Submitted:
10 March 2025
Posted:
11 March 2025
You are already at the latest version
Abstract
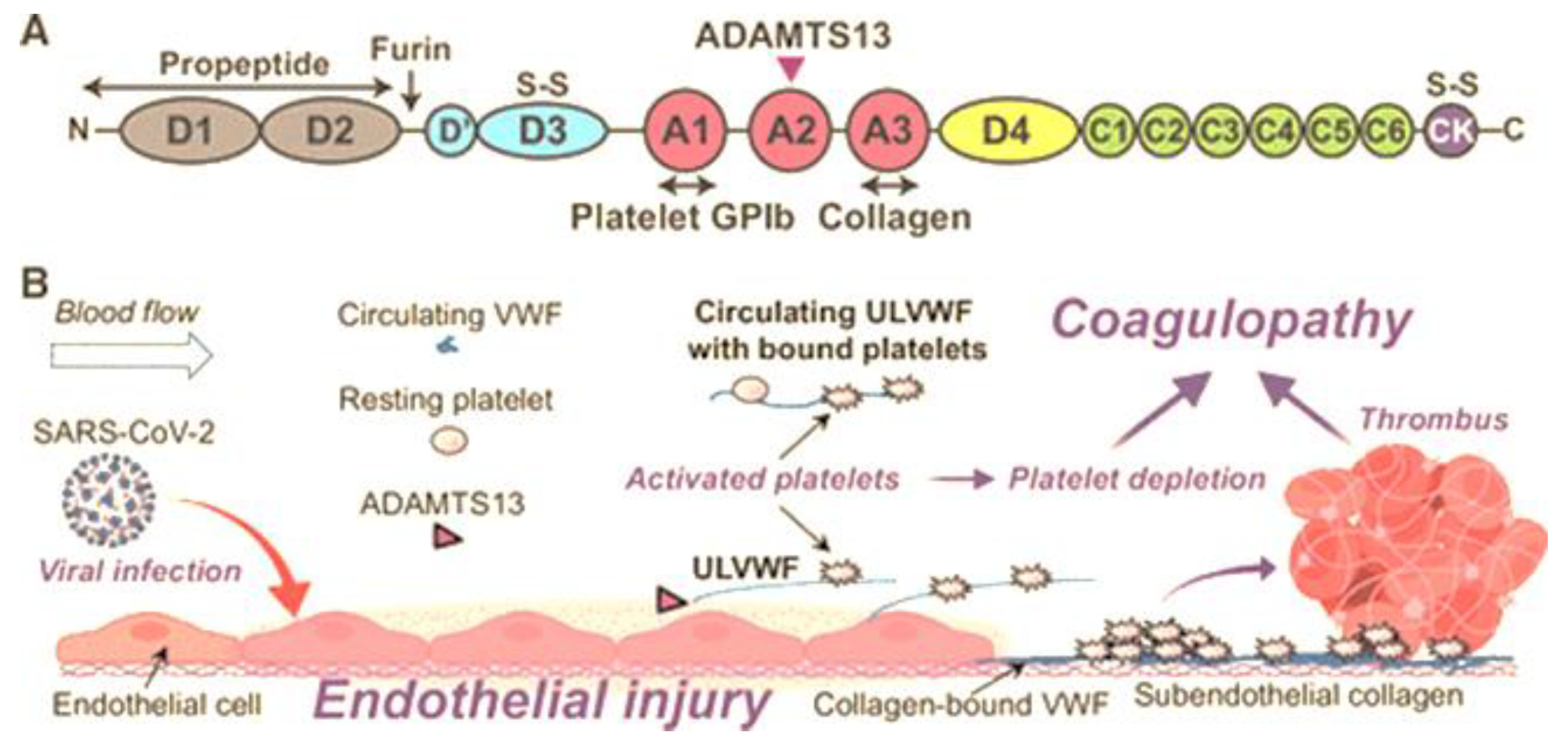
Normal pregnancy is associated with multiple changes of the coagulation and the fibrinolytic system. In contrast to a non-pregnant state, pregnancy is a hypercoagulable state where the level of vWF increases by 200–375% affecting coagulation activity. Moreover, in this hypercoagulable state of pregnancy, preeclampsia is exacerbated. ADAMTS13 cleaves the bond between Tyr1605 and Met1606 in the A2 domain of vWF, thereby reducing its molecular weight. A deficiency of ADAMTS13 originates from mutations in gene or autoantibodies formed against the protease, leading to defective enzyme production. Von Willebrand protein is critical for hemostasis and thrombosis, promoting thrombus formation by mediating adhesion of platelets and aggregation at high shear stress conditions within the vessel wall. Mutations in vWF disrupts multimer assembly, secretion and/or catabolism thereby influencing bleeding. The release of even small amounts of active ADAMTS13 protease has a profound inhibitory effect on thrombosis and inflammation, making vWF the major regulator of plasma ADAMTS13 concentration. Endothelial activation caused by HIV infection leads to the release of vWF. The SARS-CoV-2 infection promotes circulating proinflammatory cytokines, in-creasing endothelial secretion of ultra large vWF that causes an imbalance in vWF/ADAMTS13. Raised vWF levels corresponds with greater platelet adhesiveness, promoting a thrombotic tendency in stenotic vessels, leading to increased shear stress conditions. Keywords: HIV; Preeclampsia; ADAMTS13; vWF; Pregnancy.
Keywords:
1. Introduction
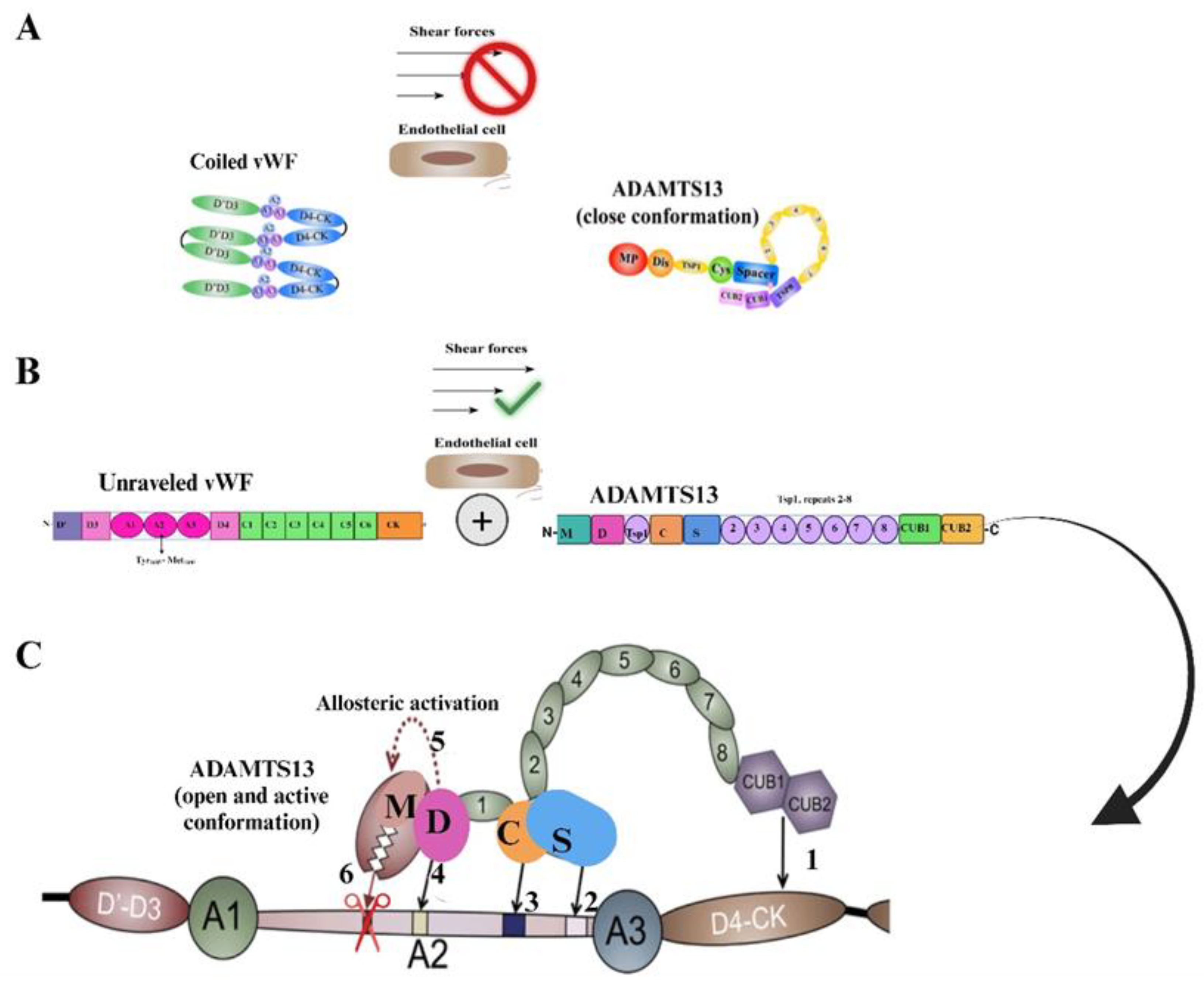
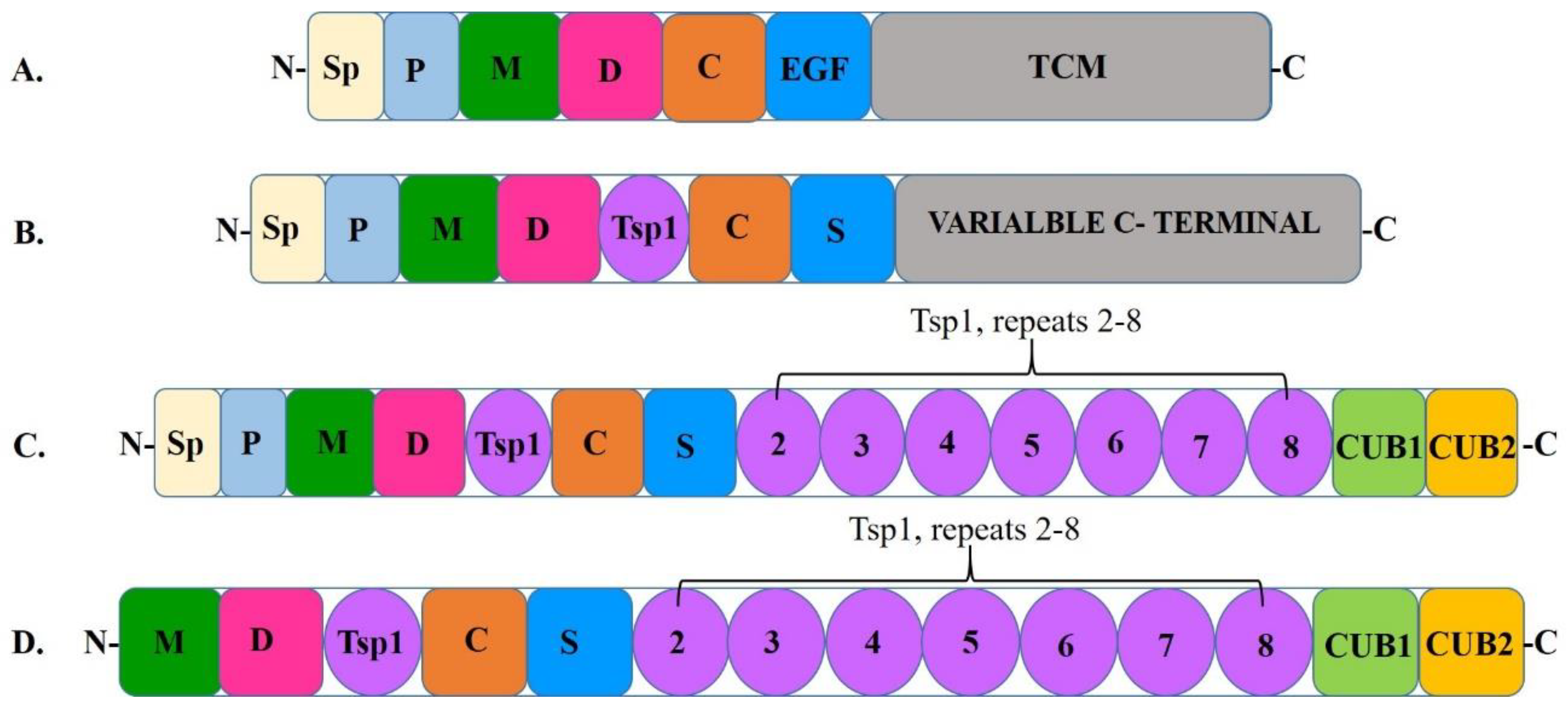
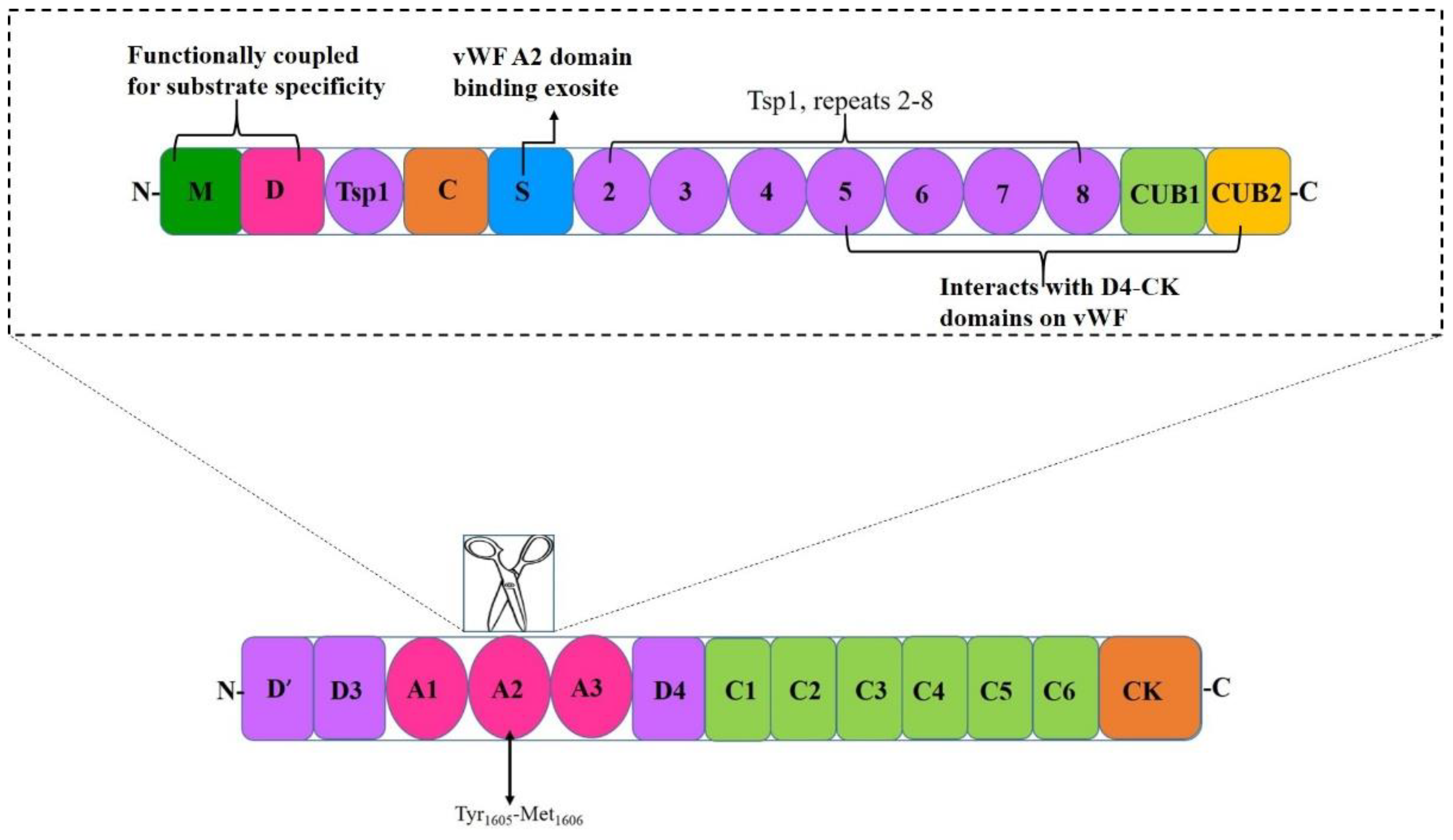
2. A Disintegrin and Metalloprotease with A Thrombospondin Motif Type 1 Member 13 (ADAMTS13)
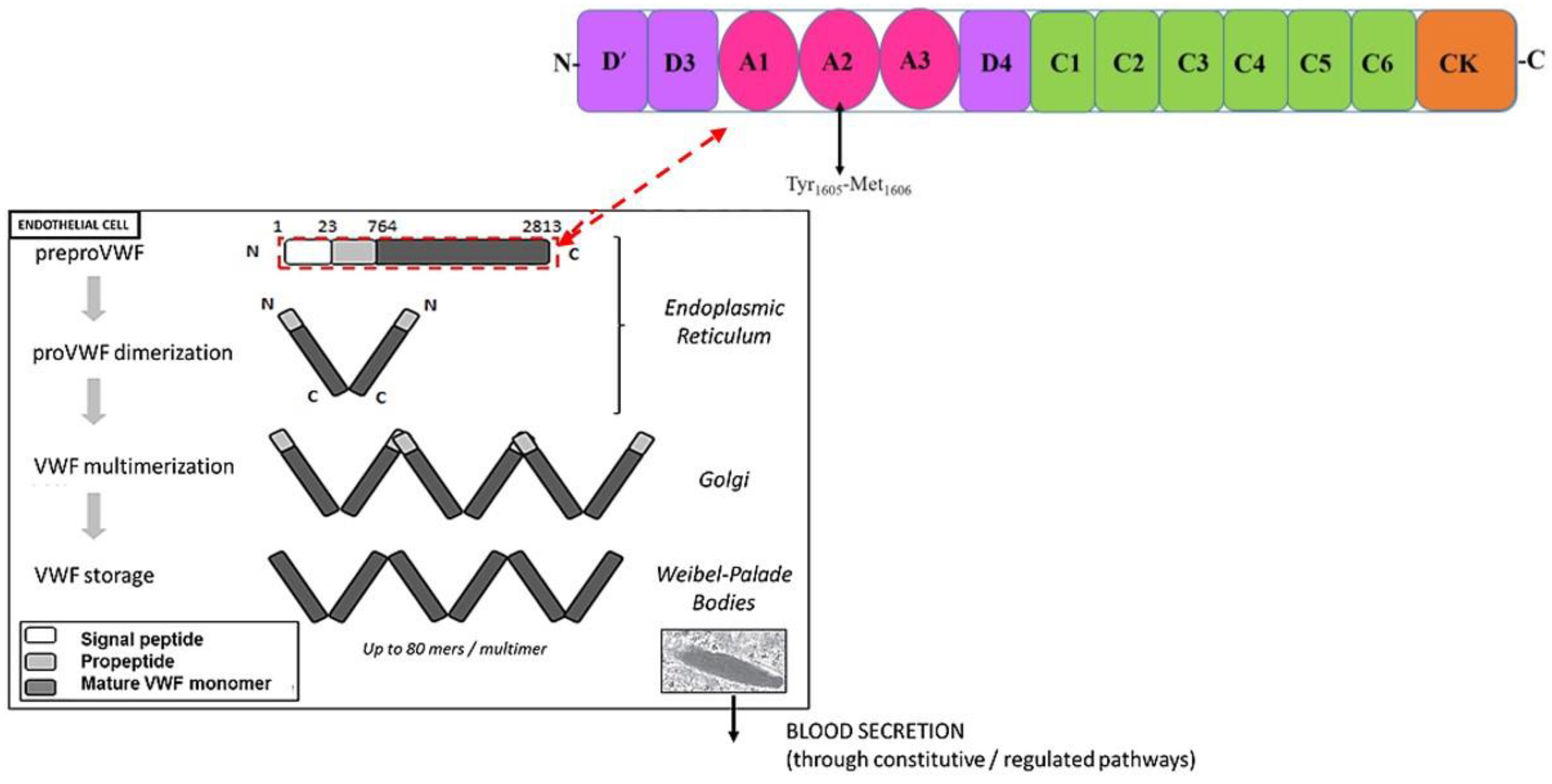
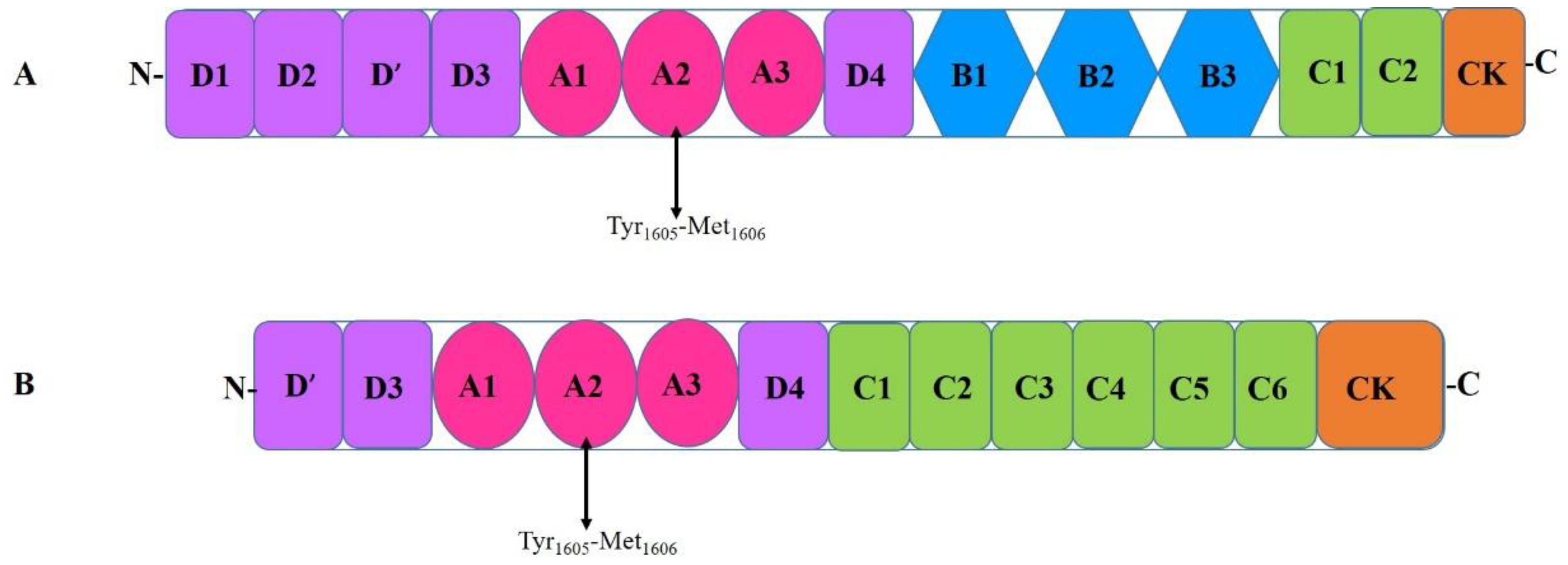
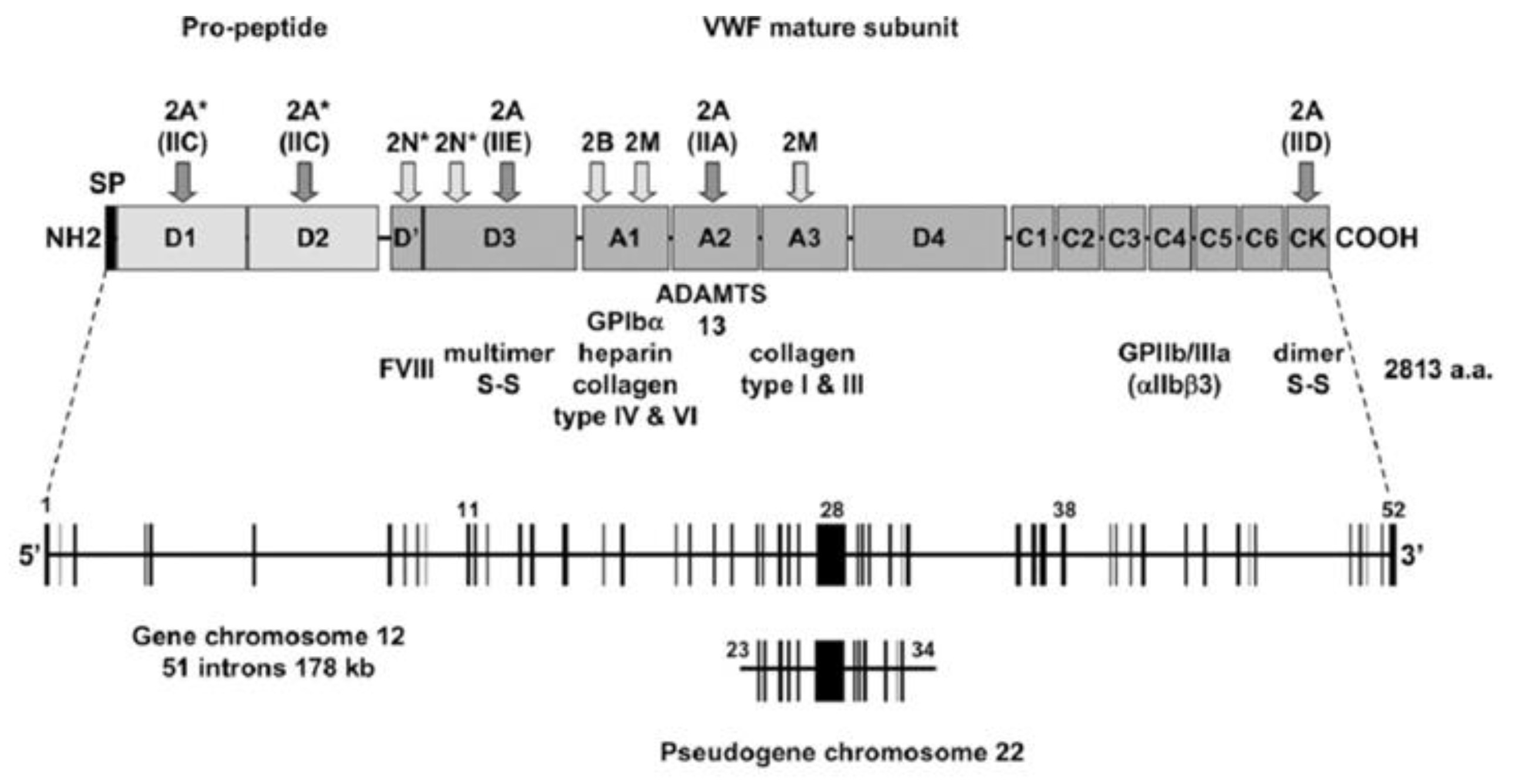
3. Von Willebrand Factor
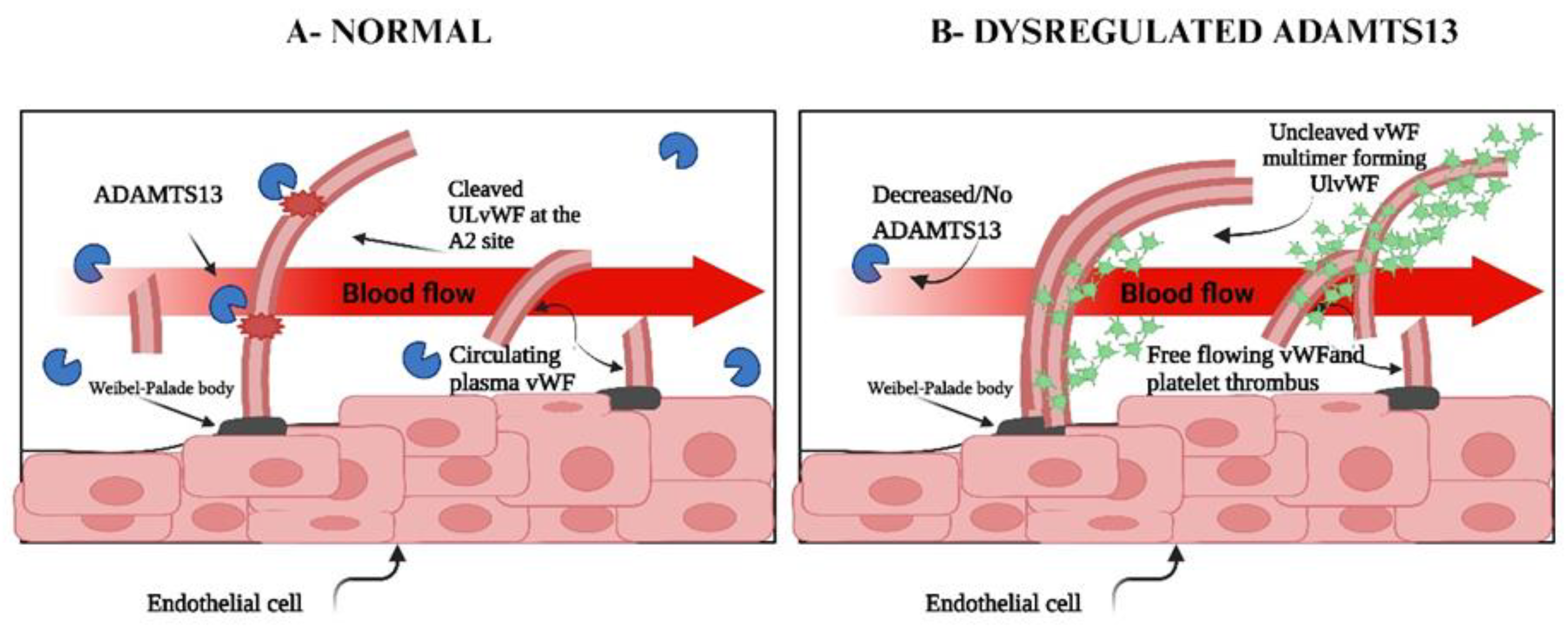
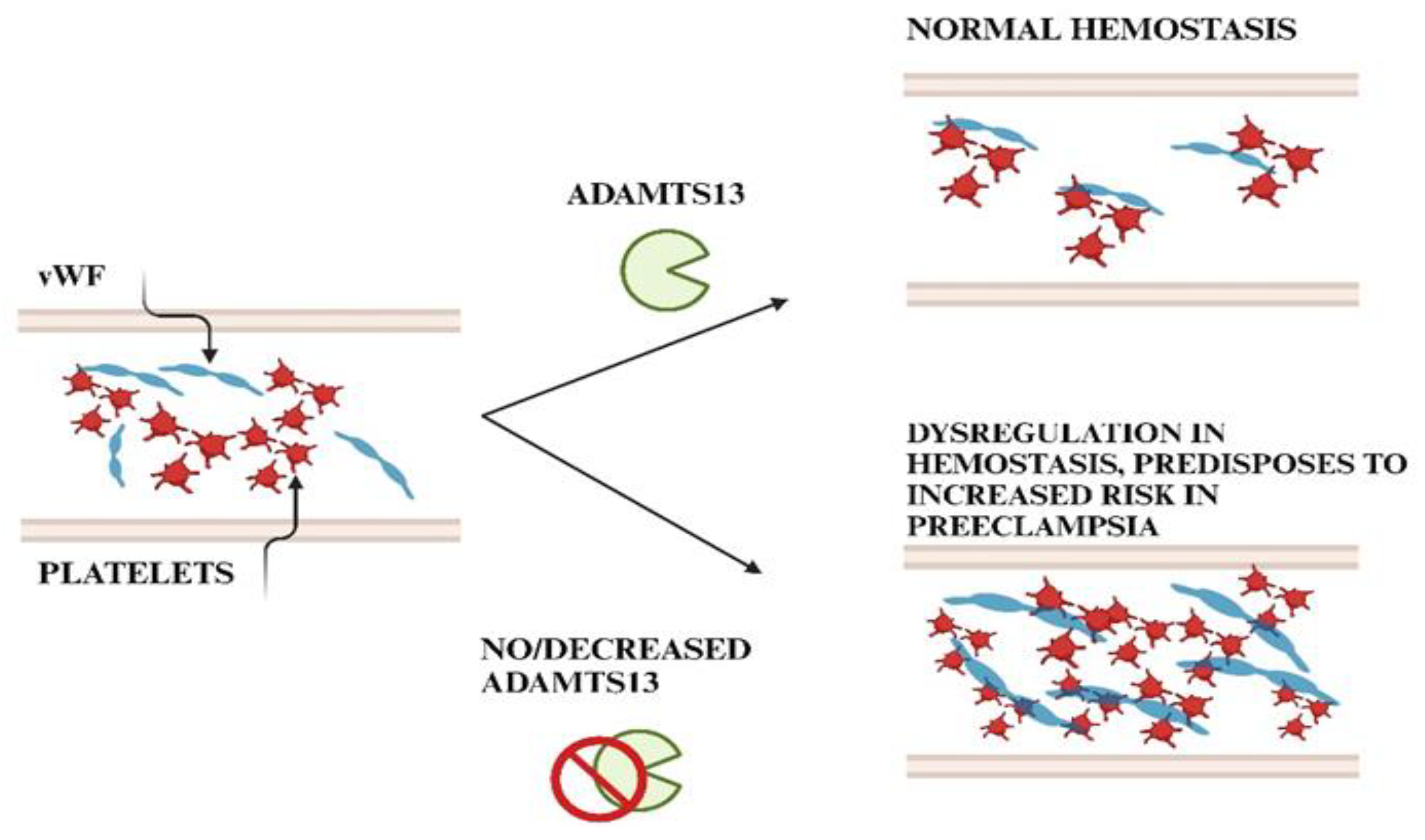
4. Interactions Between Adamts13 and Von Willebrand Factor
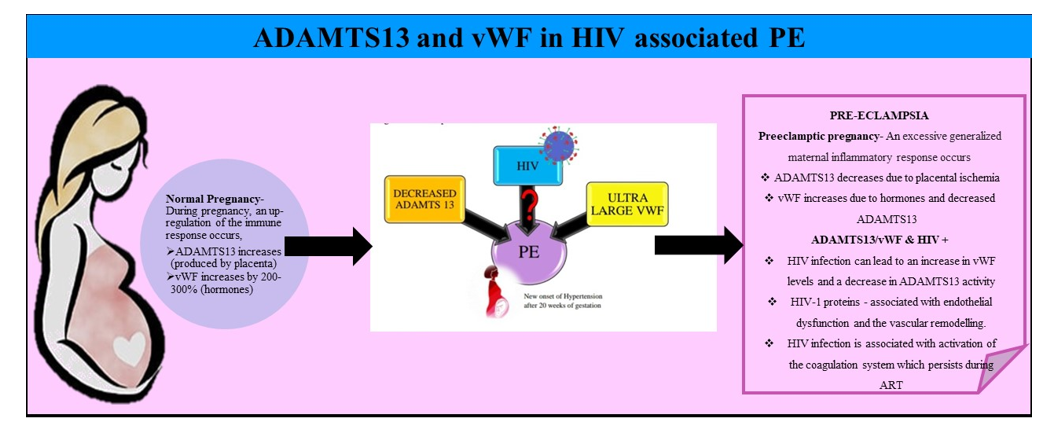
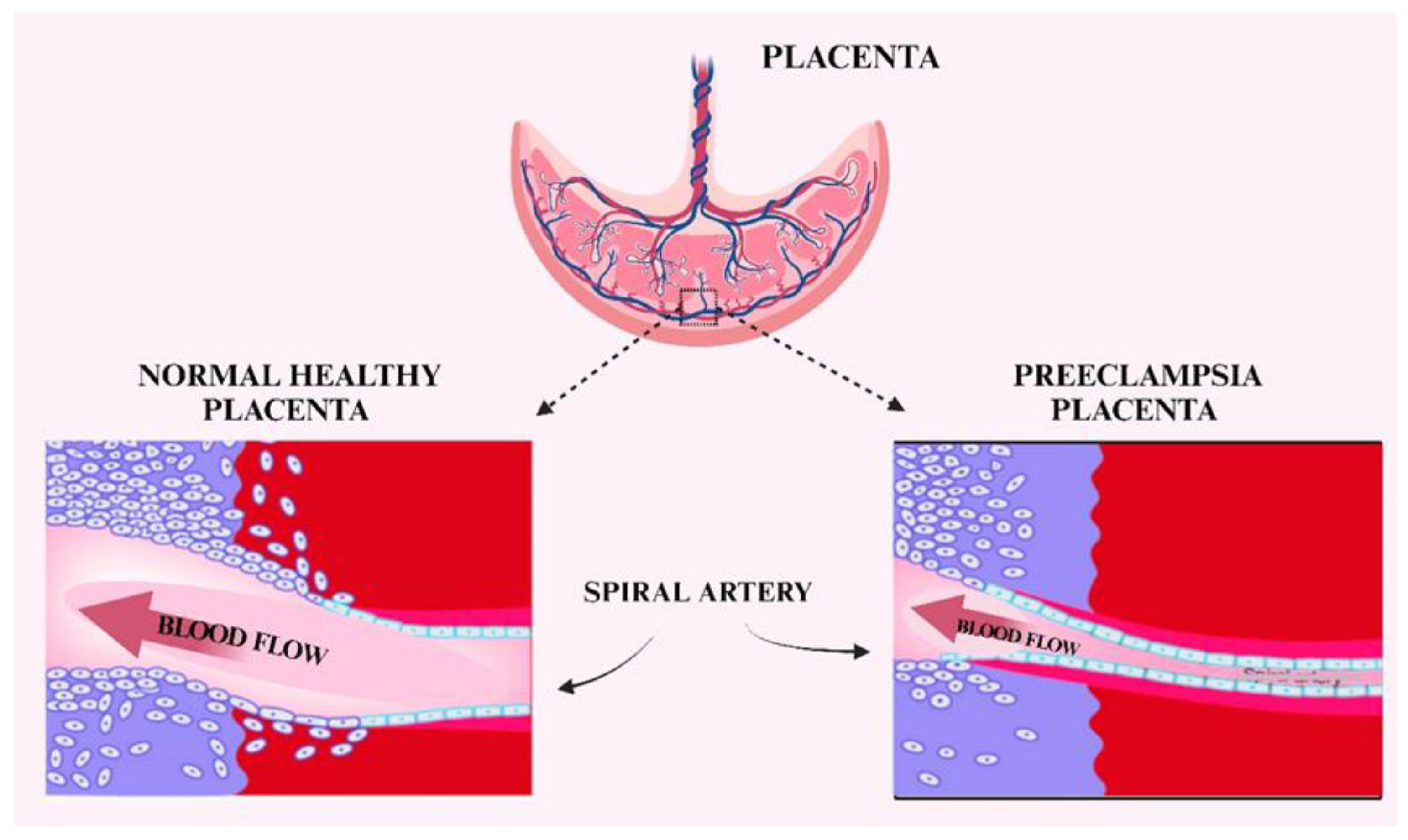
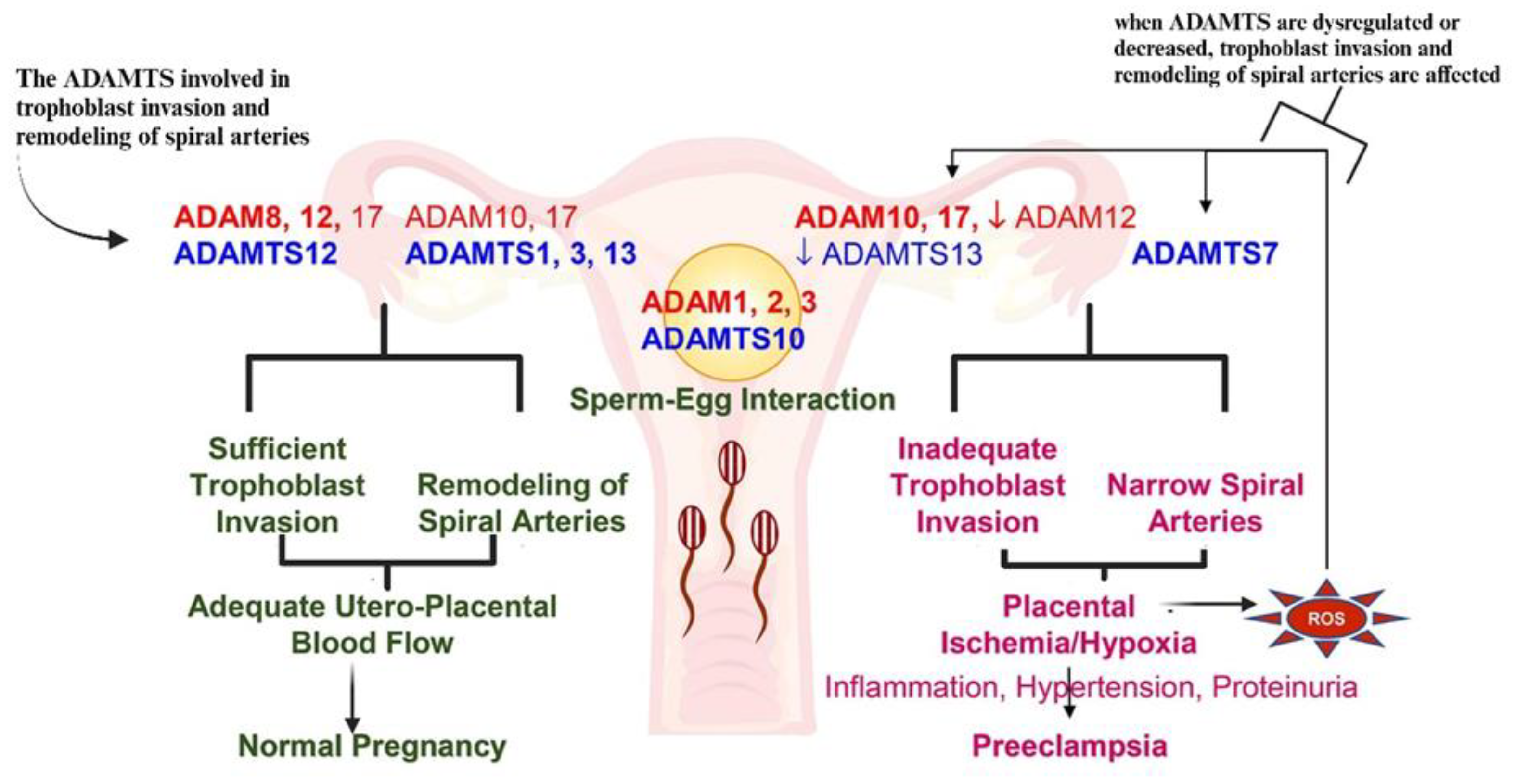
5. Preeclampsia
6. Human Immunodeficiency Virus Infection
7. The Synergy of PE and HIV Infection
8. Dysregulation of Adamts13
9. Dysregulation of Von Willebrand Factor
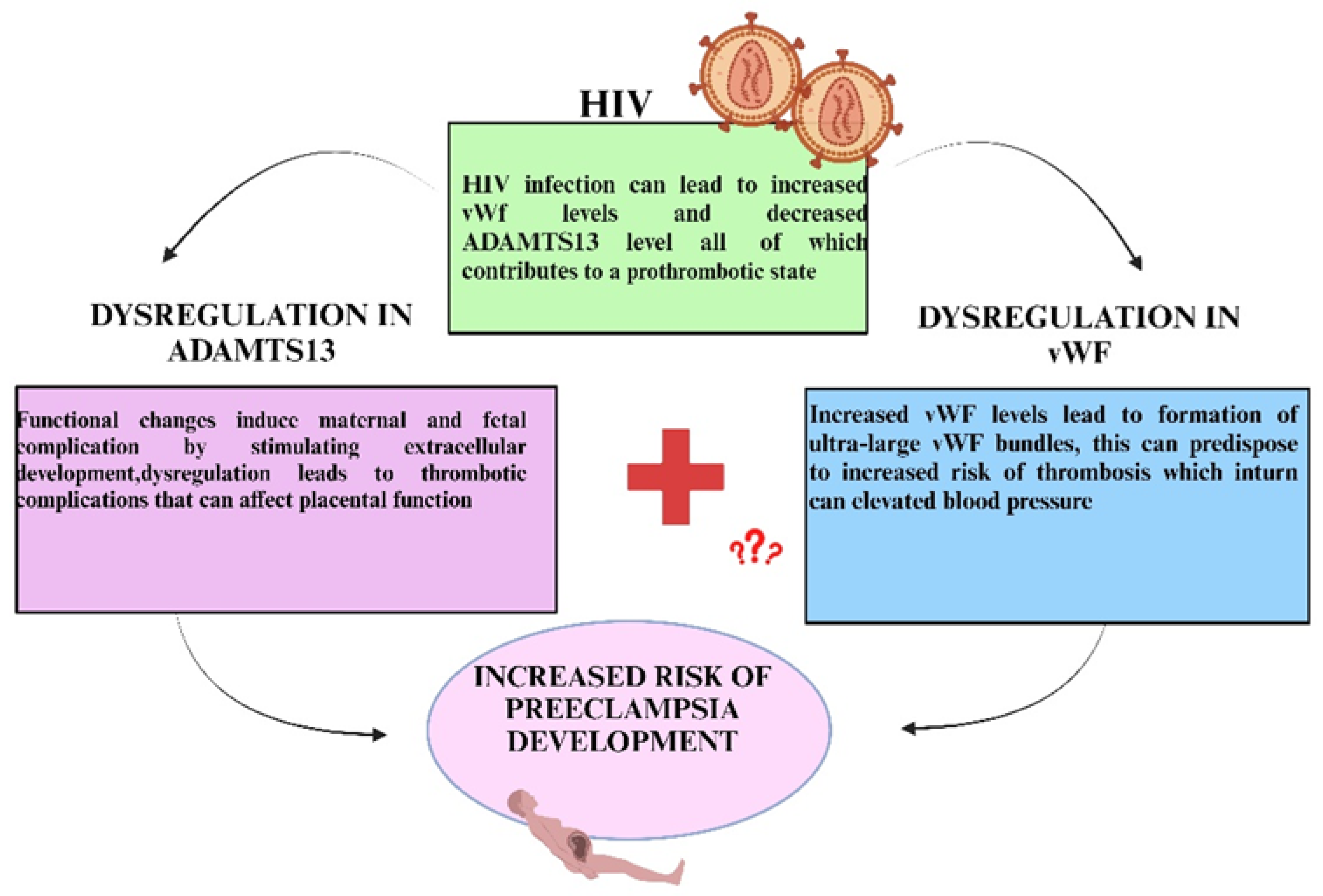
10. Adamts13 in HIV Infection
11. Von Willebrand Factor in HIV Infection
12. Von Willebrand Factor and HIV-Treatment
13. Adamts13 in Pregnancy
14. Adamts13 in Pre-Eclampsia
15. Von Willebrand Factor in Pregnancy
16. Von Willebrand Factor Levels in Pre-Eclampsia
17. Adamts13 in The Duality of PE and HIV Infection
18. Von Willebrand Factor in The Duality of PE and HIV Infection
19. Coupled Adamts13 and Von Willebrand Factor in HIV Associated Pre-Eclampsia.
20. COVID-19 In The Synergy of HIV and Preeclampsia in Relation to Adamts13 and Von Willebrand Factor
21. Conclusions
22. Future Directions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Maternal mortality. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/maternal-mortality (accessed on 16 May 2023).
- UNICEF Data. Maternal mortality rates and statistics. 21 June 2023. [Google Scholar]
- Pillay, Y.; Moodley, J. Will South Africa meet the Sustainable Development Goals target for maternal mortality by 2030? SAMJ South Afr. Med. J. 2024, 114, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Health, N.D.o. Saving Mothers: Executive Summary 2020 - 2022: Includes data for COVID-19 pandemic. Pretoria. NDoH 2023. [Google Scholar]
- Moodley, J. Deaths from hypertensive disorders of pregnancy during 2017-2019: declining trends in South Africa. In Obstetrics and Gynaecology Forum; In House Publications, 2020. [Google Scholar]
- Booker, W.A. Hypertensive disorders of pregnancy. Clin. Perinatol. 2020, 47, 817–833. [Google Scholar]
- A Magee, L.; Brown, M.A.; Hall, D.R.; Gupte, S.; Hennessy, A.; Karumanchi, S.A.; Kenny, L.C.; McCarthy, F.; Myers, J.; Poon, L.C.; et al. The 2021 International Society for the Study of Hypertension in Pregnancy classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. 2022, 27, 148–169. [Google Scholar] [CrossRef]
- Sikhosana, M.L.; Suchard, M.; Kuonza, L.; Cutland, C.; Slogrove, A.; Otwombe, K.; Motaze, N.V. Association between preeclampsia and HIV: a case-control study in urban South Africa. AJOG Glob. Rep. 2022, 2, 100056. [Google Scholar] [CrossRef]
- Dimitriadis, E.; et al. Pre-eclampsia. Nat. Rev. Dis. Primers 2023, 9, 8. [Google Scholar]
- Naicker, T.; et al. Quantitative analysis of trophoblast invasion in preeclampsia. Acta Obstet. Et Gynecol. Scand. 2003, 82, 722–729. [Google Scholar]
- Naljayan, M.V.; Karumanchi, S.A. New Developments in the Pathogenesis of Preeclampsia. Adv. Chronic Kidney Dis. 2013, 20, 265–270. [Google Scholar] [CrossRef]
- Bitsadze, V.; Bouvier, S.; Khizroeva, J.; Cochery-Nouvellon, É.; Mercier, É.; Perez-Martin, A.; Makatsariya, A.; Gris, J.-C. Early ADAMTS13 testing associates with pre-eclampsia occurrence in antiphospholipid syndrome. Thromb. Res. 2021, 203, 101–109. [Google Scholar] [CrossRef]
- Bremner, L.; Gill, C.; Seed, P.T.; Conti-Ramsden, F.; Webster, L.; Fleminger, J.; Chappell, L.C.; Shennan, A.; Bramham, K. Rule-in and rule-out of pre-eclampsia using DELFIA Xpress PlGF 1-2-3 and sFlt-1: PlGF ratio. Pregnancy Hypertens. 2021, 27, 96–102. [Google Scholar] [CrossRef]
- Qu, H.; Khalil, R.A. Role of ADAM and ADAMTS disintegrin and metalloproteinases in normal pregnancy and preeclampsia. Biochem. Pharmacol. 2022, 206, 115266. [Google Scholar] [CrossRef]
- Katz, D.; Beilin, Y. Disorders of coagulation in pregnancy. BJA: Br. J. Anaesth. 2015, 115 (suppl_2), ii75–ii88. [Google Scholar]
- Forstner, D.; Guettler, J.; Gauster, M. Changes in Maternal Platelet Physiology during Gestation and Their Interaction with Trophoblasts. Int. J. Mol. Sci. 2021, 22, 10732. [Google Scholar] [CrossRef]
- Gangakhedkar, G.R.; Kulkarni, A.P. Physiological changes in pregnancy. Indian J. Crit. Care Med. 2021, 25 (Suppl 3). [Google Scholar]
- Tadu, S.; Yerroju, K.; Gudey, S. A Comparative Study of Coagulation Profile in Normal Pregnancy, Mild Preeclampsia, and Severe Preeclampsia Patients. J. South Asian Fed. Obstet. Gynaecol. 2023, 15, 71–75. [Google Scholar]
- Erez, O.; et al. DIC in pregnancy–pathophysiology, clinical characteristics, diagnostic scores, and treatments. J. Blood Med. 2022, 21–44. [Google Scholar]
