
Submitted:
31 January 2025
Posted:
31 January 2025
You are already at the latest version
Abstract
Heart failure with preserved ejection fraction (HFpEF) remains a significant challenge in modern healthcare. It accounts for the majority of heart failure cases and their number worldwide is steadily increasing. With its high prevalence and substantial clinical impact, therapeutic strategies for HFpEF are still inadequate. This review focuses on the cardiometabolic phenotype of HFpEF which is characterized by such conditions as obesity, type 2 diabetes mellitus and hypertension. Various murine models that mimic this phenotype are discussed. Each model’ pathophysiological aspects, namely inflammation, oxidative stress, endothelial dysfunction, changes in cardiomyocyte protein function and myocardial metabolism alterations are examined in detail. Understanding these models can provide insight into the mechanisms underlying HFpEF and aid in the development of effective therapeutic interventions.
Keywords:
heart failure with preserved ejection
; murine models
; myocardial fibrosis
; diastolic dysfunction
; cardiometabolic HFpEF
1. Introduction
Heart failure (HF) with preserved ejection fraction (HFpEF) remains a significant challenge in modern healthcare due to its complex pathophysiology and limited treatment options. It accounts for the majority of all HF cases and the number of cases worldwide is steadily increasing. This rise is attributed to factors like the gradual aging of the population, the prevalence of sedentary lifestyle and the increasing incidence of cardiometabolic conditions, such as obesity, arterial hypertension and type 2 diabetes mellitus (T2DM) [1]. HFpEF is associated with a priori high morbidity and mortality comparable to HF with reduced ejection fraction (EF) (HFrEF) [2]. According to the US Get With The Guidelines-HF registry, the 5-year mortality rate in hospitalised patients with HFpEF is as high as 75%, similar to that in patients with HFrEF [3].
HFpEF is diagnosed on the basis of HF symptoms, preserved left ventricular (LV) EF (>50%) and the evidence of increased LV filling pressure/congestion with elevated brain natriuretic peptide (BNP) levels [4,5,6]. Despite the enormous clinical and social impact, therapeutic strategies for HFpEF are rather limited. This is typically a common problem owing to a gap in understanding the mechanisms underlying the development of this condition and the marked heterogeneity of its manifestations [7]. To date, regarding the drugs that have been proved to be effective in HFrEF treatment, sodium glucose transporter 2 inhibitors and mineralocorticoid receptor antagonists are the only ones that seem to improve the prognosis of patients with HFpEF by preventing HF exacerbations [8,9,10]. In addition, the majority of pharmacological agents have no potential to improve exercise capacity and quality of life in patients with HFpEF [11]. As most of these patients are elders with limited functional capacity due to natural age-related changes, there may be a significant problem in their treatment [8]. Besides, the differences in drug efficacy between the two major forms of HF result from the differences in the disease initiation mechanisms. Cardiomyocyte death for any reason (ischaemic, toxic, infectious, etc.) is the key pathological process in HFrEF as it triggers the entire cascade of subsequent structural and functional myocardial changes. Regarding HFpEF, the main pathophysiological drivers are a delayed relaxation and reduced LV compliance in which chronic low-grade myocardial inflammation appears to play a key role [12,13,14]. Additionally, the role of inflammation in the development and progression of HFpEF has been confirmed by a large body of clinical evidence. To illuminate the main manifestations in HFpEF, we offer their main characteristics as given below.
High blood levels of inflammatory cytokines. Patients with HFpEF are well known to have significantly higher blood levels of pro-inflammatory and pro-fibrotic markers, lower levels of markers of myocardial damage (high-sensitivity troponin T) and LV wall stress (N-terminal fragment of brain natriuretic hormone precursor, NT-proBNP) compared with patients with HFrEF [15,16,17,18,19]. In addition, elevated blood levels of interleukin(IL)-2 and C-reactive protein are associated with new-onset HFpEF [20,21]. Thus, the intensity of systemic inflammation significantly influences the course and prognosis of HFpEF [20,21,22,23].
Activation of endothelial cells. In patients with HFpEF, high levels of endothelial-derived molecules (intercellular adhesion molecule-1, vascular cell adhesion molecule-1, E-selectin) identified in peripheral blood and homogenates of myocardial specimens indicate endothelial cell activation [24,25,26]. Since circulating pro-inflammatory cytokines primarily affect microvascular endothelial cells, the activity of nicotinamide adenine dinucleotide phosphate oxidase (NOX), the enzyme that leads to the accumulation of reactive oxygen species (ROS) in cells, is increased [26]. This leads to uncoupling of endothelial nitric oxide synthase (eNOS) and decreased nitric oxide (NO) bioavailability. The above findings are supported by the low myocardial nitrite/nitrate concentrations in HFpEF patients [26]. Besides, the activation of endothelial cells in HFpEF is associated with structural microvascular changes, as such as rarefaction and basal membrane thickening of the microvasculature [27,28] which reduce coronary reserve and cause relative myocardial ischaemia.
Activation of circulating monocytes and monocyte infiltration of myocardium. Patients with HFpEF are characterised by increased content of pro-inflammatory monocytes in the peripheral blood, translating LV dysfunction [29]. Our recent findings represent that myocardial remodeling in HFpEF patients is associated with increased numbers of circulating intermediate monocytes and a decrease in the ratio of regulatory T cells/activated T helper cells [30]. By other research, stimulation with tumor necrosis factor (TNF) or lipopolysaccharide results in the release of large amounts of pro-inflammatory cytokines by neutrophils from HFpEF patients compared to neutrophils isolated from healthy subjects [24]. In addition, a significant accumulation of activated monocytes/macrophages migrated from bloodstream is consistently found in myocardial biopsy specimens from patients with HFpEF [25,31,32]. Chemoattractants (mainly monocyte chemotactic protein-1 (MCP-1)) are also required for the migration of monocytes from the bloodstream into the subendothelial space of the myocardium, initial key step in the inflammatory process. One must note that peripheral chemokine concentration (ССL2/MCP-1, CXCL10/ interferon-γ-induced protein 10, CCL17, CCL18) is increased in HFрEF [29]. To note, the numbers of classic/pro-inflammatory monocytes and alternative/anti-inflammatory circulating monocytes are increased in HFpEF patients. Culture of healthy donor monocytes with HFpEF patient-derived sera leads to M2-macrophage differentiation (as evidenced by the CD206 and IL-10 expression) [29]. According to the data of Bajpai G. et al, CCR2+ macrophage abundance is associated with LV remodeling [33]. In patients with HFpEF, transforming growth factor (TGF)-β-producing macrophages are detected in the myocardium. Besides, the addition of TGF-β to the culture of fibroblasts obtained from patients with HFpEF is associated with their transformation into myofibroblasts. A positive correlation between cardiac collagen as well as the number of inflammatory cells, and diastolic dysfunction suggests a direct influence of inflammation on fibrosis triggering diastolic dysfunction [25,32].
HFpEF as a cardiometabolic disorder.
The main risk factors predisposing to the development of HFpEF are pro-inflammatory conditions such as obesity, T2DM, arterial hypertension, chronic kidney disease, hypodynamia and aging [34,35,36,37,38]. They often combine (especially obesity and T2DM) exacerbating the role of each other [36,39]. As the obesity continues rising, it increases the amount of adipose tissue secreting pro-inflammatory adipokines, which provokes the development of insulin resistance and oxidation processes caused by free radicals, the production of pro-inflammatory cytokines, dysfunctional and reduced microvascular density, increased leukocyte adhesion, macrophage activation and myocardial fibrosis [2,14,40,41,42]. Insulin resistance and T2DM are rather common in HFpEF indicating a worse prognosis [43,44]. Irrespectively, T2DM leads to impaired diastolic and systolic LV function with the development of the so-called diabetic cardiomyopathy regardless of other diseases such as arterial hypertension and ischaemic heart disease [45]. Hypertension is also to some extent a pro-inflammatory state. Increased blood levels of the pro-inflammatory cytokines IL-1β, TNF, interferon-γ have been proved to confirm the risk of arterial hypertension [46]. An increase in the number of T helper 17 cells and in the concentration of cytokines produced by these cells, IL-17 and IL-23, has been reported in the blood of patients with arterial hypertension [47]. One of the key mechanisms implicated in blood pressure increase is the hyper-activation of the renin-angiotensin-aldosterone system. Besides, angiotensin II, through its type 1 receptors, promotes inflammation in the myocardium by triggering intracellular signaling pathways leading to NF-κB activation and subsequent production of pro-inflammatory cytokines, synthesis of adhesion molecules and oxidative stress [48,49].
HFpEF is a heterogeneous condition.
The main reason for the ineffectiveness of drug interventions in HFpEF is the extraordinary diversity of its manifestations in different subgroups of patients (phenotypes). These phenotypes differ significantly in their clinical and haemodynamic characteristics, mechanisms of development, prognosis and response to specific interventions [36,38,39,50]. Such heterogeneity is best assessed using sophisticated approaches (e.g. machine learning techniques). Since 2014, several research groups have applied machine learning to identify multiple clusters/phenotypes of HFpEF. To date, around two dozen phenotypes have been proposed. From study to study the phenotypes vary, but in most cases they are reduced to a small number of fairly well-defined phenotypes: cardiometabolic, left atrial myopathy phenotype, natriuretic peptide deficiency phenotype, and pulmonary hypertension/right ventricular dysfunction phenotype [51].
One of the most common HFpEF phenotypes mentioned above is cardiometabolic (or inflammatory-metabolic) [7]. In patients with this phenotype, the inflammatory-metabolic milieu of obesity and T2DM comorbidities is thought to be a major contributor to the pathophysiology of HFpEF [35]. Interestingly, this phenotype is more common in women, who are more prone to systemic inflammatory diseases than men [5,36,39,52]. Packer M. et al considered obesity and T2DM to have a greater impact on the woman heart than man heart, HFpEF being clinically more severe. However, the prognosis for women is more favourable than for men [53]. The cardiometabolic phenotype is characterised by pronounced LV hypertrophy and diastolic dysfunction, and a high incidence of adverse outcomes [39,50,51,53,54,55]. As the vast majority of HFpEF patients are overweight/obese, this phenotype is to some extent universal [56]. Therapeutic interventions for this phenotype may be useful in a vast majority of patients with HFpEF. In other words, a "one-size-fits-all" approach is being implemented, as to some extent is suggested by the success of sodium glucose transporter 2 inhibitors in HFpEF [9,56].
Animal models of HFpEF.
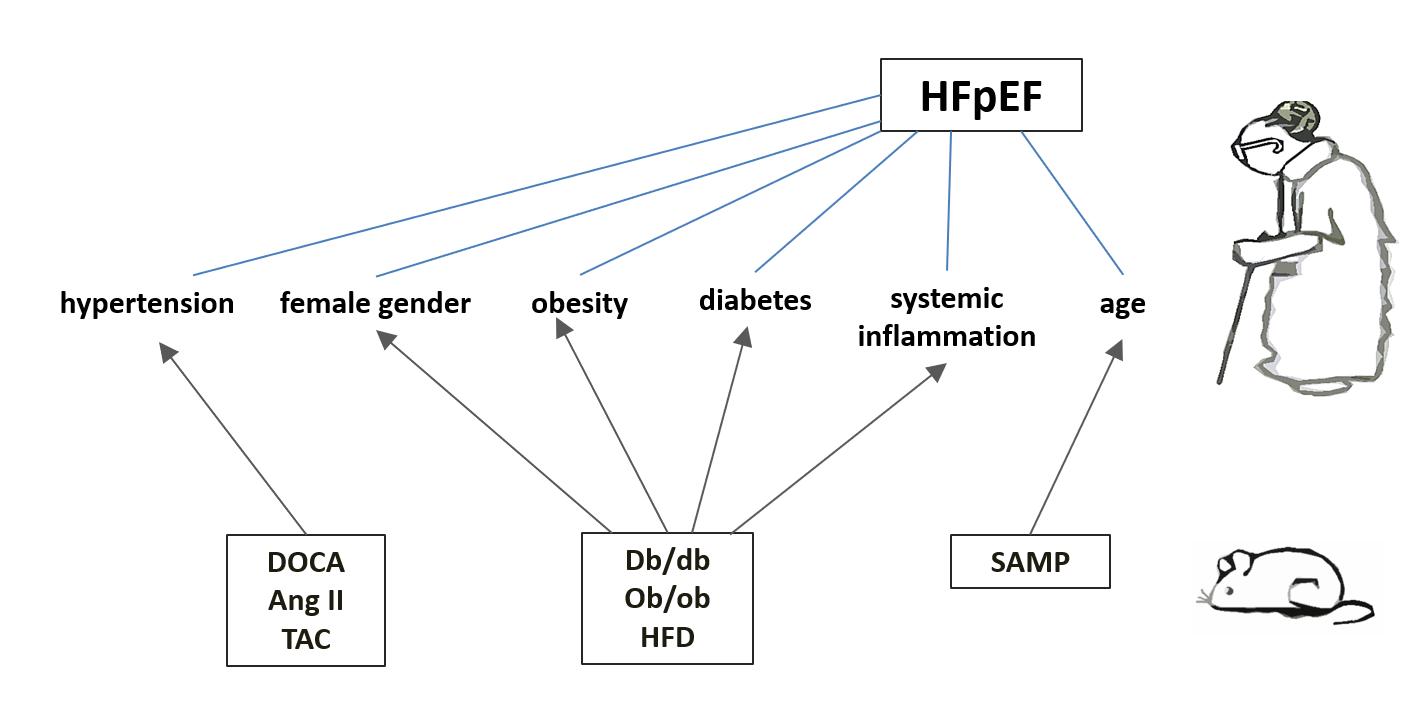
At present, the availability of suitable animal models of HFpEF throws the light on understanding the pathophysiological mechanisms of HFpEF and testing new pharmacological agents. Moreover, pharmacological or surgical approaches, genetic modifications as well as their possible combinations are commonly used in the development of preclinical models of HF [57]. Both small (rodents, guinea pigs) and medium or large (dogs, cats, pigs, monkeys) laboratory animals may be the subject of such manipulation. Yet, a large number of preclinical models that reproduce aspects of HFpEF are also available to researchers [57,58,59]. For a number of reasons, mice are most commonly used to mimic HFpEF. Firstly, from a practical point of view, mice seem to be optimal animals to carry out different researches: they are cheap to maintain, have a short breeding cycle, develop pathological changes rapidly and provide easy access to genetic manipulations. Secondly, mouse genome proves to have sufficient homology with the human genome. However, one of the major challenges of animal models is the fact that none of them are able to fully reproduce the pathophysiological diversity of human HFpEF. Additionally, the heterogeneity of HFpEF manifestations represent another difficulty to create animal models. Thus, no animal model is suitable for all patients with HFpEF, but for a certain group of patients. In addition, even numerous preclinical models of concentric LV hypertrophy have been a hallmark of HFpEF as they are unable to reproduce the complex myocardial remodeling observed in humans with HFpEF. Usually, the development of animal models of HFpEF has focused on recapitulating the pathology of a single organ (typically the heart) making it difficult to mimic the numerous comorbidities associated with HFpEF [60]. However, the last decade has witnessed the development of innovative multi-component animal models of HFpEF that are able to reproduce the multiple comorbidities and multi-organ involvement typical of HFpEF. Such multi-component models include two-hit models (mimicking two cardiometabolic stresses, that is, arterial hypertension, T2DM or obesity [61,62,63,64,65]). Although, three-hit models are also developed including age as an extra risk cardiometabolic stresses [66,67,68,69,70].
