
Submitted:
27 January 2025
Posted:
27 January 2025
You are already at the latest version
Abstract
The coronary slow flow phenomenon (CSFP) is characterized by a delayed flow of contrast medium through the coronary arteries, predominantly affecting smokers and young men with acute coronary syndrome. Over 20% of these patients experience readmission to coronary care units due to recurring chest pain. Recent advancements in sequencing technologies have boosted microbiome studies, revealing that the microbial community within the human body contributes to disease progression by releasing various substances. While the gut microbiome’s role in cardiovascular disease is well-documented, its relationship with CSFP remains under investigation. This study aims to compare the gut microbiota of CSFP patients to healthy controls. Results highlight the potential significance of the genus Gemmiger and species Anaerobutyricum in the selenium metabolism pathway, suggesting their viability as microbial indicators for CSFP, especially given the link between Selenium deficiency and cardiovascular diseases. Additionally, an increased presence of bacteria involved in producing trimethylamine (TMA), a metabolite linked to cardiovascular issues, was observed in CSFP patients. Although the study did not achieve statistical significance due to sample size limitations, findings support the potential importance of these microbial markers.
Keywords:
coronary slow flow phenomenon
; coronary health
; microbiota
1. Introduction
Coronary angiography is the gold standard diagnostic method used in the diagnosis of coronary artery disease. Although there is no severe stenosis in the epicardial coronary arteries in 1% to 5% of coronary angiographies, the flow rate of the opaque material in the coronary arteries decreases [1]. This phenomenon is called the coronary slow flow phenomenon. Coronary slow flow phenomenon is one of the causes of cardiac chest pain. This may be the reason for hospitalization due to myocardial ischemia, myocardial infarction, arrhythmias, and recurrent chest pain [2]. Although there are various theories and hypotheses about the pathophysiology of coronary slow flow, endothelial dysfunction is the strongest among them. Owing to endothelial dysfunction, the balance between the release of vasodilating nitric oxide and the release of vasoconstricting endothelin is disrupted, and the underlying cause of coronary slow flow is thought to occur [3]. Causes of endothelial dysfunction include classical causes such as advanced age, hypertension, diabetes mellitus, hyperlipidemia, smoking, genetic factors, obesity, increased CRP, and some microorganisms that cause inflammation [4]. Many viruses, including SARS-CoV-2, have been reported to cause endothelial dysfunction [5].
Next-generation sequencing provides a better understanding of the human gut microbiome and its association with diseases such as cardiovascular diseases, obesity, diabetes, cancer, and neurological disorders [6]. The gut microbiome of patients with cardiovascular diseases and healthy control individuals has been widely studied. An increasing amount of Faecalibacterium prausnitzii and a significant decrease in the population of Ruminococcus gravus were found to be correlated with heart failure [7]. Moreover, an increase in Prevotella and Klebsiella genera is a potential biomarker of hypertension [8]. In addition, Escherichia coli, Klebsiella spp., Enterobacter aerogenes, Streptococcus spp., Lactobacillus salivarius, Solobacterium moorei, Atopobium parvulum, Ruminococcus gnavus and Eggerthella lenta are positively correlated with atherosclerotic cardiovascular diseases. In contrast, Roseburia intestinalis, Faecalibacterium cf. prausnitzii, Bacteroides spp., Prevotella copri, and Alistipes shahii are negatively correlated with atherosclerosis [9]. All these associations indicate the importance of microbiome studies; however, without knowledge of microbiome pathways and their contributions to disease progression, these correlations will remain deficient.
CSFP is not a rare finding in patients undergoing routine coronary angiography. Coronary Slow Flow has been reported in 1% of patients who underwent coronary angiography with a diagnosis of acute coronary syndrome [10]. On the other hand, in another study, slow coronary flow was observed in 7% of patients who presented with chest pain and underwent angiography with the suspicion of coronary artery disease [11]. It has been associated with significant morbidity and possibly mortality. Further studies are necessary for effective treatment [12]. CSFP was first observed during angiographic imaging [13]. The thrombolysis flow rate chart in myocardial infarction is used to indicate coronary blood flow. It shows the speed of the passage of the injected contrast agent in the coronary artery and whether it is complete [14]. It is characterized by slow passage of contrast medium without stenosis in some patients with chest pain who are receiving selective coronary angiography. This phenomenon has been named the slow flow phenomenon, which was defined by Tambe in 1972 [11,15]. It is undeniable that coronary slow flow is still referred to as a phenomenon that is not an innocent entity. Unless there are prospective objective methods and studies investigating the effect of coronary slow flow on ventricular function, CSFP will remain a phenomenon for a long time, perhaps the opposite of what it deserves.
The microbial community inside the human gut affects human disease progression and disease mechanisms of action directly or indirectly. However, the associations between the gut microbiome and patients with CSFP have never been studied. In this study, we aimed to understand the potential differences between the healthy human gut microbiome and the gut microbiome of CSFP patients.
2. Materials and Methods
2.1. The Patients and Cohorts
This preliminary study was conducted on 8 adults at the age of 50 (SD ± 7), comprising four CSFP patients and four healthy controls for investigating the differences between gut microbial community of two groups. Pair matching was considered while constructing the groups to limit the possibility of independent variables such as age, diet and lifestyle. Those who had distinct lifestyle and bad habits (smoking, alcohol consumption, diet, exercise, etc.), those that recently used antibiotics, antidepression and other drugs that directly affect the microbial community changes in the gut were excluded from the study. The control group’s inclusion criteria included no association with cardiovascular diseases and an age of older than 40.
2.2. Sample Collection and Preparation
Fecal samples were collected from each participant into storage tubes that contain DNA/RNA Shield solution to protect the microbial community in fecal samples. DNA isolation was performed immediately after it arrived to laboratory. The gDNA extraction process was carried out with a ZymoBIOMICS DNA Mini-prep kit (cat. no. D4300). The concentrations and purity levels were measured with an HS dsDNA Qubit 2.0 (cat. no. Q32866) and a NanoDrop OneC (cat. no. ND-ONE-W), respectively. Isolated DNA samples were kept at -20°C for sequencing.
2.3. 16S rRNA Microbiome Sequencing Analysis
The extracted DNA samples were subjected to 16S rRNA microbiome and pathway prediction analyses. The Oxford Nanopore Technology (ONT) sequencing platform was used to perform the sequencing assay and analyze the gut microbial community of coronary slow-flow syndrome patients via 16S targeted sequencing, which covers all variable regions of the 16S rRNA gene (V1-V9). The gDNA obtained from DNA extraction was subjected to PCR for targeted 16S rRNA gene region sequencing via the ONT 16S barcoding kit (cat. no. SQK-16S024). Library preparation was performed according to the manufacturer’s instructions. Base calling conversion from fast5 signal files to fastq sequence data and adapter trimming were performed with the Guppy cli toolkit (v. 6.0.6). Quality control of each sample was carried out with FastQC (v. 0.11.2). Trimming and error correction were carried out with the bbtools cli toolkit (v. 38.97). The corrected reads were used to create consensus sequences via ncbi-magicblast (v 1.6.0). Consequently, prokaryotic annotation was carried out with blastn (v 2.13.0) to identify the microorganisms according to the NCBI 16S database (as of 27/03/2023).
2.4. Statistical Analysis
The R programming language was used for statistical analysis.[16] The Otu tables obtained from the preprocessing steps were standardized in R Studio via the total sum scaling (TSS) approach. The normality of OTU count data was evaluated using Shapiro-Wilk test. The Mann‒Whitney U test was performed for normalized out tables to determine the significant taxonomy differences between patients with CSFP and healthy subjects. Results were accepted as statistically significant if p value is less than 0.05. Significantly differentiated OTUs between control and healthy groups were visualized by violin plots generated by ggplot2 package (version 3.4.4.).
3. Results
3.1. This Gut Microbiota Differences Between CSFP Patients and Control
The 16S rRNA gene is approximately 1.5 kb long, with 9 hypervariable regions from V1 to V9. The targeted sequencing assay used all the V1 to V9 rRNA gene regions. A total of 135016 reads were generated for 8 samples, and the N50 of the reads was calculated to be 1423 bp. The average quality of the samples was Q18, and the GC content of eight samples was 53%. 16SrRNA results were evaluated at phylum, genus and species level.
