
Submitted:
25 January 2025
Posted:
27 January 2025
You are already at the latest version
Abstract
Granulomatous and amyloidogenic cardiomyopathies are infiltrative conditions that can be fatal if left untreated. Among these, cardiac amyloidosis and cardiac sarcoidosis are significant but often underdiagnosed causes of heart failure, each serving as cardiac manifestations of broader systemic diseases. Advancements in imaging techniques, along with the emergence of novel therapies—particularly for cardiac amyloidosis—have brought these conditions into sharper focus for both clinicians and researchers. The assessment of patients' cardiac and extracardiac symptoms, combined with echocardiography, is crucial in raising initial suspicion for these diseases. Cardiac magnetic resonance imaging is essential for differentiating between the two, as distinct late gadolinium enhancement patterns are observed in each condition. Additional diagnostic value is provided by positron emission tomography and Technetium-labeled nuclear scintigraphy, which can help confirm the diagnosis in the majority of patients. Early diagnosis is key to improving outcomes. Treatment strategies for these conditions differ significantly: cardiac amyloidosis is primarily managed with disease-modifying therapies for the transthyretin subtype and chemotherapy for the AL subtype, while cardiac sarcoidosis is treated with corticosteroids and immunosuppressive drugs aimed at reducing inflammation.
Keywords:
cardiac amyloidosis
; cardiac sarcoidosis
; granulomatous cardiomyopathy
; cardiac magnetic resonance
; echocardiography
; nuclear scintigraphy
; PET
Introduction
Granulomatous and amyloidogenic cardiomyopathies are infiltrative conditions that can be fatal if left untreated. Among these, cardiac amyloidosis (CA) and cardiac sarcoidosis (CS) are significant but often underdiagnosed causes of heart failure with distinct pathogenetic mechanisms, each serving as cardiac manifestations of broader systemic diseases (Figure 1). In CA, pathophysiology involves synthesis of misfolded proteins which form amyloid fibrils that infiltrate the myocardium[1]. These fibrils accumulate in the extracellular matrix of the heart, disrupting normal myocardial architecture and function. The amyloid deposition leads to stiffening of the myocardium, impairing both diastolic and systolic function, which contributes to heart failure and arrythmias. In >90% of cases, the amyloidogenesis involves two proteins, transthyretin (ATTR) and immunoglobulin light chain (AL) [3].
TTR is a tetrameric protein primarily produced in the liver that plays a role in transporting thyroxine and vitamin A. In ATTR, genetic mutations (ATTRv) or aging-related changes (ATTRwt) lead to instability in the TTR tetramer, causing dissociation into monomers that subsequently misfold and aggregate into amyloid fibrils [3]. ATTRwt commonly presents as heart failure with preserved ejection fraction (HFpEF), with patients experiencing symptoms such as dyspnea, fatigue, and exercise intolerance [4]. Other notable symptoms include peripheral edema, orthopnea, and paroxysmal nocturnal dyspnea. As many as ~1/3rd of patients hospitalized due to HFpEF are found to have ATTRwt [5]. In addition to heart failure, patients may experience arrhythmias, especially atrial fibrillation and conduction disturbances, such as bundle branch blocks or atrioventricular (AV) block (AVB), due to amyloid infiltration of the cardiac conduction system [6]. One of the distinguishing features of ATTRwt is its insidious onset, often without clear preceding risk factors for heart failure. It is also more common in men, with elderly Caucasians being particularly affected. Although ATTRwt primarily affects the heart, it can also involve other organs, albeit less frequently than the familial forms of amyloidosis.
ATTRv is a genetically heterogeneous condition, characterized by a wide range of clinical manifestations, depending on the specific mutation, as well as the affected organ systems. The disease often presents as either cardiomyopathy or neuropathy, or a combination of both. The V122I mutation is the common mutation in the United States, that behaves like ATTRwt, affecting predominantly elderly men [7]. The key phenotypic difference is that, unlike ATTRwt that affects predominantly Caucasians, V122I mutation almost exclusively affects individuals of African-American race [7,8]. The most common mutation worldwide, V30M, is caused by a methionine for valine substitution at residue 30 of the mature TTR protein, which is encoded by the TTR gene located on chromosome18q12.1, and could manifest neuropathy and/or cardiomyopathy [9]. It presents either as early-onset or late-onset disease, and while the worldwide prevalence is unknown, data is available for countries like Portugal, Japan and Sweden where the disease is endemic [9]. T60A remains the predominant genetic mutation identified in Irish patients, who typically present in the seventh decade with an already manifest neuropathy phenotype, largely predating their cardiac phenotype, which is dominated by heart failure [10].
AL is a hematologic disorder characterized by the deposition of misfolded immunoglobulin light chains as amyloid fibrils in various organs, including the kidneys, heart, liver, and nervous system. The pathogenesis of AL begins with the clonal expansion of plasma cells in the bone marrow, which produce abnormal light chains [11]. These light chains are produced in excess or in an improperly folded form and are secreted into the bloodstream. Once in circulation, the light chains can undergo misfolding, leading to the formation of amyloid fibrils. Heart is the second most commonly affected organ after the kidneys, and is the key determinant of prognosis. AL is a more aggressive disease than ATTR, with a 6-month survival from the onset of cardiac symptoms if untreated [12].
CS, a prototype of granulomatous cardiomyopathy, is a rare manifestation of sarcoidosis, characterized by the infiltration of the myocardium by noncaseating granulomas. The exact cause of CS is not fully understood but is thought to result from a complex interplay of genetic, environmental, and immunologic factors [13,14]. It is commonly associated with systemic sarcoidosis, particularly pulmonary involvement, but isolated cardiac involvement can also occur. While 5% of patients with CS are symptomatic, while upto 1/3rd have silent disease that is detected after imaging or autopsy [15]. In CS, granulomatous inflammation typically affects the interventricular septum, left ventricle (LV), and occasionally the right ventricle, leading to a variety of clinical manifestations. Arrhythmias and conduction abnormalities are more common, while heart failure and sudden cardiac death (SCD) are less common. The extent and location of myocardial involvement influence the severity of symptoms; patients with limited myocardial involvement may remain asymptomatic, while those with more widespread disease are at increased risk for significant conduction disturbances and heart failure [16]. The condition predominantly affects young adults, with a higher prevalence in individuals of African American and Northern European descent.
