Submitted:
12 February 2025
Posted:
12 February 2025
You are already at the latest version
Abstract
Iron represents a vital element needed for normal physiologic functions across the body. A vast network of transporters is involved not only in uptake but in processing, oxidation and recycling to maintain this element in a tight balance to avoid excess storage. This complex network of transporters including heme and ferroportin among many others are responsible for facilitating inter-organ and tissue iron exchange, contributing to systemic heme homeostasis. Patients with genetic diseases such as sickle disease suffer from a chronic anemia due to the presence of an abnormal hemoglobin that requires, in most instances, a lifetime of red blood cell transfusions to overcome disease crises. These transfusions over time lead to a high iron exposure that results in a profound change to the physiology of organ systems at the cellular level requiring aggressive chelation. This exposure unfortunately leads to irreversible changes to these organs from the cardiovascular system and bone marrow, to the central nervous system. In the bone marrow, heme is synthesized, mainly within developing red blood cells, iron excess leads to impairments in cell production and differentiation due to processes needing a balanced intracellular heme concentration to prevent toxicity. In light of the extensive role of iron in the body, the aim of this review is to summarize the important metabolic pathways involved in iron homeostasis across a number of cell types and organ systems while contrasting these against effects caused by iron overload.
Keywords:
Iron metabolism
; sickle cell disease
; molecular mechanisms
; transfusion
; iron overload
; metabolism
; heme
; ferroportin
1. Introduction
Iron metabolism is essential for a wide array of physiological processes, underpinning critical cellular and systemic functions. Iron is needed for DNA synthesis, electron transport, oxygen transport through synthesis of hemoglobin and myoglobin, ATP production, spermatogenesis, and enzymatic reactions needed for the electron transfer chain among many other functions [1]. Most of the iron in the body is found coupled to hemoglobin (65%), while 10% is present in non-hemoglobin components (myoglobin, enzymes and cytochromes), with the remainder in iron stores across the body [2]. It has been estimated that there is a daily loss of 1-2 mg of iron from the body or 0.05% of total body iron via shedding of the gastrointestinal lining, skin or through minimal blood loss [3]. However, these losses are balanced through regulation of iron absorption and its recycling from senescent erythrocytes [4].
In the trauma setting, increases in circulating iron occur due to acute blood loss while in genetic disorders such as those in hemoglobinopathies, i.e. sickle cell disease (SCD), are due to ineffective erythropoiesis. Whichever the case may be, despite an expansion of the erythroid progenitor pool, a continuous state of chronic stress erythropoiesis ensues, but the expanded pool of erythroid precursors is unable to generate sufficient erythrocytes [5]. The resulting anemia is accompanied by a decrease in expression of the HAMP gene encoding for hepcidin, a 25 amino acid protein synthesized by hepatocytes and to a lesser extent by the intestine and heart, that increases iron availability [5,6]. However, there can be a wide spectrum of increased iron concentrations from either acute blood loss or from exposure to blood transfusions such as in SCD patients who are chronically transfused. Physiologic iron balance is regulated by proteins known as iron regulatory proteins (IRPs) which control both concentration and its elemental functions [7,8]. Of these, IRPs 1 and 2, are essential to iron homeostasis at the cellular level, first of which occur through binding to iron response elements (IREs) and second to iron translator regions of mRNA-encoding proteins that are further involved in iron uptake, storage, and export via proteins like transferrin receptor 1 (TFR1), divalent metal transporter-1 (DMT1) and ferroproteins [3]. In this way, IRPs 1 and 2 bind to IREs when iron concentration in the body is low and dissociate from them when its concentration is high [4].
There are two states in which iron can be found, one is ferrous (Fe2+) and the second is the ferric (Fe3+) form [9]. A close balance in iron concentration is required for proper homeostasis; thus, iron is toxic when present in excess (overload) and toxicity leads to a variety of human health issues encountered frequently in the population exposed to frequent transfusions or those with genetic predisposition such as those with hereditary hemochromatosis [8]. In the diet iron is equally available in two forms: as either non-heme or as ionic-heme [10]. Both forms can be found in plant or animal sources and have distinct absorption rates closely regulated by hepcidin [11]. Plant-derived dietary heme iron is available from cereals, vegetables, legumes and fluids [12]. Gastric acid in the proximal duodenum acting on diet sources reduces via ferric reductases that subsequently mediate transport of Fe2+ across the apical membrane of enterocytes [13]. Similarly, most of the non-heme iron is in the ferric form that is reduced to ferrous before it can be absorbed. This is carried out by membrane-bound ferric reductase duodenal cytochrome B1 (DCYTB) [14]. Iron is then transported across the apical brush border membrane of intestinal epithelial cells by DMT1 capable of transporting several divalent cations including Fe2+ [15]. Iron can subsequently be stored as ferritin or trafficked as needed to the basolateral surface [14]. On this surface, Fe2+ is transported to the blood by the iron transporter ferroportin (FPN), a transmembrane domain protein, encoded by the SLC40A1 gene [15]. Along these lines, use of proton pump inhibitors which impair gastric acid production results in substantial reduction of iron absorption [2]. Notably, the most Fe3+important checkpoint for iron absorption is the proximal small bowel which regulates whole-body iron levels [16]. As a result, the interplay between intake and bioavailability establishes how this element is not only needed by the organism, but how its concentration is balanced to prevent both deficiency and toxicity.
Heme is an iron-containing tetra-pyrrole that is important in binding oxygen, globin transportation, and detoxification [14]. Heme’s functions include activation of transcription factors BACH1, nuclear-related factor-2 (NRF2), GATA 1-mediated gene expression, while working as an antioxidant to cellular stress, and regulator of cell proliferation and apoptosis through activation of the previously mentioned transcription factors [17]. Heme is also involved in regulation of the circadian rhythm and cell cycles like those mediated by mitochondrial respiration and iron-sulfur (Fe-S) clusters [18]. When hydrophobic heme is cytotoxic, it is due to generation of various reactive oxygen species (ROS), which in turn leads to enhanced oxidative stress responses [19]. As a result, heme homeostasis in an organism is tightly regulated at synthesis, import, utilization, degradation, and export [20].
Iron export from cells is as important as mechanisms of iron intake. FPN is expressed across a diverse array of cell types including macrophages, hepatocytes, placental syncytiotrophoblast cells and membranes of mature erythrocytes where it works as an iron exporter from cells to tightly regulate its concentration intracellularly and consequently throughout the body [21]. Hepcidin binds to FPN resulting in the internalization and lysosomal degradation of iron via the ubiquitin pathway [22,23]. Through this binding hepcidin regulates FPN, providing an additional layer of iron flow regulation from enterocytes and macrophages into plasma/circulation [22]. This would explain how HAMP mutations result in iron overload, as the absence of hepcidin permits constitutively high iron absorption and overall unregulated iron transport [23]. Bone Morphogenetic Protein 6 (BMP6) synthesized by hepatic sinusoidal endothelial cells [24], regulates hepcidin transcription and its overall expression [25].
Inflammation and inflammatory cytokines, mainly interleukin (IL)-6, lead to increased levels of hepcidin [26]. This is because IL-6 binding to its receptor and through activation of the JAK/STAT3 signaling pathway increases hepcidin production. These high levels of hepcidin block iron release by FPN, lowering iron concentration and making it less available in circulation [27]. On the contrary, during conditions of increased circulatory iron or cases of overload due to high iron exposure, as those seen in inflammatory states and hemoglobinopathies, the net result is decreased absorption of iron by FPN leading to slow and gradual restoration of iron homeostasis [28]. This is a tightly regulated feedback mechanism that when impaired leads to those health problems encountered by patients chronically transfused and a resulting higher iron burden.
Hypoxic conditions, most commonly anemia, decrease hepcidin production resulting in increased erythropoiesis. Hemoglobinopathies such as thalassemia, SCD, congenital dyserythropoietic anemia type I, and even myelodysplastic syndrome are characterized by lower hepcidin and compromised erythropoiesis [29]. Newly-formed erythroid precursors in these settings release growth differentiated factor-15 which further decreases hepcidin production [30,31]. Whereas in conditions of ineffective erythropoiesis due to bone marrow failure syndromes, characterized by decreased erythrocyte production, iron absorption and recycling is increased [32]. On the contrary, in situations when bone marrow activity increases iron absorption, there is a corresponding decrease in hepcidin production [33,34]. Of interest, mutations in the TFR2 gene leads to downregulation of HAMP expression and decreased hepcidin synthesis and iron accumulation [35]. Anemias have also been classified according to hepcidin levels as either high or low hepcidin [36]. When hepcidin is persistently high, further iron absorption is halted leading most commonly to iron deficiency anemia.; however, under iron excess/overload low hepcidin is more likely observed [37]. Hypoxia also plays a major role in ferroptosis (iron-mediated apoptosis). In this regard, oxygen availability, anti-oxidative factors like hypoxia-inducible factors (HIF), and NRF2 influence ferroptosis [38]. Hypoxia as seen in SCD patients during crises, increases expression of NRF2 facilitating heme oxygenase-1 (HO-1), which forms a complex with heme and NADPH-cytochrome-P450 reductase [39]. Oxygen binds to this complex, and NADPH acts as electron donor forming carbon monoxide, chelatable Fe2+ and biliverdin, thus preventing ROS formation and ferroptosis [39]. Similarly, hypoxia decreases nuclear receptor coactivator-4 NCOA4 expression that results in additional protection from ferroptosis [40].
Iron that is not directed to mitochondria for heme synthesis is stored in the ferric state coupled to ferritin [35]. This protein is a hetero-polymer composed of 24 subunits of light and heavy chains, of which the latter has ferroxidase activity [41]. In iron deficiency states, ferritin is degraded by auto phagosomes, and this ferritin autophagy is known as “ferritinophagy”. Iron release from ferritin degradation is mediated by the intracellular protein NCOA4 so that in states of iron excess/overload, iron binding sites on ferritin are saturated resulting in decreased iron binding and increased iron delivery to mitochondria [42]. Thus, NCOA4 has an essential role in iron hemostasis being upregulated in conditions of iron deficiency and downregulated in conditions of iron excess [43]. Imbalances in these regulatory systems increase risk of cell death such as in settings of infection and malignancies where higher ferroptosis rates provide iron rapidly to proliferating cells [44].
The release of heme and free iron during an acute SCD crisis is highly toxic and drives a high production of ROS, which potentiates apoptosis by inhibiting the proteasome leading to iron-dependent ferroptosis [45,46]. Importantly, iron overload and ferroptosis play essential roles in SCD progression. Targeting therapies like antioxidants are as a result helpful in preventing ferroptosis and minimizing SCD disease progression [47]. In light of these findings, the aim of this review is to present the latest advances in understanding the molecular regulation of iron metabolism, highlighting its role in cellular homeostasis and overall integration of iron-dependent systemic and cellular processes. The role of iron will be examined looking at essential regulatory networks, exploring pathological consequences of iron dysregulation and excess, while discussing its impact on specific organ systems taking SCD patients as a population deeply affected by iron overload. Beginning with the interplay between the liver and bone marrow, the analysis will extend to iron’s roles in the CNS, heart, lungs, endocrine, and reproductive systems (Figure 1). This comprehensive approach aims to establish a foundational understanding of iron metabolism across different physiological and pathological contexts.
2. Physiologic Absorption of Iron
Dietary iron exists in two forms: heme and nonheme. Nonheme iron is predominantly sourced from cereals, vegetables, legumes, and fruits, where is present in the ferric state. In contrast, heme iron, which is more abundant from animal-based sources, is absorbed more efficiently than the nonheme form. A critical early step in the absorption of nonheme iron requires the secretion of hydrochloric acid by the stomach [48]. This form of iron is solubilized by gastric acid, facilitating its reduction by ascorbic acid and other reductases that precedes its absorption by the small intestine. Gastric acid also preserves ascorbic acid in its active reduced state so that it chelates soluble iron, preventing its precipitation. However, since humans lack the ability to synthesize ascorbic acid, this must be obtained through dietary sources [48].
2.1. Absorption
Nonheme iron absorption involves a multi-step process. Initially, gastric acid and ferrireductases, such as DCYTB, reduce nonheme iron to the ferrous form in the gut lumen. Ferrous iron is subsequently transported into enterocytes by DMT1 also known as SLC11A2, or less commonly, via SLC36A1 when bound to nicotianamine [15]. Within enterocytes, iron is either stored in ferritin, utilized for cellular processes, or trafficked to the basolateral membrane. Iron is later exported to the bloodstream by FPN in a process coupled with H+ exchange and hephaestin, an iron regulatory protein that oxidizes iron to its ferric form [49]. Finally, ferric iron binds to transferrin (Tf), a liver-derived serum protein, for systemic distribution across the body [50].
2.2. Hepcidin and other Related Mediators
The liver’s hepcidin regulates iron absorption by binding to FPN, in this way blocking iron transport, while inducing FPN internalization followed by lysosomal degradation, which preferentially targets iron-replete FPN [51]. Once internalized, FPN undergoes proteasomal degradation facilitated by the E3 ubiquitin ligase RNF217, the E1 enzyme UBA6, and the adaptor protein NDFIP1 [52]. Hepcidin-mediated FPN downregulation increases enterocyte iron levels, promoting degradation of HIF-2, which reduces expression of key iron absorption genes such as DMT1 and DCYTB [53]. Hepcidin, like FPN, is itself tightly regulated to maintain iron homeostasis [54]. Its expression increases with iron overload and inflammation but decreases under states of hypoxia or heightened iron demand, such as during increased erythropoiesis. This regulation is mediated by BMP-dependent signaling pathways, primarily involving BMP2 and BMP6 produced by liver sinusoidal endothelial cells [55].
Hepatocyte surface proteins including hereditary hemochromatosis protein (HFE), hemojuvelin (HJV), TFR2, BMP receptors (ALK2, ALK3, BMPR-II), neogenin, and TMPRSS6, modulate hepcidin expression in response to BMPs and diferric Tf [56]. Of interest, inherited mutations in HFE, HJV, and TFR2 cause hereditary hemochromatosis, a condition of iron overload that physiologically impairs multiple organs such as liver, pancreas, heart, and endocrine glands. Subsequently, hepcidin expression is closely regulated by several mechanisms: BMP signaling via SMAD1/5/8 transcription factors [57], NRF2 activation under iron-induced oxidative stress [58], and erythropoietic activity mediated by the hormone erythroferrone [59].
The gut microbiome also contributes to regulation of dietary iron absorption and uptake. Intestinal microbes produce metabolites that inhibit iron absorption, thereby ensuring an adequate supply for their own metabolism [60]. For instance, 1,3-diaminopropane, derived from both bacterial and dietary sources, suppresses iron transport proteins DCYTB, DMT1, and FPN by inhibiting HIF-2 activity in enterocytes [60]. These metabolites also enhance ferritin expression, sequestering iron within enterocytes and reducing its availability and export to the bloodstream. Ultimately, iron stored in ferritin is lost when enterocytes are shed, returning it to the gut microenvironment.
2.3. Cellular Nonheme Iron Uptake
Erythroid precursors predominantly acquire circulating diferric Tf for erythropoiesis through the Tf cycle. In this process, diferric Tf binds to TFR1 and is internalized via endocytosis [35]. Endosomal acidification then releases ferric iron, which is reduced by STEAP3 and transported into the cytoplasm by DMT1 [35]. TFR1 also interacts with serum ferritin, suggesting the existence of an alternative Tf-independent iron uptake pathway [61].
Tf, remains central to our evolving understanding of iron homeostasis. Its two iron-binding sites are functionally distinct, as demonstrated by differing binding sensitivities to erythropoietin (EPO) in mice with mutations preventing iron loading in either site [62]. Additionally, TFR1 has emerging roles beyond iron transport. For instance, TFR1 deficiency in mice protects against high-fat diet-induced metabolic dysfunction by impairing intestinal lipid absorption, though the mechanisms linking adipocytes and the intestine are unclear. In adipocytes, deficiency of this receptor disrupts thermogenesis, increases insulin resistance, inflammation, and mitochondrial dysfunction, effects not replicated by dietary iron deficiency [63].
Cellular iron uptake is tightly regulated by IRPs, which control the translation of key mRNAs, including those for TFR1, DMT1, FPN, and ferritin [35]. IRPs bind to iron-responsive elements in mRNA untranslated regions (UTR), stabilizing or inhibiting translation depending on the binding site, specifically, IRP binding to the 3′ UTR enhances mRNA stability and translation; while binding to the 5′ UTR suppresses translation. Likewise, the RNA-binding protein roquin also plays a regulatory role by destabilizing TFR1 mRNA [64]. Among the IRPs, IRP1 activity is modulated by Fe-S clusters, binding RNA only when these clusters are absent, while IRP2 is regulated through degradation by the FBXL5 E3 ligase in an iron-dependent manner [35]. However, cellular iron import is not limited to the Tf cycle. During iron overload, as observed in SCD, Tf saturation leads to enhanced circulation of non-transferrin-bound iron (NTBI), a potentially toxic form of iron [35]. While the exact nature of NTBI remains unclear, its uptake by the liver and pancreas relies on SLC39A14 also known as ZRT/IRT-like (ZIP14), a protein that primarily transports manganese under normal conditions but facilitates iron transport during iron excess or overload [35].
2.4. Non Cellular Heme Intracellular Iron Trafficking, Utilization, Storage, and Recycling
A substantial portion of cellular iron is directed toward the mitochondria, where it is either stored or utilized for heme and Fe-S cluster synthesis [65]. Research has implicated multiple pathways in mitochondrial iron uptake, including through direct endosome-mitochondria contact (kiss-and-run model) and acquisition from the labile iron pool (LIP) comprising of redox-active, low-molecular-weight iron species [66]. Another important mediator in mitochondria iron regulation are the Fe-S clusters. Biogenesis of these clusters is a complex, multistep process involving sulfur donors, iron supply, and specialized proteins that facilitate cluster synthesis, transport, and incorporation into target proteins [67]. This process occurs in both mitochondria and the cytoplasm. These clusters are critical for electron transfer in the respiratory chain and function as cofactors in DNA metabolism, oxygen sensing, and other essential cellular processes. Mutations in key components of this pathway, such as heat shock cognate B, which transfers clusters to target proteins, have been linked to human diseases like congenital sideroblastic anemia [68]. Impaired lysosomal acidification can also result in disruption of cellular iron uptake, depletion of Fe-S clusters, limit mitochondrial function, and reduce overall cell viability [69]. Notably, defects in Fe-S cluster biosynthesis, such as those associated with frataxin mutations in Friedreich’s ataxia, can be mitigated during hypoxic conditions, as evidenced by improved outcomes in mice exposed to 11% oxygen [70].
Iron not used in cellular processes or directed to mitochondria is stored in ferritin. Ferritin’s heavy chains exhibit ferroxidase activity, enabling iron storage in its ferric form [35]. The delivery of iron to ferritin is mediated by poly(rC)-binding protein 1 (PCBP1), a multifunctional protein with distinct RNA- and iron-binding capabilities that also facilitates iron delivery to other iron-dependent proteins [71]. Notably, liver-specific deletion of Pcbp1 in mice results in disrupted oxidative stress, lipid peroxidation, and steatosis, highlighting its critical role in mitigating cytoplasmic iron toxicity [72]. Along these lines, a recent study revealed that exosomal release of ferritin-rich vesicles is regulated by IRP and involves the extracellular vesicle marker CD63 [73]. One of ferritin’s most important functions is mitigating oxidative damage by reducing the pool of free iron available for ROS generation. Free excess iron catalyzes Fenton reactions which result in increased production of hydroxyl radicals that damage lipids, proteins, and DNA. Thus, by storing iron as Fe³⁺ in a safe, non-reactive form, ferritin minimizes oxidative stress and protects hepatocytes from ROS-related cellular injury [72,74].
Iron mobilization from ferritin is regulated by ferritinophagy, a selective autophagy process driven by NCOA4 [42]. Its expression is upregulated during iron deficiency via hypoxia-inducible factors (HIFs) and downregulated under iron excess through proteasomal degradation [42]. NCOA4 deficiency in mice results in ferritin and iron accumulation in tissues, reduced serum iron levels, and anemia. Tissue-specific deficiencies highlight its role in erythropoiesis and hepatocyte iron mobilization during blood loss [74]. In vitro, NCOA4 deficiency disrupts mitochondrial function, further emphasizing its importance in cellular iron metabolism [74].
