
Submitted:
10 February 2025
Posted:
11 February 2025
You are already at the latest version
Abstract
Aggressive Periodontitis (AP) and Feline Chronic Gingivostomatitis (FCGS) are two oral inflammatory diseases in cats with unknown etiology. Both conditions present with severe inflammation of the oral cavity and in FCGS it is found with additional deterioration of the non-keratinized mucosa. The oral microbiome is increasingly implicated in disease progression, but little is known about shifts of the microbial community during the AP and FCGS progression. To that end, we used deep metagenomic sequencing with total RNA on three longitudinal samples of the oral microbiome in a cat first diagnosed with AP that progressed to FCGS. This deep sequencing approach revealed that increased diversity at both the genus and species-level marked the shift from AP to FCGS, including increases in Porphyromonas and Treponema species, and decreased Streptobacillus species. The metatranscriptomes were then probed for expression of antimicrobial resistance genes and virulence factors. Disease-related genes that include cheY, and ompP5 were expressed in early AP and FCGS, while others like galU were only expressed in one or the other disease state. Both genus and species-level shifts were observed along the longitudinal microbiome samples with a noted increase in species diversity in the FCGS-associated microbiome. Corroborating that functional shifts accompany taxonomic changes, the AMR and virulence factor expression similarly changed between the sampling points. Together these taxonomic and functional shifts indicate that AP and FCGS are potentially linked and may be marked by changes in the oral microbiome, which supports the development of microbial-based clinical diagnostics and therapeutics.
Keywords:
Aggressive Periodontitis
; Feline Chronic Gingivostomatitis
; oral microbiome
; metatranscriptomics
; microbial community
1. Introduction
Aggressive periodontitis (AP) and Feline Chronic Gingivostomatitis (FCGS) are two debilitating diseases of the cat oral cavity marked by progressing inflammation and deterioration leading to early tooth loss[1]. Both diseases decrease the quality of life and are prevalent, with AP affecting up 13% of juvenile cats and FCGS affecting up to 26% of adult cats [1,2]. While clinically AP and FCGS are not considered to be connected diseases, a portion of cats diagnosed with AP in early life have been later diagnosed with FCGS [1]. Both chronic diseases are important contributors to declining oral health and subsequent sequalae, but the origin of either disease remains unknown. Ongoing work supports a multifactorial cause that includes a combination of host immune status, systemic infection, and changes in the oral microbiota [3,4,5,6,7,8,9]. The lack of a known cause for either AP or FCGS has led to limited treatment options for clinicians that largely include near full mouth or full mouth tooth extractions, and medical management including pain control and immunosuppressive or modulating therapies that are often times given lifelong [10]. A deeper understanding of the suspected contributing factors, like related shifts in oral microbiome, is therefore necessary to improve treatment options and patient outcomes.
The oral microbiome is a known contributor to oral health and disease in cats and other mammals [11,12,13]. Though there is some variation between individuals, the health oral microbiota in cats is dominated by members Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria [14,15]. These microbes colonize the hard dental structures and soft tissue of the oral cavity and together help digest food particles, influence immune responses of the host, and contribute to systemic health [16,17,18]. When this delicate compositional and functional balance of is broken through antibiotic treatment, dietary changes, infection, or other influences, the host becomes susceptible to progressive oral dysfunction and disease [19,20,21]. Periodontal pathogens also contribute to the development and exacerbation of dysbiosis and disease [22,23,24,25]. While no single pathobiont has been identified for AP or for FCGS in cats, periodontal pathogens like Aggregatibacter actinomycetemcomitans, Porphymonas gingivalis and Treponema denticola have previously been associated with AP in humans [26,27]. These organisms may well play a role in cat oral health as well [15]. The association of dental pathogens to some periodontal disease and the known contributions of the oral microbiome to maintaining host health together underscore how the oral microbiome contributes to both disease and health.
Though the importance of the microbiome in driving disease is established, the underlying triggers driving the switch from a commensal organism to an antagonistic pathobiont remain unknown [28]. Given the importance of the oral microbiome in either initiating or driving the progression of AP and FGCS, characterizing community shifts over time is a necessary step towards developing a mechanistic understanding of the oral microbiome’s contribution to dental health. To that end, we used deep sequencing of total RNA from buccal swabs in a cat first diagnosed with AP and later diagnosed with FCGS. The longitudinal sampling of three time points during the progression of dental disease coupled to the deep sequencing approach provides a previously unparalleled look at the shifting oral microbiome in two understudied cat oral diseases. Taken together these microbiome snapshots across time chronicle an oral microbiome in flux and highlight that the microbial composition in the mouth changes over taxonomic levels with progressive inflammation and worsening oral lesions. Notably, the use of total RNA sequencing in this work illustrates changing microbial activity across time, via functional profiling and enrichment analyses of activities that include antimicrobial resistance (AMR) genes and virulence factors. While this work focuses on a single cat and thus requires larger studies to validate the findings at a population level, the observations here of an actively changing oral microbiome longitudinally supports the hypothesis that the oral microbiome may function as a marker for and possibly a contributor to disease status.
2. Case Description
A 7-month-old female spayed domestic short hair weighing 3.9 kg presented 4 months after being rescued from a shelter in Northern CA with suspected juvenile gingivitis. About three weeks prior to presentation, the primary care veterinarian had noted enlarged mandibular lymph nodes, moderate gingivitis with buccal mucositis and halitosis. Feline leukemia virus (FeLV) and feline calicivirus (FIV) testing was negative at that time. No evidence of oral discomfort was noted by the owner.
On presentation to a board-certified veterinary dentist, mild to moderate gingival enlargement with moderate to severe gingivitis was noted on the maxillary and mandibular premolar and molar teeth. Mild inflammation of the buccal mucosa was noted, and the mandibular lymph nodes were moderately enlarged and firm. The rest of the physical examination was unremarkable. Pre-operative blood minimum data base was unremarkable.
Anesthetized evaluation two weeks later including dental charting and intraoral dental radiographs were performed and revealed pseudo pocketing on the maxillary and mandibular premolar and molar teeth and stage 2 periodontal disease. Gingival recontouring and a periodontal treatment were performed. Extractions were not indicated at that time. Biopsy of the inflamed gingiva showed severe, chronic, erosive to proliferative lymphoplasmacytic and neutrophilic gingivitis.
