1. Introduction
Platelet glycoprotein VI (GPVI) is both an important platelet plasma membrane receptor protein and an important platelet adhesion protein [
1,
2]. It consists of two extracellular ligand-binding Ig domains, a stalk domain, a single transmembrane domain, and a cytoplasmic domain tail containing calmodulin and Src kinase binding sites [
2]. It’s signaling activity is dependent on association with Fc receptor g-chain, which has an immunoreceptor tyrosine-based activation motif (ITAM) [
3,
4,
5]. Ligand binding, for example, collagen or collagen related peptides, to GPVI leads to Src kinase dependent phosphorylation, Syk kinase activation [3.4] and initiation of an intraplatelet signaling cascade which stimulates platelet aggregation and secretion. In addition to collagen, other GPVI ligands include fibrinogen/fibrin, laminin, and other extracellular matrix proteins [
2]. The Syk kinase dependent cascade can also be activated by a second platelet ITAM sequence containing protein. C-type lectin receptor 2 (CLEC-2) who’s naturally occurring ligand podoplanin has a diverse tissue distribution and has been extensively studied in a cancer context [
6,
7].
Functionally, GPVI has important role (s) in hemostasis and thrombosis. Upon vascular injury or plaque rupture, GPVI interacts with exposed subendothelial extracellular matrix components, principally collagen [
8,
9,
10,
11], and initiates a response likely restricted to the immediate damage site. Binding to more recently discovered ligands such as fibrinogen/fibrin [
2] likely extends more generally throughout the thrombus formation process and could have a greater contribution to the activation of integrin mediated platelet aggregation. Experimentally, knockout of GPVI in mice has a mild effect on bleeding cessation, but a more significant effect on the formation of an occlusive clot [
12]. One interpretation of this outcome is that GPVI fibrinogen/fibrin interaction is a strong factor in the propagation of platelet aggregation across the multiple hundreds of micron lumen of an artery. In brief, based on these outcomes and speculations, GPVI has become a prime therapeutic target for selective modulation of thrombosis versus hemostasis.