As the cardiometabolic phenotype is to some extent a universal phenotype in HFpEF [7], rodent models that recapitulate cardiometabolic stress promise an extra opportunity to the study of HFpEF. In this review, the following murine models that are more or less consistent with the cardiometabolic phenotype of HFpEF are as follows:
1) obesity-related HFpEF (leptin-deficient ob/ob and leptin receptor-deficient db/db mutant strains; induced by high fat diet (HFD));
2) HFpEF induced by arterial hypertension (chronic subjection to deoxycorticosterone acetate (DOCA), DOCA + angiotensin II infusion, transverse aortic constriction (TAC), chronic subjection to angiotensin II, DOCA infusion + TAC);
3) Aging HFpEF (senescence-accelerated mouse strains).
For each of these models the individual pathophysiological aspects of cardiometabolic phenotype are characterized below:
1) Inflammation (production of pro-inflammatory cytokines and activation of inflammatory cells);
2) Oxidative stress (dysfunction of the antioxidant systems and overproduction of ROS, mitochondrial dysfunction);
3) Dysfunction of the NО/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) pathway (decreased PKG and eNOS activity, activation of inducible NOS (iNOS), decreased cGMP and NO production);
4) Changes in the level of phosphorylation in cardiomyocyte proteins involved in contraction and regulation of intracellular calcium waves (titin, myosin heavy chain (MHC), sarco-endoplasmic reticulum Ca2+-ATPase (SERCA), phospholamban);
5) Changes in myocardial metabolism (suppression of glycolysis, increased fatty acid uptake and oxidation, impaired processes of oxidative phosphorylation and ATP synthesis).
1. Obesity-Related Models of HFpEF
Given the high prevalence of obesity in patients with HFpEF, its increasing burden contributes to cardiovascular dysfunction.
1.1. Diabetic db/db and Obese ob/ob Mice
Db/db [BKS-Lepr/db/db/JOrlRj, BKS.Cg-Dock7m+/+Leprdb/J] and ob/ob [B6.V-Lepob/ob/JRj, B6.Cg-Lepob/J] strains are originally utilised as the models of T2DM. Both strains are based on impaired signaling of leptin which represents a small adipokine protein with hormonal properties. As it is synthesised by adipocytes, so its level in blood correlates with adipose tissue mass (it is higher in women because they have more excess adipose tissue). The leptin receptor is expressed in various tissues (heart, muscle, lung, small intestine, liver, adipose tissue, central nervous system) and is able to cross the blood-brain barrier and interacts with receptors in the hypothalamus to induce a feeling of satiety, thereby regulating appetite. It is worth noting that mutations in the leptin gene or its receptor related to the loss of function lead to an uncontrollable hunger [71]. There is also a mutation in the db/db mice gene encoding the long isoform of the leptin receptor [72]. In this mouse strain, impaired leptin signaling in the hypothalamus leads to hyperphagia and obesity accompanied by elevated levels of insulin and leptin, hyperglycaemia and the development of insulin resistance [73,74,75,76]. Experimentally, by 6-8 weeks of age, db/db mice suffer from significant hyperinsulinemia, obesity and hyperglycaemia which worsen thereafter [73,74,77,78,79,80,81,82,83,84,85,86,87,88].
Another murine strain, ob/ob, has genetic leptin deficiency which can be supplemented externally [89]. Thus, they suffer from obesity, hyperphagia, hyperglycaemia, insulin resistance and hyperinsulinemia from an early age [76,90,91,92,93,94,95,96,97]. Impaired glucose tolerance in ob/ob mice occurs at the same age as in db/db mice, at about 6-8 weeks [75].
Both db/db and ob/ob mice demonstrate numerous signs of disturbed lipid metabolism, both models being characterised by hypercholesterolaemia and increase in plasma fatty acids and triglycerides [73,75,77,79,85,86,93,97,98]. However, these changes are more pronounced in db/db strain. Moreover, only db/db mice show an increase in plasma ceramide levels [73,86]. Other data report that ob/ob mice have normal blood triglycerides levels, but liver steatosis can be elevated [95,96]. Db/db mice also demonstrate increased very-low-density lipoprotein and decreased high-density lipoprotein plasma levels [98]. In addition, db/db mice have a reduced activity of hepatic triglyceride lipase which makes them similar to people with T2DM [77]. In db/db mice, a decrease in lipoprotein lipase mRNA content and activity are observed in all tissues [77].
However, there is a conflicting evidence regarding metabolic disorders in the myocardium of these murine strains. For example, some authors have described high levels of triglycerides and cholesterol in the myocardium of db/db and ob/ob mice [76], particularly in ob/ob females [96], while no findings have been confirmed by Barouch L.A. et al [90]. In the myocardium of ob/ob female mice, increased mRNA content of lipoprotein lipase and other proteins involved in lipolysis are found, as well as an increased mRNA level of the insulin-sensitive glucose transporter GLUT-4 and the microsomal triacylglycerol-transport protein which has a protective function in the presence of lipids excess [96]. Increased expression of sodium-glucose cotransporter 1 is also observed in the myocardium of ob/ob mice of both sexes [97].
A decrease in the level of aerobic glucose oxidation and glycolysis in the myocardium is observed in both strains [75], while db/db mice also confirm an increase in the levels of acetyl coenzyme A and succinyl coenzyme A, crucial metabolites in the tricarboxylic acid cycle [73]. All these factors point to a substrate shift in the energy supply of cardiomyocytes, switching from glucose oxidation to beta-oxidation of fatty acids. In addition, there is an extra evidence of a disruption in the intracellular insulin signaling pathway in both strains. For example, db/db mice demonstrate an impaired response to insulin stimulation [73] and increased serine residue (307) phosphorylation on insulin receptor substrate-1 (IRS-1) (a protein that plays a pivotal role in insulin signaling) in cardiomyocytes, with subsequent inactivation of the insulin receptor [80]. In addition, these mice have reduced activity of SIRT [silent mating type information regulation 2 homolog 1], which is responsible for the 'beneficial' tyrosine phosphorylation of IRS-1 required for normal insulin receptor function [86]. An increase in the serine-phosphorylated IRS-1 has also been found in ob/ob mice [Hammoudi2019]. In general, impaired cardiac metabolism precedes the onset of hyperglycaemia and the development of insulin resistance in both db/db and ob/ob mice [75].
It is also worth mentioning that leptin is important for the maintenance of normal cardiac structure and function as it may have both direct and indirect (via neurohormonal regulation) antihypertrophic effects. Hence, leptin deficiency leads to LV hypertrophy [90, 92]. Ob/ob mice exhibit LV hypertrophy by 6 months of age, regardless of their weight [90]. Although weight loss by dietary restriction is associated with a reduc[90,92tion in LV hypertrophy, a much greater reduction in LV hypertrophy may be achieved by leptin administration [90]. In ob/ob mice, the cardiac response to beta-adrenergic stimulation was also reduced; administration of leptin partially restored the beta-adrenergic inotropic response [93].
In addition to HFpEF, some comorbidities consistent with the ones observed in humans are developed in these murine strains, the common comorbidities being the cardiometabolic phenotype of HFpEF which usually occurs in combination with obesity, T2DM and arterial hypertension. For example, in db/db mice, the development of arterial hypertension with age is associated with obesity and T2DM [78,80,83,87]. There is an increase in blood pressure starting with 11 weeks of age and a pronounced arterial hypertension is already present by 14 weeks of age as well. Developing arterial hypertension is associated with an increased renin-angiotensin system activity while the baroreflex is not impaired. It is also worth noting that in db/db mice, renin-angiotensin-aldosterone system blockers do not improve metabolism and insulin sensitivity, i.e. they have no effect on hyperglycaemia [78]. Unlike db/db mice, blood pressure is not increased with age and may be even decreased in ob/ob mice [90,96]. Such differences may be related to the fact that the authors of ob/ob mice studies have measured the blood pressure in rather young mice (not older than 10-11 weeks), whereas the authors performing db/db mice studies measured blood pressure in aged mice (10 to 20 weeks). Perhaps, ob/ob mice develop arterial hypertension at a more “mature” age, especially taking into account that all the changes in these mice occurred later and were less pronounced than in db/db mice.
One more critical component of the pathogenesis of HFpEF is a chronic low-grade inflammation [12] related to both db/db and ob/ob strains. It is also worth noting that the inflammation can be both systemic or myocardial one. For example, the blood levels of the pro-inflammatory cytokines TNF and IL-6 are increased in db/db mice [98] and C-reactive protein - in ob/ob mice [97]. In the myocardium of ob/ob mice, RT-qPCR reveals increased mRNA of IL-1β, IL-6, TNF, NLRP3 and caspase-1 [97], and increased expression of the TNF and MCP-1 genes in db/db mice [82]. According to other data, TNF, MCP-1, IL-1β and IL-6 mRNA content is increased in the adipose tissue but not in the myocardium of db/db mice [73]. However, no findings of any infiltration of the myocardium by macrophages are confirmed by the same authors [73]. In db/db mice, the enhanced NF-κB activity (assessed by the binding activity of free NF-κB p65 in nuclear extracts) and NF-κB p50 expression (determined by RT–qPCR and western blot) in LV tissue is detected. This was associated with increased ROS, superoxide, and peroxynitrite production by mitochondria. The addition of a NF-kB antagonist reduced the production of pro-inflammatory cytokines and protected the heart from oxidative stress, restored mitochondrial integrity and reduced myocardial dysfunction [98].
Echocardiographic and invasive haemodynamic data confirm the presence of HFpEF characteristic features in both murine strains. In db/db mice, the development of first diastolic (increased early to late mitral inflow (E/A) ratio and early mitral inflow to mitral annulus relaxation velocity (E/e′) ratio, prolonged isovolumic relaxation time (IVRT), decreased tissue Doppler e′/a′ ratio) [73,81,83,84,86,87,88,99] and subsequently systolic (reduced shortening fraction, EF, velocity of circumferential fibre shortening) LV dysfunction [76,81] is typical. Ob/ob mice also exhibit LV diastolic dysfunction (decreased E/A ratio), but there are conflicting data regarding systolic dysfunction [76,91,95,96,97]. Many studies in db/db mice [73,76,79,81,83,84,87,88,98,99] demonstrate echocardiographic and histological evidence of LV hypertrophy accompanied by the development of myocardial fibrosis. However, no confirmations were found in other studies [82,88]. Additionally, in female mice, myocardial fibrosis is much more pronounced than in male mice [79]. Some studies have reported reduced capillary density in the myocardium [83,88], while others have reported normal or even increased density [82,87]. Ob/ob mice also demonstrate the signs of LV hypertrophy (by echocardiography and post-mortem heart weighing) [76,90,93]. However, in contrast to db/db mice, histological data are scarce and ambiguous. For example, according to some sources, ob/ob mice have no or negligible myocardial fibrosis [90,91]; according to other researches, myocardial fibrosis is still present [97].
In these murine strains, the presence of LV diastolic dysfunction is confirmed by an increase in plasma BNP level, a biological marker of haemodynamic stress [87,95]. Increased expression of natriuretic peptide estimated by mRNA level has been reported in the myocardium of ob/ob mice [91]. In db/db female mice, there is a significant relative increase in myocardial BNP mRNA content with age which is not confirmed in males [79]. In contrast, another study describes reduced myocardial expression of natriuretic peptide in db/db mice [100].
It should be noted that db/db mice demonstrate sex differences in the development of HFpEF symptoms. Thus, female mice have higher blood pressure levels and greater weight gain than males. Besides, they have more pronounced LV remodeling processes (more pronounced LV hypertrophy and diastolic dysfunction) [83]. Such differences are indirectly in line with what is observed in humans where women are more likely to have severe HFpEF than men.
