
Submitted:
29 January 2025
Posted:
30 January 2025
You are already at the latest version
Abstract
In this work, we have studied the main decomposition reactions on the ground state of nitromethane (CH3NO2) with the CASPT2 approach. The energetics of the main elementary reactions of the title molecule have been analyzed on the basis of Gibbs free energies obtained from standard expressions of Statistical Thermodynamics. In addition, it is described a mapping method (orthogonalized 3D-representation) for the potential energy surfaces (PESs) by defining an orthonormal basis consisting of two Rn orthonormal vectors (n, internal degrees of freedom) that allows to obtain a set of ordered points in the plane (vector subspace) spanned by such a basis. Geometries and harmonic frequencies of all species and orthogonalized 3D-representations of the PESs have been computed with the CASPT2 approach. It is found that all of the analyzed kinetically controlled reactions of nitromethane are endergonic. For such a class of reactions, the dissociation of nitromethane into CH3 and NO2 is the process with lower acctivation energy barrier (G), that is, the C-N bond cleavage is the most favorable process. In contrast, there exists a dynamically controlled process that evolves through a roaming reaction mechanism and is an exergonic reaction at high temperatures: CH3NO2 [CH3NO2]* [CH3ONO]* CH3O + NO. The above assertions are supported by CASPT2 mappings of the potential energy surfaces (PESs) and semiclassical trajectories obtained by "on-the fly" CASSCF molecular dynamics calculations.
Keywords:
CASSCF
; CASPT2
; nitromethane
; exergonic decomposition
1. Introduction
Nitro compounds are a class of molecules that play important roles in several areas and applications, such as atmospheric chemistry, explosives, propellants or fuels. [1,2,3] For this reason, the thermal and photochemical decompositions of the simplest organic nitro compound [nitromethane (CH3NO2)] have been extensively studied both theoretically [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20] and experimentally, [18,19,20,21,22,23,24] due precisely to its molecular simplicity and the applications above mentioned. There is consensus in the literature that the weakest bond in the molecule is C-N. [6] However, in spite of the structural simplicity of nitromethane, it is not clear what is the initial reaction or the key reaction step that occurs in the chemically simplest decomposition processes, for example, when acts as an explosive without the intervention of an external reactant, or when is decomposed in IRMPD experiments where the thermal chemistry occurs in collision-free conditions. [21]
The detonation of an energetic material is a process that occurs under the stimuli of an external action that leads to a local chemical reaction releasing an enormous amount of energy which is transformed in internal energy of the material with the consequent local increasing of the temperature and pressure. [14] According to the definition of detonation, the initial local reaction is expected to be an elementary unimolecular process. Thus, in this context, detonation of CH3NO2 acting as energetic material remains a major puzzle because all of the elementary reactions of this molecule are highly endothermic (vide infra). In this work, we propose an elementary reaction, that leads to an exergonic decomposition process at high temperatures that passes through the nitro-nitrite isomerization channel via a roaming reaction and ends with the dissociation of cis-methyl nitrite into CH3O and NO. The roaming mechanism of nitromethane has been previously studied by other authors [7,8,9]; therefore, we assume, as starting hypothesis, that this process takes place in the decomposition mechanism. For the sake of completeness, all of the elementary reactions of nitromethane decomposition have been studied with the CASPT2 approximation.
A roaming reaction is a class of unimolecular reaction that was reported relatively recently. [25] Roaming reactions occur at the near-dissociation limit of the molecule where radical products are almost formed, that is, when the fragments are separated to 3-4 Å, then roaming reorientation of the fragments becomes feasible as the kinetic energy is low and, consequently, the angular forces may be comparable to the radial forces, in other words, roaming rotation of the fragments is almost a free energy process. This effect may allow the system to access a distinct reactive domain with respect to dissociation, such as abstraction or isomerization, with the consequent formation of unexpected highly vibrationally excited products. [19,26] Roaming reactions may be considered a special case of those reactions that are dynamically controlled and deviate from the minimum energy path or intrinsic reaction coordinate [27,28,29,30] and is now widely accepted as a nearly universal aspect of chemical reactivity that could make a significant contribution to product branching in unimolecular reactions. [30,31,32,33,34,35,36]
2. Methods of Calculation
The multi-configurational calculations have been carried out with the complete active space self-consistent field (CASSCF) [37,38,39,40,41,42,43] method and the multi-state second-order perturbation (MS-CASPT2) approach, [44,45] as implemented in the MOLCAS 8.4 and OpenMOLCAS 22.06 programs. [46,47,48,49] Extended relativistic ANO-RCC basis sets [50,51] have been used throughout this work by applying the contraction scheme [C,N,O]/[H]: [4s3p2d1f]/[3s2p1d]. The IPEA empirical correction has been fixed at 0.25 in all of the MS-CASPT2 calculations, equally, to avoid the inclusion of intruder states in such calculations an imaginary shift set to 0.1 has been applied.
All geometry optimizations have been performed with both CASSCF and CASPT2 methods. The characterization of all the species as minimum or transition state has been done by means of frequency calculations, analytically for CASSCF calculations and numerically for the CASPT2 ones. In addition, the Møller-Plesset (MP2) [52] and DFT/M06-2X [53] methods have been applied as they are implemented in GAUSSIAN-16, [54] in conjunction with def2-TZVPP basis sets. [55,56]
The selection of the active space to study the reactions of nitro-nitrite derivatives (X-NO2; X-=NO) consists of 14 electrons distributed in 11 orbitals, [57,58,59,60,61,62] or 16 electrons and 13 orbitals when proton migration is under study. Molecular dynamics calculations at the CASSCF level were performed with the velocity algorithm of Verlet [63,64] implemented in MOLCAS.
The analysis of molecular orbitals and molecular geometries have been done with the programs Molden [65] and Gabedit. [66] The analysis of vibrational normal modes have been performed with the MacMolplt program. [67]
Construction of the 1D-potential energy curves (PECs) have been performed by applying a linear interpolation method that uses the full space of non-redundant internal coordinates. [68,69,70,71,72]
Mapping of 2D-Potential Energy Surfaces
Mapping of 2D-potential energy surface of a reaction was sketched out in a previous work. [73] Here, we describe the procedure in detail (Figure 1). First of all, to start the mapping, three points (geometrical configurations) are required for defining the reaction domain, such points usually content critical points of the system under study, e.g., minima, transition states, surface crossings or a combination of them. Secondly, we define a common set of non-redundant internal coordinates ( for the three reference points, where correspond to internuclear distances and and are valence bond angles and dihedral angles, respectively. Third, one of the selected points is chosen as reference (0 in Figure 1A). Then, we built the linear interpolation vectors and by subtraction of the i, j non-redundant internal coordinates:
The coordinates of correspond to the following elements: non-dimensional internuclear distances ( internuclear distance in Å and ) and the differences between the and , respectively, which correspond to non-dimensional valence bond angle coordinates or dihedral angle coordinates of and in radians.
Given that we are working in internal coordinates, the relative orientation of the and vectors can be arbitrarily chosen. Thus, in this case, the relative orientation is selected in such a way that the bisector line of the angle is aligned with the Qs-axis and is orthogonal to the QR-axis (Figure 1A.1).
The next step is the normalization of the vectors and :
Thus, the addition and subtraction of the unit vectors and -with equal lengths- yields vectors and that are mutually orthogonal and are in line with the Qs-axis and QR -axis, respectively (Figure 1A.2). Normalization of and yields and that form an orthonormal basis
In order to span the configurational domains of species 1 and 2, the orthonormal vectors and are scaled by the norm of the largest vector, or . Thus, we obtain two vectors and , which are mutually orthogonal and with the same norm (Figure 1A.3) as follows:
Once the vectors and are built, in order to map the potential energy surface, it is necessary to select a step size s,
where is an arbitrary integer number that will determine the grid size of the 2D-surface. Thus, the position of any point , which represents a distortion of the reference geometry 0 ( times along the vector and times along the vector ), is given by the position vector (Figure 1A.4).
In practice, we run the mapping in horizontal mode, that is, following the interpolation vector (Figure 1A.4). As an example of the application of the mapping procedure described above, Figure 1B,C depict the 2D and 3D-representations of the potential energy surfaces of the ternary system [nitromethane:trans-methylnitrite:cis-methylnitrite]. The mapping of such a system identifies two saddle points, labelled as pTS1 and pTS4, that would connect (i) nitromethane with trans-methyl nitrite and (ii) trans-methyl nitrite with cis-methyl nitrite, respectively. Thus, each one of these saddle points has been successfully used as good starting geometries to optimize the true transition states (TS1 and TS4; vide infra) for the reactions CH3NO2 trans-CH3ONO and trans-CH3ONO cis-CH3ONO, respectively.
To finish this section, it must be remarked that the points used to mapping the PESs satisfy the condition of being contained in a plane because they are constructed from the basis vectors and that form a subspace of . In contrast, the points that would be obtained by the so-called constrained optimization method would not satisfy the above condition because they would not be in the same vector subspace.
3. Results and Discussion
3.1. Unimolecular Reactions of Nitromethane and Methyl Nitrite
The elementary unimolecular reactions of nitromethane and methyl nitrite have been widely studied by many authors with a variety of theoretical approximations,[4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20] mostly with single-determinantal representation for the electrons. In this section, with the objective of showing the accuracy and reliability of the CASPT2 theory, whose reference wave function is multi-determinantal, we have studied some of the main unimolecular reactions of nitromethane and methyl nitrite with such a theory (energetics, frequencies and geometries of stable species, intermediates and transition states), as well as where possible, CASPT2 results are compared with experimental data [74,75,76] and with MP2 and DFT calculations obtained in this work. To be specific, the reactions reported are: (i) dissociation of nitromethane into methyl and nitrogen dioxide radicals; (ii) trans-methyl nitrite dissociation into methoxy and nitric oxide radicals; (iii) nitromethane dissociation into nitrosomethane and atomic oxygen; (iv) nitro-nitrite isomerization of nitromethane leading to formation of trans-methyl nitrite; (v) proton migration in nitromethane to give aci-nitromethane [CH2N(O)OH]; (vi) proton migration in methyl nitrite to give formaldehyde and nitrosyl hydride (HNO); and (vii) trans-cis isomerization of methyl nitrite. The thermodynamics properties (enthalpies and Gibbs free energies) of such reactions computed with the expressions of Statistical Thermodynamics and obtained from three different theoretical approaches (CASPT2, MP2/HF and DFT) are compared with experimental values in Tables 1,2. In general, there exists an excellent agreement between experimental and CASPT2 calculated values for the reaction enthalpies; the major source of deviation from experimental data arises from the harmonic CASPT2 frequencies. Comparison of the dissociation and activation Gibbs energies for the tabulated reactions shows that the C-N bond cleavage is the most energetically favourable process of nitromethane. As another probe/prove of the accuracy of the CASPT2 method to dealt with this kind of compounds, the comparison of experimental [77,78,79] and calculated geometrical parameters of the species studied in this work for nitromethane, intermediates and products, is given in ESI (Tables S1-S8). Equally, in order to show that the MS-CASPT2 approach is an appropriate method to deal with dissociation reactions, [80,81,82,83,84] the potential energy surfaces of the dissociation reactions collected in Table 1 are given in Figures S1,S2, where is clearly observed as every potential energy curve reaches the asymptotic limit at the dissociation region.
