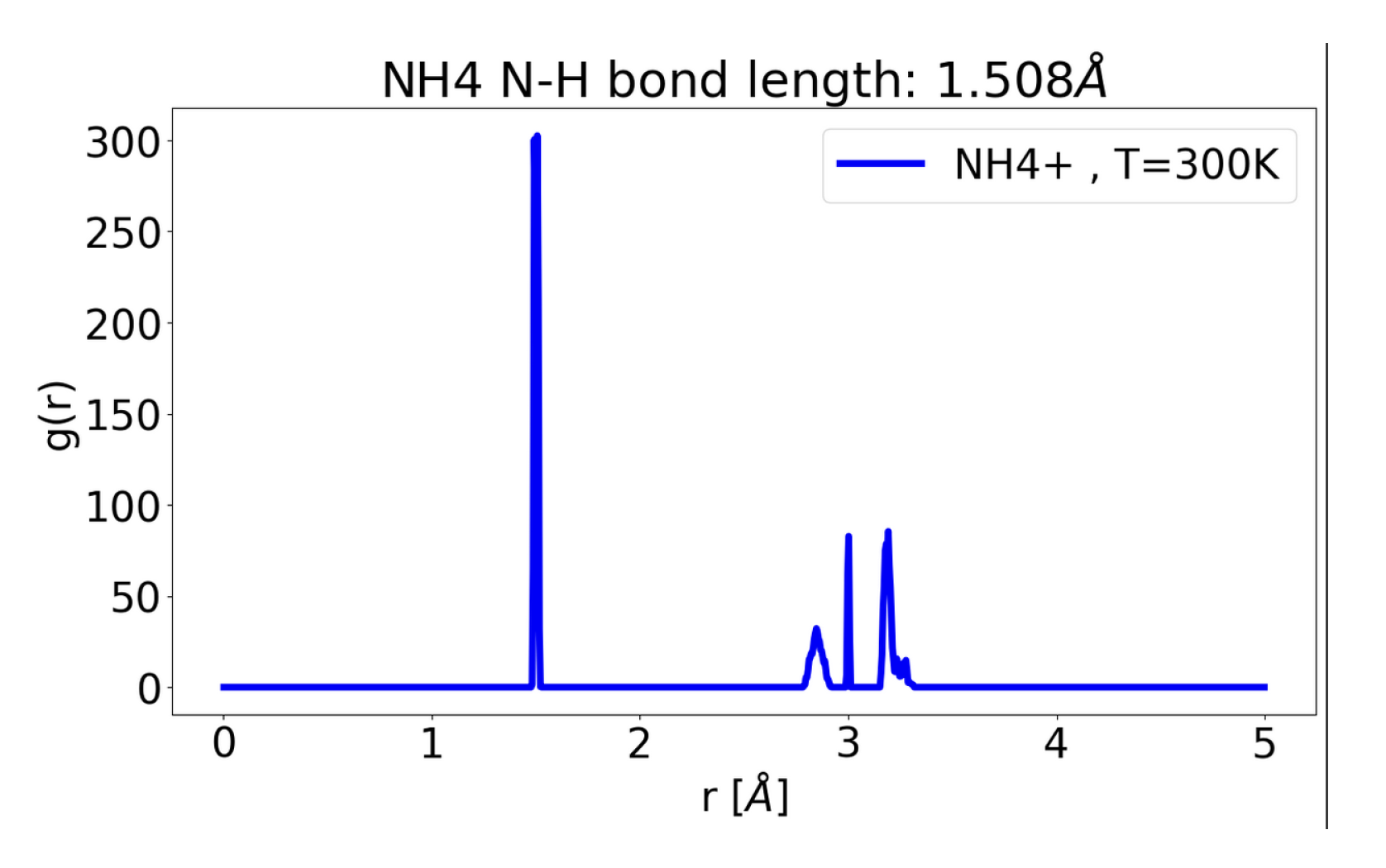
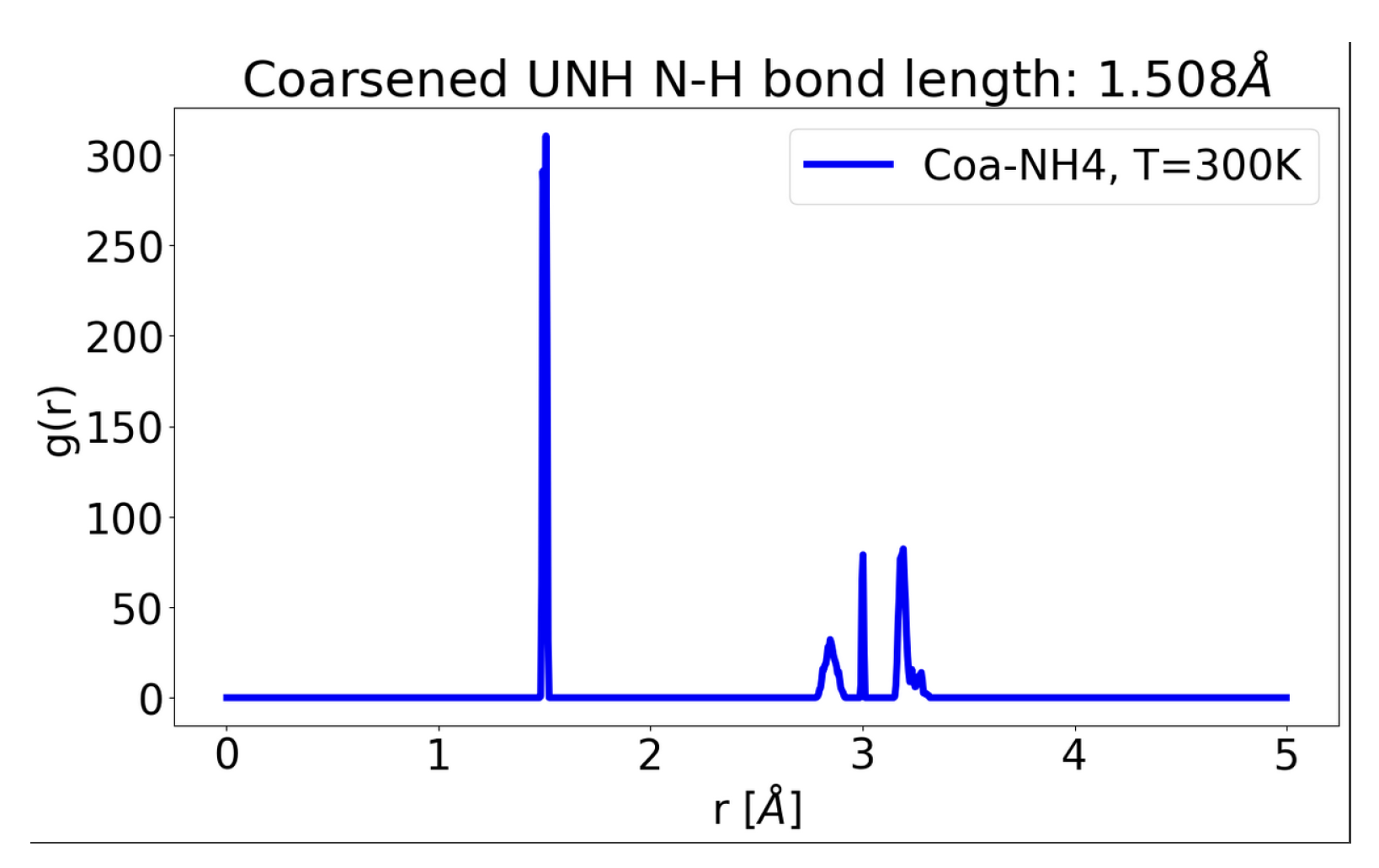
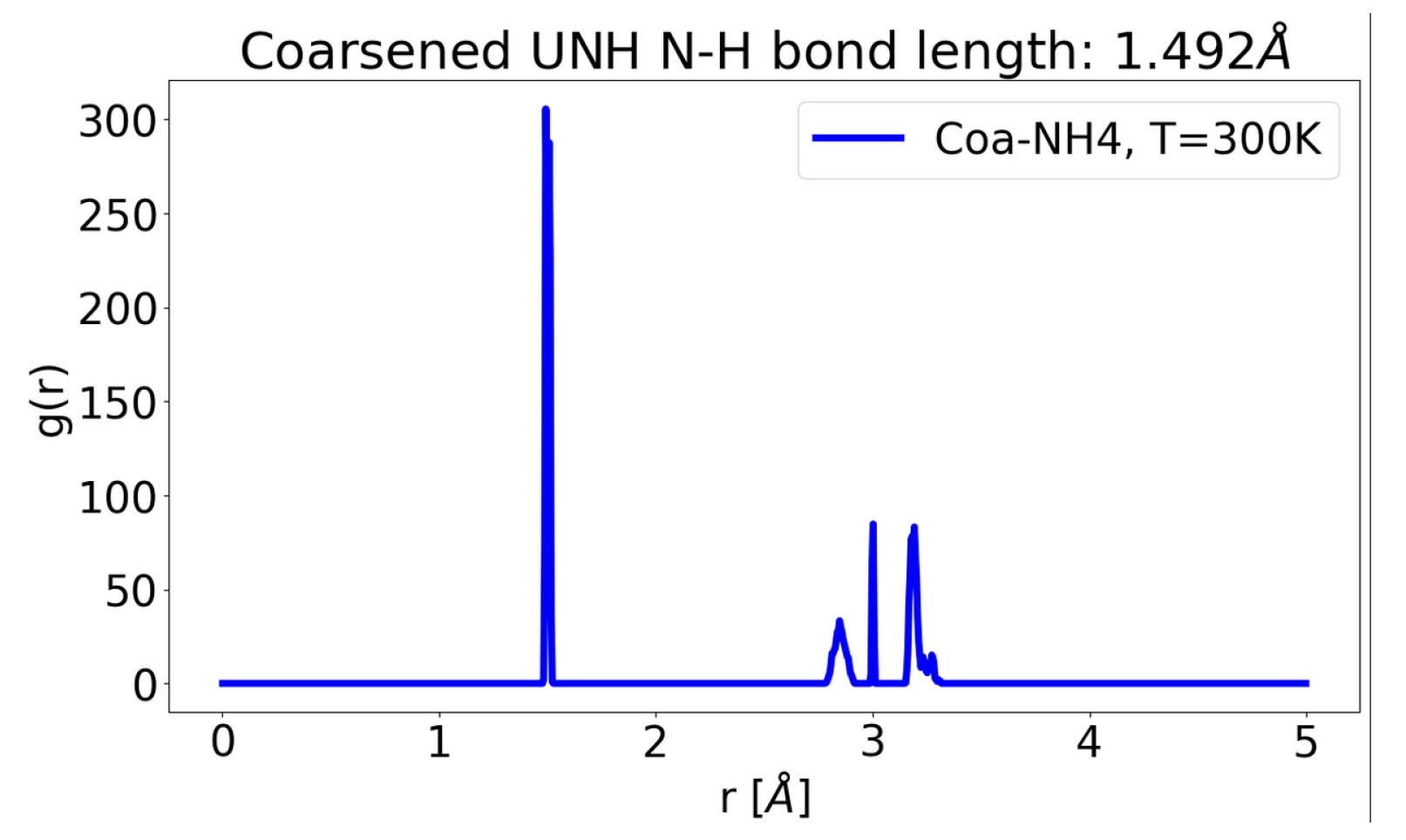
1. Quick Introduction to Pseudo-Vectors i.e Bra and Ket (Anti-Symmetric Inner Products), for GNN’s Equi-Variance Property
Quantum mechanics using Matrix, is called Matrix Mechanics, covered in great detail in almost all Quantum Mechanics text books such as Sakurai et al. [
17]. We are only going to briefly cover, relevant multi-electron spherical tensor interactions occuring at quantum level, to implement/modify/experiment Equivariant GNNs, such as Allegro, Nequip, E3NN, which deploy geometric tensors, deploying convolution filters, filtering interactions by spherical harmonics [
15,
16].
In euclidean space dot products commute, i.e. , however in quantum space represented as complex valued hilbert space , inner product , where * indicates complex conjugate (just a mathematical trick to express inversion (reflection) across real axis), and A, B are complex vectors of n dimensions, this dot product can also be represented using hermitian matrix (a complex extension of real valued symmetric matrix) as as i.e. complex square matrix is equal to its conjugate , transpose, producing real valued eigenvalues, representing position, momentum, spin, angular momentum or energy on a spectrum of Hermitian. Eigenvectors of Hermitian corresponds to quantum states associated with each measured value.