It is important that an increased cardiomyocyte residual stress induced by altered protein function increases LV myocardial stiffness. The studies report a decreased mRNA and protein content of SERCA2a in the db/db myocardium [73,79,101]. However, the other studies confirm the molecular expression of SERCA2a in these mice that does not differ from that one in healthy mice [87,88]. In ob/ob mice, the total amount of SERCA2a in the myocardium is not increased, although its decreased activity and increased oxidation is observed. In fact, this is associated with a decreased calcium ion transport from the cytoplasm to the sarcoplasmic reticulum and calcium ion accumulation in the cytoplasm [94] contributing to cardiomyocyte residual stress. A lot of studies [73,87,88] in db/db mice demonstrate that the total amount of the SERCA activity regulator protein phospholamban in the myocardium, as determined by western blotting, is comparable with the control ones, except for one study [101]. Phosphorylation of phospholamban removes its blocking effect on SERCA2a, favouring the fastest possible release of calcium ions into the sarcoplasmic reticulum and accelerating myocardial relaxation. Phosphorylation of phospholamban at serine 16 and threonine 17 has been revealed to be impaired in both db/db and ob/ob mice resulting in delayed calcium efflux, disturbed relaxation and increased LV myocardial stiffness [87,88,91]. In ob/ob mice, leptin administration restores the levels of phosphorylated phospholamban [93]. Db/db murine cardiomyocytes are characterized by an impaired response to extracellular calcium and reduced contractility [80,87,101].
Changes in titin molecules which are intracellular spring elements responsible for myocardial stretching during diastole play an important role in increasing myocardial stiffness. In db/db mice, titin changes are mainly due to its hypophosphorylation rather than oxidation or isoform shift from highly extensible N2A molecules to more 'rigid' N2B molecules [84]. In both db/db and ob/ob mice, a change in MHC expression pattern is understood as an isoform shift from a faster α-isoform to a slower β-isoform [75,94]. Such alterations in myocardial MHC isoforms can be detected in 4 week-old mice [75,91]. However, the myocardial levels of MHC-β mRNA in db/db mice is higher in males, females demonstrate a more pronounced relative increase with age [79]. Simultaneously, another study in db/db mice reports reduced mRNA content of MHC-α and MHC-β [100]. Additionally, in myocardial tissue of ob/ob mice the activity of protein kinase A which is responsible for the phosphorylation of titin and other cardiac proteins is reduced [93]. AMP-activated protein kinase levels are also reduced in the myocardium of db/db and ob/ob mice resulting in hypophosphorylation of titin and increased myocardial stiffness [73,74,82,86,88]. In contrast, no differences in the levels of the phosphorylated cardiac proteins myosin-binding protein C [cMYBP-C] and troponin I are observed in db/db mice [82,87].
Notably, mice of both strains have signs of a dysfunction in the NO-cGMP-PKG signaling pathway that plays an essential role in normal myocardial function and has numerous beneficial effects such as accelerating relaxation, reducing cardiomyocyte stiffness, inhibiting pro-hypertrophic signaling, improving mitochondrial function, etc. [102]. Endothelium-dependent vasodilation is mediated by NO which is a potent relaxing factor. In the myocardium of db/db mice, both NO and nitrate levels (reflecting NO content) are reduced in comparison to that of healthy mice [86,87]. In db/db mice, there is a switch from NO-dependent to H2O2-dependent vasodilation contributing to the impaired microvascular function [103]. The mRNA and protein content of different isoforms of NOS is altered in the myocardium of these mice, that is predominantly manifested by decreased expression of eNOS responsible for NO synthesis [74,100,103] and increased iNOS activity which is responsible for ROS generation [74,103]. Indeed, the decrease of cGMP protein level and cGMP-dependent PKG activity in the myocardium of db/db mice is also observed [84]. An indirect confirmation of the reduced NO bioavailability in db/db mice is confirmed by the disturbed myocardial perfusion observed in ex vivo and in vivo experiments [103].
The evidence of reduced NO bioavailability and impaired signaling through the intracellular NO-cGMP-PKG pathway is also confirmed in ob/ob mice. In particular, they have reduced levels of L-arginine which is a substrate for NOS, and increased levels of the NOS inhibitor asymmetric dimethylarginine [ADMA] in plasma [95]. In addition, ob/ob mice exhibit a reduced nitrate/nitrite ratio in heart homogenates which is a marker of ROS accumulation and reduced NO production [104]. However, other studies demonstrate an increased NO content in the myocardium [92]. A high level of iNOS and a low level of phosphorylated eNOS have been observed in the myocardium of ob/ob mice [92]. Unlike Guo W. et al [92], these data are not confirmed when assessing eNOS mRNA levels by RT-qPCR. The same study illustrates a decrease in the content of neuronal NOS (nNOS) in the myocardium which can be restored after leptin administration [104].
Moreover, coronary microvascular dysfunction is observed in both ob/ob and db/db mice, most likely as a consequence of a vascular tone-impaired regulation induced by reduced NO bioavailability [95]. To date, nitrosative/oxidative stress is a highly evident component of HFpEF pathophysiology [105] and its signs are found in the myocardium of both db/db and ob/ob strains. For example, an increase in NOX activity has been demonstrated in the myocardium of db/db mice. As a result, the binding of NO to superoxide (O2-) and subsequent formation of the cytotoxic reactive oxidant peroxynitrite (ONOO-) leads to protein dysfunction [73,98]. On top of that, levels of peroxynitrite are found to be elevated in the myocardium of both mouse strains [92,98]. The amount of protein carbonyls is increased in the myocardium of ob/ob mice indicating a protein damage induced by oxidative stress [94]. High level of the NOX1 and gp91phox (NOX2) subunits of NOX has been demonstrated in the myocardium of db/db mice [98], however mRNA content of the NOX2 subunit is higher in females [79]. Moreover, high expression of the membrane NOX subunits p47phox and gp91phox in ob/ob mice is revealed [94], although other studies have not confirmed this at either protein or mRNA level [104]. Nevertheless, increased activity of xanthine oxidoreductase [XOR] which is another likely source of ROS has been observed in ob/ob mice [104].
Furthermore, myocardial levels of the antioxidant glutathione (GSH), attributed to scavenging excess ROS, are reduced in db/db mice [98]. Glutathione is oxidised to glutathione disulfide (GSSG) during peroxide detoxification. A decrease in the GSH/GSSG ratio indicating high oxidative stress activity is also shown in the myocardium of ob/ob mice [94,104]. In addition, the expression of superoxide dismutase 1 and heme oxygenase 1 which provide protection against oxidative stress is increased in the myocardium of db/db mice [82]. In turn, the activity of catalase which has antioxidant properties, is increased in db/db myocardium [79].
It should be also noted that oxidative stress results in lipid peroxidation. Thus, the levels of malonic dialdehyde, the end product of lipid peroxidation, and non-esterified fatty acids which are markers of oxidative stress are increased in db/db mice [79]. Remarkably, the levels of these substances are higher in female mice indicating that females undergo more oxidative stress than males [79]. Besides, the increased levels of malonic dialdehyde are also confirmed in the myocardium of ob/ob mice [94].
In accordance with the experiments results, in the myocardium of db/db mice are observed the signs of increased endoplasmic reticulum stress in the form of increased expression of phosphorylated protein kinase RNA-like endoplasmic reticulum kinase [PERK], inositol-requiring protein-1α [IRE-1] and eukaryotic initiation factor 2 [eIF2α] [Dong2009] or the unfolded protein response in the form of increased protein concentration of GRP78/94, XBP-1 and C/EBP homologous protein [85]. Similarly, the signs of endoplasmic reticulum stress and unfolded protein response, i.e the presence of phosphorylated PERK, eIF2α and C/EBP homologous protein, are also observed in ob/ob mice [92].
It is well-known that ROS are generated in the mitochondria. In keeping with the above findings, db/db mice demonstrate the signs of mitochondrial dysfunction, mainly at the level of complex I, as evidenced by increased levels of H2O2 (produced by complex I) and superoxide in mitochondria. Thus, an increased permeability of the mitochondrial membrane contributes to the uncoupling of oxidative phosphorylation and ATP synthesis and leads to the accumulation of ROS in cardiomyocytes [86,98]. There is also evidence of a reduced enzymatic activity of complex III of the electron transport chain (the main source of O2-) in these mice [98], although this has not been fully confirmed [86].
1.2. HFD
Notably, C57BL/6J mice that are maintained on a HFD develop obesity [49,72,75,106,107,108,109,110,111,112,113,114,115,116,117,118,119] as glucose tolerance is impaired and insulin resistance results with elevated blood glucose and insulin levels [76,113,114,115,116,117,119,120]. These mice have also elevated blood levels of leptin which allows to draw some parallels with obese humans [116,118]. Williams T.D. et al highlight the development of arterial hypertension as one of the underlying conditions of this mouse model [112]. However, some authors have no confirmation of this data [120,121]. Such differences in blood pressure can be explained by the different HFD duration and the age of the mice. In all these studies, mice were switched to a HFD at the same age (5-6 weeks), but being followed for 12-15 weeks in one case [112] and for only 8 weeks in others [120,121]. It is possible that the increase in blood pressure in these mice occurs at a later age and with a longer period of time on a diet.
Yet, it is evident that HFD-fed mice show the signs of systemic and local inflammation, the most important pathophysiological mechanism in the development and progression of HFpEF. In particular, they have an increased concentration of IL-6 in plasma [118] and NF-kB content in myocardial tissue [122] as well as increased IL-1β, IL-6, matrix metalloproteinase-9, plasminogen activator inhibitor-1, adiponectin mRNA levels [Wang2012]. In fact, plasminogen activator inhibitor-1 is a key protein secreted by metabolically unhealthy visceral adipose tissue; its activation may contribute to HFpEF via accelerated aging, inflammation, visceral adiposity and impaired metabolism [123]. Moreover, histological examination demonstrates an inflammatory cell infiltration in the myocardium and liver of these mice [113].
According to echocardiography, magnetic resonance imaging and invasive haemodynamic studies, HFD mice demonstrate the signs of LV diastolic dysfunction (decreased mitral velocity E, E/A and e′/a′ ratios, increased E/e′ ratio, prolonged IVRT) after 8 weeks on a HFD. At the same time, systolic function either is not altered significantly or worsened (reduced shortening fraction and EF) [76,115,117,119,120,122]. In addition to LV diastolic dysfunction, LV hypertrophy is identified according to echocardiography, magnetic resonance imaging, post-mortem weighing and histological examination in HFD-fed mice as well as myocardial fibrosis according to histological examination [115,117,119,120,121,122]. Within this concept, some researchers have demonstrated the increased content of the pro-fibrotic factors TGF-β and phosphorylated Smad3 in the myocardium and decreased content of the anti-fibrotic factors bone morphogenetic protein-2 and phosphorylated Smad1/5 [116].
In HFD-fed mice, cardiac protein function has not been investigated as extensively as in db/db and ob/ob strains. Nevertheless, similar phenomena can be observed among the three models. For instance, the expression of SERCA2a is not significantly altered in all three models. However, the level of phosphorylated phospholamban which regulates SERCA2a and cardiac troponin T activity in the myocardium is reduced [117].
Furthermore, the signs of altered NO-cGMP-PKG signaling in the myocardium are proved in HFD-fed mice bringing this model closer to the db/db and ob/ob strains. Specifically, the content of nNOS and phosphorylated eNOS (with normal levels of total eNOS) is suppressed in HFD-fed mice [120,122]. Additionally, these mice have impaired endothelium-dependent vasodilation [118,124] and increased myocardial mRNA levels of the endothelial dysfunction markers endothelin-1 and angiotensin II type 1 receptor [116].
In addition to the db/db and ob/ob strains, HFD-fed mice also demonstrate signs of oxidative stress. For instance, they have increased levels of ROS and nitrotyrosine in the cytoplasm and mitochondria of cardiomyocytes, high content of NOX-1, NOX-2 and NOX4 in the myocardium [116,118,120,122]. To compensate, myocardial antioxidants such as superoxide dismutase may be accumulated [117]. However, in other studies, superoxide dismutase levels remain unchanged [119,122] although another antioxidant, GSH, is decreased [119].
HFD-fed mice have pathological changes in the structure of cardiomyocyte mitochondria and an increased ratio of intracellular nicotinamide adenine dinucleotide phosphate [NADH] to nicotinamide adenine dinucleotide+ [NAD+] levels in the myocardium. This is indicative of mitochondrial oxidative stress and points to disturbances in the electron transport chain as a major source of ROS in the myocardium [120]. Other research in HFD-fed mice indicates that the permeability of myocardial mitochondrial membranes remains unchanged. The expression of uncoupling protein 3 [UCP3] crucial for electron transport chain disruption and oxidative phosphorylation uncoupling is similar to controls. Additionally, oxygen consumption in isolated cardiac mitochondria is not reduced [117]. Besides myocardium, aortic protein levels of gp91phox and p47phox, subunits of NOX, are increased in these mice [118].
In obesity, HFD-fed mice exhibit a variety of metabolic disorders: plasma cholesterol, uric acid, triglycerides and fatty acids, LDL levels are elevated compared to mice fed a healthy diet [113,114,115,117,118]. Myocardial lipid accumulation and fatty deposits in the liver (signs of steatosis) are also observed in this mouse strain [113,114,115,117]. Noticeably, activation of pro-apoptotic pathways is a molecular hallmark of HFD-fed mice. In particular, an increased expression of the pro-apoptotic Bax and caspase-3 and a decrease in the anti-apoptotic B-cell lymphoma 2 [Bcl-2] as well as an increase in pro-apoptotic procaspase-9 and caspase-9 expression are found in the myocardium of these mice [116,117].
2. Hypertension-Induced HFpEF Models
It has been established that arterial hypertension is a major risk factor for HFpEF, influencing both its development and prognosis.
2.1. DOCA + High Salt Diet + Unilateral Nephrectomy [DOCA]
The DOCA model mimics the development of arterial hypertension as a result of mineralocorticoids excess reproducing only one component of HFpEF pathophysiology, specifically arterial hypertension. Given the simplicity, these mice can be employed to replicate multi-component models of HFpEF when combined with other risk factors.
