
Submitted:
11 February 2025
Posted:
12 February 2025
You are already at the latest version
Abstract
Boron-containing compounds (BCC) have emerged as potential drugs. The effects of letting act as drugs are mainly by means of action on enzymes. Moreover, no crystal structure shows binding to G-Protein coupled receptors (GPCRs), albeit if those are recognized as targets in many diseases. However, some experimental data support the effects of specific BCC on GPCRs. Objective: Updating data about the reported effects that suggest or support interactions of BCC on GPCR. Methods: Data were collected and reviewed from the National Center of Biotechnology Information, PubMed, Global Health, Embase, the Web of Science, and Google Scholar databases. Results: Some experimental reports support the interactions of BCC in several GPCRs, acting as labels, agonists, or antagonists on them. Details of interaction can be inferred by in silico and in vitro results. Albeit if not a crystal structure showing such a complex. Conclusions: The action of BCCs on GPCR is beyond a presumptive proposal. Evidence supports the interaction and actions of BCCs on GPCRs.
Keywords:
boron
; GPCR
; adrenergic
; cholinergic
; vascular
; neurological
; binding assays
1. Introduction
Boron-containing compounds (BCC) have emerged as potential drugs [1,2]. The effects of letting act as drugs are mainly by means of action on enzymes. Currently, five BCC are marketed as human drugs: Bortezomib and ixazomib acting on the proteasome, crisaborole by its action on phosphodiesterase 4 (PDE4), tavaborole on the protein synthesis by inhibiting Leucyl-tRNA synthetase in some fungi organisms, and vaborbactam, a beta-lactamase inhibitor [1].
Nowadays, many effects of BCC are reported involving the metabolism of mammals, but also effects related to the neurological and cardiovascular system functions [2,3,4,5]. Some of them are explained by suggested or supported interactions on G-Protein coupled receptors (GPCRs) [6]. The classification for GPCRs divided them, based on sequence homology, into six classes. These classes and their prototype members were as follows: Class A (rhodopsin-like), Class B (secretin receptor family), Class C (metabotropic glutamate), Class D (fungal mating pheromone receptors), Class E (cyclic AMP receptors), and Class F (frizzled/smoothened) [7]. In fact, to the best of our knowledge just some works have reported binding of BCC on GPCRs of class A; by competitive displacement of some well-known ligands to GPCRs or presumptive action on the effects of well-known GPCRs-ligands. Less data support actions of other classes or subfamilies as is sentenced below.
However, there is no evidence of specific interactions on any residue of GPCRs. Moreover, no crystal structure shows binding to GPCRs, albeit if those are recognized as targets in many diseases, and some observed effects are strongly associated with action on these targets.
2. Materials and Methods
All data were collected and reviewed from the National Center of Biotechnology Information, PubMed, Global Health, Embase, the Web of Science, Clinical trials, and Google Scholar databases. All articles were carefully revised to identify if the reported information was original or referencing any other document regarding the possible action of a BCC on GPCRs.
3. Results
3.1. Experimental Evidence of Effects of BCC on Family A of GPCRs
3.1.1. On the Catecholamine (Dopamine and Adrenergic) Receptors
The first report on this field could be the use of boron-dipyrromethene (BODIPY)-derivatives as fluorescent ligands on adrenoceptors. In 1998, Daly et al. probed a compound structurally related to prazosin (named BODIFY-FL-prazosin, quinazolinyl piperazine borate-dipyrromethene or QAPB), binding to α1-adrenoceptors with nanomolar affinity, and modifying the inositol-phosphate generation (by blocking the phenylephrine induction), then described as antagonist on that receptor [8]. Moreover, one year later his workgroup reported that confocal visualization showed that the clustered component was located mainly intracellularly. In rat basilar artery smooth muscle cells the intracellular binding sites were located in close proximity to the nuclear membrane, which is interesting because of the known distribution of receptors in some tissues [9]. In the following years later, they described the binding and distribution of adrenoceptors in aorta rat [10], and Baker et al. reported the binding to β2-adrenoceptors, but also it stimulated and increased cyclic-AMP production in CHO-K1 cells expressing the human β2-adrenoceptor. Those effects were reduced by the well-known ligand ICI118551. The authors concluded that the compound (named BODIPY-TMR CGP12177 or BODIPY-TMR-CGP) is a long-acting ligand of β1,β2-adrenoceptors, fluorescent β2-adrenoceptor agonist that can be used to label β2-adrenoceptors in the plasma membrane of living cells which expressing it [11].
In 2008, Soriano-Ursúa et al. reported the first BCC derived from salbutamol (albuterol), which induced relaxation of smooth muscle in guinea-pig trachea, this compound also showed multiple interactions on the binding site in the in-silico assays on the recent reported crystallized structures of β2-adrenoceptor [12]. After that, they reported two additional BCC salbutamol-derivatives, all with micromolar affinity in binding assays as well as higher efficacy and potency on cells expressing human receptors than its boron-free precursor and stable at biological conditions. Later, they studied the differences in the binding affinity on the guinea pig or human homologous receptors and suggested some differences in the mode of binding on the orthosteric site [13]. They described slight differences in molecular dynamic assays, particularly in the role of the boron atom, in the interactions of the compound named politerol with the β2-adrenoceptors of each species, affecting interactions and movements of fifth to seventh transmembrane domains, known to be key in receptor activation (details discussed below).
Also, the use of boron-containing clusters was reported by Louie et al. (2011) as they screened carboranes for binding to serotonin, σ, and adrenergic receptors as well as dopamine, serotonin, and norepinephrine transporters. They reported several metal-carborane derivatives, one of them acting as a probe on α-adrenoceptors with mean-Ki values ranging from 17-435 nm, while less affinity on other amine-receptors and low specific binding [14].
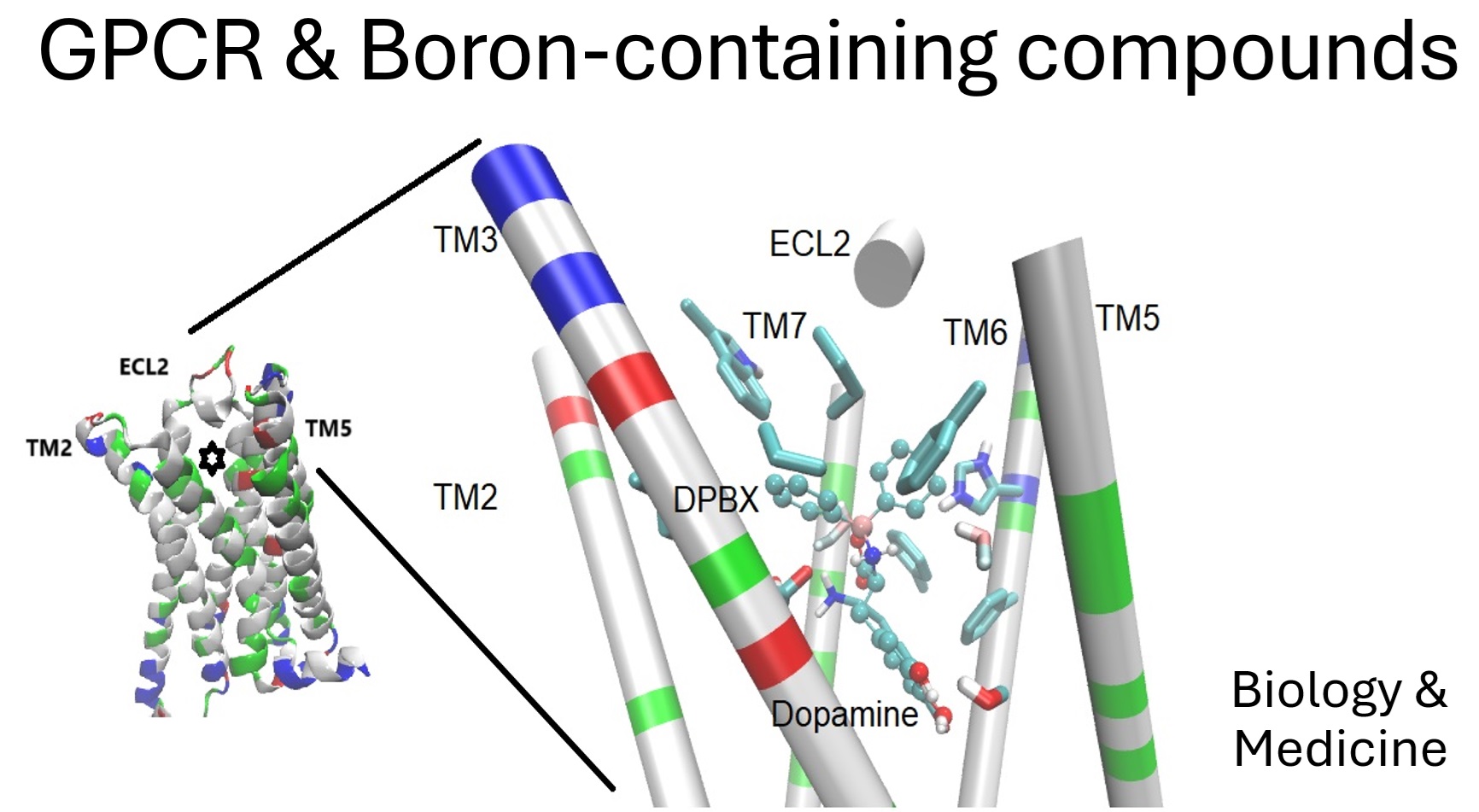
Recently, the description of a BCC exerting effects presumably through interactions on D2DR was reported (Figure 1). In fact, a boroxazolidone-derivative structurally related to levodopa was reported as limiting the neuronal damage and disrupted performance of mice administered with MPTP (a known parkinsonism inducer), while in silico assays suggest binding with submicromolar affinity. The beneficial effects were diminished by the coadministration of the well-known D2DR-antagonist risperidone [6,15].
3.1.2. On other GPCRs of Class A
Among subtypes of receptors, Class A represents the major transmembrane receptor involved in physiological processes such as vision, smell, cellular homeostasis, and specific responses for hormones and neurotransmitters. The scope of BCC is beyond the labeling of receptors; it was enhanced by observations that some BCC can modulate their conformational dynamics. The applications are expanding since this subtype of GPCRs is a key target for the development of novel pharmacological therapies due to their high distribution and functionality. Thus, recent research has demonstrated that BBCs have potential as therapeutic and diagnostic agents.
