Submitted:
24 January 2025
Posted:
24 January 2025
You are already at the latest version
Abstract
Antifungal resistance poses a critical global health threat, particularly in immuno-compromised patients. Beyond the traditional resistance mechanisms rooted in heritable and stable mutations, a distinct phenomenon known as heteroresistance has been concerned, wherein a minority of resistant fungal cells coexist within a predominantly susceptible population. It may be induced by pharmacological factors or non-pharmacological agents. The reversible nature of it presents significant clinical challenges, as it can lead to undetected resistance during standard susceptibility testing. As heteroresistance allows fungal pathogens to survive antifungal treatment, this adaptive strategy often leads to treatment failure and infection recurrence. Though extensively studied in bacteria, limited research has explored its occurrence in fungi. This review synthesizes the current findings on antifungal heteroresistance mechanisms, highlighting the clinical implications of fungal heteroresistance and the pressing need for deeper mechanism insights. We aim to combine the latest research advances in the field of antifungal heteroresistance, summarizing in detail the characteristics, inducing factors, molecular mechanisms, and clinical significance of it, and describing the similarities and differences between heteroresistance, tolerance and persistence. Further research is essential to elucidate this phenomenon and develop more effective antifungal therapies to combat fungal infections.
Keywords:
Antifungals
; Drug resistance
; Heteroresistance
; Aneuploidy
; Copy Number Variations (CNVs)
1. Introduction
Fungal infections pose a significant and growing threat to public health worldwide, particularly in immunocompromised patients [1,2]. The increasing prevalence of antifungal-resistant pathogens is complicating treatment, making fungal infection a major global health challenge [3]. Traditionally, antifungal resistance has been defined as the result of inherent and stable genetic mutations that diminish drug efficacy, with elevated MIC (Minimum Inhibitory Concentration). However, recent researches have uncovered a more complex and dynamic form of resistance: heteroresistance. Fungal heteroresistance, in which a small subpopulation of a fungal strain exhibits resistance to antifungal drugs while the majority remain susceptible, represents a significant global threat. This phenomenon complicates treatment as it can lead to treatment failure despite the apparent efficacy of the drug against most of the fungal cells, further exacerbating the public health crisis caused by fungal pathogens.
Heteroresistance was first discovered in 1947 in Haemophilus influenzae [4]. In bacteriology, it refers to the presence of a heterogeneous population with one or several subpopulations with increased levels of antibiotic resistance compared with the major population. This can be due to clonal heterogeneity during the co-infection of colonies with different levels of resistance, or microevolution of a single colony [5,6]. The heteroresistance phenotype can either be stable that do not rapidly revert to susceptible phenotype, or unstable that may affect the laboratory test results [7]. Under persistent selective pressure, it may even evolve into heritable ones, causing higher rate of treatment failure and create an antibiotic resistance reservoir [8]. As for mechanisms, they vary among different pathogens and various kinds of drugs. According to a research on Pseudomonas aeruginosa, heteroresistance to imipenem is due to biofilm formation and OprD gene mutation [9], while heteroresistance to levofloxacin is linked to elevated expression of genes involved in DNA replication and repair [10]. Another research on H. influenzae showed that imipenem heteroresistance was linked to alteration of PBP3 (penicillin-binding protein 3), slowed drug influx and efflux alterations [11]. It can also be induced by antibiotic exposure, through upregulation of stress-related pathways, etc. [12,13].
While extensively studied in bacteria, research on fungal heteroresistance remains comparatively limited. From a clinical perspective, heteroresistance has been implicated in treatment failure in murine models [14], and relative mechanisms have not been studied in sufficient depth. However, the clinical impact of heteroresistance leading to drug resistance during treatment should not be underestimated, as it may lead to prophylaxis failure, recurrent infection or relapse [15,16,17,18,19]. In this review, we synthesize current research to outline the characteristics, mechanisms, and clinical prevalence of heteroresistance in fungal pathogens, with the goal of providing a foundation for future investigations.
2. Antifungal Drugs
Fungi are an important class of human pathogenic pathogens with immune escape, intra-host environmental adaptability and multiple virulence mechanisms, posing a great threat to human health [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40]. Currently, there are four main classes of drugs approved for antifungal infection treatment: azoles, echinocandins, polyenes (Amphotericin B, AmB), and 5-fluorocytosine (5-FC). Azoles act on fungal 14-α-demethylases and inhibit ergosterol biosynthesis, leading to alterations in the permeability and metabolic state of fungal cells, which may result in growth inhibition or cell death [41,42]. Echinocandins target and inhibit the FKS gene which encodes β-1,3-glucan synthase, thereby inhibit β-1,3-glucan production and impairing fungal cells from maintaining their shape and resisting external stresses [43]. AmB targets membrane ergosterol, leading to pore formation, altered permeability, and reactive oxygen species (ROS) accumulation, all of which led to eventual fungal cell death [44,45,46]. 5-FC is a prodrug that is imported into cells by cytosine permease, which is encoded by the FCY2 gene. Once inside the cell, 5-FC is converted to 5-fluorouracil (5-FU) through the action of cytosine deaminase and uracil phosphoribosyl transferase, both encoded by the FCY1 gene, inhibiting DNA and RNA biosynthesis [47,48]. However, in recent years, there has been a concerning surge in the isolation of antifungal resistant strains, raising a critical alarm for public health.
Mechanisms associated with azole resistance mainly include mutations in targets, efflux pumps and biofilm formation etc. [49,50,51]. In Candida spp., up-regulation of the efflux transporter genes CDR1, TAC1B and MDR1 and point mutations in the target coding genes ERG11 and ERG3, such as Y132F and R398I in the ERG11 gene, have been associated with fluconazole resistance [49,51,52,53,54]. In C. neoformans, a point mutation at the serine-substituted glycine 484S site of the 14-α-demethylase leads to single-drug resistance to fluconazole [55]. In A. fumigatus, mutations in the cyp51A gene encoding 14-α-demethylase and mutations in the ABC transporter gene atrF are common azole resistance mechanisms [56].
Currently, resistance to echinocandins is mainly associated with mutations in the FKS1/FKS2 genes [49,57,58]. Other molecular mechanisms associated with echinocandin resistance have also been reported. Yu et al. found that the ADA2 gene of C. glabrata is associated with resistance to three classes of antifungals, as evidenced by the Δada2 knockout strains’ significant downregulation of their MICs [59]. Singh et al. found that deletion of seven genes, including MOH1, GPH1, CDC6, TCB1/2, DOT6, MRPL11 and SUI2, showed increased levels of resistance to echinocandins; but considering their small effect on caspofungin resistance or conjured occurrence with FKS2 mutations, it is more likely that they create a genetic background in which the FKS2 mutations are adaptive and less harmful, rather than a direct cause of resistance [60].
5-FC resistance is highly correlated with its pharmacological mechanism, i.e., mutations in any one or more of the key enzyme genes of the pyrimidine salvage pathway, which may involve mutations in the genes encoding the cytosine permease FCY2, purine FCY1 encoding the cytosine deaminase, FUR1 encoding the uracil phosphoribosyl transferase, and ADE17 [49,61]. However, other mechanisms have also been reported: Kannan et al. found that V668G substitution of a putative transcriptional activator (MRR1) led to the up-regulation of MFS7, a multidrug transporter protein that mediated azole-5-FC cross-resistance in C. lusitaniae [62], and Billmyre et al. found that deletion of the mismatch repair gene, MSH2, in C. deuterogattii also led to an elevated rate of 5-FC mutations [63].
Fungal resistance to AmB is less common but has been reported in strains such as C. auris and the C. haemulonii complex [49,64]. Resistance to AmB in both species involves a variety of mechanisms, including altered cell membrane composition, altered cellular metabolic state, altered iron homeostasis, and altered ROS metabolism [44,45,64,65]. The molecular mechanisms of resistance to AmB in C. haemulonii have been systematically discussed in the author’s previous review [66] .
