
Submitted:
20 February 2025
Posted:
20 February 2025
You are already at the latest version
Abstract
Erythrobacter, an aerobic anoxygenic photoheterotrophic bacterial genus, plays a vital role in carbon and energy cycling in marine environments. However, their phage predators remain poorly understood, with only two strains previously reported. This study isolated and characterized two novel Erythrobacter phages, vB_EauS-R34L1 (R34L1) and vB_EauS-R34L2 (R34L2), from coastal seawater. Both phages exhibit long-tailed, icosahedral morphologies and relatively narrow but slightly different host ranges. One-step growth curve analysis revealed a 160-min latent period and burst sizes of 81 and 91 PFU/cell for R34L1 and R34L2, respectively. Genomic analysis showed that the phages possess dsDNA genomes of 56,415 bp (R34L1) and 54,924 bp (R34L2), with G+C contents of 61.60% and 61.19%, respectively. Both phages harbor a suite of unique genes, including GapR and GH19, which are crucial for host interaction and ecological functionality. Blastn analysis indicated a 99.73% genome similarity between them. Taxonomic and phylogenetic analyses positioned them in a novel viral genus cluster, Eausmariqdvirus, under the family Casjensviridae, indicating a distant evolutionary relationship with known phages. Metagenomic queries suggested that R34L1- and R34L2-like phages are exclusively abundant in temperate and tropical epipelagic zones. This study expands our understanding of Erythrobacter phages and provides insights into their ecological roles in marine ecosystems.
Keywords:
Bacteriophage
; Erythrobacter
; biological features
; genome
1. Introduction
Aerobic anoxygenic photoheterotrophic bacteria (AAPB), which contain bacteriochlorophyll-a and lack light-harvesting complex II, represent photoheterotrophic microorganisms that may consume less organic carbon and be crucial to the marine carbon cycle [1-4]. Erythrobacter is the first identified AAPB and frequently detected in and isolated from nutrient-rich coastal seawaters and sediments, and its metabolism is versatile [5-10]. Studies have demonstrated that Erythrobacter species show potential applications in bioremediation of alkane contamination (e.g., E. longus and E. citreus) [11], production of yellow xanthophyll pigment (e.g. Erythrobacter sp. SDW2) [8], and production of poly-β-hydroxybutyrate, which is known as a material for degradable plastics (e.g. E. aquimaris) [12]. Erythrobacter has also shown capacities for nitrate reduction, denitrification, aesculin hydrolysis, and multiple substrates utilization, such as amino acids, carbohydrates, and fatty acids [9,13-15], and refractory dissolved organic carbon degradation, and may play an essential role in labile dissolved organic carbon acquisition for surrounding heterotrophs [4,9].
Bacteriophages (viruses that infect bacteria, phage for short) are crucial components of marine ecosystems. They could regulate bacterial abundance, community structure, and carbon sequestration efficiency [16-18]. The isolation and identification of the genomic features of phages could advance our knowledge of the ecology and evolution of their hosts. To date, nearly 80 roseophages—viruses that infect the Roseobacter lineage (a representative member of AAPB), have been identified and are suggested to provide versatility for roseobacters to adapt to changing environments quickly [19,20]. They can rapidly alter the growth and abundance of their hosts to shunt their secondary production [21]. Similarly, cyanophages (viruses that infect cyanobacteria) can mediate the mortality of cyanobacteria and affect their distribution and composition [22,23]. Beyond that, the isolation of novel phages could also contribute to understanding the ecological distribution patterns and physiological characteristics of phages in aquatic environments by combining the virome database [24-26].
To date, only two Erythrobacter phages, vB_EliS-R6L (hereafter R6L) and vB_EliS-L02 (hereafter L02) have been isolated, each exhibiting unique morphological traits and distinct ecological distribution patterns [25,27]. These phages exhibit complex interactions with their hosts, highlighting their ecological and evolutionary significance. For example, both phages contain phoH genes, which promote host phosphorus uptake in low-phosphorus environments. Additionally, phage L02 harbors several genes related to nucleotide metabolism, potentially enhancing its ability to regulate host metabolic processes. Despite these advances, our understanding of Erythrobacter phages remains fragmented, particularly concerning their morphology diversity, physiological features, ecological distribution patterns, and interactions with the host. Here, we reported two novel Erythrobacter phages, providing detailed insights into their morphology, basic physiology, genomic features, and distribution patterns, which represent a significant step forward in the study of Erythrobacter phages and offer a foundation for future research into their ecological functions and host-phage coevolutionary relationships.
2. Results
2.1. Phage Isolation and Biological Features of R34L1 and R34L2
Two phages, vB_EauS-R34L1 (hereafter R34L1) and vB_EauS-R34L2 (hereafter R34L2), infecting Erythrobacter sp. JL 475, were isolated from the Qingdao and Zhoushan coastal seawater, respectively. Both phages formed small, clear, and round plaques on the lawn of Erythrobacter sp. JL 475 with a diameter of approximately 3~4 mm. Transmission electron microscopy (TEM) analysis revealed that phages R34L1 and R34L2 both belonged to siphovirus (Figure 1A), exhibiting icosahedral capsids with average diameters of 56.7 ± 3.0 and 57.0 ± 2.1 nm, and visible long tails with average lengths of 224.6 ± 14.0 nm and 188.5 ± 3.6 nm, respectively. Additionally, chloroform treatment analysis revealed that phages R34L1 and R34L2 were not sensitive to chloroform, suggesting that these phages do not contain lipids as structural components, as lipids are typically dissolved by chloroform [28].
Both phages showed a relatively narrow host range (Table S1). Specifically, phage R34L1 could only infect Erythrobacter sp. JL 475, while phage R34L2 lysed Erythrobacter sp. JL 475, JL 967 and JL 2286. Notably, the host ranges of isolated phages differ from those of phages R6L and L02 [25,27].
The infection activities of phages R34L1 and R34L2 were characterized by a one-step growth curve derived from plaque assays. Both phages exhibited latent and rising periods of approximately 160 minutes (Figure 1B). The burst sizes reached approximately 91 and 88 plaque-forming units per infected cell (PFU/cell) for phages R34L1 and R34L2, respectively. These values are comparable to the latent period (<1~6 h) and burst size (27~1500 PFU/cell) reported for the two Erythrobacter and most Roseobacter-infecting phages [29-31].
The stability of phages R34L1 and R34L2 was evaluated across different temperatures, pH levels, and salinities. Overall, the trends in stability were similar for both phages. The activity of phages R34L1 and R34L2 was significantly affected by temperature, with a notable decrease in activity observed as the temperature increased (Figure 2A). pH stability tests revealed that the highest activity for both phages was at pH 7.5, indicating that these phages are sensitive to both acidic and alkaline conditions (Figure 2B). The salinity stability experiment demonstrated a decrease in phage viability with increasing freshwater dilution ratios, with viabilities of 52.49% and 49.58% in total freshwater for phages R34L1 and R34L2, respectively (Figure 2C). These results suggest that phages R34L1 and R34L2 may have a broad distribution range and could potentially thrive in diverse environmental conditions.
2.2. Genomic Features of R34L1 and R34L2
Phages R34L1 and R34L2 both possess dsDNA genomes, with sizes of 56,415 bp and 54,924 bp, respectively. Their G+C contents are 61.60% and 61.19%, respectively. Notably, the two genomes exhibit a high degree of similarity, with an identity of 99.73% as determined by Blastn and 99.68% by OrthoANI analysis. The G+C content of both phage genomes is slightly lower than that of their host (61.77%, GenBank accession no. NZ_CP017057) and differed from that of other isolated Erythrobacter phages (59.43% and 66.52%) (Table 1) [25,27]. This difference in G+C content may reflect distinct evolutionary adaptations or selective pressures acting on the phage genomes compared to their bacterial hosts. Bioinformatic analysis revealed that neither phage R34L1 nor R34L2 contains tRNA genes, which may also account for their relatively narrow host range [32,33].
A total of 78 open reading frames (ORFs) were predicted for phage R34L1 and 72 ORFs for phage R43L2 (Figure 3, Tables S2 and S3). Functional analysis revealed that 27 ORFs (34.62%) in phage R34L1 and 27 ORFs (37.50%) in phage R34L2 had predicted functions (Tables S2 and S3). Furthermore, the predicted ORFs between the two phages were largely consistent, with high similarity ranging from 94.57% to 100.00% as determined by Blastp analysis (Table S4).
Given the high homology and consistency observed between the two phages, we propose that phage R34L2 is a sub-strain of R34L1. Therefore, the introduction of functional genes primarily focuses on phage R34L1. The functionally related ORFs can be divided into four main groups: DNA replication and metabolism (8 ORFs), assembly and structure (17 ORFs), host lysis (ORF33, glycoside hydrolase family 19 catalytic domain-containing protein), and auxiliary metabolic genes (AMGs) (ORF28, GapR) (Figure 3). Specifically, eight ORFs were related to DNA replication and metabolism, including DNA synthesis (ORF65, ORF66, and ORF72), DNA methyltransferase (ORF64), DNA helicase-related protein (ORF62), and ligase-related proteins (ORF39, ORF63, and ORF67). Seventeen ORFs were related to assembly and structure, including tail-related proteins (ORF37, ORF38, ORF39, ORF42, ORF43, ORF44, ORF47, ORF48, and ORF49), stopper protein (ORF50), major capsid protein (ORF52), head decoration protein (ORF53), S49 family peptidase (ORF54), portal protein (ORF55), head-tail adaptor protein (ORF54), and phage packaging protein terminase (ORF57 and ORF58).
2.3. Phylogenetic Analysis and Comparative Genomic Analyses
The genomes of phages R34L1 and R34L2 were analyzed using Blastn on Genebank, and neither of them showed significant similarity to any of the uploaded sequences in the NCBI nr database, with query coverage ranging from 0% to 1% for all other alignment sequences (accessed on 15 February 2025). To assess the phylogenetic relationship of phages R34L1 and R34L2 to known phages, the ViPTree server was utilized to construct a proteomic tree based on genome sequences (Figure 4A and 4B). The results indicated that phages R34L1 and R34L2 only clustered with Sphingomonas phage Carli (OR225223.1) and Burkholderia phage BcepNazgul (NC_005091), yet exhibiting a relatively distant evolutionary relationship. Moreover, phages R34L1 and R34L2 and the taxa clustered with them were classified within the virus family “Others” (Figure 4B).
The Average Nucleotide Identity (ANI) values between R34L1 and R34L2 are higher than 95% indicating that they belonged to the same species [34]. Subsequently, the phages clustered with phages R34L1 and R34L2 were selected for further genome comparison analysis. The ANI values between R34L1 or R34L2 and other phages ranged from 58.91% to 61.37%, suggesting that phages R34L1 and R34L2 may represent a new genus, based on mechanistic demarcation criterion for phage genera (70%) (Figure 4C) [34]. Additionally, the genome similarity analysis of phages R34L1 and R34L2 with other phages, as determined by VIRIDIC, revealed quite low genomic similarity values, ranging from 4.46% to 8.54% (Figure 4D).