In this sense, it was observed that in GPR30, the administration of concentrations of 0.4 mmol/L of boric acid enhances the splenic lymphocyte proliferation and the immune response by the expression of IL-2 and IFN-γ, whereas concentrations of 40 mmol/L of the acid cause opposite effects such as cell apoptosis and described as dose-dependent behavior. The selective inhibition of the receptor with the compound G-15 confirmed that GPR30 is the key mediator of these effects [16]. Also, carborane clusters were reported as GPCR-ligands just a few years ago. Peptidic agonists modified with carborane motifs and directed to ghrelin receptor 1a (GhrR) demonstrate high chemical stability and efficient activation. It was attractive as increased expression of the ghrelin receptor on various cancer cells makes it a viable target for BNCT. They showed that the combination of synthetic, boron-modified peptide ligands with the GhrR represents a suitable system for BNCT application [17,18]. On the other hand, peptidic conjugates based in neuropeptide Y and their optimization for maximizing the carborane charge, showed a significant enhancement in the solubility of this chemical structures without modifications in the affinity to the Y1 receptor, increasing the number of BCCs for targeted therapy. Another class of peptide conjugates that incorporate 80 atoms of boron per molecule and specifically act in the gastrin-releasing peptide receptor (GRPR) was reported by Hoppenz et al. and resulted that these compounds retain affinity and selectivity to the receptor and showed a selective internalization of tumoral cells and low cytotoxic effects and can be considered their use as boron loading source in a non-invasive approach like BNCT [19]. However, another feature of the BCCs is that they act like energy reservoirs, which is the case of some BODIPYS that exhibit fluorescence and stability in acid media as reported elsewhere [20]. The use of BODIPYS in GPCRs has efficiently demonstrated that these chemical structures would be used for treatments that involve the receptor GPR54 like type 2 diabetes (T2D) in which this receptor is overexpressed. For their part, Sharma et al. described peptide–boron difluoride formazanate conjugates with the ability to label the growth hormone secretagogue receptor (GHSR-1a); this compound not just is different from the conventional BODIPY core, if not also share structure with known molecules able to modify activation of the receptor [21]. The use and activities of BCCs in adenosine receptors have been shown. Representative works are by Fernandes et al. on the A2a adenosine receptor bound by a BODIPY ligand which modify the dynamics of the receptor judged by Single-molecule Förster resonance energy transfer assays and fluorescence correlation spectroscopy [22]. But also that work reported by Bednarska-Szczepaniak et al. showed boron cluster-containing compounds with the high affinity of a BCC for the A3AR receptor, favoring their selectivity over other adenosine receptor subtypes such as A1 and A2A. This specificity is attributed to the unique property of boron clusters, which include the formation of non-covalent bonds with specific regions of the receptor. Compared to the phenyl counterparts, the boron derivatives had lower affinity but a significantly improved selectivity profile, which could be exploited in the development of more precise therapies[23].
Barron-González et al. have explored the action of a boroxazolidone tryptophan- derivative named borolatonin, that compound induces changes in behavior and the brain of male and female rats with hormonal deprivation in a similar form as melatonin does. In fact, the cognitive deficit and neuronal loss as well as the formation of amyloids and the changes in BDNF after hormonal deprivation were diminished with both melatonin or borolatonin administration [24]. The docking assays support the binding of borolatonin in a similar way to melatonin on the MT1 and MT2 melatonin receptors (Figure 2), as well as the interactions of key residues for activation of those receptors.
3.2. Effects of BCC on Family B and C of GPCRs
3.2.1. Interaction and Effects of BCC on Family B Receptors
GPCRs of the B family, also known as secretin receptors, are proteins that interact with polypeptide hormones, molecules with intercellular communication functions and proteins that closely regulate stress response and longevity. They are present in mammals, Caenorhabditis elegans, and Drosophila melanogaster, but have not been found in fungi, plants, or prokaryotes. This family stands out in having 7 transmembrane segments like other types of GPCRs [25]. In 1991, the receptor for the intestinal hormone secretin was cloned for the first time by Dr. Nagata's team, finding that the activation of the receptor regulates the concentration of intracellular cAMP by modifying the enzymatic activity of adenylate cyclase [26]. Since that date, 18 genes from Drosophila, 6 from Caenorhabditis, and more than 33 human genes have been identified that encode a member of the B family of GPCRs. Currently, there are 3 subtypes of receptors belonging to this family: B1, B2, and B3 [27].
The B1 subfamily is the largest group of the B family, they are characterized by being classic hormone receptors and having polypeptide ligands of 27 to 141 amino acid residues. The receptors included are: the Secretin Receptor, responsible for the secretion of bicarbonate, zymogens, and potassium at the pancreatic level; VPAC1 receptor, involved in T Lymphocyte differentiation, neuromodulation and circadian rhythm; Glucagon receptor, which modulates hepatic glycogenolysis, gluconeogenesis, and pancreatic insulin secretion; Receptor for Growth Hormone Releasing Hormone (GHRH), closely linked to the synthesis and release of Growth Hormone in the pituitary gland; Glucagon-Like Peptide 1 Receptor responsible for pancreatic secretion of glucagon and insulin, among others [26]. The prototype B2 subfamily receptors are the mucin-like hormone receptor (EMR1) and the leukocyte cell surface antigen CD97. On the other hand, the B3 subfamily is represented by proteins related to Methuselah present mainly in Drosophila [28].
Peptidomimetics are molecules produced by the chemical-pharmaceutical industry with the aim of emulating the structure of natural peptides, increasing affinity for receptors, and increasing bioavailability, among others [29]. On the other hand, the addition of boron to the previous peptides reduces the adverse effects, improves metabolic stability by reducing its enzymatic degradation, and increases the activity of the receptor, because boron can form covalent bonds with nucleophilic amino acid residues in the pocket binding, facilitates the formation of hydrogen bonds with extra and transmembrane residues, resulting in the improvement of the strength and stability of the interaction between ligand and receptor [30]. However, there is currently no study that assesses the impact of peptides or small molecules containing boron on family B receptors.
Therefore, their use could lead to great advances in the treatment of endocrine diseases or in the improvement of patient symptoms. As an example of potential application, secretin is synthesized and released in the S cells of the intestinal mucosa of the duodenum and jejunum in response to various luminal nutrients, resulting in the activation of secretin receptors in the pancreatic acinar cells and consequently the production of pancreatic juice rich in bicarbonate [31]. Hence, peptides or molecules containing boron that behave as agonists of secretin receptors could bring benefits in patients with pancreatic insufficiency and chronic pancreatitis, resulting in greater absorption of nutrients and reduction of clinical symptoms [29].
Similarly, the addition of boron to peptides or small molecules that activate GHRH receptors in the somatotroph cells of the pituitary could be beneficial in those patients with short stature due to problems with the synthesis and release of growth hormone. Analyzed from another perspective, molecules could also be conjugated with boron to act as antagonists of the GHRH receptor in cases of acromegaly and gigantism, which present a high concentration of growth hormone [32]. Finally, the addition of boron to polypeptides that behave as GLP-1 receptor agonists would improve the quality of life of patients suffering from T2D, because it would considerably reduce plasma glucose concentration and could help restore sensitivity from beta cells to secretagogues; and the study of some small molecules containing boron affecting incretins level deserves to be scrutinized [3,33].
Although the effects of BCC on Family B of GPCR are lacking, Tian et al. reported the ability of a BCC to label the glucagon receptor. They performed a confocal fluorescent microscopic study of the intracellular bioorthogonal labeling of the third intracellular loop (ICL3) of glucagon receptors. Their labeling strategy involves the site-specific introduction of a strained alkene amino acid into the ICL3 through genetic code expansion, followed by a highly specific inverse electron-demand Diels-Alder reaction with the fluorescent tetrazine-BODIPY probes. Those probes afforded successful fluorescent labeling of GCGR-ICL3 and yielded high background fluorescence due to its intracellular retention [34].
3.2.2. Effects of BCC on C GPCRs
The class C family has 2 unique characteristics compared to other GPCRs: a) they have a large extracellular domain, far from the seventh transmembrane domain, which has the orthosteric sites, and b) they form constitutive dimers that are characterized by having unique activation modes [35]. The receptors that constitute class C correspond to metabotropic glutamatergic receptors (mGluR), GABA-B receptor, calcium sensing receptor (CaSR), sweet taste receptor, and pheromone receptors. The CaSR, GABA-B, and mGluR receptors are the subject of study in the field of neuroscience and in the control of plasma calcium. On the other hand, the receptors related to sweet taste become relevant in the food industry because additives can be used that impact these receptors and increase the intensity of the flavor [36].
The mGluRs are divided into 3 groups. Members of the Group I (mGluR 1 and 5) couple to a Gq protein; group II (mGluR 2 and 3) and group III (mGluR 4, 6, 7, and 8) are coupled to Gi protein. All of the above are expressed profusely throughout the central nervous system and are involved in pathologies such as Parkinson's disease, Fragile X Syndrome, Alzheimer’s disease, anxiety and schizophrenia [37]. Therefore, they are striking in new studies aimed at creating new drugs that can improve the clinical picture of the aforementioned pathologies.
There is work that studies the effects of a BCC on mGluR receptors. Just a work is suggesting the activity on metabotropic glutamate receptors, belonging to the family C of GPCRs. In fact, the oxazaborolidine (BZQuin) reported as active to enhance seizures is suggested as a ligand of those receptors (specifically mGluR1, mGluR2, and mGluR7) by docking assays; in line with the observed in vivo effects and comparison with other drugs [38]. In silico (Figure 3), BZQuin binds with high affinity to mGluR1, 2, and 7 receptors, precisely to the L-glutamate binding site, with higher negative Gibbs binding energy than L-glutamate and D-aspartate. BZQuin produces a positive modulation in mGluR receptors and can be directly related to increased glutamatergic neurotransmission, increased motor activity, and increased seizure episodes. Although the above compound is a positive modulator of some mGluRs, molecules containing boron could be synthesized with the aim of providing a negative modulation to the mGluRs and providing improvement to models of Parkinson's disease, anxiety, or epilepsy. The addition of boron to allosteric molecules could bring several advantages such as increasing the potency and selectivity of a drug, improving pharmacokinetic properties, and reducing toxicity.
For its part, mGluR 5 receptors have increasing interest as targets for neurodegenerative diseases [39], they have negative and positive allosteric domains (NAM and PAM, respectively) in their transmembrane domain, which can be targets of molecules that modulate the activity of the receptor. Allosteric agonists cannot activate or inhibit a receptor themselves [40]; however, in the presence of the orthosteric ligand, they can promote the total activation or inhibition of the receptor [41]. Because the allosteric sites of mGluR 5 are more conserved than orthosteric sites, fluorescent BCCs targeting allosteric sites allow more precise localization of mGluR 5 in the nervous system.
Therefore, the use of BCCs directed at the interaction with mGluR stands out above all when used in fluorescence tests [42]. The BODIPYs great photochemical stability, high molar absorptivity, and fluorescence yield have been applied for this purpose [43,44]. The addition of BODIPY to alkyne groups can generate fluorescent molecules that interact with NAM and PAM of mGluR 5 [45]. Based on the above, allosteric mGluR 5 ligands are developed with the aim of showing improvement in various situations such as: anxiety, depression, and abuse of drugs. Thanks to the identification of allosteric sites of mGluR 5 through BCC, it has been possible to develop negative allosteric drugs such as Basimglurant, designed for the treatment of drug-resistant depression and Fragile X Syndrome [46].
Since the development of BCCs that behave as allosteric modulators is still in its initial phases, there are no studies related to their interaction with GABA-B and CaSR receptors. However, they will be key to addressing pathologies such as depression, epilepsy, hypo- and hyperparathyroidism, and therefore, the plasma control of extracellular calcium.