The patient healed from gingivoplasty uneventfully however, inflammation of the caudal mucosa continued to worsen. Re-evaluation 3 months after surgery showed persistent gingivitis of the premolar and molar teeth and worsening of the caudal oral cavity despite treatment with moderate inflammation and ulceration noted. The incisors and canine teeth were spared. The patient had gained weight and body condition score at the time was 7/9 (4.5kg). Given progression of inflammation and no response to conservative management, FCGS was discussed and partial mouth extractions were recommended and performed five months after initial presentation. Biopsy of lesional tissue from the caudal oral mucosa then showed marked multifocal to coalescing chronic neutrophilic and plasmacytic inflammation with intralesional bacterial colonies.
Extractions healed uneventfully, persistent but mild inflammation was seen two months post-operatively and then the patient was lost to follow up until two years later. Anesthetized evaluation then showed stage 2 periodontal disease in the remaining incisors and early stage 3 (30% attachment loss) in the remaining canine teeth and persistent mild inflammation in the caudal oral cavity. Great appetite, energy level and persistent over conditioning (BCS 7/9 at 5.2 kg) were noted during that visit. No plaque and calculus control was ever done at home nor was this patient treated with immunosuppressives. Onsior was prescribed after the first biopsy (6 mg PO QD for 2 days). Amoxicillin clavulanate was prescribed after extraction of premolar and molar teeth (62.5 mg PO BID for 10 days). Pain management was otherwise accomplished with buprenorphine (0.15mg PO up to TID) and gabapentin (25-50 mg PO up to TID) as needed.
The collection and study design were reviewed and approved by the University of California-Davis Institutional Animal Care and Use Committee (IACUC #22738) and signed owner consent was obtained before sampling. A swab of the caudal buccal mucosa was taken during each of three visits (1/13/21, 2/10/21, 5/26/22) and the same method for extraction ad sequencing was applied to all three swabs (Figure 1) and as described previously [8].
A cytobrush (FLOQSwabs, Coplan, Italy, EU) was used to swab the oral mucosa lateral to the palatoglossal folds, then placed in 500 μL of DNA/RNA Shield (Zymo, Irvine, CA, USA), vortexed, and stored at −20 °C. Bacterial cells were enzymatically lysed according to the protocol used by the 100K pathogen project [29], and then RNA was isolated using Trizol LS (Ambion, Austin, TX, USA) according to manufacturer instructions. RNA sequencing libraries were prepared as described previously [30,31,32], with RNA purity and integrity confirmed using TapeStation (Agilent Technologies Inc., Santa Clara, CA, USA). Sequencing libraries were constructed using the enzymatic-based KAPA HyperPlus Library Preparation kit (KK8514) (Kappa Biosystems, Wilmington, MA, USA) on a PerkinElmer Sciclone G3 (PerkinElmer Inc. Waltham, MA, USA) and sequenced on an Illumina NovaSeq S4 (Illumina, San Diego, CA, USA).
Trimmomatic (version 0.39) [33] was first used to remove low-quality sequences and sequencing adapters then sequence data quality was reviewed with FastQC (version 0.11.9) [34]. Kraken2 with a microbial reference database, using standard settings (k-mer size = 35), was used to assign taxonomy and Bracken (version 2.6.1) [35] was then used to estimate the relative proportion of respective taxa at the species level [30]. Expression of AMR genes was determined by running Trinity (v2.15.1) [36] assembled reads through the Comprehensive Antimicrobial Resistance Database (CARD, built 10 August 2023) [37]. Virulence factor expression was evaluated using the Virulence Factor Database (VFDB, built 10 August 2023) [38]. The STRING database (accessed on October 2, 2024) was used to search for gene and protein connections between AMR and virulence genes in selected oral microorganisms [39]. Shannon diversity and Bray-Curtis dissimilarity were calculated using the diversity function of the vegan package (Version 2.6-8) in R (Version 4.4.1), and subsequently plotted using Prism 10 (GraphPad, Menlo Park, CA). The correlation plot was made using R (Version 4.2.3) in tandem with Inkscape (Version 1.0) and accessed via GitHub (https://github.com/inkscape/inkscape). The Venn diagram and alluvial plot were made using ggplot (Version 3.5.1) in R (Version 4.4.1), with Adobe Illustrator (Adobe, San Jose, CA) used to reformat text placement and size. All other figures were made using BioRender (biorender.com).
3. Results
The species-level diversity in the oral microbiome of a single cat with progressive dental disease increased from early AP to the onset of FCGS (Figure 2). A total of 4,014 microbial species were found among all three sampling points. The number of species unique to each oral microbiome increased from 168 in the first AP sample, to 256 in the second AP sample and 1,192 in the FCGS sample (Figure 2A). Notably AP_1 and AP_2 samples shared 513 and 584 species with FCGS, respectively, while only sharing 66 species with each other. The increased number of unique species in the most progressed disease state suggests species-level remodeling of the oral microbial community underlies disease status as opposed to the outgrowth of a pathobiont or collapse of the community. Further supporting this observation, all three oral samples had high α- and β-diversity indices (Figure 2A, 2B). The high Shannon Diversity Index for all three microbiome samples suggest notable diversity in each microbiome and an even distribution of species abundance (AP_1 = 4.7, AP_2 = 3.8, FCGS = 4.6). Reflecting the observed increase in oral microbial diversity from inflammation limited to the periodontium (AP) to more generalized inflammation affecting mucosa in addition (FCGS), the Bray-Curtis dissimilarity index for all three samples was close to one and consistently increased from AP_1 (0.78) to AP_2 (0.85) to FCGS (0.93). Together the number of unique species in each sampling point and the general trend of increasing α- and β-diversity along the disease continuum from focal to diffuse inflammation crossing over the mucogingival line supports microbiome remodeling may be an important facet in determining disease status.
The observed increase in microbial diversity at the species level from early AP to later FCGS suggests that these inflammatory diseases are not the result of a single pathobiont and instead are likely to be initiated or exacerbated by complex community dynamics in the oral microbiome that shift over time. To better understand the dynamic changes across the disease continuum, each oral microbiome composition was assessed at both the genus and species level, then compared across time and disease classification (Figure 3). A comparison of the microbial composition in a time series revealed clear changes in the genera between the AP_1 and AP_2 samples, as well as between the second AP sample and FCGS (Figure 3A). While the oral microbiome of AP_1 contained 19 genera with total proportions above 1% each, the second AP microbiome sampled contains 13 genera with Aspergillus accounting for almost half of the community proportion. This increased Aspergillus population is concomitant with a decrease in the proportion of multiple genera including Capnocytophaga, Frederiksenia, Pasteurella, and Streptobacillus. Intriguingly, microbial community shifts at the genus-level once again from AP_2 to FCGS. In the FCGS community there was a slightly more even distribution, reflected by the high Shannon Index of 4.6, that included Campylobacter, Fusobacterium, Pasteurella, Porphymonas, and Treponema. The pattern of a diverse oral microbiome in AP1, to a markedly different composition to AP2, to a completely different composition yet once again an evenly distributed one in FCGS raises questions as to whether changes of specific species are connected to disease presentation and progressive tissue damage.
