1. Introduction
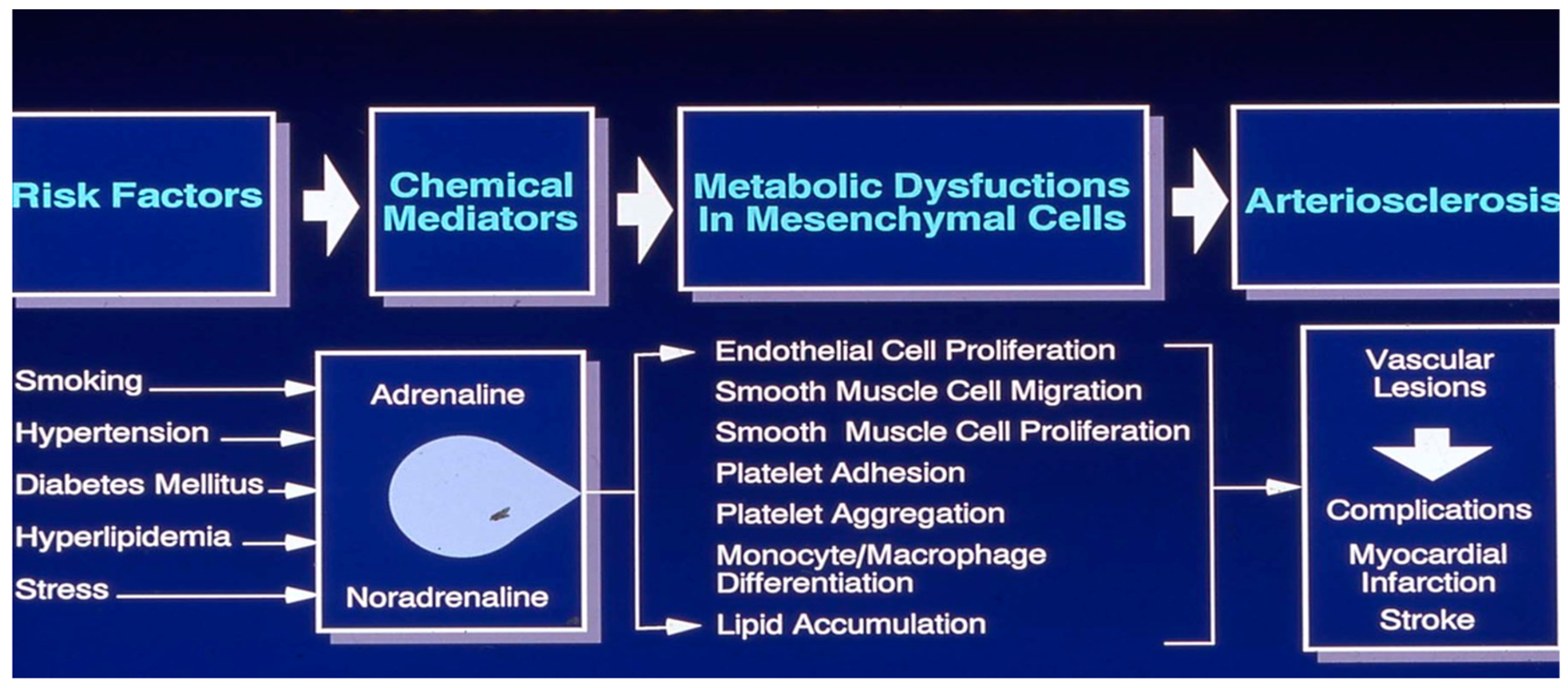
Cardiometabolic diseases accounted for more than 4.8 million deaths among the U.S. working-age population from 1990 to 2017[1]. Cardiovascular diseases (CVDs) remain the leading cause of mortality worldwide. Of the 20.5 million CVD-related deaths in 2021, approximately 80% occurred in low- and middle-income countries [2]. Much of what we know about heart disease's pathophysiology comes from the Framingham Heart Study Group, which identified traditional risk factors like high blood pressure, diabetes, and cigarette smoking. Additionally, the Framingham Heart Study (FHS) highlighted several critical risk factors contributing to heart disease: hypertension, high cholesterol—especially low-density lipoprotein (LDL) or "bad" cholesterol—smoking, obesity, physical inactivity, diabetes, family history, age, and sex [3]. Experts estimate that eliminating CVDs could increase the average lifespan by about 11 years [4].
Despite the identification of modifiable risk factors for CVD six decades ago, medical practice has been slow to evolve in managing metabolic diseases like hypertension, diabetes and obesity. Although high blood pressure, cholesterol, smoking, and inactivity were identified as risks in the 1950s and 1960s, it has taken decades for widespread adoption of these factors in medical management. Early detection and management of these risks can significantly reduce CVD incidence. Managing risk factors like hypertension, high cholesterol, smoking, and obesity remains essential to preventing heart attacks, strokes, and peripheral artery disease [5].
The INTERHEART study found that nine modifiable risk factors explain over 90% of the global risk of heart attacks across all regions and major ethnic groups, suggesting that managing these factors could prevent most premature myocardial infarctions [6]. Khera and colleagues at the Harvard University showed that even among participants with high genetic risk, a favorable lifestyle reduced the relative risk of coronary artery disease by nearly 50% [7]. Researchers at the Imperial College London reported a decline in cardiovascular mortality but an increase in diabetes mortality in high-income countries. Between 1980 and 2009, age-standardized cardiometabolic mortality fell across 26 industrialized nations, with reductions in risk factors accounting for 49% of the decline in men and 40% in women. However, no country has successfully reversed or prevented the rising incidence of vascular diseases [8].
Research underscores that managing CVD risk factors, such as high blood pressure, cholesterol, and diabetes, can decrease premature CVD mortality. Nonetheless, this reduction hasn't always resulted in a significant decline in overall CVD mortality rates,-highlighting the need for more comprehensive approaches. In 2019, experts from the National Institutes of Health concluded, “There have been substantial declines in premature CVD mortality in much of the U.S. population. However, increases in CVD mortality before age 50 years among American Indian/Alaska Native individuals, flattening rates in white people, and overall increases in deaths from hypertensive disease suggest that targeted public health interventions are needed to prevent these deaths” [9]. Valentine Fuster and colleagues advocate for a comprehensive approach, utilizing intervention opportunities across the lifespan to promote cardiovascular health, prevent risk accumulation, detect and mitigate risk, manage CVD events, and prevent disease progression and recurrence [10].
Some experts suggest that improving current CVD risk models by incorporating more risk factors could enhance treatment targeting. The Framingham Risk Score (FRS), a widely used model estimating 10-year coronary heart disease risk based on age, sex, cholesterol levels, smoking, and blood pressure, may benefit from adding new risk factors. These factors include a family history of CVD, diabetes, high-sensitivity C-reactive protein (hs-CRP), lipoprotein(a), and homocysteine. Including additional metrics such as coronary artery calcium score, cardiac troponin levels, waist circumference, LDL cholesterol, low HDL cholesterol, triglycerides, and glucose index (TGI) could offer a more comprehensive risk assessment. Though eliminating CVDs might be unrealistic, accurate individual risk assessment and preventive measures could significantly reduce cardiovascular disease rates and mortality [3].
According to the ancient systems of traditional medicines from India and Tibet, every individual is born with a unique blend of three primary energies. In Ayurveda, these energies are Vata, Pitta, and Kapha, which correspond to Loong (movement energy), Tripa (heat energy), and Baekan (cold energy) in Tibetan Medicine. These Tri-Doshas oversee various functions at the cellular, tissue, and systemic levels, ensuring that the body maintains equilibrium. Professor Alex Hankey, Dean of Academic Studies at the Institute of Ayurveda Integrative Medicine, Bengaluru, India, explains, “Vata’s membrane transport governs homeostasis, enabling cells to maintain a regulated internal environment. This constancy ensures reliable enzyme function and keeps the organism far from equilibrium with the external environment. Pitta governs energy production, providing energy-rich molecules like ATP that drive cellular metabolism. This energy, described as ‘negative energy’ in thermodynamics, is crucial to sustaining an organized and structured organism. Kapha ensures lubrication, maintaining bodily structure. Together, these energies offer the most comprehensive definition of life ever formulated” [11]. Professor Hankey also emphasizes the importance of establishing scientific validity for the Tridosha concept, particularly in a scientific era, where relevance and evidence are paramount.
Modern views on altered metabolism and its role in initiating and advancing metabolic diseases closely parallel these ancient insights. Dong and colleagues highlight that “accumulating evidence suggests multiple intricate molecular and cellular mechanisms establish and maintain metabolic memory. This includes processes like epigenetic regulation, gluconeogenesis, insulin secretion, glycosylation of end products, and oxidative stress. Together, these interconnected mechanisms form a complex network governing metabolic memory and present novel targets for the detection and intervention of metabolic diseases” [13]. In 1990, British epidemiologist David Barker proposed a hypothesis, linking intrauterine growth retardation, low birth weight, and premature birth to the development of hypertension, coronary artery disease, and non-insulin-dependent diabetes in adulthood. According to Malhotra and associates, the hypothesis has two main components [14]. First, when a fetus faces nutrient limitations, it prioritizes resource allocation to protect brain development. Second, the fetus receives cues about the post-birth environment and adapts accordingly. Barker suggested that a fetus experiencing nutrient scarcity develops a ‘thrifty’ metabolic pattern. Although the genetic regulation mechanisms responsible for this metabolic programming were unclear at that time, research has since made advancements in understanding some of the mechanisms [15].
A news release from Children’s National Health System in Washington, DC, introduces research led by physician-scientist Dr. Robert Freishtat. The findings may be transformative for early intervention and prevention of obesity-related illnesses. It is well known that there is a direct correlation between visceral adipose (belly fat) and severe health complications, such as cardiovascular disease and insulin resistance leading to diabetes. However, the precise mechanisms linking increased belly fat to these diseases were not understood until recently. Dr. Freishtat notes that as visceral fat expands, fat cells undergo a transformation and begin releasing different exosomes compared to lean adipose cells. These new exosomal messages interfere with vital biological processes, impairing the body’s ability to manage sugar and cholesterol efficiently. Dr. Freishtat likens exosomes to “biological tweets”—brief messages that facilitate intercellular communication and alter gene expression. The research team collected fat tissue samples from lean and obese female patients, using modified bead-based flow cytometry to separate, identify, and compare the exosomal miRNA released by the fat cells. These researchers believe that isolating these exosomes has opened possibilities for developing diagnostic tests and potential early intervention strategies to delay or prevent obesity-related diseases [16,17].
Despite the ancient, observation-based knowledge of Tridosha, and modern concepts such as metabolic memory and the fetal programming hypothesis of adult diseases, we still lack a comprehensive understanding of the cellular and molecular mechanisms that initiate and advance metabolic risks. These mechanisms contribute to the onset and progression of metabolic diseases, which can eventually lead to acute, occlusive arterial events. While it is known that numerous cellular and molecular events drive the progression of metabolic disorders, understanding the complex interplay between altered metabolism, inflammation, oxidative stress, and vascular dysfunction is crucial. These factors are key in the development and progression of conditions such as hypertension, obesity, type-2 diabetes, and vascular diseases. This review highlights some of the significant molecular mechanisms involved in the pathogenesis of metabolic diseases. It is a complex topic, and readers are urged to refer to original articles and reviews on this subject for additional information [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32]. The initiation of metabolic risks and disease progression primarily involves oxidative stress, chronic low-grade inflammation, disrupted glucose and lipid metabolism, impaired insulin signaling, vascular atherosclerosis, and the activation of platelet and coagulation pathways. A better understanding of these processes is essential for developing effective preventive and therapeutic strategies for metabolic disorders.
