Submitted:
19 January 2025
Posted:
20 January 2025
You are already at the latest version
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive cognitive decline and loss of neuronal integrity. Emerging evidence suggests that RhoA, Rho-associated coiled-coil kinase (ROCK), and their downstream effector molecule glycogen synthase 3β (GSK3β) interact within a complex signaling pathway (RhoA/ROCK/GSK3β) that plays a crucial role in the pathogenesis of AD. RhoA, a small GTPase, along with its downstream effector, ROCK, regulates various cellular processes, including actin cytoskeleton dynamics, apoptosis, and synaptic plasticity. GSK3β, a serine/threonine kinase plays a key role in neuronal function and AD pathology including regulation of tau phosphorylation, amyloid-beta cleavage. Overactive GSK3β has been closely linked to tau hyperphosphorylation, neurodegeneration, and the progression of AD. Thus GSK3β has been considered as a promising therapeutic target for treating AD and mitigating cognitive impairment. However, clinical trials of GSK3β in AD have faced considerable challenges due to the complexity inhibition of GSK3β and neuronal specific. In this review, we summarize the literature regarding the relationship of RhoA/ROCK and GSK3β signaling pathways in AD pathogenesis. We further discuss recent findings of sTREM2 -transgelin-2 (TG2) axis as the potential mediators of this complex pathways and provide our review on a novel targeting strategy for AD.
Keywords:
GSK3β
; RhoA
; ROCK
; RhoA/ROCK
; Alzheimer’s Disease
; sTREM2
; Transgelin-2
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by the accumulation of amyloid-beta (Aβ) peptides and neurofibrillary tangles (NFTs) induced by the hyperphosphorylation of tau proteins [1]. AD is the most common form of dementia and constitutes 60-70% of cases of senile dementia [2]. Prevalence of AD increases with age, and current treatment is mostly ineffective in preventing disease progression [3]. The etiology and pathogenesis of AD is complex and is dependent on interactions between cell types in the central nervous system (CNS) and multifaceted signaling pathways that lead to tau hyperphosphorylation [4,5,6,7,8]. Inhibiting GSK3β to activate the Wnt/β-catenin pathway has been proposed as promising therapeutic avenue and is currently under many active investigations; however, targeting the GSK3β pathway for AD has presented challenges hitherto in clinic due to small-molecule inhibitor specificity [8]. In parallel, the Ras homolog family member A (RhoA)/Rho-associated coiled-coil containing protein kinase (ROCK) has emerged as a significant regulator of AD and been proposed, as well, as a promising therapeutic avenue with unclear mechanisms being defined [9]. In this review, we summarize the to-date investigations of each of these two major pathways and present our view on the potential complex cross-talk between RhoA/ROCK and GSK3β pathways to regulate tau phosphorylation and current knowledge gap. Signaling by soluble Triggering Receptor Expressed on Myeloid Cells 2 (sTREM2) and its ligand transgelin-2 (TG2) is newly described pathway that plays a critical role in AD pathogenesis [10]. We also presented our view on how STREM2/TG2 axis may modulate RhoA/ROCK and GSK3β in regulating tau phosphorylation and serve as a viable therapeutic target.
RhoA/ROCK Pathway in Neuronal Regulation
RhoA is a small GTPase protein in the Rho family of GTPases composed of 3 isoforms, RhoA, RhoB, and RhoC [11]. RhoA transduces signals and mediates various cellular processes including cell migration, gene expression, and vesicle trafficking [12,13,14]. RhoA is abundant in smooth muscle cells, neurons, and immune cells [15,16,17]. RhoA regulates various cellular activities, including cytoskeleton modulation, cell death, mitochondrial homeostasis, autophagy, inflammation, and gene transcription [18]. In the brain, RhoA plays a role in regulating neuronal development, synaptic plasticity, and the progression of neurodegenerative diseases [16,18,19]. The RhoA/ROCK pathway specifically inhibits many of these processes [20,21]. Following brain injury, RhoA has been found to be upregulated and activated, resulting in growth cone collapse and failed axon regeneration [22,23].
RhoA activity is controlled by four main regulatory proteins: guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), guanine nucleotide dissociation inhibitors (GDIs), and GDI dissociation factors (GDFs) [24]. Over 70 distinct GEFs for Rho GTPases have been identified in mammals [25]. GEFs act as positive regulators of RhoA by exchanging GDP for GTP and activate RhoA by dissociating GDP from the GDP-RhoA complex; this unbound RhoA is subsequently able to bind to GTP [25,26]. While around 80 distinct RhoGAPs have been found in mammals, only a small number of GAPS have been shown to be specific for certain GTPases within the Rho family [24]. RhoGAPs specific for RhoA, but not RhoB or RhoC, have not yet been discovered [24,25]. GAPs act as negative regulators of RhoA by increasing the intrinsic rate of GTP hydrolysis in GTP-bound RhoA [27]. The mechanism by which the intrinsic rate of GTP hydrolysis varies across the diverse pool of GAPs, but RhoGAPs specifically have been found to contain a characteristic RhoGAP domain capable of binding to Rho GTPases and promoting GTP hydrolysis [25,26].
Unlike RhoGEFs and RhoGAPs, only three isoforms of RhoGDIs have been found in mammals [28]. GDIs act as negative regulators of RhoA by inhibiting the dissociation of guanine nucleotides (i.e., GDP) and preventing the loading of GTP to the GDP-RhoA complex [29]. However, they do not inhibit the loading of GDP/GTP to nucleotide-free RhoA [30,31]. RhoGDIs were also found to inhibit RhoA activity, preventing intrinsic and GAP-stimulated GTP hydrolysis [32]. Another function of RhoGDIs is as a chaperone for inactive RhoA. GDIs keep GTPases inactive in the cytosol, where 90-95% of Rho proteins in the cell reside at any given time [33,34]. This chaperone function enables inactive RhoA to be rapidly deployed to any membrane in the cell for activation in response to cellular signals [28]. An additional class of positive regulatory proteins of RhoA are known as GDFs. GDFs have been proposed to catalyze the dissociation of RhoGTPase-GDI complexes, thus allowing for activation of Rho GTPases [28,35].
In addition to the various types of Rho GTPase regulatory proteins, various post-translational modifications (PTMs) can regulate RhoA activity, such as prenylation and phosphorylation [18]. Prenylation is required for RhoA activation, as RhoA must be bound to cellular membranes to be activated [36]. Prenylation of RhoA results in modifications that increase protein hydrophobicity and facilitate membrane association [37]. Moreover, numerous kinases phosphorylate RhoA, the most well documented being protein kinase A (PKA) and cyclic GMP-dependent protein kinase (PKG), both of which mediate the phosphorylation of Ser188, a residue close to the prenylated cysteine residue of the C-terminal essential for RhoA membrane binding [38,39,40]. The phosphorylation negatively regulates RhoA activity by preventing dissociation of GDP-bound RhoA from RhoGDIs [41]. Ser188 phosphorylation has also been implied to inhibit RhoA binding to ROCK but not to other effector proteins, effectively inhibiting the RhoA/ROCK pathway [42].
ROCK is a key regulator of the cytoskeleton and impacts various cellular functions, such as cell shape, motility, proliferation, and gene expression [43]. ROCK has 2 isoforms: ROCK-I and ROCK-II [44]. ROCK-I is prominently expressed in non-neuronal tissues such as the liver, testis, and kidney. ROCK-II is mostly expressed in the brain and skeletal muscle [44]. ROCK contains an N-terminally located kinase domain, coiled-coil domain, and a Rho-binding domain (RBD). The switch regions of activated RhoA can bind to the RBD to activate ROCK [45]. At the C-terminus, there is a pleckstrin homology (PH) domain containing a cysteine-rich C1 domain [46]. The PH-C1 tandem has two functions. The tandem inhibits ROCK by sequestering its N-terminal kinase domain and reducing its kinase activity. The tandem also facilitates binding to membrane bilayers [47]. ROCK is the most widely studied downstream effector molecule of RhoA and is activated when bound to GTP-bound RhoA [48,49]. RhoA activates ROCK through a mechanism known as derepression, where the PH-C1 tandem-induced autoinhibition of the kinase domain is relieved, leading to an active kinase domain [50]. Other mechanisms of ROCK activation have been alleviated, such as the binding of arachidonic acid to the PH domain or cleavage of the carboxyl-terminus by granzyme B or caspase-2/3 [51,52] [Figure 1].
RhoA/ROCK signaling has been linked to AD risk factors, mainly tau hyperphosphorylation, synaptic damage, Aβ aggregation, and neuroinflammation [49]. When activated, RhoA/ROCK signaling pathways phosphorylate and activate downstream effectors that are also involved in regulating neuronal processes such as axonal guidance and regeneration and dendritic spine formation [53]. RhoA/ROCK phosphorylates various substrates, including Lim kinase (LMK), myosin light chain (MLC), and collapsing response mediator protein-2 (CRMP-2), which inhibit axonal growth [54]. Activated ROCK also stimulates actomyosin contractility by decreasing myosin phosphatase activity, MLC2 phosphorylation and resulting in spine shortening and retraction [55]. ROCK-I and ROCK-II differentially regulate actomyosin organization in influencing synaptic polarity [56]. ROCK-I is thought to be responsible for actin cytoskeleton destabilizing through regulating MLC2 phosphorylation while ROCK-II is required for stabilizing the actin cytoskeleton by regulating cofilin phosphorylation [57]. The RhoA/ROCK pathway indirectly activates GSK3β by deactivating GSK3β-inhibitory kinases such as Protein Kinase B (PKB or AKT) [58]. Both ROCK-I and ROCK-II have been shown to regulate RhoA/ROCK signaling to indirectly activate GSK3β. It was shown that inhibition of either ROCK-I or ROCK-II efficiently inactivated GSK3β and prevented subsequent tau phosphorylation [59,60]. The role of GSK3β in AD will be discussed extensively in the following sections.
GSK3β Regulation
GSK3β activation is well described to impact AD pathogenesis by regulating tau phosphorylation [61]. GSK-3 has two isoforms in mammals, GSK3α and GSK3β, which share 95% homology in the kinase domain but differ at the N and C terminal regions [62]. GSK3α has slightly longer N and C termini [62]. While both isoforms are ubiquitously expressed, GSK3β has greater abundance in the brain and has specificity for a broad range of substrates including metabolic proteins, transcription and translation factors, and cytoskeletal proteins [63,64]. GSK3β is abundantly expressed in neurons and plays essential roles in regulating neuronal development, synaptic pruning, and influencing the pathogenesis of neurodegenerative diseases, primarily AD [65]. It was observed that ventricular progenitors in the developing neocortex experience a higher expression of GSK3β that gradually declines following neuronal differentiation [66,67]. Transcripts of GSK3β have two alternative splicing forms, the short form GSK3β1 and the long form GSK3β2. The GSK3β2 is neuron-specific with a high expression during brain development that persists until adulthood. Interestingly, GSK3β2 shows less phosphorylation activity of tau at the AD-associated Ser396 epitope in comparison to GSK3β1 [68], suggesting that GSK3β2 may not be associated with AD. In addition to neurons, GSK3β is also expressed in microglia, astrocytes, and oligodendrocytes [69,70,71].
The function of GSK3β is largely mediated by its activation and inhibition through phosphorylation at the Tyr216 and Ser9 residues respectively [58,72]. Protein kinase B (PKB/Akt), located downstream of phosphatidylinositol 3-kinase (PI3K) has been shown to rapidly phosphorylate Ser9 of GSK3β both in vivo and in vitro with growth factor stimulation [58,73]. cAMP-dependent protein kinase A (PKA) and PKC have also been suggested to phosphorylate Ser9 in vivo and in vitro [74,75]. Phosphorylation of GSK3β at Tyr216 at the activating loop is thought to be by autophosphorylation, although phosphorylation by other kinases, such as the Src family kinases (SFKs) and Janus kinases (JAKs) has also been reported [76,77,78]. Further, GSK3β can be activated through the dephosphorylation of Ser9 by phosphatases such as protein phosphatase 2 (PP2A) [79]. To date, how GSK3β activity is regulated is not fully understood and remains an active area of investigation.