In the present study, we have taken an ultrastructure approach to reexamine the relative contribution to hemostasis of GPVI as an adhesion protein versus a signaling receptor. We used a mouse puncture wound structure model and compared the effect on thrombus ultrastructure of knocking out the protein and hence eliminating both its adhesion and signaling functions versus electively inhibiting it’s signaling function only through pretreatment of mice with the Syk inhibitor, Bi 1002494 [
13]. Our hypothesis was that if the predominant contribution of GPVI to hemostasis was mediated by a Syk-dependent signaling cascade then the structural effect of a Syk inhibitor and a GPVI knockout would be same. If the extracellular domains of the protein had an important platelet adhesion role, then significant comparative differences in thrombus structure would be expected. Our experimental outcomes strongly indicate that GPVI extracellular domains play a crucial role in how tight the adherence of platelets within intravascular crown of the jugular vein puncture is.
2. Results
Consistent with previous research [
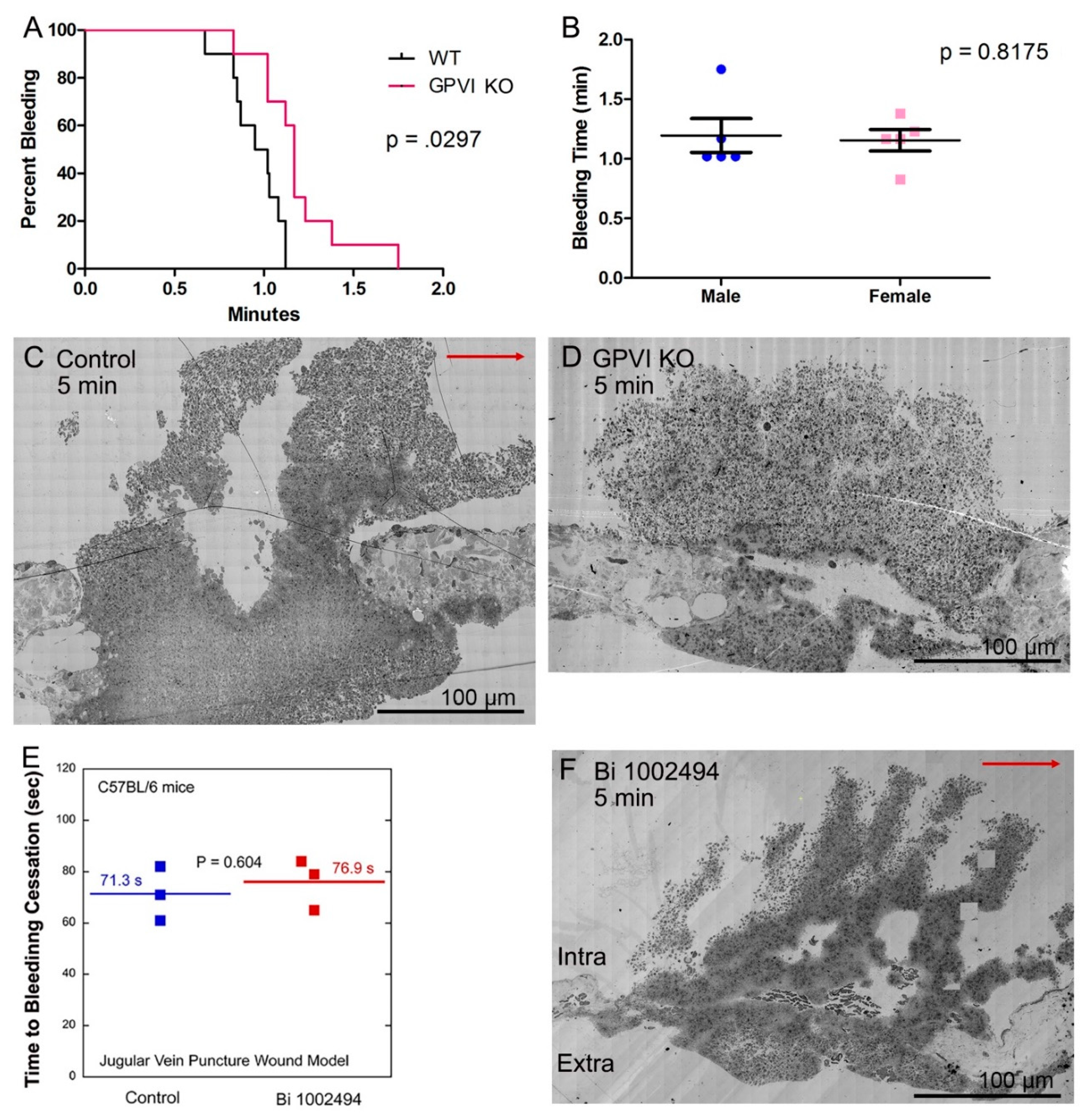
12], deletion of GPVI produces a mild bleeding defect with a 20% increase in time to bleeding cessation in a mouse jugular vein puncture wound model (
Figure 1A,B). The increased bleeding time was accompanied by two significant changes in thrombus ultrastructure. As shown in
Figure 1C, wild type, versus
Figure 1D, GPVI knockout, the GPVI knockout thrombus at 5 min post puncture was altered in both the structure of the intravascular thrombus crown and the extravascular thrombus cap which prevents further bleeding. The tightness of platelet adherence within the crown was significantly decreased. The extravascular cap of the knockout was thin compared to wild type and similar in appearance to that formed in a Syk inhibitor treated mouse (Bi 1002494, 5 min post puncture,
Figure 1F). The intravascular thrombus crown in the inhibitor treated mouse was like wild type, indicating normal platelet adhesion. Consistent with the more normal thrombus structure in Bi 1002494 treated mice, bleeding time in the Syk inhibitor treated mice was normal (