Then, the phylogenetic tree constructed by VICTOR (https://ggdc.dsmz.de/victor.php#) also suggested that phages R34L1 and R34L2 exhibit strong novelty and should be classified as a new viral genus within the family Casjensviridae, with no other known members (Figure 5A). And, the comparative genomic analysis was performed with phages R34L1, R34L2, R6L, and L02, as well as Sphingomonas phage Carli and Burkholderia phage BcepNazgul (Figure 5B). Based on BLASTx analysis results, phage R34L1 or R34L2 shares 33 and 30 similar ORFs (E-value <10-5) with Sphingomonas phage Carli, and Burkholderia phage BcepNazgul, respectively, and six and four with R6L and L02, respectively. Only one homologous ORF, the tail fiber protein, is shared among all four Erythrobacter phages with an average identity of 50.29%. However, the identities of all pairs are quite low, ranging from 23.86 to 60.76% (with an average of 39.03%), providing further evidence for the novelty of phages R34L1 and R34L2, as well as the high diversity of Erythrobacter phages.
Next, the phylogenetic relationship between phages R34L1 and R34L2 and their related phages was further analyzed using GH19 protein (ORF 33), major capsid protein (ORF 52), and portal protein (ORF 55). Firstly, the distant phylogenetic relationships with other phages indicated that phages R34L1 and R34L2 have a significant genetic distance from other known phages, further supporting their novelty (Figure 6). In the phylogenetic trees based on the portal protein and major capsid protein, phages R34L1 and R34L2 primarily clustered with Sphingomonas phage Carli, Burkholderia phage BcepNazgul, consistent with the result of the ViPTree analysis. However, in the GH19-based tree, phages R34L1 and R34L2 clustered with bacteria sequences, including Erythrobacter, Sphingomonas, Caulobacter, and Phenylobacterium, rather than with other phages. Overall, all results consistently support the classification of R34L1 and R34L2 within a novel genus, Eausmariqdvirus, within the family Casjensviridae.
2.4. Marine Ecological Distribution of R34L1 and R34L2
The biogeographical distribution patterns of phages R34L1 and R34L2 were examined across five distinct virus ecological zones (VEZs) in the Global Ocean Viromes (GOV v2.0) data set, including bathypelagic (BATHY), temperate and tropical mesopelagic (MES), temperate and tropical epipelagic (EPI), Arctic (ARC), and Antarctic (ANT). Both phages R34L1 and R34L2 were exclusively detected in temperate and tropical epipelagic zones of the ocean (Figure 7). Moreover, the relative abundances of phages R34L1 and R34L2 (log10TPM=6.51) were significantly higher than those of the reference phages and other phages analyzed using the same methodology, such as Vibrio phage vB_ValR_NF (EPI: -1.12; ARC: -0.88) [35], Shewanella phage vB_SbaS_Y11 (ARC: 0.79) [36], and Psychrobacter phage vB_PmaS_Y8A (MES: 2.24; ARC: 3.66; ANT: 2.61) [37]. This distribution is consistent with that of the previously isolated Erythrobacter phage R6L and differs slightly from that of L02 [27], which could also be sparsely distributed in open oceans and intermediate seawaters [25]. These findings suggested that Erythrobacter phages may have relatively broad distribution patterns in marine epipelagic zones, in agreement with the general distribution of Erythrobacter in the environment [1,3,5].
3. Discussion
Bacteriophages play a crucial role in marine ecosystems. They can regulate bacterial populations by infecting and killing bacteria, participate in nutrient cycling by releasing bacterial contents into the water, maintain ecosystem balance by interacting with bacteria, facilitate genetic exchange and evolution by horizontal gene transformation, and impact carbon cycling by breaking down bacteria [38-41]. Despite their importance, research on phages targeting the genus Erythrobacter in the coastal euphotic zone remains significantly limited, with only two bacteriophage strains reported to date [25,27]. This study identified two novel phages infecting Erythrobacter named R34L1 and R34L2 (R34L2 being a sub-strain of R34L1), representing a new genus of Eausmariqdvirus.
Among all 27 functionally related ORFs, eight ORFs are related to DNA replications and metabolism. Four ORFs are specifically involved in DNA synthesis, including DNA-directed DNA polymerase family A palm domain-containing protein (ORF65, R34L1), ssDNA-binding protein (ORF66), exonuclease (ORF67), and DNA primase/polymerase bifunctional N-terminal domain-containing protein (ORF72). The ssDNA-binding protein and exonuclease are involved in the proofreading and repair processes of DNA, by removing nucleotides from the end of DNA strands and cleaning damaged DNA fragments to maintain genomic stability [42]. The N-terminal domain of bifunctional DNA primase-polymerase should play a crucial role in accommodating the DNA duplex by the prim-pol domain regulating the selection of replication starting points and maintaining the stability of the replication process through its interaction with DNA polymerase [43,44]. The SAM-dependent methyltransferase (ORF64) catalyzes the transfer of methyl groups from S-adenosylmethionine (SAM) to a variety of acceptor substrates, related to proteins, DNA, and polysaccharides metabolites [45]. And, SAM-dependent methyltransferase may also be involved in BacteRiophage Exclusion (BREX) defense mechanism in the host cells [46]. BREX is a defense system in prokaryotes and can act by providing restriction-modification (R-M) systems that epigenetically modify some specific sites (methylation) in the genome of the host, resulting in the restriction-cleavage of incoming phage DNA by endonucleases because of these modifications lacking [47,48]. Thus, SAM-dependent methyltransferase may help phages evade degradation by host endonucleases. The ParB/Sulfiredoxin domain (ORF29) exhibits NTPase and DNase activities, which facilitate the separation and division of the host chromosome, thereby supporting phage replication [49,50]. The DEAD/DEAH box helicase (ORF62) regulates ATP binding and hydrolysis and plays pivotal roles in viral RNA replication and transcription [51,52]. The VRR-NUC domain (ORF63), which belongs to an ancient restriction endonuclease-like superfamily, is closely involved in phage infection and gene expression regulation and repair, ensuring the stability and integrity of phage DNA during replication [53,54].
The glycoside hydrolase family 19 catalytic domain-containing protein (ORF33) has been predicted to associate with host cell lysis [55,56]. The glycoside hydrolase family 19 (GH19 family), encompassing both chitinases and endolysins, is well-recognized for its dual functionality. This family has been extensively investigated for its potential applications in managing plant fungal infestations, facilitating the recycling of chitin-rich biomass, and combating bacteria that exhibit resistance to multiple drugs [55]. Based on the analysis by the Carbohydrate-Active EnZymes database (CAZy), ORF33 is likely to be an endolysin (EC 3.2.1.17) rather than a chitinase (EC 3.2.1.14) [57]. And, the GH19 family is highly diverse and widely distributed across plants, fungi, bacteria, and phages, exhibiting notable geographical and ecological variability [55]. However, in the GH19-based tree (Figure 6A), our phages were found to cluster with bacterial sequences, such as Erythrobacter, Sphingomonas, Caulobacter, and Phenylobacterium, instead of with other phages. This unusual clustering suggests a potential horizontal gene transfer event, where the GH19 gene of phages R34L1 and R34L2 may have been acquired from a bacterial host, specifically Erythrobacter. Meanwhile, GH19 homologs were not detected in Sphingomonas phage Carli, Burkholderia phage BcepNazgul, indicating that GH19 sequences are not universally present or well-identified in phages. As previously reported, numerous phage GH19 sequences cluster with bacterial homologs, likely evolving from insertions under the selective pressure of the co-evolutionary phage-bacteria interaction process [55]. Here, we report the first identification of GH19 sequences in AAPB-isolated phages, highlighting the dynamic nature of phage genomes and their ability to acquire genes from their bacterial hosts.
AMGs are genes carried by phages that encode proteins involved in host metabolic pathways, and these genes are believed to have been acquired from their hosts or other phages by horizontal gene transfer [58]. Studies have shown that AMGs can enhance viral replication and influence the metabolic processes of their hosts [59,60]. In our study, GapR (ORF28) was identified for the first time in the phage genome. GapR is typically a chromosome structuring protein that plays a crucial role in bacterial DNA replication [61,62]. Specifically, GapR forms a dimer-of-dimers that fully encircles overtwisted DNA, stimulating gyrase and topo IV to relax positive supercoils caused by the DNA unwinding during replication [61]. Interestingly, GapR homologs with high similarity to that of R34L1 have also been identified in the genomes of several Erythrobacter strains [63,64,65], suggesting that our phages may have acquired the GapR gene during a specific infection event.
Theoretically, variations in the host range of the tailed phages are frequently associated with the receptor-binding proteins (RBP) located at the distal end of the tail, including tail spikes, extended tail fibers, and central tail spike proteins, which are responsible for host recognition and adsorption [66,67,68]. In the amino acid sequence of all predicted function ORFs, seven point mutations were detected (Figure 8), including tail-related protein (ORF 37 of phage R34L1 and ORF 31 of phage R34L2), tip attachment protein J domain-containing protein (ORF 33 and ORF 39), phage tail length tape measure family protein (ORF 38 and ORF 44), minor tail protein Z (ORF 43 and ORF 49) and portal protein (ORF 49 and ORF 55). Additionally, two ORFs are likely associated with host adsorption: the tail fiber protein (ORF 32 and ORF 38) and the tip attachment protein J domain-containing protein [69]. The amino acid sequence similarity between the tail fiber proteins of the two phages is 100%, while the similarity between the tip attachment protein J domain-containing proteins is 99.87%. Notably, the point mutation in tip attachment protein J domain-containing protein occurs in the C terminus (Figure 8), which is known to play a significant role in determining the host range [66]. Point mutations in RBPs have been shown to lead to host range expansion in phages, by altering their ability to recognize and bind to specific bacterial receptors [70]. In our study, the tail fiber protein of phages R34L1 and R34L2 exhibited low sequence identity (48.18%) and query cover (26.51%) when compared to the adsorption-related protein (tail fiber protein, ORF 25) of phage L02. In contrast, no significant similarity was observed with the corresponding protein of phage R6L. These findings provide more evidence for the differences in host range and highlight the diversity among Erythrobacter phages. However, further investigation is needed to elucidate the factors determining the host ranges of Erythrobacter phages.
4. Materials and Methods
4.1. Isolation and Propagation of Phages
Erythrobacter sp. JL 475, isolated from seawater samples in the South China Sea at a depth of 75 m (20.00 °N, 112.00 °E) [10], was used as a host to detect phages and evaluate their physical features. All of the bacterial strains used in this study are listed in Table S1. RO medium (containing 1 g/L yeast extract, 1 g/L tryptone, and 1 g/L sodium acetate, pH 7.5) was used for bacterial cultivation [71].