Overall distribution of samples indicated that the most abundant phyla was Bacillota with 73%, followed by Bacteroidota (15.58%), Pseudomonadota (7.95%) Actinomycetota (1.28%), Thermodesulfobacteriota (1.08%) and others (1.07%) as illustrated in Figure 1. Despite the result not significant (p-value > 0.05), 5 OTUs were discovered to be explanatory for the CSFP patients: Bacillota, Calditrichota, Lentisphaerota, Pseudomonadota and Synergistota (OTU4, OTU9, OTU26, OTU32 and OTU35 at the phylum level, respectively). Almost 90% of microbial consumption was occupied by Bacillota and Bacteroidota in disease and healthy groups (74% for disease group, 71% for control group, p > 0.05). Bacillota was the most abundant phylum, followed by Bacteroidota, Pseudomonadota, (13.3% for disease group, 2.6% for control group, p > 0.05) Actinomycedota, (0.22% for disease group, 2.3% for control group, p > 0.05) Thermodesulfobacteriota (0.05% for disease group, 2.1% for control group, p > 0.05) and others (Figure 2A). Eight phyla (Acidobacteriota, Balneolota, Campylobacterota, Cyanobacteriota, Elusimicrobiota, Myxococcota, Spirochaetota, and Thermotogota) were identified as significantly more abundant in the control group compared to patients diagnosed with CSFP (p < 0.05). Principal component analysis was applied to the microbiome dataset, and the clustering of the control and patient groups was evaluated at phylum level (Figure 2B). Discriminative OTUs that are only found in control groups provide a certain aggregate.
Conversely, the Bacillota-to-Bacteroidota ratio was markedly elevated in the CSFP patient group, reaching 6.56, compared to 4.4 observed in the healthy control group. The reason behind the increase of this ratio is the decreasing amount of Bacteroidota in control group, because Bacillota populations were almost same for each group (Figure 3).
At the genus level, 44 genera were significantly different (p < 0.05, Figure 4). Notably, the genera Flavobacterium, Gracilibacillus, Novosphingobium, and Weissella exhibited significantly higher abundance in the control group compared to the patient cohort (P < .01). Moreover, Gemmiger was the only significant genus that was more abundant in patient group compared to the control group.
As a result of the KEGG pathway analysis, the Gemmiger genus, which belongs to the Bacillota phylum, was found to play a role in selenocompound metabolism. Microbial diversity undergoes dynamic alterations during the onset of a disease, progressively diminishing as the disease advances within the host organism. In our study, results revealed a significant decrease in the gut microbiome diversity of CSFP patients at the genus level (p < 0.05, Figure 5A). In addition to that, the genera Roseburia and Megasphaera were identified as key contributors to propionate production. The relative abundance of these genera showed a slight reduction in the disease group, suggesting a potential link to disease-associated metabolic alterations (p > 0.05). Only 84 distinct OTUs were observed in diseased individuals, whereas 964 distinct genera were observed in the healthy group. The common OTUs that were observed in both groups were 488. More than half of the significant genera (Absiella, Anaerobutyricum, Bacillus, Defluviitalea, Desulfofundulus, Faecalimonas, Gracilibacillus, Hespellia, Kurthia, Lachnobacterium, Lachnotalea, Lacrimispora, Maledivibacter, Paralkalibacillus, Pediococcus, Solibacillus, Sporosalibacterium, Syntrophomonas, Thermoanaerobacterium, Thermoclostridium, Traorella, Virgibacillus, Weissella, Xylanibacillus, p < 0.05) belongs to Bacillota phylum and together with Pseudomonadota they dominate genus level of control group with high diversity. Notably, we observed detailed view of decreasing in microbial diversity at species level. 1597 species were only discovered in control group, whereas 264 species only present in disease one. (Figure 5B).
Further investigation of species level analysis revealed 25 bacterial species with significant differences in abundance, with the control group exhibiting higher levels than the patient group for all but Anaerobutyricum (p < 0.05, Figure 6). Fournierella massiliensis and Gracilibacillus were the most significant species differing in each group and it is found to be more abundant in controls (p < 0.01). Subsequent analysis into pathways associated with the Anaerobutyricum genus highlighted its pivotal role in butyrate synthesis, a metabolite essential for human defense mechanisms against various diseases, including cardiovascular conditions [17].
3.2. Trimethylamine Produced from Microbial Organisms and Its Potential Effect on CSFP Disease
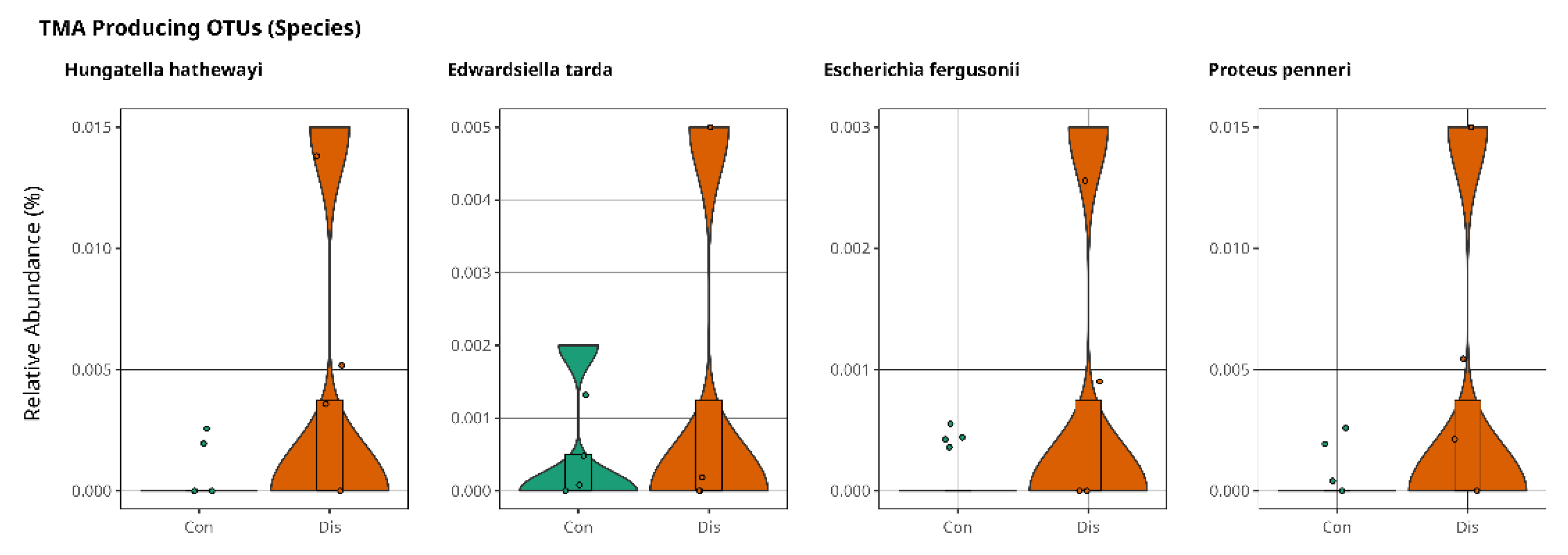
We conducted an in-depth analysis of metabolite markers implicated in the pathogenesis of cardiovascular disease, identifying elevated levels of trimethylamine N-oxide (TMAO) as a key contributor to disease progression [18]. TMAO is derived from trimethylamine (TMA), a metabolite generated by gut microbiota and subsequently oxidized in the liver. Notably, TMAO is predominantly associated with individuals consuming excessive quantities of red meat, establishing a strong correlation with the development of cardiovascular disorders [22,23]. Anaerococcus Hydrogenalis, Clostridium Asparaggiforme, Clostridium Hathewayi, Clostridium sporogenes, Edwardsiella tarda, Escherichia Fergusonii, Proteus Penneri, and Providencia Rettgeri have been reported as bacterial species found in the gut that contribute to the formation of TMA [19,21,22]. Our results support the findings of a relatively high abundance of Hungatella Hathaway, Edwardsiella tarda, Escherichia Fergusonii, and Proteus penneri in patients with CSFP disease (p > 0.05, Figure 7).