Clinical Characteristics in CA
Early diagnostic workup for CA relies heavily on clinical evaluation, including a comprehensive history and EKG. A patient’s history can provide valuable clues that help differentiate it from other heart conditions and guide further investigations. Extracardiac manifestations can particularly be useful, including carpal tunnel syndrome, spinal stenosis, hip or knee replacement, prior shoulder surgery, proteinuria, and peripheral or autonomic neuropathy causing orthostatic hypotension (Table 1). Carpal tunnel syndrome is one of the earliest features of CA, and precedes cardiac involvement by ~5-10 years [17]. Low-voltage EKG, although not pathognomonic, can be seen, which is a result of the poorly conducting amyloid fibrils infiltrating the myocardium [18]. Importantly, low voltage is detected only in ~35% of ATTR and 55% of AL patients, and therefore absence of low voltage criteria does not rule out CA [19]. It often manifests in advanced disease stage, and carries prognostic implications [19]. Low-voltage EKG can become more significant in the presence of LV ‘hypertrophy’, as this discrepancy is a peculiar feature seen in CA and strengthens suspicion for CA. Other EKG features that are reflective of the generalized infiltrative nature of this disease include include atrial fibrillation, pseudo-infarct pattern and first-degree AV block [20,21]. Atrial fibrillation is the most common arrhythmia in CA, and this high prevalence could be attributed to atrial enlargement due to high filling pressures and atrial myopathy secondary to direct amyloid infiltration [22,23]. Ventricular tachyarrhythmia (VT) and bradyarrhythmias are also seen [24,25]. On laboratory evaluation, persistent elevation of high-sensitivity troponin due to myocardial injury is almost invariably seen in CA and has prognostic implications [26]. Several mechanisms have been proposed in myocardial injury in CA, including amyloid-related causes such as amyloid precursor toxicity, amyloid interstitial infiltration, and amyloid vascular involvement, as well as non-amyloid-related causes such as diastolic and systolic dysfunction, heart failure, atherosclerotic coronary artery disease, and other supply-demand imbalances such as tachyarrhythmias and hypotension [26].
Clinical Characteristics in CS
The presence of extracardiac sarcoidosis, particularly in the lungs, raises suspicion for CS in the presence of any cardiac symptoms. In patients with CS, various types of arrhythmias have been reported, among which AVB is the most common while VT/ventricular fibrillation (VF) are the second most common initial presentation [27]. AVB results from infiltration of the intraventricular septum due to sarcoid granuloma or the scar tissue. It has been reported that SCD due to VT/VF or AVB and related complications account for 30–65% of deaths in sarcoidosis [28]. The risk of SCD is significant in CS presenting with high-grade AVB with or without VT or LV dysfunction, and the risk for SCD is high in these patients during a 5-year-follow-up [29]. Therefore, detecting cardiac involvement and an early diagnosis with treatment is one of the most important issues, especially because immunosuppressive treatment substantially enhances prognosis in such patients. As sarcoidosis is typically a disease of middle-aged persons and predominantly women, clinical suspicion of CS should be high in middle-aged with “idiopathic” higher degree AVB.
Echocardiographic Features in CA
Echocardiography is a valuable and reproducible imaging modality for identifying the structural and functional abnormalities associated with CA [30]. This infiltrative disease typically manifests as myocardial thickening with restrictive physiology and elevated filling pressures, in the absence of significant aortic stenosis or uncontrolled hypertension [30]. Characteristic findings include septal wall thickness exceeding 12 mm and a speckled appearance of the myocardium due to amyloid deposits [31,32,33]. Tissue Doppler imaging often reveals a restrictive filling pattern, with markedly reduced myocardial velocities (e', a', and s' velocities typically <5 cm/s) [30]. The presence of pericardial effusion further supports the diagnosis [30].
Biatrial enlargement is commonly observed, reflecting increased filling pressures, and all phases of left atrial strain (reservoir, conduit, and contraction phases) are significantly reduced [34,35]. Interatrial septal thickening exceeding 6 mm has been reported to have 100% specificity for diagnosing CA [32], providing a strong diagnostic clue. In addition, unexplained right ventricular free wall thickening and chamber dilatation raise suspicion for CA [32]. Speckled tracking echocardiography often reveals marked impairment in global longitudinal strain (GLS) of the left and the right ventricles, particularly at the basal and mid-segments, with relative apical sparing. This distinctive pattern produces the "cherry-on-top" appearance on the GLS bull's-eye plot, a hallmark feature of CA [31,36].
Echocardiographic Features in CS
2D-echocardiography is often the initial screening tool for CS. In a classic clinical scenario, echocardiography is typically prompted by cardiac symptoms (palpitations, chest pain, presyncope, and syncope) and/or abnormal EKG in an individual with preexisting extra-cardiac sarcoidosis [37]. The echocardiographic features in CS are often nonspecific. Any echocardiographic abnormality, when considered in the context of presence of extracardiac sarcoidosis, should raise suspicion for CS and warrant further investigation with more advanced imaging modalities. These echocardiographic abnormalities may include LV chamber dilation, mild LV wall thickening or even LV thinning in some cases, diastolic dysfunction, trivial pericardial effusions, and LV aneurysm. LV systolic dysfunction is an important feature in CS, although usually observed late in the disease course [38]. The presence of wall motion abnormalities (WMAs) in a noncoronary distribution could serve as an important indicator of cardiac involvement in CS, and could be present in upto 50% patients at the time of diagnosis of CS [39]. LV free wall and interventricular septum are most commonly involved. The most characteristic finding is thinning or scarring of the basal portion of the interventricular septum and associated akinesis, present in upto 4% of patients with a CS diagnosis [39,40].
Importantly, 2D-echocardiography demonstrates poor sensitivity in detecting early, mild, or focal CS [41]. Myocardial deformation imaging has been proposed as a sensitive tool to detect myocardial dysfunction before a decrease in LV ejection fraction (LVEF) occurs [42]. Early impairment of LV GLS reflects disruption of the myofibrils that are primarily organized longitudinally, with the greatest concentration in the subendocardial layer [43]. While CS preferentially affects the epicardial and mid-myocardial layers, patients with CS still demonstrate early impairment of LV GLS.
Echocardiographic features also have prognostic implications in CS. Patients with reduced LVEF have poorer prognosis compared to those with preserved LVEF [44]. Impaired LV has been shown to have incremental prognostic value for predicting major adverse cardiac events (defined as death, VT, heart failure hospitalization, or transplantation) in patients with systemic sarcoidosis [45]. Finally, right ventricular involvement has been associated with higher risk of VA and death, and right ventricular free wall longitudinal strain has been recognized as an important surrogate of disease activity and prognosis [46].
Cardiac Magnetic Resonance in CA
Cardiac magnetic resonance imaging (CMR) is the gold standard noninvasive modality for diagnosing CA, offering exceptional spatial resolution and comprehensive myocardial tissue characterization [47,48]. Key features include late gadolinium enhancement (LGE), T1 and T2 mapping, and extracellular volume (ECV) quantification. LGE reflects gadolinium contrast accumulation in the myocardial interstitium, with delayed washout in amyloid-laden areas, often displaying a non-coronary artery territorial distribution. Patterns of LGE include subendocardial, diffuse, focal, and transmural enhancement, with transmural involvement indicating worse prognosis. The subendocardial pattern is particularly characteristic of CA, making LGE a cornerstone of its diagnosis [49,50].
T1 mapping allows early detection of amyloid infiltration before overt myocardial thickening, with prolonged (pre contrast) T1 relaxation times serving as a diagnostic and prognostic marker [51,52,53]. The pre and post contrast T1 relaxation times measure the extent of extracellular matrix expansion, which helps differentiate CA from other fibrotic myocardial conditions and provides an objective metric for assessing disease burden and treatment response [54,55,56]. Complementing these techniques, T2 mapping quantifies myocardial edema, reflecting inflammation or active disease, and contributes additional prognostic information [57].