3.2. A Look at the so-Called Loose Transition State
Two different reaction mechanisms for nitro-nitrite isomerization of nitromethane (CH3NO2) to methyl nitrite (CH3ONO) have been proposed in the literature: one involves a so-called loose transition state (Figure 2a), in which the methyl and NO2 radicals are almost formed and well separated from each other [4] and the other mechanism passes through a so-called tight transition state in which the internuclear distances among the atoms of the C-NO2 group are shorter than in the loose one (Figure 2b). The geometries of these two types of transition states obtained in this work are given in Figure 2 and the CASSCF and CASPT2 structural parameters are collected in Table 3. It should be noted that due to the very flexible nature of the transition state given in Figure. 2a, we could not find a precise stationary point; although the convergence criteria for maximum gradient and root mean square of gradient were fulfilled; the stopping criterium for displacements was not reached.
The loose transition state has acquired a special interest in the context of the roaming reaction for the nitro-nitrite isomerization of nitromethane, [7] where is proposed a nitro-nitrite isomerization mechanism which evolves through a loose transition state or roaming saddle point (RSP). In this respect, inclusion of such an RSP seems to deviate from the original statement of the definition of roaming reaction, [25] that is, a reaction that does not pass through a conventional transition state. [25,27] For this reason, we have re-investigated these two types of transition states (loose and tight). To obtain such transition states, two different active spaces must be selected: (i) 10 electrons distributed in 7 orbitals for the loose transition state [CAS(10,7)], and (ii) 14 electrons in 11 orbitals [CAS(14,11)] for the tight one. The CASSCF molecular orbitals of the Cs symmetry equilibrium geometry of nitromethane, which are included in each one of the active spaces, are represented in Figure 3. The reduced active space (Figure 3a) comprises the following orbitals that coincide with the description of Saxon and Yoshimine:5 one O σ lone pair (nσ), one O π lone pair (nπ), two N-O σ bonds (σNO), one N-O σ∗ bond (σ∗NO), one N-O π bond (πNO) and one N-O π* (π∗NO). The larger active space (Figure 3b) comprises one O σ lone pair (nσ), one O π λone pair (nπ), two N-O σ bonds (σNO) and the two correlating σ∗ orbitals (σ∗NO), one N-O π (πNO) and the correlating N-O π* orbital (π∗NO), one C-N σ bond (σCN) and the correlating σ∗ orbital (σ∗CN), and the 2s-orbital of nitrogen (2sN) that has a strong C-N bonding character. Although both types of calculations have been done without symmetry restrictions, the CAS(14,11) wave function of the larger active space keeps the Cs symmetry properties. In contrast, as a result of the unbalanced selection of the CAS(10,7) reduced active space; it is clearly observed in the orbitals that we have obtained a symmetry breaking solution of the wave function. Symmetry breaking has an important impact on the results, generally, such results are unappropriated. For example, Figure 4 represents the 1D-potential energy surfaces that connects the equilibrium geometry of nitromethane with the geometry of the loose transition state (Figure 2a), obtained with the two active spaces, CAS(10,7) and CAS(14,11), and with the linear interpolation method. [68,69,70,71,72] The curve obtained with the CAS(10,7) space shows a discontinuity between points 21 and 22, the reason for this behaviour is orbital rotations (Figure 3) among the inactive, active and secondary spaces, what, in practice, is equivalent to a change in the wave function in passing from one point to the other on the reactive domain, which is, from our point of view, chemically unacceptable to describe any reaction. Therefore, the loose transition state represented in Figure 2a must be considered an artefact of the unappropriated CAS(10,7) active space.
Figure 5a,b depict the 2D and 3D-representations of the potential energy surfaces [CASPT2/CASSCF(14e, 11o)] that comprises the configurational domain of the ternary system [CH3NO2:Loose:cis-CH3ONO], the reference geometries correspond to the CASPT2 minimum geometries of nitromethane and cis-methyl nitrite plus the CASSCF loose transition state. It is clearly observed in such a figure that there is not any signal of a transition state (saddle point) which relates nitromethane with cis-methyl nitrite. In contrast, seems that they are related through a roaming mechanism.
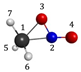
To gain more insights about the roaming structures of the nitro-nitrite isomerization of nitromethane, as in Figure 5, we have built four CASPT2 2D-potential energy surfaces for the ternary system [CH3NO2-Roaming-Structure-cis-CH3ONO] by taking different geometries for the quasi-dissociated species (Figure 6) and using a CASSCF(14, 11) reference wave function. The first surface (Figure 6a), in fact, would not correspond to a roaming intermediate because the analysis of the wave function indicates that the two fragments are well formed radicals, that is, the molecule is completely dissociated. The geometries and relative energies of the reference points (0n) are included in the graphic. It is shown that a significant change in the geometry of the reference point is accompanied by a small variation of its relative energy, that is, roaming rotation must not be a very hindered process at these points of the potential energy surface, especially if rotation is assumed to be free at the higher energy geometry (01). Figure 6b–d represent the topology of the potential surfaces for true roaming structures. What it is observed in these latter figures is that as the roaming complex is more compact with decreasing internuclear distances, the product (cis-CH3ONO) is closer to the roaming structure, in addition, the slope of the surface becomes more negative, which enhances the formation of the intermediate [cis-CH3ONO]* from the roaming structure. To finish this paragraph, the brown flat regions on the surfaces depicted in Figure 6 correspond to very high energy points arising from geometries with very close separation between atoms
To demonstrate that the roaming process leads to decomposition of nitromethane into CH3O and NO passing through the methyl nitrite intermediate, we have performed molecular dynamics calculations at the CASSCF level with the algorithm of Verlet. [63,64] The obtained results are shown in Figure 7. These calculations are started at the geometry represented in Figure 6c (03). The initial condition imposed on the dynamical calculations consists of addition of kinetic energy on the NO2 fragment as is shown in Figure 7. The pattern of formation/dissociation of cis-methyl nitrite is similar in the four calculations: (i) the nitrite molecule is formed from the roaming structure, more rapid as higher is the initial kinetic energy; (ii) the molecule lives during approximately 80 fs in the well of cis-methyl nitrite; (iii) the molecule dissociates into NO and CH3O. (iv) Excess of kinetics energy (Ekin = 80 kcal/mol) leads to a delay in the dissociation process of [cis-CH3ONO]*.
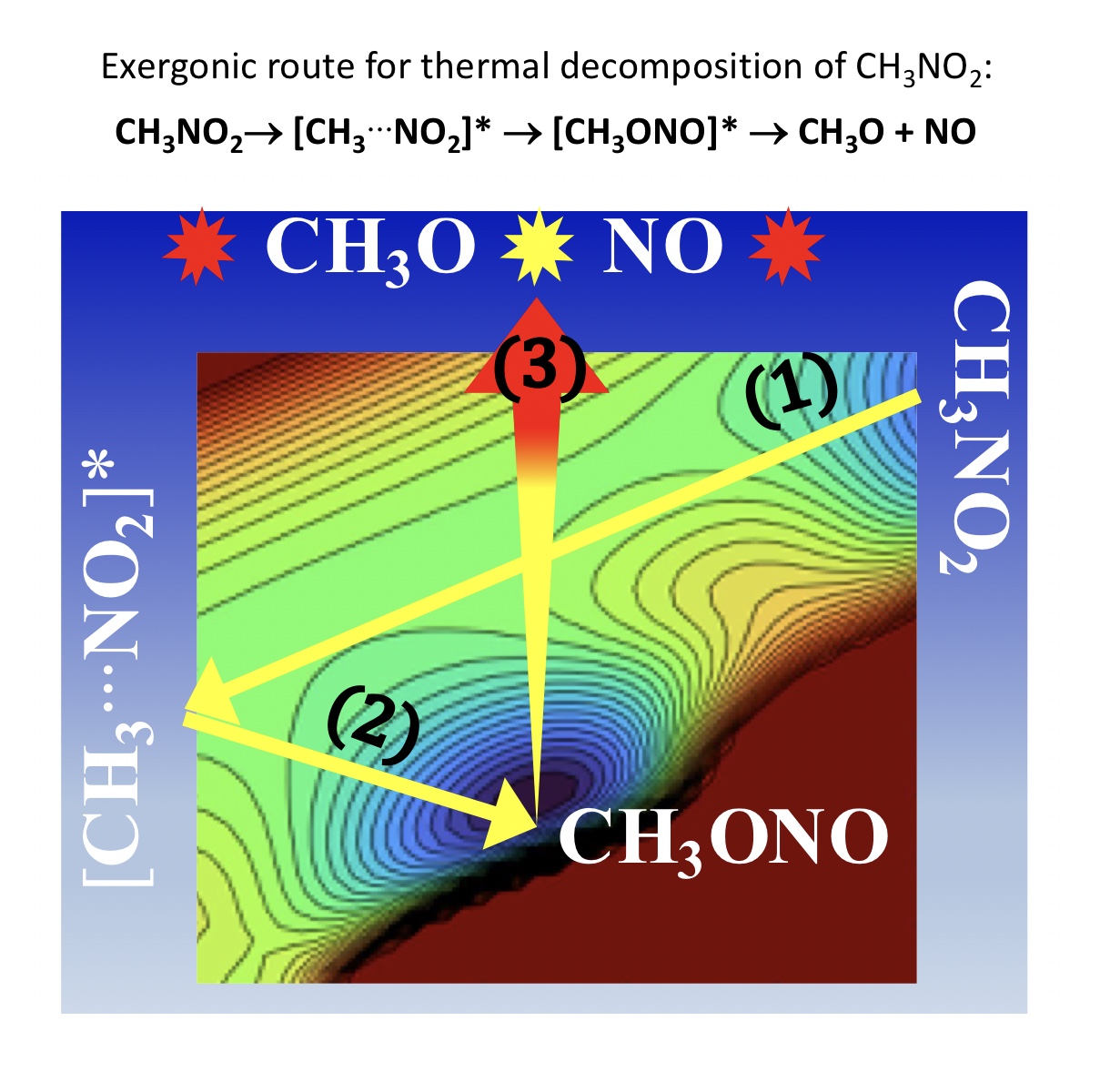
The global process starting at nitromethane is represented in equation 13
CH3NO2→ [CH3…NO2]* → [CH3ONO]* → CH3O + NO
Figure 8 collects diagrammatically the Gibbs free energies at four different temperatures of the process represented in equation 13 and Figure 6b–c. In accordance with such diagrams, the activation energy of the first step decreases as temperature increases and is the higher energy step in every case. Therefore, when the system reaches the region of the roaming rearrangement, it has accumulated enough energy to complete reaction 13. Curiously, the activation energies of the process that passes through the roaming molecular arrangement with a C-N internuclear distance of 4.0 Å (Figure 6c) are the lower ones at any temperature (Figure 8b). Gibbs free energies at different temperatures have been calculated with the standard expressions of Statistical Thermodynamics, the thermal corrections to the energies of the roaming structures have computed after projecting out the vibrational Hessian matrix the rotation-translation eigenvectors and gradient vectors. [85,86]
4. Conclusions
It is described a mapping method (orthogonalized 3D-representation) for the PESs by defining an orthonormal basis consisting of two orthonormal vectors that allows to obtain a set of ordered points in the vector subspace defined by the plane that contains the basis vectors.