Seawater samples used to isolate Erythrobacter phages were collected separately at the coast of Zhoushan and Qingdao, China, in September 2013 and February 2014, by filtering through a 0.22 μm filter membrane (Millipore, USA), respectively. Twenty milliliters of filtrate was added to 100 mL of Erythrobacter sp. JL 475 culture (mid-log phase) and incubated overnight at 30°C. The mixture was then filtered to remove bacterial cells. The filtrate was subsequently diluted serially to isolate phages using the double-agar layer method [72]. Each single plaque was purified at least three times using sodium chloride-magnesium sulfate (SM) buffer (100 mM NaCl, 50 mM Tris, 10 mM MgSO4, and 0.01% gelatin, pH 7.5) with several drops of chloroform and stored at -80°C.
For phage propagation, the phage suspension was added to the host cultures (mid-log phase) at a multiplicity of infection (MOI) of 10. The mixture was then incubated at 30°C for 12 hours with shaking. Afterward, the sample was centrifuged at 6,000×g for 10 min and filtered to remove bacterial cells. Then, the filtrate was treated with 10% (w/v) Polyethylene glycol (PEG) 8000 (containing 1 M NaCl) and incubated at 4°C overnight. The phage particles were subsequently concentrated using CsCl density gradient ultracentrifugation [73]. Finally, the purified phage particles were dialyzed twice in SM buffer to remove CsCl.
4.2. Transmission Electron Microscopy (TEM)
Phage morphology was analyzed using transmission electron microscopy (TEM). The purified phage suspension was placed on a copper grid and negatively stained with 2% uranyl acetate for 10 min. Then the grid was examined under a 120 kV TEM (JEM-2100HC transmission electron microscope, JEOL, Japan). The isolated phages were identified and classified according to the guidelines of the International Committee on Taxonomy of Viruses [74].
4.3. Chloroform Sensitivity
Filtered lysate (~109 plaque-forming units (PFU)/mL) was mixed with chloroform at a final concentration of 0%, 1%, 10%, and 50%, separately. Each mixture was then vigorously shaken for 2 minutes. After incubation at 30°C for 30 minutes, the samples were diluted and plated for phage titration using the double-layer agar method.
4.4. Host Range Test
The lytic spectra of phages were determined using the double-layer agar method. The host range of isolated phages was evaluated against 40 bacterial strains, including 20 Erythrobacter strains, 17 Citromicrobium strains, one Dinoroseobacter strain, and one Roseobacter strain. The testing was performed by spotting serial dilutions of the phage suspensions (~107, ~108, ~109 PFU/mL) onto double-layer agar plates, that had been previously inoculated with the potential host strains. The plates were then incubated at 30°C for at least 24 h. After incubation, the presence of phage plaques on the bacterial lawns was examined to determine the host range of the isolated phages.
4.5. One-step Growth Curve
A one-step growth curve was performed to determine the burst size and latent period of the isolated phages, following previously described methods [72,75]. Briefly, each phage was inoculated separately into 100 mL of the host culture (optical density at 600 nm = 0.3~0.5) at a MOI of 0.1. The mixture was incubated at 30°C for 10 min for phage adsorption. After incubation, the mixture was centrifuged at 8,000 × g for 5 min. The cell pellet was resuspended in RO medium and cultured at 30°C. The phage titer was measured every 20 min for a total of 220 minutes post-infection. Triplicate samples were serially diluted and titrated by the double-layer plaque assay method [72]. The latent period and burst size were calculated based on the ratio between the phage count at the post-burst plateau and the initial phage count [76].
4.6. Stability Characterization
To determine the effect of pH on phage stability, the isolated phages were diluted in SM buffer at different pH values (ranging from pH 5 to 9). The pH was adjusted and stabilized using 5 M HCl, 0.2 M Na₂HPO₄/NaH₂PO₄ buffer, or 5 M NaOH. The diluted phages were then incubated at 30°C for 24 hours. The thermal tolerance of the isolated phages was tested by incubating the phages in SM buffer at various temperatures (4°C, 8°C, 12°C, 16°C, 20°C, 24°C, 30°C, 32°C, 36°C, and 40°C) for 24 hours. After incubation, the phage suspensions were cooled to 4°C to estimate phage activity. The salinity stability of the isolated phages was assessed by incubating the phages in mixtures of sterile seawater (34‰) and sterile freshwater for 24 hours, with varying percentages (0% to 100%) of seawater. Phage activity was determined using the double-layer plaque assay method [72]. All the experiments were repeated at least three times.
4.7. Phage DNA Extraction, Genome Sequencing and Bioinformatics
Phage DNA was extracted using the phenol/chloroform DNA extraction method. Briefly, the CsCl-purified phage was first treated with DNase I (1 μg/mL) and Rnase A (1 μg/mL) for 30 min at 37°C to remove free DNA and RNA. Subsequently, the mixture was treated with 100 μg/mL Proteinase K and 10% SDS for 2 h at 56°C. Then, one volume of phenol:isoamyl alcohol (25:24:1) was added and centrifuged for 10 minutes at 12,000 rpm. The aqueous layer was collected and extracted with an equal volume of chloroform:isoamyl alcohol (24:1). After centrifugation for 10 min at 12,000 rpm, DNA was precipitated by adding isopropanol (1:1) and sodium acetate (10:1) for 1 h at -20°C. The mixture was then centrifuged for 10 minutes at 12,000 rpm to pellet the DNA. Next, the DNA pellet was washed twice with 70% and once with 100% ethanol. Finally, the DNA was dried and resuspended in 50 μL 10 mM Tris (pH=8.0), and concentration was measured using a Qubit fluorimeter (Life Technologies, USA).
The phage genomes were sequenced using the Illumina Miseq platform with 2 × 250 bp paired-end reads. The raw reads were assembled using CLC Genome Workbench software (43 × coverage). The quality and completeness of the assembled genomes were determined by CheckV with default parameters (Nayfach et al., 2021). The putative open reading frames (ORFs) were predicted with GeneMarkS online server (http://exon.gatech.edu/Genemark/genemarks.cgi) [77], Glimmer 3.0 (http://ccb.jhu.edu/software/glimmer/index.shtml) [78], and ORF Finder online server (https://www.ncbi.nlm.nih.gov/orffinder/). The predicted ORFs were then annotated against the GenomeNet nr-aa database (a non-redundant protein sequence database merging sequences from RefSeq, SwissProt, TrEMBL, and GenPept), with a cut-off E-value of 10-5 [79]. The final annotation of the ORFs was manually verified to ensure accuracy. The tRNA was identified using tRNAscan-SE 2.0 (http://lowelab.ucsc.edu/tRNAscan-SE/) [32,33]. The genomic structures of the two isolated phages were conducted using CGView-Circular Genome Viewer (https://proksee.ca) [80]. The sequence data of the isolated phages R34L1 and R34L2 have been deposited in the GenBank databases under access No. PQ394074 and PQ394075, separately.
4.8. Phylogenetic Analysis
Viral Proteomic Tree server (ViPTree, https://www.genome.jp/viptree/) was used to employed to generate a proteomic tree based on genome-wide sequence similarities, computed by tBLASTx [79,81]. Subsequently, the average nucleotide identity by orthology (OrthoANI) values were performed by the OAT software V0.93 [82]. The phage genome similarities were calculated using the VIRIDIC tool, which employs BLASTn with default parameters (https://rhea.icbm.uni-oldenburg.de/VIRIDIC/) [83]. The taxonomy position of the isolated phages was further investigated with Virus Classification and Tree Building Online Resource (VICTOR; https://ggdc.dsmz.de/victor.php) [84], employing the Genome-BLAST Distance Phylogeny (GBDP) method under settings recommended for prokaryotic viruses [85]. BLASTx was employed to assess the similarity of ORFs among phages R34L1 and R34L2 and their respective closest phages, with a cut-off E-value of 10-5 [86].
The neighbor-joining phylogenetic trees were constructed using glycoside hydrolase family 19 (GH19), major capsid protein, and portal protein. The seed amino acid sequences of GH19 used for phylogenetic analysis were retrieved from NCBI non-redundant protein database (https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/) and the Protein Data Bank (PDB, https://www.rcsb.org/) with a maximal E-value of 10-10 based on Orlando et al. [55]. Amino acid sequences of orthologs of the major capsid protein and portal protein were retrieved from NCBI by BLASTp with the nr protein database. Sequence alignments and phylogenetic analyses were performed using MEGA X software with 1000 bootstrap replications to assess the robustness of the tree topology [87].
4.9. Recruitment of Reads to Metagenomic Data
To evaluate the distribution of phages R34L1 and R34L2 in different marine environments, the genome of isolated phage was mapped to the Global Ocean Viromes 2.0 (GOV 2.0) [88] using minimap2 (2.17-r941) [89]. The relative abundance of the phages was compared using Transformed Per Million (TPM) mapped reads [90]. To ensure the accuracy of the analysis, several widely distributed representative phages were included as references, namely Pelagibacter phages HTVC010P, HTVC019P, and HTVC011P, and Cyanophages P-SSP7, P-SSM7, P-HM1, P-HM2, P-RSM4, S-SSM7, S-B28, and S-B05. The relative abundance of these phages was calculated using CoverM (v0.3.1) [35] and visualized by R packages through the OmicStudio platform [91].
5. Conclusions
As an important AAPB genus in the ocean, Erythrobacter plays a significant role in the oceanic carbon cycle. In this study, we isolated and characterized two novel Erythrobacter phages, R34L1 and its sub-strain R34L2, marking significant advancements in our understanding of this understudied phage group. Notably, we identified the GapR gene as an AMG within phage genomes for the first time and discovered a homolog of the GH19 family sequence in AAPB-isolated phages—a finding previously unreported in this context. Taxonomic and genomic analyses indicated that phages R34L1 and R34L2 share a distant evolutionary relationship with known phages and form a distinct viral genus cluster within the family Casjensviridae, for which we propose to name Eausmariqdvirus. Ecological distribution analysis revealed that both R34L1 and R34L2 exclusively prefer the temperate and tropical epipelagic zones of the ocean, suggesting niche-specific adaptations. Overall, this study provides more information on the poorly understood Erythrobacter phages and deepens our understanding of the phage-host interactions in complex environments.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1: Host range of the phage vB_EauS-R34L1 and vB_EauS-R34L2; Table S2: Genome annotation of phage vB_EauS-R34L1; Table S3: Genome annotation of phage vB_EauS-R34L2; Table S4: ORF similarity comparison between Phages vB_EauS-R34L1 and vB_EauS-R34L2.
Author Contributions
Conceptualization, R.Z.; methodology, L.L.; software, X.L.; validation, L.L. and X.L.; formal analysis, X. L., L.L., and W.S.; investigation, L.L.; resources, L.L.; data curation, X.L.; writing—original draft preparation, L.L. and X. L.; writing—review and editing, L.L., X.L., and Y.Y.; visualization, X.L.; supervision, N.J. and R.Z.; project administration, R.Z.; funding acquisition, N.J, L.L., and R.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (grant number 42188102), the Science and technology plan project of Beihai City (grant number 201995076), the Guangdong Major Project of Basic and Applied Basic Research (grant number 2023B0303000017), the Innovation Team Project of Universities in Guangdong Province (grant number 2023KCXTD028).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The genome sequences of phage vB_EauS-R34L1 and vB_EauS-R34L2 were deposited in the GenBank databases under access No. PQ394074 and PQ394075, separately.
