Submitted:
28 February 2025
Posted:
03 March 2025
You are already at the latest version
Abstract
Oxovanadium and zinc complexes are reported as insulin-mimetics. They inhibit several proteins including enzymes which belong to the same class of membrane sensitive phosphatases, similar in terms of general architecture and biochemistry of the active site. Borrowing from this summary, we explore the structural and electronic properties of representative oxovanadium and zinc complexes as computed in isolation and upon binding to PTEN and PTP1B phosphatases. Using crystallographic data and quantum chemistry calculations, we investigate how bonding nature and structural flexibility of the studied inhibitors affects efficiency of their binding to the active sites of the enzymes: albeit different, the two active sites represent evolutionary variant choices of the same type of biochemistry of phosphatases. As a result of our studies, we address optical responses which can be suitable for diagnostics and discuss engineering of AI assisted protein embedding to alter electronic states of metal centres which may be beneficial for biomedical and quantum information applications within the bio-spintronics of tomorrow.
Keywords:
Oxovanadium-complexes
; zinc-complexes
; Insulin-mimetics
; diabetes
; DFT
; infrared
; Raman
; circular dichroism
Introduction
Metal cations and their complexes are reported as possible medical remedies since ancient times. Perhaps, the case of cisplatin [1] can be considered to be the beginning of a systematic approach to exploring how oxidation state, coordination geometry and ligand nature determine the chemistry of biomolecular interactions with medical implications [2].
Diabetes mellitus is a family of endocrine diseases characterized by high blood sugar levels. The main forms, known as type 1 and type 2, are due to insulin insufficiency (arising from autoimmune β-cell destruction) and to decay of β-cell insulin secretion on a background of insulin resistance that involves insulin receptor malfunction, respectively. In the first case, treatment relies on insulin injection. Treatment of patients with type 2 diabetes involves bio-chemo-therapies employing insulin secretion promoters, suppression of glucose absorption and insulin resistance [3]. Improvements of the efficiency of such treatments relies on better understanding of type 2 diabetes biochemistry and on devising pharmacophores to compensate diabetes signal transduction pathways with better intelligence.
Positive effects of using metal ions for the treatment of diabetes were reported in 1899, prior to the discovery of insulin by Lyonnet and coworkers [4], who observed glucose-lowering when using sodium metavanadate [4]. In 1971, ammonium metavanadate solution injected intraperitoneally was reported to affect glucose metabolism in rats [5]. Later studies discussed vanadium (V) bioactivity to stem from the role of vanadate, , in regulation of phosphate-dependent processes, such as metabolic pathways relying on phosphatases, such as tyrosine phosphatase (PTP1B), and kinases [6,7]. Reports on the significance of ligands in V bioactivity include oxovanadium complexes with maltolate, VO(mal)2, and picolinate, VO(pic)2, which were reported as effective anti-diabetic remedies [8,9].
Zinc (Zn) cations came under focus as a possible antidiabetic agent in 1980, when ZnCl2 was reported to stimulate lipogenesis in rat adipocytes in a similar manner to the action of insulin [10], aiding both types of diabetes treatment but under high doses of ZnCl2 [11,12]. Since then, mechanisms of the insulin-mimetic action of Zn(II) have been examined in respect to glucose oxidation and lipolysis stimulation [13], glucose transport and glycogen synthesis [14], and inhibition of endogenous glycogen synthase kinase-3b [15]. As a result of these studies, maltolato-Zn(II) complex, Zn(mal)2 has been found to be potent in lowering blood glucose levels in the treatment of type 2 diabetic animals [16]. The ability of Zn compounds to increase glucose transport, glycogen synthesis, and lipogenesis; to inhibit gluconeogenesis and lipolysis, and to modulate key elements of the insulin signaling pathway [17,18] have been suggested to contribute to improve hyperglycemia and glucose homeostasis in diabetes.
Promise of effective Zn and V based organometallic anti-diabetic remedies, stimulated research towards the identification of proteins involved in interactions, the speciation of active metal states if complexes exchange ligands with bioactive species like tartrate and citrate [19,20], characterization pharmacodynamics and biocompatibility in dependence on the nature of the ligands present. For example, the lower solubility and stronger gastrointestinal irritation of VO(pic)2 with respect to VO(mal)2 stimulated further studies towards better biocompatibility [19]. In this respect, it is worth noting that natural allixin, found in garlic, provides vanadyl and Zn complexes with excellent capacity to lower high blood glucose levels in animal models in both type 1 and 2 diabetes [21,22].
Understanding mechanisms of anti-diabetic activities concerns the identification of which enzymes are affected by Zn and V complexes. As we have mentioned, early studies suggested that V systems were identified to affect metabolic pathways relying on phosphatases, like PTP1B, and kinases [6,7,23]. Comparatively, it has been reported that Zn2+ cations induce degradation of phosphatase and tensin homologue (PTEN) [24]. In addition, the overexpression of PTEN in mice impaired the insulin signalling pathway leading to insulin resistance [25]. Based on these observations, it has been proposed that Zn(alx)2 and Zn(tanm)2 affect PTEN and activate PI3K-Akt/PKB signalling [26].
Both, vanadyl and Zn2+ ions, in place of insulin, induce insulin-mimetic effects with regard to both incorporation of glucose in rat adipocytes and inhibition of free fatty acids released from the adipocytes [27,28]. These ions have been found to simultaneously act on multiple sites in the adipocytes, thus suggesting their action to be noted as an “ensemble mechanism” [29]. However, the critical action sites for V and Zn ionic species are slightly different from each other. For example, it was observed that VO(alx)2 targets IRβ and/or PTPase [30], while Zn(alx)2 and Zn(tanm)2 target PTEN [31].
The complexity of the discussed “ensemble mechanism” of anti-diabetic actions of Zn and V complexes concerns the interactions of the complexes with different enzymes, but, often, they belong to the phosphatase family, which are substrate specific and exhibit sequence homology at the catalytic site [32]. Considering the diversity of the family as a promising palette for artificial intelligence training, here, we may adopt PTP1B and PTEN as archetype phosphatase examples for introductory investigations of the interactions of V and Zn complexes with such enzymes.
PTP1B is an enzyme involved in the dephosphorylation of proteins with tyrosine residues, and, hence, it plays an essential role in many signal transduction pathways. [33,34,35]. It is known that the enzyme works as a negative regulator of insulin signal transduction [36] and hence it could be a pharmacological target for the treatment of type 2 diabetes and obesity [37,38,39]. PTP1B is composed of three domains: the catalytic domain (30–278) at the N-terminal, the regulatory domain (278–401), and the C-terminal (401–435), which is responsible for membrane binding [40]. The active site of the enzyme is located in a relatively shallow pocket: the site contains a P-loop with Cys215 and Arg221 residues, which are crucial for the dephosphorylation processes [41,42]. The P-loop is a major binding site for orthovanadate and many other inhibitors [43,44]. It is surrounded by four structural elements: the WPD loop (Trp179-Asp181), the Q-loop (Arg257-Thr263), the pTyr-loop (Asn42-Val49) and the E-loop (Leu110-Gln123). These structures are responsible for ligand binding, substrate recognition and take part in catalytic functions [45]. Crystallographic studies indicated that PTP1B exists in “open” and “closed” conformations, where the WPD loop moves out or inside to form an open or close binding pocket, respectively [41,42,46,47].
PTEN is a critical negative regulator of insulin signalling through its role in dephosphorylation of phosphatidylinositol 3,4,5-triphosphate to phosphatidylinositol 4,5-bisphosphate in the phosphoinositide 3-kinase (PI3K) pathway [48]. The enzyme has two important folds contributing to this behaviour: a DUSP domain (residues 15-185) and a C2 domain (residues 192-353). These domains are linked with three disordered segments which play a role in membrane binding and allosteric regulation of the enzyme [49]. Compared to PTP1B, the pocket of PTEN has similar depth (about 8 Å), but it is about twice as wide, 5 × 11 Å [50]. The active site of PTEN is made by three loops: the WPD loop (residues 88-98), the TI loop, which contains a Thr and Ile pair and spans residues 160-171, and the P loop with a Cys residue (C124) which is fundamental for substrate catalysis. Both the Cys and Arg residues within this loop, C124 and R130, are completely intolerant to mutations [51].
Recently, structural properties of anticancer V and antidiabetic Zn complexes bound to PTP1B were investigated using classical molecular dynamics simulations [52,53]. To the best of our knowledge, there are no reports concerning interactions of candidate metal-organic inhibitors with PTEN. Considering the lack of comparative studies of structural and electronic properties of the considered Zn and V metalorganic potential inhibitors either in solution or bound to enzymes, in the present contribution we adopt density functional theory (DFT) calculations to “refine” predictions of docking and describe structure and flexibility of the selected Zn and oxovanadium metalorganic systems reported to have antidiabetic properties. A quantum level description is particularly important in the application of the V4+ ion, featured by one unpaired d-electron.
In this work, we address structural features of the complexes, especially focusing on their binding to the considered enzymes. We then analyse optical electronic, infrared (IR) and Raman spectral properties, which can be used for experimental diagnostics. This investigation allows us to discuss what makes the investigated complexes similar and different as antidiabetic remedies. Finally, we address biospintronic opportunities to alter metal electronics via engineering of protein embedding environment, which may be attuned using Neural Network training according to diversity of phosphatases, predictions of DFT and experimental/empirical knowledge.
Methods
Structural Definition of the Complexes
In Figure 1, we report a schematic outline of the complexes considered here. In the case, when R2, R5 and R6 are hydrogen atoms, M is a V4+ cation double bonded to Oa, the ligands are pyromeconic acids or 3-hydroxy-4-pyrone that form the bis(3-hydroxy-4-pyronato) oxovanadium(IV) complex, VO(3hp)2. When R2 is a methyl moiety, the ligand is larixinic acid (or maltol, or 3-hydroxy-2-methyl-4-pyrone): with M = Zn2+, the complex is bis(maltolato)zinc(II), Zn(mal)2. Adopting a pentyl moiety for R2, a methoxy group for R5 and a methyl for R6 we represent the allixin ligand (or 3-hydroxy-5-methoxy-6-methyl-2-pentyl-4-pyrone): with oxovanadium the ligands form bis(allixinato)oxovanadium(IV), VO(alx)2. The image at the right side of Figure 1 shows that in experiment the complexes may be presented as D and L enantiomers to demonstrate no optical activity. We conducted studies for the D enantiomer mainly, unless indicated otherwise.
Dynamics of the Complexes in Vacuum and in Local Water Clusters
To characterize structural features of the systems under study, we carry out Born-Oppenheimer ab initio molecular dynamics simulations for the bis(3-hydroxy-4-pyronato) oxovanadium(IV) complex, VO(3hp)2, and the bis(maltolato) zinc(II) complex, Zn(mal)2 in vacuum and in water clusters made of 25 molecules using the CP2K program [54]. We perform NVT (constant volume, constant temperature, using a Nosé thermostat [61]) simulations in cubic boxes with side-lengths of about 17 Å (the length varies ±2 Å from system to system), under periodic boundary conditions. We employ the generalized gradient approximation and the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional [55,56]. The Grimme D3 approach is taken to account for dispersive interactions [57] and the dzvp-molpot double-ζ polarization basis [58] is used for the Geodecker-Teter-Hutter pseudopotentials [59]. A time-step of 0.5 fs is employed. In each case, we performed a thermalization phase under a velocity rescaling regime [60]: typically, thermalization in the prepared systems is reached within 300 fs. Next, we conduct NVT sampling of 1 ps. Target accuracy for the self-consistent field convergence is 10−6 hartree. The cut-off and the relative cut-off of the grid level are set to 400 and 100 Rydberg, respectively. The energy convergence threshold is set to 10−12. We conduct data analysis as well as preparation and training of convolution Neural Net using Mathematica 12, Wolfram Research Inc.
DFT Calculations
Considering the results reported earlier [62,63], we carry out DFT calculations for selected configurations using the LANL2DZ basis set for the V4+ and Zn2+ ions and the 6-31++g(d,p) basis set for all other atoms under B3LYP exchange-correlation functional [64] as implemented in the Gaussian 09 program [65]. Natural atomic orbital and natural bond orbital analysis is according to Gaussian NBO Version 3.1 [66].
To compute pre-resonance Raman intensities, we employ cphf=rdfreq instruction, as implemented under Gaussian09. The instruction allows setting the electromagnetic perturbation wavelength, which we ascribe to 530 nm, as the most commonly used wavelength in experiments. The vibrational frequencies have been scaled by a factor 0.97 (according to our FTIR measurements in metal-organic systems as we reported elsewhere [67]). We express IR and Raman spectra taking convolutions with a Lorentzian function of half-width at half-maximum of 10 cm-1. In the Supplementary Material, we present data without scaling.
To address the optical electronic properties of the considered systems, we employ time-dependent DFT (TD-DFT) using the level of theory employed for the ground state calculations. Optical absorption and circular dichroism spectra in ultraviolet-visible (UV-VIS) spectral range are expressed taking convolutions with Lorentzian line-shapes of half-width at half-maximum of 50 cm-1. While TD-DFT provides a description of the electronic excited states, typically, the results are ambiguous for transitions between the highest occupied molecular orbital and the lowest unoccupied molecular orbital for simple molecules, only. For complex species, electronic transitions may involve several molecular orbitals without a single dominant component. In order to reduce the complexity for the considered complexes, we exploit the Koopmans’ theorem [68] and search for a transformation of the density matrix [69] to consolidate electronic redistributions specific to a selected transition as the “lower” and the “upper” orbital components. Pairs of such orbital components form the so-called natural transition orbitals (NTO) [70]. By visualizing NTOs of the electronic transitions responsible for optical density in the visible spectral range, we obtain a systematic description of the electronic components governing spectral responses in the visible spectral range in dependence on the conformation and the degree of hydration.