The main elementary unimolecular reactions of nitromethane -including energetics, geometrical optimizations and calculated vibrational harmonic frequencies- have been studied with the CASPT2 method by using a well-balanced CASSCF reference wave function of 14(16) electrons distributed in 11(13) orbitals. All the kinetically controlled reactions are endergonic, being the C-N bond breaking the process with lower ΔG energy among this class of reactions. In contrast, it is found that the roaming process [CH3NO2→ [CH3…NO2]* → [CH3ONO]* → CH3O + NO] is exergonic at high temperatures, where the star symbols indicate vibrationally excited species. At this point, it is important to note that this mechanism has been previously proposed by other authors. [7,8,9] Furthermore, all the roaming structures analyzed in this work, the higher energy points in the mechanism given in equation 13, are well below the dissociation limit of nitromethane that leads to CH3 and NO2.
The so-called loose transition state has been re-investigated with two CASSCF active spaces: (i) 10 electrons distributed in 7 orbitals and (ii) 14 electrons in 10 orbitals. It is shown that the smaller active space yields artifactual results arising from an unbalanced description of the electronic structure. In fact, the selection of the active space for studying nitromethane would be straightforward after the work of Blahous et al. on NO2 radical, [87] where they pointed out that the CASSCF wave function to describe the whole configurational domain of the potential energy surface must contain 13 electrons distributed in 10 orbitals.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: CASPT2 and MP2/HF geometrical parameters of nitromethane.; Table S2: Geometrical parameters of trans-methyl nitrite (trans-CH3ONO); Table S3: Geometrical parameters of cis-methyl nitrite (cis-CH3ONO); Table S4: Geometrical parameters of the transition state (TS1) for nitromethane to methyl-nitrite isomerization; Table S5: Geometrical parameters of the transition state (TS2) for proton migration CH3NO2 → CH2N(O)OH; Table S6: Geometrical parameters of the transition state (TS3) of reaction CH3NO2 → CH2O + HNO; Table S7: Geometrical parameters of the transition state (TS4) of reaction trans-CH3ONO → cis-CH3ONO; Table S8: Geometrical parameters of nitrosomethane (CH3NO). Figure S1: MS-CASPT2 energy profiles of the linear interpolations for (a) the dissociation of CH3NO2 into CH3 and NO2; (b) the dissociation of trans-CH3ONO into CH3O and NO; (c) the dissociation of trans-CH3ONO and cis-CH3ONO into CH3 and NO2; Figure S2: . (a) MS-CASPT2 energy profiles of the dissociation of CH3NO2 into CH3NO and (O) atomic oxygen (linear interpolation in internal coordinates). Blue solid line (singlet A’); blue dotted line (singlet A”); red solid line (triplet A’); red dotted line (triplet A”). Green numbers: relative energy in kcal/mol with respect to the last point of lowest energy. (b) Calculated energetics of the isolated oxygen atom and nitrosomethane. Coordinate list: CASPT2/CASSCF/ANO-RCC Cartesian Coordinates in Å.
Funding
This research was funded by Spanish Ministry of Science and Innovation (MCIN/AEI/10. 13039/501100011033) through project PID2021-122613OB-I00.
Acknowledgments
The author thanks Dr. R. Larrosa and M. Guerrero for the technical support in running the calculations and the SCBI (Supercomputer and Bioinformatics) center of the University of Málaga (Spain) for computer resources. The author thanks the Spanish Ministry of Science and Innovation (MCIN/AEI/10. 13039/501100011033) through project PID2021-122613OB-I00.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Word, M.D.; Lo, H.A.; Boateng, D.A.; McPherson, S.L.; Gutsev, G.L.; Gutsev, L.G.; Lao, K.U.; Tibbetts, K.M. J. Phys. Chem. A 2022, 126, 879–888. [CrossRef]
- Leyva, E.; Loredo-Carrillo, S.E.; Aguilar, J. Reactions 2023, 4, 432–447. [CrossRef]
- Zhang, J.; Peng, J.; Hu, D.; Lan, Z. Phys. Chem. Chem. Phys., 2021, 23, 25597–25611. [CrossRef] [PubMed]
- McKee, M.L. J. Am. Chem. Soc., 1986, 108, 5784–5792.; McKee, M.L.. J. Phys. Chem., 1989, 93, 7365–7369.
- Saxon, R.P.; Yuoshimi, M. Can. J. Chem., 1992, 70, 572–579. [CrossRef]
- Chang, P.; Zhou, P.; Liu, J.; Yin, S. Chem. Phys. Lett., 2022, 792, 139413. [CrossRef]
- Homayoon, Z.; Bowman, J.M. J. J. Phys. Chem. A, 2013, 117, 11665–11672. (b) Homayoon, Z.; Bowman, J.M.; Dey, A.; Abeysekera, C.; Fernando, R.; Suits, A.G. Z. Phys. Chem., 2013, 227, 1267–1280. [Google Scholar]
- Zhu, R.S.; Raghunath, P.; Lin, M.C. J. Phys. Chem. A, 2013, 117, 7308–7313. [CrossRef]
- Zhu, R.S.; Lin, M.C. Chem. Phys. Lett., 2009, 478, 11–16. [CrossRef]
- Isegawa, M.; Liu, F.; Maeda, S.; Morokuma, K. J. Chem. Phys., 2014, 140, 244310. [CrossRef] [PubMed]
- Arenas, J.F.; Otero, J.C.; Peláez, D.; Soto, J. J. Chem. Phys., 2005, 122, 084324. [CrossRef] [PubMed]
- Arenas, J.F.; Otero, J.C.; Peláez, D.; Soto, J. J. Chem. Phys., 2003, 119, 7814–7823. [CrossRef]
- Sumida, M.; Kohge, Y.; Yamasaki, K.; Kohguchia, H. J. Chem. Phys., 2016, 144, 064304. [CrossRef] [PubMed]
- Li, W.-G.; Liu, Q.-J.; Liu, F.-S.; Liu, Z.-T. Phys. Chem. Chem. Phys., 2023, 25, 5613–5618. [CrossRef]
- Zheng, W.; Liu, Q.-J.; Liu, F.-S.; Liu, Z.-T. Phys. Chem. Chem. Phys., 2023, 25, 5685–5693. [CrossRef] [PubMed]
- Rice, B.M.; Sahu, S.; Owens, F.J. J. Mol. Struct. (Theochem), 2002, 583, 69–72. [CrossRef]
- Ford, J.; Seritan, S.; Zhu, X.; Sakano, M.N.; Islam, M.M.; Strachan, A.; Martínez, T.J. J. Phys. Chem. A, 2021, 125, 1447–1460. [CrossRef]
- Nelson, T.; Bjorgaard, J.; Greenfield, M.; Bolme, C.; Brown, K.; McGrane, S.; Scharff, R.J.; Tretiak, S. J. Phys. Chem. A, 2016, 120, 519–526. [CrossRef]
- Dey, A.; Fernando, R.; Abeysekera, C.; Homayoon, Z.; Bowman, J.M.; Suits, A.G. J. Chem. Phys., 2014, 140, 054305. [CrossRef]
- Annesley, C.J.; Randazzo, J.B.; Klippenstein, S.J.; Harding, L.B.; Jasper, A.W.; Georgievskii, Y.; Ruscic, B.; Tranter, R.S. J. Phys. Chem. A, 2015, 119, 7872–7893. [CrossRef] [PubMed]
- Wodtke, A.M.; Hintsa, E.J.; Lee, Y.T. J. Phys. Chem., 1986, 90, 3549–3558. [CrossRef]
- Bhattacharya, A.; Guo, Y.Q.; Bernstein, E.R. J. Chem. Phys., 2012, 136, 024321. [CrossRef] [PubMed]
- Guo, Y.Q.; Bhattacharya, A.; Bernstein, E.R. J. Phys. Chem. A, 2009, 113, 85–96. [CrossRef]
- Matsugi, A.; Shiina, H. J. Phys. Chem. A, 2017, 121, 4218–4224. [CrossRef]
- Townsend, D.; Lahankar, S.A.; Lee, S.K.; Chambreau, S.D.; Suits, A.G.; Zhang, X.; Rheinecker, J.; Harding, L.B.; Bowman, J.M. Science, 2004, 306, 1158–1161. [CrossRef] [PubMed]
- Herath, N.; Suits, A.G. J. Phys. Chem. Lett., 2011, 2, 642–647. [CrossRef]
- Suits, A.G. Annu. Rev. Phys. Chem., 2020, 71, 77–100. [CrossRef] [PubMed]
- Lopez, J.G.; Vayner, G.; Lourderaj, U.; Addepalli, S.V.; Kato, S.; W. A. deJong; Windus, T.L.; Hase, W.L. J. Am. Chem. Soc., 2007, 129, 9976–9985. [Google Scholar]
- Pomerantz, A.E.; Camden, J.P.; Chiou, A.S.; Ausfelder, F.; Chawla, N.; Hase, W.L.; Zare, R.N. J. Am. Chem. Soc., 2005, 127, 16368–16369. [CrossRef] [PubMed]
- Lourderaj, U.; Park, K.; Hase, W.L. Int. Rev. Phys. Chem., 2008, 27, 361–403. [CrossRef]
- Suits, A.G. Acc. Chem. Res., 2008, 41, 873–881. [CrossRef]
- Bowman, J.M.; Shepler, B.C. Annu. Rev. Phys. Chem., 2011, 62, 531–553. [CrossRef] [PubMed]
- Bowman, J.M.; Houston, P.L. Chem. Soc. Rev., 2017, 46, 7615–7624. [CrossRef]
- Houston, P.L.; Kable, S.H. Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16079–16082. [CrossRef]
- Harding, L.B.; Klippenstein, S.J.; Jasper, A.W. Phys. Chem. Chem. Phys., 2007, 9, 4055–4070. [CrossRef]
- Heazlewood, B.R.; Jordan, M.J.T.; Kable, S.H.; Selby, T.M.; Osborn, D.L.; Shepler, B.C.; Braams, B.J.; Bowman, J.M. Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12719–12724. [CrossRef] [PubMed]
- Roos, B.O., in Advances in Chemical Physics; Ab initio Methods in Quantum Chemistry II, ed. K. P. Lawley, John Wiley & Sons, Chichester, UK, 1987, ch. 69, p. 399. The Complete Active Space Self-Consistent Field Method and Its Applications in Electronic Structure Calculations.