Acknowledgments
We thank Prof. Yantao Liang and his laboratory at the College of Marine Life Sciences, Ocean University of China, for analyzing the recruitment of reads to metagenomic data.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kolber, Z.S.; Plumley, F.G.; Lang, A.S.; Beatty, J.T.; Blankenship, R.E.; VanDover, C.L.; Vetriani, C.; Koblizek, M.; Rathgeber, C.; Falkowski, P.G. Contribution of aerobic photoheterotrophic bacteria to the carbon cycle in the ocean. Science 2001, 292, 2492–2495. [Google Scholar] [CrossRef]
- Eiler, A. Evidence for the ubiquity of mixotrophic bacteria in the upper ocean: implications and consequences. Appl Environ Microbiol 2006, 72, 7431–7437. [Google Scholar] [CrossRef]
- Jiao, N.; Zhang, Y.; Zeng, Y.; Hong, N.; Liu, R.; Chen, F.; Wang, P. Distinct distribution pattern of abundance and diversity of aerobic anoxygenic phototrophic bacteria in the global ocean. Environ Microbiol 2007, 9, 3091–3099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, J.; Qiang, L.; Zou, L.; Zhang, Y. The research of typical microbial functional group reveals a new oceanic carbon sequestration mechanism—A case of innovative method promoting scientific discovery. Sci China Earth Sci 2016, 59, 456–463. [Google Scholar] [CrossRef]
- Shiba, T.; Simidu, U. Erythrobacter longus gen. Nov., sp. nov., an aerobic bacterium which contains bacteriochlorophyll a. Int J Syst Bacteriol 1982, 32, 211–217. [Google Scholar] [CrossRef]
- Yurkov, V.; Stackebrandt, E.; Holmes, A.; Fuerst, J.A.; Hugenholtz, P.; Golecki, J.; Gad'on, N.; Gorlenko, V.M.; Kompantseva, E.I.; Drews, G. Phylogenetic positions of novel aerobic, bacteriochlorophyll a-containing bacteria and description of Roseococcus thiosulfatophilus gen. nov., sp. nov., Erythromicrobium ramosum gen. nov., sp. nov., and Erythrobacter litoralis sp. nov. Int J Syst Bacteriol 1994, 44, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Zhang, H.; Chen, Y.; Li, Y.; Chen, Z.; Lai, Q.; Zhang, J.; Zheng, W.; Xu, H.; Zheng, T. Erythrobacter luteus sp. nov., isolated from mangrove sediment. Int J Syst Evol Microbiol 2015, 65, 2472–2478. [Google Scholar] [CrossRef]
- Jeong, S.W.; Yang, J.E.; Choi, Y.J. Isolation and characterization of a yellow xanthophyll pigment-producing marine bacterium, Erythrobacter sp. SDW2 strain, in coastal seawater. Marine Drugs 2022, 20, 73. [Google Scholar] [CrossRef]
- Zhuang, L.; Liu, Y.; Wang, L.; Wang, W.; Shao, Z. Erythrobacter atlanticus sp. nov., a bacterium from ocean sediment able to degrade polycyclic aromatic hydrocarbons. Int J Syst Evol Microbiol 2015, 65, 3714–3719. [Google Scholar] [CrossRef]
- Zheng, Q.; Lin, W.; Liu, Y.; Chen, C.; Jiao, N. A Comparison of 14 Erythrobacter genomes provides insights into the genomic divergence and scattered distribution of phototrophs. Front Microbiol 2016, 7, 984. [Google Scholar] [CrossRef]
- RoLing, W.F.; Milner, M.G.; Jones, D.M.; Lee, K.; Daniel, F.; Swannell, R.J.; Head, I.M. Robust hydrocarbon degradation and dynamics of bacterial communities during nutrient-enhanced oil spill bioremediation. Appl Environ Microb 2002, 68, 5537–5548. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, Y.S.; Alrumman, S.A.; Otaif, K.A.; Alamri, S.A.; Mostafa, M.S.; Sahlabji, T. Production and characterization of bioplastic by polyhydroxybutyrate accumulating Erythrobacter aquimaris isolated from mangrove rhizosphere. Molecules 2020, 25, 179. [Google Scholar] [CrossRef] [PubMed]
- Koblížek, M.; Béjà, O.; Bidigare, R.R.; Christensen, S.; Benitez-Nelson, B.; Vetriani, C.; Kolber, M.K.; Falkowski, P.G.; Kolber, Z.S. Isolation and characterization of Erythrobacter sp. strains from the upper ocean. Arch Microbiol 2003, 180, 327–338. [Google Scholar] [CrossRef]
- Wei, J.; Mao, Y.; Zheng, Q.; Zhang, R.; Wang, Y.-N. Erythrobacter westpacificensis sp. nov., a marine bacterium isolated from the Western Pacific. Curr Microbiol 2013, 66, 385–390. [Google Scholar] [CrossRef]
- Fang, C.; Wu, Y.; Sun, C.; Wang, H.; Cheng, H.; Meng, F.; Wang, C.; Xu, X. Erythrobacter zhengii sp. nov., a bacterium isolated from deep-sea sediment. Int J Syst Evol 2019, 69, 241–248. [Google Scholar] [CrossRef]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS microbiol rev 2004, 28, 127–181. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—major players in the global ecosystem. Nat rev microbiol 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Jiao, N.; Robinson, C.; Azam, F.; Thomas, H.; Baltar, F.; Dang, H.; Hardman-Mountford, N.J.; Johnson, M.; Kirchman, D.L.; Koch, B.P. Mechanisms of microbial carbon sequestration in the ocean—future research directions. Biogeosciences 2014, 11, 5285–5306. [Google Scholar] [CrossRef]
- Huang, X.; Lu, L.; Yu, C. Isolation and characterization of a roseophage representing a novel genus in the N4-like Rhodovirinae subfamily distributed in estuarine waters. bioRxiv 2024, 2024–10. [Google Scholar] [CrossRef]
- Zhan, Y.; Chen, F. Bacteriophages that infect marine roseobacters: genomics and ecology. Environ microbiol 2019, 21, 1885–1895. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, Y.; Chen, F.; Jiao, N. Complete genome sequence of a marine roseophage provides evidence into the evolution of gene transfer agents in alphaproteobacteria. Virol J 2011, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Proctor, L.M.; Fuhrman, J.A. Viral mortality of marine bacteria and cyanobacteria. Nature 1990, 343, 60–62. [Google Scholar] [CrossRef]
- Yang, M.; Xia, Q.; Du, S.; Zhang, Z.; Qin, F.; Zhao, Y. Genomic characterization and distribution pattern of a novel marine OM43 phage. Front Microbiol 2021, 12, 651326. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Cai, L.; Jiao, N.; Zhang, R. Isolation and characterization of the first phage infecting ecologically important marine bacteria Erythrobacter. Virol J 2017, 14, 104. [Google Scholar] [CrossRef]
- Budinoff, C.R. Diversity and activity of roseobacters and roseophage, Doctor, University of Tennessee, Knoxville, May 2012.
- Li, X.; Guo, R.; Zou, X.; Yao, Y.; Lu, L. The first cbk-like phage infecting Erythrobacter, representing a novel siphoviral genus. Front Microbiol 2022, 13, 861793. [Google Scholar] [CrossRef]
- Espejo, R.T.; Canelo, E.S. Properties of bacteriophage PM2: a lipid-containing bacterial virus. Virology 1968, 34, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, S.; Long, L.; Huang, S. Characterization and complete genome sequences of three N4-like roseobacter phages isolated from the South China Sea. Curr microbiol 2016, 73, 409–418. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, K.; Jiao, N.; Chen, F. Genome sequences of two novel phages infecting marine roseobacters. Environ Microbiol 2009, 11, 2055–2064. [Google Scholar] [CrossRef]
- Cai, L.; Ma, R.; Chen, H.; Yang, Y.; Jiao, N.; Zhang, R. A newly isolated roseophage represents a distinct member of Siphoviridae family. Virol J 2019, 16, 1–9. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: searching for tRNA genes in genomic sequences. In Gene prediction. Methods in Molecular Biology; Kollmar, M. Eds, Ed.; Humana: New York, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, Y.; Zheng, K.; Wang, Z.; Dong, Y.; Liu, Y.; Ren, L.; Wang, H.; Han, Y.; McMinn, A. Characterization and genomic analysis of phage vB_ValR_NF, representing a new viral family prevalent in the Ulva prolifera blooms. Front Microbiol 2023, 14, 1161265. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, K.; Wang, M.; Ma, K.; Ren, L.; Guo, R.; Ma, L.; Zhang, H.; Liu, Y.; Xiong, Y. Shewanella phage encoding a putative anti-CRISPR-like gene represents a novel potential viral family. Microbiol Spectr 2024, 12, e03367–e03323. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ren, L.; Liang, Y.; Zheng, K.; Guo, R.; Liu, Y.; Wang, Z.; Han, Y.; Zhang, X.; Shao, H. Psychrobacter phage encoding an antibiotics resistance gene represents a novel caudoviral family. Microbiol Spectr 2023, 11, e05335–e05322. [Google Scholar] [CrossRef]
- Focardi, A.; Ostrowski, M.; Goossen, K.; Brown, M.V.; Paulsen, I. Investigating the diversity of marine bacteriophage in contrasting water masses associated with the East Australian Current (EAC) system. Viruses 2020, 12, 317. [Google Scholar] [CrossRef]
- Luo, E.; Aylward, F.O.; Mende, D.R.; DeLong, E.F. Bacteriophage distributions and temporal variability in the ocean’s interior. MBio 2017, 8, e01903. [Google Scholar] [CrossRef]
- Brown, T.L.; Charity, O.J.; Adriaenssens, E.M. Ecological and functional roles of bacteriophages in contrasting environments: marine, terrestrial and human gut. Curr Opin in Microbiol 2022, 70, 102229. [Google Scholar] [CrossRef]
- Sanz-Gaitero, M.; Seoane-Blanco, M.; van Raaij, M.J. Structure and function of bacteriophages. In Bacteriophages; Harper, D.R., Abedon, S.T., Burrowes, B.H., McConville, M.L., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2021; pp. 19–91. [Google Scholar] [CrossRef]
- Marceau, A.H. Functions of single-strand DNA-binding proteins in DNA replication, recombination, and repair. In Single-Stranded DNA Binding Proteins: Methods and Protocols; Keck, J., Ed.; Humana: Totowa, NJ, USA, 2012; Volume 922, pp. 1–21. [Google Scholar] [CrossRef]