3. Heteroresistance, Tolerance and Persistence of Antifungal Drugs
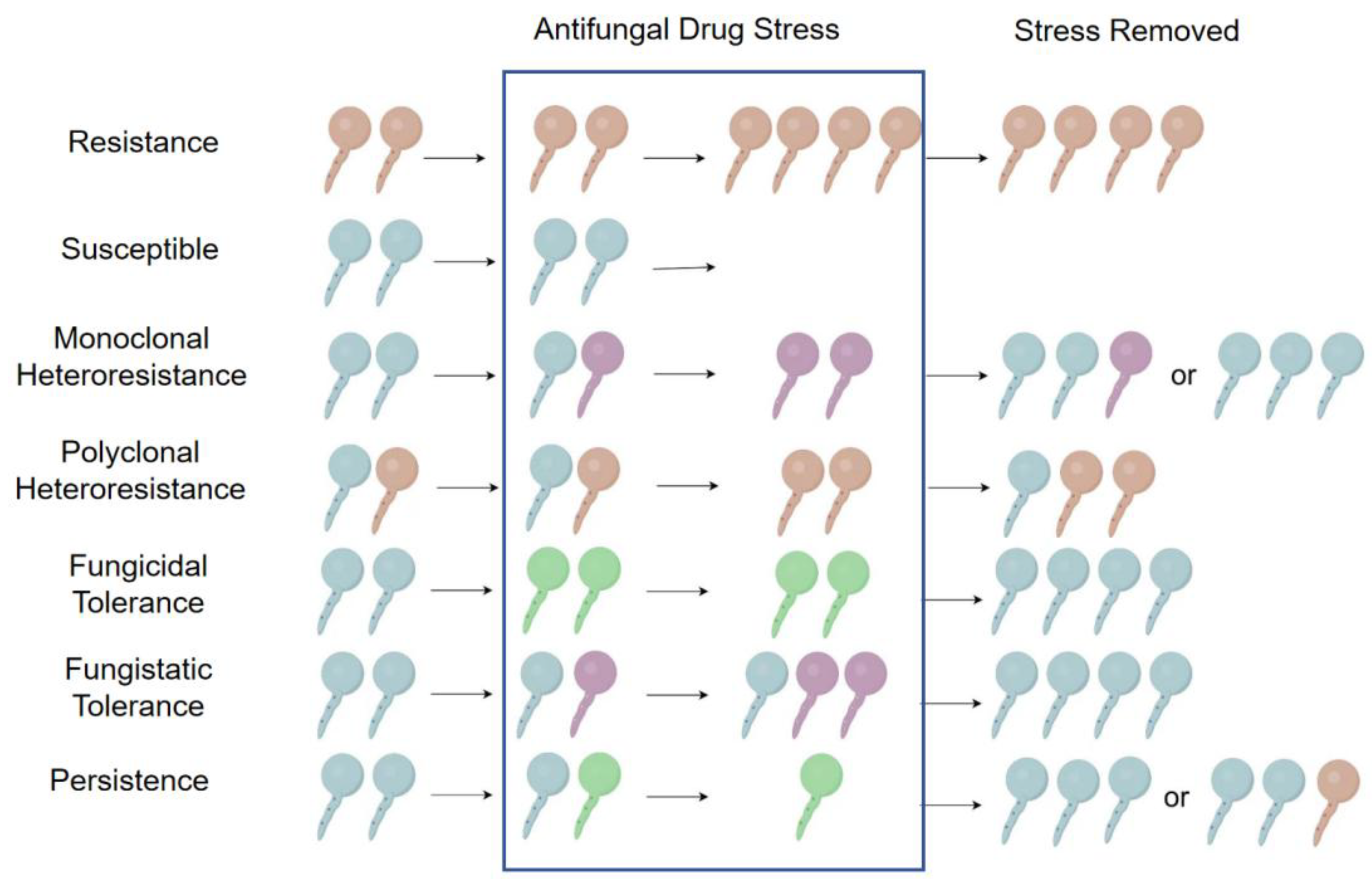
At the level of cell populations, not all survival under fungicidal concentrations of antifungals can be attributed to elevated MICs. This is when heteroresistance, tolerance and persistence come into play, while they are often confused conceptually. Here we describe the differences between the three from the perspective of the performance of cell populations under drug stress (Figure 1).
Heteroresistance , a concept first introduced in bacteria, is a form in which a minority subpopulation of resistant phenotype cells with higher MIC, sometimes as rare as one in one million cells, coexists with a majority population of susceptible cells with lower MIC [4]. It may be polyclonal or monoclonal. Polyclonal heteroresistance is related to heterogeneous consortia with genetically distinct subpopulations or to the appearance of rare resistant mutants whose frequency increases with antibiotic exposure. In turn, heteroresistance is monoclonal if present in pure clones [7]. As a variant, it is more of continuous than binary. A study on the nature of heteroresistance properties showed that incremental effects of multiple binary genetic switches, including CDR1, PDH1, PDR1 and SNQ1, etc. produced a spectrum of heteroresistance states rather than binary, in C. glabrata [67]. From the perspective of population evolution, it can be seen as a strategy that helps the fungal population survive diverse environments [68]. In vitro, heteroresistance can be obtained through antifungal induction or non-antifungal agents and artificial selection, the latter of which may get the longer-lasting phenotype [69,70]. And repeated culture in stress-free environments can abolish the heteroresistance [71]. But interestingly, this majority subpopulation can never be purified, which means there’s always a minority that is phenotypically resistant [72].
Tolerance is defined as an extension of the killing time, characterized by an increase in MDK99 (Minimum Duration of Killing 99%) time [73]. It refers to the whole population that can survive a transient exposure to antifungals at concentrations that would otherwise be lethal, without increasing antifungal MIC [74]. And some (5%-90%) can slowly proliferate [75]. This definition applies to fungicides such as echinocandins and polyenes. However, for antifungals rather than fungicidal drugs (e.g., azoles), tolerance has a different definition, i.e., the ability of a tolerant strain to overcome growth inhibition faster than a sensitive strain at drug concentrations above the MIC [76]. It can be measured through broth microdilution assay or disk diffusion assay [77]. And it is a reversible portrait, as confirmed in several fungi species or antifungals [78,79].
Persistence and persister cells have been well-studied in bacteriology [80]. For fungus, persistence is a subpopulation of genetically susceptible fungal cells that survives fungicidal concentrations of antifungals and may lead to the emergence of genetically resistant isolates [81,82,83]. This phenomenon has been observed in intra-macrophage C. glabrata cells, in which a subpopulation survives and gradually evolves heritable resistance mutations in FKS regions [81]. Sometimes “persistence” is used interchangeably with “tolerance,” as the MIC values of the fungal cell population do not change in either phenotype. Most notably, persistence is characterized by a biphasic killing curve, whereas tolerance is not [80]. In other words, “tolerance” means that all survive and some may proliferate, whereas “persistence” means that some survive but no one proliferates.
4. Mechanisms of Antifungal Tolerance
Fungal tolerance to antifungals arises from a variety of molecular mechanisms. For example, azole tolerance in Candida populations is often characterized by the ability to survive and proliferate slowly at concentrations above the minimum inhibitory concentration (MIC). This tolerance is typically temperature-independent and frequently associated with aneuploidy, which may be lost upon temperature changes or ploidy recovery [84,85]. Similarly, in C. neoformans, brain glucose has been shown to induce AmB tolerance without affecting the MIC. This process is mediated by the zinc-finger transcription factor Mig1 and involves more complex mechanisms, such as the inhibition of ergosterol synthesis and the promotion of competing compounds that contribute to antifungal tolerance [86]. And in C. neoformans, mitochondria metabolism alteration caused by cell aging drive increased ergosterol synthesis and ABC transporter upregulation and led to azole tolerance [87].
Alterations in cellular structure and metabolic levels also play a key role in antifungal tolerance. Cell wall remodeling contributes echinocandin tolerance, with changes in β-glucan synthesis or increased production of compensatory components, like chitin, strengthening the cell wall and reducing the drug’s effectiveness [88,89]. Additionally, activation of cellular stress response pathways, such as the heat shock protein (Hsp) and calcineurin pathways, has been shown to contribute to echinocandin tolerance in C. albicans [90,91]. Moreover, a single-cell transcriptomics study demonstrated that the ribosome assembly stress response supports survival under fluconazole exposure above the MIC, further highlighting the importance of stress responses in antifungal tolerance [92]. Together, these mechanisms illustrate how fungi adapt to antifungal treatments through a combination of genetic, transcriptional, and physiological changes, making effective treatment more challenging.
5. Mechanisms of Antifungal Persistence
Persistence is always linked to cell dormancy, in a state which the cellular metabolism is temporarily ceased [82,93,94]. As the fungal pathogen enters the host organism, it is often phagocytosed by immune cells, like macrophages, etc. Studies on pathogen-phagocyte interactions have revealed the inducing effect of the phagocytic intracellular environment on the formation of persisters through comparing the metabolism of planktonic cells and persisters and exerting ROS or pH-related environmental stress, which also suggest a possible role for cellular stress response in persistence [81]. Ke et al. constructed a mouse model with pulmonary infection and demonstrated that AmB-tolerant persisters are enriched in C. neoformans cells with high levels of the stationary-phase protein Sps1 and the metabolic marker ergothioneine [83]. This also provides us with the critical role of the EGT gene, which encodes the ergothioneine, in regulating the AmB susceptibility [83].