Protein Structures
For the structures of a phosphatase and tensin homolog (PTEN) and Protein Tyrosine Phosphatase 1B (PTP1B), we take structural information from crystallographic studies according to RCSB Protein Data Bank PDB ID 5BZX [71] and PDB ID 1ONZ [72]. Further, we employ the NAMD program [73] to add hydrogens atoms and to confirm the structural consistency of the proteins.
In this study, we adopt Cys residues protonated according to their common pKa, c.a. 8.5. Since we explore the consequences of most effective binding of oxovanadium complexes to the active site of PTP1B, to avoid strong electrostatic repulsion between the oxovanadium oxygen and the negatively charged thiol, we protonate PTP1B Cys215 residue. Here, it is important to note that commonly it is assumed that, due to unusually low pKa (between 4.5 and 5.5, compared to a typical Cys pKa, c.a. 8.5 [74]), deprotonated Cys215 should play the key role of a nucleophile interacting with the phosphate group. However, there is a discussion which suggests that protonation and hydration of Cys215 may be important for Michaelis complex formation [75,76,77,78].
Docking and ONIOM Studies
Docking calculations were performed using Autodock software [79] under the scoring and minimization Vina setting [80]. The docked conformations of each ligand were ranked according to binding energies. For QM/MM studies, we sorted the top-ranked structures. To refine docked structures and to express optical properties of the complexes when in protein environment, we employ two-layer ONIOM (our own n-layered integrated molecular orbital and molecular mechanics) approach [81].
Specifically, when using a two-layered approach, we adopt AMBER molecular mechanics [82] for protein and DFT setting for a complex as we describe in the DFT subsection. The step from docking to ONIOM optimization accounts full protonation of the docked structure. This may introduce strongly hindered interactions. Thus, to allow for a soft structural relaxation, we realize the first 4 optimization cycles allowing full flexibility to proximal (to the complex protein) moieties, while freezing the rest of the protein and the central metal cation. Consequently, we unlock the complex and proximal side- groups for optimization, while keeping the rest of the protein in a frozen configuration.
Results and Discussion
Dynamics of Complexes in Water Clusters
In Figure 2, we present structural and electronic properties of VO(3hp)2 and Zn(mal)2 complexes simulated in water clusters using BOMD. Here, it is important to stress that BOMD trajectories have been realized in an explicit water environment starting from energy global minimum structures for the complexes in vacuum obtained by DFT. Table 1 and Table 2 report selected properties of bonds for the selected systems computed by DFT, while Tables S1-S4 in the Supplementary Material provide an extended data set. For example, in the case of the Zn systems, DFT predicts tetrahedral geometry: for Zn(mal)2, as represented by the small cyan and dark red points in Figure 2A2, which reflect the angular values of the mainframe under tetrahedral geometry.
The results sampled along the BOMD trajectories, as presented in Figure 2, are introductory to address why the complexes demonstrate analogous antidiabetic tendencies, whereas the metals and their valency are not alike. According to the formal valencies, we may note that Mulliken charge for V4+ cation fluctuates around higher values than is the case of Zn2+ (panels D1 and D2). At the same time, polarities of the coordinated oxygen atoms of VO(3hp)2 are nearly comparable to those in the case of Zn(mal)2 complex (Figure 2B1 and Figure 2B2), while polarities of the next coordinating carbon atoms of VO(3hp)2 are slightly higher than in the case of Zn(mal)2 complex (Figure 2C1 and Figure 2C2). Here, it is interesting to compare correlations of the Mulliken charge fluctuations in both systems. Figure 2F1 and Figure 2F2 show comparable metal-oxygen, oxygen-oxygen and oxygen-carbon anticorrelation patterns. Nonuniform metal-oxygen anticorrelations (for example, M-O3 and M-O’3 covariance values are not the same) suggest not identical interactions with proximal water at opposite sides of the complexes, what is due to insufficient averaging along the finite length of the trajectories.
What is rather interesting is that, in explicit aqueous environment, Zn(mal)2 departs from its global minimum tetrahedral geometry (see the corresponding characteristic angular relations as given by small cyan and dark red points in Figure 2A2) to attract a water molecule under apical geometry: see green line trajectory in Figure 2A2 and Mulliken reading in Figure 2E2 (to parallel analogous data for VO(3hp)2 as presented in Figure 2A2), and Mulliken reading in Figure 2E2 (to parallel analogous data for VO(3hp)2 as presented in Figure 2A1 and Figure 2E1). Association of water is via sigma p-electron interactions, which are for the expense of electronic depletion of carbonyl and carboxylate moieties as the metal cations. This is well reflected if we compare ESP charges computed under DFT for tetrahedral Zn(mal)2 and Zn(mal)2·H2O (see Table 2).
Stimulated by the results of BOMD trajectories for Zn systems, we conduct additional DFT studies to address the structural properties of Zn2+ systems under coordination with explicit water molecules. Since water association with Zn(mal)2 in an explicit aqueous cluster is dynamic, computing this complex in vacuum (under DFT) cannot suggest a structure similar to that of VO(3hp)2. Only using ab initio simulation in explicit water, do we observe the coordination change from Zn(mal)2 to Zn(mal)2·H2O (where there is one water under apical coordination), namely the transformation from tetrahedral to pentahedral geometry analogous to that of VO(3hp)2. We obtain the same result when we initiate the ab initio simulation from a DFT optimized octahedral Zn complex with two water molecules on opposite sides of the ligand plane: Zn2+ loses coordination with one water molecule, keeping the second dynamically associated, and bends the ligand planes, which undergo deformation. In this respect, BOMD studies of structural electronic properties of the complexes in an explicit water cluster are indispensable to access structural realizations of Zn2+ complexes prior to and upon their initial interactions with phosphatase family members. Furthermore, BOMD predicts that both Zn and oxovanadium systems show, not only a similarity of the angular ranges spanned by their structural realizations (blue and red trajectories in Figure 2A1 and Figure 2A2), but, also, that such angular properties are featured by analogous anticorrelations (see antidiagonal character of the trajectories).
The observed similarity, however, does not mean exactly the same structural and electronical properties. Dynamic association of water due to p-electron association with Zn2+ is not as strong and stable as the V4+=O double bonding with participation of the constructive p-d electronic overlap. As a result, the green trajectory in panel A2 explores a broad angular space (comparing to that in Figure 2A1), reflecting the rather unstable nature of water coordination, while the Mulliken charge on oxygen (see Figure 2E2) is predicted to show significant fluctuations due to water OH stretching dynamics and hydrogen bonding dynamics involving other neighboring water molecules.
It is worth noting that borrowing from the relatively weak nature of the apical aqueous O association (in contrast to the strong V=O bonding in an oxo-vanadium complex), Zn systems may demonstrate unprecedented structural flexibility upon binding to a protein. Overall, a Zn complex may vary from distorted tetragonal prism to distorted tetrahedron. Such structural flexibility may play a significant role upon possible post-binding speciation [19,20] to release the ligands and the metal cation - a process where protein carboxylate moieties may be involved.
Structure upon Binding to Proteins
Inspired by BOMD and DFT structural outcomes, we carry out docking predictions to find candidate arrangements of the complexes within the space of the reaction centres, that may lead to the enzymes inhibition. In order to account accurately for the quantum nature of the metal ions and especially the peculiarity of the unpaired electron of V4+, starting from the structural poses obtained by docking realizations, we perform ONIOM QM/MM optimizations of the systems. In Figure 3, we report ball and stick representations of the optimized complex-enzyme configurations. In the Supplementary Material, we provide Gaussian log files of the optimized geometries.
QM/MM calculations predict that V complexes undergo a strong folding distortion in order to occupy the reactive centre of PTP1B. In this respect, we note the particular features of the bonding angles involving the V atom. The blue and cyan large dots in Figure 2A1 indicate smaller O3VO’4 and O’3VO4 angles in the complexes bound to the protein with respect to those observed in vacuum (small dots) and in water cluster (blue trajectory). Consistently, Figure 3A and Figure 3B present structures, where the ligand planes are under the estimated smaller angles. It is important to note that theory predicts a role of the reaction centre helix Amide 1 NH group and protonated Cys215 interactions with the apical O atom of the V systems (see dark red dotted lines in Figure 3A and Figure 3B).
In the case of the Zn systems, structural arrangements are different due to the significantly more flexible nature of the complex. Figure 3C shows Zn(mal)2 where the Angle1 O’3VO4 is quite large, giving rise to a nearly flat structure in the plane, resembling a distorted square pyramid. The Zn(alx)2 complex, in contrast, forms a nearly tetrahedral geometry (see Figure 3D). In the case of Zn systems, the reaction centre helix Amide 1 NH groups are observed to interact with the carbonyl and carboxylate O atoms of Zn(mal)2 (see Figure 3C) and with the O atom of the methoxy group of Zn(alx)2 (see Figure 3D).
In summary, the results of the QM/MM optimizations suggest that induction and hydrophobic mechanisms may play a significant role in fine fitting of the long pentyl moieties into suitable protein cavities to contribute to structural and electronic perturbations. This may be reflected in UV-VIS optical electronic properties.
Nature of Coordination Bonds
As BOMD calculations indicate similar structural and electronic tendencies of the V and Zn systems, it is thus important to address the nature of bonding in the complexes. According to the NBO analysis, in vacuum, DFT calculations predict about the same degree of ionicity of the V(12%)-O(88%) and V(29%)=O(71%) bonding for VO(3hp)2 (see Table 1) and VO(alx)2 complexes (data in the Supplementary Material). Upon protein association, ionicity of carbonyl/carboxylate bonding slightly decreases, while the V=O bond becomes more ionic. These tendencies are larger for VO(alx)2 (see Table 1 and images in Figure S1 in Supplementary Material). Slight redistributions of the bond nature as computed by NBO analysis agree with the changes of the ESP atomic charges estimated according to the DFT population analysis. We may ascribe the changes in the nature of the ligand bonding to nonpolar interactions of the long pentyls with the enzyme moieties and to polar attraction of the apical O atom to Cys215 of the PTP1B enzyme.
In comparison to bonding in oxovanadium systems, attachment of carbonyl and carboxylate O atoms to Zn2+ in both complexes, Zn(mal)2 and Zn(alx)2, is significantly more ionic Zn(5%)-O(95%) (see Table 2 and data in the Supplementary Material). While d-orbitals play a key role in bonding of the oxovanadium complexes, integrity of the Zn systems is according to sigma type sp-electronic overlaps. Effects of both, oxidation states and principal quantum numbers (nuclei charges) for the metal ions deliver comparable ionic radii and bond lengths in the two systems, but the relative ionicities of the bonds are different, as we have computed and described. In result, the main-frame structural resemblance allows oxovanadium and Zn complexes to demonstrate parallel pharmacologic properties. Nonetheless, as we have already mentioned, the latter is expected to show better structural flexibility. Here, we may add that the larger ionicity for Zn2+ bonding should contribute to stronger electrostatic interactions with moieties of a hosting protein and this may stimulate electrophilic/nucleophilic interactions with charged protein moieties to stimulate post-binding complex decomposition and speciation [19,20].
Normal Modes Analysis, VO(3hp)2
In Figure 4, we compare IR and Raman spectra of the considered complexes in vacuum and in the protein environment. The spectra highlight effects of side-groups of the pyrone moieties, the nature of the bonding to the metal cations, and structural constraints imposed by the protein. We start the normal mode analysis considering VO(3hp)2, since this complex contains the simplest ligands, namely pyromeconic acids, Figure 4A1. The normal mode 57 (at 1566 cm-1) dominates the IR spectrum. This is a stretching mode localized on both ligands. In particular, it presents a C3-O3&C3-O4 symmetric stretching accompanied by an out-of-phase C2-C3/C4-C5 symmetric stretching. We use the ampersand and the slash to indicate in-phase and out-of-phase motion, respectively. Such a motion occurs in opposite phase on the two ligands. Normal mode 58 (at 1577 cm-1) dominates the Raman spectrum. This vibration accounts for a C2-C3&C5-C6 symmetric stretching in phase on both ligands. At lower frequencies, aromatic CH in-plane bending modes 40, 41 and V=O stretching mode 42 present distinct resonances, which can be helpful to identify this molecule experimentally. Aromatic CH stretching modes dominate the spectra at the highest frequencies (above 3100 cm-1). Supplementary Material includes an archive with a corresponding Gaussian output file with normal modes analysis.
In the protein environment, 6 additional low frequency modes occur, namely, 3 translations and 3 rotations of the whole complex in respect to the protein cavity. Consequently, for example, mode 48 = 42 + 6 in Figure 4B1 presents V=O stretching of the complex, when in the protein environment. Comparing the spectra of Figure 4A1 and Figure 4B1, we note that, in general, normal modes experience blue shifts upon association with the protein. Furthermore, a change of relative IR intensities of the modes 63 and 64 (versus the corresponding modes 57 and 58, as well as degeneracy lifting at 3150 cm-1 in Figure 4B1 suggest a loss of symmetry in the complex due to distortion upon association with the protein (see description of the structural properties in the previous section). In the Supplementary Material, we report a comparison of the spectra computed for the neutral and dehydrated anion of 3-hydroxy-4-pyrone.
