
Submitted:
28 February 2025
Posted:
03 March 2025
You are already at the latest version
Abstract
The targeting of B-lymphocyte cells has emerged as one of the most pivotal strategies in the management of autoimmune diseases. This review provides an overview of protein therapeutics illustrating their direct and indirect effects on B-cells using different molecule formats. The design and format of these molecules influence their mode of action and affect their manufacturing strategies. Manufacturability should be assessed at an early stage and continuously through collaboration between discovery and development teams. Scalability evaluations should encompass not only process development and facility compatibility but also cell line development. Molecule format-specific manufacturing aspects are also reviewed to provide general considerations for productivity and quality improvements in a cost-effective manner.
Keywords:
Autoimmune disease
; biologics manufacturing
; protein therapeutics
; therapeutic target
1. Introduction
The immune system includes the innate and adaptive subsystems, which work together to defend against infection and disease. In autoimmune diseases, the immune system may malfunction, erroneously targeting its own cells, tissues, and organs. The NIH Autoimmune Disease Coordinating Committee reports that there are at least 80 types of chronic and often disabling autoimmune disorders, affecting about 24 million people in the United States (https://dpcpsi.nih.gov/sites/default/files/NIH-Triennial-Report-FY-2016-2018_Final508.pdf). The root cause remains poorly understood, but an overactive response is frequently observed in the immune network, involving humoral, cellular components, or both.
B-cells are vital for humoral immunity, but they can become pathogenic in autoimmunity.[1] Autoreactive B-cells may produce autoantibodies against the body’s tissues, present self-antigens to T-cells, or generate inflammatory cytokines. Due to their role in autoimmune disease initiation and progression,[2] B-cells have become key therapeutic targets. Strategies to target B-cells vary based on the disease mechanism and B-cell functions.[3] B-cell depletion effectively treats pemphigus and neuromyelitis optica spectrum disorder (NMOSD) [4] but not for other autoimmune diseases, such as systemic lupus erythematosus (SLE).[3] The variability in efficacy may be associated with the breadth and depth of pathogenic B-cell depletion and the diversity of B-cell functions being a driver of pathophysiology in these heterogeneous diseases. Consequently, the urgent need for further improvements is prompting collaboration between academia and the biopharmaceutical industry in this emerging field.
2. Therapeutic Targets of B-Cells in Autoimmune Disorders
Targeting B-cells can be direct or indirect (Figure 1). Direct depletion is achieved by targeting surface molecules like CD20, CD19, and BAFF-R using monoclonal antibodies. Indirect depletion involves blocking survival cytokines or regulators of activation and differentiation. Blocking B-cell-activating factor (BAFF) inhibits essential pro-survival gene expressions in B-cells and plasma cells. BAFF signals through BAFF-R, B-cell maturation antigen (BCMA), and transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) receptors. The abundance of these markers varies among B-cell lineages, impacting different signaling pathways. CD40 on B cells mediates growth and transcription regulation when bound to CD40L. Targeting CD40L inhibits stimulation and differentiation into memory B cells and plasma cells, specifically modulating activated B cells. Some targets are intracellular and accessible by small-molecule therapeutics; for instance, proteasome inhibitors like Velcade (bortezomib) treat diseases involving excessive NF-κB pathway activation. Other modalities, such as Chimeric Antigen Receptor (CAR) T-cell therapy, have also been explored to target B-cells, however, the following sections will only discuss protein therapeutics that target/interact with B-cells directly or indirectly.
3. Biologics Targeting B-Cells for Autoimmune Disease Treatments
Previous review articles have collectively provided a good reference for B-cell targeting biologics from different perspectives.[5,6,7] Table 1 summarizes the related contents with additional molecules and new updates, followed by the highlights on seven representative products. Those are directly or indirectly targeting B-cells with diverse molecule formats that may need special considerations for manufacturing.
3.1. OCREVUS (ocrelizumab) and KESIMPTA (ofatumumab) - Next Generation Anti-CD20 Monoclonal Antibodies (mAbs)
Both ocrelizumab and ofatumumab, as examples of next generation B-cell targeting mAbs, were designed to reduce immunogenicity with the former humanized and the latter fully human. Ocrelizumab binds an epitope that overlaps with rituximab’s binding site and offers enhanced antibody-dependent cell-mediated cytotoxicity (ADCC)[6] whereas ofatumumab specifically recognizes an epitope that encompasses both the small and large extracellular loops of CD20 and has a more effective complement-dependent cytotoxicity (CDC) induction and killing target cell capacity.[27] Ocrelizumab was first approved in US for the treatment of patients with relapsing remitting multiple sclerosis (RRMS) and primary progressive multiple sclerosis (PPMS) in 2017,[28] which led B cell depletion to be a mainstay of treatment for MS, and the first therapy specifically for PPMS.[29] In addition to the approvals for RRMS, PPMS, and secondary progressive MS (SPMS),[30] Ofatumumab is also under clinical investigation for RA.
3.2. UPLIZNA (inebilizumab) – Afucosylated Anti-CD19 mAb
Developed by Viela Bio and acquired by Horizon Therapeutics and more recently by Amgen, inebilizumab is a humanized IgG1 mAb targeting the extracellular loop of CD19.[31,32] It initially received FDA approval in 2020 for the treatment of NMOSD in adult patients who are seropositive for immunoglobulin G (IgG) autoantibodies against aquaporin-4 (AQP4).[4] Since then, the authorization for access to this biologic drug has been quickly expanded to include the NMOSD patients in Japan in 2021, in Germany and France in August 2022 and in Brazil in December 2022. As an afucosylated IgG1, inebilizumab has 10-fold higher binding affinity to human Fcγ receptor (FcγR) IIIA and achieves quick and potent B-cell depletion through ADCC[31]. Further clinical data showed that the beneficial outcomes could be maintained for more than 4 years in terms of the reductions in NMOSD attacks[33] and in AQP4-IgG titers. Other indications such as kidney transplant desensitization, myasthenia gravis, and IgG4-RD are also under clinical evaluation.[4] Phase 3 clinical trial for the treatment of IgG4-RD was randomized, double-blind and placebo-controlled study at 80 sites in 22 countries. Compared to placebo for its primary endpoint, this novel and steroid-sparing trial showed a statistically significant 87% reduction in the risk of IgG4-RD flare for 52 weeks; all key secondary endpoints including annualized flare rate and treatment-free and corticosteroid-free complete remission were all met.[34] In a Phase 3 trial in myasthenia gravis, inebilizumab met its primary endpoint, with a statistically significant change from baseline in Myasthenia Gravis Activities of Daily Living (MG-ADL) score (-4.2) compared with placebo (-2.2) (difference: –1.9, p<0.0001) at Week 26 for the combined study population. Inebilizumab demonstrated continued improvement through Week 26 (NCT04524273 and https://wwwext.amgen.com/newsroom/press-releases/2024/10/amgen-presents-positive-phase-3-data-for-uplizna-inebilizumabcdon-in-generalized-myasthenia-gravis-gmg-at-aanem-2024).
