Submitted:
09 March 2025
Posted:
10 March 2025
You are already at the latest version
Abstract
Sickle cell disease (SCD) is a monogenic blood disorder characterized by abnormal hemoglobin S production, which polymerizes under hypoxia conditions to produce chronic hemolysis, widespread organ damage, and vasculopathy. As a result of vaso-occlusion and ischemia-reperfusion injury, individuals with SCD have recurrent pain episodes, infection, pulmonary disease and fall victim to early death. Oxidative stress due to chronic hemolysis and the release of hemoglobin and free heme is a key driver of the clinical manifestations of SCD. The net result is the generation of reactive oxygen species that consume nitric oxide and overwhelm the antioxidant system due to a reduction in enzymes such as superoxide dismutase and glutathione peroxidase. The primary mechanism for handling cellular oxidative stress is the activation of antioxidant proteins by the transcription factor NRF2, a promising target for treatment development, given the significant role of oxidative stress in the clinical severity of SCD. In this review, we discuss the role of oxidative stress in health and the clinical complications of SCD, and the potential of NRF2 as a treatment target, offering hope for developing effective therapies for SCD. This task requires our collective dedication and focus.
Keywords:
sickle cell disease
; oxidative stress
; NRF2
; heme
; hemolysis
; red blood cells
; fetal hemoglobin
; hemoglobin S
; reactive oxygen species
1. Introduction
Sickle cell disease (SCD) is a global health problem affecting millions worldwide, mainly in sub-Saharan Africa, where 4-6 million people live with the disease [1,2]. The urgency of this problem is underscored by the fact that 90% of the 150,000 babies born annually in Nigeria will die by 5 years of age [3]. It is imperative that we change these statistics by developing robust newborn screening programs and designing safe and effective low-cost oral agents to treat children affected with SCD.
The most common genetic mutation causing SCD is the A to T transversion in the HBB gene on chromosome 11, leading to a substitution of valine for glutamic acid in the sixth codon [4] of the β-globin protein chain and the production of sickle hemoglobin S (HbS). Due to the process of hemoglobin switching at birth when the γ-globin to β-globin switch occurs during the first year of life [5], the manifestations of SCD are delayed. However, as fetal hemoglobin (HbF) decreases and HbS levels increase, the pathophysiology of SCD is observed clinically. Under low oxygen conditions, HbS forms polymer, thus producing sickled red blood cells (RBCs), vaso-occlusive episodes, and tissue damage due to ischemia [6]. Inheritance of the homozygous hemoglobin SS genotype produces sickle cell anemia, the most common subtype of hemoglobinopathies observed worldwide. Other common forms of SCD, include HbSC, HbSβ0-Thalassemia, and HbSβ+-Thalassemia, and many others, comprise this group of disorders [7].
The clinical manifestations of SCD consist of chronic hemolytic anemia and recurrent acute vaso-occlusive pain episodes that contribute to clinical severity, morbidity, and early mortality [7]. Common complications include splenic infarcts, high infection rates, dactylitis, stroke, acute chest syndrome, and pulmonary hypertension, among others. The kidneys are particularly susceptible to vaso-occlusive episodes due to functional abnormalities produced by RBCs sickling in the hypoxic microenvironment, producing focal and segmental glomerular sclerosis [8]. As more individuals with SCD survive to adulthood, the prevalence of chronic kidney disease is increasing by 15-30%, with significant mortality due to end-stage kidney disease and a lack of tissue matched transplant donors in the US.
In addition to the formation of sickled RBCs by HbS polymerization, it also triggers RBC hemolysis and the release of hemoglobin and free heme, causing inflammation, oxidative stress, and vascular–endothelial dysfunction [9,10]. Excessive free radicals produced by the release of hemoglobin cause the activation of pro-oxidant enzymes and the release of reactive oxygen species (ROS), mediating oxidative stress [2,11]. The primary function of mitochondria is the process of oxidative phosphorylation, which controls ROS production and ATP synthesis to provide cells with energy. Although normal RBCs lose mitochondria when fully mature, that is not true for individuals with SCD. Rivers et al. [12,13] demonstrated abnormal mitochondrial retention in people and mice with SCD-mediating ROS-associated hemolysis. Subsequent work from the Rivers lab showed that stress erythropoiesis contributes to mitochondrial retention and ROS generation in SCD [14].
The transcription factor NRF2 (erythroid-derived 2)-like 2) plays a crucial role in regulating the antioxidant response to oxidative stress in cells [15,16]. This factor is sequestered in the cytoplasm by Kelch-like ECH-associated protein 1 (KEAP1) and β-transducin repeat-containing protein and directed to the proteasome for degradation. However, under oxidative stress conditions NRF2 is released and translocated to the nucleus where it activates several antioxidant proteins, including heme oxygenase 1 (HMOX1), quinone oxidoreductase 1, glutamate-cysteine ligase catalytic subunit, and glutamate-cysteine ligase modifier subunit. These proteins work together to reduce cellular ROS and inflammation. Our group and others have demonstrated another role of NRF2 as a modulator of γ-globin gene transcription and HbF induction [17,18,19]. In the case of SCD, NRF2 provides a unique benefit through HbF induction to inhibit HbS polymerization and lowering oxidative stress to ameliorate clinical symptoms. Therefore, US FDA-approved oral activators of NRF2, such as dimethyl fumarate (DMF) [20,21] and simvastatin [22,23], are attractive targets for drug development in SCD.
Efforts to target oxidative stress using small oral-active chemical molecules to decrease the toxic side effects are widespread. However, despite these efforts, there remains a paucity of drugs approved for clinical treatment of oxidative stress in SCD. Hydroxyurea (HU) was the first US FDA-approved agent specifically for treating SCD, mediating several beneficial effects, including HbF induction which blocks HbS polymerization, decreasing inflammation and oxidative stress, and increasing nitric oxide production [24,25]. These benefits contribute to amelioration of pain episodes and other complications by HU in adults and children with SCD. However, there remain concerns over the long-term use of HU and its effects on fertility and secondary malignancy; the latter has not occurred in SCD after more than 25 years of clinical use [26,27]. Children living in Sub-Saharan Africa received HU at a fixed dose or with dose escalation. After an adequate evaluation period, the data safety monitoring board halted the trial when the number of clinical events was significantly lower among children receiving escalated dosing of HU when compared to a fixed dose [27]. They have fewer vaso-occlusive pain episodes, acute chest syndrome, transfusions, and hospitalizations. Laboratory tests confirmed similar toxicity in the two groups. However, there were no cases of severe neutropenia or thrombocytopenia.
Over the last 10 years, three additional agents were US FDA-approved for the treatment of SCD, including l-glutamine [28] and crizanlizumab [29,30] that indirectly target oxidative stress and voxeletor [31], which inhibits HbS polymerization to improve total hemoglobin levels. Recently, during the fall of 2024, there was an abrupt discontinuation of voxelotor due safety concerns. Extraordinary progress was made by two pharma companies in 2023 with the US FDA approval of the first two gene therapies for SCD, Casgevy (exagamglogene autotemcel) [32,33] and Lyfgenia (lovotibeglogene autotemcel) [34,35]. Whether these therapies will be curative requires long-term follow-up. However, there was a remarkable resolution of vaso-occlusive episodes, marker of chronic hemolysis, and oxidative stress levels, along with improved quality of life for individual treated with both innovative drugs. Whether there will be equal accessibility to gene therapy for individuals with SCD in the US or globally is a challenging question for healthcare providers.
In this review, we will discuss progress made in the field to understand the role of oxidative stress in human health and the pathophysiology of SCD. We will also discuss current strategies for developing small molecules and NRF2-activating drugs for treating oxidative stress. Specifically, we will explore the impact of these therapies in the field, highlighting the promising role of targeting NRF2 to improve clinical outcomes and reducing the global burden of SCD.
2. Oxidative Stress
The pathological state of oxidative stress arises from an imbalance between the production of ROS and the capacity of the antioxidant defense system to neutralize them. Under physiological conditions, ROS plays an essential role in cellular signaling and homeostasis [36]. Various natural biological processes in the human body, including respiration, digestion, alcohol and drug metabolism, and the conversion of fats into energy, generate ROS as a metabolic byproduct [37]. Moderate levels of ROS are beneficial for protecting against pathogens and wound healing activities [38]. However, excessive ROS accumulation leads to cellular and tissue damage through lipid peroxidation, protein oxidation, and DNA damage, contributing to disease pathogenesis [37,39,40,41].
Reactive oxygen species are generated from various sources in the human body, such as the Phase I cytochrome P450 metabolizing enzymes, NADPH oxidases, and the mitochondrial electron transport chain (ETC). The P450 system utilizes molecular oxygen to form superoxide anions and other ROS [42], which participate in cell signaling or contribute to oxidative stress and tissue damage [42,43]. Membrane-bound enzymes like NADPH oxidases generate superoxide in response to cell signaling activated by inflammatory stimuli. These enzymes play a crucial role in immune defenses by producing ROS to kill pathogens; however, sustained oxidative damage causes chronic disease states [44]. Other enzymatic sources of ROS include xanthine oxidase during purine metabolism and uncoupled endothelial nitric oxide synthase generating superoxide under conditions of endothelial dysfunction [45,46]. However, the primary generator of oxidative stress is mitochondria through the ETC [47]. These mechanisms regulate ROS levels to prevent oxidative stress and tissue damage [48,49]. As a physiologic mechanism, NRF2, superoxide dismutase, catalase, and glutathione peroxidase are the primary antioxidants to neutralize oxidative stress [50].
3. Electron Transport Chain
The ETC in mitochondria is crucial for energy production in eukaryotic cells. It primarily generates ATP while being a significant source of ROS generation. Mitochondria utilizes about 90% of the body’s oxygen for ATP production through oxidative phosphorylation, leading to ROS generation as a byproduct [51,52]. Under normal conditions, the NRF2-mediated antioxidant defense system neutralizes excess ROS, but when oxidative stress ensues the production exceeds its capacity to neutralize, causing cellular damage [48,49].
Glucose is a primary energy source, undergoing glycolysis in the cytoplasm to yield pyruvate, which is then oxidized to acetyl-CoA or carboxylated to oxaloacetate before entering the tricarboxylic acid cycle, generating NADH and FADH₂ for the ETC [53]. The ETC in the inner mitochondrial membrane consists of four protein complexes, with ubiquinone and cytochrome c as mobile electron carriers. A series of electron transfers through Complexes I-IV reduces molecular oxygen to water to create a proton gradient utilized for ATP synthesis [54,55].
Antioxidant enzymes such as superoxide dismutase, catalase, and glutathione peroxidase are vital for mitigating oxidative stress by oxidative phosphorylation [56]. Superoxide dismutase converts superoxide radicals into oxygen and hydrogen peroxide to protect cellular structures. Mitochondrial superoxide dismutase 2 is crucial for detoxifying mitochondrial ROS; deficiencies can worsen oxidative damage linked to disease manifestations [57,58,59]. Catalase metabolizes hydrogen peroxide into water and oxygen and is critical for protecting cells from oxidative injury in ROS-producing tissues such as the liver [60]. Glutathione peroxidase reduces hydrogen peroxide using glutathione to maintain cellular homeostasis and host defenses against oxidative damage in diseases characterized by high oxidant stress [61,62].
