Submitted:
13 February 2025
Posted:
13 February 2025
Read the latest preprint version here
Abstract
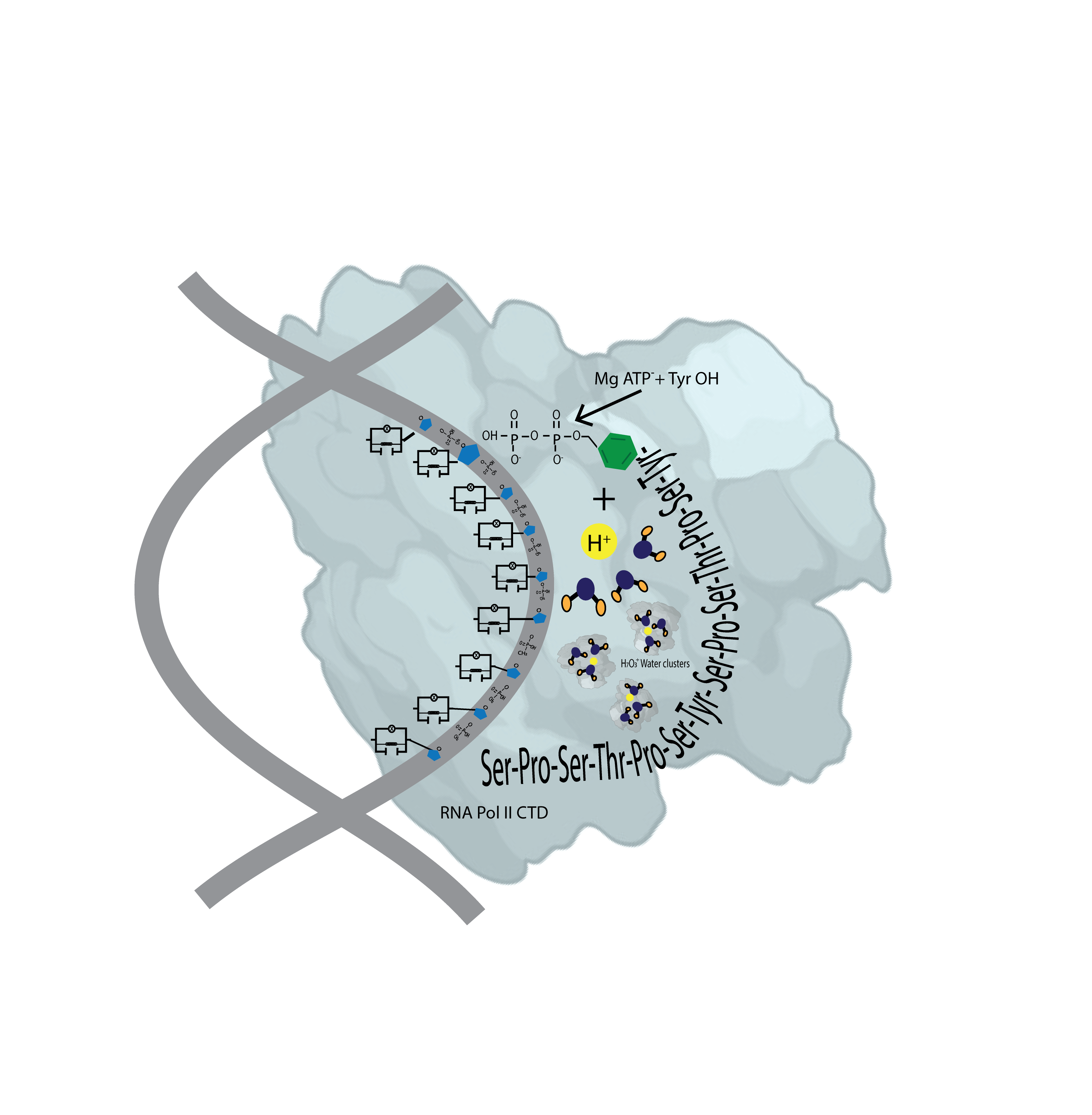
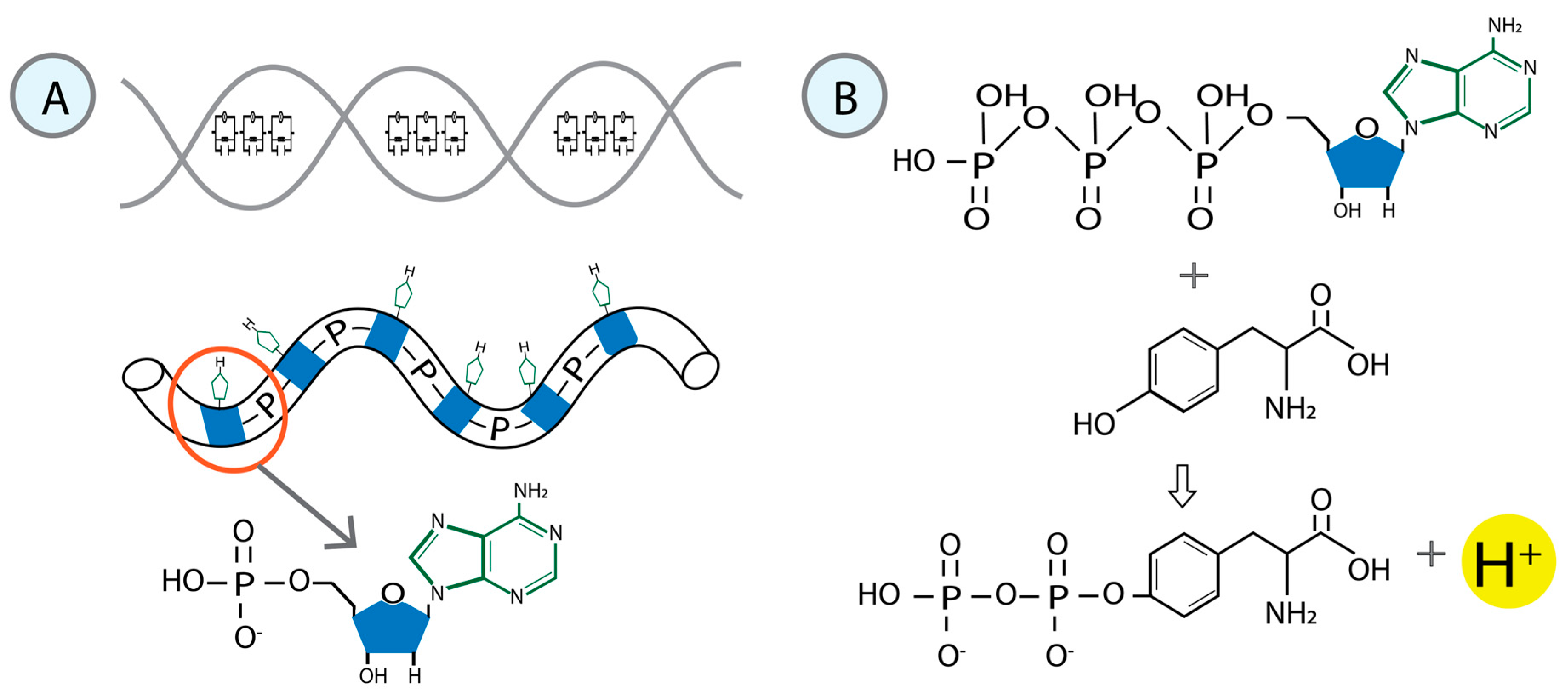
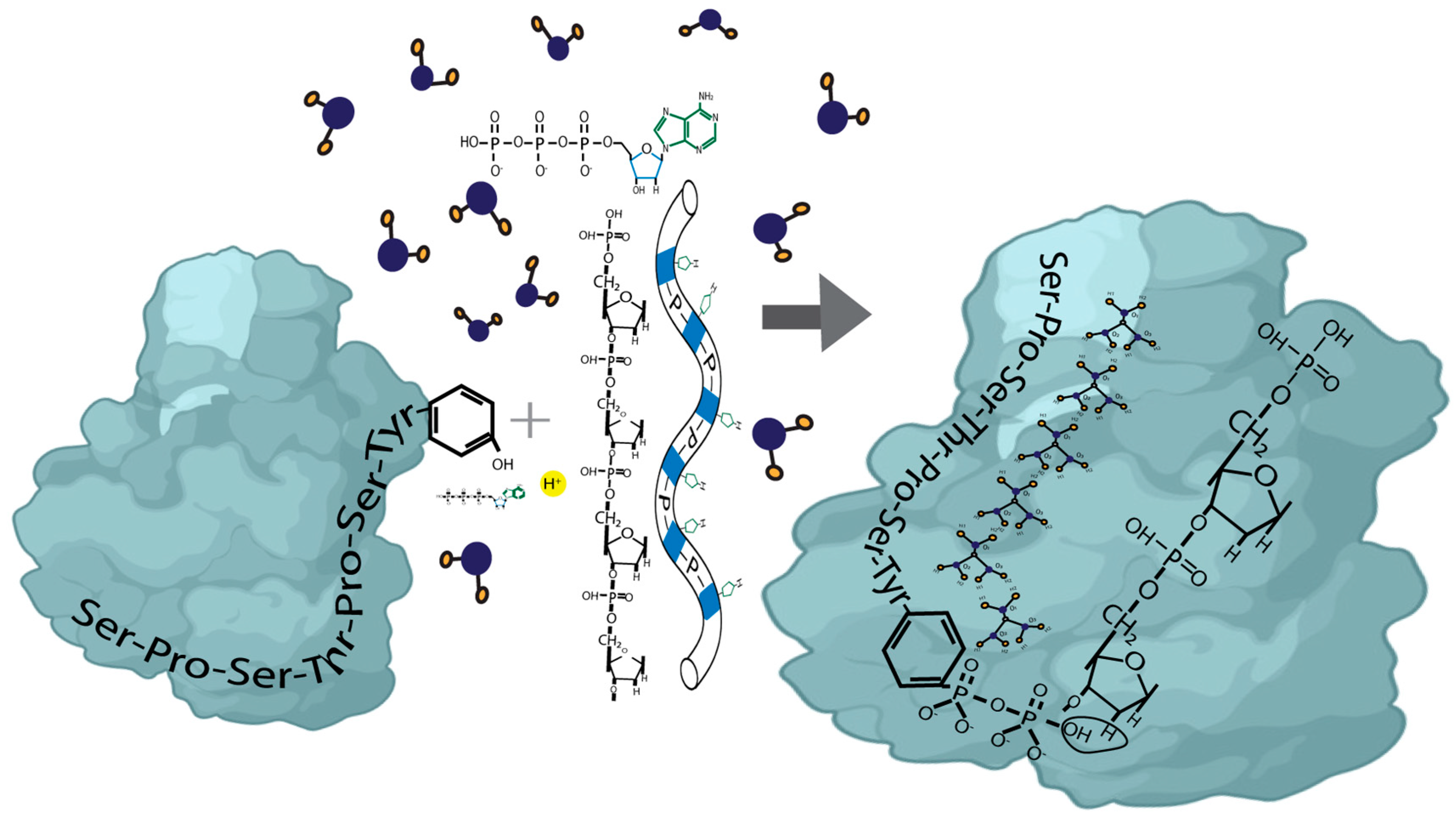
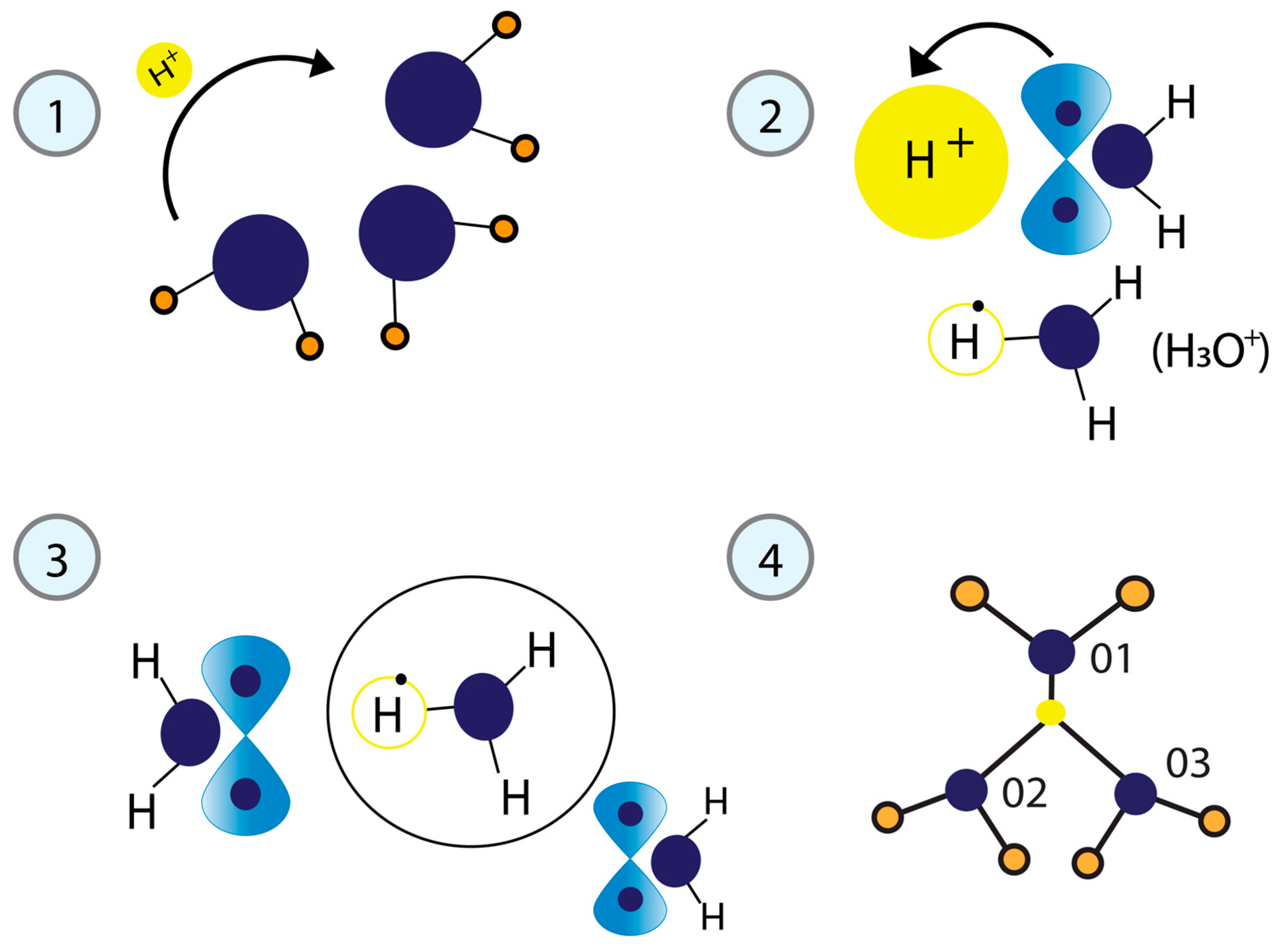
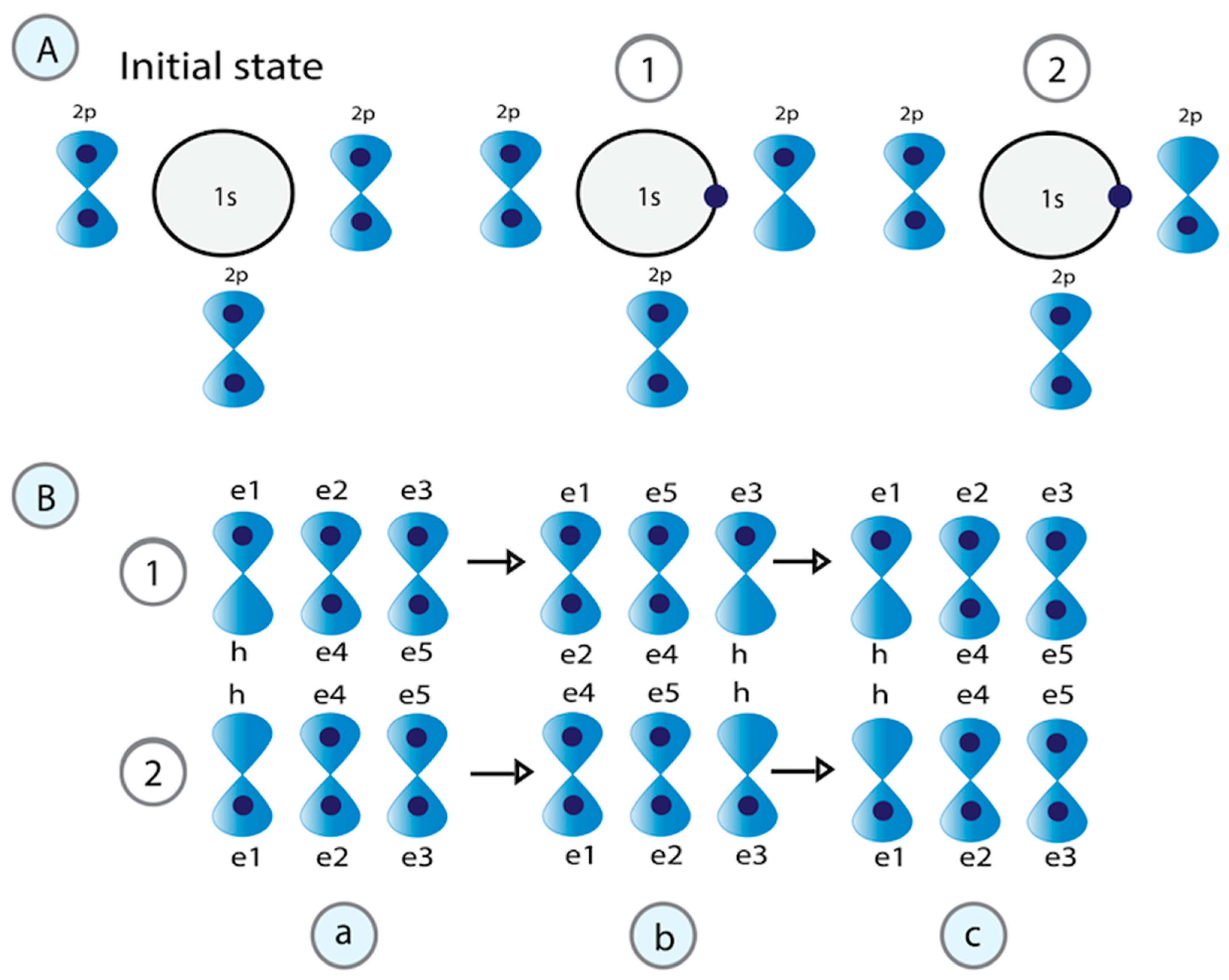
The dynamic phosphorylation of the human RNA Pol II CTD establishes a code applicable to all eukaryotic transcription processes. However, the ability of these specific post-translational modifications to convey molecular signals through structural changes remains unclear. We previously explained that each gene can be modeled as a combination of n circuits connected in parallel. RNA Pol II accesses these circuits and, through a series of pulses, matches the resonance frequency of the DNA qubits, enabling it to extract genetic information and quantum teleport it. Negatively charged phosphates react under RNA Pol II catalysis, increasing the electron density on the deoxyribose acceptor carbon. The first pulse of phosphorylation connects tyrosine to the nitrogenous base, while the subsequent pulses link the protein to molecular water through hydrogen bonds. The coupling of hydrogen proton transfer with electron transfer in water generates a supercurrent, which is explained by the correlation of pairs of the same type of fermions exchanging a boson. All these changes lead to the formation of a molecular protein-DNA-water condensate.
Keywords:
1. Introduction
2. Characterization of the Pulse During the Transcription Process
2.1. Phosphates as the Driving Force
2.2. Model of Phosphorylation Inducing Interactions Between RNA POL II And DNA
3. Water Molecules in DNA and Intrinsically Disordered Proteins
