Submitted:
07 March 2025
Posted:
10 March 2025
You are already at the latest version
Abstract
Cancer is a lethal disease worldwide that damages bodily tissues, linked with predisposing variables including inheritance, social, infections and curative measures requiring behavioral modifications. But now a days, researchers are working to find targeted drug therapy with limited side effects that are associated with previous approaches. Plant based novel drug Halofuginone (HaF), by blocking prolyl tRNA synthetase exhibits promising diverse clinical activities, against fibrosis, cancer, metabolic, and infectious disorders. However, the drug is quite neurotoxic and gastrotoxic requiring thorough investigational analyses as per preclinical studies at Phase-II clinical trials. The present study explored various databases, software and monographs to establish therapeutic aspects of this drug to address gaps by highlighting the potential impact of salt reformation on improving its properties. Hence, our study invites the researchers to look upon novel Halofuginone derivatives with other possible halogen salts like HCl to enhance pharmacokinetics and pharmacodynamics qualities of Halofuginone. Our present study summaries the possibilities of other form of Halofuginone for safe utility in pharmacotherapeutics for future research in this area, provided its toxicity concerns are adequately addressed through innovative strategies.
Keywords:
Immunosurveillance and immunomodulation
; NFK-B- P60/P65
; Prolyl-transfer-RNA-synthetase
; TGFβ-SMAD3 / AKT-MTOR
; PI3K/Akt-Bax/Bcl2
; Plasmodium falciparum
; Tumorigenesis
; Cefotiam HCl and
; Exidartinib HCl
; Cytokine IL-17
; p38-MAP-Kinase / P-ARP
1. Introduction
Cancer (Canc.) is a life threatening ailment with various predisposing factors mainly socioenvironmental, genetic, infectious diseases may be prevented through behavioural changes or vaccination to prevent infection [1]. Canc. hallmarks include evading growth suppressors, sustaining cell proliferative signalling events, tissue invasion, metastasis, angiogenesis or neo-vasculature, cell death/apoptosis (Apo.), mutated energy metabolism, and loss of immunosurveillance [2,3]. In 2018 around 18.1M new cases were diagnosed and are expected to rise to 21.6M by 2030 annually, and in 2020 nearly 10M deaths or approx. 1 in every 6 deaths or approx. 1 in every 6 deaths reported [4,5].
Global burden and prevalence were expected to increase up to 27.5M with 16.3M death rate or approx. 1 in every 6 affected by 2040 [5,6]. As per US data of 2022 new Canc. patients count was 1.9M out of which 36.33 % deaths are projected [1]. Around 46.5K Canc. deaths and 176.6K new cases were projected among the Hispanic population in the U.S. and Hawaii in 2021 accounting for 20% of deaths [1]. During lifetime, as per one estimate out of 100, about 39 women and 40 men will likely develop Canc. and may differ depending upon factors like genetics, cigarette smokers, illicit drug users and workers expose to radiations [1,7].
Prevalence varies based on organs affected such as breast Canc. 2.26M, lung Canc. 2.21M, gastric Canc. (GC) 1.089M, hepatic Canc. (Hep.ca) 0.96 M and colon Canc. by 1.93M [8]. Deadliest Canc. reported in Asia include lung, breast, GIT and liver [6]. With the highest population ratio across the globe (60%), life expectation in Asia has increased and was expected to be around 25% more by 2050 among people aged ≥60yrs [9,10,11]. Most common Canc. in young females are 13.8% lung, 10.8% breast, 10.6% colorectal, 8.6% stomach and 6.9% liver with recommendation of intensive early Canc. diagnosis, precautionary measures, and lifestyle modifications. In year 2020, 950,3710 new cases are reported with age-standardized rate of incidence was 0.169.1 % for both sexes with highest in Japan, Korea and Israel and total of 580,94,31 deaths, most commonly lung Canc. 19.2% with liver ranked at the second 0.1016% (highest in Mongolia, China and Armenia). More in males around 0.1852 %, highest lung then G.I.T, whereas for females, 0.1567 % with highest of breast then colorectal and lung. Breast Canc. in 2004-2013, rise amongst Asia-Pacific females (20-49 years) due to reproductive factors, high levels of breast exposure to oestrogen at menopause with aging, over-diagnosis of thyroid and lung Canc. with tobacco use [12,13,14,15,16,17,18,19,20]. GIT Canc. -related deaths were because of higher H. pylori level [9,21]. In Korea, more prevalent was Canc. of thyroid 27.5% during 1999-2018, and more in Japan 1985- 2010 while lesser in the Philippines and Israel in both sexes [22,23,24,25,26,27]. Canc. prevalence increased in Pakistan due to smog, population and pollution, with 19M new cases in 2020 [8].
More than 60 % clinically proven natural origin anticancer drugs are available and research and NDDA are focused on diversity and structural modification, the main foundation of new anticancer drugs [28]. Among these novel approaches, one of the plant origin drugs Halofuginone (HaF), could be a medical breakthrough as per its multiple mode of actions and associated efficacy in Canc. and non-cancerous diseases. However, the drug is in phase 2 trials because of toxicity. Through our review we will try to elucidate its mode of action, SAR, multiple targets and how we can enhance its physicochemical parameters for Canc. therapy in future for the betterment of mankind.
2. Materials and Methods
The author searched systematically, thoroughly and screen with comprehension most relevant, authentic and original group of potential studies, index, articles and writings from the Scientific literature; Scopus; Uniport; RSCB; PD; WOS; JSTOR; CNKI DATABASE; books; Electronic DATABASES; and Google Scholar; PubMed; Science Direct; Elsevier; National library of medicine Bethesda; Maryland; NCBI; The New York Academy Of Medicine; Library of Speech Rehabilitation; NY; ST. Thomas ‘s Hospital Library; Repository of Kings College London; Oxford Academic Repository; Wiley Online Library; CAS data base; PubChem; American Canc. society; IJCEP; MDP; journals including Chinese Chemical Letters; Journal of Medicinal Chemistry; Journal of Biochemistry; Sage journals from February 2023 till June 2024. Moreover, Software’s that are manifested include Adobe Photoshop 2021; Molinspiration bioactivity score v-2022.08; Chem 3D Pro 12.0 for pictorial explanations and literature review writing. We have set predetermined rules for inclusion and exclusion of applicable studies from primary studies on HaF with significant investment of time in order to ensure objectivity, free from biasness with multiple time screening to ensure high scientific quality of the selected studies, with relevance to problem of interest and maximum validity. For this purpose, we collect, precise, combined, systematically organize data and compare the evidence extracted HaF data and present it in an eloquent way and in future recommends other researchers. In order to meet set criteria and to make sense of existing knowledge on a problem of interest in our case HaF toxicity, we provided a comprehensible lens from each angle, articles included are from 1946 till 2024. Total 414 research and review articles are explored and studied for this review altogether to identify, critically analyse and detailing of all relevant literature on a Canc. in correlation to HaF, in order to derive conclusions about the mechanism of actions on various molecular genetic factors in cancerous and noncancerous diseases, currently available or under study analogues of HaF and identifying gaps in previous literature available.
2.1. Biochemistry and Structural Analogues of Halofuginone:
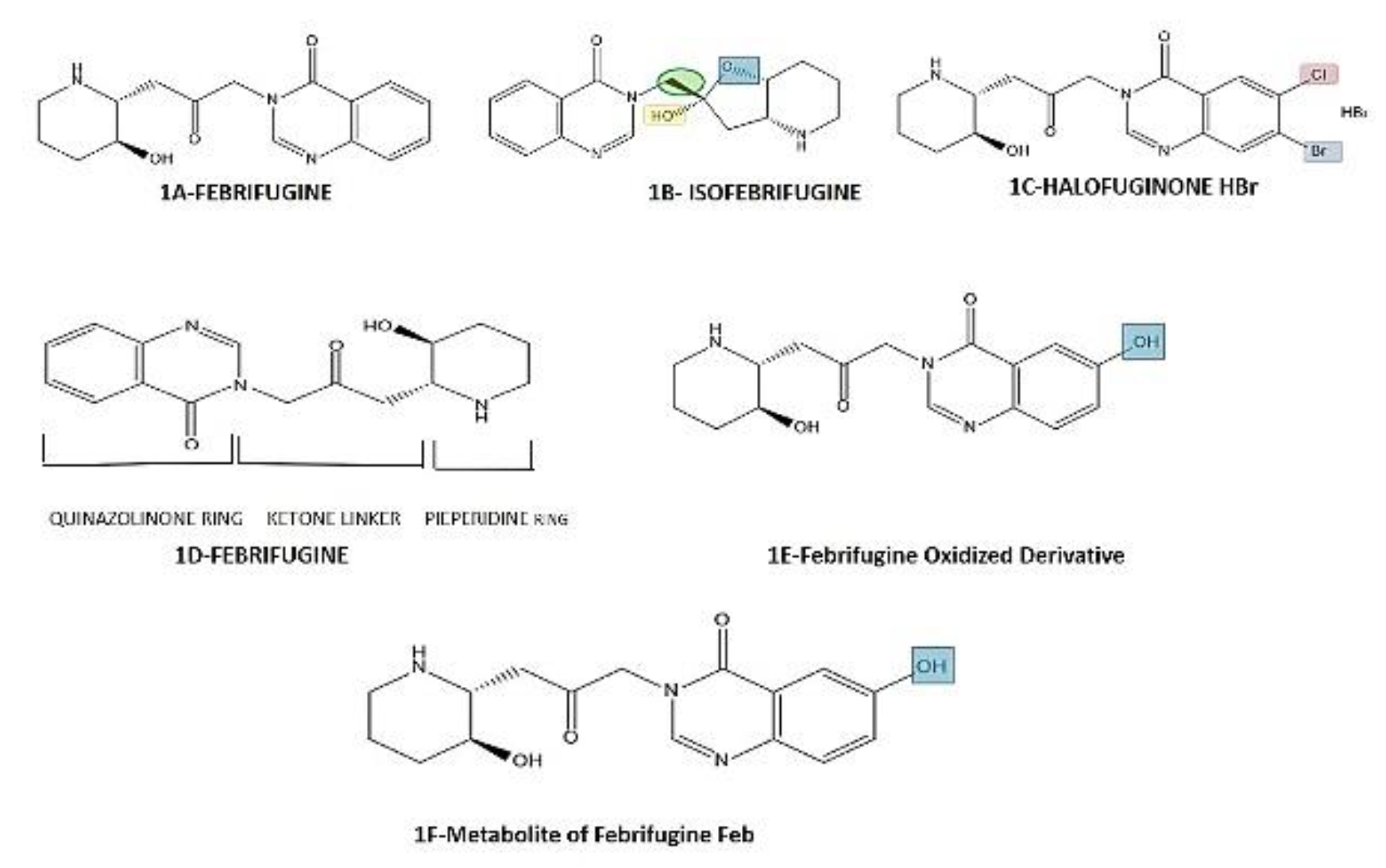
In advanced anticancer research, folk Chinese medicines particularly of natural origin (including plant, animal, fungi) have been considered promising. Among these plant origin drugs novel RNA synthesis inhibitors agents, a structural derivative of Febrifugine (Febr.) with IUPAC name 3-[3-[(2R,3S)-3-hydroxypiperidin-2-yl]-2-oxopropyl]quinazolin-4-one is Halofuginone (HaF) with IUPAC name 7-Bromo-6-chloro-3-[3-[(2S,3R)-3-hydroxy-2-piperidinyl]-2-oxopropyl]-4-quinazolinone and has been in use for over 2000 years, an analogue of quinazolinone alkaloid extracted from leaves and roots of Chinese herb ‘Dichroa febrifuga’ Hydrangeaceae family in 1948, used as an antimalarial medicine and in poultry feed and also received US-FDA certificate in early 1980s [29,30,31]. In 1967 Waletzky et al. developed a compound utilized in poultry feed as a coccidiostat in cattle and show positive outcomes for treatment of fibrotic diseases, immunosuppressive disorder like cancers, and excessive immunostimulant disorder like autoimmune diseases named as HaF ([Figure 1C) [32-37]. Quinazolinone ring containing alkaloids [32–37includes Febrifugine (2R,3S) (Figure 1A), Isofebrifugine (2S,3S) (stereoisomer of Febrifugine) (Figure 1B) also isolated from Dichroa Febrifuga, were reported along with their skeletal structures in 1967, however absolute configuration was attained after more than 40yrs of research, and another derivative Halofuginone hydrobromide discovered and synthesized by American Cyanamid Company, later on transferred to Roussel Uclaf S and under the named “Stenorol” A. who marketed it in a premix [38,39,40,41]. Following the first isolation test back in 1946 Febr. shown markedly higher activity both in vitro and in vivo than quinine, or chloroquine against several strains of malaria that was why gathered a lot of attention [38,42,43,44]. However, early findings from human trials exposed that it has several side-effects including nausea, vomiting, and lethargy resulted in creation of several analogues with significantly enhanced activity and metabolic stability, against Canc. as well [45].
Febr. structure consist of three parts: the ketone linker, the piperidine ring, and the quinazolinone ring (Figure 1D). To provide better therapeutic candidate all parts of Febr. structure have been altered. Several metabolites were detected in 2003 in mouse model and isolated [46]. Alteration in the quinazolinone ring was done by Baker et al. including mono and di substitution at the 5th, 6th , 7th, and 8th positions, showing better anticancer efficacy, by mono substitution at the 5th and 6th position and di substitution at the 6th and 7th positions against Plasmodium lophurae in ducks and similarly, further modification was done by increasing the range of substituents and altering the attached type of aromatic ring as well [47,48,49]. The oxidised quinazolinone ring derivatives (Figure 1E) have shown reduced anti-malarial activity against P. falciparum, by blocking the susceptible positions and by changing the oxidation potential of the ring making the quinazolinone group as an attractive target for modification due to metabolic susceptibility[48,49,50,51,52,53,54,55].
Shingo Hirai and Haruhisa Kikuchi, coresearchers studied and observed the Febr. analogues with the aim of conserving the strong antimalarial activity and potency in past, as it can be valuable leads for new drugs development (NDD) and for studying other related therapeutic properties such as anticancer or for scleroderma treatment. Basicity of both the nitrogen atoms at position 1 and the 1′′ of Febr. was important in conversing powerful antimalarial activity in vitro and the metabolite 4 given below (Figure 3), of Febr. with hydroxy ‘-OH’ substitution at position C-6 in place of ‘H’ at quinazolinone ring, exhibited powerful antimalarial activities. Initial data suggest that the ‘OH’ group has shown low toxicity against mammalian cells and preserves antimalarial activity against P. falciparum, seems to be viable main component for the development of novel antimalarial drugs in future due to lesser side effects then parent compound [38,56]. The oral administration of this compound with ED50 values of 15mg/kg/day for 4 and 8mg/kg/day was seen to be effective against murine malaria and single administration at 3 mg/kg intraperitoneal in mice, has no associated toxicities, such as diarrhoea or weight loss, after four successive days of therapy [38,39,40,41,46,57].
Figure 1.
(1A) Structures of Febrifugine: (1B) Isofebrifugine stereoisomer of Febrifugine; 1(C) Halofuginone Hydrobromide-halogenated derivative of Febrifugine with bromine at position 7 and chlorine at position 6 and HBr salt [38]; (1D) Structural Sections of Febrifugine[57]; (1E) Structures of Febrifugine Oxidized Derivative [46]; (1F) Structures of Metabolite of Febrifugine Febr-A(4) [46].
Figure 1.
(1A) Structures of Febrifugine: (1B) Isofebrifugine stereoisomer of Febrifugine; 1(C) Halofuginone Hydrobromide-halogenated derivative of Febrifugine with bromine at position 7 and chlorine at position 6 and HBr salt [38]; (1D) Structural Sections of Febrifugine[57]; (1E) Structures of Febrifugine Oxidized Derivative [46]; (1F) Structures of Metabolite of Febrifugine Febr-A(4) [46].
By organizational alterations at the quinazoline or piperidine groups It has been confirmed that majority of Febr. analogues have enhanced activity consisting of a modified or unmodified 4-quinazolinone moiety, but analogues with modification at the side chain attached to the N3 of the 4-quinazolinone ring have no activity, additionally a synthetic racemic Febr. was about 50% as effective as natural Febr. Therefore, the absolute position of functional groups and quinazolinone ring, localisation of ‘N’ in piperidine and ‘OH’ are essential for the anti-malarial action in Haf therefore, approved by FDA for coccidiosis treatment in chickens and turkeys in the salt form with Hydrobromine. Haf-Lactate are also used for parasitic attack in cattle[58,59,60,61,62]. Many reports on its synthesis were published as per importance of Febr., along with study on its analogues, however early synthesis methods met various difficulties in relation to stereo-chemistry, complexity and could interconvert [32,39,62,63,64]. Among the derivatives of Febr. the IC50s of HaF were detected to be the best of all [63].
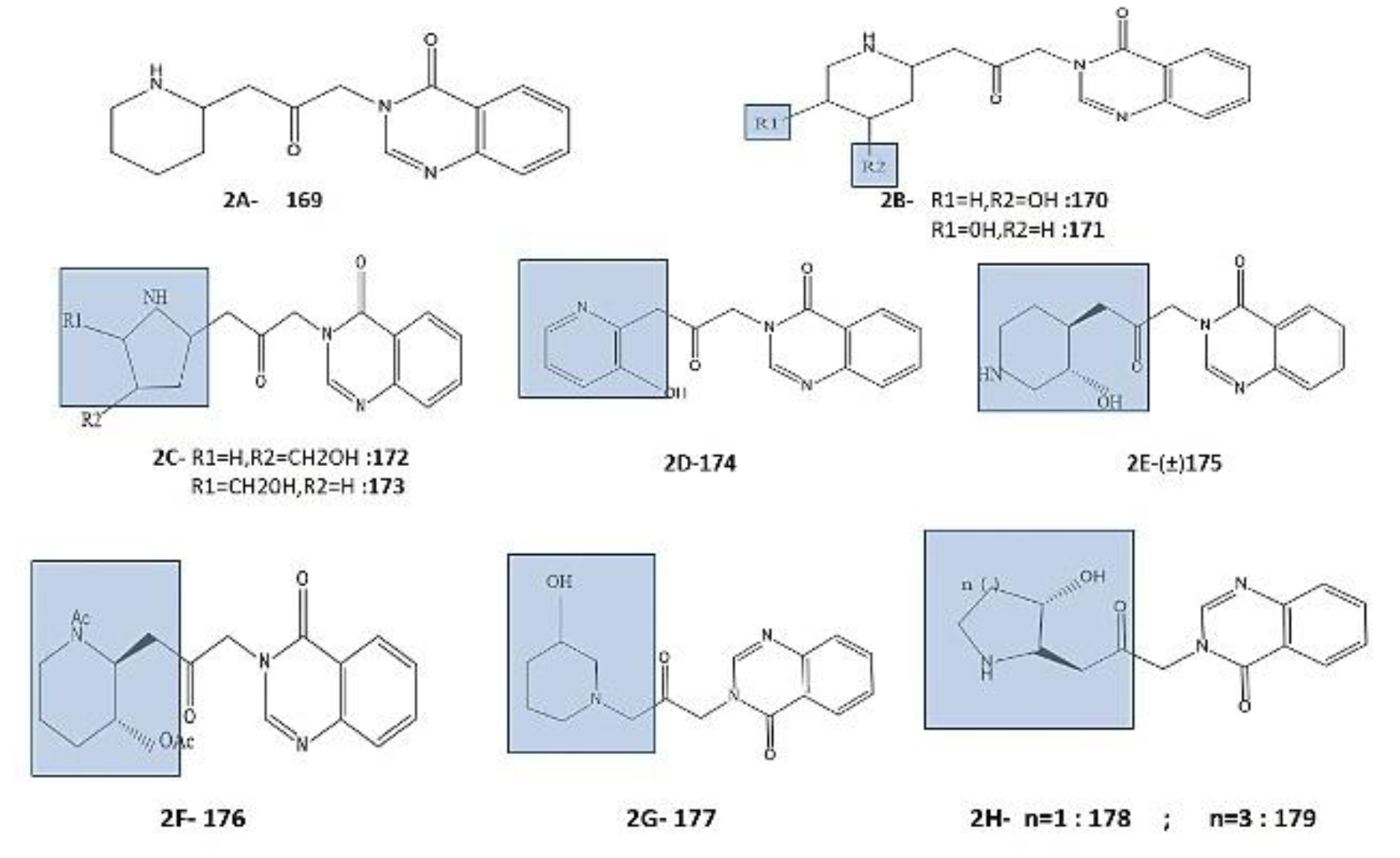
Modification of the piperidine ring during Febr. analogues manufacturing has also been considered, as studied by Baker et. al. studies he manufactured many isomers/analogues including Deoxy-Febrifugine 169 ([Figure 2A), the isomeric 4- hydroxypiperidine 170 (Figure 2B), 5-hydroxypiperidine 171 (Figure 2B), 3-hydroxymethylpyrrolidine 172 (Figure 2C), and 4-hydroxymethylpyrrolidine 173 (Figure 2C), without reporting their antimalarial activity [48,49,50,51,52,53,54,55,65,66,67,68,69,70,71]. However, Deoxyfebrifugine 169 antimalarial activity was reported by Takeuchi et al. in 1999 and synthesis was completed by Evans et al. in 2015 showing that it has reduced activity than Febr. against Plasmodium falciparum-FCR-3. after piperidine ring alteration, similar study was conducted by Cruickshank et al. in 1970 and Takeuchi by modification of piperidine analogues 175 (Figure 2E), and the pyridine-based analogue 174 (Figure 2D) respectively, against P. berghei (in mice) and P. falciparum-FCR-3 (in vitro), in which the position of the ‘N’ in the piperidine ring has been altered leading to no activity [44,48,49,50,51,52,53,54,55,65,66,67,68,69,70,71,72,73,74,75,76,77]. Similarly, with a broad scope of modifications in a piperidine ring, a huge collection of analogues has been synthesised by Oshima et al., in parent compound Febr. Including, the substitution of ‘N’ and ‘O’ in analogue 176 (Figure 2F) with acetyl substitutions, resulting in reduced activity against Plasmodium falciparum-FCR-3, and similar result was seen in analogue 177 (Figure 2G) and analogues 178 (Figure 2H) synthesised by Zhu et al. having more activity against plasmodium sp. (chloroquine-resistant strain) and analogue 179 (Figure 2H) with change in the size and the optically activity of Pyrrolidine and Azepane analogues of the piperidine ring, when compared to Febr. [49,78,79].
Figure 2.
Structures of Metabolite of Febrifugine: Piperidine analogues (2A) 169- deoxyfebrifugine; (2B) 170- the isomeric 4- hydroxypiperidine; 171-5-hydroxypiperidine; (2C) Pyrrolidine analogues; 172-3-hydroxymethylpyrrolidine; 173 -4-hydroxymethylpyrrolidine; Piperidine analogues (2D) 174; pyridine analogue (2E) 175; Piperidine and pyrrolidine analogues; (2F) 176 with N- and O-protecting groups; (2G) 177; (2H) 178 and 179 with change in the size and the optically activity of Pyrrolidine and Azepane analogues of the piperidine ring [48,49,50,51,52,53,54,55,65,66,67,68,69,70,71,78,79,80].
Figure 2.
Structures of Metabolite of Febrifugine: Piperidine analogues (2A) 169- deoxyfebrifugine; (2B) 170- the isomeric 4- hydroxypiperidine; 171-5-hydroxypiperidine; (2C) Pyrrolidine analogues; 172-3-hydroxymethylpyrrolidine; 173 -4-hydroxymethylpyrrolidine; Piperidine analogues (2D) 174; pyridine analogue (2E) 175; Piperidine and pyrrolidine analogues; (2F) 176 with N- and O-protecting groups; (2G) 177; (2H) 178 and 179 with change in the size and the optically activity of Pyrrolidine and Azepane analogues of the piperidine ring [48,49,50,51,52,53,54,55,65,66,67,68,69,70,71,78,79,80].
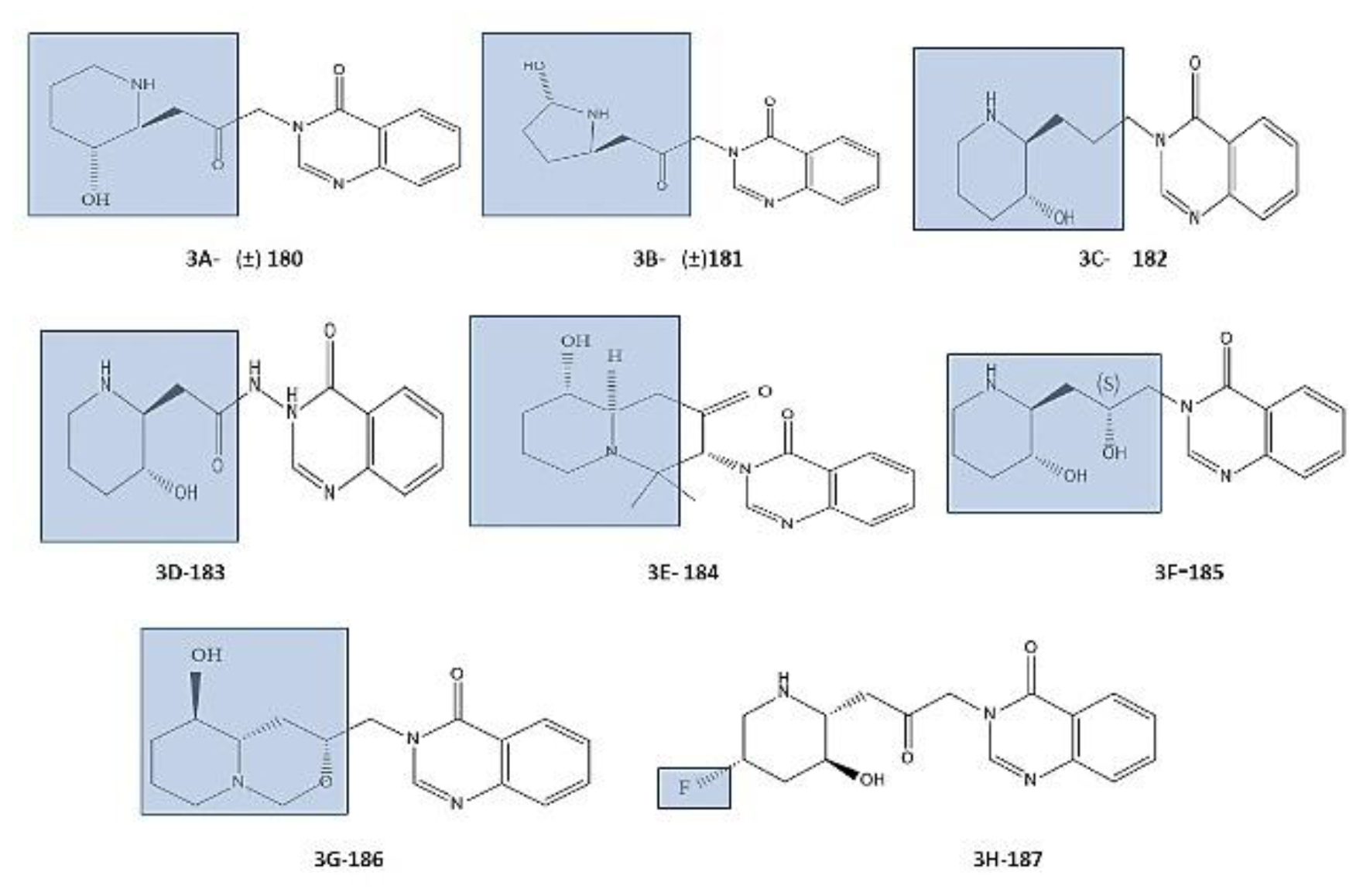
However, some analogues showed similar activity to Febr. including analogues 180 ([Figure 3A) and 181 (Figure 3B) against P. falciparum W2 and TM6 respectively [81,82,83]. Another researcher also discussed the importance of ketone linker, loss of functionality with the ketone elimination including analogue 182 (Figure 3C), replacing it with amide including analogue 183 (Figure 3D), acetone condensation analogue possessing a quinolizidine bicycle including analogue 184 (Figure 3E), showing loss/decrease in activity against Plasmodium falciparum-FCR-3. Bicyclic compound “Febrifuginol” 185 (Figure 3F) and analogue 186 (Figure 3G) with a link to the ‘OH’ and ‘N’ of the piperidine ring, showed better activity and reduce toxicity respectively [48,58,79,84]. Fluoride containing analogue of Febr. was also synthesized and studied by Liu et al. namely 5- Fluorofebrifugine 187(Figure 3H), with no reported data related to its impact on biological activity [85].