Cardiac Magnetic Resonance in CS
CMR is an excellent tool for the diagnosis and prognostication of patients with suspected CS, offering a multi-dimensional assessment of cardiac involvement that allows for a non-invasive detection of scar, biventricular function, edema, and myocardial perfusion defects. CMR offers the advantages of high spatial resolution, excellent soft-tissue contrast, and the use of non-ionizing radiation. While CMR can easily recognize morphological abnormalities such as areas of wall thinning or aneurysm, foremost diagnostic technique to detect CS by CMR relies on identifying areas of midwall and subepicardial late gadolinium enhancement (LGE) [58]. Rarely, CS can also cause subendocardial LGE, thus mimicking an infarct pattern. Patchy LGE in a noninfarct pattern is a nonspecific finding, which is also seen in other disease such as scar from previous myocarditis or from fibrosis in idiopathic cardiomyopathy, but this finding in the presence of preexisting extracardiac sarcoidosis would account for the presence of CS unless proven otherwise [58]. Features of LGE that typically favor the diagnosis of CS include multifocal involvement and involvement of the basal anteroseptum and inferoseptum, demonstrating contiguous extension into the right ventricle [59]. Transmural pattern is significantly more common in patients with LVEF of 35% or lower than in those with LVEF exceeding 35% [60]. LGE has prognostic value as well, and is associated with future cardiovascular death and VT [61].
CMR has the potential to assess the inflammatory aspect of CS. By incorporating T2-weighted imaging and T2 mapping, CMR can detect edema and inflammation [62]. Although T2-weighted CMR has been suggested as a potential alternative to positron emission tomography (PET) for detecting inflammation and tracking therapeutic response, its lower signal-to-noise ratio presents a challenge, necessitating further clinical validation [62,63]
Nuclear Imaging with Bone-Avid Tracers
Nuclear scintigraphy with technetium-labeled tracers, including 99mTc-pyrophosphate (PYP), is a key imaging modality for diagnosing ATTR [64]. This technique has demonstrated high sensitivity and specificity when combined with serum immunofixation electrophoresis and serum free light chain testing to exclude monoclonal gammopathy [65]. The use of Tc-PYP scan has significantly reduced the need for histological confirmation of ATTR in most cases, although AL still requires histological confirmation [65].
The diagnostic utility of Tc-PYP scintigraphy relies on the affinity of ATTR deposits for the radioactive tracer, which is visualized by comparing tracer uptake in the myocardium with uptake in the rib cage (bone). Grade 2 uptake (myocardial uptake equal to bone) and Grade 3 uptake (myocardial uptake greater than bone) are highly specific for ATTR in the absence of paraproteinemia [66]. Heart-to-contralateral lung (H/CL) ratio adds one more layer to the accuracy of diagnosis, although there can be discrepancy between planar image and H/CL ratio [67]. SPECT is always performed to confirm myocardial uptake, and to distinguish it from blood pooling. This noninvasive imaging modality has become an essential tool in diagnosing and differentiating ATTR from other types of CA [68].
Positron Emission Tomography
Fluorine-18 fluorodeoxyglucose (FDG) PET has become an essential imaging technique for diagnosing and evaluating CS, especially in situations where standard methods like echocardiography or MRI may not provide clear results. FDG-PET is particularly useful in detecting areas of granulomatous inflammation in the tissues. In CS, the granulomas contain activated macrophages that exhibit increased glucose metabolism, which is effectively captured by FDG, allowing for visualization of both cardiac and extracardiac disease involvement [69]. This imaging modality is highly sensitive in cases with advanced disease or active inflammation, offering valuable prognostic information regarding the extent and severity of myocardial involvement. Additionally, FDG-PET can detect myocardial inflammation that may remain undetected by conventional imaging, particularly in early or subclinical stages of the disease [69]. It is also a useful tool for differentiating between areas of active inflammation and scar tissue, which aids in monitoring treatment progress, especially during corticosteroid or immunosuppressive therapy [70]. However, while FDG-PET is a highly specific method for identifying active CS, its sensitivity can be affected by factors such as prior corticosteroid treatment, which may reduce metabolic activity, and the need for specific dietary preparation, including prolonged fasting, dietary modifications, and intravenous heparin to minimize normal myocardial glucose uptake [71]. Despite these limitations, FDG-PET continues to be a vital tool for both diagnosing and managing CS, offering critical insights into disease activity and informing therapeutic decisions.
Disease-Modifying Treatments
Therapies in ATTR
Therapies for ATTR that are FDA-approved are TTR silencers and TTR stabilizers. TTR silencers, such as RNA interference (RNAi) therapies, work by inhibiting TTR production through the degradation of TTR messenger RNA (mRNA) [72]. Patisiran, an RNAi agent, was the first of its kind to gain FDA approval for treating ATTR-related polyneuropathy. In the landmark trial comprising 225 patients underwent randomization (148 to the patisiran group and 77 to the placebo group), patisiran treatment resulted in significant improvements in neuropathy, quality of life, walking, nutritional status, and activities of daily living. [73]. There was also evidence that patisiran improved cardiac manifestations of ATTRv, as indicated by echocardiographic measures of cardiac structure and function and a reduction in NT-proBNP levels [73]. The authors proposed that the increased gait speed in patients who received patisiran may have resulted from favorable effects on both the neuropathic and cardiac aspects of the disease.
Another TTR silencer, inotersen, is an antisense oligonucleotide that targets TTR mRNA, that has demonstrated slowing of neuropathy progression in patients with ATTRv, including those with cardiac involvement [74]. The NEURO-TTR trial showed that weekly subcutaneous injections of inotersen (300 mg) were effective, though careful monitoring is required due to potential adverse effects such as glomerulonephritis, severe thrombocytopenia, and renal issues [75].
TTR stabilizers function by preventing the dissociation of TTR tetramers into monomers, a key step in the amyloid fibril formation process. A multicenter, international, double-blind, placebo-controlled, phase 3 trial, randomly assigned 441 patients with ATTR in a 2:1:2 ratio to receive 80 mg of tafamidis, 20 mg of tafamidis, or placebo for 30 months [76]. In this study, tafamidis was associated with reductions in all-cause mortality and cardiovascular-related hospitalizations and reduced the decline in functional capacity and quality of life as compared with placebo [76]. Importantly, the overall incidence and type of adverse events were similar in the tafamidis and placebo groups; the discontinuation of the trial drug owing to adverse events was less common in patients who received tafamidis than in those who received placebo, and dose reductions were uncommon and occurred more often in the placebo group [76].
Additionally, diflunisal, a nonsteroidal anti-inflammatory drug (NSAID), has demonstrated TTR-stabilizing effects and has been shown to slow the progression of familial amyloid polyneuropathy in small scale studies [77,78]. One of the key advantages of diflunisal is its cost-effectiveness. However, it should be avoided in patients with severe renal dysfunction, advanced heart failure, or those at high risk of bleeding [79].
