Submitted:
15 January 2025
Posted:
16 January 2025
You are already at the latest version
Abstract
A continuous-flow process was optimised for the perchlorination of p-cresol to the corresponding 2,4,6-trichloro-cyclohexa-2,5-dienone derivative employing trichloroisocyanuric acid as a green and safer to handle chlorinating agent. The system could furnish 200 grams of pure material within 5 hours of operation (throughput = 40 g h-1).
Keywords:
Chlorination
; Flow chemistry 2
; trichloroisocyanuric acid
1. Introduction
2,4,6-trichloro-cyclohexa-2,5-dienone derivatives have been studied as insecticides and chemotherapeutics [1,2,3,4]. These derivatives have also been used as mild active chlorinating agents for selective para-chlorination of phenols, achieving over 95% selectivity [5,6,7]. We also anticipated they may hold promise as mild chlorination sources in several new reactions such as organocatalytic and photochemical halogenations. However, there availability is currently limited and thus restricts their wider evaluation and application.
Various preparative procedures have been investigated for their preparation which consist of perchlorinating phenolic rings utilizing either elemental chlorine or other strong chlorinating agents such as N-chlorimides [8,9,10]. In 1883, Benedikt and Schmidt first reported perchlorination employing chlorine gas as oxidising agent [11]. In 1959, Mukawa et al. isolated a dienone-estradiol employing a combination of surfuryl chloride and acetic anhydride, subsequently the same group investigated the alternative usage of trichloroisocyanuric acid (TCCA) [3,12,13]. Later, in 1993, Jacquesy et al. reported a selective ipso-chlorination of phenols employing antimony pentachloride [14]. More recent methodologies have also been reported which utilise sodium hypochlorite pentahydrate, 1,3-dichloro-5,5-dimethylhydantoin (DCDMH), and NaCl/Oxone [15,16,17,18].
Our research group is interest in developing efficient environmental-friendly continuous processes. We have significant experience using the readily available, low cost and easy-to-handle reagent, TCCA, which is a potential alternative to other more toxic and unstable chlorinating agents. Furthermore, its active chlorine content of 91.5% per molecule makes TCCA a fantastic candidate for developing atom-economic processes [19,20]. The apolar characteristics inherited by the three chlorine atoms allows TCCA to be highly soluble in most common organic solvents such as ethyl acetate, methanol, acetone, and toluene, whereas its by-product (cyanuric acid) has a much lower solubility and can often be filtered following precipitation from the mixtures.
Since the beginning of this century, chemists around the world have been searching for new processing methodologies and tools to perform synthetic organic chemistry more efficiently. Flow chemistry is one of the tools that has been achieving much attention across the various hierarchies of the chemical manufacturing industry [21,22,23,24]. Performing chemical transformations through flow streams allows for increased heat and mass transfer as well as a reduced risk of dangerous reactant accumulation during process scale-ups. Better control of critical process parameters such as temperature and reaction times generate improved reaction profiles and as such has found good uptake in the processing of pharmaceutical active ingredients (API) respecting current Good Manufacturing Practice (cGMP) legislation.
Combining our interests in TCCA as a chlorinating agent and flow chemistry as a synthesis tool, we report the first continuous flow perchlorination of p-cresol employing TCCA as the chlorinating agent.