The DOCA mice subjected to pellet implantation with a gradual release of DOCA and unilateral nephrectomy after being fed a high salt diet develop arterial hypertension within 3-7 days after surgery [125,126,127,128,129]. Simultaneously, it has been observed that male mice exhibit higher blood pressure levels compared to females [130]. Blood pressure elevation is associated with kidney damage [125] being more pronounced in male mice [130]. DOCA mice also demonstrate signs of pulmonary hypertension. This is most likely secondary to heart failure (left-sided pulmonary hypertension) [125]. No differences in body weight are observed in this mouse model [131]. Although initially these mice were used exclusively for hypertension modelling, they now are often considered as one of the relevant HFpEF models. This can be explained by the fact that the heart is a typical target organ of arterial hypertension, and hypertension itself is a major risk factor for the development of HFpEF [132].
Echocardiography and invasive haemodynamic study show these mice have LV diastolic dysfunction (low e′ velocity, high E/e′ ratio and prolonged IVRT), sometimes with mild systolic dysfunction (low EF) [125,128,129,133]. Additionally, LV hypertrophy being more pronounced in male mice and myocardial fibrosis not being examined in detail in all studies are reported according to echocardiography, post-mortem weighing and histological examination [125,126,127,128,130,134]. Besides, one study reports higher TGF-β expression in male mice myocardium [130]. Some authors demonstrate lower myocardial capillary density [125], but others fail to confirm this [134].
Previously, the DOCA model considered only mechanical damage caused by volume overload of target organs including the heart. Similar to patients with arterial hypertension, these mice demonstrate signs of myocardial inflammation. For example, pro-inflammatory cytokines mRNA such as IL-1β, IL-6, IL-10, TNF, MCP-1 (only in male mice) are detected alongside macrophage accumulation in the myocardium [125,126,130]. Furthermore, these mice exhibit signs of vascular inflammation as manifested by increased IL-1β, TNF and MCP-1 mRNA content in the aortic wall [127].
In addition to the echocardiography and histological data, other signs of HFpEF have been established in this model, in particular, an increased expression of the haemodynamic stress marker atrial natriuretic peptide (ANP) in the myocardium [125,130,135]. Furthermore, male mice show a greater relative increase in ANP than females [130]. One study reported an increase in BNP expression [125], while other studies have not confirmed in other studies [130,135].
Regarding myocardial protein dysfunction, DOCA model has not been investigated in detail. However, one study has reported a down-regulation of phospholamban phosphorylation [129]. The signs of the NO-cGMP-PKG pathway dysfunction in the myocardium, i.e. decreased NO levels [136] and increased levels of oxidised tetrahydrobiopterin (a cofactor of eNOS) are confirmed in DOCA mice. As such, these alterations promote subsequent substrate uncoupling of eNOS [129] and expression of iNOS [128], and reduced expression of eNOS [136] (although the latter has not been demonstrated in all studies) [128]. Besides, low level of eNOS mRNA has also been found in the aorta [127].
As anticipated, DOCA mice demonstrate signs of oxidative stress in myocardium and vessels. In particular, the levels of superoxide and nitrotyrosine are increased in their myocardium [128,129,136]. However, myocardial mRNA levels of p22phox, gp91phox (NOX-2) and NOX-4 are not significantly altered distinguishing these mice from the above-described models [134]. Some authors suggest that NOS may be the main source of superoxide in DOCA mice [129]. Thus, in iNOS knockout mice, myocardial relaxation is less impaired, myocardial nitrotyrosine content is lower and eNOS expression tended to be higher compared to non-knockout mice. Therefore, these findings indirectly support the involvement of iNOS in ROS production [128]. In addition, superoxide levels and the expression of gp91phox and p22phox in the aorta are also increased in this murine model [127,137,138]. Some authors propose that these changes are more pronounced in male mice than in female mice which were similar to the control group in several parameters [130]. This is not exactly in line with what is observed in HFpEF patients where women with the cardiometabolic phenotype are more likely to have a higher incidence and severity of HFpEF compared to men.
This model may better suite the cardiorenal phenotype of HFpEF, characterised by high blood pressure and kidney damage which occurs equally in men and women. However, it is too early to draw any conclusions about the suitability of this model for HFpEF as it has not been well studied as a model for HFpEF. Furthermore, it should be noted that the C57BL/6 mouse strain is quite resistant to the development of arterial hypertension and kidney damage [131]. In this case, the 129/Sv murine strain is most susceptible, with arterial hypertension, LV hypertrophy and kidney damage being more pronounced than in the C57BL/6 strain [131].
2.2. Angiotensin II (Angiotensin II Infusion) [Ang II]
Ang II mice are implanted with an osmotic mini-pump that infuses angiotensin II. The development of arterial hypertension occurs within one week of starting angiotensin II infusion [138,139]. Ang II mice is also not well characterised as a model of arterial hypertension as angiotensin II infusion alone may not be sufficient to mimic arterial hypertension because of the resistance of the C57BL/6 strain to hypertensive organ damage. However, higher levels of blood pressure are observed in Ang II model compared to the DOCA model as pointed out by some authors [135].
The signs of LV diastolic dysfunction (verified by IVRT prolongation) without systolic dysfunction are described in Ang II model even with administration of low doses of angiotensin II insufficient to raise blood pressure [140]. This observation suggests that inflammation and/or metabolic disturbances, rather than mechanical or haemodynamic factors (increase in afterload and preload) are crucial in developing HFpEF. In the above experiment [140], LV filling abnormalities at low doses angiotensin II infusion may be due to its pro-inflammatory properties.
Consistent with the prior studies angiotensin II also stimulates NOX in inflammatory cells leading to ROS accumulation. Adoptive transfer of T-cells lacking the angiotensin type I receptor blunts angiotensin II-dependent hypertension and reduces aortic superoxide production [138]. Myocardial infiltration by CD68+ macrophages and increased myocardial protein expression of the pro-inflammatory cytokines TNF and IL-1β is also observed in Ang II model [139]. This provides further evidence for the involvement of inflammation in the development of HFpEF.
As in other models of HFpEF, histological examination reveals hypertrophy and fibrosis of the LV myocardium [139,140]. Furthermore, the fibrosis is more pronounced in Ang II model than in the DOCA model [135]. In addition, the expression of ANP and BNP and the MHC-α/MHC-β ratio are increased in the myocardium to a greater extent than in the DOCA mice [135]. These mice also show impaired endothelium-dependent vasodilation and increased ROS production in the aorta [138,139].
2.3. DOCA + High Salt Diet + Angiotensin II + Unilateral Nephrectomy [DOCA+Ang II]
DOCA+Ang II model has been developed in order to overcome the resistance of the C57BL/6 strain to hypertensive organ damage, in particular, kidneys. It has also been understudied and poorly characterised as a model of HFpEF, as are all hypertensive models. Obviously, many abnormalities appear to be more pronounced in DOCA+Ang II model compared to DOCA or Ang II models alone [135]. They include high blood pressure, histochemical and laboratory signs of renal damage, severity of LV hypertrophy and fibrosis, increase in myocardial expression of ANP and BNP, and MHC-β/MHC-α ratio [135].
2.4. Transverse Aortic Constriction (TAC)
TAC mice undergo ligation of the aortic arch between the bronchopulmonary trunk and the left common carotid artery to induce the development of pressure overload-induced cardiac hypertrophy and HF. Body weight of TAC mice is comparable with to that of intact animals [141]. Yet, TAC model is widely utilised to study arterial hypertension. Moreover, it is actively used as a model for HFpEF demonstrating LV filling abnormalities. The obvious disadvantage of TAC model as well as other above-mentioned models of HF induced by arterial hypertension is the reproduction of only one clinical aspect of HFpEF, specifically pressure overload. In some hypertensive models, TAC mice show signs of pulmonary oedema, fibrosis and pulmonary vasculature remodeling (according to the histological study) [142]. These changes are likely result from LV dysfunction.
Data published indicate that TAC model typically develops LV diastolic dysfunction shown by a shortened deceleration time of early diastolic mitral flow followed by systolic dysfunction indicated by reduced EF and shortening fraction [133,141,142,143,144,145,146]. It should be noted that most of these studies did not evaluate the most important diastolic parameters such as the E/A, E/e′, e′/a′ ratios and IVRT. Besides, LV hypertrophy is confirmed in TAC mice according to echocardiography, post-mortem weighing and histological examination [134,141,142,143,145,146,147].
Histological studies also conclude the development of myocardial fibrosis in TAC mice providing an increased expression of TGF-β by myocardial cells [134,141,143,145]. In addition, there is a decrease in the density of capillaries in the myocardium [134]. Increased production of ANP and BNP in myocardium is another sign of HF [143,145].
Data limited exist on contractile protein expression patterns for TAC model. In particular, an increase in MHC-β content is found indicating a shift from fast MHC-α to slower MHC-β isoform. It should be noted that these changes have also been observed in other HF models as well [143]. In addition, phosphorylation of ryanodine receptor type 2 [RyR2], the major intracellular calcium release channel in the cardiac sarcoplasmic reticulum, and the calcium transport regulatory protein phospholamban are reduced [147].
TAC model also demonstrates some evidence of NO-cGMP-PKG pathway dysfunction in the myocardium: although total eNOS and nNOS levels are comparable with the control ones, the phosphorylation of eNOS on Ser114 and nNOS on Ser1412 is reduced [141].
Additionally, this model has evidence of myocardial oxidative stress: increased ROS, decreased nitrate/nitrite ratio (a marker of increased ROS and decreased NO production), activity of the antioxidant enzyme superoxide dismutase, GSH/GSSG ratio [141,143]. Thus, this evidence suggests a depletion of antioxidant systems [145].
2.5. TAC + DOCA
TAC+DOCA mice undergo a ligation of the aortic arch followed by the implantation of a DOCA pellet. The development of diastolic dysfunction and increased LV filling pressure, but preserved systolic function is characteristic of this model [148,149,150]. Obviously, such changes are more pronounced in TAC+DOCA than in TAC or DOCA models alone [134].
3. HFpEF as a Result of Accelerated Aging (The Senescence-Accelerated Mice)
Aging is a significant risk factor for heart failure, with HFpEF becoming more common due to age-related conditions.
The SAMP8 murine strain derived from AKR/J mice is known for accelerated senescence, with an average lifespan of 9.7 months [152]. There are over a dozen accelerated aging lines, but SAMP8 is commonly used. Six-months-old SAMP8 mice exhibit early indicators of aging [153]. Unlike humans, where age correlates with arterial hypertension and HFpEF risk, blood pressure is not elevated [153]. Therefore, it was initially utilised only to study the aging process.
Despite the fact that aging affects the heart being an independent risk factor for the development of HFpEF, the limited information regarding HF pathophysiology is provided in this model. In particular, there is no data on whether myocardial proteins involved in contraction/relaxation and regulation of intracellular calcium turnover are altered.
Following echocardiography and invasive haemodynamic study, LV diastolic dysfunction (decreased E/A and e′/a′ ratios) is developed by 6 months of age in SAMP8 mice, but systolic function remains preserved [153].
According to post-mortem weight data, LV hypertrophy is proved in this strain [153,154]. Histological examination reveals myocardial fibrosis [153,155], and up-regulation of pro-fibrotic TGF-β is confirmed in heart tissue [153]. In addition, the ANP mRNA content is increased in the myocardium of SAMP8 mice [154].
As stated before, aging is associated with the development of chronic inflammation. In the SAMP8 mice, signs of inflammation can be detected in the myocardium, in particular increased mRNA levels of the pro-inflammatory cytokines TNF, IL-1 and IL-6 and increased mRNA and protein levels of the p50 and p52 subunits of NF-kB [155,156]. In contrast, other data have shown that these mice are comparable with control ones in terms of NF-kB activity and expression of TNF and MCP-1 [154].
The SAMP8 strain also demonstrates signs of myocardial NO-cGMP-PKG pathway dysfunction (down-regulation of eNOS, and up-regulation of iNOS) as well as evidence of oxidative damage of cardiomyocyte DNA [155,156]. On the other hand, there is an evidence for an elevated expression of myocardial haem oxygenases 1 and 2 which may protect cells from the pathological action of ROS [156].
Senescence-accelerated mice often develop both senescent amyloidosis and secondary AA amyloidosis [157]. They are susceptible to lymphoma, inflammation (colitis, pneumonia, abscesses), and renal failure [157]. The molecular features of SAMP8 model are a decrease in the anti-apoptotic B-cell lymphoma 2 [Bcl2] expression and an increase in the pro-apoptotic Bcl-2 associated X-protein [BAX] expression [156] as well as an increase in the activity of the pro-apoptotic caspase 3 [155,156].
Figure 1.
Pathophysiological Mechanisms in Cardiometabolic HFpEF and related murine models.