- Roos, B.O.; Taylor, P.R.; Siegbahn, P.E.M. Chem. Phys., 1980, 48, 157–173. [CrossRef]
- Roos, B.O. Int. J. Quantum Chem., 1980, 18, 175–189. [CrossRef]
- Siegbahn, P.E.M.; Almlo, J.; Heiberg, A.; Roos, B.O. J. Chem. Phys., 1981, 74, 2384–2396. [CrossRef]
- Werner, H.-J.; Meyer, W. J. Chem. Phys. 1980, 73, 2342–2356. [CrossRef]
- Werner, H.-J.; Meyer, W. J. Chem. Phys., 1981, 74, 5794–5801. [CrossRef]
- Olsen, J. Int. J. Quantum. Chem., 2011, 111, 3267–3272. [CrossRef]
- Roos, B.O.; Andersson, K.; Fu, M.P.; P. Å. Malmqvist; Serrano-Andre, L.; Pierloot, K.; Mercha, M. Adv. Chem. Phys., 1996, 93, 219–331. [Google Scholar]
- Finley, J.; Malmqvist, P.-Å.; Roos, B.O.; Serrano-Andrés, L. Chem. Phys. Lett., 1998, 288, 299–306. [CrossRef]
- MOLCAS 8. 4; Veryazov, V.; Widmark, P.-O.; Serrano-Andrés, L.; Lindh, R.; Roos, B.O. Int. J. Quantum Chem., 2004, 100, 626–635. [Google Scholar]
- MOLCAS 8. 4; Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Galván, I.F.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; Garavelli, M.; Giussani, A.; Hoyer, C.E.; Manni, G.L.; Lischka, H.; Ma, D.; Malmqvist, P.Å.; Müller, T.; Nenov, A.; Olivucci, M.; Pedersen, T.B.; Peng, D.; Plasser, F.; Pritchard, B.; Reiher, M.; Rivalta, I.; Schapiro, I.; Segarra-Martí, J.; Stenrup, M.; Truhlar, D.G.; Ungur, L.; Valentini, A.; Vancoillie, S.; Veryazov, V.; Vysotskiy, V.P.; Weingart, O.; Zapata, F.; Lindh, R. J. Comp. Chem. 2016, 37, 506–541. [Google Scholar]
- Galván, I.F.; et al. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput., 2019, 15, 5925–5964. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; et al. J. Chem. Phys., 2020, 152, 214117.
- Roos, B.O.; Lindh, R.; Malmqvist, P.-Å.; Veryazov, V.; Widmark, P.-O. J. Phys. Chem. A, 2004, 108, 2851–2858. [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.-Å.; Veryazov, V.; Widmark, P.-O. J. Phys. Chem. A, 2005, 109, 6575–6579. [CrossRef]
- Møller, C.; Plesset, M.S. Phys. Rev., 1934, 46, 618–622. [CrossRef]
- Zhao, Y.; Truhlar, D.G. Theor Chem Account, 2008, 120, 215–241. [CrossRef]
- Gaussian 16, Revision C.02, Frisch, M.J. et al. Gaussian, Inc.: Wallingford CT, 2016.
- Weigend, F. . Ahlrichs. Phys. Chem. Chem. Phys., 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Phys. Chem. Chem. Phys., 2006, 8, 1057–1065. [CrossRef] [PubMed]
- Soto, J.; Algarra, M. J. J. Phys. Chem. A 2021, 125, 9431–9437. [CrossRef] [PubMed]
- Arenas, J.F.; Otero, J.C.; Peláez, D.; Soto, J.; Serrano-Andrés, L. J. Chem. Phys., 2004, 121, 4127–4132. [CrossRef] [PubMed]
- Arenas, J.F.; Otero, J.C.; Peláez, D.; Soto, J. J. Phys. Chem. A, 2005, 109, 7172–7180. [CrossRef] [PubMed]
- Soto, J.; Peláez, D.; Otero, J.C.; Avila, F.J.; Arenas, J.F. Phys. Chem. Chem. Phys., 2009, 11, 2631–2639. [CrossRef] [PubMed]
- Zhang, J.J.; Peng, J.W.; Zhu, Y.F.; Hu, D.P.; Lan, Z.G. J. Phys. Chem. Lett., 2023, 14, 6542–6549. [CrossRef] [PubMed]
- Zhang, J.J.; Peng, J.W.; Hu, D.P.; Xu, C.; Lan, Z.G. Chin. J. Chem. Phys., 2022, 35, 451–460. [CrossRef]
- Verlet, L. Phys. Rev., 1967, 159, 98–103. [CrossRef]
- Swope, W.C.; Andersen, H.C.; Berens, P.H.; Wilson, K.R. J. Chem. Phys., 1982, 76, 637–649. [CrossRef]
- Schaftenaar, G.; Noordik, J.H. J. Comput. Aided Mol. Des., 2000, 14, 123–134. [CrossRef] [PubMed]
- Allouche, A.R. J. Comput. Chem. 2011, 32, 174–182. [CrossRef] [PubMed]
- Bode, B.M.; Gordon, M.S. J. Mol. Graphics Modell. 1998, 16, 133–138. [CrossRef]
- Soto, J.; Peláez, D.; Algarra, M. J. Chem. Phys. 2023, 158, 204301. [CrossRef] [PubMed]
- Aranda, D.; Avila, F.J.; López-Tocón, I.; Arenas, J.F.; Otero, J.C.; Soto, J. Phys. Chem. Chem. Phys. 2018, 20, 7764–7771. [CrossRef] [PubMed]
- Soto, J.; Otero, J.C.; Avila, F.J.; Peláez, D. Phys. Chem. Chem. Phys. 2019, 21, 2389–2396. [CrossRef]
- Soto, J. J. Phys. Chem. A, 2022, 126, 8372–8379. [CrossRef] [PubMed]
- Peláez, D.; Arenas, J.F.; Otero, J.C.; Soto, J. J. Chem. Phys. 2006, 125, 164311. [CrossRef]
- Soto, J.; Arenas, J.F.; Otero, J.C.; Peláez, D. J. Phys. Chem. A 2006, 110, 8221–8226. [CrossRef]
- Ruscic, B.; Pinzon, R.E.; Morton, M.L.; von Laszevski, G.; Bittner, S.J.; Nijsure, S.G.; Amin, K.A.; Minkoff, M.; Wagner, A.F. J. Phys. Chem. A 2004, 108, 9979–9997. [CrossRef]
- Ruscic, B.; Pinzon, R.E.; von Laszewski, G.; Kodeboyina, D.; Burcat, A.; Leahy, D.; Montoy, D.; Wagner, A.F. J. Phys. Conf. Ser. 2005, 16, 561–570. [CrossRef]
- Ruscic, B.; Bross, D.H. Active Thermochemical Tables (ATcT), values based on ver. 1.124 of the Thermochemical Network (2022). Available online: https://atct.anl.gov/Thermochemical Data/version 201.124/ version 1.124/.
- Cox, A.P.; Waring, S. J. Chem. Soc. Faraday Trans. II 1972, 68, 1060–1071. [CrossRef]
- Turner, P.H.; Corkill, M.J.; Cox, A.P. J. Phys. Chem. 1979, 83, 1473–1482; Veken, B.J.; Maas, R.; Guirgis, G.A.; Stidham, G.A.; Sheehan, T.G.; Durig, J.R.. J. Phys. Chem. 1990, 94, 4029–4039.
- Turner, P.H.; Cox, A.P. . Dipole Moment of Acetaldehyde. J. Chem. Soc. Faraday Trans. II 1978, 74, 533–559. [Google Scholar] [CrossRef]
- Soto, J. J. Phys. Chem. A 2023, 127, 9781–9786. [CrossRef] [PubMed]
- Soto, J.; Peláez, D.; Otero, J.C. J. Chem. Phys. 2021, 154, 044307. [CrossRef]
- Liu, M.K.; Li, J.; Li, Q.S.; Li, Z.S. Phys. Chem. Chem. Phys. 2022, 24, 6266–6273. [CrossRef]
- Mu, D.; Li, Q.S. Phys. Chem. Chem. Phys. 2023, 25, 8074–8081. [CrossRef]
- Peng, X.L.; Migani, A.; Li, Q.S.; Li, Z.S.; Blancafort, L. Phys. Chem. Chem. Phys. 2018, 20, 1181–1188. [CrossRef] [PubMed]
- Soto, J.; Algarra, M.; Peláez, D. Phys. Chem. Chem. Phys. 2022, 24, 5109–5115. [CrossRef]
- Arenas, J.F.; Marcos, J.I.; López-Tocón, I.; Otero, J.C.; Soto, J. J. Chem. Phys. 2000, 113, 2282–2289. [CrossRef]
- C. P. Blahous III; Yates, B.F.; Xie, Y.; Schaefer, H.F., III. J. Chem. Phys. 1990, 93, 8105–8109. [Google Scholar]
Figure 1.