2.5. Iron Elimination and Export
Iron excretion occurs mostly via passive, unregulated pathways, such as the shedding of intestinal and skin epithelial cells, menstruation, and minor/limited epithelial trauma. Recent studies have shed light on additional mechanisms of iron excretion using a Tf-deficient mouse model, in which Tf treatment promoted gastrointestinal iron excretion, leading to normalization of iron levels [75]. Additionally, hepatic uptake of NTBI via SLC39A14 was identified as a prerequisite for biliary excretion of ferritin-bound iron, though its role in overall iron homeostasis remains unclear [76]. Paradoxically, a study on anemic children using stable iron isotopes revealed increased iron losses during iron supplementation, potentially linked to occult gastrointestinal bleeding, however if this is not the case, the exact pathways mediating this phenomenon will require future investigation [77].
FPN (SLC40A1) is critical for exporting iron from cells involved in storage and recycling, such as enterocytes, macrophages, and hepatocytes [78]. It transports Fe²⁺ into the bloodstream, where it is oxidized to Fe³⁺ by ferroxidases like hephaestin or ceruloplasmin, enabling Tf-mediated systemic transport [79]. As mentioned previously FPN's role in iron homeostasis makes it a primary target of hepcidin regulation. This regulatory step adjusts plasma iron levels in response to systemic signals during iron sufficiency, deficiency, inflammation, or increased erythropoiesis. Elevated hepcidin during iron sufficiency or inflammation inhibits FPN, reducing iron efflux and limiting plasma iron availability; conversely, low hepcidin levels during iron deficiency or heightened erythropoietic activity allow FPN to remain active, promoting iron mobilization for essential functions like hemoglobin synthesis [80]. In hereditary hemochromatosis, hepcidin production is reduced, causing excessive FPN activity and systemic iron overload which results in oxidative stress, liver fibrosis, cirrhosis, and potentially hepatocellular carcinoma [81]. On the other hand, chronic inflammation elevates hepcidin levels, suppressing FPN activity and trapping iron within macrophages [82]. As a result, this functional iron deficiency contributes to anemia despite adequate iron stores.
2.6. Pathways Modulating Hepcidin
2.6.1. Erythropoietin-Responsive Factor Erythroferrone (ERFE)
The renal medulla, which under physiologic conditions consumes significant energy, monitors oxygen delivery by monitoring hemoglobin levels, oxygen binding, and oxygen release [59]. When oxygen delivery falls short of demand, interstitial fibroblasts detect hypoxia and produce EPO [59] and its production is closely regulated by HIF-2 [83]. In response to EPO, erythroid precursors in the bone marrow divide and mature into erythrocytes, a process requiring iron for heme and hemoglobin synthesis. Thus, iron deficiency disrupts heme and hemoglobin production and impairs erythrocyte development [84]. As a result, at baseline, erythropoiesis consumes most circulating plasma iron [36]. When erythropoiesis is stimulated—by hypoxia, blood loss, or exogenous EPO—it suppresses hepcidin which enables increased iron absorption from the diet and mobilization from stores to meet the demand for heme and hemoglobin production [85].
However, EPO does not directly suppress hepcidin but instead uses an intermediary EPO-responsive factor erythroferrone (ERFE) [86]. Once plasma iron levels and liver iron stores increase, hepcidin synthesis is stimulated by the activation of the Smad1/5/8 pathway [87]. This activation is driven by BMPs, especially the BMP2/6 heterodimer, which binds to the BMP receptor complex [87,88]. ERFE effectively suppresses this pathway through a proposed signaling with matripase-2, a serine protease encoded by the TMPRSS6 gene, which cleaves the hepcidin activator HJV [89]. However, this mechanism has been disputed by studies using TMPRSS6−/− mice, which mimic iron-refractory iron deficiency anemia, showing that elevated BMP-SMAD signaling leads to increased hepcidin production despite increased ERFE levels [90]. In this model, disruption of ERFE showed minimal impact on their hematological phenotype or hepcidin production. Furthermore, in vitro studies of hepatocytes from TMPRSS6−/− mice showed that ERFE suppresses hepcidin expression independently of TMPRSS6, even under BMP-stimulated conditions [90]. This can be due to ERFE lowering hepcidin levels via binding of BMP ligands and preventing them from interacting with their cell surface receptor ALK3 [91].
Erythroferrone Pathophysiology—Baseline Erythropoiesis and Stress Erythropoiesis
Under normal conditions, the bone marrow constantly produces red blood cells (RBCs) to replace older or damaged ones, while most of their iron content from dead/damaged cells is recycled by macrophages in the spleen and liver. When anemia or low oxygen levels occur, kidney cells sense the reduced oxygen supply and respond by increasing EPO production through HIF-2 signaling [92,93]. This higher EPO synthesis enhance the survival of RBC precursors, boost their numbers, and prompt these precursors to secrete ERFE [93]. ERFE then lowers hepcidin production in the liver resulting in increased FPN activity, allowing cells to release additional iron into the bloodstream [94]. As a result, dietary iron absorption rises, and stored iron from macrophages and hepatocytes becomes available for hemoglobin production in new RBCs. ERFE is especially critical early in the response to increased RBC demands as shown in ERFE-deficient mice, in which hepcidin suppression after blood loss or EPO treatment is delayed, slowing anemia recovery by several days compared to wild type mice [94]. When anemia develops, ERFE production increases for two main reasons, EPO stimulation causes the pool of erythroid precursor cells to expand, and second, each of these precursors increases ERFE production [95]. However, during ineffective erythropoiesis, although there are far more erythroid precursors driven by high EPO levels, most of them fail to mature into healthy RBCs. Instead, these “stalled” precursors release large amounts of ERFE, persistently lowering hepcidin and leading to iron overload [95]. This excess iron that fails to bind to Tf can then generate harmful ROS. This iron-driven toxicity subsequently damages cells particularly sensitive to high iron levels such as liver, heart, and endocrine glands, while also raising the infection risk [95].
Erythroferrone Variants
Excessive ERFE levels play an important role in iron-loading anemias. Variations in the ERFE gene alter the protein’s activity. affect how long the ERFE mRNA or protein persists, or both [96]. For example, a point mutation in the C1q domain (A260S) leads to higher ERFE RNA and protein levels than normal, even in healthy individuals [96]. This same mutation is also present in some patients with congenital dyserythropoietic anemia type II, who already have markedly increased ERFE levels due to ineffective red blood cell production [97]. In these patients, the mutation pushes ERFE RNA and protein levels even higher, causing more severe anemia [97]. The increased ERFE worsens the condition by raising iron availability for erythropoiesis, however, in doing so boosting oxidative stress in a background of already dysfunctional erythroblasts.
Erythroferrone as a Biomarker in Chronic Kidney Disease
Chronic kidney disease (CKD) often leads to anemia due to both lower EPO levels and limited iron availability [98]. This anemia has several causes, including inflammation, reduced EPO production, and poor hepcidin clearance by the damaged kidneys. Thus, current treatment guidelines from expert panels recommend regularly checking RBC counts and iron levels, and supplementing iron and EPO if patients become deficient [99,100]. Of interest, using an animal model of CKD, it was shown that removing hepcidin improved anemia suggesting that in patients whose anemia is mainly due to high hepcidin levels, future therapies that mimic or boost ERFE might prove therapeutic [98]. Although giving EPO does increase ERFE naturally, it happens more slowly in CKD mice compared to wild type [101]. Therefore, this delay reduces EPO’s effectiveness at releasing iron, suggesting that directly supplementing ERFE might help mobilize iron and improve anemia in CKD.
Myelodysplastic Syndromes and ERFE
Myelodysplastic syndromes (MDS) are blood disorders where immature blood cells fail to develop properly, often dying in the bone marrow. Over time, surviving abnormal cells gain additional mutations and progress toward leukemia. A subtype called MDS-RS (with ring sideroblasts) features immature RBCs loaded with excess iron in their mitochondria, preventing them from properly maturing and entering the bloodstream [102]. Many MDS patients—especially those with MDS-RS—have mutations in splicing factor genes, most notably SF3B1 [102,103]. Those patients with SF3B1 mutations and MDS-RS often develop systemic iron overload, even without receiving transfusions. Notably, reports have indicated the presence of mutated ERFE transcripts in cells with SF3B1 mutations encoding an ERFE protein variant with four extra amino acids, but still capable of suppressing hepcidin as effectively as native ERFE [104]. These mutated cells synthesize more ERFE than normal cells, and this higher production is linked to improved survival in SF3B1-mutant MDS patients. Thus, since ERFE is produced in erythroblasts, measuring it could prove useful in managing MDS-RS [104].
Beta-Thalassemia and ERFE
Animal models of anemias involving ineffective erythropoiesis generally exhibit markedly elevated ERFE levels, because the number of ERFE producing erythroblasts expands beyond what would be expected given the degree of anemia. For example, the Hbb(th3/+) mouse—representing a non–transfusion-dependent form of β-thalassemia—displays moderately reduced hemoglobin levels, significantly lower hepcidin during growth, and elevated plasma and liver iron concentrations [105]. This arises from β-globin haploinsufficiency, which leads to the buildup of unpaired α-globin chains. In these mice, ERFE expression in bone marrow and spleen remains consistently high (8- to 32-fold above wild-type) across all ages studied [106]. However, use of ERFE-specific neutralizing antibodies alleviates anemia in this model. This indicates that excess ERFE contributes to both iron overload and development of anemia [106,107]. Notably, antibody treatment also reduces the reticulocyte percentage, implying a possible improvement in RBC quality and lifespan using this approach. However, it remains to be determined whether ERFE inhibition in humans with β-thalassemia can sufficiently reduce iron absorption and deposition to eliminate the need for concurrent iron-chelation therapies.
2.6.2. BMP Signaling via SMAD1/5/8 Transcription Factors
The earlier mentioned BMP-SMAD pathway regulates hepcidin and this modulates iron levels through its binding of FPN inducing both its internalization and degradation. This process restricts iron absorption from the intestine and limits release from cellular stores. This signaling pathway orchestrates the transcriptional regulation of the HAMP gene, in response to systemic iron levels and other regulatory feedback signals [108]. It should become evident by now, hepcidin as the master regulator of systemic iron homeostasis, is controlled by multiple pathways converging on its transcriptional and post-transcriptional regulation to maintain a balance between iron availability and storage. Consequently, its expression is transcriptionally regulated through a feedback loop that allows hepatocytes to sense circulating iron levels. This regulation depends on several proteins on hepatocytes’ plasma membrane, including HFE, TFR2, and HJV, which collectively modulate hepcidin synthesis via the BMP-6 signaling pathway [108]. BMP-6, a member of the TGF-β superfamily, is produced in hepatocytes, with its expression upregulated by increased liver iron concentrations [109]. BMP-6 binds to BMPR and HJV, a co-receptor, initiating an intracellular signaling cascade mediated by SMAD proteins [109]. Likewise, even though BMP-6 is the primary driver to stimulate hepcidin expression, phosphorylated SMAD1, SMAD5, and SMAD8 form complexes with SMAD4 which translocate to the nucleus to also activate the HAMP gene [110]. On the other hand, hepatocytes’ TFR1 and TFR2 along with HFE, function as iron sensors to monitor Tf-bound iron (Tf-Fe). In this way, under low iron conditions HFE binds to TFR1, and when iron levels rise and Tf-Fe saturates TFR1 and HFE are displaced [110]. The freed HFE then associates with TFR2, forming an HFE/TfR2 complex which interacts with HJV, activating the BMP/SMAD pathway and promoting hepcidin production; any disruption of this regulatory mechanism, such as HFE deficiency, can result in hereditary hemochromatosis [110].
NRF2 Activation Under Iron-Induced Oxidative Stress
Under normal conditions, the transcription factor NRF2 is bound to Keap1, a protein that mediates its ubiquitination and proteasomal degradation [111]. Oxidative stress, especially from iron-induced ROS, modifies cysteine residues on Keap1, preventing NRF2 degradation and stabilizing it; this will subsequently translocate to the nucleus, where it binds to antioxidant response elements in the promoter regions of target genes [112]. These genes encode antioxidant enzymes such as heme oxygenase-1 (HO-1), NADPH quinone oxidoreductase 1, and glutamate-cysteine ligase catalytic subunit (GCLC), among others [112]. NRF2 also regulates and promotes expression of ferritin, which stores excess iron safely, and FPN, the sole mammalian iron exporter [113]. These actions decrease the LIP, mitigating ROS production and maintaining redox homeostasis. While NRF2 activation protects cells from oxidative damage, its hyperactivation in cancer cells contributes to tumor progression [114]. In particular, cancer cells, characterized by high iron demands for proliferation and metabolism, produce excessive ROS due to increased mitochondrial activity [114]. Hyperactive NRF2 counteracts ROS, supporting cancer cell survival, proliferation, and resistance to therapies such as chemotherapy and radiotherapy [112]. NRF2 also facilitates cancer cell adaptation to ferroptosis, a form of regulated cell death driven by lipid peroxidation and iron accumulation [115]. Thus, by inducing antioxidant defenses and regulating iron export, NRF2 diminishes ferroptosis sensitivity, offering a survival advantage to cancer cells [115]. As a result, inhibition of NRF2 can restore ferroptosis sensitivity, presenting a potential therapeutic strategy in malignancies.
2.6.4. IL-6 Pathway
Inflammatory cytokine interleukin-6 (IL-6) plays a pivotal role in triggering hepcidin production through the IL-6 receptor (IL-6R)/STAT3 signaling pathway [116]. During infections involving iron-dependent pathogens, inflammatory cytokines such as IL-6 are generated as part of the innate immune response, a process is primarily mediated by macrophages, which release IL-6 in response to infection and inflammation. The increased IL-6 levels activate STAT3 signaling, leading to a rise in hepcidin production and subsequent iron sequestration [117]. The IL-6R/STAT3 pathway is a vital signaling mechanism playing a crucial role in immune responses, inflammation, and physiological processes such as iron homeostasis and the acute-phase response [118]. The pathway begins with IL-6 binding to its receptor, IL-6R, which may be membrane-bound or soluble [118]. This IL-6/IL-6R complex associates with gp130, forming a hexameric receptor complex that triggers intracellular signaling. Subsequent activation of Janus kinases (JAK1 and JAK2) associated with gp130 leads to phosphorylation of tyrosine residues, creating docking sites for the transcription factor STAT3 [117]. STAT3 is subsequently phosphorylated, dimerizes, and translocates into the nucleus, where it binds to specific DNA sequences, known as STAT3-responsive elements, on the promoters of target genes [119]. Through this mechanism, STAT3 regulates the transcription of genes involved in hepcidin production, acute-phase proteins, and cellular processes such as survival, proliferation, and differentiation [117]. IL-6-induced STAT3 signaling upregulates hepcidin expression, sequestering iron to limit its availability to pathogens during infections while enhancing immune responses by promoting production of inflammatory mediators and acute-phase reactants [118]. Therefore, dysregulation of the IL-6R/STAT3 pathway is implicated in chronic inflammatory diseases, autoimmune disorders such as rheumatoid arthritis, and certain cancers [120]. Therapeutic targeting of this pathway, such as with IL-6R inhibitors may prove effective in managing these conditions.
2.6.5. ZIP14
ZIP14, a member of the ZRT/IRT-like protein family, is the primary transporter for NTBI in the liver. Located on the basolateral membrane of hepatocytes, ZIP14 facilitates NTBI uptake during iron overload, aided by its metal-binding residues and structure comprising of eight transmembrane domains [121]. NTBI internalization depends on prior iron reduction mediated by surface ferrireductases such as DCYTB and STEAP proteins, and prion protein (PrPᴰ) [122]. ZIP14 plays a crucial role in protecting extrahepatic tissues from iron toxicity by sequestering NTBI in the liver, particularly during iron overload conditions like hereditary hemochromatosis [123]. In such cases, dysregulated hepcidin and FPN activity lead to excessive iron absorption, Tf saturation, and appearance of NTBI in plasma, which ZIP14 efficiently removes [121]. However, prolonged hepatic iron accumulation due to ZIP14 activity can contribute to fibrosis, cirrhosis, and hepatocellular carcinoma if left untreated [124]. Importantly, regulation of ZIP14 expression by inflammatory cytokines like IL-6 couples increases in iron uptake during infection and chronic inflammation, highlighting its relevance in these settings [123]. ZIP14 localizes is upregulated in response to ferric ammonium citrate [121]. In contrast, DMT1 is crucial for iron acquisition during deficiency, with protein levels increasing by 200% under iron-deficient conditions, driven by elevated mRNA levels [125]. DMT1 dominates in the pancreas, where its mRNA levels are 3.8 times higher than ZIP14, supporting its role in the Tf cycle [126]. Together, these transporters enable the liver to maintain systemic iron balance: ZIP14 handles NTBI during overload, while DMT1 supports Tf-Fe iron uptake during deficiency. This adaptability underscores their therapeutic potential for addressing iron-related disorders such as hemochromatosis and iron-deficiency anemia [126].
2.6.6. Prion Protein
PrPᴰ appears to play an important role in hepatic iron uptake and storage. PrPᴰ facilitates the absorption of both Tf-Fe and NTBI in the liver through its ferrireductase activity [127], by reducing Fe³⁺ to Fe²⁺ for cellular uptake [128]. Experiments with PrP−/− mice revealed that absence of PrPᴰ significantly impairs NTBI uptake and iron storage, as demonstrated by reduced NTBI uptake and lower 59Fe-ferritin levels in these mice [128]. Additionally, PrPᴰ enhances iron transport by upregulating ZIP14 and DMT1 activity, while co-expressing ZIP14 and DMT1 modulates PrPᴰ processing, decreases full-length PrPᴰ and increases truncated forms, potentially via mechanisms involving PrPᴰ recycling or selective cleavage [128]. These findings suggest an important role of PrPᴰ in hepatic iron regulation and its interactions with known iron transport pathways.
3. Bone Marrow and Heme Iron
Heme iron bioavailability is enhanced by the alkaline pH of the small intestine, which prevents polymerization. While nonheme iron uptake mechanisms are well-characterized, heme iron absorption remains less understood but likely involves two potential mechanisms: receptor-mediated endocytosis and membrane transport [129]. Historically, the heme receptor was described as a high-affinity, pH-dependent heme-binding protein in the small intestine with activity influenced by trypsin digestion [130]. Importantly, iron deficiency was shown to enhance heme uptake and binding by duodenal cells [131]. Localization studies would reveal heme accumulation at microvilli and in endosomal compartments of duodenal cells [132]. Heme carrier protein 1, now known as the proton-coupled folate transporter (SLC46A1), was originally misidentified as an intestinal heme importer but is primarily a high-affinity, pH-dependent folate transporter [133]. Mutations in SLC46A1 lead to hereditary folate malabsorption in humans, which is treatable with folate supplementation and does not affect iron metabolism. However, the mechanism of intestinal heme transport remains generally unclear due to lack of an appropriate genetic model, as mice are inefficient heme absorbers [134]. HRG1, a four-transmembrane-domain heme transporter involved in macrophage iron recycling, is a promising candidate for intestinal heme absorption [135]. Expressed in the human small intestine, HRG1 facilitates heme import through endocytic compartments [136].
3.1. Heme Synthesis
The synthesis of 5-aminolevulinate (ALA) from succinyl-CoA and glycine, catalyzed by ALA synthase (ALAS), is the first and rate-limiting step of heme biosynthesis occurring in the mitochondrial matrix [137]. ALAS exists in two isoforms: ALAS1, expressed ubiquitously, and ALAS2, specific to erythroid cells. Interestingly, while heme tightly regulates ALAS1 at multiple levels, ALAS2 is not subject to heme-mediated regulation [137]. Heme regulates ALAS1 by destabilizing its mRNA, promoting degradation, and inhibiting both its maturation and mitochondrial import [138]. After its synthesis, ALA exits the mitochondria via the SLC25A38 gene product, though this is also involved in glycine transport and Fe-S cluster biogenesis [139,140]. In the cytosol, two ALA molecules are enzymatically combined by ALA dehydratase to form porphobilinogen but this enzyme requires an Fe-S cluster for function [141]. Four porphobilinogen units are subsequently assembled into hydroxymethylbilane by hydroxymethylbilane synthase which is first converted into uroporphyrinogen III by uroporphyrinogen synthase, followed by decarboxylation by uroporphyrinogen decarboxylase to form coproporphyrinogen III, which re-enters the mitochondria [142]. There, the latter is sequentially converted first into protoporphyrinogen IX and later to protoporphyrin IX (PPIX) by enzymes coproporphyrinogen oxidase and protoporphyrinogen oxidase, respectively. Finally, ferrochelatase (FECH) catalyzes insertion of Fe²⁺ into PPIX to produce heme on the inner mitochondrial membrane [142]. Iron delivery to FECH is mediated by mitoferrin 1 and 2; and studies have revealed that FECH interacts with ALAS and protoporphyrinogen oxidase, as well as mitochondrial α-ketoglutarate dehydrogenase, succinyl-CoA synthase, and the porphyrinogen transporter TMEM14C [143]. Additionally, FECH forms complexes with mitoferrin, ABC transporters ABCB7 and ABCB10, and heme chaperones PGRMC1 and PGRMC2 [144].