Figure 3.
Structures of truncated Analog of Febrifugine: (A), compound 180; (B), compound 181 [81,82,83]; Ketone Linker and fluorinated Analogues of Febrifugine; (C), Compound 182- loss of functionality with the removal; (D), Compound 183- replacement with an amide; (E) Compound 184- acetone condensation; (F), Compound 185 Bicyclic compound febrifuginol; (G), Compound 86- with a linkage to the ‘N’ of the piperidine ring and the ‘OH’ group; (H), compound 187- fluorofebrifugine [48,58,79,84,85].
Figure 3.
Structures of truncated Analog of Febrifugine: (A), compound 180; (B), compound 181 [81,82,83]; Ketone Linker and fluorinated Analogues of Febrifugine; (C), Compound 182- loss of functionality with the removal; (D), Compound 183- replacement with an amide; (E) Compound 184- acetone condensation; (F), Compound 185 Bicyclic compound febrifuginol; (G), Compound 86- with a linkage to the ‘N’ of the piperidine ring and the ‘OH’ group; (H), compound 187- fluorofebrifugine [48,58,79,84,85].
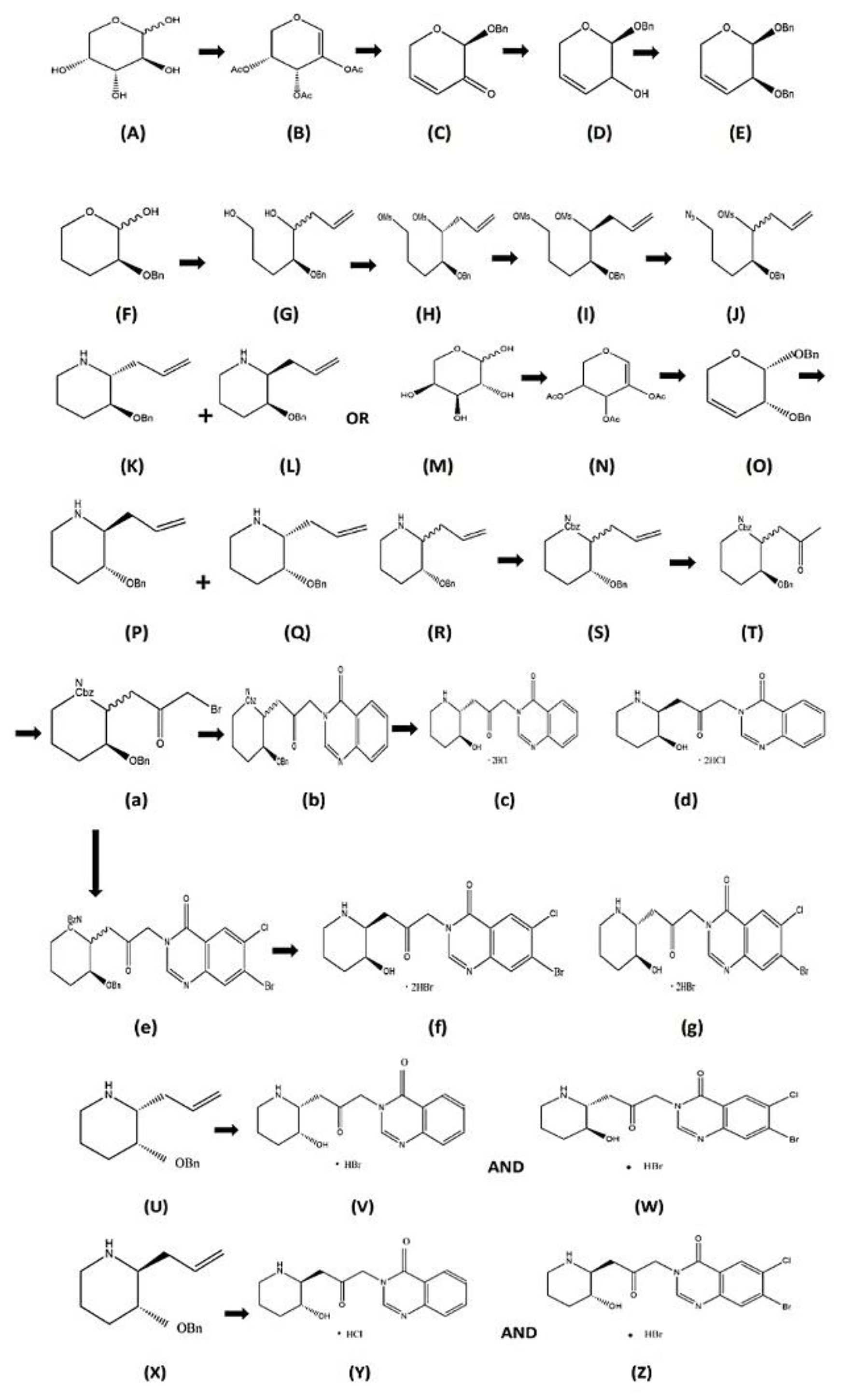
2.2. Stereoisomeric Synthesis of Halofuginone HBr Salt:
Synthesis for HBr Salt of HaF from Febr. precursor starts piperidine ring formation from chiral pool of Darabinal. By peracetylation D-arabinose changed to 2-Acetoxy-3,4-di-O-Acetylglycal, Glycosyl Bromide in presence of 33 percentage weightage of HBr/acetic acid mixture, then dehydrogenated by 1,8, di-azabicyclo undec-7-ene (DBU). Through a facial-diastereoselective glycosylation reaction dihydropyranone then formed from glycal in presence of a promoter BF3·Et2O with the use of benzyl alcohol, then ketone reduced to allyl alcohol derivative (single diastereomer) by applying Luche conditions. Then 77 % yield of cyclic hemiacetal obtained by alcohol benzylation, hydrogenolysis and alkene hydrogenation respectively. 3:7 ratio mixture of diastereomeric dimesylates obtained with use of allyl MgBr2 by facilitating the diols, which then mesylated. Azido mesylated then synthesized from primary mesylate by nucleophilic substitution in presence of sodium azide and then by Staudinger reduction convert to amine and subsequently cyclization occurs resulting in 3:7 ratio of piperidine derivatives, respectively. After the two diastereomeric piperidines were separated by silica gel column chromatography and their configurations assigned to the two stereocenters, a diastereomeric mixture of azido acetates was created by substituting cesium acetate for the stereocentre of the secondary mesylate carbon in the azido mesylate mixture. This led to the production of azido alcohols and azido mesylates, which underwent one pot reductive cyclization and produced a 7:3 ratio of piperidine derivatives, respectively. Single crystal XRD analysis verified the structure through the crystallization of the appropriate HCl salt where L-arabinose was converted to glycal ent-5 and then changed again into the dibenzylpyranose derivative ent-8 as a single enantiomer, exactly like in the synthesis of 13a and 13b and the diastereomeric piperidine compounds ent-13a and ent-13b were produced by further processes employing ent-8. In the next step was to obtain all the stereoisomers of Febr. and HaF enantiomerically from pure piperidine. The free NH protected by using Cbz-Cl, alkene group conversion occur into the corresponding methyl ketone derivatives by using Wacker oxidation which then by using TMSOTf transformed to the silyl enol ether and DIPEA, then bromination with NBS produced the α-bromo ketone respectively, undergo substitution with quinazolinone and protected (+)-isofebrifugine and (+)-febrifugine formed, eventually (+)- isofebrifugine and (+)-febrifugine gained as their di-HCl acid salt, Cbz-group undergo deprotection, the benzyl, started with refluxing the compounds in aqueous 6N hydrochloric acid, respectively. In other reaction the α- bromo ketones substituted with 7-Br-6-Cl-4-OH-quinazoline resulting in (+)-HaF derivatives, then undergo refluxing in water with 6N-HBr acid for removal of the protecting groups, (+)- iso- HaF and (+)-HaF as their di-HBr salts obtained finally. (+)- Febrifugine and (+)-HaF and their respective diastereomers are synthesised ([Figure 4) [86].
Figure 4.
Synthesis of (-)-Febrifugine and (-)-Halofuginone along with Diasterioisomers: (A) D-Arabinose; (B) 5(44%); (C) 6(79%); (D) 7(95%); (E) 8(91%); (F) 9(77%) (G) 10a;10b(69%); (H) 11a, I-11b, (J) 12a;12b(64%); (K) 13a—(L) 13b(13a:13b=3:7 -91%); (M) L-Arabinose, (N) ent-5; (O) ent-8; (P) ent-13b (Q) ent-13a; R-13a;13b (S) 16a;16b; (T) 17a;17b, (a) 18a;18b; (b) 19a;19b; (c) (+)-Isofebrifugine.2HCl(3.2HCl- 40%); (d) (+)-Febrifugine.2HCl(1.2HCl-50%); (e) 20a;20b, (f) (+)-IsoHalofuginone. 2HBr (4.2HBr- 45%); (g) (+)-Halofuginone.2HBr(2.2HBr-51%) (U) ent-13a; (V) (-)-Isofebrifugine. HCl (ent-3); (W) (-)-IsoHalofuginone.HBr (ent-4); (X) (-)-ent-13b; (Y) (-) Febrifugine.HCl (ent-1); (Z) (-)-Halofuginone -2HBr [86].
Figure 4.
Synthesis of (-)-Febrifugine and (-)-Halofuginone along with Diasterioisomers: (A) D-Arabinose; (B) 5(44%); (C) 6(79%); (D) 7(95%); (E) 8(91%); (F) 9(77%) (G) 10a;10b(69%); (H) 11a, I-11b, (J) 12a;12b(64%); (K) 13a—(L) 13b(13a:13b=3:7 -91%); (M) L-Arabinose, (N) ent-5; (O) ent-8; (P) ent-13b (Q) ent-13a; R-13a;13b (S) 16a;16b; (T) 17a;17b, (a) 18a;18b; (b) 19a;19b; (c) (+)-Isofebrifugine.2HCl(3.2HCl- 40%); (d) (+)-Febrifugine.2HCl(1.2HCl-50%); (e) 20a;20b, (f) (+)-IsoHalofuginone. 2HBr (4.2HBr- 45%); (g) (+)-Halofuginone.2HBr(2.2HBr-51%) (U) ent-13a; (V) (-)-Isofebrifugine. HCl (ent-3); (W) (-)-IsoHalofuginone.HBr (ent-4); (X) (-)-ent-13b; (Y) (-) Febrifugine.HCl (ent-1); (Z) (-)-Halofuginone -2HBr [86].
Identification of bioactive molecules and their SAR studies are critical steps to get deep insights. A huge reservoir of new chemical entities obtained from the natural products, essentially plant sources, by NDD having diverse characteristic with more efficacy [87]. Gained through drug-discovery methods including bioinformatics, HTS, computer-modelling, chemical genetics overcoming providence and HaF was example of one of the bioactive molecules [88]. Its historical background and diverse pharmacodynamics both in vivo/in vitro models were already discussed, with molecular structure C16H21O3N3, named as dichroin B after the plant origin ‘Dichroa’, most effective among several plant extracts with antimalarial actions, in poultry feed, and P. gallinaceum infection in chicks, and later renamed as Febr. [89,90]. Febr. structure, used as a lead compound in isofebrifugine synthesis with less toxicity, high efficacy chloroquine-sensitive and-resistant Plasmodium species (falciparum, vivax) and against Plasmodium species (P. lophurae, P. gallinaceum, P. cynomolgi) showed better activity, overcome GIT toxicity, including diarrhoea, vomiting and liver toxicity [41,45,50,89].
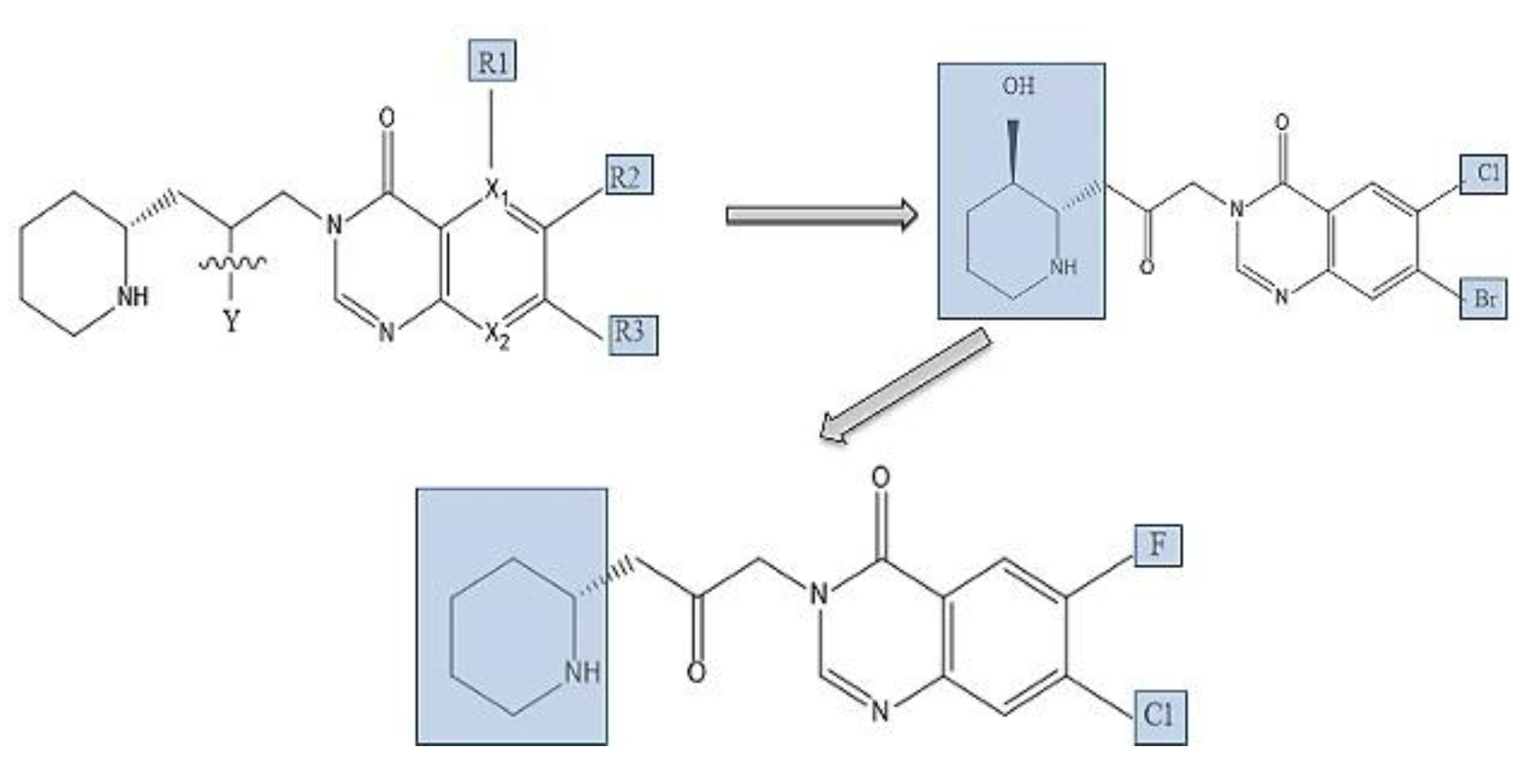
Quinazoline ring SAR study shows, Br substitution conserved the antimalarial property and depressed cytotoxicity to the host cells by replacement of vulnerable protons with a Br and Cl atom and for improving Febr.’s activity, HaF was prepared ultimately [33]. In vitro, HaF-Br salt was effective against Cryptosporidium parvum with toxicity of racemic mixture and limits the effective dose range therefore trans-enantiomers (2R,3S) -(+) more efficacious, in-vivo toxicity was more in mice than its optical antipode ((2S,3R) and (2S,3R) (−) of lactate salt, with broad therapeutic window [72,90,91]. Based on the protein constituents and analogues cellular actions, smaller halogen groups like Br and Cl at R2 and R3 positions ([Figure 5) of 4-Quinazolinone group has positive impact on potency (64 fold increase that is IC50; 0.18 μM, Kd; 30.3 nM), because in Hs-Prolyl-RS enzymes hydrophobic region for halogen atoms attachment in Sa-Prolyl-RS was minor and hydrogen or ‘F’ substitution reduce activity, elimination of the ‘OH’ of HaF enhanced antibacterial activity where single or dual substitutions with dioxole groups, −CH3, −OCH3, −CF3, −C(CH3)3, and benzene ring showed reduction in efficacy and replacement of benzene ring with a ‘N’ in heterocyclic ring; compounds became inactive [92,93]. Compound with Fl and Cl substitution has strong in vitro activity against bacterial ProRS with minimum inhibitory amount, 1−4μg/mL and better membrane permeability, with more hydrophobicity than that of HaF [94].
Figure 5.
Structures of Halofuginone Analog Br and Cl at R2 and R3 regions of the 4-Quinazolinone group increased activity, H or Fl substitution at same positions reduced the activity, elimination of the ‘OH’ of HAF improved antibacterial efficacy where other substitutions(single or dual) with ‘dioxole groups, −CH3, −OCH3, −CF3, −C(CH3)3’, benzene, showed reduced efficacies, Replacement of benzene ring with a ‘N’ in heterocyclic ring compound become inactive, Compound with Fl and Cl substitution has strong activity against bacterial ProRS and better membrane permeability, with more hydrophobicity than that of H [92,93,94,95,96,97].
Figure 5.
Structures of Halofuginone Analog Br and Cl at R2 and R3 regions of the 4-Quinazolinone group increased activity, H or Fl substitution at same positions reduced the activity, elimination of the ‘OH’ of HAF improved antibacterial efficacy where other substitutions(single or dual) with ‘dioxole groups, −CH3, −OCH3, −CF3, −C(CH3)3’, benzene, showed reduced efficacies, Replacement of benzene ring with a ‘N’ in heterocyclic ring compound become inactive, Compound with Fl and Cl substitution has strong activity against bacterial ProRS and better membrane permeability, with more hydrophobicity than that of H [92,93,94,95,96,97].
Salt forms of HaF has benefits and toxicity as well, includes HaF-HBr (C16H18BrClN3O3Br) and lactate salt for coccidiosis infection in sheep, calves, hen and turkeys [98,99,100,101]. Halogen salt with high electronegativity due to less lipophilicity more polarity, and electrostatic hydrophilicity, compounds have lesser drug distribution toward BBB because of difficulty to cross lipid-bilayer membranes. Order of decreasing electronegativity from F, Cl, Br and least of I, have impact on PK and PD aspects of HaF. Drug should have amphiphilic property to dissolve, distribute and produce pharmacological action. Interaction with amino acid of various halogens vary such as ‘Cl’ for leucine, ‘F’ for glycine, ‘Br’ for arginine and ‘I’ for lysine and for cysteine, Cl, Br and I had the lowest for proline, ‘Fl’ had a lowest and equal tendencies for the those including ‘N’ and ‘O’ atoms, as for all the halogens highest tendency shifted towards the hydrophobic residues, when side chain interactions were measured with different geometries [100,101]. Since halogens are unique elements that have a significant impact on the structure and properties are safer for more rational drug design, it is crucial to have a thorough understanding of the non-covalent molecular interactions with pharmaceutical compounds, amino acids, and nucleic acids. These interactions involve short-range electrostatic forces due to their polar nature, which is involved in charge movements, hydrogen bonding with donors along with halogenation with proteins or nucleic acids, and carbonyl groups, are all important parameters in the structural toxicity relationships and meaningful surface area ratios assessment [102,103,104].
By fluorinating 3-(3-(piperidin1-yl)-propyl)-indole and piperazinyl derivatives, increase in the metabolic stability occurred by improving receptors binding, ionic channel, or total target protein sites, and also effect on lipophilicity. However, this process can occasionally have unintended effects, such as toxicity. Other effects include increasing oral absorption, blocking compound oxidation, increasing bio-availability, and increasing binding for the 5-HT-2A receptor by fluorinating 3-(4-piperidin-3-yl)-2-phenyl-indoles at the 6-C on indole [105,106]. If the substance binds to off- target with increased affinity it will result into toxicity such as ion channel inhibitors, anti-cholesterol, antibiotics and anti-cancerous drugs [107,108]. Halogens enhance desirable effects including stability, shelf life, intestinal permeation and BBB permeability, as a vital part of pharmacophore of the newer neuroleptic drugs. Among them ‘F’ is the most frequently used, depending upon interactive compound and drugs used to treat RTIs contain ‘Br’, ‘chlorine’ in antibiotics such as chlorinated form of vancomycin or cefaclor and ‘I’ used to treat hormonal imbalance, Canc. subtypes, in imaging and structural studies, beneficial effect on affinity for oestrogen receptors by halogenation of oestradiol analogues in sequence of; Br >Cl > I, utilized for SPECT imaging in breast Canc. diagnosis and compounds, and 3-fluorotyrosyl green used as probes, check intracellular calcium concentration alterations. [109,110,111]. Halogenation is the process by which peptides lose some of their conformational stability. For instance, when a helical coiled peptide's hydrophobic surface contains leucine instead of trifluoroleucine or hexafluoroleucine, the compounds' thermal and structural stability is significantly increased [112,113].
The broad-spectrum activity of HaF has been reported, includes anti-fibrosis, anti-tumor and anti-coccidia(3mg/kg in animal feed), leishmaniasis, and toxoplasmosis as well as to inhibit the growth and proliferation of Canc. cells in vitro, reduce tumor metastasis, angiogenesis in rats, and Hep.ca in mice. and growth-promoting activities [12,13,14,15,16,17,18,19,20,114,115,116,117,118,119,120]. Primary source agents of active pharmacological potential are plant origin with less toxicity and higher efficacy, playing significant role in Canc., to avoid MDR and side effect of current chemotherapeutics. HaF, is a novel anticancer drug with multiple molecular mechanism of actions and target broad range of diseases [121].
2.3. Clinical Significance of Halofuginone- Novel tRNA Synthesis Inhibitor:
2.3.1. Antimalarial Therapy:
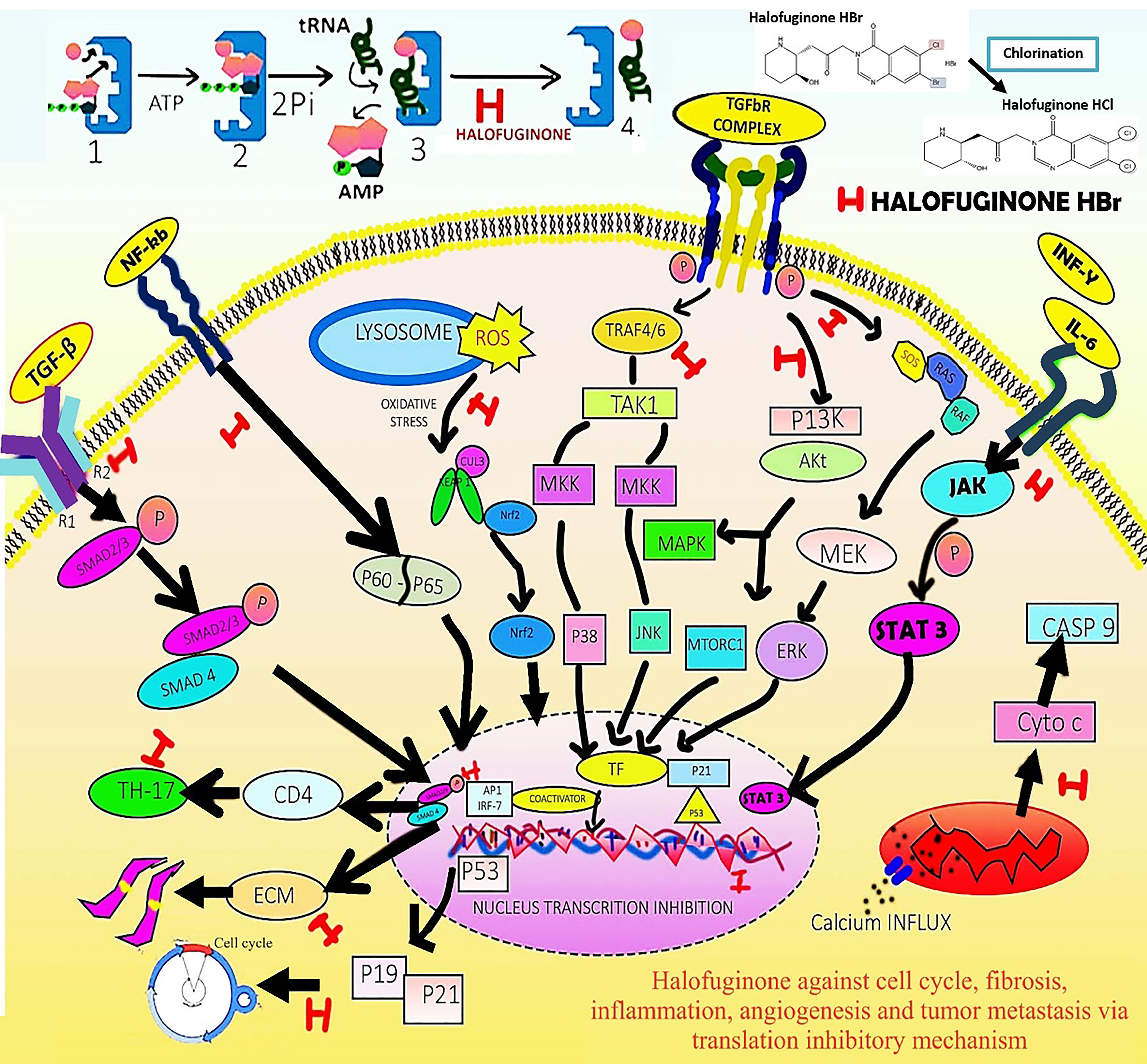
The alarmingly high rates of malaria-related death and morbidity in a number of developing nations, such as South America, Africa, and Asia, along with the rise of drug-resistant parasite strains highlight the need for innovative treatments that target parasite’s life-cycle. Febr. and the equivalents HaF directly secures onto two components of human Prolyl-RS, one of which mimics the bound amino acid (proline) and 3'-terminal region of tRNA, inhibiting prolyl-transfer-RNA-synthetase (Prolyl-RS) non-competitively by utilizing energy in form of Adenosine-TP. This causes an intracellular build-up of uncharged tRNA and reduces the availability of proline-containing proteins in the blood stage of the cell, as observed in P. falciparum, the asexual blood stage, and was resident in the parasite's cytoplasm [93,122]. The stage of the Plasmodium 's life cycle with asymptomatic liver, HaF targets the sporozoite in Hepatic-G-2 cells in infected mice at 17nM concentration and highly effective in in-vitro Plasmodium species (falciparum, berghei) growth retardation with equivalent speed on all three stages [123,124].