2. Oxidative Stress and Cardiometabolic Diseases
Mitochondria are the powerhouses of cells, responsible for generating energy. When mitochondrial function is impaired, it can lead to a range of metabolic disorders. Mitochondrial dysfunction can result in oxidative stress, inflammation, and disruption of cellular metabolism, all of which are hallmarks of metabolic diseases like metabolic syndrome, diabetes, obesity and vascular diseases [33]. Mitochondria seem to play an important role in molecular signaling and determining the state of cellular health through generation of reactive oxygen species (ROS) [34,35,36,37,38,39,40,41,42,43,44]. Cytosolic sources of reactive oxygen species include NADPH oxidases (NOX), a complex of enzymes that generate superoxide by transferring electrons from NADPH to molecular oxygen. NOX enzymes are a major source of ROS in the cardiovascular system and are tightly regulated in response to various stimuli [44]. Xanthine oxidase also generates superoxide and hydrogen peroxide during the breakdown of purines. These enzymes play a role in oxidative damage, particularly under conditions of vascular ischemia and reperfusion. Cyclooxygenases (COX) involved in the production of prostanoids can also generate ROS as by-products during the oxidation of arachidonic acid. Drug metabolizing enzymes, -Cytochrome P450 are also involved in the oxidation of organic substrates contributing to ROS production, particularly in conditions of metabolic stress or inflammation.
As to the mitochondrial source of ROS, the primary site of ATP production where leakage of electrons from complexes 1 and 11 can reduce oxygen to form superoxide, especially under conditions of high mitochondrial membrane potential or electron transport dysfunction. Another source of ROS is monoamine oxidases (MAOs) located in the outer mitochondrial membrane that degrades neurotransmitters, producing hydrogen peroxide as by-product. This endogenously produced hydrogen peroxide can be neutralized by various antioxidants, including glutathione, catalase, and superoxide dismutase, which help maintain cellular redox balance and prevent oxidative damage. A mitochondrial adaptor protein, p66shc can influence ROS production and regulate cellular responses to oxidative stress, impacting aging and promoting vascular diseases[41]. A unique NADPH oxidase isoform, NOX4 localized in the mitochondria produces hydrogen peroxide, playing a role in redox signaling and mitochondrial function. ROS signaling is intricately balanced during normal cell function, with both beneficial and harmful outcomes depending on the context and levels of ROS produced. The tight regulation of ROS is crucial for maintaining vascular homeostasis and preventing pathological manifestations and dysfunction.
An imbalance between energy production and utilization can result in defective cell metabolism, a major factor in the development of metabolic syndrome. High glucose levels lead to excessive production of reactive oxygen species (ROS), causing mitochondrial morphological changes. Moreover, disruptions in the insulin signaling pathway lead to the accumulation of lipids, lipid peroxides, and free fatty acids (FFA), contributing to various metabolic disorders. Cellular energy dysfunction (CED) is a condition where cells are unable to efficiently generate energy leading to metabolic dysregulation. CED is often associated with metabolic diseases such as diabetes, obesity and metabolic syndrome,-characterized by impaired glucose and lipid metabolism, resulting in increased blood glucose and lipid levels. Glucose and lipid metabolism are dependent on mitochondria to generate energy in cells. Mitochondrial dysfunction seems to be associated with insulin sensitivity [35,36,37,38,39]. Recent research has focused on the development of therapeutic strategies targeting mitochondrial function and oxidative stress [38]. Kim and associates from the Department of Nutrition Sciences, University of Connecticut have reviewed the therapeutic potential of bioactive components such as resveratrol, quercetin, coenzyme Q10, curcumin, and astaxanthin as enhancers of mitochondrial function [39].
Although oxidative stress has been associated with various human diseases, including cardiovascular conditions, clinical trials involving oral antioxidant therapy have demonstrated limited or no effectiveness in reducing morbidity or mortality. For example, a recent meta-analysis of 50 randomized controlled trials, which included nearly 300,000 participants, found no significant benefit of vitamin or antioxidant supplementation in preventing cardiovascular outcomes, such as cardiovascular death. Traditionally, oxidative stress research has focused heavily on the role of reactive oxygen species, particularly superoxide. However, in recent years, the understanding of oxidative stress has expanded significantly, now encompassing the genetic origins of this imbalance. Nuclear factor erythroid 2-related factor 2 (NRF2) has emerged as a critical regulator of the antioxidant response, influencing the expression of hundreds of genes [40]. Researchers from the University of Wisconsin demonstrated that activating NRF2 with Protandim mitigates salt-induced vascular dysfunction and microvascular rarefaction [39]. Their findings suggest that direct activation of NRF2-regulated enzymatic antioxidant defenses through dietary interventions may offer a more effective therapeutic strategy for preventing and treating cardiovascular disease compared to supplementation with external antioxidant compounds. Joe McCord and colleagues have further noted that oxidative stress may be linked to around 200 diseases, potentially contributing to them, though not necessarily as a direct cause in every case [40].
3. Chronic Inflammation and Cardiometabolic Diseases
Inflammation and oxidative stress are interrelated pathophysiological processes that can mutually influence one another. When one occurs, it often triggers the other, contributing significantly to the pathogenesis of many chronic metabolic diseases. Oxidative stress arises from an imbalance between the production of reactive oxygen species (ROS) and the body's ability to neutralize them, leading to cellular damage and activation of inflammatory pathways. In contrast, inflammation is the body's protective response to injury or infection, but chronic inflammation driven by persistent oxidative stress can lead to many health issues[41,42,43,44].The connection between oxidative stress and inflammation is cyclical. Oxidative stress can initiate inflammatory responses, and inflammation can further exacerbate oxidative stress, creating a feedback loop that contributes to conditions like hypertension, diabetes, obesity, vascular dysfunction and cardiovascular diseases (CVD)[32].
The pathophysiology of CVD, which includes vascular dysfunction, hypertension, atherosclerosis, and diabetes mellitus (DM), is heavily influenced by chronic inflammation. Low-grade, sustained inflammation is a key factor in insulin resistance and hyperglycemia, leading to DM and its related microvascular and macrovascular complications. Moreover, inflammation is a major promoter of atherosclerosis, accelerating CVD progression[42,43,44]. Chronic, low-grade systemic inflammation becomes more prevalent with aging and is associated with increased risk of all-cause mortality in older adults, independent of other risk factors. Thus, systemic inflammation is a crucial determinant of long-term health outcomes in this population[44]. Polyphenols, antioxidant compounds found in plant-based foods, are being explored for their potential to mitigate the effects of oxidative stress and inflammation. Further research is needed to elucidate the precise molecular mechanisms linking inflammation to metabolic disease progression, which could promote the development of effective and specific anti-inflammatory therapies[32].
Sterile inflammation, a non-infectious immune response driven by damage-associated molecular patterns (DAMPs), plays a significant role in the initiation and advancement of metabolic diseases[45]. The NOD-like receptor (NLR) family, -pyrin domain-containing 3 (NLRP3) inflammasome, is a key mediator of sterile inflammation. This protein complex regulates caspase-1 and promotes the release of interleukin (IL)-1 family of cytokines, which are implicated in cardiometabolic diseases. The inflammasome’s activation, particularly through cytokines like IL1β and IL-18, has adverse effects on cardiometabolic disease progression. The Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) provided evidence that anti-inflammatory treatment with Canakinumab, an antibody targeting inflammation, significantly reduced levels of high-sensitivity C-reactive protein (hs-CRP) and IL-6 in patients with atherosclerotic disease, independently of lipid-lowering effects[46,47]. In this context, CRP just serves as a biomarker for inflammation. However, reducing CRP alone may not necessarily lower metabolic risks or prevent cardiovascular events, as the relationship between inflammation and metabolic disease is complex.
4. Atherosclerosis and Cardiometabolic Diseases
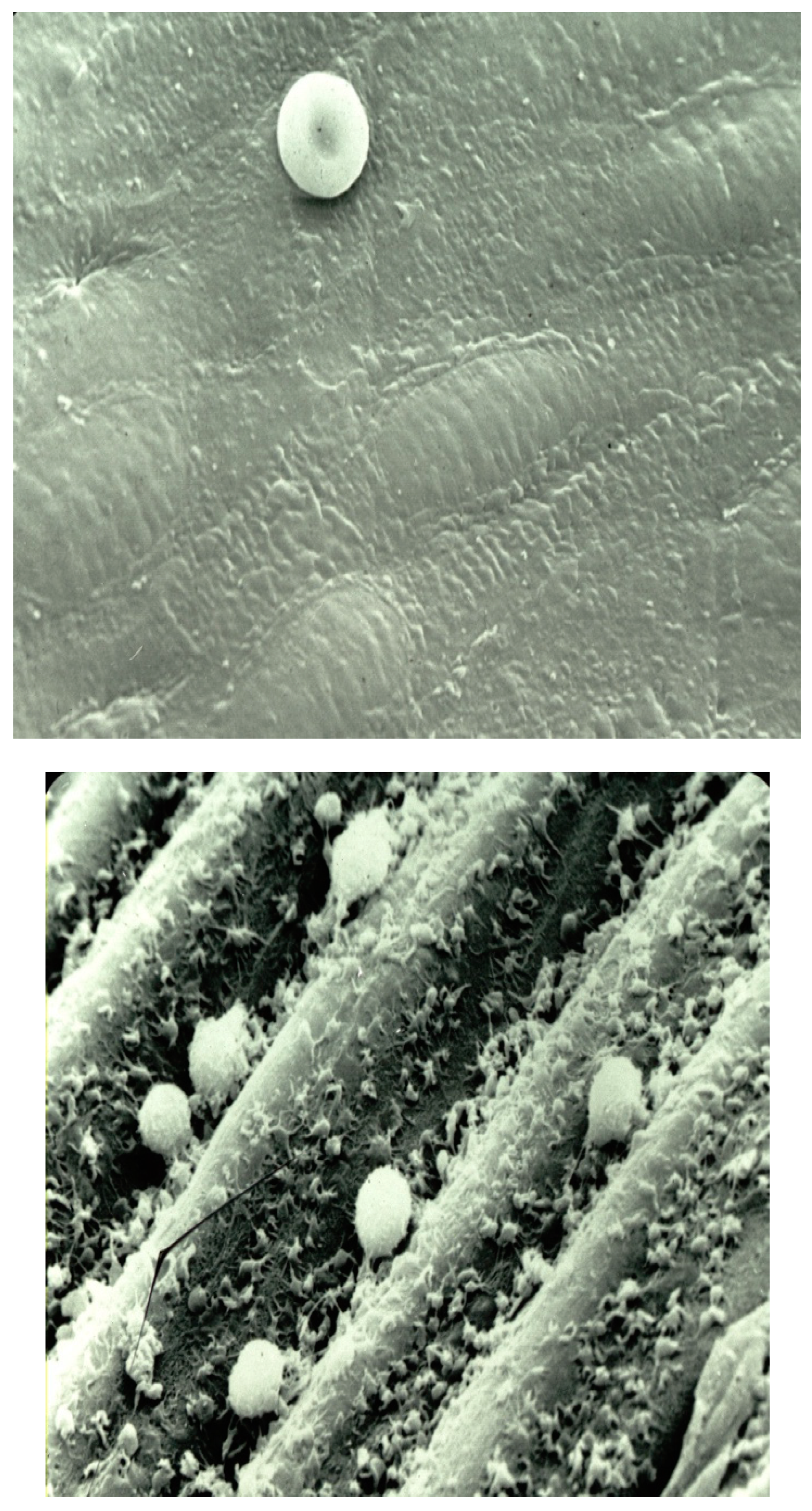
Atherosclerosis is a disease characterized by the accumulation of lipids, lipid peroxides, fibrous elements, and calcification, leading to plaque buildup on the arterial walls of blood vessels. The pathophysiology of atherosclerosis is complex process and involves multiple stages, including damage to the endothelium, which allows lipids to infiltrate the arterial wall. This damage promotes monocyte adherence to endothelial cells, where monocytes transform into macrophages. Smooth muscle cells then migrate from the media to the intima, and collagen and elastic fibers are produced. A fibrous cap forms around lipid pools, creating a mature atherosclerotic plaque. These plaques may become calcified and, upon rupture, can induce arterial occlusion, leading to ischemia and potentially causing stroke or myocardial infarction. Professor Russell Ross described atherosclerosis as an inflammatory disease in his seminal article published in the
New England Journal of Medicine [48]. He explained that injury to the endothelium results in alterations in the normal properties of endothelial cells concerning leukocyte and platelet adhesion (
Figure 1). Under normal conditions, platelets and leukocytes do not adhere to the endothelium (
Figure 1A). However, when endothelial dysfunction occurs, these cells interact with the damaged endothelium (
Figure 1B). The molecular mechanisms involved in these interactions are discussed under the section "Platelet-Vessel Wall Interactions".