3.1.1. Protoporphyrin IX
PPIX requires eight molecules of glycine (from plasma) and eight molecules of succinyl-CoA to form the tetrapyrrole macrocycle [145]. The plasma membrane glycine transporter 1 and mitochondrial transporter SLC25A38 are essential for normal erythropoiesis [146]. Deficiencies in either transporter has a negative impact on the synthesis of heme and results in anemia, specifically, mutations in the SLC25A38 gene are responsible for inherited recessive sideroblastic anemia [140,146,147].
3.1.2. Posttranscriptional Modifications
Phosphorylation, lysine acylation, and cysteine glutathionylation are key to regulating metabolic pathways and their disruption can result in disease states [148,149,150]. The [2Fe-2S] cluster in FECH is sensitive to the cell’s acidity (pH) and membrane charge, meaning it adjusts FECH's activity based on the cell's energy state and environment and in this way regulating heme synthesis coupling it to the cell's overall energy and redox balance [151].
3.1.3. Dysfunctional Heme Synthesis
Unlike iron deficiency anemia, which primarily arises from insufficient iron for heme synthesis, anemia of chronic disease/inflammation (ACD) involves dysfunctional heme synthesis among additional factors [152]. One such factor is the impact of inflammation over erythropoiesis, mediated by macrophages in erythroblastic islands that interact closely with developing erythroid cells [152]. During inflammation, macrophages upregulate the Irg1 gene, which encodes aconitate decarboxylase responsible for catalyzing the production of itaconate, an antimicrobial compound that disrupts the tricarboxylic acid cycle by inhibiting succinate dehydrogenase, leading to increased succinate [153,154,155]. While itaconate is not synthesized by erythroid cells, it is actively transported into them to be converted to itaconyl-CoA via succinyl-CoA:glutaryl-CoA transferase, which exchanges succinate in succinyl-CoA with itaconate [156]. This process reduces cellular succinyl-CoA, a critical substrate for ALAS2, the first enzyme in the heme synthesis pathway [156]. Furthermore, itaconyl-CoA directly inhibits ALAS2 as a competitive inhibitor, hereby reducing production of ALA, the initial heme precursor. Itaconyl-CoA also inhibits ketoglutarate dehydrogenase, limiting succinyl-CoA availability. This duality of itaconate—depleting succinyl-CoA and inhibiting ALAS2—reduces hemoglobin production during inflammation [156]. This resembles the role of hepcidin, which inhibits heme synthesis by regulating iron [157]. Together, itaconate and hepcidin act as complementary mechanisms to limit the production of potentially toxic intermediates (iron and protoporphyrin) during inflammation, tightly regulating heme synthesis in ACD. ALAS2 is itself regulated at both translational and post-translational levels to fine-tune heme synthesis. Under low iron conditions, apo-IRP1 binds to the 5′ UTR of ALAS2 mRNA inhibiting its translation; while when iron is plentiful, holo-IRP1 (containing an Fe–Su cluster) acts as a cytosolic aconitase that interconverts citrate and isocitrate [158]. Post-translationally, the mitochondrial protease complex CLPXP—consisting of the unfoldase CLPX and the protease CLPP—governs ALAS2 turnover, and in erythroid cells CLPX not only regulates ALAS2 levels but also influences other terminal heme synthesis enzymes (protoporphyrinogen oxidase and FECH) as well as mitochondrial iron metabolism [159].
3.2. Heme in Erythrocytes
In human bone marrow and fetal liver, hematopoietic stem cells sequentially generate burst-forming and colony-forming erythroid progenitors, then proerythroblasts, followed by basophilic, polychromatic, and orthochromatic erythroblasts that will give origin to 2.5 billion erythrocytes per second [160]. Late-stage erythroblasts undergo enucleation and organelle loss to form reticulocytes, which enter the bloodstream and mature into RBCs [161]. The transition from proerythroblast to erythroblast occurs in specialized “erythroblastic islands,” where a central nurse macrophage supports a surrounding ring of RBC precursors, a process during which developing erythrocytes demand high levels of iron to fuel heme and hemoglobin synthesis [161]. Despite the precise molecular mechanisms through which iron and heme regulate erythropoiesis being poorly understood, one key known regulatory factor in erythroid cells is the heme-regulated eIF2α kinase (HRI). This protein aligns globin mRNA translation with heme availability to ensure balanced hemoglobin synthesis [162]. Under conditions of heme deficiency, HRI becomes activated and phosphorylates eIF2α, thereby inhibiting globin mRNA translation and preventing the deleterious precipitation of unbound globins [162]. In regard to enucleation and erythroblast maturation, both are regulated by the transcription factor FOXO during which mitochondria aggregate around the nucleus—an event driven primarily by pyruvate and required for enucleation [163]. Notably, RBCs retain mitochondria even after the enucleation, allowing heme synthesis and other metabolic activities through cells lifespan.
3.3. Systemic Heme Recycling, Transport, Sequestration, Degradation, and Elimination
During erythrophagocytosis, the heme importer HRG1 on phagolysosomal membranes transports heme into the cytosol, where it is subsequently catabolized by HO-1 and HO-2 [136]. These enzymes, anchored to the endoplasmic reticulum (ER) membrane with their active sites facing the cytosol, release iron to be stored in ferritin or exported via FPN for new RBC production. Of note, HRG1, HO, and FPN are transcriptionally upregulated during erythrophagocytosis [136,164]. In vivo, deletion of HO-1 is embryonically lethal, whereas Hrg1-deficient mice survive but accumulate excess heme [165]. Double knockouts of both genes are nonviable, while partial knockout of Hrg1 in Hmox1-null mice causes 40% lethality, indicating an indispensable genetic interaction between both proteins [165]. Recent studies have identified that the cation channel PIEZO1 is an important mediator in macrophage iron homeostasis since mice with a gain-of-function (GOF) of its gene linked to hereditary xerocytosis showed late-onset iron overload [166]. A similar phenotype was observed when the mutation was restricted to macrophages, that was accompanied by elevated RBC turnover, enhanced erythropoiesis, increased erythroferrone expression, and reduced hepcidin levels [166]. In contrast, in hepatocytes expression of GOF PIEZO1 mutants increases calcium influx and activates ERK signaling, ultimately inhibiting BMP-SMAD signaling and suppressing hepcidin expression [167].
Vascular hemolysis releases free heme and hemoglobin into the bloodstream where they are predominantly cleared by acute-phase proteins [168]. Hemopexin sequesters free heme, forming a complex that is internalized via CD91/lipoprotein receptor–related protein 1 (LRP1) on hepatocytes, macrophages, and neurons, while haptoglobin binds free hemoglobin, enabling uptake through CD163-mediated endocytosis on macrophages [169]. Unlike the haptoglobin–hemoglobin complex, which is ultimately degraded, hemopexin can be recycled after delivering heme. Circulating albumin also binds free heme and forms a complex that is endocytosed via the TFR. These heme-scavenging pathways mitigate toxicity and oxidative stress, partly by activating HO-1 [169]. Hemopexin has been studied extensively in hemolytic disorders such as SCD and together with haptoglobin exhibit neuroprotective effects in ischemia and intracerebral hemorrhage [170]. Once in the liver, heme degradation is mediated by HO-1 and HO-2, the former stress-inducible while the latter is constitutively expressed [171]. Heme oxygenases are primarily expressed in macrophages of the reticuloendothelial system but can be induced in various cells under stress [172]. These enzymes cleave Fe³⁺-PPIX to produce biliverdin, carbon monoxide (CO), iron, and water [18]. Biliverdin is subsequently reduced to bilirubin by biliverdin reductase using NADPH. The iron released is stored in ferritin, while CO participates in signaling and binds iron in hemoproteins. HO-1 transcription is regulated by redox-responsive transcription factors, including NRF2 and HIFs, and is negatively controlled by BACH1, a heme-binding repressor [173]. Heme oxygenases also play a protective role in various human diseases, including cardiovascular, neurodegenerative, neoplastic, metabolic, and inflammatory disorders [174]. In conditions of severe hemolysis, such as SCD, excessive heme is released, accompanied by low levels of hemopexin [170].
Alpha-1-microglobulin acts as a secondary scavenger, directing heme to the kidneys, which are key sites for hemoglobin and myoglobin clearance [175]. Hemoglobin interacts with renal proximal tubules through endocytic receptors megalin and cubilin, with megalin mediating reabsorption under normal conditions and cubilin becoming active during hemoglobinuria [176]. However, hemoglobin is nephrotoxic, damaging tubular epithelium, impairing distal tubule function through precipitation, and causing vasoconstriction via nitric oxide scavenging. Injury to proximal tubules, which are rich in mitochondria, exacerbates damage by releasing mitochondrial cytochrome heme [176]. These processes contribute to acute kidney injury during hemolytic stress and rhabdomyolysis, which are significant causes of mortality in SCD [176].
Hemoparasites like Plasmodium crystallize heme into chemically stable hemozoin that involves histidine-rich proteins in acidic condition to mitigate toxicity in the absence of a heme degradation system [177], as well as lipids and a parasite-derived protein (PV5) [178]. Although hemozoin’s structure is well-characterized, its in vivo formation mechanisms remain unclear [178]. In Hrg1-deficient mice, hemozoin accumulates in enlarged lysosomes of macrophages that promote heme tolerance. Whether hemozoin serves as a bioavailable iron source during deficiency remains to be elucidated, but targeting its formation could offer new therapeutic options for hemolytic anemias [165]. Additionally, the lysosomal abnormalities in Hrg1-deficient mice suggest possible links between hemozoin formation and lysosomal storage disorders [165].
FLVCR1 has been identified as a plasma membrane heme exporter [179], which has been confirmed in both rat renal epithelial cells and erythroid cell lines [180]. FLVCR1-deficient mice exhibit defective erythropoiesis, mid-gestation lethality, craniofacial and limb deformities, as well as impaired sensory neuron maintenance and defective T cell development [181]. FLVCR1 also mediates heme-iron recycling by exporting heme from macrophages that engulf senescent RBCs. An isoform, FLVCR1b, localized to mitochondria, regulates erythropoiesis by exporting mitochondrial heme and its loss leads to mitochondrial heme accumulation and disrupted erythroid differentiation [182]. FLVCR1a and FLVCR1b also interact with hemopexin to facilitate heme export, but mechanisms remain to be elucidated [183]. FLVCR2 (MFSD7C), initially proposed as a cell surface heme importer, is linked to Fowler syndrome, a condition involving proliferative vasculopathy in the brain [184]. Evidence supporting its role in heme transport includes its binding to hemin-conjugated agarose and reduced heme import following FLVCR2 silencing. Cells expressing FLVCR2 exhibit enhanced heme uptake and increased sensitivity to heme toxicity; however, recently FLVCR2 has been localized to the mitochondria with a potential role in thermogenesis in response to heme [185]. Notably, FLVCR2 expression in yeast does not rescue growth in heme-deficient strains, highlighting the need for further research to clarify its localization and full function in heme transport [186].
MRP-5/ABCC5 has been identified as a heme exporter in C. elegans, localizing to the basolateral membranes of the intestinal cells [187]. As a member of the ABC transporter superfamily, its deficiency results in embryonic lethality and is associated with multidrug resistance, including roles in cancer therapy. Knockdown of mrp5 in zebrafish leads to severe anemia, highlighting its importance in heme transport. MRP-5 is found on the plasma membrane, Golgi complex, and recycling endosomes, thus supporting its involvement in heme export and delivery to hemoproteins [187]. In mammals, its homolog, MRP9, plays a compensatory role in maintaining heme homeostasis [188].
ABCG2, also known as the breast cancer resistance protein, has been suggested as a cell surface heme exporter [189]. Abcg2-deficiency causes extreme photosensitivity due to the accumulation of pheophorbide, a compound structurally similar to PPIX [142]. While ABCG2 exhibits broad substrate specificity for drugs and xenobiotics, its precise physiological substrate has not been reported [190]. However, structural studies point to having a role in transporting heme, PPIX, and other porphyrins [190]. Proteomic analysis has identified eight putative ABC transporters on RBC membranes involved in porphyrin efflux, including mitochondrial transporters ABCB6, ABCB7, ABCB8, and ABCB10 [191]. Of note, ABCB6 initially linked to coproporphyrinogen III transport in the outer mitochondrial membrane has since been found in plasma membranes, endolysosomal compartments, and exosomes of reticulocytes [192]. Specifically, ABCB6 mediates porphyrin transport, although its specific substrate is still debated [193]. On the other hand, ABCB7 restores iron homeostasis and cytochrome levels and is implicated in heme biosynthesis through interactions with FECH, and mutations in ABCB7 are associated with X-linked sideroblastic anemia and mitochondrial iron overload [194]. Similarly, ABCB10 interacts with FECH and the mitochondrial iron importer mitoferrin, further coupling ABC transporters to heme metabolism and mitochondrial function [183].
4. Central Nervous System
Iron is indispensable for CNS function, contributing to essential processes such as neuronal energy metabolism, axonal myelination, and neurotransmitter synthesis. However, its redox activity poses a dual threat, as uncontrolled iron can catalyze production of toxic free radicals that can lead to oxidative stress and neuronal damage. As a result, a tightly regulated system governs iron homeostasis within the CNS to ensure simultaneous availability and detoxification when in excess. Dysregulation of this system is increasingly recognized as a central factor in several neurodegenerative diseases. The molecular signaling pathways involved in iron regulation and their dysfunction in pathological conditions provide insights into mechanisms of disease progression and opportunities for therapeutic intervention.
4.1. Normal CNS Mechanisms of Iron Traffic and Homeostasis
Iron enters the CNS primarily through the blood-brain barrier (BBB) in either Tf-bound or non-Tf-bound forms [195]. The Tf/TFR1 complex facilitates Fe3+ uptake, where Fe3+ is reduced to Fe2+ in endosomes by STEAP3 followed by Fe2+ transport to the cytoplasm via DMT1 [196,197]. Iron is stored intracellularly in ferritin cages composed of heavy (FTH) and light (FTL) chain subunits, where it is kept in a redox-inactive ferric state to prevent oxidative damage from ROS [198,199]. Export of iron is mediated by FPN with the oxidizing assistance of ferroxidases such as ceruloplasmin and hephaestin. Hepcidin—a key modulator of CNS iron homeostasis—binds to FPN and induces its ubiquitin-mediated degradation, thereby preventing further iron release into circulation [200,201]. This multi-level regulation ensures iron availability for metabolic needs while mitigating potential toxicity.
4.2. The NRF2/GPX4 Axis in Antioxidant Defense
This axis plays a pivotal role in protecting neuronal cells from ferroptosis. NRF2 is a transcription factor sequestered in the cytoplasm under normal conditions by Keap1. During oxidative stress, NRF2 dissociates from Keap1 and enters the nucleus to induce expression of genes such as GPX4, which neutralizes lipid peroxides by reducing lipid hydroperoxides to non-toxic alcohols [199,202]. GPX4's activity relies on glutathione (GTH), whose levels and biosynthesis are also under NRF2 regulation [203]. Disruption of the NRF2/GPX4 pathway has been linked to neurological disorders such as Parkinson’s and Alzheimer’s disease; and reduction in NRF2 activity severely restricts antioxidant defenses, exacerbating neuronal vulnerability to lipid peroxidation and ferroptosis [199,204]. Elevated ROS through mechanisms like the Fenton reaction further drives the cascade of oxidative damage [197].
4.3. BMP/SMAD-Mediated Hepcidin Regulation
BMP6 secreted by liver sinusoidal endothelial cells binds to receptors on hepatocytes to induce phosphorylation of SMAD 1/5/8 proteins, which form complexes with SMAD4 that translocate to the nucleus to upregulate hepcidin expression [199,205]. Locally in the CNS, astrocytes and microglia express hepcidin in response to inflammatory cytokines such as IL-6 that activates the JAK/STAT3 signaling cascade to drive hepcidin transcription during infection or inflammation [201]. High hepcidin levels in the CNS block FPN activity, thus retaining iron within resident cells to limit free iron availability during infection [196]. On the contrary, chronic hepcidin upregulation fosters excessive intracellular iron accumulation that exacerbates iron-dependent oxidative damage and lipid peroxidation, contributing to neurodegenerative processes in diseases such as multiple sclerosis and Alzheimer’s disease [196,200].
4.4. Ferritinophagy and Iron Availability
Selective autophagic degradation of ferritin, termed ferritinophagy, provides labile iron needed for cellular functions. NCOA4 mediates the ferritin trafficking to autophagosomes for lysosomal degradation, releasing stored iron into the cytoplasm [197,206]. Dysregulation of NCOA4 expression or activity disrupts iron homeostasis and the resulting excessive ferritinophagy enhances cytoplasmic iron levels, promoting ROS production and higher lipid peroxidation and ferroptosis [198]. Conversely, insufficient ferritinophagy in conditions of iron loading impair the cell’s ability to utilize stored iron effectively exacerbating neurotoxicity. NRF2 also influences ferritinophagy by regulating NCOA4 and ferritin heavy chain expression, providing a control mechanism over intracellular iron pools [198]. This reduction in labile iron through reduced ferritin degradation has shown protective effects against ROS-mediated damage in experimental disease models.[195,203].
4.5. Mitochondrial Iron Dysregulation
Mitochondria also play a pivotal role in CNS iron metabolism, acting as sites for Fe-S cluster biosynthesis and energy generation via oxidative phosphorylation. Excessive mitochondrial iron leads to elevated ROS levels, mitochondrial membrane depolarization, and impairments in ATP production [204]. Mitochondrial proteins such as mitoferrin (responsible for mitochondrial iron import) exhibit dysregulated expression in neurodegenerative diseases, further disturbing cellular iron balance [203,204]. Since the role of mitochondria in ferroptosis is increasingly recognized, strategies that target mitochondrial iron dyshomeostasis, such as using antioxidants like MitoTEMPO have shown protective effects in preclinical models of Alzheimer’s and Parkinson’s disease [201,207].
4.6. Neuroinflammation and Iron Dysregulation
Neuroinflammation provides another layer of complexity to CNS iron regulation. Activation of microglia during inflammatory states increases hepcidin secretion, promoting iron sequestration within cells [198]. This localized retention contributes to a feed-forward mechanism of oxidative stress and neurotoxicity, particularly in disorders like multiple sclerosis and traumatic brain injury. Chronic inflammation linked to SCD, accelerates these processes by enhancing hepcidin expression and perpetuating iron imbalance [206,208].
4.7. Role of Iron in CNS aging and Neurodegenerative Diseases
Iron metabolism in the CNS is tightly regulated to support vital processes like myelination, mitochondrial function, and neurotransmitter synthesis while avoiding oxidative stress. With age, iron begins to accumulate in certain brain regions, potentially amplifying inflammation and contributing to neurodegeneration [207]. Reports indicate that iron dysregulation is a hallmark of several diseases, including both acquired conditions like Parkinson’s and Alzheimer’s diseases and congenital disorders such as pantothenate kinase-associated neurodegeneration and Friedreich’s ataxia [207,209]. For instance, excessive iron in the substantia nigra of Parkinson’s patients promotes α-synuclein aggregation and neuronal loss due to oxidative damage [210,211]. Similarly, Alzheimer’s disease pathology is associated with tightly bound iron in cortical areas where oxidative stress and ferroptosis mediate amyloid-beta toxicity and neuronal death [210]. This data highlights the critical need for balanced iron homeostasis in brain aging and pathology.
4.8. Mechanisms Underlying Iron-Induced Neurotoxicity
Iron’s capacity to transition between Fe3+ and Fe2+ oxidation states underlies its dual role as both a physiologic requirement and a threat to cells. Dysregulated iron amplifies production of ROS, which regardless of organ system oxidizes lipids, damages DNA, and disrupts protein functionality [212]. This oxidative stress not only injures neurons but also exacerbates neuroinflammatory pathways. For example, the choroid plexus of the brain, which regulates iron exchange at the blood-cerebrospinal fluid barrier, has been implicated in conditions of iron overload where iron deposition contributes to localized neurodegenerative processes [207].