3.1. Physical Model of Phosphorylation Mediating H+ Proton Transfer
3.2. Superconductor Character of Water Induced by H+ Proton Transfer
3.3. Application of the Physical-Mathematical Model for the Correlation of Pairs of the Same Type of Fermions (Two Electrons) That Exchange a Boson to Explain Superconductivity in Water
4. Discussion
5. Conclusions
References
- Adhav, V. A., Shelke, S. S., Balanarayan, P., & Saikrishnan, K. (2023). Sulfur-mediated chalcogen versus hydrogen bonds in proteins: a see-saw effect in the conformational space. QRB Discovery, 4, e5. [CrossRef]
- Banani, S. F., Lee, H. O., Hyman, A. A., & Rosen, M. K. (2017). Biomolecular condensates: organizers of cellular biochemistry. Nature Reviews Molecular Cell Biology, 18(5), 285–298. [CrossRef]
- Basile, W., Salvatore, M., Bassot, C., & Elofsson, A. (2019). Why do eukaryotic proteins contain more intrinsically disordered regions? PLOS Computational Biology, 15(7), e1007186. [CrossRef]
- Biswas, R., Tse, Y.-L. S., Tokmakoff, A., & Voth, G. A. (2016). Role of Presolvation and Anharmonicity in Aqueous Phase Hydrated Proton Solvation and Transport. The Journal of Physical Chemistry B, 120(8), 1793–1804. [CrossRef]
- Blumberger, J. (2015). Recent Advances in the Theory and Molecular Simulation of Biological Electron Transfer Reactions. Chemical Reviews, 115(20), 11191–11238. [CrossRef]
- Bogot, A., Poline, M., Ji, M., Dochain, A., Simonsson, A., Rosén, S., Zettergren, H., Schmidt, H. T., Thomas, R. D., & Strasser, D. (2024). The mutual neutralization of hydronium and hydroxide. Science, 383(6680), 285–289. [CrossRef]