- Lipps, G.; Weinzierl, A.O.; von Scheven, G.; Buchen, C.; Cramer, P. Structure of a bifunctional DNA primase-polymerase. Nat struct mol biol 2004, 11, 157–162. [Google Scholar] [CrossRef]
- Guo, H.; Li, M.; Wang, T.; Wu, H.; Zhou, H.; Xu, C.; Yu, F.; Liu, X.; He, J. Crystal structure and biochemical studies of the bifunctional DNA primase-polymerase from phage NrS-1. Biochem Bioph Res Co 2019, 510, 573–579. [Google Scholar] [CrossRef]
- Struck, A.W.; Thompson, M.L.; Wong, L.S.; Micklefield, J. S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem 2012, 13, 2642–2655. [Google Scholar] [CrossRef]
- Went, S.C.; Picton, D.M.; Morgan, R.D.; Nelson, A.; Brady, A.; Mariano, G.; Dryden, D.T.; Smith, D.L.; Wenner, N.; Hinton, J.C. Structure and rational engineering of the PglX methyltransferase and specificity factor for BREX phage defence. Nat Commun 2024, 15, 7236. [Google Scholar] [CrossRef]
- Isaev, A.; Drobiazko, A.; Sierro, N.; Gordeeva, J.; Yosef, I.; Qimron, U.; Ivanov, N.V.; Severinov, K. Phage T7 DNA mimic protein Ocr is a potent inhibitor of BREX defence. Nucleic Acids Res 2020, 48, 5397–5406. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, E.A.; Brooks, J.E. Restriction modification systems: where they are and what they do. In Bacterial genomes: physical structure and analysis; de Bruijn, F.J., Lupski, J.R., Weinstock, G.M., Eds.; Springer: Boston, MA, USA, 1998; pp. 78–92. [Google Scholar] [CrossRef]
- Maindola, P.; Raina, R.; Goyal, P.; Atmakuri, K.; Ojha, A.; Gupta, S.; Christie, P.J.; Iyer, L.M.; Aravind, L.; Arockiasamy, A. Multiple enzymatic activities of ParB/Srx superfamily mediate sexual conflict among conjugative plasmids. Nat commun 2014, 5, 5322. [Google Scholar] [CrossRef]
- Osorio-Valeriano, M.; Altegoer, F.; Steinchen, W.; Urban, S.; Liu, Y.; Bange, G.; Thanbichler, M. ParB-type DNA segregation proteins are CTP-dependent molecular switches. Cell 2019, 179, 1512–1524.e15. [Google Scholar] [CrossRef] [PubMed]
- Tanner, N.K.; Cordin, O.; Banroques, J.; Doère, M.; Linder, P. The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol cell 2003, 11, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Tuteja, R. Prokaryotic and eukaryotic DNA helicases: essential molecular motor proteins for cellular machinery. Eur J Biochem 2004, 271, 1835–1848. [Google Scholar] [CrossRef]
- Pennell, S.; Déclais, A.-C.; Li, J.; Haire, L.F.; Berg, W.; Saldanha, J.W.; Taylor, I.A.; Rouse, J.; Lilley, D.M.; Smerdon, S.J. FAN1 activity on asymmetric repair intermediates is mediated by an atypical monomeric virus-type replication-repair nuclease domain. Cell Rep 2014, 8, 84–93. [Google Scholar] [CrossRef]
- Elahi, Y.; Mazaheri Nezhad Fard, R.; Seifi, A.; Mahfouzi, S.; Saboor Yaraghi, A.A. Genome Analysis of the Enterococcus faecium Entfac.YE Prophage. Avicenna J Med Biotechnol 2022, 14, 54–60. [Google Scholar] [CrossRef]
- Orlando, M.; Buchholz, P.C.; Lotti, M.; Pleiss, J. The GH19 Engineering Database: Sequence diversity, substrate scope, and evolution in glycoside hydrolase family 19. PLoS One 2021, 16, e0256817. [Google Scholar] [CrossRef]
- Valero-Rello, A. Diversity, specificity and molecular evolution of the lytic arsenal of Pseudomonas phages: in silico perspective. Environ Microbiol 2019, 21, 4136–4150. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J Bacteriol 2002, 184, 4891–4905. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; U’Ren, J.M. Viral metabolic reprogramming in marine ecosystems. Curr Opin Microbiol 2016, 31, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.R.; Zeng, Q.; Kelly, L.; Huang, K.H.; Singer, A.U.; Stubbe, J.; Chisholm, S.W. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. PNAS 2011, 108, E757–E764. [Google Scholar] [CrossRef]
- Guo, M.S.; Haakonsen, D.L.; Zeng, W.; Schumacher, M.A.; Laub, M.T. A bacterial chromosome structuring protein binds overtwisted DNA to stimulate type II topoisomerases and enable DNA replication. Cell 2018, 175, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Tarry, M.J.; Harmel, C.; Taylor, J.A.; Marczynski, G.T.; Schmeing, T.M. Structures of GapR reveal a central channel which could accommodate B-DNA. Sci Rep 2019, 9, 16679. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Leung, P.M.; Bay, S.K.; Hugenholtz, P.; Kessler, A.J.; Shelley, G.; Waite, D.W.; Cook, P.L.; Greening, C. Metabolic flexibility allows generalist bacteria to become dominant in a frequently disturbed ecosystem. BioRxiv 2020, 2020–02. [Google Scholar] [CrossRef]
- Probst, A.J.; Elling, F.J.; Castelle, C.J.; Zhu, Q.; Elvert, M.; Birarda, G.; Holman, H.-Y.N.; Lane, K.R.; Ladd, B.; Ryan, M.C. Lipid analysis of CO2-rich subsurface aquifers suggests an autotrophy-based deep biosphere with lysolipids enriched in CPR bacteria. The ISME J 2020, 14, 1547–1560. [Google Scholar] [CrossRef]
- Aguirre, E.G.; Schwartzman, J.A. Metagenome-assembled genomes of Macrocystis-associated bacteria. Microbiol Resour Ann 2024, 13, e00715–e00724. [Google Scholar] [CrossRef]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering phage host-range and suppressing bacterial resistance through phage tail fiber mutagenesis. Cell 2019, 179, 459–469.e459. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, F.L.; Vlot, M.; De Jonge, P.A.; Dreesens, L.L.; Beaumont, H.J.; Lavigne, R.; Dutilh, B.E.; Brouns, S.J. Targeting mechanisms of tailed bacteriophages. Nat Rev Microbiol 2018, 16, 760–773. [Google Scholar] [CrossRef]
- de Jonge, P.A.; Nobrega, F.L.; Brouns, S.J.; Dutilh, B.E. Molecular and evolutionary determinants of bacteriophage host range. Trends Microbiol 2019, 27, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Wang, J. Structural mechanism of bacteriophage lambda tail’s interaction with the bacterial receptor. Nat Commun 2024, 15, 4185. [Google Scholar] [CrossRef]
- Subramanian, S.; Dover, J.A.; Parent, K.N.; Doore, S.M. Host range expansion of Shigella phage Sf6 evolves through point mutations in the tailspike. J Virol 2022, 96, e00929–e00922. [Google Scholar] [CrossRef]
- Yurkov, V.V.; Krieger, S.; Stackebrandt, E.; Beatty, J.T. Citromicrobium bathyomarinum, a novel aerobic bacterium isolated from deep-sea hydrothermal vent plume waters that contains photosynthetic pigment-protein complexes. J Bacteriol 1999, 181, 4517–4525. [Google Scholar] [CrossRef]
- Pajunen, M.; Kiljunen, S.; Skurnik, M. Bacteriophage phiYeO3-12, specific for Yersinia enterocolitica serotype O:3, is related to coliphages T3 and T7. J Bacteriol 2000, 182, 5114–5120. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Chen, Q.; Xu, Y.; Suttle, C.A.; Jiao, N. A virus infecting marine photoheterotrophic alphaproteobacteria (Citromicrobium spp.) defines a new lineage of ssDNA viruses. Front Microbiol 2018, 9, 1418. [Google Scholar] [CrossRef]
- Murphy, F.A.; Fauquet, C.M.; Bishop, D.H.; Ghabrial, S.A.; Jarvis, A.W.; Martelli, G.P.; Mayo, M.A.; Summers, M.D. Virus taxonomy: classification and nomenclature of viruses, 6th eds; Springer Science & Business Media: Berlin, Germany, 2012; pp. 1–49. [Google Scholar]
- Wu, L.T.; Chang, S.Y.; Yen, M.R.; Yang, T.C.; Tseng, Y.H. Characterization of extended-host-range pseudo-T-even bacteriophage Kpp95 isolated on Klebsiella pneumoniae. Appl Environ Microbiol 2007, 73, 2532–2540. [Google Scholar] [CrossRef]
- Yang, Y.; Cai, L.; Ma, R.; Xu, Y.; Tong, Y.; Huang, Y.; Jiao, N.; Zhang, R. A novel roseosiphophage isolated from the oligotrophic South China Sea. Viruses 2017, 9, 109. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: the viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Edwards, R. The Phage Proteomic Tree: a genome-based taxonomy for phage. J Bacteriol 2002, 184, 4529–4535. [Google Scholar] [CrossRef]
- Lee, I.; Ouk Kim, Y.; Park, S.-C.; Chun, J. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Micr 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC bioinformatics 2013, 14, 1–14. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zayed, A.A.; Conceição-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C. Marine DNA viral macro-and microdiversity from pole to pole. Cell 2019, 177, 1109–1123.e1114. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Vera Alvarez, R.; Pongor, L.S.; Mariño-Ramírez, L.; Landsman, D. TPMCalculator: one-step software to quantify mRNA abundance of genomic features. Bioinformatics 2019, 35, 1960–1962. [Google Scholar] [CrossRef]
- Lyu, F.; Han, F.; Ge, C.; Mao, W.; Chen, L.; Hu, H.; Chen, G.; Lang, Q.; Fang, C. OmicStudio: A composable bioinformatics cloud platform with real-time feedback that can generate high-quality graphs for publication. Imeta 2023, 2. [Google Scholar] [CrossRef]
Figure 1.
Isolation and growth curve of phages vB_EauS-R34L1 and vB_EauS-R34L2. (A) Transmission electron microscopy (TEM) results. (B) One-step growth curve. Error bars indicate standard deviations among triplicate samples.
Figure 1.
Isolation and growth curve of phages vB_EauS-R34L1 and vB_EauS-R34L2. (A) Transmission electron microscopy (TEM) results. (B) One-step growth curve. Error bars indicate standard deviations among triplicate samples.