There are also several key factors in the formation of persisters. The formation of persister cells does not require biofilm, but biofilm-containing persister populations are more tolerant to oxidative stress and better resist the fungicidal effects of AmB [95]. And inhibiting biofilm aids in the eradication of persister cells of C. tropicalis [96]. Also, as antifungals can cause an increased level of ROS, persister cells may possess stronger antioxidant capacity by upregulating enzymes like superoxide dismutases (SODs) or alkyl hydroperoxide reductase 1, etc. [97]. Such mechanisms can be can be confirmed inversely by SOD inhibition leading to down-regulation of persistence levels [98]. Formation of persister cells should be emphasized, as they can cause chronic or recurrent infections, complicating treatment.
6. Mechanisms of Antifungal Heteroresistance
The molecular mechanisms underlying fungal heteroresistance are diverse and involve a complex interplay of genetic, transcriptional, and physiological factors. However, they do not always include heritable changes. In many cases, heteroresistance arises through transient changes that allow the fungus to survive antifungal stress without permanent genetic changes. This adaptability complicates treatment strategies, as the resistant phenotype may not be stable and can fluctuate depending on environmental conditions.
5.1. Aneuploidy and Copy Number Variations (CNVs)
Aneuploidy is a common mechanism in heritable resistance and the most widely studied mechanism of heteroresistance [99,100]. It is the presence of an abnormal number of chromosomes, playing a crucial role in fungal survival and evolution by providing rapid adaptability in response to environmental stress, such as antifungal drug exposure [101,102]. Yang et al. found that exposure to fluconazole at subinhibitory concentrations for a short period of time (48 h) may cause C. neoformans to acquire different aneuploid chromosomes and confer heteroresistance to fluconazole and cross-resistance to 5-FC [103]. Thus, exposure to one class of antifungal can promote adaptation to similar or even more potent antifungal agents, highlighting the plasticity of the fungal genome and raising serious public health concerns. By increasing the copy number of chromosomes containing drug resistance genes, such as those encoding efflux pumps or involved in ergosterol biosynthesis, aneuploidy leads to resistance against multiple antifungals like azoles and echinocandins [67,104,105]. Also, non-antifungal agents can also induce heteroresistance. Zhang et al. found that the endoplasmic reticulum stress chemo inducer Brefeldin A (BFA) led to aneuploidy in C. neoformans: disomy in chromosome 1 led to cross-resistance to two classes of antifungal drugs, fluconazole and fluconazole and 5-FC and hypersensitivity to AmB [106]. By altering gene dosage or expression levels, aneuploidy allows fungi to quickly adjust to adverse conditions without relying on permanent genetic mutations, promoting survival in fluctuating environments [67,102,104,105,107]. Importantly, it is reversible, offering a dynamic mechanism for fungi to balance adaptation and stability, population and individuals [101,108]. As a result, aneuploidy plays a pivotal role in both fungal adaptability and the evolution of drug resistance, making it a key factor in heteroresistance and its clinical implications.
6.1.1. C. albicans
Mechanisms related to azole heteroresistance in C. albicans are diverse. Under fluconazole exposure, C. albicans rapidly evolve CNVs (Copy Number Variation) and aneuploidy in vitro [109]. Associated resistance mainly related to chr5, which has ERG11 and TAC1 gene [104,110]. TAC1 encodes a transcription regulator of ABC transporter genes on chr3 [111]. Loss of chr5 can result in enhanced susceptibility to azoles, the mechanisms of which has been clarified [110]. However, loss of chr5 can also lead to the enhanced susceptibility to AmB and increased resistance to 5-FC, the latter of which is due to the location of negative regulator (s) of anti 5-fluorocytosin on chr5 and the former of which needs to be further clarified [110]. Harrison et al. reported the appearance of “trimeras,” three connected cells composed of a mother, daughter, and granddaughter bud, after exposure to fluconazole [112]. The same morphology cannot be found in genetically resistant strains, suggests the potential role of trimeras in heteroresistance. Also, these trimeras produce genetically progeny with different chromosomes, increasing chances of developing heteroresistance [112].
C. albicans’ heteroresistance to echinocandins is related to chr2, as chr2 trisomy can be induced after exposure to caspofungin and exhibits higher echinocandin resistance, according to Yang et al. [113]. It is also related to chr5, as chr5 aneuploidy after caspofungin exposure can obtain cross-resistance to caspofungin, micafungin and anidulafungin [114]. Yang et al. found three negative regulators of echinocandin susceptibility on chr5, including CHT2 encoding a glycosylphosphatidylinositol (GPI)-dependent chitinase which is a covalently bound cell wall protein, PGA4 encoding a GPI-anchored cell surface 1,3-β-d-glucanosyltransferase, and CSU51 encoding another putative GPI-anchored protein, and two positive regulators, CNB1 encoding a regulatory subunit of calcineurin B and MID1 encoding a putative stretch-activated Ca2+ channel of the high-affinity calcium uptake system [88]. However, the exact gene copies and whether they are cumulative remain unclear, making it hard to simply summarize their specific roles in echinocandin resistance [88].
6.1.2. C. glabrata
C. glabrata has been considered a haploid and an asexual organism for decades, but recent years witnessed studies reporting the instability of clinical isolates genome, mainly due to the frequent change in ploidy forms [115]. Ploidy variation could not only promote the rapid adaptation of C. glabrata to the changing environment but also benefit the evolution of new traits, like heteroresistance. Ksiezopolska et al. found chrE aneuploidy contributes to heteroresistance after exposing clinical isolates to anidulafungin [116]. And heteroresistance obtained from environmental stress may remain even after the stress [116]. And when exposed to azoles, C. glabrata can also form “trimeras” that may be related to heteroresistance, but the exact mechanisms need further investigation [112].
6.1.3. C. parapsilosis
In C. parapsilosis, multi-center research proved that heteroresistance facilitates breakthrough infections in immunocompromised patients and may cause the prophylaxis failure [19]. However, unlike C. neoformans or itself when exposed to tunicamycin, this research couldn’t find a significant relationship between aneuploidy and heteroresistance in C. parapsilosis [19,103]. Evidence of aneuploidy in the heteroresistance are put forward by Harrison et al., elaborating C. parapsilosis can also form “trimeras” when exposed to azoles, but the exact mechanisms need further investigation [112].
6.1.4. C. auris
For highly resistant pathogen C. auris, Zhai et al. reported heteroresistance of C. auris towards echinocandins, which is the first-line treatment for C. auris infection [19]. In vitro evolution under fluconazole exposure is relatively slow, compared to C. albicans, and mainly composed of SNP, with a minority of aneuploidy [109,117]. But due to its haploid genome, SNPs may have immediate phenotypic impact [118].
6.1.5. C. neoformans and C. gattii
For Cryptococcus spp., heteroresistance means a subpopulation appears under antifungal exposure and retain the potential to grow under continuous drug stress [77]. Because of the broad definition, many studies have reported “tolerance” as cryptococcal azole heteroresistance and vice versa [119]. It is a crucial factor in the treatment failure of cryptococcosis. For C. neoformans and C. gattii, the heteroresistance to fluconazole is intrinsic and can be induced to increase [72,120]. According to a comparative study, C. gattii showed a higher heteroresistance level to fluconazole than C. neoformans [121]. For C. neoformans, heteroresistance can be reduced or inhibited by several environmental factors, including temperature, media type, growth phase, and the age of cells [122]. One of its mechanisms is associated with the multiple types of aneuploid daughter cells produced by titan cells [123,124,125]. Stone et al. carried out a genomic analysis of clinical C. neoformans strains and found a high rate of aneuploidy in heteroresistant colonies and recurrent isolates, with a predominance of chr1 disomy [126]. Strains with chr1 disomy can also be isolated from mice brain during treatment with fluconazole [127]. Ngamskulrungroj et al. demonstrated the high incidence of chr4 disomy, which may be related to SEY1 (GTPase with a role in Endoplasmic Reticulum morphology), GCS2 (ADP-ribosylation factor GTPase activating proteins) or GLO3 (ADP-ribosylation factor GTPase activating proteins) genes on it [128]. And if any of the ERG11, SEY1, GCS2 or GLO3 genes are relocated to chr3, then the frequency of chr3 disomy alternatively increases [128,129]. This is due to the presence on chr1 of ERG11, which is the drug target of fluconazole, and AFR1, which encodes the drug efflux pump. But AFR1 gene does not directly lead to heteroresistance, though [72]. Another research on C. neoformans’ ploidy found that exposure to inhibitory concentrations of fluconazole leads to diminished budding and subsequent growth while permitting nuclear events, resulting in populations with an increase in DNA content grow better in the presence of fluconazole, through which aneuploidy rates increase and the fitter survive [130]. Specific mechanisms of C. neoformans heteroresistance need to be explored, and their development may help to optimize clinical therapies for the effective elimination of drug-resistant Cryptococcus subpopulations.