To target eukaryotic Prolyl-RS and other Amino-ARS members with greater specificity, several HaF analogues were created [125, 126]. Targeting prolyl-tRNA synthetase (Pro-RS), the AARS member responsible for linking proline to tRNA-Pro [125,126in the eukaryotes within the cytoplasm and in the presence of ATP or its analogue adenylate cyclase AMP, its analogues and cocrystals have also been clinically evaluated for managing adult solid tumor, recurrent Kaposi sarcoma, fibrotic diseases, and SARS-CoV-2 inhibitor of the invasion, infection, and replication. This mechanism is exclusive to all Amino-ARS inhibitors known to date [127,128,129].
2.3.2. Antiprotozoal:
Orally administered HaF lactate, used as antiprotozoal for prevention of cryptosporidiosis in poultry flocks, cattle, calves and lambs, effective against Eimeria species (mitis) used in poultry feed for coccidiosis, treat lesions of different severity levels in gut, weight loss, diarrhoea or faecal oocyst counts, and decrease mortality rate. In young broiler chickens, by blocking parasite's entry into cecum of animal in the early stages, hinders parasite growth by disruption of schizonts and thereby hindering the division of the resulting unusual schizonts into schizoites, which are then eventually attacked by immune response [59, 130-132]. It also significantly increased body weight, reduced oocyst shedding, and prevented [133]second generation schizonts and E. tenella infections from destroying cecum by preventing the parasites from invading the hypothetical host cecal cell. HaF inhibits prolyl-tRNA synthetase in accordance wit[59,130–132h the codes of genetics and inhibits the production of proline-containing proteins and also catalyzes the connection of the relevant amino acid to the homologous tRNA molecule engaged in the aminoacylation reaction [78,124,134].
2.3.3. Antifibrotic Agent:
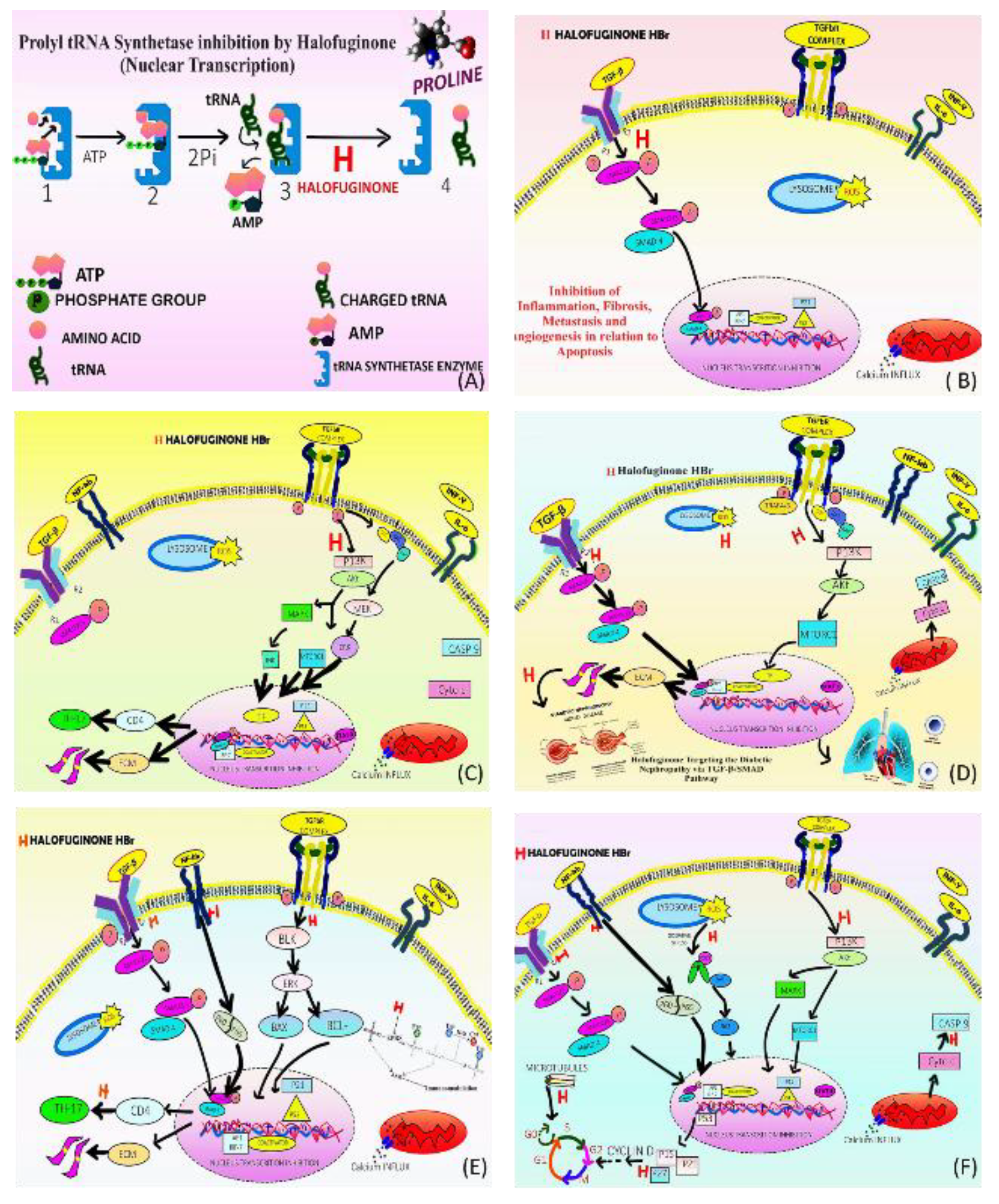
Chronic inflammation result in tissue fibrosis induced due to high concentration of transcription-growth factor-beta, Matrix-MPs, and metallo-proteinases which inhibit tissues, which regulate Extra cellular matrix (Extra-CM) turnover, destroy organ structure, function and death occur due to high levels of Extra-CM proteins, myofibroblasts, primary Extra-CM -producing cells and collagen type I. HaF, by inhibiting ProRS enzyme reduce collagen α1-gene appearance, inhibit TGFβ-induced collagen formation by blocking Smad3 phosphorylation, and reduce levels of α-smooth muscle positive cells [33,135,136,137,138]. Effective in Duchenne Muscular Dystrophy (Duchene-MD), pulmonary fibrosis, adhesions at various sites, hepatic and pancreatic fibrosis and chronic graft-versus-host patients, tight skin and effective in humans skin fibroblasts due to scleroderma [139,140,141,142,143,144,145,146].
HaF downstream Transcription-GFβ-Smad3 protein pathway in in vitro animal models by inhibiting prolyl-tRNA synthetase, activate Amino-AR response, inhibit T-helper-17 differentiation and concentration dependent Transcription-GFβ mediated Smad 2/3- Smad 4 complex phosphorylation and due to activation of Akt-MAP-kinase/ER-kinase and p38- MAP-Kinase phosphorylation pathway, thereby raised expression of the inhibitory Smad7 in fibroblasts, tumor cells, myoblasts, hepatic and pancreatic stellate cells, inhibit inflammatory reactions by blocking transition of fibroblasts to myofibrob lasts [33,64,135,136,138,145,147,148,149,150]. Additionally, the loss of normal living cell population (CD4+ T cells) impairs organ functions, inhibits pathogenesis, prevents tumor development in stomach carcinoma in relation to T-helper-17 cell division, and prevents con-canavali-A related fibrosis of hepatic cells by directly impacting T-helper-17 cell proliferation and destroying the tissue microenvironment [149,151,152,153,154,155,156].
2.3.4. Halofuginone Targeting metabolic Disorders:
HaF also found to be effective in various metabolic disorders due to antifibrosis activity. HaF diminishes hypoxia-induced pulmonary vascular remodelling, and pulmonary vasoconstriction, and also reverses pulmonary hypertension or contraction in pulmonary artery hypertensive patients. Also, it is a potent vasodilator of smooth muscle in pulmonary artery by triggering potassium channels, increases ions currents and by inhibiting the PI3-Kinase/AKT/ mamalian TOR signalling pathway, produces anti proliferative effect and by blocking voltage-gated Ca2+ channels through inhibition of E-C coupling decreases Ca2+ influx [157,158,159]. HaF by blocking transcription-GF-beta, prevents the development of diabetic nephropathy by reducing the accumulation of extracellular matrix proteins and the production of type I collagen, which is involved in fibrosis. The primary mechanism involves by blocking the expression of Transcription-GF-b type-2-receptor, which in turn inhibits the overexpression of type I collagen and fibronectin and suppresses mesangial growth in mice in the kidneys. An analysis of urine 8-OHdG levels along with dihydroethidium fluorescence supported the finding that there was a reduction in oxidative stress in the glomerulus as hyperglycemia weakens antioxidative mechanisms by releasing excessive ROS, such as hydrogen peroxide and superoxide, by glycating scavenging enzymes like superoxide dismutase (SOD). By targeting Transcription-GF-b1/SMAD 2 pathway also suppresses the development of diabetic nephropathy by halting Extra-CM deposition and decreasing oxidative stress, as diabetic nephropathy is affecting about one-third of all diabetic patients leading to end-stage renal failure, this drug could be a wonder [160].
2.3.5. Antiinflammation and Immunomodulation:
InL -17 stimulates release of other cytokines, interleukins (InL- 17A,7F,21,22,23) and has pro-inflammatory functions, involve in formation of new blood cell, thus it directly impacts on epithelium cells, endothelial, and fibroblasts production in addition to releasing chemokines and prostaglandins, also overexpressed in rheumatoid arthritis, and SLE- individuals [161]. HaF inhibits inflammatory pathogenesis and contributes to control of autoimmune and inflammatory diseases including cancer (metstasis, angiogenesis, apoptosis induction, invasion) by directly inhibiting the Transcription-GFβ-SMAD pathway and a malfunctioning CD4-T-helper-cells associated with secretion of InL-17 [155,162,163,164,165]. Also, by targeting BLK in myeloid derived suppressed cells, inhibit pathogenesis in SLE by inhibiting the nuclear translocation of p65/p50 heterodimer resulting in increasing the mRNA expression of Blk, induced suppression of the downstream E-RK signalling pathway in order to increase the Apo. of myeloid derived suppressed cells, alleviated the disease progression, indicating potent immunotherapeutic potential by targeting prolyl-transfer-RNA synthetase and amino acid starvation response [166,167,168,169]. The anticancer effects are attributed to their inhibition of STMN-1 and p53, as well as their impact on migration, mutation, metastasis, invasion, MDR, and autophagy activation, all of these promotes early-phase cell death [170,171,172,173].
Figure 6.
Halofuginone Targeting numerous pathways By inhibiting Prolyl tRNA Synthetase enzyme to prevent collagen synthesis in fibrosis, inflammation and to treat various diseases (A) Inhibiting Prolyl tRNA Synthetase; (B) As antifibrotic by inhibiting transcription growth factor beta (TGFβ)-induced collagen synthesis-TGFβ-SMAD3 pathway, reduce collagen α1(I) gene expression, blocking small mother against decapentaplegic -3 (Smad3) protein phosphorylation and conversion to excitatory Smad 2/3, and co Smad4, subsequently reduce levels of α-smooth muscle positive cells hence inhibiting transcription in nucleus and translation process in cytoplasm to collagen protein formation which is involve in fibrosis; (C) By targeting P13-AKT-MTOR pathways, TGFβ- Smad3 protein pathway in in vitro animal models, activate AAR response, inhibit T-helper-17 differentiation and concentration dependent TGFβ mediated Smad 2/3- Smad 4 complex phosphorylation and due to initiation of Akt-MAP-Kinase/ER-Kinase and p38- MAP-Kinase phosphorylation pathway where Akt is protein kinase-B, MAP-Kinase is mitogen activated protein kinase and ER-Kinase extracellular signal regulation; and raised amount of the inhibitory Smad7 in fibroblasts, tumor cells, myoblasts, hepatic and pancreatic stellate cells, block inflammatory and autoimmune harmful reactions by blocking transition (fibroblasts to myofibroblasts); (D) By blocking TGFβ-SMAD3 AND Extra-CM Pathway to inhibit Diabetic Nephropathy progression and pulmonary hypertension, reduce collagen α1(I) gene expression, inhibit TGFβ-induced collagen synthesis by blocking Smad3 hence inhibiting transcription and translation to collagen protein formation which is involve in diabetic nephropathy. As a potent vasodilator effective for pulmonary hypertension treatment of smooth muscle cells in pulmonary artery by activation of K+ channels, increases voltage gated K+-ions currents and by inhibiting the PI3K/AKT/mTOR signalling pathway, produces anti proliferative effect and by blocking voltage-gated Ca2+ channels through inhibition of E-C coupling decreases Ca2+ influx. Where Mammalian-TORC1 is mammalian target of rapamycin complex-1; (E) By inhibiting TGFβ-SMAD and T-helper-17 (TH-17) pathways to combat inflammation during autoimmune diseases and for immunomodulation reduce collagen α1(I) gene expression, inhibit TGFβ-induced collagen synthesis, reduce levels of α-smooth muscle positive cells hence inhibiting transcription and translation to collagen protein formation. By directly inhibiting a dysregulated distinct subset of cluster of differentiation (T helper cell-CD4), T helper-17 cells with cytokine IL-17, result in inhibition of inflammatory pathogenesis, and contribute to anti-inflammatory and autoimmune disease control and by inhibiting NF-kappa-b and p38-MAP-kinase pathway (in activated T-cells) [174]. Also, by targeting BLK in myeloid derived suppressed cells, inhibit pathogenesis in SLE by inhibiting the nuclear translocation of p65/p50 heterodimer resulting in increasing the mRNA expression of gene B-lymphocyte kinase (Blk), by enhancing kinase activity of Blk via direct molecular binding and also Blk induced suppression of the downstream ERK signalling pathway in order to increase the Apo.; (F) In cancer To inhibit cell-cycle progression and induce apoptosis, .target at, TGFβ-SMAD3, P13-AKT-MTOR1, P13-AKT-MAPK, CASP-9, Cell Cycle-CYCLIN D, Microtubule assembly, NFK-B-P60/P65 and Lysosome ROS-NRF-2 Pathways. by inhibiting Prolyl transfer RNA synthetase enzyme reduce collagen protein formation. By inhibiting T-helper-17, Interleukin-17, result in inhibition of inflammatory pathogenesis, Arrest cell cycle in various cancers including colonic cancer cells (G1/G0 phase) by increasing p21 and p27, lung cancer by upregulating p21, p27, Caspase-3, PARP, ROS, Bax/Bcl2 and Cyclin D1 downregulation, malignant mesothelioma by Cyclin D1 downregulation, hepatocellular carcinoma by upregulating of p15, p21, Caspase-3,9,8, PARP, and downregulating Mcl-1, cIAP1, breast cancer by targeting microRNA-31, ROS increase, by downregulating TGF-β, G2 and M phase seizure and destruction microtubules, acute promyelocytic leukaemia by upregulating p15, p21, and decreasing MYC, S phase arrest, cell death, colonic cancer by upregulating Caspase (3,9,8). Inducing Apo. in different cancers by targeting various pathways including erythroid related factor of nuclear factor-2 NRF2, and Phosphatidylinositol 3 -kinase (PI3-Kinase)/Akt signalling pathway [175].
Figure 6.
Halofuginone Targeting numerous pathways By inhibiting Prolyl tRNA Synthetase enzyme to prevent collagen synthesis in fibrosis, inflammation and to treat various diseases (A) Inhibiting Prolyl tRNA Synthetase; (B) As antifibrotic by inhibiting transcription growth factor beta (TGFβ)-induced collagen synthesis-TGFβ-SMAD3 pathway, reduce collagen α1(I) gene expression, blocking small mother against decapentaplegic -3 (Smad3) protein phosphorylation and conversion to excitatory Smad 2/3, and co Smad4, subsequently reduce levels of α-smooth muscle positive cells hence inhibiting transcription in nucleus and translation process in cytoplasm to collagen protein formation which is involve in fibrosis; (C) By targeting P13-AKT-MTOR pathways, TGFβ- Smad3 protein pathway in in vitro animal models, activate AAR response, inhibit T-helper-17 differentiation and concentration dependent TGFβ mediated Smad 2/3- Smad 4 complex phosphorylation and due to initiation of Akt-MAP-Kinase/ER-Kinase and p38- MAP-Kinase phosphorylation pathway where Akt is protein kinase-B, MAP-Kinase is mitogen activated protein kinase and ER-Kinase extracellular signal regulation; and raised amount of the inhibitory Smad7 in fibroblasts, tumor cells, myoblasts, hepatic and pancreatic stellate cells, block inflammatory and autoimmune harmful reactions by blocking transition (fibroblasts to myofibroblasts); (D) By blocking TGFβ-SMAD3 AND Extra-CM Pathway to inhibit Diabetic Nephropathy progression and pulmonary hypertension, reduce collagen α1(I) gene expression, inhibit TGFβ-induced collagen synthesis by blocking Smad3 hence inhibiting transcription and translation to collagen protein formation which is involve in diabetic nephropathy. As a potent vasodilator effective for pulmonary hypertension treatment of smooth muscle cells in pulmonary artery by activation of K+ channels, increases voltage gated K+-ions currents and by inhibiting the PI3K/AKT/mTOR signalling pathway, produces anti proliferative effect and by blocking voltage-gated Ca2+ channels through inhibition of E-C coupling decreases Ca2+ influx. Where Mammalian-TORC1 is mammalian target of rapamycin complex-1; (E) By inhibiting TGFβ-SMAD and T-helper-17 (TH-17) pathways to combat inflammation during autoimmune diseases and for immunomodulation reduce collagen α1(I) gene expression, inhibit TGFβ-induced collagen synthesis, reduce levels of α-smooth muscle positive cells hence inhibiting transcription and translation to collagen protein formation. By directly inhibiting a dysregulated distinct subset of cluster of differentiation (T helper cell-CD4), T helper-17 cells with cytokine IL-17, result in inhibition of inflammatory pathogenesis, and contribute to anti-inflammatory and autoimmune disease control and by inhibiting NF-kappa-b and p38-MAP-kinase pathway (in activated T-cells) [174]. Also, by targeting BLK in myeloid derived suppressed cells, inhibit pathogenesis in SLE by inhibiting the nuclear translocation of p65/p50 heterodimer resulting in increasing the mRNA expression of gene B-lymphocyte kinase (Blk), by enhancing kinase activity of Blk via direct molecular binding and also Blk induced suppression of the downstream ERK signalling pathway in order to increase the Apo.; (F) In cancer To inhibit cell-cycle progression and induce apoptosis, .target at, TGFβ-SMAD3, P13-AKT-MTOR1, P13-AKT-MAPK, CASP-9, Cell Cycle-CYCLIN D, Microtubule assembly, NFK-B-P60/P65 and Lysosome ROS-NRF-2 Pathways. by inhibiting Prolyl transfer RNA synthetase enzyme reduce collagen protein formation. By inhibiting T-helper-17, Interleukin-17, result in inhibition of inflammatory pathogenesis, Arrest cell cycle in various cancers including colonic cancer cells (G1/G0 phase) by increasing p21 and p27, lung cancer by upregulating p21, p27, Caspase-3, PARP, ROS, Bax/Bcl2 and Cyclin D1 downregulation, malignant mesothelioma by Cyclin D1 downregulation, hepatocellular carcinoma by upregulating of p15, p21, Caspase-3,9,8, PARP, and downregulating Mcl-1, cIAP1, breast cancer by targeting microRNA-31, ROS increase, by downregulating TGF-β, G2 and M phase seizure and destruction microtubules, acute promyelocytic leukaemia by upregulating p15, p21, and decreasing MYC, S phase arrest, cell death, colonic cancer by upregulating Caspase (3,9,8). Inducing Apo. in different cancers by targeting various pathways including erythroid related factor of nuclear factor-2 NRF2, and Phosphatidylinositol 3 -kinase (PI3-Kinase)/Akt signalling pathway [175].
2.4. Halofuginone Targeting Various Cancer Subtypes:
2.4.1. Gastric Cancer and Colorectal Cancer:
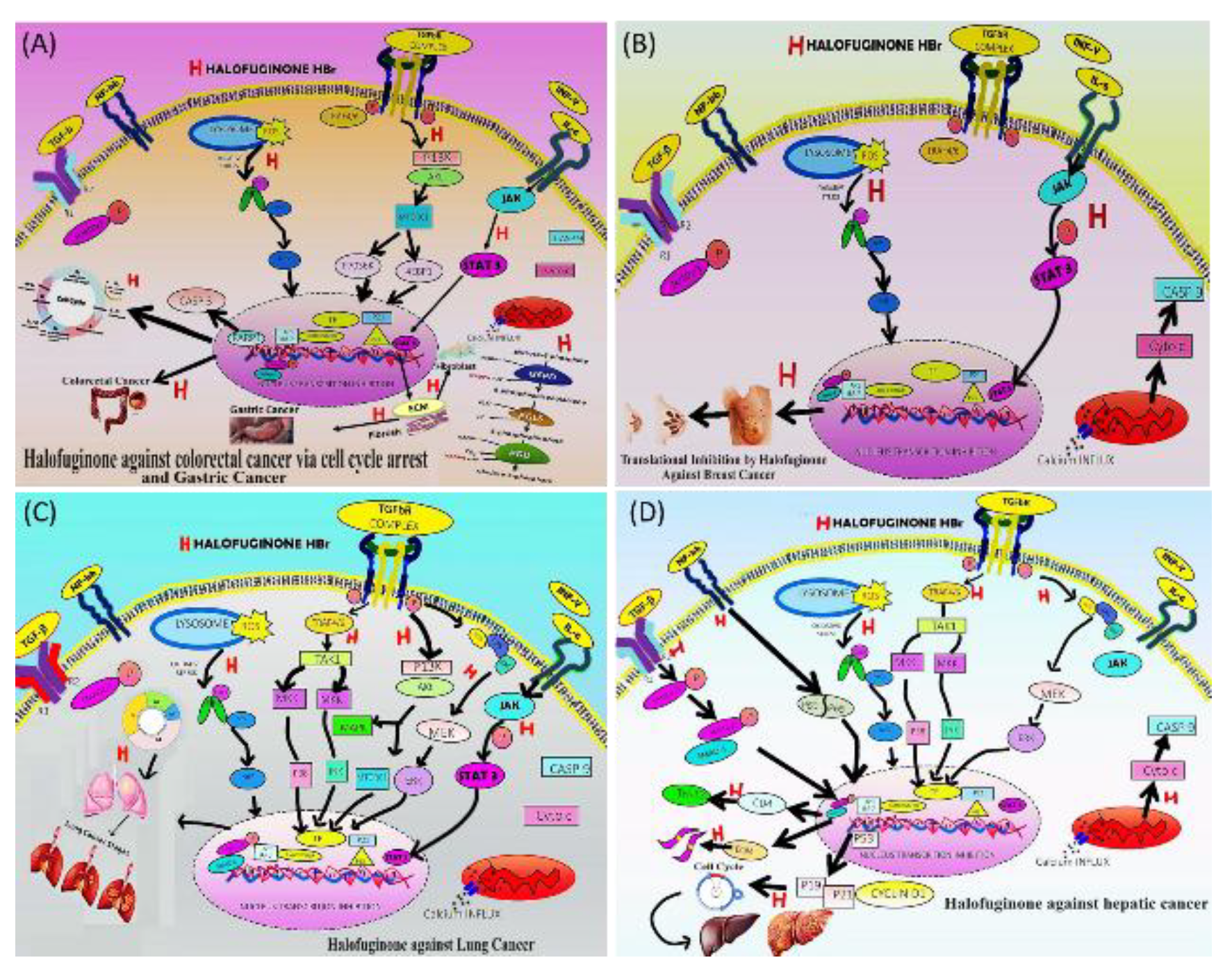
According to the GLOBOCAN statistics report in 2020, 5th most common cancer(Canc.) globally is gastric (GC), and the 4th major cause of Canc. associated mortality with more than 1M morbidity rate. Annually >1.2 M colorectal malignancy cases are also reported, involving majorly lower GIT, with more than 600K deaths rate globally and 8% mortality rate in United States, 2nd leading cause of death, with main mode of action is induced by preliminary genetic alteration, environmental factors, and lifestyles habits and A-kt-serine/threonine kinase B pathway overexpression involve in Canc. cell proliferation, glucose metabolism, transcription and cell metastasis [176,177,178].
Because of chemoresistance, the current available chemotherapy treatments are ineffective. GC sometime secondary to H. Pylori infection and associated with gastric-perforations and involving the stroma of gastric tumor Canc.-associated fibroblasts causing a huge risk, and also poor prognosis. There are limited available therapies, with main molecular target includes Signal-TAT-3 (involve in changeover of signals from receptors for several growth factors), VEG-Factor and membrane receptors of the cytokines- InL-6 family. In addition to targeting the Janus kinase/STAT route and the Extra-CM elements secreted and transformed by CAFs in stomach Canc. HaF inhibits the phosphorylation of Signal-TAT3 and the ensuing pathway, which in turn inhibits the cytotoxicity of tumor cells, results in decreased size of the tumor by preventing connections between Canc. cells and the stromal space, fibrosis and microscopic vessel density in human tumor xenografts in mice, and blockade of VEG-Factor signalling, which in turn inhibits tumor metastasis[179,180,181].