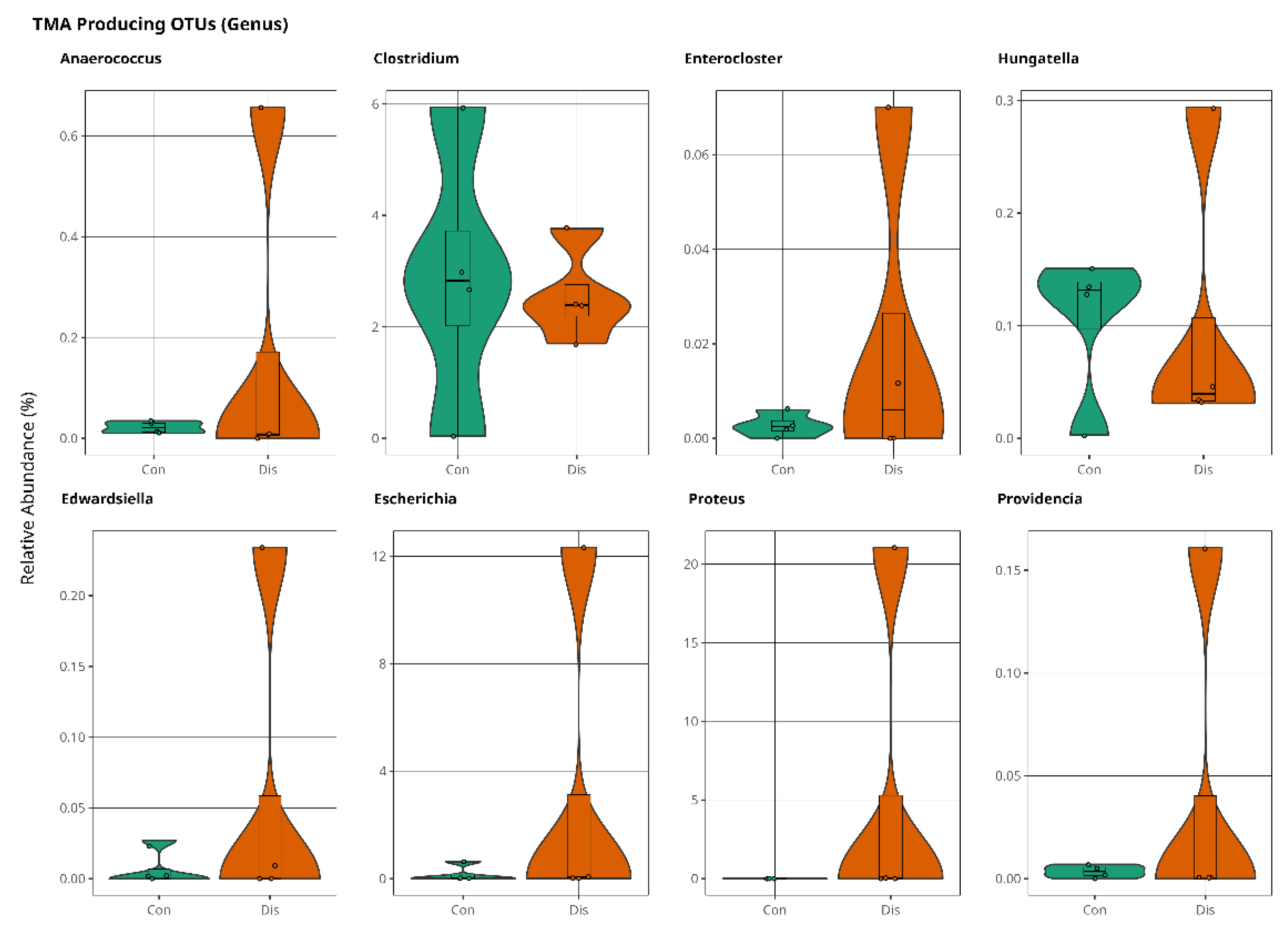
At the genus level, Anaerococcus, Enterocloster, Hungatella, Edwardsiella, Escherichia, Proteus, and Providencia were slightly more abundant in the patient samples (Figure 8). Although these results are not significant, they are promising for further investigations of the coronary slow flow phenomenon to explain disease mechanism of occurrence.
Figure 8.
The violin plot illustrates population variations of TMA-producing bacteria between control and patient samples at genus level, with no statistically significant difference (p > 0.05).
Figure 8.
The violin plot illustrates population variations of TMA-producing bacteria between control and patient samples at genus level, with no statistically significant difference (p > 0.05).
4. Discussion
Authors The gut microbiome has a strong effect on the progression of many cardiovascular diseases, such as heart failure, atherosclerosis, hypertension, myocardial fibrosis, and coronary artery diseases, by altering metabolism pathways, especially the trimethylamine N-oxide, short-chain fatty acid, and primary and secondary bile acid pathways [23]. Elucidating the microbiome and pathway interactions is very important for understanding disease occurrence and its mechanism of action. Studies focusing on this interaction aim to provide a better and early diagnosis and improve more specific treatment approaches. Many studies have been published to explain the microbiome and cardiovascular disease relationship [6,7,8,9,23,24,25]. However, the association between the microbiome and coronary slow flow phenomenon remains unclear. In our study, we analyzed the microbiome differences between patients with coronary slow flow phenomenon and healthy individuals and discovered potential pathways related to disease progression.
We analyzed the microbiome at each level and concluded with results that confirmed some findings from the literature. At the phylum level, we identified 8 significant phyla: Spirochaetota, Acidobacteriota, Campylobacterota, Elusimicrobiota, Balneolota, Thermotogota, Cyanobacteriota and Myxococcota. These findings were all more apparent in the control groups than in the patients with coronary slow flow phenomenon. Bacillota and Bacteroidota accounted for more than 90% of the microbiota in both the healthy and patient groups, but there was no significant difference between the groups. However, the ratio of Bacillota to Bacteroidota was greater in patients than in healthy subjects. It is caused by the slight decrease in Bacteroidota. However, the relative abundance of Bacillota is almost same for two groups. This ratio is elevated in patients with coronary artery disease and is closely linked to various metabolites that play a significant role in the progression of cardiovascular disease. These metabolites include phosphatidylcholine with a diacyl residue, phosphatidylcholine with an acylalkyl, sphingomyelin with an acyl residue, hematocrit, hemoglobin, cholesterol, and LDL cholesterol [23]. A study conducted by Liu Z. and colleagues reported that the increase in the ratio of Bacillota to Bacteroidota in patients with atherosclerosis in one of their studies [26]. Our findings that confirm literature propose the significant role of this Bacillota/Bacteroidota ratio to be taken into consideration for future studies.
At the genus level, microbial diversity was significantly lower in the CSFP patient group than in the healthy group. A common microbial output is that the richness of the bacterial population decreases sharply in cardiovascular diseases [27,28,29]. Diversity mostly occurs because of the reduction in beneficial bacterial populations so that pathogens occupy the gut. This leads to the generation of diseases because of the metabolites and proteins secreted by pathogens [30].
Gemmiger was found to be the most informative genus for explaining the microbiome and coronary slow flow phenomenon disease, among the 44 statistically significant genera. Gemmiger, which belongs to the phylum Bacillota, is positively associated with selenocompound metabolism [31,32]. Selenium deficiency may lead to various diseases, such as cancer, thyroid dysfunction, inflammatory bowel disease, and cardiovascular disorders. Dietary selenium is highly correlated with Bacillota [33,34]. These findings increase the strength of the suggestion that Gemmiger, which belongs to Bacillota, is a very consequential genus that causes coronary slow flow disease through selenocompound metabolism. Another study revealed that an increasing abundance of Gemmiger genera is positively associated with rheumatoid heart disease [33]. All these findings and observations of the high abundance of Gemmiger found in patients with CSFP in our study indicate the important role of this genus in cardiovascular disease occurrence. Selenium is an essential chemical for human health, and inadequate amounts of selenium may cause coronary artery diseases [35]. Furthermore, a decrease of propionate producing bacteria shouldn’t have been missed out. Because, increasing level of Acetate to Propionate ratio is considered as high-risk factor for cardiovascular disorders [36]. In our case, the relative abundance of Roseburia and Megasphaera diminished in disease group.
Another significant finding was the high prevalence of Anaerobutyricum in patients. It is a butyrate-producing bacteria that enables metabolic regulation and has anti-inflammatory, antioxidant and anti-obesity effects [37,38]. These short-chain fatty acids play important roles in cardiovascular diseases by improving cardiac function and maintaining cardiovascular hemostasis. Butyrate is a four-carbon short-chain fatty acid that has anti-inflammatory, antioxidant, antiobesity and metabolic regulatory effects. The enrichment of butyrate-producing bacteria regulates the human immune system [39]. However, there is only one species of butyrate producing bacteria because the diversity of the microbial community in patients’ guts significantly decreased. Butyrate producing bacteria belongs to the phylum of Bacillota and it is discovered that more than half of the significant genera belongs to Bacillota phyla in control groups. This finding indicates the importance of Bacillota phylum and the diversity of microbial communities in healthy individuals.