3.3. Theoretical Assays Supporting Interactions of BCC on GPCRs
In sections above it is mentioned docking assays have been reported showing the ability of BCC to reach the orthosteric site of some GPCR. Some docking procedures and molecular dynamic simulations have yielded advances in the improved mode and favorable energy of BCC in the linkage to specific GPCRs [47,48]. The following paragraphs are examples found in those studies.
Various studies involving boron cluster conjugates with biologically important low molecular weight compounds have been made and have been evaluated for their potential bioactivity. In this case, there was the question of whether the addition of a phenyl or boron cluster to adenosine derivatives would affect or modify the ligand affinity for A2A and A3 adenosine receptors, both GPCRs. The study was carried out using PatchDock, and due to the limited availability of boron parameters, they had to be changed to the Carbon atom type, and the A3 receptor had to be modeled using the Local Meta-Threading-Server, LOMETS, while the A2A crystallized structure was downloaded (PDB ID: 2YDO). It was a blind docking, and it was seen that in the case of the A2A receptor, the adenosine derivatives with the phenyl cluster had a better affinity with interactions such as that seen with amino acid residues T88, I275, while with the boron cluster there are interactions with the extracellular residues of phenylalanine and leucine in the second extracellular loop which result the cluster to be in a small pocket giving unfavorable interactions. In the case of the A3 receptor, the addition of the boron cluster helps to give a better affinity and interactions, having more interactions with transmembrane amino acids, L85, S181, Ile186, and W243, which could be due to a geometrically more fit structure and reduced mobility to the binding pocket which helps to anchor it to the inner surface of the channel [49].
Other studies were done due to the increased interest in the use of boron neutron capture therapy in cancer therapy, in this field is attractive the use of hydrophobic carborane clusters with their high boron content, stability, and chemically modifiable properties. But also, carboranes have shown their potential for other pharmaceutical applications, such as derivatives of retinoic acid on GPCR; thus, some boron clusters have been proposed as ligands on adenosine receptors A1, A2A, A2B, and A3. The A3 receptor was modeled using the LOMETS and a directed docking procedure was done to cover the intra and extracellular part of the receptor, which covered the entire extracellular part, binding pocket, and intracellular region. The boron parameters were added to the Autodock 4.2.6 parameters. With a further docking of the results, and the analogous binding of the polyhedral with a phenyl ring, it was shown that carborane clusters with adenosine and 2´-deoxyadenosine pairs do bind to the entrance of the adenosine A3 receptor in the orthosteric site, which compares the best compound with the carborane cluster against the other which has a phenyl cluster, as potential agonists leading to new potential modulation in neuroprotection, neurodegeneration, cellular proliferation and cell death [50].
In another study, an array of histamine H1 receptor ligands were synthesized and explored, where the incorporation of a boron dipyrromethene-based fluorophore to various ligands with peptide linker composition and orthosteric targeting moiety. In this case, it was seen that this fluorophore conferred a high binding affinity and that the peptide linker, which was optimized, helps to interact with key residues in the receptor. A GLIDE docking experiment was carried out with the use of the H1R crystal structure (PDB ID: 3RZE) and prepared with the Schrödinger modeling suite. The missing atoms were modeled using PROPKA and minimized using the OPLS3 force field. The proposed receptor residues interacting with the side groups were near or in the extracellular loop region of the receptor, D178, K179, K191, N443, and E447, with the theory that the interactions formed are due to the disruption of the existing water hydrogen bonds. The addition of a phosphatidylcholine-based phospholipid bilayer and posterior docking show that it had a hydrophobic effect, with the fluorophore increasing the affinity on the binding site. In addition, it was found that F440 could act as a lid for the pharmacophore to help in its binding position [51].
The exploration of ligands for monoamine receptors to treat Alzheimer's disease is increasing. Among these molecules, melatonin has been linked with an inverse correlation with the severity of neuropathology, and it has been reported to reduce the formation of β-plaques and neurofibrillary tangles. Its receptors, MT1 and MT2, are GPCRs that are involved in the cellular reduction of cAMP. An in silico study was done where both receptors were modeled from their human analogs, MT1 from 6ME5 of MT1-agomelatine, and 6ME9 from MT2-ramelteon. The ligands were agomelatine, ramelteon, melatonin and borolatonin, an adduct from tryptophan and 2-aminoethoxydiphenyl borate (2-APB). All compounds did bind to the orthosteric site, with interactions with residues T178, F179, A191, V192, F251, N255, A284, and Y285, with most interactions being through Van der Waals and hydrophobic types (Figure 2). The different residues from the homolog receptors being the tryptophan in position 254 in MT1 and 264 in MT2 appear to add interactions in the binding pockets, allowing ligands to form pi-pi interactions with side chain residues in the rat models. With regards to borolatonin, there were interactions with aromatic rings from the residues and hydrogen bonds from the amine in the indole moiety, while exposed oxygen interacting with the boron-containing ring. Interestingly, although the boron atom did not interact directly with the residues, all BCC that were studied had a higher affinity than known ligands for melatonin [52].
Another GPCR that is a good prospect to study due to the various pharmaceutical uses for its modulation is the β-2 adrenoreceptor. Taking into account previously known agonists, in this case, albuterol, boron-containing derivatives were used, in this case, BR-AEA, Boronterol, and Politerol. This was done due to previous reports that BCC demonstrated higher potency and efficacy than carbon counterparts. Thus, the human β2AR was obtained, PDB ID: 3PDS, while the guinea pig was built using MODELLER using as templates PDB ID: 3PDS and 3P0G. Thus, for the docking procedure, Autodock software was used with a blind procedure, and the calculation box was centered on the midpoint between the α-carbons of 2 conserved amino acids, D113 and S204. The three compounds did bind to the hydrophobic orthosteric site between TM3 and TM7, with their amine moiety interacting with D113 of both receptors while their tert-butyl moiety oriented to the upper segment of TM7 on the guinea pig receptor. For the human one, for BR-AEA and Boronterol, the moiety was in the middle of the crevice between TM3 and TM7 while for Politerol in the upper segment of TM3, and having hydrogen bonds with S203, S204, and S207 in TM5 in both receptors, while for the human receptor, BR-AEA, and Politerol showed fewer interactions with N293. Their binding affinities are similar to those of albuterol [13]. These results do correlate with previous studies done where a tetracoordinate boron atom was placed on salbutamol, a known β2AR agonist making 4 BCC that showed low toxicity, and the resulting compounds were bound to the guinea pig receptor. Of special interest was Politerol, which, due to the tetravalent boron atom and its negative charge, helps bind to hydroxyl moieties and key interactions to the orthosteric site, including D113, S203, S207, W286, F289, and N312 [33]. There was also a molecular dynamics simulation done where the human and guinea pig protein was used as both apo-protein and as complexes. They were embedded in a lipid bilayer membrane with CHARMM force fields at 1 ATM and at a temperature of 300 K; it was carried out by using the AMBER 12 executable for 250 ns. In this case, only politerol was used and compared against the isoproterenol and albuterol complexes. With regards to the simulations, stabilization was found after 100 ns, with only small differences with RMSD and RG values, but the RMSF values of the last 150 ns of the free system had better flexibility than the bound complexes, with the extracellular residues 170-195 and cytoplasmic residues 231 to 254 having the greater flexibility. Nevertheless, in the guinea pig results, no clear difference was found, but no difference in the cumulative RMSF than that seen in the human one, with the complexes having a bigger difference [13].
3.4. The Role of Moieties Including a Boron Atom in the Interactions on GPCRs
The role of moieties including the boron atom on the binding on proteins has been recently commented [1,2]. Moreover, Gao et al. revised the wide-gamma of possibilities of BCC enabling bioconjugation [53].
Hence, specifically on GPCR, there are some interactions and mechanisms of binding that could be particularly attractive for BCC. In this regard, it is well known the key interactions of GPCR-ligands conferring selectivity, potency, and efficacy are related with some residues identified forty years ago by punctual mutations, but in the last twenty years by advances in crystallography approach of these proteins [54]. In this sense, at least in the class A receptors, the relevance is for the residue in the 3.32 position (in the agreement with Ballesteros-Weinstein nomenclature) and that in the 7.43 position, often Aspartic acid and Asparagine, respectively, for the first contact of endogenous ligands (often an amine moiety). The second step involves the interactions to some serines in the fifth transmembrane domain. And consecutively, some interactions with toggle switches and the conformational changes in the ionic lock trigger conformational changes that modify distances among third, fifth, sixth, and seventh domains related to activation of classic and other pathways in the intracellular face of the receptor [55].
Thus, interactions of BCC could be suggested for some compounds tested in classical pharmacology evaluation. Keeping in mind the possibilities of tricoordinate (acid of Lewis) or tetracoordinate boron atom (as is found in most biological applications), as well as the dynamic nature of B–O and B–N bond formation, and a significant portion of the boron-enabled bioconjugations exhibit rapid reversibility, the interactions on GPCR could be applied to develop reversible and covalent ligands with at least one boron atom [2,54,56]. For example, in the first interaction with Asp, Glu, or Gln residues, the boronic compounds able to form iminoboronates are attractive, while the boronate ester mediated binding of biomolecules could be applied to interactions with serines in the fifth transmembrane domain. Also, regarding hydrophobic interactions with toggle switches (constituted by phenylalanine residues in the sixth domain) could be useful for arylboronic acids or carboranes[57]. Additionally, the reaction with N-terminal cysteines could be useful to enhance the binding to some cysteines, in a similar way to the previous design of some covalent full agonists [58]. Figure 4 depicts examples of putative mode of interactions on key points into or near the orthosteric site of a GPCR. Additionally, some other interactions in allosteric sites have been proposed, which expand the possibilities of BCC to act in GPCR [59].
3.5. Clinical Trials and Effects in Humans with a Presumptive Key Role of GPCRs
The specific action of a BCC on a GPCR is lacking in clinical trials. However, some effects observed in those studies could involve action on these receptors. As examples are the effects of some BCC on glioblastoma or neck and head tumours (albeit if results are not reported in clinical trials database (https://clinicaltrials.gov examples: NCT00115440, NCT00974987, NCT00062348), since the effect of some compounds as phenylalanine have been suggested by a way free of radiation in preclinical and clinical studies [60,61], while some structurally-related compounds have been reported as potentially active against hose tumors by acting on the recent described GPCR, such as GPR17 or GPR68.
Nguyen et al. (2021) explored the development of novel indole derivatives as G protein-coupled receptor 17 (GPR17) agonists for glioblastoma treatment. Using molecular docking, they screened over 6,000 indoline derivatives and identified CHBC, a phenolic Mannich base synthesized via a multicomponent Petasis borono-Mannich reaction, as a promising candidate. CHBC effectively reduced cAMP and calcium levels in glioblastoma cells, indicating successful GPR17 activation. In vitro assays revealed dose-dependent cytotoxicity with an IC₅₀ of approximately 85 μM, surpassing the effects of the known agonist MDL29,951. These findings highlight CHBC as a potential therapeutic agent targeting GPR17 in glioblastoma [62].