3.3. BENLYSTA (belimumab) – mAb Indirectly Targeting B Cells
Belimumab is a fully human mAb that does not directly target B-cells but neutralizes the biologically active soluble form of BAFF and in turn blocks the binding of this cytokine stimulator to its receptor on the involved B-cells.[16] It has been found that BAFF overexpression induces autoreactive B cells to increase autoantibody levels under autoimmune conditions. [35] Inhibition of B-cell proliferation and differentiation by belimumab represents a treatment option for adult patients with seropositive active SLE (especially musculoskeletal and cutaneous disease). Following the FDA approval of belimumab in 2011, which was the first biologic approved for SLE and the first new lupus drug in more than 50 years[16], BAFF was established as a molecular target for further therapeutic developments. TAIAI (telitacicept) is an Fc fusion protein that contains TACI amino acids 13-118 of the extracellular domain, which binds to and neutralizes BAFF and APRIL. It received approval for SLE in China in 2021[23]. Conversely, tabalumab is a humanized monoclonal antibody designed to have a broader blocking effect by neutralizing both soluble and membrane-bound BAFF. However, it did not achieve the expected clinical results in relapsing multiple sclerosis[36,37]. Furthermore, atacicept is another TACI-Ig fusion protein that binds to and blocks BAFF and APRIL[36]. Blisibimod, an Fc-conjugated peptibody, was developed as a specific inhibitor with high avidity antagonizing both soluble and membrane-bound BAFF[38]. Nonetheless, neither atacicept nor blisibimod met clinical expectations in SLE treatment, highlighting the challenges within this field[16].
3.4. Ianalumab (VAY736) – mAb Directly Targeting B Cells
Ianalumab is a fully human mAb that specifically targets BAFF-R to lyse B-cells through ADCC and interrupt B-cell maturation, proliferation and survival via blocking BAFF-mediated signaling. It has been explored for potential therapeutic effects in various autoimmune conditions including MS, SLE, LN and Sjögren’s disease, and more recently in patients with diffuse cutaneous system sclerosis. Subcutaneous administration of ianalumab in a randomized, double-blind, placebo-controlled study was well-tolerated and resulted in clinical improvements in Sjögren’s patients with statistically significant dose-response for overall disease activity.[17]
3.5. Dazodalibep (HZN4920/AMG611) – HSA-Fusion Protein Antagonizing CD40L
Dazodalibep is a novel CD40 ligand (CD40L) antagonist. It is a human serum albumin (HSA)-fusion protein possessing two Tn3 scaffolds;[23] derived from the third fibronectin type III domain of human tenascin-C. Tn3 contains Ig-like folds, structurally analogous to antibody complementarity-determining regions (CDR). The Tn3 scaffolds of dazodalibep have been engineered to confer binding specificity to CD40L.[24] Lacking an Fc domain, dazodalibep does not lead to platelet aggregation or activation.[24] As an indirect and non-depleting B-cell modulator, dazodalibep blocks CD40L on T cells interacting with CD40-expressing B cells and disrupts the overactivation of the CD40 co-stimulatory pathway. Its clinical trials for several autoimmune diseases, such as Sjögren’s disease, kidney transplant rejection and rheumatoid arthritis, are ongoing or have been completed. In addition to the positive data from the Phase 2 trial in patients with RA[22], the results from the Phase 2 trial for patients with Sjögren’s syndrome met the primary endpoint and led to the design of a Phase 3 program.[36]
3.6. PRV-3279 (formerly MGD010) - Bispecific Antibody
PRV-3279 (formerly MGD010) is a humanized dual-affinity-retargeting (DART) bispecific molecule. This antibody targets B-cell surface proteins CD32B and CD79B simultaneously. Developed by MacroGenics and licensed by Provention Bio, CD32B (FcγRIIB) is a low-affinity inhibitory receptor for IgG[43], while CD79B is part of the B cell receptor complex. Targeting CD79B alone has shown efficacy in treating autoimmune diseases in animal models[44]. Crosslinking CD32B and CD79B enhances downregulation of B-cell receptor signaling[45]. This non-depleting B-cell modulating bispecific drug aims to treat lupus and other autoimmune diseases and is currently in Phase 2a clinical trials for moderate-to-severe SLE patients.
4. Impact of Molecule Format on Process Development and Manufacturability
Manufacturability is a critical consideration to ensure the ability to manufacture a product with desired quality at optimized cost. In the context of biologics development and manufacturing (Figure 2), continued evaluation and optimization are iteratively needed to improve molecule format selection, molecule design and may extend through to the entire product lifecycle management. Such reiteration could lead to “next generation” molecule design.[37] The development and commercialization of next generation anti-CD20 antibody therapies are typical examples for these efforts.[6,28] While the most common type of molecule format used in B-cell targeting biologics is a monoclonal antibody (mAb), other molecules with more diversified formats and enhanced features have been developed along the years (Figure 3). Many biologics were originally developed for cancer treatments and have since been repurposed for autoimmune diseases. However, each new format presents its own set of challenges during the development and scale-up of the manufacturing process. For example, the production of afucosylated monoclonal antibodies may require the use of specialized host cells to achieve the desired product quality attributes.