One of the most important metabolites involved in cardiovascular disease occurrence is trimethyl-N oxide (TMAO). Increasing level of circulating TMAO has been found to be directly associated with cardiovascular disease occurrence. Some certain bacteria that live in our gut, aging and diet are main factors that play a crucial role in increasing the density of TMAO in circulatory system. Trimethyl amine (TMA) is derived from foods that contain L-carnitine, betaine, and choline by the gut microbiota and is then converted into TMAO in the liver [40,41,42], and some of gut microbes are directly involved in TMA production. These include Anaerococcus, Clostridium, Enterocloster, Hungatella, Edwardsiella, Escherichia, Proteus and Providencia at the genus level and Anaerococcus hydrogenalis, Clostridium asparaggiforme, Clostridium hathewayi, Clostridium sporogenes, Edwardsiella tarda, Escherichia fergusonii, Proteus penneri and Providencia rettgeri at the species level [20,22,43]. Our findings demonstrated that these genera were more prevalent in patients with CSFP compared to healthy controls. Additionally, the abundances of Hungatella hathewayi, Edwardsiella tarda, Escherichia fergusonii, and Proteus penneri were elevated in the CSFP group. Although these differences did not reach statistical significance, the results are noteworthy and align with existing literature, offering promising insights into future research.
Outstanding inferences were made even though we had an insufficient number of samples for analysis to reach a more specific role of the gut microbiome in occurrence of CSFP. Because CSFP is generally neglected while diagnosing as the clinicians generally check the patients for diagnosing with more common cardiac disorders. Another reason is the exclusion criteria of antibiotic usage, unfortunately we had to exclude 6 patients from study after reaching the information of drug usage history of patients. However, the results of this preliminary study were still worthwhile. Particularly, the contribution of Gemmiger at the genus level to the Selenium metabolism pathway makes it a potential microbial biomarker for coronary slow flow phenomenon disease because selenium deficiency is highly correlated with cardiovascular diseases [44,45,46]. As well as TMA is one of the most important metabolites associated with many cardiovascular diseases [47,48,49,50,51]. In our study, we successfully found the association of gut bacteria that plays a role in TMA production CSFP. Further studies with an increased number of samples are needed to clarify the microbial biomarkers and to understand the pathways that specifically explain their effects on the coronary slow flow phenomenon.
5. Conclusions
The development of next generation sequencing increased the number of studies that indicate association with microbiomes remarkably. CSFP is a significant cardio-vascular disease generally observed in acute coronary syndrome. Although many re-searchers studied this phenomenon, its association with the microbiome has remained unclear. In this study, we tried to distinguish the gut microbiome of healthy individuals from patients with CSFP. The Bacilotta/Bacteroidota ratio was higher in patients with CSFP at the phylum level. The Gemmiger genus was discovered as the most significant difference between patients and healthy individuals. TMA is one of the most important metabolites associated with cardiovascular diseases. In our study, bacteria species (Anaerococcus hydrogenalis, Clostridium asparaggiforme, Clostridium hathewayi, Clostridium sporogenes, Edwardsiella tarda, Escherichia fergusonii, Proteus penneri, and Providencia rettgeri) that take a crucial role in TMA production was found to be higher abundance in patients with CSFP. All these findings are quite promising for further studies to unravel the exact metabolite pathway of the CSFP disease.
Author Contributions
Analyzing, writing, review, and editing: The study was conceptualized by T.G., methodology was determined by T.G, Y.G., O.U.S and S.O.O., project administration was done by Y.G. T.K. was responsible for sample collection and all analysing steps, T.G. T.K., S.O.O. and Y.G. were completed writing and editing part, research was completed under supervision of T.G., Y.G., S.O.O and O.U.S., and Y.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was carried out by the foundation of Acıbadem Mehmet Ali Aydınlar University, no other external funding was received.
Data Availability Statement
Datasets are available upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CSFP | Coronary Slow Flow Phenomenon |
| TMA | Trimethylamine |
| SD | Standard Deviation |
| TSS | Total Sum Scaling |
| TMAO | Trimethyl-N-Oxide |
References
- Alvarez, C.; Siu, H. Coronary Slow-Flow Phenomenon as an Underrecognized and Treatable Source of Chest Pain: Case Series and Literature Review. J. Investig. Med. High Impact Case Rep. 2018, 6, 2324709618789194. [CrossRef]
- Hawkins, B. M.; Stavrakis, S.; Rousan, T. A.; Abu-Fadel, M.; Schechter, E. Coronary Slow Flow: – Prevalence and Clinical Correlations –. Circ. J. 2012, 76 (4), 936–942. [CrossRef]
- Vane JR, Botting RM. Secretory functions of the vascular endothelium. J Physiol Pharmacol. 1992 Sep;43(3):195-207. [PubMed]
- Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. 2005;1(3):183-98. [PubMed] [PubMed Central]
- Fosse, J. H.; Haraldsen, G.; Falk, K.; Edelmann, R. Endothelial Cells in Emerging Viral Infections. Front. Cardiovasc. Med. 2021, 8, 619690. [CrossRef]
- Zhao, Y.; Wang, Z. Gut Microbiome and Cardiovascular Disease. Curr. Opin. Cardiol. 2020, 35 (3), 207–218. [CrossRef]
- Cui, X.; Ye, L.; Li, J.; Jin, L.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; Zhou, X.; Zhang, J.; Cai, J. Metagenomic and Metabolomic Analyses Unveil Dysbiosis of Gut Microbiota in Chronic Heart Failure Patients. Sci. Rep. 2018, 8 (1), 635. [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; Zhang, W.; Weldon, R.; Auguste, K.; Yang, L.; Liu, X.; Chen, L.; Yang, X.; Zhu, B.; Cai, J. Gut Microbiota Dysbiosis Contributes to the Development of Hypertension. Microbiome 2017, 5 (1), 14. [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; Zhang, D.; Su, Z.; Fang, Z.; Lan, Z.; Li, J.; Xiao, L.; Li, J.; Li, R.; Li, X.; Li, F.; Ren, H.; Huang, Y.; Peng, Y.; Li, G.; Wen, B.; Dong, B.; Chen, J.-Y.; Geng, Q.-S.; Zhang, Z.-W.; Yang, H.; Wang, J.; Wang, J.; Zhang, X.; Madsen, L.; Brix, S.; Ning, G.; Xu, X.; Liu, X.; Hou, Y.; Jia, H.; He, K.; Kristiansen, K. The Gut Microbiome in Atherosclerotic Cardiovascular Disease. Nat. Commun. 2017, 8 (1), 845. [CrossRef]
- Wang, X.; Nie, S.-P. The Coronary Slow Flow Phenomenon: Characteristics, Mechanisms and Implications. Cardiovasc. Diagn. Ther. 2011, 1 (1).
- Mangieri, E.; Macchiarelli, G.; Ciavolella, M.; Barillà, F.; Avella, A.; Martinotti, A.; Dell’Italia, L. J.; Scibilia, G.; Motta, P.; Campa, P. P. Slow Coronary Flow: Clinical and Histopathological Features in Patients with Otherwise Normal Epicardial Coronary Arteries. Cathet. Cardiovasc. Diagn. 1996, 37 (4), 375–381. [CrossRef]
- Beltrame, J. F.; Limaye, S. B.; Horowitz, J. D. The Coronary Slow Flow Phenomenon – A New Coronary Microvascular Disorder. Cardiology 2002, 97 (4), 197–202. [CrossRef]
- Goel, P. K.; Gupta, S. K.; Agarwal, A.; Kapoor, A. Slow Coronary Flow: A Distinct Angiographic Subgroup in Syndrome X. Angiology 2001, 52 (8), 507–514. [CrossRef]
- Tambe, A. A.; Demany, M. A.; Zimmermcln, H. A.; Muscnrenhns, E.; Ohio, C. Angina Pectoris and Slow Flow Velocity of Dye in Coronary Arteries-A New Angiograpbic Finding.
- Saya, S.; Hennebry, T. A.; Lozano, P.; Lazzara, R.; Schechter, E. Coronary Slow Flow Phenomenon and Risk for Sudden Cardiac Death Due to Ventricular Arrhythmias: A Case Report and Review of Literature. Clin. Cardiol. 2008, 31 (8), 352–355. [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013.