4. Oxidative Stress in Sickle Cell Disease
Thus far, we have reviewed oxidative stress mainly in healthy people, but now we will turn our attention to explore its detrimental effects in SCD. Under hypoxic conditions, HbS polymerizes to produce unstable sickled RBCs, which undergo autoxidation, producing excessive ROS by NADPH oxidase, creating a milieu of oxidative stress [63,64,65,66]. Moreover, oxidation of HbS contributes to cell membrane and protein damage and activates lipid peroxides to accelerate intravascular hemolysis [11,67]. This characteristic sequence of events causes the release of hemoglobin, the formation of microparticles, and damage-associated molecular patterns such as Hsp-70, interleukin 33, and ATP; ultimately, from the breakdown of hemoglobin, free heme is released into plasma, causing the accumulation of ROS. Both microparticles and damage-associated molecular patterns increase blood viscosity and stimulate the formation of inflammasomes by interacting with macrophages through TLR-4 binding and activation of the NF-kB and NLRP3 signaling pathways. These activated immune cells and sickled RBCs recruit endothelial cells to expand the inflammatory niche initiating vaso-occlusion, vasculopathy, and end organ ischemic-reperfusion injury [18,68]. This toxic process contributes to cardiac dysfunction, leading to cardiomyopathy, heart failure, and pulmonary hypertension because of repeated injury to cells [69,70,71,72].
4.1. Environmental Factors
Parallel with endogenous ROS production sources, environmental stressors, including infections and aging, contribute to oxidative stress in SCD. For example, pneumonia causes an inflammatory response and triggers immune activation, generating ROS by neutrophils and macrophages. These cells produce oxidative bursts and secrete effector proteins or toxins that interfere with the translocation of the NADPH oxidase complex or signaling pathways, exacerbating endothelial dysfunction and hemolysis [73,74]. The immune response to pathogens is associated with ROS production, as immune cells such as neutrophils and macrophages utilize ROS to eliminate invading microorganisms [75]. Additionally, aging is associated with mitochondrial dysfunction, reduced antioxidant enzyme activity, and cumulative oxidative damage [76], all of which worsen the oxidative burden in SCD [77], underscoring the need for targeted antioxidant therapies for clinical management [74].
The process of vaso-occlusion and oxidative stress affects every body organ system, which is especially detrimental in SCD, where chronic oxidative stress exists. For example, cardiopulmonary complications are the leading cause of death due to diastolic heart failure and pulmonary hypertension [78]. Chronic hemolysis produces excessive pro-oxidant enzymes and HbS and free heme, which promotes free hydroxy radicals accumulation via the Fenton reaction [2], retained mitochondrial ETC activity, and RBC auto-oxidation [79,80,81].
Splenic sequestration crisis is another major complication of SCD mediated by oxidative stress that occurs when sickled RBC obstructs the draining vein in the red pulp of the spleen. This leads to enlargement of this organ [82] and disordered architecture of the white pulp. Over time, the number of splenic follicles are reduced ad replaced by fibrotic tissue from RBC congestion in the red pulp [83], and eventual ischemic necrosis and infarction i.e., autosplenectomy [84,85].
The scale of liver disease in SCD ranges from hepatocyte damage to severe liver failure associated with multiple organ failure syndrome [86,87], accounting for 3-11% of deaths in SCD [88,89,90]. Oxidative stress related to iron overload markedly increases malondialdehyde, a marker of lipid peroxidation [91], and high levels of myeloperoxidase released by activated leukocytes and neutrophils, causing endothelial dysfunction and liver injury [92]. Lastly, acute RBC intrahepatic cholestasis can occur due to sickling in the hepatic circulation, leading to ischemia and liver failure [93].
Oxidative stress-mediated endothelial dysfunction and ischemia-reperfusion injury play a key role in developing proliferative retinopathy [94], which causes vision loss and blindness in SCD. This complication affects mainly adults with SCD due to retinal ischemia with secondary neovascularization and hemorrhage, mediating retinal artery infarction and retinal detachment [95]. Reactive oxygen species inflict endothelial cell damage through oxidative reactions with membrane lipids, peptides, and nucleic acids [96,97], exposing subendothelial structures and proteins, including tissue factors, causing a hypercoagulable state [98]. Previous studies demonstrated improvement in retinal pigment epithelial cells after treatment with the oral antioxidant monomethylfumarate in SCD mice [99].
Lastly, sickle nephropathy is a significant complication of SCD, with up to 18% of affected individuals developing end-stage renal disease [100,101,102]. Oxidative stress occurs in the kidney due to high heme content and lipid peroxidation [103,104,105] producing malondialdehyde and damage to the cell membrane that plays a key role in the pathophysiology of sickle nephropathy [68]. Oxidative stress damages the endothelial lining of kidney blood vessels, leading to proteinuria, impaired glomerular filtration, hypertension, and the development of chronic kidney disease [105,106,107].
5. Murine Studies Linking Oxidative Stress to SCD Clinical Severity
Several published murine studies in the Townes SCD mouse show the involvement of oxidative stress in SCD phenotype severity through drug development. Simvastatin (Zocor) is a statin drug developed to decrease cholesterol levels in humans by inhibiting the function of hydroxymethylglutaryl-coenzyme A reductase [108,109]. This agent modulates ROS levels, activates NRF2 expression, and activates PI3K/AKT signaling. Simvastatin controls iron metabolism through HMOX1 activation [110] and glutathione reduction in the liver [111], lungs [112], and spleen. Our group recently published data on Townes mice treated with simvastatin (Table 1) showed the ability of simvastatin to mitigate HbS sickling and ROS stress levels in SCD transgenic mice. [23]. A Phase I/II clinical trial with short-term simvastatin treatment in SCD patients showed increased nitric oxide levels and decreased C-reactive protein and interleukin-6 expression. However, no effects on vascular endothelial growth factor, vascular cell adhesion molecule-1, or tissue factor protein levels were observed [22].
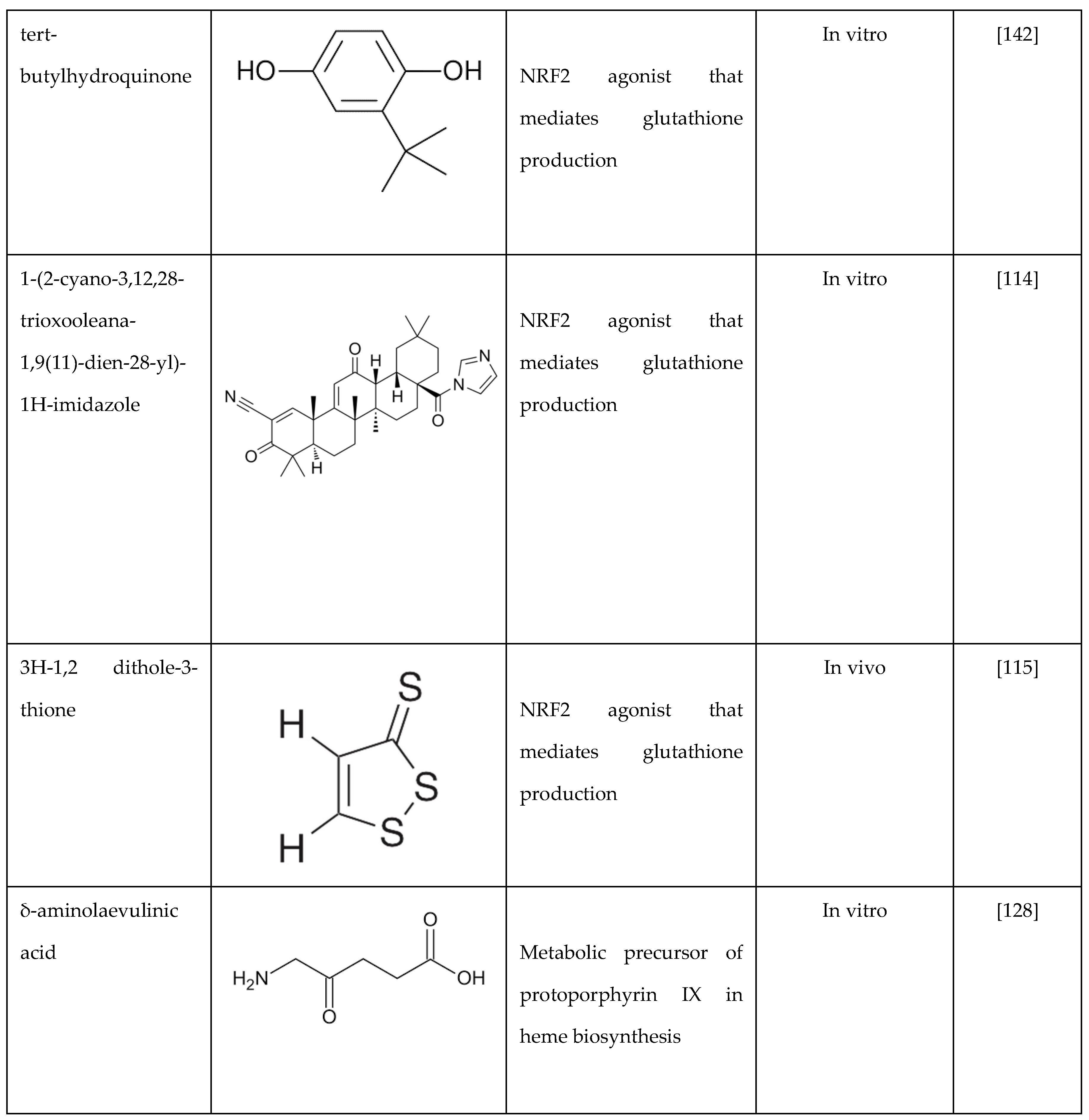
Our group also conducted studies in SCD mice treated with another NRF2 activator DMF, which inhibits the cytoplasmic sequestration by KEAP1 to release NRF2 and activation of its downstream target HMOX1 among others [21]. DMF also reduces inflammation and organ failure and improves the SCD phenotype [18,113], supported by decreased proinflammatory cytokine such as interleukin-6 and interleukin-1β, adhesion molecule, heme and oxidative stress levels. Moreover, our group showed that DMF induces HbF expression and reduced the percentage of sickle RBCs under hypoxia conditions [21]. Chronic DMF administration induced the expression of NRF2-dependent antioxidant genes to detoxify heme and suppress inflammation. On the contrary, in an SCD NRF2 knockout mouse, we showed that HMOX1 and γ-globin gene expression were silenced [114], and disease severity increased significantly [113]. These findings support the rationale of NRF2 induction as a therapeutic approach to induce HbF expression, reduce oxidative stress, and improve organ function [114].