This dot product representation using complex conjugate of Hermitian, with elements of matrix in complex conjugate form (reflection across real axis), is also known as symmetric sesquilinear form (Hermitian form). We need to incorporate right orientation in order to get correct aggregation of angular momentum of various electrons in multi-body receptive field while using GNNs so that we can preserve equivariance, while performing aggregation of forces, momentums coming from various particles residing in given 3 D receptive field, characterized as sphere of given cutoff-radius.
In order to get better predictions about energy/forcefields triggering interatomic interactions, Density field Theory (DFT) approximates N electrons in a region as N-electron Hamiltonian, simplifying it later as Hohenberg-Kohn model of N electrons, using their spatial (x,y,z) co-ordinates, using Born-Oppenheimer approximation of total energy as sum of kinetic, rotaional, vibrartional and nuclear-spin energies as:
under positive force of nuclei.
Kinetic energy
is invariant under rotation in x,y,z frame, Coulomb potentials (
are also invariant, assuming clamped nucleus Hamiltonian (nucleus becomes a fixed particle in classical mechanics sense), only hinderance towards implementing GNNs arises in modeling electron electron interactions (
), as
can be ignored as nuclei are not getting so close in MD simulations unless material is approaching plasma state, if that is what we need to model, then we need to also add Yukawa potential (
(proton, neutron interaction)), as we need to be aware of particles interacting...from Pauli’s exclusion principle, stating wavefunction must change sign when we switch order of Fermions (electron, protons etc.. we only care about electrons) involved:
where
represents fermion’s (electron) position and spin. When Bosons are involved, we get
A combined N particle wavefunction is thus represented using Slater determinants, as we can’t separate out contributions from each particle as separate wave function, due to spatial and spin component’s contributions on each particle’s spatial/spin terms:
Bra (row vector) is multiplied to Ket (column vector) representing projection operator matrix showing projection of quantum state represented by Bra vector on quantum state represented by Ket vector .
As one can see, last exterior product involving linearly independent
equating to Slater determinant as bivectors (
[
19], a plane with directional rotation, operating on other vectors (
) carrying, angular rotational information forward, at each step of exterior product computation. This exterior product, is also called Plucker embedding on Grassmannian manifold [
4]. This brings us to reason for using GNNs, as GNNs map nodes to some embedding space by learning mapping functions.
This is all well and good but how will we compute this value?
Here we need to bring separation of variables (spherical co-ordinates r, , ) and angular momentum’s projections on unit sphere (thus taming 3rd degree of freedom r as that can be linarly multiplied once we have potential energy surface value for unit sphere as ), into consideration. This projection is represented by a vector on the sphere’s surface. Vector’s length corresponds to the magnitude of the angular momentum projection along the axis defined by the vector’s direction. Direction of the vector indicates the specific axis on the sphere along which the angular momentum is projected. At Quantum scale, this projection is quantized, meaning it can only take on discrete values depending on the angular momentum quantum number (l) and its magnetic quantum number (m). In quantum mechanics, the z-component of angular momentum () is often used as the projection, and its possible values are quantized as , where m is the magnetic quantum number ranging from -ℓ to +ℓ. We also have 2 spins, spin up and spin down.
This also explains why we will need to deploy tensors to accurately represent angular momentum of various electrons (adding individual orbital and angular momentum components) of multiple electrons interacting/orbiting around nucleus...
, where m is magnetic quantum number (describing spatial orientation of given orbital),
ℓ angular momentum quantum number (describes shape of electron orbital,
ℓ = 0 s orbital,
ℓ = 1 p orbital,
ℓ = 2 d orbital etc..
ℓ can take values from 0 to n-1, where n is principal quantum number (i.e. how far electron is from nucleus)), and L is total angular momentum vector, as we will also need direction of spin, component so that projection of angular momentum on z axis, are properly computed as electron states have to follow Pauli’s exclusion principle (i.e no two electrons in atom can have same values for n (distance from nucleus),
ℓ shape of orbital, m spatial orientation and s spin [+1/2, -1/2]).
As we are modeling molecular dynamics, molecules are moving with time (translation) and these atoms are not fixed in structurally in space, molecules are rotating, inverting in 3D space. Even though we have x,y,z co-ordinates, we do not want to compute spherical versions using r, , at every instant to figure out translation, rotation and inversion status of each atom to compute what really has changed in terms of actual or forcefield or momentum etc... with respect to, our point of reference of spherical receptive field.