Monocytes are circulating leukocytes that interact with endothelial cells under various physiological and pathological conditions. This interaction is mediated through a multistep process involving a variety of cell surface adhesion molecules. At sites where the endothelium is injured or activated, patrolling monocytes promote endothelial cell proliferation by secreting growth factors such as VEGF. In turn, activated endothelial cells facilitate the recruitment and migration of monocytes. The ability of monocytes to significantly promote endothelial cell proliferation and migration plays a crucial role in processes like wound healing and vascular remodeling. This dynamic interplay is essential for maintaining vascular and tissue homeostasis and responding to injury or inflammation. Monocytes are drawn to specific areas within blood vessels or tissues by chemokines and receptors, where they transdifferentiate into macrophages in response to tissue damage or infection[49,50]. Bioactive molecules released by infiltrating macrophages, such as monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor (TNFα), attract additional monocytes and expose them to adhesion molecules.
Atherosclerosis is a complex process involving formation of glycosylated hemoglobin and advance glycated products, accumulation of lipids, inflammatory cells, and fibrous elements in the large arteries, leading to plaque formation and potential rupture. Oxidative stress, caused by an imbalance between free radical production and antioxidant defenses, can damage the endothelium, the inner lining of blood vessels, making them more susceptible to atherosclerosis. Inflammation, triggered by oxidative stress, also contributes to the development of atherosclerosis by promoting the adhesion and migration of inflammatory cells, such as macrophages, into the arterial wall. These cells ingest lipids, becoming foam cells that accumulate and contribute to the progression of vascular atherosclerosis.
The development of the lipid-rich core in atherosclerotic plaques is a complex process that begins early in lesion progression, particularly during the transition from fatty streaks to fibrous plaques[5]. These cores are often acellular, with lipid deposits primarily located in the extracellular matrix. Low-density lipoprotein (LDL) aggregates in the matrix, forming lipid-rich deposits high in sphingomyelin, a lipid that impacts signaling pathways by reducing protein kinase-C activity. Myeloperoxidase, an oxidative enzyme produced by monocytes and granulocytes, is frequently found in both the shoulder regions and the core of fibrous plaques. Additionally, cholesterol crystals are often observed, suggesting that local cell membranes may become oversaturated with cholesterol, exceeding physiological levels. Studies have demonstrated the efficacy of continuous statin therapy in controlling plaque progression and reducing plaque volume[52,53].
Oxidized lipids, particularly oxidized low-density lipoprotein (oxLDL), play a critical role in atherosclerosis development. The oxidation of LDL cholesterol (LDL-C) generates oxidized and hydroxidized fatty acid products that drive this process. OxLDL promotes the expression of adhesion molecules on cell surfaces, activating endothelial cells. Modifications to LDL particles confer atherogenic properties. Additionally, oxidized phospholipids (Lps) can interact with platelet receptors, such as protease-activated receptor 1 and CD36, providing a biological basis for their pro-aggregatory effects. Lipoprotein(a) {Lp(a)} subfractions also increase thromboxane production in endothelial cells, an effect observed in vitro studies on platelets [54,55,56,57,58,59]. Studies from our laboratory at the University of Minnesota demonstrated that cholesterol- enriched platelets produce increased levels of thromboxane on stimulation compared to the control platelets. Our studies also demonstrated that glutathione and glutathione peroxidase levels modulate platelet arachidonic acid metabolism [59,60]
Elevated Lp(a) levels are independent risk factors for atherosclerotic cardiovascular disease (CVD) and aortic stenosis; however, no effective therapies currently target these conditions. Emerging RNA therapeutics are being developed to target hepatic apolipoprotein synthesis [61]. Several trials, including the phase-3 Lp(a)-HORIZON and ASPREE studies, using aspirin as an antithrombotic agent are underway to investigate whether lowering Lp (a) levels or use of an anti-platelet agent (Aspirin) reduces CVD risk in elderly patients with elevated lipoproteins[62]. Statins have been shown to slow the progression of atherosclerosis, with evidence from significant trials, such as the Post-Coronary Artery Bypass Graft (POST-CABG) trial, the Reversal of Atherosclerosis with Aggressive Lipid Lowering (REVERSAL) trial, the ASTEROID trial (which evaluated rosuvastatin’s effect on coronary atheroma burden via intravascular ultrasound), the SATURN study (comparing rosuvastatin and atorvastatin), and the METEOR-China study (which measured intima-media thickness [63]. Professor Rao has recently published case studies suggesting that lowering LDL-cholesterol to below 33 mg/dL has likely stabilized coronary plaques in individuals despite high coronary calcium scores, with the right coronary artery having an Agatston Score of 1007 and the left circumflex artery a score of 1748. Although statin therapy appears to accelerate coronary calcification, researchers believe this calcification process may stabilize vulnerable plaques, potentially lowering the risk of acute occlusive arterial events [64].
5. Platelet -Vessel Wall Interactions
Platelet-vessel wall interactions encompass the complex and dynamic processes that occur between circulating platelets and the blood vessel wall, especially during vascular injury or disease. Platelets are not attracted to a healthy endothelium. Antithrombogenic property of the healthy endothelium is modulated by a variety of endogenously generated biomolecules including adenosine, prostacyclin, thrombomodulin, protein C, plasminogen activators, protein S and heparin- like glycosaminoglycans [32]. When a blood vessel is damaged, subendothelial structures are exposed, initiating rapid platelet responses. These include adhesion, activation, and aggregation at the injury site. While platelets circulate in a resting state under normal conditions, they promptly adhere to exposed subendothelial structures following vascular injury. This process is facilitated by adhesive proteins and platelet surface receptors that bind to extracellular matrix components like collagen and von Willebrand factor (vWF). The interaction triggers intricate signaling pathways and cellular activating mechanisms. These interactions are essential for physiological hemostasis but also play a central role in pathological processes, including thrombosis and cardiovascular disease. Understanding these mechanisms is crucial for developing therapies to mitigate platelet-mediated vascular disorders.
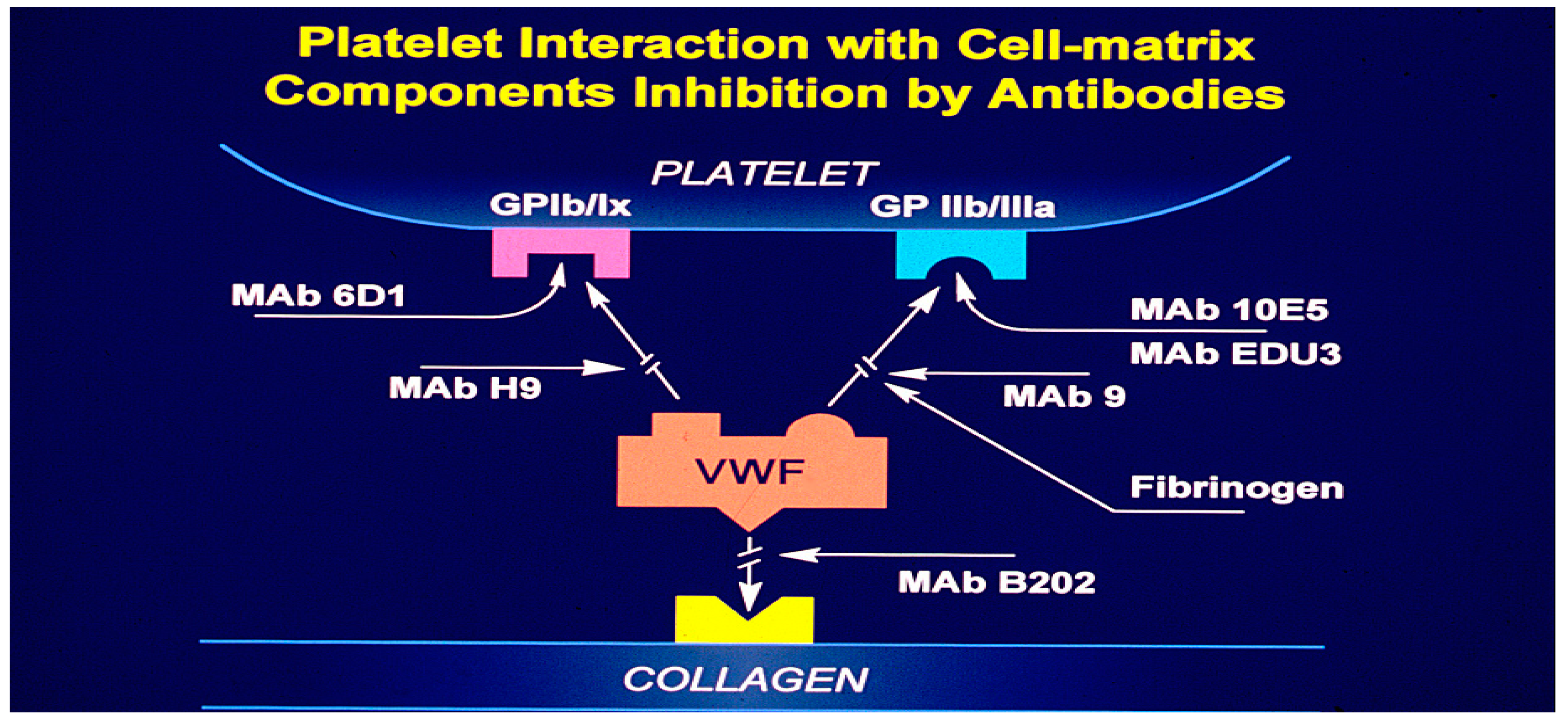
In flowing blood, platelet adhesion to the vessel wall relies on various adhesion molecules and receptors. Initially, plasma vWF binds to exposed collagen in the subendothelium, creating a substrate for platelet binding. Platelets then interact with vWF via glycoprotein Ibα (GP1bα), a component of the GP1b-V-IX receptor complex, especially under high shear forces. At lower shear rates, activated platelets utilize additional receptors, such as αIIbβ3 (GP IIb/IIIa), α2β1, and Glycoprotein IV (GP IV), to engage with vWF and collagen. Under reduced shear conditions, adhesion is further supported by other membrane-associated glycoproteins (GP I, III, IV, VI) and adhesive proteins like laminin, fibronectin, and thrombospondin. Specific binding mechanisms include interactions with laminin via α5β1, thrombospondin via GP1bα, fibrinogen via αIIbβ3, and collagen via α2β1 and GP VI. Research from the University of Minnesota highlights that currently available antiplatelet drugs fail to prevent platelet interactions with exposed subendothelium [65]. This underscores the intricate nature of these interactions, and the challenges associated with developing effective treatments. Consequently, there is significant interest in creating synthetic peptides and monoclonal antibodies to disrupt platelet-vessel wall interactions (
Figure 3) [66].