- Brandl, M., Weiss, M. S., Jabs, A., Sühnel, J., & Hilgenfeld, R. (2001). C-h⋯π-interactions in proteins. Journal of Molecular Biology, 307(1), 357–377. [CrossRef]
- Bremer, A., Farag, M., Borcherds, W. M., Peran, I., Martin, E. W., Pappu, R. V., & Mittag, T. (2022). Deciphering how naturally occurring sequence features impact the phase behaviours of disordered prion-like domains. Nature Chemistry, 14(2), 196–207. [CrossRef]
- Bubon, T. L., & Perepelytsya, S. M. (2021). Low-frequency vibrations of water molecules in DNA minor groove. The European Physical Journal E, 44(6), 84. [CrossRef]
- Bubon, T., Zdorevskyi, O., & Perepelytsya, S. (2023). Molecular dynamics study of collective water vibrations in a DNA hydration shell. European Biophysics Journal, 52(1–2), 69–79. [CrossRef]
- Burevschi, E., Alonso, E. R., & Sanz, M. E. (2020). Binding Site Switch by Dispersion Interactions: Rotational Signatures of Fenchone–Phenol and Fenchone–Benzene Complexes. Chemistry – A European Journal, 26(49), 11327–11333. [CrossRef]
- Byun, B. J., & Kang, Y. K. (2010). Conformational preferences and prolyl cis-trans isomerization of phosphorylated Ser/Thr-Pro motifs. Biopolymers, 93(4), 330–339. [CrossRef]
- Cho, W.-K., Spille, J.-H., Hecht, M., Lee, C., Li, C., Grube, V., & Cisse, I. I. (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science, 361(6400), 412–415. [CrossRef]
- Codorniu-Hernández, E., & Kusalik, P. G. (2012). Mobility Mechanism of Hydroxyl Radicals in Aqueous Solution via Hydrogen Transfer. Journal of the American Chemical Society, 134(1), 532–538. [CrossRef]
- Conti Nibali, V., D’Angelo, G., Paciaroni, A., Tobias, D. J., & Tarek, M. (2014). On the Coupling between the Collective Dynamics of Proteins and Their Hydration Water. The Journal of Physical Chemistry Letters, 5(7), 1181–1186. [CrossRef]