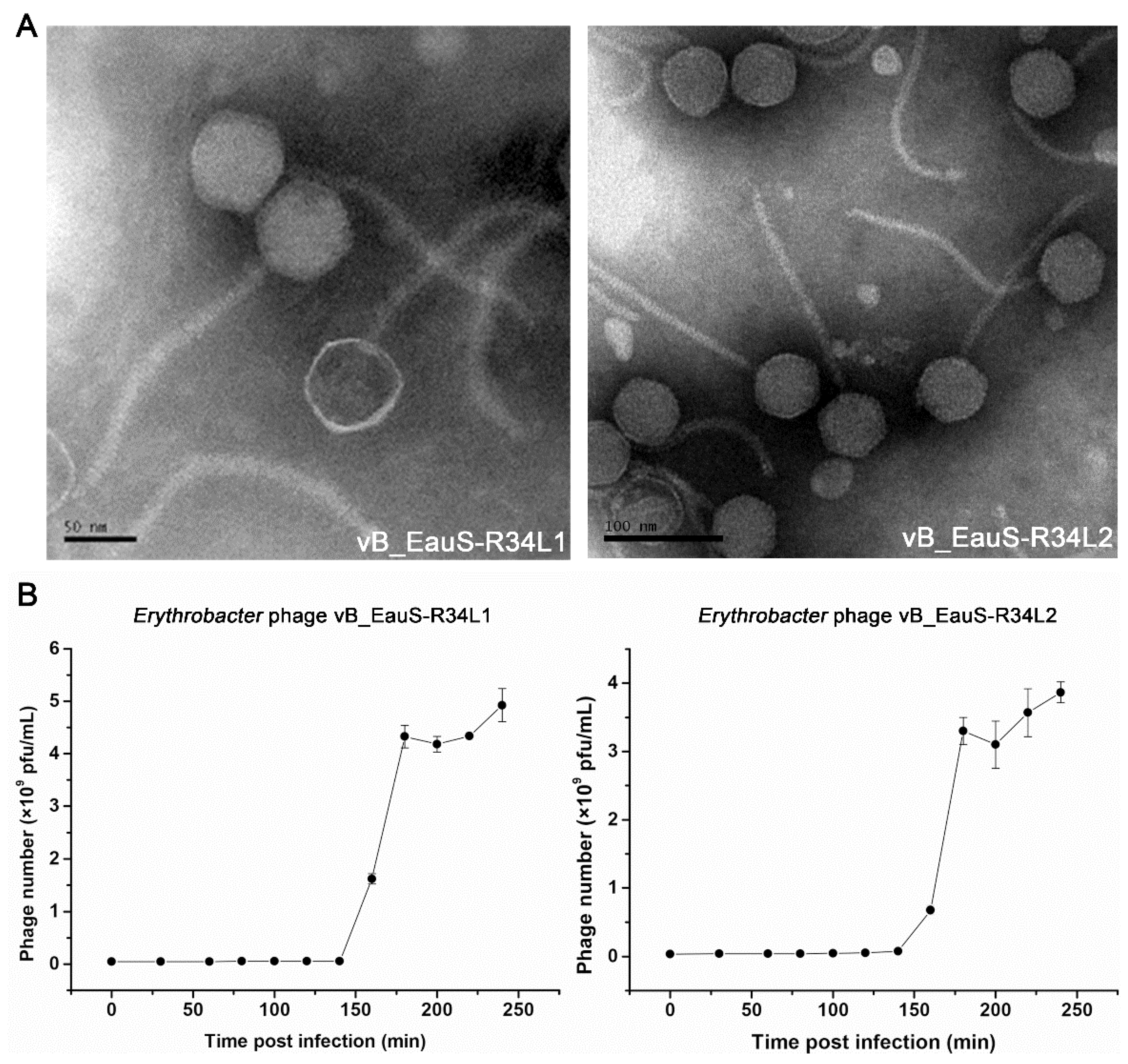
Figure 2.
Environmental stability of phages vB_EauS-R34L1 and vB_EauS-R34L2. (A) Thermal stability treated with different temperatures for 24 h. (B) pH stability treated with different pH for 24 h. (C) Salinity stability treated with different salinity conditions for 24 h. Error bars indicate standard deviations among triplicate samples. Significant differences (ANOVA test, p < 0.05) among different treatments are represented by different letters.
Figure 2.
Environmental stability of phages vB_EauS-R34L1 and vB_EauS-R34L2. (A) Thermal stability treated with different temperatures for 24 h. (B) pH stability treated with different pH for 24 h. (C) Salinity stability treated with different salinity conditions for 24 h. Error bars indicate standard deviations among triplicate samples. Significant differences (ANOVA test, p < 0.05) among different treatments are represented by different letters.
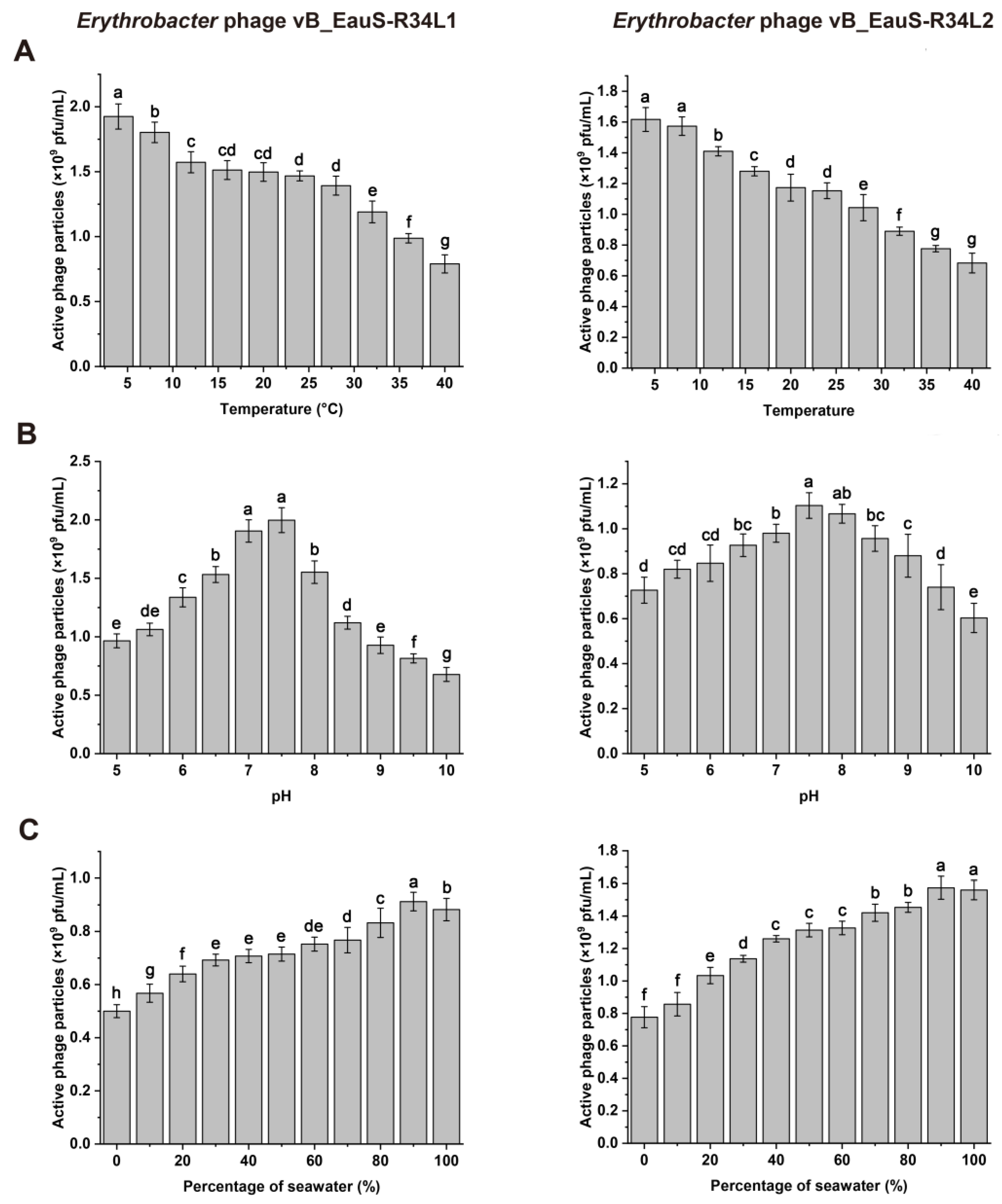
Figure 3.
Genomic structures of phages vB_EauS-R34L1 and vB_EauS-R34L2. The first and second outmost circles comprising arrows represent predicted ORFs. The third outmost circle represents the GC contents. The fourth outmost circle represents the GC-skewness values (GC-skew=(G-C)/(G +C)). The scale representing the genome size (bp) is displayed using the innermost circle.
Figure 3.
Genomic structures of phages vB_EauS-R34L1 and vB_EauS-R34L2. The first and second outmost circles comprising arrows represent predicted ORFs. The third outmost circle represents the GC contents. The fourth outmost circle represents the GC-skewness values (GC-skew=(G-C)/(G +C)). The scale representing the genome size (bp) is displayed using the innermost circle.
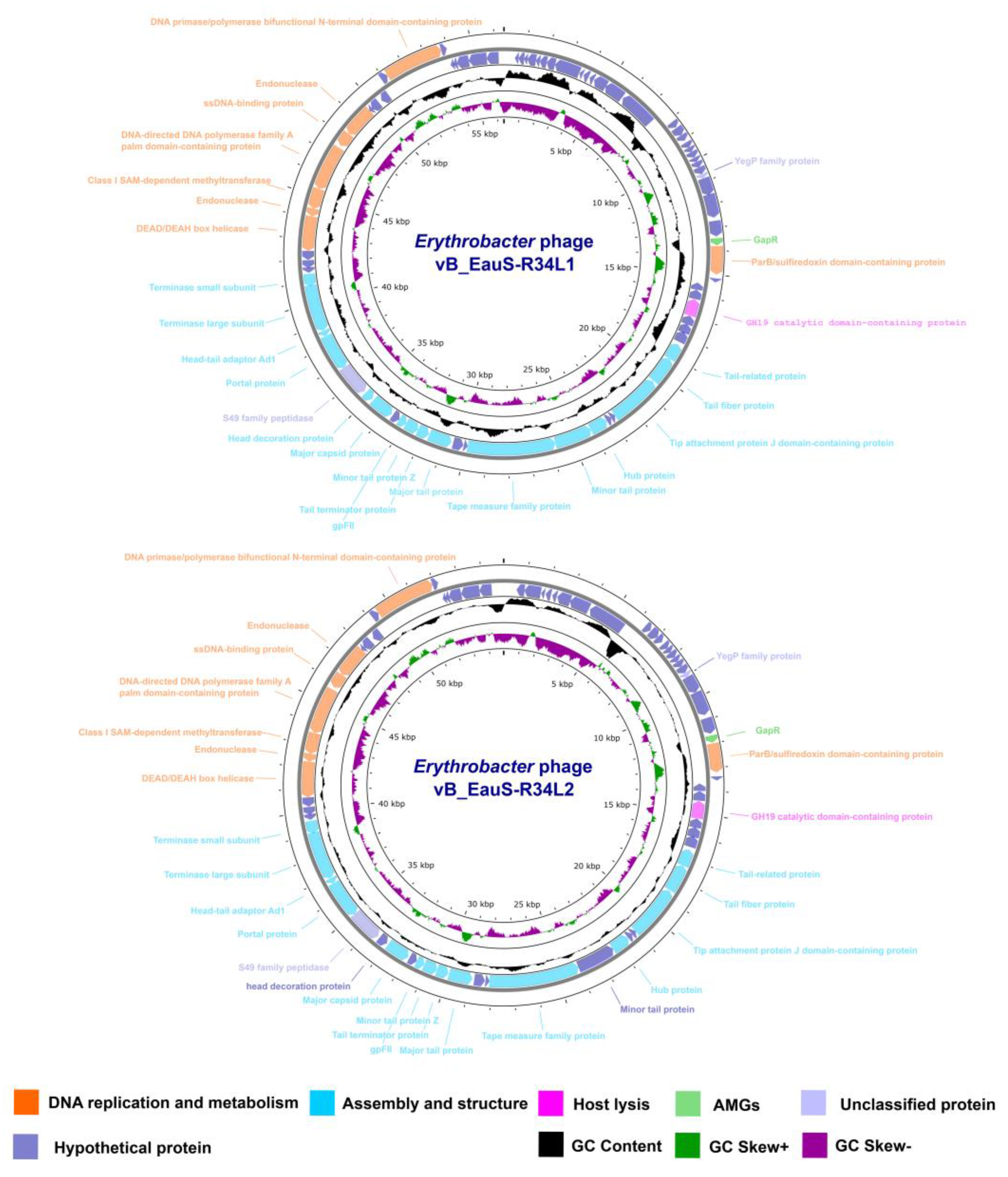
Figure 4.