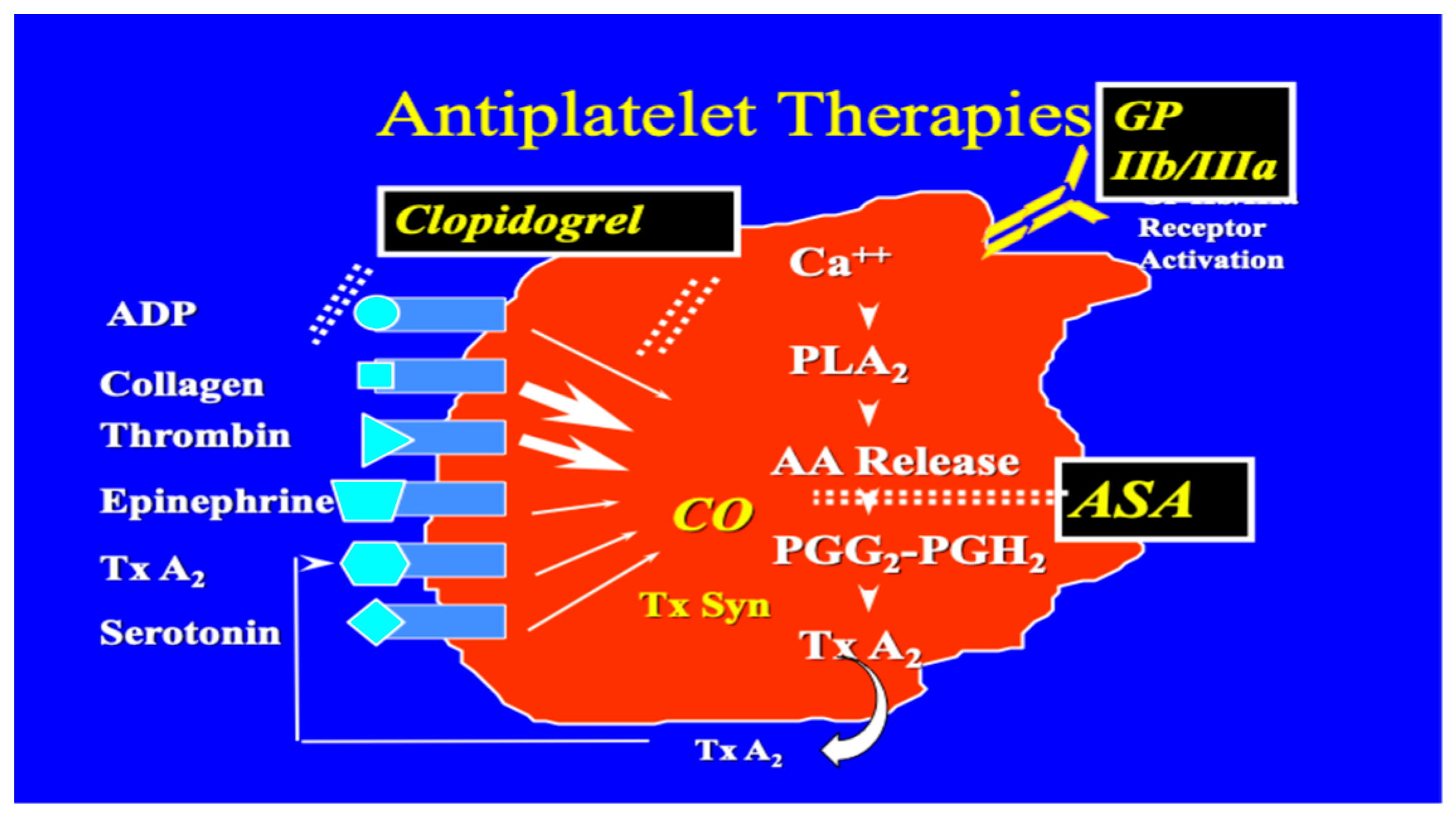
White and associates at the University of Minnesota, using ultrastructure studies demonstrated that synthetic peptides and monoclonal antibodies to the 11b/111a complex reduced the tension of platelet-rich clots, suggesting that clot tension requires the availability and interactions with a platelet receptor for polymerizing fibrin. In this study fibrinogen peptides, Arg-Gly-Asp-Ser (RGDS), Arg-Gly-Asp-Val (RGDV), Lys-Gly-Asp-Ser (KGDS), Arh-Gly-Glu-Ser (RGES) and Ala-Gly-Asp-Val (AGDV) were used as antagonists. Monoclonal antibodies used were MAB10E and C7E3. They all bound to 11b/111a and prevented platelet aggregation and fibrinogen binding to platelets [66]. Barry Coller of Rockefellers University, New York developed a monoclonal antibody that inhibits platelet function and a derivative of that antibody (Abciximab; ‘ReoPro’ ; Centocor/Eli Lilly) was approved for human use by the FDA in 1994. The EPILOG Investigators concluded that inhibition of the platelet glycoprotein 11b/111a receptor with abciximab markedly reduces the risk of acute ischemic complications in patients undergoing percutaneous coronary revascularization [67,68]. The discovery of 11b/111a receptor, which is responsible for fibrinogen recognition and platelet aggregation, facilitated the development of newer antiplatelet drugs. These drugs include abciximab (ReoPro), eptifibatide (Integrilin), and tirofiban (Aggrastat)[69].
6. Acute Occlusive Arterial Events
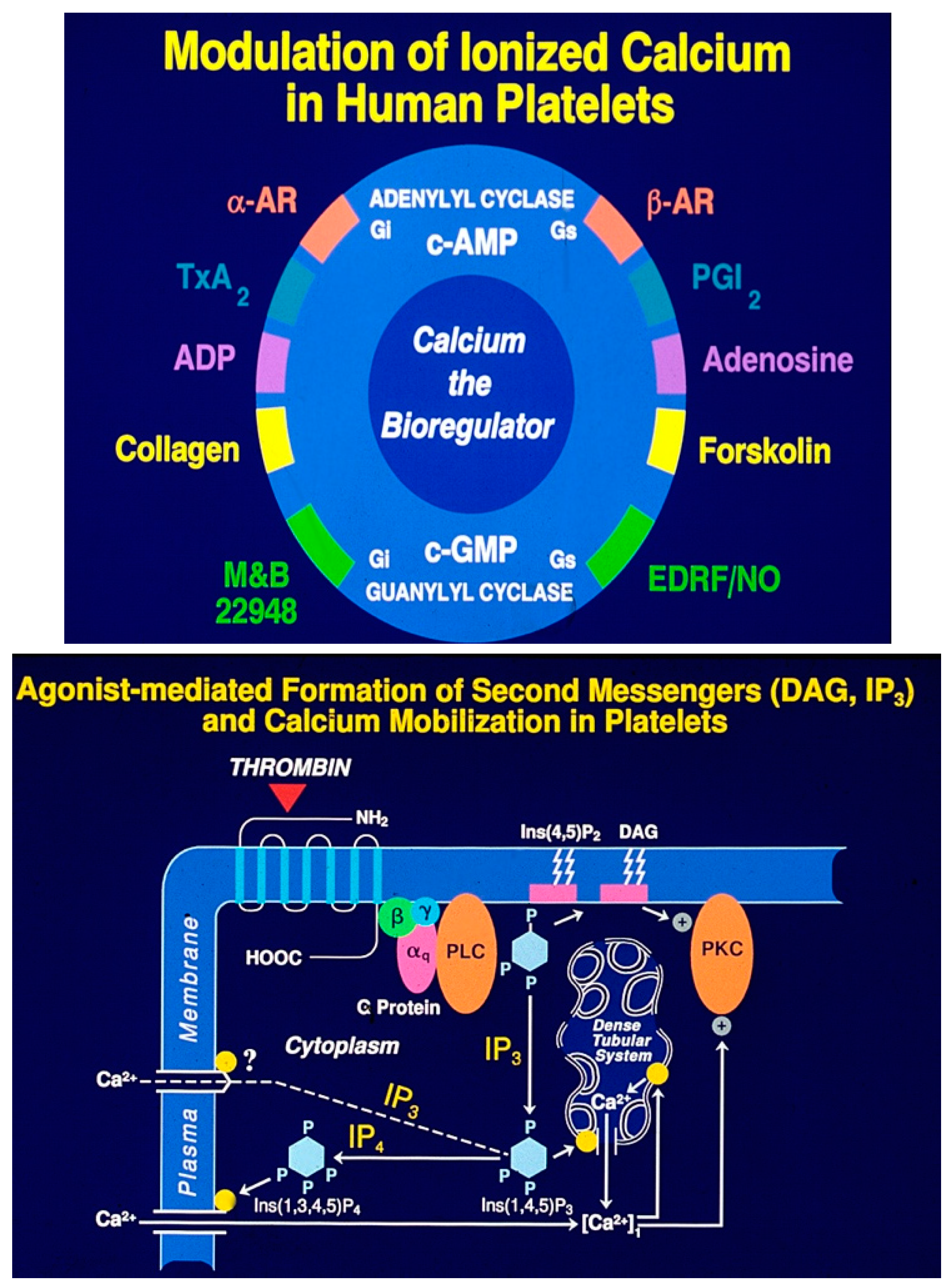
Metabolic alterations, such as oxidative stress, inflammation, vascular dysfunction, arterial narrowing, formation of vulnerable atherosclerotic plaques, and activation of platelet and coagulation pathways, initiate and promote acute occlusive arterial events. This overview does not address venous thromboembolism or microvascular dysfunction, which can lead to limb function loss or cognitive decline. In this section, we summarize our five decades of research on platelet ultrastructure, morphology, physiology, and pharmacology. While a detailed discussion of our discoveries is beyond the scope of this review, readers are encouraged to refer to a dedicated monograph on this topic [70]. Platelets interact with various soluble agonists, such as epinephrine and adenosine phosphate, insoluble extracellular matrix components, including collagen, laminin, fibronectin, and biomaterials used in medical devices. These interactions activate specific receptors and glycoprotein-rich domains (both integrins and non-integrins) on the plasma membrane, triggering intracellular effector enzymes. The regulation of platelet activation predominantly depends on the availability of membrane associated as well as free cytosolic calcium.