- Cramer, P. (2019). Organization and regulation of gene transcription. Nature, 573(7772), 45–54. [CrossRef]
- Czernek, J., Brus, J., Czerneková, V., & Kobera, L. (2023). Quantifying the Intrinsic Strength of C–H⋯O Intermolecular Interactions. Molecules, 28(11), 4478. [CrossRef]
- Dopkins, N., & Nixon, D. F. (2024). Activation of human endogenous retroviruses and its physiological consequences. Nature Reviews Molecular Cell Biology, 25(3), 212–222. [CrossRef]
- Dragan, A. I., Read, C. M., & Crane-Robinson, C. (2017). Enthalpy–entropy compensation: the role of solvation. European Biophysics Journal, 46(4), 301–308. [CrossRef]
- Dubey, V., Kumar, N., & Daschakraborty, S. (2018). Importance of Solvents’ Translational–Rotational Coupling for Translational Jump of a Small Hydrophobic Solute in Supercooled Water. The Journal of Physical Chemistry B, 122(30), 7569–7583. [CrossRef]
- Duboué-Dijon, E., Fogarty, A. C., Hynes, J. T., & Laage, D. (2016). Dynamical Disorder in the DNA Hydration Shell. Journal of the American Chemical Society, 138(24), 7610–7620. [CrossRef]
- Dutta, A., & Lazaridis, T. (2024). Classical Models of Hydroxide for Proton Hopping Simulations. The Journal of Physical Chemistry B, 128(49), 12161–12170. [CrossRef]
- Ehara, H., Yokoyama, T., Shigematsu, H., Yokoyama, S., Shirouzu, M., & Sekine, S. (2017). Structure of the complete elongation complex of RNA polymerase II with basal factors. Science, 357(6354), 921–924. [CrossRef]
- Farrell, K. M., & Zanni, M. T. (2019). Observing Aqueous Proton Transfer Dynamics. ACS Central Science, 5(7), 1114–1116. [CrossRef]