Figure 1E). Overall, these differences indicate non-signaling, Syk-independent, adherence role for the extracellular domains of GPVI in the formation of an intravascular thrombus crown.
Significantly, when examined as a time series, jugular vein puncture wound thrombi from GPVI knockout mice appeared more abnormal in structural organization at late versus early time points, 20 min versus 5 min, or 1 min. As shown in
Figure 2A, a 1 min knockout thrombus was composed primarily of tightly adherent platelets with a thick extravascular cap. While at later time points, 5 min (
Figure 2C, Supplemental
Figure 1) and 20 min (
Figure 2E), the thrombi formed had progressively thinner extravascular caps and a decreased incidence of tightly adherent platelets in the intravascular crown. Counter intuitively, this outcome suggests that the collagen binding function of GPVI is of limited importance to platelet adherence during initial steps in thrombus formation. If anything, these results indicate that the fibrinogen/fibrin binding activity of the extracellular D1/D2 domains of GPVI becomes increasingly important during thrombus formation/maturation. 2D heat mapping of platelet activation state indicated a concentration of highly activated platelets, organelle-free cytoplasm (examples demarcated by yellow dots) and cytoplasm-free, membrane ghosts (procoagulant platelets, red dots) on the periphery of intra-thrombus vaults and “fjords”, red dots). Interestingly, knockout intra-thrombus vaults appeared to be enriched in apparent procoagulant platelets at 5 min post-puncture wounding (
Figure 2C). A concentration of highly activated platelets to the peripheral surfaces of intra-thrombus vaults has been noted previously [
14].
In contrast, all three examples of jugular puncture wound thrombus formation in Syk inhibitor treated mice showed a more normal formation of an intravascular thrombus crown with extensive vaulting at 5 min post puncture. The platelet rich columns delimiting the vaults were mostly rich in tightly adherent platelets with some loosely adherent platelets coating the blood stream proximate surfaces of the columns. As noted in