Furthermore, GPR68 (Ovarian Cancer G Protein-Coupled Receptor 1 or OGR1), a proton-sensing GPCR activated by extracellular acidity, plays a pivotal role in tumor microenvironments. Although direct evidence of BCC modulation of GPR68 activity is limited, compounds like 2-APB are known to influence calcium signaling pathways, including those mediated by inositol trisphosphate (IP3). Given GPR68’s role in pH-sensing and calcium regulation, the possibility of BCCs modulating its activity presents an intriguing area for future research. Understanding the interaction between boron compounds and GPR68 could uncover novel therapeutic strategies targeting this receptor in oncology [63].
There are five boron-containing drugs approved by the FDA. In the clinical trials evaluating their use in humans, some effects could be linked to action on GPCRs. In this regard, bortezomib was developed as a dipeptide with bioactivity on the proteasome, but it also has affinity on other enzymes such as chymotrypsin and thrombin, while the affinity on GPCR which are targets for peptides was not measured [64]. However, multiple clinical trials have shown both activity on multiple neoplastic cells and adverse effects, which could involve the action on GPCR. An example of that is the study about the autonomic nervous system dysfunction after bortezomib administration (NCT01314625) with no clear results. However, the effects of this drug are clearly linked to vascular and nervous systems, with the GPCR probably involved. In fact, the incidence of autonomic neuropathy of the digestive system induced by bortezomib is around 60%, and its severity is closely related to efficacy, advanced age, constipation, diabetes, fracture/spinal cord compression in bed, and history of alcoholism. Early detection and adjustment of treatment are key for reversibility, autonomic neuropathy of the digestive system was significantly alleviated in most patients after the timely adjustment of the treatment regimen, and bortezomib could continue to be administered [65]. Also, changes in the nervous system linked to neurodegeneration could be linked to effects by binding and modulation of glutamate receptors and S1P receptors, in addition to increased oxidative stress and increased inflammatory processes [66]. Additionally, Ghelardini et al. reported that the use of a selective antagonist of GluR5 (2-methyl-6-(phenylethynyl)-pyridine) inhibits the glutamatergic system decreased development of painful peripheral chemo-neuropathy induced by BTZ [67].
Another clinical trial, NCT04566328 in phase III, compared two therapeutic regimens in patients newly diagnosed with multiple myeloma. This study evaluated a four-drug combination (daratumumab-hyaluronidase, bortezomib, lenalidomide, and dexamethasone) against a three-drug regimen (daratumumab-hyaluronidase, lenalidomide, and dexamethasone). Results demonstrated that bortezomib could inhibit cancer cell growth by blocking key enzymes required for cellular proliferation. The addition of bortezomib to the three-drug regimen enhanced its effectiveness in reducing cancer progression or preventing recurrence compared to the three-drug regimen alone; these results were compared to a previous study phase II from the same working team, clearing the results obtained [68]. Similarly, the phase III clinical trial NCT05561387 focuses on induction regimens followed by either double- or single-drug maintenance therapy for newly diagnosed multiple myeloma in patients who are not undergoing stem cell transplantation. This study specifically targets frail or intermediate-fit patients, as defined by age, comorbidities, and functional status. The trial evaluates four drugs—bortezomib, lenalidomide, dexamethasone, and daratumumab-hyaluronidase—to identify the most effective combination for controlling and shrinking multiple myeloma while preventing relapse. Each drug in the regimen plays a distinct role: bortezomib inhibits enzymes essential for cancer cell growth, lenalidomide promotes the production of healthy blood cells while inducing cancer cell death, dexamethasone reduces inflammation and modulates the immune response, and daratumumab-hyaluronidase, a monoclonal antibody, targets and interferes with cancer cell proliferation. By testing various combinations, the trial aims to optimize therapeutic outcomes for this challenging patient population.
The topical boron-based phosphodiesterase 4 (PDE4) inhibitor crisaborole has been approved by the US Food and Drug Administration (FDA) for the treatment of mild-to-moderate atopic dermatitis (AD) in patients aged two years and older. In pivotal phase III clinical trials, crisaborole demonstrated superior efficacy compared to the vehicle control, achieving the primary endpoint of clear or almost clear skin with a ≥2-grade improvement in disease severity more frequently in both children and adults. Crisaborole offers a non-steroidal treatment option for eczema, providing an alternative to corticosteroids with a favorable safety profile. [69,70] Protease-activated receptor-2 (PAR2), a class-A G protein-coupled receptor (GPCR) expressed in various tissues, including the skin, is critical in the inflammatory processes associated with AD. PAR2 promotes Th2-mediated inflammation, delays skin barrier repair, and influences keratinocyte differentiation. Additionally, it is implicated in itch and pain transmission in the skin, making it a potential target in dermatological therapies. While the therapeutic effects of crisaborole have been well-documented, its possible interaction with PAR2 remains an area of interest. A study by Peña et al. (2020) indicated the efficacy of crisaborole in reducing adverse events compared to topical steroids, suggesting a possible link to PAR2 modulation [71,72].
A preclinical study exploring the combined effects of crisaborole and vitamin D in a mouse model of allergic contact dermatitis provides further insights. This research demonstrated that the combination significantly reduced inflammation and epidermal hyperkeratosis, indicating enhanced therapeutic potential. These findings suggest a broader anti-inflammatory role for crisaborole, particularly when combined with other agents. Despite these promising findings, the precise interaction of crisaborole with GPCRs, including PAR2, remains unclear. Crisaborole’s primary mechanism of action involves PDE4 inhibition, which modulates inflammatory pathways. Although GPCRs are integral to various physiological processes related to inflammation, current evidence does not establish direct interactions between crisaborole and these receptors. In conclusion, crisaborole represents a safe and effective treatment for mild-to-moderate AD through its PDE4 inhibitory effects. Emerging evidence, including its combination with vitamin D, underscores its potential for enhanced anti-inflammatory efficacy. However, further studies are warranted to elucidate the relationship between crisaborole and GPCRs, such as PAR2, and to explore its broader implications in dermatological therapies [73,74].
The boron-based compound Talabostat (Val-boroPro) is an oral inhibitor of dipeptidyl peptidases, including fibroblast activation protein (FAP), which is highly expressed in tumor-associated fibroblasts. FAP plays a significant role in promoting the progression of colorectal cancer and other malignancies. Talabostat, the first clinical inhibitor of FAP enzymatic activity, has been evaluated for its therapeutic potential in various cancer settings [75]. In a phase II clinical trial (NCT04171219) involving patients with metastatic colorectal cancer, Talabostat demonstrated its potential as a therapeutic agent targeting FAP-expressing tumor stroma. Preclinical studies further highlighted its immunomodulatory effects. It was found to stimulate the production of cytokines and chemokines, which enhance T-cell-mediated immunity and exhibit T-cell-independent activity. For instance, in melanoma xenograft models (A375, A2058), Talabostat reduced tumor size by over 60% in immunodeficient mice, emphasizing its role as an immune-modulating agent [76] Talabostat has also been investigated in advanced non-small cell lung cancer (NSCLC). A phase II trial (NCT00080080) combined Talabostat with docetaxel to assess its efficacy in improving treatment outcomes for this challenging patient population. The study reported a modest objective response rate (ORR) with partial tumor responses in some patients. However, there was no significant improvement in progression-free survival (PFS) or overall survival (OS) compared to docetaxel monotherapy. While the combination therapy was generally well-tolerated, the lack of substantial clinical benefit limited its further development in NSCLC [77]. Despite its promising preclinical activity and immune-mediated mechanisms, Talabostat's efficacy in FAP-expressing tumors has not been directly explored in clinical trials. While it primarily acts by inhibiting dipeptidyl peptidases, its broader impact on the tumor microenvironment suggests possible indirect interactions with GPCRs [78]. Talabostat’s stimulation of cytokine and chemokine production could influence immune responses and tumor-stroma interactions mediated through GPCR signaling pathways. These indirect effects might contribute to its anti-tumor activity. Although no direct interaction between Talabostat and GPCRs has been reported, future research should investigate its role in modulating GPCR-associated pathways. Such studies could uncover novel therapeutic mechanisms and expand the scope of Talabostat’s applications, particularly in cancers characterized by GPCR dysregulation [79].
Dutogliptin is a selective inhibitor of dipeptidyl peptidase-4 (DPP-4), an enzyme that plays a crucial role in glucose metabolism by degrading incretin hormones, such as glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP). These hormones are essential regulators of insulin secretion [80]. By inhibiting DPP-4, dutogliptin extends the activity of GLP-1 and GIP, allowing them to bind to their respective GPCRs, thereby enhancing insulin secretion through GPCR-mediated pathways and improving glycemic control in patients with T2D [81]. Concerning interactions with GPCR, current scientific evidence does not indicate that dutogliptin directly interacts with or modulates GPCR function. Instead, its therapeutic effects are primarily mediated through the preservation of incretin hormone activity, which subsequently acts on their corresponding GPCR to facilitate insulin release and maintain glucose homeostasis [82]. In conclusion, dutogliptin enhances the activity of incretin hormones, which exert their effects via GPCRs to regulate insulin secretion and glucose balance. This mechanism highlights the therapeutic potential of targeting the DPP-4-incretin axis and GPCR signaling pathways in managing T2D.
4. Conclusions and Future Directions
There is increasing evidence of BCC binding, labeling, and acting on GPCR. The relevance of those advances is due to the fact that most current drugs exert their benefits through interaction with GPCRs (acting as agonists or antagonists, by means of orthosteric effects or allosteric effects, and by biased signaling). Moreover, GPCR-ligands are considered among promising drugs for treating many of the diseases with high incidence around the world, such as neurodegenerative, cardiac, pulmonary or metabolic diseases, and cancer [83] and included among small molecules recently approved for human use [84].
On the other hand, increased data are demonstrating the biological action of BCC. Also, the knowledge about the engagement of BCC in proteins is accumulating. Currently, the evidence of the action of BCC on GPCR is limited to competitive analysis in the classic pharmacological approach by using well-known ligands of GPCR, but also the theoretical approaches are in agreement with direct interaction of BCC on the reported orthosteric and allosteric sites of some GPCR. However, some evidence, such as the crystallography data of these compounds on protein complexes, is lacking, and the evidence of competition or intervention in the action of well-known ligands of GPCR (including some marketed drugs) is desirable.
Future studies will let us observe the specific role of a boron atom in each molecule probed as ligand, or the relevance in specific moieties containing it; but also if there are particular difference among the BCC interactions on GPCRs as in the crystallized ligand-protein complexes. Moreover, the use of more than a boron moiety to build new potential drugs on GPCR could be considered.
The multiple observed and potential effects of BCC after enhanced interactions on GPCR invite enhanced efforts in the design of new BCC-ligands to prevention, diagnostic and treatment of human diseases with high global burden such as those related to cancer, cardiovascular dysfunction, pulmonary maladies, neurological and metabolic diseases.
Author Contributions
Conceptualization, M.A.S.U.; methodology, M.A.S.U. and R.M.L.M.; investigation, J.MS.Q, A.B.N., M.N.R. and R.B.L.; data curation, M.A.S.U. and B.D.; writing—original draft preparation, J.MS.Q, A.B.N., M.N.R., R.B.L. and M.A.S.U.; writing—review and editing, M.A.S.U. and R.M.L.M.; funding acquisition, M.A.S.U.. All authors have read and agreed to the published version of the manuscript.