- Neave, L.; Thomas, M.; de Groot, R.; Doyle, A.J.; Singh, D.; Adams, G.; David, A.L.; Maksym, K.; Scully, M. Alterations in the von Willebrand factor/ADAMTS-13 axis in preeclampsia. J. Thromb. Haemost. 2023, 22, 455–465. [Google Scholar] [CrossRef]
- Woods, A.I.; Paiva, J.; Dos Santos, C.; Alberto, M.F.; Sánchez-Luceros, A. From the Discovery of ADAMTS13 to Current Understanding of Its Role in Health and Disease. Semin. Thromb. Hemost. 2022, 49, 284–294. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Anticoagulant SERPINs: Endogenous Regulators of Hemostasis and Thrombosis. Front. Cardiovasc. Med. 2022, 9, 878199. [Google Scholar] [CrossRef]
- Pinheiro, M.; Gomes, K.; Dusse, L. Fibrinolytic system in preeclampsia. Clin. Chim. Acta 2013, 416, 67–71. [Google Scholar] [CrossRef]
- Han, C.; Huang, P.; Lyu, M.; Dong, J. Oxidative Stress and Preeclampsia-Associated Prothrombotic State. Antioxidants 2020, 9, 1139. [Google Scholar] [CrossRef] [PubMed]
- Bloch, M.; John, M.; Smith, D.; Rasmussen, T.; Wright, E. Managing HIV-associated inflammation and ageing in the era of modern ART. HIV Med. 2020, 21, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.V.; Joseph, S.B.; Dittmer, D.P.; Mackman, N. Cardiovascular Disease and Thrombosis in HIV Infection. Arter. Thromb. Vasc. Biol. 2023, 43, 175–191. [Google Scholar] [CrossRef]
- Obeagu, E.; Obeagu, G. Platelet Dysfunction in HIV Patients: Assessing ART Risks. Elite J. Sci. Res. Rev. 2024, 2, 1–16. [Google Scholar]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef]
- Gao, W.; Anderson, P.J.; Majerus, E.M.; Tuley, E.A.; Sadler, J.E. Exosite interactions contribute to tension-induced cleavage of von Willebrand factor by the antithrombotic ADAMTS13 metalloprotease. Proc. Natl. Acad. Sci. 2006, 103, 19099–19104. [Google Scholar] [CrossRef]
- DeYoung, V.; Singh, K.; Kretz, C.A. Mechanisms of ADAMTS13 regulation. J. Thromb. Haemost. 2022, 20, 2722–2732. [Google Scholar]
- Lancellotti, S.; Basso, M.; De Cristofaro, R. Proteolytic processing of von Willebrand factor by adamts13 and leukocyte proteases. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013058. [Google Scholar] [CrossRef]
- Zheng, X.L. ADAMTS13 and von Willebrand Factor in Thrombotic Thrombocytopenic Purpura. Annu. Rev. Med. 2015, 66, 211–225. [Google Scholar] [CrossRef]
- Khemisi, M.M. Characterization of autoantibodies to ADAMTS13 in HIV-associated thrombotic thrombocytopenic purpura; University of the Free State, 2020. [Google Scholar]
- Stepanian, A.; et al. Von Willebrand factor and ADAMTS13: a candidate couple for preeclampsia pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1703–1709. [Google Scholar] [CrossRef]
- Peyvandi, F.; Lavoretano, S.; Palla, R.; Valsecchi, C.; Merati, G.; De Cristofaro, R.; Rossi, E.; Mannucci, P.M. Mechanisms of the interaction between twoADAMTS13 gene mutations leading to severe deficiency of enzymatic activity. Hum. Mutat. 2006, 27, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Joly, B.S.; Boisseau, P.; Roose, E.; Stepanian, A.; Biebuyck, N.; Hogan, J.; Provot, F.; Delmas, Y.; Garrec, C.; Vanhoorelbeke, K.; et al. ADAMTS13 Gene Mutations Influence ADAMTS13 Conformation and Disease Age-Onset in the French Cohort of Upshaw–Schulman Syndrome. Thromb. Haemost. 2018, 118, 1902–1917. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Khalil, R.A. A Disintegrin and Metalloproteinase (ADAM) and ADAM with thrombospondin motifs (ADAMTS) family in vascular biology and disease. Biochem. Pharmacol. 2019, 164, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Lumangtad, L.A.; Bell, T.W. The signal peptide as a new target for drug design. Bioorganic Med. Chem. Lett. 2020, 30, 127115. [Google Scholar] [CrossRef]
- Shelat, S.G.; Ai, J.; Zheng, X.L. Molecular Biology of ADAMTS13 and Diagnostic Utility of ADAMTS13 Proteolytic Activity and Inhibitor Assays. Semin. Thromb. Hemost. 2005, 31, 659–672. [Google Scholar] [CrossRef]
- Gao, W.; Zhu, J.; Westfield, L.A.; Tuley, E.A.; Anderson, P.J.; Sadler, J.E. Rearranging Exosites in Noncatalytic Domains Can Redirect the Substrate Specificity of ADAMTS Proteases. J. Biol. Chem. 2012, 287, 26944–26952. [Google Scholar] [CrossRef]
- Lynch, C.J.; Lane, D.A.; Luken, B.M. Control of VWF A2 domain stability and ADAMTS13 access to the scissile bond of full-length VWF. Blood 2014, 123, 2585–2592. [Google Scholar] [CrossRef]
- Zander, C.B.; Cao, W.; Zheng, X.L. ADAMTS13 and von Willebrand factor interactions. Curr. Opin. Hematol. 2015, 22, 452–459. [Google Scholar] [CrossRef]
- Zheng, X.; Chung, D.; Takayama, T.K.; Majerus, E.M.; Sadler, J.E.; Fujikawa, K. Structure of von Willebrand Factor-cleaving Protease (ADAMTS13), a Metalloprotease Involved in Thrombotic Thrombocytopenic Purpura. J. Biol. Chem. 2001, 276, 41059–41063. [Google Scholar] [CrossRef]
- Anderson, P.J.; Kokame, K.; Sadler, J.E. Zinc and Calcium Ions Cooperatively Modulate ADAMTS13 Activity. Journal of Biological Chemistry 2006, 281, 850–857. [Google Scholar] [CrossRef]
- Yang, J.; Wu, Z.; Long, Q.; Huang, J.; Hong, T.; Liu, W.; Lin, J. Insights Into Immunothrombosis: The Interplay Among Neutrophil Extracellular Trap, von Willebrand Factor, and ADAMTS13. Front. Immunol. 2020, 11, 610696. [Google Scholar] [CrossRef] [PubMed]
- Adil, S.N.; Karim, F. Thrombotic microangiopathies: role of ADAMTS-13. JPMA-J. Pak. Med. Assoc. 2012, 62, 91. [Google Scholar]
- de Groot, R.; Lane, D.A.; Crawley, J.T.B. The role of the ADAMTS13 cysteine-rich domain in VWF binding and proteolysis. Blood 2015, 125, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Takeda, S.; Kokame, K.; Takagi, J.; Miyata, T. Crystal structures of the noncatalytic domains of ADAMTS13 reveal multiple discontinuous exosites for von Willebrand factor. Proc. Natl. Acad. Sci. 2009, 106, 19274–19279. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Anderson, P.J.; Sadler, J.E. Extensive contacts between ADAMTS13 exosites and von Willebrand factor domain A2 contribute to substrate specificity. Blood 2008, 112, 1713–1719. [Google Scholar] [CrossRef]
- Markham-Lee, Z.; Morgan, N.V.; Emsley, J. Inherited ADAMTS13 mutations associated with Thrombotic Thrombocytopenic Purpura: a short review and update. Platelets 2022, 34, 2138306. [Google Scholar] [CrossRef]
- Kim, H.J.; Xu, Y.; Petri, A.; Vanhoorelbeke, K.; Crawley, J.T.B.; Emsley, J. Crystal structure of ADAMTS13 CUB domains reveals their role in global latency. Sci. Adv. 2021, 7, eabg4403. [Google Scholar] [CrossRef]
- Singh, K.; Sparring, T.; Madarati, H.; Kretz, C.A. Review of our Current Understanding of ADAMTS13 and Von Willebrand Factor in Sepsis and Other Critical Illnesses. In Biomarkers in Trauma, Injury and Critical Care; Springer: Cham, 2022. [Google Scholar]