2. Results
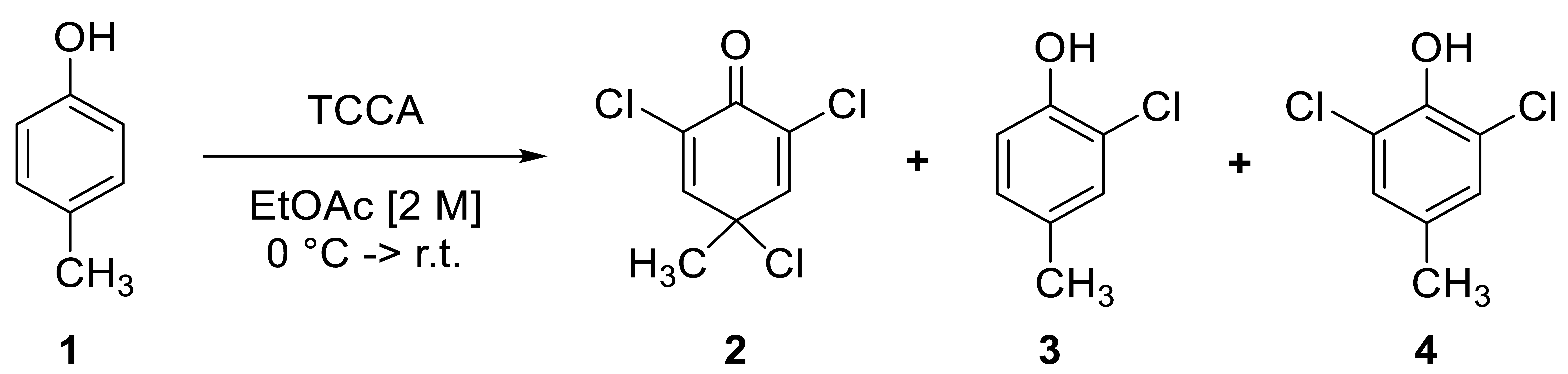
As a preliminary investigation, we decided to evaluate the batch reaction condition for the perchlorination of p-cresol (1) (Table 1). Our exploratory experiments were performed adding TCCA portion-wise at 0 °C to a 2 M solution of 1 in ethyl acetate (10 mmol scale). An increasing quantity of TCCA was investigated to evaluate the best conditions to realise complete conversion to the desired 4-methyl-2,4,6-trichloro-cyclohexa-2,5-dienone (2). As shown in table 1, when 2 or 2.2 equivalents of TCCA (6 or 6.6 mole equivalent electrophilic chlorine) were employed, perchlorination occurred only partially and if extended reaction times of more than 30 minutes were used, de-chlorination with the resultant formation of 3 and 4 was encountered (Entries 1-4). Increasing the equivalents of TCCA to 2.5 afforded the highest conversions of 2 and reducing undesired impurities (3 & 4) (Entry 5). Further increasing TCCA did not improve the conversion (Entry 6). It should be noted that each process needed to be carefully cooled at the start as a strong exothermic process was associated with the chlorination, unchecked this gave rise to a 30-38 °C temperature increase. In addition, the perchlorination produces copious amounts of white cyanuric acid precipitate which are almost completely insoluble in the EtOAc solvent system.
Having outlined a set of viable reaction conditions, we decided to convert the sequence to a flow system capable of continuously chlorinating p-cresol (1), taking into account the biphasic nature of the reaction and notable exotherm. Many techniques have been implemented to avoid solid accumulation when dealing with slurry in flow [25,26,27,28,29]. High flowrates and ultrasonic radiation have been employed many times and these can be easily adopted in a chemistry laboratory [30,31,32,33,34,35]. Furthermore, due to the high surface area of a coiled tubular flow reactor, the heat generated from the TCCA addition could be easily dissipated without the need for excessive external cooling of the reaction stream. Finally, the improved mixing, created by the turbulent flow in the reactor, should lead to an improved control and therefore selectivity reaction.
Based upon our initial batch reaction conditions, and following some scoping runs pertaining to solubiltiy limits and precipriation (avoidance of reactor blockage) we determined the following optimised procedure. A 1.5 M stock solution of p-cresol (1) in EtOAc was merged with a flow stream comprising a 0.5 M solution of TCCA in the same solvent (Scheme 1, Figure 1). A Y-shaped PEEK thru mixer (1/4-28 Y mixer .020 in thru) was used to blend the two streams. The unified stream was subsequently directed through a 10 mL PTFE coil (1.5 mm I.D.) placed in an ultrasonic bath (Ultrawave 50-60 Hz) maintained at 25 °C (Scheme 1, Figure 1). The exiting reaction mixture (69 second residence time) was collected for 5 hours in a Schott bottle and then filtered over celite. We tested the reaction mixture upon directly exiting the reactor and again sampled after standing for 1, 3 and 5 h but this showed no change in composition (GC peak area 94.6% 2, 4.5% 3 and 0.9% 4). After filtration and solvent evaporation, an orange liquid was obtained that on standing crystallised. The final product 2 could be obtained in pure form by recrystallisation (hexane/EtOAc 20:1) isolated in 86% (204 g) yield (throughput = 40 g h-1). The flow system proved robust enabling repeated runs to be performed at different scales and thus fresh material could be generated on demand in a very simple set-up.