Funding
Authors thank the support of Secretaria de Investigación y Posgrado del Instituto Politécnico Nacional, grant number Multi2303.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Any data related to the revised information or for supporting the declared sentences in this review could be requested from the corresponding author via e-mail.
Acknowledgments
Authors thank to M.Pharm. M. Emilio Cuevas-Galindo for the output files for generating the figure showing the compound BDZ-Quin docked on the mGluR1.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Grams, R.J.; Santos, W.L.; Scorei, I.R.; Abad-García, A.; Rosenblum, C.A.; Bita, A. ... & Soriano-Ursúa, M.A. The Rise of Boron-Containing Compounds: Advancements in Synthesis, Medicinal Chemistry, and Emerging Pharmacology. Chem rev. 2024, 124, 2441–2511. [Google Scholar] [CrossRef] [PubMed]
- Das, B.C.; Nandwana, N.K.; Das, S.; Nandwana, V.; Shareef, M.A.; Das, Y. ... & Evans, T. Boron chemicals in drug discovery and development: Synthesis and medicinal perspective. Molecules 2022, 27, 2615. [Google Scholar] [CrossRef]
- Soriano-Ursúa, M.A.; Cordova-Chávez, R.I.; Farfan-García, E.D.; Kabalka, G. Boron-containing compounds as labels, drugs, and theranostic agents for diabetes and its complications. World J Diabetes 2024, 15, 1060. [Google Scholar] [CrossRef]
- Barrón-González, M.; Montes-Aparicio, A.V.; Cuevas-Galindo, M.E.; Orozco-Suárez, S.; Barrientos, R.; Alatorre, A. ... & Soriano-Ursúa, M.A. Boron-containing compounds on neurons: Actions and potential applications for treating neurodegenerative diseases. J Inorg Biochem, 2023, 238, 112027. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Tripathi, N.M.; Bandyopadhyay, A. The modern role of boron as a ‘magic element’ in biomedical science: chemistry perspective. Chem Commun 2021, 57, 13629–13640. [Google Scholar] [CrossRef]
- Abad-García, A.; Ocampo-Néstor, A.L.; Das, B.C.; Farfán-García, E.D.; Bello, M.; Trujillo-Ferrara, J.G.; Soriano-Ursúa, M.A. Interactions of a boron-containing levodopa derivative on D 2 dopamine receptor and its effects in a Parkinson disease model. J. Biol Inorg Chem 2022, 27, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J. , Mordalski, S., Pándy-Szekeres, G., Esguerra, M., Mamyrbekov, A., Munk, C.,... & Gloriam, D.E. GPCRdb in 2021: integrating GPCR sequence, structure and function. Nucleic Acids Research 2021, 49, D335–D343. [Google Scholar]
- Daly, C.J.; Milligan, C.M.; Milligan, G.; Mackenzie, J.F.; McGrath, J.C. Cellular localization and pharmacological characterization of functioning alpha-1 adrenoceptors by fluorescent ligand binding and image analysis reveals identical binding properties of clustered and diffuse populations of receptors. J Pharmacol Exp Ther 1998, 286, 984–990. [Google Scholar] [CrossRef]
- McGrath, J.C.; Mackenzie, J.F.; Daly, C.J. Pharmacological implications of cellular localization of alpha1-adrenoceptors in native smooth muscle cells. J Auton Pharmacol 1999, 19, 303–310. [Google Scholar] [CrossRef]
- McGrath, J.C.; Daly, C.J. Use of fluorescent ligands and receptors to visualize adrenergic receptors. The Adrenergic Receptors: In the 21st Century; 2006; pp. 151–172. [Google Scholar]
- Baker, J.G. , Hall, I.P., & Hill, S.J. Pharmacology and direct visualisation of BODIPY-TMR-CGP: a long-acting fluorescent β2-adrenoceptor agonist. Br J Pharmacol. 2003, 139, 232–242. [Google Scholar]
- Soriano-Ursúa, M.A.; Valencia-Hernández, I.; Arellano-Mendoza, M.G.; Correa-Basurto, J.; Trujillo-Ferrara, J.G. Synthesis, pharmacological and in silico evaluation of 1-(4-di-hydroxy-3, 5-dioxa-4-borabicyclo [4.4. 0] deca-7, 9, 11-trien-9-yl)-2-(tert-butylamino) ethanol, a compound designed to act as a β2 adrenoceptor agonist. Eur J Med Chem 2009, 44, 2840–2846. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Ursúa, M.A.; Bello, M.; Hernández-Martínez, C.F.; Santillán-Torres, I.; Guerrero-Ramírez, R.; Correa-Basurto, J.; Trujillo-Ferrara, J.G. Cell-based assays and molecular dynamics analysis of a boron-containing agonist with different profiles of binding to human and guinea pig beta2 adrenoceptors. Eur Biophys J, 2019, 48, 83–97. [Google Scholar] [CrossRef]
- Louie, A.S. , Vasdev, N., & Valliant, J.F. Preparation, characterization, and screening of a high affinity organometallic probe for α-adrenergic receptors. J Med Chem, 2011, 54, 3360–3367. [Google Scholar] [PubMed]
- Aringhieri, S.; Carli, M.; Kolachalam, S.; Verdesca, V.; Cini, E.; Rossi, M.; McCormick, P.J.; Corsini, G.U.; Maggio, R.; Scarselli, M. Molecular targets of atypical antipsychotics: From mechanism of action to clinical differences. Pharmacol Ther 2018, 192, 20–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, C. , Jin, E., Deng, J., Pei, Y., Ren, M., Hu, Q.,... & Li, S. GPR30 mediated effects of boron on rat spleen lymphocyte proliferation, apoptosis, and immune function. Food Chem Toxicol., 2020, 146, 111838. [Google Scholar]
- Worm, D.J.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S.; Koebberling, J. ... & Beck-Sickinger, A.G. A stable meta-carborane enables the generation of boron-rich peptide agonists targeting the ghrelin receptor. Journal of Peptide Science 2018, 24, e3119. [Google Scholar] [CrossRef]
- Worm, D.J.; Hoppenz, P.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S. ... & Beck-Sickinger, A.G. Selective neuropeptide Y conjugates with maximized carborane loading as promising boron delivery agents for boron neutron capture therapy. J Med Chem 2019, 63, 2358–2371. [Google Scholar] [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S.; Lerchen, H.G. ... & Beck-Sickinger, A.G. A selective carborane-functionalized gastrin-releasing peptide receptor agonist as boron delivery agent for boron neutron capture therapy. J Organic Chem 2019, 85, 1446–1457. [Google Scholar] [CrossRef]
- Mendive-Tapia, L.; Miret-Casals, L.; Barth, N.D.; Wang, J.; de Bray, A.; Beltramo, M. ... & Vendrell, M. Acid-Resistant BODIPY Amino Acids for Peptide-Based Fluorescence Imaging of GPR54 Receptors in Pancreatic Islets. Angewandte Chemie 2023, 135, e202302688. [Google Scholar] [CrossRef]
- Sharma, N.; Barbon, S.M.; Lalonde, T.; Maar, R.R.; Milne, M.; Gilroy, J.B.; Luyt, L.G. The development of peptide–boron difluoride formazanate conjugates as fluorescence imaging agents. RSC advances 2020, 10, 18970–18977. [Google Scholar] [CrossRef]
- Fernandes, D.D.; Neale, C.; Gomes GN, W.; Li, Y.; Malik, A.; Pandey, A. ... & Gradinaru, C.C. Ligand modulation of the conformational dynamics of the A2A adenosine receptor revealed by single-molecule fluorescence. Scientific reports 2021, 11, 5910. [Google Scholar] [CrossRef] [PubMed]
- Bednarska-Szczepaniak, K. , Mieczkowski, A., Kierozalska, A., Saftić, D.P., Głąbała, K., Przygodzki, T.,... & Leśnikowski, Z.J. Synthesis and evaluation of adenosine derivatives as A1, A2A, A2B and A3 adenosine receptor ligands containing boron clusters as phenyl isosteres and selective A3 agonists. Eur J Med Chem., 2021, 223, 113607. [Google Scholar] [PubMed]
- Barrón-González M, Rivera-Antonio AM, Jarillo-Luna RA, et al. Borolatonin limits cognitive deficit and neuron loss while increasing proBDNF in ovariectomised rats. Fundam Clin Pharmacol. 2024, 38, 730–741. [CrossRef]
- Hollenstein, K.; de Graaf, C.; Bortolato, A.; Wang, M.W.; Marshall, F.H.; Stevens, R.C. Insights into the structure of class B GPCRs. Trends Pharmacol Sci 2014, 35, 12–22. [Google Scholar] [CrossRef]
- Karageorgos, V. , Venihaki, M., Sakellaris, S. et al. Current understanding of the structure and function of family B GPCRs to design novel drugs. Hormones. 2018, 17, 45–59. [Google Scholar] [CrossRef]
- Ishihara, T.; Nakamura, S.; Kaziro, Y.; Takahashi, T.; Takahashi, K.; Nagata, S. Molecular cloning and expression of a cDNA encoding the secretin receptor. EMBO J 1991, 10, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Ja, W.W.; Carvalho, G.B.; Madrigal, M.; Roberts, R.W.; Benzer, S. The Drosophila G protein-coupled receptor, Methuselah, exhibits a promiscuous response to peptides. Protein Sci. 2009, 18, 2203–2208. [Google Scholar] [CrossRef]