Various other compounds have been evaluated in SCD mice as NRF2 activators or their ability to reduce ROS and/or inflammatory cytokines. For example, the NRF2 activator 1-(2-cyano-3,12,28-trioxooleana-1,9(11)-dien-28-yl)-1H-imidazole (CDDO-Im) reduces inflammation and improves organ function in SCD mice [114]. Similarly, when SCD mice were treated with 3H-1,2-dithiole-3-thione, a small chemical inducer of NRF2, a decrease in acute chest syndrome and increased survival was observed [115]. In SCD mice, activated neutrophils release myeloperoxidase, which generates oxidants that impair endothelial cell function. Zhang et al. showed that inhibiting myeloperoxidase decreases myeloperoxidase-dependent oxidative stress and restores endothelial cell function [92]. The inhibition of NADPH oxidase decreases oxidative stress to improve vascular functions in SCD [116]. Lastly, ongoing hemolysis in SCD, due to hemoglobin and free heme, activates the toll-like receptor 4 that mediates ROS generation. Of note, treatment of BERK SCD mice microglial cells with toll-like receptor-4 inhibitors reversed this effect [117].
6. Targeted Therapy to Reduce Oxidative Stress in SCD
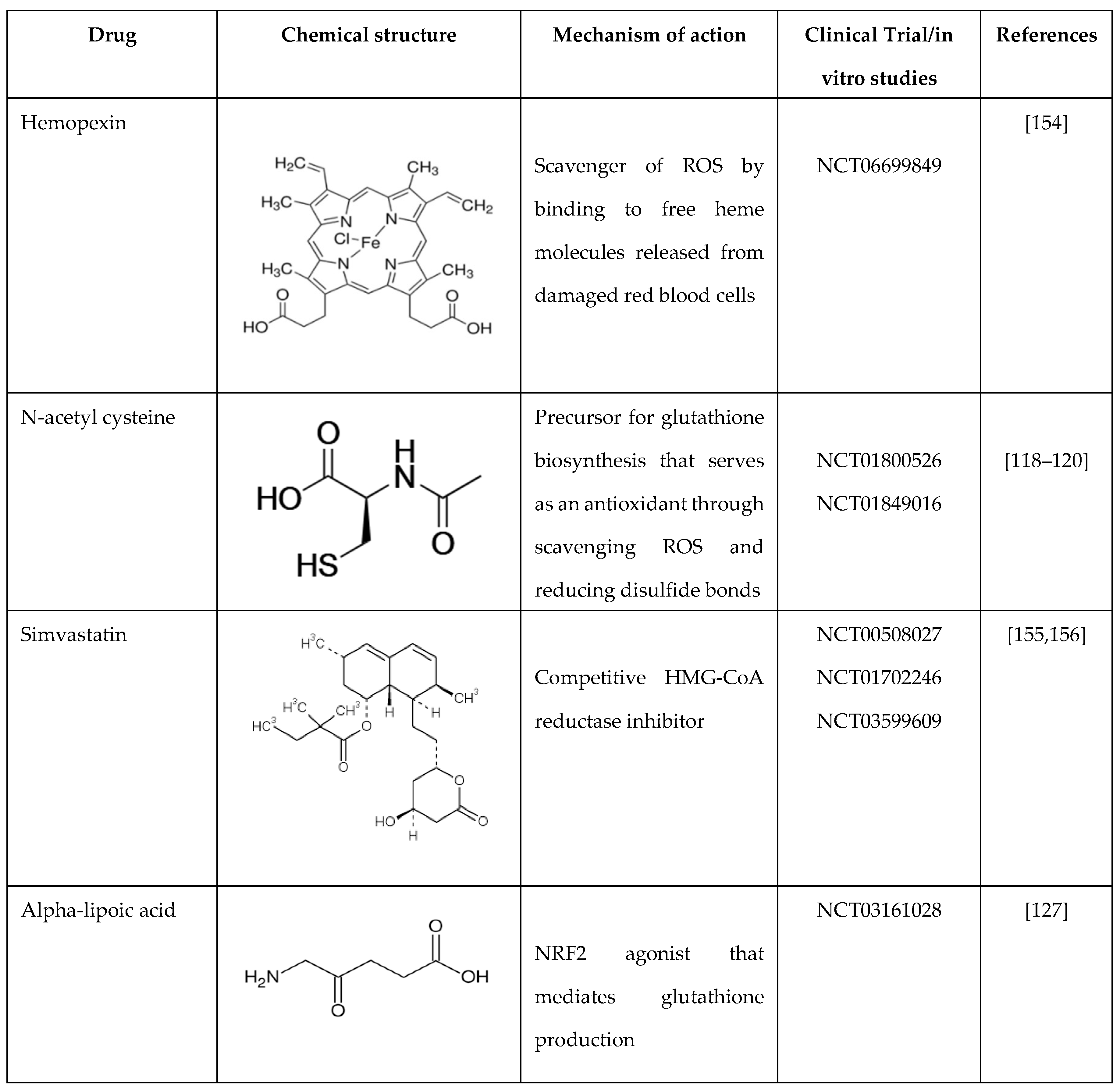
N-acetyl cysteine (NAC) provides cysteine for glutathione de novo biosynthesis and has been investigated for its effects on cellular oxidative damage, hemolysis, coagulation, and endothelial activation. We were the first to show in an early phase I trial that NAC inhibits dense RBC and irreversible sickle cell formation and restores glutathione levels toward normal in adolescents and young adults with SCD [118]. In addition, we observed a trend toward the resolution of vaso-occlusive episodes in this population after a 6-month treatment period; subsequently, other groups confirmed our findings. Nur et al. demonstrated that 6 weeks of NAC treatment increases whole blood glutathione levels and decreases plasma advanced glycation end-products and cell-free hemoglobin levels; moreover, none of the SCD patients experienced painful episodes or other significant NAC-related complications while on treatment [119]. In addition, Ozpolat et al. demonstrated that NAC increases total and free-thiol concentrations of cysteine and glutathione in blood after intravenous administration, thus relieving oxidative stresses and reducing intra-chain disulfide bonds in von Willebrand factor; however, only two SCD patients were recruited for their study [120].
Previous clinical trials demonstrated the beneficial effects of glutamine in preventing SCD vaso-occlusive crises [28]. SCD patients treated with l-glutamine showed significant increases in NADH and NAD redox potential. Furthermore, l-glutamine decreased endothelial adhesion of sickle RBCs in vitro, suggesting that clinical improvement most likely occurred through oxidative stress mechanisms. The ability of NRF2 to regulate glutamine metabolism is another reason to support the development of small molecule activators for treating SCD.
6.1. Hemopexin
Hemopexin is a crucial plasma protein that binds and transports heme for scavenging. The hemopexin levels are significantly reduced in individuals with SCD compared to healthy controls. Hemopexin was shown to prevent heme-iron loading in the cardiovascular system, thus limiting the production of ROS and the activation of adhesion molecules; on the contrary, hemopexin promoted heme recovery and detoxification by the liver mainly through the activation of HMOX1 [121,122]. Three times a week administration of hemopexin for three months produced a dose-dependent reduction in heme exposure and pulmonary hypertension while improving cardiac pressure-volume relationships and exercise tolerance in SCD mice. In addition, hemopexin administration attenuated pulmonary fibrosis and oxidative modifications in the lung and myocardium of the right ventricle [123]. More widely, hemopexin reduces endothelial exposure of P-selectin and von Willebrand factor, stimulating hepatic HMOX1 to decrease vascular inflammation and vaso-occlusion [124]. These studies demonstrate therapeutic proof-of-concept for the development of hemopexin for treating SCD.
Recently, plasma-derived human hemopexin (CSL889) was investigated in a phase 1 study to evaluate its safety, tolerability, and pharmacokinetics in SCD patients [125]. The results showed that CSL889 has an excellent safety and tolerability profile when administered as a single dose of up to 200 mg/kg in subjects in steady-state SCD status and at 60 mg/kg in subjects in an acute vaso-occlusive episode (Table 2). These results provide a strong foundation for future trials to evaluate the efficacy of hemopexin in SCD [125].
6.2. Alpha-Lipoic Acid
Alpha-lipoic acid (ALA) is another potent antioxidant drug that has been investigated in the treatment of several diseases. Recently, Simonia et al. reported that ALA reduced platelet activation and thrombus formation in SCD mice [126]. High dietary intake of ALA reduced sickle RBCs, liver fibrosis, and adhesion molecule expression, including (vascular cell adhesion molecule 1, intercellular adhesion molecule 1, and von Willebrand Factor) in multiple organs in SCD mice. Martins et al. recently evaluated the effects of ALA in sickle trait and SCD individuals. Unfortunately, ALA (200 mg) decreased glutathione peroxidase 4 activity in both groups while increasing catalase activity and reducing lipid and protein damage only in sickle trait subjects [127]. These results indicate that more studies are required to optimize the dose of ALA to produce therapeutic efficacy in SCD.
6.3. δ-Aminolevulinate
Previously, we investigated the ability of δ-aminolevulinate, the heme precursor, to activate γ-globin gene expression and its effects on cellular functions in erythroid cell systems. We demonstrated that this small molecule induced γ-globin expression at both the transcriptional and protein levels in KU812 erythroid cells [128]. Using inhibitors of the heme biosynthesis pathway, we demonstrated that heme participated in δ-aminolevulinate-mediated γ-globin transcription via NRF2 activation. These data support future studies to explore the potential of stimulating intracellular heme biosynthesis by δ-aminolevulinate or similar compounds as a novel therapeutic strategy for NRF2 activation for treating SCD and β-thalassemia.
6.4. Arginine
Arginine levels are acutely deficient in SCD patients due to consumption during vaso-occlusive pain episodes. When arginine levels are low, superoxide is produced instead of nitric oxide, reducing its bioavailability, and generating oxidative stress. Arginine recently emerged as a new treatment option for SCD since intravenous arginine was shown to improve mitochondrial function and reduce oxidative stress in children and young adults with SCD during vaso-occlusive pain episodes [129]. An ongoing phase 3 randomized controlled trial, Sickle Cell Disease Treatment with Arginine Therapy (STArT), is testing the efficacy of arginine treatment in 360 children, adolescents, and young adults in an acute vaso-occlusive pain episode [130].
7. Role of NRF2 in Globin Gene Regulation
Reactivation of HbF expression is a key therapeutic option in developing novel therapies for SCD. It is well established that HbF protein inhibits the polymerization of HbS to reverse the pathophysiology of SCD and disease severity. The FDA-approved drug HU and other small molecule compounds, such as butyrate and decitabine, were shown to induce HbF expression and inhibit HbS polymerization under hypoxic conditions to ameliorate the clinical severity of SCD [131,132,133,134]. Indeed, multiple transcription factors participate in the regulation of the five functional globin genes located in the HBB locus on chromosome 11. Interestingly, HU has been shown to remodel the γ-globin gene promoter in association with GATA1, GATA2, NFY, and BCL11A to facilitate an open chromatin structure [135], while butyrate and decitabine affect γ-globin gene transcription through modifications of the proximal promoter histone acetylation and DNA methylation levels respectively [132,133,134].
