1 mutation. 12-month PFS rate: 22.3% vs 9.4% in all patients; 26.8% and 8.2% in patients with ESR1 mutation |
128 | ||
| ELEVATE (Ib/II) | Elacestrant in combination with alpelisib, or capivasertib, or everolimus, or palbociclib, or abemaciclib, or ribociclib | ABC/MBC, progressing on one or up to two prior lines of ET (inclusion criteria differ in every study arm) | Primary: PFS, safety Secondary: ORR, DOR, CBR, OS |
130 | ||
| ADELA (III) | Elacestrant plus everolimus vs elacestrant alone | ABC patients with ESR1-mutated tumors progressing on ET plus CDK4/6i | Primary: PFS Secondary: OS, ORR, CBR, DOR, safety and quality of life |
NCT06382948 | ||
| SERENA-2 (II) | Camizestrant once daily at 75 mg, 150 mg, or 300 mg vs fulvestrant 500 mg | ABC patients progressing on at least one line of ET, and no more than one previous ET in the advanced setting | mPFS 7.2 months with camizestrant 75 mg, 7.7 with 150 mg, vs 3.7 months with fulvestrant | 131 | ||
| SERENA-6 (III) | Camizestrant plus CDK4/6i maintaining | ABC patients progressing on AI plus CDK4/6i, upon detection of ESR1m in ctDNA before clinical disease progression. | Primary end point: PFS; secondary: CT-free survival, PFS2, OS, safety. | NCT04964934 | ||
| EMBER-3 (III) | Imlunestrant vs monoET (88% fulvestrant) vs imlunestrant plus abemaciclib. |
ABC patients recurring or progressing during or after AI alone or AI plus a CDK4/6i (59.8%) | Patients with ESR1 mutations: mPFS 5.5 months with imlunestrant vs 3.8 with monoET Overall population: mPFS 5.6 months with imlunestrant vs 5.5 with monoET mPFS 9.4 months with imlunestrant plus abemaciclib vs 5.5 with imlunestrant regardless of ESR1-mutation status |
133 | ||
| acelERA Breast Cancer Study (II) | Giredestrant vs physician’s choice endocrine monotherapy (PCET) | MBC patients progressing after 1-2 lines of systemic therapy; 1 previous targeted agent, 1 line of chemotherapy and prior fulvestrant were allowed | Overall population: mPFS 5.6 months in the giredestrant arm vs 5.4 months in the PCET arm. ESR1 mutated patients: mPFS 5.3 months vs 3.5 months. |
135 | ||
| MORPHEUS-Breast cancer (I-IIb umbrella study) Cohort 1 |
Giredestrant alone vs giredestrant in combination with abemaciclib, or ipatasertib, or inavolisib, or ribociclib, or everolimus, or samuraciclib, or atezolizumab | ABC/MBC patients progressing on AI + CDK4/6i as 1st or 2nd line | Primary: ORR, safety Secondary: PFS, DCR, CBR, OS, DOR |
NCT04802759 | ||
| B) continues | ||||||
| Study (phase) | Therapy | Setting | Main endpoints | Reference | ||
| SOLAR-1 (III) | Alpelisib plus fulvestrant vs placebo plus fulvestrant | ABC patients progressing on or after AI | mOS 39.3 vs 31.4 months in the PIK3CA-mutated cohort | 137 | ||
| BYLieve (II) | Alpelisib plus fulvestrant | ABC patients with tumour PIK3CA mutation, progressing on or after previous ET, including CDK4/6i | 53.8% patients alive without disease progression at 6 months, after a median follow-up of 21.8 months, | 139 | ||
| CAPItello-291 (III) | Capivasertib plus fulvestrant vs placebo plus fulvestrant | ABC patients relapsed or progressing during or after treatment with an AI, with or without CDK4/6i | mPFS 7.2 vs 3.6 months in the overall population mPFS 7.3 vs 3.1 months in patients with AKT pathway alterations |
142 | ||
| INAVO-120 (III) | Inavolisib plus palbociclib-fulvestrant vs placebo plus palbociclib-fulvestrant | PIK3CA-mutated ABC/MBC patients relapsed during or within 12 months after the completion of adjuvant ET | mPFS 15.0 months vs 7.3 months ORR 58.4% vs 25.0% |
143 | ||
| INAVO-121 (III) | Inavolisib plus fulvestrant vs alpelisib plus fulvestrant | PIK3CA-mutated ABC/MBC patients progressing after ET plus CDK4/6i | Primary: PFS Secondary: ORR, CBR, DOR, OS |
NCT05646862 | ||