Dopamine and glutamate are neurotransmitters that have been associated with the hyperactive state of GSK3β. A study by Beaulieu et al. observed increased dopaminergic neurotransmission in dopamine transporter null (DAT-/-) mice that resulted in reduced PKB activation and subsequent GSK3β hyperactivation [80]. In the presence of glutamate, the interaction between GSK3β and glutamatergic N-methyl-D-aspartate (NMDA) receptors is believed to be dual-directional [81]. One study reported rapid dephosphorylation of the inhibitory Ser9 residue in GSK3β and simultaneous neurotoxicity in cultured hippocampal neurons following increased NMDA signaling [82]. This suggests that NMDA receptor signaling activates GSK3β. NMDA receptor antagonists such as phencyclidine and memantine have also been shown to increase Ser9 phosphorylation of GSK3β in murine models [83,84]. It was later observed that NMDA signaling activates protein phosphatase 1 (PP1), which is capable of dephosphorylating the Ser9 residue [85]. Conversely, GSK3β plays a role in the regulation of NMDA receptor surface localization and function [86]. One of the most well-known clinical inhibitors of GSK3β is lithium [87]. Its mechanism of action encompasses both direct and indirect pathways. Directly, lithium competes with Mg [2]+ ions, cofactors of GSK3β that stabilize the enzyme’s active site, thereby inhibiting its function [88]. Indirectly, lithium enhances the phosphorylation of GSK3β at the Ser9, although the exact reported mechanisms behind lithium-induced inhibition are conflicting and unclear [89,90,91]. Consistent with this notion, chronic lithium exposure has been shown to diminish NMDA receptor signaling and subsequent glutamate-induced excitotoxicity in murine cortical neurons [92,93]. These interactions highlight how GSK3β plays a fundamental role in the body’s response to the usage of psychoactive drugs.
The expression of GSK3β may be regulated epigenetically. The promoter region of GSK3β was shown to contain the cis-regulatory molecule (CRM) that binds to cell-type specific transcription factors, such as Sox2, Sox9, and Neurogenin2 (Ngn2) [94]. Using Histone ChIP-seq analysis of the GSK3β genomic region, it was shown that two histone marks, H3K4me3 and H3K27ac, exhibited open chromatin around the promoter region and exon 1 of murine GSK3β [94]. These findings are consistent with prior studies demonstrating that the GSK3β promoter region contains CCAAT/enhancer-binding proteins (C/EBP) consensus sequences, the known binding sites for transcription factors Sox 2 and Sox9, Ngn2 [94,95]. It is noteworthy that the GSK3β gene is highly conserved evolutionarily across cell types in all eukaryotic species and that GSK3β deletion was shown to cause postnatal fatality with multiple developmental defects [96,97]. These studies underscore the essential role of GSK3β in both physiological functions and disease pathogenesis.
GSK3β in Neuronal Development
GSK3β is broadly expressed throughout normal tissues and plays a role in numerous cellular functions including gene expression, stress responses, cell survival and death, cell structure, migration, metabolism, and differentiation [63,98,99]. When concentrated to the CNS, GSK3β is essential in regulating neurogenesis, neuronal differentiation, neuronal polarization, and progenitor proliferation [100,101]. It was shown that silencing of GSK3 in mice resulted in hyperproliferation of progenitor cells but a suppression of neuronal differentiation with increased cortical surface area and a thinner cortex [100,102]. It is believed that the ability of GSK3β to mediate neurogenesis, progenitor proliferation, and neuronal differentiation originates from its ability to interact with and regulate various signaling pathways including Wnt, Sonic Hedgehog (SHH), and Notch. These pathways, through interactions with GSK3β, promote the proliferation of neuronal progenitors by either inhibiting GSK3β activity or utilizing GSK3β as a regulatory molecule [103,104,105,106]. In regulating the Wnt pathway, GSK3β exhibits a dual role. Under canonical Wnt ligation, GSK3β can be inhibited by Dishevelled (Dsh), a transducer in the signaling cascade that inhibits GSK3β-mediated phosphorylation and degradation of β-catenin [107,108]. In the absence of Wnt signaling, GSK3β is able to mark β-catenin for proteasomal degradation, thereby preventing progenitor proliferation and differentiation [109]. Similarly, it has been suggested that GSK3β can function as either a positive or a negative regulator of Notch signaling [110,111,112], suggestion the cell type-specific role of GSK3β. It is understood that SK3β primarily acts as a downstream effector in the SHH pathway, although its role as either a positive or a negative regulator remains similarly cotroversial [113,114]. Nonetheless, inactivation of GSK3β by phosphorylation at Ser9 by PI3K/AKT has been shown to promote axon formation [115]. Further studies in hippocampal neurons revealed high concentrations of inactive GSK3β at the tips of newly formed axons, in correlation with the proper localization and functioning of Partitioning-Defective 3 (PAR3), a molecule that facilitates neuronal polarity and axon specification [116,117,118]. The dual role of GSK3β in neuronal regulation underscores the complexity of the molecule in neural development and a potential challenge in inhibiting GSK3β for treating AD.
RhoA/ROCK/GSK3β in Alzheimer’s Disease
The RhoA/ROCK pathway, when dysregulated, is a recognized significant contributor to tau phosphorylation and subsequent AD development [9]. Emerging evidence suggests that RhoA/ROCK may indirectly activate GSK3β, through reducing the activity of the inhibitory kinase PKB in the PI3K/AKT pathway [Figure 2]. RhoA/ROCK has been found to be able to directly phosphorylate and activate phosphatase and tensin homolog (PTEN) [119,120]. Active PTEN inhibits PI3K signaling by dephosphorylating the lipid signaling intermediate phosphatidylinositol (3,4,5)-triphosphate (PIP3) into PIP2, effectively impeding the activation of the downstream GSK3β inhibitor PKB [121,122]. Corroboratively, studies have shown that the inhibition of RhoA/ROCK leads to rapid activation of the PI3K/PKB signaling pathway [123]. Whether RhoA/ROCK is able to indirectly inhibit other GSK3β-inhibiting kinases, such as PKA and PKC, remains to be elucidated.
Upon RhoA/ROCK dysregulation, the hyperactivation of GSK3β has been well demonstrated to be a major contributor to the pathogenesis of AD, largely by phosphorylating tubulin associated units (tau) [124,125,126]. Tau is an intrinsically disordered, microtubule-associated protein predominantly expressed in neurons [127,128]. In its normal state, tau binds to and stabilizes distal axonal microtubules (MTs) [129]. The stabilizing function of tau promotes the polymerization of axonal MTs, an essential regulatory mechanism for proper axonal transport [130,131]. Tau is the substrate of many Ser/Thr kinases including GSK3β, Cdk5, MARK, PKA, CamKII, MAPK, PKC, JNK, and ROCK [132]. Tau is also the substrate of the tyrosine kinases Fyn and Abl [133]. Phosphorylation of tau at specific residues, most notably Ser202, Ser396, Ser404, Thr181, and Thr231, is thought to induce conformational changes that creative a positive feedback loop where tau can become sequentially phosphorylated at multiple sites, leading to hyperphosphorylation, although the specific relationship between these residues and AD still needs to be better defined [134,135,136,137]. Hyperphosphorylated tau exhibits a reduced affinity for MTs, which results in axonal destabilization [138]. Hyperphosphorylated tau presents aggregation-prone properties and forms neurofibrillary tangles (NFTs) which are a hallmark of AD [139,140].
While RhoA/ROCK is a known activator of neuronal GSK3β, other signaling pathways, such as Cdk5, ERK, and JNK are also key players in AD pathogenesis [132]. Cyclin-dependent kinase 5 (Cdk5) closely modulates Aβ deposition and, when hyperactivated by p25, induces aberrant Aβ cleavage and tau hyperphosphorylation [141,142]. Pharmacological inhibition of Cdk5 activation has been shown to reduce tau phosphorylation and Aβ processing and ameliorates neuronal death in p25 transgenic mice [143,144]. Mitogen-activated protein kinase (MAPK) pathways, including the extracellular signal-regulated kinase 1/2 (ERK), c- Jun N-terminal kinase (JNK), and P38 kinase (P38) pathways, have been shown influence AD development and progression [145,146]. In AD mouse models, ERK, JNK, and P38 are all found in the overactivated state in the nervous system, spine, and brain respectively [147,148,149]. While moderate ERK activation is necessary for synaptic plasticity, hyperactivated ERK has been associated with NFT formation and early-stage AD-related protein deposition, leading to impaired hippocampal function in AD patients and murine models [146,150,151,152]. Similarly, overactivated P38 has been shown to increase tau phosphorylation and favor amyloidogenic processing of the amyloid precursor protein (APP), while inhibition of JNK by D-JNKI1 is suggested to suppress synaptic shrinkage in AD patients [148,153,154]. Inhibitors of MAPK pathways can reduce Aβ deposition, neuronal apoptosis, memory impairment, and tau hyperphosphorylation, making them a favorable therapeutic target to be investigated [155,156,157].
Activated GSK3β was shown to phosphorylate tau at most of the Ser/Thr residues, which associated with tau hyperphosphorylation and AD pathogenesis [158,159]. Corroboratively, GSK3β inhibition in diverse mouse models has shown to reduce tau phosphorylation and improve cognitive impairments measured through behavioral assays [160,161]. By contrast, the overexpression of active GSK3β in murine forebrains was associated with tau hyperphosphorylation and somatodendritic tau accumulation in hippocampal neurons [162]. Interestingly, tau-deficient mice overexpressing GSK3β displayed reduced neurodegenerative symptoms and milder cognitive deficits, suggesting that the interaction between GSK3β and tau is critical for tau hyperphosphorylation and the development of AD [163]. Furthermore, postmortem examinations of brains of AD patients showed elevated levels of GSK3β in comparison to non-AD patients of the same age [164].
GSK3β has been suggested to serve as a bridge connecting Aβ and tau in the pathogenesis of AD [165]. Aβ formation has been observed to result in the activation GSK3β by activating RhoA/ROCK through the phosphorylation of the Tyr42 residue in RhoA [166]. Once activated, RhoA/ROCK is thought to activate Src, which then phosphorylates GSK3β at the Tyr216 residue [166]. The linking pathway between the presence of Aβ and activated RhoA/ROCK is not fully understood, but it is thought that Aβ oligomers bind to the p75 neurotrophin receptor (p75NTR) [167]. Paradoxically, GSK3β is also shown to regulate the production of Aβ [168,169]. Aβ is formed from successive proteolytic cleavages of APP. APP is a transmembrane protein abundantly expressed in the brain and is subject to two distinct metabolic pathways regulated by secretases [170]. The nonamyloidogenic pathway is modulated by α-secretases that cleave APP into easily-degradable fragments [171]. Conversely, the amyloidogenic pathway is mediated by a β-secretase (BACE-1) and γ-secretase complex. The amyloidogenic cleaving of APP forms an Aβ peptide that aggregates in AD brains [172]. GSK3β regulates Aβ production by phosphorylating and mediating the activation of presenilin-1 (PS1), a component of the γ–secretase complex [169]. Alternatively, GSK3β, through NF-κB overexpression, is thought to upregulate BACE-1 expression [173]. While the upregulation of BACE-1 by GSK3β increases Aβ cleavage, the effects of GSK3β phosphorylation of PS1 and the resulting modulation of γ–secretase activation on Aβ processing are unclear and are a necessary area for future investigation [161,174]. Finally, it is suggested that Aβ blocks Wnt signaling-induced GSK3β deactivation, leading to increased GSK3β activation. It is thought that Aβ can bind to the extracellular domain of the Wnt ligand receptor Frizzled (FZD), disrupting the interactions between Wnt ligands and the Low-Density Lipoprotein Receptor-Related Protein 6 (LRP6)/Fz complex [175]. Increased GSK3β activation results in an increase in Aβ cleavage and tau hyperphosphorylation, completing the positive feedback loop [175]. This positive feedback loop underscores the central role of GSK3β as a critical mediator linking Aβ production, tau hyperphosphorylation, and the amplification of pathological signaling in AD [Figure 2].