(A) Schematic representation of the orthogonalized mapping method. (B) CASPT2 contour plot (2D-representation) of the potential energy surface (PES) connecting the system the system [nitromethane:trans-methylnitrite:cis-methylnitrite]. (C) CASPT2 3D-representation of the PES of the same system. pTS1 and pTS4: saddle points on the PES. Grid size (41x41).
Figure 1.
(A) Schematic representation of the orthogonalized mapping method. (B) CASPT2 contour plot (2D-representation) of the potential energy surface (PES) connecting the system the system [nitromethane:trans-methylnitrite:cis-methylnitrite]. (C) CASPT2 3D-representation of the PES of the same system. pTS1 and pTS4: saddle points on the PES. Grid size (41x41).
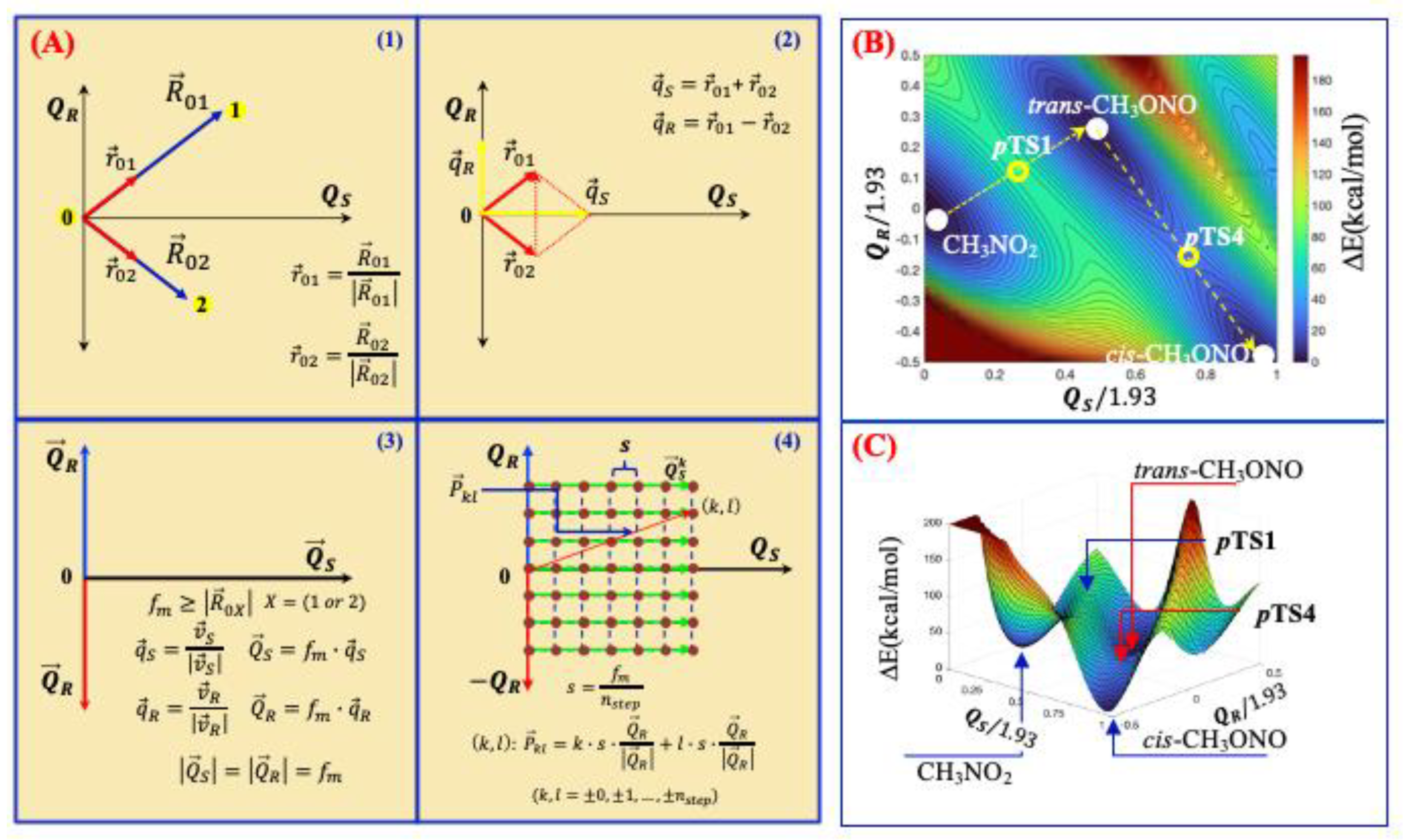
Figure 2.
CASSCF/ANO-RCC optimized geometries of (a) loose transition state [CAS(10e, 7o)] and (b) tight transition state [CAS(14e, 11o)]. Numbers correspond to main geometrical parameters in Å; arrows: transition mode.
Figure 2.
CASSCF/ANO-RCC optimized geometries of (a) loose transition state [CAS(10e, 7o)] and (b) tight transition state [CAS(14e, 11o)]. Numbers correspond to main geometrical parameters in Å; arrows: transition mode.
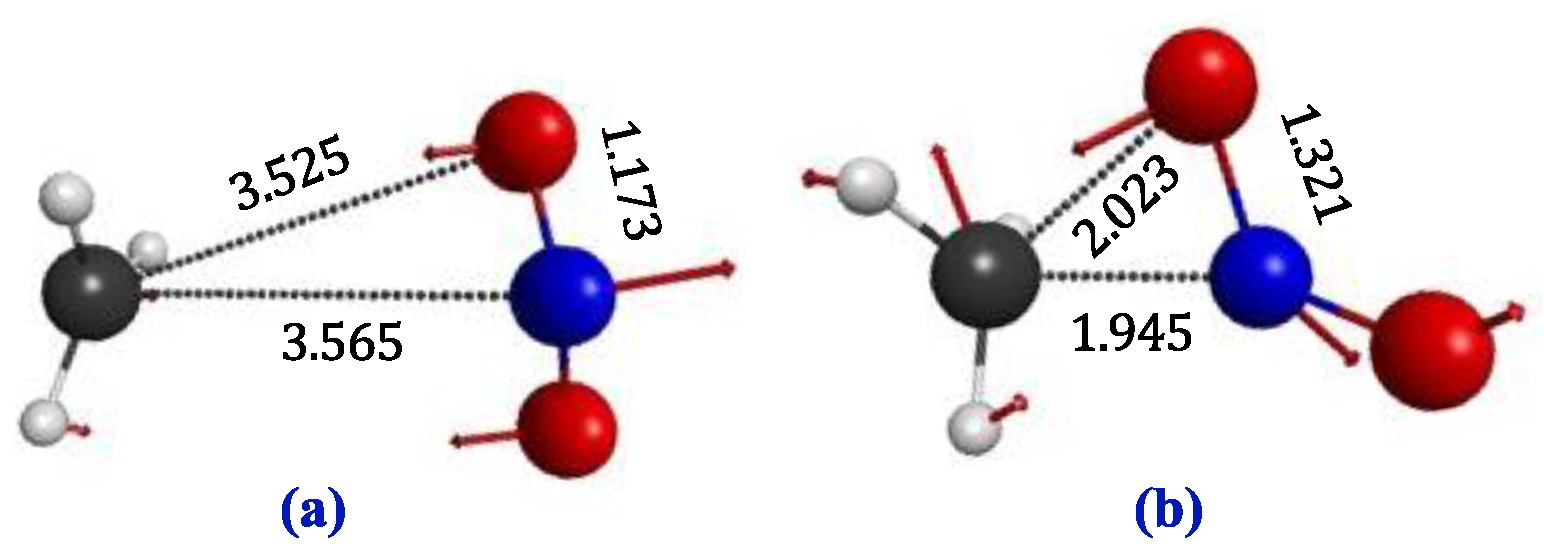
Figure 3.
CASSCF/ANO-RCC molecular orbitals. (a) CASSCF(10e,7o); (b) CASSCF(14e,11o). In parenthesis: occupation numbers.
Figure 3.
CASSCF/ANO-RCC molecular orbitals. (a) CASSCF(10e,7o); (b) CASSCF(14e,11o). In parenthesis: occupation numbers.
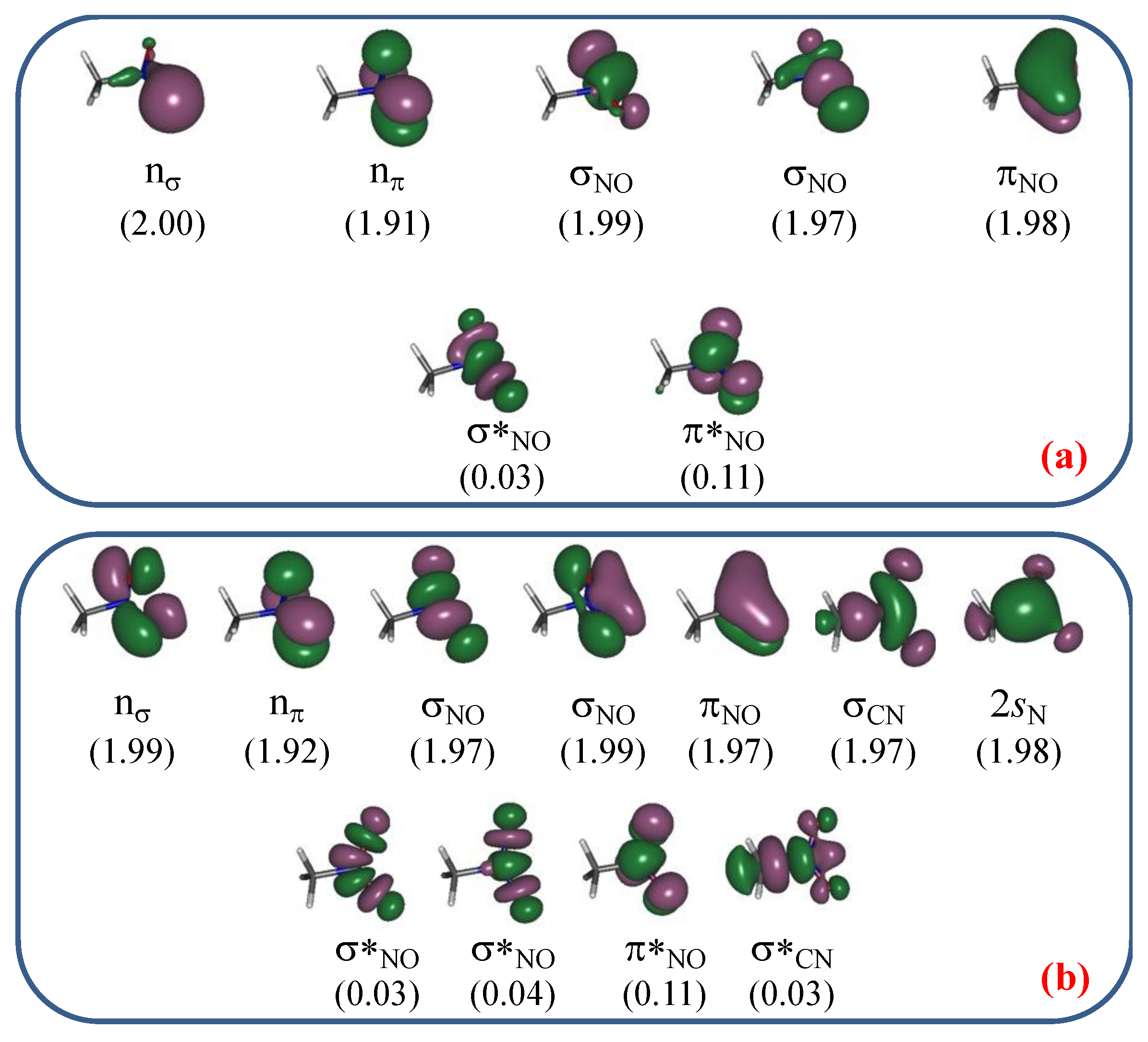
Figure 4.
