1. Introduction
The production of antigen specific antibodies represents the culmination of specific humoral immunity, with the isotype determined by the inciting antigen. IgG4 is an unusual isotype with weak Fc-dependent effector function and inability to activate complements. The function of IgG4 is unclear and is not usually involved in humoral immunity [
1]. IgE, involved in type 2 immunity and allergy, is also an uncommon immunoglobulin [
2]. Prominent increase in tissue plasma cells expressing these unusual or uncommon immunoglobulins G4 or E are present respectively in two uncommon etiologically unclear fibroinflammatory diseases IgG4RD and KD [
3,
4,
5]. It has been reported that these diseases are characterized by skewed or polarized immunoglobulin (Ig) isotype switching, with sustained Ig production of the specific isotypes [
6]. This may lead to increased IgG4- or IgE-positive plasma cells in the affected tissues, with or without elevated tissue and/or serum levels of the respective Ig isotype. The accompanying associated cellular, cytokine and chemotaxin interactions ensue in tissue inflammation and fibrosis [
6]. Both KD and IgG4RD often present as tumefactive lesions, thus confounding with true neoplasms. Increased awareness and recognition of these benign fibroinflammatory diseases is thus essential for distinction from true neoplasms for proper clinical management [
7].
There are, however, also diseases manifesting increased tissue and/or serum levels of IgG4 or IgE. Diseases with increased tissue IgG4-positive plasma cells, with/without blood IgG4 level include Rosai-Dorfman disease [
8], autoimmune atrophic gastritis [
9], sclerosing variant of mucoepidermoid carcinoma of salivary gland [
10], multicentric Castleman disease [
11,
12,
13], eosinophilic granulomatosis with polyangiitis [
14] and lymphocyte-variant hypereosinophilic syndrome [
15]. Diseases with increased tissue IgE-positive plasma cells with or without increased blood IgE include atopic asthma [
16] and atopic dermatitis [
17]. These well recognized entities will not be covered in this review on uncommon Ig-related fibroinflammatory diseases with unclear etiology with polarized Ig isotype switching. This review gears towards a probable relationship of IgG4RD with KD, based on their commonalities of polarized Ig isotype switching, overlapping clinical and histologic features. This review begins with a short recapitulation of B cell immunology, Ig heavy chain isotype switching, and characteristics of IgG4 and IgE.
2. B Cell Development
B cell development begins in the fetal liver and continues in the bone marrow where stromal cells provide cytokines and chemokines (CXCL12 and interleukin (IL)-7) for development into common lymphoid progenitor cells (CLP). CLP further develops into pro-B cells on stimulation which after sequential genetic rearrangement of immunoglobulin heavy and light chain genes, differentiate into IgM-expressing immature B cells. These immature B cells finally become IgD and IgM-coexpressing mature B cells (which occurs in the spleen in mice, but unclear in humans) and then migrate to peripheral lymphoid organs and enter into germinal centers (GC). In the GC dark zones, B cells undergo somatic hypermutation (SHM), isotype switching and clonal expansion. In the light zone, there is affinity maturation and clonal selection for B-cells with high affinity B-cell receptors (BCR). Antigen activation of B cells involves cellular interaction of antigen presenting cells (APC), B and T cells and requires two signals. Firstly, antigen coupling of BCR and secondly, activation in the T cell independent (TI) or T cell dependent (TD) manners. The TI path requires encountering antigens with repetitive epitopes (such as polysaccharide and glycolipids) leading to BCR crosslinking, and production of short-lived plasma cells and low affinity antibodies. The TD response requires antigen and T-follicular helper (Tfh) cell interaction. Both result in B cell differentiation to plasma cells which produce affinity antibodies and production of memory B cells [
18,
19].
3. Immunoglobulin (Ig) Class Switching Recombination (CSR)
CSR occurs in the GC of peripheral lymphoid organs. This involves switching of the heavy chain (HC) class from IgD or IgM of mature plasma cells to IgA, G or E, forming antibodies with different effector functions, as required for the immune response. CSR occurs by a common mechanism. There is transcription of the HC constant region DNA, induced by IL and tumor necrosis factor (TNF). This process could be TD or TI. In TD, CD40 ligand (CD40L) expressed by T follicular helper (Tfh) and T-helper 2 (Th2) cells ligates to CD40 on B cells leading to induction of HC constant region transcription and activation induced cytidine deaminase (AID), which is essential for DNA excision, ligation and recombination in CSR and SHM. The transcribed constant region contains switch regions which are excised by AID to allow the constant regions of the final Ig HC to remain and recombine. In TI, B lymphocyte stimulator (BLYs), a proliferation-inducing cytokine family (APRIL and BAFF) expressed by dendritic cells, and CD40L collaborate with IL to induce expression of AID followed by switch region excision and constant region recombination. The control of class switching is through exposure of activated B cells in a cytokine milieu produced by Tfh cells which directs the switching. In IgE class switching; Tfh13 cells, type 2 cytokines IL-4, IL-5, IL-9 and IL-13, IL-6 from activated monocytes and IL-7 from stromal cells are responsible. In IgG4 switching, Tfh4 cells and cytokines IL-4, IL-10 and IL-21 promote IgG4 production. The switching process involves the JAK/STAT, NFKB and STAT6 pathways. For IgE switching, there are alternative pathways which include corticosteroids and B cell activating EBV. There is also negative regulation of class switching through cytokines (IFN-α, IL-21), BCR, BCL6, transforming growth factor (TGFβ) [
20,
21,
22,
23,
24].
4. T Follicular Helper (Tfh) Cells
Tfh cells belong to one of the 5 types of CD4+ T helper (Th) cells, Th1, Th2, Th17, Treg (T-regulatory) and Tfh cells [
25]. Tfh cells were not well characterized until the recent decades. They are characterized by expression of CX chemokine receptor 5 (CXCR5), programed cell death protein 1 (PD-1), inducible T cell co-stimulator (ICOS), B-cell lymphoma 6 protein (Bcl-6) and IL-21. Tfh cells play a significant role in mounting humoral immunity by interacting with B cells, and is important in SHM and Ig CSR; which results in B cells with high affinity BCR and specific Ig isotype targeted at the inciting antigen with antigen specificity and required effector function. Tfh cell differentiation is a complex multistage process dependent on multiple intrinsic and extrinsic factors, with prominent participation of specific cytokines. Initiation of Tfh cell differentiation occurs when naive T cells are activated by an antigen-dependent process in an appropriate cytokine milieu. This is followed by Bcl-6, CXCR5, ICOS and PD-1 expressions. Bcl-6 is the key to Tfh cell development, which is necessary for repression of development to other Th cell subtypes. The extrinsic factors which may promote or inhibit Tfh cell differentiation are produced by other immune cells including follicular dendritic cells (DC), natural killer (NK) cells and other APC. The promoters include IL-6, IL-12, IL-21, IL-23, IL-27, activin A, TGFβ and type 1 interferon (IFN-α/β). The inhibitors are IL-2 and IL-7. Intrinsic factors include Maf, interferon regulatory factor 4 (IRF4), activation protein 1 (AP-1), basic leucine zipper transcription factor (Batf), STATs and E proteins. Tfh cell differentiation leads to cytokine skewed humoral immune responses, characterized by antigen specific and effector function defined by switched Ig isotype. According to the cytokine milieu, naive CD4+ T cells show skewed differentiation into Tfh1, Tfh2, Tfh17 and Tfr (T follicular regulatory) cells [
25,
26,
27,
28,
29,
30]. Tfh cells also exhibit plasticity [
26,
28] and are capable of transition to conventional T helper cells secreting the same cytokines.