GSK3β-induced tau hyperphosphorylation impairs axonal transport, leading to further cognitive decline [176]. Axonal transport disturbances are an early hallmark of neurological disorders including AD [176]. While GSK3β is required to phosphorylate kinesin-1 to induce axonal transport of tau, the hyperphosphorylated tau disrupts axonal trafficking by destabilizing the MT cytoskeleton, diminishing the binding between motor proteins and cargo [177]. It is suspected that hyperphosphorylated tau impairs the function of c-Jun N-terminal kinase-interacting protein 1 (JIP1), which is responsible for facilitating the binding of cargo to motor proteins, although the complete mechanism is not clear [178]. Loss of axonal transport leads to vesicular aggregation and subcellular mislocation [179,180]. Additionally, the overactivation of GSK3β is thought to decrease cholinergic function, parallel to the reductions of cholinergic neurons exhibited in AD brains [181,182]. GSK3β is able to inhibit the production of the cholinergic neurotransmitter acetylcholine (ACh) by phosphorylating and deactivating pyruvate dehydrogenase, an essential enzyme for the functioning of choline acetyltransferase (ChAT), which is responsible for synthesizing ACh [181,183]. The loss of ACh causes loss of function in cholinergic neurons, inducing further cognitive impairment [182].
GSK3β also plays an essential role in modulating cognitive functions at the presynaptic and postsynaptic levels. Within presynaptic regions, the overactivation of GSK3β hinders the exocytosis of synaptic vesicles by phosphorylating P/Q-type calcium channels and disrupting the soluble NSF attachment protein receptor (SNARE) complex formation [184]. Such inhibition significantly diminishes the presynaptic release of glutamate and the clustering of synapsin I, a protein crucial for the release of neurotransmitters, preventing synapse formation [185]. Contrastingly, at the postsynaptic level, GSK3β regulates synaptic plasticity by mediating long-term potentiation (LTP) and long-term depression (LTD) [186,187,188]. It has been shown that LTP induction inhibits GSK3β activation by an increase in phosphorylated Ser9 levels, while transgenic mice overexpressing active GSK3β showed impaired LTP, suggesting that overactive GSK3β may impair LTP in cognition-associated conditions [185,189,190]. Further, is it suggested that GSK3β activation facilitates the induction of N-methyl-D-aspartate receptor (NMDAR)-mediated LTD through mechanisms involving PP1-mediated dephosphorylation and Akt inhibition downstream of NMDAR activation, strongly suggesting that overactivated GSK3β supports the induction of abnormal levels of LTD observed in multiple neuropsychiatric disorders [190,191,192,193,194,195]. It is noteworthy that GSK3β may be involved in the cross-communication between LTP and LTD [188]. Multiple studies have shown that inhibitors of GSK3β may be able to help normalize abnormal levels of both LTP and LDP in the brain, as treatment with GSK3β inhibitors were accompanied by increased cognitive abilities in mouse models of Fragile X syndrome, and Down syndrome, and AD [191,192,193,194,195].
sTREM2 as a Regulator of RhoA/ROCK/GSK3β Signaling
Multiple studies have suggested that the solubilized form of triggering receptors expressed on myeloid cells 2 (sTREM2) can reduce Aβ accumulation and slow AD progression [9,196,197,198,199]. TREMs are a broadly expressed family of cell surface receptors. TREMs primarily act as modulators of the immune response that regulate the activation of myeloid cells, notably macrophages [200,201]. Within the family, TREM2 is the most widely studied molecule and is implicated in the pathogenesis of numerous macrophage-associated and inflammation related diseases such as AD [202]. When membrane-bound, TREM2 binds to lipids and elicits essential neuroprotective effects by downregulating the expression of pro-inflammatory cytokines, such as TNF-α, IL-1β, and NOS2, and upregulating the transcription of anti-inflammatory cytokines including IL-4, IL-10 and IL-11 [203,204,205]. Additionally, TREM2 signaling has been shown to mediate the expression of the activating toll-like receptors (TLRs) [206]. In the brain, TREM2 is highly expressed by microglia in the temporal cortex surrounding Aβ plaques [207]. The TREM2 signaling pathway is involved in the microglial response and clearance of amyloid plaques, and is critical for successful synaptic pruning in the brain [208,209]. It has been shown with microglial RNA-seq analysis that TREM2 is required to sustain the microglial response to clear or prevent Aβ plaque formation [210,211].
The ectodomain of TREM2 can be solubilized by the α-secretases disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM17 at the His157-Ser158 bond within the stalk region to form soluble TREM2 (sTREM2) [212]. Studies revealed that elevated concentrations of sTREM2 are released by microglia into cerebral spinal fluid (CSF) in AD patients, although the extracellular conditions that increase or decrease sTREM2 shedding are unsertain [213]. sTREM2 has been suggested to influence the pathogenesis of AD, but the mechanisms behind the effects are unclear. Putatively, it is believed that sTREM2 plays a protective role in AD development [214]. This notion was supported by studies that sTREM2 is able to block the aggregation and neurotoxicity of Aβ plaques in murine models [215,216,217]. Furthermore, it has been suggested that sTREM2 reduces cognitive impairments induced by tau pathologies, such as preventing the loss of hippocampal synapses in tau P301S mice [10,218]. However, one study demonstrated that the injection of sTREM2 into the brains of healthy mice increased levels of pro-inflammatory cytokine production and microglial activation and proliferation [218]. Another study has implicated that sTREM2 may act as a decoy receptor and have indirect pro-inflammatory effects by reducing the modulatory anti-inflammatory function of TREM2 signaling [219]. Although TREM2 is generally considered advantageous in mitigating AD pathogenesis, the underlying mechanisms behind its neuroprotective effects are not well understood and would be an interesting area for future investigations.
Transgelin-2 (TG2 or SM22β), an actin-binding protein, has recently been suggested to ligate sTREM2 and deactivate the RhoA/ROCK/GSK3β signaling pathway, which may explain the neuroprotective effects observed following the shedding of sTREM2 [10]. TG2 is encoded by the TAGLN2 gene and is one of three transgelin isoforms alongside TG1 and TG3 [220]. Transgelins characteristically have transformation and conformation-sensitive properties, although the full implications of such are not fully known [221]. TG2 is highly expressed in smooth muscle cells [222]. It is also found in non-smooth muscle cells, including fibroblasts, epithelial cells, and immune cells throughout the body [223,224,225]. In the brain, TG2 is expressed primarily in neurons and microglia [10,226]. Broadly, TG2 is involved in regulating actin binding and stabilization, smooth muscle contraction, cell motility, and migration [227,228,229].
Recently, an interesting study by Zhang et al., 2023, proposed that TG2 expressed on neurons may serve as a receptor for sTREM2 [10]. Affinity purification pulldown and affinity purification mass spectrometry revealed that sTREM2 and TG2 colocalized on the cell surface of hippocampal neurons from AD patients and tau P301S transgenic mice, implying that TG2 serves as a sTREM2 receptor [10]. Supportively, TG2 has recently been shown to localize at the cellular membrane and function as a regulatory receptor of the myosin cytoskeleton and airway smooth muscle [222]. It has been shown that the TG2 agonist, TSG12, induced RhoA phosphorylation at the Ser188 residue, deactivating the molecule and, by extension, inhibiting RhoA/ROCK/GSK3β signaling [166,222]. Furthermore, neuroblastoma cell lines treated with sTREM2 exhibited decreased levels of activated RhoA and elevated levels of inactive RhoA phosphorylated at the Ser188 residue [10,230,231]. Further treatment with RhoA and ROCK inhibitors, Tat-C3 and Y-27632 respectively, emulated the inhibitory effect of sTREM2 on GSK3β activation and subsequent tau phosphorylation [10]. Additional transfection of phosphorylation-resistant RhoA (RhoA S188A) into an endogenous RhoA-null background silenced the inhibitory effect of sTREM2 [10]. It was observed that sTREM2 dramatically reduced the phosphorylation of tau at S202 and S396 residues, identical sites that are targets of GSK3β phosphorylation [10,232]. Concentration-dependent sTREM2 was also observed to significantly decrease the phosphorylation of GSK3β at the Tyr216 residue, and inhibition of sTREM2 with an anti-TREM2 antibody depleted this effect, suggesting that sTREM2 is able to deactivate GSK3β through TG2 ligation [10,72]. Finally, TG2 knockdown attenuated the neuroprotective function of sTREM2 on the inhibitory and excitatory synapses in the hippocampus of P301S mice [10]. This study strongly suggests that the ligation of TG2 by sTREM2 deactivates the RhoA/ROCK pathway through inhibiting RhoA, effectively preventing GSK3β activation and successive tau hyperphosphorylation.
Although TG2 was suggested to act as a cell surface receptor, it is primarily a cytosolic protein and is not known to possess a transmembrane domain when localized at the cellular membrane [233,234]. Thus, TG2 is not capable of inducing or inhibiting a signaling cascade directly. Alternatively, it is possible that TG2 may be coupled in low affinity with an adaptor molecule in the transmembrane domain that would not be detected in an affinity pulldown assay, potentially explaining how the ligation of TG2 exhibits RhoA/ROCK inhibition [Figure 3]. Perhaps, TG2 exhibits transient, low-affinity interactions with lipid raft-resident adaptor proteins or actin-linked scaffolding molecules. Given the proximity of TG2 to cortical actin networks, it is possible that TG2 interacts with adaptor proteins via weak electrostatic forces or PTMs that mediate binding affinity [10]. Notwithstanding, the interaction between sTREM2, TG2, and the RhoA/ROCK pathway warrants further investigation.
Emerging Perspectives and Challenges
Given the pivotal role of GSK3β overactivation in tau hyperphosphorylation, many preclinical studies attempted to inhibit GSK3β signaling as a therapeutic target for AD. Inhibitors including 6-bromoindirubin-3′oxime (6BIO), Hymenialdisine (HD), CHIR98014, SB-216763, and Alsterpaullone have been tested in preclinical studies in mouse models, with decreased tau phosphorylation, Aβ deposition, inflammation, and spatial memory deficits [235,236,237,238,239]. Preclinical studies have also suggested potential offsite toxicity due to suboptimal GSK3β specificity [236]. Targeting GSK3β has advanced to clinical trials [Table 1], while some results are encouraging, they are mostly inconclusive and unsatisfactory at this moment.

Table 1.
Clinical Trials targeting GSK3β.
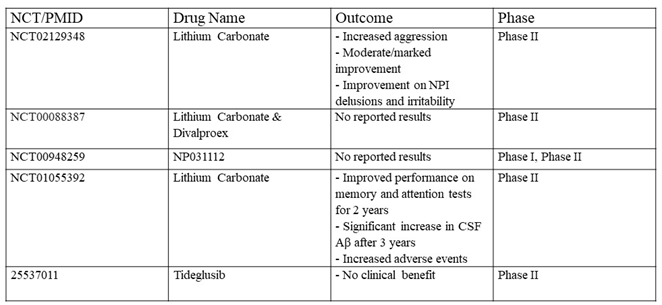
Although the therapeutic targeting of GSK3β is promising, it is still in its infancy, pending the resolution of difficulties, most notably specificity. Due to the broad expression of GSK3β and numerous physiological processes throughout the body, such as cell growth and survival, inhibition of GSK3β in general will likely lead to adverse effects. Moreover, due to the broadly conserved ATP-binding sites across the kinase family, engineering a small molecule that can effectively target specific kinases is exceedingly challenging. Nevertheless, developing a selective inhibitor to specifically target hyperactive of GSK3β in neurons may avoid such adverse effects. However, the difficulty in developing a GSK3β-specific inhibitor is further solidified by blood-brain barrier (BBB) prevention, an inhibitor must be small enough to pass through BBB and a cargo such as a nanoparticle may be considered.