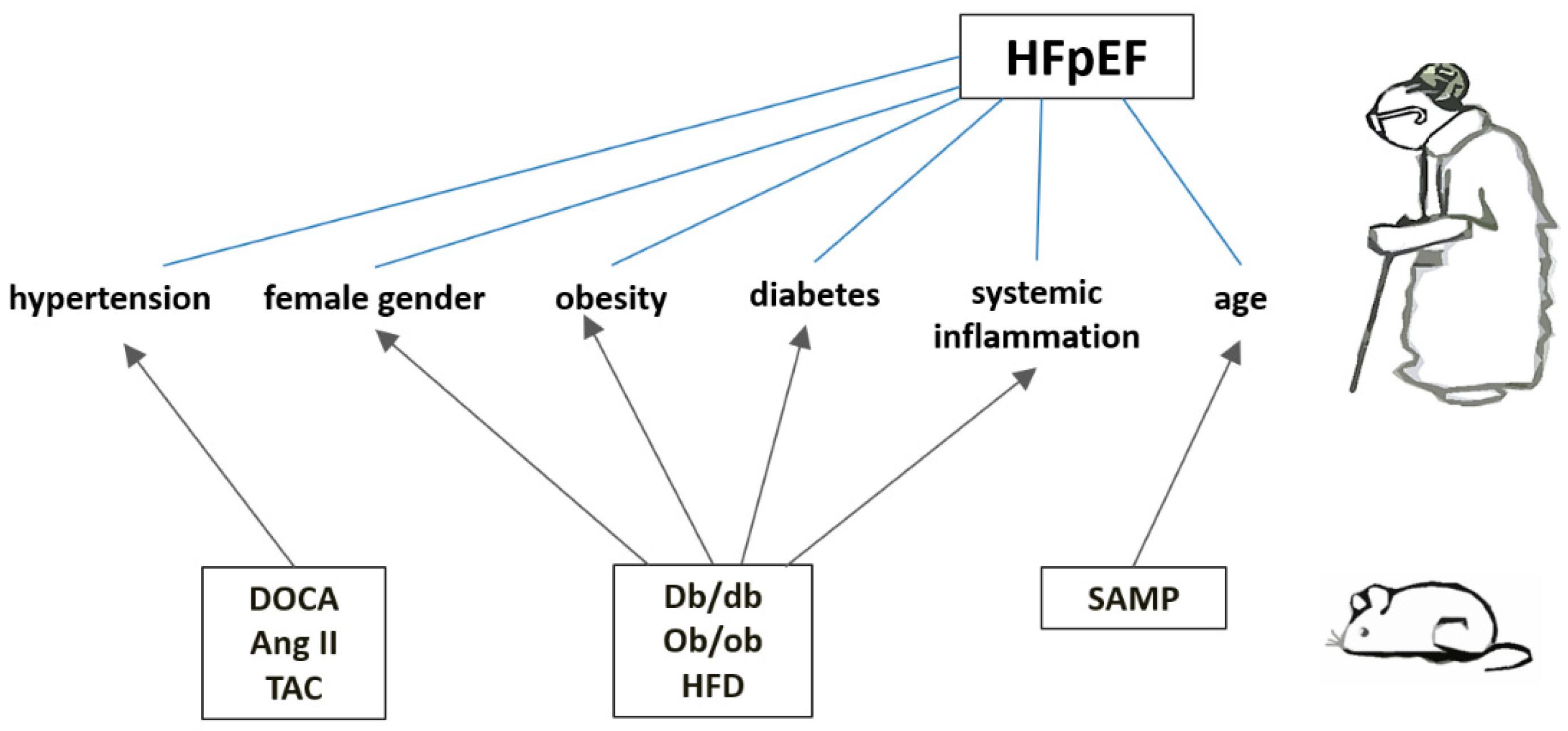
The figure illustrates the key pathophysiological mechanisms involved in the cardiometabolic phenotype of heart failure with preserved ejection fraction (HFpEF). Understanding these mechanisms is crucial for developing targeted therapeutic strategies for HFpEF. Murine models: Ang II - angiotensin II infusion; db/db - leptin receptor-deficient line; DOCA - chronic subjection to deoxycorticosterone acetate; ob/ob - leptin-deficient line; SAMP - senescence-accelerated prone strain; TAC - transverse aortic constriction.
Conclusions and Future Perspectives
In conclusion, HFpEF remains a complex and multifaceted condition with significant clinical and social impact. A better understanding of the mechanisms underlying the pathophysiology of HFpEF depends on the quality and comprehensiveness of basic, translational and clinical research. The murine models described in this review reproduce classic risk factors for the development of HFpEF, such as arterial hypertension, obesity, T2DM and aging, and will undoubtedly contribute to a better understanding of different aspects of HFpEF and help develop new treatment strategies (Figure). However, these models have several disadvantages. First, they do not replicate additional cardiac and non-cardiac impairments found in human HFpEF, such as atrial fibrillation, renal dysfunction, and chronic obstructive pulmonary disease [57]. Second, the lack of extensive characterization of certain models hinders their potential for further investigation as models for HFpEF. Additionally, several murine models may develop LV systolic dysfunction over time which is more typical of 'mono' models with isolated pressure overload or insulin resistance. It is worth noting that in complex models, LV systolic function typically remains preserved into advanced age [59].
According to data published, the db/db and ob/ob lines most accurately replicate the cardiometabolic characteristics typical of HFpEF among all the murine models. These strains represent the most well-characterized models examined in this review. Consequently, they illustrate sex differences in the onset and progression of HFpEF that closely resemble its clinical course in humans. The db/db strain shows a rapid development of pathological changes. In contrast, people with HFpEF experience a slower progression of these changes. Ob/ob mice show slower pathological changes making them more comparable to humans. Another drawback of the db/db and ob/ob strains is their infertility which creates challenges in breeding these models. In the ob/ob strain, this issue can be addressed with leptin administration. The HFD model is simpler and more accessible resembling natural conditions, but it has been studied less extensively than the db/db and ob/ob strains. Furthermore, HFD mice likely do not exhibit sex differences in pathological manifestations and thus, they may not accurately replicate the human clinical progression of HFpEF. Next, DOCA and Ang II models serve as representations of isolated arterial hypertension. Available data indicate that these models primarily involve vascular and renal systems rather than cardiac involvement; however, there are instances where inflammation is observed in the myocardium. The DOCA and Ang II models can be effectively utilised to investigate isolated arterial hypertension as a contributing factor to HFpEF. Further studies are needed to better characterize them. Aortic ligation models are the least characterized murine models for inflammation in HFpEF development. Thus, isolated aortic ligation is seen as a 'mono' model as it is neither 'natural' nor easy to perform. Although the addition of DOCA infusion to aortic ligation may have potential benefits, current data on the TAC+DOCA model is insufficient to assess its appropriateness for studying HFpEF. Finally, accelerated ageing models can study HFpEF's age-related characteristics, but they remain poorly understood. Additionally, 'genetically accelerated' aging is an artificial process, and these mice do not have T2DM, obesity, or arterial hypertension which are characteristics of patients with HFpEF. It is also noticeable that elderly patients with HFpEF and no cardiometabolic comorbidities are rarely seen in clinical practice.
Further research should explore these models and their translational potential to bridge the gap between preclinical findings and clinical applications.
Author Contributions
Conceptualization, T.A., A.O.; literature search, writing-original draft preparation, E.O., A.F.; writing - review and editing, A.F., N.R., T.A. and A.O.; supervision, T.I. and A.O.; funding acquisition, T.A. and A.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, grant number 24-75-00053.
Acknowledgments
The authors extend their gratitude to Elena Zwaan and Olga Filatova for their assistance with the English editing of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ADMA | Asymmetric dimethylarginine |
| ANP | Atrial natriuretic peptide |
| Bcl-2 | anti-apoptotic B-cell lymphoma 2 |
| BNP | Brain natriuretic peptide |
| cGMP | Cyclic guanosine monophosphate |
| cMYBP-C | Myosin-binding protein C |
| DOCA | Deoxycorticosterone acetate |
| eIF2α | Eukaryotic initiation factor 2 |
| eNOS | Endothelial nitric oxide synthase |
| GSH | Glutathione |
| GSSG | Glutathione disulfide |
| HF | Heart failure |
| HFD | High fat diet |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| IL | Interleukin |
| IRE-1 | Inositol-requiring protein-1α |
| iNOS | Inducible nitric oxide synthase |
| IRS-1 | Insulin receptor substrate-1 |
| IVRT | Isovolumic relaxation time |
| LV | Left ventricular |
| MCP-1 | Monocyte chemotactic protein-1 |
| MHC | Myosin heavy chain |
| NAD+ | Nicotinamide adenine dinucleotide phosphate |
| NADH | Nicotinamide adenine dinucleotide phosphate |
| nNOS | Neuronal nitric oxide synthase |
| NO | Nitric oxide |
| NOX | Nicotinamide adenine dinucleotide phosphate oxidase |
| NT-proBNP | N-terminal fragment of brain natriuretic hormone precursor |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| PKG | Protein kinase G |
| ROS | Reactive oxygen species |
| SAMP | Senescence-accelerated prone strain |
| SERCA | Sarco-endoplasmic reticulum Ca2+-ATPase |
| SIRT | Silent mating type information regulation 2 homolog 1 |
| T2DM | Type 2 diabetes mellitus |
| TAC | Transverse aortic constriction |
| TGF | Transforming growth factor |
| TNF | Tumor necrosis factor |
| XOR | Xanthine oxidoreductase |
References
- Vasan, R.S.; Xanthakis, V.; Lyass, A.; Andersson, C.; Tsao, C.; Cheng, S.; Aragam, J.; Benjamin, E.J.; Larson, M.G. Epidemiology of Left Ventricular Systolic Dysfunction and Heart Failure in the Framingham Study: An Echocardiographic Study Over 3 Decades. JACC Cardiovasc Imaging. 2018, 11, 1-11. [CrossRef]
- Borlaug, B.A.; Jensen, M.D.; Kitzman, D.W.; Lam, C.; Obokata, M.; Rider, O.J. Obesity and heart failure with preserved ejection fraction: new insights and pathophysiological targets. Cardiovasc Res. 2023, 118, 3434–3450. [CrossRef]
- Shah, K.S.; Xu, H.; Matsouaka, R.A.; Bhatt, D.L.; Heidenreich, P.A.; Hernandez, A.F.; Devore, A.D.; Yancy, C.W.; Fonarow, G.C. Heart Failure With Preserved, Borderline, and Reduced Ejection Fraction: 5-Year Outcomes. J Am Coll Cardiol. 2017, 70, 2476-2486. [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022, 145, e876-e894. 10.1161/CIR.0000000000001062.
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021, 42, 3599-3726. [CrossRef]
- Bozkurt, B.; Coats, A.;Tsutsui, H.; Abdelhamid, C.M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal definition and classification of heart failure: a report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. Eur J Heart Fail. 2021, 23, 352-380. [CrossRef]
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation. 2016, 134, 73-90. [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N Engl J Med. 2021, 385, 1451-1461. [CrossRef]
- Solomon, S.D.; McMurray, J.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N Engl J Med. 2022, 387, 1089-1098. [CrossRef]
- Solomon, S.D.; McMurray, J.; Vaduganathan, M.; Claggett, B.; Jhund, P.S.; Desai, A.S.; Henderson, A.D.; Lam, C.; Pitt, B.; Senni, M.; et al. Finerenone in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N Engl J Med. 2024, 391, 1475-1485. [CrossRef]
- Redfield, M.M.; Borlaug, B.A. Heart Failure With Preserved Ejection Fraction: A Review. JAMA. 2023, 329, 827-838. [CrossRef]
- Ovchinnikov, A.G.; Arefieva, T.I.; Potekhina, A.V.; Filatova, A.Y.; Ageev, F.T.; Boytsov, S.A. The Molecular and Cellular Mechanisms Associated with a Microvascular Inflammation in the Pathogenesis of Heart Failure with Preserved Ejection Fraction. Acta Naturae. 2020, 12, 40-51. [CrossRef]
- Schiattarella, G.G.; Rodolico, D.; Hill, J.A. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc Res. 2021, 117, 423–434. [CrossRef]
- Schiattarella, G.G.; Alcaide P.; Condorelli, G.; Gilette, T.G.; Heymans, S.; Jones A.; Kallikourdis, M.; Lichtman, A.; Marelli-Berg, F.; Shah, S.; et al. Immunometabolic mechanisms of heart failure with preserved ejection fraction. Nature Cardiovascular Research. 2022, 1, 211–222. [CrossRef]
- Putko, B.N.; Wang, Z.; Lo, J.; Anderson, T.; Becher, H.; Dyck, J.; Kassiri, Z.; Oudit, G.Y.; Alberta HEART Investigators. Circulating levels of tumor necrosis factor-alpha receptor 2 are increased in heart failure with preserved ejection fraction relative to heart failure with reduced ejection fraction: evidence for a divergence in pathophysiology. PLoS One. 2014, 9, e99495. [CrossRef]
- Tromp, J.; Khan, M.; Klip, I.T.; Meyer, S.; de Boer, R.A.; Jaarsma, T.; Hillege, H.; van Veldhuisen, D.J.; van der Meer, P.; Voors, A.A. Biomarker Profiles in Heart Failure Patients With Preserved and Reduced Ejection Fraction. J Am Heart Assoc. 2017, 6, e003989. [CrossRef]
- Santhanakrishnan, R.; Chong, J.P.; Ng, T.P.; Ling, L.H.; Sim, D.; Leong, K.T.; Yeo, P.S.; Ong, H.Y.; Jaufeerally, F.; Wong, R.; et al. Growth differentiation factor 15, ST2, high-sensitivity troponin T, and N-terminal pro brain natriuretic peptide in heart failure with preserved vs. reduced ejection fraction. Eur J Heart Fail. 2012, 14, 1338-1347. [CrossRef]
- Sanders-van Wijk, S.; van Empel, V.; Davarzani, N.; Maeder, M.T.; Handschin, R.; Pfisterer, M.E.; Brunner-La Rocca, H.P.; TIME-CHF investigators. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur J Heart Fail. 2015, 17, 1006-14. [CrossRef]
- Tromp, J.; Westenbrink, B.D.; Ouwerkerk, W.; van Veldhuisen, D.J.; Samani, N.J.; Ponikowski, P.; Metra, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K; et al. Identifying Pathophysiological Mechanisms in Heart Failure With Reduced Versus Preserved Ejection Fraction. J Am Coll Cardiol. 2018, 72, 1081-1090. [CrossRef]
- Carris, N.W.; Mhaskar, R.; Coughlin, E.; Bracey, E.; Tipparaju S.M., Halade, G.V. Novel biomarkers of inflammation in heart failure with preserved ejection fraction: analysis from a large prospective cohort study. BMC Cardiovasc Disord. 2022, 22, 221. [CrossRef]
- Lakhani, I.; Wong, M.V.; Hung, J.; Gong, M.; Waleed, K.B.; Xia, Y.; Lee, S.; Roever, L.; Liu, T.; Tse, G.; Leung, K.; Li, K. Diagnostic and prognostic value of serum C-reactive protein in heart failure with preserved ejection fraction: a systematic review and meta-analysis. Heart Fail Rev. 2021, 26, 1141-1150. [CrossRef]
- Alogna, A.; Koepp, K.E., Sabbah, M.; Espindola Netto, J.M.; Jensen, M.D.; Kirkland, J.L., Lam, C.; Obokata, M.; Petrie, M.C.; Ridker, P.M.; et al. Interleukin-6 in Patients With Heart Failure and Preserved Ejection Fraction. JACC Heart Fail. 2023, 11, 1549–1561. [CrossRef]
- Abernethy, A.; Raza, S.; Sun, J.L.; Anstrom, K.J.; Tracy, R.; Steiner, J.; VanBuren, P.; LeWinter, M.M. Pro-inflammatory biomarkers in stable versus acutely decompensated heart failure with preserved ejection fraction. J Am Heart Assoc. 2018, 7, e007385. [CrossRef]
- Chaar, D.; Dumont, B.L.; Vulesevic, B.; Neagoe, P-E.; Räkel, A.; White, M.; Sirois, M.G. Neutrophils and Circulating Inflammatory Biomarkers in Diabetes Mellitus and Heart Failure With Preserved Ejection Fraction. Am J Cardiol. 2022, 178, 80–88. [CrossRef]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011, 4, 44–52. [CrossRef]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312-324. [CrossRef]