5. IgE
IgE is a critical factor in allergic inflammation and type 2 immunity. It was described in 1967 [
2] and has four HC constant domains, different from 3 of IgG. It is mostly produced in the mucosa-associated lymphoid tissue [
31]. In GC of lymphoid tissue, class switch to IgE is facilitated in the TD mechanism by cytokine microenvironment of most importantly IL-13 with also IL-4, IL-21 and Tfh13 and Th2 cells. High affinity IgE is produced under influence of IL-13; and low affinity IgE produced under influence of IL-4 (as occurring in IgG4RD) [
6]. There are two types of Fc receptors for IgE: high affinity (mostly on mast cells and basophils) and low affinity (CD23) receptors, with the former having full binding avidity for IgE in the absence of antigen crosslinking of the receptor [
31]. This initiates a signal transduction cascade and downstream effects including mediator release causing tissue inflammation and damage [
31]. The high affinity Fc receptor on mast cells and basophils is a tetramer. An alternative dimeric high affinity Fc receptor occurs on eosinophils, DC, macrophages and Langerhan cells. The low affinity Fc receptor (CD23) is constitutively expressed on B cells, monocytes, eosinophils, DC, Langerhan cells and platelets. An important effector function of IgE is activation of basophil and mast cells leading to chemotaxis for eosinophil through various eotaxins, IL-4 and IL-13 [
32,
33]. On degranulation of eosinophils, various factors including major basic protein (MBP), eosinophil peroxidase (EPX), eosinophil cationic protein (ECP) and eosinophil derived neurotoxin (EDN) are released; contributing to tissue damage and inflammation [
34].
6. IgG4
IgG4 is an unusual immunoglobulin being the latest discovered type of IgG and occurs at the lowest concentration among IgGs in the human body [
35]. It has unique structural and functional properties and undergoes a continuous process of “half antibody exchange” or “Fab arm exchange” resulting in asymmetry and heterobivalency, though usually behaving as a monovalent antibody. The major structural difference between IgG1 and IgG4 lies in a few amino acids at the hinge regions of the CH2 and CH3 domains leading to flexible hinging and appearance of “half antibodies” and “ Fab arm exchange”. IgG4 consequently exhibits negligible activation of the classical complement pathway and much reduced binding to both low and high affinity Fc-gamma receptors [
35,
36,
37].
Control of IgG4 class switch in GC is mediated by a cytokine milieu produced by IL-10 expressing Tfh and Th2 cells, including IL-4, IL-10 and IL-21. IL-10 contributes by facilitating IL-4 mediated class switch to IgG4 rather than IgE [
6,
38]. The stimulating antigen that drives IgG4 production appears to be similar to that for IgE production, being allergic or atopic antigens, though findings to the contrary have been reported [
6]. The physiological role of IgG4 is enigmatic. Due to the weak affinity of its Fc fragment with inability to bind Fc receptors on effector cells, and low ability to activate complements. IgG4 blocks antibody-dependent cellular cytotoxicity, antibody-dependent cellular phagocytosis and complement dependent cytotoxicity. This bestows on IgG4 anti-inflammatory and immune evasive ability [
1].
7. Genetic Predisposition
Some work has been done on the genetics of IgG4RD. However, much less has been done or known about the genetics of KD [
39]. The following is a summary on the genetics of IgG4RD. These are genetic risk factors and not causative.
7.1. Heritability
Heritability may play a role in IgG4RD. There have been reports of the disease in 2 siblings with type 1 autoimmune pancreatitis [
40] and heritability of IgG4 levels in patients from families with autoimmune thyroiditis [
41].
7.2. HLA Genes
Various HLA genes have been reported to be associated with IgG4RD. The following are associated with disease susceptibility:
HLA-DRB1, HLA-DQB1, HLA I, HLA-B8, and the following with disease relapse:
HLA-A, HLA-C, HLA-DQB1 [
42,
43].
7.3. Non-HLA Genes
In IgG4RD, non-HLA genes have also been reported to be associated with disease susceptibility in IgG4RD, including
P2RX3, TOP1, PRSS1, SPINK1, FCRL3, FCGR, CACNA1C, CFTR, CTLA4, CXCR3, KCNA3, MLL3, TNFα
, SPINK1; with disease relapse
CACNA1C; with IgG4 related pancreatitis
CTLA4, KCNA3, FCRL3, PRSS1, with IgG4-related chronic aortitis FCGR; and with other extrapancreatic lesions
TNFα
, CXCR3, MLL3 [
42,
43].
8. Pathogenesis
The common pathogenetic mechanism of IgG4RD and KD is polarized immune stimulation, cytokine-skewed Tfh cell activation and polarized Ig isotype switching; with subsequent accompanying cellular, cytokine and chemotaxin interactions leading to tissue inflammation and fibrosis, with or without elevated plasma Ig isotype levels.
8.1. KD
On stimulation by polarized immune conditions, including unknown aberrant immune or allergic stimulation, cellular interactions involving APC, Th2 with other type 2 immune cells and B cells occur; causing a skewed cytokine milieu for development of disease specific Tfh cells. IL-13 expressing Tfh cells develop and are activated, ensuing in secretion of IL-4, IL-5 and IL-13. Polarized Ig isotype switching to IgE occurs, resulting in IgE-positive plasma cells. The cytokines and IgE rich miulieu lead to inflammation with many IgE positive plasma cells. IgE activates basophils and mast cells , releasing eotaxins for eosinophils [
32,
33]. Profibrogenic mediators including galectin-10, osteopontin are released from eosinophils. Other profibrogenic factors released from activated mast cells in the mediator milieu include IL-4, IL-13, TGFβ1, tryptase and chymase. Consequently, the characteristic histological features of ectopic GC, lymphoplasmacytic inflammation, IgE positive plasma cells, tissue eosinophilia with or without eosinophilic abscess; and fibrosis are developed [
3,
6]. Fibrosis in KD is mostly paucicellular or collagenous and patternless [
3]. KD is the sequel of type 2 immunity involving type 2 immune cells, IgE activation of basophils and mast cells, and a cytokine and cytotoxic protein milieu including IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12, IL-13, IFNα, TNF, TGFβ1, macrophage migration inhibiting factor (MIF), eosinophil released MBP1 and EPX [
3,
32,
33,
34,
44,
45,
46,
47,
48,
49].