This is where Lie Algebra (handling sequence of translation, rotation and inversion from hilbert space (allowing superposition of multiple basis states as linear combination to represent a quantum state using Linear Algebra terms such as eigenstates, infinte dimension to allow particle to move with time as dimension) to so that we can compare data with our LAMMPS values, use nearest-neighbors pairs etc.., representation theory (separating out similar representation of rotation as identical, i.e irrep) and special functions (spherical harmonics) come to rescue.
This can all only begin by understanding projection of group (Special Unitary group SU(2) on Special group of all rotations about origin SO(3).
Special here means that when we have matrix representation of mapping, determinant of the matrix is +1 (rotation) as -1 will represent reflection or loss of invariance for GNN and we will need to bring equivariance in to GNN. Groups can describe how individual actions (rotation, inversion, translation as time moves) taken together transform a molecule. Lie groups are mathematical tools representing continuous quantities as elements (i.e. there is no sudden breaks in rotational representation as molecule rotates etc..). These rotations, translation and inversion when done using irreducible representations (i.e. just like basis vectors, there is no subrepresentation for a given action exists), follow group laws of closure under addition etc. See general linear group of order 2 describing rotation matrix in 2D, using Euler’s identity
as :
Considering rotations in complex plane SO(2) is isomorphic to U(1):
Using Shur’s lemma: if group is commutative then these irreducible representations are one dimensional, in Complex space We know all imaginary terms cancel out as we do projections from Hilbert space to x,y,z i.e. as at any time t, we will always have real valued position, potential energy, momentum terms. can be exchanged with time t, this giving us full, time t dependent representaiton, of all rotations in 2 D space, as one matrix element.
Tangent space of Lie Groups (group representing rotations) at identity brings structures about multiplications, called Lie Algebra. These operations follow following rules (similar to cross product and Lie Bracket):
Where
is an ideal [
24]. Since SU(2) and SO(3) both are Perfect Lee algebra (i.e. commutator:
, a measure of commutativity, when taken for all elements producing derived algebra is equal to original algebra), we have
[
24] and
, i.e.
is anti-symmetric tensor product.