Normal Modes Analysis, Zn(mal)2
Maltol is the next structural augmentation of 3-hydroxy-4-pyrone, where methyl is the R2 residue. In Figure 4A2 and Figure 4B2, we display the IR and Raman spectra computed for the Zn(mal)2·H2O complex in vacuum and the Zn(mal)2 complex in the protein environment, respectively. In the Supplementary Material, we provide the spectra computed for the Zn(mal)2 and Zn(mal)2·2H2O complexes under tetrahedral and octahedral geometries, respectively. Concerning the Zn(mal)2·H2O complex in vacuum, theory predicts IR active modes 56 and 60 to arise from aromatic CH in-plane bending admixed with O1-C2 stretching, with methyl CH in-plane bending and of carbonyl and carboxylate stretching on both ligands, under different phase relations: see the displacement images in Figure S19 in the Supplementary Material. Mode 75 is an analogue of mode 57 in VO(3hp)2. IR activities 77 and 78 are related to motions localized on each of the two ligands, accounting for in phase C5-C6&C4-O4 symmetric stretching admixed with aromatic C-H in-plane bendings. The two localizations are due to the low symmetry of Zn(mal)2·H2O. In the high frequency spectral range, aromatic CH stretchings at 3150 cm-1 accompany CH stretchings of the methyl group within the 2900 - 3000 cm-1 spectral range. Considering the results of previous theoretical and experimental (in KBr powder) studies of the normal modes of maltol [83] and Zn(mal)2·H2O [84], in the Supplementary Material, we present computed spectra and image displacements of representative normal modes of deprotonated and protonated maltol, as well as of Zn(mal)2·H2O. Accordingly, we may ascribe the IR transitions observed at 852, 922, 1202, 1277, 1459, 1513, 1577 and 1610 cm-1 to the 42, 45, 56, 60, 68, 73, 75 and 77+78 modes, respectively. Consistently, we may ascribe Raman transitions detected at 539, 718, 1045, 1366, 1465, 1511 and 1602 cm-1 to the 31, 38+39, 53, 62, 67, 72, and 78 modes, respectively. Supplementary Material includes an archive with a corresponding Gaussian output file with normal modes analysis.
Furthermore, as one may easily note comparing Figure 4B2 and Figure 4A2 (as also observed while comparing Figure 4B1 and Figure 4A1), normal modes of Zn(mal)2 shift to the blue upon binding to the protein, also revealing some degree of stronger dispersion of the frequencies. These are the signatures of structural distortion and symmetry lowering. Our results indicate that, as observed for the modes 57 and 58 of VO(3hp)2, the modes 74 (at 1566 cm-1) and 76 (at 1611 cm-1) of Zn(mal)2 in PTEN may provide valuable experimental markers of the presence and the structural state of Zn(mal)2. The mode 74 accounts C2-C3/C4-O4 out-of-phase symmetric stretching admixed with CH bendings to localize on one of the ligands. The other mode accounts analogous vibration but to localize on the other ligands.
Normal Modes Analysis: VO(alx)2
Let us consider now the vibrational properties of the VO(alx)2 complex, where the aromatic CH moieties are replaced with methoxy and extended hydrocarbon groups, which should allow for more specific structural attuning of the complex and the protein cavity upon association. The Raman intense mode 95, at 1139.5 cm-1 (Figure 4C1), is due to V=O stretching. Given its dominant role in the spectral region around 1100 cm-1, this mode may serve as a helpful spectral marker to track the presence of such a complex. The same role could be played by the intense IR active modes 159 and 160 (at 1507 and 1508 cm-1), which are C=O antisymmetric and symmetric stretchings, respectively. Consistently, the intense IR mode 161 (at 1546.8 cm-1) is a delocalized stretching to involve both ligands as (C2-C3/C5-C6)&(C’2-C’3/C’5-C’6). Considering Raman diagnostics, theory predicts efficient scattering for the mode 162 (at 1552.7 cm-1), which accounts for (C2-C3/C5-C6)/(C’2-C’3/C’5-C’6) stretching; and the mode 163 (at 1590 cm-1), which is due to in phase (C2-C3&C5-C6)&(C’2-C’3&C’5-C’6) symmetric stretchings that involves the two ligands. In the high frequency range, CH2 and CH3 stretching modes of the side groups define a series of transitions between 2900 and 3100 cm-1. If in vitro, such transitions occur in the same spectral region of phospholipids and proteins, probably yielding band overlap thus making them ineffective as spectral markers. Supplementary Material includes an archive with a corresponding Gaussian output file with normal modes analysis.
In the protein environment, 6 additional low frequency modes are expected for VO(alx)2: 3 translations and 3 rotations of the whole complex in respect to the protein cavity. Consequently, for VO(alx)2 associated with PTP1B (see Figure 4D1), normal mode 95 + 6 = 101 (at 1068.3 cm-1) is specific to V=O stretching, but not uniquely if we compare with the case of VO(3hp)2. Specifically, in the case of VO(alx)2 in PTP1B, theory computes mixing of V=O stretching with delocalized CH2 bendings: there is another intense transition denoted as 102 in Figure 4D1 (1069.7 cm-1). Further, for the complex in the protein environment, there are two intense IR modes 166 (at 1519 cm-1) and 168 (at 1548.2 cm-1), as well as a strong Raman mode 170 (at 1586.8 cm-1) localized on the same ligand. Mode 166 is due to (C4-O4&C2-C3)/C4-C5 stretching: here, the slash indicates opposite phase relations. Mode 168 accounts (C5-C6&C3-O3&C4-O4)/C2-C3 stretching admixed with CH3 bendings. Mode 170 represents (C2-C3&C5-C6)/C4-O4 stretching admixed with CH2 and CH3 bendings. In the considered spectral region, the other ligand demonstrates only one intense Raman mode 169 (at 1581 cm-1) to account (C’2-C’3&C’5-C’6)/C’4-O’4 stretching admixed with CH2 and CH3 bendings. The results of our theoretical studies suggest great sensitivity of the complex vibrational properties to anisotropy of interactions within the embedding cavity. We believe the provided description of the nature of the most intense normal modes may allow a helpful diagnostic to address the binding process if using FTIR and Raman spectroscopy.
Normal Modes Analysis: Zn(alx)2
Considering rather effective association of a water molecule (under apical geometry) predicted for Zn complexes in water clusters using BOMD, first, we compute vibrational properties of Zn(alx)2·H2O in vacuum, where the water is under apical geometry. For this complex, a number of IR active vibrations, possibly suitable for diagnostics, are predicted: mode 109 (at 1136 cm-1) involves CH bendings of the methoxy and methyl group; vibration 163 (at 1512 cm-1) accounts (C2-C3&C4-O4)/C’4-O’4 admixed with CH and HOH bendings; mode 165 (at 1550 cm-1) presents (C2-C3/C5-C6)/(C’2-C’3/C’5-C’6) stretching which is antisymmetric within a ring and in respect to the ring of the other ligand. The Raman active mode 167 (1584 cm-1) is dominant in the middle spectral range and accounts (C2-C3&C5-C6)&(C’2-C’3&C’5-C’6) stretching, which is symmetric within a ring and in respect to the ring of the other ligand.
As in the case of VO(alx)2, for the Zn(alx)2 complex embedded in the protein environment, the normal modes of the dominant IR and Raman bands shift to the blue. A nearly degenerated doublet of IR transitions 106 and 107 to involve CH bendings of the methoxy and methyl group peak at 1146 cm-1. Mode 161 (at 1520 cm-1) accounts C2-C3&C4-O4/C4-C5 admixed with CH bendings on the same ligand. The infrared dominant peak at 1557 cm-1 is due to the mode 164 to demonstrate (C2-C3/C4-O4&C5-C6)/(C’4-O’4/C’5-C6) delocalized over both ligands to admix with their CH bendings. The dominant Raman peak at 1593 cm-1 accounts a nearly degenerated doublet of C2-C3&C5-C6 and C’2-C’3/C’5-C’6 vibrations localized on each of the ligands. Overall, the computed vibrations of the complex in PTEN demonstrate wider spectral distributions of the resonances and stronger localization tendencies reflecting lower symmetry due to structural distortions resulting from the hindered interactions with the protein.
Oxovanadium Systems UV-VIS Properties
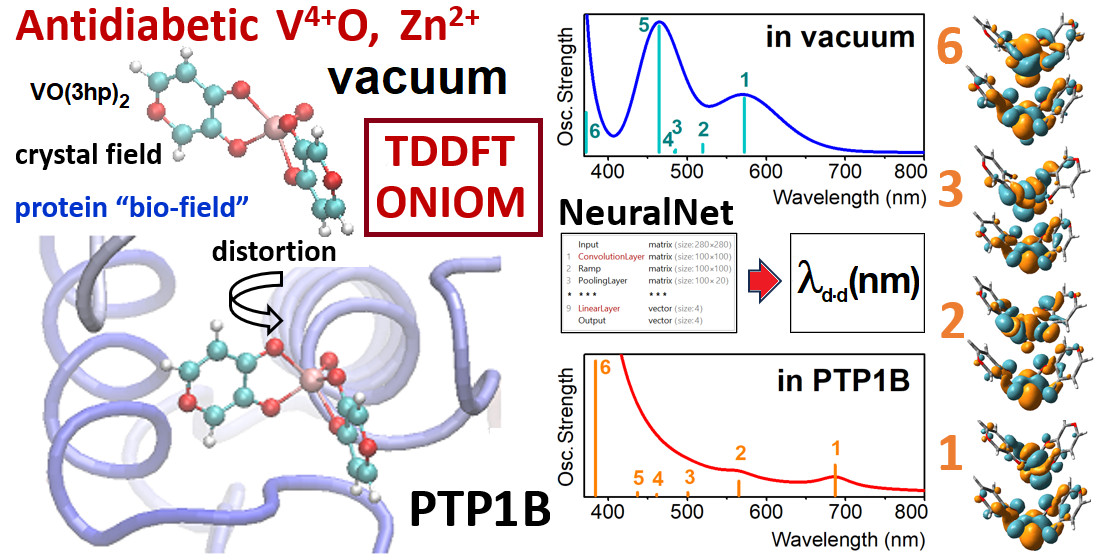
In Figure 5A and Figure 5B, we report the electronic absorption and circular dichroism (CD) spectra computed for VO(3hp)2 in vacuum and in the PTP1B protein environment. First, a significant red shift (about 10 nm) for the HOMO-LUMO and the next electronic transitions upon protein-complex association is observed: the shift is more than 100 nm for the HOMO-LUMO. NTO orbitals for the HOMO-LUMO transition of the complex in vacuum and in the protein environment, reported in Figure 6, suggest a similar electronic pattern, in which the dxy to π*(dxz-px) components (admixed with ligand p electrons) dominate. This resembles the character of the HOMO-LUMO transition computed for VO(acac)2 reported in the literature [85]. Admixture with ligand electronics is stronger for the complex that is distorted in the protein cavity. It is interesting that, while the delocalized ligand electronics determine third and fourth electronic transitions of the complex in vacuum, in the protein environment, localized ligand electronics take a lead in the fourth and fifth transitions. Calculations predict a significant perturbation of electronic properties of the complex upon interactions with phosphatase.
Additionally, in Figure 5B, using a thin dashed blue line, we display the CD spectrum obtained for the VO(3hp)2 L enantiomer in vacuum. The two CD spectra of the complex in vacuum indicate that a solution of this complex (as a racemic mixture) should not show any optical activity. In contrast, in the protein environment, a certain optical rotation is expected due to conserved geometry of the embedding cavity of the enzyme active site (note the positive rotations for the first and second transitions by the red line in Figure 5B). Our calculations suggest a role of CD spectroscopy to monitor visible spectral range d-d single spin electronic transitions and ultimately probe structural-electronic tuning of oxovanadium complexes in a protein environment.
In Figure 5C and Figure 5D, we present the electronic absorption and CD spectra of the VO(alx)2 complex in vacuum and in the PTP1B protein environment. There is an obvious similarity of the CD spectra computed for VO(3hp)2 and VO(alx)2 complexes in vacuum. This indicates that, regardless of the side groups, the relative orientations of the electronic and magnetic transition dipole vectors are quite similar. However, optical electronic properties of the complexes are rather different when embedded in the PTP1B enzyme. First, the shift of the HOMO-LUMO transition for the VO(alx)2 complex in the protein environment is larger. Second, CD spectra are significantly different. This suggests that the extended side groups of the VO(alx)2 complex introduce a “fulcrum”, that their interactions and the relative co-adjustments with the proximal protein moieties may impose additional perturbations on the structure and electronic properties of the complex when in PTP1B. Here, it is interesting to note that, while calculations anticipate that both, the nature of the VO(alx)2 transient electronic orbital components and their reordering upon association with the protein (see Figure 7) are quite similar to those predicted for VO(3hp)2 (see Figure 6), the optical activities of the two complexes are significantly different. Therefore, we may infer that selecting side groups upon synthesis and tuning protein environment (via primary sequence change or affecting environment to stimulate a different conformation) one may control frontier d-d electronic and magnetic properties of the cation in such ways that can never be achieved otherwise.
Zinc Systems UV-VIS Properties
To address how optical properties would change upon binding to a protein, for the aqueous reference we adopt Zn complexes with a single apical coordinated water such that the coordination geometry would be similar to that of square pyramidal – the most likely arrangement, according to the BOMD predictions in water clusters. Figure 8 presents the electronic absorption and CD spectra of Zn(mal)2·H2O and Zn(alx)2·H2O, and in the PTEN protein environment. In contrast to the case of oxovanadium systems, where a single d-electron determines the edge optical absorption, in the Zn systems, the computed optical transitions are due to optical transitions of the ligands. In Figures S32 and S33 of the Supplementary Material we display the corresponding NTOs. For Zn(mal)2·H2O and Zn(alx)2·H2O, the frontier transitions are in the UV, below 350 nm. These transitions involve intramolecular ligand and inter-ligand excitations. Accordingly, TDDFT for such systems predicts similar participation of water in optical electronic transitions: for example, see NTO pairs 5 and 6 in the upper sets in Figures S32 and S33 of the Supplementary Material.