8.2. IgG4RD
Under polarized immune conditions such as autoimmunity or unknown aberrant immune stimulation; complex interaction among B cells, T cells and APC leads to a cytokine milieu rich in IL-4, IL-10 and IL-21 which favors polarized IL-10 expressing Tfh cell development. The latter directs polarized Ig isotype switching to IgG4, resulting in IgG4-positive plasma cell development. These IL-10 expressing Tfh cells also show Treg cell and cytotoxic cell functions. There is recent evidence that the T cell repertoire includes CD4+ or CD8+ cytotoxic T lymphocytes (CTL) which may be autoreactive [
50]. The CTL leads to cellular apoptosis of the affected tissues, followed by tissue remodeling and fibrosis [
6]. Further, IgG4 antibody may promote pathogenesis independent of Fc signaling or complement activation [
51,
52]. In the fully developed lesions, the characteristic fibroinflammatory pathologic picture of ectopic GC, lymphoplasmacytic infiltration, abundance of IgG4-positive plasma cells and fibrosis is produced [
3,
6] Fibrosis in IgG4RD is caused by remodeling of CTL induced cellular apoptosis, as distinct from allergic fibrosis in KD [
6]. The storiform fibrosis pattern in IgG4RD is different from paucicellular patternless fibrosis in KD [
3].
9. Etiology
The etiology of IgG4RD and KD is unclear. Genetic factors may play a role but not solely accountable. The following are some possible triggering factors.
9.1. Atopic and Allergic Antigenic Stimulation
It has been observed that allergic and atopic challenges occur in both polarized IgE and IgG4 isotype switches involving type 2 immune reactions with Th2 cells and IL-4, IL-5, IL-13 [
1,
20,
53,
54,
55].
Allergy has been hypothesized to be the etiologic event in both IgG4RD and KD [
6,
49,
56,
57,
58]. While it is true for at least a proportion of KD, this may not be true for IgG4RD, as there is little evidence of Th2 cells accumulation in tissues of IgG4RFID after exclusion of cases with coexisting allergy [
6,
49,
56,
57,
58]. Allergic antigenic stimulation, however, has been demonstrated in KD where type 2 immune cells, Tfh13 cells, eosinophils, basophils, mast cells and IgE-positive B cells and mast cells are demonstrated in lesional tissues with frequent increase in plasma IgE levels [
6]. Atophic/allergic stimulation is therefore likely to be etiologically responsible for at least a proportion of KD.
9.2. Autoimmunity
There is evidence that autoimmunity may be the etiologic event in IgG4RD. Autoantibodies against self-antigens including ubiquitin ligase-associated protein [
59], carbonic anhydrase IV [
60], annexin A11 [
61], amylase α-2A [
62], galectin-3 [
63], pancreatic secretory trypsin inhibitor and trypsinogen [
50,
64,
65] have been reported. These mostly represent IgG4 isotype antibodies which are also detected in systemic lupus erythematosus (SLE) and rheumatoid arthritis. Passive transfer of purified IgG and IgG4 antibodies from IgG4RD patients induced manifestations of IgG4RD in mice. Autoreactive CD4+ and CD8+ cytotoxic T cells leading to apoptosis have also been demonstrated [
49]. Recently, an IgG4 anticytokine autoantibody against the IL-1 receptor antagonist (IL-1RA) has been identified in IgG4RD patients [
52]. This antibody promotes expression of IL-1, proinflammatory and profibrotic cytokines in fibroblast and epithelial cell lines in vitro, and is also detected in lesional tissues in IgG4RD patients. Furthermore, there is recent evidence that IgG4 directly promotes disease in an Fc-independent manner in pemphigus vulgaris and myasthenia gravis through binding to skin and muscle-specific autoantigens [