4.9. Iron Accumulation, Functional Deficiencies and Possible Therapies
Accumulated iron in the CNS does not merely reflect pathology but may also actively propagate it. In Alzheimer’s disease, functional deficiencies arise as CNS iron sequesters into non-bioavailable forms, impairing mitochondrial processes and neuronal metabolism [210]. Downregulation of FPN exacerbates iron retention, further driving oxidative stress and damage. Likewise, disruptions in genes responsible for iron transport and mitochondrial function, such as PITRM1 seen in Alzheimer’s, point to an intricate interplay between iron metabolism and neurodegeneration [210].
Advances in understanding the molecular pathways of iron dysregulation in the CNS have created avenues for targeted therapies. Enhancing NRF2 activity pharmacologically or modulating BMP/SMAD-induced hepcidin expression can recalibrate iron homeostasis and mitigate oxidative stress. Remarkably, iron chelators such as deferoxamine, have shown neuroprotective potential in reducing iron overload and preventing lipid peroxidation [199,201]. Ferritinophagy inhibitors targeting NCOA4 or strategies to restore mitochondrial iron balance further broaden the scope for therapeutic interventions. Furthermore, enhancing the antioxidant defenses of NRF2/GPX4 pathways may mitigate ferroptosis and reduce oxidative stress [212]. Along these lines, compounds that modulate ferritinophagy or regulate iron transport across the blood-brain barrier can alleviate the toxicity of iron overload [207,210]. Additionally, the interplay between trace metal toxicity (e.g., iron and manganese) and their combined impact on neural integrity could provide insights into developing new therapies [212]. The integration of these molecular insights into clinical frameworks holds promise for addressing iron-driven neurotoxicity in neurodegenerative diseases.
5. Iron Metabolism in the Cardiovascular System
5.1. Iron Uptake
Iron binds to TFR1 and is then endocytosed by clathrin protein in cardiomyocytes [213,214]. Decreases in iron levels in cardiac cells increases the expression of IRP1 and IRP2 [214]. Mice lacking TFR1 in the heart develop severe and lethal cardiomyopathy [215]. In hemochromatosis and iron overload states, iron accumulates in many organs including liver, heart, spleen, pancreas and adrenal glands. However, cardiac iron overload is a late manifestation of iron overload presentations. Of note, NTBI uptake in heart is 10-100 times lower compared to other organs [216]. It is mediated by SLC39A14, a member of the ZIP family of metal-ion transporters that is required for NTBI. However, the main role of ZIP transporters is to transport zinc [216,217]. SLC39A14 deficiency has resulted in decreased iron accumulation in liver and pancreatic cells [218]. The other NTBI transporters in heart are calcium channels, mainly L-type and T-type Ca2+ channels [121]. In iron overload states, L-type Ca2+ channels play the principal role in uptake of ferrous iron into cardiomyocytes and possibly drive the initiation of pathologic changes [219].
5.2. Hepcidin-FPN Axis and Iron Export Regulation
Hepcidin exerts its action by degrading FPN in macrophages, intestine and liver as discussed earlier [220]. In ischemic and reperfusion injuries both cellular and mitochondrial functions are deranged, leading to increased iron stores in mitochondria followed by oxidative damage of cardiac cells [221]. Thus, there is a direct association between heart failure and cellular and mitochondrial injuries. Nevertheless, the degree of increased mitochondrial iron stores depends on the type of cardiomyopathy, specifically if it is ischemic/reperfusion, secondary to hemochromatosis or chemotherapy induced [221]. Both iron deficiency and overload states are involved in development of heart failure, and dysfunctional mitochondria in these settings produce ROS that worsens iron metabolism and pathology [221].
5.3. BMP/SMAD Pathway in Hepcidin Regulation
BMPs mediate a critical role in regulating organogenesis and cardiovascular functioning [222]. In the cardiovascular system, the activated macrophages potentiate atherosclerosis by expressing BMP 2, BMP 4 and BMP 6 which are pro-atherogenic whereas BMP 9 is anti-atherogenic [222]. The BMP 6 inhibitor, LDN-193189, suppresses hepcidin and increases FPN expression in macrophages leading to decreased iron storage thus functioning as anti-atherogenic agent [223]. Along these lines, this agent has been reported to prevent atherosclerosis by inducing expression of ABC transporters when injected to an atherosclerotic mouse model, resulting in lower intracellular iron and oxidative stress [224]. This favors the use of this type of agents in clinical trials looking at atherosclerotic patients.
5.4. Ferroptosis in Cardiomyocytes
The involvement of ferroptosis in cardiovascular disease is well established [225]. Stress-induced lipid peroxidation promotes ferroptosis which plays a major role in cardiomyocyte injury during myocardial infarction, ischemic/reperfusion conditions, and heart failure [226]. Ferroptosis can be prevented by the hypoxia inducible factor through increases in iron uptake and expression of TFR1 [227]. FTH and FTL subunits vary among the different organs, for example FTL-rich ferritin is seen in liver and spleen, whereas FTH is found in heart [228]. FTH has ferroxidase activity (converts Fe2+ to Fe3+) and mice lacking FTH1 in cardiomyocytes when exposed to a high-iron diet causes increased lipid peroxidation and hypertrophic cardiomyopathy [229]. Also, FTH-deficient cardiomyocytes have reduced expression of the ferroptosis regulator SLC7A11which is a subunit of the heterodimeric cystine-glutamate antiporter that when overexpressed is associated with increased synthesis of GTH and inhibited lipid peroxidation, thus preventing ferroptosis [228,230]. This can cause injury to cardiac cells reducing the levels of SLC7A11 and GTH-peroxidase 4 levels to decrease ferroptosis [231]. In this regard, ferroptosis inhibitors ferrostatin-1 and liproxstatin have been shown to prevent ferroptosis in liver, kidney, brain and heart in mice [226]. Ferroptosis, however, is also reported to occur in sepsis-induced cardiac inflammation. So, treatment with ferrostatin-1 improves cardiac dysfunction by inhibiting the TLR4/NF-κB signaling pathway preventing cardiac injury secondary to sepsis [232].
5.5. Iron Deficiency and Heart Failure
The biomarkers fatty acid binding protein-4, growth differentiation factor-15, NT-proBNP, osteopontin, ST2 protein, tumor necrosis factor receptor-1, and TFR1 are increased in iron deficiency states whereas the paraoxonase-3 and tartrate-resistant acid phosphatase type-5 are down regulated [233]. Thus, measuring these markers could be used to confirm iron deficiency and to identify possible underlying cause(s) [233]. Along these lines, a ten-year-long study from the National Health and Nutrition Examination Survey showed that either intravenous or dietary iron intake can provide help to adult patients suffering of heart failure [234].
6. Iron Metabolism in Lungs
6.1. Iron Metabolism and TBI Uptake
the lung has both extracellular and intracellular iron distribution mechanisms [235]. to emphasize the importance of iron in lung tissue, both tf and tfr are expressed in bronchial epithelium, type ii alveolar cells, macrophages, and bronchus-associated lymphoid tissue [236]. alveolar epithelial cells and alveolar macrophages also express dmt1, an important transporter of dietary non-heme iron and endosomal iron from the tf-tfr1 complex into the cytoplasm under the regulation of irps and ires (irp/ire system) [237]. finally, pulmonary epithelial cells also have antioxidant molecules that prevent oxidative stress brought about by iron among a number of elements/molecules during conditions of inflammation and/or injury [235].
6.2. ntbi in Lungs
The key NTBI importers are DMT1, ZIP14, ZIP8, and lactoferrin receptors [238]. DMT1 and DCYTB are expressed by pulmonary epithelial cells and therefore, they are able to transport NTBI; while ZIP14, during iron overload states readily mediates NTBI uptake predominantly by hepatocytes, heart, pancreas and pulmonary epithelial cells [238]. ZIP8 (or SLC39A8) is a divalent metal ion importer expressed in lung, induced under inflammatory conditions that if absent leads to elevated spleen iron levels and hypoferremia; interestingly, ZIP8-/- mice do not develop acute lung injury in setting of markedly reduced serum iron suggesting that this receptor is involved in iron recycling [239]. Natural resistance-associated macrophage protein-1 (or SLC11A1) plays a role in NTBI uptake as well in pulmonary macrophages and neutrophils sequestering excess iron in the immune system and transporting it as needed [240]. The lactoferrin receptor LRP1 is an iron bound glycoprotein, belonging to the Tf family that has a major function iron chelation [238,241]. LRP1 is important since it actively removes iron, thus decreasing its pro-inflammatory effects [242], such as in pulmonary alveolar proteinosis when lactoferrin levels exceed Tf [243], and during bacterial and fungal infections where iron chelation may be therapeutic [244].
6.3. Hepcidin-FPN Axis (Iron Export Regulation)
Hepcidin is expressed in lower concentration in the lungs compared to other organs [245]. A hepcidin knockout model has shown that its absence led to an increase in FPN expression on epithelial cells and FTL1 mRNAs in the lung resulting in increased alveolar iron export and elevated iron levels in macrophages [246]. HAMP gene is expressed by alveolar macrophages and this can be induced by lipopolysaccharide stimulation through the upregulation of DMT1 and downregulation of FPN, suggesting there is decreased iron efflux in inflammatory states [247]. Indeed, HAMP expression increases on alveolar macrophages during inflammation and degradation of FPN decreases iron efflux, meaning that hepcidin protects the lungs against infection or inflammation by creating an iron-limited environment and decreased iron-mediated oxidative lung injury [247,248]. Iron has no effect on HAMP expression; however, it upregulates DMT1 and FPN1 expression. Therefore, iron loading of the lungs is regulated by an independent hepcidin-FPN mechanism [247,249].
6.4. Ferroptosis in the Lungs
inhaled oxidants or injured pulmonary cells lead to the formation of ros. several pulmonary diseases, most commonly acute respiratory distress syndrome are prooxidant states including increased body iron stores [238]. among the interleukins, il-4 and il-13 are key inflammatory intermediates causing robust inflammation in the lungs in response to insults such as iron [250]. slc7a11 is also downregulated by p53 causing decreases in gth synthesis, further increasing ros and ferroptosis [250]. interestingly, it has been shown that oleic acid induces acute lung injury in animals characterized by iron overload, decreased gth, and accumulation of malondialdehyde indicating that ferroptosis plays a part in this type of lung injury [251]. one of the significant changes in the lungs is that iron excess induces activation of fibroblasts causing pulmonary fibrosis through induction of the tgf-β-taz-tead signaling pathway resulting in fibroblast-to-myofibroblast conversion [252]. radiation-induced lung injury also causes ros accumulation, thus promoting ferroptosis in epithelial cells as shown by increased expression of slc7a11, gpx4 downregulation, and increases in malondialdehyde and total iron when exposed to lipopolysaccharide and ferrostatin-1 [253]. similarly, uridine phosphorylase 1 can also act as ferroptosis inhibitor via the nrf2 signaling pathway [254]. blood transfusions promote ferroptosis and increases the incidence of blood transfusion-related acute lung injury; therefore, iron chelators and anti-inflammatory drugs are helpful in this setting [250]. therefore, use of any of these inhibitors could limit ferroptosis in acute lung injury.
7. Iron Metabolism in Testis
Spermatogenesis is an iron dependent process [255], in particular Sertoli cells have high iron requirements due to their highly proliferative baseline phenotype [256]. The developing spermatocytes, however, are highly vulnerable to oxidative stress requiring tight regulation of iron metabolism. Gonads are indirectly responsive to iron overload because in diseases such as hereditary hemochromatosis and hemoglobinopathies, increased iron is deposited in gonadotrophs of the pituitary, decreasing release of gonadotrophins resulting in hypogonadotropic hypogonadism [257]. Over time this results in infertility and delayed puberty.
7.1. Transferrin and Non-Transferring Bound Iron
Extracellular or exogenous iron uptake is mainly mediated by the Tf-TFR1 pathway in Sertoli cells [258]. Testicular transferrin is also synthesized by seminiferous epithelium resulting in iron flow from the basal layer to the luminal spermatogonia [259]. Transferrin receptors present on seminiferous epithelial cells express TFR1 which readily takes in iron from the circulation through the blood testicular barrier [259]. Additionally, ceruloplasmin is also involved in this process and in oxidizing ferrous iron [260]. DMT1 is expressed mainly in the intracellular compartment of seminiferous tubules’ Sertoli cells which mediate most of the iron handling [261]. However, mature germ cells also express DMT1, but this expression is dependent upon the spermatogenesis stage since iron requirement does vary from one stage to the next [262].
7.2. Hepcidin-FPN axis (Iron Export Regulation)
Iron is stored as ferritin within cells and utilized during spermatogenesis. It is found in two sites in the testis: mitochondrial (in interstitial Leydig cells and germinal cells in seminiferous tubules) and cytosolic (interstitium) [261]. Sertoli cells actively phagocytose those spermatozoa not reaching their final differentiation stage [261]. The iron contained in the phagocytosed spermatogonia is recycled by Sertoli cells so it can be used for the next cycle of spermatogenesis. FPN present on the basolateral surface of seminiferous tubules exports excess iron from the luminal surface back to the circulation [262]. As an added safety mechanism, the “blood testicular barrier” and FPN prevent iron overload in the testis [263].
7.3. Ferroptosis in Testis
Ferroptosis is important during spermatogenesis. It has been shown that mice lacking GTH-synthetase, the final step in the synthesis of GTH, results in male animals having markedly reduced fertility [264]. Of note, with increasing age, this enzyme deficiency gets progressively worse so that the lower GTH allows for accumulation of ROS and lipid peroxidation, as result promoting ferroptosis. Consequently, ferroptosis can disrupt not only meiosis, but lead to a state of disordered acrosomal development [264].
8. Endocrine
8.1. Mechanisms of Iron Toxicity in the Endocrine System
Excess iron in endocrine tissues equally promotes oxidative stress and inflammation due to ROS formation. This oxidative stress damages cells in the pancreas, pituitary, thyroid among other glands. ROS damages the ER, leading to ER stress and activation of the unfolded protein response in endocrine glands [265]. Interestingly, NTBI often enters endocrine cells using calcium channels, where it causes cytotoxic effects by generating ROS. These oxidants mediate damage by stimulating FOXO3a, a transcription factor that upregulates ferritin, altering the LIP and disrupting hormonal synthesis [266]. Activation of NADPH-oxidase, generates superoxide radicals that amplify ongoing inflammation and cellular injury [267]. TRAF6 ubiquitin-ligase and Akt pathway inhibition by generated ROS also promote apoptosis of endocrine cells, including insulin-secreting pancreatic cells [266]. Under normal conditions, iron-dependent prolyl hydroxylases mark HIF-1α for degradation. However, in iron overload, the excess iron inhibit these hydroxylases, stabilizing HIF-1α and promoting its accumulation [268]. This stabilization lead to increased expression of genes involved in angiogenesis, glycolysis, and cell survival, potentially contributing to tumor progression and metastasis in thalassemia patients [268]. As a result, therapeutic interventions that focus on iron chelation (deferoxamine, deferasirox) and novel approaches like hepcidin mimetics and anti-sense oligonucleotides such as TMPRSS6 modulate hepcidin levels mitigating iron toxicity and reversing some endocrine complications [269]. Melatonin has also been reported to mitigate oxidative damage via its antioxidant properties, though its effects appear iron-independent [270]. Iron chelators beyond reducing intracellular iron load, improve endocrine outcomes in transfusion-dependent patients [271]. However, chronic elevation of hepcidin due to inflammatory stimuli exacerbate endocrine iron dysregulation further, causing or worsening diabetes and hypothyroidism. Therefore, strategies that combine iron chelation with hormone replacement address both systemic and endocrine sequelae of iron overload [272].
8.2. Pituitary and hypothalamus
It has been shown that Iron accumulation in the anterior pituitary gland reduces gonadotropic hormone production, causing hypogonadism and delayed puberty in patients with transfusion-dependent thalassemia and hereditary hemochromatosis [273,274]. Pituitary dysfunction is particularly common in transfusion-dependent β-thalassemia patients [275]. Evidence has shown that transfusion-dependent patients experience hypogonadotropic hypogonadism, mainly because of ferritin accumulation in the anterior pituitary, suppressing gonadotropin-level pathways especially those producing growth hormone and thyroid-stimulating hormone [276]. Hypogonadotropic hypogonadism, driven by pituitary hemosiderosis is a direct result of iron-related impairment of gonadotropin secretion [277,278]. Anatomically, the pituitary gland may have greater susceptibility to iron overload due to its intense vascularization and high metabolic activity. This is compounded by the dense expression of TFR in the pituitary making it particularly sensitive to iron [279]. Additionally, iron-induced downregulation of the hypothalamus-pituitary-gonadal axis occurs through iron deposition in gonadotrophs. This causes suppression of gonadotropic releasing hormone from the hypothalamus [280]. Furthermore, cells essential for the synthesis of luteinizing hormone and follicle-stimulating hormone, experience impaired secretion secondary to ROS-related mitochondrial dysfunction [276]. Finally, iron overload hastens cellular aging processes by shortening telomeres, further exacerbating tissue dysfunction. Not unexpectedly, impaired endocrine responses also linked to iron toxicity include thyroid dysfunction, hypoparathyroidism and adrenal insufficiency [278].
Bone deformities and growth retardation observed in iron-overloaded patients are linked to iron’s interference with GH-insulin-like growth factor-1 (IGF-1) axis [281]. Excess iron deposition in the liver and impairment of IGF-1 production together diminish the anabolic effects of GH, delaying skeletal growth and maturation [281]. Iron also disrupts osteoblast differentiation and activity through ROS-mediated inhibition of Wnt/β-catenin signaling, contributing to significant osteoporosis [281]. This iron-induced osteoporosis occurs through the upregulation of osteoclast activity and suppression of osteoblast function; this involves the RANKL/osteoprotegerin pathway, as observed in SCD and thalassemia patients [282].
8.3. Thyroid and Parathyroids
In the thyroid, iron excess reduces production of TSH , free T4, and it inhibits IGF-1 production by the liver, contributing to growth abnormalities [283,284]. Thyroid cells are particularly sensitive to ROS, as hydrogen peroxide, critical in thyroid hormone biosynthesis, amplifies oxidative injury during iron overload conditions [270], which can similarly affect the parathyroid glands [275]. High oxidative load also impairs iodide transport, an essential step for the synthesis of thyroxine (T4) and triiodothyronine [285]. This dysfunction correlates with altered levels of thyroid-stimulating hormone (TSH) and free T4 observed in individuals with chronic iron overload [286]. Furthermore, this dysregulation is also observed in diseases like juvenile hemochromatosis, which presents with early-onset hypothyroidism due to severe glandular iron deposition [287]. Central hypothyroidism is similarly associated with impaired TSH production due to iron-induced pituitary damage, as observed in children with transfusion-dependent anemia or SCD [288]. Notably, primary thyroid dysfunction from iron-induced oxidative stress and glandular inflammation commonly manifests as subclinical hypothyroidism, marked by elevated TSH and normal T4 levels [285,286]. Impaired fibroblast growth factor-23 secretion is also seen in hypoparathyroid thalassemic patients suggesting that iron overload disrupts both hormonal and mineral regulatory pathways in bone-parathyroid interactions [289]. As a result, this disruption worsens the clinical spectrum of hypocalcemia and hyperphosphatemia, characteristic of iron-overloaded states [289].