- Frutos-Puerto, S., Colín, M. J., Corchado, J. C., Sánchez, M. L., Martín, M. E., & Aguilar, M. A. (2023). Photophysical and photochemical properties of 3-hydroxyflavone in ethanol solution: Implicit vs explicit solvent models. Journal of Molecular Liquids, 381, 121783. [CrossRef]
- Gao, Y., Fang, H., Ni, K., & Feng, Y. (2022). Water clusters and density fluctuations in liquid water based on extended hierarchical clustering methods. Scientific Reports, 12(1), 8036. [CrossRef]
- Gavrilov, Y., Leuchter, J. D., & Levy, Y. (2017). On the coupling between the dynamics of protein and water. Physical Chemistry Chemical Physics, 19(12), 8243–8257. [CrossRef]
- Ghoshal, S., & Hazra, M. K. (2016). Impact of OH Radical-Initiated H2CO3 Degradation in the Earth’s Atmosphere via Proton-Coupled Electron Transfer Mechanism. The Journal of Physical Chemistry A, 120(4), 562–575. [CrossRef]
- Gibbs, C. A., Weber, D. S., & Warren, J. J. (2021). Clustering of Aromatic Amino Acid Residues around Methionine in Proteins. Biomolecules, 12(1), 6. [CrossRef]
- Gray, T. M., & Matthews, B. W. (1984). Intrahelical hydrogen bonding of serine, threonine and cysteine residues within α-helices and its relevance to membrane-bound proteins. Journal of Molecular Biology, 175(1), 75–81. [CrossRef]
- Gui, T., Fleming, C., Manzato, C., Bourgeois, B., Sirati, N., Heuer, J., Papadionysiou, I., Montfort, D. I. van, Gijzen, M. van, Smits, L. M. M., Burgering, B. M. T., Madl, T., & Schuijers, J. (2023). Targeted perturbation of signaling-driven condensates. Molecular Cell, 83(22), 4141-4157.e11. [CrossRef]
- Gutowsk, M., & Kowalczyk, S. (2016). A study of free radical chemistry: their role and pathophysiological significance. Acta Biochimica Polonica, 60, 1–16.