Additionally, CRISPR strategies showed significantly higher γ-globin expression in erythroid cells carrying mutations producing hereditary persistence of HbF reverse the phenotypes of SCD and β-thalassemia major [136,137,138]. An important finding by insertions and deletions of binding sites for the γ-globin repressors BCL11A and Leukemia/lymphoma-related factor (LRF) in the proximal promoter resulted in γ-globin gene activation and reversal of the sickle RBC phenotype [139] [140]. The corresponding mechanistic studies support the notion that HbF induction is associated with epigenetic modifications and changes in chromatin contacts within the HBB locus.
The essential transcription factor NRF2 binds to the HBB locus to alter chromatin structure and globin gene expression. NRF2 was originally identified as a DNA-binding protein in the HBB locus and later was characterized as the major regulator of oxidative stress [141]. Previous studies conducted by Lowrey and colleagues demonstrated enhanced NRF2 binding in the γ-globin promoter and HbF induction after tert-butylhydroquinone and simvastatin treatment [17]. Deletion of a critical region 100 bp upstream of the γ-globin gene transcription-start site, 50-GACAAGGC-30, abolished HbF induction by these agents. Likewise, our group investigated the ability of DMF to activate γ-globin expression in primary human erythroid progenitors through NRF2 binding in the γ-globin antioxidant response element [142]. Using JASPAR61 software, we identified 23 NRF2 consensus motifs 5’-TGAnnnnGC-3’ in the HBB locus. Subsequent studies demonstrated in vivo binding in the locus control region and proximal γ-globin promoter, supporting long-range chromatin looping to regulate globin gene expression during drug treatment [19]. Protein-protein interaction studies demonstrated that NRF2 dimerizes with small MAF proteins to bind DNA and activate gene transcription. Moreover, NRF2 competes with the transcription factor BACH1 (tBTB Domain and CNC Homolog 1), a cap ‘n’ collar protein family member, which binds the antioxidant response elements of target genes to regulate cellular oxidative stress levels [143].
7.1. BACH 1 Inhibitors
Heme binds to the transcription factor BACH1 and represses NRF2-mediated gene transcription. ASP8731 is a selective small molecule inhibitor of BACH1 [144]. In HepG2 liver cells, ASP8731 increased HMOX1 transcription, and in Townes-SCD mice, it inhibited heme-mediated microvascular stasis, activated HMOX1 and decreased hepatic NF-kB phospho-p65 protein expression. ASP8731 increased γ-globin transcription and F-cells percentages. In human erythroid progenitors generated from CD34+ stem cells, ASP8731 increased γ-globin mRNA levels and the percentage of F-cells 2-fold. These data indicate that BACH1 inhibitors may offer a new therapeutic target to treat SCD.
Another novel BACH-1 inhibitor HPP-D was developed by Attucks and colleagues at vTv Therapeutics. They showed that HPP-D promotes the nuclear export of BACH-1 and the translocation of NRF2 into the nucleus to activate HMOX1 gene expression [145]. In a subsequent study, in collaboration with vTv, we showed the ability of HPP-D to attenuate BACH-1 and enhance NRF2 protein levels while and binding in the γ-globin antioxidant response element and HBB locus control hypersensitive site 2 by ChIP assay in KU812 cells; moreover, HPP-D activated γ-globin gene transcription and HbF protein synthesis [146]. To validate the potential of HPP-D for treating SCD, an anti-sickling study was conducted in primary erythroid cells generated from individuals with SCD. Cells grown under hypoxia conditions, demonstrated a 50% reduction of the number of sickled RBC observed, supporting the ability of HPP-D to induce HbF and produce anti-sickling effects.
Subsequently, we completed preclinical studies in the Townes SCD transgenic mice to evaluate the capability of oral HPP-D to induce HbF (unpublished data). The mice were treated with HPP-D by oral gavage 5 days a week for 4 weeks, and blood was analyzed at weeks 0, 2, and 4. The drug was well-tolerated with normal behavior, weight gain, and no toxic effects on peripheral blood cell counts. From week 0 to week 4, we observed a maximal 3-fold increase in F-cells (HbF-positive RBCs) and activation of γ-globin gene transcription measured by mRNA levels; by contrast β-globin mRNA levels were not changed significantly by HPP-D. To assess the antioxidant effects of HPP-D, we measured ROS and sickle RBCs levels under hypoxic conditions, which both showed an antioxidant effect with significantly decreased levels of both parameters. These data support the possible clinical development of the BACH 1 inhibitors as another class of drugs with a dual impact of HbF induction and antioxidant properties.
7.2. Regulation by Microrna Genes
Recently, miR-144 and miR-451 (miR-144/451) were demonstrated to play a significant role in erythroid differentiation and fine-tuning gene expression during erythropoiesis [147,148]. Interestingly, in adults with SCD, higher miR-144 levels decreased NRF2 and glutathione levels and was associated with a more severe anemia [149]. Moreover, low NRF2 expression was associated with a lack of the antioxidant proteins glutamate cysteine ligase C/M and superoxide dismutase 1 [150]. To further investigate this observation, we conducted genome-wide microarray analysis to discover miRNA genes associated with HbF expression in individuals with SCD. We observed the upregulation of miR-144 in subjects with lower HbF levels than those with higher HbF levels [149]. Functional studies in adult erythroid progenitors generated from CD34+ stem cells showed NRF2 silencing by miR-144 and concomitant repression of γ-globin transcription; by contrast, treatment with antagomir that inhibits miR-144 function reversed its silencing effects on NRF2. Additional studies have shown miR-28 inhibits NRF2 through a KEAP1-indepenedent mechanism and conversely miR-153, miR-27a, and miR-142-5p down-regulate NRF2 expression [151,152,153]. These studies support miRNA-mediated mechanisms of NRF2 regulation might be fruitful mechanism to explore their role in γ-globin gene regulation.

Table 1.
Summary of drugs that target NRF2 as a mechanism of γ-globin gene activation.
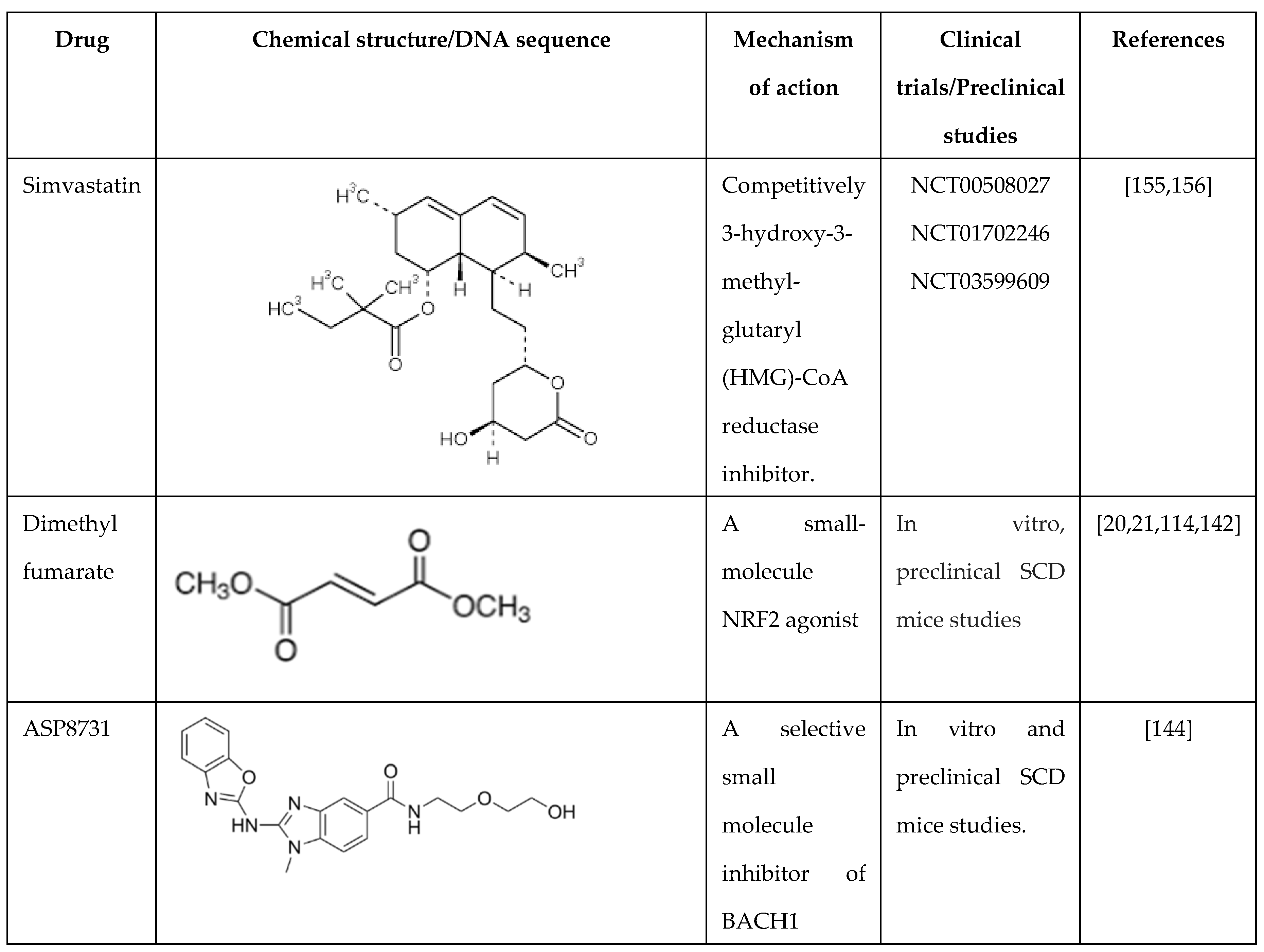
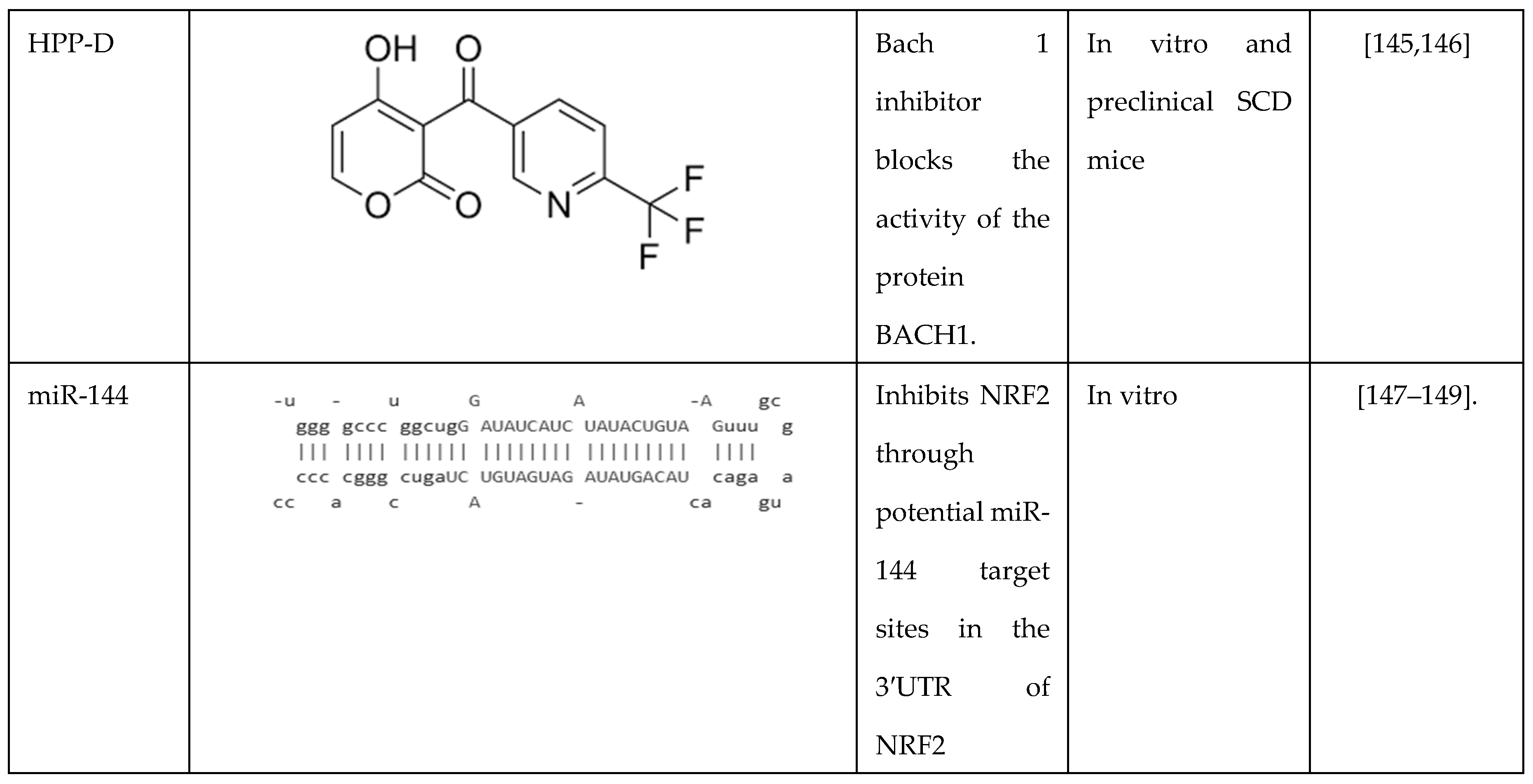
Table 2.