8.4. Pancreas
The loss of β-cell function is a critical factor in the development of diabetes in patients with hemoglobinopathies. Excessive iron deposition impairs β-cell function, leading to glucose dysregulation, insulin resistance, and overt diabetes mellitus, often termed "bronze diabetes" [290]. The resulting pancreatic siderosis is an early indicator of endocrine imbalance, aligned with hyperglycemia and dysregulated glucose metabolism [291]. The oxidative stress induced by iron overload interferes with insulin signaling pathways through modifications of insulin receptor substrates, which impairs their ability to propagate insulin signals [265]. Additionally, ROS activates stress kinases such as JNK and p38 MAPK, further inhibit insulin signaling by phosphorylating insulin substrates on serine residues [265]. Chronic ER stress also trigger apoptotic pathways, reducing β-cell mass and insulin secretion [292]. Furthermore, activation of signaling pathways protein kinase R-like ER-kinase and inositol-requiring enzyme 1 further disrupts β-cell function [293]. NTBI transporter ZIP14 facilitates parenchymal iron accumulation in pancreatic acinar cells [294]. NTBI, particularly its LIP fraction, penetrates pancreatic cells through unregulated calcium channels, amplifying intracellular iron overload and associated oxidative damage [295,296]. Of note, inflammatory responses further compound this damage, made worse in chronic settings. Excess iron activates inflammatory pathways, as shown by increased IL-1β, IL-6, inducible nitric oxide synthase expression as well as neutrophil infiltration [297]. Chronic iron overload continuously stimulates release of pro-inflammatory cytokines IL-6 and TNF-α, which promote tissue fibrosis and trigger β-cell apoptosis [298]. Chronic inflammation in iron-overloaded states also promotes development of acinar-to-ductal metaplasia in pancreatic tissues, which are precursors to neoplastic transformations often linked to increased ROS and altered p53 activity [299]. These factors equally activate pancreatic stellate cells, which drive fibrosis and acinar atrophy, leading to progressive loss of exocrine function as well [297]. Additionally, ongoing iron deposition in pancreatic islets potentiate β-cell impairment [297]. Iron deposition in the pancreas is also associated with increased hepatic insulin resistance due to decreased clearance of insulin leading to systemic hyperinsulinemia and metabolic dysfunction [298]. In response to iron overload, phenolic compounds such as ferulic acid and its derivatives enhance nuclear translocation of NRF2, upregulating antioxidant defense mechanisms within the pancreas including genes such as HO-1 and GPX4, which have protective roles against ROS-induced damage [300]. Unfortunately, mutations in the HFE gene, such as C282Y and H63D, disrupt hepcidin regulation, facilitating increased intestinal absorption of dietary iron and its diffuse deposition in organs, especially pancreas [301].
8.5. Gonads and Adrenals
ROS activates the p53 pathway causing DNA damage and p53-mediated apoptosis in endocrine cells, particularly in pituitary and testicular tissue [302]. Oxidative stress profoundly affects ovarian follicles and steroidogenesis through disrupted aromatase activity and impaired estradiol synthesis [303]. Male gonads experience Leydig cell dysfunction disrupting testosterone biosynthesis and luteinizing hormone signaling [276]. At the molecular level, iron-regulated signaling pathways include iron-mediated activation of stress-responsive pathways such as c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (MAPK), and nuclear factor kappa B (NF-κB) [281]. Once activated, these pathways trigger apoptotic signaling and cytokine production, expanding endocrine dysfunction and inflammation. Connections between iron overload and adrenal insufficiency are highlighted in patients with β-thalassemia major, where adrenal insufficiency has been detected in up to half of patients, due to iron-mediated mitochondrial damage impairing their cortisol biosynthesis pathways [304]. Adrenal dysfunction in SCD is similarly attributed to microvascular obstruction and repeated vaso-occlusive episodes, which result in ischemic injury to the small blood vessels of the adrenal glands [304]. These events, compounded by iron deposition from repeated blood transfusions, profoundly disrupt adrenal steroidogenic machinery [305]. Finally, ROS-mediated disruption of the steroidogenic acute regulatory protein impairs cholesterol transport within mitochondria, exacerbating hypocortisolism even further [293].
9. Conclusions
The role of iron in maintaining a large number of cellular functional processes is evident. The generation of transporters that establish links between sites of gastrointestinal absorption to those sites where iron is recycled such as the spleen should be seen as examples of a multilayer regulated group of pathways required to control iron intake, utilization, export, storage and recycling. Considering that some patient groups such as those with sickle cell disease are exposed to supra-physiologic quantities of this element due to blood transfusions, they exemplify the potential negative effects of iron toxicity across organ systems. Data presented stresses that additional research to get a complete understanding of the negative effects secondary to iron overload that is closer to real-time, may allow for more timely interventions, such as chelation, to be initiated to prevent the more dire complications.
Author Contributions
AT performed literature search and co-wrote the manuscript. JK performed literature search and co-wrote the manuscript. PS performed literature search and co-wrote the manuscript. RWM conceptualized the topic, performed literature search, co-wrote the manuscript, and edited the text to its final version.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data has been included in the text.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Grijota, F.J.; Toro-Roman, V.; Siquier-Coll, J.; Robles-Gil, M.C.; Munoz, D.; Maynar-Marino, M. Total Iron Concentrations in Different Biological Matrices-Influence of Physical Training. Nutrients 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J Res Med Sci 2014, 19, 164–174. [Google Scholar]
- Mercadante, C.J.; Prajapati, M.; Parmar, J.H.; Conboy, H.L.; Dash, M.E.; Pettiglio, M.A.; Herrera, C.; Bu, J.T.; Stopa, E.G.; Mendes, P.; et al. Gastrointestinal iron excretion and reversal of iron excess in a mouse model of inherited iron excess. Haematologica 2019, 104, 678–689. [Google Scholar] [CrossRef]
- Wallace, D.F. The Regulation of Iron Absorption and Homeostasis. Clin Biochem Rev 2016, 37, 51–62. [Google Scholar]
- Gupta, R.; Musallam, K.M.; Taher, A.T.; Rivella, S. Ineffective Erythropoiesis: Anemia and Iron Overload. Hematol Oncol Clin North Am 2018, 32, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Pandey, S.K.; Shah, V. Role of HAMP Genetic Variants on Pathophysiology of Iron Deficiency Anemia. Indian J Clin Biochem 2018, 33, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Prochazkova, P.; Skanta, F.; Roubalova, R.; Silerova, M.; Dvorak, J.; Bilej, M. Involvement of the iron regulatory protein from Eisenia andrei earthworms in the regulation of cellular iron homeostasis. PLoS One 2014, 9, e109900. [Google Scholar] [CrossRef]
- Ghosh, M.C.; Zhang, D.L.; Rouault, T.A. Iron misregulation and neurodegenerative disease in mouse models that lack iron regulatory proteins. Neurobiol Dis 2015, 81, 66–75. [Google Scholar] [CrossRef]
- Hinokuma, H.; Kanamori, Y.; Ikeda, K.; Hao, L.; Maruno, M.; Yamane, T.; Maeda, A.; Nita, A.; Shimoda, M.; Niimura, M.; et al. Distinct functions between ferrous and ferric iron in lung cancer cell growth. Cancer Sci 2023, 114, 4355–4364. [Google Scholar] [CrossRef]
- Coad, J.; Pedley, K. Iron deficiency and iron deficiency anemia in women. Scand J Clin Lab Invest Suppl 2014, 244, 82–89; discussion 89. [Google Scholar] [CrossRef]
- Piskin, E.; Cianciosi, D.; Gulec, S.; Tomas, M.; Capanoglu, E. Iron Absorption: Factors, Limitations, and Improvement Methods. ACS Omega 2022, 7, 20441–20456. [Google Scholar] [CrossRef] [PubMed]
- Weinborn, V.; Pizarro, F.; Olivares, M.; Brito, A.; Arredondo, M.; Flores, S.; Valenzuela, C. The Effect of Plant Proteins Derived from Cereals and Legumes on Heme Iron Absorption. Nutrients 2015, 7, 8977–8986. [Google Scholar] [CrossRef]
- Sharp, P.; Srai, S.K. Molecular mechanisms involved in intestinal iron absorption. World J Gastroenterol 2007, 13, 4716–4724. [Google Scholar] [CrossRef]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu Rev Nutr 2022, 42, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Yoshida, M.; Sakamoto, N.; Morimoto, S.; Watanabe, T.; Namba, K. Iron uptake mediated by the plant-derived chelator nicotianamine in the small intestine. J Biol Chem 2021, 296, 100195. [Google Scholar] [CrossRef] [PubMed]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am J Physiol Gastrointest Liver Physiol 2014, 307, G397–409. [Google Scholar] [CrossRef]
- Liao, R.; Bresnick, E.H. Heme as a differentiation-regulatory transcriptional cofactor. Int J Hematol 2022, 116, 174–181. [Google Scholar] [CrossRef]
- Duvigneau, J.C.; Esterbauer, H.; Kozlov, A.V. Role of Heme Oxygenase as a Modulator of Heme-Mediated Pathways. Antioxidants (Basel) 2019, 8. [Google Scholar] [CrossRef]
- Larsen, R.; Gouveia, Z.; Soares, M.P.; Gozzelino, R. Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front Pharmacol 2012, 3, 77. [Google Scholar] [CrossRef]
- Chung, J.; Chen, C.; Paw, B.H. Heme metabolism and erythropoiesis. Curr Opin Hematol 2012, 19, 156–162. [Google Scholar] [CrossRef]
- Kasvosve, I. Effect of ferroportin polymorphism on iron homeostasis and infection. Clin Chim Acta 2013, 416, 20–25. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Hepcidin and ferroportin: the new players in iron metabolism. Semin Liver Dis 2011, 31, 272–279. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Lo, E.; Yang, B.; Korolnek, T.; Hamza, I.; Ward, D.M.; Kaplan, J. The role of ubiquitination in hepcidin-independent and hepcidin-dependent degradation of ferroportin. Cell Metab 2011, 14, 635–646. [Google Scholar] [CrossRef]
- Charlebois, E.; Fillebeen, C.; Presley, J.; Cagnone, G.; Lisi, V.; Lavallee, V.P.; Joyal, J.S.; Pantopoulos, K. Liver sinusoidal endothelial cells induce BMP6 expression in response to non-transferrin-bound iron. Blood 2023, 141, 271–284. [Google Scholar] [CrossRef]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2005, 2, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. Anemia of inflammation: the cytokine-hepcidin link. J Clin Invest 2004, 113, 1251–1253. [Google Scholar] [CrossRef]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef]
- Nicolas, G.; Bennoun, M.; Porteu, A.; Mativet, S.; Beaumont, C.; Grandchamp, B.; Sirito, M.; Sawadogo, M.; Kahn, A.; Vaulont, S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci U S A 2002, 99, 4596–4601. [Google Scholar] [CrossRef]
- Gafter-Gvili, A.; Schechter, A.; Rozen-Zvi, B. Iron Deficiency Anemia in Chronic Kidney Disease. Acta Haematol 2019, 142, 44–50. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. The role of hepcidin in iron metabolism. Acta Haematol 2009, 122, 78–86. [Google Scholar] [CrossRef]
- Tamary, H.; Shalev, H.; Perez-Avraham, G.; Zoldan, M.; Levi, I.; Swinkels, D.W.; Tanno, T.; Miller, J.L. Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Blood 2008, 112, 5241–5244. [Google Scholar] [CrossRef] [PubMed]
- Rivella, S. Ineffective erythropoiesis and thalassemias. Curr Opin Hematol 2009, 16, 187–194. [Google Scholar] [CrossRef]
- Gardenghi, S.; Grady, R.W.; Rivella, S. Anemia, ineffective erythropoiesis, and hepcidin: interacting factors in abnormal iron metabolism leading to iron overload in beta-thalassemia. Hematol Oncol Clin North Am 2010, 24, 1089–1107. [Google Scholar] [CrossRef]
- Li, H.; Ginzburg, Y.Z. Crosstalk between Iron Metabolism and Erythropoiesis. Adv Hematol 2010, 2010, 605435. [Google Scholar] [CrossRef]
- Mleczko-Sanecka, K.; Silvestri, L. Cell-type-specific insights into iron regulatory processes. Am J Hematol 2021, 96, 110–127. [Google Scholar] [CrossRef]
- Camaschella, C.; Pagani, A.; Nai, A.; Silvestri, L. The mutual control of iron and erythropoiesis. Int J Lab Hematol 2016, 38 Suppl 1, 20–26. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br J Haematol 2016, 172, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Liang, Y.; Zhang, C. Ferroptosis Regulated by Hypoxia in Cells. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Luu Hoang, K.N.; Anstee, J.E.; Arnold, J.N. The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front Immunol 2021, 12, 658315. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Brune, B. A graphical journey through iron metabolism, microRNAs, and hypoxia in ferroptosis. Redox Biol 2022, 54, 102365. [Google Scholar] [CrossRef]
- Bellelli, R.; Federico, G.; Matte, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep 2016, 14, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lozovatsky, L.; Sukumaran, A.; Gonzalez, L.; Jain, A.; Liu, D.; Ayala-Lopez, N.; Finberg, K.E. NCOA4 is regulated by HIF and mediates mobilization of murine hepatic iron stores after blood loss. Blood 2020, 136, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Hoelzgen, F.; Nguyen, T.T.P.; Klukin, E.; Boumaiza, M.; Srivastava, A.K.; Kim, E.Y.; Zalk, R.; Shahar, A.; Cohen-Schwartz, S.; Meyron-Holtz, E.G.; et al. Structural basis for the intracellular regulation of ferritin degradation. Nat Commun 2024, 15, 3802. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Del Rey, M.Q.; Kapner, K.S.; Zhang, H.; Gikandi, A.; Malcolm, C.; Poupault, C.; Kuljanin, M.; John, K.M.; Biancur, D.E.; et al. NCOA4-Mediated Ferritinophagy Is a Pancreatic Cancer Dependency via Maintenance of Iron Bioavailability for Iron-Sulfur Cluster Proteins. Cancer Discov 2022, 12, 2180–2197. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, V.; Lima, J.; Oliveira, G.F.; Oliveira, Y.S.; Getachew, B.; Nekhai, S.; Aschner, M.; Tizabi, Y. Ferroptosis as an emerging target in sickle cell disease. Curr Res Toxicol 2024, 7, 100181. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; SharathBabu, B.N.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S.; Mugesh, G. The Role of Reactive Oxygen Species and Ferroptosis in Heme-Mediated Activation of Human Platelets. ACS Chem Biol 2018, 13, 1996–2002. [Google Scholar] [CrossRef]
- Rodrigue, C.M.; Arous, N.; Bachir, D.; Smith-Ravin, J.; Romeo, P.H.; Galacteros, F.; Garel, M.C. Resveratrol, a natural dietary phytoalexin, possesses similar properties to hydroxyurea towards erythroid differentiation. Br J Haematol 2001, 113, 500–507. [Google Scholar] [CrossRef]
- Schubert, M.L. Physiologic, pathophysiologic, and pharmacologic regulation of gastric acid secretion. Curr Opin Gastroenterol 2017, 33, 430–438. [Google Scholar] [CrossRef]
- Pan, Y.; Ren, Z.; Gao, S.; Shen, J.; Wang, L.; Xu, Z.; Yu, Y.; Bachina, P.; Zhang, H.; Fan, X.; et al. Structural basis of ion transport and inhibition in ferroportin. Nat Commun 2020, 11, 5686. [Google Scholar] [CrossRef]
- Yu, Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.; Wu, Q.; Fang, X.; Duan, L.; et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef]
- Billesbolle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 2020, 586, 807–811. [Google Scholar] [CrossRef]
- Traeger, L.; Wiegand, S.B.; Sauer, A.J.; Corman, B.H.P.; Peneyra, K.M.; Wunderer, F.; Fischbach, A.; Bagchi, A.; Malhotra, R.; Zapol, W.M.; et al. UBA6 and NDFIP1 regulate the degradation of ferroportin. Haematologica 2022, 107, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.J.; Das, N.K.; Ramakrishnan, S.K.; Jain, C.; Jurkovic, M.T.; Wu, J.; Nemeth, E.; Lakhal-Littleton, S.; Colacino, J.A.; Shah, Y.M. Hepatic hepcidin/intestinal HIF-2alpha axis maintains iron absorption during iron deficiency and overload. J Clin Invest 2019, 129, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Colucci, S.; Marques, O.; Altamura, S. 20 years of Hepcidin: How far we have come. Semin Hematol 2021, 58, 132–144. [Google Scholar] [CrossRef]
- Xiao, X.; Dev, S.; Canali, S.; Bayer, A.; Xu, Y.; Agarwal, A.; Wang, C.Y.; Babitt, J.L. Endothelial Bone Morphogenetic Protein 2 (Bmp2) Knockout Exacerbates Hemochromatosis in Homeostatic Iron Regulator (Hfe) Knockout Mice but not Bmp6 Knockout Mice. Hepatology 2020, 72, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Enns, C.A.; Jue, S.; Zhang, A.S. Hepatocyte neogenin is required for hemojuvelin-mediated hepcidin expression and iron homeostasis in mice. Blood 2021, 138, 486–499. [Google Scholar] [CrossRef]
- Wang, C.Y.; Xiao, X.; Bayer, A.; Xu, Y.; Dev, S.; Canali, S.; Nair, A.V.; Masia, R.; Babitt, J.L. Ablation of Hepatocyte Smad1, Smad5, and Smad8 Causes Severe Tissue Iron Loading and Liver Fibrosis in Mice. Hepatology 2019, 70, 1986–2002. [Google Scholar] [CrossRef]
- Lim, P.J.; Duarte, T.L.; Arezes, J.; Garcia-Santos, D.; Hamdi, A.; Pasricha, S.R.; Armitage, A.E.; Mehta, H.; Wideman, S.; Santos, A.G.; et al. Nrf2 controls iron homeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat Metab 2019, 1, 519–531. [Google Scholar] [CrossRef]
- Srole, D.N.; Ganz, T. Erythroferrone structure, function, and physiology: Iron homeostasis and beyond. J Cell Physiol 2021, 236, 4888–4901. [Google Scholar] [CrossRef]
- Das, N.K.; Schwartz, A.J.; Barthel, G.; Inohara, N.; Liu, Q.; Sankar, A.; Hill, D.R.; Ma, X.; Lamberg, O.; Schnizlein, M.K.; et al. Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab 2020, 31, 115–130 e116. [Google Scholar] [CrossRef]
- Montemiglio, L.C.; Testi, C.; Ceci, P.; Falvo, E.; Pitea, M.; Savino, C.; Arcovito, A.; Peruzzi, G.; Baiocco, P.; Mancia, F.; et al. Cryo-EM structure of the human ferritin-transferrin receptor 1 complex. Nat Commun 2019, 10, 1121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Funcke, J.B.; Zi, Z.; Zhao, S.; Straub, L.G.; Zhu, Y.; Zhu, Q.; Crewe, C.; An, Y.A.; Chen, S.; et al. Adipocyte iron levels impinge on a fat-gut crosstalk to regulate intestinal lipid absorption and mediate protection from obesity. Cell Metab 2021, 33, 1624–1639 e1629. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pan, X.; Pan, G.; Song, Z.; He, Y.; Zhang, S.; Ye, X.; Yang, X.; Xie, E.; Wang, X.; et al. Transferrin Receptor 1 Regulates Thermogenic Capacity and Cell Fate in Brown/Beige Adipocytes. Adv Sci (Weinh) 2020, 7, 1903366. [Google Scholar] [CrossRef]