CASPT2 profiles of the linear interpolations in internal coordinates connecting minimum of nitromethane (Cs) with the loose structure (C1): (a) CASPT2/CASSCF(10o,7e); (b) CASPT2/CASSCF(14e,11o).
Figure 4.
CASPT2 profiles of the linear interpolations in internal coordinates connecting minimum of nitromethane (Cs) with the loose structure (C1): (a) CASPT2/CASSCF(10o,7e); (b) CASPT2/CASSCF(14e,11o).
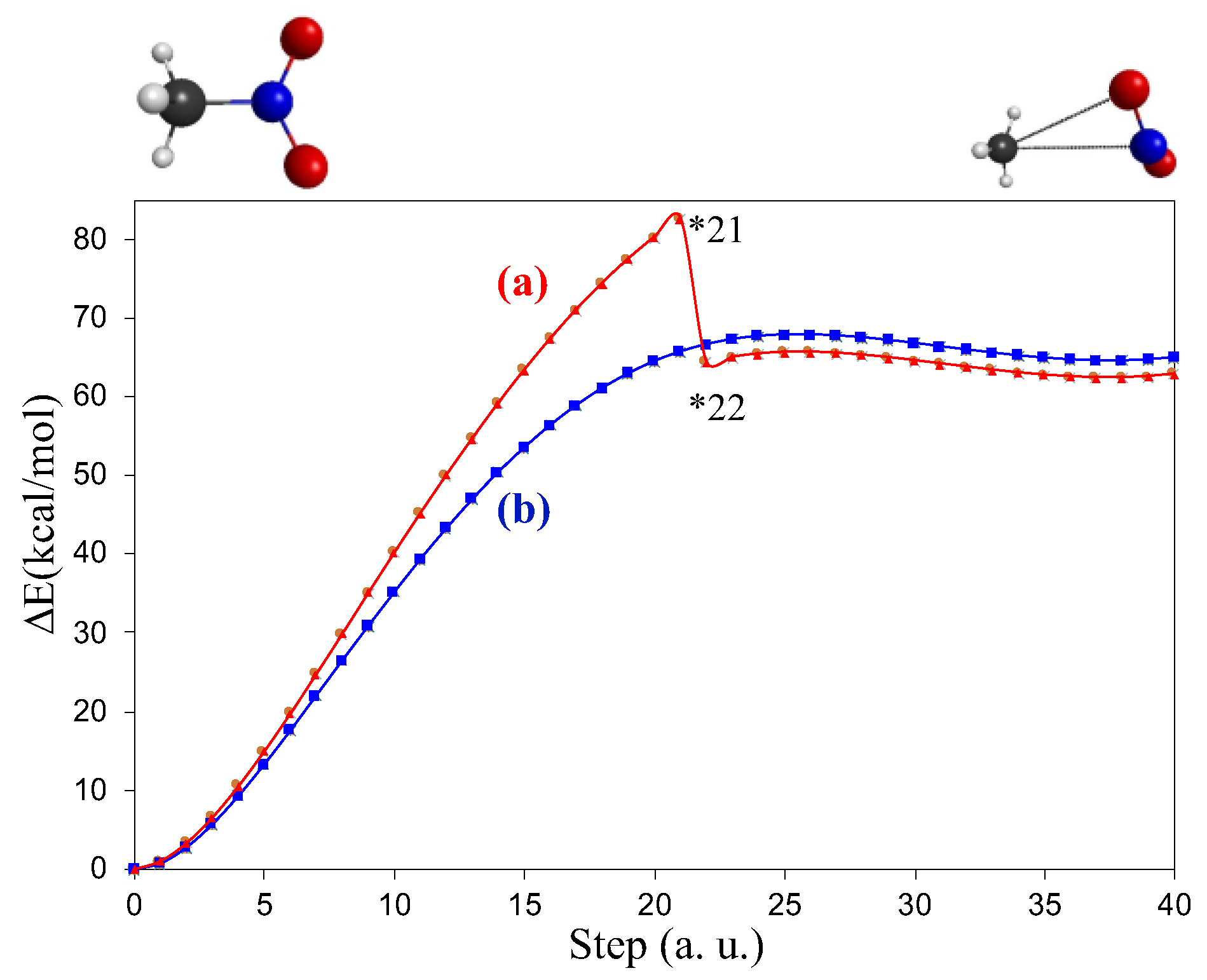
Figure 5.
Mapping of the potential energy surface of the ternary system [CH3NO2:Loose:cis-CH3ONO]. (a) 3D-representation of potential energy (b) 2D-representation of potential energy. CASPT2 from CASSCF(14,11)/ANO-RCC reference wave function. Grid size (41x41).
Figure 5.
Mapping of the potential energy surface of the ternary system [CH3NO2:Loose:cis-CH3ONO]. (a) 3D-representation of potential energy (b) 2D-representation of potential energy. CASPT2 from CASSCF(14,11)/ANO-RCC reference wave function. Grid size (41x41).
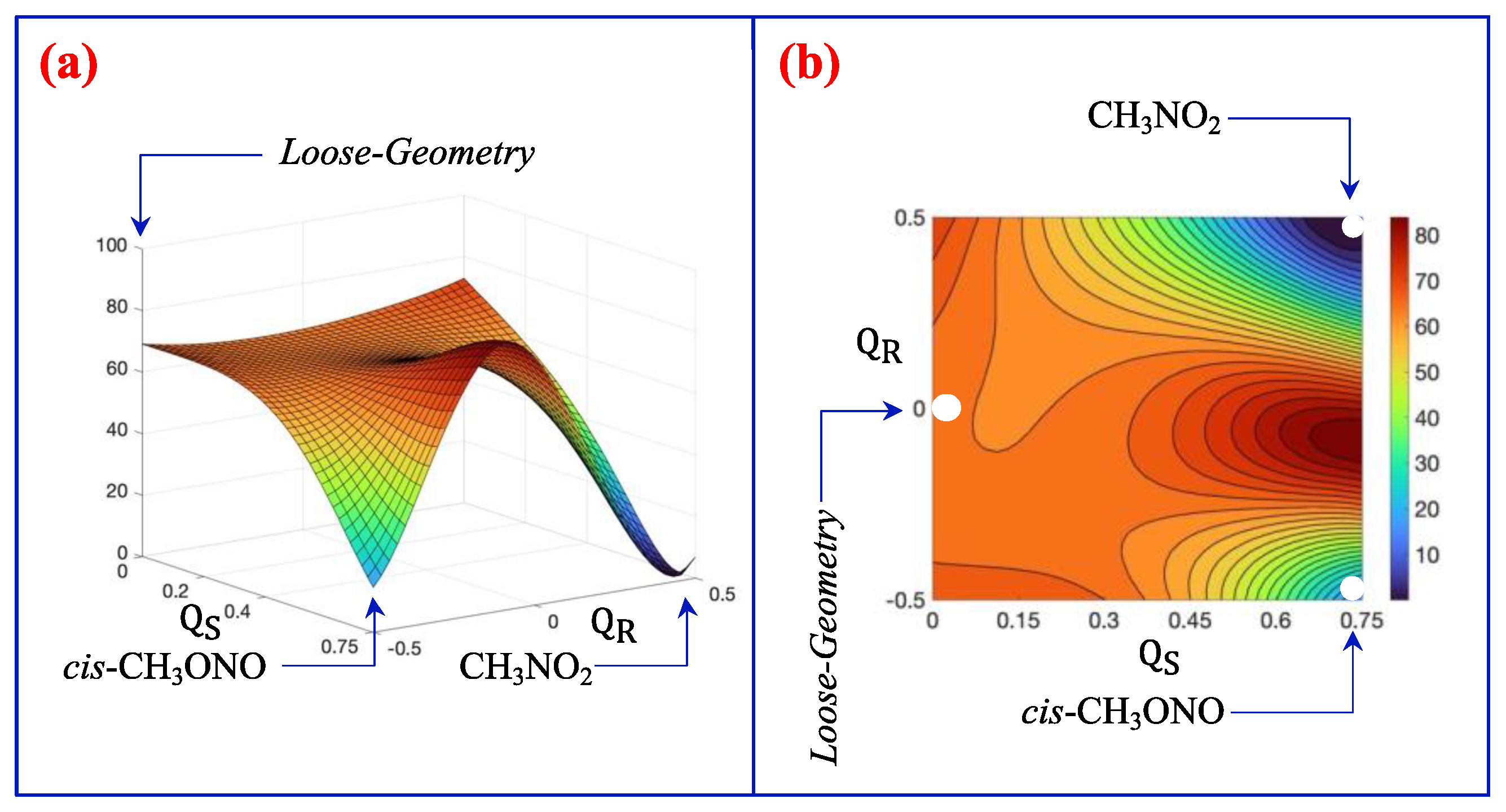
Figure 6.