Modeling SO(3), Moving frame of reference, with time t, along with rotation along z axis:
In
, one can represent molecular dynamics by using translations, rotations along x,y,z and inversions with moving frame of reference as combinations of transformations such as for rotation we can combine above 3 transformations in successive order[
18].
Changing x,y,z to spherical co-ordinates using unit sphere r=1, we get x=
, y =
and z =
. Rotation group SO(3) will have no effect on r, thus we can represent a function
which can represent any point, on unit sphere given parametrs
and
, this also means any rotation SO(3) can also be represented by
as
work as basis vectors for entire
Hilbert space. Solving laplacian
, produces this well known solution [
17] of
[
25,
26]. This shows how entire set of rotations, translations and inversions can be represented by spherical harmonics function parameterized by
ℓ, m wrt to time t, by deploying consecutive translatios, rotations, inversions.
where:
This representation
is
dimensional irrep. (irreducible representation) of SO(3). Laplace-Beltrami operator
on
in Hilbert space acts similar to Laplace operator in Euclidean space. This means Casimir operator
, not representing L square, just a symbol for angular momentum ∈
[
25], for representation of SO(3) on unit sphere
is the 2nd order differential operator of form[
25]:
Multi-particle interaction involving angular momentum:
Wigner-Eckart Theorm [
10] states:
A tensor operator
and two states of angular momenta
ℓ and
(for electron, spin is 1/2, which may align with orbital angular momentum I (s orbital, p orbital etc..) or not), there exists a constant (Clebsch Gordon coeff.) that for all (projections of angular momenta (
ℓ,
) along z axis), m,m’ and q, following holds:
Here
is not a matrix, this is called Wigner-3j [
11] representation, showing interaction of angular momentum of electron in electron cloud of shape,
ℓ, and its spatial location m, with another electron located in electron cloud of shape k, and spatial. location q, resulting in angular momentum (angular and spin) projected on to electron cloud of shape
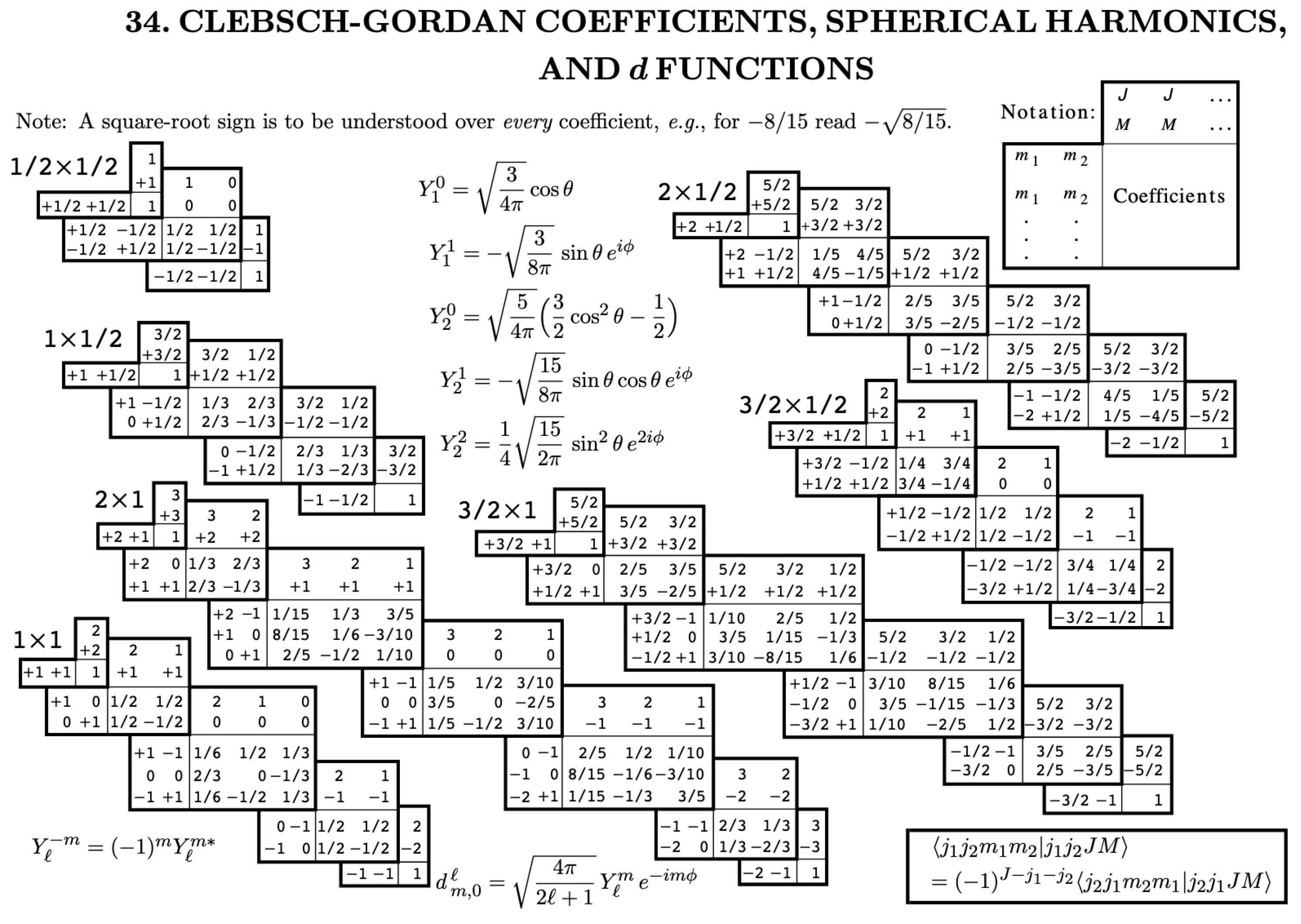
and location m’, as we sum these angular and spin part of angular momentum, separately. Clebsch-Gordon coefficients can be looked up from standard tables [
12].
needs to be computed only once for entire set of m,m’,q values ranging from
,
and
, saving us lot of repeated computation for multiple spatial locations represented by magnetic quantum numbers m,q,m’.
Figure 1.
Clebsch Gordon Coefficients, Imgsrc : Particle Data Group K. Hagiwara et al.. [
12]
Figure 1.