- Deforche, L.; Roose, E.; Vandenbulcke, A.; Vandeputte, N.; Feys, H.B.; Springer, T.A.; Mi, L.Z.; Muia, J.; Sadler, J.E.; Soejima, K.; et al. Linker regions and flexibility around the metalloprotease domain account for conformational activation of ADAMTS-13. J. Thromb. Haemost. 2015, 13, 2063–2075. [Google Scholar] [CrossRef]
- South, K.; et al. Conformational activation of ADAMTS13. Proc. Natl. Acad. Sci. 2014, 111, 18578–18583. [Google Scholar] [CrossRef]
- Avdonin, P.; et al. Von Willebrand Factor in Health and Disease. Biochem. (Mosc.) Suppl. Ser. A Membr. Cell Biol. 2021, 15, 201–218. [Google Scholar] [CrossRef]
- van der Vorm, L.N.; et al. Effects of plasmin on von Willebrand factor and platelets: a narrative review. TH Open 2018, 2, e218–e228. [Google Scholar] [PubMed]
- von Krogh, A.-S.; et al. ADAMTS13 gene variants and function in women with preeclampsia: a population-based nested case-control study from the HUNT study. Thromb. Res. 2015, 136, 282–288. [Google Scholar]
- Gyselaers, W. Preeclampsia Is a Syndrome with a Cascade of Pathophysiologic Events. J. Clin. Med. 2020, 9, 2245. [Google Scholar] [CrossRef]
- Rauch, A.; Susen, S.; Zieger, B. Acquired von Willebrand Syndrome in Patients With Ventricular Assist Device. Front. Med. 2019, 6, 7. [Google Scholar] [CrossRef]
- Zenner, H.L. The biogenesis of Weibel-Palade bodies; University of London: London, UK, 2007. [Google Scholar]
- Harris, N.S.; Pelletier, J.P.; Marin, M.J.; Winter, W.E. Von Willebrand factor and disease: a review for laboratory professionals. Crit. Rev. Clin. Lab. Sci. 2021, 59, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, Z. Von Willebrand factor, platelets and endothelial cell interactions. J. Thromb. Haemost. 2003, 1, 1335–1342. [Google Scholar]
- Chen, J.; Chung, D.W. Inflammation, von Willebrand factor, and ADAMTS13. Blood J. Am. Soc. Hematol. 2018, 132, 141–147. [Google Scholar]
- Goodeve, A.C. The genetic basis of von Willebrand disease. Blood Rev. 2010, 24, 123–134. [Google Scholar] [CrossRef]
- Atiq, F.; O’donnell, J.S. Novel functions for von Willebrand factor. Blood 2024, 144, 1247–1256. [Google Scholar] [CrossRef]
- Lenting, P.J.; Christophe, O.D.; Denis, C.V. von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood J. Am. Soc. Hematol. 2015, 125, 2019–2028. [Google Scholar] [CrossRef]
- Crawley, J.T.B.; de Groot, R.; Xiang, Y.; Luken, B.M.; Lane, D.A. Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor. Blood 2011, 118, 3212–3221. [Google Scholar] [CrossRef]
- Fujimura, Y.; Holland, L.Z. COVID-19 microthrombosis: unusually large VWF multimers are a platform for activation of the alternative complement pathway under cytokine storm. Int. J. Hematol. 2022, 115, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Leksa, N.C.; Chhabra, E.S.; Arndt, J.W.; Lu, Q.; Knockenhauer, K.E.; Peters, R.T.; Springer, T.A. The von Willebrand factor D′D3 assembly and structural principles for factor VIII binding and concatemer biogenesis. Blood 2019, 133, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Halvorsen, K.; Zhang, C.Z.; Wong, W.P.; Springer, T.A. Mechanoenzymatic Cleavage of the Ultralarge Vascular Protein von Willebrand Factor. Science 2009, 324, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Auton, M.; Zhu, C.; Cruz, M.A. The Mechanism of VWF-Mediated Platelet GPIbα Binding. Biophys. J. 2010, 99, 1192–1201. [Google Scholar] [CrossRef]
- Cortes, G.A.; Moore, M.J.; El-Nakeep, S. Physiology, von Willebrand factor. 2020. [Google Scholar]
- Flood, V.H.; Schlauderaff, A.C.; Haberichter, S.L.; Slobodianuk, T.L.; Jacobi, P.M.; Bellissimo, D.B.; Christopherson, P.A.; Friedman, K.D.; Gill, J.C.; Hoffmann, R.G.; et al. Crucial role for the VWF A1 domain in binding to type IV collagen. Blood 2015, 125, 2297–2304. [Google Scholar] [CrossRef]
- Petri, A.; Kim, H.J.; Xu, Y.; de Groot, R.; Li, C.; Vandenbulcke, A.; Vanhoorelbeke, K.; Emsley, J.; Crawley, J.T.B. Crystal structure and substrate-induced activation of ADAMTS13. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Romijn, R.A.; Westein, E.; Bouma, B.; Schiphorst, M.E.; Sixma, J.J.; Lenting, P.J.; Huizinga, E.G. Mapping the Collagen-binding Site in the von Willebrand Factor-A3 Domain. J. Biol. Chem. 2003, 278, 15035–15039. [Google Scholar] [CrossRef]
- Zhou, Y.-F.; Springer, T.A. Highly reinforced structure of a C-terminal dimerization domain in von Willebrand factor. Blood 2014, 123, 1785–1793. [Google Scholar] [CrossRef]
- Miyata, T. Autoantibody-resistant ADAMTS13 variant. Blood J. Am. Soc. Hematol. 2021, 137, 2575–2576. [Google Scholar]
- Shahidi, M. Thrombosis and von Willebrand factor. In Thrombosis and Embolism: from Research to Clinical Practice: Volume 1; Springer: Cham, 2017; pp. 285–306. [Google Scholar]
- Jakobi, A.J.; Mashaghi, A.; Tans, S.J.; Huizinga, E.G. Calcium modulates force sensing by the von Willebrand factor A2 domain. Nat. Commun. 2011, 2, 385. [Google Scholar] [CrossRef]
- Lynch, C. Unfolding of the von Willebrand factor A2 domain. 2017. [Google Scholar]
- Sukumar, S.; BLämmle; Cataland, S.R. Thrombotic thrombocytopenic purpura: pathophysiology, diagnosis, and management. J. Clin. Med. 2021, 10, 536. [Google Scholar] [CrossRef] [PubMed]
- Jikamo, B.; Adefris, M.; Azale, T.; Alemu, K. Incidence, trends and risk factors of preeclampsia in sub-Saharan Africa: a systematic review and meta-analysis. PAMJ One Heal. 2023, 11. [Google Scholar] [CrossRef]
- Feroz, A.S.; Afzal, N.; Seto, E. Exploring digital health interventions for pregnant women at high risk for pre-eclampsia and eclampsia in low-income and-middle-income countries: a scoping review. BMJ Open 2022, 12, e056130. [Google Scholar] [CrossRef]
- Poon, L.C.; Shennan, A.; Hyett, J.A.; Kapur, A.; Hadar, E.; Divakar, H.; McAuliffe, F.; da Silva Costa, F.; von Dadelszen, P.; McIntyre, H.D.; et al. The International Federation of Gynecology and Obstetrics (FIGO) initiative on pre-eclampsia: A pragmatic guide for first-trimester screening and prevention. Int. J. Gynaecol. Obstet. Off. Organ Int. Fed. Gynaecol. Obstet. 2019, 145 (Suppl. S1), 1–33. [Google Scholar] [CrossRef]
- Yang, N.; et al. Expression profiles and functions of ferroptosis-related genes in the placental tissue samples of early-and late-onset preeclampsia patients. BMC Pregnancy and Childbirth 2022, 22, 87. [Google Scholar] [PubMed]
- Aziz, A.; Mose, J.C. The Differences of Characteristic, Management, Maternal and Perinatal Outcomes among Early and Late Onset Preeclampsia. OALib 2016, 3, 1–7. [Google Scholar] [CrossRef]
- Redman, C.W.; Staff, A.C. Preeclampsia, biomarkers, syncytiotrophoblast stress, and placental capacity. Am. J. Obstet. Gynecol. 2015, 213, S9.e1–S9.e4. [Google Scholar] [CrossRef]