52]. One other recent finding is the possible role of IL-35 which stimulates Th9 cells to produce IL-9. IL-9 in turn stimulates IgG production with IgG4 predominance [
66]. Further, IgG4RD patients with a family history of autoimmune diseases show younger IgG4 RD disease onset and exhibit higher frequency of antinuclear antibodies [
67]. Autoimmunity is therefore responsible for at least a proportion of IgG4RFID.
9.3. Other Causes of Aberrant Antigenic Stimulation
Infections including Gram-positive bacteria (such as
Staphylococcus aureus) and
Mycobacterium tuberculosis infections have been reported to be related to IgG4RD [
43]. Environmental factors related to blue collar occupations, such as solvents and industrial gasses, may also play a possible role in IgGRD [
43]. Unknown causes of aberrant antigenic stimulation, however, may be responsible for most cases of IgG4RD and KD.
10. Clinicopathologic Features of Kimura Disease
KD was first described in 1937 by the Chinese researcher Jin [
68] and 11 years later by Kimura [
69], whose name became entrenched in the literature as the disease eponym. KD affects mostly young to middle aged Asian subjects, though uncommonly non-Asians are also involved [
4,
70,
71,
72]. KD produces tumor-like lesions mostly of superficial locations, namely subcutaneous masses, enlarged salivary and lacrimal glands, and regional lymph nodes [
4,
68,
69,
70,
71,
72,
73]. Histologically, KD is characterized by florid lymphoid follicular hyperplasia (ectopic lymphoid follicles), lymphoplasmacytic and eosinophilic infiltration with frequent eosinophilic abscesses and profuse stromal fibrosis [
3,
4,
68,
69,
70,
71,
72,
73]. In the lymph node, KD shows in addition vascularized GC, GC proteinaceous deposits and necrosis, polykaryocytes and GC reticular IgE deposits [
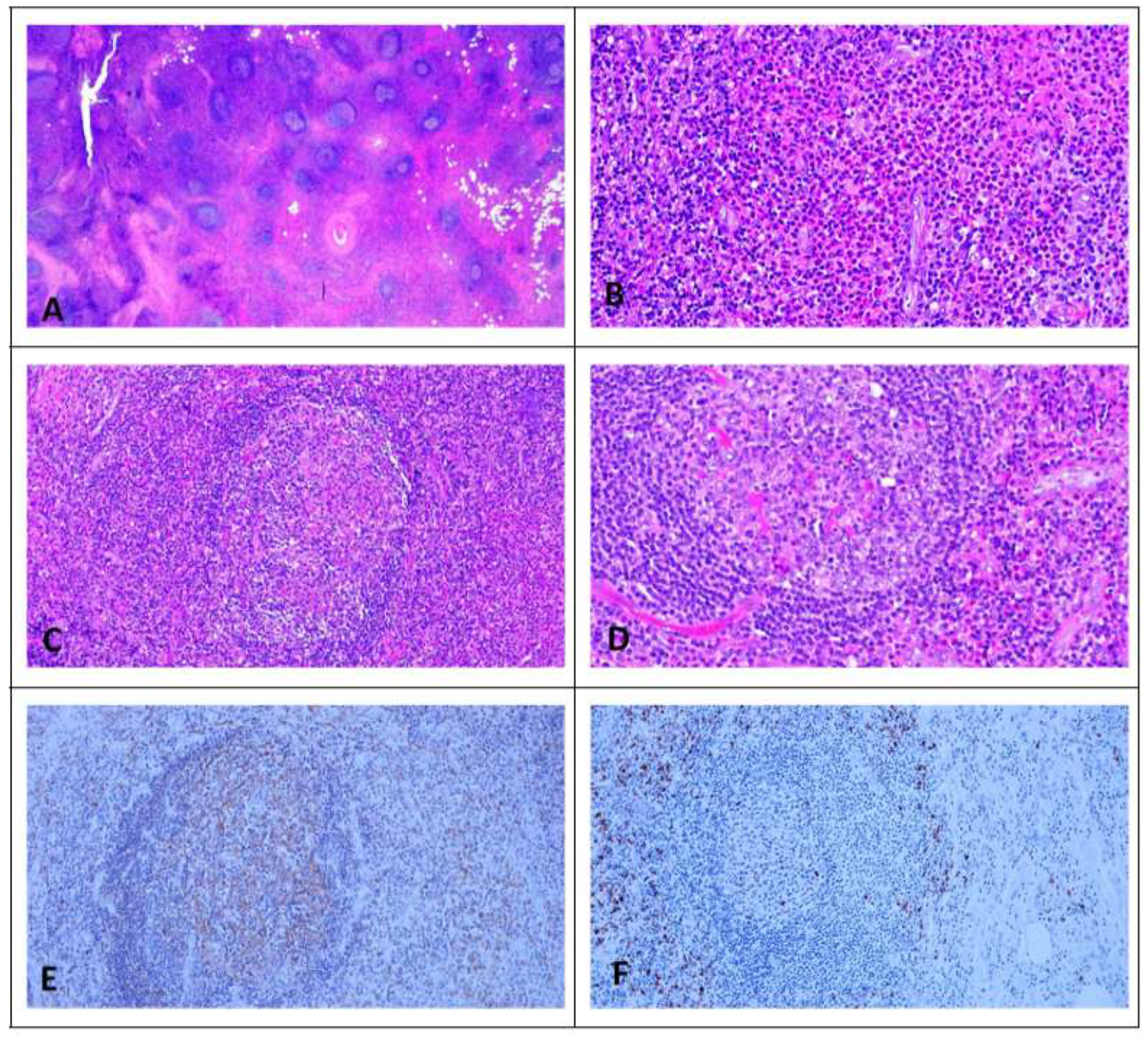
74] (
Figure 1). KD involving lymph nodes of multiple regions [
75], multiple skin sites [
76] and renal involvement [
77] have also been reported, though rare. There is a high incidence of peripheral blood eosinophilia and elevated serum IgE. Serum IgG4 may also be raised [
78]. Treatment is by surgical excision, local or systemic corticosteroids, local radiotherapy or immunosuppressant agents [
79].
11. Clinicopathologic Features of IgG4-Related Disease
IgG4RFID, masquerading as eosinophilic angiocentric fibrosis, inflammatory pseudotumor, or the eponymic diseases Kuttner’s tumor, Mikulicz’s syndrome, Osmond’s disease and Riedel’s thyroiditis in the old literature [
5]; was initially introduced in the early 2000 as sclerosing pancreatitis or autoimmune pancreatitis associated with raised serum IgG4 or tissue IgG4-positive plasma cells [
80,
81]. With increased recognition, there is a proliferation of reports on IgG4RD [
7], culminating in the consensus statement on the pathology of IgG4RD in 2012 [
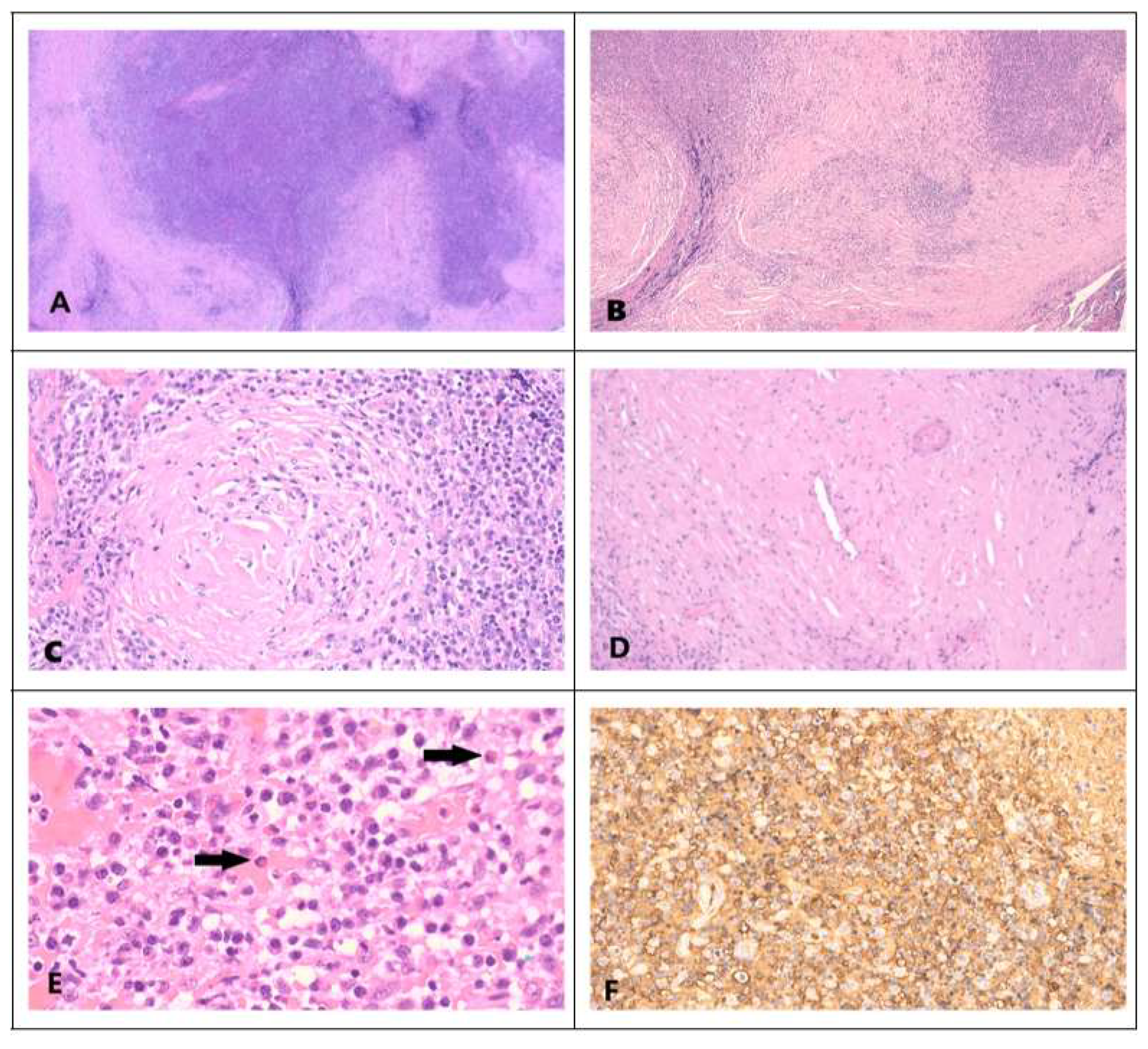
82]. The latter details 3 possible characteristic histological features of IgG4RD. (1) Dense lymphoplasmacytic infiltrate, (2) fibrosis, often storiform in character, and (3) obliterative phlebitis. With regard to IgG4+ plasma cell count, it ranges from 10 to 200 cells/HPF; depending on the organ involved. An elevated IgG4+/IgG+ cell ratio of >40% (>50% for aorta specimens) is also necessary (
Figure 2). Accordingly, there are 3 diagnostic categories. (1) histologically highly suggestive of IgG4RD, with 2 of the 3 characteristic histologic features and the required IgG4+ plasma cell count. (2) probable histologic features of IgG4RD with only one characteristic histological feature and the required IgG4+ plasma cell count. (3) Insufficient histopathologic evidence of IgG4RD, when features of neither category (1) nor (2) are met.