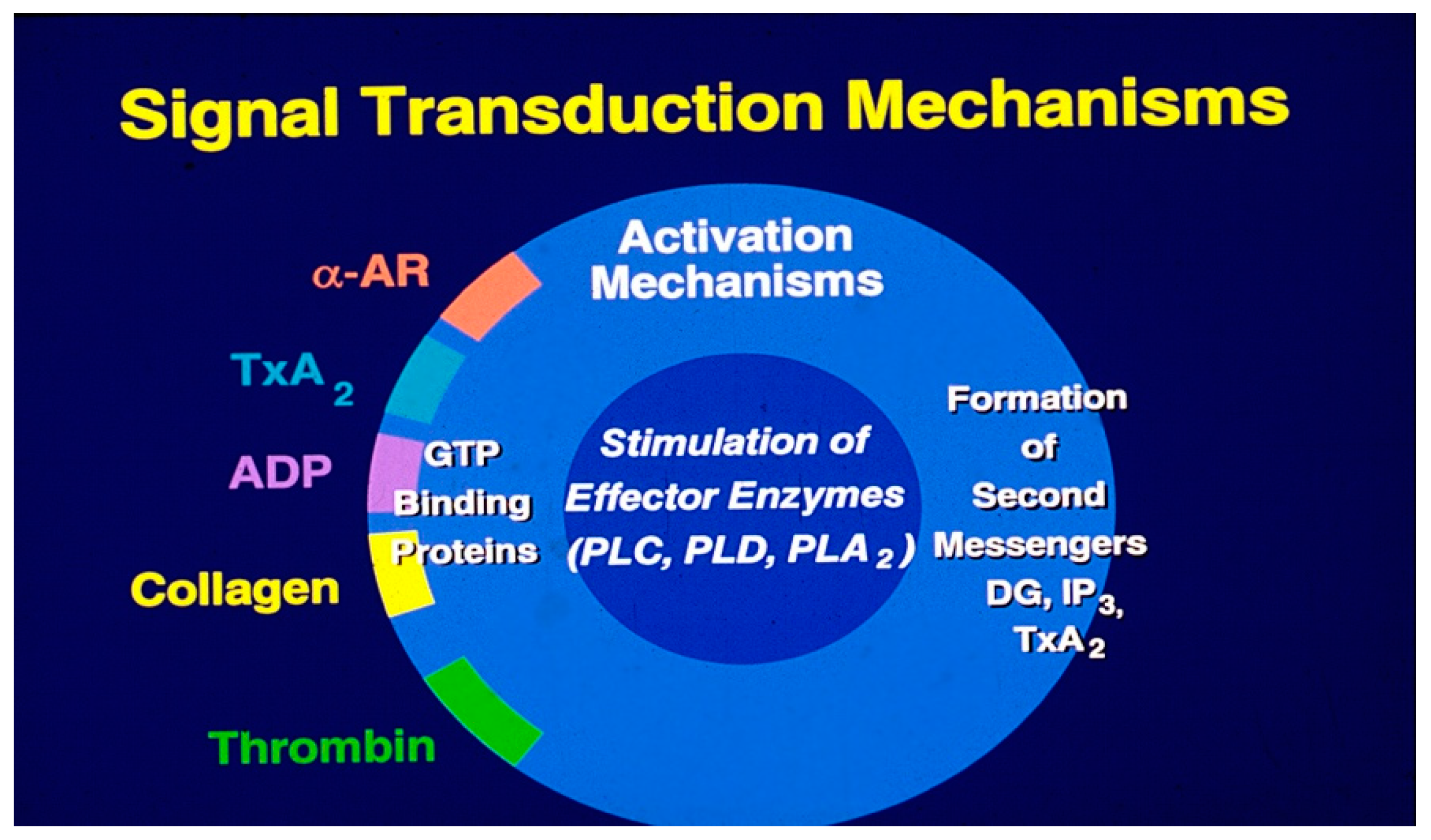
Ionized calcium acts as the primary bioregulator, with numerous biochemical mechanisms modulating the availability of free cytosolic calcium [71]. Signal transduction begins when agonists bind to specific receptors, leading to the stimulation of effector enzymes via transmembrane GTP-binding proteins (
Figure 4 A). Key enzymes that regulate calcium levels via secondary messengers include phospholipase C, phospholipase A
2, phospholipase D, adenyl cyclases, and guanyl cyclases. Phospholipase-C activation results in the hydrolysis of phosphatidylinositol trisphosphate, generating the secondary messengers 1, 2-diacylglycerol (1, 2-DG) and inositol trisphosphate (IP3) (
Figure 4B). Signal transduction mechanisms induced by antagonists are similar to the agonist-induced transmembrane signaling. Platelet antagonists act at the membrane receptors, induce transmembrane signals that result in the formation of second messengers, cyclic AMP (cAMP) and cyclic GMP (cGMP) (
Figure 4A). These second messengers lower cytosolic calcium levels and thereby limit the availability of free calcium needed for platelet activation, leading to the assembly of actin, contraction of cytoskeletal proteins and secretion of granule contents [19,23].
Diglyceride induces activation of protein kinase C (
Figure 5A) whereas IP
3 mobilizes cytosolic calcium (
Figure 5B). The activation of Protein Kinase C (PKC) leads to the translocation of cytosolic enzyme to membranes. This is a critical step in the signal transduction pathway. Upon activation, PKC undergoes a conformational change that enables it to interact with membranes rich in diacylglycerol (DAG) and calcium ions. We followed Protein kinase C activation and translocation to the membranes using Phorbol myristate and mobilization of cytosolic calcium using calcium-specific Fura-2 [72,73]. Elevation of cytosolic calcium leads to the activation of Phospholipase A
2 and release of arachidonic acid from the membranes. Free arachidonic acid is transformed into novel transient pro-aggregatory molecules, Prostaglandin (PG) PGG
2 and PGH
2 by cyclooxygenase (COX-1). These metabolites are further converted to a proaggregatory Thromboxane A
2 by thromboxane synthetase. Thromboxane A
2 acts on specific receptors and initiates the activation process again similar to the action of other agonists (
Figure 6).
Endothelial dysfunction is a hallmark of atherosclerosis. This impairs the normal protective functions of the endothelium, leading to a pro-inflammatory and pro-thrombotic state. Vulnerable atherosclerotic plaques are characterized by a thin fibrous cap, a large lipid-rich necrotic core, and inflammatory cells [74]. These unstable plaques are more prone to rupture, which can trigger acute events like myocardial infarction or stroke. In addition, activated platelets also contribute to the progression of vascular dysfunction and thrombosis. Dysfunctional endothelium and activated platelets promote the coagulation cascade, further destabilizing the plaque. Factors like inflammation, endothelial senescence, and the presence of molecules like S100A8/A9, matrix metalloproteins, (MMP-7, MMP-9), ANGPTL2, endogdlin, annexin V, serum homocysteine and apoptic microparticles influence prothrombotic events. All these molecules contribute to the development of vascular dysfunction. In summary, the complex interplay between endothelial dysfunction, vulnerable plaque formation, and platelet hyperfunction are key drivers of acute vascular events like heart attacks and strokes [74,75,76,77,78].
Aspirin (ASA) became the first effective antiplatelet drug in the 20th century. It is still widely used today to prevent cardiovascular complications caused by atherothrombosis, although it also increases the risk of major bleeding. Aspirin has been used as an antiplatelet drug for over a century, building on its long history as a painkiller and antipyretic, dating back thousands of years [79]. Aspirin acetylates the serine residue of the COX-1 enzyme. This prevents the production of prostaglandins and thromboxanes [80]. Newer drugs of this class include Prasugrel a novel thienopyridine similar to Clopidogrel that exerts its antiplatelet effects binding to the P2Y12 receptor for adenosine diphosphate (ADP), thereby preventing platelet aggregation [81]. Ticagrelor on the other hand is a cyclopentyl-triazolo-pyrimidine, a reversible inhibitor of the P2Y12 receptor. Guidelines suggest the use of these newer antiplatelet drugs as part of dual antiplatelet therapy with ASA in acute coronary syndromes. We have described other class of inhibitors such as peptides and antibodies (ReoPro, Epifibatide and Tirofiban) to various cell matrix components in an earlier section. When considering other antithrombotic therapies, recent guidelines recommend more potent P2Y12 inhibitors like ticagrelor and prasugrel over abciximab in stable patients. For instance, ticagrelor, an active drug providing rapid and potent inhibition of platelet aggregation, has shown a significant reduction in mortality and rates of major adverse cardiac events (MACE) compared to clopidogrel. In contrast, abciximab's advent was marked in the era of balloon angioplasty, showcasing its role in reducing myocardial infarction (MI) rates during PCI. It is now often reserved for high-risk scenarios or “bail-out” situations.
7. Discussions
Cardiometabolic diseases, which include conditions like ischemic heart disease and hypertensive heart disease, have seen a significant rise in mortality and prevalence globally in recent decades. Deaths from ischemic heart disease have increased by more than 1.4 million globally, which is the second- largest rise in any income group in terms of absolute numbers. The global prevalence of hypertensive heart disease has risen steadily over the last 3 decades, as have the total number of deaths, There are also significant racial and ethnic disparities in the prevalence of diagnosed heart disease and diabetes. Overall, deaths from cardiovascular disease, which includes many cardiometabolic conditions jumped globally from 12.1 million in 1990 to 20.5 million in 2021, a surge of around 60%. [82]. According to a study conducted in 21 high-middle and low-income countries, a small number of modifiable risk factors seems to be the cause of premature death due to CVDs [83]. Adding additional risk factors to the Framingham risk score seems to improve coronary heart risk scores [84]. A recent study validated four cardiovascular risk prediction models and concluded that we need better models to predict CVD risk in a multiethnic populations [85]. A recent Finnish study,- Kuopio Ischemic Heart Disease Study (KIHD), reported that four health behaviors (physical activity, diet, nicotine exposure and sleep health) and four health factors (BMI, Lipids, blood glucose, and blood pressure) computed as ‘Life’s Essential (LE8) influenced the CVD risk and not the venous thrombosis risk [86].
Oxidative stress and chronic inflammation are recognized as significant contributors to the risk of metabolic diseases. While they may not directly cause these conditions, they can exacerbate them by promoting insulin resistance, metabolic syndrome, and other related disorders [87,88]. Research highlights the critical role of the microvasculature in maintaining vascular and neural health by delivering oxygen and nutrients to endothelial cells. The endothelium, which lines the circulatory system, plays a vital role in this process. However, increased oxidative stress, excessive lipid oxidation, and nitric oxide depletion are major pathological mechanisms that disrupt endothelial function. This dysfunction fosters platelet and leukocyte interactions with the endothelium, further fueling inflammation, impairing signal transduction pathways, altering cytoskeletal structure, and disrupting intercellular communication. Addressing oxidative stress and inflammation in the microvasculature is essential to improving treatment outcomes for clinical complications associated with microvascular diseases, such as those affecting the lower limbs and cerebrovascular regions. Identifying the underlying mechanisms that drive oxidative stress and chronic sterile inflammation in these areas is vital for developing effective prevention and therapeutic strategies.
Oxidative stress and inflammation play a significant role in promoting vascular atherosclerosis [89]. Atherosclerosis is a condition characterized by the buildup of plaque in the arterial walls, leading to restricted blood flow. Oxidative stress, caused by an imbalance between free radicals and antioxidants, damages the endothelial lining of blood vessels, making them more susceptible to inflammation. Chronic inflammation in the arterial walls accelerates the progression of atherosclerosis, increasing the risk of cardiovascular events such as heart attacks and strokes. High levels of LDL cholesterol can contribute to the formation of this plaque, while also increasing the risk of atherosclerotic complications such as heart attacks and strokes. LDL's ability to transport cholesterol to the arteries contributes to the development of atherosclerosis, making it a key factor in the progression of the disease [90]. Clinical trials have consistently demonstrated the effectiveness of lowering cholesterol and Lp(a) levels in preventing the risk of cardiovascular diseases (CVDs). By reducing LDL (bad) cholesterol and Lp(a), a type of lipoprotein-associated with an increased risk of CVDs, individuals can significantly decrease their likelihood of developing conditions such as coronary artery disease, heart attack, and stroke. Studies have shown that statins and other cholesterol-lowering medications can lower Lp(a) levels, thereby reducing CVD risk [91].
Newer gene editing techniques are being evaluated for the treatment of hypercholesteremia in animal models [92]. A research paper by Minjares, Wu, and Wang has explored the relationship between oxidative stress and microRNAs in endothelial cells under metabolic disorders [93]. Lowering both cholesterol and lipoprotein (a)(Lp(a) levels can contribute to stabilizing vulnerable plaques in the arteries. Reducing these substances helps to decrease the size of the lipid core with the plaque and thicken the fibrous cap, making it less prone to rupture and causing an acute vascular event [94,95]. Zerlasiran is a small-interfering RNA (siRNA) being studied in a Phase 2 clinical trial. The trial is focused on its potential to target lipoprotein(a), a type of lipoprotein that can contribute to cardiovascular disease when elevated. Lipoprotein(a) is a complex protein composed of an apolipoprotein(a) component, and a B100 component, which can increase the risk of atherosclerotic cardiovascular disease. A clinical trial evaluated the safety and efficacy of Zerlasiran in reducing lipoprotein(a) levels in patients with elevated levels [96]. A recent publication in JAMA indicates that Zerlasiran was well tolerated and reduced lipoprotein (a) by 60% and in some cases up to 90% from the baseline in some dosages in patients with elevated Lp(a). By suppressing an LPA gene Zerlasiran (N-acetyl galactosamine conjugated small RNA) lowers Lp(a), and thereby is expected to reduce the risk of heart disease.