Clebsch Gordon Coefficients, Imgsrc : Particle Data Group K. Hagiwara et al.. [
12]
Representation of Rotation using Spherical Harmonics Basis
Here
is spherical tensor operator, qth element rank k (k dimensional). Since we want to represent these molecule’s angular momentums in 3D space (
or
), where atoms can be in any direction and moving, we need to have a single irrep representation, for various permutations [
23] of rotations, inversions and translations filtering out equivalnet permutations (i.e. irrep., irreducible representation). This is direct in our case as Spherical Harmonics form a basis for irrep. of the rotation group, allowing us to liearly transform functions as combination of spherical harmonics bases
.
This is where
Wigner-d matrices [
20]and d-functions come into picture..
Just like matrices of trignometric function in
, are representing rotations around x,y,z axis (shown above), which can be combined to represent motion using Lie Algebra (SO(3) group) rules, spherical harmonics rotations,
are governed by Wigner-D matrix [
21], defined as:
where
Wigner-(small)d-matrix is again defined as diagonal matrix, using basis as
:
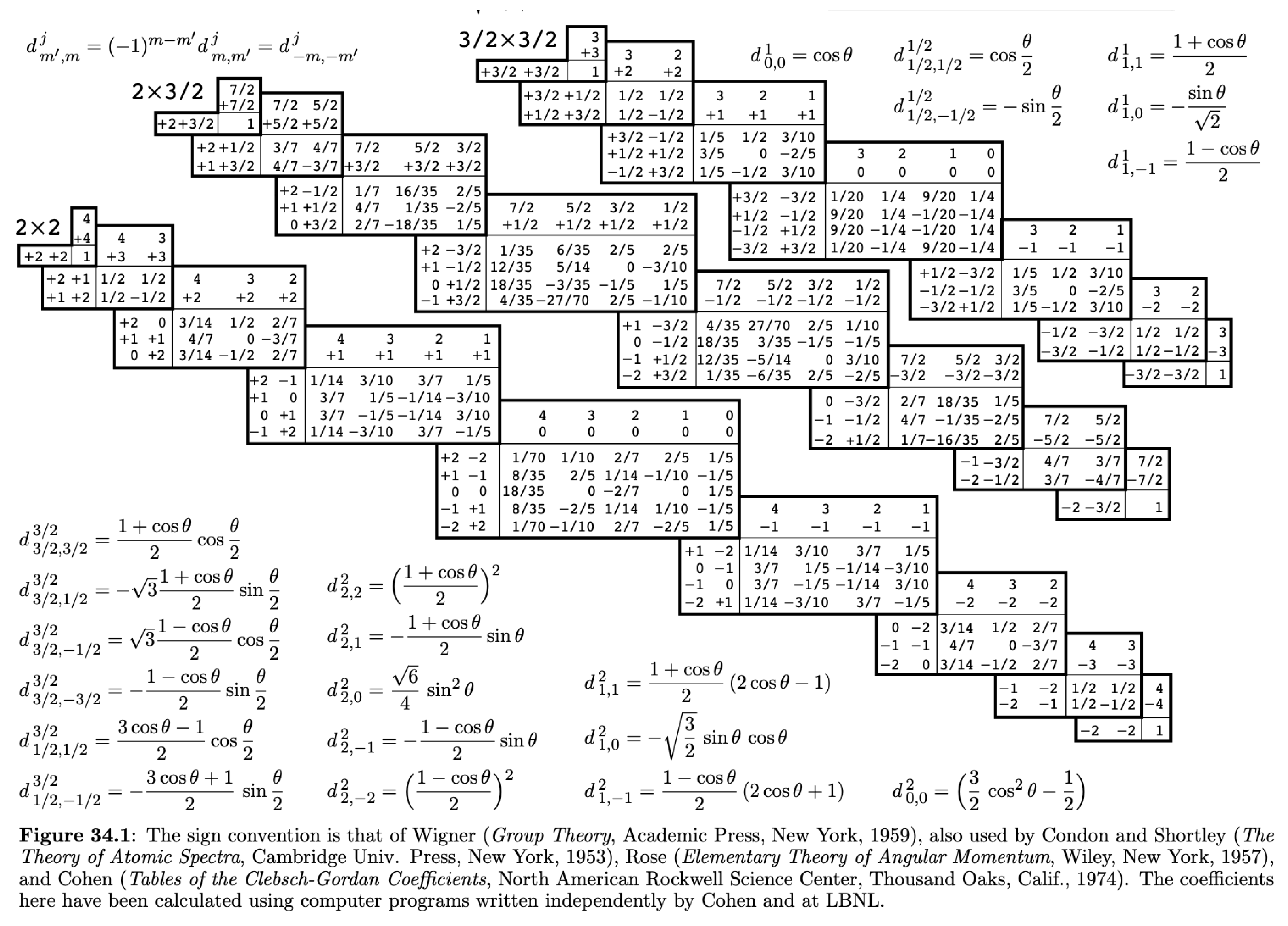
These Wigner-D matrices can also be combined like multiplication in Lie Algebra (matrix Lie Algebra SO(3) group), * above indicates that we should use complex-conjugate of an element from Wigner-D Matrix. Just like Clebsch Gordon coefficients table, we have table of d-Function at lower half of same page defined, requiring us to get these rotations by performing simple summation for
[
12].