Given the aforementioned challenges of targeting GSK3β in treating AD, targeting upstream pathways leading to GSK3β activation in neurons may be offer new treatment avenues. The intricate signaling network of RhoA/ROCK/GSK3β establishes it as a significant and promising immunotherapeutic target for treating AD. The immensely wide evolutionary conservation of GSK3β suggests that the GSK3β gene is essential for regulating synaptic function. Dysregulation of this pathway has been shown to largely contribute to tau hyperphosphorylation, neuroinflammation, and synaptic dysfunction, leading to cognitive decline. While targeting the molecules of this pathway for therapeutic treatment of AD appears to be propitious, much remains to be understood about the complex functions and signaling cascades involved, especially in the context of disease progression. The RhoA/ROCK/GSK3β pathway intersects with multiple cellular processes, and its modulatory functions have been shown to be highly context-dependent. Future research is imperative to fully understand the mechanisms behind this pathway and its influence on AD.
While sTREM2 has been suggested to be a promising immunotherapeutic target, the participation of sTREM2 in AD pathogenesis seems highly complex based on many controversial findings. How sTREM2 plays a role in AD pathogenesis must be fully understood before therapeutic interventions. Given that TG2 has recently been demonstrated to have a positive role in reducing AD development [10], strategies to increase TG2 expression in neurons may be a viable therapeutic angle.
Author Contributions
Conceptualization, M.M.M.; Investigation, M.M.M. and J.E.Y.; Project administration, M.M.M. and H.D.; Writing—Original Draft, M.M.M. and J.E.Y.; Writing—Review and Editing, M.M.M. and H.D.; Supervision, M.M.M. and H.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–97. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Perestelo-Perez, L.; Westman, E.; et al. Meta-Review of CSF Core Biomarkers in Alzheimer’s Disease: The State-of-the-Art after the New Revised Diagnostic Criteria. Front Aging Neurosci 2014, 6, 47. [Google Scholar] [CrossRef]
- Weaver, D.F. Thirty Risk Factors for Alzheimer’s Disease Unified by a Common Neuroimmune-Neuroinflammation Mechanism. Brain Sci 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Creekmore, B.C.; Watanabe, R.; Lee, E.B. Neurodegenerative Disease Tauopathies. Annu Rev Pathol 2024, 19, 345–370. [Google Scholar] [CrossRef] [PubMed]
- Govindarajulu, M.; Ramesh, S.; Beasley, M.; et al. Role of cGAS-Sting Signaling in Alzheimer’s Disease. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zheng, T.; Li, W.; et al. IL-33/ST2 signaling pathway and Alzheimer’s disease: A systematic review and meta-analysis. Clin Neurol Neurosurg 2023, 230, 107773. [Google Scholar] [CrossRef]
- Xiong, J.; Zhang, Z.; Ye, K. C/EBPbeta/AEP Signaling Drives Alzheimer’s Disease Pathogenesis. Neurosci Bull 2023, 39, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Sai Varshini, M.; Aishwarya Reddy, R.; Thaggikuppe Krishnamurthy, P. Unlocking hope: GSK-3 inhibitors and Wnt pathway activation in Alzheimer’s therapy. J Drug Target 2024, 32, 909–917. [Google Scholar] [CrossRef]
- Cai, R.; Wang, Y.; Huang, Z.; et al. Role of RhoA/ROCK signaling in Alzheimer’s disease. Behav Brain Res 2021, 414, 113481. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, L.; Yang, J.; et al. Soluble TREM2 ameliorates tau phosphorylation and cognitive deficits through activating transgelin-2 in Alzheimer’s disease. Nat Commun 2023, 14, 6670. [Google Scholar] [CrossRef] [PubMed]
- Eckenstaler, R.; Hauke, M.; Benndorf, R.A. A current overview of RhoA, RhoB, and RhoC functions in vascular biology and pathology. Biochem Pharmacol 2022, 206, 115321. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, K.; Chen, M. Dynamic functions of RhoA in tumor cell migration and invasion. Small GTPases 2013, 4, 141–7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zheng, Y. Cell type-specific signaling function of RhoA GTPase: lessons from mouse gene targeting. J Biol Chem 2013, 288, 36179–88. [Google Scholar] [CrossRef]
- Symons, M.; Rusk, N. Control of vesicular trafficking by Rho GTPases. Curr Biol 2003, 13, R409–18. [Google Scholar] [CrossRef] [PubMed]
- Loirand, G.; Guerin, P.; Pacaud, P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res 2006, 98, 322–34. [Google Scholar] [CrossRef]
- Luo, L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci 2000, 1, 173–80. [Google Scholar] [CrossRef] [PubMed]
- van Helden, S.F.; Anthony, E.C.; Dee, R.; et al. Rho GTPase expression in human myeloid cells. e: PLoS One 7, 4256; e3. [Google Scholar]
- Schmidt, S.I.; Blaabjerg, M.; Freude, K.; et al. RhoA Signaling in Neurodegenerative Diseases. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Tolias, K.F.; Duman, J.G.; Um, K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol 2011, 94, 133–48. [Google Scholar] [CrossRef]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci 2014, 8, 314. [Google Scholar] [CrossRef]
- Mulherkar, S.; Tolias, K.F. RhoA-ROCK Signaling as a Therapeutic Target in Traumatic Brain Injury. 2020; 9. [Google Scholar]
- Fournier, A.E.; Takizawa, B.T.; Strittmatter, S.M. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci 2003, 23, 1416–23. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, X.M. RhoA/Rho kinase in spinal cord injury. Neural Regen Res 2016, 11, 23–7. [Google Scholar]
- Schaefer, A.; Reinhard, N.R.; Hordijk, P.L. Toward understanding RhoGTPase specificity: structure, function and local activation. Small GTPases 2014, 5, 6. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 2007, 129, 865–77. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The ’invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol 2011, 12, 493–504. [Google Scholar] [CrossRef] [PubMed]
- DerMardirossian, C.; Bokoch, G.M. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol 2005, 15, 356–63. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Melendez, J.; Baumann, J.M.; et al. Loss of RhoA in neural progenitor cells causes the disruption of adherens junctions and hyperproliferation. Proc Natl Acad Sci U S A 2011, 108, 7607–12. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Kikuchi, A.; Ohga, N.; et al. Purification and characterization from bovine brain cytosol of a novel regulatory protein inhibiting the dissociation of GDP from and the subsequent binding of GTP to rhoB p20, a ras p21-like GTP-binding protein. J Biol Chem 1990, 265, 9373–80. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.J.; Maru, Y.; Leonard, D.; et al. A GDP dissociation inhibitor that serves as a GTPase inhibitor for the Ras-like protein CDC42Hs. Science 1992, 258, 812–5. [Google Scholar] [CrossRef] [PubMed]
- Boulter, E.; Garcia-Mata, R.; Guilluy, C.; et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol 2010, 12, 477–83. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.D.; Kiosses, W.B.; Schwartz, M.A. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J 1999, 18, 578–85. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Aivazian, D. Targeting Rab GTPases to distinct membrane compartments. Nat Rev Mol Cell Biol 2004, 5, 886–96. [Google Scholar] [CrossRef] [PubMed]
- Solski, P.A.; Helms, W.; Keely, P.J.; et al. RhoA biological activity is dependent on prenylation but independent of specific isoprenoid modification. Cell Growth Differ 2002, 13, 363–73. [Google Scholar]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; et al. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J Biol Chem 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.; Gesbert, F.; Delespine-Carmagnat, M.; et al. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J 1996, 15, 510–9. [Google Scholar] [CrossRef]
- Ellerbroek, S.M.; Wennerberg, K.; Burridge, K. Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem 2003, 278, 19023–31. [Google Scholar] [CrossRef]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; et al. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem 2000, 275, 21722–9. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Huang, F.; Lum, H. PKA inhibits RhoA activation: a protection mechanism against endothelial barrier dysfunction. L: Am J Physiol Lung Cell Mol Physiol 284, 2003; -80. [Google Scholar]
- Nusser, N.; Gosmanova, E.; Makarova, N.; et al. Serine phosphorylation differentially affects RhoA binding to effectors: implications to NGF-induced neurite outgrowth. Cell Signal 2006, 18, 704–14. [Google Scholar] [CrossRef]
- Riento, K.; Ridley, A.J. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 2003, 4, 446–56. [Google Scholar] [CrossRef]
- Nakagawa, O.; Fujisawa, K.; Ishizaki, T.; et al. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett 1996, 392, 189–93. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T.; Maekawa, M.; Fujisawa, K.; et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J 1996, 15, 1885–93. [Google Scholar] [CrossRef] [PubMed]
- Dvorsky, R.; Blumenstein, L.; Vetter, I.R.; et al. Structural insights into the interaction of ROCKI with the switch regions of RhoA. J Biol Chem 2004, 279, 7098–104. [Google Scholar] [CrossRef]
- Wen, W.; Liu, W.; Yan, J.; et al. Structure basis and unconventional lipid membrane binding properties of the PH-C1 tandem of rho kinases. J Biol Chem 2008, 283, 26263–73. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, K.; Fujita, A.; Ishizaki, T.; et al. Identification of the Rho-binding domain of p160ROCK, a Rho-associated coiled-coil containing protein kinase. J Biol Chem 1996, 271, 23022–8. [Google Scholar] [CrossRef]
- Koch, J.C.; Tatenhorst, L.; Roser, A.E.; et al. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol Ther 2018, 189, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Seto, M.; Noma, K. Rho kinase (ROCK) inhibitors. J Cardiovasc Pharmacol 2007, 50, 17–24. [Google Scholar] [CrossRef]
- Feng, J.; Ito, M.; Ichikawa, K.; et al. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem 1999, 274, 37385–90. [Google Scholar] [CrossRef] [PubMed]
- Sapet, C.; Simoncini, S.; Loriod, B.; et al. Thrombin-induced endothelial microparticle generation: identification of a novel pathway involving ROCK-II activation by caspase-2. Blood 2006, 108, 1868–76. [Google Scholar] [CrossRef]
- Schmandke, A.; Schmandke, A.; Strittmatter, S.M. ROCK and Rho: biochemistry and neuronal functions of Rho-associated protein kinases. Neuroscientist 2007, 13, 454–69. [Google Scholar] [CrossRef]
- Fujita, Y.; Yamashita, T. Axon growth inhibition by RhoA/ROCK in the central nervous system. Front Neurosci 2014, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Koleske, A.J. Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu Rev Neurosci 2010, 33, 349–78. [Google Scholar] [CrossRef] [PubMed]
- Newell-Litwa, K.A.; Badoual, M.; Asmussen, H.; et al. ROCK1 and 2 differentially regulate actomyosin organization to drive cell and synaptic polarity. J Cell Biol 2015, 210, 225–42. [Google Scholar] [CrossRef]
- Shi, J.; Wu, X.; Surma, M.; et al. Distinct roles for ROCK1 and ROCK2 in the regulation of cell detachment. Cell Death Dis, 2013; 4, e483. [Google Scholar]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–9. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Shirafuji, N.; Yen, S.H.; et al. Rho-kinase ROCK inhibitors reduce oligomeric tau protein. Neurobiol Aging 2020, 89, 41–54. [Google Scholar] [CrossRef]
- Moreira, N.; Tamarozzi, E.R.; Lima, J.; et al. Novel Dual AChE and ROCK2 Inhibitor Induces Neurogenesis via PTEN/AKT Pathway in Alzheimer’s Disease Model. 2022; 23. [Google Scholar]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; et al. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J 1990, 9, 2431–8. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Woodgett, J.R. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 2003, 116, 1175–86. [Google Scholar] [CrossRef] [PubMed]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 2001, 65, 391–426. [Google Scholar] [CrossRef] [PubMed]
- Draffin, J.E.; Sanchez-Castillo, C.; Fernandez-Rodrigo, A.; et al. GSK3alpha, not GSK3beta, drives hippocampal NMDAR-dependent LTD via tau-mediated spine anchoring. e: EMBO J 40, 1055; e13. [Google Scholar]