In colorectal Canc. PI3-Kinase/A-kt/m-TOR signalling activated through phosphorylation, signalling downstream factors, follows by regulation of cellular process in Canc. cells and regulate mammalian-target of rapamycin complex-1 through phosphorylation at Ser-2448, hence this pathway along with high-grade intraepithelial neoplasia overexpressed in Canc. cell of colon majorly involving glands (carcinoma) [182,183,184]. Also colon mutation involves glucose breakdown to pentose-phosphate (pathways for NADPH) in malignant cells was regulated by p70-S6K synthesis of lipid/sterol involve in glucose metabolism, producing redox state and promotes Canc. proliferation, and via reactive oxygen compounds (ROS), the pentose-phosphate-pathway enables Canc. cells escape death. Another well-studied m-TORC-1 downstream factors include 4EBP1, which deactivates the eukaryotic translation initiation factor 4E(eIF4E) [185,186]. HaF has shown the anticancer activity both outside body and in vivo in colorectal Canc. by the suppression of A-kt/ m-TOR-C-1 signalling and glucose metabolism along with lipid profiles by constraining G-6P-D, suppressing glucose-transporter GLU-T-1 and hexo-kinase-2 in glycolysis, glucose-derived Tri-C-Acid cycle flux in Canc. cells hence, inhibited cell growth, release ROS, mitochondrial dysfunction, reduced CRC cell viability and induced Apo.[187]. Arresting cell cycle, in the G1/G0 phase of the Colorectal Canc. cells, causing a reduction in Nictoniamide-AD-PH as a result of the deactivation of glucose-6P-dehydrogenase. Causing Canc. cell death by increasing the amount of Cas-3 and separated P-ARP due to the generation of ROS [188,189,190,191,192].
2.4.2. Breast Cancer:
Breast Canc. (B.Canc.) one of the commonest sybtype worldwide in 2020, total of 2.3M new cases emergence, in both sexes accounts for 1 in every 8, representing a quarter of all cancerous cases in females. Principally in transitioning countries with an estimated 68.5K women mortality rate in 2020 corresponding to 16% Canc. deaths because of insufficient public health awareness. But improvement appeared due to presentation of the Global Breast Canc. Initiative (by WHO), early diagnosis and adequate therapeutic guidelines, reducing the disease global burden [6,193,194]. B.Canc. on the basis of results from immuno-histo-chemistry and in-situ-hybridization testing categorized as human epidermal-growth factor-receptor 2 positive (HER2) or HER2 negative as per the 2018 American Society of Clinical Oncology and National Comprehensive Canc. Network scoring guidelines, and 80% are categorized as HER2-negative, nearly 60% fall into HER2-negative category [195,196,197,198].
One of the two major complex transcription regulators is signal-transducer/activator of transcription-3, (which includes Signal-TAT-1,2,3,4,5a, 5b, and 6), mediates the transfer of signals from the membrane to the nucleus and is a necessary gene transcription component. Other one transcription regulator is erythroid derived-Nuclear factor-2, contribute in a diversity of physiological processes and overexpressed in various Canc. subtype and help in tumor progression [138,194,199,200]. In Canc., overactive Signal-TAT-3 promotes cell growth while inhibiting cell death, persistent signalling increases tumorigenesis caused by increasing apoptotic genes inhibition, majorly Bcl-2, Mcl-1, and cyclins cell cycle regulatory including D1 or D2, and downregulating of endogenous of negative regulators [199,200,201,202]. Excessive activation of Nuclear-F-2 in malignant cells facilitates survival by activating a non-canonical pathway and interacts with Signal-TAT-3 signalling pathways, and influences Canc. progression [175,203,204].According to another study, Signal-TAT-3 boosted Nuclear-F-2 expression as a result of interleukin (InL) -6 secreted by stellate cells in pancreas, which caused Extracellular-MT traits and cause metastasis [205]. Signal-TAT-3 and Nuclear-F2 complex form, binds to the InL -23A gene promoter region, produces protein, which bind to InL-23A receptors in BL- B.Canc. - cells, signals for cell division [205].
HaF suppress Nuclear-F-2 and Signal-TAT3 viable B.Canc. cells, impede the progression. As both Nuclear-F-2 and Signal-TAT-3 higher level manifested in B.Canc., fast development of Canc. occur because of InL-23-A release, a common proinflammatory cytokine expression induction, because Nuclear-2 form a complex with Signal-TAT-3 (Y705 phosphorylated dimeric type) with worse survival rate of patients resulting in tumor growth and metastasis including B.Canc.[205,206].
2.4.3. Lung Cancer:
Almost highest morbidity and mortality rate are associated with lung-derived Canc., and difficult to treat, in both sexes worldwide, sub categorized as non-small-lung-Canc., meso-thelioma arises from the malignant transformation in pleural cells, pericardial cells, or tunica-vaginalis [207,208]. Main molecular mode of action is Mitogen activation of Epithelial-GF-R which by phosphorylation, activate several cancerous signalling pathways including Signal-TAT-3, PI-3-Kinase/Akt/m-TOR-1 and RAS/RAF/ME-Kinase/ER-Kinase1/2 involved in disrupted cell growth and survival. JN-Kinase and p38-MAP-Kinase pathways activate in response to chemical stresses, induce immune response, and cell death [209].
HaF inhibit lung Canc. by inhibiting various pathways including Transcription-GF-β signalling pathways, ER-Kinase-1/2, JN-Kinase and p-38-MAP-Kinase pathways, PI3-Kinase/AKT, NF-κB signal pathway and Signal-TAT-3, it has suppressed cancerous growth, metastasis, cell proliferation and induce apoptosis (Apo.) [148,210,211,212,213]. Stimulates apoptotic cascade in lung-derived Canc. cells by increasing the Bax : Bcl-2 ratio, activating cas-3 with concomitant Poly-AR-P cleavage, and blocking the activation of Signal-TAT-3, a downstream effector of Epithelial-GF-R and ER-Kinase1/2 and pathways. This is done via p-38/MAP-Kinase pathway [214]. HaF produces a dose-related decrease in the lifespan of cells, produce selective G1/S cell cycle arrest in cancerous versus non-malignant cell types [215].
2.4.4. Hepatocarcinoma:
One of the most commonly diagnosed Canc. worldwide is liver Canc. maybe secondary to chronic liver disease (CLDs) with estimated 830K morbidity rate in the year 2020 as per IARC GLOBOCAN Canc. statistics and ranks at the second of all malignancy-related mortality of liver Canc., in China this is the most common type of carcinoma, accounting for approximately 90% of the total, induces persisting fibrotic tissues formation/ liver destruction, result in cirrhosis development ultimately damaging the normal liver structure, recruits inflammatory cells after tissue injury and activation of hepatic-stellate cells [216,217,218]. Liver is responsible for all kinds of significant metabolic functions; thus, liver Canc. certainly causes greater troubles, and its treatment is extremely problematic as it has rich blood supply and close association with other digestive organs. For patients with advanced inoperable liver Canc., no current treatment as such available therefore researchers are working on plant extracts to find novel treatment, among them one is HaF with significant anti-tumor effect [32,219].
One of the studies reported that, therapeutic promising drug HaF has beneficial effects in fibrotic disease, upon oral administration at dose of 10ppm, by inducing suppressive effect on liver fibrosis by decreasing the levels of α-SM-A, tissue inhibitors of metallo-proteinase-2 and Smad-3, thereby reducing the severity of liver fibrosis, immunomodulatory effects induction and production of nuclear- factor-kB. Levels of chemical mediators altered mainly, inflammatory cytokines such as TNF-α, InL -6 and InL-1β in hyaluronic acid, procollagen III and Transcription-GF-β1 in the serum and collagen-I in the liver tissue, as Con-A injected into the tail vein resulting in in the serum AS-transferase and alanine amino-transferase levels increased [220,221,222]. Another study reported that HaF could inhibit growth of hepatocellular carcinoma Hep-3B cell line and Hep-G2 cell line both in vitro and in vivo with the subsequent molecular mechanism of liver Canc. suppression was; by arresting the cell cycle ( G0-G1 and S phase), by up-regulating the amount of p15 and p21 which are negative cell cycle regulatory proteins, by selective inhibition of CDK4/6 activity by p15 and while p21 is a cell cycle kinase essential for G1 and S phase. Hence HaF inhibit CDK2, CDK1 and CDK4/6 activity, p21 and tumor cell progression and induced Hep-G2 Apo.by activating caspase pathway proteins including Poly-AR-P, caspase-3, 8 and 9 level are increased, which often produce cleaved active products through the action of upstream proteases and down-regulate anti Apo. protein Mcl-1 and c-IAP1 expression in hepatocellular carcinoma cells [148,223,224,225,226,227,228]. HaF also inhibit ME-Kinase/ER-Kinase and upregulate Janus-Kinase pathways by increasing phosphorylation level in hepatocellular carcinoma cells in liver Canc. subsequently inducing tumor suppression [229,230].
Figure 7.
Halofuginone targeting multiple cancers; (A) Colorectal cancer: Inhibiting P13-AKT-MTOR, JAK-STAT, CASP-3 and Lysosome ROS-NRF-2 Pathways, inhibiting inflammation, cell cycle progression in in the G1/G0 phase, also inhibit glucose metabolism and suppress lipid profiles. Anti-cancer activity both in vitro and in vivo by suppression of Akt/ mTORC1 signalling pathway and also inhibit glucose metabolism by inhibiting G6PD, suppressing glucose transporter G-LUT-1 and hexokinase-2 in glycolysis, reduced glucose-derived T.C.A-cycle-flux mediated by Ak-t/m-TOR-C-1 signalling pathway in cancer cells hence, inhibited cell division, releases R.O.S., mitochondrial dysfunction, reduced CRC cell viability and induced apoptosis (Apo.) that is a programmed cell death with reduced level of NAD-PH due to glucose-6-PD inactivation in pentose-phosphate reaction and also in the G.1/G.0 phase of the CRC cells, a dose-dependent-arrest, inhibiting cell proliferation in cell cycle progression, and induces Apo.by increasing level of cleaved-Caspase-3 and cleaved-PARP due to ROS generation; Gastric cancer: Via targeting Janus kinase and signal transducer of activated transcription (JAK-STAT) and extracellular matrix protein Extra-CM Protein Pathways in inhibit transcription of genes involve in cancer cell cycle progression, reduce tumor volume and induce Apo.; (B) Breast cancer: Targeting JAK/STAT, CASP-9 and Lysosome ROS-NRF-2 Pathways, intracellular NRF2 signalling triggered because of Pancreatic-S-Cells release Interleukin-6 binds to its receptor and activation of Janus-K/Signal-TAT-3 signalling, resulting in nuclear-factor -2 (NRF-2) downstream EMT-related transcription factors inducing EMT in Panc-1 cells by inducing expression of E-MT related marker genes, to induce Apo., inhibit cell cycle progression by inhibiting growth differentiation, maturation and metastasis; (C) Lung cancer: Induces cell Apo., inhibit metastasis, fibrosis, growth, differentiation and inhibit inflammation cell cycle progression by targeting P13-AKT M-TOR, JAK-Signal-TAT, Cell Cycle, MEK-ER-Kinase, MK-Kinase-JN-Kinase-P-38 and Lysosome ROS-NR-F-2 Pathways, through targeting and inhibiting activation of STAT-3- a downstream effector cascade protein of endothelial growth factor receptor E-GF-R and erythroid nuclear factor ER-K1/2 and pathways and via protein 38 and mitogen-activated-protein kinase-38/MAP-Kinase induces the Apo. cascade through decreasing Bcl-2 and Bax up regulation, caspase-3 activation with Poly-ADP-RP cleavage and produces reduction in cell viability, cell retard and G1/S cell cycle-arrest cancerous cell types; (D) Liver cancer: Induces Apo. and suppress cell cycle growth differentiation by inhibiting TGFβ-SMAD3, T-helper-17, CASP-9, Extra-CM, Cell Cycle progression- G0/G1 and S phase, MEK-ERK, MKK-JNK-P38, NFK-B and Lysosome ROS-NRF-2 Pathways, as antifibrotic, by inhibiting Prolyl transfer RNA synthetase enzyme reduce collagen α1(I) gene expression, inhibit transcription growth factor beta (TGFβ)- Smad-3 protein phosphorylation and conversion to excitatory Smad 2/3, and co Smad4, subsequently collagen protein formation which is involve in fibrosis. By directly inhibiting a dysregulated distinct subset of T helper CD4 cells (T-helper-17) cells with Interleukin-17, result in inhibition of inflammatory pathogenesis, and result into autoimmune disease control by inhibiting nuclear factor-kappa-b (NF-Kb) and p38-mapk pathway in activated T-cells. Arrest cell cycle at the G-1/G-0 phase, by increasing p21 and p27, ROS, and Cyclin D1 downregulation, malignant mesothelioma by Cyclin D1 downregulation, hepatocellular carcinoma by upregulating of p15, p21, Caspase(3,9,8), and downregulating cIAP1, ROS increase, by downregulating TGF-β, phase cell cycle phases arrest at G2/M and microtubules disruption, by increasing p(15,21), and decreasing MYC, S-phase arrest, Apo., Inducing Apo. by targeting various pathways including nuclear factor -2 (NRF-2), and Phosphatidyl-inositol 3 -kinase (PI3-K)/Akt signalling pathway [231].
Figure 7.
Halofuginone targeting multiple cancers; (A) Colorectal cancer: Inhibiting P13-AKT-MTOR, JAK-STAT, CASP-3 and Lysosome ROS-NRF-2 Pathways, inhibiting inflammation, cell cycle progression in in the G1/G0 phase, also inhibit glucose metabolism and suppress lipid profiles. Anti-cancer activity both in vitro and in vivo by suppression of Akt/ mTORC1 signalling pathway and also inhibit glucose metabolism by inhibiting G6PD, suppressing glucose transporter G-LUT-1 and hexokinase-2 in glycolysis, reduced glucose-derived T.C.A-cycle-flux mediated by Ak-t/m-TOR-C-1 signalling pathway in cancer cells hence, inhibited cell division, releases R.O.S., mitochondrial dysfunction, reduced CRC cell viability and induced apoptosis (Apo.) that is a programmed cell death with reduced level of NAD-PH due to glucose-6-PD inactivation in pentose-phosphate reaction and also in the G.1/G.0 phase of the CRC cells, a dose-dependent-arrest, inhibiting cell proliferation in cell cycle progression, and induces Apo.by increasing level of cleaved-Caspase-3 and cleaved-PARP due to ROS generation; Gastric cancer: Via targeting Janus kinase and signal transducer of activated transcription (JAK-STAT) and extracellular matrix protein Extra-CM Protein Pathways in inhibit transcription of genes involve in cancer cell cycle progression, reduce tumor volume and induce Apo.; (B) Breast cancer: Targeting JAK/STAT, CASP-9 and Lysosome ROS-NRF-2 Pathways, intracellular NRF2 signalling triggered because of Pancreatic-S-Cells release Interleukin-6 binds to its receptor and activation of Janus-K/Signal-TAT-3 signalling, resulting in nuclear-factor -2 (NRF-2) downstream EMT-related transcription factors inducing EMT in Panc-1 cells by inducing expression of E-MT related marker genes, to induce Apo., inhibit cell cycle progression by inhibiting growth differentiation, maturation and metastasis; (C) Lung cancer: Induces cell Apo., inhibit metastasis, fibrosis, growth, differentiation and inhibit inflammation cell cycle progression by targeting P13-AKT M-TOR, JAK-Signal-TAT, Cell Cycle, MEK-ER-Kinase, MK-Kinase-JN-Kinase-P-38 and Lysosome ROS-NR-F-2 Pathways, through targeting and inhibiting activation of STAT-3- a downstream effector cascade protein of endothelial growth factor receptor E-GF-R and erythroid nuclear factor ER-K1/2 and pathways and via protein 38 and mitogen-activated-protein kinase-38/MAP-Kinase induces the Apo. cascade through decreasing Bcl-2 and Bax up regulation, caspase-3 activation with Poly-ADP-RP cleavage and produces reduction in cell viability, cell retard and G1/S cell cycle-arrest cancerous cell types; (D) Liver cancer: Induces Apo. and suppress cell cycle growth differentiation by inhibiting TGFβ-SMAD3, T-helper-17, CASP-9, Extra-CM, Cell Cycle progression- G0/G1 and S phase, MEK-ERK, MKK-JNK-P38, NFK-B and Lysosome ROS-NRF-2 Pathways, as antifibrotic, by inhibiting Prolyl transfer RNA synthetase enzyme reduce collagen α1(I) gene expression, inhibit transcription growth factor beta (TGFβ)- Smad-3 protein phosphorylation and conversion to excitatory Smad 2/3, and co Smad4, subsequently collagen protein formation which is involve in fibrosis. By directly inhibiting a dysregulated distinct subset of T helper CD4 cells (T-helper-17) cells with Interleukin-17, result in inhibition of inflammatory pathogenesis, and result into autoimmune disease control by inhibiting nuclear factor-kappa-b (NF-Kb) and p38-mapk pathway in activated T-cells. Arrest cell cycle at the G-1/G-0 phase, by increasing p21 and p27, ROS, and Cyclin D1 downregulation, malignant mesothelioma by Cyclin D1 downregulation, hepatocellular carcinoma by upregulating of p15, p21, Caspase(3,9,8), and downregulating cIAP1, ROS increase, by downregulating TGF-β, phase cell cycle phases arrest at G2/M and microtubules disruption, by increasing p(15,21), and decreasing MYC, S-phase arrest, Apo., Inducing Apo. by targeting various pathways including nuclear factor -2 (NRF-2), and Phosphatidyl-inositol 3 -kinase (PI3-K)/Akt signalling pathway [231].
2.5. Therapeutic and Clinical Potential of Halofuginone:
To check efficacy of HaF various trials are done both for Canc. and non-cancerous diseases in patients. Phase-I-studies of HaF to monitor the toxicities (D-LTs) and the maximum dose that can be tolerated (MTD), Phase II oral HaF studies to check systemic therapy response in patients with superficial-transitional cell-bladder carcinoma, with result showing favourable safety and tolerability profile in patients. Topically administered HaF Phase II study in patients with Acquired-I-Syndrome-related Kaposi sarcoma, serial Kaposi sarcoma biopsy assessing treatment effects on angiogenic predisposing factors and nuclear antigen-1 of Kaposi sarcoma linked with herpes-virus-latency (no effect on matrix metalloproteinase-2 (MMP-2)) and with significant heterogeneity and a decline in collagen type-I level significantly in HaF treated models [32,232]. Studies are done on solid tumor in patients, the acute Maximum Tolerated Dose=3.5milligram/day of HaF, with nausea and vomiting as Dose Limiting Toxicity, and the dose for phase II trials suggested was 0.5 mg Once-Daily. (Table 1) [30,233,234].

Table 1.
Clinical trials of Halofuginone in humans.
| Disease | Clinical trials | Active ingredient | Route | Dose | Dlt | Reference |
| Kaposi sarcoma, renal cancer. Rectum cancer, melanomas and Non-small-cell lung-cancer | Phase II and PK | Halofuginone |
Topical | 0.01% w/w ointment, twice daily | No side effect | [232] |
|
Advanced solid tumours |
Phase I and PK Studies | Halofuginone |
Oral | 0.5-3.5 mg/D | Nausea, vomiting, fatigue | [235] |
|
Chronic graft vs host disease Scleroderma |
Phase I and PKStudies | Halofuginone |
Topical cream | 0.1% | No side effect | [236] |
| Covid-19 | Phase II | Halofuginone | Oral | 0.5mg, 1 mg and placebo OD for 10 days | Well tolerated | [237] |
2.6. Effect of Halide Salt Formulations on Safety and Efficacy of Halofuginone:
In AIDS-related Kaposi sarcoma and scleroderma trials, HaF was previously used in humans, including local application on the skin of chronic-Gversus-HD patients, which resulted in reduces collagen α1(I) gene activity and collagen amount, as skin biopsies three months afterwards showed improvements in neck movement, without challenges and without side effects. Additionally, in solid-tumors patients, oral administration of HaF for three months significantly reduced the mean total skin score. Effective plasma levels can be reached safely of HaF tablets at doses from 0.5-3.5 mg/D and the 3.5 mg/D as maximum-tolerable dose with the side effects including excessive nausea, emesis, and tiredness and for chronic administration the suggested dose of 0.5 mg/day along with the necessity of antiemetic to control dose-limiting toxicities [32,143,232,236,237]. HaF in a SR-formulation is being examined to avoid dose-limiting-toxicities, as well as its tolerability, and PK in Duchene-MD individuals with OD and multiple increasing doses. HaF, combined with in-vivo-biological assays and chemistry modification approaches such as salt formations, will result in a novel and much-needed collection of drug candidates as a lead structure with greater specificity targeting various biological and pharmacological actions, improved tolerance, and efficacy.
As, we have already discussed that HBr and Lactate salts of HaF are available but possess GIT toxicity and hepatotoxicity hence not approved for use in human. HaF is basic drug with poor safety and efficacy profile in humans. Various techniques for alteration of drug physiochemical properties have previously been studied, including use of inorganic elements such as (Na+, K+), organic cations such as tromethamine, erbumine, prodrugs, salts with phosphate ion, carboxylic acids, and by the halide moiety [238,239,240]. Therefore, to alter the physico-chemical properties of poorly hydrophilic basic drugs, salification that is protonation by reacting acid with relevant bases, then adding the API cationic-functional groups and the anionic counterion and is one of the most attractive tools. Eventually substituted with halides contributing as a counterion up to 32% including hydrochloric, hydrobromic, and hydroiodic acids approved by orange book analysis from 1939 to 2022 over the last nine decades which showed approval of approx. six hundred novel drugs salts. The objective of salification is to modify the molecule such that the biological activity of poorly soluble active pharmaceutical ingredient with improved pharmacokinetics parameters including absorption, bioavailability, and physicochemical properties including dissociation constant, and is the most dominant, most preferred approach, practicable, ascendable method for delivering drugs but must ‘not affect active ingredients safety and toxicity profile [241].
Detailed study previously showed that hydrochloric acid salts conquered among all halide counterions in the descending sequence from highest of Chlorine contributing to approx. 29% then Bromine, Fluorine and lowest of Iodine. As one study revealed that methamphetamine HCl ([Figure 8-A) is the 1st halide salt by utilizing the halogen acids, dominates over other salts. Despite their corrosive and dangerous nature (minimized by the salification process), the development of novel drugs significantly affected by salification as it modulates the biopharmaceutical properties playing a vital role for poorly-soluble active pharmaceutical ingredients by modifying several drug characteristics [242,243,244]. Halide drug salts formation help in improving the solubility, dissolution, absorption, the stability of the drug (such as Pexidartinib HCl) and bioavailability for peroral administration, hygroscopicity, manufacturing and processing. Suitable for parenteral management (Cefotiam HCl) and to identify alternate formulations (SR and IR formulations of imipramine pamoate and HCl salt) [242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257,258,259]. Molecule pKa and counter-ion help in salt formation, between the drug and counterion the pKa difference (ΔpKa) decides the formation of cocrystal (Figure 10) [260].
Figure 8.
Structural examples of HCl salt containing drugs approved by FDA to elucidate importance and benefits of HCl formation of drugs for the recommendation of Halofuginone HCl salt formation in future: (A), Methamphetamine HCl; (B), Pexidartinib HCl; (C), Cefotiam HCl.
Figure 8.
Structural examples of HCl salt containing drugs approved by FDA to elucidate importance and benefits of HCl formation of drugs for the recommendation of Halofuginone HCl salt formation in future: (A), Methamphetamine HCl; (B), Pexidartinib HCl; (C), Cefotiam HCl.
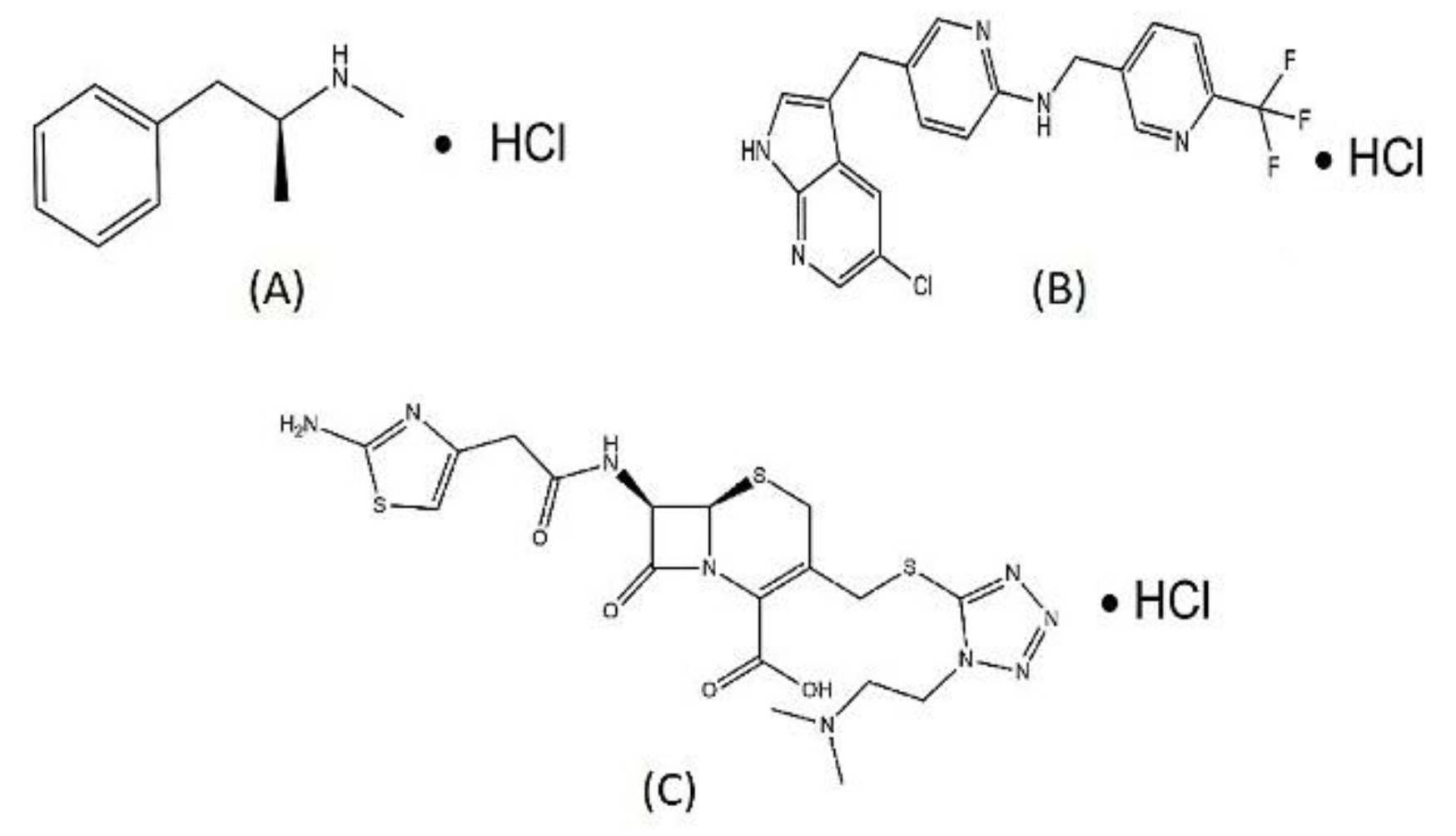
Afterwards methamphetamine salts, iodide salts are rare approx. 0.2% (echothiopate iodide). In last 90yrs hydrochlorides account for major drug salt form available. The FDA database indicates that by creating salt screening techniques, the use of strong acids as anions may be reduced, however it is difficult to avoid strong acids as a counterion, but precise control over the stoichiometric proportion of acid and base will prevent acid hazards. There have been about 171 HCl salts currently in use. According to earlier research, medications with at least one heterocyclic structure, such as amine-N-group-containing drugs like Prazosin HCl (antihypertensive), Hydralazine HCl salt (antihypertensive), and tripelenamine (an antihistaminic agent) that is absorbed quickly from the GIT are available as HCl salts [(Figure 9) [242,243,244,261].