- Sansone, M.; Sarno, L.; Saccone, G.; Berghella, V.; Maruotti, G.M.; Migliucci, A.; Capone, A.; Martinelli, P. Risk of Preeclampsia in Human Immunodeficiency Virus–Infected Pregnant Women. Obstet. Gynecol. 2016, 127, 1027–1032. [Google Scholar] [CrossRef]
- Meazaw, M.W.; Chojenta, C.; Muluneh, M.D.; Loxton, D. Systematic and meta-analysis of factors associated with preeclampsia and eclampsia in sub-Saharan Africa. PLOS ONE 2020, 15, e0237600. [Google Scholar] [CrossRef]
- Ari, N.C.; Novembriany, Y.E.; Norlina, S.; Mariyana, M.; Sari, D.P.; Intarti, W.D. Preeclampsia and the Associated Risk Factors Among Pregnant Women in Indonesia: a Literature Review. Path Sci. 2024, 10, 1001–1012. [Google Scholar] [CrossRef]
- Demissie, M.; Molla, G.; Tayachew, A.; Getachew, F. Risk factors of preeclampsia among pregnant women admitted at labor ward of public hospitals, low income country of Ethiopia; case control study. Pregnancy Hypertens. 2022, 27, 36–41. [Google Scholar] [CrossRef]
- Brosens, I.; Puttemans, P.; Benagiano, G. Placental bed research: I. The placental bed: from spiral arteries remodeling to the great obstetrical syndromes. Am. J. Obstet. Gynecol. 2019, 221, 437–456. [Google Scholar] [CrossRef]
- Gathiram; Moodley, J. Pre-eclampsia: its pathogenesis and pathophysiolgy: review articles. Cardiovasc. J. Afr. 2016, 27, 71–78. [Google Scholar]
- Valenzuela, F.J.; Pérez-Sepúlveda, A.; Torres, M.J.; Correa, P.; Repetto, G.M.; Illanes, S.E. Pathogenesis of Preeclampsia: The Genetic Component. J. Pregnancy 2011, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Boyajian, T. Preeclampsia in HIV Positive Pregnant Women on Highly Active Anti-Retroviral Therapy: A matched cohort study. 2010. [Google Scholar]
- Karumanchi, S.A. Angiogenic factors in preeclampsia: from diagnosis to therapy. Hypertension 2016, 67, 1072–1079. [Google Scholar] [PubMed]
- Fowkes, F.J.I.; Draper, B.L.; Hellard, M.; Stoové, M. Achieving development goals for HIV, tuberculosis and malaria in sub-Saharan Africa through integrated antenatal care: barriers and challenges. BMC Med. 2016, 14, 1–10. [Google Scholar] [CrossRef]
- Paudel, S.; et al. Comorbidities and factors associated with health-related quality of life among people living with HIV/AIDS in Gandaki Province of Nepal. 2024. [Google Scholar]
- Zuma, K.; Simbayi, L.; Zungu, N.; Moyo, S.; Marinda, E.; Jooste, S.; North, A.; Nadol, P.; Aynalem, G.; Igumbor, E.; et al. The HIV Epidemic in South Africa: Key Findings from 2017 National Population-Based Survey. Int. J. Environ. Res. Public Heal. 2022, 19, 8125. [Google Scholar] [CrossRef]
- Murewanhema, G.; Musuka, G.; Moyo, P.; Moyo, E.; Dzinamarira, T. HIV and adolescent girls and young women in sub-Saharan Africa: A call for expedited action to reduce new infections. IJID Reg. 2022, 5, 30–32. [Google Scholar] [CrossRef]
- Woldesenbet, S.; et al. Recent HIV infection among pregnant women in the 2017 antenatal sentinel cross–sectional survey, South Africa: Assay–based incidence measurement. PloS ONE 2021, 16, e0249953. [Google Scholar]
- Mthembu, M.H. The role of Endothelin-1 in HIV associated pre-eclampsia. 2019. [Google Scholar]
- UNAIDS. Global HIV & AIDS statistics — Fact sheet. 2023. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 17 July 2023).
- Huang, G.; Takeuchi, Y.; Korobeinikov, A. HIV evolution and progression of the infection to AIDS. J. Theor. Biol. 2012, 307, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Zayas, J.P.; Mamede, J.I. HIV infection and spread between Th17 cells. Viruses 2022, 14, 404. [Google Scholar] [CrossRef] [PubMed]
- Schapkaitz, E.; Libhaber, E.; Jacobson, B.F.; Meiring, M.; Büller, H.R. von Willebrand factor propeptide-to-antigen ratio in HIV-infected pregnancy: Evidence of endothelial activation. J. Thromb. Haemost. 2021, 19, 3168–3176. [Google Scholar] [CrossRef]
- Naicker, T.; Govender, N.; Abel, T.; Naidoo, N.; Moodley, M.; Pillay, Y.; Singh, S.; Khaliq, O.P.; Moodley, J. HIV Associated Preeclampsia: A Multifactorial Appraisal. Int. J. Mol. Sci. 2021, 22, 9157. [Google Scholar] [CrossRef]
- Margaritis, M. Endothelial dysfunction in HIV infection: experimental and clinical evidence on the role of oxidative stress. Ann. Res. Hosp. 2019, 3, 7. [Google Scholar] [CrossRef]
- Yee, L.M.; Jacobson, D.L.; Haddad, L.B.; Jao, J.; Powis, K.M.; Kacanek, D.; Zash, R.; Diperna, A.; Chadwick, E.G. Evaluating the association of antiretroviral therapy and immune status with hypertensive disorders of pregnancy among people with HIV. AIDS 2023, 37, 1715–1723. [Google Scholar] [CrossRef]
- Chilaka, V.N.; Konje, J.C. HIV in pregnancy–An update. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 256, 484–491. [Google Scholar]
- Schank, M.; Zhao, J.; Moorman, J.P.; Yao, Z.Q. The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells 2021, 10, 174. [Google Scholar] [CrossRef]
- Montessori, V.; Press, N.; Harris, M.; Akagi, L.; Montaner, J.S.G. Adverse effects of antiretroviral therapy for HIV infection. Cmaj 2004, 170, 229–238. [Google Scholar]
- Nel, J.; Dlamini, S.; Meintjes, G.; Burton, R.; Black, J.M.; Davies, N.E.; Hefer, E.; Maartens, G.; Mangena, P.M.; Mathe, M.T.; et al. Southern African HIV Clinicians Society guidelines for antiretroviral therapy in adults: 2020 update. South. Afr. J. HIV Med. 2020, 21, 39. [Google Scholar] [CrossRef]
- Gray, G.E.; McIntyre, J.A. HIV and pregnancy. Bmj 2007, 334, 950–953. [Google Scholar] [PubMed]
- Cerveny, L.; Murthi; Staud, F. HIV in pregnancy: Mother-to-child transmission, pharmacotherapy, and toxicity. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166206. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, N.; Moodley, J.; Naicker, T. Maternal endothelial dysfunction in HIV-associated preeclampsia comorbid with COVID-19: a review. Hypertens. Res. 2021, 44, 386–398. [Google Scholar] [CrossRef]
- Alkema, L.; Chou, D.; Hogan, D.; Zhang, S.; Moller, A.-B.; Gemmill, A.; Fat, D.M.; Boerma, T.; Temmerman, M.; Mathers, C.; et al. Global, regional, and national levels and trends in maternal mortality between 1990 and 2015, with scenario-based projections to 2030: a systematic analysis by the UN Maternal Mortality Estimation Inter-Agency Group. Lancet 2016, 387, 462–474. [Google Scholar] [CrossRef]
- Michalczyk, M.; Celewicz, A.; Celewicz, M.; Woźniakowska-Gondek, P.; Rzepka, R. The Role of Inflammation in the Pathogenesis of Preeclampsia. Mediat. Inflamm. 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Aneman, I.; Pienaar, D.; Suvakov, S.; Simic, T.P.; Garovic, V.D.; McClements, L. Mechanisms of Key Innate Immune Cells in Early- and Late-Onset Preeclampsia. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Pillay, Y.; Moodley, J.; Naicker, T. The role of the complement system in HIV infection and preeclampsia. Inflamm. Res. 2019, 68, 459–469. [Google Scholar] [CrossRef]