- Ahn, J.M.; Han, S.Y.; Murage, E.; Beinborn, M. Rational Design of Peptidomimetics for Class B GPCRs: Potent Non-Peptide GLP-1 Receptor Agonists. In: Valle, S.D., Escher, E., Lubell, W.D. (eds) Peptides for Youth. Advances in Experimental Medicine and Biology, 2009, 611, 125–126 Springer, New York, NY. [Google Scholar] [CrossRef]
- Tan, J.; Grouleff, J.J.; Jitkova, Y.; Diaz, D.B.; Griffith, E.C.; Shao, W.; Bogdanchikova, A.F.; Poda, G.; Schimmer, A.D.; Lee, R.E.; Yudin, A.K. De Novo Design of Boron-Based Peptidomimetics as Potent Inhibitors of Human ClpP in the Presence of Human ClpX. J Med Chem 2019, 62, 6377–6390. [Google Scholar] [CrossRef]
- Afroze, S.; Meng, F.; Jensen, K.; McDaniel, K.; Rahal, K.; Onori, P.; Gaudio, E.; Alpini, G.; Glaser, S.S. The physiological roles of secretin and its receptor. Ann Transl Med. 2013, 1, 29. [Google Scholar] [CrossRef]
- Kopchick, J.J.; Parkinson, C.; Stevens, E.C.; Trainer, P.J. Growth hormone receptor antagonists: discovery, development, and use in patients with acromegaly. Endocr Rev. 2002, 23, 623–646. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Ursúa, M.A.; Arias-Montaño, J.A.; Correa-Basurto, J.; Hernández-Martínez, C.F.; López-Cabrera, Y.; Castillo-Hernández, M.C. ... & Trujillo-Ferrara, J.G. Insights on the role of boron containing moieties in the design of new potent and efficient agonists targeting the β2 adrenoceptor. Bioorg Med Chem Letters 2015, 25, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Fang, M.; Lin, Q. Intracellular bioorthogonal labeling of glucagon receptor via tetrazine ligation. Bioorg Med Chem, 2021, 43, 116256. [Google Scholar] [CrossRef]
- Youssef, E. , Berry-Kravis, E., Czech, C. et al. Effect of the mGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacol. 2018, 43, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Urwyler S. Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacol Rev 2011, 63, 59–126. [CrossRef]
- Nicoletti, F. , Di Menna, L., Iacovelli, L., Orlando, R., Zuena, A.R., Conn, P.J.,... & Joffe, M.E. GPCR interactions involving metabotropic glutamate receptors and their relevance to the pathophysiology and treatment of CNS disorders. Neuropharmacology, 2023, 235, 109569. [Google Scholar]
- Cuevas-Galindo, M.E.; Rubio-Velázquez, B.A.; Jarillo-Luna, R.A.; Padilla-Martínez, I.I.; Soriano-Ursúa, M.A.; Trujillo-Ferrara, J.G. Synthesis, In Silico, In Vivo, and Ex Vivo Evaluation of a Boron-Containing Quinolinate Derivative with Presumptive Action on mGluRs. Inorganics 2023, 11, 94. [Google Scholar] [CrossRef]
- Wang, J.; He, Y.; Chen, X.; Huang, L.; Li, J.; You, Z.; Xie, F. Metabotropic glutamate receptor 5 (mGluR5) is associated with neurodegeneration and amyloid deposition in Alzheimer’s disease: A [18F] PSS232 PET/MRI study. Alzheimer's Res Ther., 2024, 16, 9. [Google Scholar] [CrossRef]
- Shpakov, A.O. Allosteric Regulation of G-Protein-Coupled Receptors: From Diversity of Molecular Mechanisms to Multiple Allosteric Sites and Their Ligands. Int J Mol Sci 2023, 24, 6187. [Google Scholar] [CrossRef]
- Budgett, R.F.; Bakker, G.; Sergeev, E.; Bennett, K.A.; Bradley, S.J. Targeting the Type 5 Metabotropic Glutamate Receptor: A Potential Therapeutic Strategy for Neurodegenerative Diseases? Front Pharmacol 2022, 13, 893422. [Google Scholar] [CrossRef]
- Kim, J.H. , Marton, J., Ametamey, S.M., & Cumming, P. A review of molecular imaging of glutamate receptors. Molecules 2020, 25, 4749. [Google Scholar] [PubMed]
- Wu, Y.; Zhang, B.; Xu, H.; He, M.; Deng, X.; Zhang, L. ... & Sun, W. The chronological evolution of fluorescent GPCR probes for bioimaging. Coordination Chem Rev., 2023, 480, 215040. [Google Scholar] [CrossRef]
- Soave, M.; Briddon, S.J.; Hill, S.J.; Stoddart, L.A. Fluorescent ligands: Bringing light to emerging GPCR paradigms. Br J Pharmacol 2020, 177, 978–991. [Google Scholar] [CrossRef]
- Fernández-Dueñas, V.; Qian, M.; Argerich, J.; Amaral, C.; Risseeuw, M.D.; Van Calenbergh, S.; Ciruela, F. Design, synthesis and characterization of a new series of fluorescent metabotropic glutamate receptor type 5 negative allosteric modulators. Molecules, 2020, 25(7), 1532. 2020, 25, 1532. [Google Scholar]
- Kampen, S.; Rodríguez, D.; Jørgensen, M.; Kruszyk-Kujawa, M.; Huang, X.; Collins Jr, M. ... & Carlsson, J. Structure-based discovery of negative allosteric modulators of the metabotropic glutamate receptor 5. ACS Chem Biol. 2022, 17, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Dogan, E.E. Computational bioactivity analysis and bioisosteric investigation of the approved breast cancer drugs proposed new design drug compounds: increased bioactivity coming with silicon and boron. Lett Drug Des Discov 2021, 18, 551–561. [Google Scholar] [CrossRef]
- Bello, M. Advances in theoretical studies on the design of single boron atom compounds. Curr Pharm Des 2018, 24, 3466–3475. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, M.; Bednarska, K. , Lesnikowski, Z. J. Comparative Study of Carborane-and Phenyl-Modified Adenosine Derivatives as Ligands for the A2A and A3 Adenosine Receptors Based on a Rigid in Silico Docking and Radioligand Replacement Assay, Molecules, 2018, 23, 1–21. [Google Scholar]
- Bednarska-Sczepaniak, K.; Mieczkowski, A.; Kierozalska, A.; Saftic, D.P. ; Glabala,K.; Przygodzki, T.; Stanczyk, L.; Karolczak, K.; Watala, C.; Rao, H.; Gao, Z.G.; Jacobson, K.A.; Lesnikowski, Z.J. Synthesis and Evaluation of adenosine derivatives as A1, A2A, A2B, and A3 Adenosine Receptor Ligands containing Boron Clusters as phenyl isosteres and selective A3 Agonists. Eur. J. Med. Chem. 2021, 223, 113607. [Google Scholar]
- Kok, Z.Y.; Stoddart, L.A.; Mistry, S.J.; Mocking, T.A.M.; Vischer, H.F.; Leurs, R.; Hill, S.J.; Mistry, S.N.; Kellam, B. Optimization of Peptide Linker-Based Fluorescent Ligands for the Histamine H1 Receptor, J Med Chem. , 2022, 65, 8258–8288. [Google Scholar]
- Barron-Gonzalez, M.; Rosales-Hernandez, M.C.; Abad-Garcia, A.; Ocampo-Nestor, A.L.; Santiago-Quintana, J.M.; Perez-Capistran, T.; Trujillo-Ferrara, J.G.; Padilla-Martinez, I.I.; Farfan-Garcia, E.D.; Soriano-Ursua, M.A. Synthesis, In silico, and Biological Evaluation of a Borinic Tryptophan-Derivative That Induces Melatonin-like Amelioration of Cognitive Deficit in Male Rat. Int J Mol Sci. 2022, 23, 3229. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Kong, L.; Gao, J. Boron enabled bioconjugation chemistries. Chem Soc Rev. 2024, 53(24), 11888–11907. [Google Scholar] [CrossRef]
- Mafi A, Kim SK, Goddard WA 3rd. The mechanism for ligand activation of the GPCR-G protein complex. Proc Natl Acad Sci U S A. 2022, 119, e2110085119. [CrossRef]
- Papasergi-Scott, M.M.; Pérez-Hernández, G.; Batebi, H.; Gao, Y.; Eskici, G.; Seven, A.B.; Panova, O.; Hilger, D.; Casiraghi, M.; He, F.; Maul, L.; Gmeiner, P.; Kobilka, B.K.; Hildebrand, P.W.; Skiniotis, G. Time-resolved cryo-EM of G-protein activation by a GPCR. Nature. 2024, 629(8014), 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Diaz, D.B.; Yudin, A.K. The versatility of boron in biological target engagement. Nat Chem 2017, 9, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Coghi, P.; Li, J.; Hosmane, N.S.; Zhu, Y. Next generation of boron neutron capture therapy (BNCT) agents for cancer treatment. Medicinal Research Reviews 2023, 43, 1809–1830. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.A.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.G.; Choi, H.J.; Devree, B.T.; Sunahara, R.K.; Chae, P.S.; Gellman, S.H.; Dror, R.O.; Shaw, D.E.; Weis, W.I.; Caffrey, M.; Gmeiner, P.; Kobilka, B.K. Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature. 2011, 469(7329), 236–40. [Google Scholar] [CrossRef]