- Li, S.; Mattar, P.; Zinyk, D.; et al. GSK3 temporally regulates neurogenin 2 proneural activity in the neocortex. J Neurosci 2012, 32, 7791–805. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.X.; Wang, X.L.; Chen, J.Q.; et al. Differential Roles of Glycogen Synthase Kinase 3 Subtypes Alpha and Beta in Cortical Development. Front Mol Neurosci 2017, 10, 391. [Google Scholar] [CrossRef] [PubMed]
- Saeki, K.; Machida, M.; Kinoshita, Y.; et al. Glycogen synthase kinase-3beta2 has lower phosphorylation activity to tau than glycogen synthase kinase-3beta1. Biol Pharm Bull 2011, 34, 146–9. [Google Scholar] [CrossRef] [PubMed]
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell Signal 2009, 21, 264–73. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. Glycogen synthase kinase-3 regulates inflammatory tolerance in astrocytes. Neuroscience 2010, 169, 1063–70. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Shao, C.Y.; Xu, S.M.; et al. GSK3beta promotes the differentiation of oligodendrocyte precursor cells via beta-catenin-mediated transcriptional regulation. Mol Neurobiol 2014, 50, 507–19. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.; Nikolakaki, E.; Plyte, S.E.; et al. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J 1993, 12, 803–8. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Vandenheede, J.R.; et al. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. 2: Biochem J 303 ( Pt 1), 1994; -6. [Google Scholar]
- Fang, X.; Yu, S.X.; Lu, Y.; et al. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci U S A 2000, 97, 11960–5. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.F.; van den Bosch, M.T.; Hunter, R.W.; et al. Dual regulation of glycogen synthase kinase 3 (GSK3)alpha/beta by protein kinase C (PKC)alpha and Akt promotes thrombin-mediated integrin alphaIIbbeta3 activation and granule secretion in platelets. J Biol Chem 2013, 288, 3918–28. [Google Scholar] [CrossRef]
- Cole, A.; Frame, S.; Cohen, P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J 2004, 377, 249–55. [Google Scholar] [CrossRef] [PubMed]
- Goc, A.; Al-Husein, B.; Katsanevas, K.; et al. Targeting Src-mediated Tyr216 phosphorylation and activation of GSK-3 in prostate cancer cells inhibit prostate cancer progression in vitro and in vivo. Oncotarget 2014, 5, 775–87. [Google Scholar] [CrossRef]
- Shi, W.; Xu, C.; Gong, Y.; et al. RhoA/Rock activation represents a new mechanism for inactivating Wnt/beta-catenin signaling in the aging-associated bone loss. Cell Regen 2021, 10, 8. [Google Scholar] [CrossRef]
- Chu, D.; Tan, J.; Xie, S.; et al. GSK-3beta is Dephosphorylated by PP2A in a Leu309 Methylation-Independent Manner. J Alzheimers Dis 2016, 49, 365–75. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Sotnikova, T.D.; Yao, W.D.; et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A 2004, 101, 5099–104. [Google Scholar] [CrossRef]
- Barr, J.L.; Unterwald, E.M. Glycogen synthase kinase-3 signaling in cellular and behavioral responses to psychostimulant drugs. Biochim Biophys Acta Mol Cell Res 2020, 1867, 118746. [Google Scholar] [CrossRef]
- Luo, H.R.; Hattori, H.; Hossain, M.A.; et al. Akt as a mediator of cell death. Proc Natl Acad Sci U S A 2003, 100, 11712–7. [Google Scholar] [CrossRef] [PubMed]
- De Sarno, P.; Bijur, G.N.; Zmijewska, A.A.; et al. In vivo regulation of GSK3 phosphorylation by cholinergic and NMDA receptors. Neurobiol Aging 2006, 27, 413–22. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Tzavara, E.T.; Carruthers, R.; et al. Diverse psychotomimetics act through a common signaling pathway. Science 2003, 302, 1412–5. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, E.; Habas, A.; Yang, P.; et al. A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J Biol Chem 2005, 280, 37526–35. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xiong, Z.; Chen, P.; et al. beta-amyloid impairs the regulation of N-methyl-D-aspartate receptors by glycogen synthase kinase 3. Neurobiol Aging 2014, 35, 449–59. [Google Scholar] [CrossRef]
- Chatterjee, D.; Beaulieu, J.M. Inhibition of glycogen synthase kinase 3 by lithium, a mechanism in search of specificity. Front Mol Neurosci 2022, 15, 1028963. [Google Scholar] [CrossRef] [PubMed]
- Ryves, W.J.; Harwood, A.J. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun 2001, 280, 720–5. [Google Scholar] [CrossRef]
- Kirshenboim, N.; Plotkin, B.; Shlomo, S.B.; et al. Lithium-mediated phosphorylation of glycogen synthase kinase-3beta involves PI3 kinase-dependent activation of protein kinase C-alpha. J Mol Neurosci 2004, 24, 237–45. [Google Scholar] [CrossRef]
- Mora, A.; Sabio, G.; Risco, A.M.; et al. Lithium blocks the PKB and GSK3 dephosphorylation induced by ceramide through protein phosphatase-2A. Cell Signal 2002, 14, 557–62. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Sabio, G.; Gonzalez-Polo, R.A.; et al. Lithium inhibits caspase 3 activation and dephosphorylation of PKB and GSK3 induced by K+ deprivation in cerebellar granule cells. J Neurochem 2001, 78, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, R.; Hough, C.; Nakazawa, T.; et al. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem 2002, 80, 589–97. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Hough, C.J.; Chuang, D.M. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc Natl Acad Sci U S A 1998, 95, 2642–7. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Gotoh, H.; Kiyonari, H.; et al. Cell Type-Specific Transcriptional Control of Gsk3beta in the Developing Mammalian Neocortex. Front Neurosci 2022, 16, 811689. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Wang, G.L.; Shi, X.; et al. The age-associated decline of glycogen synthase kinase 3beta plays a critical role in the inhibition of liver regeneration. Mol Cell Biol 2009, 29, 3867–80. [Google Scholar] [CrossRef] [PubMed]
- Rylatt, D.B.; Aitken, A.; Bilham, T.; et al. Glycogen synthase from rabbit skeletal muscle. Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur J Biochem 1980, 107, 529–37. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; et al. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J 2001, 359, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Johnson, G.V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Kim, W.Y.; Wang, X.; Wu, Y.; et al. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci 2009, 12, 1390–7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Guo, W.; Liang, X.; et al. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 2005, 120, 123–35. [Google Scholar]
- Ahn, J.; Jang, J.; Choi, J.; et al. GSK3beta, but not GSK3alpha, inhibits the neuronal differentiation of neural progenitor cells as a downstream target of mammalian target of rapamycin complex1. Stem Cells Dev 2014, 23, 1121–33. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Hevner, R.F. Fibroblast growth factor signaling in development of the cerebral cortex. Dev Growth Differ 2009, 51, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Machold, R.; Hayashi, S.; Rutlin, M.; et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron 2003, 39, 937–50. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.; Gaiano, N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat Neurosci 2005, 8, 709–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; et al. Wnt/beta-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Lee, S.H.; Kim, H.; et al. Direct inhibition of GSK3beta by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta-catenin signaling. e: PLoS One 3, 4046. [Google Scholar]
- Wu, G.; Huang, H.; Garcia Abreu, J.; et al. Inhibition of GSK3 phosphorylation of beta-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. e: PLoS One 4, 4926. [Google Scholar]
- Aberle, H.; Bauer, A.; Stappert, J.; et al. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 1997, 16, 3797–804. [Google Scholar] [CrossRef]
- Guha, S.; Cullen, J.P.; Morrow, D.; et al. Glycogen synthase kinase 3 beta positively regulates Notch signaling in vascular smooth muscle cells: role in cell proliferation and survival. Basic Res Cardiol 2011, 106, 773–85. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, L.; Ingles-Esteve, J.; Aguilera, C.; et al. Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J Biol Chem 2003, 278, 32227–35. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Conner, S.D. Glycogen synthase kinase 3beta inhibition enhances Notch1 recycling. Mol Biol Cell 2018, 29, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Tempe, D.; Casas, M.; Karaz, S.; et al. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol Cell Biol 2006, 26, 4316–26. [Google Scholar] [CrossRef]
- Ocasio, J.K.; Bates, R.D.P.; Rapp, C.D.; et al. GSK-3 modulates SHH-driven proliferation in postnatal cerebellar neurogenesis and medulloblastoma. 2019. [Google Scholar]
- Dill, J.; Wang, H.; Zhou, F.; et al. Inactivation of glycogen synthase kinase 3 promotes axonal growth and recovery in the CNS. J Neurosci 2008, 28, 8914–28. [Google Scholar] [CrossRef]
- Shi, S.H.; Cheng, T.; Jan, L.Y.; et al. APC and GSK-3beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr Biol 2004, 14, 2025–32. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; et al. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–49. [Google Scholar] [CrossRef] [PubMed]
- Hapak, S.M.; Rothlin, C.V.; Ghosh, S. PAR3-PAR6-atypical PKC polarity complex proteins in neuronal polarization. Cell Mol Life Sci 2018, 75, 2735–2761. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, X.; Wang, Z.; et al. Regulation of PTEN by Rho small GTPases. Nat Cell Biol 2005, 7, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Meili, R.; Sasaki, A.T.; Firtel, R.A. Rho Rocks PTEN. Nat Cell Biol 2005, 7, 334–5. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol 2009, 4, 127–50. [Google Scholar] [CrossRef] [PubMed]
- Yoeli-Lerner, M.; Chin, Y.R.; Hansen, C.K.; et al. Akt/protein kinase b and glycogen synthase kinase-3beta signaling pathway regulates cell migration through the NFAT1 transcription factor. Mol Cancer Res 2009, 7, 425–32. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.F.; Viswambharan, H.; Barandier, C.; et al. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol 2002, 22, 8467–77. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys Acta Mol Cell Res 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem 2008, 104, 1433–9. [Google Scholar] [CrossRef] [PubMed]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; et al. GSK3beta and Tau Protein in Alzheimer’s Disease and Epilepsy. Front Cell Neurosci 2020, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Biernat, J.; von Bergen, M.; et al. Sites of tau important for aggregation populate beta-structure and bind to microtubules and polyanions. J Biol Chem 2005, 280, 24978–86. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; et al. Structural polymorphism of 441-residue tau at single residue resolution. e: PLoS Biol 7, 2009; e34. [Google Scholar]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; et al. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 1975, 72, 1858–62. [Google Scholar] [CrossRef]
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J Cell Biol 1986, 103, 2739–46. [Google Scholar] [CrossRef]
- Santarella, R.A.; Skiniotis, G.; Goldie, K.N.; et al. Surface-decoration of microtubules by human tau. J Mol Biol 2004, 339, 539–53. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.; Hanger, D.P.; Miller, C.C.; et al. The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol 2013, 4, 83. [Google Scholar] [CrossRef]
- Derkinderen, P.; Scales, T.M.; Hanger, D.P.; et al. Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci 2005, 25, 6584–93. [Google Scholar] [CrossRef]
- Neddens, J.; Temmel, M.; Flunkert, S.; et al. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol Commun 2018, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; von Bernhardi, R.; Godoy, J.A.; et al. Phosphorylated tau potentiates Abeta-induced mitochondrial damage in mature neurons. Neurobiol Dis 2014, 71, 260–9. [Google Scholar] [CrossRef] [PubMed]
- Bielska, A.A.; Zondlo, N.J. Hyperphosphorylation of tau induces local polyproline II helix. Biochemistry 2006, 45, 5527–37. [Google Scholar] [CrossRef] [PubMed]