Research conducted at the Thrombosis Research Laboratory of the Lillehei Heart Institute, University of Minnesota, has significantly advanced our understanding of the cellular and molecular mechanisms underlying platelet activation [70]. A new phase in ultrastructural investigation began in the early 1960s under the leadership of Prof. White, who emphasized the importance of platelet membrane systems in physiology and function [98]. His work highlighted the role of the dense tubular system (DTS) as a key site for calcium sequestration, a critical bioregulator. Additionally, the DTS was identified as the location of enzymes responsible for fatty acid metabolism and prostaglandin synthesis [96,97,98,99]. Further studies from this laboratory explored the role of various calcium sources, actin and actin-binding proteins, the phosphorylation of cytoskeletal proteins, and the modulation of membrane-associated receptors by agonists and antagonists in platelet activation -signal transduction- coupling (71, 97-108]. A unique phenomenon termed "Membrane Modulation," mediated by alpha-adrenergic signals, was also discovered, showing its capacity to restore the responsiveness of drug-induced refractory platelets to agonists [108]. The research also revealed the effects of cholesterol loading and glutathione depletion on platelet hyperfunction [59,60]. In a groundbreaking study, it was demonstrated that in a drug-induced hyperglycemia model, arachidonic acid metabolism shifts toward a prothrombotic state. Remarkably, pancreatic islet cell transplantation in this model not only normalized blood glucose levels but also restored a balanced arachidonic acid metabolism [109].
Our understanding of the cellular and molecular mechanisms underlying the initiation of cardiometabolic risks and the progression of metabolic diseases such as hypertension, type- 2 diabetes, obesity, and vascular disorders has advanced significantly in recent years. Insights from these foundational studies have led to the development of innovative interventions for managing these chronic conditions at the individual level. However, addressing metabolic diseases like hypertension, diabetes, and obesity on a population level remains a formidable challenge. There is an urgent need for more effective educational, preventive, and early diagnostic strategies to combat these diseases. Such efforts should focus on promoting healthy lifestyle choices, enhancing access to healthcare services, and raising awareness about the risks and consequences of these conditions. Achieving these goals requires a comprehensive approach involving government policies, community-driven programs, and individual actions. This multi-pronged strategy can help reduce the societal and individual burden of metabolic diseases. The South Asian Society on Atherosclerosis and Thrombosis (SASAT) has dedicated its efforts to developing educational and preventive initiatives in this field [110]. Over the years, we have organized more than a dozen international conferences on these and related topics across India and other countries. Additionally, we have collaborated with international experts to publish several monographs on these subjects [111,112,113,114,115,116,117,118,119,120,121]. As a professional society, we strongly believe in our role as an independent educational institution committed to informing healthcare professionals and the public at large [122].
The development of messenger RNA (mRNA) vaccines is a prime example of the rapid advancements in interventions for public health challenges. The therapeutic use of mRNA and noncoding RNAs has fueled great hope to combat a wide range of incurable diseases, including type-2 diabetes and vascular disease risks [123,124]. The advancement of gene editing technologies, such as CRISPR/Cas9, and computational genomics have greatly contributed to the development of modern therapeutic approaches [125]. Gene editing enables precise modifications to an organism's genome, while computational genomics provides the tools to analyze and interpret large amounts of genetic data. This synergy has led to breakthroughs in gene therapy, personalized medicine, and cancer treatment, offering new hope for patients with previously incurable diseases. Furthermore, these technologies have also enabled the discovery of new therapeutic targets and the development of more effective treatments. Large language models, like Evo, have the potential to revolutionize the interpretation of biological sequence data [126]. Alpha Fold3 was published recently [127]. According to the authors, this can predict not just the structures of protein complexes, but also how proteins interact with other kind of molecules. This represents a major step in our understanding how biomolecules interact with one another. These technologies represent a significant advancement in our ability to comprehend and engineer biological processes across various modalities. By leveraging advanced algorithms and computational power, these models can analyze complex biological data, identify patterns, and provide insights that were previously inaccessible.
8. Conclusions
The rise of cardiometabolic diseases, hypertension, type-2 diabetes, obesity, including ischemic heart disease, is driven by modifiable risk factors, lifestyle changes, and disparities in healthcare access. Key mechanisms, such as oxidative stress, chronic inflammation, and endothelial dysfunction, play critical roles in the pathogenesis of cardiometabolic conditions. Advances in understanding these mechanisms have shed light on the interplay between lipid oxidation, nitric oxide depletion, and vascular health, emphasizing the need for therapeutic strategies that target both oxidative stress and inflammation to mitigate disease progression. Atherosclerosis remains a primary contributor to CVD, with LDL cholesterol and lipoprotein(a) being major risk factors. New approaches, such as statins and emerging therapies like Zerlasiran, demonstrate promise in reducing cholesterol and stabilizing plaques. Innovations in gene editing and RNA-based treatments offer hope for addressing hypercholesterolemia and other metabolic disorders. The role of platelets in vascular dysfunction and thrombosis has also been explored extensively. Research into platelet activation, calcium regulation, and arachidonic acid metabolism has advanced our understanding of prothrombotic states, with potential implications for diabetes and hyperglycemia-related complications. Despite these advances, there is a disconnect between the discoveries and their application in the diagnosis and management or CVD risks. According to the recently elected president of the Atherosclerosis Society, Professor Peter Libby, the lag time from garnering scientific evidence to changing the clinical practice may endure an average of 17 years (IAS Bi-Monthly News Letter Nov-Dec 2024). We hope that reviews like this will shorten this lag time from laboratory discovery to the bench by few years.
Despite these advancements, challenges persist in addressing cardiometabolic diseases on a population level. A multi-faceted approach, involving government policy, community-driven initiatives, and individual actions, is necessary to promote education, early diagnosis, and healthy lifestyle choices. Professional societies, such as SASAT, have made significant contributions through international collaborations and educational initiatives to raise awareness and combat these conditions. Emerging technologies, such as mRNA therapeutics, CRISPR/Cas9 gene editing, and computational genomics, provide groundbreaking opportunities for personalized medicine and targeted therapies. These innovations hold promise for addressing previously untreatable diseases and paving the way for future advancements in managing cardiometabolic health. In summary, while significant progress has been made in understanding and addressing cardiometabolic diseases, continued investment in research, preventive strategies, and equitable healthcare delivery is essential to reduce the global burden of these chronic conditions.
Author Contributions
This is an overview of cardiometabolic risks as it refers to the initiation and progress of metabolic diseases such as hypertension, type-2 diabetes, obesity and vascular diseases leading to the development of acute occlusive cardiovascular events. A. C. T. and G. H. R. R. have conceptualized and developed this essay. All authors have read and agreed to the published version of the manuscript.
Funding
Gundu H. R. Rao extends his thanks and gratitude to the National Institutes of Heart, Blood and Lung Institute of the National Institutes of Health (NIH) USA, for their continued backing of our collaborative studies at the University of Minnesota from 1970-2000. He also extends heartfelt gratitude to the National Science Foundation (NSF), USA (1980), the United Nations Development Program (UNDP)(1990-1993), and the International Society on Thrombosis and Hemostasis for their generous financial support.
Data Availability Statements
Not Applicable.
Acknowledgments
Professor Gundu H. R. Rao is extremely grateful to the Department of Laboratory Medicine and Pathology, Lillehei Heart Institute, University of Minnesota, for their unwavering support in our research on thrombosis and hemostasis for more than four decades. He would also like to express his deep appreciation to the late Professor James G White of the University of Minnesota for his invaluable mentorship. Additionally, he extends his thanks and gratitude to the National Heart, Blood, and Lung Institute (NHLBI) of the National Institutes of Health (NIH) for their continued financial backing of our studies from 1970 to 2000. Furthermore, he expresses his sincere appreciation to the International Society on Thrombosis and Hemostasis (ISTH), USA, for their financial assistance to the South Asian Society on Atherosclerosis and Thrombosis (SASAT) from 1992 to 2000 for international educational initiatives in India. He also expresses his thanks to the National Science Foundation (NSF), USA, and the United Nations Development Program (UNDP), for providing travel grants to visit India for developing bilateral research projects from 1992-2000.
References
- Shah, N.S.; Molsberry, R.; Rana, J.S.; Capewell, S.; O’Flaherty, M.; Carnethon, M.; Lloyd-Jones, D.M.; Khan, S.S. Heterogenous trends in burden of heart disease mortality by subtypes in the United States, 199-2018: Observational analysis of vital statistics. Brit Med. J. 2020, 370, m2688. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, M.; Perel, P.; Taylor, S.; Kabudula, C.; Bixbym, H.; Gaziano, T.A.; McGhie, D.V.; Mwangi, J.; Pervan, B.; Narual, J.; et al. The Heart of the World. Glob Heart. 2024, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Abohelwa, M.; Kopel, J.; Shurmur, S.; Ansari, M.; Awasthi, Y.; Awasthi, S. The Framingham study of cardiovascular disease risks and stress -defenses: A Historical Review. J Vasc. Dis. 2023, 2, 122–164. [Google Scholar] [CrossRef]
- Kannel, W.B.; Gordan, T. Evaluation of cardiovascular risk in the elderly. The Framingham Study. Bull N.Y. Acad Sci. 1978, 54, 573–591. [Google Scholar]
- Rippe, J.M. Lifestyle strategies for risk factor reduction, prevention, and treatment of cardiovascular disease. Am J Lifestyle Med. 2018, 13, 204–212. [Google Scholar] [CrossRef]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dnas, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infraction in 52 countries (the INTEHEART study): Case-control study. Lancet 2004, 364, 937–52. [Google Scholar] [CrossRef]
- Khera, A.V.; Emdin, C.A.; Drake, I.; Natarajan, P.; Bick, A.G.; Cook, N.C.; Chasman, D.; Baber, U.; Mehran, R.; Rader, D.J. Genetic risk, adherence to a healthy lifestyle, and coronary artery disease. N Engl. J Med 2016, 375, 2349–2358. [Google Scholar] [CrossRef]
- Di Cesare, M.; Bennett, J.E.; Best, N.; Stevens, G.A.; Danaei, G.; Ezzati, M. The contributions of risk factor trends to cardiometabolic mortality in 26 industrialized countries. Int J Epidemiol. 2013, 42, 838–848. [Google Scholar] [CrossRef]
- Chen, Y.; Freedman, D.F.; Albert, P.S.; Huxley, R.R.; Shields, M.S.; Withrow, D.R.; Spillane, S.; Powel-Willey, T.M.; de Gonzalez, A.B. Association of cardiovascular disease with premature mortality in the United States. JAMA Cardiol. 2019, 4, 1230–1238. [Google Scholar] [CrossRef]
- Fuster, C.; Kelly, B.B. (Eds.) Institute of Medicine. In Promoting Cardiovascular Health in the Developing World: A critical challenge to achieve global health; The National Academies Press: Washington DC, 2010; ISBN 978-0-309-14774-3. [Google Scholar] [CrossRef]
- Hankey, A. Establishing the scientific validity of Tridosha Part 1: Doshas, Subdoshas and Dosha Prakrithis. Ancient Sci of Life 2010, 29, 6–18. [Google Scholar]