2D- and 3D-representions of the CASPT2/CASSCF(14, 11) potential energy surfaces of the ternary system [CH3NO2:Roaming-Structure:cis-CH3ONO] at different starting geometries 0n. ΔEe: electronic energy variation in passing from 0n to 0n+1. Grid size (41x41).
Figure 6.
2D- and 3D-representions of the CASPT2/CASSCF(14, 11) potential energy surfaces of the ternary system [CH3NO2:Roaming-Structure:cis-CH3ONO] at different starting geometries 0n. ΔEe: electronic energy variation in passing from 0n to 0n+1. Grid size (41x41).
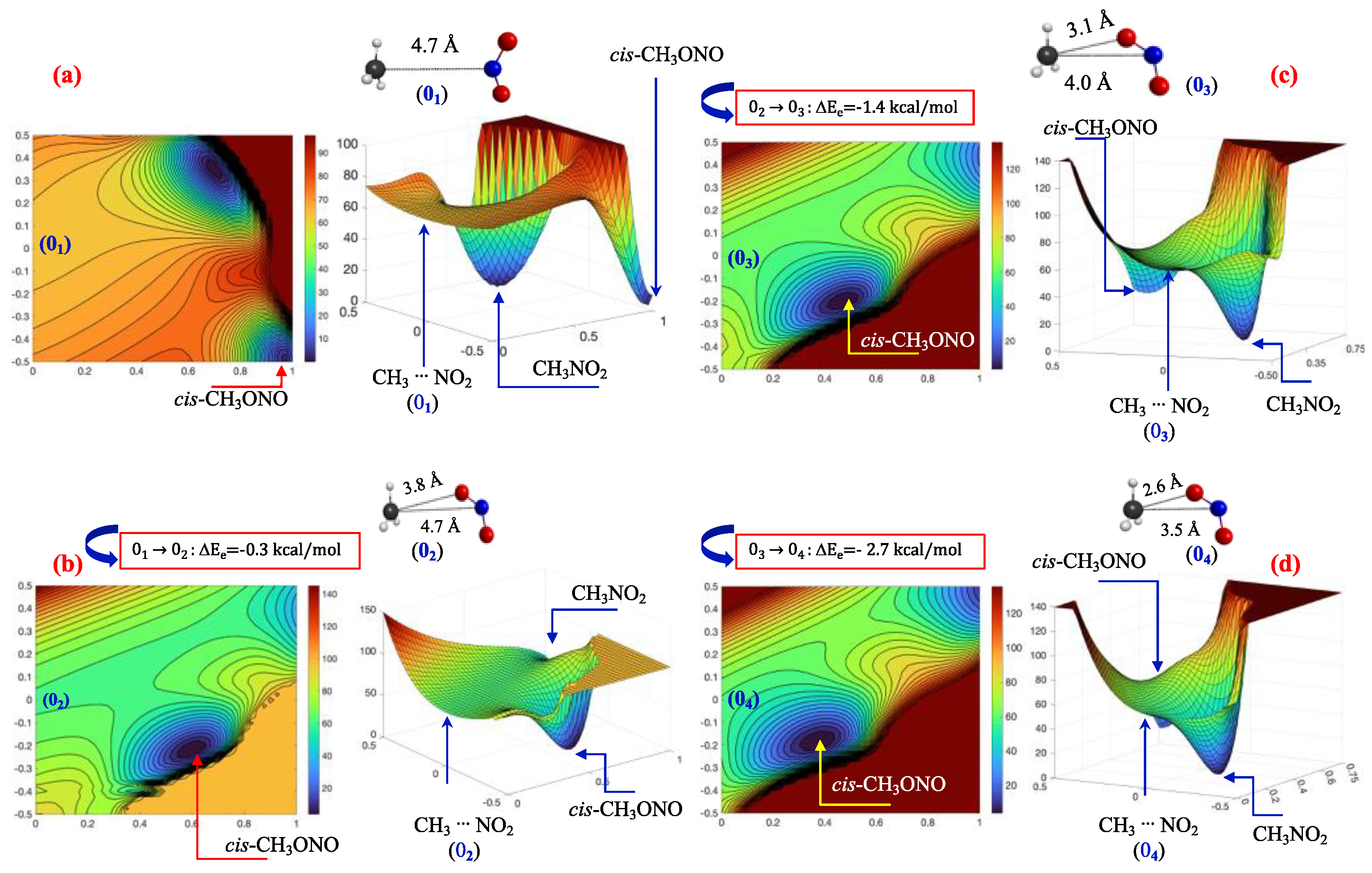
Figure 7.
(a) CASSCF energy profiles as time function starting at the roaming geometry given in Figure 6c with different kinetic energies (inset). (b) Selected molecular rearrangements for the trajectory with initial condition Ekin=22 kcal/mol. (c) End point (240 fs) in each trajectory calculation. Rectangles in (a) indicate displacements in y-axis to avoid overlapping of the curves.
Figure 7.
(a) CASSCF energy profiles as time function starting at the roaming geometry given in Figure 6c with different kinetic energies (inset). (b) Selected molecular rearrangements for the trajectory with initial condition Ekin=22 kcal/mol. (c) End point (240 fs) in each trajectory calculation. Rectangles in (a) indicate displacements in y-axis to avoid overlapping of the curves.
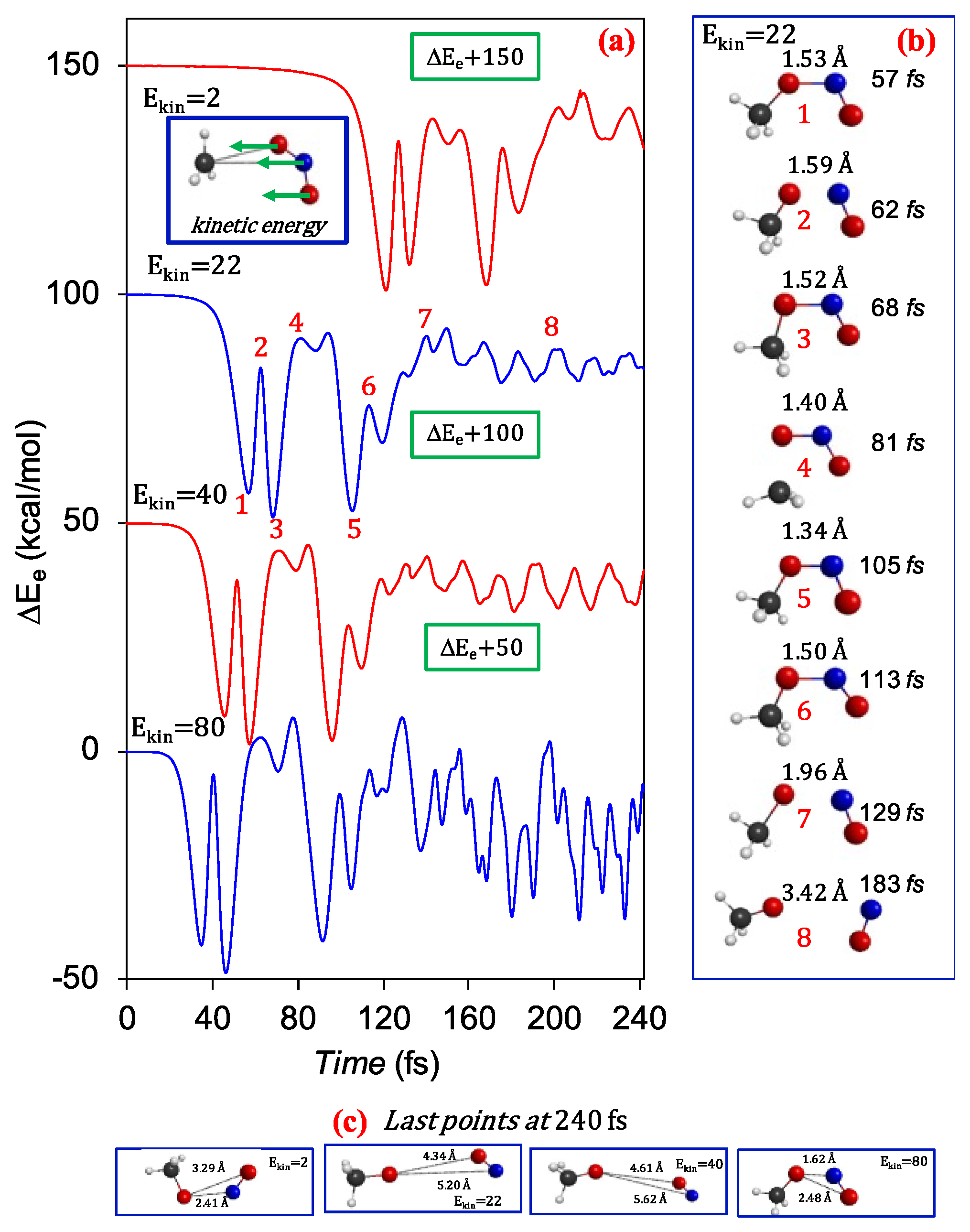
Figure 8.
Schematic representation of the Gibbs energy profiles for the process (i) CH3NO2→ (ii) [CH3…NO2]* → (iii) [CH3ONO]* → (iv) CH3O + NO (equation 13) at different temperatures and geometries included in Figure 6b,c.
Figure 8.
Schematic representation of the Gibbs energy profiles for the process (i) CH3NO2→ (ii) [CH3…NO2]* → (iii) [CH3ONO]* → (iv) CH3O + NO (equation 13) at different temperatures and geometries included in Figure 6b,c.