- Cantrelle, F.X.; Loyens, A.; Trivelli, X.; et al. Phosphorylation and O-GlcNAcylation of the PHF-1 Epitope of Tau Protein Induce Local Conformational Changes of the C-Terminus and Modulate Tau Self-Assembly Into Fibrillar Aggregates. Front Mol Neurosci 2021, 14, 661368. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 1984, 259, 5301–5. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dexheimer, T.; Sui, D.; et al. Hyperphosphorylated tau aggregation and cytotoxicity modulators screen identified prescription drugs linked to Alzheimer’s disease and cognitive functions. Sci Rep 2020, 10, 16551. [Google Scholar] [CrossRef]
- Alonso, A.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med 1996, 2, 783–7. [Google Scholar] [CrossRef]
- Lee, M.S.; Kwon, Y.T.; Li, M.; et al. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–4. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.C.; Zhou, Y.; Shen, Y.; et al. A survey of Cdk5 activator p35 and p25 levels in Alzheimer’s disease brains. FEBS Lett 2002, 523, 58–62. [Google Scholar] [CrossRef]
- Liu, F.; Su, Y.; Li, B.; et al. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett 2003, 547, 193–6. [Google Scholar] [CrossRef]
- Zheng, Y.L.; Kesavapany, S.; Gravell, M.; et al. A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J 2005, 24, 209–20. [Google Scholar] [CrossRef]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J 1995, 9, 726–35. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lee, H.G.; Raina, A.K.; et al. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 2002, 11, 270–81. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.G. The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev Neurosci 2006, 17, 619–34. [Google Scholar] [CrossRef] [PubMed]
- Sclip, A.; Tozzi, A.; Abaza, A.; et al. c-Jun N-terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. e: Cell Death Dis 5, 1019. [Google Scholar]
- Gourmaud, S.; Paquet, C.; Dumurgier, J.; et al. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: links to cognitive decline. J Psychiatry Neurosci 2015, 40, 151–61. [Google Scholar] [CrossRef]
- Kelleher, R.J., 3rd; Govindarajan, A.; Tonegawa, S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 2004, 44, 59–73. [Google Scholar] [CrossRef]
- Pei, J.J.; Braak, H.; An, W.L.; et al. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer’s disease. Brain Res Mol Brain Res 2002, 109, 45–55. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R.; Carmona, M.; et al. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol 2001, 11, 144–58. [Google Scholar] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; et al. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol 2014, 2, 873–7. [Google Scholar] [CrossRef]
- Tan, J.L.; Li, Q.X.; Ciccotosto, G.D.; et al. Mild oxidative stress induces redistribution of BACE1 in non-apoptotic conditions and promotes the amyloidogenic processing of Alzheimer’s disease amyloid precursor protein. e: PLoS One 8, 6124; e6. [Google Scholar]
- Igaz, L.M.; Winograd, M.; Cammarota, M.; et al. Early activation of extracellular signal-regulated kinase signaling pathway in the hippocampus is required for short-term memory formation of a fear-motivated learning. Cell Mol Neurobiol 2006, 26, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.; Ammit, A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–8. [Google Scholar] [CrossRef]
- Yenki, P.; Khodagholi, F.; Shaerzadeh, F. Inhibition of phosphorylation of JNK suppresses Abeta-induced ER stress and upregulates prosurvival mitochondrial proteins in rat hippocampus. J Mol Neurosci 2013, 49, 262–9. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Montano, J.R.; Moreno, F.J.; Avila, J.; et al. Lithium inhibits Alzheimer’s disease-like tau protein phosphorylation in neurons. FEBS Lett 1997, 411, 183–8. [Google Scholar] [CrossRef]
- Hong, M.; Chen, D.C.; Klein, P.S.; et al. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem 1997, 272, 25326–32. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, A.; Rosi, M.C.; Grossi, C.; et al. Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mutant mice. e: PLoS One 5, 1438; e2. [Google Scholar]
- Ly, P.T.; Wu, Y.; Zou, H.; et al. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest 2013, 123, 224–35. [Google Scholar] [CrossRef]
- Lucas, J.J.; Hernandez, F.; Gomez-Ramos, P.; et al. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 2001, 20, 27–39. [Google Scholar] [CrossRef]
- Gomez de Barreda, E.; Perez, M.; Gomez Ramos, P.; et al. Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol Dis 2010, 37, 622–9. [Google Scholar] [CrossRef]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol 2007, 33, 43–55. [Google Scholar] [CrossRef]
- Hernandez, F.; Gomez de Barreda, E.; Fuster-Matanzo, A.; et al. GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol 2010, 223, 322–5. [Google Scholar] [CrossRef]
- Cap, K.C.; Jung, Y.J.; Choi, B.Y.; et al. Distinct dual roles of p-Tyr42 RhoA GTPase in tau phosphorylation and ATP citrate lyase activation upon different Abeta concentrations. Redox Biol 2020, 32, 101446. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Zagrebelsky, M.; Korte, M.; et al. Signaling via the p75 neurotrophin receptor facilitates amyloid-beta-induced dendritic spine pathology. Sci Rep 2020, 10, 13322. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, Y.; Zhao, B. Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Curr Alzheimer Res 2012, 9, 864–79. [Google Scholar] [CrossRef]
- Uemura, K.; Kuzuya, A.; Shimozono, Y.; et al. GSK3beta activity modifies the localization and function of presenilin 1. J Biol Chem 2007, 282, 15823–32. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- de Paula, V.J.R.; Guimaraes, F.M.; Diniz, B.S.; et al. Neurobiological pathways to Alzheimer’s disease: Amyloid-beta, TAU protein or both? Dement Neuropsychol 2009, 3, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, X.; Xia, W.; et al. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front Mol Neurosci 2020, 13, 137. [Google Scholar] [CrossRef]
- Twomey, C.; McCarthy, J.V. Presenilin-1 is an unprimed glycogen synthase kinase-3beta substrate. FEBS Lett 2006, 580, 4015–20. [Google Scholar] [CrossRef]
- Sofola, O.; Kerr, F.; Rogers, I.; et al. Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer’s disease. e: PLoS Genet 6, 1001. [Google Scholar]
- Magdesian, M.H.; Carvalho, M.M.; Mendes, F.A.; et al. Amyloid-beta binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/beta-catenin signaling. J Biol Chem 2008, 283, 9359–68. [Google Scholar] [CrossRef] [PubMed]
- Berth, S.H.; Lloyd, T.E. Disruption of axonal transport in neurodegeneration. 2023. [Google Scholar]
- LaPointe, N.E.; Morfini, G.; Pigino, G.; et al. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res 2009, 87, 440–51. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D. ; Gotz J: Phosphorylated Tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J Biol Chem 2009, 284, 20909–16. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Seereeram, A.; Byers, H.L.; et al. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J 2008, 22, 3186–95. [Google Scholar] [CrossRef] [PubMed]
- Soutar, M.P.; Kim, W.Y.; Williamson, R.; et al. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J Neurochem 2010, 115, 974–83. [Google Scholar] [CrossRef]
- Zhao, L.; Chu, C.B.; Li, J.F.; et al. Glycogen synthase kinase-3 reduces acetylcholine level in striatum via disturbing cellular distribution of choline acetyltransferase in cholinergic interneurons in rats. Neuroscience 2013, 255, 203–11. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Duan, L.; Xu, W.; et al. Nerve growth factor metabolic dysfunction contributes to sevoflurane-induced cholinergic degeneration and cognitive impairments. Brain Res 2019, 1707, 107–116. [Google Scholar] [CrossRef]
- Wang, Y.; Tian, Q.; Liu, E.J.; et al. Activation of GSK-3 disrupts cholinergic homoeostasis in nucleus basalis of Meynert and frontal cortex of rats. J Cell Mol Med 2017, 21, 3515–3528. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.Q.; Liu, D.; Hu, J.; et al. GSK-3 beta inhibits presynaptic vesicle exocytosis by phosphorylating P/Q-type calcium channel and interrupting SNARE complex formation. J Neurosci 2010, 30, 3624–33. [Google Scholar] [CrossRef]
- Zhu, L.Q.; Wang, S.H.; Liu, D.; et al. Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J Neurosci 2007, 27, 12211–20. [Google Scholar] [CrossRef]
- Fan, X.; Zhao, Z.; Wang, D.; et al. Glycogen synthase kinase-3 as a key regulator of cognitive function. Acta Biochim Biophys Sin (Shanghai) 2020, 52, 219–230. [Google Scholar] [CrossRef]
- Sharma, S.; Taliyan, R. Neuroprotective role of Indirubin-3′-monoxime, a GSKbeta inhibitor in high fat diet induced cognitive impairment in mice. Biochem Biophys Res Commun 2014, 452, 1009–15. [Google Scholar] [CrossRef] [PubMed]
- Peineau, S.; Bradley, C.; Taghibiglou, C.; et al. The role of GSK-3 in synaptic plasticity. S: Br J Pharmacol 153 Suppl 1, 2008; -37. [Google Scholar]
- Hooper, C.; Markevich, V.; Plattner, F.; et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci 2007, 25, 81–6. [Google Scholar] [CrossRef] [PubMed]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007, 53, 703–17. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Schoenfeld, B.P.; Bell, A.J.; et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res 2011, 1380, 106–19. [Google Scholar] [CrossRef] [PubMed]
- Franklin, A.V.; King, M.K.; Palomo, V.; et al. Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol Psychiatry 2014, 75, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Hoeffer, C.A.; Capetillo-Zarate, E.; et al. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One, 2010; 5. [Google Scholar]
- Li, X.H.; Lv, B.L.; Xie, J.Z.; et al. AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol Aging 2012, 33, 1400–10. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A.; Greco, B.; Ghezzi, D.; et al. Lithium rescues synaptic plasticity and memory in Down syndrome mice. J Clin Invest 2013, 123, 348–61. [Google Scholar] [CrossRef]
- Ewers, M.; Biechele, G.; Suarez-Calvet, M.; et al. Higher CSF sTREM2 and microglia activation are associated with slower rates of beta-amyloid accumulation. e: EMBO Mol Med 12, 1230; e8. [Google Scholar]
- Franzmeier, N.; Suarez-Calvet, M.; Frontzkowski, L.; et al. Higher CSF sTREM2 attenuates ApoE4-related risk for cognitive decline and neurodegeneration. Mol Neurodegener 2020, 15, 57. [Google Scholar] [CrossRef]
- Biel, D.; Suarez-Calvet, M.; Hager, P.; et al. sTREM2 is associated with amyloid-related p-tau increases and glucose hypermetabolism in Alzheimer’s disease. e: EMBO Mol Med 15, 1698; e7. [Google Scholar]
- Ewers, M.; Franzmeier, N.; Suarez-Calvet, M.; et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci Transl Med, 2019; 11. [Google Scholar]
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol 2000, 164, 4991–5. [Google Scholar] [CrossRef]
- Filipello, F.; Morini, R.; Corradini, I.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. 9: Immunity 48, 2018; e8. [Google Scholar]
- Dardiotis, E.; Siokas, V.; Pantazi, E.; et al. A novel mutation in TREM2 gene causing Nasu-Hakola disease and review of the literature. 1: Neurobiol Aging 53, 2017; e22. [Google Scholar]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; et al. Pattern recognition by TREM-2: binding of anionic ligands. J Immunol 2003, 171, 594–9. [Google Scholar] [CrossRef]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 2005, 201, 647–57. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Prinz, M.; Stagi, M.; et al. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. e: PLoS Med 4, 2007. [Google Scholar]
- Hamerman, J.A.; Jarjoura, J.R.; Humphrey, M.B.; et al. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol 2006, 177, 2051–5. [Google Scholar] [CrossRef] [PubMed]
- Winfree, R.L.; Seto, M.; Dumitrescu, L.; et al. TREM2 gene expression associations with Alzheimer’s disease neuropathology are region-specific: implications for cortical versus subcortical microglia. Acta Neuropathol 2023, 145, 733–747. [Google Scholar] [CrossRef]
- Ennerfelt, H.; Frost, E.L.; Shapiro, D.A.; et al. SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell 185:4135‐4152 e22, 2022.