4.1. Afucosylated mAb
Antibody glycosylation changes can significantly modulate its effector function.[38] When B-cell depletion is an intended effect, afucosylated glycoform is desirable for a therapeutic mAb, where afucosylation strongly increases IgG affinity to FcγRIIIa and thus ADCC activity.[32,39,40]. Controlling glycoforms in manufacturing is challenging. Optimizing cell culture conditions can involve using small molecules to inhibit recombinant protein fucosylation[51]. However, a more efficient approach is using host cell lines with altered fucosylation processing (Table 2). These hosts may lack the ability to add fucose during glycosylation or have a shunted GDP-fucose pathway. As an example of FDA-approved afucosylated mAb drugs, UPLIZNA is produced by a proprietary FUT8-/- CHO cell line.[31,32] In addition to these CHO cell lines, other host cells for afucosylated mAb productions are also reported; rat hybridoma YB2/0 cells (ATCC® CRL1662™) intrinsically have lower levels of the FUT8 mRNA[41,42] and EB66 (derived from duck embryonic stem cells) have naturally reduced fucose content in the cells.[40]
4.2. Fusion Protein
Since the approval of Enbrel® (etanercept) for RA in 1998,[50] fusion proteins have emerged as one of the most used molecule formats in autoimmune disease treatments. However, their unique structures that are often derived from different cellular locations or cell types may lead to expression issues. Fusion protein heterogeneity in isoelectric point, charge densities and hydrophobicity could further pose challenges in manufacturing. The linkers between individual modules and their orientation are typically designed based on functional requirements. However, these elements must undergo rigorous evaluation for structural stability and quality attributes to ensure manufacturability. This initial assessment is crucial for effectively minimizing challenges related to structural integrity. For example, O-glycosylation site elimination in linker regions was found to confer Fc fusion proteins with more homogenous glycans and less aggregative propensity.[51] Rearrangement of the disulfide bonding pattern was demonstrated to not only reduce aggregate formation but also improve pharmacokinetic properties.[52]
As in other types of biologics process development processes, platform approach can be a good starting point, followed by product-specific improvement iteration. Fusion protein glycoforms can be more complex than standard mAbs, necessitating optimization of medium compositions. Bioreactor conditions such as temperature shifts and harvest timing must also be evaluated for their impact on glycan attributes.[53,54] Medium optimization for glycosylation and sialylation can be achieved by adjusting key component concentrations[55,56] or adding specific additives.[57,58] Hydrocortisone and dexamethasone enhance Fc-fusion protein sialylation,[57] while LongR3, an insulin-like growth factor-I analogue, increases sialic acid content and reduces asialylated N-glycan species.[58]
Fusion proteins may form aggregates during upstream cultivation and downstream purification. Disulfide bond-containing fusion protein aggregation can be minimized through balancing redox equivalents in media and culture conditions[59] or regulating glutathione reductase activity by glucocorticoid receptor agonists.[60] The pH dependency of fusion domain interactions and conformational stabilities, where a low pH may induce aggregate formation, would require downstream processing to avoid acidic elution from a capture column[61] and the use of low pH for virus inactivation.
4.3. Bispecific Antibody
Bispecifics can generally be manufactured using the same process as those for mAbs due to their structural similarities. However, variations in module formats can lead to differences in bispecifics. The manufacturing challenges include mispairing, high aggregation, and fragment formation, which necessitate early identification of sequence liabilities and the associated expression systems to ensure correct assembly of different polypeptide chains.[62] Domain crossover, Fc heterodimerization, and common light chain strategies in protein engineering have been developed to design bispecifics with a lower tendency for mispairing and enhanced recombinant product stability.
A high producer cell line that pairs the desired heterodimer is probably the key to allow standard upstream and downstream processes to be utilized for bispecifics production. For cell line development, titrations of separate gene constructs for transfection can increase the likelihood of correct protein assembly. Alternatively, different strengths of promoters should be used if all genes of interests are built into a single expression vector. Titer assay that measures the desired heterodimer form should be applied for rigorous clone screening.
As with standard mAb production, optimization of medium composition and culture conditions can affect product quality. To minimize aggregate formation and to maximize the yield of desired heterodimer, attention would particularly be paid to balancing redox reactions in media such as fine-tuning the concentrations of cysteine and metal ions.[63] In combination with closely monitoring overall quality attributes, temperature shifting is one of the most used strategies to improve process robustness and reduce product structural-related impurity formation.[64] To facilitate removal of unwanted homodimers, the resin MabSelect SuRe™ pcc that has a small bead size and decreased binding avidity can be applied to the protein A matrix for high-resolution purification.[65]
5. Manufacturing Scalability
Scalability assessment ensures that a process can be executed consistently across different scales with minimal parameter adjustments. This often includes tech transfer, facility fit analysis, and engineering runs, focusing on vessel geometry, mixing, and gassing. When lactate accumulation is higher and cell viability declines faster in large-scale cultures, optimizing agitation and oxygenation is usually the first step. Trace metal concentrations, particularly copper, may also need adjustment to prevent issues.[66,67,68]
Despite significant efforts for process scaling-up, discussions on the scalability of cell lines for manufacturing are less common. Accelerated timelines for cell line development, driven by new technologies and automation, can lead to genetic instability and increased sensitivity to environmental stress,[69] potentially inducing epigenetic modifications, lactate accumulation and even cell death.[68,70] Addition of copper has been found to be effective in mitigating some of these issues but may not address all underlying causes.
Increased selection pressure during seed train expansion has shown to improve production performance and scalability in CHO cultures without altering gene integration or copy number.[71] Lower lactate concentrations and enhanced re-maturation were observed, suggesting that re-cloning underperforming cell lines can enhance productivity and scalability.
6. Closing Remarks
B-cell-targeting biologics are innovative and successful new therapies for autoimmune diseases. Further improvements in clinical efficacy and safety would rely on continued efforts to discover new targets and to develop new molecule formats that specifically remove or inactivate pathogenic effector B cells while preserving regulatory B-cells for maintaining immune surveillance and minimizing adverse effects. While boosting potencies through avidity, bispecific format allows distinct targets to be simultaneously engaged and potentially increases the selectivity on certain populations of B-cells. Product manufacturability needs to be evaluated as early as possible and iteratively with the inputs from process development teams. Selecting the right expression system and cell line based on the molecule format-specific needs provides an upfront advantage for maximizing titer while maintaining product quality attributes, which in turn simplifies later purification and formulation steps. Platform approach using standard mAb processes can be a good starting point, but it should be complemented with a well-characterized and molecule-specific toolbox for further optimization to ensure high quality and cost-effective manufacturing. The toolbox contains various levelers of medium compositions including additives, resin selections, and in-process controls, but more importantly it is empowered by deep mechanistic understanding. New technologies are on the horizon. We need continued learning, innovation, and exploration.
Author Contributions
Conceptualization, Y.Q.; Literature Search and Analysis, Y.Q. and T.M.; Writing – Original Draft Preparation, Y.Q.; Writing – Review & Editing, Y.Q. and T.M.
Funding
This study received no external funding.
Conflicts of Interest
Y.Q. and T.M. are employees of Amgen Inc. and own stock.
Acknowledgments
The authors would like to thank Natalia Gomez for her continued management support and manuscript review and valuable suggestions.