Figure 2.
d-Functions to compute Wigner-d, Wigner-D, Imgsrc : Particle Data Group K. Hagiwara et al.. [
12]
Figure 2.
d-Functions to compute Wigner-d, Wigner-D, Imgsrc : Particle Data Group K. Hagiwara et al.. [
12]
Samilarly inversion or parity is defined as[
21], used in e3nn as :
Co-ordinates conversion :
Conversion from
space in
terms to
is not needed as
,
terms are the same. r term can be scaled, this was the reason for selecting a sphere of unit size. See Table of Spherical Harmonics below for few
ℓ [
22].
Figure 3.
Spherical harmonics (Real part), Imgsrc : Wikipidea [
22]
Figure 3.
Spherical harmonics (Real part), Imgsrc : Wikipidea [
22]
GNN - Equivariant Euclidean Neural Nets E3nn [
14] came to represent Euclidean Neural Networks with spherical tensors to represent multi-particle interactions, preserving symmetry (including parity) in an equivariant way, using standard convolution neural nets (CNN) defined for r unit sphere as:
Scalability of Density Function Theory, Hartree-Fock
For n electrons and N nuclei, using 3 spatial co-ordinates for each electron going into Shrodinger’s wave equation PDE, pinning nucleus as stationary, dimensional space for even small molecules such as Benzene (12 nuclei, 42 electron, each with x,y,z co-ordinates), grows at rate. We can ignore nuclei momentum as it rotates slower that electron, ignore cross-terms coming out of Shrodinger’s equations representing electron, nuclei interactions (not removing Coulomb potential altogether but keeping at as stationary value at some places) to further simplify... but still we end up with lot of variables. Modeling drug molecule efficacy in Cell’s protein rich environment, where drug molecule will interact with RNA, DNA molecules become prohibitively expensive.
Allegro - Learning Local Equivariant Representations [15]:
Any Message passing GNN, trying to model electron interactions, still needs to model potential energy surface (PES) at distance R of nuceli, assuming nuclear potential is average of nuclear-electron and inter-nuclear potentials so that GNN still predicts atomic-structure properties that are better than force-fields based methods, which ignore quantum level interactions and just compute forces among particles using x,y,z co-ordinates. This message passing increases exponentially with respect to cutoff radius, as we increase layers of our NN at each node (point cloud representation in our case). For 3 layer GNN and cut-off radius of 4 Angstrom (carrying 20 Ammonium ions), we will end up consolidating 4*3 = 12 Angstrom worth of space at each atom, carrying 533 ammonium ions. If strictly local scheme of Allegro is used, we will have
= 16 times less aggregation, at each atom [
15]. We preferred to build on top of Allegro for this reason, as we had limited compute resources (just 1 A100 GPU).
Our approach, SPICE Dataset[3] that has Charge, use dominant aggrgation locally: We gather messages but only allowed most dominant force to propogate for datasets such as SPICE, which has fore-fields and charge. Allegro uses SUM for aggregation which resulted in less than perfect predictions in our case where we limited training to only 100 epochs using SPICE dataset. Allegro team suggests 7 days of training using similar compute resource (A100 GPU) [
15].
Finally, one can also construct a graph of Hilbert spaces [
5] of regions (molecules interacting with other molecules), passing/summing information across nodes (used in Loop quantum gravity paper by ChunJun Cao et al.), which can be coarsened by cutting edges and approximating various nodes as one supernode passing back information to all previously cut edges, as it was happening before the cut, using
for shaded region below.