- Hassanali, A., Giberti, F., Cuny, J., Kühne, T. D., & Parrinello, M. (2013). Proton transfer through the water gossamer. Proceedings of the National Academy of Sciences, 110(34), 13723–13728. [CrossRef]
- Henninger, J. E., Oksuz, O., Shrinivas, K., Sagi, I., LeRoy, G., Zheng, M. M., Andrews, J. O., Zamudio, A. V., Lazaris, C., Hannett, N. M., Lee, T. I., Sharp, P. A., Cissé, I. I., Chakraborty, A. K., & Young, R. A. (2021). RNA-Mediated Feedback Control of Transcriptional Condensates. Cell, 184(1), 207-225.e24. [CrossRef]
- Huang, L., Matta, C. F., Wallace, S., Massa, L., & Bernal, I. (2015). A unique trapping by crystal forces of a hydronium cation displaying a transition state structure. Comptes Rendus. Chimie, 18(5), 511–515. [CrossRef]
- Hubbard, E. E., & Julian, R. R. (2025). Unravelling the favorability of radical-directed xn-H2O dissociation at serine and threonine. International Journal of Mass Spectrometry, 507, 117363. [CrossRef]
- Johnson, J. L., Yaron, T. M., Huntsman, E. M., Kerelsky, A., Song, J., Regev, A., Lin, T.-Y., Liberatore, K., Cizin, D. M., Cohen, B. M., Vasan, N., Ma, Y., Krismer, K., Robles, J. T., van de Kooij, B., van Vlimmeren, A. E., Andrée-Busch, N., Käufer, N. F., Dorovkov, M. V., … Cantley, L. C. (2023). An atlas of substrate specificities for the human serine/threonine kinome. Nature, 613(7945), 759–766. [CrossRef]
- Joshi, A., Avni, A., Walimbe, A., Rai, S. K., Sarkar, S., & Mukhopadhyay, S. (2024). Hydrogen-Bonded Network of Water in Phase-Separated Biomolecular Condensates. The Journal of Physical Chemistry Letters, 15(30), 7724–7734. [CrossRef]
- Kamerlin, S. C. L., Sharma, P. K., Prasad, R. B., & Warshel, A. (2013). Why nature really chose phosphate. Quarterly Reviews of Biophysics, 46(1), 1–132. [CrossRef]
- Kaufman, N. E. M., Dhingra, S., Jois, S. D., & Vicente, M. da G. H. (2021). Molecular Targeting of Epidermal Growth Factor Receptor (EGFR) and Vascular Endothelial Growth Factor Receptor (VEGFR). Molecules, 26(4), 1076. [CrossRef]
- Kier, L. B. (2021). Proton Hopping in Living Systems. Current Computer-Aided Drug Design, 17(3), 333–336. [CrossRef]
- Kim, Y., Joachimiak, G., Bigelow, L., Babnigg, G., & Joachimiak, A. (2016). How Aromatic Compounds Block DNA Binding of HcaR Catabolite Regulator. Journal of Biological Chemistry, 291(25), 13243–13256. [CrossRef]
- Krone, M. W., Travis, C. R., Lee, G. Y., Eckvahl, H. J., Houk, K. N., & Waters, M. L. (2020). More Than π–π–π Stacking: Contribution of Amide−π and CH−π Interactions to Crotonyllysine Binding by the AF9 YEATS Domain. Journal of the American Chemical Society, 142(40), 17048–17056. [CrossRef]
- Kubyshkin, V., & Mykhailiuk, P. K. (2024). Proline Analogues in Drug Design: Current Trends and Future Prospects. Journal of Medicinal Chemistry, 67(22), 20022–20055. [CrossRef]
- Kubyshkin, V., & Rubini, M. (2024). Proline Analogues. Chemical Reviews, 124(13), 8130–8232. [CrossRef]
- Lassila, J. K., Zalatan, J. G., & Herschlag, D. (2011). Biological Phosphoryl-Transfer Reactions: Understanding Mechanism and Catalysis. Annual Review of Biochemistry, 80(1), 669–702. [CrossRef]
- Limbach, H.-H. (1991). NMR Studies of Elementary Steps of Multiple Proton and Deuteron Transfers in Liquids, Crystals, and Organic Glasses. In Intermolecular Forces (pp. 281–295). Springer Berlin Heidelberg. [CrossRef]
- Liu, Y., Li, Y., Wang, Y., Lin, C., Zhang, D., Chen, J., Ouyang, L., Wu, F., Zhang, J., & Chen, L. (2022). Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. Journal of Hematology & Oncology, 15(1), 89. [CrossRef]
- Lyons, H., Veettil, R. T., Pradhan, P., Fornero, C., De La Cruz, N., Ito, K., Eppert, M., Roeder, R. G., & Sabari, B. R. (2023). Functional partitioning of transcriptional regulators by patterned charge blocks. Cell, 186(2), 327-345.e28. [CrossRef]
- Mahmudov, K. T., Kopylovich, M. N., Guedes da Silva, M. F. C., & Pombeiro, A. J. L. (2017). Non-covalent interactions in the synthesis of coordination compounds: Recent advances. Coordination Chemistry Reviews, 345, 54–72. [CrossRef]
- Martin, E. W., Holehouse, A. S., Peran, I., Farag, M., Incicco, J. J., Bremer, A., Grace, C. R., Soranno, A., Pappu, R. V., & Mittag, T. (2020). Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science, 367(6478), 694–699. [CrossRef]
- Matyushov, D. V. (2023). Reorganization energy of electron transfer. Physical Chemistry Chemical Physics, 25(11), 7589–7610. [CrossRef]
- McDermott, M. L., Vanselous, H., Corcelli, S. A., & Petersen, P. B. (2017). DNA’s Chiral Spine of Hydration. ACS Central Science, 3(7), 708–714. [CrossRef]
- Meng, X., & Qi, J. (2023). Manipulating Tyrosine Phosphorylation by Heterobifunctional Small Molecules. ACS Central Science, 9(8), 1512–1514. [CrossRef]
- Mohan, N., Vijayalakshmi, K. P., Koga, N., & Suresh, C. H. (2010). Comparison of aromatic NH···π, OH···π, and CH···π interactions of alanine using MP2, CCSD, and DFT methods. Journal of Computational Chemistry, 31(16), 2874–2882. [CrossRef]
- Mukherjee, S., Mondal, S., Acharya, S., & Bagchi, B. (2018). DNA Solvation Dynamics. The Journal of Physical Chemistry B, 122(49), 11743–11761. [CrossRef]