- Geldenhuys, J.; Rossouw, T.M.; Lombaard, H.A.; Ehlers, M.M.; Kock, M.M. Disruption in the Regulation of Immune Responses in the Placental Subtype of Preeclampsia. Front. Immunol. 2018, 9, 1659. [Google Scholar] [CrossRef]
- Valencia-Ortega, J.; Zárate, A.; Saucedo, R.; Hernández-Valencia, M.; Cruz, J.G.; Puello, E. Placental Proinflammatory State and Maternal Endothelial Dysfunction in Preeclampsia. Gynecol. Obstet. Investig. 2018, 84, 12–19. [Google Scholar] [CrossRef]
- Govender, S. The role of complement components C2 and C5a in HIV associated preeclampsia. 2020. [Google Scholar]
- Naicker, T.; Phoswa, W.N.; Onyangunga, O.A.; Gathiram, P.; Moodley, J. Angiogenesis, Lymphangiogenesis, and the Immune Response in South African Preeclamptic Women Receiving HAART. Int. J. Mol. Sci. 2019, 20, 3728. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; et al. Examining chronic inflammation, immune metabolism, and T cell dysfunction in HIV infection. Viruses 2024, 16, 219. [Google Scholar] [CrossRef] [PubMed]
- Hileman, C.O.; Funderburg, N.T. Inflammation, immune activation, and antiretroviral therapy in HIV. Curr. Hiv/Aids Rep. 2017, 14, 93–100. [Google Scholar] [PubMed]
- Abel, T.; Moodley, J.; Khaliq, O.P.; Naicker, T. Vascular Endothelial Growth Factor Receptor 2: Molecular Mechanism and Therapeutic Potential in Preeclampsia Comorbidity with Human Immunodeficiency Virus and Severe Acute Respiratory Syndrome Coronavirus 2 Infections. Int. J. Mol. Sci. 2022, 23, 13752. [Google Scholar] [CrossRef]
- Jamaluddin, M.; Lin, P.; Yao, Q.; Chen, C. QS419. Non-Nucleoside Reverse Transcriptase Inhibitor Efavirenz Increases Monolayer Permeability of Human Coronary Artery Endothelial Cells. J. Surg. Res. 2009, 151, 301. [Google Scholar] [CrossRef]
- Singh, A.; et al. In silico Studies on N-(Pyridin-2-yl) Thiobenzamides as NNRTIs against Wild and Mutant HIV-1 Strains. Philipp. J. Sci. 2018, 147, 37–46. [Google Scholar]
- Franzese, O.; Barbaccia, M.L.; Bonmassar, E.; Graziani, G. Beneficial and Detrimental Effects of Antiretroviral Therapy on HIV-Associated Immunosenescence. Chemotherapy 2018, 63, 64–75. [Google Scholar] [CrossRef]
- Korencak, M.; Byrne, M.; Richter, E.; Schultz, B.T.; Juszczak, P.; Ake, J.A.; Ganesan, A.; Okulicz, J.F.; Robb, M.L.; Reyes, B.d.L.; et al. Effect of HIV infection and antiretroviral therapy on immune cellular functions. J. Clin. Investig. 2019, 4. [Google Scholar] [CrossRef]
- Herschhorn, A.; Hizi, A. Retroviral reverse transcriptases. Cell. Mol. Life Sci. 2010, 67, 2717–2747. [Google Scholar]
- Bastos, M.M.; Costa, C.C.; Bezerra, T.C.; Silva, F.d.C.d.; Boechat, N. Efavirenz a nonnucleoside reverse transcriptase inhibitor of first-generation: Approaches based on its medicinal chemistry. Eur. J. Med. Chem. 2016, 108, 455–465. [Google Scholar] [CrossRef]
- Pillay, S.; Naicker, T. Morphometric image analysis of vascular endothelial growth factor receptor-3 in preeclamptic, HIV infected women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 253, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Voshavar, C. Protease Inhibitors for the Treatment of HIV/AIDS: Recent Advances and Future Challenges. Curr. Top. Med. Chem. 2019, 19, 1571–1598. [Google Scholar] [CrossRef]
- Castro-Guillén, J.L.; García-Gasca, T.; Blanco-Labra, A. Protease inhibitors as anticancer agents. New Approaches Treat. Cancer 2010, 91–124. [Google Scholar]
- E Dunk, C.; Serghides, L. Protease inhibitor-based antiretroviral therapy in pregnancy: effects on hormones, placenta, and decidua. Lancet HIV 2022, 9, e120–e129. [Google Scholar] [CrossRef]
- Kala, S.; Dunk, C.; Acosta, S.; Serghides, L. Periconceptional exposure to lopinavir, but not darunavir, impairs decidualization: a potential mechanism leading to poor birth outcomes in HIV-positive pregnancies. Hum. Reprod. 2020, 35, 1781–1796. [Google Scholar] [CrossRef]
- Trivedi, J.; Mahajan, D.; Jaffe, R.J.; Acharya, A.; Mitra, D.; Byrareddy, S.N. Recent Advances in the Development of Integrase Inhibitors for HIV Treatment. Curr. HIV/AIDS Rep. 2020, 17, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, G.; Merlini, E.; Sinigaglia, E.; Iannotti, N.; Bai, F.; Savoldi, A.; Tincati, C.; Carpani, G.; Bini, T.; Monforte, A.D. Immune Reconstitution in HIV+ Subjects on Lopinavir/Ritonavir-Based HAART According to the Severity of Pre-Therapy CD4+. Curr. HIV Res. 2012, 10, 597–605. [Google Scholar] [CrossRef]
- Assink, K.; Schiphorst, R.; Allford, S.; Karpman, D.; Etzioni, A. Mutation analysis and clinical implications of von Willebrand factor–cleaving protease deficiency. Kidney Int. 2003, 63, 1995–1999. [Google Scholar] [CrossRef] [PubMed]
- Feys, H. Deckmyn, and K. Vanhoorelbeke, Insights into thrombotic thrombocytopenic purpura by monoclonal antibody-based analysis of the Von Willebrand factor cleaving protease, ADAMTS-13. 2006. [Google Scholar]
- Ferrari, B.; Cairo, A.; Pontiggia, S.; Mancini, I.; Masini, L.; Peyvandi, F. Congenital and acquired ADAMTS13 deficiency: Two mechanisms, one patient. J. Clin. Apher. 2014, 30, 252–256. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, A.; Lourés, E.; Rodríguez-Trillo, Á.; Costa-Pinto, J.; García-Rivero, A.; Batlle-López, A.; Batlle, J.; López-Fernández, M.F. Inherited ADAMTS13 deficiency (Upshaw-Schulman syndrome): A short review. Thromb. Res. 2014, 134, 1171–1175. [Google Scholar] [CrossRef]
- Rank, C.U.; Hovinga, J.K.; Taleghani, M.M.; Lämmle, B.; Gøtze, J.P.; Nielsen, O.J. Congenital thrombotic thrombocytopenic purpura caused by new compound heterozygous mutations of theADAMTS13gene. Eur. J. Haematol. 2013, 92, 168–171. [Google Scholar] [CrossRef]
- Bashir, B.A.; Mohamed, M.H.; Hussain, M.A.; Osman, W.; Mothana, R.A.; Hasson, S. Trends of Coagulation Parameters in Human Immunodeficiency Virus Patients. Medicina 2023, 59, 1826. [Google Scholar] [CrossRef]
- Uemura, M.; Fujimura, Y.; Ko, S.; Matsumoto, M.; Nakajima, Y.; Fukui, H. Pivotal role of ADAMTS13 function in liver diseases. Int. J. Hematol. 2010, 91, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, K.I.; Meyer, D.; Holcomb, D.D.F.; Kames, J.; Hamasaki-Katagiri, N.; Katneni, U.K.; Hunt, R.C.; Ibla, J.C.; Kimchi-Sarfaty, C. Synonymous ADAMTS13 variants impact molecular characteristics and contribute to variability in active protein abundance. Blood Adv. 2022, 6, 5364–5378. [Google Scholar] [CrossRef]