Summary of drugs that decrease oxidative stress by different mechanisms as treatments of sickle cell disease.
Table 2.
Summary of drugs that decrease oxidative stress by different mechanisms as treatments of sickle cell disease.
8. Future Directions
Targeting oxidative stress through NRF2 activation represents a promising future direction for drug discovery in treating SCD since this transcription factor plays a pivotal role in cellular defenses against oxidative stress by activating the transcription of antioxidant genes and enzymes. As the hypoxia-reperfusion injury and inflammatory characteristic of SCD lead to increased oxidative damage, enhancing NRF2 activity should mitigate these effects and improve clinical outcomes. However, the translation of NRF2-targeted therapies from preclinical studies to clinical application remains limited. This gap is mainly due to funding limitations to conduct clinical trials and the number of investigators with the expertise to lead these endeavors to rigorously evaluate the safety and efficacy of NRF2 activators in SCD.
Inter-individual variability poses another clinical challenge in harnessing NRF2 activators and antioxidant small molecules as a treatment strategy for SCD. Genetic factors, such as polymorphisms in NRF2 or related antioxidant genes, can influence steady-state oxidative stress levels and the response to activators. Additionally, environmental exposures, dietary habits, and lifestyle choices significantly modulate oxidative stress responses, leading to variability in treatment outcomes. Understanding these influences is crucial to tailor NRF2-targeted therapies and developing personalized treatment plans. Future studies should focus on characterizing these factors within diverse patient populations to maximize the efficacy of NRF2 interventions in managing oxidative stress associated with SCD.
9. Conclusions
Sickle cell disease is a common genetic disorder caused by a single-point mutation in the HBB gene, leading to HbS synthesis, which polymerizes under hypoxic conditions. The net result is a chronic intravascular hemolytic anemia, producing a severe oxidative stress state due to the release of hemoglobin and free heme into the plasma. Coupled with RBCs sickling, vaso-occlusion, and ischemia-reperfusion damage to internal organs, individuals with SCD suffer high morbidity and mortality. Therefore, reducing oxidative stress and enhancing HbF levels is an efficacious strategy to develop novel therapies for SCD.
As we move into the era where we have multiple FDA-approved drugs, we can start to develop combination regimens and take a multi-pronged treatment approach, especially with novel small molecules that act by different molecular and cellular mechanisms. We reviewed the most promising agents that either lower ROS and/or induce HbF to produce a synergistic effect on ameliorating the pathophysiology of SCD. Thus far, the most efficacious clinically tested combination has been HU and voxeletor, but the latter was recently removed from the market. Fortunately, we know that NRF2 is the major regulator of oxidative stress and serves as a trans-activator of the γ-globin gene promoter with the potential as a single agent capable of increasing HbF and dampening oxidative stress. Several drugs are ripe for the picking that merit clinical safety testing in individuals with SCD, such as the FDA-approved small molecule dimethyl fumarate, among others, in the pipeline.
Agents with anti-inflammatory properties are also desirable to inhibit the release of cytokines that cause tissue damage. Hydroxyurea is an effective therapy for SCD; however, some individuals do not respond, and there are concerns over long-term safety and adverse effects. Hydroxyurea has been used for over 25 years and has improved survival and the quality of life for individuals with SCD. Although drug development has progressed, there remains a need for expanded preclinical testing in SCD mice to grow the repertoire of drugs available for clinical trials in SCD. Lastly, from the perspective of a global lens, we must develop drugs that are safe, efficacious, and low-cost, making them affordable to individuals around the world suffering with the bulk of SCD in African, India, and South America.
Author Contributions
Authors, including AS-D, CDP, XZ, and BSP made substantial contributions to the conception of each section of the article, reviewed the entire paper, and made corrections to the text. BSP supervised the writing and submission of the manuscript. All authors contributed to drafting and reviewing the work critically for important intellectual content and have reviewed and approved the submitted manuscript. All authors agree to be accountable for all aspects of the work to ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
Grant number 1R01 HL149365-01A1 funded this work to BSP.
Acknowledgments
We want to thank the Georgia Cancer Center for administrative support.
Conflicts of Interest
The authors have no relevant conflict of interest to report.
References
- Royal, C.D.M.; Babyak, M.; Shah, N.; Srivatsa, S.; Stewart, K.A.; Tanabe, P.; Wonkam, A.; Asnani, M. Sickle cell disease is a global prototype for integrative research and healthcare. Adv Genet (Hoboken) 2021, 2, e10037. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Rees, D.C.; DeBaun, M.R.; Nnodu, O.; Ranque, B.; Thompson, A.A.; Ware, R.E.; Abboud, M.R.; Abraham, A.; Ambrose, E.E.; et al. Defining global strategies to improve outcomes in sickle cell disease: a Lancet Haematology Commission. Lancet Haematol 2023, 10, e633–e686. [Google Scholar] [CrossRef] [PubMed]
- Goia, F.; Tesi, U.; Gilardino, M.O.; Gasperini, S.; Carezzana, G. [Clinical evaluation of the therapeutic efficacy of cephalexin]. Minerva Stomatol 1986, 35, 749–752. [Google Scholar] [PubMed]
- Stamatoyannopoulos, G. Human hemoglobin switching. Science 1991, 252, 383. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, F.A.; Hofrichter, J.; Eaton, W.A. Kinetics of sickle hemoglobin polymerization. I. Studies using temperature-jump and laser photolysis techniques. J Mol Biol 1985, 183, 591–610. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, G. The molecular basis of hemoglobin disease. Annu Rev Genet 1972, 6, 47–70. [Google Scholar] [CrossRef]
- Sharpe, C.C.; Thein, S.L. Sickle cell nephropathy - a practical approach. Br J Haematol 2011, 155, 287–297. [Google Scholar] [CrossRef]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Potoka, K.P.; Gladwin, M.T. Vasculopathy and pulmonary hypertension in sickle cell disease. Am J Physiol Lung Cell Mol Physiol 2015, 308, L314–L324. [Google Scholar] [CrossRef]
- Nur, E.; Biemond, B.J.; Otten, H.M.; Brandjes, D.P.; Schnog, J.J.; Group, C.S. Oxidative stress in sickle cell disease; pathophysiology and potential implications for disease management. Am J Hematol 2011, 86, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Tumburu, L.; Ghosh-Choudhary, S.; Seifuddin, F.T.; Barbu, E.A.; Yang, S.; Ahmad, M.M.; Wilkins, L.H.W.; Tunc, I.; Sivakumar, I.; Nichols, J.S.; et al. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood 2021, 137, 3116–3126. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, R.; Vazquez, B.A.; Thiruppathi, M.; Ganesh, B.B.; Ibanez, V.; Cui, S.; Engel, J.D.; Diamond, A.M.; Molokie, R.E.; DeSimone, J.; et al. Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp Hematol 2017, 50, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, A.; Alejandro, M.; Kanu, A.; Zekaryas, N.; Horneman, H.; Hong, L.K.; Vinchinsky, E.; Lavelle, D.; Diamond, A.M.; Molokie, R.E.; et al. Reticulocyte mitochondrial retention increases reactive oxygen species and oxygen consumption in mouse models of sickle cell disease and phlebotomy-induced anemia. Exp Hematol 2023, 122, 55–62. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Suzuki, T.; Takahashi, J.; Yamamoto, M. Molecular Basis of the KEAP1-NRF2 Signaling Pathway. Mol Cells 2023, 46, 133–141. [Google Scholar] [CrossRef]
- Macari, E.R.; Schaeffer, E.K.; West, R.J.; Lowrey, C.H. Simvastatin and t-butylhydroquinone suppress KLF1 and BCL11A gene expression and additively increase fetal hemoglobin in primary human erythroid cells. Blood 2013, 121, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Oseghale, A.R.; Nicole, L.H.; Li, B.; Pace, B.S. Mechanisms of NRF2 activation to mediate fetal hemoglobin induction and protection against oxidative stress in sickle cell disease. Exp Biol Med (Maywood) 2019, 244, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, B.; Pace, B.S. NRF2 mediates gamma-globin gene regulation and fetal hemoglobin induction in human erythroid progenitors. Haematologica 2017, 102, e285–e288. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xi, C.; Ward, A.; Takezaki, M.; Shi, H.; Peterson, K.R.; Pace, B.S. NRF2 mediates gamma-globin gene regulation through epigenetic modifications in a beta-YAC transgenic mouse model. Exp Biol Med (Maywood) 2020, 245, 1308–1318. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Pace, B.; Gupta, D.; Sturtevant, S.; Li, B.; Makala, L.; Brittain, J.; Moore, N.; Vieira, B.F.; Thullen, T.; et al. Dimethyl fumarate increases fetal hemoglobin, provides heme detoxification, and corrects anemia in sickle cell disease. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, C.; Jacob, E.; Styles, L.; Kuypers, F.; Larkin, S.; Vichinsky, E. Simvastatin reduces vaso-occlusive pain in sickle cell anaemia: a pilot efficacy trial. Br J Haematol 2017, 177, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Xi, C.; Palani, C.; Takezaki, M.; Shi, H.; Horuzsko, A.; Pace, B.S.; Zhu, X. Simvastatin-Mediated Nrf2 Activation Induces Fetal Hemoglobin and Antioxidant Enzyme Expression to Ameliorate the Phenotype of Sickle Cell Disease. Antioxidants (Basel) 2024, 13. [Google Scholar] [CrossRef]