3. Materials and Methods
All solvents were purchased from Fisher Scientific and used without further purification. Substrates, their precursors and reagents were purchased from Fluorochem. 1H-NMR spectra were recorded on Bruker Avance-400 instrument and are reported relative to residual solvent: CDCl3 (δ 7.26 ppm). 13C-NMR spectra were recorded on the same instruments and are reported relative to CDCl3 (δ 77.16 ppm). Data for 1H-NMR are reported as follows: chemical shift (δ/ ppm) (multiplicity, coupling constant (Hz), integration). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, s, br = broad singlet, app. = apparent. Data for 13C-NMR are reported in terms of chemical shift (δC/ ppm). IR spectra were obtained using a Perkin Elmer Spectrum Two UATR Two FT-IR Spectrometer (neat, ATR sampling) with the intensities of the characteristic signals being reported as weak (w, <20% of tallest signal), medium (m, 21-70% of tallest signal) or strong (s, >71% of tallest signal).
Low resolution gas chromatography mass spectrometry (GC-MS) was performed on a Shimadzu QP2010-Ultra equipped with an Rxi-5Sil MS column (0.15 µm x 10 m x 0.15 mm) in EI mode. Reactions were conducted in flow using Vapourtec SF-10 as peristaltic pumps, along with 0.5-1.5 mm PTFE tubing. A PEEK 1/4-28 Y mixer 0.020 in thru was employed. All the connectors tubing were 1/4” OD. The ultrasonic cleaning bath employed is a Ultrawave U300H.
For TLC, Sigma Aldrich glass-backed plates were used, and visualisation was performed using UV-irradiation and KMnO4 stain. Organic solutions were concentrated under reduced pressure using a Buchi rotary evaporator and hi-vacuum was achieved using an Edwards RV5 pump and Schlenk line.
Procedure for the continuous perchlorination of p-cresol (1).
Pumping of the solutions was performed using two independently controlled Vapourtec SF10 Laboratory pumps.
A 1.5 M solution of p-cresol in AcOEt (flow rate = 2.5 mL min-1) was merged with a solution 0.5 M of TCCA in AcOEt (flow rate = 6.25 mL min-1) and progressed into a 0.5 mm PTA coil reactor (Volume = 10 mL) placed into an ultrasonic bath maintained at 25 °C. The heterogenous mixture was collected in a Schott bottle for a fixed period. The mixture was filtered through celite and the solvent evaporated under vacuum. The orange liquid residue was solubilised in a 20:1 Hexane:AcOEt mixture and left at −20 °C overnight to furnish an off white crystalline product.
2,4,6-trichloro-4-methylcyclohexa-2,5-dien-1-one (2): 1H NMR (400 MHz, CDCl3) δ 7.14 (s, 2H), 1.91 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.93, 145.48, 131.18, 60.94, 29.98. FT-IR νmax 3058 (CH, w), 2989 (CH, w), 1681 (C=O, s), 1599 (s), 1446 (m), 1320 (m), 1038 (s), 908 (s), 769 (s), 654 (s). GC-MS Rt 3.77 min, m/z 176.0 [M-Cl]+. m.p. 110.9–112.8 °C.