- Gaither, J.B.S.; E Lammi, G.; Li, J.L.; Gordon, D.M.; Kuck, H.C.; Kelly, B.J.; Fitch, J.R.; White, P. Synonymous variants that disrupt messenger RNA structure are significantly constrained in the human population. GigaScience 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Jacobi, P.M.; Emmer, B.T.; Kretz, C.A.; Ozel, A.B.; McGee, B.; Kimchi-Sarfaty, C.; Ginsburg, D.; Li, J.Z.; Desch, K.C. Genetic variants in ADAMTS13 as well as smoking are major determinants of plasma ADAMTS13 levels. Blood Adv. 2017, 1, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Huang, D.; Kondo, Y.; Jiang, M.; Ma, Z.; Zhou, L.; Su, J.; Bai, X.; Ruan, C.; Wang, Z.; et al. Novel mutations in ADAMTS13 CUB domains cause abnormal pre-mRNA splicing and defective secretion of ADAMTS13. J. Cell. Mol. Med. 2020, 24, 4356–4361. [Google Scholar] [CrossRef]
- Alwan, F.; Vendramin, C.; Liesner, R.; Clark, A.; Lester, W.; Dutt, T.; Thomas, W.; Gooding, R.; Biss, T.; Watson, H.G.; et al. Characterization and treatment of congenital thrombotic thrombocytopenic purpura. Blood 2019, 133, 1644–1651. [Google Scholar] [CrossRef]
- Batsuli, G.; Kouides, P. Rare Coagulation Factor Deficiencies (Factors VII, X, V, and II). Hematol. Clin. North Am. 2021, 35, 1181–1196. [Google Scholar] [CrossRef]
- Patton, L.L. Bleeding and clotting disorders. Burket’s oral medicine: diagnosis and treatment, 10th ed.; Hamilton (ON): BC Decker, 2003; pp. 454–477. [Google Scholar]
- Sadler, J.E. von Willebrand factor: two sides of a coin. J. Thromb. Haemost. 2005, 3, 1702–1709. [Google Scholar] [CrossRef]
- Casari, C.; Berrou, E.; Lebret, M.; Adam, F.; Kauskot, A.; Bobe, R.; Desconclois, C.; Fressinaud, E.; Christophe, O.D.; Lenting, P.J.; et al. von Willebrand factor mutation promotes thrombocytopathy by inhibiting integrin αIIbβ3. J. Clin. Investig. 2013, 123, 5071–5081. [Google Scholar] [CrossRef] [PubMed]
- Meiring, M.; Webb, M.; Goedhals, D.; Louw, V. HIV-associated Thrombotic Thrombocytopenic Purpura — What We Know So Far. Eur. Oncol. Haematol. 2012, 8, 89. [Google Scholar] [CrossRef]
- Plautz, W.E.; et al. ADAMTS13: origins, applications, and prospects. Transfusion 2018, 58, 2453–2462. [Google Scholar] [CrossRef]
- Graham, S.M.; Chen, J.; Le, J.; Ling, M.; Chung, D.W.; Liles, W.C.; López, J.A. Von Willebrand Factor Adhesive Activity and ADAMTS13 Protease Activity in HIV-1-Infected Men. Int. J. Med Sci. 2019, 16, 276–284. [Google Scholar] [CrossRef]
- Younas, M.; et al. Immune activation in the course of HIV-1 infection: causes, phenotypes and persistence under therapy. HIV Med. 2016, 17, 89–105. [Google Scholar] [PubMed]
- Arildsen, H.; et al. Endothelial dysfunction, increased inflammation, and activated coagulation in HIV-infected patients improve after initiation of highly active antiretroviral therapy. HIV Med. 2013, 14, 1–9. [Google Scholar]
- Singh, S.; Moodley, J.; Naicker, T. Differential expression of the angiotensin receptors (AT1, AT2, and AT4) in the placental bed of HIV-infected preeclamptic women of African ancestry. Hypertens. Res. 2023, 46, 1970–1982. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E. Platelets in HIV: a guardian of host defence or transient reservoir of the virus? Front. Immunol. 2021, 12, 649465. [Google Scholar]
- Funderburg, N.T. Markers of coagulation and inflammation often remain elevated in ART-treated HIV-infected patients. Curr. Opin. HIV AIDS 2014, 9, 80–86. [Google Scholar] [CrossRef]
- Ceccarelli, G.; et al. What happens to cardiovascular system behind the undetectable level of HIV viremia? AIDS Res. Ther. 2016, 13. [Google Scholar]
- Lunghi, L.; et al. Control of human trophoblast function. Reprod. Biol. Endocrinol. 2007, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Godbole, G.; Modi, D. Decidual control of trophoblast invasion. Am. J. Reprod. Immunol. 2016, 75, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Feng, Y.; Li, X.; Li, W.; Fan, L.; Liu, J.; Zeng, X.; Chen, K.; Chen, X.; Zhou, X.; et al. Expression of ADAMTS13 in Normal and Abnormal Placentae and Its Potential Role in Angiogenesis and Placenta Development. Arter. Thromb. Vasc. Biol. 2017, 37, 1748–1756. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2021, 23, 146. [Google Scholar] [CrossRef]
- Martin, K.; Borgel, D.; Lerolle, N.; Feys, H.B.; Trinquart, L.; Vanhoorelbeke, K.; Deckmyn, H.; Legendre, P.; Diehl, J.-L.; Baruch, D. Decreased ADAMTS-13 (A disintegrin-like and metalloprotease with thrombospondin type 1 repeats) is associated with a poor prognosis in sepsis-induced organ failure*. Crit. Care Med. 2007, 35, 2375–2382. [Google Scholar] [CrossRef]
- Scully, M.; Thomas, M.; Underwood, M.; Watson, H.; Langley, K.; Camilleri, R.S.; Clark, A.; Creagh, D.; Rayment, R.; Mcdonald, V.; et al. Thrombotic thrombocytopenic purpura and pregnancy: presentation, management, and subsequent pregnancy outcomes. Blood 2014, 124, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Rodansky, E.S.; Smith, J.K.; Rodgers, G.M. ADAMTS13 promotes angiogenesis and modulates VEGF-induced angiogenesis. Microvasc. Res. 2012, 84, 109–115. [Google Scholar] [CrossRef]
- Urra, M.; Lyons, S.; Teodosiu, C.G.; Burwick, R.; Java, A. Thrombotic Microangiopathy in Pregnancy: Current Understanding and Management Strategies. Kidney Int. Rep. 2024, 9, 2353–2371. [Google Scholar] [CrossRef]
- Oglak, S.C.; Obut, M. Expresión de ADAMTS13 y PCNA en las Placentas de Madres Diabéticas Gestacionales. Int. J. Morphol. 2021, 39, 38–44. [Google Scholar] [CrossRef]
- Aref, S.; Goda, H. Increased VWF antigen levels and decreased ADAMTS13 activity in preeclampsia. Hematology 2013, 18, 237–241. [Google Scholar] [CrossRef]
- Claus, R.; et al. The balance between von-Willebrand factor and its cleaving protease ADAMTS13: biomarker in systemic inflammation and development of organ failure? Curr. Mol. Med. 2010, 10, 236–248. [Google Scholar]
- Karim, F.; Adil, S.N.; Afaq, B.; Haq, A.U. Deficiency of ADAMTS-13 in pediatric patients with severe sepsis and impact on in-hospital mortality. BMC Pediatr. 2013, 13, 44. [Google Scholar] [CrossRef] [PubMed]
- Dap, M.; Romiti, J.; Dolenc, B.; Morel, O. Thrombotic thrombocytopenic purpura and severe preeclampsia: a clinical overlap during pregnancy and a possible coexistence. J. Gynecol. Obstet. Hum. Reprod. 2022, 51, 102422. [Google Scholar] [CrossRef] [PubMed]
- Berube, C.; Dietrich, B. Ask the Hematologists: Von Willebrand Disease and Pregnancy. Hematol. 2017, 14. [Google Scholar] [CrossRef]
- Pacheco, L.D.; Costantine, M.M.; Saade, G.R.; Mucowski, S.; Hankins, G.D.; Sciscione, A.C. von Willebrand disease and pregnancy: a practical approach for the diagnosis and treatment. Am. J. Obstet. Gynecol. 2010, 203, 194–200. [Google Scholar] [CrossRef]