- Shi, X.-R.; Chen, B.-Y.; Lin, W.-Z.; Li, Y.-L.; Wang, Y.-L.; Liu, Y.; Huang, J.-J.; Zhang, W.-W.; Ma, X.-X.; Shao, S.; Li, R.-G.; Duan, S.-Z. Microbiota in Gut, Oral Cavity, and Mitral Valves Are Associated With Rheumatic Heart Disease. Front. Cell. Infect. Microbiol. 2021, 11, 643092. [CrossRef]
- Amiri, P.; Hosseini, S. A.; Ghaffari, S.; Tutunchi, H.; Ghaffari, S.; Mosharkesh, E.; Asghari, S.; Roshanravan, N. Role of Butyrate, a Gut Microbiota Derived Metabolite, in Cardiovascular Diseases: A Comprehensive Narrative Review. Front. Pharmacol. 2022, 12, 837509. [CrossRef]
- Thomas, M. S.; Fernandez, M. L. Trimethylamine N-Oxide (TMAO), Diet and Cardiovascular Disease. Curr. Atheroscler. Rep. 2021, 23 (4), 12. [CrossRef]
- Liu, Y.; Dai, M. Trimethylamine N-Oxide Generated by the Gut Microbiota Is Associated with Vascular Inflammation: New Insights into Atherosclerosis. Mediators Inflamm. 2020, 2020, 1–15. [CrossRef]
- Zheng, Y.; He, J.-Q. Pathogenic Mechanisms of Trimethylamine N-Oxide-Induced Atherosclerosis and Cardiomyopathy. Curr. Vasc. Pharmacol. 2022, 20 (1), 29–36. [CrossRef]
- Rath, S.; Rud, T.; Pieper, D. H.; Vital, M. Potential TMA-Producing Bacteria Are Ubiquitously Found in Mammalia. Front. Microbiol. 2020, 10, 2966. [CrossRef]
- Rath, S.; Heidrich, B.; Pieper, D. H.; Vital, M. Uncovering the Trimethylamine-Producing Bacteria of the Human Gut Microbiota. Microbiome 2017, 5 (1), 54. [CrossRef]
- Rahman, Md. M.; Islam, F.; -Or-Rashid, Md. H.; Mamun, A. A.; Rahaman, Md. S.; Islam, Md. M.; Meem, A. F. K.; Sutradhar, P. R.; Mitra, S.; Mimi, A. A.; Emran, T. B.; Fatimawali; Idroes, R.; Tallei, T. E.; Ahmed, M.; Cavalu, S. The Gut Microbiota (Microbiome) in Cardiovascular Disease and Its Therapeutic Regulation. Front. Cell. Infect. Microbiol. 2022, 12, 903570. [CrossRef]
- Sawicka-Smiarowska, E.; Bondarczuk, K.; Bauer, W.; Niemira, M.; Szalkowska, A.; Raczkowska, J.; Kwasniewski, M.; Tarasiuk, E.; Dubatowka, M.; Lapinska, M.; Szpakowicz, M.; Stachurska, Z.; Szpakowicz, A.; Sowa, P.; Raczkowski, A.; Kondraciuk, M.; Gierej, M.; Motyka, J.; Jamiolkowski, J.; Bondarczuk, M.; Chlabicz, M.; Bucko, J.; Kozuch, M.; Dobrzycki, S.; Bychowski, J.; Musial, W. J.; Godlewski, A.; Ciborowski, M.; Gyenesei, A.; Kretowski, A.; Kaminski, K. A. Gut Microbiome in Chronic Coronary Syndrome Patients. J. Clin. Med. 2021, 10 (21), 5074. [CrossRef]
- Liu, Z.; Li, J.; Liu, H.; Tang, Y.; Zhan, Q.; Lai, W.; Ao, L.; Meng, X.; Ren, H.; Xu, D.; Zeng, Q. The Intestinal Microbiota Associated with Cardiac Valve Calcification Differs from That of Coronary Artery Disease. Atherosclerosis 2019, 284, 121–128. [CrossRef]
- Ahmad, A. F.; Dwivedi, G.; O’Gara, F.; Caparros-Martin, J.; Ward, N. C. The Gut Microbiome and Cardiovascular Disease: Current Knowledge and Clinical Potential. Am. J. Physiol.-Heart Circ. Physiol. 2019, 317 (5), H923–H938. [CrossRef]
- Astudillo, A. A.; Mayrovitz, H. N. The Gut Microbiome and Cardiovascular Disease. Cureus 2021. [CrossRef]
- Toya, T.; Corban, M. T.; Marrietta, E.; Horwath, I. E.; Lerman, L. O.; Murray, J. A.; Lerman, A. Coronary Artery Disease Is Associated with an Altered Gut Microbiome Composition. PLOS ONE 2020, 15 (1), e0227147. [CrossRef]
- Yoshida, N.; Yamashita, T.; Hirata, K. Gut Microbiome and Cardiovascular Diseases. Diseases 2018, 6 (3), 56. [CrossRef]
- Liu, H.; Chen, X.; Hu, X.; Niu, H.; Tian, R.; Wang, H.; Pang, H.; Jiang, L.; Qiu, B.; Chen, X.; Zhang, Y.; Ma, Y.; Tang, S.; Li, H.; Feng, S.; Zhang, S.; Zhang, C. Alterations in the Gut Microbiome and Metabolism with Coronary Artery Disease Severity. Microbiome 2019, 7 (1), 68. [CrossRef]
- Ferreira, R. L. U.; Sena-Evangelista, K. C. M.; De Azevedo, E. P.; Pinheiro, F. I.; Cobucci, R. N.; Pedrosa, L. F. C. Selenium in Human Health and Gut Microflora: Bioavailability of Selenocompounds and Relationship With Diseases. Front. Nutr. 2021, 8, 685317. [CrossRef]
- Zhang, Y.; Meng, S.; Yu, Y.; Bi, L.; Tian, J.; Zhang, L. Associations of Dietary Selenium Intake with the Risk of Chronic Diseases and Mortality in US Adults. Front. Nutr. 2024, 11, 1363299. [CrossRef]
- Cai, J.; Su, W.; Chen, X.; Zheng, H. Advances in the Study of Selenium and Human Intestinal Bacteria. Front. Nutr. 2022, 9, 1059358. [CrossRef]
- Weng, Y. J.; Gan, H. Y.; Li, X.; Huang, Y.; Li, Z. C.; Deng, H. M.; Chen, S. Z.; Zhou, Y.; Wang, L. S.; Han, Y. P.; Tan, Y. F.; Song, Y. J.; Du, Z. M.; Liu, Y. Y.; Wang, Y.; Qin, N.; Bai, Y.; Yang, R. F.; Bi, Y. J.; Zhi, F. C. Correlation of Diet, Microbiota and Metabolite Networks in Inflammatory Bowel Disease. J. Dig. Dis. 2019, 20 (9), 447–459. [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165 (6), 1332–1345. [CrossRef]
- Flores-Mateo, G.; Navas-Acien, A.; Pastor-Barriuso, R.; Guallar, E. Selenium and Coronary Heart Disease: A Meta-Analysis. Am. J. Clin. Nutr. 2006, 84 (4), 762–773. [CrossRef]
- Hu, T.; Wu, Q.; Yao, Q.; Jiang, K.; Yu, J.; Tang, Q. Short-Chain Fatty Acid Metabolism and Multiple Effects on Cardiovascular Diseases. Ageing Res. Rev. 2022, 81, 101706. [CrossRef]