IgG4RD involves multiple organs and tissues, including superficial and deep seated; in contrast to KD where superficial sites are primarily involved. The myriad involved sites include pancreas, hepatobiliary system, liver, retroperitoneum, mesentery, mediastinum, aorta, lung, pleura, kidney and urinary tract, central nervous system, thyroid, prostate, seminal vesicles, maxillary sinus, nasal septum, paranasal sinus and pericardium. The involved superficial sites overlap with those of KD including orbit, lacrimal gland, salivary gland, skin and breast [
7,
83]. The disease is tumefactive and often confused with true tumors. In the pancreas, pancreatectomy and Whipple’s operation is not infrequently performed for tumor-like lesions caused by IgG4RD [
84,
85]. However, true malignancies including lymphoma, pancreatic ductal adenocarcinoma, salivary duct carcinoma, pulmonary adenocarcinoma, gastro intestinal clear cell sarcoma have been described in the backdrop of IgG4RD [
7]. It is therefore important to thoroughly examine IgG4RD involved tissues to exclude malignancies in suspicious cases. Treatment of IgG4RD does not require radical surgery and steroids are usually effective. Promising results have also been reported with anti-CD20 (rituximab) treatment [
43].
12. KDVs IgG4RD
Increased awareness and proliferation of studies on IgG4RD and KD uncovered significant differences, similarities and overlap in clinical, histological, immunological and hematological features of the 2 diseases [
3,
86,
87].
12.1. Similarities and Overlapping Features
There are many common features, including those considered to be characteristic of either IgG4RD or KD. In KD, storiform fibrosis, obliterative phlebitis, non-obliterative phlebitis, tissue IgG4-positive plasma cells >50/HPF and tissue IgG4/IgG-positive plasma cell ratio >40%, which are characteristic of IgG4RD, can occur. In IgG4RD, tissue eosinophilia, eosinophilic abscess germinal centers eosinophilic deposits, vascularized germinal centers, tissue IgE reticular staining in germinal center, and tissue IgE-positive plasma cells >10/HPF, which are characteristic of KD also can occur in IgG4RD. Despite being common overlapping features, there are statistically significant differences in manifestations of these features between IgG4RD and KD [
3]. In daily practice, however, these overlapping though statistically different features may cause confusion and misdiagnosis in individual cases. There have, therefore, been proposals on criteria and features for inclusion and exclusion of IgG4RD [
82,
88,
89,
90,
91]. According to the 2019 American College of Rheumatology/European League against Rheumatism classification criteria, peripheral blood eosinophilia is a strong exclusion criterion and storiform fibrosis is a robust inclusion criteria for IgG4RD [
88]. The presence of common overlapping features between IgG4RD and KD is not surprising, considering that both are pathogenetically related to polarized Ig isotype class switching and that IL-4 in both conditions can induce both IgG4 and IgE isotype switching [
6].
12.2. Differences
12.2.1. Epidemiology
12.2.1.1. Race
Both diseases are more common in Asians [
3,
43].
12.2.1.2. Patient age
KD affects younger (mean 30 years) while IgG4RD affects older (mean 59 years) subjects [
3,
43].
12.2.1.3. Patient sex
Though males are more frequently affected in both IgG4RD and KD, male sex dominance is more prevalent in KD [
3]. In IgG4-related sialadenitis and dacryoadenitis,the female sex has been reported to be more prevalent [
43]. A mild female dominance in IgG4RD has also been reported in a study based on the US population [
92].
12.2.2. Anatomical Sites Involved
There is a more significant difference in involved anatomical sites between IgG4RD and KD. IgG4RD is a multisystem disease with more frequent multi-organ involvement, including deep seated organs. This compares with KD which predominantly affects superficial tissues in the head and neck region [
3,
7]. This may be related to frequent increase in circulating Tfh2 (cTfh2) cells in IgG4RD [
93]. cTfh cells may home to GC of secondary and tertiary lymphoid organs, thus facilitating IgG4 isotype switching and development of IgG4RD in deep seated organs. Increase in cTfh cells has not been reported in KD, though further studies are required to validate this hypothesis. Multiple anatomic sites involvement in IgG4RD may be related to the possible role of autoimmunity in this disease, simulating other autoimmune diseases.
12.2.3. Storiform Fibrosis
This is significantly different between IgG4RD and KD. It frequently occurs in IgG4RFID and is among the important diagnostic criteria of the disease [
3,
77,
88]. However, it may infrequently occur in KD where fibrosis is mostly collagenous and patternless [
3].
12.2.4. Tissue Eosinophilia, Eosinophilic Abscess, GC Proteinaceous/IgE Deposits and GC Vascularization
These histological features are diagnostically distinctive and frequent in KD, though they may occur with lower frequency in IgG4RD [
3,
74].
13. Conclusions
Recent advances in Immunology have unraveled the complex interaction of immune cells, cytokines, chemotaxins and cell mediators in B cell development and differentiation. This unravels the mechanism of producing antigen specific Ig with desired effector functions by isotype switching. The involvement of Tfh cells has also been more recently unfolded, followed by immense interest and work on their roles in humoral immunity in the last decade. There are two conditions of unclear etiology characterized by polarized Ig isotype switching to IgG4 and IgE, namely IgG4RD and KD respectively. They exhibit a similar pathogenetic mechanism of cytokine-skewed Tfh cell directed polarized Ig isotype switching. The resulting cytokine, chemotaxin and cellular milieu cause inflammation and fibrosis characteristic of these diseases. Fibrosis in KD is of allergic type, mediated by type 2 immune cytokines and mediators from eosinophils; while that of IgG4RD is more the sequel of remodeling of cellular apoptosis caused by cytotoxic cells. Though the etiology of both diseases is mostly nebulous, there is evidence of allergic stimulation being responsible in at least some KD and autoimmunity in some IgG4RD. These triggering events may be operative in the backdrop of genetic factors. Despite significant differences with regard to anatomic distribution of the disease, pattern of fibrosis and degree of eosinophil infiltration, many overlapping features occur in both conditions. This necessitates strict adherence to defined diagnostic criteria for accurate distinction. Accurate recognition of these frequently tumefactive fibroinflammatory diseases is essential for differential diagnosis from true neoplasms and avoidance of over treatment. As both conditions show similar pathogenesis through isotype switching and histological features of inflammation and fibrosis, it is proposed that they are both regarded as Ig-related fibroinflammatory diseases, thus connecting the ancient KD with the contemporary IgG4RD.