- Connell, G.J.; Abasiri, I.M.; Chaney, E.H. A temporal difference in the stabilization of two mRNAs with a 3' iron-responsive element during iron deficiency. RNA 2023, 29, 1117–1125. [Google Scholar] [CrossRef]
- Gao, J.; Zhou, Q.; Wu, D.; Chen, L. Mitochondrial iron metabolism and its role in diseases. Clin Chim Acta 2021, 513, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Nag, S.; Mason, A.B.; Barroso, M.M. Endosome-mitochondria interactions are modulated by iron release from transferrin. J Cell Biol 2016, 214, 831–845. [Google Scholar] [CrossRef]
- Braymer, J.J.; Freibert, S.A.; Rakwalska-Bange, M.; Lill, R. Mechanistic concepts of iron-sulfur protein biogenesis in Biology. Biochim Biophys Acta Mol Cell Res 2021, 1868, 118863. [Google Scholar] [CrossRef]
- Crispin, A.; Guo, C.; Chen, C.; Campagna, D.R.; Schmidt, P.J.; Lichtenstein, D.; Cao, C.; Sendamarai, A.K.; Hildick-Smith, G.J.; Huston, N.C.; et al. Mutations in the iron-sulfur cluster biogenesis protein HSCB cause congenital sideroblastic anemia. J Clin Invest 2020, 130, 5245–5256. [Google Scholar] [CrossRef]
- Weber, R.A.; Yen, F.S.; Nicholson, S.P.V.; Alwaseem, H.; Bayraktar, E.C.; Alam, M.; Timson, R.C.; La, K.; Abu-Remaileh, M.; Molina, H.; et al. Maintaining Iron Homeostasis Is the Key Role of Lysosomal Acidity for Cell Proliferation. Mol Cell 2020, 77, 645–655 e647. [Google Scholar] [CrossRef]
- Ast, T.; Meisel, J.D.; Patra, S.; Wang, H.; Grange, R.M.H.; Kim, S.H.; Calvo, S.E.; Orefice, L.L.; Nagashima, F.; Ichinose, F.; et al. Hypoxia Rescues Frataxin Loss by Restoring Iron Sulfur Cluster Biogenesis. Cell 2019, 177, 1507–1521 e1516. [Google Scholar] [CrossRef]
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The new role of poly (rC)-binding proteins as iron transport chaperones: Proteins that could couple with inter-organelle interactions to safely traffic iron. Biochim Biophys Acta Gen Subj 2020, 1864, 129685. [Google Scholar] [CrossRef]
- Patel, S.J.; Protchenko, O.; Shakoury-Elizeh, M.; Baratz, E.; Jadhav, S.; Philpott, C.C. The iron chaperone and nucleic acid-binding activities of poly(rC)-binding protein 1 are separable and independently essential. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Yanatori, I.; Richardson, D.R.; Dhekne, H.S.; Toyokuni, S.; Kishi, F. CD63 is regulated by iron via the IRE-IRP system and is important for ferritin secretion by extracellular vesicles. Blood 2021, 138, 1490–1503. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, M.; Furuya, N.; Saiki, S.; Amo, T.; Imamichi, Y.; Hattori, N. Iron Supply via NCOA4-Mediated Ferritin Degradation Maintains Mitochondrial Functions. Mol Cell Biol 2019, 39. [Google Scholar] [CrossRef]
- Koleini, N.; Shapiro, J.S.; Geier, J.; Ardehali, H. Ironing out mechanisms of iron homeostasis and disorders of iron deficiency. J Clin Invest 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, M.; Conboy, H.L.; Hojyo, S.; Fukada, T.; Budnik, B.; Bartnikas, T.B. Biliary excretion of excess iron in mice requires hepatocyte iron import by Slc39a14. J Biol Chem 2021, 297, 100835. [Google Scholar] [CrossRef]
- Speich, C.; Wegmuller, R.; Brittenham, G.M.; Zeder, C.; Cercamondi, C.I.; Buhl, D.; Prentice, A.M.; Zimmermann, M.B.; Moretti, D. Measurement of long-term iron absorption and loss during iron supplementation using a stable isotope of iron ((57) Fe). Br J Haematol 2021, 192, 179–189. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Deshpande, C.N.; Xin, V.; Lu, Y.; Savage, T.; Anderson, G.J.; Jormakka, M. Large scale expression and purification of secreted mouse hephaestin. PLoS One 2017, 12, e0184366. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef]
- Pietrangelo, A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 2010, 139, 393–408, 408 e391-392. [Google Scholar] [CrossRef] [PubMed]
- Langer, A.L.; Ginzburg, Y.Z. Role of hepcidin-ferroportin axis in the pathophysiology, diagnosis, and treatment of anemia of chronic inflammation. Hemodial Int 2017, 21 Suppl 1, S37–S46. [Google Scholar] [CrossRef]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest 2007, 117, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Handelman, G.J.; Levin, N.W. Iron and anemia in human biology: a review of mechanisms. Heart Fail Rev 2008, 13, 393–404. [Google Scholar] [CrossRef]
- Frazer, D.M.; Inglis, H.R.; Wilkins, S.J.; Millard, K.N.; Steele, T.M.; McLaren, G.D.; McKie, A.T.; Vulpe, C.D.; Anderson, G.J. Delayed hepcidin response explains the lag period in iron absorption following a stimulus to increase erythropoiesis. Gut 2004, 53, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Diaz, V.; Recalcati, S.; Buratti, P.; Samaja, M.; Dey, S.; Noguchi, C.T.; Gassmann, M.; Cairo, G. Erythropoietin's inhibiting impact on hepcidin expression occurs indirectly. Am J Physiol Regul Integr Comp Physiol 2015, 308, R330–335. [Google Scholar] [CrossRef]
- Wang, C.Y.; Xu, Y.; Traeger, L.; Dogan, D.Y.; Xiao, X.; Steinbicker, A.U.; Babitt, J.L. Erythroferrone lowers hepcidin by sequestering BMP2/6 heterodimer from binding to the BMP type I receptor ALK3. Blood 2020, 135, 453–456. [Google Scholar] [CrossRef]
- Srole, D.N.; Jung, G.; Waring, A.J.; Nemeth, E.; Ganz, T. Characterization of erythroferrone structural domains relevant to its iron-regulatory function. J Biol Chem 2023, 299, 105374. [Google Scholar] [CrossRef]
- Ramsay, A.J.; Hooper, J.D.; Folgueras, A.R.; Velasco, G.; Lopez-Otin, C. Matriptase-2 (TMPRSS6): a proteolytic regulator of iron homeostasis. Haematologica 2009, 94, 840–849. [Google Scholar] [CrossRef]
- Aschemeyer, S.; Gabayan, V.; Ganz, T.; Nemeth, E.; Kautz, L. Erythroferrone and matriptase-2 independently regulate hepcidin expression. Am J Hematol 2017, 92, E61–E63. [Google Scholar] [CrossRef]
- Arezes, J.; Foy, N.; McHugh, K.; Sawant, A.; Quinkert, D.; Terraube, V.; Brinth, A.; Tam, M.; LaVallie, E.R.; Taylor, S.; et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood 2018, 132, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev 2013, 27, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Hodges, V.M.; Rainey, S.; Lappin, T.R.; Maxwell, A.P. Pathophysiology of anemia and erythrocytosis. Crit Rev Oncol Hematol 2007, 64, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 2014, 46, 678–684. [Google Scholar] [CrossRef]
- Vento, S.; Cainelli, F.; Cesario, F. Infections and thalassaemia. Lancet Infect Dis 2006, 6, 226–233. [Google Scholar] [CrossRef]
- Andolfo, I.; Rosato, B.E.; Marra, R.; De Rosa, G.; Manna, F.; Gambale, A.; Iolascon, A.; Russo, R. The BMP-SMAD pathway mediates the impaired hepatic iron metabolism associated with the ERFE-A260S variant. Am J Hematol 2019, 94, 1227–1235. [Google Scholar] [CrossRef]
- Iolascon, A.; Andolfo, I.; Russo, R. Congenital dyserythropoietic anemias. Blood 2020, 136, 1274–1283. [Google Scholar] [CrossRef]
- Hanudel, M.R.; Rappaport, M.; Chua, K.; Gabayan, V.; Qiao, B.; Jung, G.; Salusky, I.B.; Ganz, T.; Nemeth, E. Levels of the erythropoietin-responsive hormone erythroferrone in mice and humans with chronic kidney disease. Haematologica 2018, 103, e141–e142. [Google Scholar] [CrossRef]
- Summary of Recommendation Statements. Kidney Int Suppl (2011) 2012, 2, 283–287. [CrossRef]
- Chapter 1: Diagnosis and evaluation of anemia in CKD. Kidney Int Suppl (2011) 2012, 2, 288–291. [CrossRef]
- Noonan, M.L.; Clinkenbeard, E.L.; Ni, P.; Swallow, E.A.; Tippen, S.P.; Agoro, R.; Allen, M.R.; White, K.E. Erythropoietin and a hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHDi) lowers FGF23 in a model of chronic kidney disease (CKD). Physiol Rep 2020, 8, e14434. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes with ring sideroblasts (MDS-RS) and MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T) - "2021 update on diagnosis, risk-stratification, and management". Am J Hematol 2021, 96, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Keating, M.; Estrov, Z. The significance of spliceosome mutations in chronic lymphocytic leukemia. Leuk Lymphoma 2013, 54, 1364–1366. [Google Scholar] [CrossRef]
- Bondu, S.; Alary, A.S.; Lefevre, C.; Houy, A.; Jung, G.; Lefebvre, T.; Rombaut, D.; Boussaid, I.; Bousta, A.; Guillonneau, F.; et al. A variant erythroferrone disrupts iron homeostasis in SF3B1-mutated myelodysplastic syndrome. Sci Transl Med 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Frazer, D.M.; Wilkins, S.J.; Mirciov, C.S.; Dunn, L.A.; Anderson, G.J. Hepcidin independent iron recycling in a mouse model of beta-thalassaemia intermedia. Br J Haematol 2016, 175, 308–317. [Google Scholar] [CrossRef]
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood 2015, 126, 2031–2037. [Google Scholar] [CrossRef] [PubMed]
- Arezes, J.; Foy, N.; McHugh, K.; Quinkert, D.; Benard, S.; Sawant, A.; Frost, J.N.; Armitage, A.E.; Pasricha, S.R.; Lim, P.J.; et al. Antibodies against the erythroferrone N-terminal domain prevent hepcidin suppression and ameliorate murine thalassemia. Blood 2020, 135, 547–557. [Google Scholar] [CrossRef]
- Xiao, X.; Alfaro-Magallanes, V.M.; Babitt, J.L. Bone morphogenic proteins in iron homeostasis. Bone 2020, 138, 115495. [Google Scholar] [CrossRef]
- Fillebeen, C.; Wilkinson, N.; Charlebois, E.; Katsarou, A.; Wagner, J.; Pantopoulos, K. Hepcidin-mediated hypoferremic response to acute inflammation requires a threshold of Bmp6/Hjv/Smad signaling. Blood 2018, 132, 1829–1841. [Google Scholar] [CrossRef]
- Gao, J.; Chen, J.; Kramer, M.; Tsukamoto, H.; Zhang, A.S.; Enns, C.A. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab 2009, 9, 217–227. [Google Scholar] [CrossRef]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of Nrf2. Curr Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Jin, Y.; Yao, G.; Zhao, H.; Qiao, P.; Wu, S. Nrf2 Is a Potential Modulator for Orchestrating Iron Homeostasis and Redox Balance in Cancer Cells. Front Cell Dev Biol 2021, 9, 728172. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid Redox Signal 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol 2019, 23, 101107. [Google Scholar] [CrossRef]
- Li, B.; Gong, J.; Sheng, S.; Lu, M.; Guo, S.; Zhao, X.; Zhang, H.; Wang, H.; Tian, Z.; Tian, Y. Increased hepcidin in hemorrhagic plaques correlates with iron-stimulated IL-6/STAT3 pathway activation in macrophages. Biochem Biophys Res Commun 2019, 515, 394–400. [Google Scholar] [CrossRef]
- Schmidt, P.J. Regulation of Iron Metabolism by Hepcidin under Conditions of Inflammation. J Biol Chem 2015, 290, 18975–18983. [Google Scholar] [CrossRef]
- Ward, R.J.; Crichton, R.R.; Taylor, D.L.; Della Corte, L.; Srai, S.K.; Dexter, D.T. Iron and the immune system. J Neural Transm (Vienna) 2011, 118, 315–328. [Google Scholar] [CrossRef]
- Carballo, M.; Conde, M.; El Bekay, R.; Martin-Nieto, J.; Camacho, M.J.; Monteseirin, J.; Conde, J.; Bedoya, F.J.; Sobrino, F. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem 1999, 274, 17580–17586. [Google Scholar] [CrossRef]
- Yu, C.; Cao, J.; Wang, L.; Yang, Y.; Ni, Y.; Wang, J. Measuring the bioactivity of anti-IL-6/anti-IL-6R therapeutic antibodies: presentation of a robust reporter gene assay. Anal Bioanal Chem 2018, 410, 7067–7075. [Google Scholar] [CrossRef]
- Knutson, M.D. Non-transferrin-bound iron transporters. Free Radic Biol Med 2019, 133, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.N.; Rangel, M. The (Bio)Chemistry of Non-Transferrin-Bound Iron. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci U S A 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Physiology of iron transport and the hemochromatosis gene. Am J Physiol Gastrointest Liver Physiol 2002, 282, G403–414. [Google Scholar] [CrossRef] [PubMed]
- Yanatori, I.; Kishi, F. DMT1 and iron transport. Free Radic Biol Med 2019, 133, 55–63. [Google Scholar] [CrossRef]
- Nam, H.; Wang, C.Y.; Zhang, L.; Zhang, W.; Hojyo, S.; Fukada, T.; Knutson, M.D. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: implications for tissue iron uptake in iron-related disorders. Haematologica 2013, 98, 1049–1057. [Google Scholar] [CrossRef]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J Alzheimers Dis 2013, 35, 541–552. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Haldar, S.; Qian, J.; Beserra, A.; Suda, S.; Singh, A.; Hopfer, U.; Chen, S.G.; Garrick, M.D.; Turner, J.R.; et al. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic Biol Med 2015, 84, 322–330. [Google Scholar] [CrossRef]
- West, A.R.; Oates, P.S. Mechanisms of heme iron absorption: current questions and controversies. World J Gastroenterol 2008, 14, 4101–4110. [Google Scholar] [CrossRef]
- Grasbeck, R.; Kouvonen, I.; Lundberg, M.; Tenhunen, R. An intestinal receptor for heme. Scand J Haematol 1979, 23, 5–9. [Google Scholar] [CrossRef]
- Korolnek, T.; Hamza, I. Like iron in the blood of the people: the requirement for heme trafficking in iron metabolism. Front Pharmacol 2014, 5, 126. [Google Scholar] [CrossRef]
- Wyllie, J.C.; Kaufman, N. An electron microscopic study of heme uptake by rat duodenum. Lab Invest 1982, 47, 471–476. [Google Scholar]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Fillebeen, C.; Gkouvatsos, K.; Fragoso, G.; Calve, A.; Garcia-Santos, D.; Buffler, M.; Becker, C.; Schumann, K.; Ponka, P.; Santos, M.M.; et al. Mice are poor heme absorbers and do not require intestinal Hmox1 for dietary heme iron assimilation. Haematologica 2015, 100, e334–337. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, A.; Rao, A.U.; Amigo, J.; Tian, M.; Upadhyay, S.K.; Hall, C.; Uhm, S.; Mathew, M.K.; Fleming, M.D.; Paw, B.H.; et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 2008, 453, 1127–1131. [Google Scholar] [CrossRef]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab 2013, 17, 261–270. [Google Scholar] [CrossRef]
- Zheng, J.; Shan, Y.; Lambrecht, R.W.; Donohue, S.E.; Bonkovsky, H.L. Differential regulation of human ALAS1 mRNA and protein levels by heme and cobalt protoporphyrin. Mol Cell Biochem 2008, 319, 153–161. [Google Scholar] [CrossRef]
- Munakata, H.; Sun, J.Y.; Yoshida, K.; Nakatani, T.; Honda, E.; Hayakawa, S.; Furuyama, K.; Hayashi, N. Role of the heme regulatory motif in the heme-mediated inhibition of mitochondrial import of 5-aminolevulinate synthase. J Biochem 2004, 136, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet 2009, 41, 651–653. [Google Scholar] [CrossRef]
- Lunetti, P.; Damiano, F.; De Benedetto, G.; Siculella, L.; Pennetta, A.; Muto, L.; Paradies, E.; Marobbio, C.M.; Dolce, V.; Capobianco, L. Characterization of Human and Yeast Mitochondrial Glycine Carriers with Implications for Heme Biosynthesis and Anemia. J Biol Chem 2016, 291, 19746–19759. [Google Scholar] [CrossRef]
- Liu, G.; Sil, D.; Maio, N.; Tong, W.H.; Bollinger, J.M., Jr.; Krebs, C.; Rouault, T.A. Heme biosynthesis depends on previously unrecognized acquisition of iron-sulfur cofactors in human amino-levulinic acid dehydratase. Nat Commun 2020, 11, 6310. [Google Scholar] [CrossRef] [PubMed]
- Severance, S.; Hamza, I. Trafficking of heme and porphyrins in metazoa. Chem Rev 2009, 109, 4596–4616. [Google Scholar] [CrossRef] [PubMed]
- Donegan, R.K.; Moore, C.M.; Hanna, D.A.; Reddi, A.R. Handling heme: The mechanisms underlying the movement of heme within and between cells. Free Radic Biol Med 2019, 133, 88–100. [Google Scholar] [CrossRef]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 2019, 104, 1756–1767. [Google Scholar] [CrossRef]
- Medlock, A.E.; Dailey, H.A. New Avenues of Heme Synthesis Regulation. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Murray, J.P.; Prykhozhij, S.V.; Dufay, J.N.; Steele, S.L.; Gaston, D.; Nasrallah, G.K.; Coombs, A.J.; Liwski, R.S.; Fernandez, C.V.; Berman, J.N.; et al. Glycine and Folate Ameliorate Models of Congenital Sideroblastic Anemia. PLoS Genet 2016, 12, e1005783. [Google Scholar] [CrossRef]
- Garcia-Santos, D.; Schranzhofer, M.; Bergeron, R.; Sheftel, A.D.; Ponka, P. Extracellular glycine is necessary for optimal hemoglobinization of erythroid cells. Haematologica 2017, 102, 1314–1323. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Willmore, W.G. S-glutathionylation reactions in mitochondrial function and disease. Front Cell Dev Biol 2014, 2, 68. [Google Scholar] [CrossRef]
- Newman, J.C.; He, W.; Verdin, E. Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for metabolic disease. J Biol Chem 2012, 287, 42436–42443. [Google Scholar] [CrossRef]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab 2013, 18, 920–933. [Google Scholar] [CrossRef]
- McDonagh, B.; Pedrajas, J.R.; Padilla, C.A.; Barcena, J.A. Thiol redox sensitivity of two key enzymes of heme biosynthesis and pentose phosphate pathways: uroporphyrinogen decarboxylase and transketolase. Oxid Med Cell Longev 2013, 2013, 932472. [Google Scholar] [CrossRef]
- Davis, S.L.; Littlewood, T.J. The investigation and treatment of secondary anaemia. Blood Rev 2012, 26, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, B.; Doczi, J.; Csete, D.; Kacso, G.; Ravasz, D.; Adams, D.; Kiss, G.; Nagy, A.M.; Horvath, G.; Tretter, L.; et al. Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by LPS-induced Irg1 expression in cells of murine macrophage lineage. FASEB J 2016, 30, 286–300. [Google Scholar] [CrossRef]
- Marcero, J.R.; Cox, J.E.; Bergonia, H.A.; Medlock, A.E.; Phillips, J.D.; Dailey, H.A. The immunometabolite itaconate inhibits heme synthesis and remodels cellular metabolism in erythroid precursors. Blood Adv 2021, 5, 4831–4841. [Google Scholar] [CrossRef]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Kafina, M.D.; Paw, B.H. Intracellular iron and heme trafficking and metabolism in developing erythroblasts. Metallomics 2017, 9, 1193–1203. [Google Scholar] [CrossRef]
- Rondelli, C.M.; Perfetto, M.; Danoff, A.; Bergonia, H.; Gillis, S.; O'Neill, L.; Jackson, L.; Nicolas, G.; Puy, H.; West, R.; et al. The ubiquitous mitochondrial protein unfoldase CLPX regulates erythroid heme synthesis by control of iron utilization and heme synthesis enzyme activation and turnover. J Biol Chem 2021, 297, 100972. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef]
- Manwani, D.; Bieker, J.J. The Erythroblastic Island. In Red Cell Development, Bieker, J.J., Ed.; Current Topics in Developmental Biology; Elsevier Inc.: 2008; Volume 82, pp. 23-53.