ORCID
Yueming Qian: https: //orcid.org/0000-0001-7340-6421.
References
- Kurosaki T. B-lymphocyte biology. Immunol Rev. Sep 2010;237(1):5-9. [CrossRef]
- Townsend MJ, Monroe JG, Chan AC. B-cell targeted therapies in human autoimmune diseases: an updated perspective. Immunol Rev. Sep 2010;237(1):264-83. [CrossRef]
- Barnas JL, Looney RJ, Anolik JH. B cell targeted therapies in autoimmune disease. Curr Opin Immunol. Dec 2019;61:92-99. [CrossRef]
- Frampton JE. Inebilizumab: First Approval. Drugs. Aug 2020;80(12):1259-1264. [CrossRef]
- Merino-Vico A, Frazzei G, van Hamburg JP, Tas SW. Targeting B cells and plasma cells in autoimmune diseases: From established treatments to novel therapeutic approaches. Eur J Immunol. Jan 2023;53(1):e2149675. [CrossRef]
- Du FH, Mills EA, Mao-Draayer Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto Immun Highlights. Nov 16 2017;8(1):12. [CrossRef]
- Huda R. New Approaches to Targeting B Cells for Myasthenia Gravis Therapy. Front Immunol. 2020;11:240. [CrossRef]
- Gurcan HM, Keskin DB, Stern JN, Nitzberg MA, Shekhani H, Ahmed AR. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol. Jan 2009;9(1):10-25. [CrossRef]
- Kanatas P, Stouras I, Stefanis L, Stathopoulos P. B-Cell-Directed Therapies: A New Era in Multiple Sclerosis Treatment. Can J Neurol Sci. May 16 2022:1-10. [CrossRef]
- Rubbert-Roth A. TRU-015, a fusion protein derived from an anti-CD20 antibody, for the treatment of rheumatoid arthritis. Curr Opin Mol Ther. Feb 2010;12(1):115-23.
- Robinson WH, Fiorentino D, Chung L, et al. Cutting-edge approaches to B-cell depletion in autoimmune diseases. Front Immunol. 2024;15:1454747. [CrossRef]
- Arbitman L, Furie R, Vashistha H. B cell-targeted therapies in systemic lupus erythematosus. J Autoimmun. Oct 2022;132:102873. [CrossRef]
- Subklewe M, Magno G, Gebhardt C, et al. Application of blinatumomab, a bispecific anti-CD3/CD19 T-cell engager, in treating severe systemic sclerosis: A case study. Eur J Cancer. Jun 2024;204:114071. [CrossRef]
- Li J, Li M, Wu D, Zhou J, Leung SO, Zhang F. SM03, an anti-human CD22 monoclonal antibody, for active rheumatoid arthritis: a phase II, randomized, double-blind, placebo-controlled study. Rheumatology (Oxford). May 5 2022;61(5):1841-1848. [CrossRef]
- Geh D, Gordon C. Epratuzumab for the treatment of systemic lupus erythematosus. Expert Rev Clin Immunol. Apr 2018;14(4):245-258. [CrossRef]
- Runkel L, Stacey J. Lupus clinical development: will belimumab’s approval catalyse a new paradigm for SLE drug development? Expert Opin Biol Ther. Apr 2014;14(4):491-501. [CrossRef]
- Bowman SJ, Fox R, Dorner T, et al. Safety and efficacy of subcutaneous ianalumab (VAY736) in patients with primary Sjogren’s syndrome: a randomised, double-blind, placebo-controlled, phase 2b dose-finding trial. Lancet. Jan 8 2022;399(10320):161-171. [CrossRef]
- Furie RA, Bruce IN, Dorner T, et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology (Oxford). Nov 3 2021;60(11):5397-5407. [CrossRef]
- Kahaly GJ, Stan MN, Frommer L, et al. A Novel Anti-CD40 Monoclonal Antibody, Iscalimab, for Control of Graves Hyperthyroidism-A Proof-of-Concept Trial. J Clin Endocrinol Metab. Mar 1 2020;105(3)doi:10.1210/clinem/dgz013.
- Fisher BA, Szanto A, Ng W-F, Bombardieri M, Gergely P. Assessment of the anti-CD40 antibody iscalimab in patients with primary Sjögren’s syndrome: a multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. The Lancet Rheumatology. 2020;2(3):11. [CrossRef]
- Visvanathan S, Daniluk S, Ptaszynski R, et al. Effects of BI 655064, an antagonistic anti-CD40 antibody, on clinical and biomarker variables in patients with active rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase IIa study. Ann Rheum Dis. Jun 2019;78(6):754-760. [CrossRef]
- Kivitz A. A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Mechanistic Insight and Dosage Optimization Study of the Efficacy and Safety of Dazodalibep (VIB4920/HZN4920) in Patients with Rheumatoid Arthritis Having Inadequate Response to Conventional/Biological DMARDs. Arthritis Rheumatol. 2022;74(9).
- Oganesyan V, Ferguson A, Grinberg L, et al. Fibronectin type III domains engineered to bind CD40L: cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of two complexes. Acta Crystallogr Sect F Struct Biol Cryst Commun. Sep 2013;69(Pt 9):1045-8. [CrossRef]
- Karnell JL, Rieder SA, Ettinger R, Kolbeck R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv Drug Deliv Rev. Feb 15 2019;141:92-103. [CrossRef]
- Dhillon S. Telitacicept: First Approval. Drugs. Sep 2021;81(14):1671-1675. [CrossRef]
- Bracewell C, Isaacs JD, Emery P, Ng WF. Atacicept, a novel B cell-targeting biological therapy for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. Jul 2009;9(7):909-19. [CrossRef]
- Robak T, Robak E. New anti-CD20 monoclonal antibodies for the treatment of B-cell lymphoid malignancies. BioDrugs. Feb 1 2011;25(1):13-25. [CrossRef]
- Frampton JE. Ocrelizumab: First Global Approval. Drugs. Jun 2017;77(9):1035-1041. [CrossRef]
- McCool R, Wilson K, Arber M, et al. Systematic review and network meta-analysis comparing ocrelizumab with other treatments for relapsing multiple sclerosis. Mult Scler Relat Disord. Apr 2019;29:55-61. [CrossRef]
- Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N Engl J Med. Aug 6 2020;383(6):546-557. [CrossRef]
- Herbst R, Wang Y, Gallagher S, et al. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J Pharmacol Exp Ther. Oct 2010;335(1):213-22. [CrossRef]
- Gallagher S, Turman S, Yusuf I, et al. Pharmacological profile of MEDI-551, a novel anti-CD19 antibody, in human CD19 transgenic mice. Int Immunopharmacol. Jul 2016;36:205-212. [CrossRef]
- Rensel M, Zabeti A, Mealy MA, et al. Long-term efficacy and safety of inebilizumab in neuromyelitis optica spectrum disorder: Analysis of aquaporin-4-immunoglobulin G-seropositive participants taking inebilizumab for ⩾4 years in the N-MOmentum trial. Mult Scler. May 2022;28(6):925-932. [CrossRef]
- Stone JH, Khosroshahi A, Zhang W, et al. Inebilizumab for Treatment of IgG4-Related Disease. N Engl J Med. Nov 14 2024;doi:10.1056/NEJMoa2409712.