Authorship
The author conceived and conceptualized the paper, did literature research, curated information and data, wrote the initial draft, finalized and approved the submitted manuscript.
Financial support
No financial support was obtained.
Patient consent
Not applicable.
Ethics approval
Not applicable, as this is a review article.
Data availability statement
Not relevant.
Acknowledgments
The author thanked Ms Yvonne Chan for assistance in preparing the manuscript.
Conflicts of Interest
The author declares no competing interests.
Abbreviations
| AID: Activation induced cytidine deaminase |
IFN: Interferon |
| AP-1 :activation protein-1 |
Ig: Immunoglobulin |
| Batf: Basic leucine zipper transcription factor |
IgG4RD: Immunoglobulin G4 related disease |
| Bcl6: B-cell lymphoma 6 protein |
IL: Interleukin |
| BCR: B-cell receptor |
IL-1RA: IL-1 receptor antagonist |
| BLys: B lymphocyte stimulator |
IRF4: Interferon regulatory factor 4 |
| CD40L: CD40 ligand |
KD: Kimura disease |
| CLP: Common lymphoid progenitor cells |
LC: Light chain |
| CSR: Class switching recombination |
MBP: Major basic protein |
| CTL: Cytotoxic T lymphocytes |
PD-1: Programed cell death protein-1 |
| CXCR5: CX chemokine receptor 5 |
SHM: Somatic hypermutation |
| EDN: Eosinophil derived neurotoxin |
TD:T-cell dependent |
| EPX: Eosinophil peroxidase |
Tfh: T follicular helper |
| Fc: Fragment crystallizable |
TGFB1: Transforming growth factor B1 |
| GC: Germinal center |
Th2: T helper 2 |
| HC: Heavy chain |
TI: T-cell independent |
| ICOS: Inducible T-cell costimulator |
TNF: Tumor necrosis factor |
References
- Cresioli S, Correa I, Kargiannis P, Davies AM, Sutton BJ, Nestle FO et al. IgG4 characteristics and functions in cancer immunity. Curr Allergy Asthma Rep 2016;16:7. [CrossRef]
- Ishizaka K, Ishizaka T. Identification of gamma E-antibodies as a carrier of reaginic activity. J Immunol 1967;99:1187-98.
- Wang X, Ng CS, Yin W. A comparative study of Kimura’s disease and IgG4-related disease: similarities, differences and overlapping features. Histopathology 2021;79:801-9.
- Chen H, Thompson LDR, Auguilera NSI, Abbondanzo SLl. Kimura’s disease. A clinicopathologic study of 21 cases. Am J Surg Pathol 2004;28:505-13. [CrossRef]
- Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Ann Rev Pathol 2014;9:315-17.
- Munemura R, Maehara T, Murakami Y, KogaR, Aoyagi R, Kaneko Net al. Distinct disease-specific Tfh cell populations in 2 different fibrotic diseases: IgG4-related disease and Kimura disease. J Allergy Clin Immunol 2022;150:440-55. [CrossRef]
- Cheuk W, Chan JKC. IgG4-related sclerosing disease. A critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010;17:303-32.
- Kuo TT, Chen TC, Lee LY, Lu P.. IgG4-positive plasma cells in cutaneous Rosai-Dorfman disease: an additional immunohistochemical features and possible relationship to IgG4-related sclerosing disease. J Cutan Pathol 2009;36:1069-73. [CrossRef]
- Bedeir AS, Lash RH, Lash JG, Ray MB. Significant increase in IgG4+ plasma cells in gastric biopsy specimens from patients with pernicious anemia. J Clin Pathol 2010;63:999-1001. [CrossRef]
- Tian W, Yakirevich EMA, MatosoA, Gnepp D. IgG4 (+) plasma cells in sclerosing variant of mucoepidermoid carcinoma. A J Surg Pathol Pathol 2012;36:973-79. [CrossRef]
- Nishikori A, Nishimura MF, Nishimura Y, Notohara K, Satou A, Moriyama M et al. Investigation of IgG4-positive cells in idiopathic multicentric Castleman disease and validation of the 2020 exclusion criteria for IgG4-related disease. Pathol Int 2022;72:43-52. [CrossRef]
- Sasaki T, Akiyama M, Kaneoko Y, Mori T, Yasuoka H, Suzuki K et. Distinct features distinguishing IgG4-rated disease from multicentric Castleman’s disease. RMD Open 2017;3:e000432. [CrossRef]
- Sasaki T, Akiyama M, Kanedo Y, Takeuchi T. Immunoglobulin G4-related disease and idiopathic multicentric Castleman’s disease: confusable immune-related disorders. Rheumatology 2022;61:490-501.
- Kubo S, Kanada R, Nawata A, Miyazaki Y,Kawabe A, Hanami K et al. Eosinophilic granulomatosis with polyangiitis exhibits T cell activation and IgG4 immune response in the tissue, comparison with IgG4-related disease. RMD Open 2022;8:e002086. [CrossRef]
- Carruthers MN, Park S, Slack GW, Dalal BI, Skinnider BF, Schaeffer DF et al. IgG4-related disease and lymphocyte variant hypereosinophilic syndrome: a comparative case series. Eur J Haematol 2017;98:378-87. [CrossRef]
- Gong F, Zheng T, Zhou P. T follicular helper cell subsets and the associated cytokine IL-21 in the pathogenesis and therapy of asthma. Front Immunol 2019;10:2918.
- Boothe DW, Tarbox JA, Tarbox MB. Atopic dermatitis: pathophysiology. In: Fortson E, Feldman S, Strowd L (eds) Management of atopic dermatitis. Advances in experimental medicine and biology, vol 1027. Springer, Cham. [CrossRef]
- Tsui DY, Hung KH, Chang CW, Liu K. Regulatory mechanisms of B cell responses and the implication in B cell-related disease. J Biomed Sci 2019;26:64.
- Chi X, Li Y, Qiu X. V(D) J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunol 2020;160:233-47.
- Bacharier LB, Jabara H, Geha RS. Molecular Mechanisms of Immunoglobulin E regulation. Arch Allergy Immunol 1998;115:257-69.
- Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Ann Rev Immunol 2000;26:261-92.
- Duarte J. Functional switching. Nat Immunol 2016;17:S12.