Therapies in AL
The treatment of AL primarily focuses on targeting the underlying plasma cell dyscrasia responsible for the production of amyloid fibrils. The first-line approach typically involves chemotherapy regimens designed to reduce the abnormal production of light chains. This includes the use of proteasome inhibitors such as bortezomib or carfilzomib, which block the degradation of proteins and reduce the accumulation of amyloidogenic light chains [80]. Immunomodulatory drugs like lenalidomide and pomalidomide are also commonly used to inhibit the growth of plasma cells [80]. Additionally, monoclonal antibodies such as daratumumab, which target plasma cell surface antigens, can be employed [80]. For eligible patients, autologous stem cell transplantation may offer a potential curative option, particularly in those with good organ function and response to induction therapy. Treatment response is closely monitored through measures such as serum and urine immunofixation electrophoresis, as well as free light chain levels, with a goal of achieving a complete or very good partial response [81]. In cases involving cardiac involvement, reductions in NT-proBNP levels are targeted to assess and improve cardiac function [82]. NT-proBNP is a biomarker that is directly modulated by amyloid light-chains and is universally accepted by AL specialists as a surrogate end point for survival [82].
Therapies in CS
The treatment of CS involves a combination of immunosuppressive therapies, arrhythmia management, and heart failure management, tailored to the severity and manifestations of the disease. Corticosteroids, primarily prednisone, are the cornerstone of therapy, aiming to reduce inflammation and granuloma formation in the heart [83]. In cases of refractory disease or where steroid-sparing is needed, other immunosuppressive agents such as methotrexate, azathioprine, or mycophenolate mofetil may be used. For patients with significant arrhythmias, such as VT, antiarrhythmic drugs like amiodarone can be employed, and implantable cardioverter-defibrillators may be recommended for those at high risk of SCD [84]. In addition, standard heart failure treatments, including ACE inhibitors, beta-blockers, and diuretics, are used to manage symptoms and improve cardiac function in patients with heart failure. Monitoring the disease’s progression with imaging techniques like CMR and PET is essential for assessing treatment. In some cases, intravenous immunoglobulin (IVIG) or tumor necrosis factor-alpha (TNF-α) inhibitors may be considered for patients who do not respond adequately to conventional therapies [85]. As treatment can vary based on disease activity and organ involvement, a multidisciplinary approach, including cardiology, pulmonology, and immunology specialists, is crucial for optimal management.
Conclusions
In summary, granulomatous (specifically CS) and amyloidogenic cardiomyopathies both have overlapping clinical features, including heart failure and arrhythmias. Distinct differences in extracardiac "red flag" symptoms, echocardiographic features, and advanced imaging are key to distinguishing between the two conditions. In CA, echocardiography typically shows thickened heart walls, whereas in CS, there may be wall thinning with or without LV chamber dilation. CMR further differentiates the two, with CA often exhibiting global subendocardial and transmural LGE, while CS tends to show patchy, more localized areas of LGE. PET and Tc-PYP imaging further aid in diagnosis, with Tc-PYP uptake being a key marker for ATTR deposition in the absence of paraproteinemia and PET demonstrating active inflammation in CS. Disease-specific treatment for CS focuses on immunosuppressive therapies to reduce inflammation, while CA treatment involves stabilizing and/or silencing TTR protein in ATTR and chemotherapy or stem cell transplant for AL. Timely diagnosis and appropriate management are crucial for improving outcomes in both conditions.
Funding
Dr. Bukhari is supported by NIH grant T32HL007227
Conflicts of Interest
The author has no relevant disclosures related to this work
References
- Masri A, Bukhari S, Eisele YS, Soman P. Molecular Imaging of Cardiac Amyloidosis. J Nucl Med. 2020, 61, 965–970. [Google Scholar] [CrossRef]
- Bukhari S, Khan SZ, Ghoweba M, Khan B, Bashir Z. Arrhythmias and Device Therapies in Cardiac Amyloidosis. J Clin Med. 2024, 13, 1300. [Google Scholar] [CrossRef]
- Bashir Z, Younus A, Dhillon S, Kasi A, Bukhari S. Epidemiology, diagnosis, and management of cardiac amyloidosis. J Investig Med. 2024, 72, 620–632. [Google Scholar] [CrossRef]
- Bukhari, S. Cardiac amyloidosis: state-of-the-art review. J Geriatr Cardiol. 2023, 20, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Bukhari S, Oliveros E, Parekh H, Farmakis D. Epidemiology, Mechanisms, and Management of Atrial Fibrillation in Cardiac Amyloidosis. Curr Probl Cardiol. 2023, 48, 101571. [Google Scholar] [CrossRef] [PubMed]
- Bukhari SB, Nieves A, Eisele R, Follansbee Y, Soman WP. Clinical Predictors of positive 99mTc-99m pyrophosphate scan in patients hospitalized for decompensated heart failure. J Nucl Med. 2020, 61, 659. [Google Scholar]
- Buxbaum J, Jacobson DR, Tagoe C, Alexander A, Kitzman DW, Greenberg B, Thaneemit-Chen S, Lavori P. Transthyretin V122I in African Americans with congestive heart failure. J Am Coll Cardiol. 2006, 47, 1724–1725.
- Bukhari SF, Brownell S, Eisele A, Soman YS. Race-specific phenotypic and genotypic comparison of patients with Transthyretin Cardiac Amyloidosis. J Am Coll Cardiol. 2021, 77, 675. [Google Scholar] [CrossRef]
- Waddington-Cruz M, Wixner J, Amass L, Kiszko J, Chapman D, Ando Y; THAOS investigators. Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol Ther. 2021, 10, 753–766.
- Hewitt K, Starr N, Togher Z, Sulong S, Morris JP, Alexander M, Coyne M, Murphy K, Giblin G, Murphy SM, et al. Spectrum of hereditary transthyretin amyloidosis due to T60A(p.Thr80Ala) variant in an Irish Amyloidosis Network. Open Heart. 2024, 11, e002906. [Google Scholar] [CrossRef]
- Al Hamed R, Bazarbachi AH, Bazarbachi A, Malard F, Harousseau JL, Mohty M. Comprehensive Review of AL amyloidosis: some practical recommendations. Blood Cancer J. 2021, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Palladini G, Milani P. Diagnosis and Treatment of AL Amyloidosis. Drugs. 2023, 83, 203–216. [Google Scholar] [CrossRef]
- Trachtenberg BH, Hare JM. Inflammatory Cardiomyopathic Syndromes. Circ Res. 2017, 121, 803–818. [Google Scholar] [CrossRef]
- Newman LS, Rose CS, Bresnitz EA, Rossman MD, Barnard J, Frederick M, Terrin ML, Weinberger SE, Moller DR, McLennan G, et al; ACCESS Research Group A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med. 2004, 170, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Iwai K, Tachibana T, Takemura T, Matsui Y, Kitaichi M, Kawabata Y. Pathological studies on sarcoidosis autopsy. I. Epidemiological features of 320 cases in Japan. Acta Pathol Jpn.