6.2. Alterations in Gene Expression
Efflux pumps, such as those belonging to the ABC transporter and major facilitator superfamily (MFS), play a crucial role in antifungal resistance by actively expelling drugs from the fungal cell. In heteroresistant subpopulations, the overexpression of efflux pumps can confer a temporary survival advantage. Stone et al. carried out a genomic analysis of clinical C. neoformans strains and found a high rate of aneuploidy in heteroresistant colonies and recurrent isolates, with a predominance of chr1 disomy and upregulated activity of efflux pumps [126]. Marr et al. carried out molecular researches on one C. albicans strain and demonstrated the induction effect of fluconazole on heteroresistance, marked with elevated mRNA level of ABC superfamily CDR genes [107]. As mRNA level can represent the expression of one or more genes, this coincides with the continuous characteristic of C. glabrata [67], suggesting that heteroresistance in C. albicans may also be a continuous variable. However, further research is needed.
6.3. Environmental Stress Induction
In addition to specific antifungals that can induce aneuploidy, non-pharmacological stressors can also have an impact on antifungal heteroresistance and even cross-heteroresistance. Although resistance phenotype is not stable due to the intrinsic instability of aneuploidy, such studies still suggest the vital role of external inducing factors and genomic instability in drug resistance in C. neoformans. “Titanization” in C. neoformans also plays a role in formation of aneuploidy. Gerstein et al. found that during cryptococcosis treatment, newly emerged fungal cells -- polyploid titan cells -- produced daughter cells that were more resistant to fluconazole and thus adapted to the host environment, and that a single titan mother cell was able to produce multiple types of aneuploid daughter cells, which contributes to the survival rate of the progeny under different environmental stresses [124]. This process is associated with intracellular ROS accumulation and mitochondrial responses [125]. For C. neoformans, heteroresistance can be reduced or inhibited by several environmental factors, including temperature, media type, growth phase, and the age of cells [122]. And from these can we speculate that stress response pathways may play a role in the generation of antifungal heteroresistance. Bosch et al. demonstrated that environmental stress, such as nitrogen limitation commonly encountered in the natural habitat of the fungus, increases the resistance of C. neoformans to AmB and fluconazole, and increases the frequency of heterogeneous resistance to fluconazole [70]. For A. fumigatus, clinically, long-term itraconazole treatment may cause decreased susceptibility. And progressive itraconazole exposure in labs can reproduce such phenomenon, independent of the mutation in cyp51A gene [131]. This is called secondary resistance, suggesting the existence of heteroresistance. However, exact mechanisms need to be further explored.

Table 1.
Heteroresistance Mechanisms in Common Fungal Pathogens.
| Types | Species | Antifungals | Mechanisms | Related Components | References |
|---|---|---|---|---|---|
| Aneuploidy and CNVs | C. albicans | Fluconazole | Chr5 disomy | ERG11 and TAC1 | [104,110] |
| C. albicans | 5-Flucytosine | Loss of chr5, due to the location of negative regulator (s) of anti 5-FC | None. | [110] | |
| C. albicans | Fluconazole | “Trimeras,” three connected cells composed of a mother, daughter, and granddaughter bud | None | [112] | |
| C. albicans | Echinocandins | Chr2 trisomy | RNR1, RNR21 | [113] | |
| C. albicans | Echinocandins (caspofungin, micafungin and anidulafungin) | Chr5 aneuploidy after caspofungin exposure can obtain cross-resistance | Three negative regulators CHT2, PGA4 and CSU51, and two positive regulators, CNB1 and MID1. | [88,114] | |
| C. glabrata | Echinocandins (anidulafungin) |
ChrE aneuploidy contributes to heteroresistance after exposing clinical isolates to anidulafungin. | None | [116] | |
| C. glabrata | Azoles | Incremental effects of these multiple binary genetic switches | CDR1, PDH1, PDR1 and SNQ1 | [67] | |
| C. glabrata | Azoles | Formation of “trimeras” | None | [112] | |
| C. parapsilosis | Azoles | Formation of “trimeras” | None | [112] | |
| C. auris | Azoles (fluconazole) |
Genome changes mainly composed of SNP, with a minority of aneuploidy. But due to its haploid genome, SNPs may have immediate phenotypic impact. | SNPs | [109,117,118] | |
| C. neoformans | Cross-resistance to 5-FC and Fluconazole | Chr1 disomy | ERG11, AFR1 | [106] | |
| C. neoformans | Fluconazole | Overexpression of AFR1 on chr1 and GEA2 on chr3 | AFR1, GEA2 | [106] | |
| C. neoformans | Azoles (fluconazole) |
Titan cells that produce multiple types of aneuploid daughter cells | None | [123,124,125] | |
| C. neoformans | Azoles (fluconazole) |
Chr1 disomy | ERG11, AFR1 | [126] | |
| C. neoformans | Azoles (fluconazole) |
Chr4 disomy | SEY1, GCS2, GLO3 | [128] | |
| C. neoformans | Azoles (fluconazole) |
Chr3 disomy caused by gene relocation | ERG11, SEY1, GCS2, GLO3 | [128,129] | |
| Alterations in Gene Expression | C. albicans | Azoles | Elevation of mRNA | ATP Binding Cassette superfamily CDR genes | [107] |
| C. neoformans | Azoles (fluconazole) |
Up-regulated activity of efflux pumps | AFR1 | [126] | |
| Environmental Stress | C. neoformans | Polyene (AMB) and Azoles (fluconazole) | Nitrogen limitation | None | [70] |
| C. neoformans | Azoles | Temperature, media type, growth phase, and the age of cells | None | [122] |
7. Clinical Relevance of Antifungal Heteroresistance
7.1. Outcomes of Antifungal Heteroresistance
Heteroresistance poses significant challenges in the clinical management of fungal infections. In bacteria, heteroresistance may proceed during antibiotic therapy, leading to changes in clinical tests and treatment failures and even create an antimicrobial resistance reservoir [8,132]. Similarly, the transient nature of antifungal heteroresistance allows pathogenic fungus to survive conventional antifungal treatment, even when most of the population is inhibited. Prior to the onset of infection, this can facilitate breakthrough infection or result in prophylaxis failure [19]; for diagnosed fungal infections, it may lead to persistent infections that require longer or more aggressive treatment regimens [15,16,17,18]. Despite initial susceptibility to the drug, heteroresistant subpopulations can expand during therapy, partially caused by antifungal exposure, characterized molecularly as emergence of resistance-related genetic mutations and clinically as relapse or progression of the infection [81,101,102,103].
Up to now, there are no approved first-line therapies for antifungal heteroresistance. However, a combination of drugs or dosage escalation may aid in alleviating treatment failure. One research on C. neoformans showed that combining 5-FC with fluconazole may effectively reduce heteroresistance and treatment failure, suggesting the importance of combined therapy [126]; another research suggested the feasibility of fluconazole dose escalation and combination therapy in the treatment of cryptococcal meningitis but also revealed limitations in combination options that only 5-FC was comprehensively available [133]. To make things worse, antifungal heteroresistance can also contribute to cross-resistance between different classes of antifungal drugs. For example, in C. albicans chr5 aneuploidy caused simply by caspofungin can result in cross-resistance to caspofungin, micafungin and anidulafungin [114], and in C. neoformans, chr1 disomy may confer cross-resistance to both azoles and 5-FC [106]. This cross-resistance makes treatment options even more restrictive. Even if there is a suitable combination therapy or escalation dose therapy, both can increase the risk of toxicity and adverse effects. And in-depth study of the mechanisms of heteroresistance will help to develop low-toxicity combination therapies or new drugs.
7.2. Diagnosis of Antifungal Heteroresistance
Conventional antifungal susceptibility testing methods, such as broth microdilution and disk diffusion assays, may not detect heteroresistant subpopulations [134]. These tests typically measure the MIC for the bulk population, potentially overlooking small subpopulations that can survive higher drug concentrations. As a result, infections caused by heteroresistant strains may be mistakenly classified as susceptible, leading to inappropriate therapy.