To evaluate the importance of specific species in the two disease presentations, AP and FCGS, the two AP samples were aggregated and compared to the FCGS microbiome in a correlation plot (Figure 3B). Multiple microbes were shared in approximately equal proportion across the two disease microbiomes including Porphyromonas gingivalis, Treponema denticola, Pasteurella multocida, and Fusobacterium gastrosuis. The relatively dense cloud of 1,805 points clustered around the center diagonal line suggests notable overlap in species present in the AP and FCGS microbiomes and that the proportions of these species are similar between the two diseases.
To assess whether changes in antimicrobial resistance gene (AMR) or virulence gene expression accompanied the noted microbial membership remodeling across time, the assembled transcripts for all three sampling points were examined with AMR and virulence factor databases (Figure 4). The AMR and virulence factor paralleled the observations of changing community composition across time, with AP_1 expressing primarily capsule remodeling factors, AP_2 with no significant hit, and FCGS expressing primarily motility related genes. In AP_1, a total of nine different AMR and virulence-related genes were found in the metatranscriptome. These genes were primarily related to microbial membrane remodeling, lipid and capsule production, including galU, lpxA, lpxC, and neuA. In FCGS there were five genes found, including lpxC and ompP5-like AP1. Unique to FCGS was the expression of the flagella-relate genes cheY, fliN, and fliQ. Intriguingly the AP_2 metatranscriptome had no identified transcripts related to AMR or virulence genes, even with a lower identity threshold of 80% that allows for sequence diversity between species. The expression of capsule and membrane-remodeling genes in the AP_1 microbiome suggests an active and ongoing response to environmental stressors while the flagella-related genes expressed in FCGS support that oral microbes could be actively moving through the deteriorating tissues and binding oral structures.
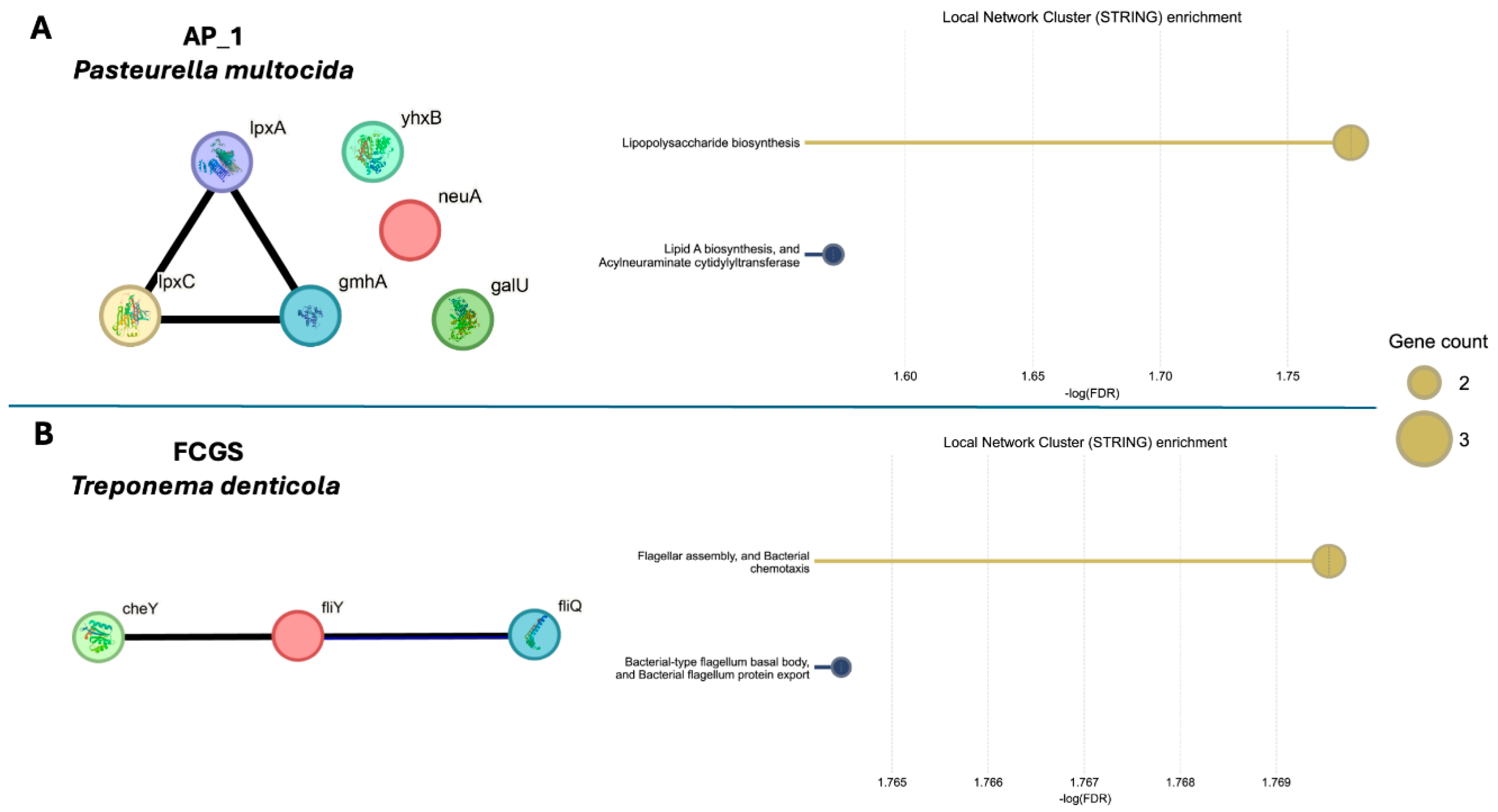
Altered microbial activity, in concert with the observed taxonomic changes, may contribute to the progression of oral inflammation and disease. To illustrate the changing microbial activity between early AP and FCGS, the respective virulence factors found in each condition (Figure 4) were analyzed for protein-protein interactions and network cluster enrichment and known protein associations using STRING (Figure 5). As abundant organisms in their respective microbiomes, Pasteurella multocida was used as the genetic background for the STRING enrichment analysis in AP1 and Treponema denticola was used as background for the FCGS STRING enrichment analysis. Membrane remodeling, from lipopolysaccharide biosynthesis (-log10FDR = 1.8) and lipid A biosynthesis and sialyation activity (-log10FDR = 1.6), was significantly enriched in the virulence factors found in AP_1. Contrastingly, the virulence factors found in the FCGS timepoint showed enrichment for the motility related pathways of flagellar assembly and chemotaxis (-log10FDR = 1.8) and flagellum body and flagellum protein export (-log10FDR = 1.8). Though only covering the annotated virulence factors and thus narrow in scope, the notable difference between the type of enriched pathways in AP and FCGS suggests the microbiome alters microbial stress responses in ways unique to each disease state and community membership. Enrichment analysis of the AP_1 virulence factors suggests a need for increased stress tolerance in the microbiome through increased membrane construction and modification. Contrastingly, the virulence factors found in FCGS support motility in the microbiome and potentially indicate bacteria may be moving in the oral cavity or through the deteriorating dental structures.