| BOLERO-2 (III) | Everolimus plus exemestane vs placebo plus exemestane | Patients who had recurrence or progression on NSAI | mPFS 6.9 months vs 2.8 months | 35 | ||
| Retrospective | Everolimus plus ET | MBC patients progressing on palbociclib | mPFS 4.2 months, mOS 18.7 months | 146 | ||
| Retrospective | Everolimus plus exemestane | MBC progressing on an NSAI alone or in association to a CDK4/6i (40%) | No significant difference in mPFS (3.6 vs. 4.2 months) or mOS (15.6 vs. 11.3 months) between patients who had received prior CDK4/6is and those who had not | 147 | ||
| IPATunity-150 (Ib) | Ipatasertib plus fulvestrant plus palbociclib | Patients relapsed during adjuvant ET who had not previously received a CDK4/6i | ORR 45%, mDOR 9.6 months | 144 | ||
| FINER (III) | Ipatasertib plus fulvestrant vs placebo plus fulvestrant | ABC patients progressing on AI plus CDK4/6i | Primary: PFS Secondary: PFS in PIK3CA/AKT1/PTEN altered and non-altered cohorts, RR, DOR, CBR, OS, TSST, safety |
NCT04650581 | ||
| VIKTORIA-1 (III) | Gedatolisib plus fulvestrant with or without palbociclib vs standard-of-care ET | ABC/MBC patients progressing on AI plus CDK4/6i | Primary: PFS Secondary: OS, ORR, DOR, TTR, CBR |
NCT05501886 | ||
| C) continues | ||||||
| Study (phase) | Therapy | Setting | Main endpoints | Reference | ||
| Destiny-Breast-04 (III) | TDX-d vs physician’s choice CT. | HR+/HER2low and HR-/HER2low MBC patients progressing after CT for metastatic disease or within 6 months after completing adjuvant CT; HR+ patients must have received at least 1 line of ET | mPFS in HR+ cohort 10.1 vs 5.4 months | 152 | ||
| Destiny-Breast-06 (III) | TDX-d vs physician’s choice CT | ER+/HER2 low or ultralow MBC patients progressing after one or more lines of ET and no previous CT for MBC | HER2-low disease: mPFS 13.2 months vs 8.1 HER2-ultralow disease: mPFS 13.2 months vs 8.3 |
153-154 | ||
| TROPiCS-02 (III) | Sacituzumab-Govitecan vs physician's CT | MBC patients progressing after at least 2 prior systemic CT regimens for metastatic disease. Patients must have received at least one taxane, at least one ET, and at least one CDK4/6i | mPFS 5.5 vs 4.0 months | 157-158 | ||
| TROPION-Breast-01 (III) | Dato-DXd vs ICCT (eribulin or vinorelbine or capecitabine or gemcitabine) | ABC patients progressing on ET and after having received 1‒2 prior lines of CT | mPFS 6.9 vs 4.9 months At 12 months, 25.5% of patients in the Dato-DXd arm versus 14.6% in the ICCT arm were progression-free OS data immature |
160-161 | ||
| D) continues | ||||||
| Study (phase) | Therapy | Setting | Main endpoints | Reference | ||
| MAINTAIN (II) | Stop CDK4/6i, switch ET (fulvestrant or exemestane) and randomized to receive ribociclib or placebo. | MBC patients progressing during ET and CDK4/6i (86.5% palbociclib and 11.7% ribociclib) | mPFS 5.29 months in patients assigned to switched ET plus ribociclib, 2.76 in patients switched ET plus placebo |
125 | ||
| PACE (II) | Fulvestrant (F) vs fulvestrant plus palbociclib (F + P) vs fulvestrant plus palbociclib and avelumab (F + P + A) | MBC patients progressing on previous AI plus CDK4/6i (90.9% palbociclib) | mPFS: 4.8 months on F, 4.6 months on F + P, 8.1 on F + P + A The difference in PFS with F + P and F + P + A versus F was greater among patients with baseline ESR1 and PIK3CA alteration |
162 | ||
| PALMIRA (II) | Palbociclib (P) maintenance plus second–line ET | ABC patients progressing on first-line palbociclib plus ET (AI or fulvestrant) | PFS was 4.2 months (95% CI 3.5–5.8) in the P+ET vs. 3.6 months (95% CI 2.7–4.2) in the ET arm (hazard ratio 0.8, 95% CI 0.6–1.1, p=0.206). 6-month PFS rate was 40.9% and 28.6% for P+ET and ET, respectively. Among 138 pts with measurable disease, no significant differences were observed in ORR (6.4% vs. 2.3%) or CBR (33.0% vs. 29.5%) for P+ET and ET, respectively |
163 | ||
| Post-MONARCH (III) |