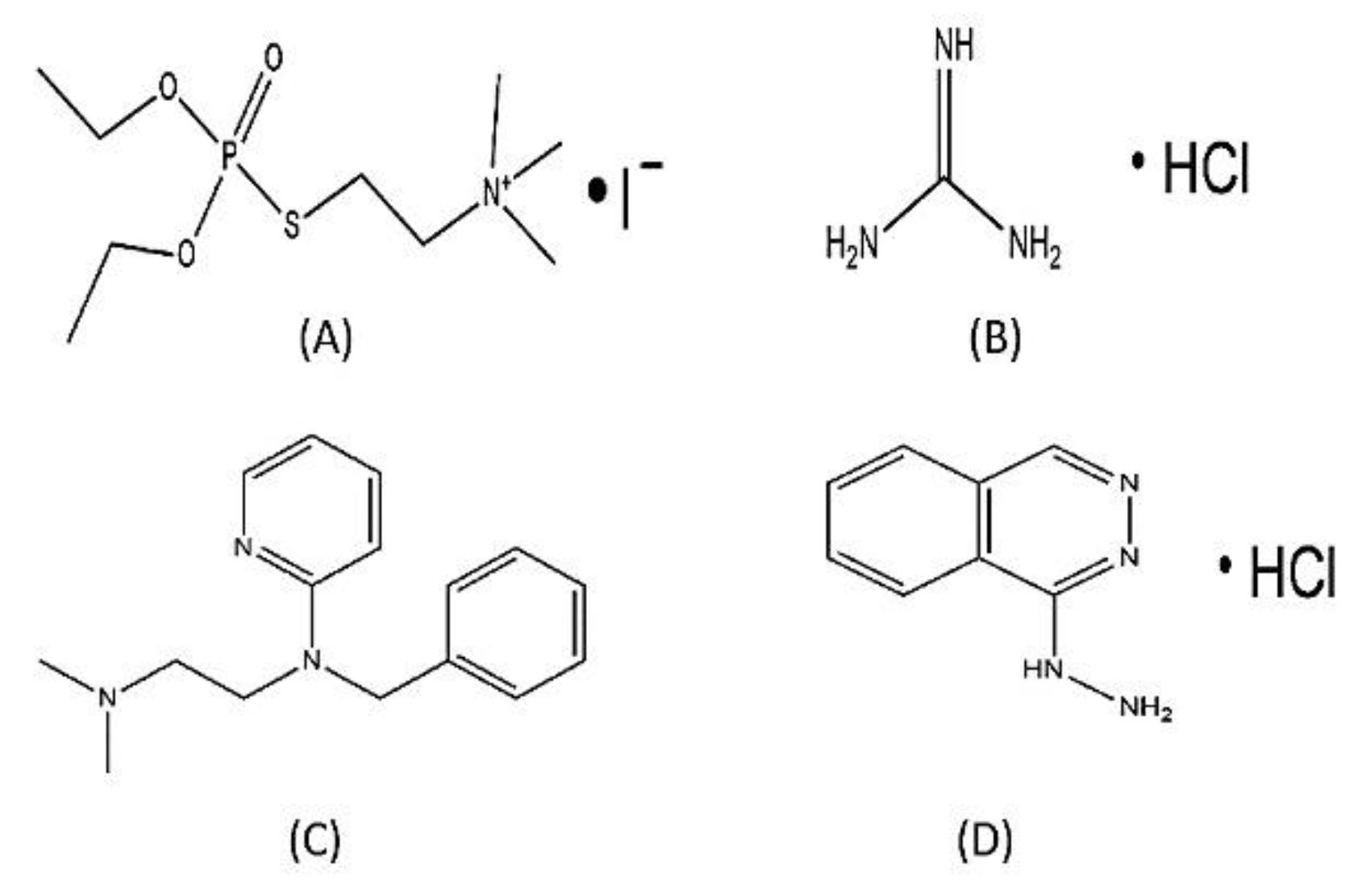
Figure 9.
Structural examples of Halogen Containing Drugs Approved by FDA to elucidate importance and benefits of Halogen salt formation of drugs for recommendation of Halofuginone HCl salt in future: (A)-Echothiopate Iodide; (B)- Guanidine HCl; (C)- Tripelennamine; (D)-Hydralazine HCl.
Figure 9.
Structural examples of Halogen Containing Drugs Approved by FDA to elucidate importance and benefits of Halogen salt formation of drugs for recommendation of Halofuginone HCl salt in future: (A)-Echothiopate Iodide; (B)- Guanidine HCl; (C)- Tripelennamine; (D)-Hydralazine HCl.
Similarly previous studies shows the importance of hydrochloride salts as FDA approved hydrochloride salt of phenothiazine in 1952, has better hydrophilicity, Triflupromazine HCl salt antipsychotic drug, free base of thioridazine hydrochloride salt (clinically used since 1962) with improve hydrophilicity, Fluphenazine HCl with better physicochemical and biopharmaceutical properties (antipsychotic agent), propiomazine HCl has higher solubility and in the case of the theoclate salt with longer duration of action, promethazine hydrochloride with high hydrophilicity, thus HCl salts seems to be the healthier choice [262,263,264,265]. Similarly, an organic base Quinoline has the HCl salts including, Ciprofloxacin HCl is clinically used with better solubility, without salt drug has low solubility and permeability. Mefloquine HCl with improved solubility (an antimalarial drug), and Moxifloxacin HCl an anti-infective, with improved stability [266,267]. FDA approved HCl salts of Piperazine scaffold beneficial with better solubility, present in almost 13 drugs, including Pentazocine HCl (FDA approved) opioid pain killer with highly water-soluble approx. 30 mg/mL , antipsychotic drug ziprasidone, Azelastine HCl salt is a phthalazine for allergic reactions, Flavoxate HCl (was developed and approved in 1970) muscle relaxant with improve water solubility and stability, trazodone HCl solubility improved, raloxifene HCl BCS class II drug (FDA approved) treat osteoporosis and donepezil HCl ([Figure 10) [268,269,270,271,272].
Figure 10.
Structural examples of HCl Salt Containing Drugs Approved by FDA To elucidate importance and benefits of HCl salt formation of drugs for the recommendation of Halofuginone HCl salt in future: (A) Phenothiazine HCl; (B) Triflupromazine HCl; (C) Thioridazine; (D) Donepezil HCl; (E) Fluphenazine HCl; (F) Propiomazine HCl; (G) Mefloquine HCl; (H) Ciprofloxacin HCl (I) Moxifloxacin HCl; (J) Pentazocine HCl (K) Azelastine HCl (L) Trazodone HCl (M) Raloxifene HCl; (N) Flavoxate HCl; (0) Ziprasidone HCl.
Figure 10.
Structural examples of HCl Salt Containing Drugs Approved by FDA To elucidate importance and benefits of HCl salt formation of drugs for the recommendation of Halofuginone HCl salt in future: (A) Phenothiazine HCl; (B) Triflupromazine HCl; (C) Thioridazine; (D) Donepezil HCl; (E) Fluphenazine HCl; (F) Propiomazine HCl; (G) Mefloquine HCl; (H) Ciprofloxacin HCl (I) Moxifloxacin HCl; (J) Pentazocine HCl (K) Azelastine HCl (L) Trazodone HCl (M) Raloxifene HCl; (N) Flavoxate HCl; (0) Ziprasidone HCl.
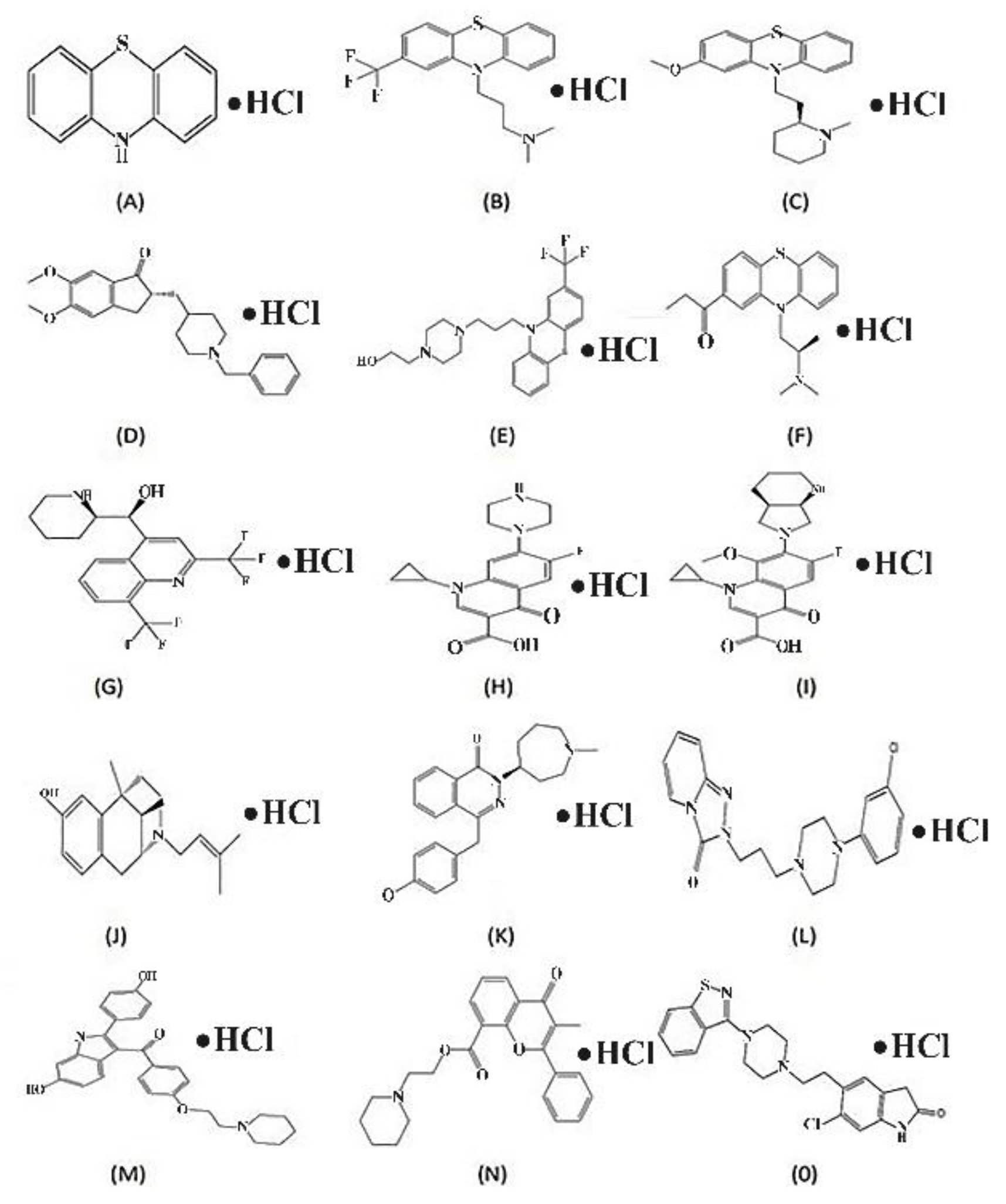
Numerous FDA-approved drugs containing Chloride and bromide salts with improve solubility and stability, for drugs having quaternary nitrogen, including Pralidoxime chloride (FDA approved in 1964) and iodide (undergoes rapid deterioration) for treating organophosphorus poisoning and echothiophate iodide salt that is an organic phosphorous cholinesterase inhibitor for the treatment of glaucoma with improve water solubility of >500 mg/mL and stability [273,274]. Among the quaternary ammonium nitrogen-containing alkaloid class of drugs the bromide counterion commonly used with improve solubility and bioavailability as poorly soluble drug also cant diffuse cross blood brain barrier so salt form can transport across lipid bilayers such as HaF-HBr salt has been in use for past few decades for malaria and topically in scleroderma other examples including Tropane alkaloids, such as Hyoscine comprising tertiary nitrogen (for motion sickness) leading to the discovery of other drugs salt as well including ipratropium HBr and tiotropium HBr (for COPD and asthma as a nasal spray), Methscopolamine bromide (approved for peptic ulcers treatment), Pyridostigmine bromide (for myasthenia gravis), Galantamine hydrobromide is a tertiary alkaloid with improve stability and numerous quaternary-N-containing molecules for several indications ([Figure 11) [275,276,277,278,279,280]. However very few drugs contain HBr salts that is why their use is reduced due to chronic toxicity of bromide ions, such as it causes gastric issues and CNS side effects including confusion and hallucinations. Similar side effects can be seen with HaF HBr [281].
Figure 11.
Structural examples of drugs containing Halogens; Chlorine, Bromine, Iodine and Fluorine substitutions: (A) Pralidoxime Chloride; (B) echothiopate iodide; (C) Ipratropium Bromide; (D)Tiotropium HBr; (E) Galantamine HBr; (F) Pyridostigmine HBr.
Figure 11.
Structural examples of drugs containing Halogens; Chlorine, Bromine, Iodine and Fluorine substitutions: (A) Pralidoxime Chloride; (B) echothiopate iodide; (C) Ipratropium Bromide; (D)Tiotropium HBr; (E) Galantamine HBr; (F) Pyridostigmine HBr.
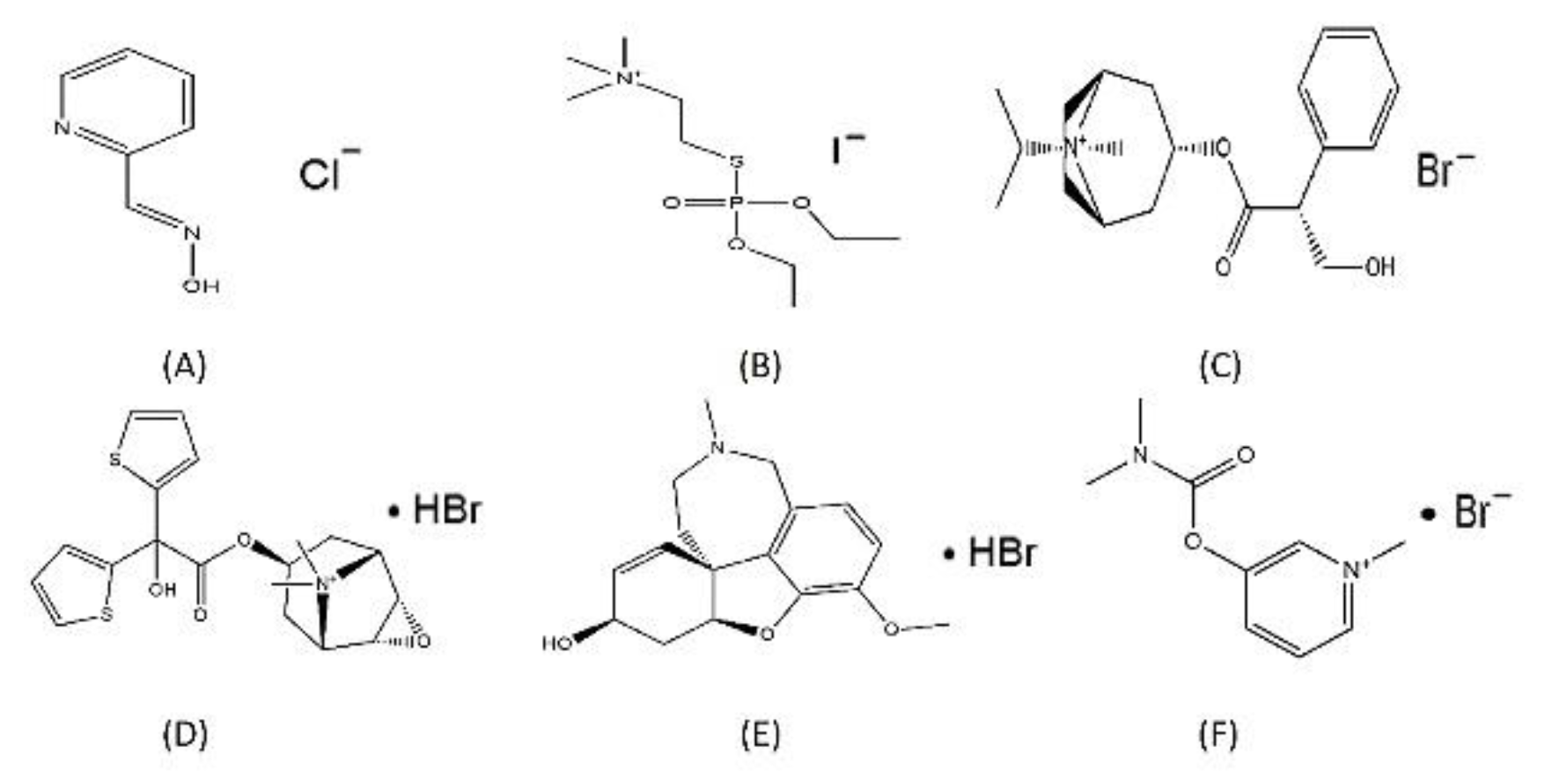
2.7. HCl Conjugate Formation of Halofuginone:
Approximately 70% drugs are in salt forms documented by numerous researchers as they contain ionizable groups improving their solubility and permeability, which is related to the ionized form of molecules because they are more polar and their dissociation constant is representative of corresponding either, acidic profile (pKa less) or basic nature (pKa more) of molecules [282]. Salt formation process also employed for drug discovery and development, done by performing salt screening in order to avoid toxicity. For basic molecules such as HaF, it forms cation with the decrease in the pH and contain ‘N’ (enhancing ‘basicity’ to the molecule) and remain in ionized form, upon interaction with an anion remain in solubilized state. However, stable basic-drugs salts obtained with strong- acids due to very low pKa values such as Hydro-Cl, Hydro-Br and Hydro-I. During salt screening ΔpKa, (more than 3), ionization site for salification in basic drugs provided by nitrogen in the structure, with strong inorganic-acids (in comparison to carboxylic-acids) more stable salts obtained, and tests of halides as counterions are vital for counterion selection and are observed very closely. Hydrochloride salts possess chloride ions, are endogenous, easy to prepare, plentiful, and the most frequently occurring especially along with sodium salts. Therefore, in the future, there is need of HaF halides salt formation with hydrochloride despite halogen acids possess restrictions due to their corrosive nature, although with specially considering the drug precipitation, common ion effect, humid conditions (due to the volatile nature might be converted to mono-HCl) and pH microenvironment while selecting the drug salts [(Figure 12). We propose the use of HCl salts of HaF as due to the high Cl content with approx. 0.1–0.15 M it have restricted solubility or precipitate out in the gastric environment, as per conversion to free base in the stomach. And HCl addition will influence various aspects of HaF including improvement in solubility in numerous solvents, dissolution profile, absorption, formulation development, administration (in tablet, syrup, capsules, suspension parenteral) with enhanced compatibility with excipients, stability during storage in various environmental conditions, improving shelf life, effectiveness and bioavailability (as it is converted to amorphized form) and upon peroral administration, drug dose dumping occur due to high dissolution in initial stages and drug concentration surpasses its equilibrium solubility [283,284,285,286]. To overcome the risk associated, rigorous process controls may suggest a solution and ultimately help in ideal drug discovery with acceptable high standard properties.
Figure 12.
Halofuginone HCl.
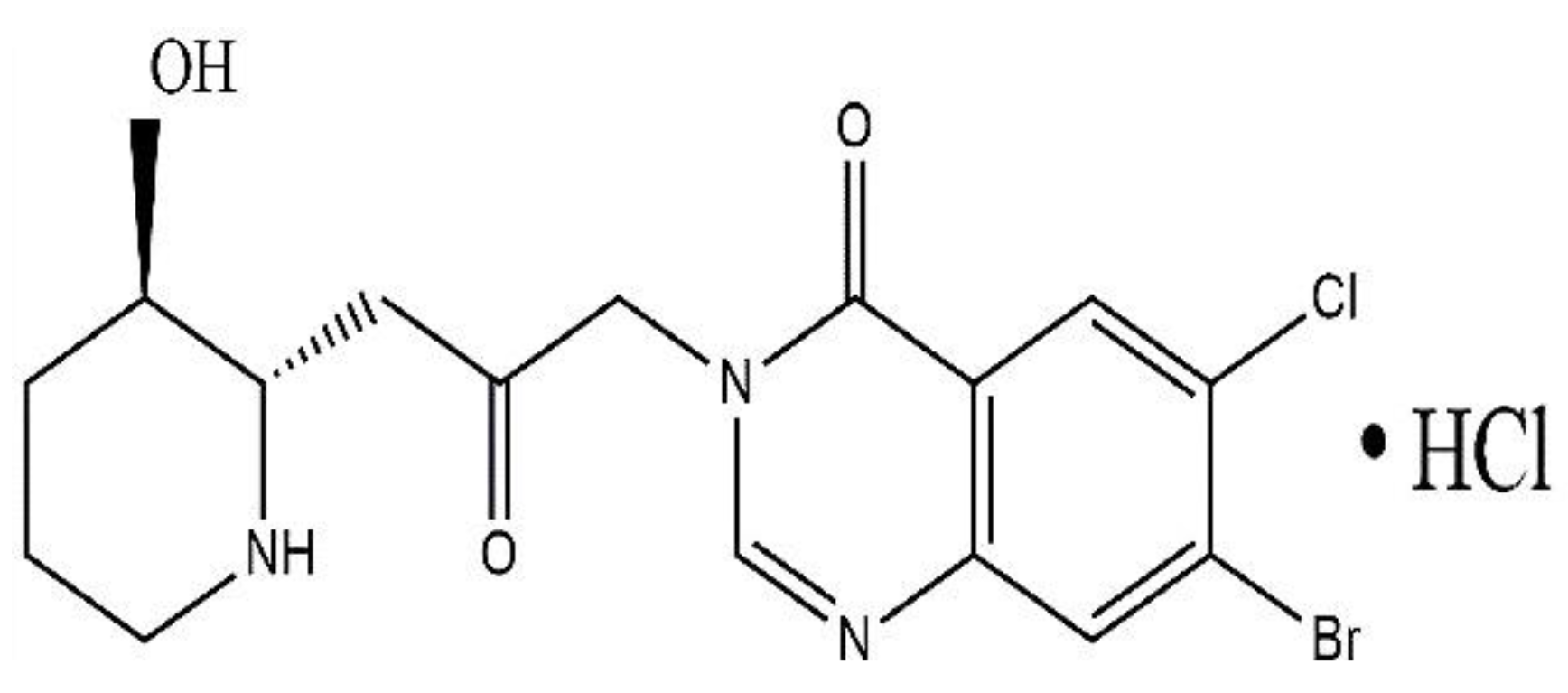
2.8. Stereoisomeric Synthesis of Halofuginone HCl Salt and Its Projected Properties:
Through our studies we are recommending the formation of HaF-HCl Salt because it will have important and beneficial expected properties to address broad range of diseases with improved bioavailability, efficacy and less toxic adverse effects. By using Molinspiration, a Batch Property Calculation Toolkit mib, which help in molecular processing of large number of molecules at processing speed of 10,000 molecules/minute, and also in property calculation, written in Java, used in a batch mode in order to retrieve through web interface directly on our intranet. As a result, we obtained a calculated molecular descriptor of HaF-HCl salt. We used SMILES a line notation system for individual drugs describing chemical species structure by using short ASCII strings and associated chemicals are calculated by ChemAxon. ‘O=C(C[C@H]1NCCC[C@H]1O)Cn3cnc2cc(Cl)c(Cl)cc2c3=O’ is the smile for given structure of HaF with two chlorine molecules [286]. When we run it on Molinspiration bioactivity score- v2022.08, ligand energy would be for receptors GPCR, Ion channel modulator, Kinase inhibitor, nuclear receptor ligand, Protease inhibitor, Enzyme inhibitor were 0.23, 0.03, -0.09, -0.43, 0.3 and 0.36 respectively. The maximum stability is for kinase receptors and minimal for enzyme inhibitor, lower the binding energy between ligand and receptor more stable is complex, as large amount of the energy released on binding of a ligand to the protein receptor, greater will be the propensity of the ligand to associate with that protein. Molinspiration also calculate the LogP (octanol/water partition coefficient) by the very robust method as a sum of fragment-based contributions and correction factors and can practically process almost all organic and most organometallic molecules. Molecular polar surface area TPSA - a sum of fragment contributions is also calculated based on the procedure published by Ertl et al. by considering O- and N- centred polar fragments by PSA used to characterize descriptor; drug absorption, including BBB penetration, intestinal availability, bioavailability and Caco-2 permeability [287]. Molinspiration based on group contributions calculate molecule volume by fitting sum of fragment contributions to "real" 3D volume for a training set of about twelve thousand, mostly drug-like molecules, fully augmented by the semi-empirical AM1 method of 3D molecular geometries for a training set. Lipinski used "Rule of 5" for formulating properties, is set of simple molecular descriptors, named as such because of the border values are 5, 500, 2*5, and 5 and it states, that most "drug-like" molecules have number of hydrogen bond acceptors <= 10, logP <= 5, molecular weight <= 500 and hydrogen bond number of donors <= 5 and molecules not complying with any of these rules, may have bioavailability issue also. A simple topological parameter NROTB (Number of Rotatable Bonds), is a measure of molecular flexibility and descriptor of drug oral bioavailability defined as any single non-ring bond, bounded to nonterminal heavy (i.e., non-hydrogen) atom, not considering amide C-N bonds because of their high rotational energy barrier ([Figure 13) [243,288].
Figure 13.
Proposed structure of Halofuginone HCl (DL-trans-7-chloro-6-chloro-3-[3-(3-hydroxy-2-piperidyl)-acetonyl]-4(3H)-quinazolinone hydrobromide).
Figure 13.
Proposed structure of Halofuginone HCl (DL-trans-7-chloro-6-chloro-3-[3-(3-hydroxy-2-piperidyl)-acetonyl]-4(3H)-quinazolinone hydrobromide).