- Modrego, J.; Ortega-Hernández, A.; Goirigolzarri, J.; Restrepo-Córdoba, M. A.; Bäuerl, C.; Cortés-Macías, E.; Sánchez-González, S.; Esteban-Fernández, A.; Pérez-Villacastín, J.; Collado, M. C.; Gómez-Garre, D. Gut Microbiota and Derived Short-Chain Fatty Acids Are Linked to Evolution of Heart Failure Patients. Int. J. Mol. Sci. 2023, 24 (18), 13892. [CrossRef]
- Canyelles, M.; Borràs, C.; Rotllan, N.; Tondo, M.; Escolà-Gil, J. C.; Blanco-Vaca, F. Gut Microbiota-Derived TMAO: A Causal Factor Promoting Atherosclerotic Cardiovascular Disease? Int. J. Mol. Sci. 2023, 24 (3), 1940. [CrossRef]
- Zheng, Y.; He, J.-Q. Pathogenic Mechanisms of Trimethylamine N-Oxide-Induced Atherosclerosis and Cardiomyopathy. Curr. Vasc. Pharmacol. 2022, 20 (1), 29–36. [CrossRef]
- Zhang, Y.; Wang, Y.; Ke, B.; Du, J. TMAO: How Gut Microbiota Contributes to Heart Failure. Transl. Res. 2021, 228, 109–125. [CrossRef]
- Rath, S.; Heidrich, B.; Pieper, D. H.; Vital, M. Uncovering the Trimethylamine-Producing Bacteria of the Human Gut Microbiota. Microbiome 2017, 5 (1), 54. [CrossRef]
- Chen, H.-C.; Liu, Y.-W.; Chang, K.-C.; Wu, Y.-W.; Chen, Y.-M.; Chao, Y.-K.; You, M.-Y.; Lundy, D. J.; Lin, C.-J.; Hsieh, M. L.; Cheng, Y.-C.; Prajnamitra, R. P.; Lin, P.-J.; Ruan, S.-C.; Chen, D. H.-K.; Shih, E. S. C.; Chen, K.-W.; Chang, S.-S.; Chang, C. M. C.; Puntney, R.; Moy, A. W.; Cheng, Y.-Y.; Chien, H.-Y.; Lee, J.-J.; Wu, D.-C.; Hwang, M.-J.; Coonen, J.; Hacker, T. A.; Yen, C.-L. E.; Rey, F. E.; Kamp, T. J.; Hsieh, P. C. H. Gut Butyrate-Producers Confer Post-Infarction Cardiac Protection. Nat. Commun. 2023, 14 (1), 7249. [CrossRef]
- Shimada, B. K.; Alfulaij, N.; Seale, L. A. The Impact of Selenium Deficiency on Cardiovascular Function. Int. J. Mol. Sci. 2021, 22 (19), 10713. [CrossRef]
- Zhang, C.; Zeng, Q.; Liu, X.; He, Q.; Zhang, J.; Zhao, S.; Hu, H. Association of Blood Selenium Levels with Diabetes and Heart Failure in American General Adults: A Cross-Sectional Study of NHANES 2011–2020 Pre. Biol. Trace Elem. Res. 2024, 202 (8), 3413–3424. [CrossRef]
- Leszto, K.; Biskup, L.; Korona, K.; Marcinkowska, W.; Możdżan, M.; Węgiel, A.; Młynarska, E.; Rysz, J.; Franczyk, B. Selenium as a Modulator of Redox Reactions in the Prevention and Treatment of Cardiovascular Diseases. Antioxidants 2024, 13 (6), 688. [CrossRef]
- Jaworska, K.; Hering, D.; Mosieniak, G.; Bielak-Zmijewska, A.; Pilz, M.; Konwerski, M.; Gasecka, A.; Kapłon-Cieślicka, A.; Filipiak, K.; Sikora, E.; Hołyst, R.; Ufnal, M. TMA, A Forgotten Uremic Toxin, but Not TMAO, Is Involved in Cardiovascular Pathology. Toxins 2019, 11 (9), 490. [CrossRef]
- He, S.; Jiang, H.; Zhuo, C.; Jiang, W. Trimethylamine/Trimethylamine-N-Oxide as a Key Between Diet and Cardiovascular Diseases. Cardiovasc. Toxicol. 2021, 21 (8), 593–604. [CrossRef]
- Roncal, C.; Martínez-Aguilar, E.; Orbe, J.; Ravassa, S.; Fernandez-Montero, A.; De Pipaon, G. S.; Ugarte, A.; Estella-Hermoso De Mendoza, A.; Rodriguez, J. A.; Fernández-Alonso, S.; Fernández-Alonso, L.; Oyarzabal, J.; Paramo, J. A. Trimethylamine (Tma) And Trimethylamine-N-Oxide (Tmao) As Predictors Of Cardiovascular Mortality In Peripheral Artery Disease. Atherosclerosis 2019, 287, e233. [CrossRef]
- He, Y.; Chen, S.; Xue, Y.; Lu, H.; Li, Z.; Jia, X.; Ning, Y.; Yuan, Q.; Wang, S. Analysis of Alterations in Intestinal Flora in Chinese Elderly with Cardiovascular Disease and Its Association with Trimethylamine. Nutrients 2024, 16 (12), 1864. [CrossRef]
Figure 1.
The total relative abundance of each phylum provides a comprehensive overview of the overall distribution within the dataset.
Figure 1.
The total relative abundance of each phylum provides a comprehensive overview of the overall distribution within the dataset.
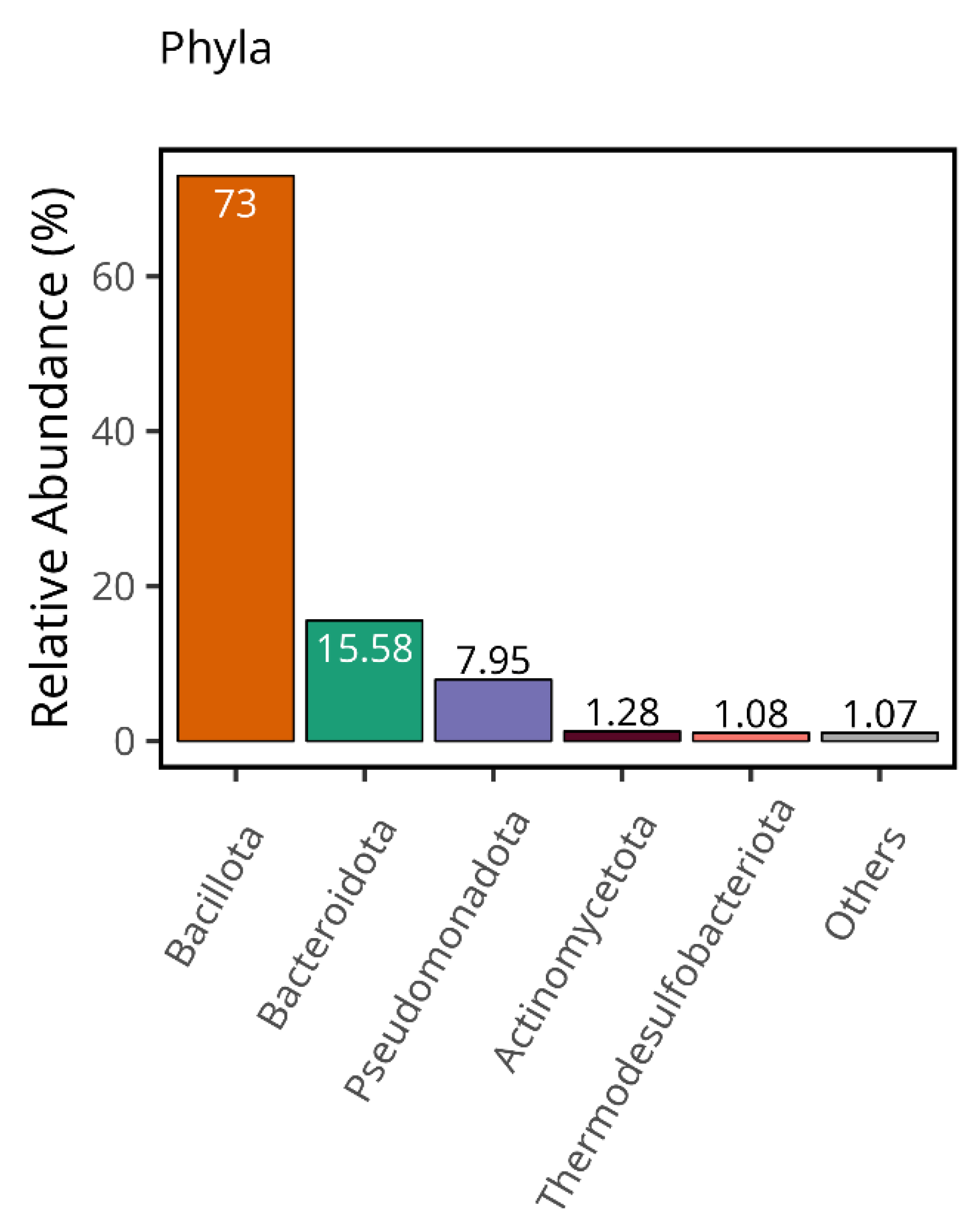
Figure 2.
A) The stacked bar plot indicates the relative abundance of phyla in the control and patient groups. “Control-AVE” refers to the average of the whole control group phylum, and “Disease-AVE” indicates the average of the whole disease group phylum. (Those that have relative abundance value less than 1% were classified as others) B) PCA results present the clustered healthy individuals (Control) and CSFP patient groups (Disease) and OTUs that provide the discrimination of both groups in a visual format. The green colors show healthy samples, and the red ones demonstrate the patient.
Figure 2.
A) The stacked bar plot indicates the relative abundance of phyla in the control and patient groups. “Control-AVE” refers to the average of the whole control group phylum, and “Disease-AVE” indicates the average of the whole disease group phylum. (Those that have relative abundance value less than 1% were classified as others) B) PCA results present the clustered healthy individuals (Control) and CSFP patient groups (Disease) and OTUs that provide the discrimination of both groups in a visual format. The green colors show healthy samples, and the red ones demonstrate the patient.