- Zhang Y, Tian J, Xiao F, et al. B cell-activating factor and its targeted therapy in autoimmune diseases. Cytokine Growth Factor Rev. Apr 2022;64:57-70. [CrossRef]
- St Clair EW, Baer AN, Ng WF, et al. CD40 ligand antagonist dazodalibep in Sjogren’s disease: a randomized, double-blinded, placebo-controlled, phase 2 trial. Nat Med. Jun 2024;30(6):1583-1592. [CrossRef]
- Chen Z, Qian Y, Song Y, et al. Design of next-generation therapeutic IgG4 with improved manufacturability and bioanalytical characteristics. MAbs. Jan-Dec 2020;12(1):1829338. [CrossRef]
- Thomann M, Reckermann K, Reusch D, Prasser J, Tejada ML. Fc-galactosylation modulates antibody-dependent cellular cytotoxicity of therapeutic antibodies. Mol Immunol. May 2016;73:69-75. [CrossRef]
- Shields RL, Lai J, Keck R, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. Jul 26 2002;277(30):26733-40. [CrossRef]
- Olivier S, Jacoby M, Brillon C, et al. EB66 cell line, a duck embryonic stem cell-derived substrate for the industrial production of therapeutic monoclonal antibodies with enhanced ADCC activity. MAbs. Jul-Aug 2010;2(4):405-15. [CrossRef]
- Kilmartin JV, Wright B, Milstein C. Rat monoclonal antitubulin antibodies derived by using a new nonsecreting rat cell line. J Cell Biol. Jun 1982;93(3):576-82. [CrossRef]
- Shinkawa T, Nakamura K, Yamane N, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. Jan 31 2003;278(5):3466-73. [CrossRef]
- Ripka J, Adamany A, Stanley P. Two Chinese hamster ovary glycosylation mutants affected in the conversion of GDP-mannose to GDP-fucose. Arch Biochem Biophys. Sep 1986;249(2):533-45. [CrossRef]
- Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng. Sep 5 2004;87(5):614-22. [CrossRef]
- Malphettes L, Freyvert Y, Chang J, et al. Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnol Bioeng. Aug 1 2010;106(5):774-83. [CrossRef]
- Chan KF, Shahreel W, Wan C, et al. Inactivation of GDP-fucose transporter gene (Slc35c1) in CHO cells by ZFNs, TALENs and CRISPR-Cas9 for production of fucose-free antibodies. Biotechnol J. Mar 2016;11(3):399-414. [CrossRef]
- von Horsten HH, Ogorek C, Blanchard V, et al. Production of non-fucosylated antibodies by co-expression of heterologous GDP-6-deoxy-D-lyxo-4-hexulose reductase. Glycobiology. Dec 2010;20(12):1607-18. [CrossRef]
- Ferrara C, Brunker P, Suter T, Moser S, Puntener U, Umana P. Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of Golgi enzyme localization domain and co-expression of heterologous beta1, 4-N-acetylglucosaminyltransferase III and Golgi alpha-mannosidase II. Biotechnol Bioeng. Apr 5 2006;93(5):851-61. [CrossRef]
- Kanda Y, Imai-Nishiya H, Kuni-Kamochi R, et al. Establishment of a GDP-mannose 4,6-dehydratase (GMD) knockout host cell line: a new strategy for generating completely non-fucosylated recombinant therapeutics. J Biotechnol. Jun 30 2007;130(3):300-10. [CrossRef]
- Nanda S, Bathon JM. Etanercept: a clinical review of current and emerging indications. Expert Opin Pharmacother. May 2004;5(5):1175-86. [CrossRef]
- Song Y, Qian Y, Huang Z, Khattak SF, Li ZJ. Computational insights into O-glycosylation in a CTLA4 Fc-fusion protein linker and its impact on protein quality attributes. Comput Struct Biotechnol J. 2020;18:3925-3935. [CrossRef]
- Way JC, Lauder S, Brunkhorst B, et al. Improvement of Fc-erythropoietin structure and pharmacokinetics by modification at a disulfide bond. Protein Eng Des Sel. Mar 2005;18(3):111-8. [CrossRef]
- Trummer E, Fauland K, Seidinger S, et al. Process parameter shifting: Part I. Effect of DOT, pH, and temperature on the performance of Epo-Fc expressing CHO cells cultivated in controlled batch bioreactors. Biotechnol Bioeng. Aug 20 2006;94(6):1033-44. [CrossRef]
- Hossler P, Khattak SF, Li ZJ. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology. Sep 2009;19(9):936-49. [CrossRef]
- Ha TK, Lee GM. Effect of glutamine substitution by TCA cycle intermediates on the production and sialylation of Fc-fusion protein in Chinese hamster ovary cell culture. J Biotechnol. Jun 20 2014;180:23-9. [CrossRef]
- Taschwer M, Hackl M, Hernandez Bort JA, et al. Growth, productivity and protein glycosylation in a CHO EpoFc producer cell line adapted to glutamine-free growth. J Biotechnol. Jan 20 2012;157(2):295-303. [CrossRef]
- Jing Y, Qian Y, Li ZJ. Sialylation enhancement of CTLA4-Ig fusion protein in Chinese hamster ovary cells by dexamethasone. Biotechnol Bioeng. Oct 15 2010;107(3):488-96. [CrossRef]
- Qian Y, Lewis AM, Sidnam SM, et al. LongR3 enhances Fc-fusion protein N-linked glycosylation while improving protein productivity in an industrial CHO cell line. Research. Process Biochemistry. 2017;53:9. [CrossRef]
- Ying J, Borys BC, Samiksha N, et al. Identification of cell culture conditions to control protein aggregation of IgG fusion proteins expressed in Chinese hamster ovary cells. Process Biochemistry. 2012;47(1):6. [CrossRef]
- Qian Y, Jing Y, Li ZJ. Glucocorticoid receptor-mediated reduction of IgG-fusion protein aggregation in Chinese hamster ovary cells. Biotechnol Prog. Sep-Oct 2010;26(5):1417-23. [CrossRef]
- Shukla AA, Gupta P, Han X. Protein aggregation kinetics during Protein A chromatography. Case study for an Fc fusion protein. J Chromatogr A. Nov 9 2007;1171(1-2):22-8. [CrossRef]
- Wang Q, Chen Y, Park J, et al. Design and Production of Bispecific Antibodies. Antibodies (Basel). Aug 2 2019;8(3)doi:10.3390/antib8030043.