- Birnie DH, Nery PB, Ha AC, Beanlands RS. Cardiac Sarcoidosis. J Am Coll Cardiol. 2016, 68, 411–421. [Google Scholar] [CrossRef]
- Donnelly JP, Hanna M, Sperry BW, Seitz WH Jr. Carpal Tunnel Syndrome: A Potential Early, Red-Flag Sign of Amyloidosis. J Hand Surg Am. 2019, 44, 868–876. [Google Scholar] [CrossRef]
- Teresi L, Trimarchi G, Liotta P, Restelli D, Licordari R, Carciotto G, Francesco C, Crea P, Dattilo G, Micari A, et al. Electrocardiographic Patterns and Arrhythmias in Cardiac Amyloidosis: From Diagnosis to Therapeutic Management-A Narrative Review. J Clin Med. 2024, 13, 5588. [Google Scholar] [CrossRef] [PubMed]
- Cipriani A, De Michieli L, Porcari A, Licchelli L, Sinigiani G, Tini G, Zampieri M, Sessarego E, Argirò A, Fumagalli C, et al. Low QRS Voltages in Cardiac Amyloidosis: Clinical Correlates and Prognostic Value. JACC CardioOncol. 2022, 4, 458–470. [Google Scholar] [CrossRef]
- Bukhari SM, Shpilsky S, Nieves D, Bashir R, Soman Z. Development and validation of a diagnostic model and scoring system for transthyretin cardiac amyloidosis. J Investig Med. 2021, 69, 1071–1072. [Google Scholar]
- Bukhari SM, Shpilsky S, Nieves D, Soman R. Amyloidosis prediction score: a clinical model for diagnosing Transthyretin Cardiac Amyloidosis. J Card Fail. 2020, 26, 33. [Google Scholar] [CrossRef]
- Bukhari S, Barakat AF, Eisele YS, Nieves R, Jain S, Saba S, Follansbee WP, Brownell A, Soman P. Prevalence of Atrial Fibrillation and Thromboembolic Risk in Wild-Type Transthyretin Amyloid Cardiomyopathy. Circulation. 2021, 143, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Bukhari S, Khan SZ, Bashir Z. Atrial Fibrillation, Thromboembolic Risk, and Anticoagulation in Cardiac Amyloidosis: A Review. J Card Fail. 2023, 29, 76–86. [Google Scholar] [CrossRef]
- Bukhari S, Khan B. Prevalence of ventricular arrhythmias and role of implantable cardioverter-defibrillator in cardiac amyloidosis. J Cardiol. 2023, 81, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Bukhari S, Kasi A, Khan B. Bradyarrhythmias in Cardiac Amyloidosis and Role of Pacemaker. Curr Probl Cardiol. 2023, 48, 101912. [Google Scholar] [CrossRef]
- De Michieli L, Cipriani A, Iliceto S, Dispenzieri A, Jaffe AS. Cardiac Troponin in Patients With Light Chain and Transthyretin Cardiac Amyloidosis: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2024, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Willy K, Dechering DG, Reinke F, Bögeholz N, Frommeyer G, Eckardt L. The ECG in sarcoidosis - a marker of cardiac involvement? Current evidence and clinical implications. J Cardiol. 2021, 77, 154–159. [Google Scholar] [CrossRef]
- Roberts WC, McAllister HA Jr, Ferrans VJ. Sarcoidosis of the heart. A clinicopathologic study of 35 necropsy patients (group 1) and review of 78 previously described necropsy patients (group 11). Am J Med. 1977, 63, 86–108.
- Nordenswan HK, Lehtonen J, Ekström K, Kandolin R, Simonen P, Mäyränpää M, Vihinen T, Miettinen H, Kaikkonen K, Haataja P, et al. Outcome of Cardiac Sarcoidosis Presenting With High-Grade Atrioventricular Block. Circ Arrhythm Electrophysiol. 2018, 11, e006145. [Google Scholar] [CrossRef]
- Cuddy SAM, Chetrit M, Jankowski M, Desai M, Falk RH, Weiner RB, Klein AL, Phelan D, Grogan M: Practical Points for Echocardiography in Cardiac Amyloidosis. J Am Soc Echocardiogr 2022, 35, A31–A40. [CrossRef]
- Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans A et al: ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 2 of 2-Diagnostic Criteria and Appropriate Utilization. Circ Cardiovasc Imaging.
- Liang S, Liu Z, Li Q, He W, Huang H: Advance of echocardiography in cardiac amyloidosis. Heart Fail Rev, 1345.
- Bashir Z, Chen EW, Tori K, Ghosalkar D, Aurigemma GP, Dickey JB, Haines P: Insight into different phenotypic presentations of heart failure with preserved ejection fraction. Prog Cardiovasc Dis 2023, 79, 80–88. [CrossRef]
- Minamisawa M, Inciardi RM, Claggett B, Cuddy SAM, Quarta CC, Shah AM, Dorbala S, Falk RH, Matsushita K, Kitzman DW et al: Left atrial structure and function of the amyloidogenic V122I transthyretin variant in elderly African Americans. Eur J Heart Fail 2021, 23, 1290–1295. [CrossRef]
- Monte IP, Faro DC, Trimarchi G, de Gaetano F, Campisi M, Losi V, Teresi L, Di Bella G, Tamburino C, de Gregorio C: Left Atrial Strain Imaging by Speckle Tracking Echocardiography: The Supportive Diagnostic Value in Cardiac Amyloidosis and Hypertrophic Cardiomyopathy. J Cardiovasc Dev Dis 2023, 10.
- Bravo PE, Fujikura K, Kijewski MF, Jerosch-Herold M, Jacob S, El-Sady MS, Sticka W, Dubey S, Belanger A, Park MA et al: Relative Apical Sparing of Myocardial Longitudinal Strain Is Explained by Regional Differences in Total Amyloid Mass Rather Than the Proportion of Amyloid Deposits. JACC Cardiovasc Imaging 2019, 12, 1165–1173.