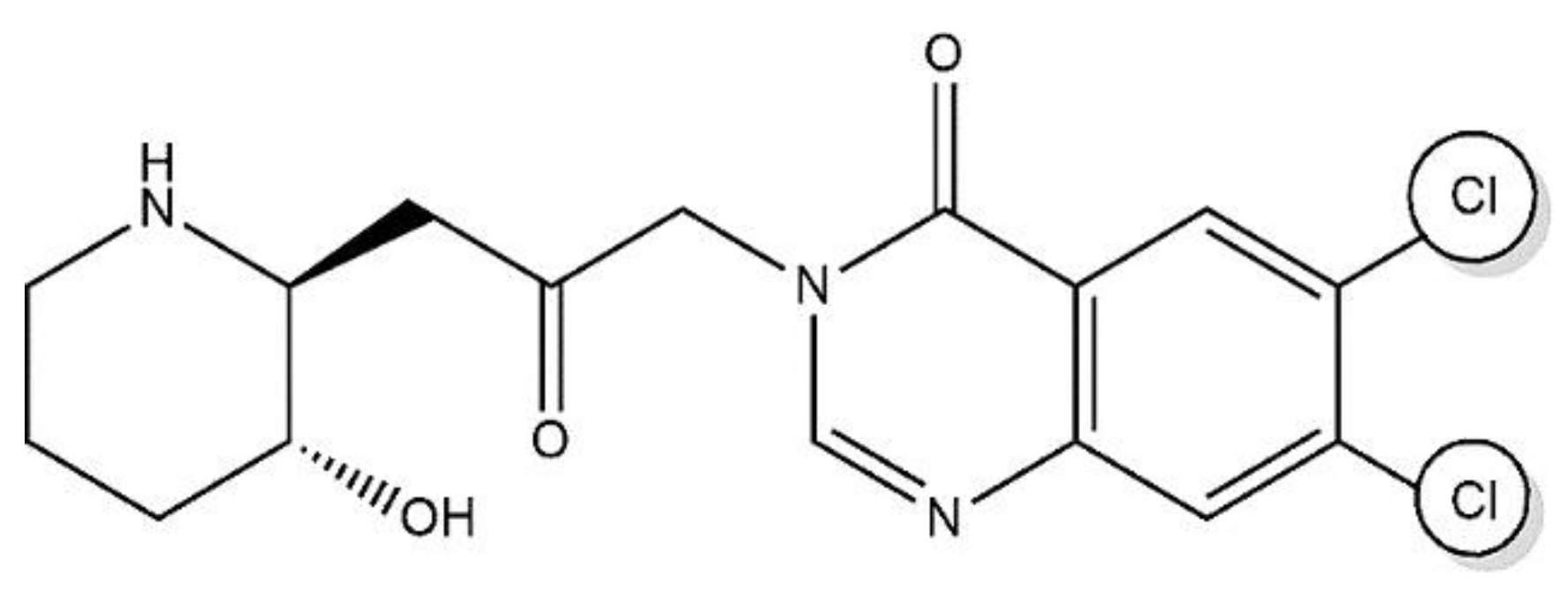
3. Discussion
Among community of less than 85years of age, cancer (Canc.) is 2nd leading cause of death and each year the American Cancer Society, estimates the numbers of new Canc. cases and deaths in the United States and compiles the most recent data on population- based Canc. occurrence. In current year 2024, >20M new cases and >6M deaths estimated in U.S., but because of restriction to tobacco smoking, earlier detection and better treatment options, the Canc. mortality rate continued to decline through 2021, averting over 4 million deaths since 1991. However, during 2015 to 2019 incidence rates increased annually such as for B.Canc., pancreas, and uterine corpus Canc. by 0.6%–1%, and by 2%–3% annually for prostate, liver, kidney, and oral Canc., for melanoma, for cervical and colorectal Canc. incidence rates also increased [289,290].
It is of great significance to develop new anticancer agents with less toxicity and better efficacy. Since with the turn in century substantial progresses in outcomes for Canc. patients can be seen with the drastic variations on Canc. treatment landscape and changing trend in the approval of Canc. therapeutic products, with multiple molecular target and mechanism of action by FDA, currently in United States. New therapeutic approaches for Canc. with targeted drug delivery are still burning topic of interest for researchers worldwide [291], some of these trends were studied in various scientific works by researchers as one of these studies emphasize on kinase inhibitors and immune checkpoint inhibitors as the dominant product class and the second most approvals on the basis of indications but due to some limitations can’t have access to the market until 2011. Others such trends include approvals for population, predicted by biomarkers and the tumour site agnostic emergence. One of the studies emphasize on these novel trends and new therapeutic approaches on Canc. treatment and their impact on future Canc. therapeutic products development with minimum toxicity [292].
The current study focused on the novel RNA synthetase inhibitor HaF which is a natural alkaloid Febr. derivative obtained from Chang Shan plant a Chinese herb, previously used in malaria and for coccidiosis. Current study guides on HaF structure activity relationship, chemical substitutions previously studied, effect in multiple organs, tissues, understanding underlying molecular mechanisms and molecular targets in various Canc. cell lines and non-cancerous diseases as it could play a vital role in novel anticancer classes of drugs and had shown positive impact but with some adverse effects which needed to be addressed. HaF by inhibiting prolyl-transfer RNA synthetase enzyme involve in proline containing protein synthesis. As we already discussed above HaF has broad scope of therapeutic activities and subsequent molecular targets, as studied in various literatures studies including antiprotozoal effect [293], anti-fibrotic studies [294], nanohydrogels as antibacterial [295,296,297], anti-viral such as is SARS-COV2 [298,299,300], anti-malarial [301], anti-inflammatory effect by targeting Nuclear-F-kB and p-38-MAP-Kinase in activated T cells, proliferation of fibroblast in rheumatoid-A disease induced by TN-Factor-alpha [36,162,226,302], immunomodulatory effect in various studies with different targets [166,303], antifungal effect [304,305], neurodegeneration protectant [306], antiarthritic effect [307], in retinopathy [308], osteoclast-genesis inhibitor [309], pulmonary hypertension [157] and cardioprotective [310] and many more therapeutic implications have been studied in which HaF has shown promising effects and used in market as an anti- coccidiosis agent in animal husbandry [311,312]. Also, its used in Covid-19 patients who are not hospitalized, has been studied in humans in phase-II clinical trials but require further evaluations [47,86,94,237,313].
Potential anticancer activity of HaF has already been studied and documented in variety of Canc. cell lines over the last decades and proven effective in a variety of Canc. both in vitro as well as in vivo multiple animal models and by targeting various molecular therapeutic targets for tumor suppression as discussed above, including for cancers of; stomach [314,315], mesoporous nanoplatform for prostate [315], bladder, pancreatic ductal adenocarcinoma [316], glioma [317], squamous cell [317,318], uterine leiomyoma [319], breast [320], nanohydrogels in canine memory carcinoma [321,322], AML [323], colorectal [324], hepatic [325], and lung adenocarcinoma [221] with combination therapies with various therapeutic agents and to overcome drug resistance to chemotherapy, HaF has shown promising effects [215,233,317,326,327].
In order to address HaF toxicity various formulations such as TPGS polymeric micelles or nano-formulations are studied but still drug is in phase 2 clinical trials in humans [328,329]. Therefore through current study we are emphasizing on preparation of novel formulation of HaF with HCl salt, as salt formation having several beneficial impacts on drug PK and PD parameters as studied by various researchers therefore before formulation development appropriate salt selection has direct impact on modifying the features of the potential target drug substance physicochemical properties with better patient compliance as it has positive effects on melting point, hygroscopicity, chemical stability, bioavailability, as well as manufacturability, aqueous solubility, dissolution rate, pH, crystallization and mechanical properties and its final properties are comparable to standard pre-formulations and should comply with the primary route of administration and dosage form. We used Molinspiration for property based virtual screening of our novel molecules parameters as it is a useful tool to pick potential drug candidates, we find out drug binding energy, Log P, and other useful and important properties of novel HaF with two chlorine atoms. As studied previously that HCl salt formation increased stability of API of antidepressant compound CGP6085 [330,331]. HCl salt enhance solubility of weakly basic drugs as it has a low counterion pKa, and recrystallisation from organic solvents is straightforward and is by far the most frequent choice due to simple availability and physiological reason, also form stable complex [248,332,333].
Figure 14.
Halofuginone inhibit inflammation and fibrosis, have direct or indirect impact at various molecular levels to treat wide range of diseases by targeting TGFβ-SMAD3, T-helper-17, P13-AKT-MTOR1, P13-AKT-MAPK, CASP-9, CASP-3, Extra-CM, Cell Cycle, MEK-ERK, MKK-JNK-P38, NFK-B- P60/P65 and Lysosome ROS-NRF-2, JAK-STAT, Pathways.
Figure 14.
Halofuginone inhibit inflammation and fibrosis, have direct or indirect impact at various molecular levels to treat wide range of diseases by targeting TGFβ-SMAD3, T-helper-17, P13-AKT-MTOR1, P13-AKT-MAPK, CASP-9, CASP-3, Extra-CM, Cell Cycle, MEK-ERK, MKK-JNK-P38, NFK-B- P60/P65 and Lysosome ROS-NRF-2, JAK-STAT, Pathways.
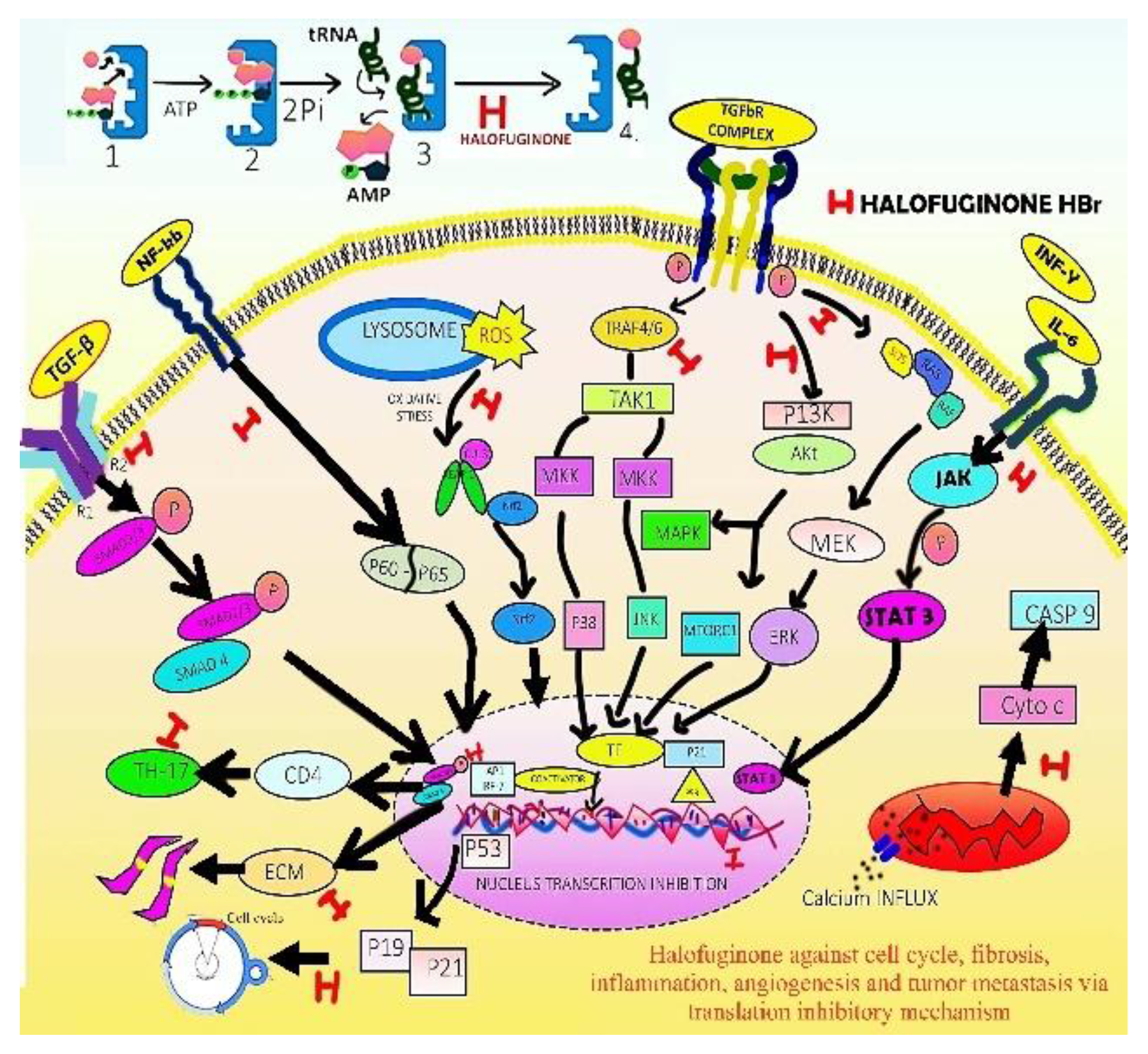
4. Conclusions
Cancer is leading cause of mortality globally and considered as the life-threatening disease reigning champion, and for achieving operative and efficient results from cancer therapy it’s important to understand the significance of early diagnosis and better novel treatment options with limited toxicity and maximum efficacy for positive subsequent outcomes on quality of life at community level. There are various natural, synthetic and semisynthetic compounds under study for novel targeted drug in therapy with limited toxicity as compared to chemotherapy induced side effects. Among these is Halofuginone, derivative of Febr. obtained from Chinese herb, which has multiple beneficial impact in various diseases due to inhibitory effect on exaggerated inflammatory immune response and also effect Treg cells, cytokines production at transcription and translation level and has various targets along with cell cycle arrest or Canc. predictive biomarkers but limited in use due to GIT toxicity and physiochemical characteristics [414]. So, in this article we emphasize on preparation of HCl salt formulation of this novel entity to minimize associated side effects and enhanced efficacy. With context to the pharmacokinetic (Absorption, Distribution, Metabolism, Elimination), pharmacodynamic and biopharmaceutical properties of Halofuginone as well. As the good choice of salt has direct impact on active pharmaceutical ingredient solubility, stability, bioavailability, therapeutic drug concentration achievement, efficacy, onset of action and other biopharmaceutical parameters, so regulatory guidelines can influence and should be considered while selecting best form of salt with maximum benefits as, well which meet safety, efficacy and quality regulatory standard as per the recommended guidelines and decision should be made according to requirement and site of administration of drug.
In order to optimize the therapeutic potential of Halofuginone, a number of important factors need to be considered for a safer and less toxic medication in the future. Thus, Halofuginone ought to be considered in subsequent research for additional assessment through modification or alternative substitutions at various positions and formulations, such as the effect of HCl salt formulation on the safety and efficacy profile of Halofuginone.
Author Contributions
(AMM) data curation, primary draft; (GJK) conceptualization, project administration, supervision, final draft and funding and is corresponding author; (JB) resources, software and funding; (AAq) investigation, validation and funding; (TM) formal analysis and primary draft; (AAy) data curation, resources; (TA) review and editing; (YL) resources; (YD) review and editing.
Funding
This work was supported by Large Research Project (LRP) [grant number RGP2/399/45]
Data Availability Statement
No new data were created.
Acknowledgments
The authors extend their appreciation to the Dean of Research and Graduate Studies at King Khalid University.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Siegel, R.L.; et al. Cancer statistics, 2022. CA: A Cancer J. Clin. 2022; 72. [Google Scholar]
- Zeng, Y.; et al. Fisetin micelles precisely exhibit a radiosensitization effect by inhibiting PDGFRβ/STAT1/STAT3/Bcl-2 signaling pathway in tumor. Chinese Chemical Letters 2024, 109734. [Google Scholar] [CrossRef]
- Li, X.; et al. Recent advances in enhancing reactive oxygen species based chemodynamic therapy. Chin. Chem. Lett. 2022, 33, 2213–2230. [Google Scholar] [CrossRef]
- Khan, G.J.; et al. Effects of Curcuma longa L. and Piper nigrum L. Against Methicillin Resistant Staphylococcus aureus and Infectious Angiogenesis. J. Biobased Mater. Bioenergy 2024, 18, 303–314. [Google Scholar] [CrossRef]
- Lu, C.-L.; et al. Traditional Chinese medicine in cancer care: an overview of 5834 randomized controlled trials published in Chinese. Integr. Cancer Ther. 2021, 20, 15347354211031650. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Miller, K.D.; et al. Cancer statistics for the US Hispanic/Latino population, 2021. CA: A Cancer J. Clin. 2021, 71, 466–487. [Google Scholar] [CrossRef]
- Ali, A.; et al. The burden of cancer, government strategic policies, and challenges in Pakistan: A comprehensive review. Front. Nutr. 2022, 9, 940514. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; et al. Cancer incidence and mortality in Asian countries: A trend analysis. Cancer Control 2022, 29, 10732748221095955. [Google Scholar] [CrossRef]
- Curado, M.-P.; et al. Cancer incidence in five continents, Volume IX. 2007: IARC Press, International Agency for Research on Cancer.
- Ferlay, J.; et al. QOL comparison of PG and TG 415. Int J Cancer 2010, 127, 2893–917. [Google Scholar] [CrossRef]
- Gopal, S.; Sharpless, N.E. Cancer as a global health priority. Jama 2021, 326, 809–810. [Google Scholar] [CrossRef]
- Dhakal, R.; et al. A scoping review on the Status of female breast Cancer in Asia with a special focus on Nepal. Breast Cancer: Targets Ther. 2022, 229–246. [Google Scholar]
- Shoemaker, M.L.; et al. Differences in breast cancer incidence among young women aged 20–49 years by stage and tumor characteristics, age, race, and ethnicity, 2004–2013. Breast Cancer Res. Treat. 2018, 169, 595–606. [Google Scholar] [PubMed]
- Lim, J.W. The changing trends in live birth statistics in Korea, 1970 to 2010. Korean J. Pediatr. 2011, 54, 429. [Google Scholar] [PubMed]
- Park, C.Y.; Lim, J.-Y.; Park, H.-Y. Age at natural menopause in Koreans: secular trends and influences thereon. Menopause 2018, 25, 423–429. [Google Scholar] [PubMed]
- Lee, J.C.; et al. A study on the menstruation of Korean adolescent girls in Seoul. Korean J. Pediatr. 2011, 54, 201. [Google Scholar] [PubMed]
- Li, M.; Maso, L.D.; Vaccarella, S. Global trends in thyroid cancer incidence and the impact of overdiagnosis. Lancet Diabetes Endocrinol. 2020, 8, 468–470. [Google Scholar] [PubMed]
- Harris, J.E. Cigarette smoking among successive birth cohorts of men and women in the United States during 1900–1980. J. Natl. Cancer Inst. 1983, 71, 473–479. [Google Scholar]
- Sharma, R.; Rakshit, B. Global burden of cancers attributable to tobacco smoking, 1990–2019: an ecological study. EPMA J. 2023, 14, 167–182. [Google Scholar]
- Rana, B.; et al. Physical activity behaviour change in black prostate cancer survivors: a qualitative study using the Behaviour Change Wheel. Support. Care Cancer 2024, 32, 154. [Google Scholar]
- Hong, S.; et al. Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2017. Cancer Res. Treat. : Off. J. Korean Cancer Assoc. 2020, 52, 335–350. [Google Scholar]
- Katanoda, K.; et al. An updated report on the trends in cancer incidence and mortality in Japan, 1958–2013. Jpn. J. Clin. Oncol. 2015, 45, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Keinan-Boker, L.; et al. Time trends in the incidence and mortality of ovarian cancer in Ireland, Northern Ireland, and Israel, 1994–2013. Int. J. Gynecol. Cancer 2017, 27. [Google Scholar] [CrossRef]
- Lavy, R.; et al. Incidence trends and mortality rates of gastric cancer in Israel. Gastric Cancer 2013, 16, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Vaccarella, S.; et al. The impact of diagnostic changes on the rise in thyroid cancer incidence: a population-based study in selected high-resource countries. Thyroid 2015, 25, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; et al. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: the increasing incidence of thyroid cancer. Endocr. Pract. 2015, 21, 686–696. [Google Scholar] [CrossRef]
- Newman, D.J. Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J. Med. Chem. 2008, 51, 2589–2599. [Google Scholar] [CrossRef]
- Du, H.; et al. Advances in anticoccidial drug halofuginone. Heilongjiang Anim. Husb. Vet. J. 2013, 3, 29–31. [Google Scholar]
- De Jonge, M.; et al. Phase I and pharmacokinetic study of halofuginone, an oral quinazolinone derivative in patients with advanced solid tumours. Eur. J. Cancer 2006, 42, 1768–1774. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.-S.; et al. Ch'ang Shan, a Chinese antimalarial herb. Science 1946, 103, 59–59. [Google Scholar]
- McLaughlin, N.P.; Evans, P.; Pines, M. The chemistry and biology of febrifugine and halofuginone. Bioorganic Med. Chem. 2014, 22, 1993–2004. [Google Scholar]
- Pines, M.; Spector, I. Halofuginone—the multifaceted molecule. Molecules 2015, 20, 573–594. [Google Scholar] [CrossRef]
- Waletzky, E.; Berkelhammer, G.; Kantor, S. Method for Treating Coccidiosis With Quinazolinones. US332 0124 (A), 1967.
- Liu, Y.; et al. Ultrasensitive detection of IgE levels based on magnetic nanocapturer linked immunosensor assay for early diagnosis of cancer. Chin. Chem. Lett. 2022, 33, 1855–1860. [Google Scholar] [CrossRef]
- Zeng, S.; et al. Halofuginone inhibits TNF-α-induced the migration and proliferation of fibroblast-like synoviocytes from rheumatoid arthritis patients. Int. Immunopharmacol. 2017, 43, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; et al. Application of nanotechnology in CAR-T-cell immunotherapy. Chin. Chem. Lett. 2023, 34, 107747. [Google Scholar] [CrossRef]
- Koepfli, J.; Mead, J.; Brockman, J.A. Jr. An alkaloid with high antimalarial activity from Dichroa Febrifuga1. J. Am. Chem. Soc. 1947, 69, 1837–1837. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; et al. Catalytic asymmetric synthesis of antimalarial alkaloids febrifugine and isofebrifugine and their biological activity. J. Org. Chem. 1999, 64, 6833–6841. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, W.; Gerald, B.; Sidney, K. Method for treating coccidiosis with quinazolinones. 1967, Google Patents.