For heteroresistance, the population analysis profile (PAP) assay is recommended as the golden standard [135]. PAP has been proved to efficiently test the heteroresistance to azoles and echinocandins in C. albicans, C. haemulonii, C. tropicalis, C. glabrata, C. parapsilosis, C. neoformans and S. cerevisiae, etc. [18]. Single-cell assays can also be used in heteroresistance rest. These include techniques such as flow cytometry, single-cell RNA sequencing (scRNA-seq), and microfluidics-based assays, allowing for the analysis of gene expression, cell viability, and drug tolerance in individual fungal cells [136,137]. Though the protocol is lengthier, it may reveal a wide spectrum of adaptation mechanisms [18]. And Time-kill assays, conventionally used to monitor the survival of fungal cells over time in the presence of an antifungal drug, can be used to detect heteroresistance, too. By measuring the rate of cell death at various time points, this method can identify delayed growth or survival of heteroresistant subpopulations that might not be detected by static MIC assays, and even evaluate the kinetics of heteroresistance and assess whether heteroresistant cells eventually adapt to drug pressure, which makes it widely applicable clinically [138,139,140]. In C. neoformans, it has been used to study azole and AmB heteroresistance, revealing that subpopulations of cells can survive for prolonged periods despite the presence of high drug concentrations [70]. These make time-kill assays a valuable tool in understanding heteroresistance dynamics.
8. Conclusions
Heteroresistance is a reversible form of antifungal resistance that fluctuates under varying conditions, primarily driven by genome aneuploidy and changes in gene expression. Unlike bacterial resistance, fungal heteroresistance remains largely underexplored, with most research focusing on azole resistance. A unique characteristic of heteroresistance is its inducibility—resistant phenotypes can emerge in response to antifungal exposure or other factors, leading to reduced drug efficacy over time.
Molecularly, aneuploidy and CNVs play a crucial role, influencing mechanisms such as gene loss, gene amplification, modifications of antifungal-binding sites, and upregulation of efflux pumps. These genetic changes enhance fungal adaptability to antifungal pressure. One point worth exploring is that CNVs can be stably inherited and may occur in both resistance and heteroresistance, which means it may contribute greatly to the evolution of antifungal resistance. Understanding the evolutionary and molecular mechanisms of heteroresistance is crucial for predicting and controlling clinical resistance.
Antifungal heteroresistance poses a significant clinical challenge, as it can develop during treatment, leading to therapeutic failure and recurrent infections. While escalating drug dosages or using combination therapies may sometimes be effective, these approaches come with limited options and increased toxicity risks. Therefore, the development of novel antifungal agents is critical, offering more therapeutic choices and combination possibilities. Additionally, gaining deeper insights into the specific mechanisms of heteroresistance could inform the development of innovative treatment strategies targeting multiple pathways, ultimately improving patient outcomes and reducing recurrence.
Author Contributions
Y.S., Y.L. (Yi Li) and Q.Y. devised the project and wrote the draft of the manuscript; T.S., Y.L. (Yingxing Li) and Y.X. edited the draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National High Level Hospital Clinical Research Funding [2022-PUMCH-C-052].
Data Availability Statement
This is a review article, and no new data were created or analyzed. All data discussed in this study are publicly available in the references cited.
Acknowledgments
We apologize to all colleagues whose work could not be cited in this Review owing to space constraints.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MIC | Minimum Inhibitory Concentration |
| CNVs | Copy Number Variations |
| AmB | Amphotericin B |
| 5-FC | 5-fluorocytosine |
| 5-FU | 5-fluorouracil |
| ROS | Reactive Oxygen Species |
| MDK99 | Minimum Duration of Killing 99% |
| SOD | Superoxide Dismutases |
| chr | Chromosome |
| GPI | Glycosylphosphatidylinositol |
| PAP | Population Analysis Profile |
| scRNA-seq | Single-Cell RNA Sequencing |
| SNP | Single Nucleotide Polyphorism |
| ABC | ATP-Binding Cassette |
References
- Spallone, A. and I.S. Schwartz, Emerging Fungal Infections. Infect Dis Clin North Am, 2021. 35(2): p. 261-277. [CrossRef]
- Lockhart, S.R. and J. Guarner, Emerging and reemerging fungal infections. Semin Diagn Pathol, 2019. 36(3): p. 177-181. [CrossRef]
- Fisher, M.C., et al., Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science, 2018. 360(6390): p. 739-742. [CrossRef]
- Alexander, H.E. and G. Leidy, Mode of action of streptomycin on type b Hemophilus influenzae: II. Nature of resistant variants. The Journal of experimental medicine, 1947. 85(6): p. 607. [CrossRef]
- Zheng, C., et al., Mixed Infections and Rifampin Heteroresistance among Mycobacterium tuberculosis Clinical Isolates. J Clin Microbiol, 2015. 53(7): p. 2138-47. [CrossRef]
- Liu, Q., et al., Within patient microevolution of Mycobacterium tuberculosis correlates with heterogeneous responses to treatment. Sci Rep, 2015. 5: p. 17507. [CrossRef]
- Andersson, D.I., H. Nicoloff, and K. Hjort, Mechanisms and clinical relevance of bacterial heteroresistance. Nat Rev Microbiol, 2019. 17(8): p. 479-496. [CrossRef]
- Kon, H., et al., Prevalence and Clinical Consequences of Colistin Heteroresistance and Evolution into Full Resistance in Carbapenem-Resistant Acinetobacter baumannii. Microbiol Spectr, 2023. 11(3): p. e0509322. [CrossRef]
- Xu, Y., et al., Mechanisms of Heteroresistance and Resistance to Imipenem in Pseudomonas aeruginosa. Infect Drug Resist, 2020. 13: p. 1419-1428. [CrossRef]
- Li, W.R., et al., Pseudomonas aeruginosa heteroresistance to levofloxacin caused by upregulated expression of essential genes for DNA replication and repair. Front Microbiol, 2022. 13: p. 1105921. [CrossRef]
- Cherkaoui, A., et al., Imipenem heteroresistance in nontypeable Haemophilus influenzae is linked to a combination of altered PBP3, slow drug influx and direct efflux regulation. Clin Microbiol Infect, 2017. 23(2): p. 118.e9-118.e19. [CrossRef]
- Adams-Sapper, S., A. Gayoso, and L.W. Riley, Stress-Adaptive Responses Associated with High-Level Carbapenem Resistance in KPC-Producing Klebsiella pneumoniae. J Pathog, 2018. 2018: p. 3028290. [CrossRef]
- Fukuzawa, S., et al., High prevalence of colistin heteroresistance in specific species and lineages of Enterobacter cloacae complex derived from human clinical specimens. Ann Clin Microbiol Antimicrob, 2023. 22(1): p. 60. [CrossRef]
- Xiong, Y., et al., Impact of Carbapenem Heteroresistance Among Multidrug-Resistant ESBL/AmpC-Producing Klebsiella pneumoniae Clinical Isolates on Antibiotic Treatment in Experimentally Infected Mice. Infect Drug Resist, 2021. 14: p. 5639-5650. [CrossRef]
- Kotilea, K., et al., Antibiotic resistance, heteroresistance, and eradication success of Helicobacter pylori infection in children. Helicobacter, 2023. 28(5): p. e13006. [CrossRef]
- Zhang, F., et al., Carbapenem-resistant K. pneumoniae exhibiting clinically undetected amikacin and meropenem heteroresistance leads to treatment failure in a murine model of infection. Microb Pathog, 2021. 160: p. 105162. [CrossRef]