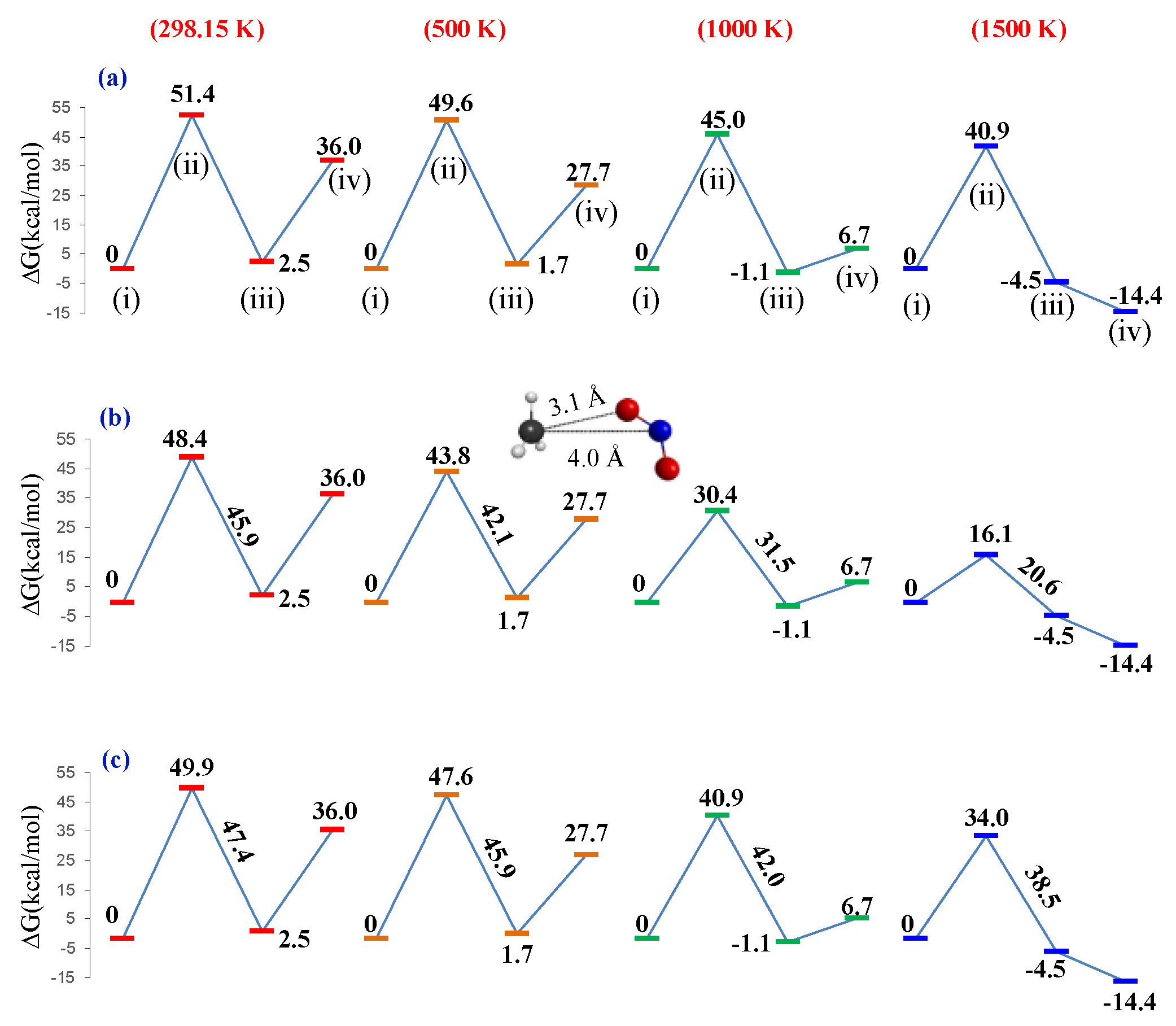

Table 1.
Energetics (kcal/mol) of the dissociation reactions of nitromethane and methyl nitrite.
| Reaction | Method | ΔdGa | ΔdEe,b | ΔdHc |
| CH3NO2(g) → CH3(g) + NO2(g) | CASPT2 | 65.95 | ||
| (0 K) | 58.76 | 58.76 | ||
| [59.16]d | ||||
| (298.15 K) | 50.83 | 60.10 | ||
| [61.00] | ||||
| MP2/HF | 66.61 | |||
| (0 K) | 63.71 | 63.71 | ||
| (298.15 K) | 56.70 | 65.06 | ||
| M06-2X | 67.93 | |||
| (0 K) | 60.51 | 60.51 | ||
| (298.15 K) | 53.35 | 61.93 | ||
| t-CH3ONO(g) →CH3O(g)+NO(g) | CASPT2 | 46.48 | ||
| (0 K) | 42.60 | 42.60 | ||
| [41.11] | ||||
| (298.15 K) | 33.13 | 43.99 | ||
| [42.32] | ||||
| MP2/HF | 51.10 | |||
| (0 K) | 49.28 | 49.28 | ||
| (298.15 K) | 39.79 | 50.69 | ||
| M06-2X | 42.63 | |||
| (0 K) | 37.86 | 37.86 | ||
| (298.15 K) | 28.31 | 39.40 | ||
| CH3NO2(g)→CH3NO(g)+O(3P)(g) | CASPT2 | 99.53 | ||
| (0 K) | 95.32 | 95.32 | ||
| [92.83] | ||||
| (298.15 K) | 88.19 | 96.47 | ||
| [94.35] | ||||
| MP2/HF | 104.88 | |||
| (0 K) | 100.44 | 100.44 | ||
| (298.15 K) | 93.46 | 101.57 | ||
| M06-2X | 96.82 | |||
| (0 K) | 92.53 | 92.53 | ||
| (298.15 K) | 85.47 | 93.68 |
aGibbs free energy. bElectronic energy. cEnthalpy. dIn square brackets: values from https://atct.anl.gov/Thermochemical Data Refs. [74,75,76].
Table 2.
Energetics (kcal/mol) of the rearrangement and proton migration reactions of nitromethane and methyl nitrite.
Table 2.
Energetics (kcal/mol) of the rearrangement and proton migration reactions of nitromethane and methyl nitrite.
| Reaction | ΔaGa | ΔaEeb | ΔaHc | ΔrHd | |
| CH3NO2(g) t-CH3ONO(g) | CASPT2 | 69.25 | |||
| (0 K) | 66.31 | 66.31 | 2.55 | ||
| [1.99]e | |||||
| (298.15 K) | 66.66 | 66.42 | 2.66 | ||
| [2.45] | |||||
| MP2 | 71.84 | ||||
| (0 K) | 69.19 | 69.19 | 6.10 | ||
| (298.15 K) | 69.91 | 69.12 | 5.91 | ||
| M06-2X | 73.15 | ||||
| (0 K) | 70.77 | 70.77 | 2.10 | ||
| (298.15 K) | 71.33 | 70.77 | 2.44 | ||
| CH3NO2(g) CH2N(O)OH(g) | CASPT2 | 66.54 | |||
| (0 K) | 63.01 | 63.01 | 12.88 | ||
| (298.15 K) | 64.98 | 62.54 | 12.76 | ||
| MP2 | 66.35 | ||||
| (0 K) | 62.86 | 62.86 | 16.32 | ||
| (298.15 K) | 62.86 | 62.95 | 16.20 | ||
| M06-2X | 65.66 | ||||
| (0 K) | 62.19 | 62.19 | 12.52 | ||
| (298.15 K) | 63.25 | 61.77 | 12.33 | ||
| t-CH3ONO(g) CH2O(g) + HNO(g) | CASPT2 | 43.80 | |||
| (0 K) | 38.76 | 38.76 | 14.34 | ||
| [13.62] | |||||
| (298.15 K) | 38.90 | 38.68 | 15.97 | ||
| [14.85] | |||||
| MP2 | 41.94 | ||||
| (0 K) | 37.69 | 37.69 | 13.42 | ||
| (298.15 K) | 37.27 | 37.99 | 15.06 | ||
| M06-2X | 49.92 | ||||
| (0 K) | 45.88 | 45.88 | 14.92 | ||
| (298.15 K) | 46.73 | 45.54 | 16.06 | ||
| t-CH3ONO(g) c-CH3ONO(g) | CASPT2 | 12.12 | |||
| (0 K) | 11.62 | 11.62 | -0.86 | ||
| [-0.74] | |||||
| (298.15 K) | 11.47 | 12.28 | -0.60 | ||
| [-0.70] | |||||
| MP2 | 11.26 | ||||
| (0 K) | 10.63 | 10.63 | -1.11 | ||
| (298.15 K) | 10.42 | 10.75 | -0.94 | ||
| M06-2X | 10.95 | ||||
| (0 K) | 10.48 | 10.48 | -1.29 | ||
| (298.15 K) | 11.46 | 9.99 | -1.66 |
aGibbs free energy. bElectronic energy. cEnthalpy. dIn square brackets: values from https://atct.anl.gov/Thermochemical Data Refs. [74,75,76].
Table 3.
CASSCF and CASPT2 geometrical parameters of the loose and tight (TS1) transition states for nitromethane to methyl-nitrite isomerization Figure 2.
Table 3.
CASSCF and CASPT2 geometrical parameters of the loose and tight (TS1) transition states for nitromethane to methyl-nitrite isomerization Figure 2.
 | ||||||
| Internal Coord.a,b,c |
CAS1 Loose |
CAS2 Tight |
CASPT2 Tight |
Ld CAS1 |
Td CAS2 |
Td CASPT2 |
| R2,1 | 3.565 | 1.945 | 1.890 | 3426 | 3435 | 3300 |
| R3,2 | 1.173 | 1.321 | 1.317 | 3425 | 3405 | 3258 |
| A3,2,1 | 78.6 | 73.8 | 72.0 | 3245 | 3263 | 3126 |
| R4,2 | 1.173 | 1.202 | 1.202 | 1921 | 1592 | 1531 |
| A4,2,1 | 92.5 | 151.2 | 144.0 | 1524 | 1558 | 1506 |
| Dh4,2,1,3 | -134.0 | 121.1 | 114.1 | 1523 | 1538 | 1461 |
| R5,1 | 1.070 | 1.069 | 1.077 | 1479 | 1360 | 1280 |
| A5,1,2 | 107.4 | 99.6 | 98.9 | 819 | 1118 | 1106 |
| Dh5,1,2,4 | 42.3 | -149.2 | -148.3 | 236 | 960 | 933 |
| R6,1 | 1.070 | 1.071 | 1.082 | 106 | 919 | 916 |
| A6,1,2 | 76.3 | 91.0 | 91.0 | 80 | 711 | 721 |
| Dh6,1,2,5 | 117.6 | 115.7 | 115.0 | 65 | 461 | 494 |
| R7,1 | 1.070 | 1.069 | 1.081 | 38 | 178 | 214 |
| A7,1,2 | 88.8 | 117.6 | 120.7 | 13 | 131 | 143 |
| Dh7,1,2,5 | -121.2 | -125.8 | -126.9 | 52 i | 1066 i | 990 i |
| R3,1 | 3.525 | 2.023 | 1.942 | |||
aR: internuclear distance in Å; bA valence bond angle; cDh: dihedral angle. dHarmonic frequencies. CAS1: CAS(10,7); CAS2:CAS(14,11).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