- Taylor, C.M.; Kasztan, M.; Sedaka, R.; Molina, P.A.; Dunaway, L.S.; Pollock, J.S.; Pollock, D.M. Hydroxyurea improves nitric oxide bioavailability in humanized sickle cell mice. Am J Physiol Regul Integr Comp Physiol 2021, 320, R630–R640. [Google Scholar] [CrossRef] [PubMed]
- King, S.B. Nitric oxide production from hydroxyurea. Free Radic Biol Med 2004, 37, 737–744. [Google Scholar] [CrossRef]
- Tshilolo, L.; Tomlinson, G.; Williams, T.N.; Santos, B.; Olupot-Olupot, P.; Lane, A.; Aygun, B.; Stuber, S.E.; Latham, T.S.; McGann, P.T.; et al. Hydroxyurea for Children with Sickle Cell Anemia in Sub-Saharan Africa. N Engl J Med 2019, 380, 121–131. [Google Scholar] [CrossRef]
- John, C.C.; Opoka, R.O.; Latham, T.S.; Hume, H.A.; Nabaggala, C.; Kasirye, P.; Ndugwa, C.M.; Lane, A.; Ware, R.E. Hydroxyurea Dose Escalation for Sickle Cell Anemia in Sub-Saharan Africa. N Engl J Med 2020, 382, 2524–2533. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med 2018, 379, 226–235. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med 2017, 376, 429–439. [Google Scholar] [CrossRef]
- Migotsky, M.; Beestrum, M.; Badawy, S.M. Recent Advances in Sickle-Cell Disease Therapies: A Review of Voxelotor, Crizanlizumab, and L-glutamine. Pharmacy (Basel) 2022, 10. [Google Scholar] [CrossRef]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med 2019, 381, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Locatelli, F.; Sharma, A.; Bhatia, M.; Mapara, M.; Molinari, L.; Wall, D.; Liem, R.I.; Telfer, P.; Shah, A.J.; et al. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N Engl J Med 2024, 390, 1649–1662. [Google Scholar] [CrossRef] [PubMed]
- Philippidis, A. CASGEVY Makes History as FDA Approves First CRISPR/Cas9 Genome Edited Therapy. Hum Gene Ther 2024, 35, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kanter, J.; Walters, M.C.; Krishnamurti, L.; Mapara, M.Y.; Kwiatkowski, J.L.; Rifkin-Zenenberg, S.; Aygun, B.; Kasow, K.A.; Pierciey, F.J., Jr.; Bonner, M.; et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N Engl J Med 2022, 386, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Herring, W.L.; Gallagher, M.E.; Shah, N.; Morse, K.C.; Mladsi, D.; Dong, O.M.; Chawla, A.; Leiding, J.W.; Zhang, L.; Paramore, C.; et al. Cost-Effectiveness of Lovotibeglogene Autotemcel (Lovo-Cel) Gene Therapy for Patients with Sickle Cell Disease and Recurrent Vaso-Occlusive Events in the United States. Pharmacoeconomics 2024, 42, 693–714. [Google Scholar] [CrossRef]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants (Basel) 2024, 13. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front Physiol 2020, 11, 694. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev 2014, 94, 329–354. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of aging. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Haghi Aminjan, H.; Abtahi, S.R.; Hazrati, E.; Chamanara, M.; Jalili, M.; Paknejad, B. Targeting of oxidative stress and inflammation through ROS/NF-kappaB pathway in phosphine-induced hepatotoxicity mitigation. Life Sci 2019, 232, 116607. [Google Scholar] [CrossRef] [PubMed]
- Veith, A.; Moorthy, B. Role of Cytochrome P450s in the Generation and Metabolism of Reactive Oxygen Species. Curr Opin Toxicol 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Stading, R.; Chu, C.; Couroucli, X.; Lingappan, K.; Moorthy, B. Molecular role of cytochrome P4501A enzymes inoxidative stress. Curr Opin Toxicol 2020, 20-21, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Hartlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol 2013, 1, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: in Physiology and in Disease States. Am J Biomed Sci Res 2022, 15, 153–177. [Google Scholar] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Sawicki, K.T.; Chang, H.C.; Ardehali, H. Role of heme in cardiovascular physiology and disease. J Am Heart Assoc 2015, 4, e001138. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 1973, 134, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol 2020, 37, 101674. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr Med Chem 2009, 16, 1266–1277. [Google Scholar] [CrossRef]
- Cogliati, S.; Lorenzi, I.; Rigoni, G.; Caicci, F.; Soriano, M.E. Regulation of Mitochondrial Electron Transport Chain Assembly. J Mol Biol 2018, 430, 4849–4873. [Google Scholar] [CrossRef]
- Bindoli, A.; Rigobello, M.P. Principles in redox signaling: from chemistry to functional significance. Antioxid Redox Signal 2013, 18, 1557–1593. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid Redox Signal 2017, 27, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Lyon, C.J.; Xia, X.; Liu, J.Z.; Tangirala, R.K.; Yin, F.; Boyadjian, R.; Bikineyeva, A.; Pratico, D.; Harrison, D.G.; et al. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res 2009, 104, e42–e54. [Google Scholar] [CrossRef]
- Rasheed, Z. Therapeutic potentials of catalase: Mechanisms, applications, and future perspectives. Int J Health Sci (Qassim) 2024, 18, 1–6. [Google Scholar]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim Biophys Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P.; Morgan, W.T.; Eaton, J.W.; Hedlund, B.E. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc Natl Acad Sci U S A 1988, 85, 237–241. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107. [Google Scholar] [CrossRef]
- MacKinney, A.; Woska, E.; Spasojevic, I.; Batinic-Haberle, I.; Zennadi, R. Disrupting the vicious cycle created by NOX activation in sickle erythrocytes exposed to hypoxia/reoxygenation prevents adhesion and vasoocclusion. Redox Biol 2019, 25, 101097. [Google Scholar] [CrossRef]
- Wang, Q.; Zennadi, R. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- De Franceschi, L.; Bertoldi, M.; Matte, A.; Santos Franco, S.; Pantaleo, A.; Ferru, E.; Turrini, F. Oxidative stress and beta-thalassemic erythroid cells behind the molecular defect. Oxid Med Cell Longev 2013, 2013, 985210. [Google Scholar] [CrossRef]
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Donkor, E.S.; Campbell, A.D. Oxidative Profile of Patients with Sickle Cell Disease. Med Sci (Basel) 2019, 7. [Google Scholar] [CrossRef]
- Jeong, E.M.; Liu, M.; Sturdy, M.; Gao, G.; Varghese, S.T.; Sovari, A.A.; Dudley, S.C., Jr. Metabolic stress, reactive oxygen species, and arrhythmia. J Mol Cell Cardiol 2012, 52, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Sachdev, V.; Jison, M.L.; Shizukuda, Y.; Plehn, J.F.; Minter, K.; Brown, B.; Coles, W.A.; Nichols, J.S.; Ernst, I.; et al. Pulmonary hypertension is a risk factor for death in patients with sickle cell disease. N Engl J Med 2004, 350, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Vichinsky, E. Pulmonary complications of sickle cell disease. N Engl J Med 2008, 359, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med 2002, 8, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol 2017, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Conran, N.; Belcher, J.D. Inflammation in sickle cell disease. Clin Hemorheol Microcirc 2018, 68, 263–299. [Google Scholar] [CrossRef] [PubMed]
- Bhol, N.K.; Bhanjadeo, M.M.; Singh, A.K.; Dash, U.C.; Ojha, R.R.; Majhi, S.; Duttaroy, A.K.; Jena, A.B. The interplay between cytokines, inflammation, and antioxidants: mechanistic insights and therapeutic potentials of various antioxidants and anti-cytokine compounds. Biomed Pharmacother 2024, 178, 117177. [Google Scholar] [CrossRef]
- Judge, S.; Jang, Y.M.; Smith, A.; Hagen, T.; Leeuwenburgh, C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J 2005, 19, 419–421. [Google Scholar] [CrossRef]
- Akhter, M.S.; Hamali, H.A.; Rashid, H.; Dobie, G.; Madkhali, A.M.; Mobarki, A.A.; Oldenburg, J.; Biswas, A. Mitochondria: Emerging Consequential in Sickle Cell Disease. J Clin Med 2023, 12. [Google Scholar] [CrossRef]
- Wood, K.C.; Gladwin, M.T.; Straub, A.C. Sickle cell disease: at the crossroads of pulmonary hypertension and diastolic heart failure. Heart 2020, 106, 562–568. [Google Scholar] [CrossRef]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The Worst Things in Life are Free: The Role of Free Heme in Sickle Cell Disease. Front Immunol 2020, 11, 561917. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Zhang, P.; Abdulla, F.; Nguyen, P.; Killeen, T.; Xu, P.; O’Sullivan, G.; Nath, K.A.; et al. Control of Oxidative Stress and Inflammation in Sickle Cell Disease with the Nrf2 Activator Dimethyl Fumarate. Antioxid Redox Signal 2017, 26, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Brousse, V.; Buffet, P.; Rees, D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol 2014, 166, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, M.; Fuligni, F.; Santoro, L.; Sabattini, E.; Ichino, M.; De Vito, R.; Zucchetta, P.; Colombatti, R.; Sainati, L.; Gamba, P.; et al. Spleen histology in children with sickle cell disease and hereditary spherocytosis: hints on the disease pathophysiology. Hum Pathol 2017, 60, 95–103. [Google Scholar] [CrossRef]