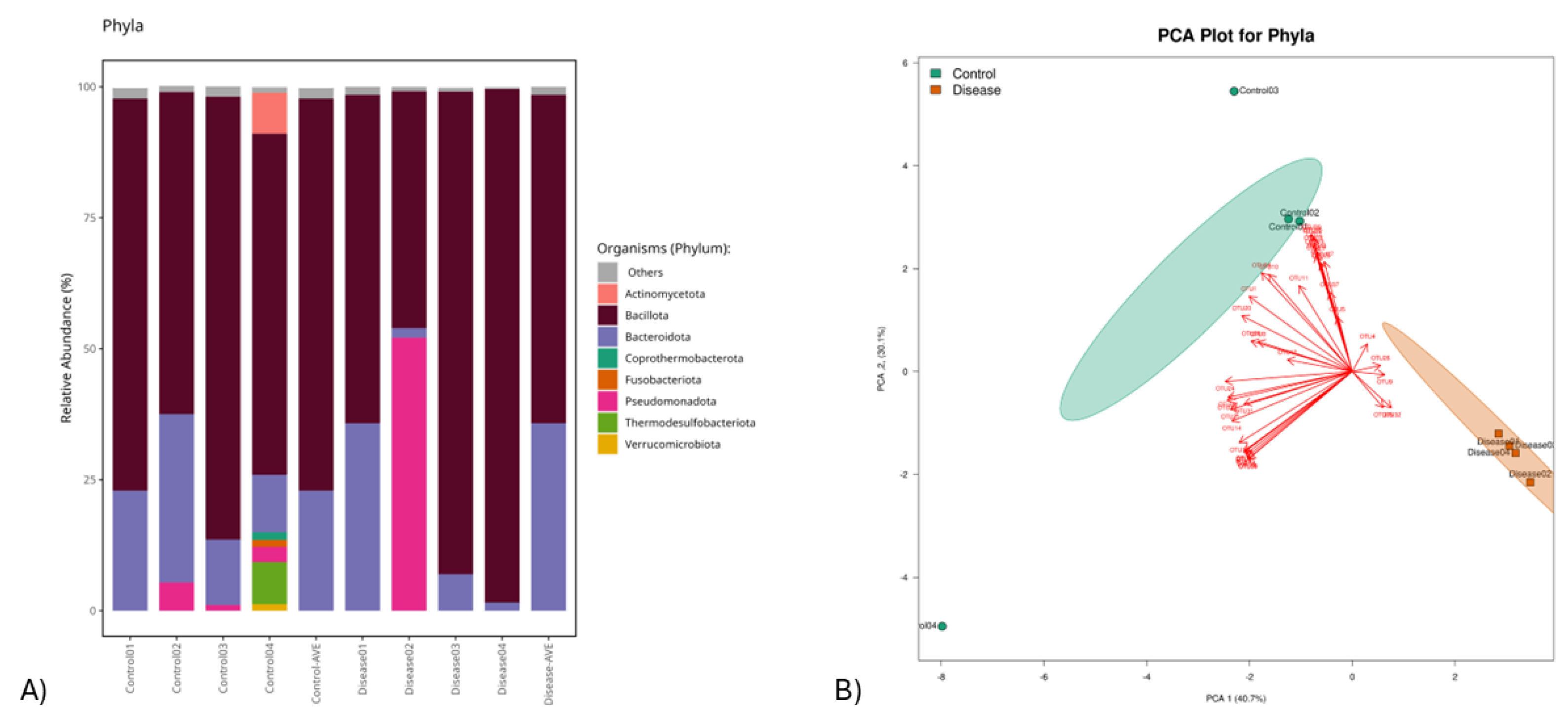
Figure 3.
Boxplots illustrate the relative abundances of the phyla Bacillota and Bacteroidota, with the green plots representing healthy individuals in the control group (Con) and the orange plots depicting patients diagnosed with CSFP disease (Dis). (p > 0.05).
Figure 3.
Boxplots illustrate the relative abundances of the phyla Bacillota and Bacteroidota, with the green plots representing healthy individuals in the control group (Con) and the orange plots depicting patients diagnosed with CSFP disease (Dis). (p > 0.05).
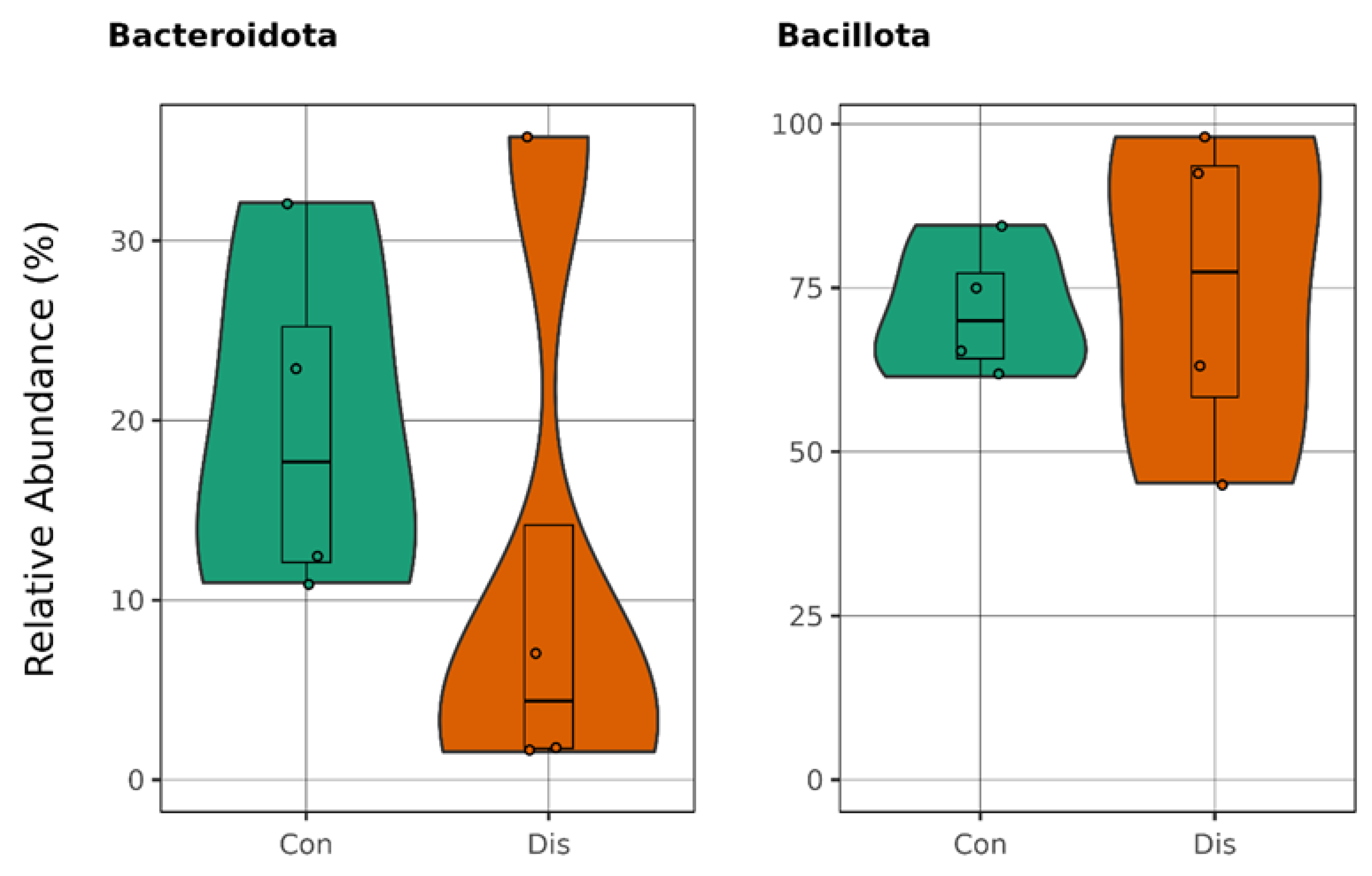
Figure 4.
The figure highlights all genera exhibiting significant differences between healthy controls (Con) and patients with CSFP disease (Dis) (p < 0.05).
Figure 4.
The figure highlights all genera exhibiting significant differences between healthy controls (Con) and patients with CSFP disease (Dis) (p < 0.05).