- Mohammed, S.F.; Hussain, S.F.; Mirzoev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015, 131, 550-559. [CrossRef]
- Srivaratharajah, K.; Coutinho, T.; deKemp, R.; Liu, P.; Haddad, H.; Stadnick, E.; Davies, R.A.; Chih, S.; Dwivedi, G.; Guo, A.; et al. Reduced Myocardial Flow in Heart Failure Patients With Preserved Ejection Fraction. Circ Heart Fail. 2016, 9, e002562. [CrossRef]
- Glezeva, N.; Voon, V.; Watson, C.; Horgan, S.; McDonald, K.; Ledwidge, M.; Baugh J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: evidence of M2 macrophage activation in disease pathogenesis. J Card Fail. 2015, 21, 167–177. [CrossRef]
- Ovchinnikov, A.; Filatova, A.; Potekhina, A.; Arefieva, T.; Gvozdeva, A.; Ageev, F.; Belyavskiy, E. Blood Immune Cell Alterations in Patients with Hypertensive Left Ventricular Hypertrophy and Heart Failure with Preserved Ejection Fraction. J Cardiovasc Dev Dis. 2023, 10, 310. [CrossRef]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Zhen Joan Lee, Y.; Riley, S.J., Subramanya, V.; Brown, E.; Danielle Hopkins, C.; et al. Endomyocardial Biopsy Characterization of Heart Failure With Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020, 8, 712–724. [CrossRef]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Muñoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. 2018, 215, 423-440. [CrossRef]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf. M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018, 24, 1234-1245. [CrossRef]
- Bronzwaer, J.; Paulus, W.J. Diastolic and systolic heart failure: Different stages or distinct phenotypes of the heart failure syndrome? Curr Heart Fail Rep. 2009, 6, 281–286. [CrossRef]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure With Preserved Ejection Fraction. J Am Coll Cardiol. 2013, 62, 263–271. [CrossRef]
- Shah, S.J.; Katz, D.H.; Selvaraj, S.; Burke, M.A.; Yancy, C.W.; Cheorghiade, M.; Bonow, R.O.; Huang, C-C.; Deo, R.C. Phenomapping for Novel Classification of Heart Failure With Preserved Ejection Fraction. Circulation. 2015, 131, 269–279. [CrossRef]
- Kao, D.P.; Lewsey, J.D.; Anand, I.S.; Massie, B.M.; Zile, M.R.; Carson, P.E.; McKelvie, R.S.; Komajda, M.; McMurray, J.; Lindenfels, J. Characterization of subgroups of heart failure patients with preserved ejection fraction with possible implications for prognosis and treatment response. Eur J Heart Fail. 2015, 17, 925–935. [CrossRef]
- Shah, S.J.; Katz, D.H.; Deo, R.C. Phenotypic Spectrum of Heart Failure with Preserved Ejection Fraction. Heart Fail Clin. 2014, 10, 407–418. [CrossRef]
- Teramoto, K.; Katherine Teng, T-H.; Chandramouli, C.; Tromp, J., Sakata, Y.; Lam C.S. Epidemiology and Clinical Features of Heart Failure with Preserved Ejection Fraction. Card Fail Rev. 2022, 8, e27. [CrossRef]
- Mancuso P. The role of adipokines in chronic inflammation. Immunotargets Ther. 2016, 5, 47-56. [CrossRef]
- Lam, C.S. Diabetic cardiomyopathy: An expression of stage B heart failure with preserved ejection fraction. Diab Vasc Dis Res. 2015, 12, 234–238. [CrossRef]
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ Res. 2020, 126, 1501–1525. [CrossRef]
- Jackson, A.M.; Rørth, R.; Liu, J.; Kristensen, S.L.; Anand, I.S.; Claggett, B.L.; Cleland, J.; Chopra, V.K.; Desai, A.S.; Ge, J.; et al. Diabetes and pre-diabetes in patients with heart failure and preserved ejection fraction. Eur J Heart Fail. 2022, 24, 497–509. [CrossRef]
- Lehrke, M.; Marx, N. Diabetes Mellitus and Heart Failure. Am J Med. 2017, 130, S40–S50. [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.; Schalkwijk, C.G.; Bronzwaer, J.; Diamant, M.; et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. 2008, 117, 43–51. [CrossRef]
- Plante, T.B.; Juraschek, S.P.; Howard, G.; Howard, V.J.; Tracy, R.P., Olson, N.C.; Judd, S.E., Mukaz D.K.; Zakai, N.A.; Long D.L.; Cushman, M. Cytokines, C-Reactive Protein, and Risk of Incident Hypertension in the REGARDS Study. Hypertension. 2024, 81, 1244–1253. [CrossRef]
- Wang, Z.; Wang, J.; Yang, P.; Song, X.; Li, Y. Elevated Th17 cell proportion, related cytokines and mRNA expression level in patients with hypertension-mediated organ damage: a case control study. BMC Cardiovasc Disord. 2022, 22, 257. [CrossRef]
- Cantero-Navarro, E.; Fernández-Fernández, B.; Ramos, A.M.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Sánchez-Niño, M.D.; Sanz, A.B.; Ruiz-Ortega, M.; Ortiz, A. Renin-angiotensin system and inflammation update. Mol Cell Endocrinol. 2021, 529, 111254. [CrossRef]
- Xiao, L.; Harrison, D.G. Inflammation in Hypertension. Can J Cardiol. 2020, 36, 635–647. [CrossRef]
- Cohen, J.B.; Schrauben, S.J.; Zhao, L.; Basso, M.D.; Cvijic, M.E.; Li, Z.; Yarde, M.; Wang, Z.; Bhattacharya, P.T.; Chirinos, D.A.; et al. Clinical Phenogroups in Heart Failure With Preserved Ejection Fraction: Detailed Phenotypes, Prognosis, and Response to Spironolactone. JACC Heart Fail. 2020, 8, 172-184. [CrossRef]
- Galli, E.; Bourg, C.; Kosmala, W.; Oger, E.; Donal, E. Phenomapping Heart Failure with Preserved Ejection Fraction Using Machine Learning Cluster Analysis: Prognostic and Therapeutic Implications. Heart Fail Clin. 2021, 17, 499-518. [CrossRef]
- Lam, C.; Donal, E.; Kraigher-Krainer, E.; Vasan, R.S. Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur J Heart Fail. 2011, 13, 18–28. [CrossRef]
- Packer, M.; Lam, C.; Lund, L.H.; Maurer, M.S.; Borlaug, B.A. Characterization of the inflammatory-metabolic phenotype of heart failure with a preserved ejection fraction: a hypothesis to explain influence of sex on the evolution and potential treatment of the disease. Eur J Heart Fail. 2020, 22, 1551–1567. [CrossRef]
- Segar, M.W.; Patel, K.V.; Ayers, C.; Basit, M.; Tang, W.; Willett, D.; Berry, J.; Grodin, J.L.; Pandey, A. Phenomapping of patients with heart failure with preserved ejection fraction using machine learning-based unsupervised cluster analysis. Eur J Heart Fail. 2020, 22, 148-158. [CrossRef]
- Woolley, R.J.; Ceelen, D.; Ouwerkerk, W.; Tromp, J.; Figarska, S.M.; Anker, S.D.; Dickstein, K.; Filippatos, G.; Zannad, F.; Metra, M.; et al. Machine learning based on biomarker profiles identifies distinct subgroups of heart failure with preserved ejection fraction. Eur J Heart Fail. 2021, 23, 983-991. [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Shahzeb Khan, M.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; et al. Baseline characteristics of patients with heart failure with preserved ejection fraction in the EMPEROR-Preserved trial. Eur J Heart Fail. 2020, 22, 2383-2392. [CrossRef]
- Riehle, C.; Bauersachs, J. Small animal models of heart failure. Cardiovasc Res. 2019, 115, 1838-1849. [CrossRef]
- Miyagi, C.; Miyamoto, T.; Kuroda, T.; Karimov, J.H.; Starling, R.C.; Fukamachi, K. Large animal models of heart failure with preserved ejection fraction. Heart Fail Rev. 2022, 27, 595-608. [CrossRef]
- Gao, S.; Liu, X.P.; Li, T.T.; Chen, L.; Feng, Y.P.; Wang, Y.K.; Yin, Y.J.; Little, P.J.; Wu, X.Q.; Xu, S.W.; Jiang, X.D. Animal models of heart failure with preserved ejection fraction (HFpEF): from metabolic pathobiology to drug discovery. Acta Pharmacol Sin. 2024, 45, 23-35. [CrossRef]
- Lourenço, A.P.; Leite-Moreira, A.F.; Balligand, J.L.; Bauersachs, J.; Dawson, D.; de Boer, R.A.; de Windt, L.J.; Falcão-Pires, I.; Fontes-Carvalho, R.; Franz, S.; et al. An integrative translational approach to study heart failure with preserved ejection fraction: a position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur J Heart Fail. 2018, 20, 216-227. [CrossRef]
- Schauer, A.; Draskowski, R.; Jannasch, A.; Kirchhoff, V.; Goto, K.; Männel, A.; Barthel. P.; Augstein, A.; Winzer. E.; Tugtekin, M.; et al. ZSF1 rat as animal model for HFpEF: Development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail. 2020, 7, 2123-2134. [CrossRef]
- Murase, T.; Hattori, T.; Ohtake, M.; Abe, M.; Amakusa, Y.; Takatsu, M.; Murohara, T.; Nagata K. Cardiac remodeling and diastolic dysfunction in DahlS.Z-Lepr(fa)/Lepr(fa) rats: a new animal model of metabolic syndrome. Hypertens Res. 2012, 35, 186-193. [CrossRef]
- Schwarzl, M.; Hamdani, N.; Seiler, S.; Alogna, A.; Manninger, M.; Reilly, S.; Zirngast, B.; Kirsch, A.; Steendijk, P.; Verderber, J.; et al. A porcine model of hypertensive cardiomyopathy: implications for heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2015, 309, H1407-18. [CrossRef]
- Sharp, T.E. 3rd; Scarborough, A.L.; Li, Z.; Polhemus, D.J.; Hidalgo, H.A.; Schumacher, J.D.; Matsuura, T.R.; Jenkins, J.S.; Kelly, D.P.; Goodchild, T.T.; Lefer, DJ. Novel Göttingen Miniswine Model of Heart Failure With Preserved Ejection Fraction Integrating Multiple Comorbidities. JACC Basic Transl Sci. 2021, 6, 154-170. [CrossRef]
- Wang, L.; Halliday, G.; Huot, J.R.; Satoh, T.; Baust, J.J.; Fisher, A.; Cook, T.; Hu, J.; Avolio, T.; Goncharov, D.A.; et al. Treatment With Treprostinil and Metformin Normalizes Hyperglycemia and Improves Cardiac Function in Pulmonary Hypertension Associated With Heart Failure With Preserved Ejection Fraction. Arterioscler Thromb Vasc Biol. 2020, 40, 1543-1558. [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ Res. 2021, 128, 232-245. [CrossRef]
- Withaar, C.; Meems, L.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Silljé, H.; Schouten, E.M.; Dokter, M.M.; Voors, A.A.; Westenbrink, B.D.; Lam, C.; de Boer, R.A. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc Res. 2021, 117, 2108-2124. [CrossRef]
- Gevaert, A.B.; Shakeri, H.; Leloup, A.J.; Van Hove, C.E.; De Meyer, G.; Vrints, C.J.; Lemmens, K.; Van Craenenbroeck, E.M. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ Heart Fail. 2017, 10, e003806. [CrossRef]
- Li, Y.; Kubo, H.; Yu, D.; Yang, Y.; Johnson, J.P.; Eaton, D.M.; Berretta, R.M.; Foster, M.; McKinsey, T.A.; Yu, J.; et al. Combining three independent pathological stressors induces a heart failure with preserved ejection fraction phenotype. Am J Physiol Heart Circ Physiol. 2023, 324, H443-H460. [CrossRef]
- Kelley, R.C.; Betancourt. L.; Noriega, A.M.; Brinson, S.C.; Curbelo-Bermudez, N.; Hahn, D.; Kumar, R.A.; Balazic, E.; Muscato, D.R.; Ryan, T.E.; et al. Skeletal myopathy in a rat model of postmenopausal heart failure with preserved ejection fraction. J Appl Physiol. 2022, 132, 106-125. [CrossRef]
- Vilariño-García, T.; Polonio-González, M.L.; Pérez-Pérez, A.; Ribalta, J.; Arrieta, F.; Aguilar, M.; Obaya, J.C.; Gimeno-Orna, J.A.; Iglesias, P.; Navarro, J.; et al. Role of Leptin in Obesity, Cardiovascular Disease, and Type 2 Diabetes. Int J Mol Sci. 2024, 25, 2338. [CrossRef]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; Moore, K.J.; Breitbart, R.E.; et al. Evidence That the Diabetes Gene Encodes the Leptin Receptor: Identification of a Mutation in the Leptin Receptor Gene in db/db Mice. Cell. 1996, 84, 491–495. [CrossRef]
- Mori, J.; Patel, V.B.; Alrob, O.A.; Basu, R.; Altamimi, T.; Desaulniers, J.; Wagg, C.S.; Zamaneh, K.; Lopaschuk, G.D.; Oudit, G.Y. Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ Heart Fail. 2014, 7, 327–339. [CrossRef]
- Vecoli, C.; Cao, J.; Neglia, D.; Inoue, K.; Sodhi, K.; Vanells, L.; Gabrielson, K.K.; Bedja, D.; Paolocci, N.; L’abbate, A.; Abraham. N.G. Apolipoprotein A-I mimetic peptide L-4F prevents myocardial and coronary dysfunction in diabetic mice. J Cell Biochem, 2011, 112, 2616–2626. [CrossRef]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Jeong Yun, U.; Cooksey, R.C.; Litwin, S.E.; Dale Abel, E. Reduced Cardiac Efficiency and Altered Substrate Metabolism Precedes the Onset of Hyperglycemia and Contractile Dysfunction in Two Mouse Models of Insulin Resistance and Obesity. Endocrinology. 2005, 146, 5341–5349. [CrossRef]
- Ge, F.; Hu, C.; Hyodo, E.; Arai, K.; Zhou, S.; Lobdell 4th, H.; Walewski, J.L.; Homma, S.; Berk, P.D. Cardiomyocyte Triglyceride Accumulation and Reduced Ventricular Function in Mice with Obesity Reflect Increased Long Chain Fatty Acid Uptake and De Novo Fatty Acid Synthesis. J Obes. 2012, 2012, 205648. [CrossRef]
- Kobayashi, K.; Forte, T.M.; Taniguchi, S.; Ishida, B.Y.; Oka, K.; Chan, L. The db/db mouse, a model for diabetic dyslipidemia: Molecular characterization and effects of western diet feeding. Metabolism. 2000, 49, 22–31. [CrossRef]
- Senador, D.; Kanakamedala, K.; Irigoyen, M.C.; Morris, M.; Elased, K.M. Cardiovascular and autonomic phenotype of db / db diabetic mice. Exp Physiol. 2009, 94, 648–658.