- Warren, B.B.; Moyer, G.C.; Manco-Johnson, M.J. Hemostasis in the pregnant woman, the placenta, the fetus, and the newborn infant. In Seminars in thrombosis and hemostasis; Thieme Medical Publishers, Inc., 2023. [Google Scholar]
- Prochazka, M.; Procházková, J.; Lubušký, M.; Pilka, R.; Úlehlová, J.; Michalec, I.; Polák, P.; Kacerovský, M.; Slavik, L. Markers of endothelial activation in preeclampsia. Clin. Lab. 2015, 61, 39–46. [Google Scholar]
- Sadler, B.; Castaman, G.; O'Donnell, J.S. von Willebrand disease and von Willebrand factor. Haemophilia 2022, 28, 11–17. [Google Scholar]
- Deng, L.; Bremme, K.; O Hansson, L.; Blombäck, M. Plasma levels of von Willebrand factor and fibronectin as markers of persisting endothelial damage in preeclampsia. Obstet. Gynecol. 1994, 84, 941–945. [Google Scholar]
- Petca, A.; Miron, B.C.; Pacu, I.; Dumitrașcu, M.C.; Mehedințu, C.; Șandru, F.; Petca, R.-C.; Rotar, I.C. HELLP Syndrome—Holistic Insight into Pathophysiology. Medicina 2022, 58, 326. [Google Scholar] [CrossRef]
- Boustani, P.; Eslamian, L.; Nurzadeh, M.; Marsosi, V.; Ghaemi, M. HELLP syndrome complicated by ischemic colitis: A case report. Clin. Case Rep. 2023, 11, e7557. [Google Scholar] [CrossRef]
- Lewandowska, M.; et al. A Rare Case of HELLP Syndrome with Hematomas of Spleen and Liver, Eclampsia, Severe Hypertension and Prolonged Coagulopathy—A Case Report. Int. J. Environ. Res. Public Health 2022, 19, 7681. [Google Scholar] [CrossRef] [PubMed]
- Bakrania, B.A.; et al. Preeclampsia: linking placental ischemia with maternal endothelial and vascular dysfunction. Compr. Physiol. 2020, 11, 1315. [Google Scholar]
- Shah, D.A.; Khalil, R.A. Bioactive factors in uteroplacental and systemic circulation link placental ischemia to generalized vascular dysfunction in hypertensive pregnancy and preeclampsia. Biochem. Pharmacol. 2015, 95, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001, 413, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Bongers, T.; de Bruijne, E.; Dippel, D.; de Jong, A.; Deckers, J.; Poldermans, D.; de Maat, M.; Leebeek, F. Lower levels of ADAMTS13 are associated with cardiovascular disease in young patients. Atherosclerosis 2009, 207, 250–254. [Google Scholar] [CrossRef]
- Larkin, D.; de Laat, B.; Jenkins, P.V.; Bunn, J.; Craig, A.G.; Terraube, V.; Preston, R.J.S.; Donkor, C.; Grau, G.E.; van Mourik, J.A.; et al. Severe Plasmodium falciparum Malaria Is Associated with Circulating Ultra-Large von Willebrand Multimers and ADAMTS13 Inhibition. PLOS Pathog. 2009, 5, e1000349. [Google Scholar] [CrossRef]
- Rafaqat, S.; Khalid, A.; Riaz, S. Irregularities of Coagulation in Hypertension. Curr. Hypertens. Rep. 2023, 25, 271–286. [Google Scholar] [CrossRef]
- Venou, T.M.; Varelas, C.; Gardikioti, A.; Vetsiou, E.; Klonizakis, P.; Daniilidis, A.; Christodoulou, I.; Koravou, E.-E.; Koutra, M.; Mavrikou, I.; et al. P1698: increased complement activation and decreased adamts13 activity in patients with pre-eclampsia compared to healthy pregnancies. HemaSphere 2022, 6, 1579–1580. [Google Scholar] [CrossRef]
- Agbani, E.O.; Skeith, L.; Lee, A. Preeclampsia: Platelet procoagulant membrane dynamics and critical biomarkers. Res. Pr. Thromb. Haemost. 2023, 7, 100075. [Google Scholar] [CrossRef]
- Marincowitz, C.; Genis, A.; Goswami, N.; De Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular endothelial dysfunction in the wake of HIV and ART. FEBS J. 2019, 286, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Balta, S. Endothelial dysfunction and inflammatory markers of vascular disease. Curr. Vasc. Pharmacol. 2021, 19, 243–249. [Google Scholar]
- Allie, S. The role of von Willebrand factor and its cleaving protease, ADAMTS13, in young patients with HIV-related stroke; University of Cape Town, 2013. [Google Scholar]
- Fourie, C.; Van Rooyen, J.; Pieters, M.; Conradie, K.; Hoekstra, T.; Schutte, A. Is HIV-1 infection associated with endothelial dysfunction in a population of African ancestry in South Africa? Cardiovasc. J. Afr. 2011, 22, 134–140. [Google Scholar] [CrossRef]
- Schneider, D.J. Factors Contributing to Increased Platelet Reactivity in People with Diabetes. Diabetes Care 2009, 32, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Platelet activation and prothrombotic mediators at the nexus of inflammation and atherosclerosis: Potential role of antiplatelet agents. Blood Rev. 2021, 45, 100694. [Google Scholar] [CrossRef]
- Tsai, H.-M. Pathophysiology of thrombotic thrombocytopenic purpura. Int. J. Hematol. 2010, 91, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hulstein, J.J.J.; Heimel, P.J.V.R.; Franx, A.; Lenting, P.J.; Bruinse, H.W.; Silence, K.; DE Groot, P.G.; Fijnheer, R. Acute activation of the endothelium results in increased levels of active von Willebrand factor in hemolysis, elevated liver enzymes and low platelets (HELLP) syndrome. J. Thromb. Haemost. 2006, 4, 2569–2575. [Google Scholar] [CrossRef]
- Jr, J.R.; Bõze, T.; Derzsy, Z.; Cervenak, L.; Makó, V.; Gombos, T.; Udvardy, M.L.; Hársfalvi, J.; Prohászka, Z.; Molvarec, A. Increased plasma von Willebrand factor antigen levels but normal von Willebrand factor cleaving protease (ADAMTS13) activity in preeclampsia. Thromb. Haemost. 2009, 101, 305–311. [Google Scholar] [CrossRef]
- Louw, S.; Gounden, R.; Mayne, E.S. Thrombotic thrombocytopenic purpura (TTP)-like syndrome in the HIV era. Thromb. J. 2018, 16, 1–7. [Google Scholar] [CrossRef]
- David, M.; Naicker, T. The complement system in preeclampsia: a review of its activation and endothelial injury in the triad of COVID-19 infection and HIV-associated preeclampsia. Obstet. Gynecol. Sci. 2023, 66, 253–269. [Google Scholar] [CrossRef]
- Govender, S.; David, M.; Naicker, T. Is the Complement System Dysregulated in Preeclampsia Comorbid with HIV Infection? Int. J. Mol. Sci. 2024, 25, 6232. [Google Scholar] [CrossRef] [PubMed]
- Francisci, D.; et al. HIV type 1 infection, and not short-term HAART, induces endothelial dysfunction. Aids 2009, 23, 589–596. [Google Scholar] [CrossRef]
- Brill, A.; Suidan, G.; Wagner, D. Hypoxia, such as encountered at high altitude, promotes deep vein thrombosis in mice. J. Thromb. Haemost. 2013, 11, 1773–1775. [Google Scholar] [CrossRef] [PubMed]
- Novoyatleva, T.; Kojonazarov, B.; Owczarek, A.; Veeroju, S.; Rai, N.; Henneke, I.; Böhm, M.; Grimminger, F.; Ghofrani, H.A.; Seeger, W.; et al. Evidence for the Fucoidan/P-Selectin Axis as a Therapeutic Target in Hypoxia-induced Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 1407–1420. [Google Scholar] [CrossRef]
- Ruggeri, Z.M. The role of von Willebrand factor in thrombus formation. Thromb. Res. 2007, 120, S5–S9. [Google Scholar] [CrossRef] [PubMed]