Figure 1F, the extravascular cap producing bleeding cessation was thin with some loosely adherent platelets apparent in these Syk treated mice. Heat mapping platelet activation state in the thrombi revealed at absence of free-procoagulant platelets (red dots) within vaults as in the case of GPVI knockout mice (
Figure 2C, Supplemental
Figure 1 versus
Figure 3B,D,F).
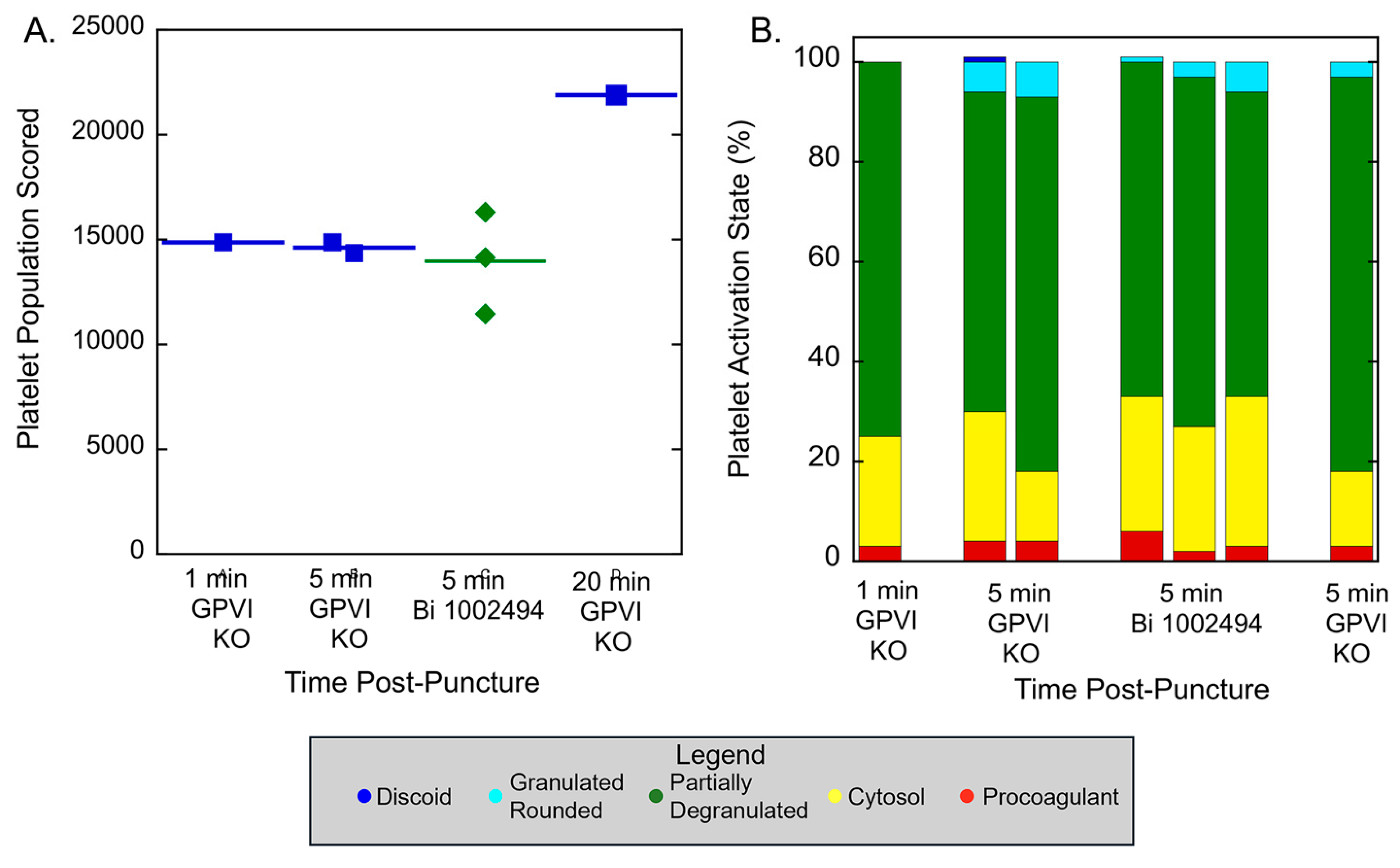
Quantitatively, as shown in
Figure 4A, platelet abundance in time-matched, 5-min puncture wound thrombi from GPVI knockout and Syk inhibitor treated mice was similar. The size of the 20-min thrombus example in the GPI knockout was almost 22,000 platelets, a value almost 50% higher than that found at earlier times points. This value is almost twice that reported earlier for control, wild type C57Bl/6 mice [
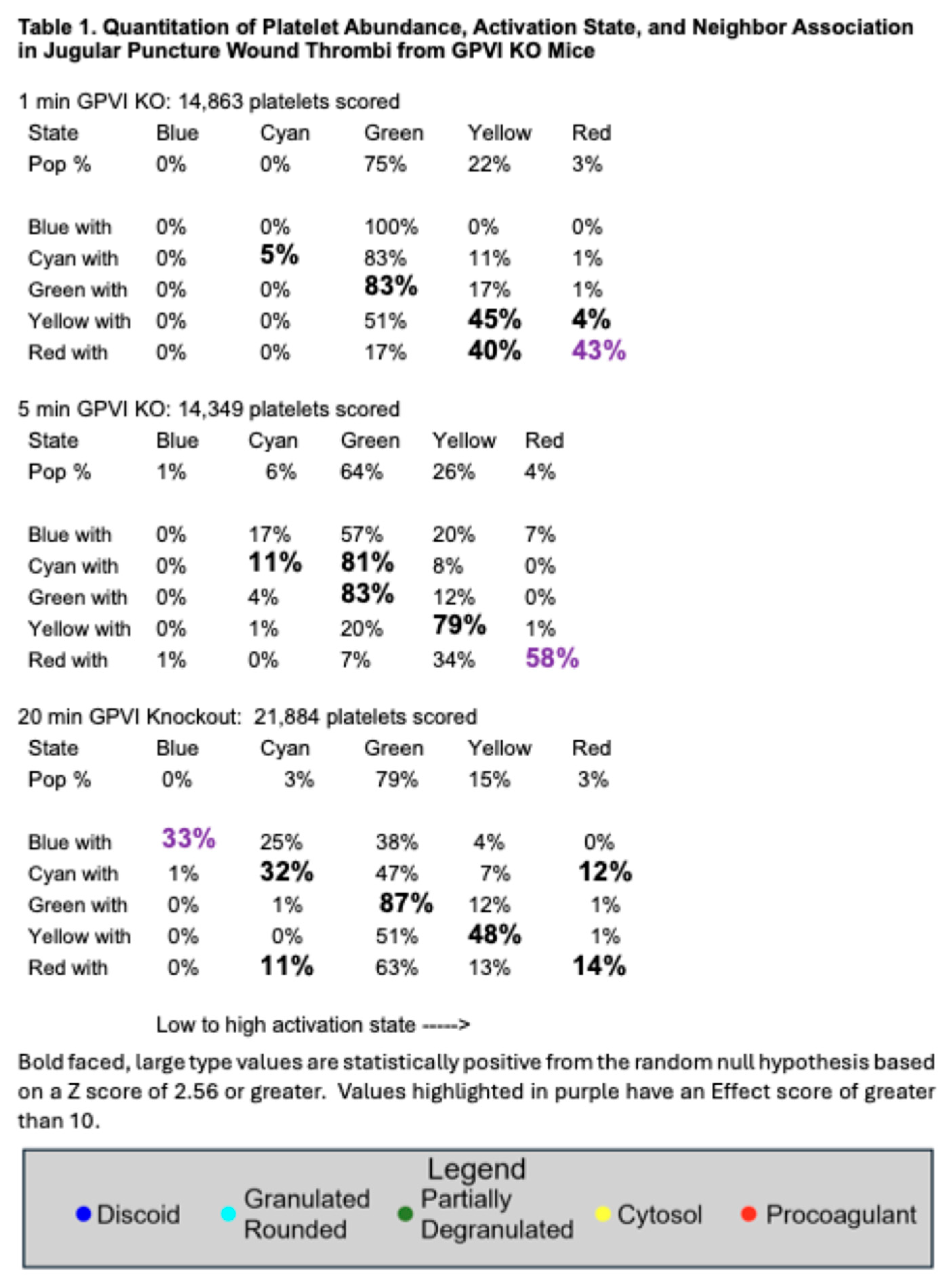
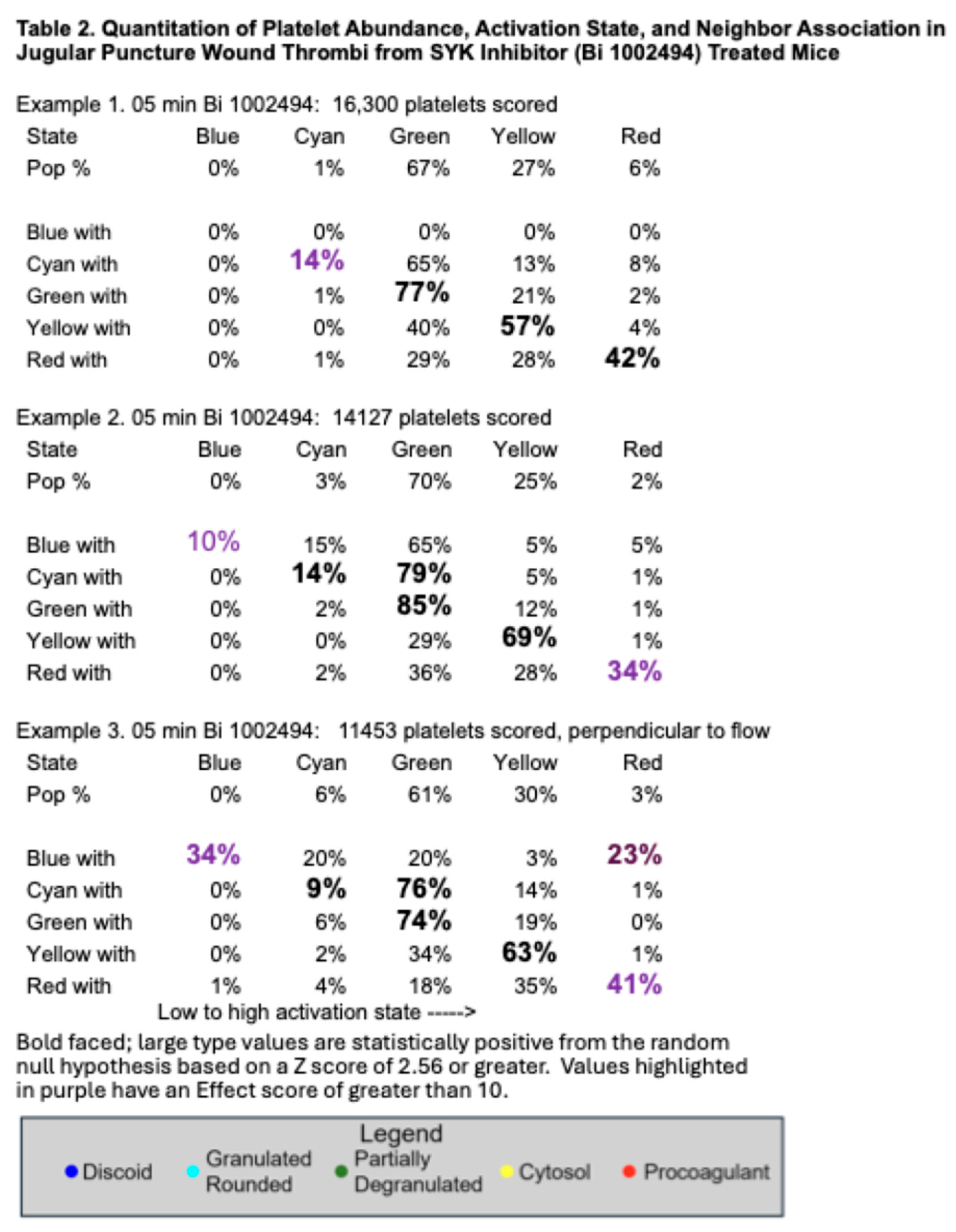
17]. In brief, the data provide quantitative evidence that GPVI is not required for platelet recruitment to a forming, hemostatic, jugular vein, puncture wound thrombus. Scoring of individual platelet profile activation state based on morphological criteria showed little to no difference in distribution between the various states based on how GPVI function was disrupted (Table 1 and Table 2;
Figure 4B), an indication that activation state in more dependent on Syk kinase signaling than is platelet adherence. In comparison to control, wild type C57Bl/6 jugular puncture wound thrombi [
14], the distribution of platelet activation state in displayed in GPVI function disrupted thrombi was shifted towards partially degranulated platelets (green). In a final quantitative comparison, we compared platelet neighbor pairings across the two different GPVI perturbant conditions. In these comparisons, platelet pairing within a 2 mm radius was determined. As shown in Table 1 and Table 2, like paired with like, more than predicted by chance, the null hypothesis; partially degranulated platelets (green) had a high probability of pairing with one another, a tendency that was more marked than in wild type thrombi [
14].