- Kargarpour Kamakoli, M., et al., Evaluation of the impact of polyclonal infection and heteroresistance on treatment of tuberculosis patients. Sci Rep, 2017. 7: p. 41410. [CrossRef]
- Gautier, C., E.I. Maciel, and I.V. Ene, Approaches for identifying and measuring heteroresistance in azole-susceptible Candida isolates. Microbiol Spectr, 2024. 12(4): p. e0404123. [CrossRef]
- Zhai, B., et al., Antifungal heteroresistance causes prophylaxis failure and facilitates breakthrough Candida parapsilosis infections. Nat Med, 2024. [CrossRef]
- Ponde, N.O., et al., Candida albicans biofilms and polymicrobial interactions. Crit Rev Microbiol, 2021. 47(1): p. 91-111. [CrossRef]
- Dahiya, S., et al., Candida auris and Nosocomial Infection. Curr Drug Targets, 2020. 21(4): p. 365-373. [CrossRef]
- Geremia, N., et al., Candida auris as an Emergent Public Health Problem: A Current Update on European Outbreaks and Cases. Healthcare (Basel), 2023. 11(3). [CrossRef]
- Lyman, M., et al., Worsening Spread of Candida auris in the United States, 2019 to 2021. Ann Intern Med, 2023. 176(4): p. 489-495. [CrossRef]
- Bing, J., et al., Candida auris-associated hospitalizations and outbreaks, China, 2018-2023. Emerg Microbes Infect, 2024. 13(1): p. 2302843. [CrossRef]
- Devrim, İ., et al., Outcome of Candida Parapsilosis Complex Infections Treated with Caspofungin in Children. Mediterr J Hematol Infect Dis, 2016. 8(1): p. e2016042. [CrossRef]
- Chang, Y.C. and K.J. Kwon-Chung, Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol Cell Biol, 1994. 14(7): p. 4912-9. [CrossRef]
- Liu, O.W., et al., Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell, 2008. 135(1): p. 174-88. [CrossRef]
- Janbon, G., et al., Cas1p is a membrane protein necessary for the O-acetylation of the Cryptococcus neoformans capsular polysaccharide. Mol Microbiol, 2001. 42(2): p. 453-67. [CrossRef]
- Hamed, M.F., et al., Clinical and pathological characterization of Central Nervous System cryptococcosis in an experimental mouse model of stereotaxic intracerebral infection. PLoS Negl Trop Dis, 2023. 17(1): p. e0011068. [CrossRef]
- Imanishi-Shimizu, Y., et al., A capsule-associated gene of Cryptococcus neoformans, CAP64, is involved in pH homeostasis. Microbiology (Reading), 2021. 167(6). [CrossRef]
- Araújo, G.R.S., et al., Ultrastructural Study of Cryptococcus neoformans Surface During Budding Events. Front Microbiol, 2021. 12: p. 609244. [CrossRef]
- de Sousa, H.R., et al., Faster Cryptococcus Melanization Increases Virulence in Experimental and Human Cryptococcosis. J Fungi (Basel), 2022. 8(4). [CrossRef]
- Erives, V.H., et al., Methamphetamine Enhances Cryptococcus neoformans Melanization, Antifungal Resistance, and Pathogenesis in a Murine Model of Drug Administration and Systemic Infection. Infect Immun, 2022. 90(4): p. e0009122. [CrossRef]
- Baker, R.P. and A. Casadevall, Reciprocal modulation of ammonia and melanin production has implications for cryptococcal virulence. Nat Commun, 2023. 14(1): p. 849. [CrossRef]
- Evans, R.J., et al., Cryptococcal phospholipase B1 is required for intracellular proliferation and control of titan cell morphology during macrophage infection. Infect Immun, 2015. 83(4): p. 1296-304. [CrossRef]
- Hamed, M.F., et al., Phospholipase B Is Critical for Cryptococcus neoformans Survival in the Central Nervous System. mBio, 2023. 14(2): p. e0264022. [CrossRef]
- Santangelo, R., et al., Role of extracellular phospholipases and mononuclear phagocytes in dissemination of cryptococcosis in a murine model. Infect Immun, 2004. 72(4): p. 2229-39. [CrossRef]
- Goich, D., et al., Gcn2 rescues reprogramming in the absence of Hog1/p38 signaling in C. neoformans during thermal stress. bioRxiv, 2024. [CrossRef]
- Chadwick, B.J., et al., Discovery of CO(2) tolerance genes associated with virulence in the fungal pathogen Cryptococcus neoformans. Nat Microbiol, 2024. [CrossRef]
- van de Veerdonk, F.L., et al., Aspergillus fumigatus morphology and dynamic host interactions. Nat Rev Microbiol, 2017. 15(11): p. 661-674. [CrossRef]
- Odds, F.C., S.L. Cheesman, and A.B. Abbott, Suppression of ATP in Candida albicans by imidazole and derivative antifungal agents. Sabouraudia, 1985. 23(6): p. 415-24.
- Saag, M.S. and W.E. Dismukes, Azole antifungal agents: emphasis on new triazoles. Antimicrob Agents Chemother, 1988. 32(1): p. 1-8. [CrossRef]
- Patil, A. and S. Majumdar, Echinocandins in antifungal pharmacotherapy. J Pharm Pharmacol, 2017. 69(12): p. 1635-1660. [CrossRef]
- Mesa-Arango, A.C., L. Scorzoni, and O. Zaragoza, It only takes one to do many jobs: Amphotericin B as antifungal and immunomodulatory drug. Front Microbiol, 2012. 3: p. 286. [CrossRef]
- Shivarathri, R., et al., Comparative Transcriptomics Reveal Possible Mechanisms of Amphotericin B Resistance in Candida auris. Antimicrob Agents Chemother, 2022. 66(6): p. e0227621. [CrossRef]
- Perlin, D.S., R. Rautemaa-Richardson, and A. Alastruey-Izquierdo, The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect Dis, 2017. 17(12): p. e383-e392. [CrossRef]
- Polak, A. and H.J. Scholer, Mode of action of 5-fluorocytosine and mechanisms of resistance. Chemotherapy, 1975. 21(3-4): p. 113-30. [CrossRef]
- Gleason, M.K. and H. Fraenkel-Conrat, Biological consequences of incorporation of 5-fluorocytidine in the RNA of 5-fluorouracil-treated eukaryotic cells. Proc Natl Acad Sci U S A, 1976. 73(5): p. 1528-31. [CrossRef]
- Jacobs, S.E., et al., Candida auris Pan-Drug-Resistant to Four Classes of Antifungal Agents. Antimicrob Agents Chemother, 2022. 66(7): p. e0005322. [CrossRef]
- Oliveira, N.K., et al., Novel ABC Transporter Associated with Fluconazole Resistance in Aging of Cryptococcus neoformans. J Fungi (Basel), 2022. 8(7). [CrossRef]
- Rogers, T.R., et al., Molecular mechanisms of acquired antifungal drug resistance in principal fungal pathogens and EUCAST guidance for their laboratory detection and clinical implications. J Antimicrob Chemother, 2022. 77(8): p. 2053-2073. [CrossRef]
- Štefánek, M., et al., Comparative Analysis of Two Candida parapsilosis Isolates Originating from the Same Patient Harbouring the Y132F and R398I Mutations in the ERG11 Gene. Cells, 2023. 12(12). [CrossRef]
- Pristov, K.E. and M.A. Ghannoum, Resistance of Candida to azoles and echinocandins worldwide. Clin Microbiol Infect, 2019. 25(7): p. 792-798. [CrossRef]
- Fattouh, N., et al., Molecular mechanism of fluconazole resistance and pathogenicity attributes of Lebanese Candida albicans hospital isolates. Fungal Genet Biol, 2021. 153: p. 103575. [CrossRef]
- Bosco-Borgeat, M.E., et al., Amino acid substitution in Cryptococcus neoformans lanosterol 14-α-demethylase involved in fluconazole resistance in clinical isolates. Rev Argent Microbiol, 2016. 48(2): p. 137-42. [CrossRef]
- Diaz-Guerra, T.M., et al., A point mutation in the 14alpha-sterol demethylase gene cyp51A contributes to itraconazole resistance in Aspergillus fumigatus. Antimicrob Agents Chemother, 2003. 47(3): p. 1120-4. [CrossRef]
- Garcia-Effron, G., et al., A naturally occurring proline-to-alanine amino acid change in Fks1p in Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis accounts for reduced echinocandin susceptibility. Antimicrob Agents Chemother, 2008. 52(7): p. 2305-12. [CrossRef]
- Healey, K.R. and D.S. Perlin, Fungal Resistance to Echinocandins and the MDR Phenomenon in Candida glabrata. J Fungi (Basel), 2018. 4(3). [CrossRef]
- Yu, S.J., Y.L. Chang, and Y.L. Chen, Deletion of ADA2 Increases Antifungal Drug Susceptibility and Virulence in Candida glabrata. Antimicrob Agents Chemother, 2018. 62(3). [CrossRef]