Upon association of Zn complexes with PTEN proteins, significant red shifts (larger than 150 nm) for the HOMO-LUMO and next to them optical transitions (compare red and blue line spectra in Figure 8A and Figure 8C) are observed. This suggests both, electronic interactions of the complexes with the protein environment and significant structural perturbation upon binding. Blue line spectra in Figure 8B and Figure 8D present CD dispersions for the selected enantiomers computed in vacuum. In aqueous environment, where racemic mixtures are expected, one cannot observe optical activity for such complexes. This is the same conclusion reached for the oxovanadium systems (see discussion of Figure 5B). Instead, optical activity of the complexes in the protein environment is expected (red lines in Figure 8B and Figure 8D). Since the optical activity of the helical and β-sheet structural components of enzymes is detected below 240 nm, the relatively red shifted optical activity (above 300 nm) predicted for Zn systems (due to ligand electronics) may provide a valuable diagnostic to address interactions and binding. In Figures S32 and S33 of the Supplementary Material we report NTO electronic components of the considered Zn2+ systems in the protein environment. Binding to the protein induces a slightly stronger localization for the red edge transitions, consistent with structural distortion.
Comparison of the electronic spectra of Zn and V complexes in vacuum and in the protein environment indicates that these complexes may undergo significant spectral alterations upon binding to enzymes. This is particularly evident and interesting in the case of the V systems, where the protein environment shifts the computed d-d transitions into the near IR. This result suggests a new opportunity in bioengineering of novel electro-optical and biospintronic applications using either crystallised or surface-oriented proteins with embedded complexes. Specifically, computed optical properties suggest that using circularly polarized light it may be possible to engineer spin-polarized electronic excitations to detect spin polarization as an electrical signal [86,87]. This is particularly attractive because of the structural diversity of protein families, and possible modifications of enzymes (by single amino-acid editing) and can be assisted by artificial intelligence to secure unique cavities to tune d-electron transitions to spectral ranges not attainable so far. From this perspective, exploring docking implications for electronics of polyoxidovanadates [47,88] and systems alike may open a new frontier in multi-spin biospintronics.
Finally, here, it is important to note that nearly a century ago Hans Bethe introduced crystal field theory to explain colors in transition metal complexes [89]. The considered cases of tuning d-d electronic transitions of the oxovanadium complexes may be considered as “bio-field” electronic engineering, where anisotropy of the embedding cavity is predictable according to primary sequence, and variable according to the secondary structure of an adopted protein or by editing the cavity upon single amino acid substitutions. In the Supplementary Material, we describe an example of a convolution Neural net to train on Coulomb forces (between atoms of the reactive centres and of the complexes) and the wavelengths of d-d electronic resonances computed using QM/MM for the purpose, such that one may anticipate changes of d-d electronics, for example, upon a single amino acid substitution that perturbs Coulomb interactions: The Supplementary Material includes an archive with Mathematica codes for Neural Net training and application. While our example offers training using wavelengths of the computed d-d transitions, one may adopt relations with more general definitions of a crystal field [90].
Conclusions
Using Born-Oppenheimer molecular dynamics simulations, DFT, and QM/MM approach integrated to DFT, we address structural and electronic properties of insulin-mimetic oxovanadium and Zn complexes in vacuum, in water clusters and when bound to PTEN and PTP1B phosphatases. In aqueous environment, Zn(mal)2 departs from its global minimum tetrahedral geometry to bind a water molecule softly under apical geometry. Association of water is via sigma p-electron interactions, which are at the expense of electronic depletion on the carbonyl and carboxylate oxygens, as wells as on the metal cation. Since water association with Zn(mal)2 in water cluster is dynamic, computing this complex in vacuum (under DFT) cannot lead to a structure similar to that of VO(3hp)2. Only using BOMD simulations in explicit water, we do observe a structural change from the tetrahedral geometry of Zn(mal)2 to Zn(mal)2·H2O, an arrangement analogous to that of VO(3hp)2. In this respect, a dynamical approach to the structural and electronic properties of the complexes in water clusters, as allowed by BOMD simulations, would seem to be indispensable to access structural realizations of Zn2+ complexes.
For both, zinc and oxovanadium systems, DFT predicts effective interactions with the reaction centre helix Amide 1 NH group, and proximal residues. Employing NBO analysis, we evaluated similar degree of ionicity of the coordination bonding in VO(3hp)2 and for VO(alx)2 complexes, as computed in vacuum. Comparatively, upon protein association, ionicity of carbonyl/carboxylate bonding decreases, while the V=O bond becomes more ionic. In comparison, coordinating Zn2+ in both complexes, Zn(mal)2 and Zn(alx)2, are significantly more ionic. While d-orbitals play the key role in bonding of the oxovanadium complexes, integrity of the zinc systems is according to sigma type sp-electronic overlaps. Effects of both, oxidation states and principal quantum numbers (nuclei charges) for the metal ions deliver comparable ionic radiuses and bond lengths in the two systems, but the relative ionicities of the bonds are different as we have computed and described. In result, the main-frame structural resemblance provides oxovanadium and zinc complexes that demonstrate parallel pharmacological properties. At the same time, results of theoretical studies predict the zinc complexes to demonstrate better structural flexibility, what may lead to different post-binding speciation processes.
Assisted with normal mode analysis, we characterize vibrations, which may be suitable for experimental verification of structural states of the considered complexes and efficiency of binding. TDDFT studies indicate significant alterations of optical electronic properties of the complexes upon association with the enzymes. This is particularly evident and interesting in the case of the vanadium systems, where the protein environment shifts the computed d-d transitions into the near Infrared. In contrast to the case of oxovanadium systems, optical transitions computed for Zn2+ molecular systems are due to optical transitions of the ligands. Considering the results of our theoretical studies we discuss engineering of AI assisted protein embedding to alter electronic states of metal centres, which may be beneficial for biomedical and quantum information applications within the biospintronics of tomorrow.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. a directory named SUPPLEMENTARY_MATERIAL will be created, which contains all supporting information to the article. In this directory, several files/subdirectories will be found, as described in the following. The file supplementary_material.pdf with following information: (1) nature of bonding orbital components; (2) structural and electronic properties using NBO analysis; (3) IR and Raman spectra as well as displacement images for the normal modes of the complexes and ligands; (4) optical electronic spectra computed using TDDFT; (5) NTO transitions; (6) description of a convolution Neural Net to train on Coulomb forces (between atoms of the reactive centres and the complexes) and the wavelengths of d-d electronic resonances computed using QM/MM for the purpose such that one may anticipate changes of d-d electronics (a pilot code is provided in the AI.zip archive file). The Directory XYZ reporting files with the structural arrangements of the following systems obtained from QM/MM energy minimization: VO(3hp)2 and VO(alx)2 in the PTP1B protein environment, Zn(mal)2 and Zn(alx)2 in the PTEN protein environment. The Directory LOG reporting Gaussian output files with normal modes analysis for selected complexes and ligands. The Directory AI reporting an archive with a Mathematica code to exemplify Neural Net training and application for the estimate of d-d electronic transitions in complex-protein systems (examples, related to VO(3hp)2 and VO(alx)2 in the PTP1B protein environment, are reported). All directories contain a README.txt file describing the contents. Refs. [83,84] have been cited in the supplementary material.pdf file.
Author Contributions
V.V.V.: Conceptualization, methodology, visualization, and writing original draft. V.V.V. C.C.P and R.C.: Computations. R.C. and C.C.P.: Review and editing. C.C.P.: Funding acquisition.
Funding
AFOSR FA9550-16-1-0213, and FA9550-20-1-0206; Air Force Office of Scientific Research, 875 N. Randolph, Ste.325, Arlington Virginia, 22203 USA.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study is available from the authors on reasonable request.
Acknowledgments
Dr. Carole C. Perry gratefully acknowledges funding from AFOSR FA9550-16-1-0213, and FA9550-20-1-0206. Dr. Victor V. Volkov and Dr. Carole C. Perry are thankful to Dr. Fernando R. Clemente and Dr. Douglas J. Fox (Gaussian Inc., Wallingford, USA) for stimulating discussions. Dr. Victor V. Volkov is a self-funded researcher living on savings collected during USA Air Force Research program at Nottingham Trent University, 2016-2020.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
V, vanadium; Zn, zinc; PTP1B, protein tyrosine phosphatase; PTEN, phosphatase and tensin homologue; PI3K, phosphoinositide 3-kinase; Cys, cysteine; Arg, arginine; Trp, tryptophan; Pro, proline; Asp, aspartic acid; Thr, threonine; Asn, asparagine; Val, valine; VO(3hp)2, bis(3-hydroxy-4-pyronato) oxovanadium(IV) complex; VO(pic)2, bis(picolinate) oxovanadium(IV); Zn(mal)2, bis(maltolato)zinc(II); VO(alx)2, bis(allixinato)oxovanadium(IV); VO(acac)2, vanadyl acetylacetonate; ONIOM, our own n-layered integrated molecular orbital and molecular mechanics; IR, infrared; DFT, density functional theory; TD-DFT, time-dependent density functional theory; MD, molecular dynamics; QM/MM, quantum mechanics/molecular mechanics; BOMD, Born-Oppenheimer molecular dynamics; crbn, carbonyl; crbx, carboxylate; HOMO, highest occupied molecular orbital; LUMO, lowest unoccupied molecular orbital; UV-VIS, ultraviolet-visible; CD, circular dichroism; NTO, natural transition orbitals.
References
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: a new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Kilpin, K.J.; Dyson, P.J. Enzyme inhibition by metal complexes: concepts, strategies and applications. Chem. Sci. 2013, 4, 1410–1419. [Google Scholar] [CrossRef]
- Diagnosis and classification of diabetes mellitus. Diabetes Care. American Diabetes Association, Diabetes Care 2006, 29, S43–S48.
- Lyonnet, B.; Martz, X.; Martin, E. L’emploi therapeutique des derives du. vanadium. La Presse Med. 1899, 1, 191–192. [Google Scholar]
- Meeks, M.J.; Landolt, R.R.; Kessler, W.V.; Born, G.S. Effect of vanadium on metabolism of glucose in the rat. J. Pharmacol. Sci. 1971, 60, 482–483. [Google Scholar] [CrossRef]
- McLauchlan, C.C.; Hooker, J.D.; Jones, M.A.; Dymon, Z.; Backhus, E.A.; Greiner, B.A.; Dorner, N.A.; Youkhana, M.A.; Manus, L.M. J. Inorg. Biochem. 2010, 104, 274-281.