- Denis, C.V.; André, P.; Saffaripour, S.; Wagner, D.D. Defect in regulated secretion of P-selectin affects leukocyte recruitment in von Willebrand factor-deficient mice. Proc. Natl. Acad. Sci. 2001, 98, 4072–4077. [Google Scholar] [CrossRef] [PubMed]
- Obeagu, E.; Obeagu, G. P-Selectin Expression in HIV-Associated Coagulopathy: Implications for Treatment. Elite J. Haematol. 2024, 2, 25–41. [Google Scholar]
- Palalioglu, R.M.; Erbiyik, H.I. Evaluation of maternal serum SERPINC1, E-selectin, P-selectin, RBP4 and PP13 levels in pregnancies complicated with preeclampsia. J. Matern. Neonatal Med. 2023, 36, 2183472. [Google Scholar] [CrossRef]
- Cho, J.-S.; Ouriel, K. Differential Thrombogenicity of Artery and Vein: the Role of von Willebrand Factor. Ann. Vasc. Surg. 1995, 9, 60–70. [Google Scholar] [CrossRef]
- Mancini, I.; et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 2021, 19, 513–521. [Google Scholar] [CrossRef]
- Theilen, L.H.; Campbell, H.D.; Mumford, S.L.; Purdue-Smithe, A.C.; Sjaarda, L.A.; Perkins, N.J.; Radoc, J.G.; Silver, R.M.; Schisterman, E.F. Platelet activation and placenta-mediated adverse pregnancy outcomes: an ancillary study to the Effects of Aspirin in Gestation and Reproduction trial. Am. J. Obstet. Gynecol. 2020, 223, 741.e1–741.e12. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, P.; Han, C.; Li, J.; Liu, L.; Zhao, Z.; Gao, Y.; Qin, Y.; Xu, Q.; Yan, Y.; et al. Association of placenta-derived extracellular vesicles with pre-eclampsia and associated hypercoagulability: a clinical observational study. BJOG: Int. J. Obstet. Gynaecol. 2020, 128, 1037–1046. [Google Scholar] [CrossRef]
- Barbera, L.K.; Kamis, K.F.; Rowan, S.E.; Davis, A.J.; Shehata, S.; Carlson, J.J.; Johnson, S.C.; Erlandson, K.M. HIV and COVID-19: review of clinical course and outcomes. HIV Clin. Trials 2024, 22, 102–118. [Google Scholar] [CrossRef]
- Tran, M.; Alessandrini, V.; Lepercq, J.; Goffinet, F. Risk of preeclampsia in patients with symptomatic COVID-19 infection. J. Gynecol. Obstet. Hum. Reprod. 2022, 51, 102459. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; McKinnon, T.A.J.; Zhang, X.F. Contribution of the von Willebrand factor/ADAMTS13 imbalance to COVID-19 coagulopathy. Am. J. Physiol. Circ. Physiol. 2021, 322, H87–H93. [Google Scholar] [CrossRef]
- Serrano, B.; Bonacina, E.; Garcia-Ruiz, I.; Mendoza, M.; Garcia-Manau, P.; Garcia-Aguilar, P.; Gil, J.; Armengol-Alsina, M.; Fernández-Hidalgo, N.; Sulleiro, E.; et al. Confirmation of preeclampsia-like syndrome induced by severe COVID-19: an observational study. Am. J. Obstet. Gynecol. MFM 2022, 5, 100760. [Google Scholar] [CrossRef]
- Coronado-Arroyo, J.C.; Concepción-Zavaleta, M.J.; Zavaleta-Gutiérrez, F.E.; Concepción-Urteaga, L.A. Is COVID-19 a risk factor for severe preeclampsia? Hospital experience in a developing country. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 256, 502–503. [Google Scholar] [CrossRef]
- Zhang, Q.; Bignotti, A.; Yada, N.; Ye, Z.; Liu, S.; Han, Z.; Zheng, X.L. Dynamic Assessment of Plasma von Willebrand Factor and ADAMTS13 Predicts Mortality in Hospitalized Patients with SARS-CoV-2 Infection. J. Clin. Med. 2023, 12, 7174. [Google Scholar] [CrossRef]

| ART CLASS | EFFECTS | REFERENCE |
|---|---|---|
| Nucleoside/Nucleotide Reverse Transcriptase Inhibitors (NRTIs) | Zidovudine and Stavudine, have been shown to induce mitochondrial toxicity and oxidative stress in platelets, leukopenia, elevation of liver enzyme levels, elevation of lactic acid level, Abacavir- Hypersensitivity reactions such as fever, rash, myalgia, arthralgia, malaise |
[113]. [114]. [27]. |
| Non-nucleoside Reverse Transcriptase Inhibitors (NNRTIs) | Efavirenz and Nevirapine- induce hepatic enzyme induction, alter platelet metabolism, central nervous system toxicity, and psychosis, rash. | [113]. [114]. [27]. |
| Protease Inhibitors (PIs) | Impairing platelet function Induce endothelial dysfunction Alter the balance of pro- and anti-thrombotic factors Gastrointestinal upset, rash Indinavir- nephrolithiasis, hypertension |
[113]. [114]. [27]. |
| Integrase strand transfer Inhibitors (INSTIs) | Raltegravir and Dolutegravir is associated with changes in lipid metabolism, endothelial function, gastrointestinal upset, hepatitis. | [114]. [27]. |
| ART CLASS | MECHANISM OF ACTION | REFERENCE |
|---|---|---|
| Non-nucleoside reverse transcriptase inhibitor (NNRTIs) | They bind in a non-competitive way to HIV-1 reverse transcriptase enzyme and inhibit the conversion of viral RNA into DNA. Restores immune response and are elevated during oxidative stress. Dysregulates NF-κB transcription factors hence decrease MMP-9 and VEGF expression. Dysregulates immunoexpression of angiopoietin, endoglin and PlGF. Decreases tight junction proteins such as claudin-1, occludin, zonula occluden-1 and junctional adhesion molecule-1 which increases vascular permeability. |
[129] [130] [131] [132] [117]. |
| Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) | These directly block HIV-1 reverse transcriptase enzyme from converting viral RNA into DNA. They reconstitute immune response. Decreases endothelial cell proliferation, migration via defective tyrosine kinase receptor and VEGFR-2 signalling. Exacerbates mitochondrial oxidative stress, and this increase in ROS generation pre-empts trophoblast apoptosis and thus predisposing PE and/or IUGR development. |
[133] [134] [135] [117] |
| Protease Inhibitors (PIs) | Inhibit HIV-1 protease, inhibiting the transformation of immature HIV particles to mature HIV particles. They restore immune response They deplete uNK cells. However, they lower progesterone in trophoblast cells hence impeding invasion following decreased expression of the transcription factor STAT3. This leads to a dysregulated uterine decidualization, incomplete trophoblast cell invasion and defective spiral artery remodelling. They also decrease VEGF, PlGF, angiopoietin-2, interferon-gamma, and MMP-9 in decidual cells They decrease endothelial cell proliferation, migration and causes defective tyrosine kinase receptor and VEGFR-2 signalling. Moreover, PIs also elevate mitochondrial oxidative stress which leads to increased ROS generation elevating trophoblast apoptosis and predisposing PE and/or IUGR development. |
[136] [137]. [117] [138] [139] |
| Integrase strand transfer Inhibitors (INSTIs) | Prevents HIV replication by blocking integrase which is used to insert viral DNA into the host CD4 cell. | [140] |
| HAART | Based on immune reconstitution. HAART dysregulates NF-κB transcription factors hence decreases MMP and VEGF expression. HAART also is implicated in an increase in sFlt-1 and sEng with concomitant decrease in PlGF and VCAM-1 expression. |
[141] [125] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