X-ray: (CIF: CCDC 2415243): Crystal Data for C7H5Cl3O (M = 211.46 g/mol): monoclinic, space group P21. a = 5.3071(5) Å, b = 10.6875(10) Å, c = 7.7006(7) Å, α/°: 90. β/°:105.038(4). γ/°:90. Volume/Å3: 421.82(7). Z: 2. ρcalcg/cm3: 1.665. μ/mm-1: 1.019. F(000): 212.0. Crystal size/mm3: 0.246 × 0.152 × 0.078. Radiation MoKα: (λ = 0.71073). 2Θ range for data collection/°: 5.478 to 61.044. Index ranges: -7 ≤ h ≤ 7, -15 ≤ k ≤ 15, -10 ≤ l ≤ 10. Reflections collected: 8893. Independent reflections: 2571 [Rint = 0.0295, Rsigma = 0.0305]. Data/restraints/parameters: 2571/1/120. Goodness-of-fit on F2: 1.063. Final R indexes [I>=2σ (I)]: R1 = 0.0238, wR2 = 0.0563. Final R indexes [all data]: R1 = 0.0290, wR2 = 0.0577. Largest diff. peak/hole / e Å-3: 0.29/-0.23. Flack parameter: -0.02(3).
The 1H NMR characterization and indicative IR signals for compound 2 match well with those previous reported via an alternative synthetic procedure.[37] However, the determined melting point of 89-90 °C [crystallized from petroleum ether (b.p. 30-60 °C)][37] did not match our findings 110.9–112.8 °C [crystallized from 20:1 Hexane:AcOEt]. Although the additional obtained 13C NMR and x-ray single crystal structure provided further evidence for the authentication of the material obtained.
4. Conclusions
The described procedure is operationally simple and readily scalable offering easy access to multigram quantities (40 g h-1, 86% isolated yield) of 2,4,6-trichloro-cyclohexa-2,5-dienone (2) for the first time in a safe and much improved yield compared to the existing literature [9,36]. We also believe that the general approach should also be readily adaptable to other derivatives.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, I.R.B and G.G.; methodology, G.G and T.Q.H; formal analysis, G.G.; investigation, G.G, I.R.B. and T.Q.H; data curation, G.G and T.Q.H;.; writing—original draft preparation, G.G. and I.R.B.; supervision and project administration, I.R.B; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Leftwick, A.P. Parnell E.W., New pesticides derived from cyclohexadiene, OA5232A, 1981. https://patents.google.com/patent/OA5232A/en?oq=OA5232A.
- Takegawa, B. , Steroid Compound, JPH0267296A, 1988. https://patents.google.com/patent/JPH0267296A/en?oq=JPH0267296.
- Mukawa, F. , 10β-Chloro-17β-hydroxyestra-1,4-dien-3-one and its related compounds. J. Chem. Soc., Perkin Trans. 1988, 1, 457–460. [Google Scholar] [CrossRef]
- Kaiser, K.L.E. , Niculescu S. P., On the PNN modeling of estrogen receptor binding data for carboxylic acid esters and organochlorine compounds, Water Qual. Res. J. Canada. 2001, 36, 619–630. [Google Scholar] [CrossRef]
- Kato, T. , Ichinose I. , Selective bromination of polyenes by 2,4,4,6-tetrabromocyclohexa-2,5-dienone, J. Chem. Soc. Perkin Trans. 1980, 1, 1051–1056. [Google Scholar] [CrossRef]
- Guy, A.; Lemaire, M.; Guetté, J.P.; Halogenation regioselective en serie, a.r.o.m.a.t.i.q.u.e.-I.I. Chloration des naphtols et de leurs ethers a l’aide de reactifs mettant en jeu des interactions du type donneur-accepteur. Tetrahedron 1982, 38, 2347–2354. [Google Scholar] [CrossRef]