- Rehder, D. The potentiality of vanadium in medicinal applications. Future Med. Chem. 2012, 4, 1823–1837. [Google Scholar] [CrossRef]
- Thompson, K.H.; Liboiron, B.D.; Sun, Y.; Bellman, K.D.D.; Setyawati, I.A.; Patrick, B.O.; Karunaratne, V.; Rawji, G.; Wheeler, J.; Sutton, K.; et al. Preparation and characterization of vanadyl complexes with bidentate maltol-type ligands; in vivo comparisons of anti-diabetic therapeutic potential. J. Biol. Inorg. Chem. 2003, 8, 66–74. [Google Scholar] [CrossRef]
- Sakurai, H.; Fujii, K.; Watanabe, H.; Tamura, H. Orally active and long-term acting insulin-mimetic vanadyl complex: Bis(picolinato)oxovanadium(IV). Biochem. Biophys. Res. Commun. 1995, 214, 1095–1101. [Google Scholar] [CrossRef]
- Coulston, L.; Dandona, P. Insulin-like effect of zinc on adipocytes. Diabetes 1980, 29, 665–667. [Google Scholar] [CrossRef]
- Shisheva, A.; Gefel, D.; Schechter, Y. Insulin-like effects of zinc ion in vitro and in vivo. Diabetes 1992, 41, 982–988. [Google Scholar] [CrossRef]
- Chen, M.D.; Liou, S.J.; Lin, P.Y.; Yang, V.C.; Alexander, P.S.; Lin, W.H. Effects of zinc supplementation on the plasma glucose level and insulin activity in genetically obese (ob/ob) mice. Biol. Trace Elem. Res. 1998, 61, 301–311. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Contoreggi, C.S. The mechanism of the insulinlike effects of ionic zinc. J. Biol. Chem. 1982, 257, 4362–4368. [Google Scholar] [CrossRef]
- Ezaki, O. IIb group metal ions (Zn2+, Cd2+, Hg2+) stimulate glucose transport activity by post-insulin receptor kinase mechanism in rat adipocytes. J. Biol. Chem. 1989, 264, 16118–16122. [Google Scholar] [CrossRef] [PubMed]
- Ilouz, R.; Kaidanovich, O.; Gurwitz, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3b by bivalent zinc ions: insight into the insulin-mimetic action of zinc. Biochem. Biophys. Res. Commun. 2002, 295, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Adachi, Y. The pharmacology of the insulinomimetic effect of zinc complexes. Bio. Metals 2005, 18, 319–323. [Google Scholar] [CrossRef]
- Wu, W.; Silbajoris, R.A.; Whang, Y.E.; Graves, L.M.; Bromberg, P.A.; Samet, J.M. p38 and EGF receptor kinase-mediated activation of the phosphatidylinositol 3-kinase/Akt pathway is required for Zn2+-induced cyclooxygenase-2 expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L883–L889. [Google Scholar] [CrossRef]
- Basuki, W.; Hiromura, M.; Sakurai, H. J. Inorg. Insulinomimetic Zn complex (Zn(opt)2) enhances insulin signaling pathway in 3T3-L1 adipocytes. Biochem. 2007, 101, 692–699. [Google Scholar]
- Gundhla, I.Z.; Ugirinema, V.; Walmsley, R.S.; Mnonopi, N.O.; Hosten, E.; Betz, R.; Frost, C.L.; Tshentu, Z.R. pH-metric chemical speciation modeling and in vitro anti-diabetic studies of bis[(imidazolyl)carboxylato]oxovanadium(IV) complexes. J. Inorg. Biochem. 2015, 145, 11–18. [Google Scholar] [CrossRef]
- Sanna, D.; Ugone, V.; Serra, M.; Garribba, E. Speciation of potential anti-diabetic vanadium complexes in real serum samples. J. Inorg. Biochem. 2017, 173, 52–65. [Google Scholar] [CrossRef]
- Ellis, B.L.; Duhme, A.K.; Hider, R.C.; Hossain, M.B.; Rizvi, S.; van der Helm, D. Synthesis, physicochemical properties, and biological evaluation of hydroxypyranones and hydroxypyridinones: novel bidentate ligands for cell-labeling. J. Med. Chem. 1996, 39, 3659–3670. [Google Scholar] [CrossRef]
- Hiromura, M.; Adachi, Y.; Machida, M.; Hattori, M.; Sakurai, H. Glucose lowering activity by oral administration of bis(allixinato)oxidovanadium(iv) complex in streptozotocin-induced diabetic mice and gene expression profiling in their skeletal muscles. Metallomics 2009, 1, 92–100. [Google Scholar] [CrossRef]
- Yuan, C.; Lu, L.; Gao, X.; Wu, Y.; Guo, M.; Li, Y.; Fu, X.; Zhu, M. Ternary Oxovanadium(IV) Complexes of ONO-Donor Schiff Base and Polypyridyl Derivatives as Protein Tyrosine Phosphatase Inhibitors: Synthesis, Characterization, and Biological Activities. J. Biol. Inorg. Chem. 2009, 14, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, X.; Zhang, W.; Reed, W.; Samet, J.M.; Whang, Y.E.; Ghio, A.J. Zinc-induced PTEN Protein Degradation through the Proteasome Pathway in Human Airway Epithelial Cells. J. Biol. Chem. 2003, 278, 28258–28263. [Google Scholar] [CrossRef] [PubMed]
- Lazar, D.F.; Saltiel, A.R. Lipid phosphatases as drug discovery targets for type 2 diabetes. Nat. Rev. Drug Discovery 2006, 5, 333–342. [Google Scholar] [CrossRef]
- Sakurai, H.; Katoh, A.; Kiss, T.; Jakusch, T.; Hattori, M. Metallo–allixinate complexes with anti-diabetic and anti-metabolic syndrome activities. Metallomics 2010, 2, 670–682. [Google Scholar] [CrossRef]
- Goldwaser, I.; Qian, S.; Gershonov, E.; Fridkin, M.; Schechter, Y. Organic vanadium chelators potentiate vanadium-evoked glucose metabolism in vitro and in vivo: establishing criteria for optimal chelators. Mol. Pharmacol. 2000, 58, 738–746. [Google Scholar] [CrossRef]
- Kawabe, K.; Yoshikawa, Y.; Yasui, H.; Sakurai, H. Possible mode of action for insulinomimetic activity of vanadyl(IV) compounds in adipocytes. Life Sci. 2006, 78, 2860–2866. [Google Scholar] [CrossRef]
- Sakurai, H.; Katoh, A.; Yoshikawa, Y. Chemistry and biochemistry of insulin-mimetic vanadium and zinc complexes. Trial for treatment of diabetes mellitus. Bull. Chem. Soc. Jpn. 2006, 79, 1645–1664. [Google Scholar] [CrossRef]
- Hiromura, M.; Nakayama, A.; Adachi, Y.; Doi, M.; Sakurai, H. Action mechanism of bis(allixinato)oxovanadium(IV) as a novel potent insulin-mimetic complex: regulation of GLUT4 translocation and FoxO1 transcription factor. J. Biol. Inorg. Chem. 2007, 12, 1275–1287. [Google Scholar] [CrossRef]
- Nakayama, A.; Hiromura, M.; Adachi, Y.; Sakurai, H. Molecular mechanism of antidiabetic zinc-allixin complexes: regulations of glucose utilization and lipid metabolism. J. Biol. Inorg. Chem. 2008, 13, 675–684. [Google Scholar] [CrossRef]
- Sacco, F.; Perfetto, L.; Castagnoli, L.; Cesareni, G. The human phosphatase interactome: An intricate family portrait. FEBS Letters 2012, 586, 2732–2739. [Google Scholar] [PubMed]
- Lessard, L.; Stuible, M.; Tremblay, M.L. The Two Faces of PTP1B in Cancer. Biochim. Biophys. Acta 2010, 1804, 613–619. [Google Scholar] [PubMed]
- Bononi, A.; Agnoletto, C.; De Marchi, E.; Marchi, S.; Patergnani, S.; Bonora, M.; Giorgi, C.; Missiroli, S.; Poletti, F.; Rimessi, A.; Pinton, P. Protein Kinases and Phosphatases in the Control of Cell Fate. Enzym. Res. 2011, 2011, 329098. [Google Scholar]
- Sivaganesh, V.; Sivaganesh, V.; Scanlon, C.; Iskander, A.; Maher, S.; Lê, T.; Peethambaran, B. Protein Tyrosine Phosphatases: Mechanisms in Cancer. Int. J. Mol. Sci. 2021, 22, 12865. [Google Scholar] [CrossRef]
- Ahmad, F.; Li, P.M.; Meyerovitch, J.; Goldstein, B.J. Osmotic loading of neutralizing antibodies demonstrates a role for protein-tyrosine phosphatase 1B in negative regulation of the insulin action pathway. J. Biol. Chem. 1995, 270, 20503–20508. [Google Scholar] [CrossRef]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; McDowell, R.S.; Hansen, S.K. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730. [Google Scholar] [CrossRef]
- Koren, S.; Fantus, I.G. Inhibition of the Protein Tyrosine Phosphatase PTP1B: Potential Therapy for Obesity, Insulin Resistance and Type-2 Diabetes Mellitus. Best Prac. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. [Google Scholar] [CrossRef]
- Eleftheriou, P.; Geronikaki, A.; Petrou, A. PTP1b Inhibition, A Promising Approach for the Treatment of Diabetes Type II. Curr. Top. Med. Chem. 2019, 19, 246–263. [Google Scholar]
- Villamar-Cruz, O.; Loza-Mejía, M.A.; Arias-Romero, L.E.; Camacho-Arroyo, I. Recent advances in PTP1B signalling in metabolism and cancer. Biosci. Rep. 2021, 41, BSR20211994. [Google Scholar]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal structure of human protein tyrosine phosphatase 1B. Science 1994, 263, 1397–1404. [Google Scholar]
- Jia, Z.; Barford, D.; Flint, A.J.; Tonks, N.K. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 1995, 268, 1754–1758. [Google Scholar] [CrossRef] [PubMed]
- Kamerlin, S.C.; Rucker, R.; Boresch, S. A targeted molecular dynamics study of WPD loop movement in PTP1B. Biochem. Biophys. Res. Commun. 2006, 345, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Brandao, T.A.; Hengge, A.C.; Johnson, S.J. Insights into the reaction of protein-tyrosine phosphatase 1B: Crystal structures for transition state analogs of both catalytic steps. J. Biol. Chem. 2010, 21, 15874–15883. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Xie, L.; Yang, F.; Wang, W.; Zhou, Q.; Xiang, M.; Zhou, S.; Lv, W.; Jia, Y.; Pokhrel, L.; et al. Recent advance on PTP1B inhibitors and their biomedical applications. Eur. J. Med. Chem. 2020, 199, 112376. [Google Scholar] [CrossRef]
- Moise, G.; Gallup, N.M.; Alexandrova, A.N.; Hengge, A.C.; Johnson, S.J. Conservative Tryptophan Mutants of the Protein Tyrosine Phosphatase YopH Exhibit Impaired WPD-Loop Function and Crystallize with Divanadate Esters in Their Active Sites. Biochem. 2015, 54, 6490–6500. [Google Scholar] [CrossRef]
- Shen, R.; et al. Insights into the importance of WPD-loop sequence for activity and structure in protein tyrosine phosphatases. Chem. Sci. 2022, 13, 13524–13540. [Google Scholar] [CrossRef]
- Li, Y.Z.; Di Cristofano, A.; Woo, M. Metabolic Role of PTEN in Insulin Signaling and Resistance. Cold Spring Harb Perspect Med. 2020, 10, a036137. [Google Scholar] [CrossRef]
- Masson, G.R.; Williams, R.L. Structural Mechanisms of PTEN Regulation. Cold Spring Harb Perspect Med. 2020, 10, a036152. [Google Scholar] [CrossRef]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef]
- Mighell, T.L.; Evans-Dutson, S.; O’Roak, B.J. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am. J. Hum. Genet. 2018, 102, 943–955. [Google Scholar] [CrossRef]
- Kostrzewa, T.; Jonczyk, J.; Drzezdzon, J.; Jacewicz, D.; Górska-Ponikowska, M.; Kołaczkowski, M.; Kuban-Jankowska, A. Synthesis, In Vitro, and Computational Studies of PTP1B Phosphatase Inhibitors Based on Oxovanadium(IV) and Dioxovanadium(V) Complexes. Mol. Sci. 2022, 23, 7034. [Google Scholar] [CrossRef] [PubMed]
- Parvaiz, N.; Abro, A.; Azam, S.S. Three-state dynamics of zinc(II) complexes yielding significant antidiabetic targets. J. Mol. Graph. Model. 2024, 127, 108665. [Google Scholar] [CrossRef] [PubMed]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package-Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 125, 114105. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Modern Theoretical Chemistry. Plenum: New York, NY, USA, 1977, Volume 3, pp. 1-28.
- Wedig, U.; Dolg, M.; Stoll, H.; Preuss, H. Quantum Chemistry: The Challenge of Transition Metals and Coordination Chemistry. Springer: Berlin/Heidelberg, Germany, 1986, pp. 79–89.
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01. Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Glendening, E.D., Reed, A.E., Carpenter, J.E. and Weinhold, F. NBO Version 3.1. Gaussian Inc., Pittsburgh, 2003.
- Volkov, C.C.; Chelli, R.; Perry, C.C. Cu(Proline)2 Complex: A Model of Bio-Copper Structural Ambivalence. Molecules 2022, 27, 5846. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Amos, A.T.; Hall, G.G. Single determinant wave functions. Proc. Royal. Soc. A 1961, 263, 483–493. [Google Scholar]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Lee, C.U.; Hahne, G.; Hanske, J.; Bange, T.; Bier, D.; Rademacher, C.; Hennig, S.; Grossmann, T.N. Redox Modulation of PTEN Phosphatase Activity by Hydrogen Peroxide and Bisperoxidovanadium Complexes. Angew. Chem. 2015, 54, 13796–13800. [Google Scholar] [CrossRef]
- Liu, G.; Szczepankiewicz, B.G.; Pei, Z.; Janowich, D.A.; Xin, Z.; Hadjuk, P.J.; Abad-Zapatero, C.; Liang, H.; Hutchins, C.W.; Fesik, S.W.; Ballaron, S.J.; Stashko, M.A.; Lubben, T.; Mika, A.K.; Zinker, B.A.; Trevillyan, J.M.; Jirousek, M.R. Discovery and Structure-Activity Relationship of Oxalylarylaminobenzoic Acids as Inhibitors of Protein Tyrosine Phosphatase 1B. J. Med. Chem. 2003, 46, 2093–2103. [Google Scholar] [CrossRef]
- Phillips, J.C.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar]
- Tautz, L.; Critton, D.A.; Grotegut, S. Protein Tyrosine Phosphatases: Structure, Function, and Implication in Human Disease. Phosphatase Modul. 2013, 1053, 179–221. [Google Scholar]
- Wilson, D.P.; et al. Structure-based optimization of protein tyrosine phosphatase 1B inhibitors: from the active site to the second phosphotyrosine binding site. J. Med. Chem. 2007, 50, 4681–4698. [Google Scholar] [CrossRef]
- Özcan, A. Effect of protonation states of catalytically important residus and active site water molecules on PTP1B conformation. Diploma, 2008.
- Özcan, A.; Olmez, E.O.; Alakent, B. Effects of protonation state of Asp181 and position of active site water molecules on the conformation of PTP1B. Proteins 2013, 81, 788–804. [Google Scholar] [CrossRef] [PubMed]
- Gapsys, V.; Perez-Benito, L.; Aldeghi, M.; Seeliger, D.; van Vlijmen, H.; Tresadern, G.; de Groot, B. L. Large scale relative protein ligand binding affinities using non-equilibrium alchemy. Chem. Sci. 2020, 11, 1140. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dapprich, S.; Komáromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A New ONIOM Implementation in Gaussian 98. 1. The Calculation of Energies, Gradients and Vibrational Frequencies and Electric Field Derivatives. J. Mol. Struct. (Theochem) 1999, 462, 1–21. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Case, D.A.; Singh, U.C.; Ghio, C.; Alagona, G.; Profeta, S.; Weiner, P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar] [CrossRef]
- Zborowski, K.; Grybos, R.; Proniewicz, L.M. Structure modification of maltol (3-hydroxy-2-methyl-4H-pyran-4-one) upon cation and anion formation studied by vibrational spectroscopy and quantum-mechanical calculations. Vib. Spec. 2007, 43, 344–350. [Google Scholar] [CrossRef]
- Parajón-Costa, B.S.; Baran, E.J. Vibrational spectra of bis(maltolato)zinc(II), an interesting insulin mimetic agent. Spectrochim. Acta Part A: Mol. and Biomol. Spec. 2013, 113, 337–339. [Google Scholar] [CrossRef]
- Van Stappen, C.; Maganas, D.; DeBeer, S.; Bill, E.; Neese, F. Investigations of the Magnetic and Spectroscopic Properties of V(III) and V(IV) Complexes. Inorg. Chem. 2018, 57(11), 6421–6438. [Google Scholar] [CrossRef]
- Hirohata, A.; Takanashi, K. Future perspectives for spintronic devices. J. Phys. D: Appl. Phys. 2014, 47, 193001. [Google Scholar] [CrossRef]
- Gupta, R.; Chinnasamy, H.V.; Sahu, D.; Matheshwaran, S.; Sow, C.; Chandra Mondal, P. Spin-dependent electrified protein interfaces for probing the CISS effect. J. Chem. Phys. 2023, 159, 024708. [Google Scholar] [CrossRef] [PubMed]
- Feder, D.; Gahan, L.R.; McGeary, R.P.; Guddat, L.W.; Schenk, G. The binding mode of an ADP analog to a metallohydrolase mimics the likely transition state. Chembiochem. 2019, 20, 1536–1540. [Google Scholar] [CrossRef] [PubMed]
- Bethe, H. Termaufspaltung in Kristallen. Annalen der Physik 1929, 395, 133–208. [Google Scholar] [CrossRef]
- Wood, J.S.; Green, P.T. Ligand Field Theory for Pentacoordinate Molecules. II. A Crystal Field—Spin-Orbit Coupling Treatment of the d1, d3, d6, and d8 Configurations in Trigonal-Bipyramidal Molecules and the Magnetic Properties of E Ground Terms. Inorganic Chemistry 1969, 8, 491–497. [Google Scholar] [CrossRef]
Figure 1.