Qualitatively, one would expect that any adhesive or signaling role of collagen through binding to GPVI would be most obvious at the platelet collagen adventitial interface. This expectation is confirmed in
Figure 5A where collagen proximal platelets in a 5 min wild type thrombus were often degranulated and nearly devoid of cytoplasmic contents [see also, 15]. However, cytosol loss did not occur in 5 min thrombi from either GPVI knockout mice or Syk inhibitor treated mice (
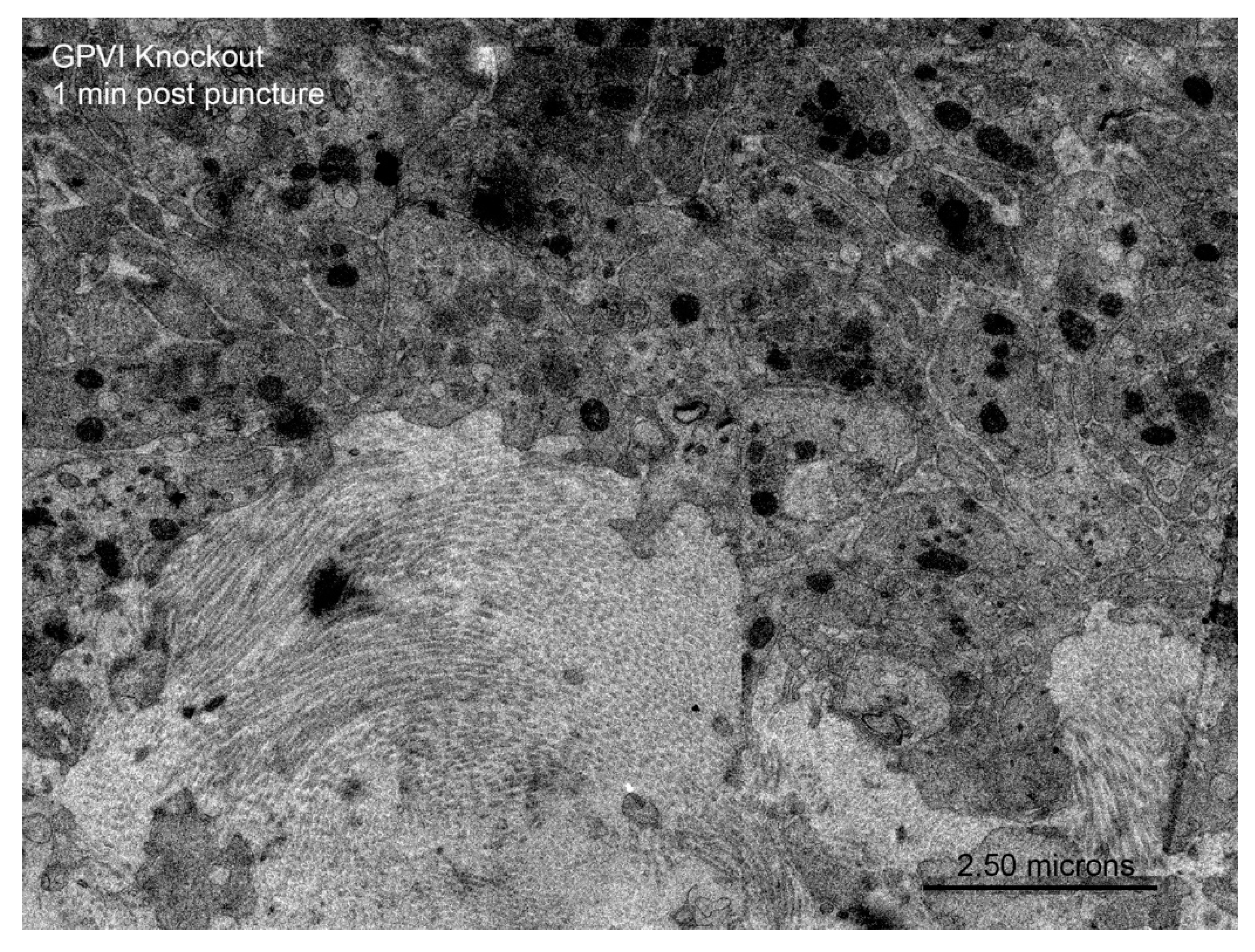
Figure 5 B,C). In both cases, platelet-platelet adherence appeared normal. Similar results were observed at an early, 1 min time point for the GPVI knockout (
Figure 6). Overall, our results strongly indicate a dependence of platelet activation state on collagen-associated Syk dependent signaling but provide little to no evidence for collagen-associated platelet-platelet adhesion. Note that dabigatran inhibition of thrombin activity produces a similar inhibition of platelet cytosol loss at the adventitial interface. These suggest that cytosol loss is dependent on multiple signaling pathways and may well be a quantitative parameter that is determined by overall signaling intensity.
3. Discussion
Taking an ultrastructural approach to reexamine the in vivo role of platelet glycoprotein (GP)VI in thrombus structure and bleeding cessation, we found that GPVI knockout had significantly greater effects on bleeding cessation times and mouse, jugular vein, puncture wound thrombus formation than did treatment with the Syk inhibitor Bi 1002494 [
13]. In agreement with previous results in other bleeding assays, GPVI knockout gave a mild, but significant prolongation of bleeding times while the Syk inhibitor had no effect on bleeding cessation time. We attribute these differences to the greater effect of the GPVI knockout on thrombus structure. Both the protein knockout and Bi 1002494 inhibited cytoplasm loss from platelets at the collagen-rich adventitia interface, a near terminal step in a platelet activation sequence, indicating a Syk dependence. Significantly, this inhibition had no effect on bleeding time in the Syk treatment case indicating that that this activation state is not required for bleeding cessation. Moreover, neither perturbation had any effect on platelet-platelet adherence at the adventitial interface. Importantly both GPVI knockout and Bi 1002494 yielded at 5 min post puncture a thinned and less tightly adherent extravascular cap, the structural element producing bleeding cessation, indicating a greater sensitivity of the ultrastructural assay than the more common bleeding time assay. Importantly, the wound thrombus formed in the GPVI knockout appeared to be much more porous containing extensive intravascular areas of loosely adherent platelets that were found at 5- and 20-min post puncture to protrude into the extravascular bleeding cessation cap. We project that the steps leading to this outcome are the cause of the longer bleeding times in the GPVI knockout mouse.