- Wang, S.; Sudan, R.; Peng, V.; et al. TREM2 drives microglia response to amyloid‐beta via SYK‐dependent and ‐independent pathways. Cell 185:4153‐4169 e19, 2022.
- Wang, S.; Mustafa, M.; Yuede, C.M.; et al. Anti‐human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med 217, 2020.
- Mathys, H.; Adaikkan, C.; Gao, F.; et al. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep 2017, 21, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Thornton, P.; Sevalle, J.; Deery, M.J.; et al. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol Med 2017, 9, 1366–1378. [Google Scholar] [CrossRef]
- Feuerbach, D.; Schindler, P.; Barske, C.; et al. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci Lett 2017, 660, 109–114. [Google Scholar] [CrossRef]
- Raha, A.A.; Henderson, J.W.; Stott, S.R.; et al. Neuroprotective Effect of TREM-2 in Aging and Alzheimer’s Disease Model. J Alzheimers Dis 2017, 55, 199–217. [Google Scholar] [CrossRef]
- Vilalta, A.; Zhou, Y.; Sevalle, J.; et al. Wild-type sTREM2 blocks Abeta aggregation and neurotoxicity, but the Alzheimer’s R47H mutant increases Abeta aggregation. J Biol Chem 2021, 296, 100631. [Google Scholar] [CrossRef]
- Kober, D.L.; Stuchell-Brereton, M.D.; Kluender, C.E.; et al. Functional insights from biophysical study of TREM2 interactions with apoE and Abeta(1‐42). Alzheimers Dement, 2020.
- Lessard, C.B.; Malnik, S.L.; Zhou, Y.; et al. High‐affinity interactions and signal transduction between Abeta oligomers and TREM2. EMBO Mol Med 10, 2018.
- Zhong, L.; Xu, Y.; Zhuo, R.; et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat Commun 2019, 10, 1365. [Google Scholar] [CrossRef]
- Piccio, L.; Buonsanti, C.; Cella, M.; et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 2008, 131, 3081–91. [Google Scholar] [CrossRef]
- Camoretti-Mercado, B.; Forsythe, S.M.; LeBeau, M.M.; et al. Expression and cytogenetic localization of the human SM22 gene (TAGLN). Genomics 1998, 49, 452–7. [Google Scholar] [CrossRef] [PubMed]
- Shapland, C.; Hsuan, J.J.; Totty, N.F.; et al. Purification and properties of transgelin: a transformation and shape change sensitive actin-gelling protein. J Cell Biol 1993, 121, 1065–73. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Xu, Y.D.; Peng, L.L.; et al. Transgelin‐2 as a therapeutic target for asthmatic pulmonary resistance. Sci Transl Med 10, 2018.
- Elsafadi, M.; Manikandan, M.; Dawud, R.A.; et al. Transgelin is a TGFbeta-inducible gene that regulates osteoblastic and adipogenic differentiation of human skeletal stem cells through actin cytoskeleston organization. e: Cell Death Dis 7, 2321. [Google Scholar]
- Na, B.R.; Kim, H.R.; Piragyte, I.; et al. TAGLN2 regulates T cell activation by stabilizing the actin cytoskeleton at the immunological synapse. J Cell Biol 2015, 209, 143–62. [Google Scholar] [CrossRef]
- Na, B.R.; Jun, C.D. TAGLN2-mediated actin stabilization at the immunological synapse: implication for cytotoxic T cell control of target cells. BMB Rep 2015, 48, 369–70. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Lee, H.S.; Lee, K.S.; et al. An Essential Role for TAGLN2 in Phagocytosis of Lipopolysaccharide-activated Macrophages. Sci Rep 2017, 7, 8731. [Google Scholar] [CrossRef]
- Jo, S.; Kim, H.R.; Mun, Y.; et al. Transgelin-2 in immunity: Its implication in cell therapy. J Leukoc Biol 2018, 104, 903–910. [Google Scholar] [CrossRef]
- Yuan, H.K.; Lu, J.; Wang, X.L.; et al. The Effects of a Transgelin-2 Agonist Administered at Different Times in a Mouse Model of Airway Hyperresponsiveness. Front Pharmacol 2022, 13, 873612. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Park, J.S.; Park, J.H.; et al. Cell-permeable transgelin-2 as a potent therapeutic for dendritic cell-based cancer immunotherapy. J Hematol Oncol 2021, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Islam, R.; Cho, J.Y.; et al. Regulation of RhoA GTPase and various transcription factors in the RhoA pathway. J Cell Physiol 2018, 233, 6381–6392. [Google Scholar] [CrossRef] [PubMed]
- Forget, M.A.; Desrosiers, R.R.; Gingras, D.; et al. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J 2002, 361, 243–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liang, Z.; Shi, J.; et al. PKA modulates GSK-3beta- and cdk5-catalyzed phosphorylation of tau in site- and kinase-specific manners. FEBS Lett 2006, 580, 6269–74. [Google Scholar] [CrossRef]
- Yin, L.M.; Ulloa, L.; Yang, Y.Q. Transgelin-2: Biochemical and Clinical Implications in Cancer and Asthma. Trends Biochem Sci 2019, 44, 885–896. [Google Scholar] [CrossRef]
- Gez, S.; Crossett, B.; Christopherson, R.I. Differentially expressed cytosolic proteins in human leukemia and lymphoma cell lines correlate with lineages and functions. Biochim Biophys Acta 2007, 1774, 1173–83. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Qiao, A.; Fan, G.H. Indirubin-3′-monoxime rescues spatial memory deficits and attenuates beta-amyloid-associated neuropathology in a mouse model of Alzheimer’s disease. Neurobiol Dis 2010, 39, 156–68. [Google Scholar] [CrossRef]
- Meijer, L.; Thunnissen, A.M.; White, A.W.; et al. Inhibition of cyclin-dependent kinases, GSK-3beta and CK1 by hymenialdisine, a marine sponge constituent. Chem Biol 2000, 7, 51–63. [Google Scholar] [CrossRef]
- Hildebrand, M.; Krause, W.; Fabian, H.; et al. Pharmacokinetics of iloprost in patients with chronic renal failure and on maintenance haemodialysis. Int J Clin Pharmacol Res 1990, 10, 285–92. [Google Scholar] [PubMed]
- Hu, S.; Begum, A.N.; Jones, M.R.; et al. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis 2009, 33, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Selenica, M.L.; Jensen, H.S.; Larsen, A.K.; et al. Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br J Pharmacol 2007, 152, 959–79. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Activation pathways of ROCK. (A) Rho GTPase-dependent activation of ROCK through binding to the RBD. (B) Rho GTPase-independent activation of ROCK through Caspase 3 or Granzyme B cleavage of the carboxyl terminal region of ROCK. Images A and B were adapted from Julian, L., & Olson, M. F. (2014). Rho-associated coiled-coil containing kinases (ROCK): Structure, regulation, and functions. Small GTPases, 5(2). https://doi.org/10.4161/sgtp.29846.
Figure 1.
Activation pathways of ROCK. (A) Rho GTPase-dependent activation of ROCK through binding to the RBD. (B) Rho GTPase-independent activation of ROCK through Caspase 3 or Granzyme B cleavage of the carboxyl terminal region of ROCK. Images A and B were adapted from Julian, L., & Olson, M. F. (2014). Rho-associated coiled-coil containing kinases (ROCK): Structure, regulation, and functions. Small GTPases, 5(2). https://doi.org/10.4161/sgtp.29846.
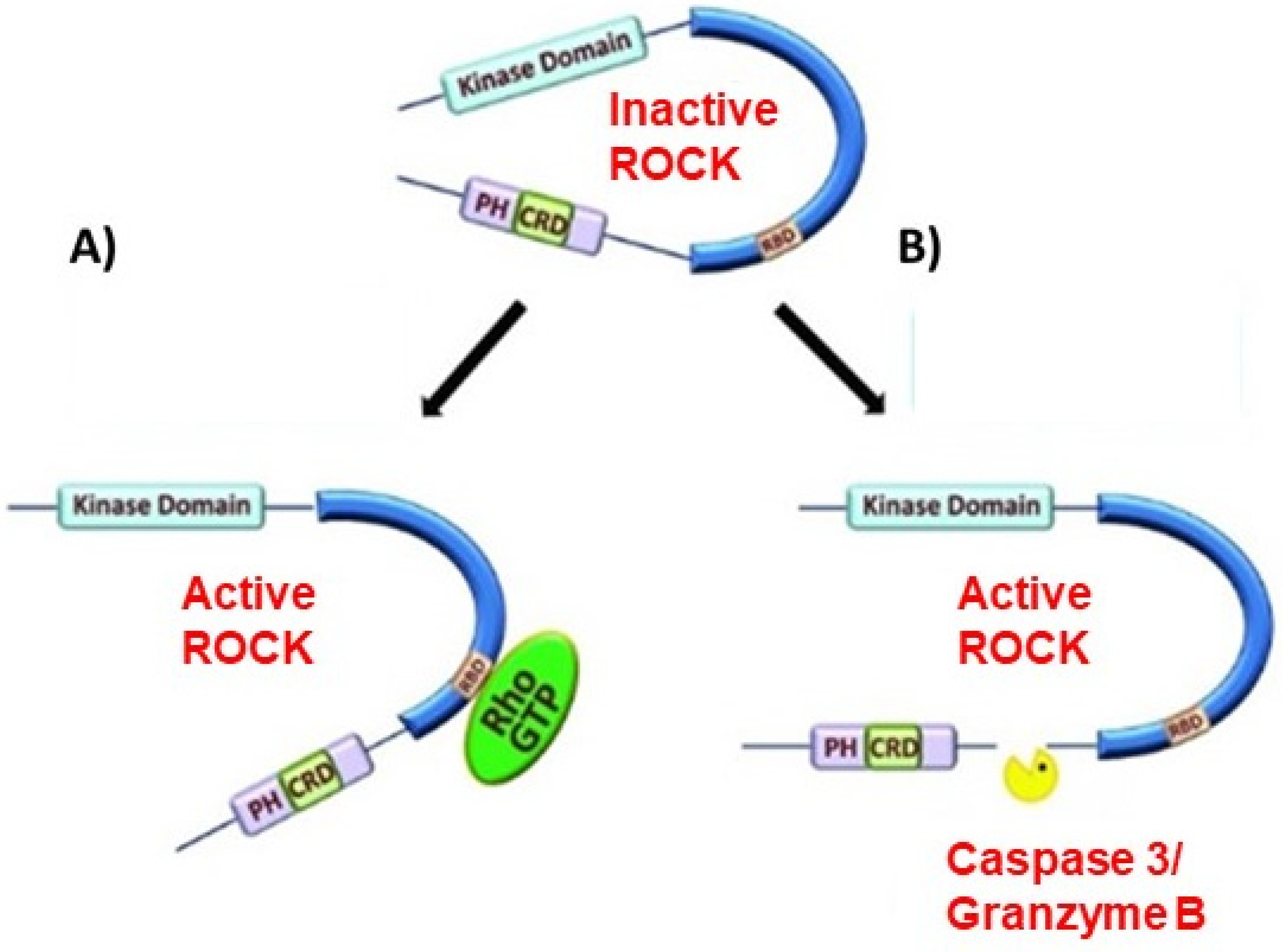
Figure 2.