- Nakane, K., Nagasawa, H., Fujimura, C., Koyanagi, E., Tomoshige, S., Ishikawa, M., & Sato, S. (2022). Switching of Photocatalytic Tyrosine/Histidine Labeling and Application to Photocatalytic Proximity Labeling. International Journal of Molecular Sciences, 23(19), 11622. [CrossRef]
- Nishio, M., Umezawa, Y., Fantini, J., Weiss, M. S., & Chakrabarti, P. (2014). CH–π hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys., 16(25), 12648–12683. [CrossRef]
- Nott, T. J., Petsalaki, E., Farber, P., Jervis, D., Fussner, E., Plochowietz, A., Craggs, T. D., Bazett-Jones, D. P., Pawson, T., Forman-Kay, J. D., & Baldwin, A. J. (2015). Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Molecular Cell, 57(5), 936–947. [CrossRef]
- Onaka, S. (2024). Periodic arrangements of tetrahedra having appearances similar to that of the Boerdijk–Coxeter helix. Scientific Reports, 14(1), 18260. [CrossRef]
- Pappu, R. V., Cohen, S. R., Dar, F., Farag, M., & Kar, M. (2023). Phase Transitions of Associative Biomacromolecules. Chemical Reviews, 123(14), 8945–8987. [CrossRef]
- Patikoglou, G. A., Kim, J. L., Sun, L., Yang, S.-H., Kodadek, T., & Burley, S. K. (1999). TATA element recognition by the TATA box-binding protein has been conserved throughout evolution. Genes & Development, 13(24), 3217–3230. [CrossRef]
- Pei, G., Lyons, H., Li, P., & Sabari, B. R. (2024). Transcription regulation by biomolecular condensates. Nature Reviews Molecular Cell Biology. [CrossRef]
- Perets, E. A., Konstantinovsky, D., Santiago, T., Videla, P. E., Tremblay, M., Velarde, L., Batista, V. S., Hammes-Schiffer, S., & Yan, E. C. Y. (2024). Beyond the “spine of hydration”: Chiral SFG spectroscopy detects DNA first hydration shell and base pair structures. The Journal of Chemical Physics, 161(9). [CrossRef]
- Popov, I., Zhu, Z., Young-Gonzales, A. R., Sacci, R. L., Mamontov, E., Gainaru, C., Paddison, S. J., & Sokolov, A. P. (2023). Search for a Grotthuss mechanism through the observation of proton transfer. Communications Chemistry, 6(1), 77. [CrossRef]
- Qiao, H., Xia, M., Cheng, Y., Zhou, J., Zheng, L., Li, W., Wang, J., & Fang, P. (2023). Tyrosine-targeted covalent inhibition of a tRNA synthetase aided by zinc ion. Communications Biology, 6(1), 107. [CrossRef]
- Riera Aroche, R., Ortiz García, Y. M., Martínez Arellano, M. A., & Riera Leal, A. (2024). DNA as a perfect quantum computer based on the quantum physics principles. Scientific Reports, 14(1), 11636. [CrossRef]
- Riera Aroche, R., Ortiz García, Y. M., Sánchez Moreno, E. C., Enriquez Cervantes, J. S., Machado Sulbaran, A. C., & Riera Leal, A. (2024). DNA Gene’s Basic Structure as a Nonperturbative Circuit Quantum Electrodynamics: Is RNA Polymerase II the Quantum Bus of Transcription? Current Issues in Molecular Biology, 46(11), 12152–12173. [CrossRef]
- Robertson, A. J., Wilson, A. L., Burn, M. J., Cliff, M. J., Popelier, P. L. A., & Waltho, J. P. (2021). The Relationship between Enzyme Conformational Change, Proton Transfer, and Phosphoryl Transfer in β-Phosphoglucomutase. ACS Catalysis, 11(21), 12840–12849. [CrossRef]
- Rodríguez-Molina, J. B., West, S., & Passmore, L. A. (2023). Knowing when to stop: Transcription termination on protein-coding genes by eukaryotic RNAPII. Molecular Cell, 83(3), 404–415. [CrossRef]
- Roy, S., Lessing, J., Meisl, G., Ganim, Z., Tokmakoff, A., Knoester, J., & Jansen, T. L. C. (2011). Solvent and conformation dependence of amide I vibrations in peptides and proteins containing proline. The Journal of Chemical Physics, 135(23). [CrossRef]
- Sabari, B. R., Dall’Agnese, A., Boija, A., Klein, I. A., Coffey, E. L., Shrinivas, K., Abraham, B. J., Hannett, N. M., Zamudio, A. V., Manteiga, J. C., Li, C. H., Guo, Y. E., Day, D. S., Schuijers, J., Vasile, E., Malik, S., Hnisz, D., Lee, T. I., Cisse, I. I., … Young, R. A. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science, 361(6400). [CrossRef]
- Savinkova, L. K., Sharypova, E. B., & Kolchanov, N. A. (2023). On the Role of TATA Boxes and TATA-Binding Protein in Arabidopsis thaliana. Plants, 12(5), 1000. [CrossRef]
- Schwarz, M., Rizzo, S., Paz, W. E., Kresinsky, A., Thévenin, D., & Müller, J. P. (2022). Disrupting PTPRJ transmembrane-mediated oligomerization counteracts oncogenic receptor tyrosine kinase FLT3 ITD. Frontiers in Oncology, 12. [CrossRef]
- Singh, A. K., Wen, C., Cheng, S., & Vinh, N. Q. (2021). Long-range DNA-water interactions. Biophysical Journal, 120(22), 4966–4979. [CrossRef]
- Singh, A., Raj, P., Dubowski, J. J., & Singh, N. (2018). ATP Induced Modulation in π–π Stacking Interactions in Pyrene Based Zinc Complexes: Chemosensor Study and Quantitative Investigation of Apyrase Activity. Crystal Growth & Design, 18(8), 4320–4333. [CrossRef]
- Singh, S., Tian, W., Severance, Z. C., Chaudhary, S. K., Anokhina, V., Mondal, B., Pergu, R., Singh, P., Dhawa, U., Singha, S., & Choudhary, A. (2023). Proximity-inducing modalities: the past, present, and future. Chemical Society Reviews, 52(16), 5485–5515. [CrossRef]
- Tereshko, V., Minasov, G., & Egli, M. (1999). A “Hydrat-Ion” Spine in a B-DNA Minor Groove. Journal of the American Chemical Society, 121(15), 3590–3595.