Phylogenetic and genomic comparison analysis. (A) Determination of taxa and host group of phages vB_EauS-R34L1 and vB_EauS-R34L2 by a proteomic tree using ViPTree server. The colored rings represent the virus family (inner ring) and host group (outer ring). The red star marks the position of phages vB_EauS-R34L1 and vB_EauS-R34L2, and the green star marks the position of phages vB_ElisS-R6L and vB_EliS-L02. (B) Phylogenetic relationship of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. The left and right color bars indicate the taxonomic virus family and host group, respectively. (C) Heatmap showing OrthoANI values of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. (D) The intergenomic comparison of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. The heatmap generated by VICDIC shows the intergenomic similarity values (right half) and alignment indicators (left half).
Figure 4.
Phylogenetic and genomic comparison analysis. (A) Determination of taxa and host group of phages vB_EauS-R34L1 and vB_EauS-R34L2 by a proteomic tree using ViPTree server. The colored rings represent the virus family (inner ring) and host group (outer ring). The red star marks the position of phages vB_EauS-R34L1 and vB_EauS-R34L2, and the green star marks the position of phages vB_ElisS-R6L and vB_EliS-L02. (B) Phylogenetic relationship of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. The left and right color bars indicate the taxonomic virus family and host group, respectively. (C) Heatmap showing OrthoANI values of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. (D) The intergenomic comparison of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. The heatmap generated by VICDIC shows the intergenomic similarity values (right half) and alignment indicators (left half).
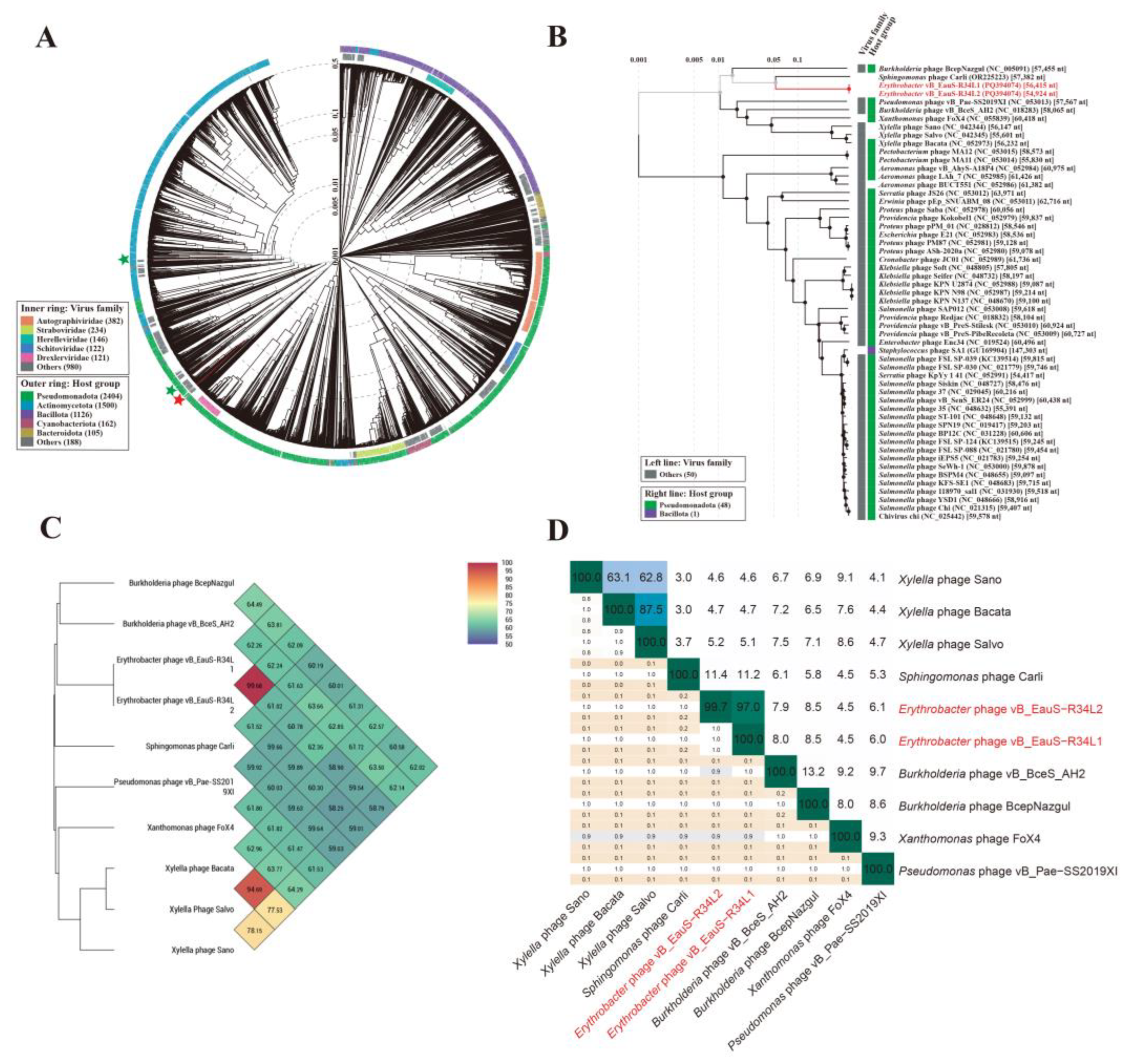
Figure 5.
Phylogenetic analysis based on whole genome and conserved proteins. (A) Phylogenetic analysis of whole-genome sequences of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. Annotations, including species, genus, and family cluster were predicted by VICTOR, with different colors, which are different in classification. (B) Genomic comparison between Erythrobacter phages vB_EauS-R34L1, vB_EauS-R34L2, vB_EliS-R6L and vB_EliS-L02, Sphingomonas phage Carli, and Burkholderia phage BcepNazgul. Arrows present the ORFs. The direction of each arrow represents the direction of transcription. Genome regions showing similarity were searched using tBLASTX and matches satisfying length and E-value (<10-5) cutoffs were indicated by the rectangle according to the color scale on the top.
Figure 5.
Phylogenetic analysis based on whole genome and conserved proteins. (A) Phylogenetic analysis of whole-genome sequences of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. Annotations, including species, genus, and family cluster were predicted by VICTOR, with different colors, which are different in classification. (B) Genomic comparison between Erythrobacter phages vB_EauS-R34L1, vB_EauS-R34L2, vB_EliS-R6L and vB_EliS-L02, Sphingomonas phage Carli, and Burkholderia phage BcepNazgul. Arrows present the ORFs. The direction of each arrow represents the direction of transcription. Genome regions showing similarity were searched using tBLASTX and matches satisfying length and E-value (<10-5) cutoffs were indicated by the rectangle according to the color scale on the top.
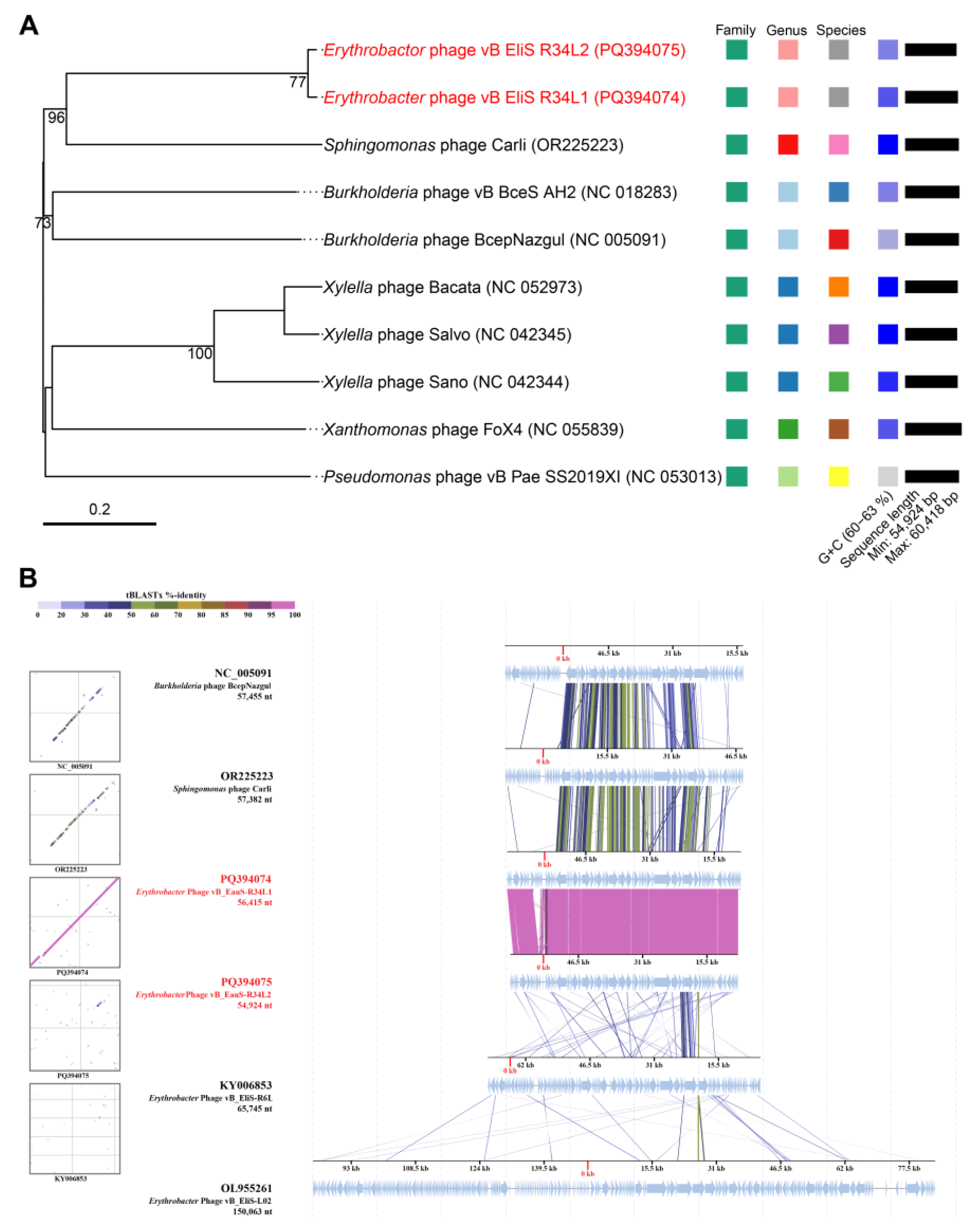
Figure 6.