Signaling pathways directly and indirectly linked to RhoA/ROCK-mediated activation of GSK3β and positive feedback loop between Aβ deposition and GSK3β activation. (1) ROCK phosphorylates and activates PTEN, a phosphatase that inhibits the PI3K/AKT signaling pathway. PTEN dephosphorylates PIP3 into PIP2, preventing the activation of PKB (AKT). PKB inhibits GSK3β by Ser9 phosphorylation. Inhibition of PKB by RhoA/ROCK activates GSK3β; (2) Aβ ligation of p75NTR activates RhoA by Tyr42 phosphorylation. Activated RhoA/ROCK phosphorylates and activates Src at the Tyr416 residue. Activated Src phosphorylates and activates GSK3β at the Tyr216 residue, subsequently activating IKK. IKK phosphorylates IκB at Ser32/36, marking it for proteasomal degradation and impeding NF-κB inhibition. NF-κB increases BACE-1 expression which increases the activity of amyloidogenic processing of APP into Aβ. Newly processed Aβ oligomers bind to p75NTR and complete the positive feedback cycle; (3) Aβ binds to the extracellular domains of FZD and LRP6, preventing Wnt ligand interactions with the LRP6/FZD complex and successive inhibition of GSK3β. It is noteworthy that GSK3β is capable of phosphorylating PS1 and the Ser353 and 357 residues. However, the exact effect on Aβ processing is unclear; (4) Overactivation of GSK3β induces AD pathogenesis such as Aβ deposition, tau hyperphosphorylation, neurotic plaque formation, and cognitive decline.
Figure 2.
Signaling pathways directly and indirectly linked to RhoA/ROCK-mediated activation of GSK3β and positive feedback loop between Aβ deposition and GSK3β activation. (1) ROCK phosphorylates and activates PTEN, a phosphatase that inhibits the PI3K/AKT signaling pathway. PTEN dephosphorylates PIP3 into PIP2, preventing the activation of PKB (AKT). PKB inhibits GSK3β by Ser9 phosphorylation. Inhibition of PKB by RhoA/ROCK activates GSK3β; (2) Aβ ligation of p75NTR activates RhoA by Tyr42 phosphorylation. Activated RhoA/ROCK phosphorylates and activates Src at the Tyr416 residue. Activated Src phosphorylates and activates GSK3β at the Tyr216 residue, subsequently activating IKK. IKK phosphorylates IκB at Ser32/36, marking it for proteasomal degradation and impeding NF-κB inhibition. NF-κB increases BACE-1 expression which increases the activity of amyloidogenic processing of APP into Aβ. Newly processed Aβ oligomers bind to p75NTR and complete the positive feedback cycle; (3) Aβ binds to the extracellular domains of FZD and LRP6, preventing Wnt ligand interactions with the LRP6/FZD complex and successive inhibition of GSK3β. It is noteworthy that GSK3β is capable of phosphorylating PS1 and the Ser353 and 357 residues. However, the exact effect on Aβ processing is unclear; (4) Overactivation of GSK3β induces AD pathogenesis such as Aβ deposition, tau hyperphosphorylation, neurotic plaque formation, and cognitive decline.
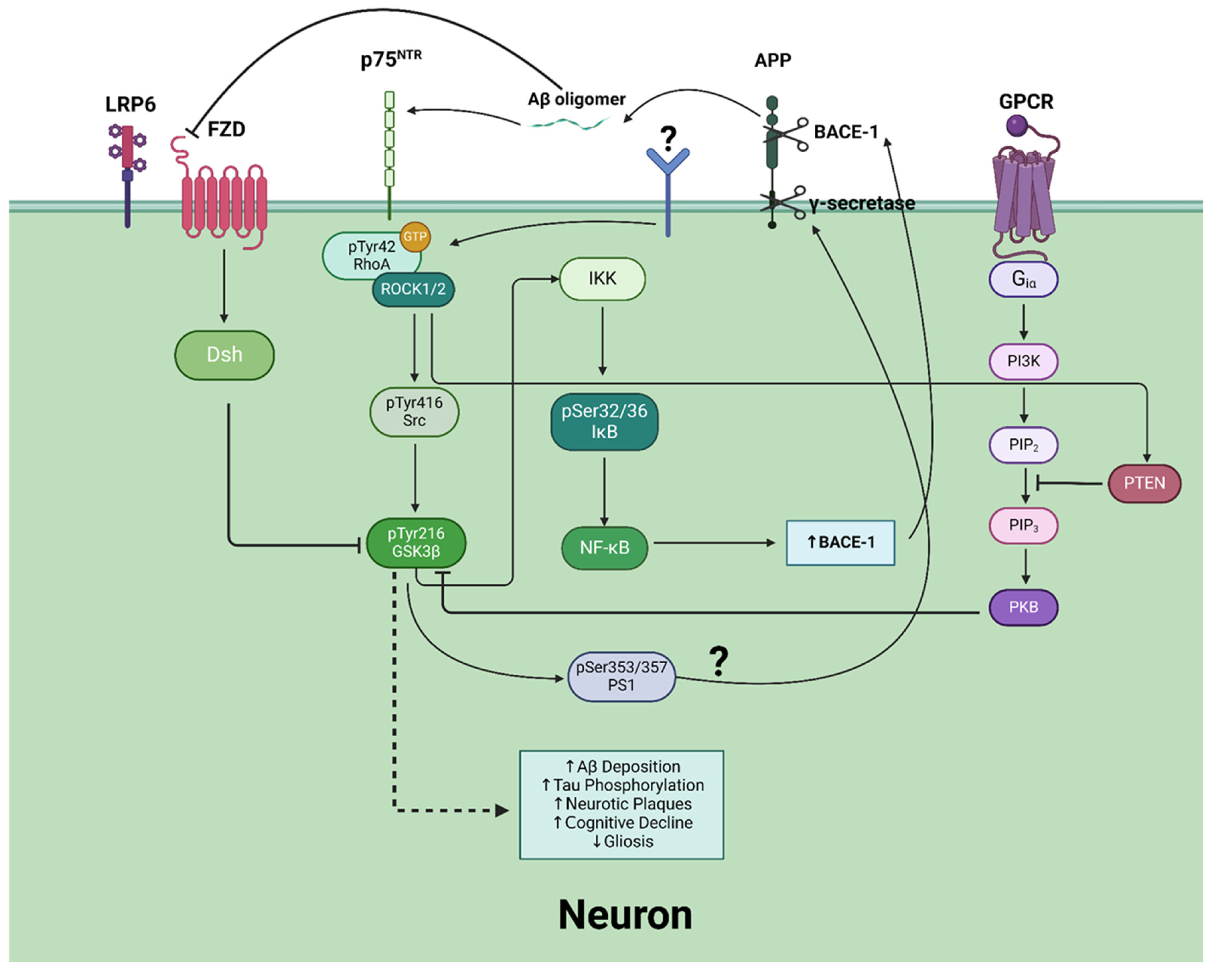
Figure 3.
Signaling pathway depicting the potential for TG2 to serve as a sTREM2 receptor and inhibit the RhoA/ROCK/GSK3β pathway. A potential adaptor molecule is shown, as TG2 lacks a transmembrane domain. However, what this adaptor protein is, and if there truly is an adaptor protein altogether, is still under investigation.
Figure 3.
Signaling pathway depicting the potential for TG2 to serve as a sTREM2 receptor and inhibit the RhoA/ROCK/GSK3β pathway. A potential adaptor molecule is shown, as TG2 lacks a transmembrane domain. However, what this adaptor protein is, and if there truly is an adaptor protein altogether, is still under investigation.
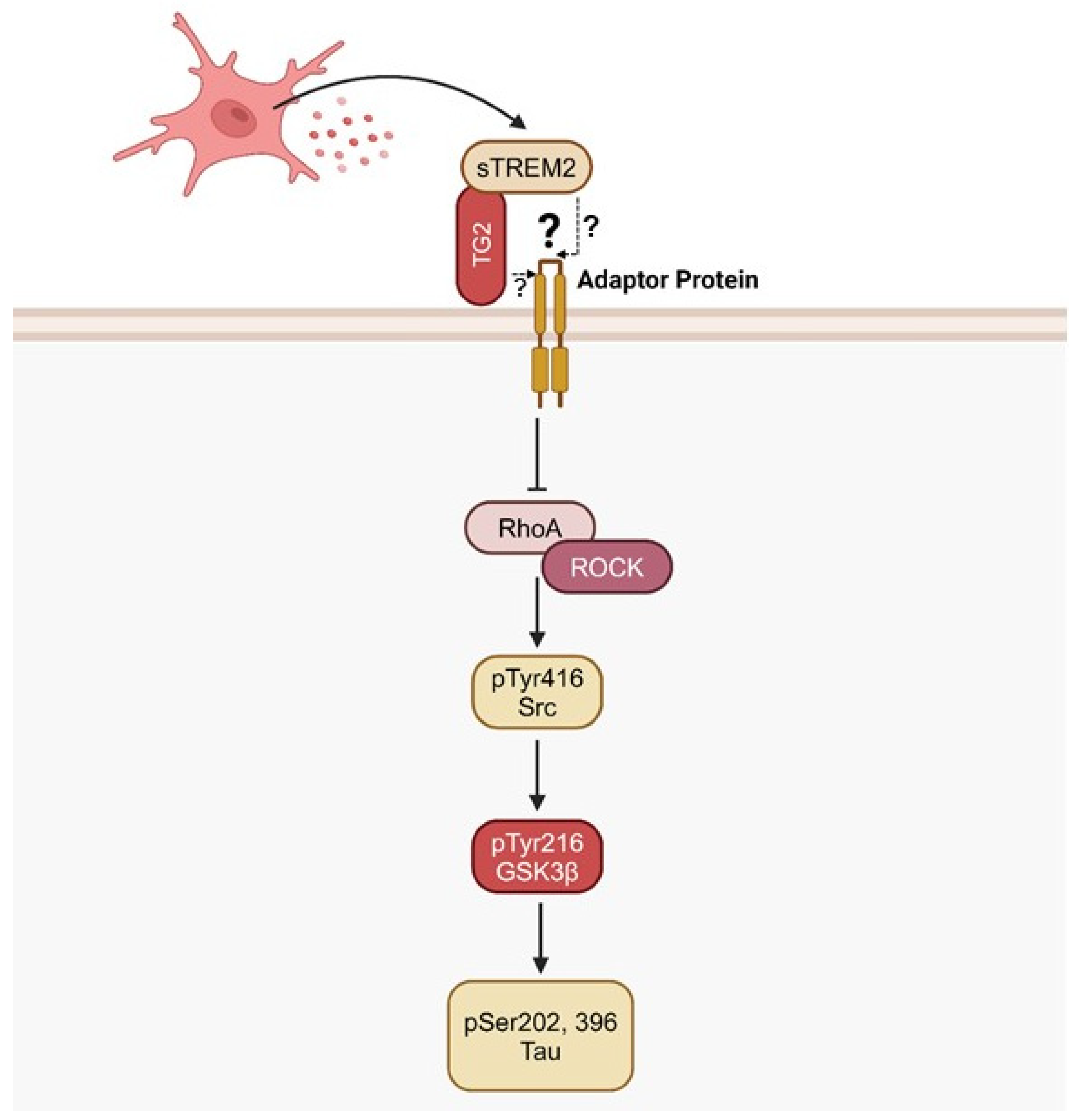
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