- Purdie JL, Kowle RL, Langland AL, Patel CN, Ouyang A, Olson DJ. Cell culture media impact on drug product solution stability. Biotechnol Prog. Jul 8 2016;32(4):998-1008. [CrossRef]
- Gomez N, Wieczorek A, Lu F, et al. Culture temperature modulates half antibody and aggregate formation in a Chinese hamster ovary cell line expressing a bispecific antibody. Biotechnol Bioeng. Dec 2018;115(12):2930-2940. [CrossRef]
- Tustian AD, Laurin L, Ihre H, Tran T, Stairs R, Bak H. Development of a novel affinity chromatography resin for platform purification of bispecific antibodies with modified protein a binding avidity. Biotechnol Prog. May 2018;34(3):650-658. [CrossRef]
- Brantley T, Moore B, Grinnell C, Khattak S. Investigating trace metal precipitation in highly concentrated cell culture media with Pourbaix diagrams. Biotechnol Bioeng. Oct 2021;118(10):3888-3897. [CrossRef]
- Qian Y, Khattak SF, Xing Z, et al. Cell culture and gene transcription effects of copper sulfate on Chinese hamster ovary cells. Biotechnol Prog. Jul 2011;27(4):1190-4. [CrossRef]
- Qian Y, Xing Z, Lee S, et al. Hypoxia influences protein transport and epigenetic repression of CHO cell cultures in shake flasks. Biotechnol J. Nov 2014;9(11):1413-24. [CrossRef]
- Qian Y, Sowa SW, Aron KL, et al. New insights into genetic instability of an industrial CHO cell line by orthogonal omics. Biochemical Engineering Journal. 2020;164:12. [CrossRef]
- Qian Y, Rehmann MS, Qian NX, et al. Hypoxia and transforming growth factor-beta1 pathway activation promote Chinese Hamster Ovary cell aggregation. Biotechnol Bioeng. Apr 2018;115(4):1051-1061. [CrossRef]
- Tian J, He Q, Oliveira C, et al. Increased MSX level improves biological productivity and production stability in multiple recombinant GS CHO cell lines. Eng Life Sci. Mar 2020;20(3-4):112-125. [CrossRef]
Figure 1.
Direct and indirect targets of B cells by biologics. The surface proteins such as CD19 and CD20 that can be targeted directly whereas the interactions between the surface protein and its ligand can be direct or indirect. Bars under molecular targets represent surface protein expressions at different stages of B-cell development. B = B-cells; BAFF-R = B-cell-activating factor receptor; TACI = transmembrane activator and calcium modulator and cyclophilin ligand interactor; BCMA = B-cell maturation antigen.
Figure 1.
Direct and indirect targets of B cells by biologics. The surface proteins such as CD19 and CD20 that can be targeted directly whereas the interactions between the surface protein and its ligand can be direct or indirect. Bars under molecular targets represent surface protein expressions at different stages of B-cell development. B = B-cells; BAFF-R = B-cell-activating factor receptor; TACI = transmembrane activator and calcium modulator and cyclophilin ligand interactor; BCMA = B-cell maturation antigen.
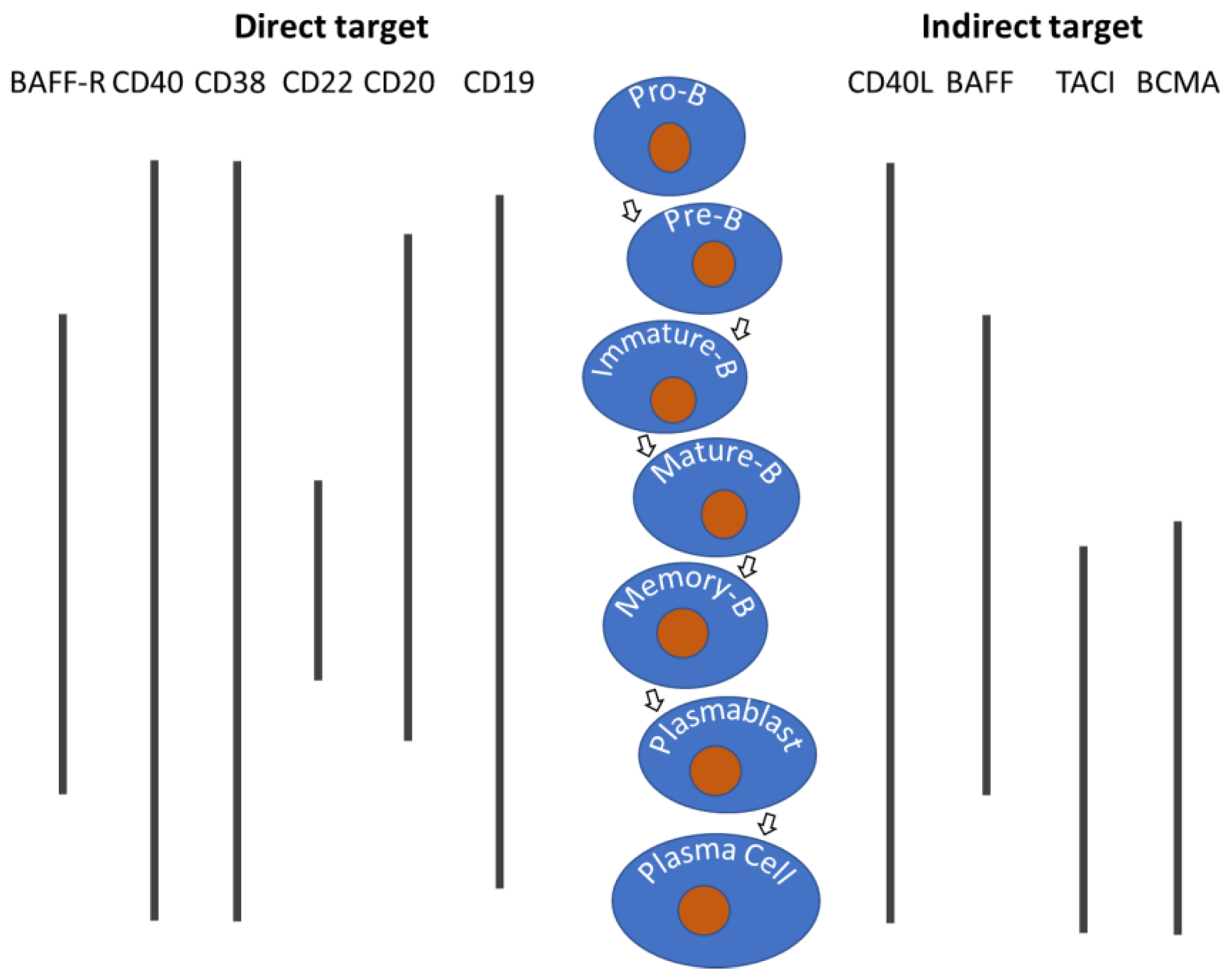
Figure 2.