- Mamaghani, M. , Zolfigol M. A., Shojaei M., N-Chloro-2,3,4,4,5,6-hexachlorocyclohexa-2,5-dienylideneamine as a mild and highly regioselective chlorinating reagent. Synth. Commun. 2002, 32, 735–740. [Google Scholar] [CrossRef]
- Fischer, A. , Henderson G. N., Ipso chlorination of 4-alkylphenols. Formation of 4-alkyl-4-chlorocyclohexa-2,5-dienones. Can. J. Chem. 1979, 57, 552–557. [Google Scholar] [CrossRef]
- Bergquist, K.-E. , Nilsson A. , Ronlán A., Norin T., Hjeds H., Electrophilic Chlorination of 4-Methylphenols with Molecular Chlorine. Synthesis of Dimethoxy Aromatics by Methanolysis of 4-Chloro-4-methylcyclohexa-2,5-dienones. Acta Chem. Scand. 1982, 36b, 675–683. [Google Scholar] [CrossRef]
- Antinori, G.; Baciocchi, E.; Illuminati, G. ; Non-conventional paths in electrophilic aromatic reactions Part, V.I. Chlorination of 3,5-dichloro-2,4,6-trimethylanisole and related compounds. J. Chem. Soc. B Phys. Org. 1969, 373. [Google Scholar] [CrossRef]
- Benedikt, R. , Schmidt M. V., Notizen über Halogenderivate. Monatshefte Für Chemie. 1983, 4, 604–609. [Google Scholar] [CrossRef]
- Mukawa, F. , Suzuki T. , Ishibashi M., Yamada F., Estrogen and androgen receptor binding affinity of 10β-chloro-estrenen derivatives, J. Steroid Biochem. 1988, 31, 867–870. [Google Scholar] [CrossRef]
- Mukawa, F. The anomalous chlorination of estradiol 17β-acetate with isocyanuric chloride, Tetrahedron Lett. 1959, 1, 17–20. [Google Scholar] [CrossRef]
- Ferron, B. , Jacquesy JC., Jouannetaud M.P., Karam O., Coustard J.M., Ipso-chlorination of 4-alkylphenols ethers a novel route to 4-chlorocyclohexa-2,5-dienones., Tetrahedron Lett. 1993, 34, 2949–2952. [Google Scholar] [CrossRef]
- Uyanik, M. , Sasakura N, Kuwahata M., Ejima Y., Ishihara K., Practical oxidative dearomatization of phenols with sodium hypochlorite pentahydrate, Chem. Lett. 2015, 44, 381–383. [Google Scholar] [CrossRef]
- Zhang, Z. , Sun Q, Xu D., Xia C., Sun W., Direct halogenative dearomatization of 2-naphthols by NXS (X = Cl, Br) in water, Green Chem. 2016, 18, 5485–5492. [Google Scholar] [CrossRef]
- Yin, Q. , Wang SG., Liang X.W., Gao D.W., Zheng J., You S.L., Organocatalytic asymmetric chlorinative dearomatization of naphthols, Chem. Sci. 2015, 6, 4179–4183. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M. , Sahara N, Ishihara K., Regioselective Oxidative Chlorination of Arenols Using NaCl and Oxone, Euro. J. Org. Chem. 2019, 2019, 27–31. [Google Scholar] [CrossRef]
- Tilstam, U. , Weinmann H, Trichloroisocyanuric Acid: A Safe and Efficient Oxidant, Org. Process Res. Dev. 2002, 6, 384–393. [Google Scholar] [CrossRef]
- Gaspa, S. , Carraro M, Pisano L., Porcheddu A., De Luca L., Trichloroisocyanuric Acid: a Versatile and Efficient Chlorinating and Oxidizing Reagent, Euro. J. Org. Chem. 2019, 2019, 3544–3552. [Google Scholar] [CrossRef]
- Gambacorta, G. , Sharley JS., Baxendale I.R., A comprehensive review of flow chemistry techniques tailored to the flavours and fragrances industries, Beilstein J. Org. Chem. 2021, 17, 1181–1312. [Google Scholar] [CrossRef] [PubMed]