- Chen, J.J.; Zhang, S. Heme-regulated eIF2alpha kinase in erythropoiesis and hemoglobinopathies. Blood 2019, 134, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Menon, V.; Qiu, J.; Arif, T.; Renuse, S.; Lin, M.; Nowak, R.; Hartmann, B.; Tzavaras, N.; Benson, D.L.; et al. Mitochondrial localization and moderated activity are key to murine erythroid enucleation. Blood Adv 2021, 5, 2490–2504. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, P.; Vijayan, V.; Gueler, F.; Immenschuh, S. Interplay of Heme with Macrophages in Homeostasis and Inflammation. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Pek, R.H.; Yuan, X.; Rietzschel, N.; Zhang, J.; Jackson, L.; Nishibori, E.; Ribeiro, A.; Simmons, W.; Jagadeesh, J.; Sugimoto, H.; et al. Hemozoin produced by mammals confers heme tolerance. Elife 2019, 8. [Google Scholar] [CrossRef]
- Ma, S.; Dubin, A.E.; Zhang, Y.; Mousavi, S.A.R.; Wang, Y.; Coombs, A.M.; Loud, M.; Andolfo, I.; Patapoutian, A. A role of PIEZO1 in iron metabolism in mice and humans. Cell 2021, 184, 969–982 e913. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, I.; Rosato, B.E.; Manna, F.; De Rosa, G.; Marra, R.; Gambale, A.; Girelli, D.; Russo, R.; Iolascon, A. Gain-of-function mutations in PIEZO1 directly impair hepatic iron metabolism via the inhibition of the BMP/SMADs pathway. Am J Hematol 2020, 95, 188–197. [Google Scholar] [CrossRef]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: allies against heme toxicity from hemoglobin not contenders. Front Physiol 2015, 6, 187. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front Pharmacol 2014, 5, 61. [Google Scholar] [CrossRef]
- Ashouri, R.; Fangman, M.; Burris, A.; Ezenwa, M.O.; Wilkie, D.J.; Dore, S. Critical Role of Hemopexin Mediated Cytoprotection in the Pathophysiology of Sickle Cell Disease. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11. [Google Scholar] [CrossRef]
- McMahon, M.; Ding, S.; Acosta-Jimenez, L.P.; Frangova, T.G.; Henderson, C.J.; Wolf, C.R. Measuring in vivo responses to endogenous and exogenous oxidative stress using a novel haem oxygenase 1 reporter mouse. J Physiol 2018, 596, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.V.; Sapochnik, D.; Garcia Sola, M.; Coso, O. Regulation of the Expression of Heme Oxygenase-1: Signal Transduction, Gene Promoter Activation, and Beyond. Antioxid Redox Signal 2020, 32, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Yachie, A. Heme Oxygenase-1 Deficiency and Oxidative Stress: A Review of 9 Independent Human Cases and Animal Models. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Balla, J.; Zarjou, A. Heme Burden and Ensuing Mechanisms That Protect the Kidney: Insights from Bench and Bedside. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Gburek, J.; Verroust, P.J.; Willnow, T.E.; Fyfe, J.C.; Nowacki, W.; Jacobsen, C.; Moestrup, S.K.; Christensen, E.I. Megalin and cubilin are endocytic receptors involved in renal clearance of hemoglobin. J Am Soc Nephrol 2002, 13, 423–430. [Google Scholar] [CrossRef]
- Coronado, L.M.; Nadovich, C.T.; Spadafora, C. Malarial hemozoin: from target to tool. Biochim Biophys Acta 2014, 1840, 2032–2041. [Google Scholar] [CrossRef]
- Matz, J.M.; Drepper, B.; Blum, T.B.; van Genderen, E.; Burrell, A.; Martin, P.; Stach, T.; Collinson, L.M.; Abrahams, J.P.; Matuschewski, K.; et al. A lipocalin mediates unidirectional heme biomineralization in malaria parasites. Proc Natl Acad Sci U S A 2020, 117, 16546–16556. [Google Scholar] [CrossRef]
- Khan, A.A.; Quigley, J.G. Heme and FLVCR-related transporter families SLC48 and SLC49. Mol Aspects Med 2013, 34, 669–682. [Google Scholar] [CrossRef]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef]
- Philip, M.; Funkhouser, S.A.; Chiu, E.Y.; Phelps, S.R.; Delrow, J.J.; Cox, J.; Fink, P.J.; Abkowitz, J.L. Heme exporter FLVCR is required for T cell development and peripheral survival. J Immunol 2015, 194, 1677–1685. [Google Scholar] [CrossRef]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J Clin Invest 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Kalailingam, P.; Wang, K.Q.; Toh, X.R.; Nguyen, T.Q.; Chandrakanthan, M.; Hasan, Z.; Habib, C.; Schif, A.; Radio, F.C.; Dallapiccola, B.; et al. Deficiency of MFSD7c results in microcephaly-associated vasculopathy in Fowler syndrome. J Clin Invest 2020, 130, 4081–4093. [Google Scholar] [CrossRef]
- Li, Y.; Ivica, N.A.; Dong, T.; Papageorgiou, D.P.; He, Y.; Brown, D.R.; Kleyman, M.; Hu, G.; Chen, W.W.; Sullivan, L.B.; et al. MFSD7C switches mitochondrial ATP synthesis to thermogenesis in response to heme. Nat Commun 2020, 11, 4837. [Google Scholar] [CrossRef]
- Yuan, X.; Protchenko, O.; Philpott, C.C.; Hamza, I. Topologically conserved residues direct heme transport in HRG-1-related proteins. J Biol Chem 2012, 287, 4914–4924. [Google Scholar] [CrossRef]
- Korolnek, T.; Zhang, J.; Beardsley, S.; Scheffer, G.L.; Hamza, I. Control of metazoan heme homeostasis by a conserved multidrug resistance protein. Cell Metab 2014, 19, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Chambers, I.G.; Kumar, P.; Lichtenberg, J.; Wang, P.; Yu, J.; Phillips, J.D.; Kane, M.A.; Bodine, D.; Hamza, I. MRP5 and MRP9 play a concerted role in male reproduction and mitochondrial function. Proc Natl Acad Sci U S A 2022, 119. [Google Scholar] [CrossRef]
- Jonker, J.W.; Buitelaar, M.; Wagenaar, E.; Van Der Valk, M.A.; Scheffer, G.L.; Scheper, R.J.; Plosch, T.; Kuipers, F.; Elferink, R.P.; Rosing, H.; et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci U S A 2002, 99, 15649–15654. [Google Scholar] [CrossRef]
- Chambers, I.G.; Willoughby, M.M.; Hamza, I.; Reddi, A.R. One ring to bring them all and in the darkness bind them: The trafficking of heme without deliverers. Biochim Biophys Acta Mol Cell Res 2021, 1868, 118881. [Google Scholar] [CrossRef]
- Bryk, A.H.; Wisniewski, J.R. Quantitative Analysis of Human Red Blood Cell Proteome. J Proteome Res 2017, 16, 2752–2761. [Google Scholar] [CrossRef]
- Krishnamurthy, P.C.; Du, G.; Fukuda, Y.; Sun, D.; Sampath, J.; Mercer, K.E.; Wang, J.; Sosa-Pineda, B.; Murti, K.G.; Schuetz, J.D. Identification of a mammalian mitochondrial porphyrin transporter. Nature 2006, 443, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zhang, S.; Tian, M.; Zhang, L.; Guo, R.; Zhuo, W.; Yang, M. Molecular insights into the human ABCB6 transporter. Cell Discov 2021, 7, 55. [Google Scholar] [CrossRef]
- Lill, R.; Kispal, G. Mitochondrial ABC transporters. Res Microbiol 2001, 152, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Grubic Kezele, T.; Curko-Cofek, B. Age-Related Changes and Sex-Related Differences in Brain Iron Metabolism. Nutrients 2020, 12. [Google Scholar] [CrossRef]
- Zierfuss, B.; Wang, Z.; Jackson, A.N.; Moezzi, D.; Yong, V.W. Iron in multiple sclerosis - Neuropathology, immunology, and real-world considerations. Mult Scler Relat Disord 2023, 78, 104934. [Google Scholar] [CrossRef]
- Qin, D.; Li, D.; Wang, C.; Guo, S. Ferroptosis and central nervous system demyelinating diseases. J Neurochem 2023, 165, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Garringer, H.J.; Irimia, J.M.; Li, W.; Goodwin, C.B.; Richine, B.; Acton, A.; Chan, R.J.; Peacock, M.; Muhoberac, B.B.; Ghetti, B.; et al. Effect of Systemic Iron Overload and a Chelation Therapy in a Mouse Model of the Neurodegenerative Disease Hereditary Ferritinopathy. PLoS One 2016, 11, e0161341. [Google Scholar] [CrossRef]
- Li, Y.; Sun, M.; Cao, F.; Chen, Y.; Zhang, L.; Li, H.; Cao, J.; Song, J.; Ma, Y.; Mi, W.; et al. The Ferroptosis Inhibitor Liproxstatin-1 Ameliorates LPS-Induced Cognitive Impairment in Mice. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Hu, X.; Xu, Y.; Xu, H.; Jin, C.; Zhang, H.; Su, H.; Li, Y.; Zhou, K.; Ni, W. Progress in Understanding Ferroptosis and Its Targeting for Therapeutic Benefits in Traumatic Brain and Spinal Cord Injuries. Front Cell Dev Biol 2021, 9, 705786. [Google Scholar] [CrossRef]
- Cui, J.; Guo, X.; Li, Q.; Song, N.; Xie, J. Hepcidin-to-Ferritin Ratio Is Decreased in Astrocytes With Extracellular Alpha-Synuclein and Iron Exposure. Front Cell Neurosci 2020, 14, 47. [Google Scholar] [CrossRef]
- Wu, T.; Liang, X.; Liu, X.; Li, Y.; Wang, Y.; Kong, L.; Tang, M. Induction of ferroptosis in response to graphene quantum dots through mitochondrial oxidative stress in microglia. Part Fibre Toxicol 2020, 17, 30. [Google Scholar] [CrossRef]
- Chen, S.; Chen, Y.; Zhang, Y.; Kuang, X.; Liu, Y.; Guo, M.; Ma, L.; Zhang, D.; Li, Q. Iron Metabolism and Ferroptosis in Epilepsy. Front Neurosci 2020, 14, 601193. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.Y.; Huang, B.Y.; Nie, H.F.; Chen, X.Y.; Zhou, Y.; Yang, T.; Cheng, S.W.; Mei, Z.G.; Ge, J.W. The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases. Brain Sci 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Bardestani, A.; Ebrahimpour, S.; Esmaeili, A.; Esmaeili, A. Quercetin attenuates neurotoxicity induced by iron oxide nanoparticles. J Nanobiotechnology 2021, 19, 327. [Google Scholar] [CrossRef]
- Huang, S.; Li, S.; Feng, H.; Chen, Y. Iron Metabolism Disorders for Cognitive Dysfunction After Mild Traumatic Brain Injury. Front Neurosci 2021, 15, 587197. [Google Scholar] [CrossRef]
- Wimmer, I.; Scharler, C.; Kadowaki, T.; Hillebrand, S.; Scheiber-Mojdehkar, B.; Ueda, S.; Bradl, M.; Berger, T.; Lassmann, H.; Hametner, S. Iron accumulation in the choroid plexus, ependymal cells and CNS parenchyma in a rat strain with low-grade haemolysis of fragile macrocytic red blood cells. Brain Pathol 2021, 31, 333–345. [Google Scholar] [CrossRef]
- Slone, J.D.; Yang, L.; Peng, Y.; Queme, L.F.; Harris, B.; Rizzo, S.J.S.; Green, T.; Ryan, J.L.; Jankowski, M.P.; Reinholdt, L.G.; et al. Integrated analysis of the molecular pathogenesis of FDXR-associated disease. Cell Death Dis 2020, 11, 423. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lucas, S.; Proudman, D.; Nellesen, D.; Paulose, J.; Sheehan, V.A. Burden of central nervous system complications in sickle cell disease: A systematic review and meta-analysis. Pediatr Blood Cancer 2022, 69, e29493. [Google Scholar] [CrossRef]
- LeVine, S.M.; Tsau, S.; Gunewardena, S. Exploring Whether Iron Sequestration within the CNS of Patients with Alzheimer's Disease Causes a Functional Iron Deficiency That Advances Neurodegeneration. Brain Sci 2023, 13. [Google Scholar] [CrossRef]
- Porras, C.A.; Rouault, T.A. Iron Homeostasis in the CNS: An Overview of the Pathological Consequences of Iron Metabolism Disruption. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Chen, P.; Totten, M.; Zhang, Z.; Bucinca, H.; Erikson, K.; Santamaria, A.; Bowman, A.B.; Aschner, M. Iron and manganese-related CNS toxicity: mechanisms, diagnosis and treatment. Expert Rev Neurother 2019, 19, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol 2023, 20, 7–23. [Google Scholar] [CrossRef]
- Kozlowska, B.; Sochanowicz, B.; Kraj, L.; Palusinska, M.; Kolsut, P.; Szymanski, L.; Lewicki, S.; Kruszewski, M.; Zaleska-Kociecka, M.; Leszek, P. Clinical and Molecular Aspects of Iron Metabolism in Failing Myocytes. Life (Basel) 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep 2015, 13, 533–545. [Google Scholar] [CrossRef]
- Noetzli, L.J.; Papudesi, J.; Coates, T.D.; Wood, J.C. Pancreatic iron loading predicts cardiac iron loading in thalassemia major. Blood 2009, 114, 4021–4026. [Google Scholar] [CrossRef]
- Liu, Q.; Azucenas, C.; Mackenzie, B.; Knutson, M. Metal-Ion Transporter SLC39A14 Is Required for Cardiac Iron Loading in the Hjv Mouse Model of Iron Overload. Blood 2021, 138, 758–758. [Google Scholar] [CrossRef]
- Jenkitkasemwong, S.; Wang, C.Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.S.; Hojyo, S.; Fukada, T.; Knutson, M.D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab 2015, 22, 138–150. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Trivieri, M.G.; Khaper, N.; Liu, P.P.; Backx, P.H. Role of L-type Ca2+ channels in iron transport and iron-overload cardiomyopathy. J Mol Med (Berl) 2006, 84, 349–364. [Google Scholar] [CrossRef]
- Grammer, T.B.; Scharnagl, H.; Dressel, A.; Kleber, M.E.; Silbernagel, G.; Pilz, S.; Tomaschitz, A.; Koenig, W.; Mueller-Myhsok, B.; Marz, W.; et al. Iron Metabolism, Hepcidin, and Mortality (the Ludwigshafen Risk and Cardiovascular Health Study). Clin Chem 2019, 65, 849–861. [Google Scholar] [CrossRef]
- Sawicki, K.T.; De Jesus, A.; Ardehali, H. Iron Metabolism in Cardiovascular Disease: Physiology, Mechanisms, and Therapeutic Targets. Circ Res 2023, 132, 379–396. [Google Scholar] [CrossRef]
- Morrell, N.W.; Bloch, D.B.; ten Dijke, P.; Goumans, M.J.; Hata, A.; Smith, J.; Yu, P.B.; Bloch, K.D. Targeting BMP signalling in cardiovascular disease and anaemia. Nat Rev Cardiol 2016, 13, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Trebicka, E.; Fu, Y.; Ellenbogen, S.; Hong, C.C.; Babitt, J.L.; Lin, H.Y.; Cherayil, B.J. The bone morphogenetic protein-hepcidin axis as a therapeutic target in inflammatory bowel disease. Inflamm Bowel Dis 2012, 18, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Saeed, O.; Otsuka, F.; Polavarapu, R.; Karmali, V.; Weiss, D.; Davis, T.; Rostad, B.; Pachura, K.; Adams, L.; Elliott, J.; et al. Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler Thromb Vasc Biol 2012, 32, 299–307. [Google Scholar] [CrossRef]
- Chen, Z.; Yan, Y.; Qi, C.; Liu, J.; Li, L.; Wang, J. The Role of Ferroptosis in Cardiovascular Disease and Its Therapeutic Significance. Front Cardiovasc Med 2021, 8, 733229. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Zhang, S.; Zhou, X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics 2021, 11, 3052–3059. [Google Scholar] [CrossRef]
- Gao, X.; Hu, W.; Qian, D.; Bai, X.; He, H.; Li, L.; Sun, S. The Mechanisms of Ferroptosis Under Hypoxia. Cell Mol Neurobiol 2023, 43, 3329–3341. [Google Scholar] [CrossRef]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ Res 2020, 127, 486–501. [Google Scholar] [CrossRef]
- Gozzelino, R.; Soares, M.P. Coupling heme and iron metabolism via ferritin H chain. Antioxid Redox Signal 2014, 20, 1754–1769. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond) 2018, 38, 12. [Google Scholar] [CrossRef]
- Liu, G.; Xie, X.; Liao, W.; Chen, S.; Zhong, R.; Qin, J.; He, P.; Xie, J. Ferroptosis in cardiovascular disease. Biomed Pharmacother 2024, 170, 116057. [Google Scholar] [CrossRef]
- Xiao, Z.; Kong, B.; Fang, J.; Qin, T.; Dai, C.; Shuai, W.; Huang, H. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered 2021, 12, 9367–9376. [Google Scholar] [CrossRef]
- van der Wal, H.H.; Grote Beverborg, N.; Dickstein, K.; Anker, S.D.; Lang, C.C.; Ng, L.L.; van Veldhuisen, D.J.; Voors, A.A.; van der Meer, P. Iron deficiency in worsening heart failure is associated with reduced estimated protein intake, fluid retention, inflammation, and antiplatelet use. Eur Heart J 2019, 40, 3616–3625. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Yang, L.; Zheng, L. Association between dietary intake of iron and heart failure among American adults: data from NHANES 2009-2018. BMC Nutr 2024, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wessling-Resnick, M. The Role of Iron Metabolism in Lung Inflammation and Injury. J Allergy Ther 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Heilig, E.A.; Thompson, K.J.; Molina, R.M.; Ivanov, A.R.; Brain, J.D.; Wessling-Resnick, M. Manganese and iron transport across pulmonary epithelium. Am J Physiol Lung Cell Mol Physiol 2006, 290, L1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, V.; Nemeth, E.; Kim, A. Iron in Lung Pathology. Pharmaceuticals (Basel) 2019, 12. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Mumby, S.; Adcock, I.M.; Choi, A.M.K.; Chung, K.F.; Quinlan, G.J. The "Iron"-y of Iron Overload and Iron Deficiency in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 2017, 196, 1103–1112. [Google Scholar] [CrossRef]
- Zhang, V.; Jenkitkasemwong, S.; Liu, Q.; Ganz, T.; Nemeth, E.; Knutson, M.D.; Kim, A. A mouse model characterizes the roles of ZIP8 in systemic iron recycling and lung inflammation and infection. Blood Adv 2023, 7, 1336–1349. [Google Scholar] [CrossRef]
- Ghio, A.J.; Carter, J.D.; Richards, J.H.; Richer, L.D.; Grissom, C.K.; Elstad, M.R. Iron and iron-related proteins in the lower respiratory tract of patients with acute respiratory distress syndrome. Crit Care Med 2003, 31, 395–400. [Google Scholar] [CrossRef]