- Birnie DH, Sauer WH, Bogun F, Cooper JM, Culver DA, Duvernoy CS, Judson MA, Kron J, Mehta D, Cosedis Nielsen J, et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm. 2014, 11, 1305–1323. [Google Scholar]
- Murtagh G, Laffin LJ, Beshai JF, Maffessanti F, Bonham CA, Patel AV, Yu Z, Addetia K, Mor-Avi V, Moss JD, et al. Prognosis of Myocardial Damage in Sarcoidosis Patients With Preserved Left Ventricular Ejection Fraction: Risk Stratification Using Cardiovascular Magnetic Resonance. Circ Cardiovasc Imaging. 2016, 9, e003738. [Google Scholar]
- Nabeta T, Kitai T, Naruse Y, Taniguchi T, Yoshioka K, Tanaka H, Okumura T, Sato S, Baba Y, Kida K, et al. Risk stratification of patients with cardiac sarcoidosis: the ILLUMINATE-CS registry. Eur Heart J. 2022, 43, 3450–3459. [Google Scholar] [CrossRef] [PubMed]
- Tanizawa K, Handa T, Nagai S, Yokomatsu T, Ueda S, Ikezoe K, Ogino S, Hirai T, Izumi T. Basal interventricular septum thinning and long-term left ventricular function in patients with sarcoidosis. Respir Investig. 2022, 60, 385–392. [Google Scholar] [CrossRef]
- Youssef G, Beanlands RS, Birnie DH, Nery PB. Cardiac sarcoidosis: applications of imaging in diagnosis and directing treatment. Heart. 2011, 97, 2078–2087. [Google Scholar] [CrossRef]
- Joyce E, Ninaber MK, Katsanos S, Debonnaire P, Kamperidis V, Bax JJ, Taube C, Delgado V, Ajmone Marsan N. Subclinical left ventricular dysfunction by echocardiographic speckle-tracking strain analysis relates to outcome in sarcoidosis. Eur J Heart Fail. 2015, 17, 51–62. [Google Scholar] [CrossRef]
- Buckberg G, Hoffman JI, Mahajan A, Saleh S, Coghlan C. Cardiac mechanics revisited: the relationship of cardiac architecture to ventricular function. Circulation. 2008, 118, 2571–2587. [Google Scholar] [CrossRef]
- Kusano K, Ishibashi K, Noda T, Nakajima K, Nakasuka K, Terasaki S, Hattori Y, Nagayama T, Mori K, Takaya Y, et al. Prognosis and Outcomes of Clinically Diagnosed Cardiac Sarcoidosis Without Positive Endomyocardial Biopsy Findings. JACC Asia. 2021, 1, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Sperry BW, Ibrahim A, Negishi K, Negishi T, Patel P, Popović ZB, Culver D, Brunken R, Marwick TH, Tamarappoo B. Incremental Prognostic Value of Global Longitudinal Strain and 18F-Fludeoxyglucose Positron Emission Tomography in Patients With Systemic Sarcoidosis. Am J Cardiol. 2017, 119, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Albakaa NK, Sato K, Iida N, Yamamoto M, Machino-Ohtsuka T, Ishizu T, Ieda M. Association between right ventricular longitudinal strain and cardiovascular events in patients with cardiac sarcoidosis. J Cardiol. 2022, 80, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Bashir Z, Musharraf M, Azam R, Bukhari S. Imaging modalities in cardiac amyloidosis. Curr Probl Cardiol. 2024, 49, 102858. [Google Scholar] [CrossRef]
- Korthals D, Chatzantonis G, Bietenbeck M, Meier C, Stalling P, Yilmaz A: CMR-based T1-mapping offers superior diagnostic value compared to longitudinal strain-based assessment of relative apical sparing in cardiac amyloidosis. Sci Rep 2021, 11, 15521. [CrossRef]
- Lin L, Li X, Feng J, Shen KN, Tian Z, Sun J, Mao YY, Cao J, Jin ZY, Li J et al: The prognostic value of T1 mapping and late gadolinium enhancement cardiovascular magnetic resonance imaging in patients with light chain amyloidosis. J Cardiovasc Magn Reson 2018, 20, 2. [CrossRef]
- Fontana M, Pica S, Reant P, Abdel-Gadir A, Treibel TA, Banypersad SM, Maestrini V, Barcella W, Rosmini S, Bulluck H et al: Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation 2015, 132, 1570–1579. [CrossRef]
- Karamitsos TD, Piechnik SK, Banypersad SM, Fontana M, Ntusi NB, Ferreira VM, Whelan CJ, Myerson SG, Robson MD, Hawkins PN et al: Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 2013, 6, 488–497. [CrossRef]
- Banypersad SM, Fontana M, Maestrini V, Sado DM, Captur G, Petrie A, Piechnik SK, Whelan CJ, Herrey AS, Gillmore JD et al: T1 mapping and survival in systemic light-chain amyloidosis. Eur Heart J 2015, 36, 244–251. [CrossRef]
- Martinez-Naharro A, Kotecha T, Norrington K, Boldrini M, Rezk T, Quarta C, Treibel TA, Whelan CJ, Knight DS, Kellman P et al: Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovasc Imaging 2019, 12, 810–819.
- Pan JA, Kerwin MJ, Salerno M: Native T1 Mapping, Extracellular Volume Mapping, and Late Gadolinium Enhancement in Cardiac Amyloidosis: A Meta-Analysis. JACC Cardiovasc Imaging 2020, 13, 1299–1310. [CrossRef] [PubMed]
- Martinez-Naharro A, Abdel-Gadir A, Treibel TA, Zumbo G, Knight DS, Rosmini S, Lane T, Mahmood S, Sachchithanantham S, Whelan CJ et al: CMR-Verified Regression of Cardiac AL Amyloid After Chemotherapy. JACC Cardiovasc Imaging 2018, 11, 152–154. [CrossRef]
- Olausson E, Wertz J, Fridman Y, Bering P, Maanja M, Niklasson L, Wong TC, Fukui M, Cavalcante JL, Cater G, et al. Diffuse myocardial fibrosis associates with incident ventricular arrhythmia in implantable cardioverter defibrillator recipients. medRxiv [Preprint]. 2: 16, 2023.
- Kotecha T, Martinez-Naharro A, Treibel TA, Francis R, Nordin S, Abdel-Gadir A, Knight DS, Zumbo G, Rosmini S, Maestrini V et al: Myocardial Edema and Prognosis in Amyloidosis. J Am Coll Cardiol 2018, 71, 2919–2931.