Structural layout for the considered Zn and oxovanadium complexes. Apical oxygen atom Oa is absent in the case of Zn systems computed in vacuum. When in aqueous environment, theory anticipates departure of the Zn complex from its tetrahedral geometry and association with a water molecule with oxygen to take Oa position. Considered cases: R2 = R5 = R6 =H for pyromeconic acids (or 3-hydroxy-4-pyrone) to form, for example, bis(3-hydroxy-4-pyronato)oxovanadium, VO(3hp)2; R2 = CH3 for larixinic acid (or maltol, or 3-hydroxy-2-methyl-4-pyrone) to form, for example, bis(maltolato)zinc(II), Zn(mal)2; R2 = C4H8CH3, R5 = OCH3, R6 = CH3 for allixin ligand (or 3-hydroxy-5-methoxy-6-methyl-2-pentyl-4-pyrone) to bring under attention, for example, bis(allixinato)oxovanadium(IV), VO(alx)2.
Figure 1.
Structural layout for the considered Zn and oxovanadium complexes. Apical oxygen atom Oa is absent in the case of Zn systems computed in vacuum. When in aqueous environment, theory anticipates departure of the Zn complex from its tetrahedral geometry and association with a water molecule with oxygen to take Oa position. Considered cases: R2 = R5 = R6 =H for pyromeconic acids (or 3-hydroxy-4-pyrone) to form, for example, bis(3-hydroxy-4-pyronato)oxovanadium, VO(3hp)2; R2 = CH3 for larixinic acid (or maltol, or 3-hydroxy-2-methyl-4-pyrone) to form, for example, bis(maltolato)zinc(II), Zn(mal)2; R2 = C4H8CH3, R5 = OCH3, R6 = CH3 for allixin ligand (or 3-hydroxy-5-methoxy-6-methyl-2-pentyl-4-pyrone) to bring under attention, for example, bis(allixinato)oxovanadium(IV), VO(alx)2.
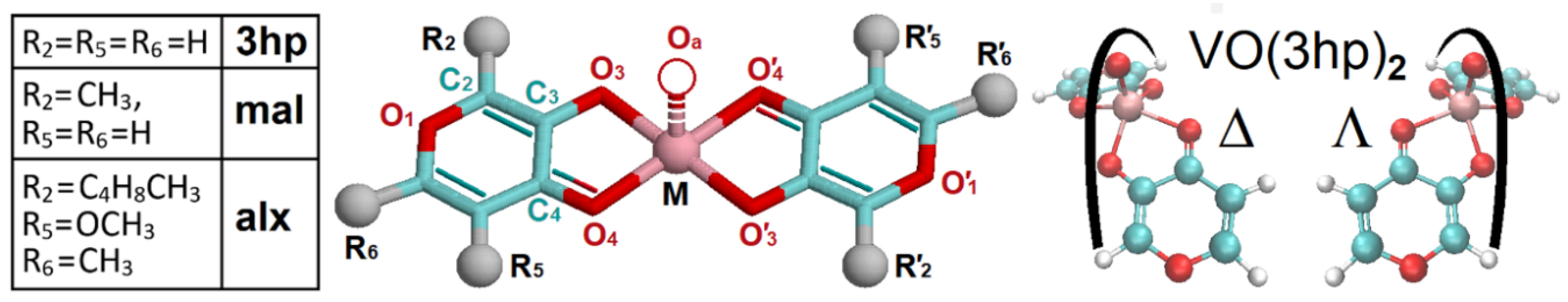
Figure 2.
Dynamic properties of VO(3hp)2 and Zn(mal)2 complexes. A1: Angle2 versus Angle1 variance along trajectory for O4VO’4 versus O3VO’3 (red line), O3VO’4 versus O’3VO4 (blue line), O’3VO3 versus OaVO3 (green line) for VO(3hp)2 in water cluster computed using BOMD. Dark red, cyan and dark green small points indicate angular realizations computed for the complex in vacuum using DFT. Dark red and cyan large points indicate angular realizations computed for VO(3hp)2 complex at PTP1B using QM/MM. Red and blue large points indicate angular realizations computed for VO(alx)2 complex at PTP1B using QM/MM. B1-E1: Mulliken charge for O4 (dark cyan) and O3 (magenta) atom, for C4 (blue) and C3 (magenta) atom, for Vanadium cation, and for Oa atom, respectively, as computed for VO(3hp)2 in water cluster using BOMD. Thick and thin lines present results for the atoms which belong to different ligands. F1: covariance matrix for Mulliken charges for VO(3hp)2 atoms O3, O4, O’4, O’3, M=V, C4, C3 counted from 1 to 7 along the plot axes. Panels A2 - F2 present analogous data for Zn(mal)2: E2 presents Mulliken charge for oxygen atom of the water nearest to Zn2+. Dark red and cyan large points indicate angular realizations computed for Zn(mal)2 complex at PTEN using QM/MM. Red and blue large points indicate angular realizations computed for Zn(alx)2 complex at PTP1B using QM/MM.
Figure 2.
Dynamic properties of VO(3hp)2 and Zn(mal)2 complexes. A1: Angle2 versus Angle1 variance along trajectory for O4VO’4 versus O3VO’3 (red line), O3VO’4 versus O’3VO4 (blue line), O’3VO3 versus OaVO3 (green line) for VO(3hp)2 in water cluster computed using BOMD. Dark red, cyan and dark green small points indicate angular realizations computed for the complex in vacuum using DFT. Dark red and cyan large points indicate angular realizations computed for VO(3hp)2 complex at PTP1B using QM/MM. Red and blue large points indicate angular realizations computed for VO(alx)2 complex at PTP1B using QM/MM. B1-E1: Mulliken charge for O4 (dark cyan) and O3 (magenta) atom, for C4 (blue) and C3 (magenta) atom, for Vanadium cation, and for Oa atom, respectively, as computed for VO(3hp)2 in water cluster using BOMD. Thick and thin lines present results for the atoms which belong to different ligands. F1: covariance matrix for Mulliken charges for VO(3hp)2 atoms O3, O4, O’4, O’3, M=V, C4, C3 counted from 1 to 7 along the plot axes. Panels A2 - F2 present analogous data for Zn(mal)2: E2 presents Mulliken charge for oxygen atom of the water nearest to Zn2+. Dark red and cyan large points indicate angular realizations computed for Zn(mal)2 complex at PTEN using QM/MM. Red and blue large points indicate angular realizations computed for Zn(alx)2 complex at PTP1B using QM/MM.
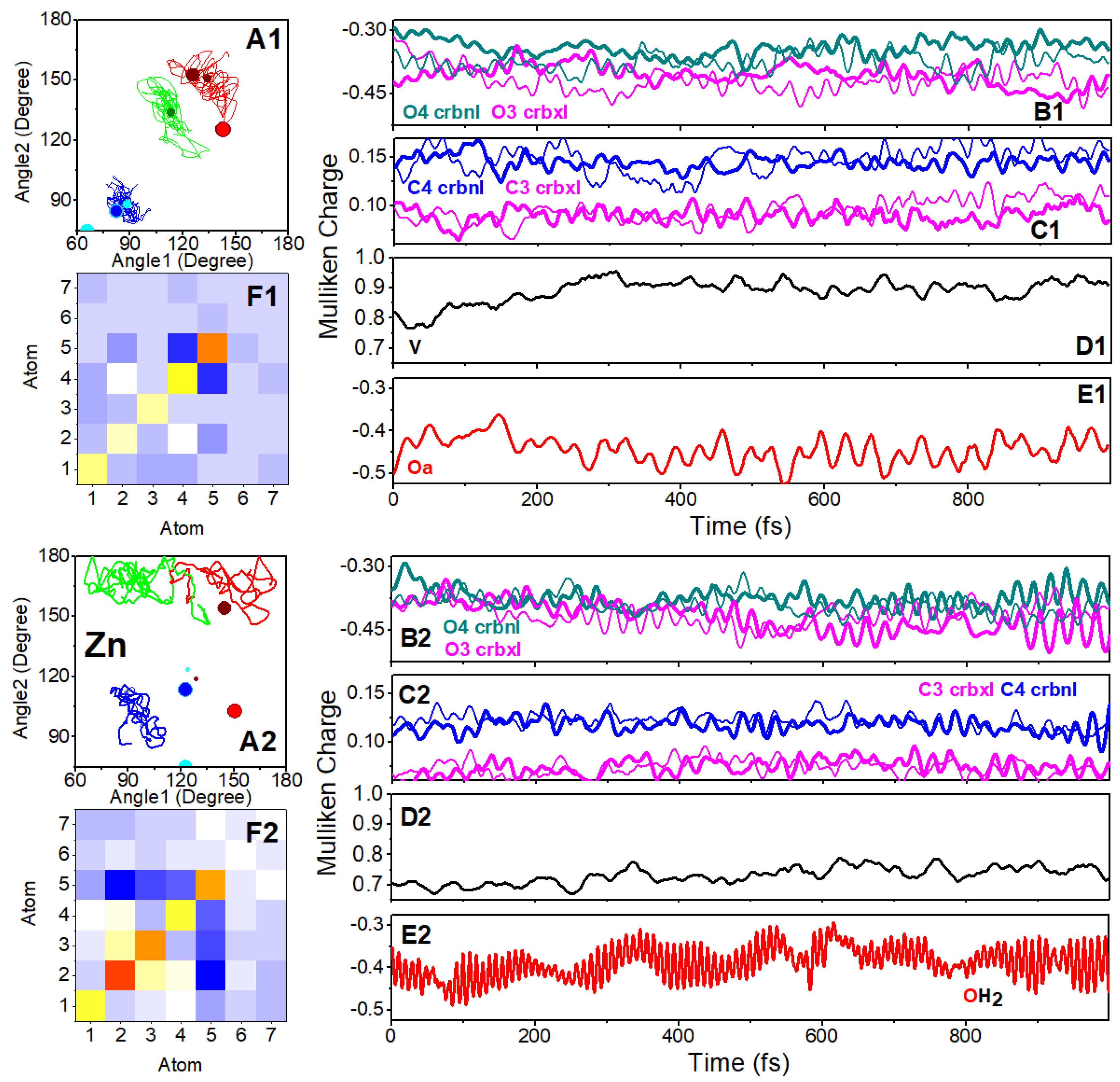
Figure 3.
Ball and stick representations of the structural arrangements of the complex-enzyme systems obtained by docking realizations followed by ONIOM QM/MM optimizations. A,B: VO(3hp)2 and VO(alx)2 at PTP1B reactive site, respectively. C,D: Zn(mal)2 and Zn(alx)2 at PTEN reactive site, respectively. For the sake of clarity, only the protein residues interacting with the complexes are reported in the diagrams. Tubular representation is adopted for the other parts of the protein.
Figure 3.
Ball and stick representations of the structural arrangements of the complex-enzyme systems obtained by docking realizations followed by ONIOM QM/MM optimizations. A,B: VO(3hp)2 and VO(alx)2 at PTP1B reactive site, respectively. C,D: Zn(mal)2 and Zn(alx)2 at PTEN reactive site, respectively. For the sake of clarity, only the protein residues interacting with the complexes are reported in the diagrams. Tubular representation is adopted for the other parts of the protein.