General workflow for biologics development and manufacturing. Continued assessment and optimization are iteratively needed. Different molecule formats may have different impacts on process development and manufacturing.
Figure 2.
General workflow for biologics development and manufacturing. Continued assessment and optimization are iteratively needed. Different molecule formats may have different impacts on process development and manufacturing.
Figure 3.
Trend in molecule formats of B-cell-targeting biologics launched or clinically trialed for autoimmune diseases. With better understanding of the field and technology advancement, more diversified formats and enhanced features have been developed with time. Many molecules were proven successful in oncology and are being introduced for autoimmune disease treatment. Approximate timeline in the diagram is for the time when those biologics entered the clinical trials for autoimmune indications.
Figure 3.
Trend in molecule formats of B-cell-targeting biologics launched or clinically trialed for autoimmune diseases. With better understanding of the field and technology advancement, more diversified formats and enhanced features have been developed with time. Many molecules were proven successful in oncology and are being introduced for autoimmune disease treatment. Approximate timeline in the diagram is for the time when those biologics entered the clinical trials for autoimmune indications.
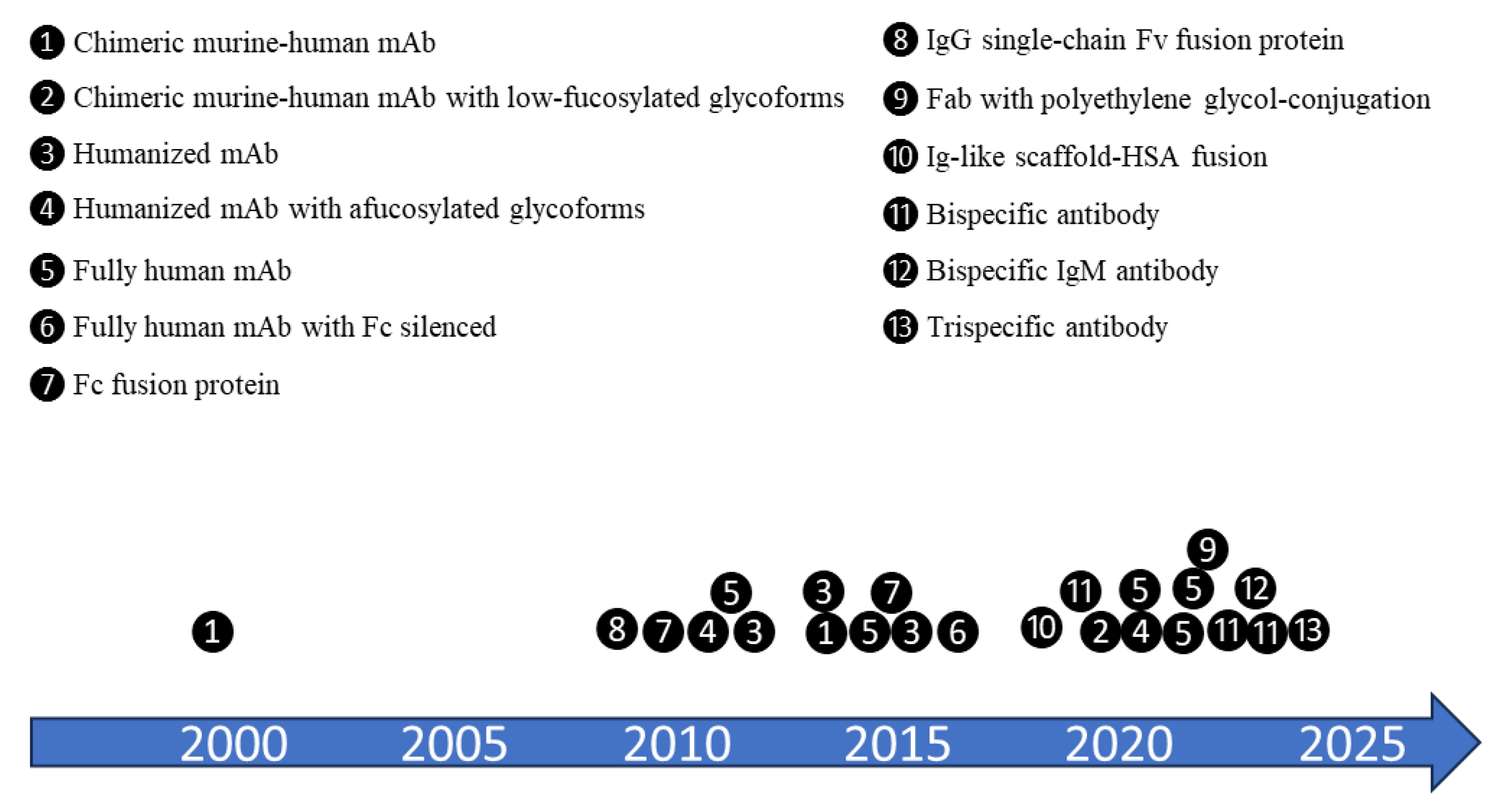

Table 1.