- Ben Khaled, M.; Ouederni, M.; Mankai, Y.; Rekaya, S.; Ben Fraj, I.; Dhouib, N.; Kouki, R.; Mellouli, F.; Bejaoui, M. Prevalence and predictive factors of splenic sequestration crisis among 423 pediatric patients with sickle cell disease in Tunisia. Blood Cells Mol Dis 2020, 80, 102374. [Google Scholar] [CrossRef] [PubMed]
- Al-Rimawi, H.S.; Abdul-Qader, M.; Jallad, M.F.; Amarin, Z.O. Acute splenic sequestration in female children with sickle cell disease in the North of Jordan. J Trop Pediatr 2006, 52, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, R.; Bandyopadhyay, S.K.; Dutta, A. Sickle cell hepatopathy. Indian J Pathol Microbiol 2008, 51, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Owen, C.; Chopra, S. Sickle cell hepatopathy. Hepatology 2001, 33, 1021–1028. [Google Scholar] [CrossRef]
- Darbari, D.S.; Kple-Faget, P.; Kwagyan, J.; Rana, S.; Gordeuk, V.R.; Castro, O. Circumstances of death in adult sickle cell disease patients. Am J Hematol 2006, 81, 858–863. [Google Scholar] [CrossRef]
- Hamideh, D.; Alvarez, O. Sickle cell disease related mortality in the United States (1999-2009). Pediatr Blood Cancer 2013, 60, 1482–1486. [Google Scholar] [CrossRef]
- Lacaille, F.; Allali, S.; de Montalembert, M. The Liver in Sickle Cell Disease. J Pediatr Gastroenterol Nutr 2021, 72, 5–10. [Google Scholar] [CrossRef]
- Badria, F.A.; Ibrahim, A.S.; Badria, A.F.; Elmarakby, A.A. Curcumin Attenuates Iron Accumulation and Oxidative Stress in the Liver and Spleen of Chronic Iron-Overloaded Rats. PLoS One 2015, 10, e0134156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Weihrauch, D.; Jones, D.W.; Jing, X.; Shi, Y.; Gourlay, D.; Oldham, K.T.; Hillery, C.A.; Pritchard, K.A., Jr. Inhibition of myeloperoxidase decreases vascular oxidative stress and increases vasodilatation in sickle cell disease mice. J Lipid Res 2013, 54, 3009–3015. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Fasano, R.M. Management of Patients with Sickle Cell Disease Using Transfusion Therapy: Guidelines and Complications. Hematol Oncol Clin North Am 2016, 30, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Hebbel, R.P.; Steinberg, M.H.; Gladwin, M.T. Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol 2009, 84, 618–625. [Google Scholar] [CrossRef]
- Do, B.K.; Rodger, D.C. Sickle cell disease and the eye. Curr Opin Ophthalmol 2017, 28, 623–628. [Google Scholar] [CrossRef]
- Beetsch, J.W.; Park, T.S.; Dugan, L.L.; Shah, A.R.; Gidday, J.M. Xanthine oxidase-derived superoxide causes reoxygenation injury of ischemic cerebral endothelial cells. Brain Res 1998, 786, 89–95. [Google Scholar] [CrossRef]
- Hamer, I.; Wattiaux, R.; Wattiaux-De Coninck, S. Deleterious effects of xanthine oxidase on rat liver endothelial cells after ischemia/reperfusion. Biochim Biophys Acta 1995, 1269, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Solovey, A.; Gui, L.; Key, N.S.; Hebbel, R.P. Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest 1998, 101, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Promsote, W.; Powell, F.L.; Veean, S.; Thounaojam, M.; Markand, S.; Saul, A.; Gutsaeva, D.; Bartoli, M.; Smith, S.B.; Ganapathy, V.; et al. Oral Monomethyl Fumarate Therapy Ameliorates Retinopathy in a Humanized Mouse Model of Sickle Cell Disease. Antioxid Redox Signal 2016, 25, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.J.; Jennette, J.C. Sickle cell nephropathy. Adv Nephrol Necker Hosp 1994, 23, 133–147. [Google Scholar]
- Saborio, P.; Scheinman, J.I. Sickle cell nephropathy. J Am Soc Nephrol 1999, 10, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.T.; Pham, P.C.; Wilkinson, A.H.; Lew, S.Q. Renal abnormalities in sickle cell disease. Kidney Int 2000, 57, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P. The sickle erythrocyte in double jeopardy: autoxidation and iron decompartmentalization. Semin Hematol 1990, 27, 51–69. [Google Scholar]
- Nath, K.A.; Grande, J.P.; Haggard, J.J.; Croatt, A.J.; Katusic, Z.S.; Solovey, A.; Hebbel, R.P. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol 2001, 158, 893–903. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr Nephrol 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Juncos, J.P.; Grande, J.P.; Croatt, A.J.; Hebbel, R.P.; Vercellotti, G.M.; Katusic, Z.S.; Nath, K.A. Early and prominent alterations in hemodynamics, signaling, and gene expression following renal ischemia in sickle cell disease. Am J Physiol Renal Physiol 2010, 298, F892–899. [Google Scholar] [CrossRef]
- Jin, Q.; Liu, T.; Qiao, Y.; Liu, D.; Yang, L.; Mao, H.; Ma, F.; Wang, Y.; Peng, L.; Zhan, Y. Oxidative stress and inflammation in diabetic nephropathy: role of polyphenols. Front Immunol 2023, 14, 1185317. [Google Scholar] [CrossRef]
- Goedeke, L.; Fernandez-Hernando, C. Regulation of cholesterol homeostasis. Cell Mol Life Sci 2012, 69, 915–930. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Hong, E.M.; Kim, M.; Kim, J.H.; Jang, J.; Park, S.W.; Byun, H.W.; Koh, D.H.; Choi, M.H.; Kae, S.H.; et al. Simvastatin induces heme oxygenase-1 via NF-E2-related factor 2 (Nrf2) activation through ERK and PI3K/Akt pathway in colon cancer. Oncotarget 2016, 7, 46219–46229. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Feng, Y.; Cui, R.; Qiu, M.; Zhang, J.; Liu, C. Simvastatin protects against acetaminophen-induced liver injury in mice. Biomed Pharmacother 2018, 98, 916–924. [Google Scholar] [CrossRef]
- Pritchard, K.A.; Ou, J.; Ou, Z.; Shi, Y.; Franciosi, J.P.; Signorino, P.; Kaul, S.; Ackland-Berglund, C.; Witte, K.; Holzhauer, S.; et al. Hypoxia-induced acute lung injury in murine models of sickle cell disease. Am J Physiol Lung Cell Mol Physiol 2004, 286, L705–L714. [Google Scholar] [CrossRef]
- Zhu, X.; Xi, C.; Thomas, B.; Pace, B.S. Loss of NRF2 function exacerbates the pathophysiology of sickle cell disease in a transgenic mouse model. Blood 2018, 131, 558–562. [Google Scholar] [CrossRef]
- Keleku-Lukwete, N.; Suzuki, M.; Otsuki, A.; Tsuchida, K.; Katayama, S.; Hayashi, M.; Naganuma, E.; Moriguchi, T.; Tanabe, O.; Engel, J.D.; et al. Amelioration of inflammation and tissue damage in sickle cell model mice by Nrf2 activation. Proc Natl Acad Sci U S A 2015, 112, 12169–12174. [Google Scholar] [CrossRef]
- Ghosh, S.; Hazra, R.; Ihunnah, C.A.; Weidert, F.; Flage, B.; Ofori-Acquah, S.F. Augmented NRF2 activation protects adult sickle mice from lethal acute chest syndrome. Br J Haematol 2018, 182, 271–275. [Google Scholar] [CrossRef]
- Musicki, B.; Liu, T.; Sezen, S.F.; Burnett, A.L. Targeting NADPH oxidase decreases oxidative stress in the transgenic sickle cell mouse penis. J Sex Med 2012, 9, 1980–1987. [Google Scholar] [CrossRef]
- Lei, J.; Paul, J.; Wang, Y.; Gupta, M.; Vang, D.; Thompson, S.; Jha, R.; Nguyen, J.; Valverde, Y.; Lamarre, Y.; et al. Heme Causes Pain in Sickle Mice via Toll-Like Receptor 4-Mediated Reactive Oxygen Species- and Endoplasmic Reticulum Stress-Induced Glial Activation. Antioxid Redox Signal 2021, 34, 279–293. [Google Scholar] [CrossRef]
- Pace, B.S.; Shartava, A.; Pack-Mabien, A.; Mulekar, M.; Ardia, A.; Goodman, S.R. Effects of N-acetylcysteine on dense cell formation in sickle cell disease. Am J Hematol 2003, 73, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Nur, E.; Brandjes, D.P.; Teerlink, T.; Otten, H.M.; Oude Elferink, R.P.; Muskiet, F.; Evers, L.M.; ten Cate, H.; Biemond, B.J.; Duits, A.J.; et al. N-acetylcysteine reduces oxidative stress in sickle cell patients. Ann Hematol 2012, 91, 1097–1105. [Google Scholar] [CrossRef]
- Hasan Tahsin Ozpolat, J.C. , Xiaoyun Fu, Shelby A Cate, Jennie Le, Minhua Ling, Colette Norby, Dominic W Chung, Barbara A. Konkle, Jose A. Lopez. A Pilot Study of High-Dose N-Acetylcysteine Infusion in Patients with Sickle Cell Disease. 2021.
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Abdulla, F.; Zhang, P.; Nguyen, H.; Nguyen, P.; Killeen, T.; Miescher, S.M.; Brinkman, N.; et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS One 2018, 13, e0196455. [Google Scholar] [CrossRef] [PubMed]
- Buehler, P.W.; Swindle, D.; Pak, D.I.; Ferguson, S.K.; Majka, S.M.; Karoor, V.; Moldovan, R.; Sintas, C.; Black, J.; Gentinetta, T.; et al. Hemopexin dosing improves cardiopulmonary dysfunction in murine sickle cell disease. Free Radic Biol Med 2021, 175, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Gentinetta, T.; Belcher, J.D.; Brugger-Verdon, V.; Adam, J.; Ruthsatz, T.; Bain, J.; Schu, D.; Ventrici, L.; Edler, M.; Lioe, H.; et al. Plasma-Derived Hemopexin as a Candidate Therapeutic Agent for Acute Vaso-Occlusion in Sickle Cell Disease: Preclinical Evidence. J Clin Med 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Shore, P.M.; B.J.B.; Kesse-Adu, R.; Wahab, E.; Desai, P.C.; Boucher, A.; Eleftheriou, P.; Rijneveld, A.; De Castro, L.M.; Bergmann, S.; Kato, G.J.; Jochems, J.; Wilson, F.; Jung, K.; Gordeuk, V.R. Phase 1 Study of the Safety and Pharmacokinetics of CSL889 (Hemopexin) in Adults with SCD. Journal of Sickle Cell Disease 2024, 1.