- Cao, X.; Ren, Y.; Lu, Q.; Wang, K.; Wu, Y.; Wang, Y.; Zhang, Y.; Cui, X.S.; Yang, Z.; Chen, Z. Lactoferrin: A glycoprotein that plays an active role in human health. Front Nutr 2022, 9, 1018336. [Google Scholar] [CrossRef]
- Rosa, L.; Cutone, A.; Lepanto, M.S.; Paesano, R.; Valenti, P. Lactoferrin: A Natural Glycoprotein Involved in Iron and Inflammatory Homeostasis. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Stonehuerner, J.G.; Richards, J.H.; Crissman, K.M.; Roggli, V.L.; Piantadosi, C.A.; Carraway, M.S. Iron homeostasis and oxidative stress in idiopathic pulmonary alveolar proteinosis: a case-control study. Respir Res 2008, 9, 10. [Google Scholar] [CrossRef]
- Mateos, F.; Gonzalez, C.; Dominguez, C.; Losa, J.E.; Jimenez, A.; Perez-Arellano, J.L. Elevated non-transferrin bound iron in the lungs of patients with Pneumocystis carinii pneumonia. J Infect 1999, 38, 18–21. [Google Scholar] [CrossRef]
- Zumerle, S.; Mathieu, J.R.; Delga, S.; Heinis, M.; Viatte, L.; Vaulont, S.; Peyssonnaux, C. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood 2014, 123, 3646–3650. [Google Scholar] [CrossRef]
- Deschemin, J.C.; Mathieu, J.R.R.; Zumerle, S.; Peyssonnaux, C.; Vaulont, S. Pulmonary Iron Homeostasis in Hepcidin Knockout Mice. Front Physiol 2017, 8, 804. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.B.; Callaghan, K.D.; Ghio, A.J.; Haile, D.J.; Yang, F. Hepcidin expression and iron transport in alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2006, 291, L417–425. [Google Scholar] [CrossRef]
- Yang, F.; Wang, X.; Haile, D.J.; Piantadosi, C.A.; Ghio, A.J. Iron increases expression of iron-export protein MTP1 in lung cells. Am J Physiol Lung Cell Mol Physiol 2002, 283, L932–939. [Google Scholar] [CrossRef]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: expression and regulation. Biochim Biophys Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef]
- Qu, M.; Zhang, H.; Chen, Z.; Sun, X.; Zhu, S.; Nan, K.; Chen, W.; Miao, C. The Role of Ferroptosis in Acute Respiratory Distress Syndrome. Front Med (Lausanne) 2021, 8, 651552. [Google Scholar] [CrossRef]
- Zhou, H.; Li, F.; Niu, J.Y.; Zhong, W.Y.; Tang, M.Y.; Lin, D.; Cui, H.H.; Huang, X.H.; Chen, Y.Y.; Wang, H.Y.; et al. Ferroptosis was involved in the oleic acid-induced acute lung injury in mice. Sheng Li Xue Bao 2019, 71, 689–697. [Google Scholar] [PubMed]
- Pei, Z.; Qin, Y.; Fu, X.; Yang, F.; Huo, F.; Liang, X.; Wang, S.; Cui, H.; Lin, P.; Zhou, G.; et al. Inhibition of ferroptosis and iron accumulation alleviates pulmonary fibrosis in a bleomycin model. Redox Biol 2022, 57, 102509. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett 2020, 25, 10. [Google Scholar] [CrossRef]
- Lai, K.; Song, C.; Gao, M.; Deng, Y.; Lu, Z.; Li, N.; Geng, Q. Uridine Alleviates Sepsis-Induced Acute Lung Injury by Inhibiting Ferroptosis of Macrophage. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Gabrielsen, J.S.; Lamb, D.J.; Lipshultz, L.I. Iron and a Man's Reproductive Health: the Good, the Bad, and the Ugly. Curr Urol Rep 2018, 19, 60. [Google Scholar] [CrossRef]
- Leandri, R.; Power, K.; Buonocore, S.; De Vico, G. Preliminary Evidence of the Possible Roles of the Ferritinophagy-Iron Uptake Axis in Canine Testicular Cancer. Animals (Basel) 2024, 14. [Google Scholar] [CrossRef] [PubMed]
- El Osta, R.; Grandpre, N.; Monnin, N.; Hubert, J.; Koscinski, I. Hypogonadotropic hypogonadism in men with hereditary hemochromatosis. Basic Clin Androl 2017, 27, 13. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, S.R.; Griswold, M.D. Localization of transferrin and transferrin receptors in rat testes. Biol Reprod 1984, 31, 195–203. [Google Scholar] [CrossRef]
- Morales, C.; Sylvester, S.R.; Griswold, M.D. Transport of iron and transferrin synthesis by the seminiferous epithelium of the rat in vivo. Biol Reprod 1987, 37, 995–1005. [Google Scholar] [CrossRef]
- Yuan, W.; Sun, Z.; Ji, G.; Hu, H. Emerging roles of ferroptosis in male reproductive diseases. Cell Death Discov 2023, 9, 358. [Google Scholar] [CrossRef]
- Jabado, N.; Canonne-Hergaux, F.; Gruenheid, S.; Picard, V.; Gros, P. Iron transporter Nramp2/DMT-1 is associated with the membrane of phagosomes in macrophages and Sertoli cells. Blood 2002, 100, 2617–2622. [Google Scholar] [CrossRef]
- Griffin, K.P.; Ward, D.T.; Liu, W.; Stewart, G.; Morris, I.D.; Smith, C.P. Differential expression of divalent metal transporter DMT1 (Slc11a2) in the spermatogenic epithelium of the developing and adult rat testis. Am J Physiol Cell Physiol 2005, 288, C176–184. [Google Scholar] [CrossRef] [PubMed]
- Leichtmann-Bardoogo, Y.; Cohen, L.A.; Weiss, A.; Marohn, B.; Schubert, S.; Meinhardt, A.; Meyron-Holtz, E.G. Compartmentalization and regulation of iron metabolism proteins protect male germ cells from iron overload. Am J Physiol Endocrinol Metab 2012, 302, E1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Cheng, Y.; Wang, X.; Yang, X.; Liu, M.; Liu, J.; Liu, S.; Wang, H.; Zhang, A.; Li, R.; et al. Gss deficiency causes age-related fertility impairment via ROS-triggered ferroptosis in the testes of mice. Cell Death Dis 2023, 14, 845. [Google Scholar] [CrossRef]
- De Sanctis, V.; Daar, S.; Soliman, A.; Tzoulis, P.; Yassin, M.; Kattamis, C. A retrospective study of glucose homeostasis, insulin secretion, sensitivity/resistance in non- transfusion-dependent beta-thalassemia patients (NTD- beta Thal): reduced beta-cell secretion rather than insulin resistance seems to be the dominant defect for glucose dysregulation (GD). Acta Biomed 2023, 94, e2023262. [Google Scholar] [CrossRef]
- Halon-Golabek, M.; Borkowska, A.; Herman-Antosiewicz, A.; Antosiewicz, J. Iron Metabolism of the Skeletal Muscle and Neurodegeneration. Front Neurosci 2019, 13, 165. [Google Scholar] [CrossRef]
- Mathew, M.; Sivaprakasam, S.; Phy, J.L.; Bhutia, Y.D.; Ganapathy, V. Polycystic ovary syndrome and iron overload: biochemical link and underlying mechanisms with potential novel therapeutic avenues. Biosci Rep 2023, 43. [Google Scholar] [CrossRef]
- Yu, X.; Peng, Y.; Nie, T.; Sun, W.; Zhou, Y. Diabetes and two kinds of primary tumors in a patient with thalassemia: a case report and literature review. Front Oncol 2023, 13, 1207336. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, J.; Tarhini, A.; Bou-Fakhredin, R.; Saliba, A.N.; Cappellini, M.D.; Taher, A.T. Non-Transfusion-Dependent Thalassemia: An Update on Complications and Management. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Stepniak, J.; Rynkowska, A.; Karbownik-Lewinska, M. Membrane Lipids in the Thyroid Comparing to Those in Non-Endocrine Tissues Are Less Sensitive to Pro-Oxidative Effects of Fenton Reaction Substrates. Front Mol Biosci 2022, 9, 901062. [Google Scholar] [CrossRef]
- Chalmers, A.W.; Shammo, J.M. Evaluation of a new tablet formulation of deferasirox to reduce chronic iron overload after long-term blood transfusions. Ther Clin Risk Manag 2016, 12, 201–208. [Google Scholar] [CrossRef]
- Ngim, C.F.; Lai, N.M.; Hong, J.Y.; Tan, S.L.; Ramadas, A.; Muthukumarasamy, P.; Thong, M.K. Growth hormone therapy for people with thalassaemia. Cochrane Database Syst Rev 2020, 5, CD012284. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.C.; Vishnu, P.; Sanchez, W.; Aqel, B.; Riegert-Johnson, D.; Seaman, L.A.K.; Bowman, A.W.; Rivera, C.E. Diagnosis and Management of Genetic Iron Overload Disorders. J Gen Intern Med 2018, 33, 2230–2236. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.C.; Lee, B.S.; Tang, S.O.; Loh, E.W.; Ng, S.C.; Tan, X.Y.; Ahmad Noordin, M.N.; Ong, G.B.; Chew, L.C. Iron burden and endocrine complications in transfusion-dependent thalassemia patients In Sarawak, Malaysia: a retrospective study. Med J Malaysia 2024, 79, 281–287. [Google Scholar]
- De Sanctis, V.; Soliman, A.T.; Canatan, D.; Elsedfy, H.; Karimi, M.; Daar, S.; Rimawi, H.; Christou, S.; Skordis, N.; Tzoulis, P.; et al. An ICET- A survey on Hypoparathyroidism in Patients with Thalassaemia Major and Intermedia: A preliminary report. Acta Biomed 2018, 88, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Yaghobi, M.; Miri-Moghaddam, E.; Majid, N.; Bazi, A.; Navidian, A.; Kalkali, A. Complications of Transfusion-Dependent beta-Thalassemia Patients in Sistan and Baluchistan, South-East of Iran. Int J Hematol Oncol Stem Cell Res 2017, 11, 268–272. [Google Scholar]
- Shah, R.; Shah, A.; Badawy, S.M. An evaluation of deferiprone as twice-a-day tablets or in combination therapy for the treatment of transfusional iron overload in thalassemia syndromes. Expert review of hematology 2023, 16, 81–94. [Google Scholar] [CrossRef]
- Mahwi, T.O.; Rashid, Z.G.; Ahmed, S.F. Hypogonadism among patients with transfusion-dependent thalassemia: a cross-sectional study. Annals of Medicine and Surgery 2023, 85, 3418–3422. [Google Scholar] [CrossRef]
- De Sanctis, V.; Soliman, A.T.; Yassin, M.A.; Di Maio, S.; Daar, S.; Elsedfy, H.; Soliman, N.; Kattamis, C. Hypogonadism in male thalassemia major patients: pathophysiology, diagnosis and treatment. Acta Biomed 2018, 89, 6–15. [Google Scholar] [CrossRef]
- Di Maio, S.; Marzuillo, P.; Mariannis, D.; Christou, S.; Ellinides, A.; Christodoulides, C.; de Sanctis, V. A Retrospective Long-Term Study on Age at Menarche and Menstrual Characteristics in 85 Young Women with Transfusion-Dependent beta-Thalassemia (TDT). Mediterr J Hematol Infect Dis 2021, 13, e2021040. [Google Scholar] [CrossRef]
- Arab-Zozani, M.; Kheyrandish, S.; Rastgar, A.; Miri-Moghaddam, E. A Systematic Review and Meta-Analysis of Stature Growth Complications in beta-thalassemia Major Patients. Ann Glob Health 2021, 87, 48. [Google Scholar] [CrossRef]
- Giordano, P.; Urbano, F.; Lassandro, G.; Faienza, M.F. Mechanisms of Bone Impairment in Sickle Bone Disease. Int J Environ Res Public Health 2021, 18. [Google Scholar] [CrossRef] [PubMed]
- Atmakusuma, T.D.; Hasibuan, F.D.; Purnamasari, D. The Correlation Between Iron Overload and Endocrine Function in Adult Transfusion-Dependent Beta-Thalassemia Patients with Growth Retardation. J Blood Med 2021, 12, 749–753. [Google Scholar] [CrossRef]
- Carsote, M.; Vasiliu, C.; Trandafir, A.I.; Albu, S.E.; Dumitrascu, M.C.; Popa, A.; Mehedintu, C.; Petca, R.C.; Petca, A.; Sandru, F. New Entity-Thalassemic Endocrine Disease: Major Beta-Thalassemia and Endocrine Involvement. Diagnostics (Basel) 2022, 12. [Google Scholar] [CrossRef]
- De Sanctis, V.; Soliman, A.T.; Canatan, D.; Yassin, M.A.; Daar, S.; Elsedfy, H.; Di Maio, S.; Raiola, G.; Corrons, J.V.; Kattamis, C. Thyroid Disorders in Homozygous beta-Thalassemia: Current Knowledge, Emerging Issues and Open Problems. Mediterr J Hematol Infect Dis 2019, 11, e2019029. [Google Scholar] [CrossRef]
- D'Aprile, S.; Denaro, S.; Pavone, A.M.; Giallongo, S.; Giallongo, C.; Distefano, A.; Salvatorelli, L.; Torrisi, F.; Giuffrida, R.; Forte, S.; et al. Anaplastic thyroid cancer cells reduce CD71 levels to increase iron overload tolerance. J Transl Med 2023, 21, 780. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Risco, M.B.; Mendez, M.; Moreno-Carralero, M.I.; Lopez-Moreno, A.M.; Vagace-Valero, J.M.; Moran-Jimenez, M.J. Juvenile Hemochromatosis due to a Homozygous Variant in the HJV Gene. Case Rep Pediatr 2022, 2022, 7743748. [Google Scholar] [CrossRef]
- A, A.H.; M, S.E.-F.; A, M.A.E.-E. Study of Adrenal Functions using ACTH stimulation test in Egyptian children with Sickle Cell Anemia: Correlation with Iron Overload. Int J Hematol Oncol Stem Cell Res 2015, 9, 60–66. [Google Scholar]
- Tangngam, H.; Mahachoklertwattana, P.; Poomthavorn, P.; Chuansumrit, A.; Sirachainan, N.; Chailurkit, L.; Khlairit, P. Under-recognized Hypoparathyroidism in Thalassemia. J Clin Res Pediatr Endocrinol 2018, 10, 324–330. [Google Scholar] [CrossRef]
- Mousa, S.O.; Abd Alsamia, E.M.; Moness, H.M.; Mohamed, O.G. The effect of zinc deficiency and iron overload on endocrine and exocrine pancreatic function in children with transfusion-dependent thalassemia: a cross-sectional study. BMC Pediatr 2021, 21, 468. [Google Scholar] [CrossRef]
- Li, J.; Wang, P.; Li, X.; Wang, Q.; Zhang, J.; Lin, Y. Cost-Utility Analysis of four Chelation Regimens for beta-thalassemia Major: a Chinese Perspective. Mediterr J Hematol Infect Dis 2020, 12, e2020029. [Google Scholar] [CrossRef]
- de Sanctis, V.; Soliman, A.T.; Daar, S.; Tzoulis, P.; Di Maio, S.; Kattamis, C. Long-Term Follow-up of beta-Transfusion-Dependent Thalassemia (TDT) Normoglycemic Patients with Reduced Insulin Secretion to Oral Glucose Tolerance Test (OGTT): A Pilot Study. Mediterr J Hematol Infect Dis 2021, 13, e2021021. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.T.; Lim, S.L.; Goh, A.S. Prevalence of endocrine complications in transfusion dependent thalassemia in Hospital Pulau Pinang: A pilot study. Med J Malaysia 2020, 75, 33–37. [Google Scholar] [PubMed]
- Papanikolaou, G.; Pantopoulos, K. Systemic iron homeostasis and erythropoiesis. IUBMB Life 2017, 69, 399–413. [Google Scholar] [CrossRef]
- Pinto, V.M.; Forni, G.L. Management of iron overload in beta-thalassemia patients: clinical practice update based on case series. International Journal of Molecular Sciences 2020, 21, 8771. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Karmakar, S.; Asthana, A.; Ashok, A.; Desai, V.; Baksi, S.; Singh, N. Transport of non-transferrin bound iron to the brain: implications for Alzheimer’s disease. Journal of Alzheimer's Disease 2017, 58, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zhao, J.; Xiong, Q.; Yu, H.; Du, H. Secondary iron overload induces chronic pancreatitis and ferroptosis of acinar cells in mice. Int J Mol Med 2023, 51. [Google Scholar] [CrossRef]
- Meloni, A.; Pistoia, L.; Gamberini, M.R.; Ricchi, P.; Cecinati, V.; Sorrentino, F.; Cuccia, L.; Allo, M.; Righi, R.; Fina, P.; et al. The Link of Pancreatic Iron with Glucose Metabolism and Cardiac Iron in Thalassemia Intermedia: A Large, Multicenter Observational Study. J Clin Med 2021, 10. [Google Scholar] [CrossRef]
- Canali, S.; Wang, C.Y.; Zumbrennen-Bullough, K.B.; Bayer, A.; Babitt, J.L. Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am J Hematol 2017, 92, 1204–1213. [Google Scholar] [CrossRef]
- Kose, T.; Sharp, P.A.; Latunde-Dada, G.O. Phenolic Acids Rescue Iron-Induced Damage in Murine Pancreatic Cells and Tissues. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Hattori, Y.; Kato, H.; Kato, A.; Tatsumi, Y.; Kato, K.; Hayashi, H. A Japanese female with chronic mild anemia and primary iron overloading disease. Nagoya J Med Sci 2020, 82, 579–583. [Google Scholar] [CrossRef]
- Mostafa, G.G.; Zahran, F.E.; Omer, S.A.; Ibrahim, A.; Elhakeem, H. The Effect of Serum Ferritin Level on Gonadal, Prolactin, Thyroid Hormones, and Thyroid Stimulating Hormone in Adult Males with Sickle Cell Anemia. J Blood Med 2020, 11, 27–32. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Daar, S.; Di Maio, S. Adverse events during testosterone replacement therapy in 95 young hypogonadal thalassemic men. Acta Biomed 2019, 90, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Evangelidis, P.; Venou, T.M.; Fani, B.; Vlachaki, E.; Gavriilaki, E.; on behalf of the International Hemoglobinopathy Research, N. Endocrinopathies in Hemoglobinopathies: What Is the Role of Iron? Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Mandava, M.; Lew, J.; Tisdale, J.F.; Limerick, E.; Fitzhugh, C.D.; Hsieh, M.M. Thyroid and Adrenal Dysfunction in Hemoglobinopathies Before and After Allogeneic Hematopoietic Cell Transplant. J Endocr Soc 2023, 7, bvad134. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Organ systems requiring iron for normal function and affected by iron overload. Shown are pituitary-hypothalamic axis, central nervous system, pulmonary system, heart, bone marrow, liver, pancreas, small bowel, kidneys, thyroid/endocrine.
Figure 1.
Organ systems requiring iron for normal function and affected by iron overload. Shown are pituitary-hypothalamic axis, central nervous system, pulmonary system, heart, bone marrow, liver, pancreas, small bowel, kidneys, thyroid/endocrine.
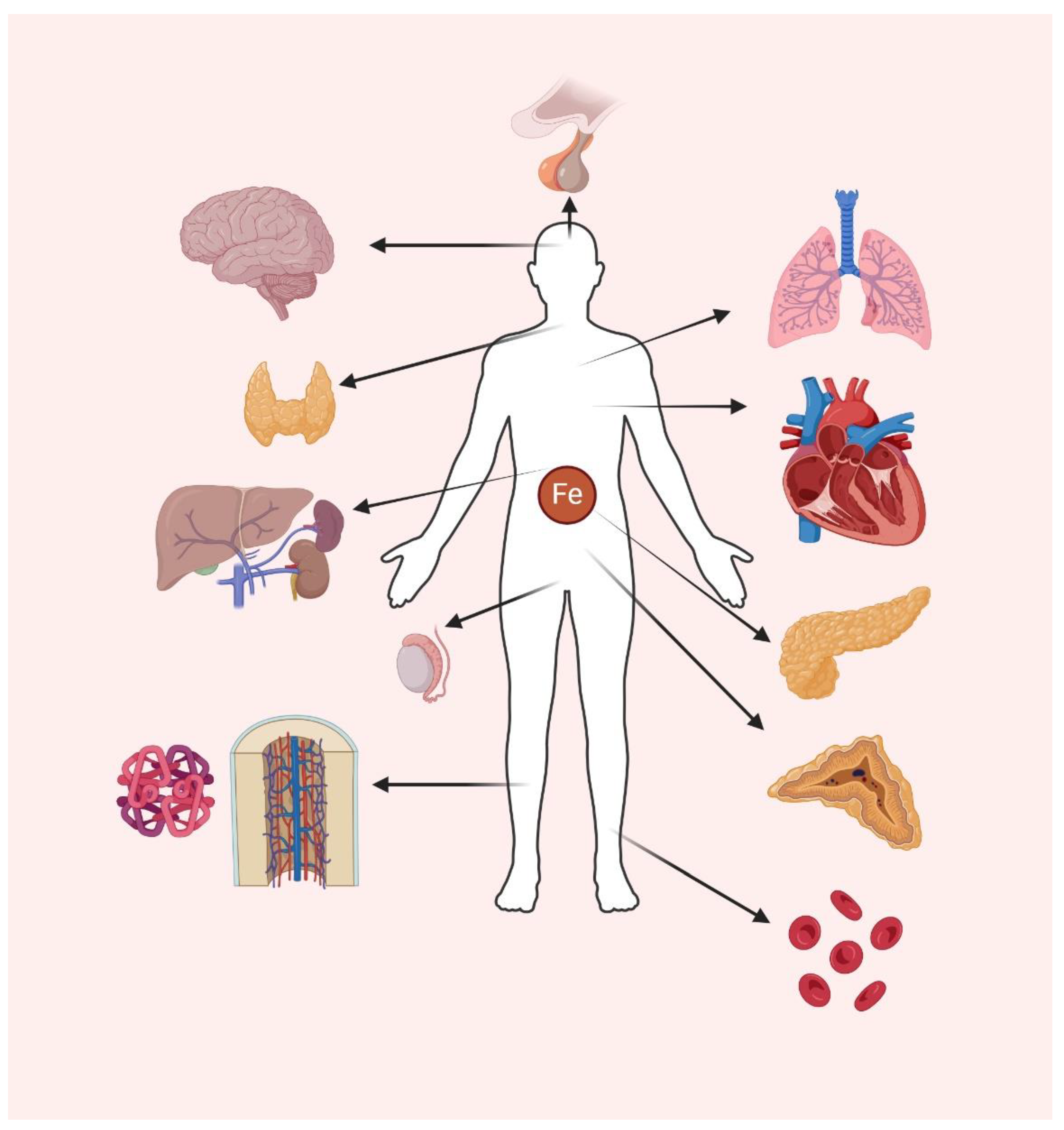
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