- Sagmeister, P. , Williams JD., Hone C.A., Kappe C.O., Laboratory of the future: A modular flow platform with multiple integrated PAT tools for multistep reactions, React. Chem. Eng. 2019, 4, 1571–1578. [Google Scholar] [CrossRef]
- Webb, D. , Jamison TF., Continuous flow multi-step organic synthesis, Chem. Sci. 2010, 1, 675–680. [Google Scholar] [CrossRef]
- Pastre, J.C. , Browne DL., Ley S.V., Flow chemistry syntheses of natural products, Chem. Soc. Rev. 2013, 42, 8849–8869. [Google Scholar] [CrossRef]
- Browne, D.L. , Deadman BJ., Ashe R., Baxendale I.R., Ley S.V., Continuous Flow Processing of Slurries: Evaluation of an Agitated Cell Reactor, Org. Process Res. Dev. 2011, 15, 693–697. [Google Scholar] [CrossRef]
- Deadman, B.J. , Browne DL., Baxendale I.R., Ley S.V., Back Pressure Regulation of Slurry-Forming Reactions in Continuous Flow, Chem. Eng. Technol. 2015, 38, 259–264. [Google Scholar] [CrossRef]
- Sharma, M.K. , Suru A., Joshi A., Kulkarni A.A., A Novel Flow Reactor for Handling Suspensions: Hydrodynamics and Performance Evaluation, Ind. Eng. Chem. Res. 2020, 59, 16462–16472. [Google Scholar] [CrossRef]
- Bianchi, P. , Williams JD., Kappe C.O., Oscillatory flow reactors for synthetic chemistry applications, J. Flow Chem. 2020, 10, 475–490. [Google Scholar] [CrossRef]
- Doyle, B.J. , Gutmann B, Bittel M., Hubler T., Macchi A., Roberge D.M., Handling of Solids and Flow Characterization in a Baffleless Oscillatory Flow Coil Reactor. , Ind. Eng. Chem. Res. 2020, 59, 4007–4019. [Google Scholar] [CrossRef]
- Hartman, R.L. , Naber JR., Zaborenko N., Buchwald S.L., Jensen K.F., Overcoming the Challenges of Solid Bridging and Constriction during Pd-Catalyzed C−N Bond Formation in Microreactors, Org. Process Res. Dev. 2010, 14, 1347–1357. [Google Scholar] [CrossRef]
- Schoenitz, M. , Grundemann L, Augustin W., Scholl S., Fouling in microstructured devices: A review, Chem. Commun. 2015, 51, 8213–8228. [Google Scholar] [CrossRef] [PubMed]
- Wu, K. , Kuhn S, Strategies for solids handling in microreactors, Chim. Oggi/Chemistry Today. 2014, 32, 62–66. [Google Scholar]
- Dong, Z. , Zhao S, Zhang Y., Yao C., Yuan Q., Chen G., Mixing and residence time distribution in ultrasonic microreactors, AIChE J. 2017, 63, 1404–1418. [Google Scholar] [CrossRef]
- Rivas D., F. , Kuhn S, Synergy of Microfluidics and Ultrasound: Process Intensification Challenges and Opportunities, Top. Curr. Chem. 2016, 374, 1–30. [Google Scholar] [CrossRef]
- Dong, Z. , Delacour C, Carogher K.M., Udepurkar A.P., Kuhn S., Continuous ultrasonic reactors: Design, mechanism and application, Materials 2020, 13, 344–1. [Google Scholar] [CrossRef]
- Husain, S. , Kifayatullah, M., Phenol-dienone rearrangement in the chlorination of p-cresol with tert-butyl hypochlorite: Indian. J. Chem., Section B: Org. Chem. Incl. Med. Chem. 1985, 24B, 711–714. [Google Scholar]
- Chip, G. K. , Grossert, J. S. Can. J. Chem. 1972, 50, 1233–1240. [Google Scholar] [CrossRef]
Scheme 1.
Setup employed for the preparation of 2 under flow conditions.