- Tian, T., Chu, X.-Y., Yang, Y., Zhang, X., Liu, Y.-M., Gao, J., Ma, B.-G., & Zhang, H.-Y. (2019). Phosphates as Energy Sources to Expand Metabolic Networks. Life, 9(2), 43. [CrossRef]
- Trivedi, R., & Nagarajaram, H. A. (2022). Intrinsically Disordered Proteins: An Overview. International Journal of Molecular Sciences, 23(22), 14050. [CrossRef]
- Tyburski, R., Liu, T., Glover, S. D., & Hammarström, L. (2021). Proton-Coupled Electron Transfer Guidelines, Fair and Square. Journal of the American Chemical Society, 143(2), 560–576. [CrossRef]
- Ubersax, J. A., & Ferrell Jr, J. E. (2007). Mechanisms of specificity in protein phosphorylation. Nature Reviews Molecular Cell Biology, 8(7), 530–541. [CrossRef]
- Uversky, V. N. (2015). The intrinsic disorder alphabet. III. Dual personality of serine. Intrinsically Disordered Proteins, 3(1), e1027032. [CrossRef]
- Vanaja, A., & Yella, V. R. (2022). Delineation of the DNA Structural Features of Eukaryotic Core Promoter Classes. ACS Omega, 7(7), 5657–5669. [CrossRef]
- Vernon, R. M., & Forman-Kay, J. D. (2019). First-generation predictors of biological protein phase separation. Current Opinion in Structural Biology, 58, 88–96. [CrossRef]
- Vervoort, S. J., Devlin, J. R., Kwiatkowski, N., Teng, M., Gray, N. S., & Johnstone, R. W. (2022). Targeting transcription cycles in cancer. Nature Reviews Cancer, 22(1), 5–24. [CrossRef]
- Villegas, J. A., Heidenreich, M., & Levy, E. D. (2022). Molecular and environmental determinants of biomolecular condensate formation. Nature Chemical Biology, 18(12), 1319–1329. [CrossRef]
- Visser, B. S., Lipiński, W. P., & Spruijt, E. (2024). The role of biomolecular condensates in protein aggregation. Nature Reviews Chemistry, 8(9), 686–700. [CrossRef]
- Wood, K., Plazanet, M., Gabel, F., Kessler, B., Oesterhelt, D., Tobias, D. J., Zaccai, G., & Weik, M. (2007). Coupling of protein and hydration-water dynamics in biological membranes. Proceedings of the National Academy of Sciences, 104(46), 18049–18054. [CrossRef]
- Yamauchi, O., & Odani, A. (1985). Structure-stability relationship in ternary copper(II) complexes involving aromatic amines and tyrosine or related amino acids. Intramolecular aromatic ring stacking and its regulation through tyrosine phosphorylation. Journal of the American Chemical Society, 107(21), 5938–5945. [CrossRef]
- Yasuhara, T., Xing, Y.-H., Bauer, N. C., Lee, L., Dong, R., Yadav, T., Soberman, R. J., Rivera, M. N., & Zou, L. (2022). Condensates induced by transcription inhibition localize active chromatin to nucleoli. Molecular Cell, 82(15), 2738-2753.e6. [CrossRef]
- Yu, C.-C., Chiang, K.-Y., Okuno, M., Seki, T., Ohto, T., Yu, X., Korepanov, V., Hamaguchi, H., Bonn, M., Hunger, J., & Nagata, Y. (2020). Vibrational couplings and energy transfer pathways of water’s bending mode. Nature Communications, 11(1), 5977. [CrossRef]
- Yu, Q., & Bowman, J. M. (2020). Tracking Hydronium/Water Stretches in Magic H3O+(H2O)20 Clusters through High-level Quantum VSCF/VCI Calculations. The Journal of Physical Chemistry A, 124(6), 1167–1175. [CrossRef]
- Zhang, C., Tang, C., & Jiao, N. (2012). Recent advances in copper-catalyzed dehydrogenative functionalization via a single electron transfer (SET) process. Chemical Society Reviews, 41(9), 3464. [CrossRef]
- Zhang, J., Hao, M., Zhang, D., Zhang, X., Guo, S., Wang, B., Xiao, J., Gao, Y., & Li, X. (2023). Enhanced Polyacrylamide Degradation via OH Radical-Initiated Single-Electron Transfer. ACS Omega, 8(49), 46589–46597. [CrossRef]
- Zhang, L., & Reilly, J. P. (2009). Radical-driven dissociation of odd-electron peptide radical ions produced in 157 nm photodissociation. Journal of the American Society for Mass Spectrometry, 20(7), 1378–1390. [CrossRef]
- Zhu, Y., Alqahtani, S., & Hu, X. (2021). Aromatic Rings as Molecular Determinants for the Molecular Recognition of Protein Kinase Inhibitors. Molecules, 26(6), 1776. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