The comparative structural outcomes provide direct visual evidence GPVI acts as both a signaling protein and a late acting, major adhesive protein in normal hemostasis. Contrary to expectation for a protein originally discovered as the major platelet collagen receptor [
8,
9,
10,
11]. we found that the adhesive effect of GPVI became prominent not at the early time point of 1 min but rather later, 5 and 20 min, and was most pronounced intravascularly in the thrombus crown, a site distant from the collagen rich adventitia. We attribute this outcome to the importance of the fibrinogen/fibrin binding activity of the extracellular D1/D2 domains of GPVI [for review, see 2]. This outcome lends further support to the longstanding dogma for a dominant role of the collagen/von Willebrand factor/platelet glycoprotein Ib-IX axis in primary platelet adhesion [
15,
16,
17,
18,
19,
20,
21]. The structural consequences of Syk-dependent inhibition were chiefly manifested on the structure of the platelet-rich extravascular thrombus cap and had no effect on bleeding time. This minor outcome is a strong indicator of the limited role of Syk dependent signaling in hemostasis. In brief, our studies suggest that major adhesive role of GPVI in thrombus formation is as a fibrinogen/fibrin binder rather than as a collagen binder. For an overview model of our ultrastructural outcomes, see
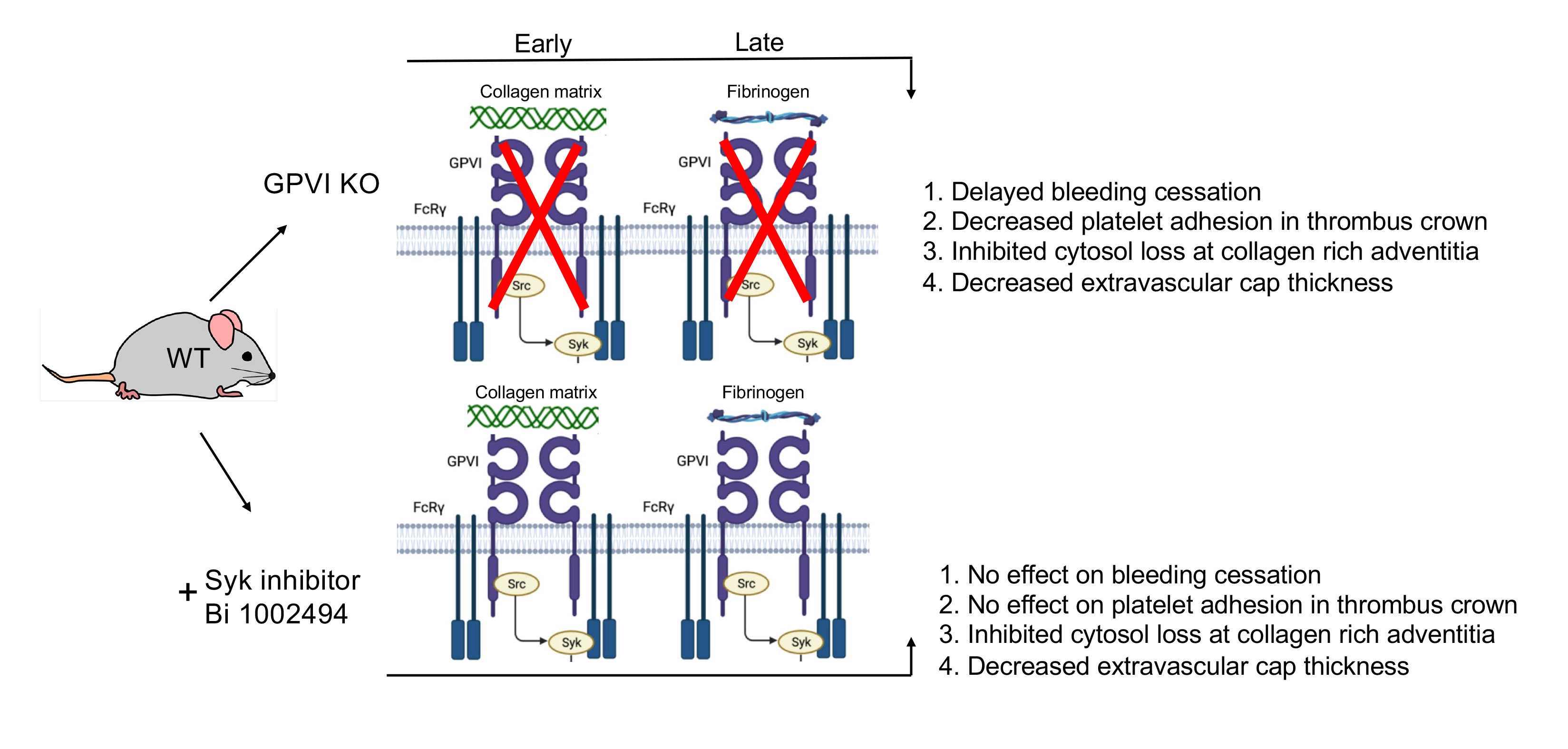
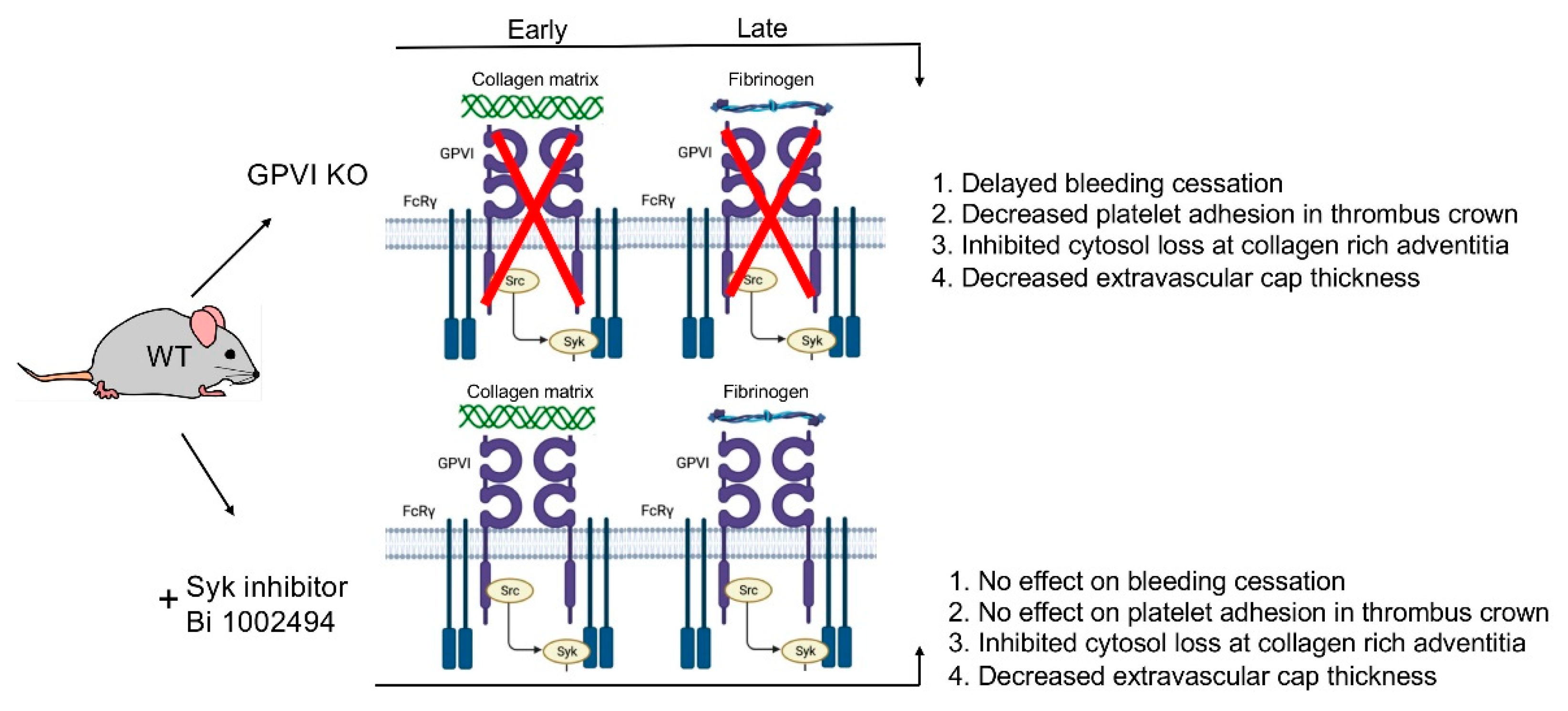
Figure 7.
In conclusion, platelet glycoprotein VI has been long considered to be a therapeutic target for controlling thrombus because of its mid effects on hemostasis [
12,
22,
23,
24,
25,
26]. Our ultrastructural outcomes indicate that targeting the extracellular domains of GPVI could well have more consequences for hemostasis than inhibiting GPVI’s Syk-dependent intracellular signaling role. Syk targeting could well be the safer route, albeit of untested ultrastructural consequences for occlusive clotting.
4. Materials and Methods
4.1. Mice and Reagents
All animal usage was approved by the relevant local Institutional Animal Care and Use Committees. Wild-type C57BL/6 or GPVI
-/- male and female mice ([
12], 8-12 weeks old) were used in equal numbers across the individual data sets. All reagents were of reagent grade and listed previously [
27,
28,
29].
4.2. Bi 1002494 Spleen Tyrosine Kinase (Syk) Inhibitor Treatment
Mice were treated with Bi 1002494 Syk inhibitor as described earlier [
13]. In brief, Bi1002494 was dissolved in deionized water and administered (100 mg/kg body weight) twice (15 hours and 1 hour before clot induction) by oral gavage [
13].
4.3. Thrombus Preparation and Electron Microscopy [27,28,29]
Jugular vein wounding was done with a 30 G needle (312 mm nominal diameter) and thrombi were fixed in situ at 1, 5, and 20 min, post injury, with 4% paraformaldehyde. For WA-TEM, samples were processed for plastic embedding as previously described and stained with uranyl acetate and lead citrate post-embedding [