- Dong, H.; Sun, Y.; Nie, L.; Cui, A.; Zhao, P.; Leung, W.K.; Wang, Q. Metabolic Memory: Mechanisms and diseases. Nature Signal Transduction and targeted therapy 2024. [Google Scholar] [CrossRef] [PubMed]
- Krug, E.G. Trends in diabetes: Sounding the alarm. The Lancet 2016, 387, 1485–1486. [Google Scholar] [CrossRef]
- Malhotra, N.; Malhotra, J.; Bora, N.M.; Bora, R.; Malhotra, K. Fetal origin of adult disease. Donald School J. Ultrasound Obstet. Gynecol. 2014, 8, 164–177. [Google Scholar]
- Yajnik, C.S. Confession of a thin-fat- Indian. Euro. J. Clin Nutr. 2018, 72, 69–473. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, S.C.; Nadler, E.P.; Pillai, D.K.; Hubal, M.J.; Wang, Z.; Wang, M.W.; Gordish-Dressman, H.; Koeck, E.; Sevilla, S.; Wiles, A.W. Pillai DK Adipocyte-derived exosomal miRNA: A novel mechanism for obesity-related disease. Ped. Res. 2015, 77, 447–454. [Google Scholar] [CrossRef]
- Camussi, G.; Deregibus, M.-C.; Bruno, S.; Grange, C.; Fonsato, V.; Ciro, T. Exosome/macrovesicle-mediated epigenetic reprogramming of cells. Am J. Cancer Res 2011, 1, 98–100. [Google Scholar] [PubMed]
- Rao, G.H.R. Fetal Origin of Adult Diseases: Micronutrient and microRNA Interventions. Endocrinol. Met. Res. 2019, 4.3, 7–16. [Google Scholar]
- Rao, G.H.R. Role of cyclic AMP and cyclic GMP as modulators of platelet cytosolic calcium. J. Clin & Prevent Cardiol. 2016, 5, 99. [Google Scholar]
- Rao, G.H.R. Prevention or reversal of cardiometabolic diseases. J. Clin Prevent Cardiol. 2018, 7, 22–28. [Google Scholar] [CrossRef]
- Rao, G.H.R.; Gandhi, P.G.; Sharma, V. Clinical complications of type-2 diabetes mellitus in South Asians and Chinese Populations: An Overview. J Diab. & Metab 2014, 5, 1000420. [Google Scholar] [CrossRef]
- Rao, G.H.R.; Thethi, I.; Fareed, J. Vascular disease: Obesity and excess weight as modulators of risk. Exp Rev of Cardiovasc Ther. 2011, 994, 525–534. [Google Scholar] [CrossRef]
- Rao, G.H.R. Cellular Signaling Pathways and Vascular Dysfunctions. J Cardiol Cardiovasc Ther. 2018, 12, 55844. [Google Scholar] [CrossRef]
- Rao, G.H.R. Global syndemic of metabolic disease; Editorial Comments. J Daib and Clin Res. 2019, 1, 2–4. [Google Scholar] [CrossRef]
- Rao, G.H.R. Diabetes and cardiovascular disease in South Asians: A global perspective. J Clin and Prevent Cardiol. 2018, 7, 161–167. [Google Scholar] [CrossRef]
- Rao, G.H.R. Coronavirus disease and acute vascular events. Clin Appl Thromb Hemost. Editorial. 2020, 26. [Google Scholar] [CrossRef]
- Rao, G.H.R. Number one Killer: Vascular Disease. Ann Clin Diab and Endocrinol. 2018, 1, 1008. [Google Scholar]
- Rao, G.H.R. Risk prediction, assessment, and management of type-2 diabetes. EC Endocrinol Met. Res. 2018, 3. [Google Scholar] [CrossRef]
- Agrawal, A.; Rao, G.H.R. Novel approaches for early diagnosis and prevention of cardiometabolic diseases. J Clin and Prevent Cardiol. 2023, 121, 23–36. [Google Scholar] [CrossRef]
- Rao, G.H.R. Obesity is a unique metabolic disease: An Update. EC Clin. And Med Case Rept. 2023, 68, 01–11. [Google Scholar]
- Amit, R.T.; Rao, G.H.R. Inflammation: Is it a healer, Confounder, or a Promoter of Cardiometabolic Risks? Biomolecules 2024, 14, 948. [Google Scholar] [CrossRef] [PubMed]
- Hamilos, M.; Petousis, S.; Parthenakis, F. Interaction between platelets and endothelium: From Pathophysiology to new therapeutic options. Cardiovasc Diagn Ther. 2018, 8, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim Biophys Acta Mol Basis Dis 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. & Mol Med. 2019, 51, 1–13. [Google Scholar]
- Hu, F.; Liu, F. Mitochondrial stress: A bridge between mitochondrial dysfunction and metabolic diseases? Cellular signaling. 2011, 23, 1528–1533. [Google Scholar] [CrossRef]
- Gastaldi, G.; Giacobino, J.P.; Ruiz, J. Metabolic syndrome, a mitochondrial disease? Revue Medicale Suisse. 2008, 1387–1388,1390–1381. [Google Scholar]
- Maassen, J.A.; LM, T.H.; Van Essen, E.; Heine, R.J.; Nijpels, G.; Jahangir, T.R.S.; Raap, A.K.; Janssen, G.M.; Lemkes, H.H. Mitochondrial diabetes: Molecular mechanisms and clinical presentation. Diabetes 2004, 53, S103–109. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.-K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders. A step towards mitochondrial based therapeutic strategies. Biochim Biophys Acta 2016, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Kim, M.-B.; Lee, J.; Lee, J.-Y. Targeting mitochondrial dysfunction for the prevention and treatment of metabolic disease by bioactive food components. J Lipid Atheroscler. 2024, 13, 306–327. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, K.S.; McCord, J.M. Oxidative stress In health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp of Med. 2011, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Galimov, E.R. The Role of p66shc in oxidative stress and apoptosis. Acta Nat 2010, 2, 44–51. [Google Scholar] [CrossRef]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Paolmieri, V.O.; Portincasa, P.; Moschetta, A.; Palasciano, G. Oxidative stress- induced risk factors associated with the metabolic syndrome. J. Nutr. Biochem. 2008, 8, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Rad. Biol Med. 2004, 43, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Ying, F.; Sweeney, G. Sterile inflammation and the NLRP3 inflammasome in cardiometabolic disease. Biomed J. 2023, 46, 100624. [Google Scholar] [CrossRef] [PubMed]
- Suffee, N.; Le Goff, W.; Chen, J. Editorial: Cardiometabolic diseases and inflammatory responses. Front. Immunol. 2024, 1384022. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, E.H.; Ballantyne, C.; Fonesca, F.; +21 for the CANTOS Trial Group. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N. Engl J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis- An inflammatory disease. N Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Jebari-Benslaiman, S.; Galicia-Garcia, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandebroek, K.; Benito-Vicente, A.; Martin, C. Pathophysiology of Atherosclerosis. Int J Mol Sci 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Medrano-Bosch, M.; Simon-Codina, B.; Jimenez, W.; Edelman, R.E.; Meglar-Lesmes, P. Monocyte-endothelial cell interactions in vascular and tissue remodeling. Front Immunol 2023, 14, 1196033. [Google Scholar] [CrossRef]
- Guyton, R.J.; Klemp, F.K. Development of the lipid-rich core in human atherosclerosis. Arteriosclerosis, Thrombosis and Vasc. Biol. 1996, 16, 4–11. [Google Scholar] [CrossRef]
- Wu, X.; Liu, X.; Liu, T.; Tian, W.; Dun, Y.-J. Effects of different statins application methods on plaques in patients with coronary atherosclerosis. World J. Clin Cases 2012, 9, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chang, H.; Sung, J.; Park, H.; Heo, R.; Rizvi, A.; Lin, F.Y.; Kumar, A.; Hadamitzky, M.; Kim, J.Y. Effects of statins on coronary atherosclerotic plaques: The PARADIGM Study. JACC Cardiovasc. Imaging 2018, 11, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Weisser, B.; Locher, R.; Graff, J.; Moser4, R.; Sachinidis, A.; Vetter, W. Low density lipoprotein subfractions increase thromboxane formation in endothelial cells. Biochim Biophs Res. Commun. 1993, 192, 1245–50. [Google Scholar] [CrossRef] [PubMed]
- Vagimigli, L. Lipid peroxidation and antioxidant protection. Biomolecules 2023, 13, 1291. [Google Scholar] [CrossRef] [PubMed]
- Baumer, Y.; Irei, J.; Boisvert, W.A. Cholesterol crystals in the pathogenesis of atherosclerosis. Nat. Rev. Cardiol. 2024. [CrossRef] [PubMed]
- Bhatia, H.S.; Becker, R.C.; Leibundgut, G.; Patell, M.; Lacaze, P.; Tonkin, A.; Narula, J.; Tsimikas, S. Lipoprotein (a), platelet function and cardiovascular disease. Nat Rev Cardiol. 2024, 21, 299–311. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikigorov, G.N.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Oveview of OxLDL and its impact on cardiovascular health: Focus on Atherosclerosis. Front Pharmacol. Sec. Ethnopharmacology. 2020, 613780. [Google Scholar]
- Stuart, M.; Gerrard, J.M.; White, J.G. Effect of cholesterol on production of thromboxane B2 by platelets in vitro. Bew Engl J. Med. 1980, 302, 6–10. [Google Scholar] [CrossRef]
- Hill, T.D.; White, J.G.; Rao, G.H. Role of glutathione and glutathione peroxidase in human platelet arachidonic acid metabolism. Prostaglandins 1989, 38, 21–32. [Google Scholar] [CrossRef]
- Tsimikas, S.; Moriarty, P.M.; Stores, E.S. Emerging therapeutics to lower blood levels of Lp(a): JACC Focus Seminar2/4. J. Am. Coll Cardiol. 2021, 77, 1576–1589. [Google Scholar] [CrossRef]
- Bhatia, H.S.; Trainor, P.; Carlisle, S.; Tsai, M.Y.; Criqui, M.H.; DeFulippis, A.; Tsimikas, S. Aspirin and cardiovascular risk in individuals with elevated lipoprotein (a)L The Multi-ethnic study if atherosclerosis. J Am Heart Assoc. 2024, 13, e033562. [Google Scholar] [CrossRef]
- Yang, C.; Wu, Y.; Qian, J.; Li, J. Landscape of statin as a cornerstone in atherosclerotic disease. Rev Cardiovasc. Med 2023, 24, 373. [Google Scholar] [CrossRef]
- Rao, G.H.R. Early diagnosis of risks and management of cardiometabolic diseases: A case study. Open Access J. Cardiol. 2024, 121, 23–36. [Google Scholar]
- Rao, G.H.R. Influence of anti-platelet drugs on platelet-vessel wall interactions. Prost. Leukotriene. and Med. 1980, 5, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Burk, D.L.; White, J.G. The effect of peptide and monoclonal antibodies that bind to platelet glycoprotein 11b-111a complex on the development of clot tension. Blood 1989, 73, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- The EPILOG Investigators. Platelet glycoprotein 11b/111a receptor blockade and low-dose heparin during percutaneous coronary revascularization. New Engl J. Med. 1997, 336, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Coller, B.S.; Scudder, L.E.; Beer, J.; Gold, H.K.; Folts, J.D.; Cavagnaro, J.; Jordan, R.; Wagner, C.; Luliucci, J.; Knight, D. Monoclonal antibodies to platelet glycoprotein 11b/111a as antithrombotic agents. Ann N.Y. Acad Sci. 1991, 614, 193–213. [Google Scholar] [CrossRef] [PubMed]
- Shlansky-Goldberg, R. Platelet aggregation inhibitors for use in peripheral vascular interventions: What can we learn from the experience in the coronary arteries? J. Vasc and Int Radiol. 2002, 13, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.H.R. Manual of Platelet Morphology, Physiology and Pharmacology; J.P Medical Publishers: New Delhi, India, 2019; ISBN 978-93-5270-202-2. [Google Scholar]
- Rao, G.H.R.; Gerrard, J.M.; Cohen, I.; Witkop, C.J.; White, J.G. Fiskum, G., Ed.; Origin and role of calcium in platelet activation-contraction -secretion -coupling. In Cell Calcium Metabolism; GWUMC Department of Biochemistry Annual Spring Symposia; Springer: Boston, MA, 1989; pp. 411–425. [Google Scholar] [CrossRef]
- Sharma, N.; Norman-O’Guin, K.; Shafit-Zagardo, B. Phorbol-1`2-myristate-13-acetate(PMA) and inhibitors of protein kinase C alter glial fibrillary acidic protein (GFAO)mRNA levels. Glia 1991, 4, 572–9. [Google Scholar] [CrossRef]
- Rao, G.H.R.; Peller, J.D.; White, J.G. Measurement of ionized calcium in blood platelets with a new generation calcium indicator. Biochem, Biophys Res, Commun. 1985, 132, 652–657. [Google Scholar] [CrossRef]
- Zhang, J. Biomarkers of endothelial activation and dysfunction in cardiovascular diseases. Rev. Cardiovasc Med. 2022, 23, 73. [Google Scholar] [CrossRef]
- Uehara, Y. Special Issue “Lipids in Atherosclerosis”. Biomolecules (ISSN 2218-273X). Biomacromolecules: Lipids 2023. [Google Scholar]
- Orekhov, A.; Sobenin, I.A. Special Issue “Atherosclerosis and Related Diseases: Particular Focus on Molecular Biology”. Biomolecules(ISSN 2218-273X). Mol Med. 2019. [Google Scholar]
- Scicali, R. Special Issue “ Emerging Circulating Biomarkers in Atherosclerosis: From Molecular Mechanisms to therapeutic strategies”. Biomolecules (ISSN2218-273X). Mol Biol. 2021. [Google Scholar]
- Uehara, Y. Special Issue “Lipids in Atherosclerosis”. Biomolecules (ISSN 2218-273X). Biomacromolecules: Lipids 2023. [Google Scholar]
- Montinari, M.R.; Minelli, S.; Caterina, R. The first 3500 years of aspirin history from its roots- A concise summary. Vascul Pharmacol. 2019, 113, 18. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K. Aspirin and other cyclooxygenase inhibitors: New therapeutic insights. Semni. Vasc Med. 2003, 3, 107–112. [Google Scholar]
- Chiua, S.; Nishi, C. New antiplatelet agents for cardiovascular disease. CMAJ 2013, 185, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- World Heart Report. Confronting the World’s Number One Killer. 2023. Available online: https://world-heart-federation.org/wp-content/uploads/World-Heart-Report-2023.pdf.