4. Discussion
Shifts in the oral microbiome composition and function have been associated with the onset and progression of multiple periodontal diseases [21,40,41,42,43]. Considering this, diseases without an established etiology, like AP and FCGS, warrant a deeper investigation into potential microbial connections. Identifying microbial signatures of disease progression or potentially causative community structures is necessary for the development of effective treatments. The current lack of effective treatments make AP and FCGS clinically challenging to manage for veterinarians and severely decrease the quality of life for patients [44]. Connecting the oral microbiome composition and function to disease status is therefore an important step in developing better clinical approaches to treating AP and FCGS.
AP and FCGS are treated as separate diseases of the oral cavity, but 7% of cats 2 years old or younger included in a retrospective study first diagnosed with AP went on to later develop FCGS [1]. The connection between these two diseases is not well-understood, nor is the origin of either [1,8,45]. To that end this case study provides insight into how the oral microbiome may link AP and FCGS in time and potentially how shifts in the microbial community can mark or contribute to disease progression. This case describes a single cat first diagnosed with AP and later with FCGS was sampled with a caudal buccal swab at three timepoints encompassing two within the diagnosis of AP and one after the diagnosis of FCGS. This work combined longitudinal samples with progressing disease using total RNA sequencing revealed a dynamic shift in both microbiome composition and virulence related activity.
The sampling of a single microbiome over time in a cat revealed a changing microbial community with markedly different compositions at the genus and species level during progressive inflammatory disease in the oral cavity. Interestingly, while two of these microbiomes were sampled during the same diagnosis of AP, they were distinctly different in their composition. The first AP sample included species of Capnocytophaga, Fusobacterium, Leptotrichia, Pasteurella, and Streptobacillus. Contrastingly, the second sample of the AP associated microbiome was dominated by Aspergillus and saw the rise of other fungal grouping Kluveromyces, along with increased bacterial membership of species from Porphyromonas and Prevotella. The microbiome community shifted once again in the FCGS sampling point, where Treponema and Porphyromonas were the two most dominant genera. This shifting profile both within the same diagnosis and to severe chronic disease highlights the complex and dynamic nature of the oral microbiome in the context of inflammatory disease progression.
The importance of considering the entire microbial community rather than just a few pathogens is further evidenced by the fact that, while Porphyromonas was present in both the AP and FCGS samples, its relative activity varied greatly between the two states. Similarly, Treponema, a genus associated with periodontal disease [46], only became dominant in FCGS. These findings suggest that the entire consortium of microbes, rather than a single or small group of "keystone" pathogens, play a critical role in driving the transition to a dysbiotic state and the progression of inflammatory disease in the oral cavity [47]. Clinically this indicates the need for a holistic consideration of the oral microbiome in treatment for inflammatory conditions, rather than a focus on a small set of pathogens [48,49].
In addition to profiling taxonomic shifts, the assembled transcripts from each microbiome were examined using a virulence factor database and an AMR database to reveal microbial functional changes associated with disease progression. The first sampled AP microbiome expressed genes related to membrane remodeling, including lipid synthesis and glycosylation. For instance, galU was found in the AP_1 sample and is a known contributor to virulence in multiple organisms through its role in modifying lipopolysaccharide (LPS) and connection to biofilm formation [50,51]. Relatedly drivers of LPS component lipidA, lpxA and lpxC, were also expressed in periodontitis. The expression of virulence factors primarily related to membrane modifications and subsequently to biofilm formation support the ongoing hypothesis that biofilm activity contributes to the deterioration of oral tissues and structures as seen in periodontitis [52]. This observation of potential biofilm formation in AP is congruent with the notable microbial diversity observed in the AP microbiome. Previous work in human periodontitis revealed increasing diversity was connected to progressing disease in part due to the expansion of biofilm niches in deepening gingival pockets [53,54]. While the samples in this case study were from the mucosa and not the sub gingival compartment like the aforementioned human periodontal work, a parallel process of mucosal deterioration may be taking place in the cat oral tissue. Biofilms that contribute to worsening disease can be initiated by commensal oral organisms like Streptococcus oralis, which are early colonizers of the dental structure [55,56]. These early colonizers change the local environment, releasing metabolites and polysaccharide matrices that recruit other oral microbes, include periodontal pathogens like T. denticola and P. gingivalis [54,57]. This complex community structure on the surface of host structures then utilizes the host tissues as metabolic substrates and ultimately contributes to dental decay [52,54]. The manual disruption of oral biofilms in one study improved periodontal outcomes in humans with periodontitis and led to decreased diversity in the oral microbiome, further supporting that biofilms are contributing factors to dental decay [54]. Thus, the observation of diverse microbiomes and biofilm-related gene expression in AP supports microbial activity is contributing to worsening dental disease in cats.
The expression of these biofilm and membrane-related virulence factors in the first AP microbiome is in contrast with the motility-related genes expressed by the FCGS-associated microbiome. CheY, fliN and fliQ were all expressed in the FCGS microbiome and are involved in flagellar activity with fliN and fliQ contributing flagellar building blocks and cheY transmitting chemotaxis signals to direct movement [58]. The expression of such motility related genes in the FCGS is an interesting observation in the context of previous work highlighting the role of flagella in instigating oral inflammation and systemic disease in host tissues [27,59,60,61]. The periplasmic flagella of oral pathogen T. denticola has been shown to initiate inflammation through activation of the innate immune system via interaction with toll-like receptor 2 (TLR2) on host cells [62] . Other work in T. denticola has shown flagella to be important for bacterial penetration and thus successful infection of the host epithelium, while non-motile or chemotaxis-deficient counterparts were unable to invade host tissue [61]. The immunogenic nature of flagellar structure in conjunction with their importance for successful host colonization suggest the expression of these functions in the FCGS microbiome may be a notable finding from this study. FCGS results in the degradation of tissues and thus release of host proteins and other compounds which may act as chemotaxis signals, inviting motile oral microbes to colonize the deteriorating host tissues and structures [63].