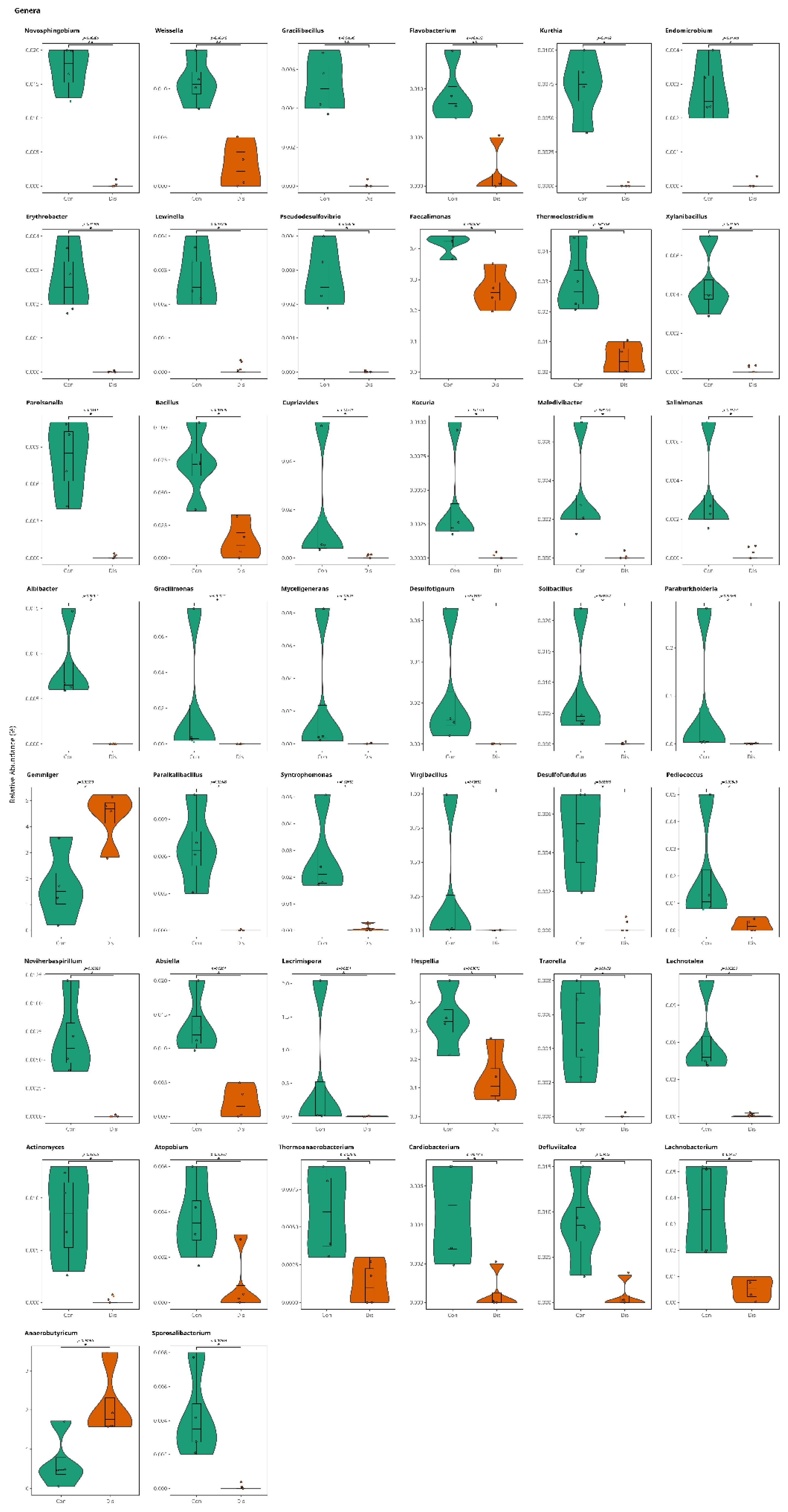
Figure 5.
A) The Venn diagram depicts the distribution of OTUs across the genus level. and species levels. The green section represents control samples, while the red section corresponds to patient samples. B) This visualization illustrates the percentile of both group-specific OTUs and those shared between the groups, providing a detailed overview of the dataset's composition at species level.
Figure 5.
A) The Venn diagram depicts the distribution of OTUs across the genus level. and species levels. The green section represents control samples, while the red section corresponds to patient samples. B) This visualization illustrates the percentile of both group-specific OTUs and those shared between the groups, providing a detailed overview of the dataset's composition at species level.
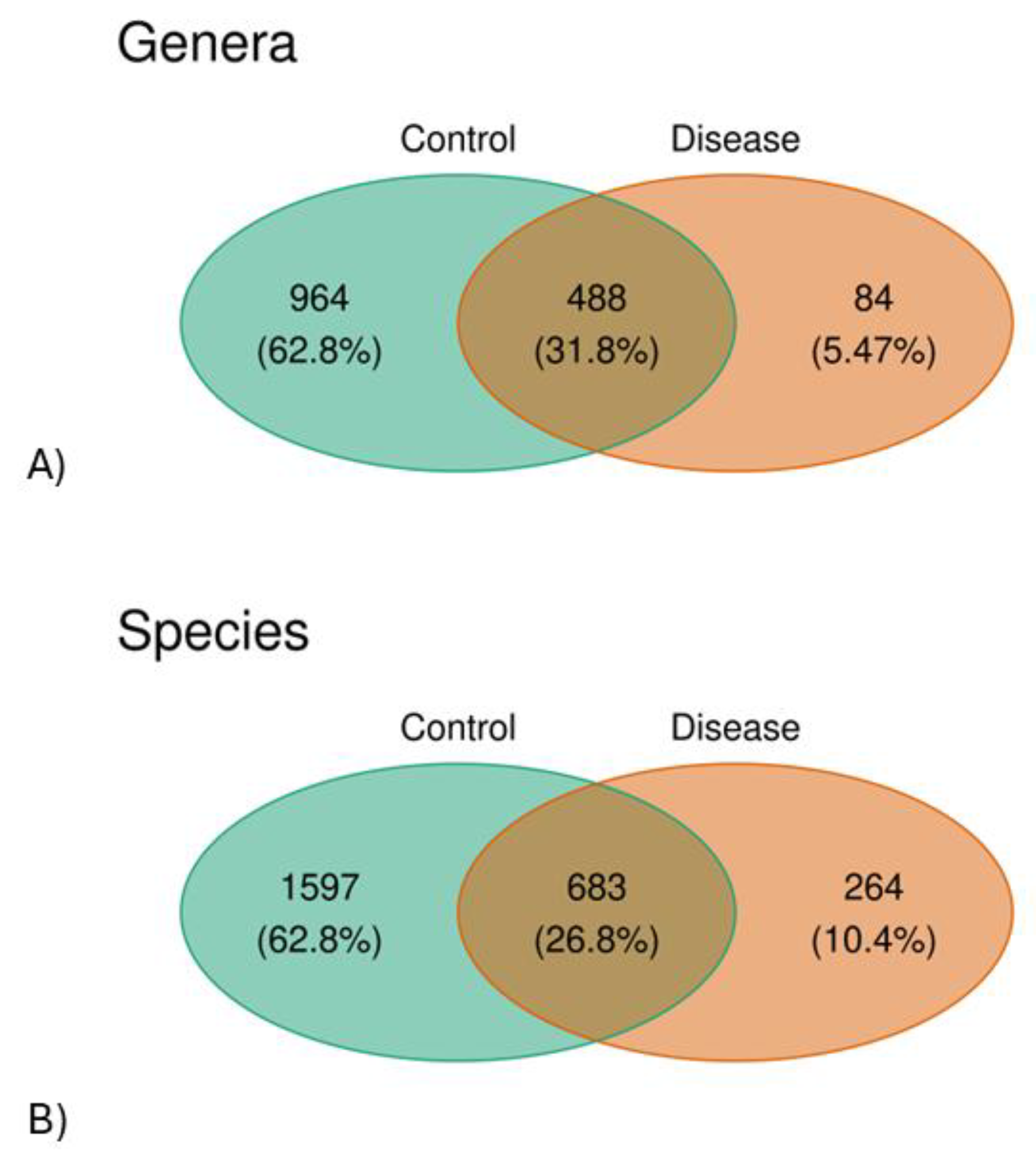
Figure 6.
The figure demonstrates the relative abundance of statistically significant bacterial species of healthy controls (Con) and patients with CSFP disease (Dis) (p < 0.05).
Figure 6.
The figure demonstrates the relative abundance of statistically significant bacterial species of healthy controls (Con) and patients with CSFP disease (Dis) (p < 0.05).

Figure 7.
Violinplots illustrate the relative abundance of bacterial species directly involved in TMA production for comparison of control and patient groups, with no statistically significant differences observed (p > 0.05).
Figure 7.
Violinplots illustrate the relative abundance of bacterial species directly involved in TMA production for comparison of control and patient groups, with no statistically significant differences observed (p > 0.05).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