27,
28].
Automatically montaged images were collected at 3.185 nm XY pixel size using SerialEM software (version 3.6, 32-bit) and visualized with 3DMOD software (version 4.9.13). Fine image blending was done with eTomo software (version 4.11.12). Image visualization was done with IMOD software (version 4.11). iMac Pro computers (MacOs 10.14, 5K display) were used, and images were displayed at various zoom factors ranging from 2% to 100%. The raw images were as large as 130,000 by 90,000 pixels. All software is from the Mastronarde group at
https://www.colorado.edu/mcdb/resources/mastronarde-group.
4.3. Datasets
All datasets will be deposited and publicly available as raw images upon acceptance of this manuscript. All mapping analysis, resulting quantitative analysis, and mapping imagery are unique and original to the present work.
Manually Annotated Platelet Activation State Heat Mapping and Nearest Neighbor Analysis [
14]
Montaged WA-TEM images recorded in 24-bit .mrc format were blended into a single image using eTomo (IMOD software package), binned (~5 × 5) in 3DMOD, and converted to 8-bit grayscale images with FIJI software. Platelet activation states were annotated in the binned images. and displayed in 8-bit color using iVision-Mac software (32-bit software, BioVision Technologies, Inc., Exton, Pennsylvania). From the WA-TEM images, platelets were classified into five groups based on their morphology: discoid (blue); rounded, granulated (cyan); partially degranulated (green); cytosol-rich, degranulated, but containing mitochondria (yellow); and devoid of internal contents (red). On the images, each platelet was scored, marked with a point in its center (centroid) which was given a unique identification number, and its X, Y coordinates were recorded. Platelet scoring was done manually by outcome blinded scorers. The ‘Measure Segments’ command in iVision was used to evaluate the distributions of platelet states in the region surrounding each individual platelet. For each centroid, the state of all adjacent centroids, within a 2 µm radius, was tabulated. This was repeated iteratively for each platelet centroid in each field. The clustering of different platelet classes was calculated using the average numbers of each class associated with a given centroid and expressing that as a percentage of the total for that class. Pairing percentages were tabulated for each class in a series of rows reading from left to right for a given activation with then the individual pairings arranged column wise. Each individual row adds up to 100% as the full set of pairing for that activation state. Bold-faced, large font pairing values indicate statistically significant localized co-clustering of that activation state (Table 1 and Table 2). Calculation formulas can be revealed by clicking on the Excel spreadsheet example.