The exploration here of changing microbial composition and virulent activity in the oral microbiome of a cat with expanding oral inflammation provides multiple findings that can guide future work in this area. Though limited to a single cat as a case study, the results of this study suggest taxonomic differences are associated with progressing inflammation, that it remains to be determined whether these changes instigate or follow disease in a larger study. Similarly, the identification of different microbiome virulence factor profiles in AP and FCGS support that function, especially in connection to biofilm and motility, is an important facet when evaluating the connection between a shifting microbiome and host health. Collectively, this work provides a foundation for future work investigating how inflammatory diseases of the oral cavity are connected to the microbial consortia in the oral cavity.
5. Conclusions
Longitudinal sampling of the oral microbiome during progressing AP on to the diagnosis of FCGS in a single cat revealed a dynamic microbiome with distinct profiles at the genus and species level. Streptobacillus species were most dominant in the first AP sample followed by a notable increase in Aspergillus at the second AP timepoint with Porphyromonas and Treponema as the predominant genera in the FCGS microbiome. In concert with the changing composition, the expression of virulence factors also changed through the progression of disease. Genes primarily related to microbial membrane composition and modification were expressed in the first AP community, while motility related virulence factors were the predominate virulence function in the FCGS profile. The longitudinal profiling of both the oral microbiome composition and virulence profile across time and in conjunction with progressive oral inflammation support the microbiome is either responding to or inciting disease in the host. While further work is necessary to confirm these results at a population level, the observations here support that changing microbial composition and function mark disease progression from AP to FCGS and that development of clinically relevant microbial markers for oral disease state is possible.
Author Contributions
C.A.S. analyzed the data, created the visualizations, and wrote and edited the original manuscript. M.S.-R. conceptualized the work, collected samples, and wrote and edited the manuscript. R.P. analyzed the data, contributed to the visualizations, and edited the manuscript. B.C.W. conceptualized the work and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The project described was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant number UL1 TR001860. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Institutional Review Board Statement
All study procedures were reviewed and approved by the University of California-Davis Institutional Animal Care and Use Committee 19881 (4 May 2017), 19170 (28 January 2016), 18476 (21 November 2014).
Informed Consent Statement
We received the owner’s consent for all sampling procedures.
Data Availability Statement
Sequencing data generated and analyzed in this study can be found at the 100K Pathogen Project on NCBI SRA under BioProject PRJNA1136879.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Soltero-Rivera, M.; et al. Clinical, radiographic and histopathologic features of early-onset gingivitis and periodontitis in cats (1997-2022) . J Feline Med Surg 2023, 25, 1098612X221148577. [Google Scholar] [CrossRef] [PubMed]
- Soltero-Rivera, M.; Goldschmidt., S.; Arzi, B. Feline chronic gingivostomatitis current concepts in clinical management. J Feline Med Surg 2023, 25, 1098612X231186834. [PubMed]
- Dolieslager, S.M.; et al. The influence of oral bacteria on tissue levels of Toll-like receptor and cytokine mRNAs in feline chronic gingivostomatitis and oral health . Vet Immunol Immunopathol 2013, 151, 263–274. [Google Scholar] [CrossRef]
- Dolieslager, S.M.; et al. Identification of bacteria associated with feline chronic gingivostomatitis using culture-dependent and culture-independent methods . Vet Microbiol 2011, 148, 93–98. [Google Scholar] [CrossRef]
- Older, C.E.; et al. Influence of the FIV Status and Chronic Gingivitis on Feline Oral Microbiota . Pathogens 2020, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Peralta, S.; et al. Transcriptomic signatures of feline chronic gingivostomatitis are influenced by upregulated IL6 . Sci Rep 2023, 13, 13437. [Google Scholar] [CrossRef]
- Perry, R.; Tutt, C. Periodontal disease in cats: back to basics--with an eye on the future. J Feline Med Surg 2015, 17, 45–65.
- Shaw, C.A.; et al. Case Report: Inflammation-Driven Species-Level Shifts in the Oral Microbiome of Refractory Feline Chronic Gingivostomatitis. Bacteria 2025, 4, 1. [CrossRef]
- Soltero-Rivera, M.; et al. Feline Chronic Gingivostomatitis Diagnosis and Treatment through Transcriptomic Insights . Pathogens 2024, 13, 192. [Google Scholar] [CrossRef]
- Winer, J.N.; Arzi, B.; Verstraete, F.J. Therapeutic Management of Feline Chronic Gingivostomatitis: A Systematic Review of the Literature. Front Vet Sci 2016, 3, 54.
- Davis, E.M. , Gene Sequence Analyses of the Healthy Oral Microbiome in Humans and Companion Animals. J Vet Dent 2016, 33, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Duran-Pinedo, A.E.; Frias-Lopez, J. Beyond microbial community composition: functional activities of the oral microbiome in health and disease. Microbes and Infection 2015, 17, 505–516.