- Bowden, M.A.; Tesch, G.H.; Julius, T.; Rosli, S.; Love, J.E.; Ritchie, R.H. Earlier onset of diabesity-Induced adverse cardiac remodeling in female compared to male mice. Obesity (Silver Spring). 2015, 23, 1166–1177. [CrossRef]
- Dong, F.; Ren, J. Adiponectin improves cardiomyocyte contractile function in db/db diabetic obese mice. Obesity (Silver Spring). 2009, 17, 262–268. [CrossRef]
- Semeniuk, L.M.; Kryski, A.J.; Severson, D.L. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db -hGLUT4 mice. Am J Physiol Heart Circ Physiol. 2002, 283, H976–H982. [CrossRef]
- Daniels, A.; van Bilsen, M.; Janssen, B.; Brouns, A.E.; Cleutjens, J.; Roemen, T.; Schaart, G.; van der Velden J.; van der Vusse, G.J.; van Nieuwenhoven, F.A. Impaired cardiac functional reserve in type 2 diabetic db/db mice is associated with metabolic, but not structural, remodelling. Acta Physiologica. 2010, 200, 11–22. [CrossRef]
- Alex, L.; Russo, I.; Holoborodko, V.; Frangogiannis, N.G. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2018, 315, H934–H949. [CrossRef]
- Hamdani, N.; Hervent, A-S.; Vandekerckhove, L.; Matheeussen, V.; Demolder, M.; Baerts, L.; De Meester, I.; Linke, W.A.; Paulus, W.J.; Keulenaer, G. Left ventricular diastolic dysfunction and myocardial stiffness in diabetic mice is attenuated by inhibition of dipeptidyl peptidase 4. Cardiovasc Res. 2014, 104, 423–431. [CrossRef]
- Zhao, R.; Xie, X.; Le, K.; Li, W.; Moghadasian, M.H.; Beta, T.; Shen, G.X. Endoplasmic reticulum stress in diabetic mouse or glycated LDL-treated endothelial cells: protective effect of Saskatoon berry powder and cyanidin glycans. J Nutr Biochem. 2015, 26, 1248–1253. [CrossRef]
- Koka, S.; Aluri, H.S.; Xi, L.; Lesnefsky, E.; Kukreja, R.C. Chronic inhibition of phosphodiesterase 5 with tadalafil attenuates mitochondrial dysfunction in type 2 diabetic hearts: potential role of NO/SIRT1/PGC-1α signaling. Am J Physiol Heart Circ Physiol. 2014, 306, H1558-68. [CrossRef]
- Monma, Y.; Shindo, T.; Eguchi, K.; Kurosawa, R.; Kagaya, Y.; Ikumi, Y.; Ichijo, S.; Nakata, T.; Miyata, S.; Matsumoto, A.; Sato, H.; Miura, M.; Kanai, H.; Shimokawa, H. Low-intensity pulsed ultrasound ameliorates cardiac diastolic dysfunction in mice: a possible novel therapy for heart failure with preserved left ventricular ejection fraction. Cardiovasc Res. 2021, 117, 1325–1338. [CrossRef]
- Reil, J.-C.; Honl, M.; Reil, G-H.; Granzier, H.L.; Kratz, M.T.; Kazakov, A.; Fries, P.; Müller, A.; Lenski, M.; Custodis, F.; et al. Heart rate reduction by If-inhibition improves vascular stiffness and left ventricular systolic and diastolic function in a mouse model of heart failure with preserved ejection fraction. Eur Heart J. 2013, 34, 2839–2849. [CrossRef]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J Hered. 1950, 41, 317–318. [CrossRef]
- Barouch, L.A.; Berkowitz, D.E.; Harrison, R.W.; O’Donell, C.P.; Hare, J.M. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. 2003, 108, 754–759. [CrossRef]
- Hammoudi, N.; Jeong, D.; Singh, R.; Farhat, A.; Komajda, M.; Mayoux, E.; Hajjar, R.; Lebeche, D. Empagliflozin Improves Left Ventricular Diastolic Dysfunction in a Genetic Model of Type 2 Diabetes. Cardiovasc Drugs Ther. 2017, 31, 233–246. [CrossRef]
- Guo, W.; Jiang, T.; Lian, C.; Wang, H.; Zheng, Q.; Ma, H. QKI deficiency promotes FoxO1 mediated nitrosative stress and endoplasmic reticulum stress contributing to increased vulnerability to ischemic injury in diabetic heart. J Mol Cell Cardiol. 2014, 75, 131–140. [CrossRef]
- Minhas, K.M.; Khan, S.A.; Raju, S.; Phan, A.C.; Gonzalez, D.R.; Skaf, M.W.; Lee, K.; Tejani, A.D.; Saliaris, A.P.; Barouch, L.A.; et al. Leptin repletion restores depressed {beta}-adrenergic contractility in ob/ob mice independently of cardiac hypertrophy. J Physiol. 2005, 565, 463–474. [CrossRef]
- Li, S-Y.; Yang, X.; Ceylan-Isik, A.F.; Du, M.; Sreejayan, N.; Ren, J. Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia. 2006, 49, 1434–1446. [CrossRef]
- Adingupu, D.D.; Göpel 2, S.O.; Grönros, J.; Behrendt, M.; Sotak, M.; Miliotis, T.; Dahlqvist, U.; Gan, L-M.; Jönsson-Rylander, A-C. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob-/- mice. Cardiovasc Diabetol. 2019, 18, 16. [CrossRef]
- Christoffersen, C.; Bollano, E.; Lindegaard, M.; Bartels, E.D.; Goetze, J.P.; Andersen. C.B.; Nielsen, L.B. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003, 144, 3483–3490. [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H-C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc Drugs Ther. 2017, 31, 119–132. [CrossRef]
- Mariappan, N.; Elks, C.M.; Sriramula, S.; Guggilam, A.; Liu, Z.; Borkhsenious, O.; Francis, J. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc Res. 2010, 85, 473–483. [CrossRef]
- Methawasin, M.; Strom, J.; Borkowski, T.; Hourani, Z.; Runyan, R.; Smith 3rd J.E.; Granzier, H. Phosphodiesterase 9a Inhibition in Mouse Models of Diastolic Dysfunction. Circ Heart Fail. 2020, 13, e006609. [CrossRef]
- Broderick, T.L.; Parrott, C.R.; Wang, D.; Jankowski, M.; Gutkowska, J. Expression of cardiac GATA4 and downstream genes after exercise training in the db/db mouse. Pathophysiology. 2012, 19, 193–203. [CrossRef]
- Belke, D.D.; Swanson, E.A.; Dillmann, W.H. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes. 2004, 53, 3201–3208. [CrossRef]
- Lugnier, C.; Meyer, A.; Charloux, A.; Andrès, E.; Gény, B.; Talha, S. The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure. J Clin Med. 2019, 8, 1746. [CrossRef]
- Juguilon, C.; Wang, Z.; Wang, Y.; Enrick, M.; Jamaiyar, A.; Xu, Y.; Gadd, J.; Chen, C-L.W.; Pu, A.; Kolz, C.; et al. Mechanism of the switch from NO to H2O2 in endothelium-dependent vasodilation in diabetes. Basic Res Cardiol. 2022, 117, 2. [CrossRef]
- Saraiva, R.M.; Minhas, K.M.; Zheng, M.; Pitz, E.; Treuer, A.; Gonzalez, D.; Schuleri, K.H.; Vandegaer, K.M.; Barouch, L.A.; Hare, J.M. Reduced neuronal nitric oxide synthase expression contributes to cardiac oxidative stress and nitroso-redox imbalance in ob/ob mice. Nitric Oxide. 2007, 16, 331–338. [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019, 568, 351-356. [CrossRef]
- Cantero-Navarro, E.; Fernández-Fernández, B.; Ramos, A.M.; Rayego-Mateos, S.; Rodrigues-Diez, R.P.; Dolores Sánchez-Niño, M.; Sanz, A.B.; Ruiz-Ortega, M.; Ortiz, A. Renin-angiotensin system and inflammation update. Mol Cell Endocrinol. 2021, 529, 111254. [CrossRef]
- van Heerebeek, L.; Borbély, A.; Niessen, H.; Bronzwaer, J.; van der Velden, J.; Stienen, G.; Linke, W.A.; Laarman, G.J.; Paulus, W.J. Myocardial Structure and Function Differ in Systolic and Diastolic Heart Failure. Circulation. 2006, 113, 1966–1973.. [CrossRef]
- Linke, W.A.; Hamdani, N. Gigantic Business: titin properties and function through thick and thin. Circ Res. 2014, 114, 1052–1068. [CrossRef]
- Greene, S.J.; Gheorghiade, M.; Borlaug, B.A.; Pieske, B.; Vaduganathan, M.; Burnett Jr, J.C.; Roessig, L.; Stasch, J-P.; Solomon, S.D.; Paulus, W.J.; Butler, J. The cGMP Signaling Pathway as a Therapeutic Target in Heart Failure With Preserved Ejection Fraction. J Am Heart Assoc. 2013, 2, e000536. [CrossRef]
- Münzel, T.; Gori, T.; Keaney Jr, J.F.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J. 2015, 36, 2555–2564. [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F., Begieneman, M.; Bronzwaer, J.; van der Velden, J.; Stienen, G.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012, 126, 830–839. [CrossRef]
- Williams, T.D.; Chambers, J.B.; Roberts, L.M.; Henderson, R.P.; Overton, J.M. Diet-induced obesity and cardiovascular regulation in C57BL/6J mice. Clin Exp Pharmacol Physiol. 2003, 30, 769–778. [CrossRef]
- Basheer, S.; Malik, I.R.; Awan F.R.; Sughra, K.; Roshan, S.; Khalil, A.; Iqbal, M.J.; Parveen, Z. Histological and Microscopic Analysis of Fats in Heart, Liver Tissue, and Blood Parameters in Experimental Mice. Genes (Basel). 2023, 14, 515.. [CrossRef]
- Benetti, E.; Mastrocola, R.; Vitarelli, G.; Cutrin. J.C.; Nigro, D.; Chiazza, F.; Mayoux, E.; Collino, M.; Fantozzi, R. Empagliflozin Protects against Diet-Induced NLRP-3 Inflammasome Activation and Lipid Accumulation. Journal of Pharmacology and Experimental Therapeutics. 2016, 359, 45–53. [CrossRef]
- Zhou, X.; Li, Z.; Qi, M.; Zhao, P.; Duan, Y.; Yang, G.; Yuan, L. Brown adipose tissue-derived exosomes mitigate the metabolic syndrome in high fat diet mice. Theranostics. 2020, 10, 8197–8210. [CrossRef]
- Wang, H.-T.; Liu, C-F.; Tsai, T-H.; Chen, Y-L.; Chang, H-W.; Tsai, C-Y.; Leu, S.; Zhen, Y-Y.; Chai, H-T.; Chung, S-Y.; et al. Effect of obesity reduction on preservation of heart function and attenuation of left ventricular remodeling, oxidative stress and inflamma-tion in obese mice. J Transl Med. 2012, 10, 145. [CrossRef]
- Abdurrachim, D.; Ciapaite, J.; Wessels, B.; Nabben, M.; Luiken, J.; Nicolay, K.; Prompers, J.J. Cardiac diastolic dysfunction in high-fat diet fed mice is associated with lipotoxicity without impairment of cardiac energetics in vivo. Biochim Biophys Acta. 2014, 1842, 1525–1537. [CrossRef]
- Steven, S.; Dib, M.; Hausding, M.; Kashani, F.; Oelze, M.; Kröller-Schön, S.; Hanf, A.; Daub, S.; Roohani, S.; Gramlich, Y.; et al. CD40L controls obesity-associated vascular inflammation, oxidative stress, and endothelial dysfunction in high fat diet-treated and db/db mice. Cardiovasc Res. 2018, 114, 312–323. [CrossRef]
- Li, W.; Tang, R.; Ouyang, S.; Ma, F.; Liu, Z.; Wu, J. Folic acid prevents cardiac dysfunction and reduces myocardial fibrosis in a mouse model of high-fat diet-induced obesity. Nutr Metab (Lond). 2017, 14, 68. [CrossRef]