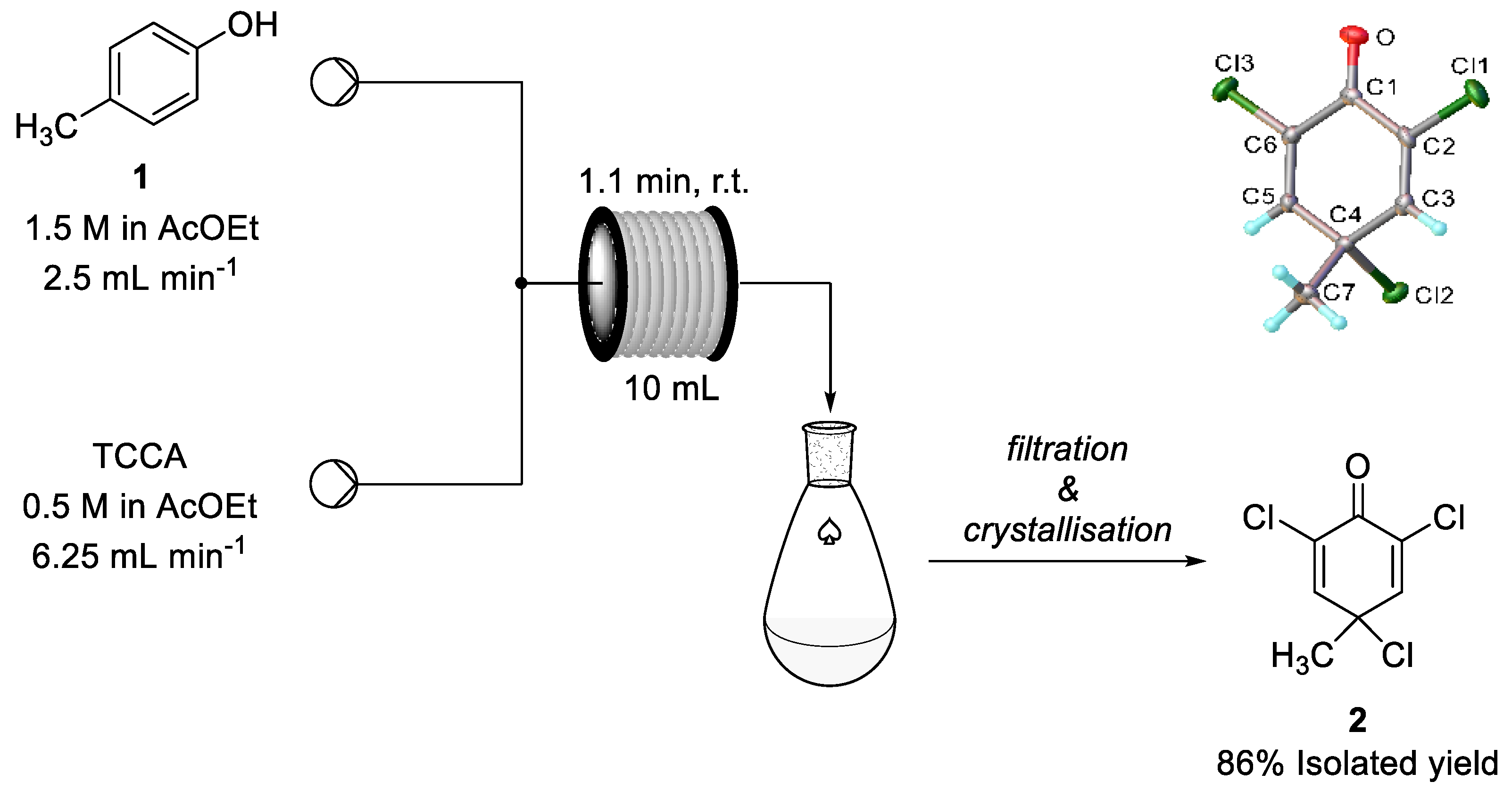
Figure 1.
Photo of the setup employed for the perchlorination of p-cresol to 2.


Table 1.
Screening of reaction conditions to perform an efficient perchlorination of p-cresol (1).
 | |||||
|---|---|---|---|---|---|
| Entry1 | TCCA (equiv.) | Residence Time (min) | GC-MS yield (%)2 3 4 | ||
| 1 | 2 | 20 | 57.7 | 42.3 | - |
| 2 | 2 | 30 | 75.0 | 25.0 | - |
| 3 | 2 | 60 | 67.0 | 20.9 | 11.9 |
| 4 | 2.2 | 30 | 75.0 | 25.0 | - |
| 5 | 2.5 | 30 | 84.9 | 9.4 | 5.7 |
| 6 | 2.7 | 30 | 83.7 | 16.3 | - |
1 The experiments were carried out on a 10 mmol scale.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