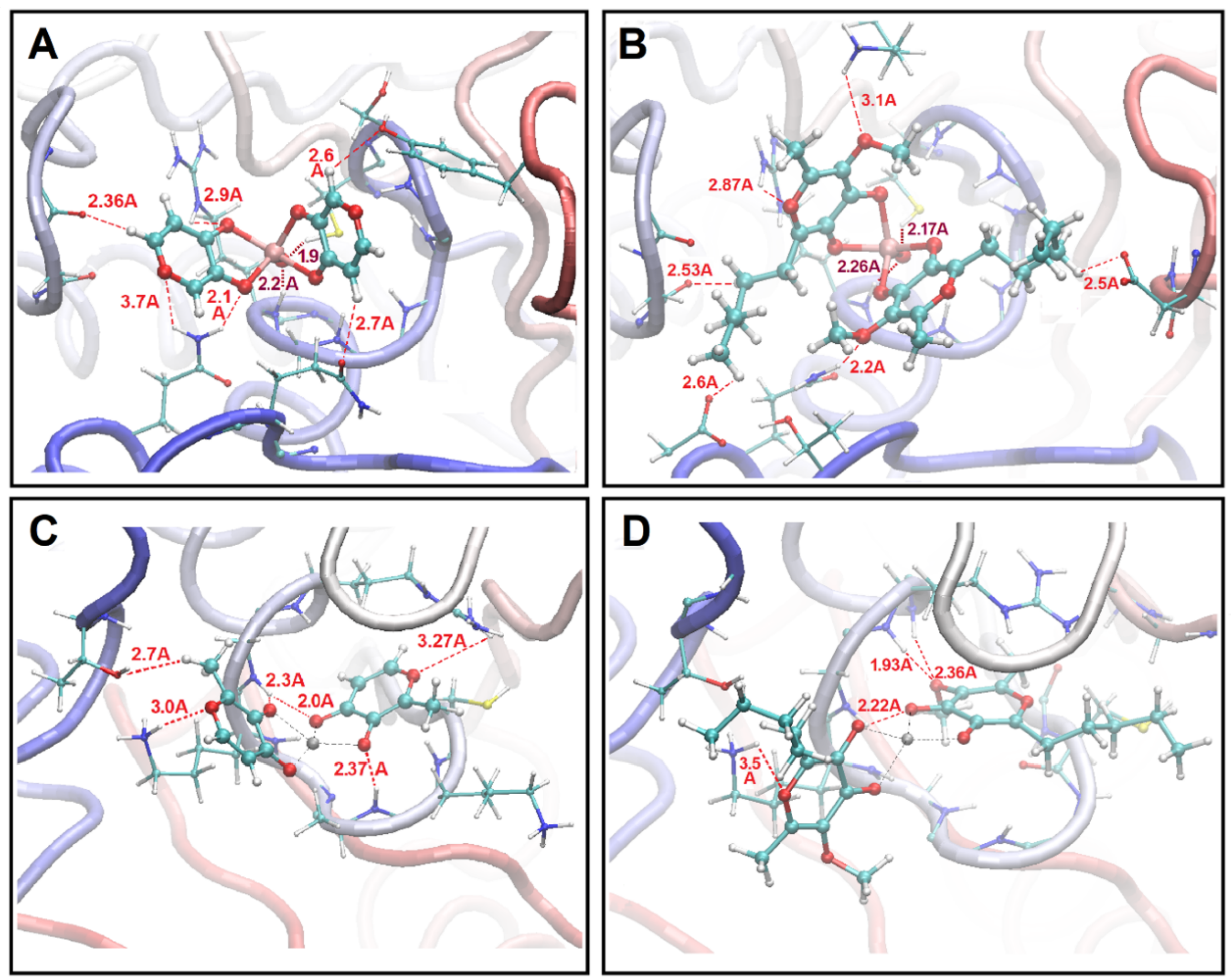
Figure 4.
IR (red line) and Raman (blue line) spectra computed for: VO(3hp)2 in vacuum (A1), VO(3hp)2 in the PTP1B protein environment (B1), VO(alx)2 in vacuum (C1), VO(alx)2 in the PTP1B protein environment (D1), Zn(mal)2·H2O in vacuum (A2), Zn(mal)2 in the PTEN protein environment (B2), Zn(alx)2·H2O in vacuum (C2), and Zn(alx)2 in the PTEN protein environment (D2). Spectra are normalized, while frequencies are scaled by the factor 0.97. Raman spectra are computed accounting excitation wavelength at 532 nm. All bands in the spectra are convoluted with a Lorentzian function of 10 cm-1 half-width at half-maximum. Resonances of OH stretching modes are out of the adopted spectral range.
Figure 4.
IR (red line) and Raman (blue line) spectra computed for: VO(3hp)2 in vacuum (A1), VO(3hp)2 in the PTP1B protein environment (B1), VO(alx)2 in vacuum (C1), VO(alx)2 in the PTP1B protein environment (D1), Zn(mal)2·H2O in vacuum (A2), Zn(mal)2 in the PTEN protein environment (B2), Zn(alx)2·H2O in vacuum (C2), and Zn(alx)2 in the PTEN protein environment (D2). Spectra are normalized, while frequencies are scaled by the factor 0.97. Raman spectra are computed accounting excitation wavelength at 532 nm. All bands in the spectra are convoluted with a Lorentzian function of 10 cm-1 half-width at half-maximum. Resonances of OH stretching modes are out of the adopted spectral range.
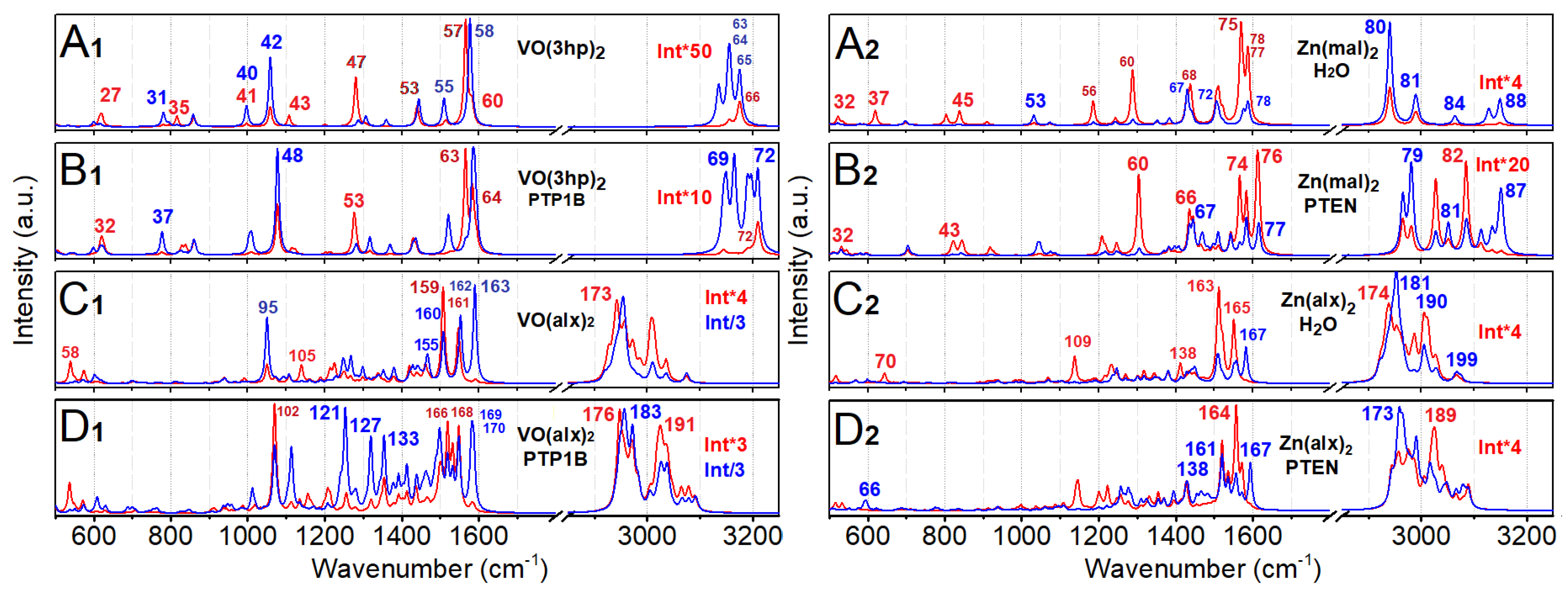
Figure 5.
Optical electronic properties of the VO(3hp)2 and VO(alx)2 complexes. A: Optical absorption spectra computed for VO(3hp)2 in vacuum (blue line) and in the PTP1B protein environment (red line); cyan and orange vertical lines specify the contributions of the single electronic transitions. B: Optical CD spectra computed for VO(3hp)2 in vacuum (blue line) and in the PTP1B protein environment (red line); cyan and orange vertical lines specify the contributions of the single electronic transitions. Dashed blue line represents the optical CD spectrum of VO(3hp)2 L enantiomer: if a racemic mixture the system should not demonstrate optical rotation. Panels C and D show the same data computed for the VO(alx)2 complex in vacuum and in the PTP1B protein environment. We indicate wavelengths of the transitions, where d electronics dominate.
Figure 5.
Optical electronic properties of the VO(3hp)2 and VO(alx)2 complexes. A: Optical absorption spectra computed for VO(3hp)2 in vacuum (blue line) and in the PTP1B protein environment (red line); cyan and orange vertical lines specify the contributions of the single electronic transitions. B: Optical CD spectra computed for VO(3hp)2 in vacuum (blue line) and in the PTP1B protein environment (red line); cyan and orange vertical lines specify the contributions of the single electronic transitions. Dashed blue line represents the optical CD spectrum of VO(3hp)2 L enantiomer: if a racemic mixture the system should not demonstrate optical rotation. Panels C and D show the same data computed for the VO(alx)2 complex in vacuum and in the PTP1B protein environment. We indicate wavelengths of the transitions, where d electronics dominate.
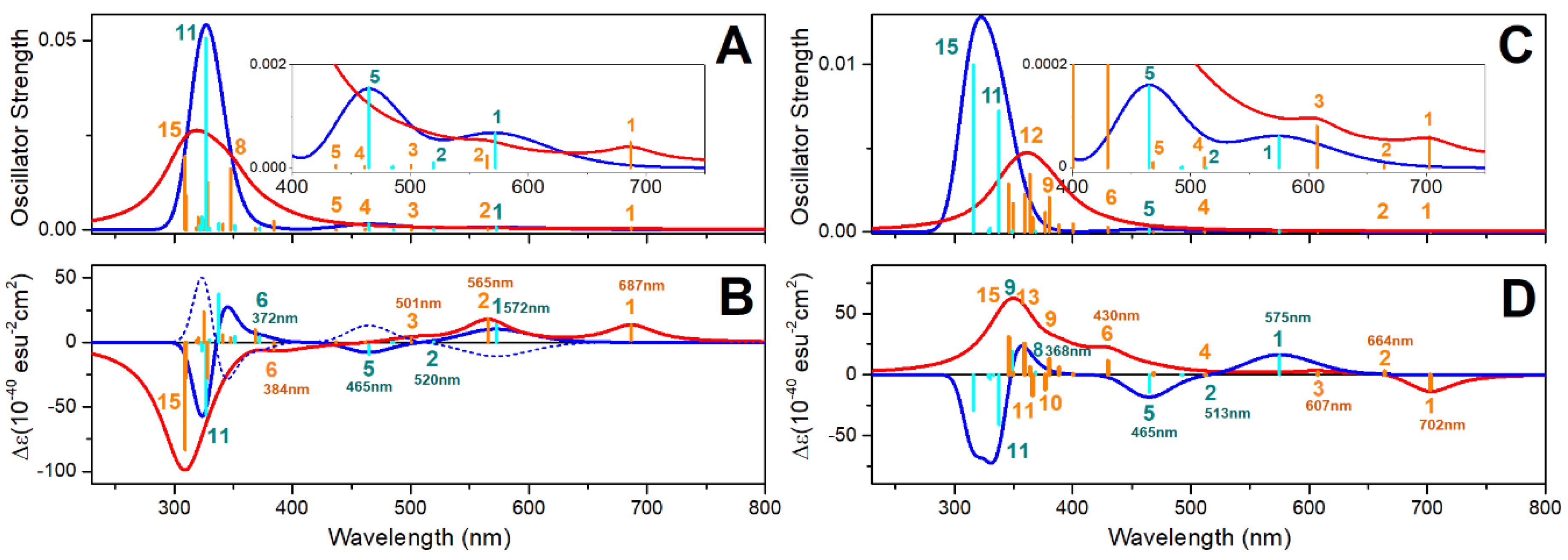
Figure 6.
Selected NTO pairs for VO(3hp)2 in vacuum (top) and in the PTP1B protein environment (bottom).
Figure 6.
Selected NTO pairs for VO(3hp)2 in vacuum (top) and in the PTP1B protein environment (bottom).
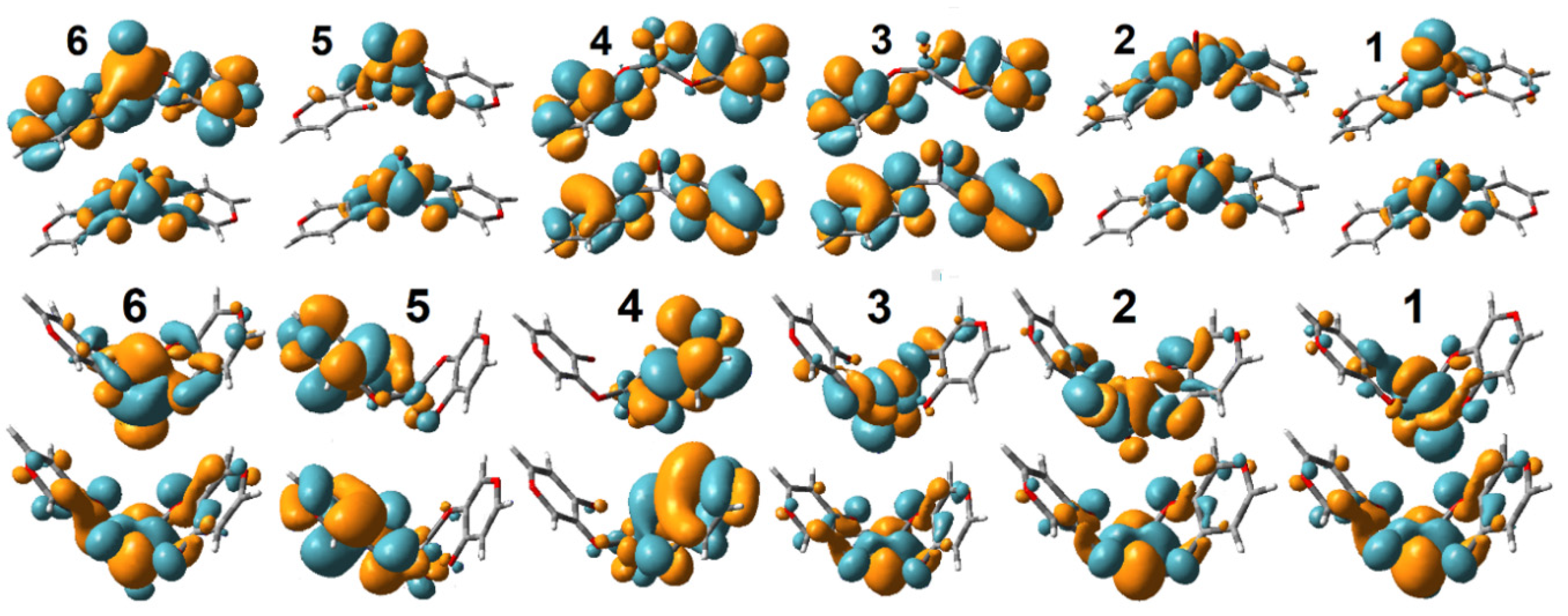
Figure 7.