- Davis, E.M.; Weese, J.S. Oral Microbiome in Dogs and Cats: Dysbiosis and the Utility of Antimicrobial Therapy in the Treatment of Periodontal Disease. Vet Clin North Am Small Anim Pract 2022, 52, 107–119. [PubMed]
- Harris, S.; et al. A Pyrosequencing Investigation of Differences in the Feline Subgingival Microbiota in Health, Gingivitis and Mild Periodontitis . PLoS One 2015, 10, e0136986. [Google Scholar] [CrossRef]
- Rodrigues, M.X.; et al. The subgingival microbial community of feline periodontitis and gingivostomatitis: characterization and comparison between diseased and healthy cats . Sci Rep 2019, 9, 12340. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol 2018, 16, 745–759. [PubMed]
- Moreno, C.M.; et al. Immunomodulatory role of oral microbiota in inflammatory diseases and allergic conditions . Front Allergy 2023, 4, 1067483. [Google Scholar] [CrossRef]
- Barbour, A.; et al. Metabolites of the oral microbiome: important mediators of multikingdom interactions . FEMS Microbiology Reviews 2022, 46, fuab039. [Google Scholar] [CrossRef]
- Cheng, X.; et al. Effects of Antibiotic Use on Saliva Antibody Content and Oral Microbiota in Sprague Dawley Rats . Front Cell Infect Microbiol 2022, 12, 721691. [Google Scholar] [CrossRef]
- Adler, C.J.; et al. Diet may influence the oral microbiome composition in cats . Microbiome 2016, 4, 23. [Google Scholar] [CrossRef]
- Lee, Y.H.; et al. Progress in Oral Microbiome Related to Oral and Systemic Diseases: An Update . Diagnostics (Basel) 2021, 11, 1283. [Google Scholar] [CrossRef] [PubMed]
- Bui, F.Q.; et al. Association between periodontal pathogens and systemic disease . Biomed J 2019, 42, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Mysak, J.; et al. Porphyromonas gingivalis: major periodontopathic pathogen overview . J Immunol Res 2014, 2014, 476068. [Google Scholar] [CrossRef]
- Santonocito, S.; et al. A Cross-Talk between Diet and the Oral Microbiome: Balance of Nutrition on Inflammation and Immune System's Response during Periodontitis . Nutrients 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Sedghi, L.M.; Bacino, M.; Kapila, Y.L. Periodontal Disease: The Good, The Bad, and The Unknown. Front Cell Infect Microbiol 2021, 11, 766944.
- Tan, K.H.; et al. Porphyromonas gingivalis and Treponema denticola exhibit metabolic symbioses . PLoS Pathog 2014, 10, e1003955. [Google Scholar] [CrossRef]
- Meuric, V.; et al. Treponema denticola improves adhesive capacities of Porphyromonas gingivalis . Mol Oral Microbiol 2013, 28, 40–53. [Google Scholar] [CrossRef]
- Nibali, L. Aggressive Periodontitis: microbes and host response, who to blame? Virulence 2015, 6, 223–228. [Google Scholar] [CrossRef]
- Kong, N.; et al. Production and analysis of high molecular weight genomic DNA for NGS pipelines using Agilent DNA extraction kit (p/n 200600) . Agilent Technologies Application Note. doi 2013, 10. [Google Scholar]
- Basbas, C.; et al. Unveiling the microbiome during post-partum uterine infection: a deep shotgun sequencing approach to characterize the dairy cow uterine microbiome . Anim Microbiome 2023, 5, 59. [Google Scholar] [CrossRef]
- Garzon, A.; et al. WGS of intrauterine E . coli from cows with early postpartum uterine infection reveals a non-uterine specific genotype and virulence factors. mBio 2024, 15, e0102724. [Google Scholar]
- Beck, K.L.; et al. Monitoring the microbiome for food safety and quality using deep shotgun sequencing . NPJ Sci Food 2021, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [PubMed]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. 2010; Available from: https://www.bioinformatics.babraham.ac.
- Lu, J.; et al. Bracken: estimating species abundance in metagenomics data . PeerJ Computer Science 2017, 3, e104. [Google Scholar] [CrossRef]
- Grabherr, M.G.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome . Nat Biotechnol 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database . Nucleic Acids Res 2023, 51(D1), D690–D699. [Google Scholar] [CrossRef]
- Yang, J.; et al. VFDB 2008 release: an enhanced web-based resource for comparative pathogenomics . Nucleic Acids Research 2008, 36 (suppl 1), D539–D542. [Google Scholar] [CrossRef]
- Szklarczyk, D.; et al. STRING v10: protein-protein interaction networks, integrated over the tree of life . Nucleic Acids Res 2015, 43(Database issue), D447–52. [Google Scholar] [CrossRef]
- Liu, B.; et al. Deep sequencing of the oral microbiome reveals signatures of periodontal disease . PLoS One 2012, 7, e37919. [Google Scholar] [CrossRef]
- Lenartova, M.; et al. The Oral Microbiome in Periodontal Health . Front Cell Infect Microbiol 2021, 11, 629723. [Google Scholar] [CrossRef]
- Matsha, T.E.; et al. Oral Microbiome Signatures in Diabetes Mellitus and Periodontal Disease . J Dent Res 2020, 99, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Munoz Navarro, C.; et al. Analysis of the Oral Microbiome in a Patient with Cardiofaciocutaneous Syndrome and Severe Periodontal Disease: Impact of Systemic Antibiotic Therapy . Antibiotics (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.E.S. , Feline oral inflammation: diagnosis and options for treatment. Companion Animal 2024, 29, 112–118. [Google Scholar] [CrossRef]
- Shaw, C.; et al. Feline Gingivostomatitis Results from Bacterial Species Switching in the Oral Microbiome . BMC Microbiome 2024.
- Antezack, A.; et al. New putative periodontopathogens and periodontal health-associated species: A systematic review and meta-analysis . J Periodontal Res 2023, 58, 893–906. [Google Scholar] [CrossRef]
- Siddiqui, R.; et al. The increasing importance of the oral microbiome in periodontal health and disease . Future Sci OA 2023, 9, FSO856. [Google Scholar] [CrossRef] [PubMed]
- Sudhakara, P.; et al. Oral Dysbiotic Communities and Their Implications in Systemic Diseases . Dent J (Basel) 2018, 6. [Google Scholar] [CrossRef]
- Mohanty, R.; et al. Red complex: Polymicrobial conglomerate in oral flora: A review . J Family Med Prim Care 2019, 8, 3480–3486. [Google Scholar]
- Yamaguchi-Kuroda, Y.; et al. Porphyromonas gingivalis diffusible signaling molecules enhance Fusobacterium nucleatum biofilm formation via gene expression modulation . J Oral Microbiol 2023, 15, 2165001. [Google Scholar] [CrossRef]
- Cools, F.; et al. Streptococcus pneumoniae galU gene mutation has a direct effect on biofilm growth, adherence and phagocytosis in vitro and pathogenicity in vivo . Pathog Dis 2018, 76. [Google Scholar] [CrossRef]
- Lasserre, J.F.; Brecx, M.C.; Toma, S. Oral Microbes, Biofilms and Their Role in Periodontal and Peri-Implant Diseases. Materials (Basel) 2018, 11. [Google Scholar] [CrossRef]
- Shi, M.; et al. The Subgingival Microbiome of Periodontal Pockets With Different Probing Depths in Chronic and Aggressive Periodontitis: A Pilot Study . Front Cell Infect Microbiol 2018, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Johnston, W.; et al. Mechanical biofilm disruption causes microbial and immunological shifts in periodontitis patients . Sci Rep 2021, 11, 9796. [Google Scholar] [CrossRef] [PubMed]
- Ingendoh-Tsakmakidis, A.; et al. Commensal and pathogenic biofilms differently modulate peri-implant oral mucosa in an organotypic model . Cell Microbiol 2019, 21, e13078. [Google Scholar] [CrossRef]
- Mark Welch, J.L.; et al. Biogeography of a human oral microbiome at the micron scale . Proceedings of the National Academy of Sciences 2016, 113, E791–E800. [Google Scholar] [CrossRef] [PubMed]
- Bacali, C.; et al. Oral Microbiome: Getting to Know and Befriend Neighbors, a Biological Approach . Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Aizawa, S.-I. and T. Minamino, Chapter 6 - Flagella, in Molecular Medical Microbiology (Third Edition), Y.-W. Tang; et al. Editors. 2024, Academic Press. p. 97-126.