Statistical significance was calculated as shown in the Excel spreadsheet example presented in Supplemental Materials. In brief, experimental values and values expected from the null hypothesis of random platelet pairing were compared and those having a positive standard error greater than 2.56 were considered significant. For the sake of simplicity only values having a positive standard error. i.e., greater than expected from random are highlighted. Effect value (purple highlight in Tables) were considered significant if 10 or greater. Between ~11,000 and ~22,000 platelet profiles were evaluated for each image set.
Figure 1.
Bleeding effects and thrombus structure at 5 min post puncture wounding. All thrombi, darker, platelet rich structures (C,D,F), are arranged as intravascular side to top, extravascular side to bottom, flow left to right. All bleeding times (A,B,E) are for the jugular vein puncture wound model.
Figure 1.
Bleeding effects and thrombus structure at 5 min post puncture wounding. All thrombi, darker, platelet rich structures (C,D,F), are arranged as intravascular side to top, extravascular side to bottom, flow left to right. All bleeding times (A,B,E) are for the jugular vein puncture wound model.
Figure 2.
Kinetics of thrombus formation, GPVI knockout mice, raw and platelet activation state mapped (red/yellow high activation and blue discoid shaped platelets; cyan, green in between, based on morphology, granule presence, cytoplasm). Thrombi, darker, platelet rich structures are arranged as intravascular side to top, extravascular side to bottom, blood flow within the vein left to right. An example of high activation platelets within vault region of a 5-min thrombus is emphasized in C by heat mapping only high activation platelets (yellow and red) compared to D, five color heat mapping of all activation states.
Figure 2.
Kinetics of thrombus formation, GPVI knockout mice, raw and platelet activation state mapped (red/yellow high activation and blue discoid shaped platelets; cyan, green in between, based on morphology, granule presence, cytoplasm). Thrombi, darker, platelet rich structures are arranged as intravascular side to top, extravascular side to bottom, blood flow within the vein left to right. An example of high activation platelets within vault region of a 5-min thrombus is emphasized in C by heat mapping only high activation platelets (yellow and red) compared to D, five color heat mapping of all activation states.
Figure 3.
Effect of SYK inhibition on thrombus morphology at 5 min post puncture raw and platelet activation state mapped (red/yellow high activation and blue discoid shaped platelets; cyan, green in between, based on morphology, granule presence, cytoplasm). Thrombi, darker, platelet rich structures are arranged as intravascular side to top, extravascular side to bottom, blood flow within the vein left to right. An example of high activation platelets within vault region of a 5-min thrombus is emphasized in C by heat mapping only high activation platelets (yellow and red) compared to D, five color heat mapping of all activation states.
Figure 3.
Effect of SYK inhibition on thrombus morphology at 5 min post puncture raw and platelet activation state mapped (red/yellow high activation and blue discoid shaped platelets; cyan, green in between, based on morphology, granule presence, cytoplasm). Thrombi, darker, platelet rich structures are arranged as intravascular side to top, extravascular side to bottom, blood flow within the vein left to right. An example of high activation platelets within vault region of a 5-min thrombus is emphasized in C by heat mapping only high activation platelets (yellow and red) compared to D, five color heat mapping of all activation states.
Figure 4.
GPVI knockout and SYK inhibitor treatment have a similar effect on thrombus size and distribution of thrombus recruited platelets between different morphologically scored activation states. These outcomes are compared in the text to the size of platelet number in no drug treated jugular vein thrombi and the distribution of these platelets between activation state.
Figure 4.
GPVI knockout and SYK inhibitor treatment have a similar effect on thrombus size and distribution of thrombus recruited platelets between different morphologically scored activation states. These outcomes are compared in the text to the size of platelet number in no drug treated jugular vein thrombi and the distribution of these platelets between activation state.
Figure 5.
Both GPVI knockout and Bi 1002494 treatment platelet cytosol loss at the thrombus/collagen-rich adventitia interface. A, WT, asterisks mark examples of platelet cytosol loss. B,C. Such loss was not observed with either GPVI perturbation. A. collagen to left side; B. collagen to ride hand side; and C, collagen to lower left. Collagen shows as bundled fibers or dots depending on orientation within the plane of the section.
Figure 5.
Both GPVI knockout and Bi 1002494 treatment platelet cytosol loss at the thrombus/collagen-rich adventitia interface. A, WT, asterisks mark examples of platelet cytosol loss. B,C. Such loss was not observed with either GPVI perturbation. A. collagen to left side; B. collagen to ride hand side; and C, collagen to lower left. Collagen shows as bundled fibers or dots depending on orientation within the plane of the section.
Figure 6.
Inhibition of platelet cytosol loss in a GPVI knockout jugular puncture thrombus, 1 min post wounding. Remaining organelles in the platelet cytoplasm are primarily mitochondria. Note: mitochondrial cisternae are not prominent at this magnification.
Figure 6.
Inhibition of platelet cytosol loss in a GPVI knockout jugular puncture thrombus, 1 min post wounding. Remaining organelles in the platelet cytoplasm are primarily mitochondria. Note: mitochondrial cisternae are not prominent at this magnification.
Figure 7.
Data based model of the role of GPVI in the formation of a vein puncture wound thrombus. Four traits were scored on the basis of morphology in montaged wide-area transmission electron micrographs (WA-TEM).
Figure 7.
Data based model of the role of GPVI in the formation of a vein puncture wound thrombus. Four traits were scored on the basis of morphology in montaged wide-area transmission electron micrographs (WA-TEM).