- Smedema JP, Snoep G, van Kroonenburgh MP, van Geuns RJ, Dassen WR, Gorgels AP, Crijns HJ. Evaluation of the accuracy of gadolinium-enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005, 45, 1683–1690. [Google Scholar] [CrossRef]
- Kuo L, Han Y, Mui D, Zhang Y, Chahal A, Schaller RD, Frankel DS, Marchlinski FE, Desjardins B, Nazarian S. Diagnostic Specificity of Basal Inferoseptal Triangular Late Gadolinium Enhancement for Identification of Cardiac Sarcoidosis. JACC Cardiovasc Imaging. 2019, 12, 2574–2576. [Google Scholar] [CrossRef] [PubMed]
- Watanabe E, Kimura F, Nakajima T, Hiroe M, Kasai Y, Nagata M, Kawana M, Hagiwara N. Late gadolinium enhancement in cardiac sarcoidosis: characteristic magnetic resonance findings and relationship with left ventricular function. J Thorac Imaging. 2013, 28, 60–66. [Google Scholar] [CrossRef]
- Hulten E, Agarwal V, Cahill M, Cole G, Vita T, Parrish S, Bittencourt MS, Murthy VL, Kwong R, Di Carli MF, Blankstein R. Presence of Late Gadolinium Enhancement by Cardiac Magnetic Resonance Among Patients With Suspected Cardiac Sarcoidosis Is Associated With Adverse Cardiovascular Prognosis: A Systematic Review and Meta-Analysis. Circ Cardiovasc Imaging. 2016, 9, e005001. [Google Scholar]
- Crouser ED, Ono C, Tran T, He X, Raman SV. Improved detection of cardiac sarcoidosis using magnetic resonance with myocardial T2 mapping. Am J Respir Crit Care Med. 2014, 189, 109–112. [Google Scholar] [CrossRef]
- Amano Y, Tachi M, Tani H, Mizuno K, Kobayashi Y, Kumita S. T2-weighted cardiac magnetic resonance imaging of edema in myocardial diseases. ScientificWorldJournal. 2012, 2012, 194069. [Google Scholar]
- Bukhari S, Bashir Z. Diagnostic Modalities in the Detection of Cardiac Amyloidosis. J Clin Med. 2024, 13, 4075. [Google Scholar] [CrossRef]
- Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016, 133, 2404–2412. [Google Scholar] [CrossRef]
- Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, Leone O, Farsad M, Ciliberti P, Bacchi-Reggiani L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005, 46, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Bukhari S, Masri A, Ahmad S, Eisele YS, Brownell A, Soman P. Discrepant Tc-99m PYP Planar grade and H/CL ratio: Which correlates better with diffuse tracer uptake on SPECT? Journal of Nuclear Medicine May 2020, 61, 1633. [Google Scholar]
- Masri A, Bukhari S, Ahmad S, Nieves R, Eisele YS, Follansbee W, Brownell A, Wong TC, Schelbert E, Soman P. Efficient 1-Hour Technetium-99 m Pyrophosphate Imaging Protocol for the Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Cardiovasc Imaging. 2020, 13, e010249. [Google Scholar] [CrossRef] [PubMed]
- Chareonthaitawee P, Beanlands RS, Chen W, Dorbala S, Miller EJ, Murthy VL, Birnie DH, Chen ES, Cooper LT, Tung RH, et al. Joint SNMMI-ASNC expert consensus document on the role of 18F-FDG PET/CT in cardiac sarcoid detection and therapy monitoring. J Nucl Cardiol. 2017, 24, 1741–1758. [Google Scholar] [CrossRef]
- Divakaran S, Stewart GC, Lakdawala NK, Padera RF, Zhou W, Desai AS, Givertz MM, Mehra MR, Kwong RY, Hedgire SS, et al. Diagnostic Accuracy of Advanced Imaging in Cardiac Sarcoidosis. Circ Cardiovasc Imaging. 2019, 12, e008975. [Google Scholar] [CrossRef]
- Blankstein R, Waller AH. Evaluation of Known or Suspected Cardiac Sarcoidosis. Circ Cardiovasc Imaging. 2016, 9, e000867. [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Crooke ST, Wang S, Vickers TA, Shen W, Liang XH. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Planté-Bordeneuve V, Barroso FA, Merlini G, Obici L, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, et al; ATTR-ACT Study Investigators Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, Heneghan MA, Gorevic PD, Litchy WJ, Wiesman JF, et al; Diflunisal Trial Consortium Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim M, Saint Croix GR, Lacy S, Fattouh M, Barillas-Lara MI, Behrooz L, Mechanic O. The use of diflunisal for transthyretin cardiac amyloidosis: a review. Heart Fail Rev. 2022, 27, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda S. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015, 22, 79–83. [Google Scholar] [CrossRef]
- Hasib Sidiqi M, Gertz MA. Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm 2021. Blood Cancer J. 2021, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Muchtar E, Dispenzieri A, Gertz MA, Kumar SK, Buadi FK, Leung N, Lacy MQ, Dingli D, Ailawadhi S, Bergsagel PL, et al. Treatment of AL Amyloidosis: Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) Consensus Statement 2020 Update. Mayo Clin Proc. 2021, 96, 1546–1577. [Google Scholar] [CrossRef]
- Merlini G, Lousada I, Ando Y, Dispenzieri A, Gertz MA, Grogan M, Maurer MS, Sanchorawala V, Wechalekar A, Palladini G, et al. Rationale, application and clinical qualification for NT-proBNP as a surrogate end point in pivotal clinical trials in patients with AL amyloidosis. Leukemia. 2016, 30, 1979–1986. [Google Scholar] [CrossRef]
- Baughman RP, Valeyre D, Korsten P, Mathioudakis AG, Wuyts WA, Wells A, Rottoli P, Nunes H, Lower EE, Judson MA, et al. ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J. 2021, 58, 2004079. [Google Scholar] [CrossRef]
- Yada H, Soejima K. Management of Arrhythmias Associated with Cardiac Sarcoidosis. Korean Circ J. 2019, 49, 119–133. [Google Scholar] [CrossRef]
- Baker MC, Sheth K, Witteles R, Genovese MC, Shoor S, Simard JF. TNF-alpha inhibition for the treatment of cardiac sarcoidosis. Semin Arthritis Rheum. 2021, 50, 546–552, Erratum in: Semin Arthritis Rheum. 2021;51:1390.. [Google Scholar]
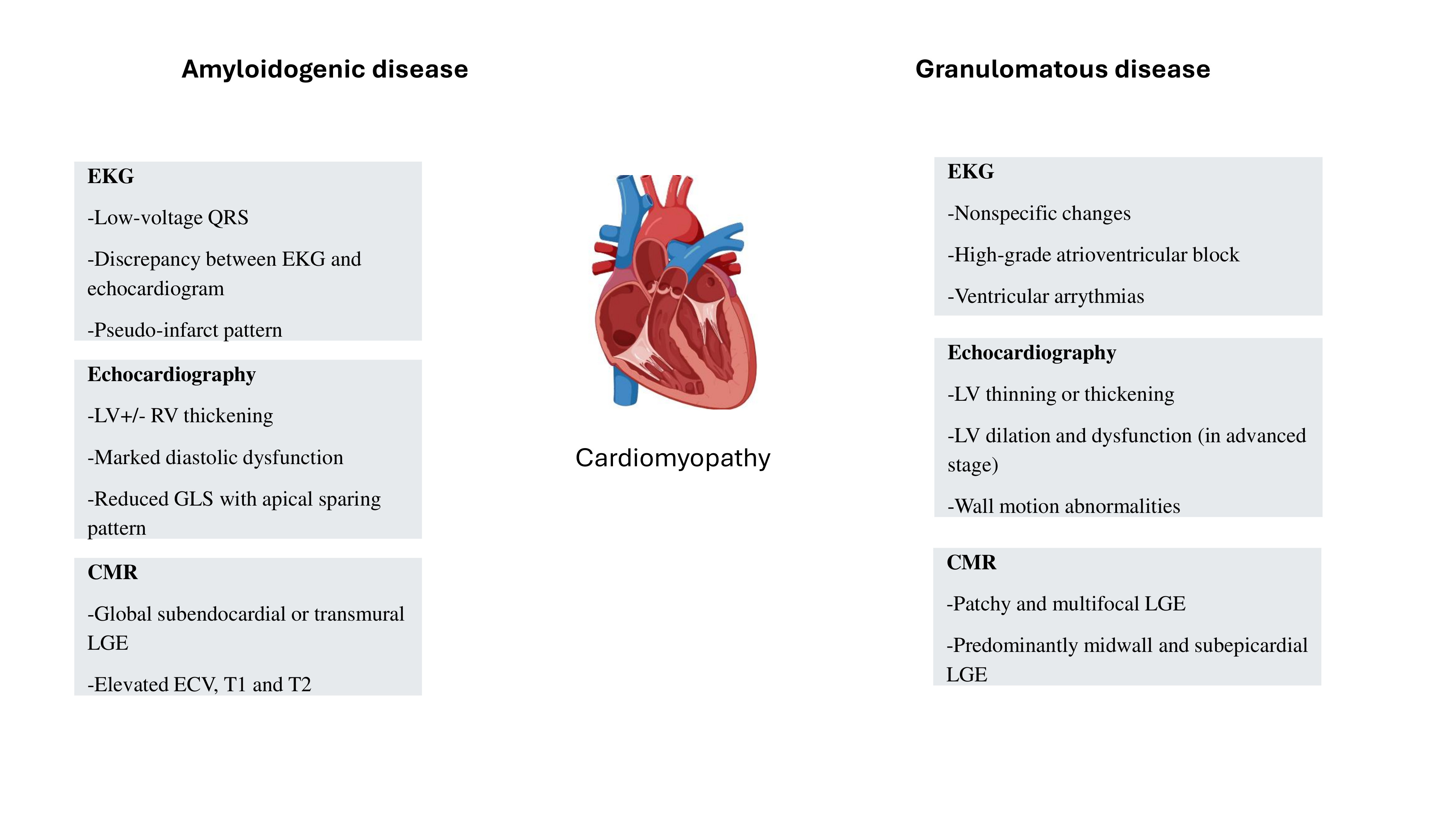
Figure 1.