- Scannapieco, F.A.; Kornman, K.S.; Coykendall, A.L. Observation of fimbriae and flagella in dispersed subgingival dental plaque and fresh bacterial isolates from periodontal disease. J Periodontal Res 1983, 18, 620–633.
- Goetting-Minesky, M.P.; Godovikova, V.; Fenno, J.C. Approaches to Understanding Mechanisms of Dentilisin Protease Complex Expression in Treponema denticola. Front Cell Infect Microbiol 2021, 11, 668287.
- Lux, R.; et al. Motility and chemotaxis in tissue penetration of oral epithelial cell layers by Treponema denticola . Infect Immun 2001, 69, 6276–6283. [Google Scholar] [CrossRef]
- Ruby, J.; et al. Activation of the Innate Immune System by Treponema denticola Periplasmic Flagella through Toll-Like Receptor 2 . Infect Immun 2018, 86. [Google Scholar] [CrossRef]
- Zhou, B.; Szymanski, C.M.; Baylink, A. Bacterial chemotaxis in human diseases. Trends Microbiol 2023, 31, 453–467.
Figure 1.
Schematic overview of the extraction and processing of three oral swabs.
Figure 2.
Species-level diversity increases from early aggressive periodontitis (AP_1 and AP_2) through severe dental disease (FCGS). (A) Venn diagram illustrating the unique number of species and number of shared species found in each sampling point. (B) Alpha diversity, as indicated by Shannon Diversity Index, at each sampling point and (C) beta diversity illustrated by Bray-Curtis.
Figure 2.
Species-level diversity increases from early aggressive periodontitis (AP_1 and AP_2) through severe dental disease (FCGS). (A) Venn diagram illustrating the unique number of species and number of shared species found in each sampling point. (B) Alpha diversity, as indicated by Shannon Diversity Index, at each sampling point and (C) beta diversity illustrated by Bray-Curtis.
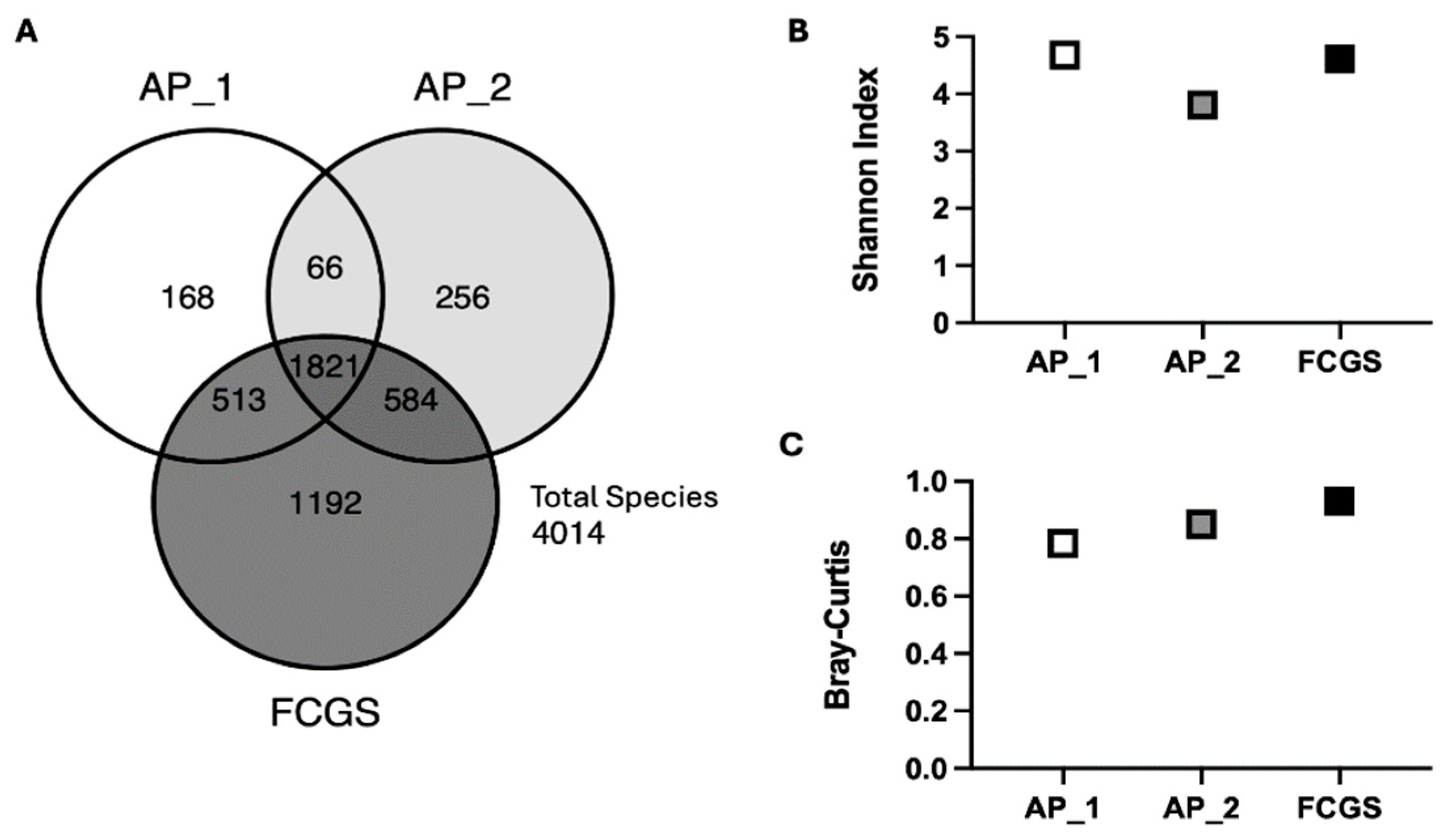
Figure 3.