- Oudinet C, Braikia F, Dauba A, Khamlichi AA. Mechanism and regulation of class switch recombination by IgH transcriptional control elements. Adv Immunol 2020;147:89-137.
- Dauba A, Khamlichi AA. Long-range control of class switch recombination by transcriptional regulatory elements. Front Immunol 2021;12:738216.
- Nurieva RI, Chung Y. Understanding the development and function of T follicular helper cells. Cell Molecular Immunol 2010;7:190-7.
- Olatunde AC, Hale JS, Lamb TJ. Cytokine-skewed Tfh cells: functional consequences for B cell help. Trends Immunol 2021;42:536-50.
- Crotty S. T follicular helper cell differentiation, function and roles in disease. Immunity 2014;41:529-42.
- Read KA, Powell MD, Oestreich KJ. T follicular helper cell programming by cytokine-mediated events. Immunology 2016;149:253-61.
- Krishnaswamy JK, Alsen S, Yrlid U, Eeisenbarth SC, Williams A. Determination of T follicular helper cell fate by dendritic cells. Front Immunol 2018;9:2169.
- Crotty S. T follicular helper cell biology: a decade of discovery and diseases Immunity 2019;50:1132-48.
- Rosenwasser LJ. Mechanisms of IgE inflammation. Curr Allergy Asthma Rep 2011;11:178-83. [CrossRef]
- Iype J, Fux M. Basophils orchestrating eosinophils’ chemotaxis and function in allergic inflammation. Cells 2021;10:895.
- Kampen GT, Stafford S, Adachi T, Jinquan T, Quan S, Grant JA et al. Eotaxin induced degranulation and chemotaxis of eosinophils through the activation of ERK2 and P38 mitogen-activated protein kinases. Blood 2000;95:1911-17. [CrossRef]
- Fettrelet T, Gigon L, Karaulov A, Yousefi S, Simon H. The enigma of eosinophil degranulation. Int J Mol Sci 2021;22:7091. [CrossRef]
- Nirula A, Glaser SM, Kalled SL, Taylor FR. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol 2011;23:119-24. Erratum in: Curr Opin Rheumatol 2011;23:227. Taylor, Frederick R [corrected to Taylor, Frederick RL PMID21124094]. [CrossRef]
- Aalberse RC, Stapel SO, Schumann J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy 2009;39:469-77. Epub 2009 Feb 13. PMID 19222496. [CrossRef]
- Aalberse R. The role of IgG antibodies in allergy and immunotherapy. Allergy 2011;66(supp 95):28-30.
- Jeannin P, Lecoanet S, Delneste Y, Gauchat JF, Bonnefoy JY. IgE versus IgG4 production can be differentially regulated by IL-10. J Immunol 1999;160:3555-61.
- Wu X, Wang A, Zhang S, Wang X, Guo P, Zhu W, Jiao Y, Zhou J, Zhang W, Peng L, Duan M, Fei Y. Multiomic landscape of immune pathogenesis in Kimura’s disease. iSci 2023;26:106559. [CrossRef]
- Watanabe T, Maruyama M, Ito T, Kanai K, Oguchi T, Muraki T et al. Two siblings with type 1 autoimmune pancreatitis. Intern Med 2013;52:895-99. [CrossRef]
- Outschooru IM, Talor MV, Burek CL, Hoffman WH, Rose NR. Heritability analysis of IgG4 antibodies in autoimmune thyroid disease. Autoimmunity 2014;47:320-26.
- Ishikawa Y, Terao C. Genetic analysis of IgG4-related disease. Mod Rheumatol 2020;30:17-23. [CrossRef]
- Floreani A, Okazaki K, Uchida K,Gershwin ME. IgG4-related disease:changing epidemiology and new thoughts on a multisystem disease. J Transl Autoimmun 2021;4:100074. [CrossRef]
- Aoki A, Hirahara K, Kiuchi M, Nakayama T. Eosinophils: cells known for over 140 years with broad and new functions. Allergol Int 2021;70:3-8.
- Kita H, Gleich G. Eosinophils and IgE receptors: a continuing controversy. Blood 1997;89:3497-3501.
- Pritam P, Manna S, Sahu A, Swain SS, Ramchandani S, Bissoyi S et al. Eosinophils: a central player in modulating pathological complexity in asthma. Allergol Immunopathol 2021;49:191-207. [CrossRef]
- Simon H-U. The eosinophil and its role in physiology and disease: news and views. Sem Immunopathol 2021;43:291-3. [CrossRef]
- Bozza MT, Lintomen L, Kitoko JZ, Paiva CN, Olsen PC. The role of MIF on eosinophil biology and eosinophilic inflammation. Clin Rew Allergy Immunol 2020;58:15-24.
- Choi JW, Lee MH, Fujii T. Relationship between neutrophil gelatinase-associated lipocalin, eosinophil cationic protein, cytokines, and atopic sensitization in patients with allergic disease. Bio Med Ref Int 2022;6564706.
- Mattoo H, Mahajan VS, Machara T, Deshpande V, Della Torre E, Wallace ZS et al. Clonal expansion of CD4+ cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol 2016;138:825-38. [CrossRef]
- Shiokawa M, Kodama Y, Kuriyama K, Yoshimura K, Tomono Y, Uza N et al. Pathogenicity of IgG in patients with IgG4-related disease. Gut 2016;65:1322-32. [CrossRef]
- Jarrell JA, Baker MC, Perugino CA, Liu H, Bloom MS, Maehara T et al. Neutralizing anti-IL-1 receptor antagonist autoantibodies induce inflammatory and fibrotic mediators in IgG4-related disease. J Allergy Clin Immunol 2022;149:358-68. [CrossRef]
- Geha RS, Jabara HH, Brodeur SR. The regulation of immunoglobulin E class-switch recombination. Nature Rev Immunol 2003;3:721-32.
- Coffman RL, Lebman DA, Rothman P. Mechanism and regulation of immunoglobulin isotype switching. In: Advances in Immunology, Dixon FJ (eds) Academic Press 1993,54:229-70.
- Yanagihara Y. Regulatory mechanism of immunoglobulin E synthesis by human B cells. Clin Exp Allergy Rew 2006:6:101-5.
- Maehara T, Moriyama M, Nakashima H, Miyake K, Hayashida J, Tanaka A et al. Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Ann Rheum Dis 2012:71:2011-19. [CrossRef]
- Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia and IgE elevation in IgG4-related disease. Allergy 2014;69:269-72.
- Mattoo H, Della Torre E, Mahajan VS, Stone JH. Circulating Th2 memory cells in IgG4-related disease are restricted to a defined subset of subjects with atopy. Allergy 2014;69:379-402.
- Frulloni L, Lunardi C, Simmone R, Dolcino M, Scattolini C, Falconi M et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Eng J Med 2009;36:2135-42. [CrossRef]
- Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut 2005;54:274-81.