- Stivala, S.; Gobbato, S.; Bonetti, N.; Camici, G.G.; Luscher, T.F.; Beer, J.H. Dietary alpha-linolenic acid reduces platelet activation and collagen-mediated cell adhesion in sickle cell disease mice. J Thromb Haemost 2022, 20, 375–386. [Google Scholar] [CrossRef]
- Martins, V.D.; Manfredini, V.; Peralba, M.C.; Benfato, M.S. Alpha-lipoic acid modifies oxidative stress parameters in sickle cell trait subjects and sickle cell patients. Clin Nutr 2009, 28, 192–197. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, X.; Yu, A.; Ward, C.M.; Pace, B.S. delta-Aminolevulinate induces fetal hemoglobin expression by enhancing cellular heme biosynthesis. Exp Biol Med (Maywood) 2019, 244, 1220–1232. [Google Scholar] [CrossRef]
- Morris, C.R.; Brown, L.A.S.; Reynolds, M.; Dampier, C.D.; Lane, P.A.; Watt, A.; Kumari, P.; Harris, F.; Manoranjithan, S.; Mendis, R.D.; et al. Impact of arginine therapy on mitochondrial function in children with sickle cell disease during vaso-occlusive pain. Blood 2020, 136, 1402–1406. [Google Scholar] [CrossRef]
- Rees, C.A.; Brousseau, D.C.; Cohen, D.M.; Villella, A.; Dampier, C.; Brown, K.; Campbell, A.; Chumpitazi, C.E.; Airewele, G.; Chang, T.; et al. Sickle Cell Disease Treatment with Arginine Therapy (STArT): study protocol for a phase 3 randomized controlled trial. Trials 2023, 24, 538. [Google Scholar] [CrossRef]
- Perrine, S.P.; Ginder, G.D.; Faller, D.V.; Dover, G.H.; Ikuta, T.; Witkowska, H.E.; Cai, S.P.; Vichinsky, E.P.; Olivieri, N.F. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N Engl J Med 1993, 328, 81–86. [Google Scholar] [CrossRef]
- Atweh, G.F.; Sutton, M.; Nassif, I.; Boosalis, V.; Dover, G.J.; Wallenstein, S.; Wright, E.; McMahon, L.; Stamatoyannopoulos, G.; Faller, D.V.; et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood 1999, 93, 1790–1797. [Google Scholar] [PubMed]
- Molokie, R.; Lavelle, D.; Gowhari, M.; Pacini, M.; Krauz, L.; Hassan, J.; Ibanez, V.; Ruiz, M.A.; Ng, K.P.; Woost, P.; et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: A randomized phase 1 study. PLoS Med 2017, 14, e1002382. [Google Scholar] [CrossRef] [PubMed]
- Saunthararajah, Y.; Molokie, R.; Saraf, S.; Sidhwani, S.; Gowhari, M.; Vara, S.; Lavelle, D.; DeSimone, J. Clinical effectiveness of decitabine in severe sickle cell disease. Br J Haematol 2008, 141, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Hu, T.; Ho, M.H.; Wang, Y.; Yu, M.; Patel, N.; Pi, W.; Choi, J.H.; Xu, H.; Ganapathy, V.; et al. Hydroxyurea differentially modulates activator and repressors of gamma-globin gene in erythroblasts of responsive and non-responsive patients with sickle cell disease in correlation with Index of Hydroxyurea Responsiveness. Haematologica 2017, 102, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, J.; Tan, Y.; Beyer, A.I.; Xie, F.; Muench, M.O.; Kan, Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and beta-thalassemia. Proc Natl Acad Sci U S A 2016, 113, 10661–10665. [Google Scholar] [CrossRef]
- Venkatesan, V.; Christopher, A.C.; Rhiel, M.; Azhagiri, M.K.K.; Babu, P.; Walavalkar, K.; Saravanan, B.; Andrieux, G.; Rangaraj, S.; Srinivasan, S.; et al. Editing the core region in HPFH deletions alters fetal and adult globin expression for treatment of beta-hemoglobinopathies. Mol Ther Nucleic Acids 2023, 32, 671–688. [Google Scholar] [CrossRef]
- Antoniani, C.; Meneghini, V.; Lattanzi, A.; Felix, T.; Romano, O.; Magrin, E.; Weber, L.; Pavani, G.; El Hoss, S.; Kurita, R.; et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human beta-globin locus. Blood 2018, 131, 1960–1973. [Google Scholar] [CrossRef]
- Martyn, G.E.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.M.; Funnell, A.P.W.; et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet 2018, 50, 498–503. [Google Scholar] [CrossRef]
- Weber, L.; Frati, G.; Felix, T.; Hardouin, G.; Casini, A.; Wollenschlaeger, C.; Meneghini, V.; Masson, C.; De Cian, A.; Chalumeau, A.; et al. Editing a gamma-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv 2020, 6. [Google Scholar] [CrossRef]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Macari, E.R.; Lowrey, C.H. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood 2011, 117, 5987–5997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, H.; Zhao, M.; Chang, C.; Lu, Q. The Bach Family of Transcription Factors: A Comprehensive Review. Clin Rev Allergy Immunol 2016, 50, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Nataraja, S.; Abdulla, F.; Zhang, P.; Chen, C.; Nguyen, J.; Ruan, C.; Singh, M.; Demes, S.; Olson, L.; et al. The BACH1 inhibitor ASP8731 inhibits inflammation and vaso-occlusion and induces fetal hemoglobin in sickle cell disease. Front Med (Lausanne) 2023, 10, 1101501. [Google Scholar] [CrossRef] [PubMed]
- Attucks, O.C.; Jasmer, K.J.; Hannink, M.; Kassis, J.; Zhong, Z.; Gupta, S.; Victory, S.F.; Guzel, M.; Polisetti, D.R.; Andrews, R.; et al. Induction of heme oxygenase I (HMOX1) by HPP-4382: a novel modulator of Bach1 activity. PLoS One 2014, 9, e101044. [Google Scholar] [CrossRef]
- Palani, C.D.; Zhu, X.; Alagar, M.; Attucks, O.C.; Pace, B.S. Bach1 inhibitor HPP-D mediates gamma-globin gene activation in sickle erythroid progenitors. Blood Cells Mol Dis 2024, 104, 102792. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wu, F.; Yang, L.; Wang, F.; Zhang, T.; Deng, X.; Zhang, X.; Yuan, X.; Yan, Y.; Li, Y.; et al. miR-144/451 inhibits c-Myc to promote erythroid differentiation. FASEB J 2020, 34, 13194–13210. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chao, R.; Zhang, Y.; Wang, P.; Gong, X.; Liang, D.; Wang, Y. CAP1, a target of miR-144/451, negatively regulates erythroid differentiation and enucleation. J Cell Mol Med 2021, 25, 2377–2389. [Google Scholar] [CrossRef]
- Li, B.; Zhu, X.; Ward, C.M.; Starlard-Davenport, A.; Takezaki, M.; Berry, A.; Ward, A.; Wilder, C.; Neunert, C.; Kutlar, A.; et al. MIR-144-mediated NRF2 gene silencing inhibits fetal hemoglobin expression in sickle cell disease. Exp Hematol 2019, 70, 85–96. [Google Scholar] [CrossRef]
- Mann, G.E. Nrf2-mediated redox signalling in vascular health and disease. Free Radic Biol Med 2014, 75 (Suppl. 1), S1. [Google Scholar] [CrossRef]
- Milan, K.L.; Gayatri, V.; Kriya, K.; Sanjushree, N.; Vishwanathan Palanivel, S.; Anuradha, M.; Ramkumar, K.M. MiR-142-5p mediated Nrf2 dysregulation in gestational diabetes mellitus and its impact on placental angiogenesis. Placenta 2024, 158, 192–199. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, L.; Lu, Y.; Zhang, M.; Zhang, Z.; Wang, K.; Lv, J. Down-regulation of microRNA-142-5p attenuates oxygen-glucose deprivation and reoxygenation-induced neuron injury through up-regulating Nrf2/ARE signaling pathway. Biomed Pharmacother 2017, 89, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Teimouri, M.; Hosseini, H.; Shabani, M.; Koushki, M.; Noorbakhsh, F.; Meshkani, R. Inhibiting miR-27a and miR-142-5p attenuate nonalcoholic fatty liver disease by regulating Nrf2 signaling pathway. IUBMB Life 2020, 72, 361–372. [Google Scholar] [CrossRef]
- BJ Biemond, B.S., F Wilson, J Jochems, LM Lindqvist, K Jung, T Gentinetta, S Costin, GJ Kato, P Eleftheriou, H Fok, E Wahab, PM Leung, J Sharif, A Boucher, R Fitzgerald, R Keese-Adu, R Azbell, D Liles, S Bergmann, S Lanzkron, V Gordeuk. A PHASE 1 STUDY OF CSL888 (HEMOPEXIN) IN ADULT PATIENTS WITH SICKLE CELL DISEASE. Hemasphere 2023, 7. [CrossRef]
- Hoppe, C.; Kuypers, F.; Larkin, S.; Hagar, W.; Vichinsky, E.; Styles, L. A pilot study of the short-term use of simvastatin in sickle cell disease: effects on markers of vascular dysfunction. Br J Haematol 2011, 153, 655–663. [Google Scholar] [CrossRef]
- Haffner, S.M.; Alexander, C.M.; Cook, T.J.; Boccuzzi, S.J.; Musliner, T.A.; Pedersen, T.R.; Kjekshus, J.; Pyorala, K. Reduced coronary events in simvastatin-treated patients with coronary heart disease and diabetes or impaired fasting glucose levels: subgroup analyses in the Scandinavian Simvastatin Survival Study. Arch Intern Med 1999, 159, 2661–2667. [Google Scholar] [CrossRef]
- Melo, D.; Ferreira, F.; Teles, M.J.; Porto, G.; Coimbra, S.; Rocha, S.; Santos-Silva, A. Catalase, Glutathione Peroxidase, and Peroxiredoxin 2 in Erythrocyte Cytosol and Membrane in Hereditary Spherocytosis, Sickle Cell Disease, and beta-Thalassemia. Antioxidants (Basel) 2024, 13. [Google Scholar] [CrossRef]
- Oh, J.Y.; Bae, C.Y.; Kasztan, M.; Pollock, D.M.; Russell, R.T.; Lebensburger, J.; Patel, R.P. Peroxiredoxin-2 recycling is slower in denser and pediatric sickle cell red cells. FASEB J 2022, 36, e22267. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; Federti, E.; Tibaldi, E.; Di Paolo, M.L.; Bisello, G.; Bertoldi, M.; Carpentieri, A.; Pucci, P.; Iatchencko, I.; Wilson, A.B.; et al. Tyrosine Phosphorylation Modulates Peroxiredoxin-2 Activity in Normal and Diseased Red Cells. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, S.U.; Kwon, T.H.; Lee, D.S.; Ha, H.L.; Park, D.S.; Woo, E.J.; Lee, S.H.; Kim, J.M.; Chae, H.B.; et al. Peroxiredoxin II is essential for preventing hemolytic anemia from oxidative stress through maintaining hemoglobin stability. Biochem Biophys Res Commun 2012, 426, 427–432. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