Selected NTO pairs for VO(alx)2 in vacuum (top) and in the PTP1B protein environment (bottom).
Figure 7.
Selected NTO pairs for VO(alx)2 in vacuum (top) and in the PTP1B protein environment (bottom).
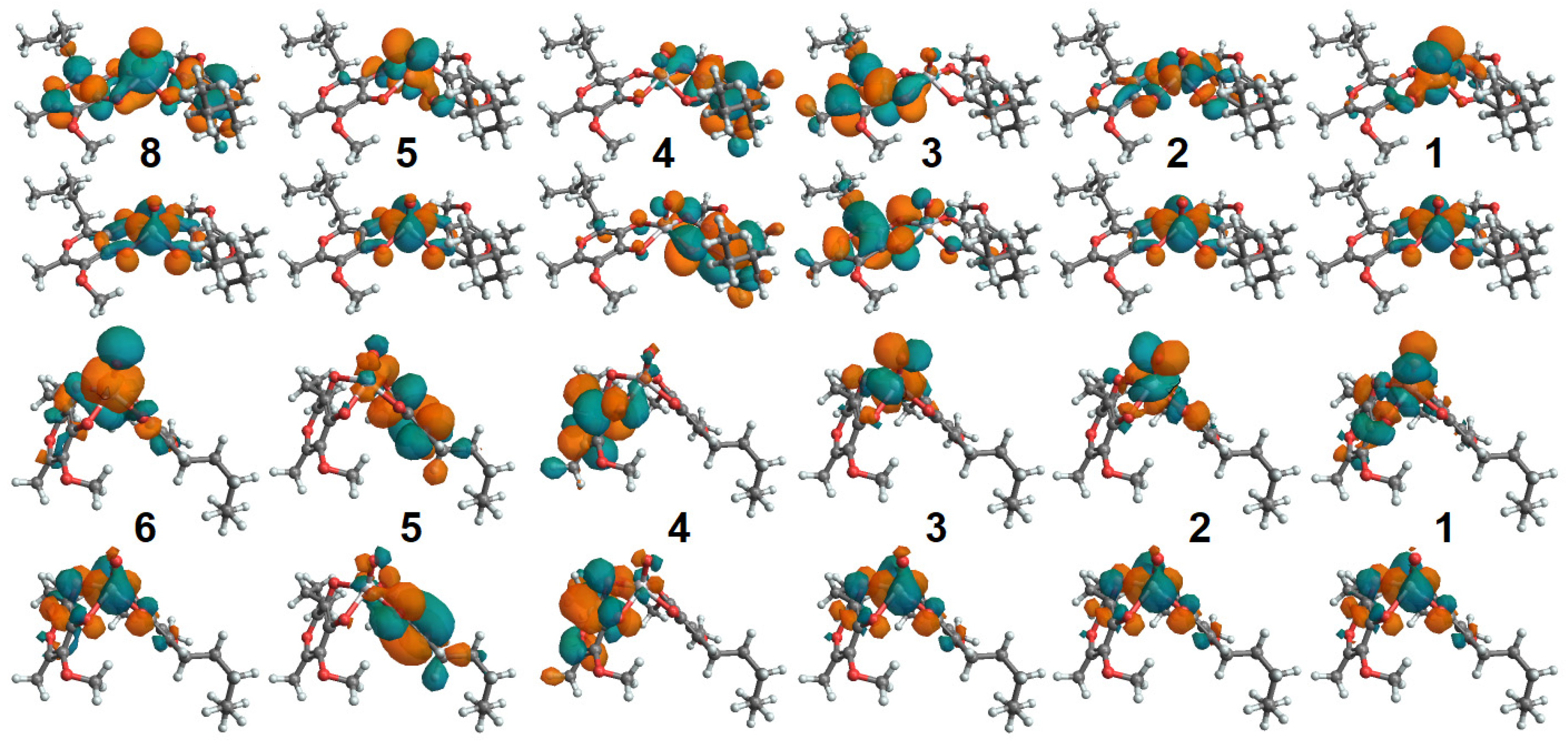
Figure 8.
Optical electronic properties of Zn(mal)2 and Zn(alx)2 systems. A: Optical absorption spectra computed for Zn(mal)2·H2O (blue line) in vacuum and when associated with PTEN protein (red line); cyan and orange lines specify contributions of resonances. B: Optical rotation spectra computed for Zn(mal)2·H2O (blue line) in vacuum and for Zn(mal)2 associated with PTEN protein (red line); cyan and orange lines specify contributions of resonances. If a racemic mixture the system should not demonstrate optical rotation. Panels C and D present analogous data for Zn(alx)2·H2O in vacuum, and for Zn(alx)2 associate with PTEN protein.
Figure 8.
Optical electronic properties of Zn(mal)2 and Zn(alx)2 systems. A: Optical absorption spectra computed for Zn(mal)2·H2O (blue line) in vacuum and when associated with PTEN protein (red line); cyan and orange lines specify contributions of resonances. B: Optical rotation spectra computed for Zn(mal)2·H2O (blue line) in vacuum and for Zn(mal)2 associated with PTEN protein (red line); cyan and orange lines specify contributions of resonances. If a racemic mixture the system should not demonstrate optical rotation. Panels C and D present analogous data for Zn(alx)2·H2O in vacuum, and for Zn(alx)2 associate with PTEN protein.
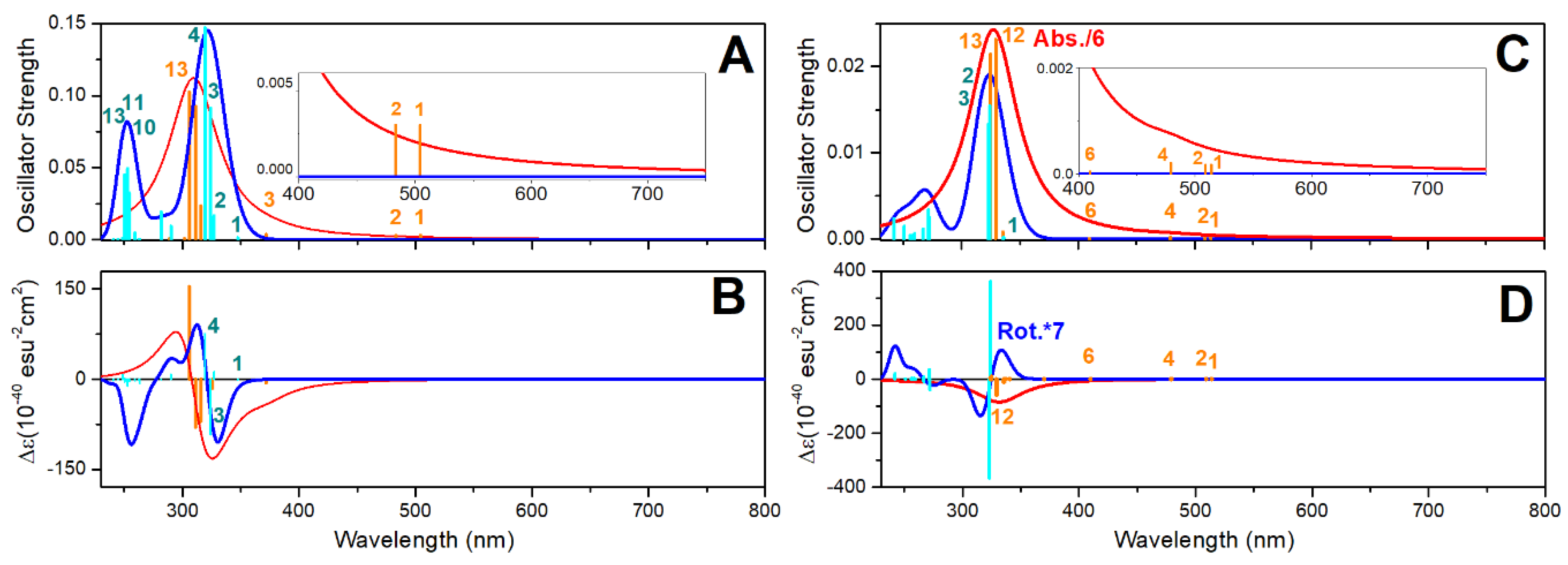

Table 1.
Properties of VO(3hp)2 complex using NBO analysis. Supplementary Material includes the corresponding data for VO(alx)2 complexes: here, carbonyl and carboxylate are noted as crbn and crbx.
Table 1.
Properties of VO(3hp)2 complex using NBO analysis. Supplementary Material includes the corresponding data for VO(alx)2 complexes: here, carbonyl and carboxylate are noted as crbn and crbx.
| bond | Length Å | ESP charges | oxygen contribution | metal contribution |
| hp: V-Ocrbn | 2.05 | -0.5985, -0.5997 | 88.27% s(23.53%) p3.25(76.42%) | 11.73% s(19.76%) p0.96(18.91%) d3.10(61.33%) |
| hp: V-Ocrbx | 1.97 | -0.5478, -0.5493 | 87.66% s(26.00%) p2.84(73.95%) | 12.34% s(21.40%) p1.39(29.77%) d2.28(48.83%) |
| hp: V=O | 1.5790 | V: 1.4057 O: -0.5655 |
71.49% s(18.16%) p4.50(81.74%) | 28.51% s(13.17%) p0.00( 0.04%) d6.59(86.79%) |
| 77.11% p1.00(99.86%) | 22.89% p1.00(32.16%) d2.11(67.84%) | |||
| P-hp: V-Ocrbn | 2.04, 207 | -0.5923, -0.5681 | 88.10% s(23.74%) p3.21(76.21%) | 11.90% s(18.66%) p1.40(26.49%) d2.79(54.14%) |
| P-hp: V-Ocrbx | 1.97, 1.96 | -0.5183, -0.5494 | 86.12% s(25.27%) p2.95(74.68%) | 13.88% s(20.64%) p1.25(25.81%) d2.59(53.55%) |
| P-hp: V=O | 1.57968 | V: 1.292 Oa: -0.6463 |
72.92% s(21.05%) p3.75(78.88%) | 27.08% s(18.98%) p0.01(0.23%) d4.26(80.78%) |
| 77.49% p1.00 (99.85%) | 22.51% p1.00 (30.53%) d2.10(69.39%) | |||
Table 2.
Properties of Zn(mal)2 and Zn(mal)2·H2O complexes using NBO analysis. Supplementary Material includes the corresponding data for Zn(alx)2 complexes.
Table 2.
Properties of Zn(mal)2 and Zn(mal)2·H2O complexes using NBO analysis. Supplementary Material includes the corresponding data for Zn(alx)2 complexes.
| bond | Length Å | ESP charges | oxygen contribution | metal contribution |
| mal: Zn-Ocrbn | 2.07 | -0.72; -0.74 Zn: 1.441 |
95.62% s(14.83%) p5.74(85.10%) | 4.38% s(22.66%) p3.39(76.73%) d0.03(0.62%) |
| mal: Zn-Ocrbx | 1.99 | 94.81% s(16.71%) p4.98(83.23%) | 5.19% s(27.22%) p2.65(72.08%) d0.03(0.70%) | |
| mal: Zn-O7crbn | 2.10 | O7;18: -0.71; -0.65 O9;11: -0.57;-0.67 Zn: 1.25 Oa: -0.81 |
96.18% s(15.58%) p5.41(84.36%) | 3.82% s(20.82%) p3.14(65.29%) d0.67(13.89%) |
| mal: Zn-O18crbn | 2.08 | 95.97% s(16.29%) p5.13(83.65%) | 4.03% s(22.66%) p2.88(65.30%) d0.53(12.04%) | |
| mal: Zn-O9crbx | 2.03 | 96.03% s(17.82%) p4.61(82.13%) | 3.97% s(23.36%) p2.13(49.82%) d1.15(26.81%) | |
| mal: Zn-O11crbx | 2.08 | 96.67% s(16.97%) p4.89(82.98%) | 3.33% s(19.41%) p2.67(51.81%) d1.48(28.78%) | |
| mal: Zn-Oa | 2.23 | 97.64% s(30.23%) p2.31(69.72%) | 2.36% s(13.77%) p4.97(68.48%) d1.29(17.75%) | |
| P-mal:Zn-O7crbn | 2.02121 | O7;O16:-0.69;-0.82 O9; O23:-0.67;-0.64 Zn 1.34 |
95.62% s(14.05%) p6.11(85.88%) | 4.38% s(25.69%) p2.83(72.61%) d0.07(1.70%) |
| P-mal:Zn-O16crbn | 2.24524 | 97.40% s(8.40%) p10.90(91.53%) | 2.60% s(14.56%) p5.72(83.32%) d0.15(2.12%) | |
| P-mal:Zn-O9crbx | 2.05197 | 96.16% s(12.15%) p7.22(87.79%) | 3.84% s(25.85%) p2.79(72.08%) d0.08(2.06%) | |
| P-mal:Zn-O23crbx | 1.94144 | 95.21% s(14.87%) p5.72(85.06%) | 4.79% s(33.55%) p1.92(64.55%) d0.06(1.90%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