- Jeong, E.; Chung, J.; Liu, H.; Go, Y.; Gladstein, S.; Farzaneh-Far, A.; Lewandowski, E.D.; Dudley Jr, S.C. Role of Mitochondrial Oxidative Stress in Glucose Tolerance, Insulin Resistance, and Cardiac Diastolic Dysfunction. J Am Heart Assoc. 2016, 5, e003046. [CrossRef]
- Carbone, S.; Mauro, A.G.; Mezzaroma, E.; Kraskauskas, D.; Marchetti, C.; Buzzetti, R.; Van Tassell, B.W.; Abbate, A.; Toldo, S. A high-sugar and high-fat diet impairs cardiac systolic and diastolic function in mice. Int J Cardiol. 2015, 198, 66–69. [CrossRef]
- Roche, C.; Besnier, M.; Cassel, R.; Harouki, N.; Coquerel, D.; Guerrot, D.; Nicol, L.; Loizon, E.; Remy-Jouet, I.; Morisseau, C.; et al. Soluble epoxide hydrolase inhibition improves coronary endothelial function and prevents the development of cardiac alterations in obese insulin-resistant mice. Am J Physiol Heart Circ Physiol. 2015, 308, H1020-9. [CrossRef]
- Heinzel, F.R.; Shah, S.J. The future of heart failure with preserved ejection fraction: Deep phenotyping for targeted therapeutics. Herz. 2022, 47, 308-323. [CrossRef]
- Xia, N.; Weisenburger, S.; Koch, E.; Burkart, M.; Reifenberg, G.; Förstermann, U.; Li, H. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br J Pharmacol. 2017, 174, 3443–3453. [CrossRef]
- Smart, C.D.; Fehrenbach, D.J.; Wassenaar, J.W.; Agrawal, V.; Fortune, N.L.; Dixon, D.D.; Cottam, M.A.; Hasty, A.H.; Hemnes, A.R.; Doran, A.C.; Gupta, D.K.; Madhur, M.S. Immune profiling of murine cardiac leukocytes identifies triggering receptor expressed on myeloid cells 2 as a novel mediator of hyper-tensive heart failure. Cardiovasc Res. 2023, 119, 2312–2328. [CrossRef]
- Yan, W.; Bi, H-L.; Liu, L-X.; Li, N-N.; Liu, Y.; Du, J.; Wang, H-Z.; Li, H-H. Knockout of immunoproteasome subunit β2i ameliorates cardiac fibrosis and inflammation in DOCA/Salt hypertensive mice. Biochem Biophys Res Commun. 2017, 490, 84–90. [CrossRef]
- Cai, R.; Hao, Y.; Liu, Y-Y.; Huang, L.; Yao, Y.; Zhou, M-S. Tumor Necrosis Factor Alpha Deficiency Improves Endothelial Function and Cardiovascular Injury in Deoxycorticosterone Acetate/Salt-Hypertensive Mice. Biomed Res Int. 2020, 2020, 1–10. [CrossRef]
- Sun, Y.; Carretero, O.A.; Xu, J.; Rhaleb, N-E.; Wang, F.; Lin, C.; Yang, J.J.; Pagano, P.J.; Yang, X-P. Lack of inducible NO synthase reduces oxidative stress and enhances cardiac response to isoproterenol in mice with deoxycorticosterone acetate-salt hypertension. Hypertension. 2005, 46, 1355–1361. [CrossRef]
- Silberman, G.A.; Fan, T-H.M.; Liu, H.; Jiao, Z.; Xiao, H.D.; Lovelock, J.D.; Boulden, B.M.; Widder, J.; Fredd, S.; Bernstein, K.E.; et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010, 121, 519–528. [CrossRef]
- Karatas, A.; Hegner, B.; Windt, L.J.; Luft, F.C.; Schubert, C.; Gross, V.; Akashi, Y.J.; Gürgen, D.; Kintscher, U.; da Costa Goncalves, A.C.; et al. Deoxycorticosterone acetate-salt mice exhibit blood pressure-independent sexual dimorphism. Hypertension. 2008, 51, 1177–1183. [CrossRef]
- Hartner, A.; Cordasic, N.; Klanke, B.; Veelken, R.; Hilgers, K.F. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol Dial Transplant. 2003, 18, 1999–2004. [CrossRef]
- Lin, Y.; Fu, S.; Yao, Y.; Li, Y.; Zhao, Y.; Luo, L. Heart failure with preserved ejection fraction based on aging and comorbidities. J Transl Med. 2021, 19, 291. [CrossRef]
- Schnelle, M.; Catibog, N.; Zhang, M.; Nabeebaccus, A.A.; Anderson, G.; Richards, D.A,; Sawyer, G.; Zhang, X.; Toischer, K.; Hasenfuss, G.; Monaghan, M.J.; Shah, A.M. Echocardiographic evaluation of diastolic function in mouse models of heart disease. J Mol Cell Cardiol. 2018, 114, 20–28. [CrossRef]
- Mohammed, S.F.; Ohtani, T.; Korinek, J.; Lam, C.; Larsen, K.; Simari, R.D.; Valenchik, M.L.; Burnett Jr, J.C.; Redfiled, M.M. Mineralocorticoid accelerates transition to heart failure with preserved ejection fraction via “nongenomic effects”. Circulation. 2010, 122, 370–378. [CrossRef]
- Kirchhoff, F.; Krebs, C.; Abdulhag, U.N.; Meyer-Schwesinger, C.; Maas, R.; Helmchen, U.; Hilgers, K.F.; Wolf, G.; Stahl, R.; Wenzel, U. Rapid development of severe end-organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int. 2008, 73, 643–650. [CrossRef]
- Wang, Q.; Euy-Myoung, J.; Liu, H.; Gu, L.; Dudley, S.C.; Yu, J. Astragaloside IV improves left ventricular diastolic dysfunction in hypertensive mice by increasing the phosphorylation of endothelial nitric oxide synthase. J Hypertens. 2016, 34, e48–e49. [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. Journal of Clinical Investigation. 2003, 111, 1201–1209. [CrossRef]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007, 204, 2449–2460. [CrossRef]
- Tian, Y.; Luo, J.; Xu, Q.; Liu, Y.; Cai, R.; Zhou, M-S. Macrophage depletion protects against endothelial dysfunction and cardiac remodeling in angiotensin II hypertensive mice. Clin Exp Hypertens. 2021, 43, 699–706. [CrossRef]
- Regan, J.A.; Mauro, A.G.; Carbone, S.; Marchetti, C.; Gill, R.; Mezzaroma, E.; Raleigh, J.V.; Salloum, F.N.; Van Tassel, B.W.; Abbate, A.; Toldo, S. A mouse model of heart failure with preserved ejection fraction due to chronic infusion of a low subpressor dose of angiotensin II. Am J Physiol Heart Circ Physiol. 2015, 309, H771-8. [CrossRef]
- Wang, B.; Xu, M.; Li, W.; Li, X.; Zheng, Q.; Niu, X. Aerobic exercise protects against pressure overload-induced cardiac dysfunction and hypertrophy via β3-AR-nNOS-NO activation. PLoS One. 2017, 12, e0179648. [CrossRef]
- Chen, Y.; Guo, H.; Xu, D.; Xu, X.; Wang, H.; Hu, X.; Lu, Z.; Kwak, D.; Xu, Y.; Gunther, R.; et al. Left ventricular failure produces profound lung remodeling and pulmonary hypertension in mice: heart failure causes severe lung disease. Hypertension. 2012, 59, 1170–1178. [CrossRef]
- An, D.; Zeng, Q.; Zhang, P.; Ma, Z.; Zhang, H.; Liu, Z.; Li, J.; Ren, H.; Xu, D. Alpha-ketoglutarate ameliorates pressure overload-induced chronic cardiac dysfunction in mice. Redox Biol. 2021, 46, 102088. [CrossRef]
- Tsujita, Y.; Kato, T.; Sussman, M.A. Evaluation of left ventricular function in cardiomyopathic mice by tissue Doppler and color M-mode Doppler echocardiography. Echocardiography. 2005, 22, 245–253. [CrossRef]
- Lu, Z.; Xu, X.; Hu, X.; Zhu, G.; Zhang, P.; van Deel. E.D.; French, J.P.; Fassett, J.T.; Oury, T.D.; Bache, R.J.; Chen, Y. Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension. 2008, 51, 19–25. [CrossRef]
- Zi, M.; Stafford, N.; Prehar, S.; Baudoin, F.; Oceandy, D.; Wang, X.; Bui, T.; Shaheen, M.; Neyses, L.; Cartwright, E.J. Cardiac hypertrophy or failure? - A systematic evaluation of the transverse aortic constriction model in C57BL/6NTac and C57BL/6J substrains. Curr Res Physiol. 2019, 1, 1–10. [CrossRef]
- Baier, M.J.; Klatt, S.; Hammer, K.P.; Maier, L.S.; Rokita, A.G. Ca2+/calmodulin-dependent protein kinase II is essential in hyperacute pressure overload. J Mol Cell Cardiol. 2020, 138, 212–221. [CrossRef]
- Slater, R.E.; Strom, J.G.; Methawasin, M.; Liss, M.; Gotthardt, M.; Sweitzer, N.; Granzier, H.L. Metformin improves diastolic function in an HFpEF-like mouse model by increasing titin compliance. J Gen Physiol. 2019, 151, 42–52. [CrossRef]
- Qiu, Z.; Fan, Y.; Wang, Z.; Huang, F.; Li, Z.; Sun, Z.; Hua, S.; Jin, W.; Chen, Y. Catestatin Protects Against Diastolic Dysfunction by Attenuating Mitochondrial Reactive Oxygen Species Generation. J Am Heart Assoc. 2023, 12, e029470. [CrossRef]
- Methawasin, M., Granzier, H. Experimentally Increasing the Compliance of Titin Through RNA Binding Motif-20 (RBM20) Inhibition Improves Diastolic Function In a Mouse Model of Heart Failure With Preserved Ejection Fraction. Circulation. 2016, 134, 1085–1099. [CrossRef]
- Dimitriadis, K.; Theofilis, P.; Koutsopoulos, G.; Pyrpyris, N.; Beneki, E.; Tatakis, F.; Tsioufis, P.; Chrysohoou, C.; Fragkoulis, C.; Tsioufis K. The role of coronary microcirculation in heart failure with preserved ejection fraction: An unceasing odyssey. Heart Fail Rev. 2025, 30, 75-88. [CrossRef]
- Marie, A.; Larroze-Chicot. P.; Cosnier-Pucheu, S.; Gonzalez-Gonzalez, S. Senescence-accelerated mouse prone 8 (SAMP8) as a model of age-related hearing loss. Neurosci Lett. 2017, 656, 138-143. [CrossRef]
- Reed, A.L.; Tanaka, A.; Sorescu, D.; Liu, H.; Jeong, E-M.; Strudy, M.; Walp, E.R.; Dudley Jr, S.C.; Sutliff, R.L. Diastolic dysfunction is associated with cardiac fibrosis in the senescence-accelerated mouse. Am J Physiol Heart Circ Physiol. 2011, 301, H824–H831. [CrossRef]
- Rodriguez-Calvo, R.; Serrano, L.; Barroso, E.; Coll, T.; Palomer, X.; Camins, A.; Sánchez, R.M.; Alegret, M.; Merlos, M.; Pallàs, M.; et al. Peroxisome Proliferator-Activated Receptor Down-Regulation Is Associated With Enhanced Ceramide Levels in Age-Associated Cardiac Hypertrophy. J Gerontol A Biol Sci Med Sci. 2007, 62, 1326–1336. [CrossRef]
- Giridharan, V.V.; Karupppagounder, V.; Arumugam, S.; Nakamura, Y.; Guha, A.; Barichello, T.; Quevedo, J.; Watanabe, K.; Konishi, T.; Thandavarayan, R.A. 3,4-Dihydroxybenzalacetone (DBL) Prevents Aging-Induced Myocardial Changes in Senescence-Accelerated Mouse-Prone 8 (SAMP8) Mice. Cells. 2020, 9, 597. [CrossRef]
- Forman, K.; Vara, E.; García, C.; Kireev, R.; Cuesta, S.; Escames, G.; Tresquerres, J. Effect of a Combined Treatment With Growth Hormone and Melatonin in the Cardiological Aging on Male SAMP8 Mice. J Gerontol A Biol Sci Med Sci. 2011, 66A, 823–834. [CrossRef]
- Takeda, T.; Matsushita, T.; Kurozumi, M.; Takemura, K.; Higuchi, K.; Hosokawa, M. Pathobiology of the senescence-accelerated mouse (SAM). Exp Gerontol. 1997, 32, 117–127. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