- Yusuf, S.; Joseph, P.; Rangarajan, S.; Islam, S.; Mente, A.; Brauer, M.; Kutty, M.R.; Gupta, R.; Wielgosz, A. Modifiable risk factors, cardiovascular disease mortality in 155, 722 individuals from 21 high-, middle-, and low-income countries. Lancet 2019, 395, 795–808. [Google Scholar] [CrossRef]
- Hu, G.; Root, M.M.; Duncan, A.W. Adding multiple risk factors improves Framingham heart disease risk scores. Vasc. Heath and Risk Management 2014, 10, 557–562. [Google Scholar]
- Bhuiyan, A.; Govindaiah, A.; Smith, R.T. External validation of four cardiovascular risk prediction models. J of Clin Cardiol. 2024, 5, 73–80. [Google Scholar] [CrossRef]
- Isozor, N.M.; Laukkanen, J.A.; Voutilainen, A.; Bensenor, I.M.; Kunutsor, S.K. Life’s essential 8 is associated with atherosclerotic cardiovascular disease but not venous thromboembolism in men: A prospective cohort study. Ann Med. 2023, 55, 223894. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oleze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstadter, J.; Kroller-Schon, S.; Munzel, T. Vascular inflammation and oxidative stress: Major triggers for cardiovascular disease. Oxidative stress and Cellular Longevity 2019, 7092151. [Google Scholar] [CrossRef]
- Crimi, E.; Ignarro, L.J.; Napoli, C. Microcirculation and Oxidative stress. Free Radic Res. 2007, 41, 1364–75. [Google Scholar] [CrossRef] [PubMed]
- Batty, M.; Bennett, M.R.; Yu, E. The role of oxidative stress in atherosclerosis. Cells 2022, 11, 3843. [Google Scholar] [CrossRef]
- Lu, Y.; Cui, X.; Zhang, L.; Wang, X.; Zhen, Q.; Liu, G.; Tain, K.; Lim, K.S.; Charles, C.J.; Zhang, J.; Tang, J. The functional role of lipoproteins in arthrosclerosis: Novel directions for diagnosis and targeting therapy. Aging Dis. 2022, 13, 491–520. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Deng, Q.; Hao, Y.; Han, L.; Jia, P.; Zhou, P.; Hao, Y.; Wang, Z.; Zhao, W.; Qi, Y.; et al. Effectiveness pf treat-to-target cholesterol-lowering interventions on cardiovascular disease and all-cause mortality risk in the community-dwelling population” a target trial emulation. Nat. Commun 2024, 15, 9922. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.; Van Eck, M. Gene editing for the treatment of Hypercholesteremia. Curr Atheroscler Rep. 2024, 26, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Minjares, M.; Wu, M.; Wang, J.M. Oxidative stress and microRNAs in endothelial cells under metabolic disorders. Cells 2023, 12, 1341. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, Y.; Daghem, M.; Tzolos, E.; Meah, N.; Doris, M.K.; Moss, A.J.; Kwiecinski, J.; Kroon, J.; Nurmohamed, N.S.; van der Harst, P. Association of lipoprotein(a) with atherosclerotic plaque progression. J Am Coll. Cardiol. 2022, 79, 223–233. [Google Scholar] [CrossRef]
- Chiorescu, R.M.; Mocan, M.; Inceu, A.L.; Buda, A.P.; Blendea, D.; Vlaicu, S.I. Vulnerable atherosclerotic plaque: Is there a molecular signature? Int J Mol Sci 2022, 23, 13638. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wang, Q.; Nicholls, S.J.; Navar, A.M.; Ray, K.K.; Schwarts, G.G.; Szarek, M.; Stores, E.S.G.; Troquay, R.; Jannick, A.X. Zeasiran- A small-interfering RNA targeting lipoprotein (a): A Phase 2 randomized clinical trial. JAMA 2024. [Google Scholar] [CrossRef]
- White, J.G. Identification of the platelet secretion in the electron microscope. Ser. Haematol. 1973, 6, 429–59. [Google Scholar]
- White, J.G. Interaction of Membrane systems in blood platelets. Am J. Pathol. 1972, 66, 295–312. [Google Scholar]
- Gerrard, J.N.; White, J.G.; Rao, G.H.R.; Townsend, D. Localization of platelet prostaglandin production in the platelet dense tubular system. Am J. Pathol. 1976, 83, 283–298. [Google Scholar]
- Gerrard, J.M.; White, J.G.; Rai, G.R.; Krivit, W.; Witkop, C.J. Labile aggregation stimulating substance)LASS): The factor from storage pool deficient platelets correct defective aggregation and release of aspirin treated normal platelets. Br J. Haematol. 1975, 29, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Statland, B.E.; Heagan, B.M.; White, J.G. Uptake of calcium by platelet relaxing factor. Nature 1969, 223, 521–522. [Google Scholar] [CrossRef]
- Escolar, G.; Krumwiede, M.; White, J.G. Organization of actin cytoskeleton of resting and activated platelet sin suspension. Am J Pathol. 1986, 123, 86–94. [Google Scholar] [PubMed]
- Rao, G.H.R.; White, J.G. Disaggregation and reaggregation of ‘irreversibly’ aggregated platelets; A method for more complete evaluation of anti-platelet drugs. Platelets and Thrombosis 1985, 16, 425–434. [Google Scholar] [CrossRef] [PubMed]
- White, J.G.; Rao, G.H.R.; Gerrard, J.M. Platelet stimulus activation coupling: A frequently fractured chain of events. In The Regulation of Coagulation; Taylor, M., Ed.; Elsevier Inc.: New York, 1980. [Google Scholar]
- Cox, C.; Carrol, R.C.; White, J.G.; Rao, G.H.R. Recycling of platelet phosphorylation and cytoskeletal assembly. J. Cell Biol. 1985, 98, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.H.R. Signal Transduction, second messengers and platelet pharmacology. Pharmacol. (Life Sci) 1994, 13, 39–44. [Google Scholar]
- Rao, G.H.R. Signal transduction, second messengers, and platelet function. J. Lab Clin Med. 1993, 12, 18–20. [Google Scholar]
- Rao, G.H.R.; White, J.G. Epinephrine-induced platelet membrane modulation. In The Platelet Amine Storage; Meyer, K.M., Barnes, C.D., Eds.; CRC Press: Boca Raton, FL, USA, 1992. [Google Scholar]
- Gerrard, J.M.; Stuart, M.J.; Rao, G.H.R.; Steffes, M.W.; Mauer, S.M.; Brown, D.M.; White, J.G. Alteration in the balance of prostaglandins and thromboxane synthesis in diabetic rats. J. Lab Clin Med. 1980, 95, 950–958. [Google Scholar] [PubMed]
- Rao, G.H.R. Contributions of the South Asian Society on Atherosclerosis and Thrombosis and the Indian Society on Atherosclerosis Research, to our understanding of atherosclerosis and Thrombosis. J. Clin & Prevent Cardiol. 2016, 5, 67–72. [Google Scholar]
- Rao, G.H.R. Handbook of Platelet Physiology and Pharmacology; Kluwer Academic Publishers: Boston, 1999. [Google Scholar]
- Rao, G.H.R.; Kakkar, V.J. Coronary Artery Disease in South Asians: Epidemiology, Risk Factors, Prevention; Jaypee Medical Publishers: New Delhi, India, 2001. [Google Scholar]
- Rao, G.H.R.; Thanikachalam, R.S. Coronary Artery Disease: Risk Factors, Pathophysiology and Prevention; Jaypee Medical Publishers: New Delhi, India, 2005. [Google Scholar]
- Rao, G.H.R.; Jagannathan, L.; Eastlund, T. Handbook of Blood Banking and Transfusion Medicine; Jaypee Medical Publishers: New Delhi, India, 2006. [Google Scholar]
- Mohan, V.; Rao, G.H.R. Diabetes Mellitus (Type-2): Epidemiology, Risk Management and Prevention; Jaypee Medical Publishers: New Delhi, India, 2007. [Google Scholar]
- Rao, G.H.R.; Kalidoki, E.; Leong, W.; Fareed, J. Management of Antithrombolytic and Thrombolytic Therapy; Kontentworkx, India, 2014; ISBN 978-93-83988-01-3. [Google Scholar]
- Rao, G.H.R. ; Handbook of Coronary Artery Disease; MacMillan Medical Communications, Springer Healthcare: New Delhi, 2017. [Google Scholar]
- Rao, G.H.R. Clinical Handbook of Coronary Artery Disease; Jaypee Medical Publishers: New Delhi, India, 2020; ISBN 978-9389-188-301. [Google Scholar]
- Mohan, V.; Shekar, M.A.; Rao, G.H.R. Current Trends in Diabetes: Focus on South Asians; Jaypee Medical Publishers: New Delhi, 2021. [Google Scholar]
- Rao, G.H.R. Cardiometabolic Diseases: Molecular Mechanisms, Risk Factors and Prevention; Elsevier: The Netherlands, 2024. [Google Scholar]
- Rao, G.H.R.; Das, U.N. Insights into Cardiometabolic Diseases. Biomolecules 2024. Available online: https://www.mdpi.com/journal/biomolecules/special_issues/09A829ZF9W.
- Rao, G.H.R. Role of Professional Society as Freestanding Educational Institution. World Heart Journal 2025, in press. [Google Scholar]
- Iqbal, S.M.; Rosen, A.M.; Edwards, D.; Bolio, A.; Larson, H.J.; Servin, M.; Rudowitz, M.; Carfi, A.; Ceddia, F. Opportunities and challenges to implementing mRNA-based vaccines and medicine: Lessons from COVID-19. Front Publ. Health 2024, 12, 149265. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, H. Emerging roles of ncRNA in Type-2 Diabetes Mellitus: From mechanisms to Drug Delivery. Biomolecules 2024, 14, 1364. [Google Scholar] [CrossRef]
- Ansori, A.N.M.; Antonius, Y.; Susilo, R.K.; Hayaza, S.; Kharisma, V.D.; Parikeist, A.A.; Zainul, R.; Jakhmola, V.; Saklani, T.; Rebezov, M.; et al. Application of CRISPR-Cas9 genome editing technology in various fields: A review. Narra J. 2023, 3, e184. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, E.; Poli, M.; Durrant, M.G.; Kang, B.; Kartekar, D.; Li, D.; Bartie, L.J.; Thomas, A.W.; King, S.H.; Brixi, G.; et al. Sequence modeling and design from molecular to genome scale with Evo. Science 2024, 386. [Google Scholar] [CrossRef]
- Editorial. AlphaFold3-why did Nature publish it Without its code? Nature 2024, 629, 728. [Google Scholar] [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).