- De Smet, P.A. The role of plant-derived drugs and herbal medicines in healthcare. Drugs 1997, 54, 801–840. [Google Scholar] [CrossRef]
- Koepfli, J.; Mead, J.; Brockman, J.A. Alkaloids of Dichroa febrifuga. I. Isol. Degrad. Stud.. J. Am. Chem. Soc. 1949, 71, 1048–1054. [Google Scholar] [CrossRef]
- Jang, C.; et al. Pharmacology of Ch ‘ang Shan (Dichroa febrifuga), a Chinese antimalarial herb. Nature 1948, 161, 400–401. [Google Scholar] [CrossRef]
- Hewitt, R.I.; et al. An antimalarial alkaloid from Hydrangea. XIII. The effects of various synthetic quinazolones against Plasmodium lophurae in ducks. Am. J. Trop. Med. Hyg. 1952, 1, 768–72. [Google Scholar] [CrossRef] [PubMed]
- Coatney, G.R.; et al. Studies in human malaria. XXV. Trial of febrifugine, an alkaloid obtained from Dichroa febrifuga lour. against the Chesson strain of Plasmodium vivax. J. Natl. Malar. Soc. 1950, 9, 183–6. [Google Scholar]
- Hirai, S.; et al. Metabolites of febrifugine and its synthetic analogue by mouse liver S9 and their antimalarial activity against Plasmodium malaria parasite. J. Med. Chem. 2003, 46, 4351–4359. [Google Scholar] [CrossRef] [PubMed]
- Chien, P.-L.; Cheng, C.-C. Structural modification of febrifugine. Some methylenedioxy analogs. J. Med. Chem. 1970, 13, 867–870. [Google Scholar] [CrossRef]
- Kikuchi, H.; et al. Exploration of a new type of antimalarial compounds based on febrifugine. J. Med. Chem. 2006, 49, 4698–4706. [Google Scholar] [CrossRef]
- Baker, B.; et al. An antimalarial alkaloid from hydrangea. xvi. synthesis of 5-, 6-, 7-, and 8-derivatives with two different substituents. J. Org. Chem. 1952, 17, 157–163. [Google Scholar] [CrossRef]
- Baker, B.; McEVOY, F.J. An Antimalarial Alkaloid from Hydrangea. XXIII. Synth. By Pyridine Approach. II. J. Org. Chem. 1955, 20, 136–142. [Google Scholar]
- Baker, B.; et al. An antimalarial alkaloid from hydrangea. XVIII. Derivatives of 4-pyrimidone. The Journal of Organic Chemistry 1953, 18, 133–137. [Google Scholar]
- Baker, B.; et al. an antimalarial alkaloid from hydrangea. XVII. some 5-substituted derivatives. J. Org. Chem. 1952, 17, 164–176. [Google Scholar] [CrossRef]
- Baker, B.; et al. An antimalarial alkaloid from hydrangea. XV. Synthesis of 5-, 6-, 7-, and 8-derivatives with two identical substituents. J. Org. Chem. 1952, 17, 149–156. [Google Scholar] [CrossRef]
- Baker, B.; et al. An antimalarial alkaloid from Hydrangea. XIV. Synthesis of 5-, 6-, 7-, and 8-monosubstituted derivatives. J. Org. Chem. 1952, 17, 141–148. [Google Scholar]
- Gao, P.; et al. Chemical proteomic profiling with photoaffinity labeling strategy identifies antimalarial targets of artemisinin. Chin. Chem. Lett. 2023, 34, 108296. [Google Scholar]
- Marapaka, A.K.; et al. Development of peptidomimetic hydroxamates as PfA-M1 and PfA-M17 dual inhibitors: Biological evaluation and structural characterization by cocrystallization. Chin. Chem. Lett. 2022, 33, 2550–2554. [Google Scholar]
- Smullen, S. N.P. McLaughlin, and P. Evans, Chemical synthesis of febrifugine and analogues. Bioorganic Med. Chem. 2018, 26, 2199–2220. [Google Scholar]
- Takaya, Y.; et al. New type of febrifugine analogues, bearing a quinolizidine moiety, show potent antimalarial activity against Plasmodium malaria parasite. J. Med. Chem. 1999, 42, 3163–3166. [Google Scholar]
- Zhang, D.-F.; et al. Anticoccidial effect of halofuginone hydrobromide against Eimeria tenella with associated histology. Parasitol. Res. 2012, 111, 695–701. [Google Scholar] [PubMed]
- Peeters, J.; et al. Specific serum and local antibody responses against Cryptosporidium parvum during medication of calves with halofuginone lactate. Infect. Immun. 1993, 61, 4440–4445. [Google Scholar] [CrossRef]
- Daugschies, A.; Gässlein, U.; Rommel, M. Comparative efficacy of anticoccidials under the conditions of commercial broiler production and in battery trials. Vet. Parasitol. 1998, 76, 163–171. [Google Scholar]
- Taniguchi, T.; Ogasawara, K. A diastereocontrolled synthesis of (+)-febrifugine: a potent antimalarial piperidine alkaloid. Org. Lett. 2000, 2, 3193–3195. [Google Scholar] [CrossRef]
- Samant, B.S.; Sukhthankar, M.G. Synthesis and comparison of antimalarial activity of febrifugine derivatives including halofuginone. Med. Chem. 2009, 5, 293–300. [Google Scholar]
- McLaughlin, N.P.; Evans, P. Dihydroxylation of vinyl sulfones: Stereoselective synthesis of (+)-and (−)-febrifugine and halofuginone. J. Org. Chem. 2010, 75, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.; et al. An antimalarial alkaloid from hydrangea. Ix. Synthesis of 3-[β-keto-γ-(4-hydroxy-2-piperidyl) propyl]-4-quinazolone, an isomer. J. Org. Chem. 1952, 17, 97–108. [Google Scholar]
- Baker, B.; et al. An antimalarial alkaloid from hydrangea. v. some 3-(β-keto-sec-aminoalkyl)-4-quinazolones. J. Org. Chem. 1952, 17, 52–57. [Google Scholar]
- Baker, B.; et al. An antimalarial alkaloid from hydrangea. vii. 3-[β-keto-γ-(5-hydroxy-2-piperidyl) propyl]-4-quinazolone, an isomer. J. Org. Chem. 1952, 17, 68–76. [Google Scholar]
- Baker, B.; et al. An antimalarial alkaloid from hydrangea. vi. a second synthesis of 3-[β-keto-γ-(2-piperidyl)-propyl]-4-quinazolone. J. Org. Chem. 1952, 17, 58–67. [Google Scholar]
- Baker, B.; et al. An antimalarial alkaloid from hydrangea. Viii. Attempted synthesis of 3-[β-keto-γ-(4-hydroxy-2-piperidyl) propyl]-4-quinazolone by the diketone approach. J. Org. Chem. 1952, 17, 77–96. [Google Scholar]
- Baker, B.; Schaub, R.E.; Williams, J.H. An antimalarial alkaloid from hydrangea. x. synthesis of 3-[β-keto-γ-(4-methyl-2-pyrrolidyl) propyl]-4-quinazolone. J. Org. Chem. 1952, 17, 109–115. [Google Scholar]
- Baker, B.; SCHAUB, R.E.; WILLIAMS, J.H. An antimalarial alkaloid from hydrangea. Xi. Synthesis of 3-[β-keto-γ-(3-and 4-hydroxymethyl-2-pyrrolidyl) propyl]-4-quinazolones. J. Org. Chem. 1952, 17, 116–131. [Google Scholar]
- Linder, M.R.; et al. (2R, 3S)-(+)-and (2S, 3R)-(−)-Halofuginone lactate: Synthesis, absolute configuration, and activity against Cryptosporidium parvum. Bioorganic Med. Chem. Lett. 2007, 17, 4140–4143. [Google Scholar]
- Katoh, M.; et al. Stereocontrolled synthesis of a potent antimalarial alkaloid,(+)-febrifugine. Tetrahedron Lett. 2004, 45, 6221–6223. [Google Scholar]
- Zaidan, R.K.; Smullen, S.; Evans, P. Asymmetric synthesis of (+)-and (−)-deoxyfebrifugine and deoxyhalofuginone. Tetrahedron Lett. 2015, 56, 6433–6435. [Google Scholar]
- Fishman, M.; Cruickshank, P.A. Febrifugine antimalarial agents. I. Pyridine Analog. Febrifugine. J. Med. Chem. 1970, 13, 155–156. [Google Scholar] [PubMed]
- Takeuchi, Y.; et al. Synthesis and antimalarial activity of febrifugine derivatives. Chem. Pharm. Bull. 2001, 49, 721–725. [Google Scholar]
- Kim, H.-S. Synthesis and antimalarial activity of dl-deoxyfebrifugine. Heterocycles 1999, 51, 1869–1875. [Google Scholar]
- Kikuchi, H.; et al. Synthesis of febrifugine derivatives and development of an effective and safe tetrahydroquinazoline-type antimalarial. Eur. J. Med. Chem. 2014, 76, 10–19. [Google Scholar] [PubMed]
- Kikuchi, H.; et al. Potent antimalarial febrifugine analogues against the plasmodium malaria parasite. J. Med. Chem. 2002, 45, 2563–2570. [Google Scholar]
- Takeuchi, Y.; et al. Re-revision of the stereo structure of piperidine lactone, an intermediate in the synthesis of febrifugine. Chem. Pharm. Bull. 2002, 50, 1011–1012. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; et al. Identification and characterization of potential antioxidant components in Isodon amethystoides (Benth.) Hara tea leaves by UPLC-LTQ-Orbitrap-MS. Food Chem. Toxicol. 2021, 148, 111961. [Google Scholar]
- Zhu, S.; et al. Febrifugine analogue compounds: synthesis and antimalarial evaluation. Bioorganic Med. Chem. 2012, 20, 927–932. [Google Scholar] [CrossRef]
- Zhu, S.; et al. Synthesis and evaluation of 4-quinazolinone compounds as potential antimalarial agents. Eur. J. Med. Chem. 2010, 45, 3864–3869. [Google Scholar]
- Mai, H.D.T.; et al. Synthesis of febrifuginol analogues and evaluation of their biological activities. Tetrahedron Lett. 2014, 55, 7226–7228. [Google Scholar]
- Wu, L.; Chen, P.; Liu, G. Pd (II)-catalyzed aminofluorination of alkenes in total synthesis 6-(R)-fluoroswainsonine and 5-(R)-fluorofebrifugine. Org. Lett. 2016, 18, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Perali, R.S.; Bandi, A. Stereoselective total synthesis of all the stereoisomers of (+)- and (−)-febrifugine and halofuginone. Tetrahedron Lett. 2020, 61, 152151. [Google Scholar] [CrossRef]
- Ziegler, S.; et al. Target identification for small bioactive molecules: finding the needle in the haystack. Angew. Chem. Int. Ed. 2013, 52, 2744–2792. [Google Scholar] [CrossRef]
- Kang, Y.B.; et al. Bioactive molecules: current trends in discovery, synthesis, delivery and testing. IeJSME 2013, 7 (Suppl. S1), S32–S46. [Google Scholar]
- Kuehl Jr, F.A. C.F. Spencer, and K. Folkers, Alkaloids of Dichroa Febrifuga Lour. J. Am. Chem. Soc. 1948, 70, 2091–2093. [Google Scholar] [CrossRef]
- Brickwedde, L.H.; et al. An Appeal. Science 1946, 103, 59–59. [Google Scholar] [CrossRef]
- Shahiduzzaman, M.; et al. Combination of cell culture and quantitative PCR for screening of drugs against Cryptosporidium parvum. Vet. Parasitol. 2009, 162, 271–277. [Google Scholar] [CrossRef]
- Rock, F.L.; et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. science 2007, 316, 1759–1761. [Google Scholar] [CrossRef]
- Ibba, M.; Söll, D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef]
- Cheng, B.; et al. Structure-Guided Design of Halofuginone Derivatives as ATP-Aided Inhibitors Against Bacterial Prolyl-tRNA Synthetase. J. Med. Chem. 2022, 65, 15840–15855. [Google Scholar] [PubMed]
- Allport, J.; et al. Efficacy of mupirocin, neomycin and octenidine for nasal Staphylococcus aureus decolonisation: a retrospective cohort study. Antimicrob. Resist. Infect. Control 2022, 11, 1–8. [Google Scholar]
- Tao, J. and P. Schimmel, Inhibitors of aminoacyl-tRNA synthetases as novel anti-infectives. Expert Opin. Investig. Drugs 2000, 9, 1767–1775. [Google Scholar]
- Williamson, D.A.; Carter, G.P.; Howden, B.P. Current and emerging topical antibacterials and antiseptics: agents, action, and resistance patterns. Clin. Microbiol. Rev. 2017, 30, 827–860. [Google Scholar] [CrossRef] [PubMed]
- Kaduk, J.A.; Gates-Rector, S.; Blanton, T.N. Crystal structure of halofuginone hydrobromide, C16H18BrClN3O3Br. Powder Diffr. 2023, 38, 7–14. [Google Scholar] [CrossRef]
- Committee, A.M. Determination of halofuginone hydrobromide in medicated animal feeds. Analyst 1983, 108, 1252–1256. [Google Scholar]
- Hu, H.; et al. Therapeutic poly(amino acid)s as drug carriers for cancer therapy. Chin. Chem. Lett. 2023, 34, 107953. [Google Scholar]
- Zhang, J.; Yao, Q.; Liu, Z. A novel synthesis of the efficient anti-coccidial drug halofuginone hydrobromide. Molecules 2017, 22, 1086. [Google Scholar] [CrossRef]
- Steiner, T. Hydrogen-bond distances to halide ions in organic and organometallic crystal structures: up-to-date database study. Acta Crystallogr. Sect. B: Struct. Sci. 1998, 54, 456–463. [Google Scholar]
- Kortagere, S.; Ekins, S.; Welsh, W.J. Halogenated ligands and their interactions with amino acids: implications for structure–activity and structure–toxicity relationships. J. Mol. Graph. Model. 2008, 27, 170–177. [Google Scholar]
- Thallapally, P.K.; Nangia, A. A Cambridge Structural Database analysis of the C–H⋯ Cl interaction: C–H⋯ Cl− and C–H⋯ Cl–M often behave as hydrogen bonds but C–H⋯ Cl–C is generally a van der Waals interaction. CrystEngComm 2001, 3, 114–119. [Google Scholar] [CrossRef]
- Rowley, M.; et al. 3-(4-Fluoropiperidin-3-yl)-2-phenylindoles as high affinity, selective, and orally bioavailable h5-HT2A receptor antagonists. J. Med. Chem. 2001, 44, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- van Niel, M.B.; et al. Fluorination of 3-(3-(piperidin-1-yl) propyl) indoles and 3-(3-(piperazin-1-yl) propyl) indoles gives selective human 5-HT1D receptor ligands with improved pharmacokinetic profiles. J. Med. Chem. 1999, 42, 2087–2104. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, S.B.; et al. Discovery of 1-(4-fluorophenyl)-(3 R)-[3-(4-fluorophenyl)-(3 S)-hydroxypropyl]-(4 S)-(4-hydroxyphenyl)-2-azetidinone (SCH 58235): a designed, potent, orally active inhibitor of cholesterol absorption. J. Med. Chem. 1998, 41, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-J.; et al. Fluorine substitution can block CYP3A4 metabolism-dependent inhibition: identification of (S)-N-[1-(4-fluoro-3-morpholin-4-ylphenyl) ethyl]-3-(4-fluorophenyl) acrylamide as an orally bioavailable KCNQ2 opener devoid of CYP3A4 metabolism-dependent inhibition. J. Med. Chem. 2003, 46, 3778–3781. [Google Scholar] [PubMed]
- Thomas, E.L.; et al. Oxidation of Bromide by the Human Leukocyte Enzymes Myeloperoxidase and Eosinophil Peroxidase: FORMATION OF BROMAMINES (∗). J. Biol. Chem. 1995, 270, 2906–2913. [Google Scholar] [CrossRef]
- Ali, H.; Rousseau, J.; Van Lier, J.E. 7. alpha.-Methyl-and 11. beta.-ethoxy-substitution of iodine-125-labeled [125I] 16. alpha.-iodoestradiol: effect on estrogen receptor-mediated target tissue uptake. J. Med. Chem. 1993, 36, 264–271. [Google Scholar] [CrossRef]
- Bae, J.H.; et al. Crystallographic evidence for isomeric chromophores in 3-fluorotyrosyl-green fluorescent protein. ChemBioChem 2004, 5, 720–722. [Google Scholar] [CrossRef]
- Tang, Y.; et al. Stabilization of coiled-coil peptide domains by introduction of trifluoroleucine. Biochemistry 2001, 40, 2790–2796. [Google Scholar] [CrossRef]
- Lee, K.-H.; et al. Fluorous effect in proteins: de novo design and characterization of a four-α-helix bundle protein containing hexafluoroleucine. Biochemistry 2004, 43, 16277–16284. [Google Scholar] [CrossRef]
- Lamora, A.; et al. Anticancer activity of halofuginone in a preclinical model of osteosarcoma: inhibition of tumor growth and lung metastases. Oncotarget 2015, 6, 14413. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; et al. Halofuginone attenuates osteoarthritis by inhibition of TGF-β activity and H-type vessel formation in subchondral bone. Ann. Rheum. Dis. 2016, 75, 1714–1721. [Google Scholar] [CrossRef]
- Anderson, A.; et al. Analysis of the anti-coccidial drug, halofuginone, in chicken tissue and chicken feed using high-performance liquid chromatography. J. Chromatogr. A 1981, 212, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Pines, M. Halofuginone for fibrosis, regeneration and cancer in the gastrointestinal tract. World J. Gastroenterol. : WJG 2014, 20, 14778. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.L.; et al. Halofuginone induces the apoptosis of breast cancer cells and inhibits migration via downregulation of matrix metalloproteinase-9. Int. J. Oncol. 2014, 44, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.-H.; et al. DT-13 attenuates human lung cancer metastasis via regulating NMIIA activity under hypoxia condition. Oncol. Rep. 2016, 36, 991–999. [Google Scholar] [CrossRef]
- Duan, H.; et al. Computational pharmacology and bioinformatics to explore the potential mechanism of Schisandra against atherosclerosis. Food Chem. Toxicol. 2021, 150, 112058. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Rodríguez, J.J.; et al. Plant antimicrobial peptides as potential anticancer agents. BioMed Res. Int. 2015. [Google Scholar] [CrossRef]
- Zhou, H.; et al. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature 2013, 494, 121–124. [Google Scholar] [CrossRef]
- Jiang, S.; et al. Antimalarial activities and therapeutic properties of febrifugine analogs. Antimicrob. Agents Chemother. 2005, 49, 1169–1176. [Google Scholar] [CrossRef]
- Derbyshire, E.R.; Mazitschek, R.; Clardy, J. Characterization of Plasmodium liver stage inhibition by halofuginone. ChemMedChem 2012, 7, 844–849. [Google Scholar] [PubMed]
- Guo, J.; et al. Structure-guided optimization and mechanistic study of a class of quinazolinone-threonine hybrids as antibacterial ThrRS inhibitors. Eur. J. Med. Chem. 2020, 207, 112848. [Google Scholar]
- Tye, M.A.; et al. Elucidating the path to Plasmodium prolyl-tRNA synthetase inhibitors that overcome halofuginone resistance. Nat. Commun. 2022, 13, 4976. [Google Scholar] [PubMed]
- Fingleton, B.; Matrisian, L.M. Matrix metalloproteinases as targets for therapy in Kaposi sarcoma. Curr. Opin. Oncol. 2001, 13, 368–373. [Google Scholar] [PubMed]
- Jain, V.; et al. Structural and functional analysis of the anti-malarial drug target prolyl-tRNA synthetase. J. Struct. Funct. Genom. 2014, 15, 181–190. [Google Scholar]
- Sandoval, D.R.; et al. The prolyl-tRNA synthetase inhibitor halofuginone inhibits SARS-CoV-2 infection. bioRxiv, 2021: p. 2021.03. 22.436522.
- Fitz-Coy, S.; Edgar, S. Pathogenicity and control of Eimeria mitis infections in broiler chickens. Avian Diseases, 1992: p. 44-48.
- Silverlås, C.; Björkman, C.; Egenvall, A. Systematic review and meta-analyses of the effects of halofuginone against calf cryptosporidiosis. Prev. Vet. Med. 2009, 91, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; et al. A novel therapeutic vaccine based on graphene oxide nanocomposite for tumor immunotherapy. Chin. Chem. Lett. 2022, 33, 4089–4095. [Google Scholar] [CrossRef]
- Wang, W.; et al. Targeted intervention of natural medicinal active ingredients and traditional Chinese medicine on epigenetic modification: possible strategies for prevention and treatment of atherosclerosis. Phytomedicine 2024, 122, 155139. [Google Scholar]
- De Waele, V.; et al. Control of cryptosporidiosis in neonatal calves: use of halofuginone lactate in two different calf rearing systems. Prev. Vet. Med. 2010, 96, 143–151. [Google Scholar] [CrossRef]
- Nelson, E.F.; et al. Halofuginone down-regulates Smad3 expression and inhibits the TGFbeta-induced expression of fibrotic markers in human corneal fibroblasts. Mol. Vis. 2012, 18, 479. [Google Scholar]
- Pines, M. Targeting TGFβ signaling to inhibit fibroblast activation as a therapy for fibrosis and cancer: effect of halofuginone. Expert Opin. Drug Discov. 2008, 3, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Pines, M.; Nagler, A. Halofuginone: a novel antifibrotic therapy. Gen. Pharmacol. : Vasc. Syst. 1998, 30, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Zhai, K.; et al. Ginsenoside Rg1 ameliorates blood–brain barrier disruption and traumatic brain injury via attenuating macrophages derived exosomes miR-21 release. Acta Pharm. Sin. B 2021, 11, 3493–3507. [Google Scholar] [CrossRef] [PubMed]
- Pines, M.; et al. Reduction in dermal fibrosis in the tight-skin (Tsk) mouse after local application of halofuginone. Biochem. Pharmacol. 2001, 62, 1221–1227. [Google Scholar] [CrossRef]
- Levi-Schaffer, F.; et al. Inhibition of collagen synthesis and changes in skin morphology in murine graft-versus-host disease and tight skin mice: effect of halofuginone. J. Investig. Dermatol. 1996, 106, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Nagler, A.; et al. Reduction in pulmonary fibrosis in vivo by halofuginone. Am. J. Respir. Crit. Care Med. 1996, 154, 1082–1086. [Google Scholar] [CrossRef]
- Nagler, A.; et al. Suppression of hepatocellular carcinoma growth in mice by the alkaloid coccidiostat halofuginone. Eur. J. Cancer 2004, 40, 1397–1403. [Google Scholar] [CrossRef]
- Nagler, A.; et al. Halofuginone-an inhibitor of collagen type I synthesis-prevents postoperative formation of abdominal adhesions. Ann. Surg. 1998, 227, 575–582. [Google Scholar] [CrossRef]
- Pines, M.; et al. Halofuginone, a specific inhibitor of collagen type I synthesis, prevents dimethylnitrosamine-induced liver cirrhosis. J. Hepatol. 1997, 27, 391–398. [Google Scholar] [CrossRef]
- Zion, O.; et al. Inhibition of transforming growth factor β signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancreas 2009, 38, 427–435. [Google Scholar] [CrossRef]
- Huebner, K.D.; et al. Functional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginone. Am. J. Physiol. -Heart Circ. Physiol. 2008, 294, H1550–H1561. [Google Scholar] [CrossRef] [PubMed]
- Fromes, Y.; et al. Inhibition of Fibrosis and Improvement of Function of the Myopathic Hamster Cardiac Muscle by Halofuginone. Exp. Clin. Cardiol. 2014, 20, 2351–2383. [Google Scholar]
- Roffe, S.; et al. Halofuginone inhibits Smad3 phosphorylation via the PI3K/Akt and MAPK/ERK pathways in muscle cells: effect on myotube fusion. Exp. Cell Res. 2010, 316, 1061–1069. [Google Scholar] [PubMed]
- Spector, I.; et al. Involvement of host stroma cells and tissue fibrosis in pancreatic tumor development in transgenic mice. PLoS One 2012, 7, e41833. [Google Scholar] [CrossRef] [PubMed]
- Gnainsky, Y.; et al. Halofuginone, an inhibitor of collagen synthesis by rat stellate cells, stimulates insulin-like growth factor binding protein-1 synthesis by hepatocytes. J. Hepatol. 2004, 40, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.S. Helper (TH1, TH2, TH17) and regulatory cells (Treg, TH3, NKT) in rheumatoid arthritis. Reumatol. Clin. 2009, 5, 1–5. [Google Scholar]
- Guo, J.; et al. Discovery of novel tRNA-amino acid dual-site inhibitors against threonyl-tRNA synthetase by fragment-based target hopping. Eur. J. Med. Chem. 2020, 187, 111941. [Google Scholar]
- Pietrella, D.; et al. Th17 cells and IL-17 in protective immunity to vaginal candidiasis. PloS One 2011, 6, e22770. [Google Scholar]
- Khan, Z.M.; Vanderberg, J.P. Specific inflammatory cell infiltration of hepatic schizonts in BALB/c mice immunized with attenuated Plasmodium yoelii sporozoites. Int. Immunol. 1992, 4, 711–718. [Google Scholar] [CrossRef]
- Liang, J.; et al. The Effect of Antifibrotic Drug Halofugine on Th17 Cells in Concanavalin A-Induced Liver Fibrosis. Scand. J. Immunol. 2014, 79, 163–172. [Google Scholar] [CrossRef]
- Wong, G.L.-H. Prediction of fibrosis progression in chronic viral hepatitis. Clin. Mol. Hepatol. 2014, 20, 228. [Google Scholar] [CrossRef]
- Jain, P.P.; et al. Halofuginone, a promising drug for treatment of pulmonary hypertension. Br. J. Pharmacol. 2021, 178, 3373–3394. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; et al. Fighting hypoxia to improve photodynamic therapy-driven immunotherapy: Alleviating, exploiting and disregarding. Chinese Chemical Letters, 2024: p. 109957.
- Khan, G.J.; et al. Non-Muscle Myosin IIC as a Prognostic and Therapeutic Target in Cancer. J. Biomed. Nanotechnol. 2024, 20, 438–456. [Google Scholar] [CrossRef]
- Sato, S.; et al. Halofuginone prevents extracellular matrix deposition in diabetic nephropathy. Biochem. Biophys. Res. Commun. 2009, 379, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Shabgah, A.G.; Fattahi, E.; Shahneh, F.Z. Interleukin-17 in human inflammatory diseases. Adv. Dermatol. Allergol. /Postępy Dermatol. I Alergol. 2014, 31, 256–261. [Google Scholar]
- Leiba, M.; et al. Halofuginone inhibits NF-κB and p38 MAPK in activated T cells. J. Leukoc. Biol. 2006, 80, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.J.; et al. Thymus as Incontrovertible Target of Future Immune Modulatory Therapeutics. Endocr. Metab. Immune Disord. Drug Targets 2024. [Google Scholar] [CrossRef]
- Tajdin, F.; et al. Antibiotic therapy in pyogenic meningitis in paediatric patients. J. Coll. Physicians Surg. Pak. 2013, 23, 703–707. [Google Scholar]
- Chen, K.; et al. The protective effects of a D-tetra-peptide hydrogel adjuvant vaccine against H7N9 influenza virus in mice. Chin. Chem. Lett. 2023, 34, 107446. [Google Scholar] [CrossRef]
- Ni, J.; et al. Halofuginone ameliorates systemic lupus erythematosus by targeting Blk in myeloid-derived suppressor cells. Int. Immunopharmacol. 2023, 114, 109487. [Google Scholar] [CrossRef]
- Wu, B.W. Molecular Mechanism of the Immunomodulatory Drug Halofuginone. 2016: Harvard University.
- Chen, L.; et al. Biomaterial-induced macrophage polarization for bone regeneration. Chin. Chem. Lett. 2023, 34, 107925. [Google Scholar] [CrossRef]
- Zhai, K.; et al. Extracellular vesicle-derived miR-146a as a novel crosstalk mechanism for high-fat induced atherosclerosis by targeting SMAD4. J. Adv. Res. 2024. [Google Scholar]
- Wang, S.-F.; et al. Autophagy modulators from traditional Chinese medicine: Mechanisms and therapeutic potentials for cancer and neurodegenerative diseases. J. Ethnopharmacol. 2016, 194, 861–876. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; et al. The pro-apoptotic role of autophagy in breast cancer. Br. J. Cancer 2014, 111, 309–317. [Google Scholar] [CrossRef]
- Zocchi, M.R.; Poggi, A. Targeting the microenvironment in hematological malignancies: how to condition both stromal and effector cells to overcome cancer spreading. Curr. Med. Chem. 2011, 18, 5172–5173. [Google Scholar]
- Li, H.; et al. DT-13, a saponin monomer of dwarf lilyturf tuber, induces autophagy and potentiates anti-cancer effect of nutrient deprivation. Eur. J. Pharmacol. 2016, 781, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; et al. Emerging nanomedicine and prodrug delivery strategies for the treatment of inflammatory bowel disease. Chin. Chem. Lett. 2022, 33, 4449–4460. [Google Scholar]
- Zhai, K.-f.; et al. Salicin from Alangium chinense ameliorates rheumatoid arthritis by modulating the Nrf2-HO-1-ROS pathways. J. Agric. Food Chem. 2018, 66, 6073–6082. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.; Jung, K.-W.; Won, Y.-J. Colorectal cancer mortality in Hong Kong of China, Japan, South Korea, and Singapore. World J. Gastroenterol. : WJG 2013, 19, 979. [Google Scholar]
- Koseoglu, S.; et al. AKT1, AKT2 and AKT3-dependent cell survival is cell line-specific and knockdown of all three isoforms selectively induces apoptosis in 20 human tumor cell lines. Cancer Biol. Ther. 2007, 6, 755–762. [Google Scholar] [CrossRef]
- Francipane, M.G.; Lagasse, E. mTOR pathway in colorectal cancer: an update. Oncotarget 2014, 5, 49. [Google Scholar] [PubMed]
- Ucaryilmaz Metin, C.; Ozcan, G. Comprehensive bioinformatic analysis reveals a cancer-associated fibroblast gene signature as a poor prognostic factor and potential therapeutic target in gastric cancer. BMC Cancer 2022, 22, 692. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.S.; Klostergaard, J. Chapter 7 - STAT3 Signaling in Cancer: Small Molecule Intervention as Therapy?, in Anti-Angiogenesis Drug Discovery and Development, R. Atta ur and M.I. Choudhary, Editors. 2014, Elsevier. p. 216-267.