- Singh-Babak, S.D., et al., Global analysis of the evolution and mechanism of echinocandin resistance in Candida glabrata. PLoS Pathog, 2012. 8(5): p. e1002718. [CrossRef]
- Delma, F.Z., et al., Molecular Mechanisms of 5-Fluorocytosine Resistance in Yeasts and Filamentous Fungi. J Fungi (Basel), 2021. 7(11). [CrossRef]
- Kannan, A., et al., Comparative Genomics for the Elucidation of Multidrug Resistance in Candida lusitaniae. mBio, 2019. 10(6). [CrossRef]
- Billmyre, R.B., et al., 5-fluorocytosine resistance is associated with hypermutation and alterations in capsule biosynthesis in Cryptococcus. Nat Commun, 2020. 11(1): p. 127. [CrossRef]
- Silva, L.N., et al., Unmasking the Amphotericin B Resistance Mechanisms in Candida haemulonii Species Complex. ACS Infect Dis, 2020. 6(5): p. 1273-1282. [CrossRef]
- Schatzman, S.S., et al., Copper-only superoxide dismutase enzymes and iron starvation stress in Candida fungal pathogens. J Biol Chem, 2020. 295(2): p. 570-583. [CrossRef]
- Huang, Y., et al., Insight into Virulence and Mechanisms of Amphotericin B Resistance in the Candida haemulonii Complex. J Fungi (Basel), 2024. 10(9). [CrossRef]
- Ben-Ami, R., et al., Heteroresistance to Fluconazole Is a Continuously Distributed Phenotype among Candida glabrata Clinical Strains Associated with In Vivo Persistence. mBio, 2016. 7(4). [CrossRef]
- Levy, S.F., N. Ziv, and M.L. Siegal, Bet hedging in yeast by heterogeneous, age-correlated expression of a stress protectant. PLoS Biol, 2012. 10(5): p. e1001325. [CrossRef]
- Claudino, A.L., et al., Mutants with heteroresistance to amphotericin B and fluconazole in Candida. Braz J Microbiol, 2009. 40(4): p. 943-51. [CrossRef]
- Bosch, C., et al., Nitrogen concentration affects amphotericin B and fluconazole tolerance of pathogenic cryptococci. FEMS Yeast Res, 2020. 20(2). [CrossRef]
- Ferreira, G.F., et al., Heteroresistance to Itraconazole Alters the Morphology and Increases the Virulence of Cryptococcus gattii. Antimicrob Agents Chemother, 2015. 59(8): p. 4600-9. [CrossRef]
- Sionov, E., et al., Heteroresistance to fluconazole in Cryptococcus neoformans is intrinsic and associated with virulence. Antimicrob Agents Chemother, 2009. 53(7): p. 2804-15. [CrossRef]
- Balaban, N.Q., et al., Publisher Correction: Definitions and guidelines for research on antibiotic persistence. Nat Rev Microbiol, 2019. 17(7): p. 460. [CrossRef]
- Kester, J.C. and S.M. Fortune, Persisters and beyond: mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit Rev Biochem Mol Biol, 2014. 49(2): p. 91-101. [CrossRef]
- Berman, J. and D.J. Krysan, Drug resistance and tolerance in fungi. Nat Rev Microbiol, 2020. 18(6): p. 319-331. [CrossRef]
- Chen, L., et al., Confronting antifungal resistance, tolerance, and persistence: Advances in drug target discovery and delivery systems. Adv Drug Deliv Rev, 2023. 200: p. 115007. [CrossRef]
- Lyons, N. and J. Berman, Protocols for Measuring Tolerant and Heteroresistant Drug Responses of Pathogenic Yeasts. Methods Mol Biol, 2023. 2658: p. 67-79. [CrossRef]
- Rasouli Koohi, S., et al., Identification and Elimination of Antifungal Tolerance in Candida auris. Biomedicines, 2023. 11(3). [CrossRef]
- Rosenberg, A., et al., Antifungal tolerance is a subpopulation effect distinct from resistance and is associated with persistent candidemia. Nat Commun, 2018. 9(1): p. 2470. [CrossRef]
- Moldoveanu, A.L., J.A. Rycroft, and S. Helaine, Impact of bacterial persisters on their host. Curr Opin Microbiol, 2021. 59: p. 65-71. [CrossRef]
- Arastehfar, A., et al., Macrophage internalization creates a multidrug-tolerant fungal persister reservoir and facilitates the emergence of drug resistance. Nat Commun, 2023. 14(1): p. 1183. [CrossRef]
- Lewis, K., Persister cells. Annu Rev Microbiol, 2010. 64: p. 357-72. [CrossRef]
- Ke, W., et al., Fungicide-tolerant persister formation during cryptococcal pulmonary infection. Cell Host Microbe, 2024. 32(2): p. 276-289.e7. [CrossRef]
- Yang, F., et al., Antifungal Tolerance and Resistance Emerge at Distinct Drug Concentrations and Rely upon Different Aneuploid Chromosomes. mBio, 2023. 14(2): p. e0022723. [CrossRef]
- Yang, F., et al., Tunicamycin Potentiates Antifungal Drug Tolerance via Aneuploidy in Candida albicans. mBio, 2021. 12(4): p. e0227221. [CrossRef]
- Chen, L., et al., Brain glucose induces tolerance of Cryptococcus neoformans to amphotericin B during meningitis. Nat Microbiol, 2024. 9(2): p. 346-358. [CrossRef]
- Yoo, K., et al., With age comes resilience: how mitochondrial modulation drives age-associated fluconazole tolerance in Cryptococcus neoformans. mBio, 2024. 15(9): p. e0184724. [CrossRef]
- Yang, F., et al., Tolerance to Caspofungin in Candida albicans Is Associated with at Least Three Distinctive Mechanisms That Govern Expression of FKS Genes and Cell Wall Remodeling. Antimicrob Agents Chemother, 2017. 61(5). [CrossRef]
- Prasetyoputri, A., et al., The Eagle Effect and Antibiotic-Induced Persistence: Two Sides of the Same Coin? Trends Microbiol, 2019. 27(4): p. 339-354. [CrossRef]
- Singh, S.D., et al., Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog, 2009. 5(7): p. e1000532. [CrossRef]
- Iyer, K.R., N. Robbins, and L.E. Cowen, The role of Candida albicans stress response pathways in antifungal tolerance and resistance. iScience, 2022. 25(3): p. 103953. [CrossRef]
- Dumeaux, V., et al., Candida albicans exhibits heterogeneous and adaptive cytoprotective responses to antifungal compounds. Elife, 2023. 12. [CrossRef]
- Defraine, V., M. Fauvart, and J. Michiels, Fighting bacterial persistence: Current and emerging anti-persister strategies and therapeutics. Drug Resist Updat, 2018. 38: p. 12-26. [CrossRef]
- Lewis, K., Persister cells, dormancy and infectious disease. Nat Rev Microbiol, 2007. 5(1): p. 48-56. [CrossRef]
- da Silva, M.A., et al., Oxidative Imbalance in Candida tropicalis Biofilms and Its Relation With Persister Cells. Front Microbiol, 2020. 11: p. 598834. [CrossRef]
- Dasilva, M.A., et al., Synergistic activity of gold nanoparticles with amphotericin B on persister cells of Candida tropicalis biofilms. J Nanobiotechnology, 2024. 22(1): p. 254. [CrossRef]
- El Meouche, I., et al., Drug tolerance and persistence in bacteria, fungi and cancer cells: Role of non-genetic heterogeneity. Transl Oncol, 2024. 49: p. 102069. [CrossRef]
- Zou, P., et al., Antifungal Activity, Synergism with Fluconazole or Amphotericin B and Potential Mechanism of Direct Current against Candida albicans Biofilms and Persisters. Antibiotics (Basel), 2024. 13(6). [CrossRef]
- Ning, Y., et al., Copy number variants of ERG11: mechanism of azole resistance in Candida parapsilosis. Lancet Microbe, 2024. 5(1): p. e10. [CrossRef]
- Fan, X., et al., Tandem gene duplications contributed to high-level azole resistance in a rapidly expanding Candida tropicalis population. Nat Commun, 2023. 14(1): p. 8369. [CrossRef]
- Tsai, H.J. and A. Nelliat, A Double-Edged Sword: Aneuploidy is a Prevalent Strategy in Fungal Adaptation. Genes (Basel), 2019. 10(10). [CrossRef]
- Vande Zande, P., X. Zhou, and A. Selmecki, The Dynamic Fungal Genome: Polyploidy, Aneuploidy and Copy Number Variation in Response to Stress. Annu Rev Microbiol, 2023. 77: p. 341-361. [CrossRef]
- Yang, F., et al., Adaptation to Fluconazole via Aneuploidy Enables Cross-Adaptation to Amphotericin B and Flucytosine in Cryptococcus neoformans. Microbiol Spectr, 2021. 9(2): p. e0072321. [CrossRef]
- Selmecki, A., A. Forche, and J. Berman, Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science, 2006. 313(5785): p. 367-70. [CrossRef]