- Hubers LM, Vos H, Schuurman AR, Erken R, Oude Elferink RP, Burgering B et al. Annexin A11 is targeted by IgG4 and IgG1antibodies in IgG4-related disease. Gut 2018;67:728-35. [CrossRef]
- Endo T, Takizawa S, Tanaka S, Takahashi M, Fujii H, Kamisawa T et al. Amylase alpha-2A autoantibodies: novel marker of autoimmune pancreatitis and fulminant type 1 diabetes. Diabetes 2009;58:732-7. [CrossRef]
- Perugino CA, Al Salem SB, Mattoo H, Della-Torre E, Mahajan V, Ganesh G et al. Identification of galectin-3 as an autoantigen in patients with IgG4-related disease. J Allergy Clin Immunol 2019;143:736-45. [CrossRef]
- Asada M, Nishio A, Uchida K, Kido M, Ueno S, Uza N et al. Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas 2006;33:20-6. [CrossRef]
- Lohr JM, Faissner R, Koczan D, Ofsky R, Kaderali L, Kleeff J et al. Autoantibodies against the exocrine pancreas in autoimmune pancreatitis: gene and protein expression profiling and immunoassays identify pancreatic enzymes as a major target of the inflammatory process. Am J Gastroenterol 2010;105:2060-7.
- Zhang J, Lian M, Li B, Gao L, Tanaka T, You Z et al. Interleukin-35 promotes Th9 cell differentiation in IgG4-related disorders : experimental data and review of the literature. Clin Rev Allergy Immunol 2021;60:132-5. [CrossRef]
- Sun R, Liu Z, Lu H, Peng Y, Li J, Nie Y et al. Potential impact of autoimmune disease family in IgG4-related disease: a retrospective cohort study. RMDOpen 2023;9:e002865. [CrossRef]
- Jin X, Shi T. Eosinophilic lymphogranuloma: a report of 7 cases similar to Mikulicz’s disease. Zhonghua Yixue Zhazhi (in Chinese) 1937;23:681-99.
- Kimura T, Yoshimura S, Ishikawa E. On the unusual granulation combined with hyperplastic changes of lymphatic tissues (in Japanese). Trans Soc Pathol Jpn 1948;37:179-80.
- Abuel-Haja M, Hurford MT. Kimura’s disease. Arch Pathol Lab Med 2007;131:650-1.
- Chen H, Thompson LDR, Auguilera NSI, Abbondanzo SL. Kimura’s disease. A clinicopathologic study of 21 cases. Am J Surg Pathol 2004;28:505-13. [CrossRef]
- Daaleman TP, Woodroof J, Kimura’s disease presenting as subcutaneous facial plaque in an African American. Cutis 2000;66:201-4.
- Li T-J, Chen X-M, Wang S-Z, Fan M-W, Semba I, Kitano M. Kimura’s disease: a clinicopathologic study of 54 Chinese patients. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1996;82:549-55.
- Hui PK, Chan JKC, Ng CS, Kung IT, Gwi E. Lymphadenopathy of Kimura’s disease. Am J Surg Pathol 1989;1:177-86. [CrossRef]
- Liu YC, Liu SC, Xu J, Xu XC, Wang MY. An unusual case of systemic lymphadenopathy - Kimura’s disease. J Inflam Res 2023;16:701-5.
- Yang BD, Kiao HL, Wang MH, Long QY, Zhong LL, Liu ZM et al. Kimura’s disease successfully affecting multiple body parts: a case-based literature review. BMC Opthalmol 2022;22::154. [CrossRef]
- Liu C, Hu W, Chen H, Tang Z, Zeng C, Liu Z et al. Clinical and pathological study of Kimura’s disease with renal involvement. J. Nephrol 2008;21:517-25.
- Liu L, Cheng Y, Fang Z, Kong JP, Wu XD, Zhang Z. Kimura's disease or IgG4-related disease? A case-based review. Clin Rheumatol 2015;34:385-9. [CrossRef]
- Ma XR, Xin SJ, Onyang TX, Ma YT, Chen WY, Chang ML. Successful treatment of Kimura’s disease with leflunomide and methylprednisolone: a case report. In J Clin Exp Med 2014;7:2219-22.
- Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Eng J Med 2001;344:732-8. [CrossRef]
- Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003;38982-4. [CrossRef]
- Deshpande V, Zea Y, Chan JKC, Yi EE, Sato Y, Yoshino T et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012;25:1181-92. [CrossRef]
- Stone JH, Zen Y, Deshpande V. IgG4-related disease. New Eng J Med 2012;366:539-51.
- Nehring P, Przybytkowski A. Think twice before operating on a pancreatic mass: could it be IgG4-related disease? Lancet 2020;395:816. doi org/10.1016/S0140-6736(20)30169-0.
- Saavedra-Perez D, Vaquero EC, Ayuso JR, Fernandez-Cruz L. Autoimmune pancreatitis: a surgical dilemma. Cir Esp (English ed.) 2014;92:645-53. [CrossRef]
- Kottler D, Barete S, Quereux G, Ingen-Housz-Oro S, Fraitag S, Ortonne N et al. Retrospective multicentric study of 25 Kimura disease patients: emphasis on therapeutics and shared features with cutaneous IgG4-related disease. Dermatol 2015;231:367-77. [CrossRef]
- Chang SY, Lee CC, Chang ML, Teng WC, Hsiao CY, Yu HH et al. Comparison of clinical manifestations and pathology between Kimura disease and IgG4-related disease: a report of two cases and literature review. J Clin Med 2022;11:6887. [CrossRef]
- Wallace ZS, Naden RP, Chari S, Choi HK, Della-Torre E, Dicaire J et al. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-related disease. Am Rheum Dis 2020;79:77-87. [CrossRef]
- Nishikori A, Nishimura MF, Nishimura Y, Notohara K, Satou A, Moriyama M et al. Investigation of IgG4-positive cells in idiopathic multicentric Castleman disease and validation of the 2020 exclusion criteria for IgG4-related disease. Pathol Int 2022;72:43-52. [CrossRef]
- Satou A, Notohara K, Zen Y, Nakamura S, Yoshino T, Okazaki K et l. Clinicopathological differential diagnosis of IgG4-related disease: a historical review and a proposal of the criteria for excluding mimickers of IgG4-related diseases. Pathol Int 2020;1:1-12. [CrossRef]
- Umehara H, Okazaki K, Kawa S, Takahashi H, Goto H, Matsui S et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol 2021;31:529-33. [CrossRef]
- Wallace ZS, Miles G, Smolkina E, Petruski-Ivleva N, Madziva D, Cook C et al. Incidence, prevalence and mortality of IgG4-related disease in the USA: a claims-based analysis of commercially insured adults. Ann Rheumat Dis 2023;82:957-62.
- Mitsuhiro A, Katsuya S, Yamaoka K, Yasuoka H, Takeshita M, Kaneko Y et al. Number of circulating follicular helper 2 T cells correlates with IgG4 and interleukin-4 levels and plasmablast numbers in IgG4-related disease. Arthritis Rheumatol 2015;65:2476-81. [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).