- Khan, G.J.; et al. Versatility of cancer associated fibroblasts: commendable targets for anti-tumor therapy. Curr. Drug Targets 2018, 19, 1573–1588. [Google Scholar]
- Zhang, Y.-J.; et al. mTOR signaling pathway is a target for the treatment of colorectal cancer. Ann. Surg. Oncol. 2009, 16, 2617–2628. [Google Scholar] [PubMed]
- Sun, Q.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. 2011, 108, 4129–4134. [Google Scholar] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. science 2009, 324, 1029–1033. [Google Scholar]
- Yalcin, S.; et al. ROS-mediated amplification of AKT/mTOR signalling pathway leads to myeloproliferative syndrome in Foxo3−/− mice. EMBO J. 2010, 29, 4118–4131. [Google Scholar] [CrossRef]
- Düvel, K.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; et al. A selective HK2 degrader suppresses SW480 cancer cell growth by degrading HK2. Chin. Chem. Lett. 2024, 35, 109264. [Google Scholar]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar]
- Roberts, D.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; et al. Manganese superoxide dismutase deficiency triggers mitochondrial uncoupling and the Warburg effect. Oncogene 2015, 34, 4229–4237. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; et al. Design, synthesis and biological evaluation of novel phthalazinone acridine derivatives as dual PARP and Topo inhibitors for potential anticancer agents. Chin. Chem. Lett. 2020, 31, 404–408. [Google Scholar]
- Zhang, X.; et al. Synthesis and biological evaluation of piperidyl benzimidazole carboxamide derivatives as potent PARP-1 inhibitors and antitumor agents. Chin. Chem. Lett. 2020, 31, 136–140. [Google Scholar]
- Anderson, B.O.; et al. The Global Breast Cancer Initiative: a strategic collaboration to strengthen health care for non-communicable diseases. Lancet Oncol. 2021, 22, 578–581. [Google Scholar] [PubMed]
- Huyen, C.T.T.; et al. Chemical constituents from Cimicifuga dahurica and their anti-proliferative effects on MCF-7 breast cancer cells. Molecules 2018, 23, 1083. [Google Scholar] [CrossRef]
- Gennari, A.; et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer☆. Ann. Oncol. 2021, 32, 1475–1495. [Google Scholar]
- Viale, G.; et al. Retrospective study to estimate the prevalence and describe the clinicopathological characteristics, treatments received, and outcomes of HER2-low breast cancer. ESMO Open 2023, 8, 101615. [Google Scholar]
- Schalper, K.A.; et al. A retrospective population-based comparison of HER2 immunohistochemistry and fluorescence in situ hybridization in breast carcinomas: impact of 2007 American Society of Clinical Oncology/College of American Pathologists criteria. Arch. Pathol. Lab. Med. 2014, 138, 213–219. [Google Scholar]
- Gao, Y.; et al. DT-13 inhibits breast cancer cell migration via non-muscle myosin II-A regulation in tumor microenvironment synchronized adaptations. Clin. Transl. Oncol. 2020, 22, 1591–1602. [Google Scholar]
- Sonnenblick, A.; et al. STAT3 activation in HER2-positive breast cancers: Analysis of data from a large prospective trial. Int. J. Cancer 2021, 148, 1529–1535. [Google Scholar] [PubMed]
- Yuan, K.; et al. Complement C3 overexpression activates JAK2/STAT3 pathway and correlates with gastric cancer progression. J. Exp. Clin. Cancer Res. 2020, 39, 1–15. [Google Scholar]
- Hu, F.; et al. The autophagy-independent role of BECN1 in colorectal cancer metastasis through regulating STAT3 signaling pathway activation. Cell Death Dis. 2020, 11, 304. [Google Scholar] [PubMed]
- You, W.; et al. IL-26 promotes the proliferation and survival of human gastric cancer cells by regulating the balance of STAT1 and STAT3 activation. PLoS One 2013, 8, e63588. [Google Scholar]
- Tajmohammadi, I.; et al. Identification of Nrf2/STAT3 axis in induction of apoptosis through sub-G 1 cell cycle arrest mechanism in HT-29 colon cancer cells. J. Cell. Biochem. 2019, 120, 14035–14043. [Google Scholar] [PubMed]
- Lau, A.; et al. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol. 2013, 33, 2436–2446. [Google Scholar] [PubMed]
- Kim, S.-J.; et al. Interaction of Nrf2 with dimeric STAT3 induces IL-23 expression: Implications for breast cancer progression. Cancer Lett. 2021, 500, 147–160. [Google Scholar] [CrossRef]
- Sheng, S.; et al. Aberrant expression of IL-23/IL-23R in patients with breast cancer and its clinical significance. Mol. Med. Rep. 2018, 17, 4639–4644. [Google Scholar]
- Betti, M.; et al. Genetic predisposition for malignant mesothelioma: A concise review. Mutat. Res. /Rev. Mutat. Res. 2019, 781, 1–10. [Google Scholar]
- Yap, T.A.; et al. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar]
- Sui, X.; et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Juárez, P.; et al. Halofuginone inhibits TGF-β/BMP signaling and in combination with zoledronic acid enhances inhibition of breast cancer bone metastasis. Oncotarget 2017, 8, 86447. [Google Scholar] [CrossRef]
- Popov, Y.; et al. Halofuginone induces matrix metalloproteinases in rat hepatic stellate cells via activation of p38 and NFκB. J. Biol. Chem. 2006, 281, 15090–15098. [Google Scholar] [CrossRef] [PubMed]
- Carlson, T.J.; et al. Halofuginone-induced amino acid starvation regulates Stat3-dependent Th17 effector function and reduces established autoimmune inflammation. J. Immunol. 2014, 192, 2167–2176. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; et al. DT-13 inhibits cancer cell migration by regulating NMIIA indirectly in the tumor microenvironment. Oncol. Rep. 2016, 36, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; et al. The potential mechanism of Isodon suzhouensis against COVID-19 via EGFR/TLR4 pathways. Food Sci. Hum. Wellness 2024. [Google Scholar] [CrossRef]
- Demiroglu-Zergeroglu, A.; et al. Anticarcinogenic effects of halofuginone on lung-derived cancer cells. Cell Biol. Int. 2020, 44, 1934–1944. [Google Scholar] [CrossRef] [PubMed]
- Perepelyuk, M.; et al. Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am. J. Physiol. -Gastrointest. Liver Physiol. 2013, 304, G605–G614. [Google Scholar] [CrossRef]
- Ferlay, J.; et al. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Gao, J.; et al. Rural–urban, sex variations, and time trend of primary liver cancer incidence in China, 1988–2005. Eur. J. Cancer prevention 2013, 22, 448–454. [Google Scholar] [CrossRef]
- Qi, J.S.; Wang, W.H.; Li, F.Q. Combination of interventional adenovirus-p53 introduction and ultrasonic irradiation in the treatment of liver cancer. Oncol. Lett. 2015, 9, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; et al. Preventive effect of halofuginone on concanavalin A-induced liver fibrosis. PLoS One 2013, 8, e82232. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; et al. Total flavonoid extract from Dracocephalum moldavica L. improves pulmonary fibrosis by reducing inflammation and inhibiting the hedgehog signaling pathway. Phytother. Res. 2023, 37, 2745–2758. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; et al. Determination of the anti-oxidative stress mechanism of Isodon suzhouensis leaves by employing bioinformatic and novel research technology. ACS Omega 2023, 8, 3520–3529. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013, 27, 1101–1114. [Google Scholar] [CrossRef]
- Zhao, H.; et al. Expression of the p12 subunit of human DNA polymerase δ (Pol δ), CDK inhibitor p21WAF1, Cdt1, cyclin A, PCNA and Ki-67 in relation to DNA replication in individual cells. Cell Cycle 2014, 13, 3529–3540. [Google Scholar] [CrossRef]
- Zheng, B.a.; Chai, R.; Yu, X. Downregulation of NIT2 inhibits colon cancer cell proliferation and induces cell cycle arrest through the caspase-3 and PARP pathways. Int. J. Mol. Med. 2015, 35, 1317–1322. [Google Scholar] [CrossRef]
- Xu, W.; Liu, L.; Hornby, D. c-IAP1 binds and processes PCSK9 protein: linking the c-IAP1 in a TNF-α pathway to PCSK9-mediated LDLR degradation pathway. Molecules 2012, 17, 12086–12101. [Google Scholar] [CrossRef]
- Wong, J.C.; Bathina, M.; Fiscus, R.R. Cyclic GMP/protein kinase G type-Iα (PKG-Iα) signaling pathway promotes CREB phosphorylation and maintains higher c-IAP1, livin, survivin, and Mcl-1 expression and the inhibition of PKG-Iα kinase activity synergizes with cisplatin in non-small cell lung cancer cells. J. Cell. Biochem. 2012, 113, 3587–3598. [Google Scholar]
- Jilany Khan, G.; et al. TGF-β1 causes EMT by regulating N-Acetyl glucosaminyl transferases via downregulation of non muscle myosin II-A through JNK/P38/PI3K pathway in lung cancer. Curr. Cancer Drug Targets 2018, 18, 209–219. [Google Scholar] [CrossRef]
- Rwibasira Rudinga, G.; Khan, G.J.; Kong, Y. Protease-activated receptor 4 (PAR4): a promising target for antiplatelet therapy. Int. J. Mol. Sci. 2018, 19, 573. [Google Scholar] [CrossRef]
- Zhai, K.; et al. Protective effects of Isodon Suzhouensis extract and glaucocalyxin A on chronic obstructive pulmonary disease through SOCS3–JAKs/STATs pathway. Food Front. 2023, 4, 511–523. [Google Scholar]
- Wang, X.; et al. Interdependent and independent multidimensional role of tumor microenvironment on hepatocellular carcinoma. Cytokine 2018, 103, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; et al. Phase ii aids malignancy consortium trial of topical halofuginone in aids-related kaposi’s sarcoma. Journal of acquired immune deficiency syndromes (1999) 2011, 56, 64. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; et al. Halofuginone for cancer treatment: A systematic review of efficacy and molecular mechanisms. J. Funct. Foods 2022, 98, 105237. [Google Scholar]
- Khan, G.J.; et al. Pharmacological effects and potential therapeutic targets of DT-13. Biomed. Pharmacother. 2018, 97, 255–263. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, M.J.A.; et al. Phase I and pharmacokinetic study of halofuginone, an oral quinazolinone derivative in patients with advanced solid tumours. Eur. J. Cancer 2006, 42, 1768–1774. [Google Scholar] [CrossRef]
- Pines, M.; et al. Halofuginone to treat fibrosis in chronic graft-versus-host disease and scleroderma. Biol. Blood Marrow Transplant. 2003, 9, 417–425. [Google Scholar] [CrossRef]
- Tomazini, B.M.; et al. Halofuginone for non-hospitalized adult patients with COVID-19 a multicenter, randomized placebo-controlled phase 2 trial. The HALOS trial. Plos One 2024, 19, e0299197. [Google Scholar] [CrossRef]
- Bharate, S.S. Modulation of biopharmaceutical properties of acidic drugs using cationic counterions: A critical analysis of FDA-approved pharmaceutical salts. Int. J. Pharm. 2021, 607, 120993. [Google Scholar] [CrossRef]
- Fulmali, A.; Bharate, S.S. Phosphate moiety in FDA-approved pharmaceutical salts and prodrugs. Drug Dev. Res. 2022, 83, 1059–1074. [Google Scholar] [PubMed]
- Bharate, S.S. Carboxylic acid counterions in FDA-approved pharmaceutical salts. Pharm. Res. 2021, 38, 1307–1326. [Google Scholar] [PubMed]
- Kramer, S.F.; Flynn, G.L. Solubility of Organic Hydrochlorides. J. Pharm. Sci. 1972, 61, 1896–1904. [Google Scholar]
- Serajuddin, A.T. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [PubMed]
- Lipinski, C.A.; et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar]
- Berge, S.M.; Bighley, L.D.; Monkhouse, D.C. Pharmaceutical salts. J. Pharm. Sci. 1977, 66, 1–19. [Google Scholar]
- Hossain Mithu, M.S.; et al. Advanced methodologies for pharmaceutical salt synthesis. Cryst. Growth Des. 2021, 21, 1358–1374. [Google Scholar]
- Bharate, S.S. Recent developments in pharmaceutical salts: FDA approvals from 2015 to 2019. Drug Discov. Today 2021, 26, 384–398. [Google Scholar]
- Bharate, S.S.; Vishwakarma, R.A. Impact of preformulation on drug development. Expert Opin. Drug Deliv. 2013, 10, 1239–1257. [Google Scholar] [CrossRef]
- Bastin, R.J.; Bowker, M.J.; Slater, B.J. Salt selection and optimisation procedures for pharmaceutical new chemical entities. Org. Process Res. Dev. 2000, 4, 427–435. [Google Scholar]
- Kumar, L.; Amin, A.; Bansal, A.K. An overview of automated systems relevant in pharmaceutical salt screening. Drug Discov. Today 2007, 12, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; et al. Salt screening and characterization of ciprofloxacin. Acta Crystallogr. Sect. B: Struct. Sci. Cryst. Eng. Mater. 2016, 72, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Thorson, M.R.; et al. A microfluidic platform for pharmaceutical salt screening. Lab A Chip 2011, 11, 3829–3837. [Google Scholar] [CrossRef]
- Kojima, T.; et al. Crystalline form information from multiwell plate salt screening by use of Raman microscopy. Pharm. Res. 2006, 23, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.E.; et al. Unraveling complexity in the solid form screening of a pharmaceutical salt: Why so many forms? Why so few? Cryst. Growth Des. 2017, 17, 5349–5365. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-Q.T. Salt screening and selection: new challenges and considerations in the modern pharmaceutical research and development paradigm. Developing solid oral dosage forms,.
- Barron, K.; et al. Comparative evaluation of the in vitro effects of hydralazine and hydralazine acetonide on arterial smooth muscle. Br. J. Pharmacol. 1977, 61, 345. [Google Scholar] [CrossRef]
- Banerji, U.; et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin. Cancer Res. 2010, 16, 1613–1623. [Google Scholar] [CrossRef]
- Borsiczky, B.; et al. The effect of clopidogrel besylate and clopidogrel hydrogensulfate on platelet aggregation in patients with coronary artery disease: a retrospective study. Thromb. Res. 2012, 129, 700–703. [Google Scholar] [CrossRef]
- Biggadike, K. Fluticasone furoate/fluticasone propionate–different drugs with different properties. Clin. Respir. J. 2011, 5, 183. [Google Scholar] [CrossRef]
- Mei, H.; et al. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. [Google Scholar] [CrossRef]
- Aitipamula, S.; et al. Polymorphs, salts, and cocrystals: what’s in a name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar]
- Hays, D.P.; Johnson, B.F.; Perry, R. Prolonged hallucinations following a modest overdose of tripelennamine. Clin. Toxicol. 1980, 16, 331–333. [Google Scholar] [PubMed]
- Bjerre, C.; Björk, E.; Camber, O. Bioavailability of the sedative propiomazine after nasal administration in rats. Int. J. Pharm. 1996, 144, 217–224. [Google Scholar] [CrossRef]
- Crombie, K.B.; Cullen, L.F. Propiomazine Hydrochloride, in Analytical Profiles of Drug Substances. 1973, Elsevier. p. 439-466.
- Florey, K. Triflupromazine Hydrochloride, in Analytical Profiles of Drug Substances, K. Florey, Editor. 1973, Academic Press. p. 523-550.
- Rifkin, A.; et al. Fluphenazine decanoate, fluphenazine hydrochloride given orally, and placebo in remitted schizophrenics: I. Relapse rates after one year. Arch. Gen. Psychiatry 1977, 34, 43–47. [Google Scholar]
- Surov, A.O.; et al. Pharmaceutical salts of ciprofloxacin with dicarboxylic acids. Eur. J. Pharm. Sci. 2015, 77, 112–121. [Google Scholar]
- Al Omari, M.M.; et al. Moxifloxacin hydrochloride. Profiles of drug substances, excipients and related methodology 2014, 39, 299–431. [Google Scholar] [PubMed]
- Wilson, T.D. Pentazocine, in Analytical Profiles of Drug Substances, K. Florey, Editor. 1984, Academic Press. p. 361-416.
- Miller, R.R. Clinical effects of pentazocine in hospitalized medical patients. J. Clin. Pharmacol. 1975, 15, 198–205. [Google Scholar] [PubMed]
- Goldberg, Y. Flavoxate Hydrochloride, in Analytical Profiles of Drug Substances and Excipients, H.G. Brittain, Editor. 2001, Academic Press. p. 77-115.
- Elhesaisy, N.; Swidan, S. Trazodone loaded lipid core poly (ε-caprolactone) nanocapsules: development, characterization and in vivo antidepressant effect evaluation. Sci. Rep. 2020, 10, 1964. [Google Scholar]
- Hettche, H.; et al. Salts of azelastine having improved solubility. 1999, Google Patents.
- Bariakar, U.V.; Patel, U.N. Pralidoxime Chloride, in Analytical Profiles of Drug Substances, K. Florey, Editor. 1988, Academic Press. p. 533-569.
- Wahl, J.W.; Tyner, G.S. Echothiophate Iodide*: The Effect of 0.0625% Solution on Blood Cholinesterase. Am. J. Ophthalmol. 1965, 60, 419–425. [Google Scholar]
- Aquilonius, S.-M.; et al. Pharmacokinetics and oral bioavailability of pyridostigmine in man. Eur. J. Clin. Pharmacol. 1980, 18, 423–428. [Google Scholar] [CrossRef]
- Brown, D.R.; Holtzman, S.G. Opiate antagonists: central sites of action in suppressing water intake of the rat. Brain Res. 1981, 221, 432–436. [Google Scholar] [CrossRef]
- Heinrich, M.; Teoh, H.L. Galanthamine from snowdrop—the development of a modern drug against Alzheimer’s disease from local Caucasian knowledge. J. Ethnopharmacol. 2004, 92, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Reichl, R. New thienylcarboxylic acid esters of aminoalcohols, their quarternary products, their preparation and use of the compounds. 1999, Google Patents.
- Banholzer, R.; Pfrengle, W.; Sieger, P. Novel tiotropium salts, process for the preparation and pharmaceutical compositions thereof. 2005, Google Patents.
- Lenke, D.; et al. Basically substituted benzyl phthalazone derivatives, acid salts thereof and process for the production thereof. 1974, Google Patents.
- Van Leeuwen, F.R.; Sangster, B.; Hildebrandt, A.G. The toxicology of bromide ion. CRC Crit. Rev. Toxicol. 1987, 18, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. In silico prediction of blood–brain barrier permeation. Drug Discov. Today 2003, 8, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.P.; Holm, R.; De Diego, H.L. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef]
- Elder, D.P.; et al. The utility of sulfonate salts in drug development. J. Pharm. Sci. 2010, 99, 2948–2961. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-L.; et al. Preformulation investigation I: relation of salt forms and biological activity of an experimental antihypertensive. J. Pharm. Sci. 1972, 61, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A. Invalid SMILES are beneficial rather than detrimental to chemical language models. Nat. Mach. Intell. 2024, 1–12. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Veber, D.F.; et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA: A Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.J.; et al. Understanding and responsiveness level about cervical cancer and its avoidance among young women of Pakistan. Asian Pac. J. Cancer Prev. 2014, 15, 4877–4883. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; et al. Recent progress in theranostic microbubbles. Chin. Chem. Lett. 2023, 34, 108137. [Google Scholar] [CrossRef]
- Scott, E.C.; et al. Trends in the approval of cancer therapies by the FDA in the twenty-first century. Nat. Rev. Drug Discov. 2023, 22, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.; Sharma, A. Prospects of halofuginone as an antiprotozoal drug scaffold. Drug Discov. Today 2022, 27, 2586–2592. [Google Scholar] [CrossRef]
- Luo, Y.; et al. The role of halofuginone in fibrosis: more to be explored? J. Leukoc. Biol. 2017, 102, 1333–1345. [Google Scholar] [CrossRef]
- Zuo, R.-n.; et al. -n.; et al. Using halofuginone–silver thermosensitive nanohydrogels with antibacterial and anti-inflammatory properties for healing wounds infected with Staphylococcus aureus. Life Sci. 2024, 339, 122414. [Google Scholar] [CrossRef]
- Cheng, S.; et al. Adhesive chitosan-based hydrogel assisted with photothermal antibacterial property to prompt mice infected skin wound healing. Chin. Chem. Lett. 2023, 34, 108276. [Google Scholar] [CrossRef]
- Khan, G.J.; et al. Ciprofloxacin: The frequent use in poultry and its consequences on human health. Prof. Med. J. 2015, 22, 001–005. [Google Scholar] [CrossRef]
- Sun, X.-H.; Fu, J.; Sun, D.-Q. Halofuginone alleviates acute viral myocarditis in suckling BALB/c mice by inhibiting TGF-β1. Biochem. Biophys. Res. Commun. 2016, 473, 558–564. [Google Scholar] [CrossRef]
- García-Rodríguez, I.; et al. Assessment of the broad-spectrum host targeting antiviral efficacy of halofuginone hydrobromide in human airway, intestinal and brain organotypic models. Antivir. Res. 2024, 222, 105798. [Google Scholar] [PubMed]
- Liu, Y.; et al. Extensible and swellable hydrogel-forming microneedles for deep point-of-care sampling and drug deployment. Chin. Chem. Lett. 2023, 34, 108103. [Google Scholar]
- Jain, V.; et al. Structure of prolyl-tRNA synthetase-halofuginone complex provides basis for development of drugs against malaria and toxoplasmosis. Structure 2015, 23, 819–829. [Google Scholar] [PubMed]
- Sevilla, L.M.; Pérez, P. Glucocorticoids and Glucocorticoid-Induced-Leucine-Zipper (GILZ) in Psoriasis. Front Immunol 2019, 10, 2220. [Google Scholar] [PubMed]
- Wang, J.; et al. Halofuginone functions as a therapeutic drug for chronic periodontitis in a mouse model. Int. J. Immunopathol. Pharmacol. 2020, 34, 2058738420974893. [Google Scholar]
- Fehr, M.; et al. Quinazolinone Alkaloid Febrifugine and Its Analogues To Control Phytopathogenic Diseases Caused by Oomycete Fungi. Eur. J. Org. Chem. 2022, 2022, e202101522. [Google Scholar]
- Zhang, S.; et al. Anti-phytophthora activity of halofuginone and the corresponding mode of action. J. Agric. Food Chem. 2022, 70, 12364–12371. [Google Scholar]
- Kunimi, H.; et al. A novel HIF inhibitor halofuginone prevents neurodegeneration in a murine model of retinal ischemia-reperfusion. Int. J. Mol. Sci. 2019, 20, 3171. [Google Scholar] [CrossRef]
- Zhu, J.; et al. Inhibition of IL-17 signaling in macrophages underlies the anti-arthritic effects of halofuginone hydrobromide: Network pharmacology, molecular docking, and experimental validation. BMC Complement. Med. Ther. 2024, 24, 105. [Google Scholar]
- Miwa, Y.; et al. Halofuginone prevents outer retinal degeneration in a mouse model of light-induced retinopathy. Plos One 2024, 19, e0300045. [Google Scholar]
- Wu, X.; et al. Halofuginone Inhibits Osteoclastogenesis and Enhances Osteoblastogenesis by Regulating Th17/Treg Cell Balance in Multiple Myeloma Mice with Bone Lesions. Indian J. Hematol. Blood Transfus. 2024, 1–8. [Google Scholar]
- Yan, G.; et al. Integrated Stress Response Potentiates Ponatinib-Induced Cardiotoxicity. Circ. Res. 2024. [Google Scholar]
- Ma, X.; et al. Nanoparticles-based lateral flow strip for halofuginone ultrasensitive detection in chicken and beef. Food Biosci. 2024, 104100. [Google Scholar]
- Semukunzi, H.; et al. IDH mutations associated impact on related cancer epidemiology and subsequent effect toward HIF-1α. Biomed. Pharmacother. 2017, 89, 805–811. [Google Scholar]
- Cheng, H.; et al. Halofugine prevents cutaneous graft versus host disease by suppression of Th17 differentiation. Hematology 2012, 17, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Ozmen, E.; Demir, T.D.; Ozcan, G. Cancer-associated fibroblasts: protagonists of the tumor microenvironment in gastric cancer. Front. Mol. Biosci. 2024, 11, 1340124. [Google Scholar]
- Tossetta, G.; et al. Role of natural and synthetic compounds in modulating NRF2/KEAP1 signaling pathway in prostate cancer. Cancers 2023, 15, 3037. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.; et al. Nuclear Factor Erythroid 2–Related Factor 2 Depletion Sensitizes Pancreatic Cancer Cells to Gemcitabine via Aldehyde Dehydrogenase 3a1 Repression. J. Pharmacol. Exp. Ther. 2021, 379, 33–40. [Google Scholar] [PubMed]
- Lv, T.; et al. Halofuginone enhances the anti-tumor effect of ALA-PDT by suppressing NRF2 signaling in cSCC. Photodiagnosis Photodyn. Ther. 2022, 37, 102572. [Google Scholar]
- Pan, Y.; et al. An injectable mPEG-PDLLA microsphere/PDLLA-PEG-PDLLA hydrogel composite for soft tissue augmentation. Chin. Chem. Lett. 2022, 33, 2486–2490. [Google Scholar]
- Lin, J.; Wu, X. Halofuginone inhibits cell proliferation and AKT/mTORC1 signaling in uterine leiomyoma cells. Growth Factors 2022, 40, 212–220. [Google Scholar] [PubMed]
- Zhang, J.; et al. Theranostic mesoporous platinum nanoplatform delivers halofuginone to remodel extracellular matrix of breast cancer without systematic toxicity. Bioeng. Transl. Med. 2023, 8, e10427. [Google Scholar]
- Zuo, R.; et al. Halofuginone-guided nano-local therapy: Nano-thermosensitive hydrogels for postoperative metastatic canine mammary carcinoma with scar removal. International Journal of Pharmaceutics: X 2024, 100241.
- Cai, C.; et al. In situ wound sprayable double-network hydrogel: Preparation and characterization. Chin. Chem. Lett. 2022, 33, 1963–1969. [Google Scholar]
- de Figueiredo-Pontes, L.L.; et al. Halofuginone has anti-proliferative effects in acute promyelocytic leukemia by modulating the transforming growth factor beta signaling pathway. PLoS One 2011, 6, e26713. [Google Scholar]
- Yu, Z.; et al. 5-Fluorouracil Combined with Rutaecarpine Synergistically Suppresses the Growth of Colon Cancer Cells by Inhibiting STAT3. Drug Des. Dev. Ther. 2023, 993–1006. [Google Scholar]
- Gao, Y.; et al. Polysaccharide-based supramolecular drug delivery systems mediated via host-guest interactions of cucurbiturils. Chin. Chem. Lett. 2021, 32, 949–953. [Google Scholar]
- Li, H.; et al. Halofuginone sensitizes lung cancer organoids to cisplatin via suppressing PI3K/AKT and MAPK signaling pathways. Front. Cell Dev. Biol. 2021, 9, 773048. [Google Scholar]
- Gong, R.-H.; et al. Cell death mechanisms induced by synergistic effects of halofuginone and artemisinin in colorectal cancer cells. Int. J. Med. Sci. 2022, 19, 175. [Google Scholar]
- Panda, H.; et al. Halofuginone micelle nanoparticles eradicate Nrf2-activated lung adenocarcinoma without systemic toxicity. Free Radic. Biol. Med. 2022, 187, 92–104. [Google Scholar]
- Zuo, R.; et al. Orally administered halofuginone-loaded TPGS polymeric micelles against triple-negative breast cancer: enhanced absorption and efficacy with reduced toxicity and metastasis. Int. J. Nanomed. 2022, 2475–2491. [Google Scholar]
- Stahl, P.; Skinner, F. Pharmaceutical aspects of the api salt form. Pharmaceutical Salts: Properties, Selection, and Use, 2nd ed.; Stahl, PH, Wermuth, CG, Eds, 2011: p. 85-124.
- Peresypkin, A.; et al. Discovery of a stable molecular complex of an API with HCl: a long journey to a conventional salt. J. Pharm. Sci. 2008, 97, 3721–3726. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; et al. Revealing the synergistic mechanism of multiple components in compound fengshiding capsule for rheumatoid arthritis therapeutics by network pharmacology. Curr. Mol. Med. 2019, 19, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Gould, P.L. Salt selection for basic drugs. Int. J. Pharm. 1986, 33, 201–217. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