- Gohar, A.A., et al., Expression Patterns of ABC Transporter Genes in Fluconazole-Resistant Candida glabrata. Mycopathologia, 2017. 182(3-4): p. 273-284. [CrossRef]
- Zhang, Z.H., et al., Aneuploidy underlies brefeldin A-induced antifungal drug resistance in Cryptococcus neoformans. Front Cell Infect Microbiol, 2024. 14: p. 1397724. [CrossRef]
- Marr, K.A., et al., Rapid, transient fluconazole resistance in Candida albicans is associated with increased mRNA levels of CDR. Antimicrob Agents Chemother, 1998. 42(10): p. 2584-9. [CrossRef]
- Beach, R.R., et al., Aneuploidy Causes Non-genetic Individuality. Cell, 2017. 169(2): p. 229-242.e21. [CrossRef]
- Todd, R.T. and A. Selmecki, Expandable and reversible copy number amplification drives rapid adaptation to antifungal drugs. Elife, 2020. 9. [CrossRef]
- Sah, S.K., J.J. Hayes, and E. Rustchenko, The role of aneuploidy in the emergence of echinocandin resistance in human fungal pathogen Candida albicans. PLoS Pathog, 2021. 17(5): p. e1009564. [CrossRef]
- Coste, A.T., et al., TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell, 2004. 3(6): p. 1639-52. [CrossRef]
- Harrison, B.D., et al., A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol, 2014. 12(3): p. e1001815. [CrossRef]
- Yang, F., et al., Aneuploidy Enables Cross-Adaptation to Unrelated Drugs. Mol Biol Evol, 2019. 36(8): p. 1768-1782. [CrossRef]
- Husain, F., et al., Candida albicans Strains Adapted to Caspofungin Due to Aneuploidy Become Highly Tolerant under Continued Drug Pressure. Microorganisms, 2022. 11(1). [CrossRef]
- Zheng, Q., et al., Ploidy Variation and Spontaneous Haploid-Diploid Switching of Candida glabrata Clinical Isolates. mSphere, 2022. 7(4): p. e0026022. [CrossRef]
- Ksiezopolska, E., et al., Long-term stability of acquired drug resistance and resistance associated mutations in the fungal pathogen Nakaseomyces glabratus (Candida glabrata). Front Cell Infect Microbiol, 2024. 14: p. 1416509. [CrossRef]
- Burrack, L.S., et al., Genomic Diversity across Candida auris Clinical Isolates Shapes Rapid Development of Antifungal Resistance In Vitro and In Vivo. mBio, 2022. 13(4): p. e0084222. [CrossRef]
- Anderson, J.B., C. Sirjusingh, and N. Ricker, Haploidy, diploidy and evolution of antifungal drug resistance in Saccharomyces cerevisiae. Genetics, 2004. 168(4): p. 1915-23. [CrossRef]
- Moreira, I.M.B., et al., Investigation of fluconazole heteroresistance in clinical and environmental isolates of Cryptococcus neoformans complex and Cryptococcus gattii complex in the state of Amazonas, Brazil. Med Mycol, 2022. 60(3). [CrossRef]
- Yamazumi, T., et al., Characterization of heteroresistance to fluconazole among clinical isolates of Cryptococcus neoformans. J Clin Microbiol, 2003. 41(1): p. 267-72. [CrossRef]
- Varma, A. and K.J. Kwon-Chung, Heteroresistance of Cryptococcus gattii to fluconazole. Antimicrob Agents Chemother, 2010. 54(6): p. 2303-11. [CrossRef]
- Altamirano, S., C. Simmons, and L. Kozubowski, Colony and Single Cell Level Analysis of the Heterogeneous Response of Cryptococcus neoformans to Fluconazole. Front Cell Infect Microbiol, 2018. 8: p. 203. [CrossRef]
- Cao, C., et al., Ubiquitin proteolysis of a CDK-related kinase regulates titan cell formation and virulence in the fungal pathogen Cryptococcus neoformans. Nat Commun, 2022. 13(1): p. 6397. [CrossRef]
- Gerstein, A.C., et al., Polyploid titan cells produce haploid and aneuploid progeny to promote stress adaptation. mBio, 2015. 6(5): p. e01340-15. [CrossRef]
- García-Barbazán, I., et al., Accumulation of endogenous free radicals is required to induce titan-like cell formation in Cryptococcus neoformans. mBio, 2024. 15(1): p. e0254923. [CrossRef]
- Stone, N.R., et al., Dynamic ploidy changes drive fluconazole resistance in human cryptococcal meningitis. J Clin Invest, 2019. 129(3): p. 999-1014. [CrossRef]
- Sionov, E., Y.C. Chang, and K.J. Kwon-Chung, Azole heteroresistance in Cryptococcus neoformans: emergence of resistant clones with chromosomal disomy in the mouse brain during fluconazole treatment. Antimicrob Agents Chemother, 2013. 57(10): p. 5127-30. [CrossRef]
- Ngamskulrungroj, P., et al., Characterization of the chromosome 4 genes that affect fluconazole-induced disomy formation in Cryptococcus neoformans. PLoS One, 2012. 7(3): p. e33022. [CrossRef]
- Sionov, E., et al., Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog, 2010. 6(4): p. e1000848. [CrossRef]
- Altamirano, S., et al., Fluconazole-Induced Ploidy Change in Cryptococcus neoformans Results from the Uncoupling of Cell Growth and Nuclear Division. mSphere, 2017. 2(3). [CrossRef]
- Escribano, P., et al., In vitro acquisition of secondary azole resistance in Aspergillus fumigatus isolates after prolonged exposure to itraconazole: presence of heteroresistant populations. Antimicrob Agents Chemother, 2012. 56(1): p. 174-8. [CrossRef]
- Chen, Y., et al., Lesion Heterogeneity and Long-Term Heteroresistance in Multidrug-Resistant Tuberculosis. J Infect Dis, 2021. 224(5): p. 889-893. [CrossRef]
- Hope, W., et al., Fluconazole Monotherapy Is a Suboptimal Option for Initial Treatment of Cryptococcal Meningitis Because of Emergence of Resistance. mBio, 2019. 10(6). [CrossRef]
- Banerjee, T., et al., Comparative evaluation of colistin broth disc elution (CBDE) and broth microdilution (BMD) in clinical isolates of Pseudomonas aeruginosa with special reference to heteroresistance. Indian J Med Microbiol, 2024. 47: p. 100494. [CrossRef]
- El-Halfawy, O.M. and M.A. Valvano, Antimicrobial heteroresistance: an emerging field in need of clarity. Clin Microbiol Rev, 2015. 28(1): p. 191-207. [CrossRef]
- Zhu, Y., et al., Single-Cell Phenotypic Analysis and Digital Molecular Detection Linkable by a Hydrogel Bead-Based Platform. ACS Appl Bio Mater, 2021. 4(3): p. 2664-2674. [CrossRef]
- Dai, Y., et al., Plasmonic Colloidosome-Coupled MALDI-TOF MS for Bacterial Heteroresistance Study at Single-Cell Level. Anal Chem, 2020. 92(12): p. 8051-8057. [CrossRef]
- Mezcord, V., et al., Induced Heteroresistance in Carbapenem-Resistant Acinetobacter baumannii (CRAB) via Exposure to Human Pleural Fluid (HPF) and Its Impact on Cefiderocol Susceptibility. Int J Mol Sci, 2023. 24(14). [CrossRef]
- Wang, Y., et al., Heteroresistance Is Associated With in vitro Regrowth During Colistin Treatment in Carbapenem-Resistant Klebsiella pneumoniae. Front Microbiol, 2022. 13: p. 868991. [CrossRef]
- Tian, Y., et al., Combined effect of Polymyxin B and Tigecycline to overcome Heteroresistance in Carbapenem-Resistant Klebsiella pneumoniae. Microbiol Spectr, 2021. 9(2): p. e0015221. [CrossRef]
Figure 1.
(By Figdraw). Explanation of resistance, susceptible, tolerance, persistence and heteroresistance from a cell population perspective. The state of cell proliferation is represented by the number of cells in the figure. Assorted colors indicate different genotypes and phenotypes, which are orange (genetically stable resistance), blue (susceptible), purple (genetically unstable or phenotypically resistance), green (phenotypically tolerant).
Figure 1.
(By Figdraw). Explanation of resistance, susceptible, tolerance, persistence and heteroresistance from a cell population perspective. The state of cell proliferation is represented by the number of cells in the figure. Assorted colors indicate different genotypes and phenotypes, which are orange (genetically stable resistance), blue (susceptible), purple (genetically unstable or phenotypically resistance), green (phenotypically tolerant).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