- Peter, S.; Siragusa, L.; Thomas, M.; Palomba, T.; Cross, S.; O’Boyle, N.M. ... & De Graaf, C. Comparative Study of Allosteric GPCR Binding Sites and Their Ligandability Potential. J Chem Inf Model 2024, 64, 8176–8192. [Google Scholar] [CrossRef]
- Bailey, S.F.; Kabalka, G.W.; Fuhr, J.E. In Vitro Effects of Boron-Containing Compounds upon Glioblastoma Cells. Proc Soc Exp Biol Med. 1997, 216(3), 452–455. [Google Scholar] [CrossRef]
- Turkez, H.; Arslan, M.E.; Tatar, A.; Mardinoglu, A. Promising potential of boron compounds against Glioblastoma: In Vitro antioxidant, anti-inflammatory and anticancer studies. Neurochem Int., 2021, 149, 105137. [Google Scholar] [CrossRef]
- Nguyen, P.; Doan, P.; Rimpilainen, T.; Konda Mani, S.; Murugesan, A.; Yli-Harja, O. ... & Kandhavelu, M. Synthesis and preclinical validation of novel indole derivatives as a GPR17 agonist for glioblastoma treatment. J Med Chem 2021, 64, 10908–10918. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Kauffman, M. Development of the proteasome inhibitor Velcade™(Bortezomib). Cancer investigation 2004, 22, 304–311. [Google Scholar] [CrossRef]
- Wiley, S.Z. , Sriram, K., Salmerón, C., & Insel, P.A. GPR68: an emerging drug target in cancer. International journal of molecular sciences 2019, 20, 559. [Google Scholar]
- Zhao, W.; Liu, J.; Wang, L.; Wang, W. Study on autonomic neuropathy of the digestive system caused by bortezomib in the treatment of multiple myeloma. Hematology 2023, 28, 2210907. [Google Scholar] [CrossRef]
- Yamamoto, S.; Egashira, N. Pathological mechanisms of bortezomib-induced peripheral neuropathy. Int J Mol Sci. 2021, 22, 888. [Google Scholar] [CrossRef] [PubMed]
- Ghelardini, C.; Menicacci, C.; Cerretani, D.; Bianchi, E. Spinal administration of mGluR5 antagonist prevents the onset of bortezomib induced neuropathic pain in rat. Neuropharmacology. 2014, 86, 294–300. [Google Scholar] [CrossRef]
- Kumar, S.; Flinn, I.; Richardson, P.G.; Hari, P.; Callander, N.; Noga, S.J.; Stewart, A.K.; Turturro, F.; Rifkin, R.; Wolf, J.; Estevam, J.; Mulligan, G.; Shi, H.; Webb, I.J.; Rajkumar, S.V. Randomized, multicenter, phase 2 study (EVOLUTION) of combinations of bortezomib, dexamethasone, cyclophosphamide, and lenalidomide in previously untreated multiple myeloma. Blood 2012, 119, 4375–4382. [Google Scholar] [CrossRef] [PubMed]
- Kitzen, J.M.; Pergolizzi Jr, J.V.; Taylor Jr, R.; Raffa, R.B. Crisaborole and Apremilast: PDE4 Inhibitors with Similar Mechanism of Action, Different Indications for Management of Inflammatory Skin Conditions. Pharmacol Pharm 2018, 09, 357–381. [Google Scholar] [CrossRef]
- Paller, A.S.; Tom, W.L.; Lebwohl, M.G.; Blumenthal, R.L.; Boguniewicz, M.; Call, R.S.; Eichenfield, L.F.; Forsha, D.W.; Rees, W.C.; Simpson, E.L.; Spellman, M.C.; Stein Gold, L.F.; Zaenglein, A.L.; Hughes, M.H.; Zane, L.T.; Hebert, A.A. Efficacy and safety of crisaborole ointment, a novel, nonsteroidal phosphodiesterase 4 (PDE4) inhibitor for the topical treatment of atopic dermatitis (AD) in children and adults. J Am Acad Dermatol 2016, 75, 494–503.e6. [Google Scholar] [CrossRef]
- Fan, M.; Fan, X.; Lai, Y.; Chen, J.; Peng, Y.; Peng, Y.; Xiang, L.; Ma, Y. Protease-Activated Receptor 2 in inflammatory skin disease: current evidence and future perspectives. Front Immunol, 2024, 15, 1448952. [Google Scholar] [CrossRef]
- Peña, S.M.; Oak AS, W.; Smith, A.M.; Mayo, T.T.; Elewski, B.E. Topical crisaborole is an efficacious steroid-sparing agent for treating mild-to-moderate seborrhoeic dermatitis. J Eur Acad Dermatol Venereol, 2020, 34, e809–e812. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I.; Kirsner, R.S.; Margolis, D.J.; Tharp, M.; Myers, D.E.; Annis, K.; Graham, D.; Zang, C.; Vlahos, B.L.; Sanders, P. Efficacy and safety of crisaborole ointment, 2%, in participants aged ≥45 years with stasis dermatitis: Results from a fully decentralized, randomized, proof-of-concept phase 2a study. J Am Acad Dermatol, 2024, 90, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chen, Z.R.; Ding, H.J.; Feng, J. Molecular and cellular mechanisms of itch sensation and the anti-itch drug targets. Acta Pharmacologica Sinica 2024, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.C. Talabostat. Exp Opin Invest Drugs 2007, 16, 1459–1465. [Google Scholar] [CrossRef]
- Redman, B.G.; Ernstoff, M.S.; Gajewski, T.F.; Cunningham, C.; Lawson, D.H.; Gregoire, L.; Haltom, E.; Uprichard, M.J. Phase 2 trial of talabostat in stage IV melanoma. J Clin Oncol 2005, 23, 7570–7570. [Google Scholar] [CrossRef]
- Eager, R.M. , Cunningham, C.C., Senzer, N., Richards, D.A., Raju, R.N., Jones, B., Uprichard, M., & Nemunaitis, J. Phase II Trial of Talabostat and Docetaxel in Advanced Non-small Cell Lung Cancer. Clin Oncol 2009, 21, 464–472. [Google Scholar] [CrossRef]
- Dogan, E.E. Computational Bioactivity Analysis and Bioisosteric Investigation of the Approved Breast Cancer Drugs Proposed New Design Drug Compounds: Increased Bioactivity Coming with Silicon and Boron. Lett Drug Des Discov 2021, 18, 551–561. [Google Scholar] [CrossRef]
- Juillerat-Jeanneret, L.; Tafelmeyer, P.; Golshayan, D. Regulation of Fibroblast Activation Protein-α Expression: Focus on Intracellular Protein Interactions. J Med Chem 2021, 64, 14028–14045. [Google Scholar] [CrossRef]
- Hoque, M.; Ali, S.; Hoda, M. Current status of G-protein coupled receptors as potential targets against type 2 diabetes mellitus. Int J Biol Macromol., 2018, 118, 2237–2244. [Google Scholar] [CrossRef]
- Johnson KM, S. Dutogliptin, a dipeptidyl peptidase-4 inhibitor for the treatment of type 2 diabetes mellitus. Curr Opin Invest Drugs 2010, 11, 455–463. [Google Scholar]
- Li, J. , Klemm, K., Marie O’Farrell, A., Guler, H.P., Cherrington, J.M., Schwartz, S., & Boyea, T. (2010). Evaluation of the potential for pharmacokinetic and pharmacodynamic interactions between dutogliptin, a novel DPP4 inhibitor, and metformin, in type 2 diabetic patients. Curr Med Res Opin 2010, 26, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G protein-coupled receptors (GPCRs): advances in structures, mechanisms, and drug discovery. Sign Transduct Target Ther 2024, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.R.; Seng, D.J.; Xu, Y.; Zhang, Y.D.; Zhou, W.J.; Jia, Y.Y.; Yuan, S. A comprehensive review of small molecule drugs approved by the FDA in 2023: Advances and prospects. Eur J Med Chem, 2024, 276, 116706. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The D2 Dopamine receptor (D2DR) as the target of a BCC. The transmembrane segments of the D2DR (on the left) and a dopa-derivative docked in its orthosteric site (marked with a star). Details of the interactions (on the right), segments of the transmembrane helix are colored by residue type, and the sidechain of residues interacting with the BCC (DPBX, in sticks and balls representation) are in licorice representation.
Figure 1.
The D2 Dopamine receptor (D2DR) as the target of a BCC. The transmembrane segments of the D2DR (on the left) and a dopa-derivative docked in its orthosteric site (marked with a star). Details of the interactions (on the right), segments of the transmembrane helix are colored by residue type, and the sidechain of residues interacting with the BCC (DPBX, in sticks and balls representation) are in licorice representation.
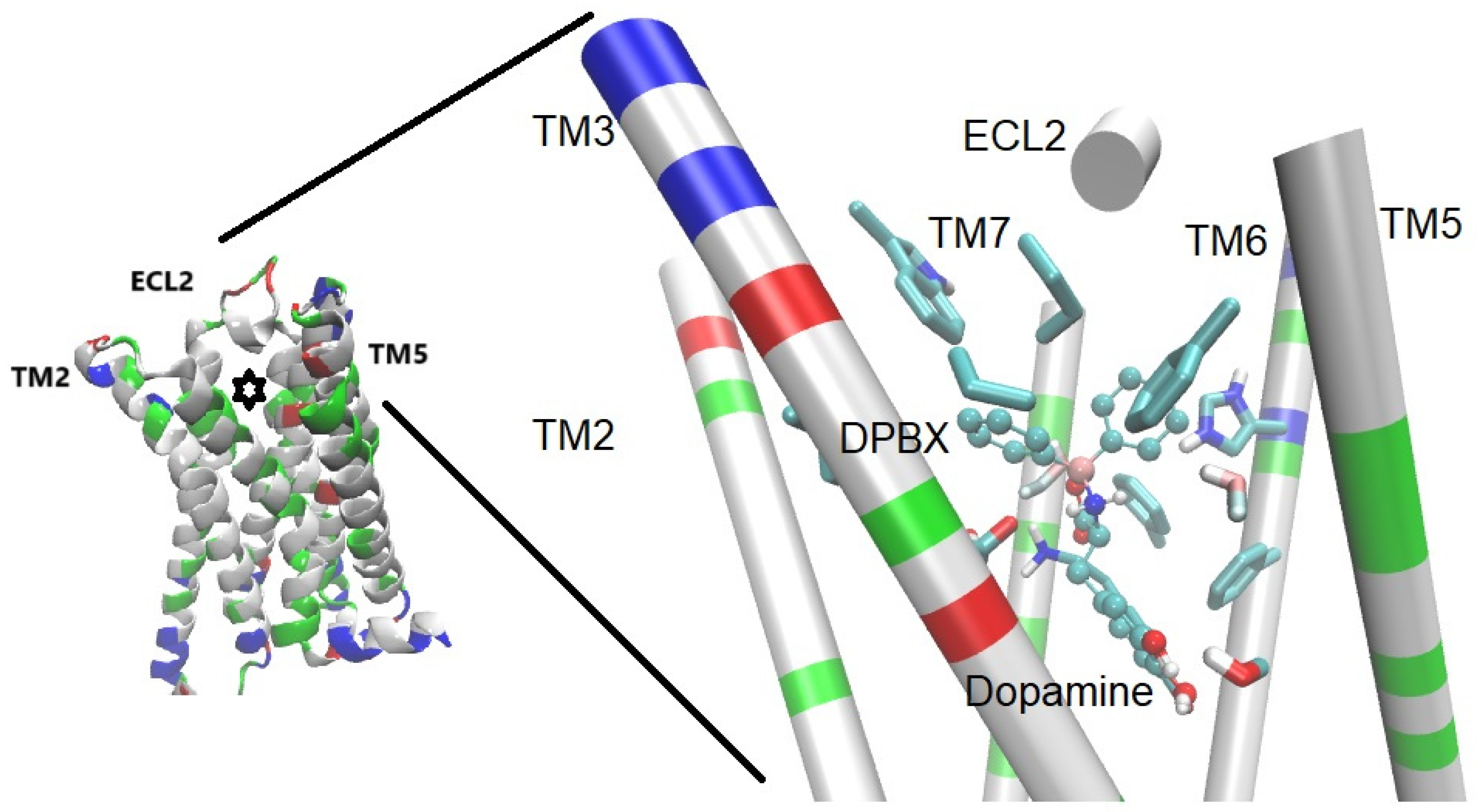
Figure 2.
The MT1 receptor with borolatonin (a BCC, structurally related to melatonin) docked in the orthosteric site (marked with a star). On the right, close-up to the binding pocket, the sidechain of three residues considered key in the orthosteric binding site are depicted and labeled as reference.
Figure 2.
The MT1 receptor with borolatonin (a BCC, structurally related to melatonin) docked in the orthosteric site (marked with a star). On the right, close-up to the binding pocket, the sidechain of three residues considered key in the orthosteric binding site are depicted and labeled as reference.