B-cell targeted biologics in autoimmune diseases.
| Target | Drug Name | Molecule Format / Features | Autoimmune Indications | References |
|---|---|---|---|---|
| CD20 | RITUXAN (Rituximab) | Chimeric murine-human IgG1k mAb / targeting CD20 on pro-B cells and all mature B cells, but not long-lived plasma or plasmablast cells. | Approved: RA, GPA, MPA, PV Clinical trials: ITP, MG |
[8] |
| OCREVUS (ocrelizumab) | Humanized mAb / with afucosylated glycoforms enhancing ADCC | Approved: RRMS and PPMS | [9] | |
| BRIUMVI (ublituximab) | Chimeric murine-human IgG mAb / with low-fucosylated glycoforms enhancing ADCC | Approved: RRMS, CIS, SPMS | [9] | |
| KESIMPTA (ofatumumab) | Fully human monoclonal antibody / first B-cell-targeting therapy that is intended for self-administration at home | Approved: RRMS, CIS, SPMS Clinical trial: RA |
[9] | |
| Veltuzumab | Humanized mAb / epratuzumab framework and rituximab CDRs | FDA granted orphan status designation for ITP and pemphigus Clinical trial: RA |
[6] | |
| TRU-015 | Fully human IgG fusion protein / a single-chain Fv specific for CD20 linked to human IgG1 hinge, CH2, and CH3 domains but devoid of CH1 and CL domains | Clinical trials: active seropositive RA on a stable background of methotrexate | [10] | |
| Mosunetuzumab | Bispecific antibody / IgG, anti- CD20 and anti-CD3 | Clinical trials: SLE |
[11] | |
| Imvotamab | Bispecific antibody / IgM, anti-CD20 and anti-CD3 | Clinical trials: RA, SLE |
[11] | |
| CD19 | UPLIZNA (inebilizumab) | Humanized IgG1k mAb / with afucosylated glycoforms enhancing ADCC | Approved: NMOSD with AQP4-IgG+ Clinical trials: GM, IgG4-RD |
[4] |
| Obexelimab | Bispecific antibody / simultaneously binds CD19 and FcγRIIb, resulting in down regulation of B cell activity | Clinical trials: GM; IgG4-RD | [12] | |
| Blinatumomab | Bispecific antibody / anti-CD19 and anti-CD3 | Clinical trials: RA, system sclerosis |
[13] | |
| PIT565 | Trispecific antibody / anti-CD19, anti-CD3, and anti-CD2 | Clinical trials: SLE |
NCT06335979 | |
| CD22 | SM03 | Chimeric murine-human mAb / targeting the extracellular portion of CD22 | Clinical trials: SLE, RA | [14] |
| Epratuzumab | Humanized mAb / targeting CD22 with modest ADCC activity | Clinical trials: SLE | [15] | |
| CD38 | Daratumumab | Fully human mAb / targeting CD38 on long-lived plasma cells | Clinical trials: SLE | [12] |
| BAFF/BAFF-R | BENLYSTA (belimumab) | Fully human mAb / neutralizing biologically active soluble form of BAFF | Approved: SLE and lupus nephritis | [16] |
| Ianalumab (VAY736) |
Fully human mAb / antagonizing BAFF-R | Clinical trials: MS, SLE, Sjögren’s syndrome, Diffuse Cutaneous Systemic Sclerosis | [17] | |
| CD40/CD40L | Dapirolizumab pegol | Fab / polyethylene glycol-conjugated, anti-CD40L, lacking the Fc-portion to avoid platelet activation | Clinical trials: SLE | [18] |
| Iscalimab (CFZ533) | Fully human mAb / Fc-silenced, antagonizing CD40 | Clinical trials: Graves disease (GD); Sjögren’s syndrome | [19,20] | |
| BI 655064 | Humanized mAb / anti-CD40 blocking CD40-CD40L interaction | Clinical trials: RA | [21] | |
| Dazodalibep (AMG611, HZN-4920) |
Ig-like scaffold-HSA fusion protein / Tn3 scaffolds derived from the 3rd fibronectin type III domain of human tenascin-C, structurally analogous to antibody CDRs and functionally blocking CD40-CD40L interaction | Clinical trials: RA, Sjögren’s syndrome | [22,23,24] | |
| BAFF/APRIL | TAIAI (Telitacicept) |
Fc fusion protein / fused with extracellular domain (amino acids 13-118) of TACI binding to and neutralizing BAFF and APRIL | Approved: SLE (in China) Clinical trials: IgA nephropathy, MS, RA, MG, Sjögren’s syndrome |
[25] |
| Atacicept | Fc fusion protein / fused with extracellular domain (amino acids 30-110) of TACI binding to and neutralizing BAFF and APRIL | Clinical trials: SLE, RA, IgA nephropathy | [26] |
mAb = monoclonal antibody; RA = rheumatoid arthritis; GPA = Granulomatosis with Polyangitis; MPA = Microscopic Polyangitis; PV = Pemphigus Vulgaris; ITP = idiopathic thrombocytopenic purpura; MG = myasthenia gravis; GCA = giant cell arteritis; SSc-ILD = systemic sclerosis-interstitial lung disease; PJIA = polyarticular juvenile idiopathic arthritis (PJIA); SJIA = systemic juvenile idiopathic arthritis; MS = Multiple sclerosis (MS); CIS = clinically isolated syndrome; RRMS = relapsing-remitting MS; PPMS = primary progressive MS; SPMS = secondary progressive MS; IgG4-RD = IgG4-related diseases.
Table 2.
CHO variant cell lines for afucosylated mAb productions.
| Cell line | Affected Biosynthesis Pathway | Reference |
|---|---|---|
| CHO Lec13 (Pro-Lec13.6a) | Natural deficiency in endogenous GDP-mannose 4,6-dehydratase (GMD) | [39,43] |
| CHO-DG44 FUT8-/- (BioWa) | FUT8 knockout by homologous recombination | [44] Patent# US6946292B2 |
| CHO-K1 FUT8-/- | FUT8 deletion by ZFN | [45] |
| CHO-gmt3 (CHO-glycosylation mutant3) | GDP-fucose transporter (SLC35C1) inactivation | [46] |
| CHO-RMD | Heterologous expression of GDP-6 deoxy-d-lyxo-4-hexulose reductase (RMD) in the cytosol of CHO cells to deflect the GDP-fucose de novo pathway | [47] |
| CHO-GnT-III | Overexpressed GnTIII catalyzes the formation of a bisecting GlcNAc to reduce Fc core fucosylation |
[48] |
| CHO-SM | GDP-fucose 4,6-dehydratase (GMD) knockout, which makes the cell unable to produce intracellular GDP-fucose and fucosylated glycoproteins in the absence of L-fucose | [49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