Key features of granulomatous and amyloidogenic cardiomyopathies. LV- left ventricle; RV- right ventricle; CMR- cardiac magnetic resonance imaging; LGE- late gadolinium enhancement; ECV- extracellular volume.
Figure 1.
Key features of granulomatous and amyloidogenic cardiomyopathies. LV- left ventricle; RV- right ventricle; CMR- cardiac magnetic resonance imaging; LGE- late gadolinium enhancement; ECV- extracellular volume.
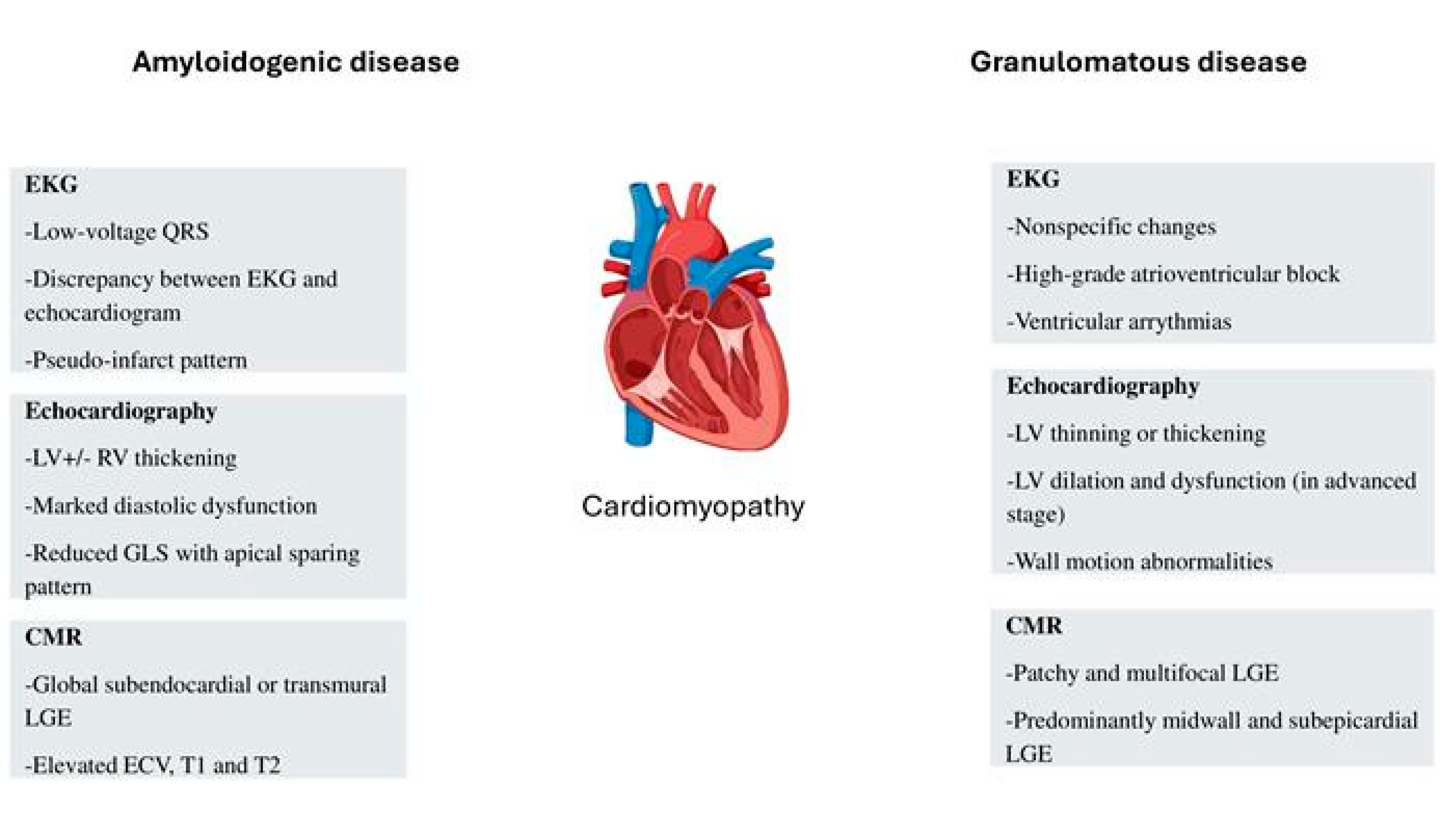

Table 1.
Extracardiac involvement of transthyretin cardiac amyloidosis, immunoglobulin light-chain amyloidosis and cardiac sarcoidosis.
Table 1.
Extracardiac involvement of transthyretin cardiac amyloidosis, immunoglobulin light-chain amyloidosis and cardiac sarcoidosis.
| Transthyretin cardiac amyloidosis | Immunoglobulin light-chain cardiac amyloidosis | Cardiac sarcoidosis |
|---|---|---|
| Carpal tunnel syndrome | Carpal tunnel syndrome | Mediastinal/hilar lymph nodes |
| Lumbar spinal stenosis | Lumbar spinal stenosis | Subcutaneous lymph nodes |
| Non-traumatic biceps tendon rupture | Non-traumatic biceps tendon rupture | Deep lymph nodes |
| Hip/knee replacement | Hip/knee replacement | Liver |
| Peripheral neuropathy | Peripheral neuropathy | Pulmonary |
| Orthostatic hypotension | Orthostatic hypotension | Spleen |
| Autonomic dysfunction | Autonomic dysfunction | Bone |
| Gastroparesis | Muscle | |
| Macroglossia | Skin | |
| Nephrotic syndrome | ||
| Hepatic amyloidosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