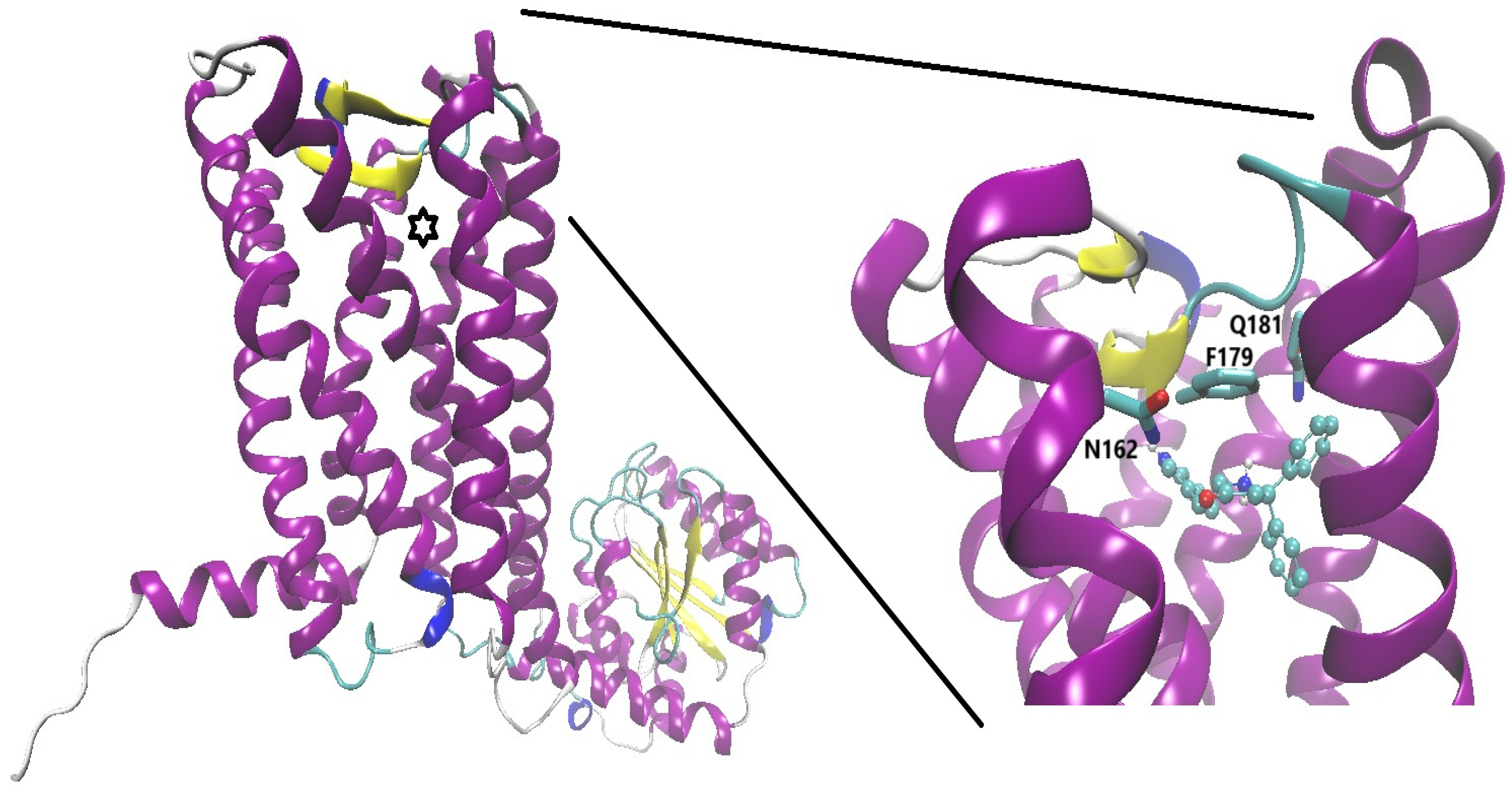
Figure 3.
A quinolinic-oxazaborolidine derivative (BDZ-quin represented as sticks and balls and colored by atom type, pink for boron atom) on the orthosteric site of mGluR1 (PDB CODE: 3KS9, originally crystallized in complex with the antagonist LY341495). The side chains of three putative key residues are depicted in licorice representation and labeled.
Figure 3.
A quinolinic-oxazaborolidine derivative (BDZ-quin represented as sticks and balls and colored by atom type, pink for boron atom) on the orthosteric site of mGluR1 (PDB CODE: 3KS9, originally crystallized in complex with the antagonist LY341495). The side chains of three putative key residues are depicted in licorice representation and labeled.
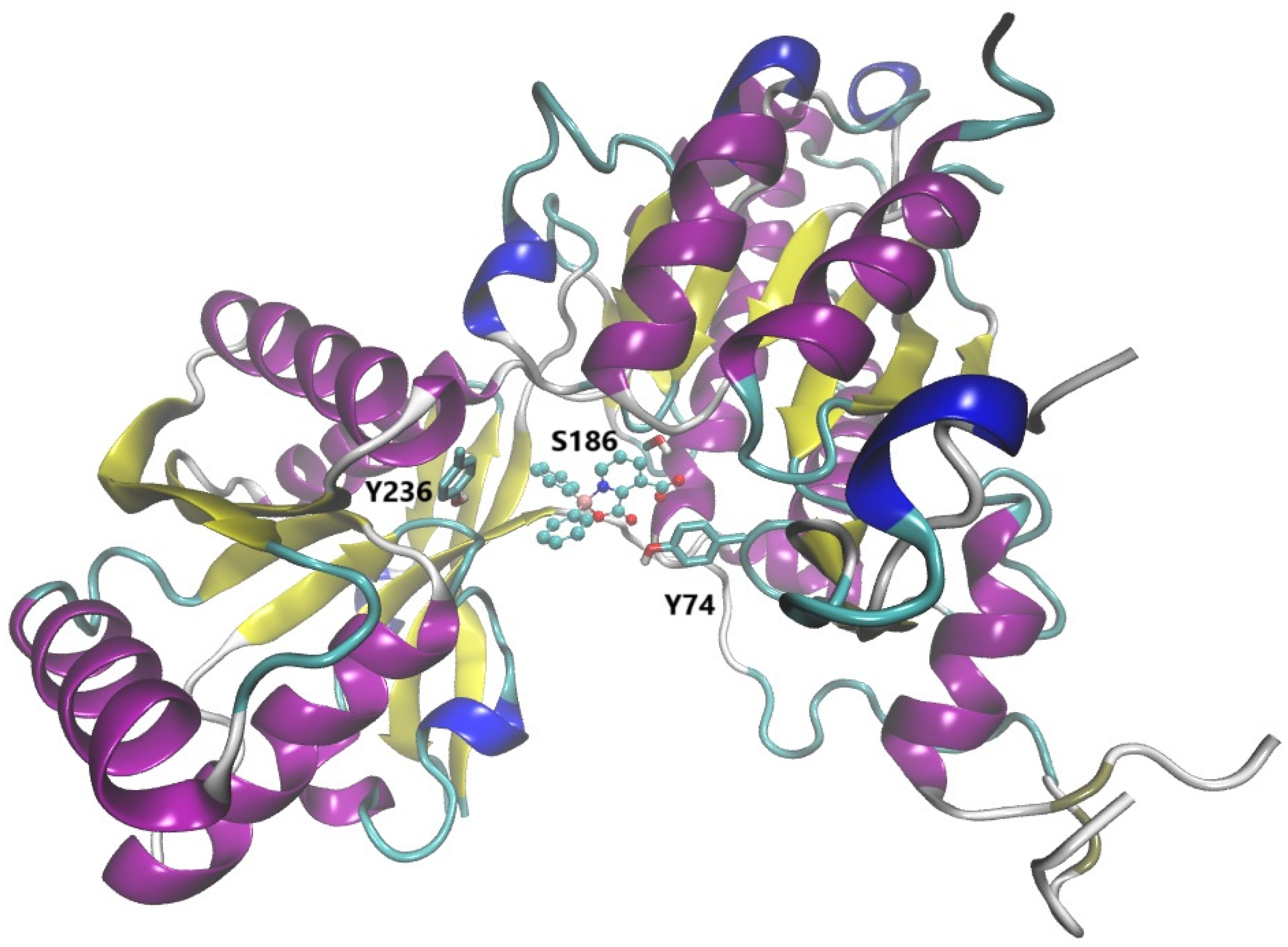
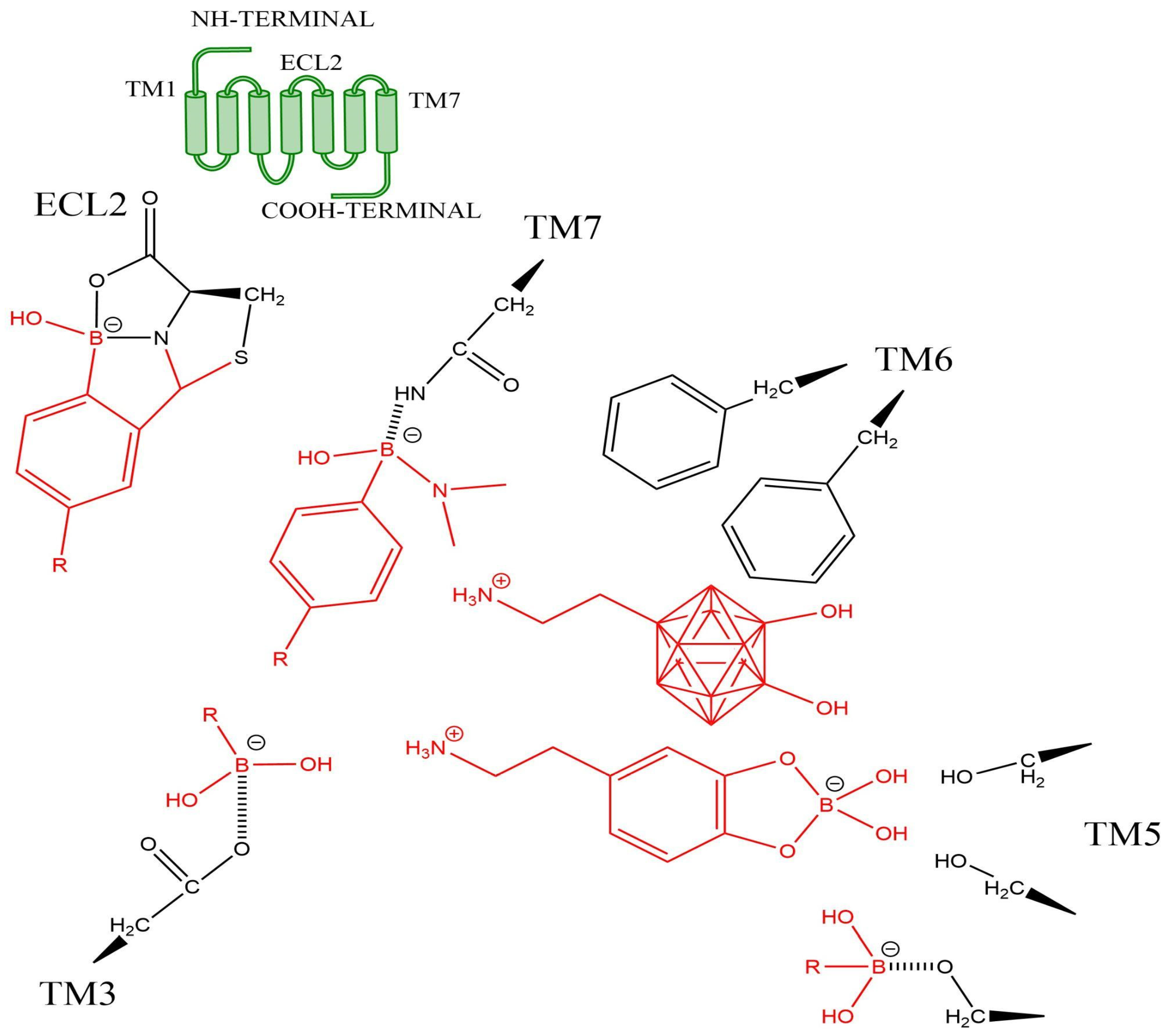
Figure 4.
Hypothetical interactions of some Boron-containing compounds (or moieties, in red) on some key residues in the orthosteric site of a GPCR (the sidechain of key residues as black structures). The scheme is based on a catecholamine (class A) GPCR. The polyhedral shape represents a carborane core.
Figure 4.
Hypothetical interactions of some Boron-containing compounds (or moieties, in red) on some key residues in the orthosteric site of a GPCR (the sidechain of key residues as black structures). The scheme is based on a catecholamine (class A) GPCR. The polyhedral shape represents a carborane core.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