Abemaciclib + fulvestrant vs placebo + fulvestrant | MBC patients progressing on prior CDK4/6i plus AI or recurred on/after adjuvant CDK4/6i + ET | mPFS 6.0 versus 5.3 months, regardless of ESR1 or PIK3CA mutations | 164 | ||
| E) | ||||||
| Study (phase) | Therapy | Setting | Main endpoints | Reference | ||
| Opera-01 (III) | Palazestrant vs standard ET (fulvestrant or an AI). | ABC patients progressing after 1 or 2 prior lines of standard ET including a CDK 4/6i | Primary: dose selection, PFS Secondary: OS, ORR, CBR, DOR, safety in patients with and without ESR1mutation |
NCT06016738 | ||
| ELAINE-2 (II) | Lasofoxifene plus abemaciclib | ESR1-mutated MBC patients progressing on prior ET including CDK4/6i | mPFS 56.0 weeks PFS rates at 6, 12, and 18 months:76.1%, 56.1%, and 38.8%, respectively. CBR at 24 weeks 65.5% |
170 | ||
| ELAINE-3 | Lasofoxifene plus abemaciclib vs fulvestrant plus abemaciclib | ABC patients with ≥1 acquired ESR1 mutation, progressing on AI plus palbociclib or ribociclib; ≤1 line of chemotherapy in the advanced/metastatic setting | Primary: PFS Secondary: ORR, OS, CBR, DOR, TTR, safety |
NCT05696626 | ||
| VERITAC (I-II) | Vepdegestrant | MBC patients pre-treated with antiestrogens plus CDK 4/6is | CBR up to 38.9% and 54.5% in the overall population and in patients with ESR1-mutated tumors respectively | 180-181 | ||
| I-II (NCT04072952) | Vepdegestrant plus palbociclib | Pretreated MBC patients | CBR 63% in ITT population, 72.4% in ESR1 mutant patients | 182 | ||
| VERITAC-2 (III) | Vepdegestrant vs fulvestrant | Patients progressing after 1st line ET plus CDK4/6i | Primary: PFS in the ITT population and ESR1 mutation-positive subpopulation. Secondary: OS, RR, safety |
NCT05654623 | ||
| II (NCT02463032) | Enobosarm 9 mg or 18 mg daily | Pretreated MBC patients | CBR of 32% and 29% depending on the dose used. ORR 48% and 0% in patients with more or less than 40% AR staining respectively | 189 | ||
| ENABLAR-2 (2-staged, phase III) | Enobosarm +/- Abemaciclib | AR+ER+HER2- MBC patients progressing after ET + Palbociclib or Ribociclib | PFS, OS, ORR, CBR | 190 | ||
| Multimodular I-II (NCT03363893) | Samuraciclib (dose escalation) plus fulvestrant | MBC patients progressing after an AI plus a CDK4/6i | CBR 36% | 118 | ||
| SUMIT-BS (II) | Samuraciclib plus fulvestrant versus fulvestrant alone | ABC/MBC patients progressing after AI plus CDK4/6i | Primary: CBR Secondary: ORR, DOR, PFS, safety |
NCT05963984 | ||
| SUMIT-ELA (Ib-II) |
Samuraciclib plus elacestrant | ABC/MBC patients progressing after AI plus CDK4/6i | Primary: dose, PFS Secondary: ORR, CBR, DOR, safety |
NCT05963997 | ||
|
Retrospective observational study |
ET in combination with cyclic interferon beta and interleukin-2 | Endocrine dependent MBC patients (also see text) | mPFS 33 months mOS 81 months |
202 | ||
ER+: estrogen receptor positive; HER2-: epidermal growth factor receptor 2 negative; MBC: metastatic breast cancer; ABC: advanced breast cancer; ET: endocrine therapy; AI: aromatase inhibitor; NSAI: non-steroidal aromatase inhibitor; CDK4/6i: cyclin-dependent kinases 4 and 6 inhibitor; ctDNA: circulating tumor DNA; CT: chemotherapy; ESR1: ESR1 gene which encodes ER-alpha; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; Akt: protein kinase B; PTEN: phosphatase and tensin homolog; TDX-d: trastuzumab deruxtecan; Dato-DXd: Datopotamab deruxtecan; AR: androgen receptor; m: median; PFS: progression free survival; OS: overall survival; ORR: overall response rate; DOR: duration of response; CBR: clinical benefit rate; DCR: disease control rate; TTR: time to response; PFS2: time to second progression event; TSST: time to commencement of subsequent line of systemic therapy or death; IC: investigator’s choice; ITT: intention-to-treat.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