The neighbor-joining phylogenetic tree based on amino acid sequences. (A) Phylogenetic analysis using glycoside hydrolase family 19 catalytic domain-containing protein of phages vB_EauS-R34L1 and vB_EauS-R34L2 with seed sequences retrieved from NCBI non-redundant protein database and Protein Data Bank. (B) Phylogenetic analysis using the portal protein of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. (C) Phylogenetic analysis using major capsid protein of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. Bootstrap values were based on 1000 replicates.
Figure 6.
The neighbor-joining phylogenetic tree based on amino acid sequences. (A) Phylogenetic analysis using glycoside hydrolase family 19 catalytic domain-containing protein of phages vB_EauS-R34L1 and vB_EauS-R34L2 with seed sequences retrieved from NCBI non-redundant protein database and Protein Data Bank. (B) Phylogenetic analysis using the portal protein of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. (C) Phylogenetic analysis using major capsid protein of phages vB_EauS-R34L1 and vB_EauS-R34L2 with their closest relatives. Bootstrap values were based on 1000 replicates.

Figure 7.
Relative abundance of phages vB_EauS-R34L1 and vB_EauS-R34L2 compared to the abundances of oceanic representative phages. Relative abundances are expressed by TPM (transcripts per million) values and described with log10 transformation. Left, the heatmap of relative abundances of different bacteriophages in different viral ecological zones (VEZs). Right, distribution patterns of bacteriophages in five VEZs with TPM values normalized by the number of databases of each VEZ. BATHY, bathypelagic; MES, temperate and tropical mesopelagic; EPI, temperate and tropical epipelagic; ARC, Arctic; ANT, Antarctic.
Figure 7.
Relative abundance of phages vB_EauS-R34L1 and vB_EauS-R34L2 compared to the abundances of oceanic representative phages. Relative abundances are expressed by TPM (transcripts per million) values and described with log10 transformation. Left, the heatmap of relative abundances of different bacteriophages in different viral ecological zones (VEZs). Right, distribution patterns of bacteriophages in five VEZs with TPM values normalized by the number of databases of each VEZ. BATHY, bathypelagic; MES, temperate and tropical mesopelagic; EPI, temperate and tropical epipelagic; ARC, Arctic; ANT, Antarctic.
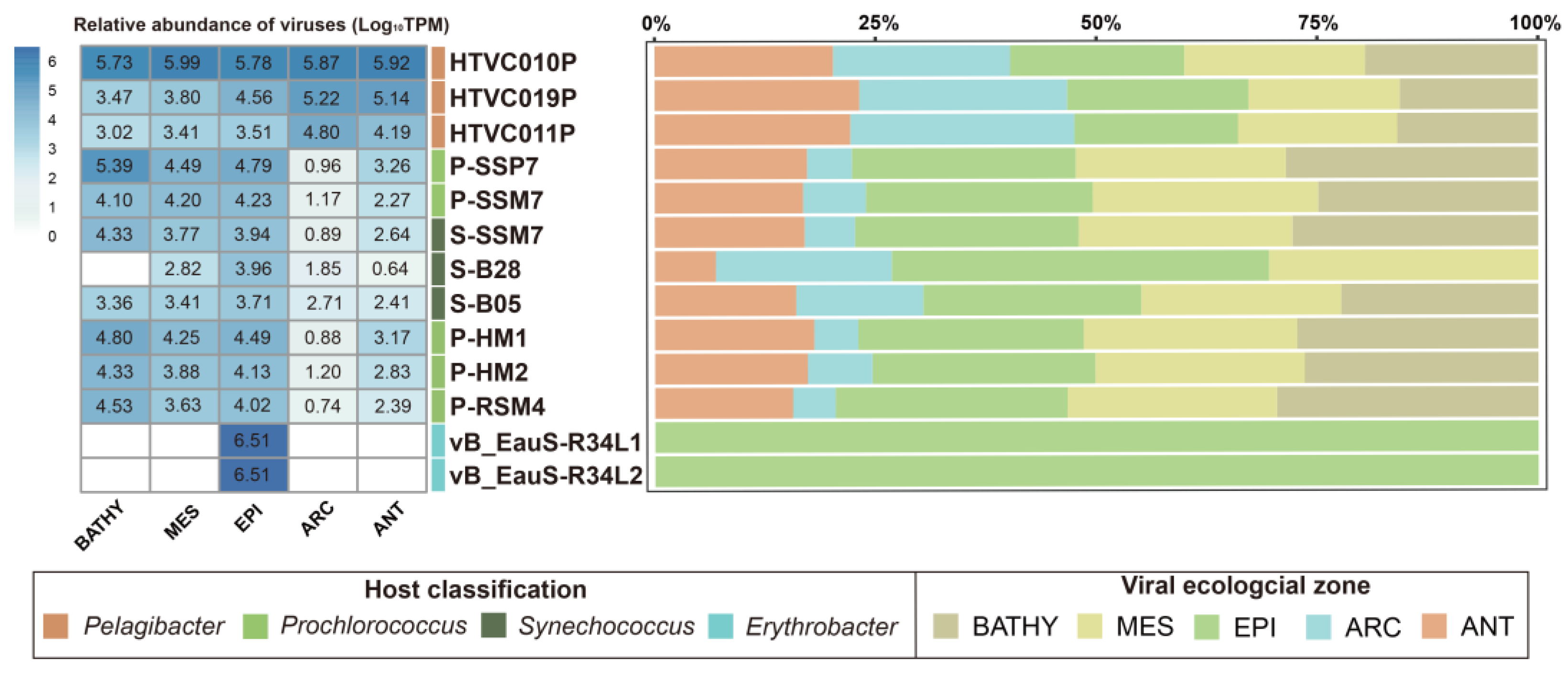
Figure 8.
The point mutations of phage tail-related genes between phages vB_EauS-R34L1 and vB_EauS-R34L2.
Figure 8.
The point mutations of phage tail-related genes between phages vB_EauS-R34L1 and vB_EauS-R34L2.
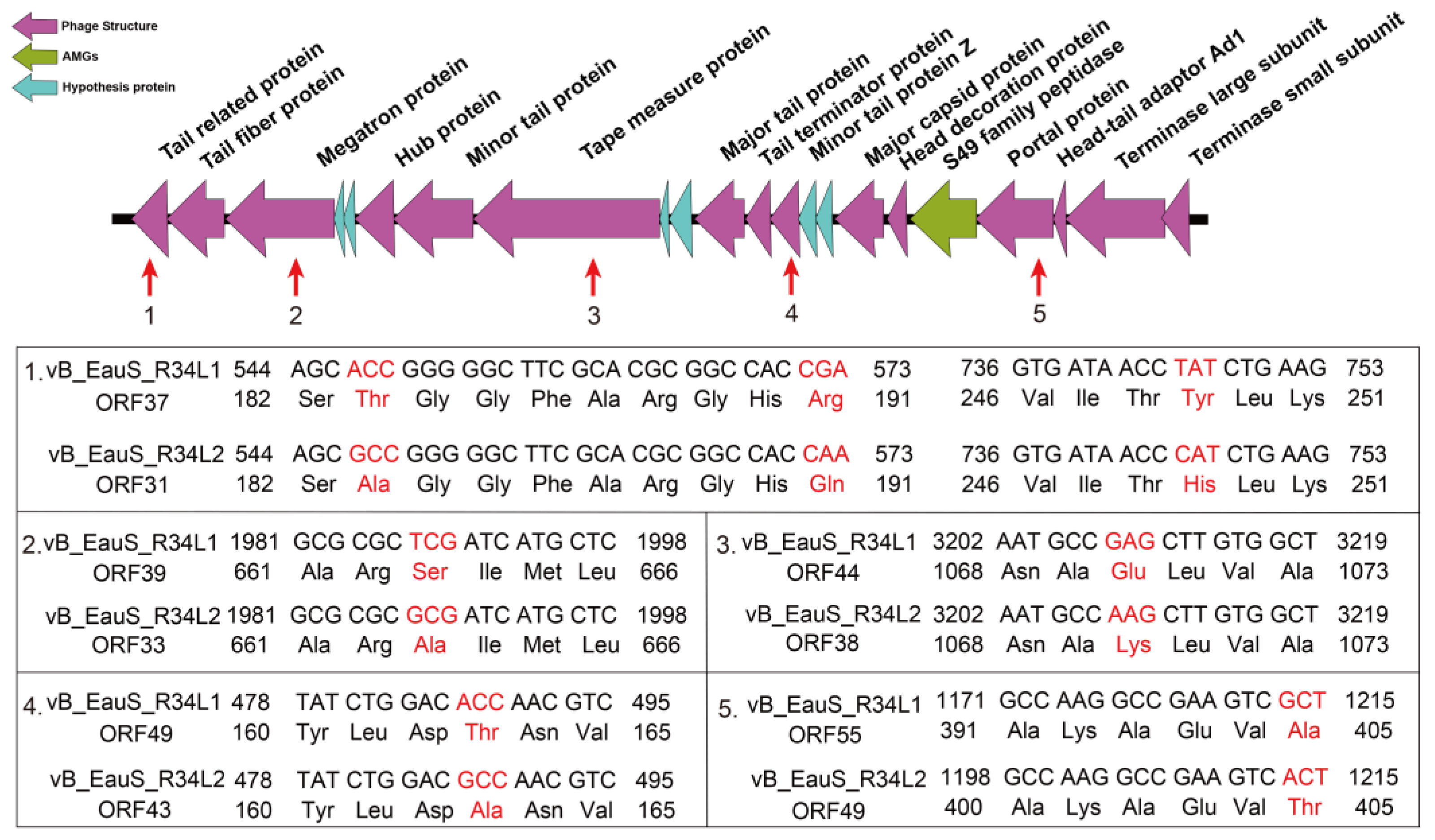

Table 1.
Genomic features of isolated Erythrobacter phages.
| Phage name | Genome size (bp) | G+C% | ORFs | tRNA | Accession no. | Reference |
|---|---|---|---|---|---|---|
| vB_EauS-R34L1 | 56,415 | 61.60 | 78 | 0 | PQ394074 | This study |
| vB_EauS-R34L2 | 54,924 | 61.19 | 72 | 0 | PQ394075 | This study |
| vB_EliS-R6L | 65,675 | 66.52 | 108 | 0 | KY006853 | [25] |
| vB_ElisS-L02 | 150,063 | 59.43 | 61 | 29 | OL955261 | [27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