The oral microbiome shows compositional changes across time and disease status at both the genus and species levels. (A) Alluvial plot illustrating changes in microbial proportions with the bars indicating genus-level composition and the alluvia in-between displaying the dynamic shifts between each sampling point. The ‘Mixed’ box contains any genus with less that 1% total proportion. (B) Correlation plot showing the overlapping and unique species by disease, with AP_1 and AP_2 aggregated into a single community profile. Each dot represents an individual species with the centerline indicating equal proportion in each group.
Figure 3.
The oral microbiome shows compositional changes across time and disease status at both the genus and species levels. (A) Alluvial plot illustrating changes in microbial proportions with the bars indicating genus-level composition and the alluvia in-between displaying the dynamic shifts between each sampling point. The ‘Mixed’ box contains any genus with less that 1% total proportion. (B) Correlation plot showing the overlapping and unique species by disease, with AP_1 and AP_2 aggregated into a single community profile. Each dot represents an individual species with the centerline indicating equal proportion in each group.
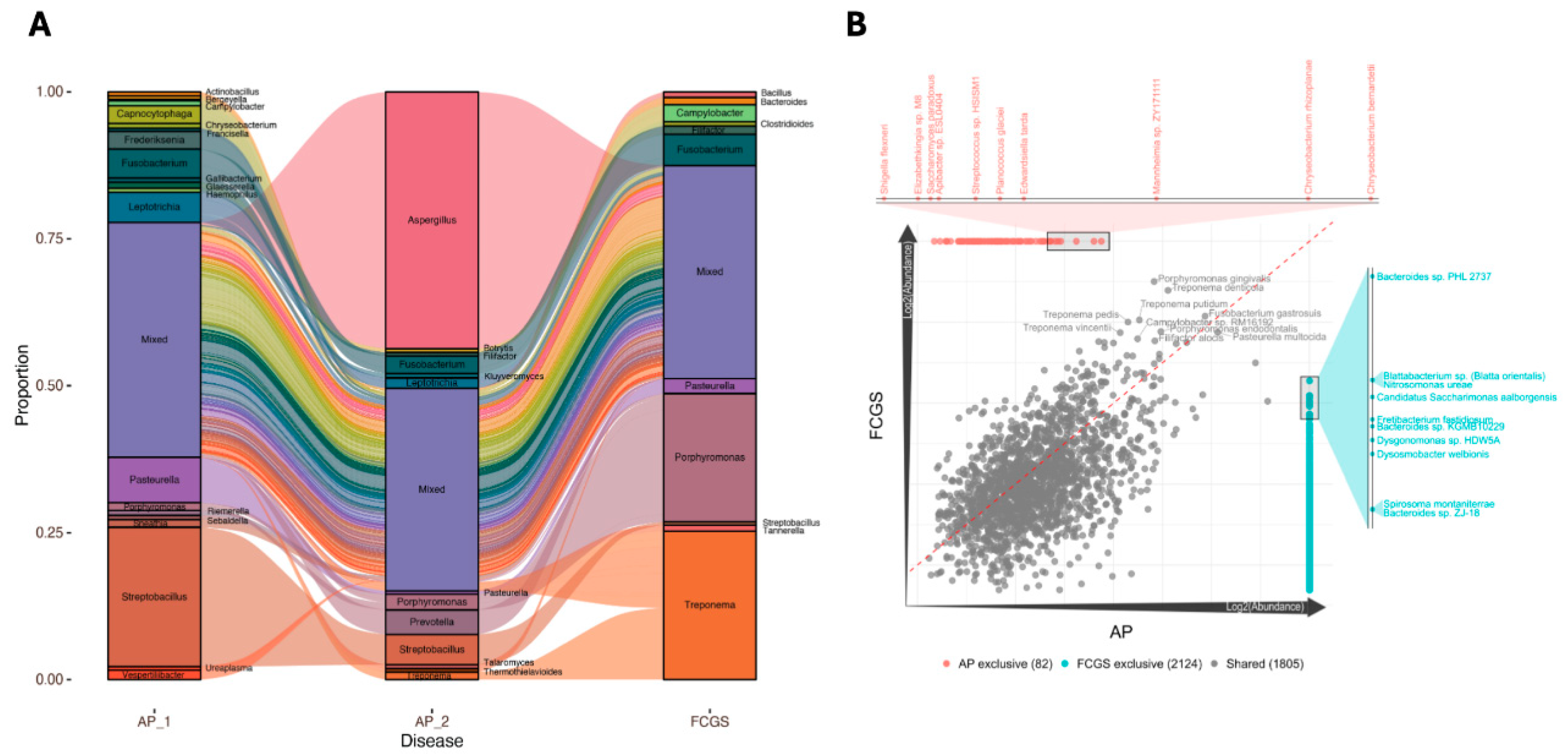
Figure 4.
Expression of AMR and virulence factors differs between early AP and FCGS. Assembled transcripts from each community were run through CARD and VFDB with an identity cut-off of 80%. Data are displayed as presence or absence for each gene hit in each sample.
Figure 4.
Expression of AMR and virulence factors differs between early AP and FCGS. Assembled transcripts from each community were run through CARD and VFDB with an identity cut-off of 80%. Data are displayed as presence or absence for each gene hit in each sample.

Figure 5.
STRING enrichment of virulence factors support functional differences between the microbiomes of AP_1 and FCGS. The STRING Database was used to perform a Local Network Cluster enrichment to determine functional profiles of the virulence factors found in each sample. One abundant organism in each sample was used as the genetic background for enrichment with (A) Pasteurella multocida used for AP_1 and (B) Treponema denticola for FCGS. AP_2 had no virulence factor hits and so was excluded from this analysis. Each circle labeled with the gene name represents a virulence factor with the connecting lines indicating gene cooccurrence, gene fusions, gene neighborhood or experimentally determined protein-protein interactions.
Figure 5.
STRING enrichment of virulence factors support functional differences between the microbiomes of AP_1 and FCGS. The STRING Database was used to perform a Local Network Cluster enrichment to determine functional profiles of the virulence factors found in each sample. One abundant organism in each sample was used as the genetic background for enrichment with (A) Pasteurella multocida used for AP_1 and (B) Treponema denticola for FCGS. AP_2 had no virulence factor hits and so was excluded from this analysis. Each circle labeled with the gene name represents a virulence factor with the connecting lines indicating gene cooccurrence, gene fusions, gene neighborhood or experimentally determined protein-protein interactions.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



